
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Transcriptome Analysis of Derris fordii and Derris elliptica to Identify Potential Genes Involved in Rotenoid Biosynthesis
1 Guangxi Institute of Botany, Guangxi Zhuangzu Autonomous Region and the Chinese Academy of Sciences, Guilin, 541006, China
2 Guangxi Key Laboratory of Plant Conservation and Restoration Ecology in Karst Terrain, Guangxi Institute of Botany, Guangxi Zhuangzu Autonomous Region and the Chinese Academy of Sciences, Guilin, 541006, China
* Corresponding Author: Lunfa Guo. Email:
(This article belongs to the Special Issue: Transcriptional Regulation and Signal Transduction Networks in Plant Growth, Development, Morphogenesis, and Environmental Responses)
Phyton-International Journal of Experimental Botany 2025, 94(1), 123-136. https://doi.org/10.32604/phyton.2025.059598
Received 12 October 2024; Accepted 27 November 2024; Issue published 24 January 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Derris fordii and Derris elliptica belong to the Derris genus of the Fabaceae family, distinguished by their high isoflavonoid content, particularly rotenoids, which hold significance in pharmaceuticals and agriculture. Rotenone, as a prominent rotenoid, has a longstanding history of use in pesticides, veterinary applications, medicine, and medical research. The accumulation of rotenoids within Derris plants adheres to species-specific and tissue-specific patterns and is also influenced by environmental factors. Current research predominantly addresses extraction techniques, pharmacological applications, and pesticide formulations, whereas investigations into the biosynthesis pathway and regulatory mechanism of rotenoids remain relatively scarce. In this study, we observed notable differences in rotenone content across the roots, stems, and leaves of D. fordii, as well as within the roots of D. elliptica. Utilizing RNA sequencing (RNA-seq), we analyzed the transcriptomes and expression profiles of unigenes from these four tissues, identifying a total of 121,576 unigenes. Differentially expressed genes (DEGs) across four comparison groups demonstrated significant enrichment in the phenylpropanoid and flavonoid biosynthesis pathways. Key unigenes implicated in the rotenoid biosynthesis pathway were identified, with PAL, C4H, CHS, CHI, IFS, and HI4OMT playing critical roles in D. fordii, while IFS and HI4OMT were determined to be essential for rotenoid biosynthesis in D. elliptica. These findings enhance our understanding of the biosynthesis mechanism of rotenoids in Derris species. The unigenes identified in this study represent promising candidates for future investigations aimed at validating their roles in rotenoid biosynthesis.Keywords
Supplementary Material
Supplementary Material FileAbbreviations
| RNA-Seq | RNA sequencing |
| DEGs | Differentially expressed genes |
| PAL | Phenylalanine ammonia-lyase |
| C4H | Trans-cinnamate 4-monooxygenase |
| 4CL | 4-coumarate-CoA ligase |
| CHS | Chalcone synthase |
| CHI | Chalcone isomerase |
| IFS | Isoflavone synthase |
| HI4OMT | Isoflavone O-methyltransferase |
| I3’H | Hydroxylase |
Species of Derris are acknowledged as traditional medicinal plants and are extensively used as natural pesticides [1,2]. There are over 70 identified species within the Derris genus, including 25 species and two varieties documented in China [3]. These plants are rich in isoflavonoids, particularly rotenoids, which find applications in agriculture, aquaculture, veterinary medicine, and pharmaceuticals [4]. D. fordii exhibits adaptability to diverse environmental conditions and is predominantly found in the south-central and southeastern regions of China. Its root extracts comprise compounds like rotenone, 12α-hydroxy rotenone, 6-methoxyflavone, and quercetin, exhibiting effectiveness against larvae of Aedes albopictus and adults of Myzus persicae [5]. Historically, D. fordii has been employed for medicinal and agricultural purposes in China [6]. Conversely, D. elliptica, native to India and the Malay Peninsula, was introduced to China in the 1950s due to its high rotenone content and continues to be cultivated today [7]. Previous studies on Derris have primarily concentrated on phylogenetic relationships [8,9], pharmacological properties [10], and cultivation practices [11]. Research has confirmed that the accumulation of rotenoids exhibits variability across different tissues [11], species [1], and environmental conditions [12]. However, investigations into the biosynthesis and regulatory mechanisms of rotenoids are still nascent, requiring further research and a concerted effort to enhance related studies.
Phenylpropanoid biosynthesis is one of the most extensively researched secondary metabolic pathways in plants, yielding over 8000 metabolites essential for plant development and plant-environment interactions [13]. Flavonoid and isoflavonoid biosynthesis are significant branches of this pathway. The biosynthesis of rotenoids occurs in four stages: the chalcone phase, flavanone/isoflavone phase, hydroxylation/methoxylation phase, and the final rotenoid phase [14]. Initially, phenylalanine is deaminated by phenylalanine ammonia-lyase (PAL) to produce p-coumaric acid, which is subsequently catalyzed by trans-cinnamate 4-monooxygenase (C4H) and 4-coumarate-CoA ligase (4CL) to form p-coumaroyl-CoA. In the second stage, p-coumaroyl-CoA is processed by chalcone synthase (CHS) and chalcone isomerase (CHI) to generate flavanones (such as liquiritigenin or naringenin) [15,16], which are further transformed into isoflavones by the action of isoflavone synthase (IFS) and 2-hydroxydihydroisoflavone dehydratase (HIDH) [17,18]. The third stage involves the conversion of isoflavones through the action of isoflavone O-methyltransferase (HI4OMT) and isoflavone 3’-hydroxylase (I3’H) to produce calycosin [19,20], followed by additional methoxylation and hydroxylation steps. Prenylation and cyclization processes culminate in the synthesis of the final rotenoid [14,21]. Previous research suggested that the dehydrogenation process in the conversion of rot-2’-enonic acid to rotenone was catalyzed by a CYP450 enzyme [21], whereas deguelin cyclase, which catalyzes the conversion of rot-2’-enonic acid to deguelin, is considered a dioxygenase [22,23]. Despite this understanding, there is still a lack of research concentrating on the genes that encode the enzymes participating in the prenylation and cyclization phases of the rotenoid biosynthesis pathway.
Despite the advancement of sequencing technology, which has enabled the sequencing of numerous plant genomes, genomic and transcriptomic data remain scant for many plant groups. RNA-seq has become a pivotal tool for analyzing differential gene expression and enhancing our understanding of genomic function [24]. In species lacking complete genome sequences, de novo transcriptome analysis can provide comprehensive insights into fundamental biological, molecular, and cellular processes [25]. Significant discoveries regarding terpenoid biosynthesis in Chamaemelum nobile [26] and the identification of candidate genes in shikonin biosynthesis pathways in Lithospermum officinale [27] have been facilitated by de novo assembly and comparative transcriptome analysis, respectively. Transcriptomic investigations provide crucial information on gene expression regulation and help in identifying promising candidate genes involved in isoflavonoid biosynthesis [28]. Notable progress has also been made in elucidating the rotenoid biosynthetic pathway in Mirabilis himalaica through integrated transcriptomic and metabolomic, or transcriptomic and proteomic analyses [29,30].
Current research on the molecular biology of D. fordii and D. elliptica is rather limited, with the available data mainly comprised of chloroplast sequences retrieved from the NCBI database. Although isoflavonoids represent the primary bioactive compounds in Derris species, no specific isoflavonoid-related genes within this genus have been identified yet. This study initially quantified the rotenone content in the roots, stems, and leaves of D. fordii, as well as in the roots of D. elliptica. Subsequent transcriptome sequencing and analysis of these four tissue types facilitated the identification of key genes involved in the rotenoid biosynthesis pathway. The aim of this research was to elucidate a foundational understanding of isoflavonoid biosynthetic pathways (specifically rotenoids) in plants and to offer a genetic resource for the molecular breeding of Derris species.
Lignified branches of D. fordii and D. elliptica were obtained from the experimental field of the Guangxi Institute of Botany in Guilin, China, and sectioned into 15–20 cm segments. The branches were immersed in a 300 mg/L ABT solution (China-ABT, Beijing, China) for 5 min prior to being diagonally planted in a sand bed. Following the development of young leaves and roots, the plants were transferred to the field in April, arranged in beds 100 cm wide (including furrows) with two rows per bed and a spacing of 40 cm × 50 cm. In February of the subsequent year, samples of roots, stems, and leaves from D. fordii (Df_root, Df_stem, Df_leaf) and roots from D. elliptica (De_root) were harvested for analysis using high-performance liquid chromatography (HPLC), RNA sequencing, and quantitative real-time PCR (qRT-PCR).
2.2 Isoflavonoid Extraction and HPLC Determination
Each sample, consisting of nine plants, was dehydrated at 70°C for 10 h, then ground and sieved through a 100-mesh filter. The extraction and measurement method of rotenone was derived from previous studies [5,31] with slight modifications. A measured amount of the powder was placed in a conical flask, to which 50 mL of methanol was added. The mixture was weighed, soaked overnight for 12 h, refluxed in a water bath at 80°C for 1 h, cooled, and adjusted to its original weight with methanol. The solution was then shaken and filtered through a 0.45 μm membrane. For quantitative analysis of rotenone, the Agilent 1200 HPLC system (Agilent Technologies, Santa Clara, CA, USA) was utilized. The column employed was an Agilent HC-C18 (250 mm × 4.6 mm, 5 μm), with a flow rate of 1.0 mL min−1, column temperature set at 30°C, and an injection volume of 10 μL. The mobile phase consisted of 0.1% phosphate and acetonitrile (48:52, v/v). The rotenone content (% dry weight) was determined by comparing the peak areas of the sample and the rotenone standard at 299 nm.
2.3 Library Construction and RNA Sequencing
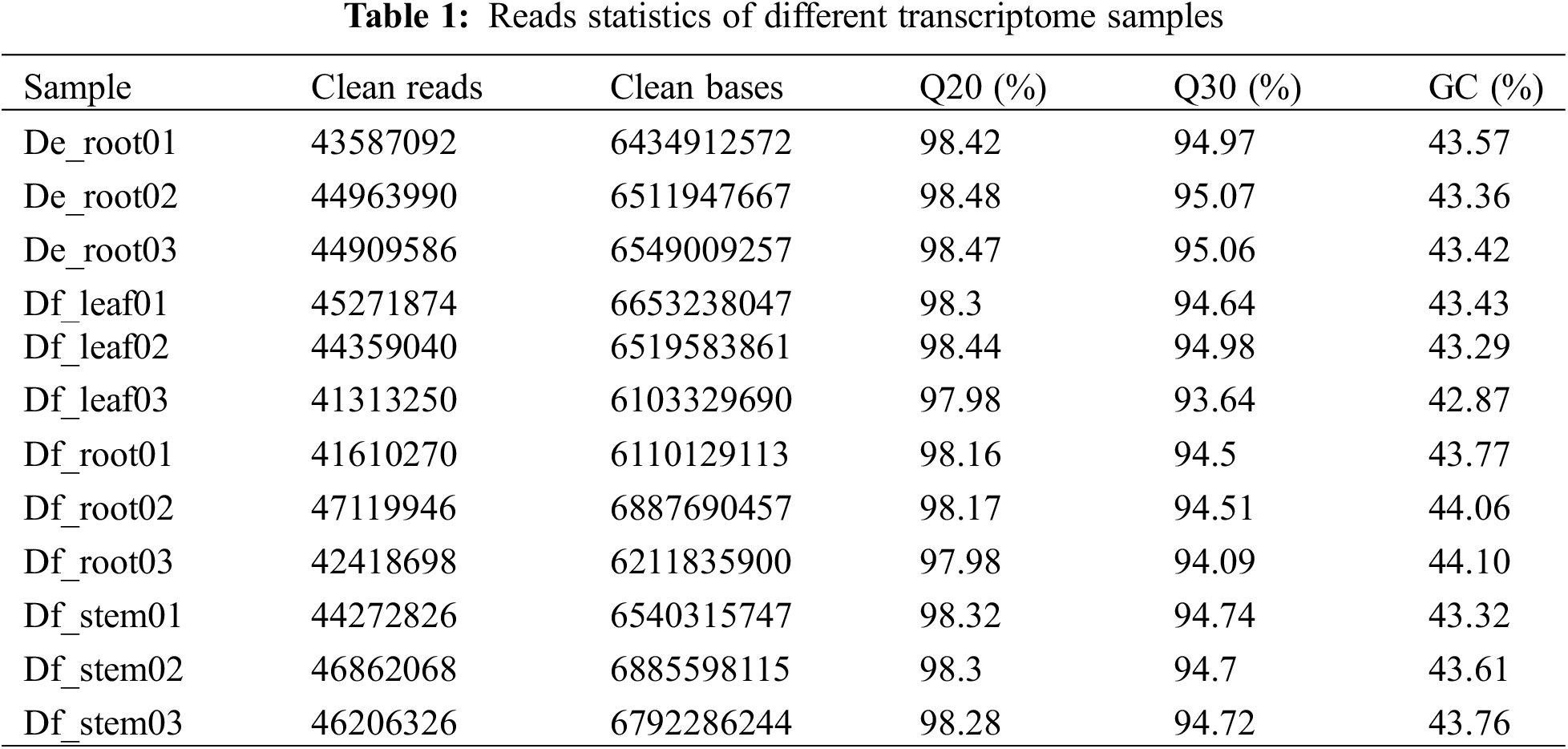
Four groups (Df_root, Df_stem, Df_leaf, and De_root), each consisting of three replicate samples, were collected, and total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Quality and quantity assessments of the RNA were performed with a NanoDrop2000 (Thermo Scientific, Waltham, MA, USA), while RNA integrity was verified using agarose gel electrophoresis and Agilent2100 Nano analysis. After quality assessment, mRNA was enriched using Oligo (dT) magnetic beads, and cDNA library synthesis was conducted using the TruSeq RNA sample preparation kit (Illumina, San Diego, CA, USA). After synthesizing the second strand of cDNA, it was purified using the QIAQuick PCR kit (QIAGEN, Hilden, Germany). The cDNA was subsequently terminally repaired by adding base A and sequencing adapters, followed by selection through agarose gel electrophoresis. The final fragments underwent PCR amplification, and the twelve libraries were sequenced on a HiSeq 4000 sequencer (Illumina, San Diego, CA, USA) at Majorbio company (Shanghai, China).
2.4 De novo Assembly, Expression Quantification, and DEGs Identification
Raw sequencing reads were trimmed and quality-checked using fastp [32] with default settings. Clean data were then subjected to de novo assembly using Trinity [33] and were analyzed via the Majorbio Cloud Platform [34]. Functional annotations were carried out by comparing homologous sequences against six databases: NCBI_NR, Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Swiss-Prot, eggNOG, and Pfam. Transcript expression levels were calculated using the transcripts per million reads (TPM) and fragments per kilobase million (FPKM) methods, with RSEM [35] employed for gene abundance quantification. Differential expression analysis was performed using DESeq2 [36], with genes demonstrating a log2FC ≥ 1 and adjusted p values < 0.05 being classified as DEGs. Subsequently, functional enrichment analyses for KEGG [34] were conducted to identify which DEGs were significantly enriched in specific metabolic pathways, applying an adjusted p value threshold of <0.05 in relation to the entire transcriptome. Expression profiles of unigenes associated with the biosynthesis of rotenoids were evaluated using TBtools software [37].
The DEGs were validated through qRT-PCR of nine candidate unigenes involved in the rotenoid biosynthesis pathway. cDNA was synthesized from isolated RNA using a cDNA reverse transcription kit (Takara, Otsu, Japan). Primers for the selected genes (Table S1) were designed using Primer3web [38]. SYBR qPCR Master Mix (Vazyme, Nanjing, China) was utilized for the qRT-PCR of the nine unigenes, with actin serving as the housekeeping gene. The fold changes of these genes were calculated via the 2−ΔΔCT method [39]. Finally, the correlation between qRT-PCR relative expression and RNA Seq (FPKM) data was assessed.
3.1 Rotenone Content in D. fordii and D. elliptica
Root, stem, and leaf samples from D. fordii (Df_root, Df_stem, Df_leaf) and D. elliptica (De_root) (Fig. 1a) were analyzed for rotenone concentration via HPLC. In D. fordii, the rotenone concentrations were 0.17% in roots, 0.04% in stems, and 0.018% in leaves. In contrast, D. elliptica’s roots exhibited a significantly higher rotenone content of 7.73% (Fig. 1b, Fig. S1). These findings indicate notable variations in rotenone levels across different tissues in D. fordii and between the two species, suggesting distinct isoflavonoid biosynthesis pathways in Derris plants.
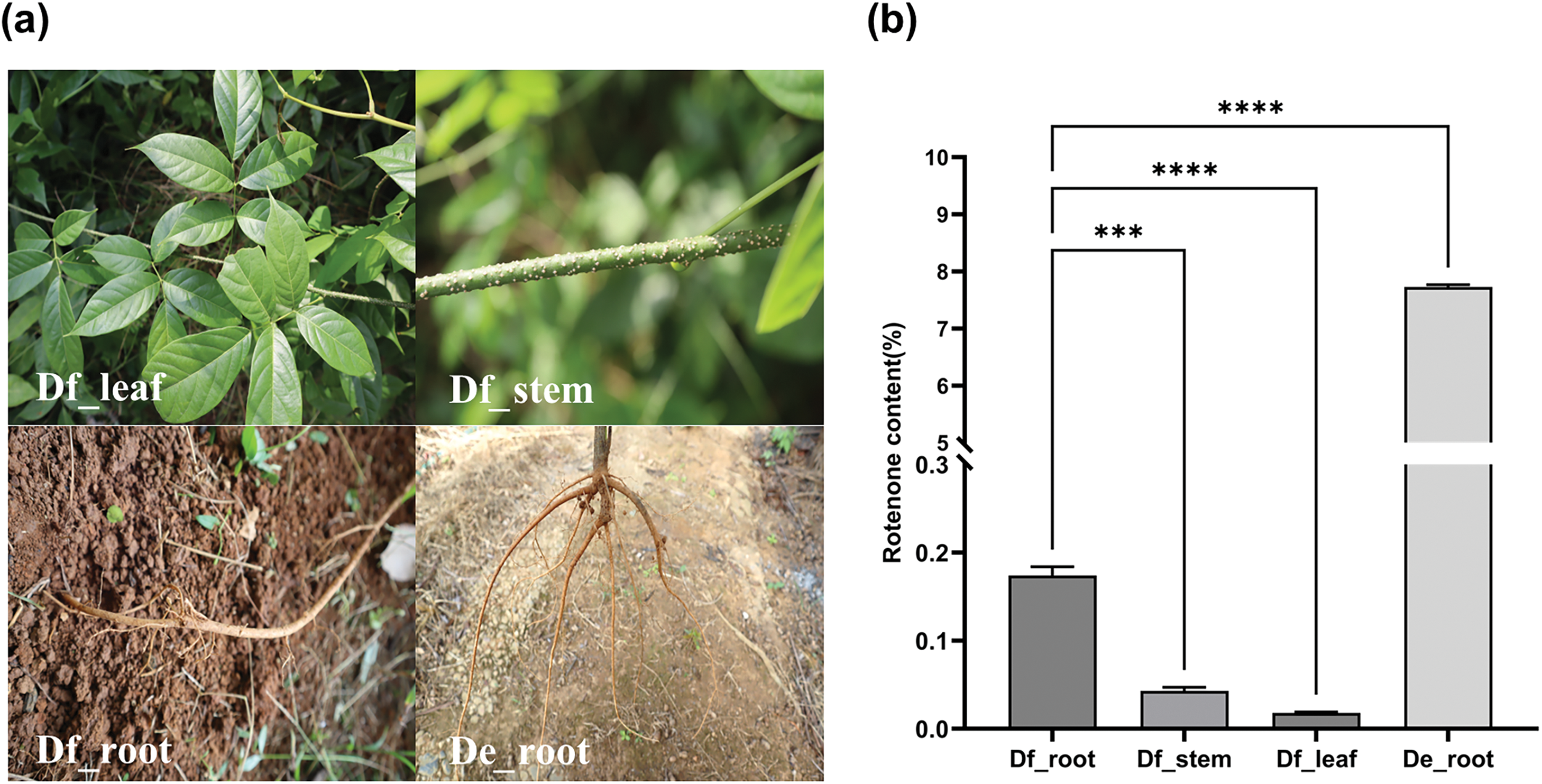
Figure 1: Phenotypes (a) and rotenone concentrations across the four tissue types (b). Asterisks (*** and ****) denote statistically significant differences, as determined by ANOVA followed by Tukey’s test (p < 0.05)
3.2 De novo Assembly and Functional Classification of Unigenes
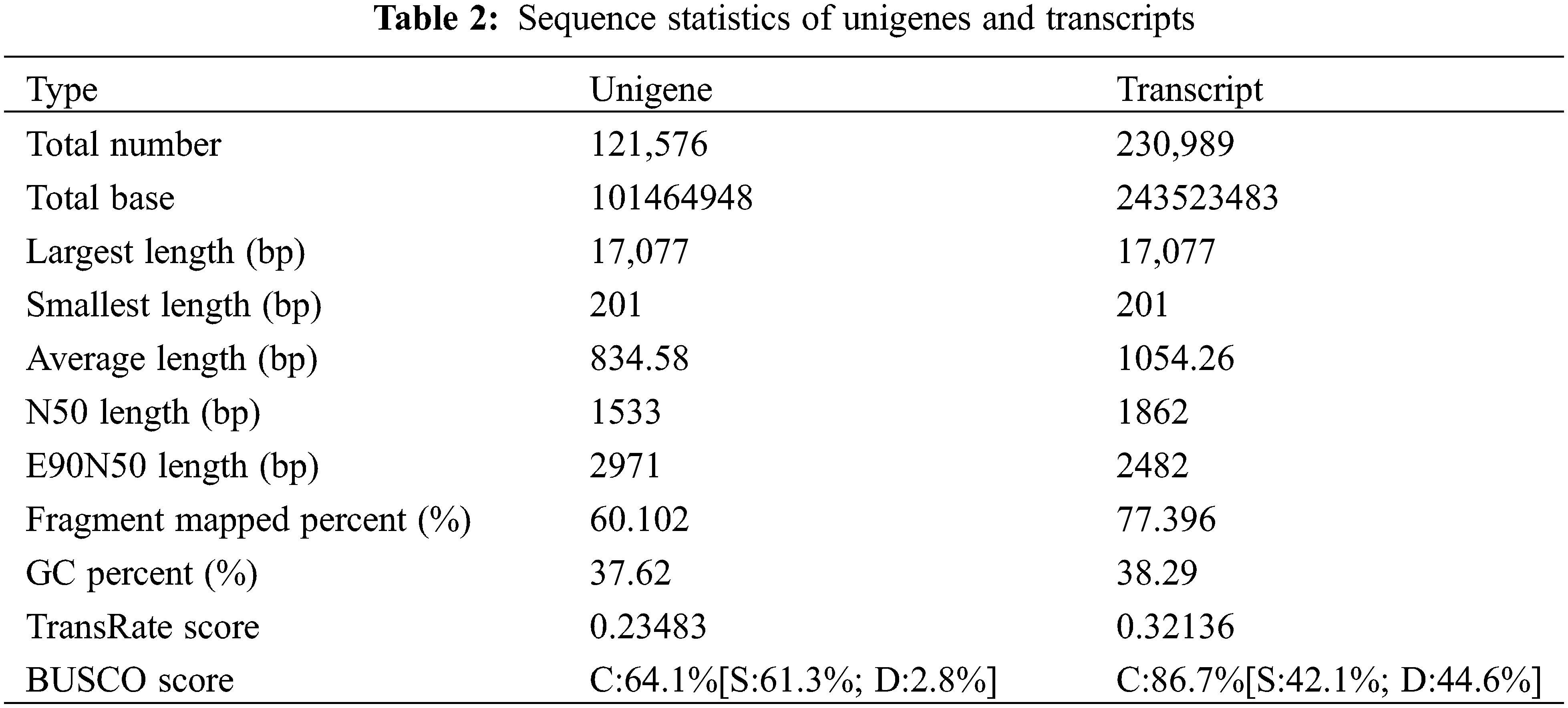
To establish a transcriptome database for D. fordii and D. elliptica, 12 cDNA libraries were sequenced, yielding 78.20 G of clean data. Table 1 summarizes the sequencing quality, with effective data volumes ranging from 6.10 to 6.88 G, Q30 values between 93.64% and 95.07%, and GC content from 42.87% to 44.10%. The clean data were assembled using Trinity, resulting in 121,576 unigenes, with an average length of 834 bp and an N50 length of 1533 bp. BUSCO analysis indicated 64.1% completeness in the assembly unigenes (Table 2). The expression levels of the identified unigenes are detailed in Table S2.
Upon excluding non-expressed genes, a total of 120,475 expressed unigenes were contrasted against six databases for annotation, attaining mappings of 38.40%, 17.25%, 36.75%, 44.42%, 27.92%, 25.64%, and 45.25% to the GO, KEGG, eggNOG, NR, Swiss-Prot, and Pfam database. This process resulted in 54,509 annotated unigenes (Table S3). The species distribution in the NR database revealed the highest similarity to Abrus precatorius (14,976 unigenes; 27.87%), followed by Spatholobus suberectus (6590 unigenes; 12.27%) and Cajanus cajan (5978 unigenes; 11.13%) (Fig. S2). GO and KEGG classifications were performed on the unigenes (Fig. S3), with 46,258 unigenes categorized into functional groups: “molecular function,” “cellular component,” and “biological process.” The predominant GO terms included “binding,” “catalytic activity,” “cellular process,” and “metabolic process”. KEGG pathway analysis categorized 20,778 unigenes into 132 pathways, with 491 unigenes linked to the other secondary metabolism and 279 unigenes associated with phenylpropanoid biosynthesis (Table S4).
3.3 Differential Expression Analysis of Unigenes across Tissues of D. fordii and Roots of D. elliptica
Principal Component Analysis (PCA) has revealed that the first two principal components accounted for 68.43% of the variation, indicating high consistency among intra-group samples (Fig. 2a). Venn analysis has identified 956, 8169 and 9375 specifically expressed genes in the roots, stems, and leaves of D. fordii, respectively. In comparison, the roots of D. elliptica exhibited the highest number of specifically expressed genes, totaling 31,286 (Fig. 2b). DEGs were identified using specific screening criteria: 6180 were upregulated and 4682 downregulated in the Df_leaf vs. Df_stem comparison (Fig. 2c); 6329 upregulated and 6469 downregulated in Df_leaf vs. Df_root (Fig. 2d); 1946 upregulated and 3083 downregulated in Df_stem vs. Df_root (Fig. 2e); and the comparison of Df_root vs. De_root revealed the most DEGs, with 19,730 upregulated and 11,904 downregulated (Fig. 2f). These findings underscore significant functional differences among roots, stems and leaves in D. fordii, as well as notable functional variability in roots between D. fordii and D. elliptica.
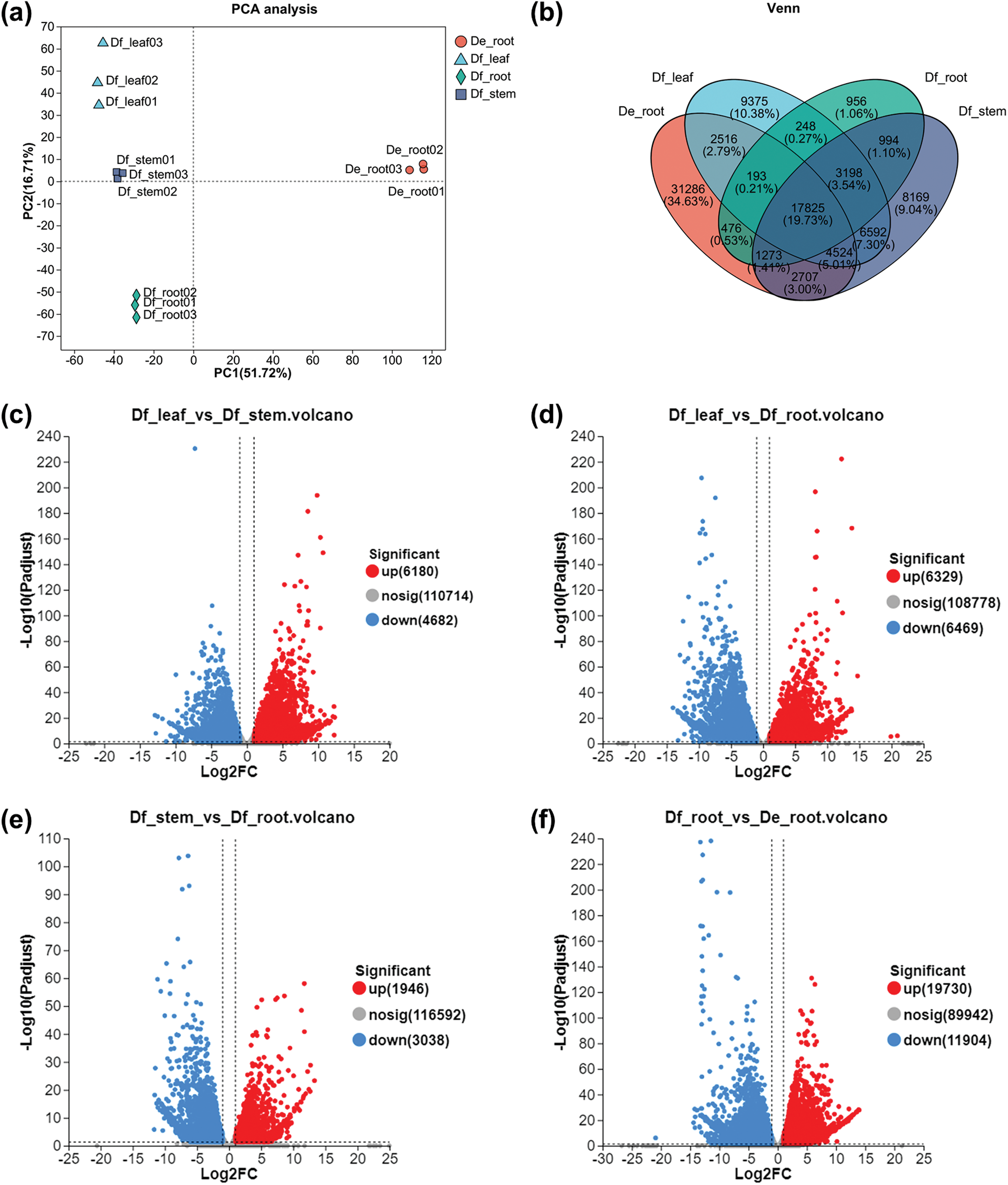
Figure 2: Analysis of transcriptome data and differential gene expression. (a) PCA analysis of 12 samples based on the complete set of unigenes. (b) Venn diagram of unigenes in four tissue types. (c) Volcano plots depicting up-regulated and down-regulated DEGs for Df_leaf vs. Df_stem comparison. (d) Volcano plots depicting up-regulated and down-regulated DEGs for Df_leaf vs. Df_root comparison. (e) Volcano plots depicting up-regulated and down-regulated DEGs for Df_stem vs. Df_root comparison. (f) Volcano plots depicting up-regulated and down-regulated DEGs for Df_root vs. De_root comparison. The former group acted as the control in all four comparisons
KEGG pathway analysis may further elucidate the biological functions associated with the DEGs. In the Df_leaf vs. Df_stem comparison, the significant DEGs were concentrated in phenylpropanoid biosynthesis (ko00940), photosynthesis (ko00195), and flavonoid biosynthesis (ko00941) (Fig. 3a and Table S5). The Df_leaf vs. Df_root comparison showed enrichment in photosynthesis (ko00195), phenylpropanoid biosynthesis (ko00940), photosynthesis-antenna proteins (ko00196), starch and sucrose metabolism (ko00500), zeatin biosynthesis (ko00908), and flavonoid biosynthesis (ko00941) (Fig. 3b and Table S5). The Df_stem vs. Df_root comparison also highlighted pathways relevant to photosynthesis (ko00195), phenylpropanoid biosynthesis (ko00940), photosynthesis-antenna proteins (ko00196), and flavonoid biosynthesis (ko00941) (Fig. 3c and Table S5). In the Df_root vs. De_root comparison, key pathways included phenylpropanoid biosynthesis (ko00940), flavonoid biosynthesis (ko00941) and purine metabolism (ko00230) (Fig. 3d and Table S5). Notably, DEGs linked to phenylpropanoid and flavonoid biosynthesis were significantly enriched across all four comparisons (Fig. S4a). These findings underscore the prominence of the phenylpropanoid metabolism as a prominent secondary metabolic pathway in D. fordii and D. elliptica.
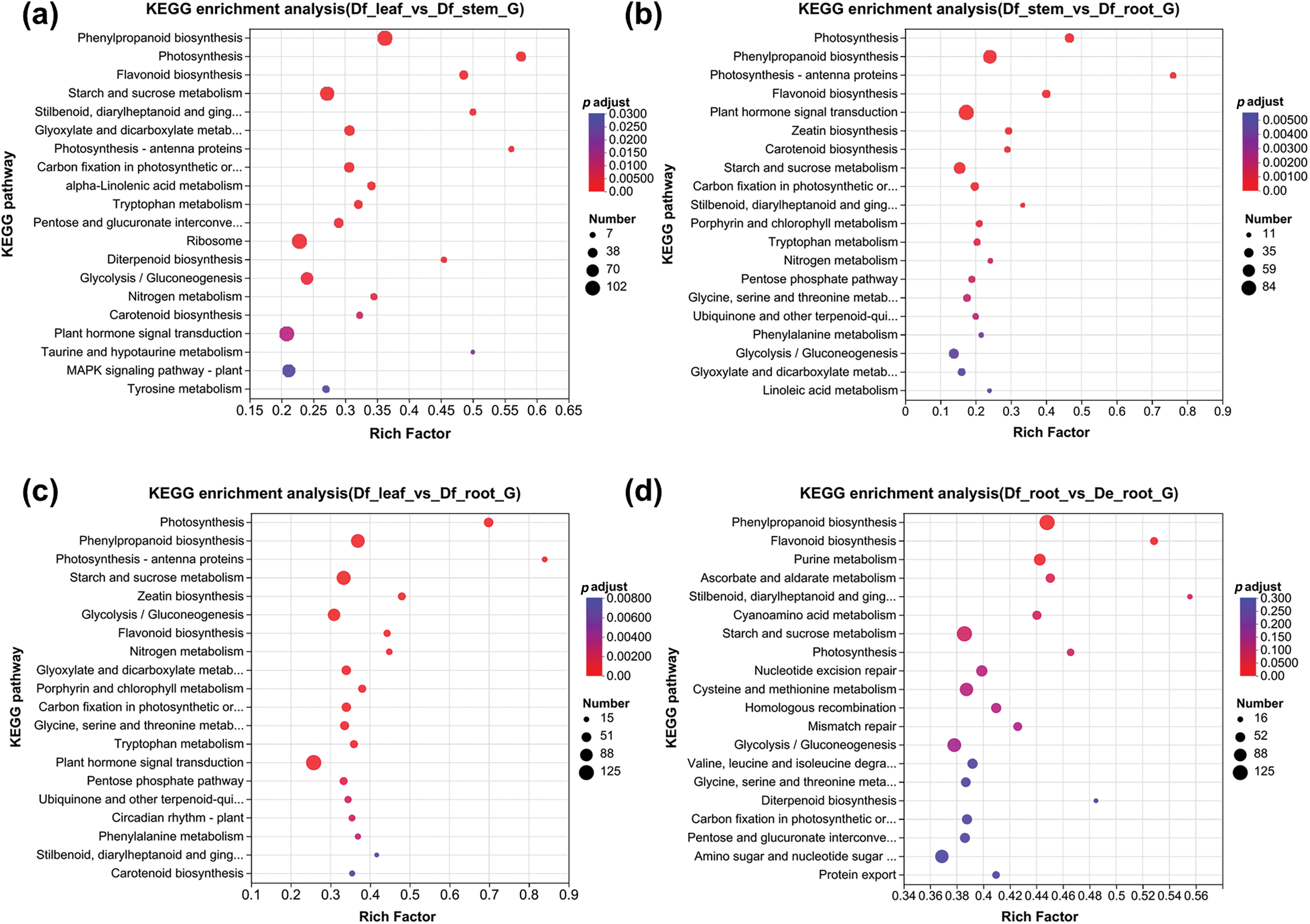
Figure 3: KEGG pathway enrichment analysis of DEGs. (a) KEGG enrichment analysis of DEGs in the Df_leaf vs. Df_stem group; (b) KEGG enrichment analysis of DEGs in the Df_leaf vs. Df_root group; (c) KEGG enrichment analysis of DEGs in the Df_stem vs. Df_root group; (d) KEGG enrichment analysis of DEGs in the Df_root vs. De_root group
3.4 Identification of Unigenes Potentially Involved in Rotenoid Biosynthesis
Respectively, a total of 279, 70, and 42 unigenes were annotated as participating in phenylpropanoid (ko00940), flavonoid (ko00941), and isoflavonoid biosynthesis (ko00943) pathways (Table S6), respectively. Amongst these, 38 unigenes encoding key enzymes were identified, including PAL (5 unigenes), C4H (3 unigenes), 4CL (8 unigenes), CHS (5 unigenes), CHI (3 unigenes), IFS (4 unigenes), HIDH (2 unigenes), HI4OMT (6 unigenes), and I3’H (2 unigenes) (Table S7). A heatmap was constructed to illustrate the expression patterns of unigenes related to the rotenoid biosynthesis pathway (Fig. 4). In D. fordii, most unigenes encoding PAL, C4H, CHS, CHI, IFS, and HI4OMT exhibited elevated expression levels in roots, while more unigenes encoding 4CL were highly expressed in leaves relative to roots and stems. On the contrary, in D. elliptica, unigenes for C4H, CHS, and CHI were expressed at lower levels compared to roots of D. fordii, whereas IFS and HI4OMT demonstrated higher expression in the roots of D. elliptica. These results indicate that the unigenes encoding key enzymes involved in rotenoid biosynthesis may vary between the two species.
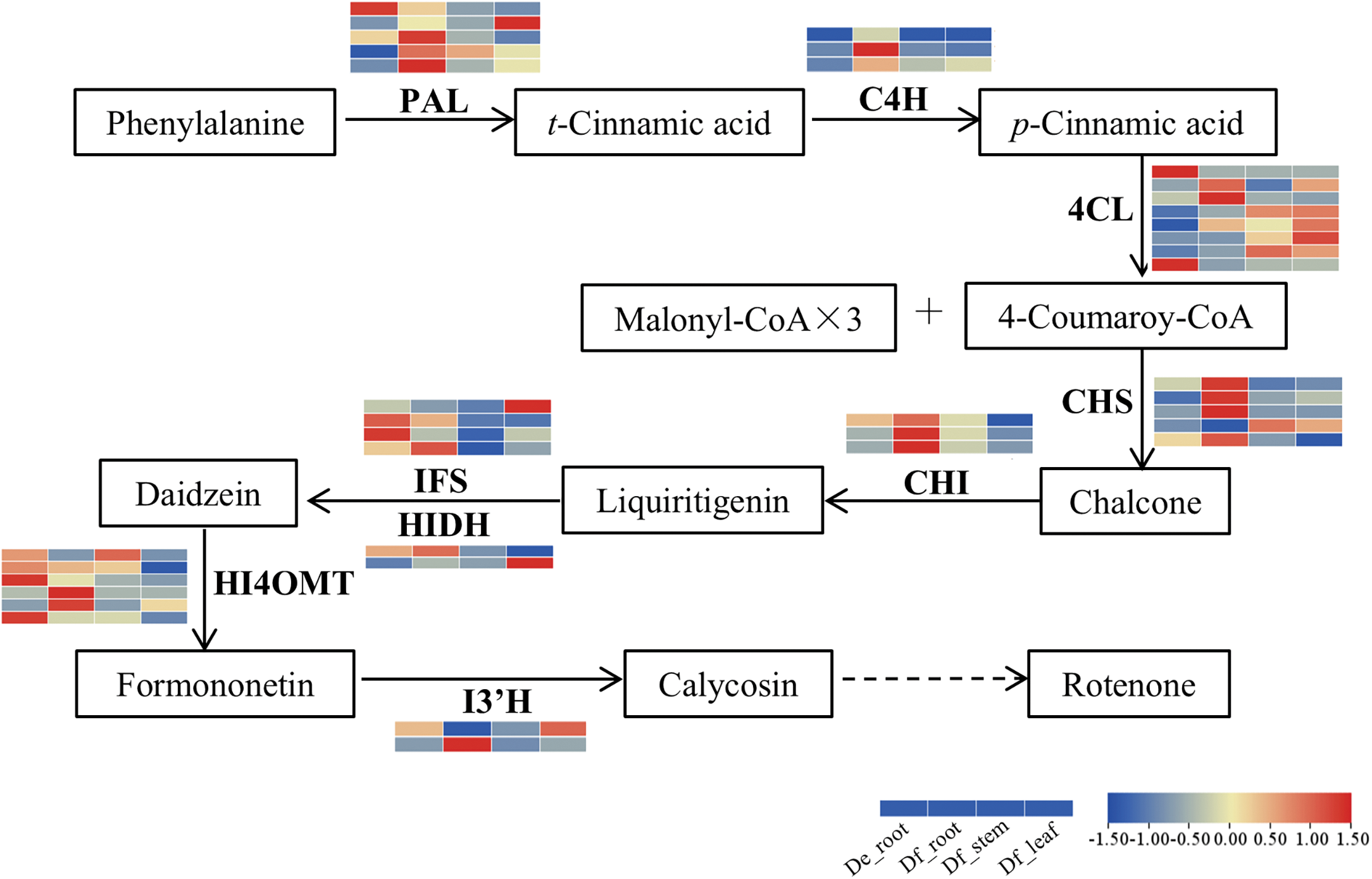
Figure 4: Expression profiles of DEGs associated with the rotenoid biosynthesis pathway. The heatmap value represents the expression levels as log2(FPKM+1)
In this study, we also identified a total of 4 unigenes encoding prenyltransferase, 199 unigenes encoding CYP450 enzymes, and 94 unigenes for dioxygenases (Table S8). These genes potentially serve as candidates for the late stages of the rotenoid biosynthetic pathway.
3.5 Verification of Transcriptome Data
To validate the reliability of transcriptome sequencing results, nine DEGs were selected for quantitative reverse transcription PCR (qRT-PCR) verification (Table S1). The DEGs selected included those related to isoflavonoid biosynthesis: PAL, C4H, 4CL, CHS, CHI, IFS, HIDH, HI4OMT, and I3’H. The expression trends of the nine DEGs demonstrated general consistency between qRT-PCR and RNA-seq findings, thereby confirming the reliability of the transcriptomic data, as presented in Fig. 5.
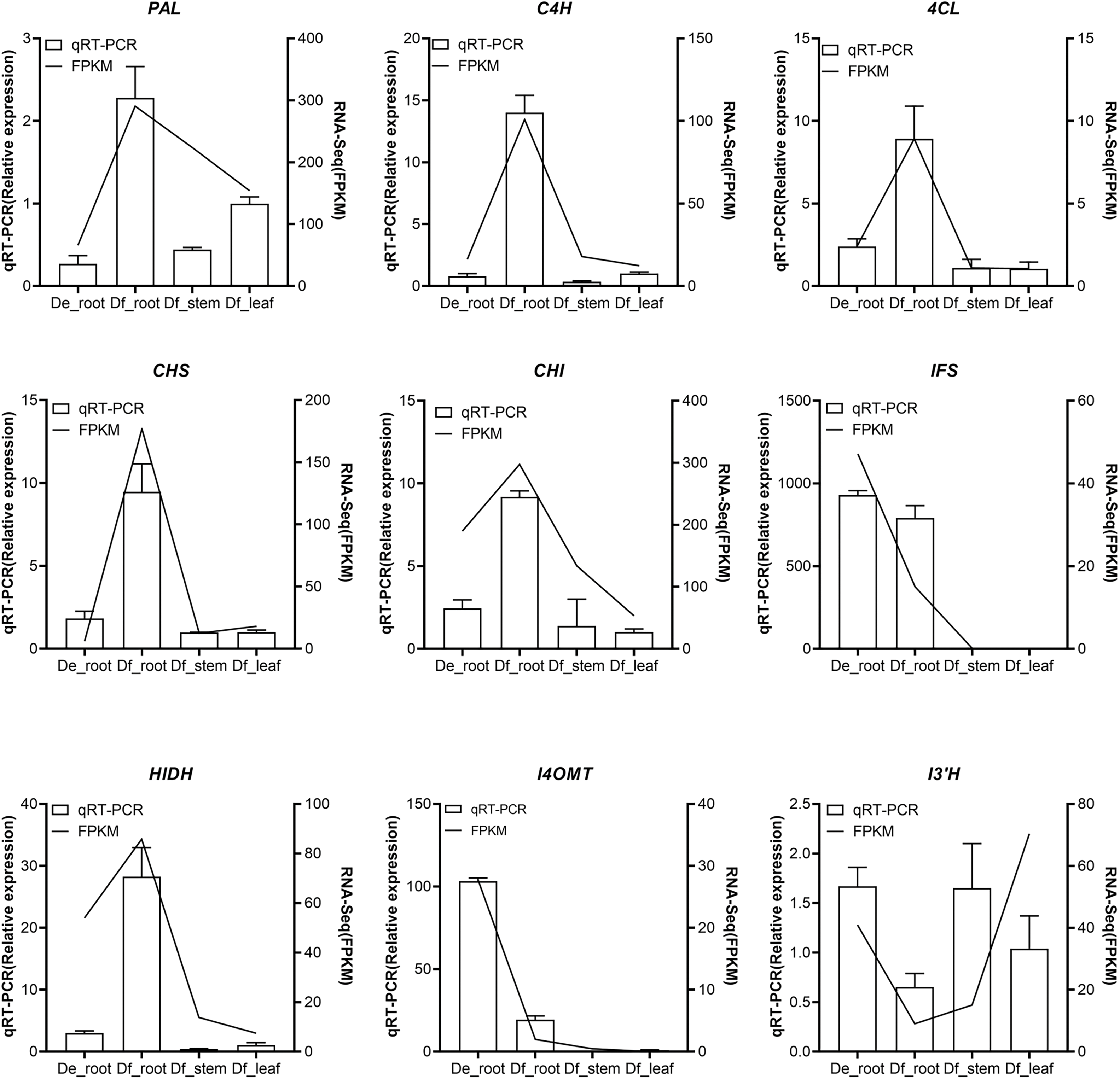
Figure 5: Quantitative real-time PCR (qRT-PCR) analysis for the validation of expression levels of nine genes involved in the rotenoid biosynthesis pathway
Rotenoids represent a significant subclass of isoflavonoids, with a wide variety being isolated from Derris species. Among these, rotenone is recognized as a key bioactive compound initially extracted from D. elliptica, and represents the most prevalent rotenoid in the roots of this species [14]. Our research quantified rotenone levels across three tissues of D. fordii, demonstrating that the root tissue harbored significantly higher concentrations than the stems or leaves (Fig. 1b). Previously, we observe similar accumulation patterns of rotenone were observed in the roots, stems, and leaves of D. elliptica and Derris montana [11]. The disparity in distribution observed could be relate to the differential expression of key enzymes involved in rotenoid accumulation, which are predominantly expressed in the roots, while other tissues show minimal or absent expression. Analysis of root tissues revealed that rotenone accumulation in D. elliptica was over 45-fold higher than in D. fordii (Fig. 1b). This discovery suggests substantial differences in the genes encoding vital enzymes and regulatory elements associated with the rotenoid biosynthesis pathway exist between the two species.
Transcriptome sequencing is a valuable tool for elucidating the key mechanisms underlying growth and development, particularly in plant species with limited genomic data [40]. This method has been employed to identify novel genes involved in secondary metabolism pathways and to explore the molecular mechanisms governing isoflavonoid biosynthesis [41,42]. To date, no published transcriptomic data are available for Derris species. In this study, we performed transcriptome analyses on the root, stem, and leaf tissues of D. fordii, and found that the enrichment pathways of DEGs varied across tissues, reflecting their distinct biological functions. In terms of energy metabolism, pathways related to photosynthesis, photosynthesis-antenna proteins, carbon fixation in photosynthetic organisms, and nitrogen metabolism were observed to have a higher number of down-regulated genes in the roots and stems when compared to the leaves (Fig. S4b), emphasizing the role of leaves in meeting the energy requirements for the growth and development of D. fordii; conversely, a larger number of up-regulated genes in the phenylpropanoid and flavonoid biosynthesis pathways were observed in the roots and stems than in the leaves (Fig. S4b), which suggests a higher accumulation of phenylpropanoid metabolites in the roots and stems tissues. Notably, a higher number of genes related to phenylpropanoid and flavonoid biosynthesis were up-regulated in the roots of D. fordii as compared with those of D. elliptica, underscoring the significant differences in phenylpropanoid metabolism between the two species.
Although isoflavonoids act as the primary bioactive compounds in D. fordii and D. elliptica, the relationship between the expression of structural genes involved in isoflavonoid biosynthesis and their accumulation patterns remains largely unclear. Previous studies have demonstrated that the expression patterns of the CHS and CHI genes correspond with rotenoid accumulation across various tissues in M. himalaica [29]. Additionally, overexpression of CHS and CHI genes has been shown to enhance rotenoid levels in transgenic hairy roots [43]. Our transcriptomic analysis has identified 38 putative structural unigenes linked to the rotenoid biosynthesis pathway (Fig. 4). Consistent with the HPLC results, which indicated the highest rotenone concentrations in the roots of D. fordii compared to its stems and leaves, we observed increased expression of unigenes encoding PAL, C4H, CHS, CHI, IFS, and HI4OMT in the roots relative to other tissues (Figs. 1, 4, and 5). Notably, all CHI-encoded genes were highly expressed in the roots, followed by the stems, with the lowest expression in the leaves, reflecting a strong positive correlation with rotenone content across these tissues. These findings suggest that the upregulation of these genes might promote rotenoid accumulation in D. fordii, with CHI genes playing a particularly significant role.
Studies have shown that the content of intermediates is lower during the chalcone and flavonoid/isoflavone phases; however, notable intermediates, such as 7,2’-dihydroxy-4’,5’-dimethoxyisoflavone in the hydroxylation/methoxylation phase and (±)-9-demethylmunduserone in the rotenoid phase, have been found to be abundant in the roots of D. elliptica [14]. This finding suggests that the accumulation of rotenoids in the roots of D. elliptica is primarily governed by the expression of enzyme-coding genes active in the latter stages of the rotenoid biosynthesis pathway. In our comparison of Df_root vs. De_root, unigenes for C4H, CHS, and CHI were expressed at lower levels, whereas more unigenes for IFS and HI4OMT showed higher expression in D. elliptica. The low expression levels of unigenes for C4H, CHS, and CHI correlate with the lower intermediate contents during the chalcone and flavonoid/isoflavone phases. Furthermore, the elevated expression of unigenes for IFS and HI4OMT correlates positively with heightened rotenone levels observed in the roots of D. elliptica, indicating that IFS and HI4OMT play critical roles in the rotenoid biosynthesis pathway of this species.
This study provides insight into the transcriptomic landscape of D. fordii (root, leaf, stem) and D. elliptica (root), facilitating the identification of candidate genes involved in rotenoid biosynthesis. The findings from this RNA-seq analysis will advance our understanding of the molecular mechanisms underlying isoflavonoid biosynthesis in Derris plants, with the identified unigenes serving as valuable candidates for further investigation into rotenoid biosynthesis. Moreover, the transcriptomic data will enable exploration of additional, less understood metabolic pathways within this genus.
Acknowledgement: Not applicable.
Funding Statement: This work was funded by the financial support (Guangxi Science and Technology Base and Talent Special Fund, Project No. AD21220130; Guangxi Key Laboratory of Plant Conservation and Restoration Ecology in Karst Terrain, Project No. 20-065-7; Guangxi Institute of Botany Fund, Project No. 21014).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Yanlin Pan and Lunfa Guo; experiments conduct: Yanlin Pan, Xingui Wang, Yibin Zhang and Hongbo Qin; data analysis and draft manuscript preparation: Yanlin Pan. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All data have been incorporated into the article and its supplementary material. The RNA-seq raw data were submitted to NCBI under the accession nos. SAMN43805039, SAMN43805040, SAMN43805041, SAMN43805042, SAMN43805043, SAMN43805044, SAMN43805045, SAMN43805046, SAMN43805047, SAMN43805048, SAMN43805049 and SAMN43805050.
Ethics Approval: No part of this research involved human or animal samples.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://doi.org/10.32604/phyton.2024.059598.
References
1. Yang XY, Ma RJ, Wang LQ, Li J. Advances in research on chemical constituents and pharmacological activities of genus Derris. Nat Prod Res Dev. 2013;25(1):117–28 (In Chinese). doi:10.16333/j.1001-6880.2013.01.028. [Google Scholar] [CrossRef]
2. Enrique EB Jr. A review on tubli plant used as organic pesticide: input toward sustainable agriculture. Int J Res Appl Sci Biotechnol. 2021;8(1):107–15. doi:10.31033/ijrasb.8.1.12. [Google Scholar] [CrossRef]
3. Wei Z. Derris. In: Editorial Committee of Chinese Flora, Chinese Academy of Sciences. Flora of China. Beijing, China: Science Press; 1994. p. 191–212 (In Chinese). [Google Scholar]
4. Praveen Kumar PK, Priyadharshini A, Muthukumaran S. A review on rotenoids: purification, characterization and its biological applications. Mini-Rev Med Chem. 2021;21(13):1734–46. doi:10.2174/1389557521666210217092634. [Google Scholar] [PubMed] [CrossRef]
5. Ou YG, Xiao ZX, Zou K, Ding WB, Li GH. Study on insecticidal ingredients from the root barks of Derris fordii Oliv. Hunan Agric Sci. 2015;8:93–5 (In Chinese). doi:10.16498/j.cnki.hnnykx.2015.08.029. [Google Scholar] [CrossRef]
6. Liu QF, Zhang BJ, Yin FM, Zhang X, Hu Z, Xie J, et al. Fungistatic activity of the extracts from the twigs and leaces of Derris fordii against plant pathogenic fungi. J Trop Biol. 2022;13(3):227–34. doi:10.15886/j.cnki.rdswxb.2022.03.004. [Google Scholar] [CrossRef]
7. Li H, Geng S. Assessment of population genetic diversity of Derris elliptica (Fabaceae) in China using microsatellite markers. Ind Crops Prod. 2015;73:9–15. doi:10.1016/j.indcrop.2015.04.023. [Google Scholar] [CrossRef]
8. Sirichamorn Y, Adema FA, Roos MC, Van Welzen PC. Molecular and morphological phylogenetic reconstruction reveals a new generic delimitation of Asian Derris (Fabaceaereinstatement of Solori and synonymisation of Paraderris with Derris. Taxon. 2014;63(3):522–38. doi:10.12705/633.13. [Google Scholar] [CrossRef]
9. Song ZQ, Pan B. Transfer of Millettia pachycarpa and M. entadoides to Derris (Fabaceaesupported by morphological and molecular data. Phytotaxa. 2022;531(3):230–48. doi:10.11646/phytotaxa.531.3.4. [Google Scholar] [CrossRef]
10. Goel B, Tripathi N, Bhardwaj N, Sahu B, Jain SK. Therapeutic potential of genus Pongamia and Derris: phytochemical and bioactivity. Mini-Rev Med Chem. 2021;21(8):920–51. doi:10.2174/1389557520999201124211846. [Google Scholar] [PubMed] [CrossRef]
11. Pan YL, Guo LF, Wang XG, Lou SM, Zhang YB, Qing HB. Screening the excellent species of genus Derris and studies on its induction and cultivation. Guihaia. 2023;43(2):390–8 (In Chinese). doi:10.11931/guihaia.gxzw202112034. [Google Scholar] [CrossRef]
12. Zhang TY, Xu HH, Huang JG, Zhang JL, Zhao Y. Variations of rotenone in different growth stage of plants and regions. J South China Agric Univ. 2006;27(3):48–50 (In Chinese). doi:10.7671/j.issn.1001-411X.2006.03.013. [Google Scholar] [CrossRef]
13. Dong NQ, Lin HX. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J Integr Plant Biol. 2021;63(1):180–209. doi:10.1111/jipb.13054. [Google Scholar] [PubMed] [CrossRef]
14. Li P, Chen YY, Xie QR, Xu YZ, Li Z, Li Y, et al. Spatiotemporal visualization of the synthesis and accumulation of rotenone in Derris elliptica roots using mass spectrometry imaging. Advanced Agrochem. 2023;2(4):340–8. doi:10.1016/j.aac.2023.07.002. [Google Scholar] [CrossRef]
15. Feinbaum RL, Ausubel FM. Transcriptional regulation of the Arabidopsis thaliana chalcone synthase gene. Mol Cell Biol. 1988;8(5):1985–92. doi:10.1128/mcb.8.5.1985-1992.1988. [Google Scholar] [PubMed] [CrossRef]
16. Shirley BW, Hanley S, Goodman HM. Effects of ionizing radiation on a plant genome: analysis of two Arabidopsis transparent testa mutations. Plant Cell. 1992;4(3):333–47. doi:10.1105/tpc.4.3.333. [Google Scholar] [PubMed] [CrossRef]
17. Steele CL, Gijzen M, Qutob D, Dixon RA. Molecular characterization of the enzyme catalyzing the aryl migration reaction of isoflavonoid biosynthesis in soybean. Arch Biochem Biophys. 1999;367(1):146–50. doi:10.1006/abbi.1999.1238. [Google Scholar] [PubMed] [CrossRef]
18. Akashi T, Aoki T, Ayabe SI. Molecular and biochemical characterization of 2-hydroxyisoflavanone dehydratase. Involvement of carboxylesterase-like proteins in leguminous isoflavone biosynthesis. Plant Physiol. 2005;137(3):882–91. doi:10.1104/pp.104.056747. [Google Scholar] [PubMed] [CrossRef]
19. Akashi T, Sawada Y, Shimada N, Sakurai N, Aoki T, Ayabe SI. cDNA cloning and biochemical characterization of S-adenosyl-L-methionine: 2, 7, 4′-trihydroxyisoflavanone 4′-O-methyltransferase, a critical enzyme of the legume isoflavonoid phytoalexin pathway. Plant Cell Physiol. 2003;44(2):103–12. doi:10.1093/pcp/pcg034. [Google Scholar] [PubMed] [CrossRef]
20. Liu CJ, Huhman D, Sumner LW, Dixon RA. Regiospecific hydroxylation of isoflavones by cytochrome p45081E enzymes from Medicago truncatula. Plant J. 2003;36(4):471–84. doi:10.1046/j.1365-313X.2003.01893.x. [Google Scholar] [PubMed] [CrossRef]
21. Crombie L, Whiting DA. Review article number 135 biosynthesis in the rotenoid group of natural products: applications of isotope methodology. Phytochemistry. 1998;49(6):1479–507. doi:10.1016/S0031-9422(98)00178-2. [Google Scholar] [PubMed] [CrossRef]
22. Crombie L, Rossiter JT, Van Bruggen N, Whiting DA. Deguelin cyclase, a prenyl to chromen transforming enzyme from Tephrosia vogellii. Phytochemistry. 1992;31(2):451–61. doi:10.1016/0031-9422(92)90016-J. [Google Scholar] [CrossRef]
23. Prescott AG, John P. Dioxygenases: molecular structure and role in plant metabolism. Annu Rev Plant Biol. 1996;47(1):245–71. doi:10.1146/annurev.arplant.47.1.245. [Google Scholar] [PubMed] [CrossRef]
24. Stark R, Grzelak M, Hadfield J. RNA sequencing: the teenage years. Nat Rev Genet. 2019;20(11):631–56. doi:10.1038/s41576-019-0150-2. [Google Scholar] [PubMed] [CrossRef]
25. Raghavan V, Kraft L, Mesny F, Rigerte L. A simple guide to de novo transcriptome assembly and annotation. Briefings Bioinf. 2022;23(2):bbab563. doi:10.1093/bib/bbab563. [Google Scholar] [PubMed] [CrossRef]
26. Liu XM, Wang XH, Chen ZX, Ye JB, Liao YL, Zhang WW, et al. De novo assembly and comparative transcriptome analysis: novel insights into terpenoid biosynthesis in Chamaemelum nobile L. Plant Cell Rep. 2019;38:101–16. doi:10.1007/s00299-018-2352-z. [Google Scholar] [PubMed] [CrossRef]
27. Rai A, Nakaya T, Shimizu Y, Rai M, Nakamura M, Suzuki H, et al. De novo transcriptome assembly and characterization of Lithospermum officinale to discover putative genes involved in specialized metabolites biosynthesis. Planta Med. 2018;84(12/13):920–34. doi:10.1055/a-0630-5925. [Google Scholar] [PubMed] [CrossRef]
28. Wang C, Xu N, Cui S. Comparative transcriptome analysis of roots, stems, and leaves of Pueraria lobata (Willd.) Ohwi: identification of genes involved in isoflavonoid biosynthesis. PeerJ. 2021;9:e10885. doi:10.7717/peerj.10885. [Google Scholar] [PubMed] [CrossRef]
29. Gu L, Zhang ZY, Quan H, Li MJ, Zhao FY, Xu YJ, et al. Integrated analysis of transcriptomic and metabolomic data reveals critical metabolic pathways involved in rotenoid biosynthesis in the medicinal plant Mirabilis himalaica. Mol Genet Genomics. 2018;293:635–47. doi:10.1007/s00438-017-1409-y. [Google Scholar] [PubMed] [CrossRef]
30. Gu L, Zheng WL, Li MJ, Quan H, Wang JM, Wang FJ, et al. Integrated analysis of transcriptomic and proteomics data reveals the induction effects of rotenoid biosynthesis of Mirabilis himalaica caused by UV-B radiation. Int J Mol Sci. 2018;19(11):3324. doi:10.3390/ijms19113324. [Google Scholar] [PubMed] [CrossRef]
31. Hu DD, Gu L, Liu GL. Determination of rotenone in Derris elliptica by reversed-phase high performance liquid chromatography. J Pharm Pract Serv. 2007;25(3):159–61 (In Chinese). [Google Scholar]
32. Chen SF, Zhou YQ, Chen YR, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90. doi:10.1093/bioinformatics/bty560. [Google Scholar] [PubMed] [CrossRef]
33. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52. doi:10.1038/nbt.1883. [Google Scholar] [PubMed] [CrossRef]
34. Ren Y, Yu G, Shi CP, Liu LM, Guo Q, Han C, et al. Majorbio cloud: a one-stop, comprehensive bioinformatic platform for multiomics analyses. iMeta. 2022;1(2):e12. doi:10.1002/imt2.12. [Google Scholar] [PubMed] [CrossRef]
35. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12:1–16. doi:10.1186/1471-2105-12-323. [Google Scholar] [PubMed] [CrossRef]
36. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:1–21. doi:10.1186/s13059-014-0550-8. [Google Scholar] [PubMed] [CrossRef]
37. Chen CJ, Xu Y, Li JW, Wang X, Zeng ZH, Xu J, et al. TBtools-II: a “one for all, all for one” bioinformatics platform for biological big-data mining. Mol Plant. 2023;16(11):1733–42. [Google Scholar] [PubMed]
38. Koressaar T, Lepamets M, Kaplinski L, Raime K, Andreson R, Remm M. Primer3_masker: integrating masking of template sequence with primer design software. Bioinformatics. 2018;34(11):1937–8. doi:10.1093/bioinformatics/bty036. [Google Scholar] [PubMed] [CrossRef]
39. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–8. doi:10.1006/meth.2001.1262. [Google Scholar] [PubMed] [CrossRef]
40. Guo JD, Huang Z, Sun JL, Cui XM, Liu Y. Research progress and future development trends in medicinal plant transcriptomics. Front Plant Sci. 2021;12:691838. doi:10.3389/fpls.2021.691838. [Google Scholar] [PubMed] [CrossRef]
41. Suntichaikamolkul N, Tantisuwanichkul K, Prombutara P, Kobtrakul K, Zumsteg J, Wannachart S, et al. Transcriptome analysis of Pueraria candollei var. mirifica for gene discovery in the biosyntheses of isoflavones and miroestro. BMC Plant Biol. 2019;19:581. doi:10.1186/s12870-019-2205-0. [Google Scholar] [PubMed] [CrossRef]
42. Wang C, Zhu J, Liu M, Yang QS, Wu JW, Li ZG. De novo sequencing and transcriptome assembly of Arisaema heterophyllum Blume and identification of genes involved in isoflavonoid biosynthesis. Sci Rep. 2018;8(1):17643. doi:10.1038/s41598-018-35664-1. [Google Scholar] [PubMed] [CrossRef]
43. Lan XZ, Quan H, Xia XL, Yin WL, Zheng WL. Molecular cloning and transgenic characterization of the genes encoding chalcone synthase and chalcone isomerase from the Tibetan herbal plant Mirabilis himalaica. Biotechnol Appl Biochem. 2016;63(3):419–26. doi:10.1002/bab.1376. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools