
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Gene-Specific Effects of Three Cry Transgenes on Rhizosphere Microbiota in Catalpa bungei
1
Jiangsu Key Laboratory for Conservation and Utilization of Plant Resources, Institute of Botany, Jiangsu Province & Chinese Academy of Sciences, Nanjing, China
2
Jiangsu Engineering Research Center for Landscape Plant Resources and Germplasm Innovation, Nanjing, China
3
State Key Laboratory of Tree Genetics and Breeding, Experimental Center of Forestry in North China, National Permanent Scientific Research Base for Warm Temperate Zone Forestry of Jiulong Mountain in Beijing, Chinese Academy of Forestry, Beijing, China
4
State Key Laboratory of Tree Genetics and Breeding, Key Laboratory of Tree Breeding and Cultivation of State Forestry Administration, Research Institute of Forestry, Chinese Academy of Forestry, Beijing, China
* Corresponding Authors: Peng Wang. Email: ; Naiwei Li. Email:
# These authors contributed equally to this work
(This article belongs to the Special Issue: Advances in Molecular Insights of Plant Secondary Metabolites: Biosynthesis, Regulation, and Applications)
Phyton-International Journal of Experimental Botany 2026, 95(2), 7 https://doi.org/10.32604/phyton.2026.072636
Received 31 August 2025; Accepted 05 January 2026; Issue published 28 February 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Catalpa bungei, a fast-growing timber tree, is threatened by the lepidopteran pest Omphisa plagialis. Previous studies in our laboratory successfully generated transgenic C. bungei lines overexpressing Cry genes (Cry1Ab, Cry2A, and Cry9-2) that exhibited resistance to O. plagialis, but their potential impact on soil bacterial communities remains unclear. In this study, we analyzed nine transgenic C. bungei lines (three independent lines for each Cry gene) to characterize their rhizosphere bacterial communities using high-throughput sequencing of the 16S ribosomal DNA (rDNA) V4–V5 regions. A total of 628 amplicon sequence variants (ASVs) were shared among all transgenic and wild-type (WT) lines, forming a stable core microbiome dominated by Proteobacteria, Bacteroidota, Acidobacteriota, and Actinobacteriota. Alpha diversity showed no significant differences, while beta diversity revealed minor but distinct compositional shifts. Cry1Ab lines exhibited higher abundances of fast-growing taxa, particularly Proteobacteria and Bacteroidota; Cry2A lines displayed intermediate profiles, whereas Cry9-2 lines were nearly indistinguishable from WT communities. Linear discriminant analysis of the effect size revealed significant enrichment of taxa such as Burkholderiaceae and Ralstonia in the Cry1Ab rhizosphere, in contrast to the higher abundance of Chloroflexi in the WT. Functional predictions indicated consistent metabolic pathways across all treatments, suggesting strong ecological redundancy. This study demonstrates minimal impact on rhizosphere microbial communities in transgenic C. bungei plants. The Cry9-2 construct exhibited superior environmental stability, whereas the Cry1Ab construct caused only slight but ecologically acceptable shifts. These findings support the ecological safety of Bt-transgenic C. bungei and identify Cry9-2 as a particularly favorable candidate for forestry applications. This comparative evaluation of three Cry genes in a tree species provides a framework for future gene-specific biosafety assessments in woody plants.Keywords
Supplementary Material
Supplementary Material FileTransgenic plants that overexpress Bacillus thuringiensis (Bt) insecticidal crystal (Cry) proteins have been widely used in agriculture and have achieved remarkable success in controlling various insect pests [1,2,3,4]. The deployment of Bt Cry genes in transgenic plants, particularly in economically significant species such as Gossypium hirsutum [5,6], Zea mays [7,8], and Oryza sativa [9,10], has significantly reduced pest damage and reliance on chemical insecticides, thereby generating substantial economic and environmental benefits. As an increasing number of Bt transgenic plant varieties have been developed and commercialized, increasing attention has been directed toward their potential biosafety and ecological impacts. It is particularly crucial to ensure that the expanded application of Bt technology does not inadvertently affect non-target organisms or disrupt essential ecosystem processes [11,12].
Bt Cry proteins represent a diverse family of proteins classified into more than 70 subgroups, each exhibiting distinct target specificity [13]. Notable representatives such as Cry1Ab, Cry2A, and Cry9-2, which belong to different functional classes of Bt toxins, were selected for further investigation. Cry1Ab targets lepidopteran borers and has been successfully expressed in rice and other crops without adverse effects on natural enemies [14]. Cry2A (e.g., Cry2Aa/Cry2Ab) is active against both lepidopteran and dipteran pests [15] and has been incorporated into cotton, sugarcane, and rice to enhance resistance [16,17,18]. Cry9-2, a Cry9A-type toxin with a distinct binding mode, exhibits a broader activity spectrum and is considered a promising candidate for resistance management [19,20]. Significant research and development efforts have been dedicated to these Cry genes, resulting in numerous insect-resistant plant varieties and providing valuable insights into their pest control efficacy.
Beyond agronomic benefits, potential environmental implications of Bt plants warrant continued assessment. Soil microorganisms play key roles in nutrient cycling, organic matter decomposition, and plant growth promotion [21]. Cry proteins released into soil through root exudates, pollen, or decomposing residues may interact with rhizosphere microbes [22]. Recent studies highlight the role of root exudates in shaping rhizosphere microbiota and mediating plant–microbe interactions [23], which may also influence the ecological responses of Cry protein-producing transgenic plants. Although previous studies on Bt maize and Bt poplar reported minimal effects on soil microbial structure and enzyme activity [24,25,26], new Bt events and host species still require case-by-case biosafety evaluations.
Catalpa bungei is a fast-growing hardwood tree that is highly valued in China owing to its high-quality timber and ornamental appeal [27]. However, its cultivation is increasingly threatened by the catalpa shoot borer (Omphisa plagialis), whose larvae burrow into young shoots, resulting in gall formation, wilting of the shoots, and the potential death of the plant. Our previous study established a reliable overexpression system for Bt Cry genes in C. bungei and confirmed the insect resistance of transgenic lines to O. plagialis under both laboratory and field conditions [28]. Although these insect-resistant lines provide an effective strategy for pest control, their ecological safety must be rigorously evaluated before deployment. Cry1Ab, Cry2A, and Cry9-2 proteins belong to different functional classes and exhibit distinct receptor-binding properties [13], yet no study has compared their potential ecological impacts within the same woody host. Such gene-specific comparisons are essential, as Cry constructs may differ in transcript accumulation, protein stability, or plant physiological responses, potentially leading to subtle and gene-dependent microbial outcomes [18,29]. Evaluating these three Cry genes in parallel therefore provides a rare opportunity to disentangle transgene-specific effects from host or environmental influences and strengthens the mechanistic foundation for biosafety assessments of Bt trees.
Here, we investigated the ecological safety of these Bt-transgenic C. bungei lines by comparing their rhizosphere bacterial communities with those of wild-type (WT) plants using 16S rDNA (V4–V5) sequencing. We hypothesized that Cry1Ab, Cry2A, and Cry9-2 would exert distinct but limited effects on rhizosphere microbial communities, providing a comparative basis for ecological safety evaluation.
Three transgenic lines of C. bungei, each carrying a distinct Cry gene (Cry1Ab, Cry2A, or Cry9-2), were used in this study. The accession NJQ301 (wild type) was collected from the Catalpa resource nursery of the Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing, China (32°3.50′ N, 118°49.77′ E). Embryogenic calli derived from NJQ301 were used for plant transformation. All transgenic lines were generated using Agrobacterium tumefaciens EHA105, which harbors the plant expression vector pCAMBIA2300, containing the CaMV 35S promoter [28]. Nine lines with confirmed Cry gene expression were selected: Cry1.1, Cry1.2, Cry1.3 (Cry1Ab), Cry2.1, Cry2.2, Cry2.3 (Cry2A), Cry9.1, Cry9.2, and Cry9.3 (Cry9-2). WT lines served as controls (Fig. S1). All transgenic and WT lines were two-year-old tissue culture seedlings planted in a field in Liuhe, Nanjing (Jiangsu Province, China), at a spacing of 1 × 3 m, under standard field management conditions.
The experiment followed a randomized block design, with each Cry genotype established as an independent plot containing multiple trees. Three individual trees per genotype were randomly selected within each plot as biological replicates for rhizosphere sampling and subsequent microbial and molecular analyses.
In July 2023, rhizosphere soil samples were collected from each of the nine transgenic lines, as well as from WT plants. For each plant, soil samples were collected from four directions around the root zone, at depths of 10–15 cm. The samples were combined and stored at −80°C until DNA extraction. Each genotype was sampled in triplicate. All samples were collected during the same growing season under uniform field management to minimize seasonal and environmental variability.
Total bacterial genomic DNA was extracted from the rhizosphere soil using a TIANGEN DNA extraction kit (DP336, TIANGEN Biotech, Beijing, China), according to the manufacturer’s instructions. The concentration and purity of DNA were assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). DNA was diluted to a final concentration of 1 ng/μL with sterile water. The V4–V5 region of the 16S rDNA gene was amplified using primers 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-CCGTCAATTCCTTTGAGTTT-3′) on a GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA, USA). Each PCR reaction was conducted using the Phusion® High-Fidelity PCR Master Mix (New England Biolabs, Ipswich, MA, USA) for a total of 30 cycles. Successful amplification was confirmed by 2% agarose gel electrophoresis. PCR products were purified using a magnetic bead-based cleanup method and quantified using a Qubit fluorometer (Thermo Fisher Scientific). Equal molar concentrations of the PCR products were combined, and sequencing libraries were prepared using the TruSeq® DNA PCR-Free Kit (Illumina, San Diego, CA, USA). Sequencing was conducted using the Illumina NovaSeq 6000 platform in paired-end 250 bp mode by Novogene Co., Ltd. (Beijing, China).
2.4 Sequence Processing and Bioinformatics Analysis
Paired-end reads were demultiplexed based on unique barcodes and merged using FLASH (Version 1.2.11). Primer sequences were trimmed with Cutadapt, and quality filtering was conducted using fastp (Version 0.23.1) with a Phred quality score cutoff of Q30, discarding reads shorter than 200 bp after trimming. Chimeric sequences were identified and removed using the UCHIME algorithm, referencing the SILVA 138.1 database.
Denoising, dereplication, and amplicon sequence variant (ASV) construction were performed using the DADA2 plugin in QIIME2 (version 2022.2). Taxonomic classification was conducted using the feature-classifier plugin with the SILVA 138.1 reference database. Phylogenetic trees were generated through the phylogeny plugin using FastTree within QIIME2. To control for unequal sequencing depth, samples were rarefied to 40,000 reads per sample, corresponding to the minimum sequencing depth among all libraries. Alpha and beta diversity analyses were performed with the diversity plugin in QIIME2. All plugins were used as included in the QIIME2 (version 2022.2) distribution, and their versions correspond to those built into this release.
Visualization of the results was conducted in R (version 4.0.3) using vegan, ggplot2, and ade4 packages. Functional predictions of the bacterial communities were performed using PICRUSt2 (version 2.3.0), and differential taxa were identified using the linear discriminant analysis of effect size (LEfSe) algorithm with default parameters and visualization scripts.
3.1 Sequencing Quality and Coverage
Illumina sequencing of 16S rDNA amplicons, targeting the V4–V5 hypervariable regions and conducted on the NovaSeq PE250 platform, yielded high coverage of bacterial communities in the rhizosphere samples. Each sample yielded an average of 72,600 quality-filtered reads, and the rarefaction curves approached asymptotes, indicating that the sequencing depth was sufficient. Similarly, the number of observed features (ASVs) plateaued as the number of reads increased, reinforcing the adequacy of our sequencing effort to represent community richness (Fig. S2).
3.2 ASV-Based Core Microbiome and Alpha Diversity Metrics
Venn diagram analysis revealed both shared and unique ASVs among the three transgenic C. bungei genotypes (Cry1Ab, Cry2A, and Cry9-2) and WT (Fig. 1A–C). The Cry9-2 lines exhibited the highest number of ASVs shared with the WT (1067), followed by Cry2A (925) and Cry1Ab (856). This indicates slight variations in rhizosphere composition among the different Cry types. Across all samples, 628 ASVs were identified in all ten lines (nine transgenic and one WT), representing the core microbiome of the C. bungei rhizospheres (Fig. 1D). This core set was derived from ASVs consistently detected across all biological replicates and genotypes. The high ASV overlap among all genotypes suggests that the rhizosphere microbiota is primarily shaped by shared host traits and environmental factors rather than by transgene type. As root exudates serve as the main carbon and energy sources for soil bacteria, the consistent community structure likely reflects similar exudation profiles across the different Cry lines. These shared ASVs likely reflect a stable microbial community associated with the roots of C. bungei under field conditions.
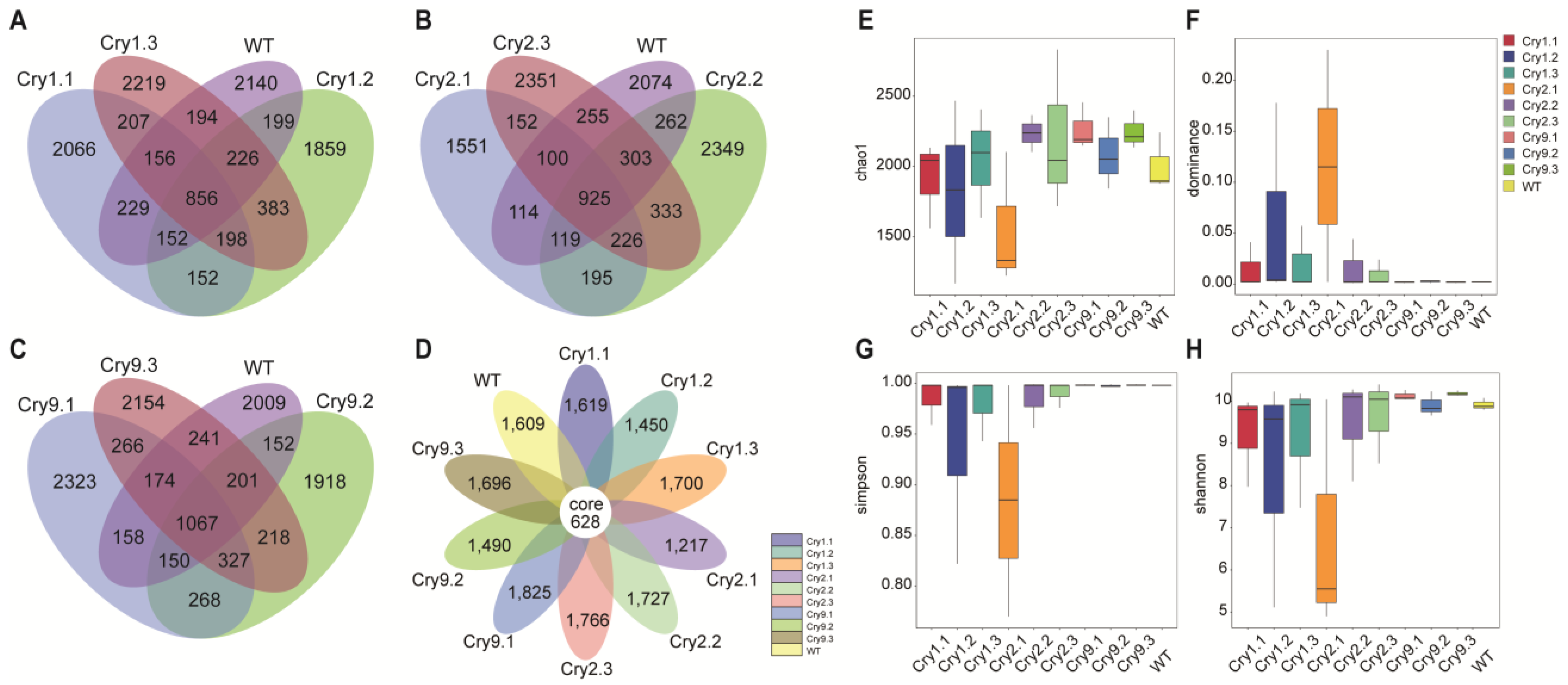
Figure 1: Comparison of rhizosphere bacterial community richness and diversity between transgenic and WT C. bungei lines. (A–C) Venn diagrams showing the number of shared and unique ASVs among WT and transgenic C. bungei lines Cry1Ab (A), Cry2A (B), and Cry9-2 (C). (D) Petal diagram illustrating the number of unique and core ASVs across all the C. bungei genotypes. (E–H) Boxplots showing alpha diversity indices calculated from the 16S rDNA sequencing data: (E) Chao1 richness estimator, (F) dominance index, (G) Simpson’s diversity index, and (H) Shannon’s diversity index. Statistical comparisons were performed using the Kruskal–Wallis test, followed by Tukey’s Honestly Significant Difference post-hoc analysis.
To evaluate the potential effects of different Cry transgenic lines on rhizosphere microbial diversity, we calculated alpha diversity indices, including Chao1, Shannon, Simpson, and Dominance (Fig. 1E–H). Alpha diversity analysis revealed that the rhizosphere bacterial communities of all transgenic and WT C. bungei lines exhibited similar richness and diversity, with no statistically significant differences between the genotypes. Chao1 richness estimates averaged approximately 2000 ASVs, with three Cry9-2 lines demonstrating the most stable values and slightly higher richness than the WT. Cry2A lines exhibited greater heterogeneity, with Cry2.1 demonstrating the lowest richness (<1500) and broader dispersion. Shannon diversity indices remained close to 10.0 across most samples, except for Cry2.1. A value of 6.0 indicates a potential, albeit non-significant, decline in diversity. Simpson and Dominance indices were generally consistent across the lines, except Cry2.1, which exhibited reduced evenness and increased dominance. However, the Kruskal–Wallis and post-hoc Tukey tests confirmed that none of these differences was statistically significant (p > 0.05) (Table S1–S8). These results suggested that the insertion of Cry1Ab, Cry2A, or Cry9-2 did not significantly alter the alpha diversity of the rhizosphere bacterial communities in C. bungei.
3.3 Community Phylogeny and Phylum Distribution
Representative ASV sequences from the top 100 most abundant bacterial genera were annotated and aligned for phylogenetic analysis. A phylogenetic tree was constructed to visualize the structure of the rhizosphere microbial communities at the genus level across all samples (Fig. 2A). The analysis indicated that although some compositional differences were present among the ten lines, the overall phylogenetic structure remained largely consistent. Genera from the dominant phyla, including Proteobacteria, Bacteroidota, Acidobacteriota, and Actinobacteriota, were distributed across various clades and exhibited relatively high abundances. These groups represented the core bacterial assemblages in the rhizosphere of C. bungei.
To assess shifts in the dominant bacterial phyla among the C. bungei genotypes, we constructed ternary plots using the top ten most abundant phyla based on their relative abundances. These plots illustrate the microbial distribution patterns among the representative Cry transgenic lines and WT plants (Fig. 2B and Fig. S3). The most dominant phyla were clustered near the centers of the ternary plots, indicating broadly similar phylum-level community compositions among the various combinations of Cry transgenic and WT Catalpa. Proteobacteria and Acidobacteriota were the predominant phyla across all the groups, forming a stable core of the rhizosphere microbiota.
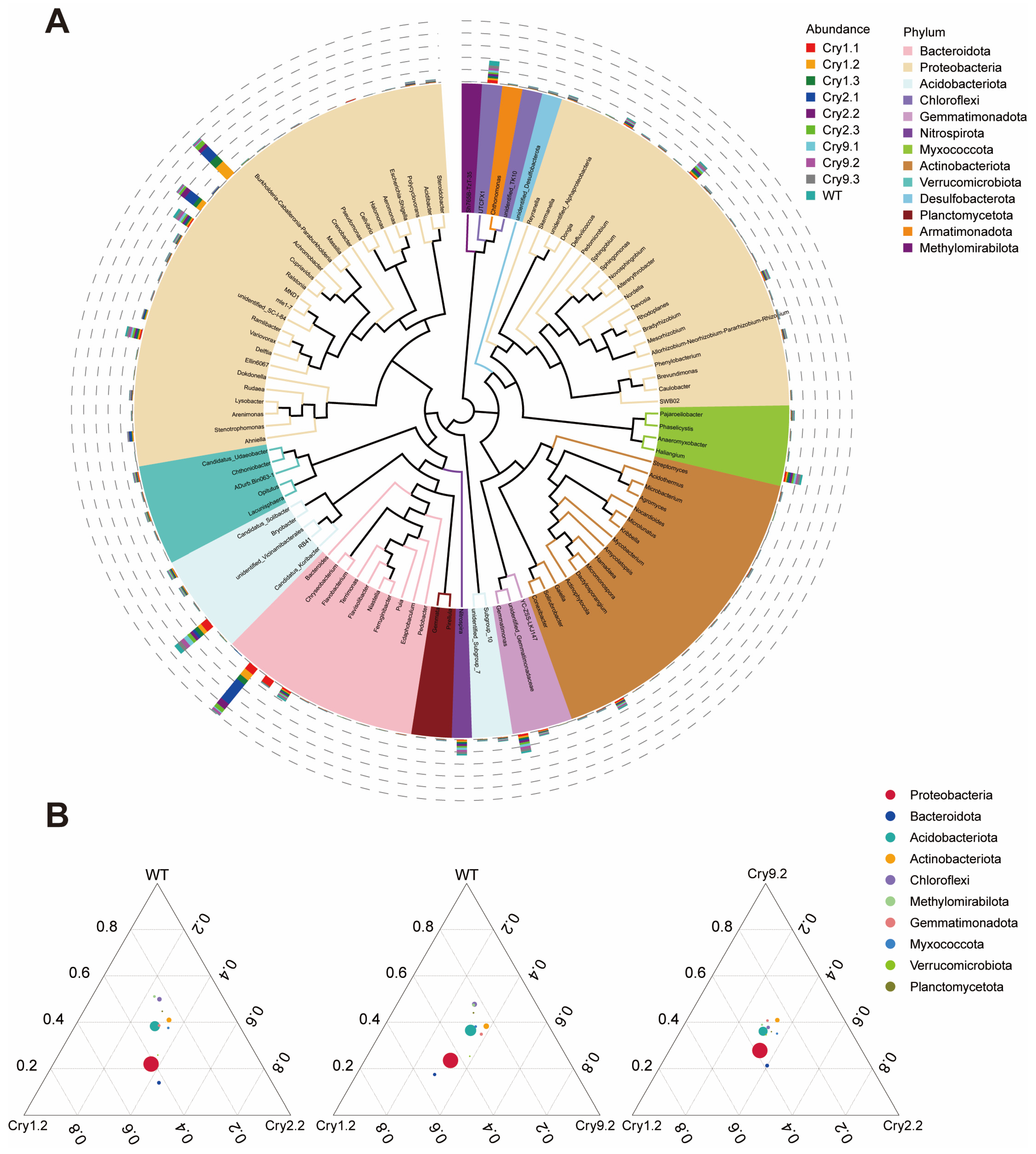
Figure 2: Taxonomic structure and phylum-level distribution patterns of rhizosphere bacterial communities. (A) Phylogenetic tree of the top 100 most abundant bacterial genera across all the samples. Each wedge represents a genus colored according to its affiliated phylum. The outer bars indicate the relative abundance of each genus in different C. bungei genotypes. (B) Ternary plots showing the relative distribution of the top 10 bacterial phyla among the transgenic C. bungei lines and WT. Each circle represents one phylum, with its position indicating proportional abundance across the three groups in each plot (Cry1.2 vs Cry2.2 vs WT, etc.).
Comparisons with the WT revealed that Proteobacteria exhibited directional shifts toward the Cry1Ab and Cry2A genotypes, indicating a potential enhancement of their dominance under transgenic conditions. Similarly, Bacteroidota and Verrucomicrobiota exhibited biased distributions toward the Cry lines, suggesting potential genotype-associated enrichment. In contrast, the microbial profiles of the Cry9-2 lines were similar to those of WT, exhibiting less pronounced shifts across multiple phyla. This suggests a relatively mild effect on the microbial community structure.
A separate ternary comparison of Cry1.1, Cry2.1, Cry9.1, and WT revealed genotype-specific patterns (Fig. S3B). Bacteroidota were significantly enriched in Cry1.1 and Cry2.1, clustering closely with these genotypes, suggesting potential gene-dependent effects. In contrast, Patescibacteria appeared to be associated with the WT, indicating a higher abundance and ecological preference in the non-transgenic rhizosphere. The phylum distribution of Cry9.1 closely resembles that of WT, reinforcing the notion that Cry9.1 has a relatively neutral or low-interference effect on the microbial composition of the rhizosphere.
3.4 Beta Diversity Reveals Community Similarities
To evaluate the similarities and differences in rhizosphere microbial communities across various Catalpa genotypes, we analyzed the relative abundance profiles at the phylum level and integrated the results using UPGMA clustering based on weighted UniFrac distances (Fig. 3A).
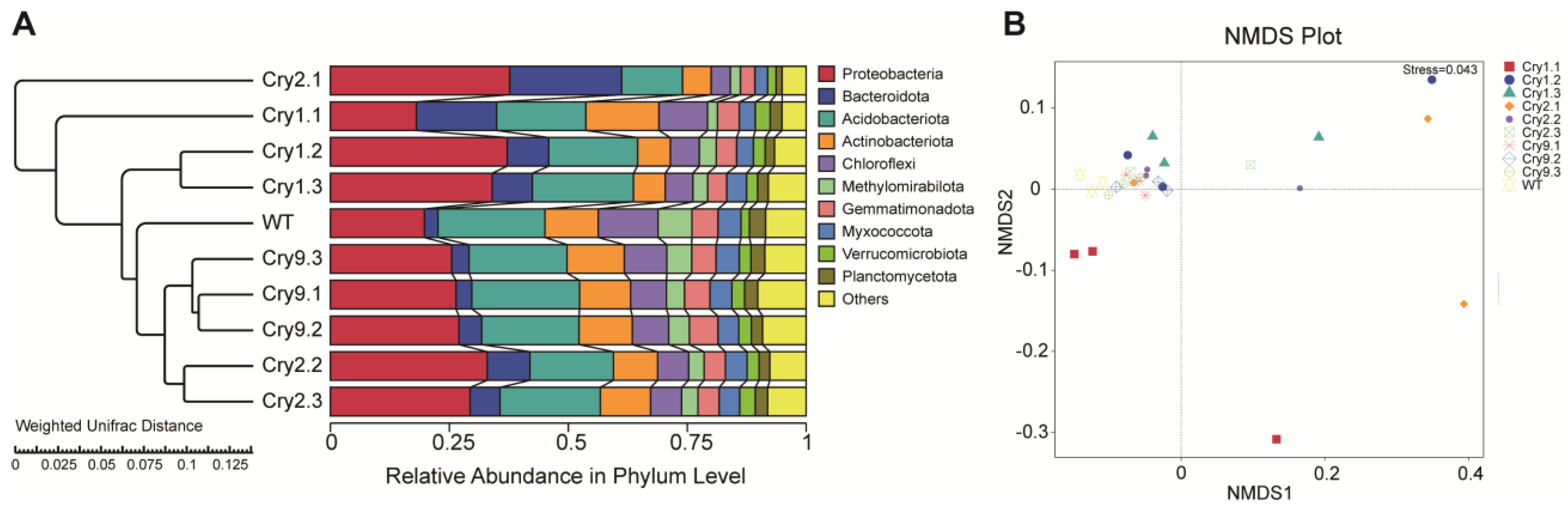
Figure 3: Beta diversity analysis of rhizosphere bacterial communities in transgenic and WT C. bungei. (A) UPGMA clustering based on weighted UniFrac distances at the phylum level, integrating relative abundance profiles. (B) NMDS ordination of weighted UniFrac distances showing tight clustering of samples from all groups, with no distinct separation, indicating broadly similar rhizosphere community structures between the transgenic and WT C. bungei.
The stacked bar plots indicate that the overall phylum-level composition of the bacterial communities was largely similar between the transgenic and WT lines. The dominant phyla included Proteobacteria, Bacteroidota, Acidobacteriota, and Actinobacteriota, which consistently constituted the core of the Catalpa rhizosphere microbiota. However, minor genotype-associated shifts in relative abundance were observed, suggesting that transgene insertion may exert subtle compositional influences.
In particular, the Cry1.1 line showed relatively higher proportions of Bacteroidota and Actinobacteriota, suggesting potential selective enrichment, while Cry2.1 exhibited modest increases in Proteobacteria and Bacteroidota. Although these patterns imply potential gene-related trends, they did not reach statistical significance (p > 0.05), indicating biological tendencies rather than statistically confirmed effects. In contrast, minor phyla, such as Chloroflexi, Methylomirabilota, Gemmatimonadota, and Planctomycetota, exhibited relatively stable abundances across treatments, showing only weak genotype-associated variations.
UPGMA clustering based on weighted UniFrac distances revealed consistent community structure across most genotypes. The WT and Cry9-2 lines (Cry9.1, Cry9.2, and Cry9.3) clustered together, indicating high similarity in relative abundance and minimal disturbance from Cry9-2 expression. Cry2.2 and Cry2.3 also grouped near WT, suggesting only minor effects of Cry2A on the community structure, whereas Cry2.1 formed a slightly separated branch, reflecting modest divergence. In contrast, the Cry1Ab lines (Cry1.1, Cry1.2, and Cry1.3) were the most distant from the WT, exhibiting the greatest UniFrac distance among all treatments.
NMDS ordination based on weighted UniFrac distances revealed that samples from all treatment lines were closely clustered in a two-dimensional space, showing no clear separation. This indicates that the phylum-level community structure remained largely consistent (Fig. 3B). This finding was further supported by the Multiple Response Permutation Procedure analysis, which indicated that pairwise comparisons yielded p-values ranging from 0.10 to 0.20 (Table 1). These results did not meet the conventional significance threshold (p < 0.05), indicating that, although some trends in microbial composition were apparent among the treatments, the differences were not statistically significant.
Table 1: Multiple Response Permutation Procedure Verification of Community Differences between Transgenic and WT Lines.
| Group | A | Sig. | Group | A | Sig. |
|---|---|---|---|---|---|
| Cry1.1-Cry2.1 | 0.126 | 0.2 | Cry1.3-Cry9.2 | 0.056 | 0.1 |
| Cry1.1-Cry2.2 | 0.093 | 0.1 | Cry1.3-Cry9.3 | 0.074 | 0.1 |
| Cry1.1-Cry2.3 | 0.088 | 0.1 | Cry1.3-WT | 0.126 | 0.1 |
| Cry1.1-Cry9.1 | 0.091 | 0.1 | Cry2.1-Cry9.1 | 0.117 | 0.1 |
| Cry1.1-Cry9.2 | 0.1 | 0.1 | Cry2.1-Cry9.2 | 0.103 | 0.3 |
| Cry1.1-Cry9.3 | 0.096 | 0.1 | Cry2.1-Cry9.3 | 0.134 | 0.1 |
| Cry1.1-WT | 0.112 | 0.1 | Cry2.1-WT | 0.16 | 0.1 |
| Cry1.2-Cry2.1 | −0.045 | 0.8 | Cry2.2-Cry9.1 | 0.032 | 0.1 |
| Cry1.2-Cry2.2 | −0.021 | 0.5 | Cry2.2-Cry9.2 | 0.025 | 0.2 |
| Cry1.2-Cry2.3 | −0.021 | 0.6 | Cry2.2-Cry9.3 | 0.019 | 0.1 |
| Cry1.2-Cry9.1 | 0.036 | 0.1 | Cry2.2-WT | 0.109 | 0.1 |
| Cry1.2-Cry9.2 | 0.034 | 0.1 | Cry2.3-Cry9.1 | 0.02 | 0.1 |
| Cry1.2-Cry9.3 | 0.061 | 0.1 | Cry2.3-Cry9.2 | 0.024 | 0.2 |
| Cry1.2-WT | 0.085 | 0.1 | Cry2.3-Cry9.3 | 0.037 | 0.1 |
| Cry1.3-Cry2.1 | 0.001 | 0.3 | Cry2.3-WT | 0.104 | 0.1 |
| Cry1.3-Cry2.2 | −0.005 | 0.5 | Cry9.1-WT | 0.103 | 0.1 |
| Cry1.3-Cry2.3 | −0.004 | 0.5 | Cry9.2-WT | 0.102 | 0.1 |
| Cry1.3-Cry9.1 | 0.041 | 0.1 | Cry9.3-WT | 0.083 | 0.1 |
3.5 Differential Taxa Identified by LefSe
To further clarify the differences in rhizosphere microbial composition among the various Cry transgenic C. bungei lines and WT, LEfSe was used to identify taxa that were significantly enriched in specific groups and to highlight potential microecological shifts.
Differential abundance analysis revealed that distinct bacterial taxa were enriched in the Cry2.1, Cry9.2, and WT lines (Linear Discriminant Analysis [LDA] score > 4) (Fig. 4). The enriched taxa in the WT lines consistently included Chloroflexi, Methylomirabilota, Anaerolineae, Anaerolineales, and Methylomirabilia, which are primarily associated with the degradation of organic matter and complex carbon metabolism. In contrast, the Cry2.1 lines exhibited significant enrichment of copiotrophic taxa, such as Weeksellaceae, Flavobacteriales, Chryseobacterium, and several taxa within Proteobacteria, including Ralstonia and Burkholderiaceae. Many of these taxa are copiotrophic, root-associated bacteria that respond to increased carbon availability and include both potential plant symbionts and opportunistic pathogens. Their enrichment likely reflects localized adjustments in root exudation profiles rather than broad-scale alterations in community structure. Similar trends were observed in the comparison of Cry1.2 with WT (Fig. S4).
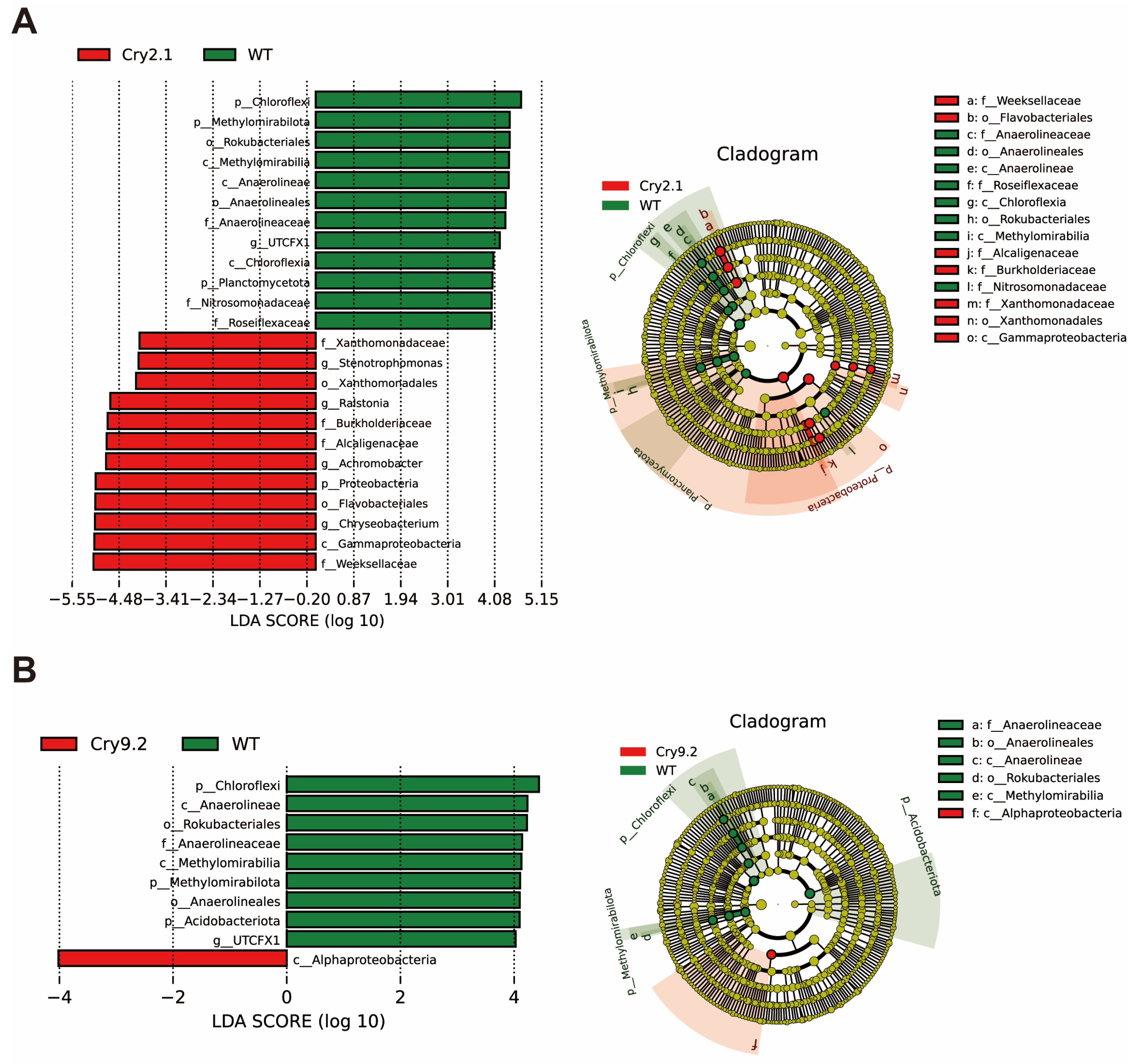
Figure 4: LEfSe analysis of rhizosphere bacterial taxa differentially enriched in Cry2.1 and Cry9.2 lines relative to WT. Histogram of LDA scores (log10) showing bacterial taxa significantly enriched in transgenic lines (Cry2.1 and Cry9.2, red) or WT (green) compared with their respective controls (LDA score > 4.0). Cladogram showing the phylogenetic positions of differentially abundant taxa. Red nodes indicate enrichment in transgenic lines, and green nodes indicate enrichment in WT. (A) Comparison of Cry2.1 and WT samples revealed some differentially abundant taxa, including enrichment of Burkholderiaceae, Ralstonia, and Weeksellaceae in Cry2.1 and Chloroflexi and Methylomirabilota in WT. (B) Cry9.2 and WT showed fewer differentially enriched taxa, with WT samples containing more anaerobic decomposers and Cry9.2 showing slight enrichment in Alphaproteobacteria.
Comparatively, Cry9.2 exhibited fewer differentially enriched taxa than Cry2.1 when compared with WT, with only Alphaproteobacteria showing significant enrichment. The cladogram visualizations further illustrated the significant taxonomic shifts in Cry2.1, which were primarily concentrated within Proteobacteria, in contrast to the less pronounced differences observed for Cry9.2.
However, given the limited sample size and inherent variability in soil microbial communities, these taxonomic patterns should be interpreted with caution as indicative rather than definitive differences. Overall, these findings indicate that Cry9.2 maintained greater stability in rhizosphere microbial communities than Cry2.1 and Cry1.2. Collectively, these findings suggest that although specific taxa and phylogenetic clades responded to transgenic treatments, no significant changes were observed in the overall structure of the rhizosphere microbial community.
3.6 Functional Potential Predicted by PICRUSt2
Functional annotations from multiple databases (Kyoto Encyclopedia of Genes and Genomes [KEGG], Clusters of Orthologous Groups [COG], Enzyme Commission numbers [EC], Protein families [PFAM], and The Institute for Genomic Research protein families [TIGRFAM]) consistently indicated that the predicted metabolic potential of rhizosphere microbiota was largely conserved between the transgenic and WT Catalpa lines. At the KEGG pathway level, the relative abundance profiles exhibited minimal variation across all groups. No pathway exhibited significant loss or enrichment in any of the transgenic lines compared to that in the WT. A substantial proportion of the predicted functions were classified as “Others”, reflecting the presence of numerous low-abundance and diverse metabolic pathways in the rhizosphere microbiome (Fig. 5A).
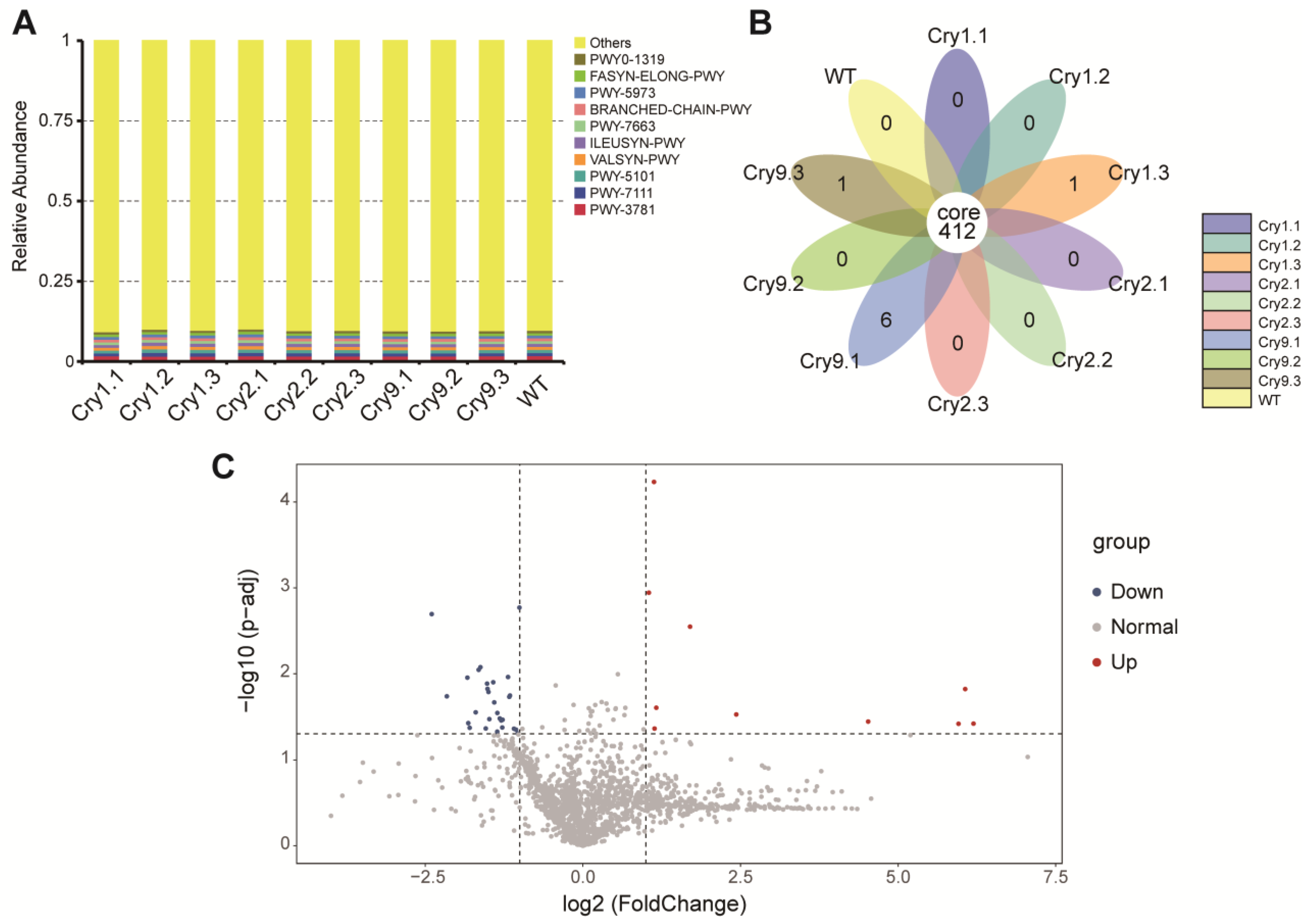
Figure 5: Functional prediction of rhizosphere bacterial communities in Cry-transgenic and WT C. bungei. (A) Relative abundance of the predicted KEGG pathways in each treatment group. (B) Petal diagram showing the number of shared and unique pathways among all the groups. (C) Volcano plot of Enzyme Commission number–based differential abundance between Cry2.1 and WT samples.
To evaluate the degree of functional redundancy and conservation, a petal diagram was constructed to illustrate the shared and unique pathways across different treatments. A total of 412 pathways were identified among all Catalpa lines, highlighting a significant degree of functional overlap and ecological consistency (Fig. 5B).
Further analysis was conducted to evaluate whether the Cry2.1 transgene influenced the microbial enzymatic function. EC-level annotations from PICRUSt2 were compared between Cry2.1 and WT using a t-test. The resulting volcano plot indicated that only a few enzyme categories were significantly upregulated (red) or downregulated (blue), whereas the majority exhibited no significant changes (gray). This pattern indicated that Cry2.1 did not significantly change the predicted enzymatic functions of rhizosphere microbial communities under field conditions (Fig. 5C). These minor variations were not clustered within any specific metabolic pathway, suggesting that no directional functional shifts occurred.
Given that PICRUSt2 provides predictive rather than directly measured functional profiles, these results should be interpreted as indicative rather than conclusive. Overall, no statistically significant or biologically consistent functional changes were detected, supporting the stability of rhizosphere functional potential across all transgenic C. bungei lines.
4.1 Rhizosphere Community Stability in Cry-Transgenic Catalpa
Our findings revealed that the rhizosphere microbial diversity and community structure of nine transgenic C. bungei lines, each expressing one of three different Cry genes (Cry1Ab, Cry2A, or Cry9-2), remained largely stable and showed no significant disruption compared with that of the WT. All transgenic lines exhibited a significant number of ASVs in common with the WT, thereby forming a shared “core microbiome”. For instance, 628 ASVs were detected across all groups, indicating that transgenic Catalpa did not significantly alter the fundamental structure of the rhizosphere microbial community. The resilience of the microbial community suggests that critical soil functions, such as nutrient cycling and organic matter decomposition, are likely to be preserved [30,31,32].
However, shifts in the relative abundance of specific microbial taxa were observed. For instance, the phyla Proteobacteria and Bacteroidota were more abundant in the transgenic lines than in the WT plants. These phyla respond rapidly to available carbon sources, indicating that transgenic plants may modify their root exudation profiles and increase carbon availability in the rhizosphere [33]. Root exudates serve as key carbon and signaling sources for soil microbes [34], and recent evidence indicates that genetic modifications can alter both their chemical composition and secretion dynamics, thereby modulating microbial recruitment and metabolism in the rhizosphere [35,36]. Consequently, the introduction of Cry genes may indirectly influence microbial assembly by modifying nutrient dynamics in the rhizosphere, thereby favoring fast-growing taxa such as Proteobacteria and Bacteroidota.
This hypothesis is supported by previous studies on Bt cotton, which found an increase in the microbial utilization of amino acids and sugars [37]. Furthermore, minor variations in microbial composition among the treatments may affect plant-microbe interactions. LEfSe analysis further supported these observations, revealing enrichment of several root-associated taxa in Cry1Ab and Cry2A lines and decomposer-related groups in WT. Such distribution patterns suggest that Cry gene expression may subtly shift rhizosphere nutrient dynamics, favoring fast-growing or root-interactive bacteria while slightly reducing taxa linked to organic matter degradation [38,39,40,41].
Overall, the observed changes in microbial communities were limited in magnitude and primarily involved shifts in the abundance of specific taxa. These shifts may indicate subtle adjustments in rhizosphere carbon allocation and plant-microbe interactions, while the overall community structure and core functions remain intact.
4.2 Consistency with Previous Studies on Bt Plant Biosafety
Our findings are consistent with those of numerous studies, indicating that Bt transgenic plants have a minimal impact on soil microbial communities, showing no significant differences in diversity or structure compared with non-Bt controls [22,42,43,44,45]. For instance, a four-year monitoring study of Cry1Ab maize demonstrated no detectable changes in rhizosphere bacterial diversity or composition compared to non-transgenic controls [46]. Similarly, research on Cry1Ac cotton has demonstrated no abnormal microbial shifts in the soil [47].
Our alpha diversity results further supported this observation, showing no significant differences in richness or evenness between the Cry lines and WT. Even in studies reporting microbial responses to Bt plants, the observed effects were typically minor, transient, and outweighed by plant developmental stages or environmental factors. For instance, the growth stage of maize had a greater influence on rhizosphere bacterial composition than the transgenic status [48], and although Cry2Ab proteins were detected in the rhizosphere of Bt maize, soil conditions exerted a more pronounced influence on microbial structure [44]. Consistent with this, recent multi-year field investigations on Bt maize and residue management reported low persistence of Cry proteins in soil and only transient, non-significant microbial shifts, reinforcing that environmental variables outweigh transgene effects [26].
Although several studies have reported shifts in functional microbial groups, such as altered bacteria-to-fungi ratios in long-term Bt poplar plantations, these changes did not lead to a reduction in enzyme activity or nutrient cycling [49]. Collectively, the current study contributes to the growing body of evidence suggesting that Cry transgenic plants do not cause significant disruptions in rhizosphere microbial diversity or structure, thereby supporting the relative biosafety of Bt transgenic plants in soil ecosystems.
4.3 Gene-Specific Microbial Responses and Biosafety Implications
The three Cry constructs displayed a clear gene-specific gradient in their influence on rhizosphere microbiota. Cry9-2 lines were the most similar to the WT, sharing the largest number of ASVs (1067) and showing the fewest unique taxa. Cry2A lines exhibited intermediate similarity, whereas Cry1Ab lines shared the fewest ASVs with the WT (856), indicating the greatest divergence. Weighted UniFrac clustering further supported this pattern: Cry9-2 lines consistently grouped with the WT, Cry2A lines were moderately separated, and Cry1Ab lines were positioned at the greatest distance. Together, these quantitative comparisons reveal a consistent gradient of community similarity (Cry9-2 ≈ WT > Cry2A > Cry1Ab).
Despite this gradient, none of the differences reached statistical significance in multivariate tests, underscoring that the overall impact of all three Cry constructs remained weak. Nevertheless, taxonomic contrasts were detectable. Cry1Ab and, to a lesser extent, Cry2A lines showed modest enrichment of several copiotrophic Proteobacteria and Bacteroidota taxa, including members of Burkholderiaceae and Ralstonia, which are known to respond rapidly to increases in carbon availability. In contrast, Cry9-2 lines exhibited minimal changes and no consistent enrichment of such indicator taxa.
These patterns allow us to propose a working model for the observed gene-specific responses. Cry genes differ in their expression stability and protein accumulation in planta, and such differences may subtly modulate root physiology and exudation profiles. Slight increases in readily utilizable carbon or shifts in defense-related metabolites could favor copiotrophic bacterial taxa, producing the weak but detectable compositional shifts observed in Cry1Ab and Cry2A lines. By comparison, Cry9-2 likely produces lower microbial exposure, possibly due to reduced protein accumulation, differences in temporal expression, or faster degradation. These features together may help minimize downstream effects on community assembly. Importantly, PICRUSt2 analysis showed that major metabolic pathways were conserved across all genotypes, indicating strong functional redundancy that likely buffers these compositional differences. It should be emphasized that this working model represents a plausible interpretation based on existing evidence rather than a directly tested mechanism in the present study.
From a biosafety perspective, all nine Cry-transgenic lines exhibited stable α-diversity and shared a core microbiome of 628 ASVs, suggesting that the rhizosphere microbial community remained largely resilient under the field conditions tested. Among them, the Cry9-2 lines showed the weakest taxonomic and functional divergence from the wild type, indicating that this transgene may be considered a comparatively stable and low-impact option for insect-resistant forestry within the scope of this study. Although no significant functional disruptions were detected, these findings should be interpreted with caution, and continued multi-year monitoring and cross-species assessments will be essential to validate the robustness of these gene-specific patterns and to strengthen biosafety frameworks for transgenic woody plants.
Future research integrating multi-site and multi-year field trials, quantitative Cry protein assays, and metabolomic profiling of root exudates will be essential to determine whether the weak gene-specific microbial patterns observed here are consistent across diverse environmental contexts and to identify the physiological drivers underlying these responses. Such efforts will further refine biosafety evaluations for Bt-transgenic forestry species and deepen mechanistic understanding of plant-microbe interactions in transgenic trees.
Acknowledgement:
Funding Statement: This research was funded by the Chinese Academy of Forestry-Special funds for basic scientific research service expenses of the central level public welfare research institutes (Grant No. CAFYBB2020QD001), the National Natural Science Foundation of China (Grant Nos. 32101550, 32271917), Jiangsu Agricultural Science and Technology Innovation Fund (Grant No. CX(24)3052), National Forestry and Grassland Administration’s Center for Science and Technology Development Projects (Grant No. KJZXSA202202).
Author Contributions: Conceptualization, Peng Wang and Naiwei Li; Methodology, Fenni Lv and Peng Wang; Formal analysis, Xiaofeng Mao, Fenni Lv, Shaofeng Li, Peng Wang and Naiwei Li; Investigation, Fenni Lv, Lulu Gao, Donglai Liu, Binpeng Wu and Yanan Wu; Data curation, Xiaofeng Mao; Writing—original draft, Xiaofeng Mao; Writing—review & editing, Fenni Lv and Peng Wang; Visualization, Xiaofeng Mao, Lulu Gao, Donglai Liu, Binpeng Wu and Yanan Wu; Supervision, Shaofeng Li, Wenjun Ma, Peng Wang and Naiwei Li; Funding acquisition, Wenjun Ma and Peng Wang. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: The original data presented in the study are openly available in China National GeneBank DataBase (CNGBdb) at https://doi.org/10.26036/CNP0007687.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest.
Supplementary Materials: The supplementary material is available online at https://www.techscience.com/doi/10.32604/phyton.2026.072636/s1.
References
1. Tabashnik BE, Carrière Y. Surge in insect resistance to transgenic crops and prospects for sustainability. Nat Biotechnol. 2017;35(10):926–35. doi:10.1038/nbt.3974. [Google Scholar] [CrossRef]
2. Xiao Y, Wu K. Recent progress on the interaction between insects and Bacillus thuringiensis crops. Philos Trans R Soc Lond B Biol Sci. 2019;374(1767):20180316. doi:10.1098/rstb.2018.0316. [Google Scholar] [CrossRef]
3. Gassmann AJ, Reisig DD. Management of insect pests with Bt crops in the United States. Annu Rev Entomol. 2023;68:31–49. doi:10.1146/annurev-ento-120220-105502. [Google Scholar] [CrossRef]
4. Ragasruthi M, Balakrishnan N, Murugan M, Swarnakumari N, Harish S, Sharmila DJS. Bacillus thuringiensis (Bt)-based biopesticide: navigating success, challenges, and future horizons in sustainable pest control. Sci Total Environ. 2024;954:176594. doi:10.1016/j.scitotenv.2024.176594. [Google Scholar] [CrossRef]
5. Siddiqui HA, Asif M, Asad S, Naqvi RZ, Ajaz S, Umer N, et al. Development and evaluation of double gene transgenic cotton lines expressing Cry toxins for protection against chewing insect pests. Sci Rep. 2019;9(1):11774. doi:10.1038/s41598-019-48188-z. [Google Scholar] [CrossRef]
6. Quan Y, Wu K. Managing practical resistance of lepidopteran pests to Bt cotton in China. Insects. 2023;14(2):179. doi:10.3390/insects14020179. [Google Scholar] [CrossRef]
7. Wang FH, Han LY, Jiang QP, Jiao P, Liu JQ, Liu SY, et al. Functional analysis of transgenic cry1Ah-1 maize. Microb Pathog. 2023;185:106455. doi:10.1016/j.micpath.2023.106455. [Google Scholar] [CrossRef]
8. Zhang Z, Yang X, Wang W, Wu K. Insecticidal effects of transgenic maize Bt-Cry1Ab, bt-Vip3Aa, and bt-Cry1Ab+Vip3Aa against the oriental armyworm, Mythimna separata (walker) in southwest China. Toxins. 2024;16(3):134. doi:10.3390/toxins16030134. [Google Scholar] [CrossRef]
9. Jiang Y, Ling L, Zhang L, Wang K, Cai M, Zhan M, et al. Transgenic Bt (Cry1Ab/Ac) rice lines with different genetic backgrounds exhibit superior field performance under pesticide-free environment. Field Crops Res. 2016;193:117–22. doi:10.1016/j.fcr.2016.03.014. [Google Scholar] [CrossRef]
10. Xu C, Cheng J, Lin H, Lin C, Gao J, Shen Z. Characterization of transgenic rice expressing fusion protein Cry1Ab/Vip3A for insect resistance. Sci Rep. 2018;8(1):15788. doi:10.1038/s41598-018-34104-4. [Google Scholar] [CrossRef]
11. Liu J, Liang YS, Hu T, Zeng H, Gao R, Wang L, et al. Environmental fate of Bt proteins in soil: transport, adsorption/desorption and degradation. Ecotoxicol Environ Saf. 2021;226:112805. doi:10.1016/j.ecoenv.2021.112805. [Google Scholar] [CrossRef]
12. Wang J, Zhang X, Liu H. Impact of Bt applicatins on soil micrbial communities. Mol Soil Biol. 2024;15(4):183–92. [Google Scholar]
13. Pardo-López L, Soberón M, Bravo A. Bacillus thuringiensis insecticidal three-domain Cry toxins: mode of action, insect resistance and consequences for crop protection. FEMS Microbiol Rev. 2013;37(1):3–22. doi:10.1111/j.1574-6976.2012.00341.x. [Google Scholar] [CrossRef]
14. Xu X, Han Y, Wu G, Cai W, Yuan B, Wang H, et al. Field evaluation of effects of transgenic cry1Ab/cry1Ac, cry1C and cry2A rice on Cnaphalocrocis medinalis and its arthropod predators. Sci China Life Sci. 2011;54(11):1019–28. doi:10.1007/s11427-011-4234-2. [Google Scholar] [CrossRef]
15. Meng M, Shen C, Lin M, Jin J, Chen W, Zhang X, et al. Characterization of the individual domains of the Bacillus thuringiensis Cry2Aa implicates Domain I as a possible binding site to Helicoverpa armigera. J Invertebr Pathol. 2024;205:108129. doi:10.1016/j.jip.2024.108129. [Google Scholar] [CrossRef]
16. Gao S, Yang Y, Xu L, Guo J, Su Y, Wu Q, et al. Particle bombardment of the cry2A gene cassette induces stem borer resistance in sugarcane. Int J Mol Sci. 2018;19(6):1692. doi:10.3390/ijms19061692. [Google Scholar] [CrossRef]
17. Iqbal A, Ali MA, Ahmed S, Hassan S, Shahid N, Azam S, et al. Engineered resistance and risk assessment associated with insecticidal and weeds resistant transgenic cotton using Wister rat model. Sci Rep. 2022;12:2518. doi:10.1038/s41598-022-06568-y. [Google Scholar] [CrossRef]
18. Hu Y, Tian C, Feng Y, Ma W, Zhang Y, Yang Q, et al. Transgenic early japonica rice: integration and expression characterization of stem borer resistance Bt gene. Gene. 2024;927:148753. doi:10.1016/j.gene.2024.148753. [Google Scholar] [CrossRef]
19. Marchetti E, Alberghini S, Battisti A, Squartini A, Baronio P, Dindo ML. Effects of conventional and transgenic Bacillus thuringiensisgalleriae toxin on Exorista larvarum (Diptera: Tachinidae), a parasitoid of forest defoliating Lepidoptera. Biocontrol Sci Technol. 2009;19(5):463–73. doi:10.1080/09583150902807535. [Google Scholar] [CrossRef]
20. Li X, Miyamoto K, Takasu Y, Wada S, Iizuka T, Adegawa S, et al. ATP-binding cassette subfamily a member 2 is a functional receptor for Bacillus thuringiensis Cry2A toxins in Bombyx mori, but not for Cry1A, Cry1C, Cry1D, Cry1F, or Cry9A toxins. Toxins. 2020;12(2):104. doi:10.3390/toxins12020104. [Google Scholar] [CrossRef]
21. Gomis-Cebolla J, Berry C. Bacillus thuringiensis as a biofertilizer in crops and their implications in the control of phytopathogens and insect pests. Pest Manag Sci. 2023;79(9):2992–3001. doi:10.1002/ps.7560. [Google Scholar] [CrossRef]
22. Li Y, Wang C, Ge L, Hu C, Wu G, Sun Y, et al. Environmental behaviors of Bacillus thuringiensis (Bt) insecticidal proteins and their effects on microbial ecology. Plants. 2022;11(9):1212. doi:10.3390/plants11091212. [Google Scholar] [CrossRef]
23. Shafi Z, Shahid M. Root exudates as molecular architects shaping the rhizobacterial community: A review. Rhizosphere. 2025;36:101212. doi:10.1016/j.rhisph.2025.101212. [Google Scholar] [CrossRef]
24. Li Z, Bu N, Chen X, Cui J, Xiao M, Song Z, et al. Soil incubation studies with Cry1Ac protein indicate no adverse effect of Bt crops on soil microbial communities. Ecotoxicol Environ Saf. 2018;152:33–41. doi:10.1016/j.ecoenv.2017.12.054. [Google Scholar] [CrossRef]
25. Zuo L, Yang R, Zhen Z, Liu J, Huang L, Yang M. A 5-year field study showed no apparent effect of the Bt transgenic 741 poplar on the arthropod community and soil bacterial diversity. Sci Rep. 2018;8:1956. doi:10.1038/s41598-018-20322-3. [Google Scholar] [CrossRef]
26. Zhang C, Lv X, Liang X, Peng P, Feng Y. Effects of continuous return of Bt corn straw on soil nutrients, enzyme activities, and microbial communities. Agronomy. 2024;14(11):2737. doi:10.3390/agronomy14112737. [Google Scholar] [CrossRef]
27. Yang J, Wang S, Huang Z, Guo P. The complete chloroplast genome sequence of Catalpa bungei (Bignoniaceae): a high-quality timber species from China. Mitochondrial DNA Part B. 2020;5(4):3854–5. doi:10.1080/23802359.2020.1841581. [Google Scholar] [CrossRef]
28. Lv F, Wang P, Zhang E, Ma L, Gao L, Yang R, et al. Efficient transformation of Catalpa bungei shows crystal genes conferring resistance to the shoot borer Omphisa plagialis. Front Plant Sci. 2021;12:777411. doi:10.3389/fpls.2021.777411. [Google Scholar] [CrossRef]
29. Dhanaraj AL, Willse AR, Kamath SP. Stability of expression of Cry1Ac and Cry2Ab2 proteins in Bollgard-II hybrids at different stages of crop growth in different genotypes across cropping seasons and multiple geographies. Transgenic Res. 2019;28(1):33–50. doi:10.1007/s11248-018-0102-1. [Google Scholar] [CrossRef]
30. Bardgett RD, Wardle DA. Herbivore-mediated linkages between aboveground and belowground communities. Ecology. 2003;84(9):2258–68. doi:10.1890/02-0274. [Google Scholar] [CrossRef]
31. Bulgarelli D, Schlaeppi K, Spaepen S, Ver Loren van Themaat E, Schulze-Lefert P. Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol. 2013;64:807–38. doi:10.1146/annurev-arplant-050312-120106. [Google Scholar] [CrossRef]
32. Sun Y, Chen L, Zhang S, Miao Y, Zhang Y, Li Z, et al. Plant interaction patterns shape the soil microbial community and nutrient cycling in different intercropping scenarios of aromatic plant species. Front Microbiol. 2022;13:888789. doi:10.3389/fmicb.2022.888789. [Google Scholar] [CrossRef]
33. Ling N, Wang T, Kuzyakov Y. Rhizosphere bacteriome structure and functions. Nat Commun. 2022;13:836. doi:10.1038/s41467-022-28448-9. [Google Scholar] [CrossRef]
34. He M, Zhong X, Xia Y, Xu L, Zeng Q, Yang L, et al. Long-term nitrogen addition exerts minor effects on microbial community but alters sensitive microbial species in a subtropical natural forest. Forests. 2023;14(5):928. doi:10.3390/f14050928. [Google Scholar] [CrossRef]
35. Upadhyay SK. Relevance of cross talk between root exudates, hormones, and root-associated microbes in developing sustainable phytoremediation strategies: a comprehensive review. Physiol Mol Biol Plants. 2025;31(10):1629–49. doi:10.1007/s12298-025-01593-3. [Google Scholar] [CrossRef]
36. Upadhyay SK, Kumar P, Jain D. Understanding the mechanistic insight and relevance of root hair-driven rhizobia for developing climate-smart crops. Plant Sci. 2026;362:112779. doi:10.1016/j.plantsci.2025.112779. [Google Scholar] [CrossRef]
37. Zhang M, Feng M, Xiao L, Song X, Ding G, Yang W. Persistence of Cry1Ac protein from transgenic Bt cotton cultivation and residue returning in fields and its effect on functional diversity of soil microbial communities. Pedosphere. 2019;29(1):114–22. doi:10.1016/S1002-0160(17)60475-2. [Google Scholar] [CrossRef]
38. Carrión VJ, Cordovez V, Tyc O, Etalo DW, de Bruijn I, de Jager VCL, et al. Involvement of Burkholderiaceae and sulfurous volatiles in disease-suppressive soils. ISME J. 2018;12(9):2307–21. doi:10.1038/s41396-018-0186-x. [Google Scholar] [CrossRef]
39. Pal G, Saxena S, Kumar K, Verma A, Sahu PK, Pandey A, et al. Endophytic Burkholderia: multifunctional roles in plant growth promotion and stress tolerance. Microbiol Res. 2022;265:127201. doi:10.1016/j.micres.2022.127201. [Google Scholar] [CrossRef]
40. Lan J, Wang S, Wang J, Qi X, Long Q, Huang M. The shift of soil bacterial community after afforestation influence soil organic carbon and aggregate stability in karst region. Front Microbiol. 2022;13:901126. doi:10.3389/fmicb.2022.901126. [Google Scholar] [CrossRef]
41. Fu X, Huang Y, Fu Q, Qiu Y, Zhao J, Li J, et al. Critical transition of soil microbial diversity and composition triggered by plant rhizosphere effects. Front Plant Sci. 2023;14:1252821. doi:10.3389/fpls.2023.1252821. [Google Scholar] [CrossRef]
42. Zhang YJ, Xie M, Wu G, Peng DL, Yu WB. A 3-year field investigation of impacts of Monsanto’s transgenic Bt-cotton NC 33B on rhizosphere microbial communities in northern China. Appl Soil Ecol. 2015;89:18–24. doi:10.1016/j.apsoil.2015.01.003. [Google Scholar] [CrossRef]
43. Li P, Li Y, Shi J, Yu Z, Pan A, Tang X, et al. Impact of transgenic Cry1Ac + CpTI cotton on diversity and dynamics of rhizosphere bacterial community of different root environments. Sci Total Environ. 2018;637:233–43. doi:10.1016/j.scitotenv.2018.05.013. [Google Scholar] [CrossRef]
44. Xu X, Liu X, Li F, Hao C, Sun H, Yang S, et al. Impact of insect-resistant transgenic maize 2A-7 on diversity and dynamics of bacterial communities in rhizosphere soil. Plants. 2023;12(10):2046. doi:10.3390/plants12102046. [Google Scholar] [CrossRef]
45. Fazal A, Yang M, Han H, Lu G, Hao C, Lai X, et al. Impact of dual Bt-transgenic maize (2A7) on soil microbial communities and enzyme activities: a comparative study with control variety Z58. Environ Res. 2024;263:120006. doi:10.1016/j.envres.2024.120006. [Google Scholar] [CrossRef]
46. Barriuso J, Valverde JR, Mellado RP. Effect of Cry1Ab protein on rhizobacterial communities of Bt-maize over a four-year cultivation period. PLoS One. 2012;7(4):e35481. doi:10.1371/journal.pone.0035481. [Google Scholar] [CrossRef]
47. Xie M, Zhang YJ, Peng DL, Li Q, Hu XP, Zhang ZR. No significant impact of transgenic Cry1Ab/1Ac cotton on rhizosphere-soil enzyme activities and bacterial communities. Agron J. 2017;109(4):1271–9. doi:10.2134/agronj2016.10.0618. [Google Scholar] [CrossRef]
48. Wang Y, Zhang M, Li S, Li P, Lang Z. Effects of insect-resistant maize HGK60 on community diversity of bacteria and fungi in rhizosphere soil. Plants. 2022;11(21):2824. doi:10.3390/plants11212824. [Google Scholar] [CrossRef]
49. Lebedev V, Lebedeva T, Tikhonova E, Shestibratov K. Assessing impacts of transgenic plants on soil using functional indicators: twenty years of research and perspectives. Plants. 2022;11(18):2439. doi:10.3390/plants11182439. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools