
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Research Advances on the Mechanisms and Clinical Outcomes of Hyperglycemia in Pregnancy Leading to Congenital Heart Disease in Offspring
1 School of Pediatrics, Hainan Medical University, Haikou, China
2 Hainan Women and Children’s Medical Center, Hainan Medical University, Hainan Academy of Medical, Sciences, Haikou, China
* Corresponding Author: Renwei Chen. Email:
Structural and Congenital Heart Disease 2026, 21(1), 7 https://doi.org/10.32604/schd.2026.075858
Received 10 November 2025; Accepted 06 March 2026; Issue published 31 March 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Hyperglycemia in pregnancy (HIP) is an important independent risk factor for congenital heart disease (CHD) in offspring. With an increasing number of women of childbearing age experiencing gestational hyperglycemia, the impact of an intrauterine hyperglycemic environment on fetal development has drawn significant attention. However, the teratogenic mechanisms underlying its effects on cardiac development remain incompletely understood. This review systematically analyzes relevant literature to summarize its underlying mechanisms and key findings: A hyperglycemic environment disrupts cardiac neural crest cell migration, differentiation, and the proliferation/apoptosis balance of cardiomyocytes by inducing oxidative stress, endoplasmic reticulum stress, and inflammatory responses during critical embryonic developmental stages (cardiac ridge and heart tube formation). The core molecular mechanisms involve abnormalities in signaling pathways such as Wnt, TGFβ, and Notch, as well as dysregulation of epigenetic controls including histone modifications and DNA methylation. Concurrently, in-depth research into the molecular mechanisms by which HIP contributes to CHD provides direction for developing early warning biomarkers (such as specific miRNAs and epigenetic markers) and potential therapeutic targets. It also offers insights into advancing intervention from pregnancy to the preconception stage and enabling proactive, lifecycle management of maternal and fetal risks through multidisciplinary collaboration.Keywords
Congenital Heart Disease (CHD) is one of the most common birth defects, referring to structural abnormalities in the heart or major blood vessels that develop during fetal development. In recent years, the global prevalence of CHD has shown an upward trend: during the period 1970–1974, the average prevalence was 4.547 per thousand people, rising to 9.410 per thousand people by 2010–2017. The prevalence of CHD has shown an upward trend, with China being one of the regions with the heaviest global burden of CHD [1,2]. With the widespread adoption of screening technologies and advancements in treatment, the survival rates of children with CHD have significantly improved; however, prognosis varies significantly among individuals: mild cases typically have a favorable outcome, with patients leading normal lives after treatment into adulthood. In contrast, complex forms of CHD (such as single-ventricle circulation or tetralogy of Fallot) often require multiple surgeries or long-term interventions, imposing a heavy financial burden on families and society. Therefore, preventing CHD at its source is of paramount importance.
The occurrence of CHD is the result of multiple factors, including genetic mutations, chromosomal abnormalities, environmental exposures, and maternal health conditions. Among these, maternal glucose metabolism plays a crucial role in fetal growth and development during pregnancy. Beginning in early pregnancy, an intrauterine hyperglycemic environment may impair myocardial cell growth and proliferation, thereby altering cardiac structure and function [3,4]. These cardiac functional alterations may persist, exerting both short- and long-term adverse effects on offspring health. For instance, myocardial hypertrophy induced by an intrauterine hyperglycemic environment may progressively lead to reduced left ventricular ejection fraction, atrial fibrillation, sudden arrhythmic death, and heart failure [5]. Furthermore, if the critical period of fetal cardiac development is compromised by adverse intrauterine conditions, the cardiovascular system may undergo adaptive remodeling, potentially increasing the risk of heart disease in adulthood [6].
In 2019, the global diabetes population was estimated at 463 million (prevalence rate of 9.3%). This figure is projected to rise to 578 million (10.2%) by 2030 and could reach 700 million (10.9%) by 2045 [7]. With the rising global prevalence of diabetes, the incidence of hyperglycemic pregnancy in women of childbearing age continues to increase. It is estimated that approximately 15%–25% of pregnancies worldwide are affected by hyperglycemia, indicates that some fetuses are exposed to unfavorable intrauterine environments for extended periods. This persistent exposure to hyperglycemia is considered a primary teratogenic factor in diabetic embryopathy [8,9]. Despite continuous improvements in gestational blood glucose management, the incidence of adverse offspring outcomes associated with hyperglycemia remains persistently high, and the impact of an intrauterine hyperglycemic environment on fetal development has drawn significant attention; however, its teratogenic mechanism on cardiac development remains incompletely elucidated. This review systematically analyzes relevant literature to explore the effects of hyperglycemia in pregnancy on fetal cardiac development, elucidate its potential association with offspring cardiac health, summarize the characteristics of cardiac function changes, and summarize existing clinical interventions. This study aims to provide a reference for preconception and prenatal healthcare management, with the goal of achieving early identification and individualized intervention for CHD to improve offspring health outcomes.
2 Definition and Classification of Hyperglycemia in Pregnancy
Hyperglycemia in Pregnancy (HIP) is one of the most common metabolic disorders during pregnancy, During pregnancy, in addition to meeting its own metabolic demands, the mother must provide additional energy to support fetal development. Simultaneously, multiple hormones secreted by the mother and placenta (such as thyroid hormones and prolactin) jointly regulate metabolic changes to ensure the smooth progression of pregnancy. These changes can lead to decreased insulin sensitivity, resulting in insulin resistance (IR) accompanied by insufficient insulin secretion, which in turn causes elevated blood glucose levels [10].
HIP includes the following types: (1) Pregestational Diabetes Mellitus (PGDM) complicated by pregnancy (including type 1 and type 2 diabetes complicated by pregnancy); (2) prediabetes [including Impaired Fasting Glucose (IFG) and Impaired Glucose Tolerance (IGT)]; and (3) Gestational Diabetes Mellitus (GDM) (defined as new-onset glucose metabolism abnormalities during pregnancy in individuals with normal glucose metabolism prior to pregnancy) [11]. PGDM and GDM are the most common types of diabetes.
2.1 Hyperglycemia in Pregnancy Caused by Pre-Existing Diabetes
PGDM is primarily classified into type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM). T1DM is an autoimmune disorder in which the immune system attacks pancreatic beta cells, leading to insufficient insulin secretion. It most commonly occurs during childhood or adolescence, although it can also develop in adulthood. T2DM is often associated with obesity and is characterized by insulin resistance and pancreatic β-cell dysfunction. Patients exhibit a low insulin processing index, rendering them unable to effectively counteract insulin resistance, which leads to hyperglycemia. Compared with healthy pregnant women of the same body mass index (BMI), pregnant women with T2DM exhibit upregulation of placental glucose, amino acid, and fatty acid transporter expression, which affects fetal nutrient metabolism and growth and development [12,13].
2.2 Hyperglycemia in Pregnancy Caused by Gestational Diabetes
GDM refers to impaired glucose tolerance that first occurs or is detected during pregnancy, typically diagnosed between 24 and 28 weeks of gestation through fasting blood glucose and an oral glucose tolerance test (OGTT) [13,14]. GDM is predominantly a transient condition during pregnancy, often resolving postpartum, and accounts for 90%–95% of all cases of diabetes in pregnancy [15].
During normal pregnancy, maternal fasting blood glucose levels decrease due to fetal glucose utilization, while insulin resistance increases (compared to pre-pregnancy levels) to ensure adequate glucose supply to the fetus. As pregnancy progresses, placental hormones (such as prolactin, human placental lactogen, and human placental growth hormone) and pro-inflammatory cytokines further exacerbate insulin resistance during the mid-to-late stages of pregnancy [14]. This promotes endogenous gluconeogenesis and lipolysis, leading to elevated blood glucose and free fatty acid levels. GDM patients develop hyperglycemia due to insufficient pancreatic beta cell function to compensate for insulin resistance during pregnancy.
3 Association between Gestational Hyperglycemia and Fetal Cardiac Development
3.1 Efects of Hyperglycemia in Pregnancy on Cardiac Development
Throughout cardiac morphogenesis, the molecular processes regulating cell migration, induction, and patterning are tightly controlled. Adverse maternal factors may disrupt developmental pathways, leading to CHD [16].
The early gestational period (particularly, weeks 3–8) represents the most critical phase. This stage marks the emergence of the heart from the primordial state, establishing its fundamental structure and function. It is also the most sensitive period, susceptible to internal and external factors that may lead to CHD [17]. Glucose serves as the primary energy source required for cardiac development and is simultaneously one of the most crucial environmental factors for normal fetal heart development. During pregnancy, maternal serum glucose crosses the placenta into the fetal circulation via glucose transporters (GLUTs), resulting in a close correlation between maternal blood glucose concentration and fetal blood glucose levels. Maternal hyperglycemia during pregnancy can alter maternal-placental and fetal-placental blood flow, potentially triggering molecular responses that lead to CHD. Concurrently, placental dysfunction may induce inflammation and oxidative stress, thereby disrupting the signaling cascades that influence various aspects of fetal development, including cardiac development [18].
A hyperglycemic environment can induce metabolic disorders in the fetus, induce oxidative stress, and subsequently promote genetic alterations and disrupt normal apoptosis patterns in cardiac cells. This is particularly evident in the neural crest cells, which are critical components of cardiac development. These cells undergo epithelial-to-mesenchymal transition and migrate to various locations within the body. The migration of cardiac neural crest (CNC) cells to the heart is essential for separating the primitive single cardiac outflow tract into the aorta and pulmonary artery, as well as for the formation of the aortic arch [18,19]. Previous studies have revealed that the migration of the CNC and the formation of the septum in the cardiac outflow tract depend on the regulation of gene expression by factors such as Pax3, Bmp4, and Msx. These genes play a crucial role in the separation of the cardiac tube during normal cardiac development. For example, in mouse embryo experiments, Pax3 expression begins to be suppressed at embryonic day 8.5 under hyperglycemic conditions; subsequently, neural epithelial cells undergo apoptosis, and the incidence of neural tube defects (NTDs) increases compared to embryos from non-diabetic pregnancies. Furthermore, studies indicate that downregulation of Bmp4, Msx1, and Pax3 gene expression impairs epithelial-mesenchymal transition (EMT) during cardiac development, affects myocardial proliferation, disrupts conical structures, and impacts the development and/or migration of cardiac neural crest cells, leading to defects in the atrioventricular septum and cardiac outflow tract [18,19,20]. Experimental studies indicate that fetuses exposed to hyperglycemia via direct infusion of glucose into the left uterine artery during late pregnancy exhibit significantly greater interventricular septal thickness and myocardial proliferation than untreated controls. This demonstrates that even transient hyperglycemic exposure induces excessive growth/hypertrophy of the interventricular septum, suggesting that a hyperglycemic environment at any stage of gestation adversely affects the developing heart [21,22,23,24].
3.2 Differential Effects of Hyperglycemia in Pregnancy on the Heart
Elevated glucose levels are considered a major teratogenic factor during pregnancy, potentially leading to various cardiac structural abnormalities with a broad range of phenotypes, including cardiac ring anomalies, truncus arteriosus, and septal defects. Most CHD can be classified into four types: ventricular septal defect, truncus arteriosus, obstructive outflow tract defects of the left or right ventricle, and lateral defects (left-right abnormalities), with ventricular septal defect accounting for nearly half of all CHD cases [16].
Research indicates that the timing of hyperglycemic injury during cardiac development is critical for CHD phenotypes [25,26,27]. The critical period for cardiac development spans gestational weeks 3 to 8. After this phase, the heart’s basic structure is established, transitioning into stages of growth, maturation, and functional refinement. During early pregnancy, the hyperglycemic environment in PGDM surrogate mothers directly impairs neural crest cell proliferation, migration, and survival, leading to abnormal septation of the conus arteriosus—the core mechanism underlying outflow tract defects [18,19,20]. Upregulation of pro-apoptotic genes in a hyperglycemic intrauterine environment leads to excessive apoptosis in the endocardial cushion or septal margin tissues, resulting in failure of the closure process. Furthermore, early embryos exposed to a high-sugar environment produce large amounts of reactive oxygen species, leading to DNA damage and abnormal histone modifications. This disrupts the stable expression of key genes associated with cardiac development, such as PAX3 and TGF-β family genes [28]. Studies have indicated that the incidence of CHD among the offspring of mothers with type 1 and type 2 diabetes is comparable. However, offspring of mothers with type 1 diabetes exhibit the highest risk of conotruncal anomalies and atrioventricular septal defects (AVSD), while offspring of mothers with type 2 diabetes are more prone to ectopic and left ventricular outflow tract obstruction anomalies [29]. In contrast, the incidence of CHD in newborns exposed to GDM is lower, suggesting that the timing of hyperglycemic injury during cardiac development is critical for the CHD phenotype, as GDM typically occurs after the critical period of cardiac morphogenesis [16]. However, regardless of the type of diabetes, the intrauterine hyperglycemic environment during pregnancy remains a common feature, and the mechanism leading to CHD remains a key research focus.
4 Mechanisms by Which Hyperglycemia in Pregnancy Affects Fetal Cardiac Development
It is well known that the heart develops from diverse cell lineages, including two mesoderm-derived cardiac precursor cells (CPCs), the first and second heart fields (FHF, SHF), and pluripotent neural crest cells [30]. This complex organogenesis process relies on the precise spatiotemporal proliferation, migration, differentiation, and integration of different cell types. Even minor disturbances in any step can lead to malformations. Through multi-level validation in animal experiments and human observational studies, previous research has gradually revealed the mechanisms underlying hyperglycemic cardiac toxicity. Over the past 3 to 5 years, the emergence of cutting-edge technologies such as single-cell RNA sequencing (scRNA-seq) and organoid models has fundamentally transformed our understanding of the mechanisms by which HIP induces fetal cardiac malformations. scRNA-seq revealed that high glucose environments do not affect all cardiac progenitor cells, but selectively target specific subpopulations, leading to imbalanced proportions and erroneous fate determination. For example, maternal diabetes alters Alx3-positive forepart of the heart region cells through epigenetic mechanisms, leading to disruption of anterior-posterior patterning [31]. In another zebrafish embryo experiment, RNA sequencing was performed to examine gene changes in the experimental group, revealing significant alterations in 556 upregulated genes and 1118 downregulated genes in the high-glucose group [32]. Furthermore, organoid models eliminate embryonic redundancy and compensatory mechanisms under specific conditions, enabling highly accurate in vitro simulation of the self-organization, chamber formation, and beating of the early human heart. This provides a direct, controllable model for studying human-specific developmental processes and hyperglycemic toxicity. Studies have demonstrated that organoid models successfully reproduce phenotypes such as high-glucose-induced ventricular wall thinning and arrhythmia [33,34]. However, the pathophysiological mechanisms underlying HIP-induced birth defects remain complex and require further investigation to elucidate [35]. This study summarizes the following primary mechanisms: (Fig. 1).
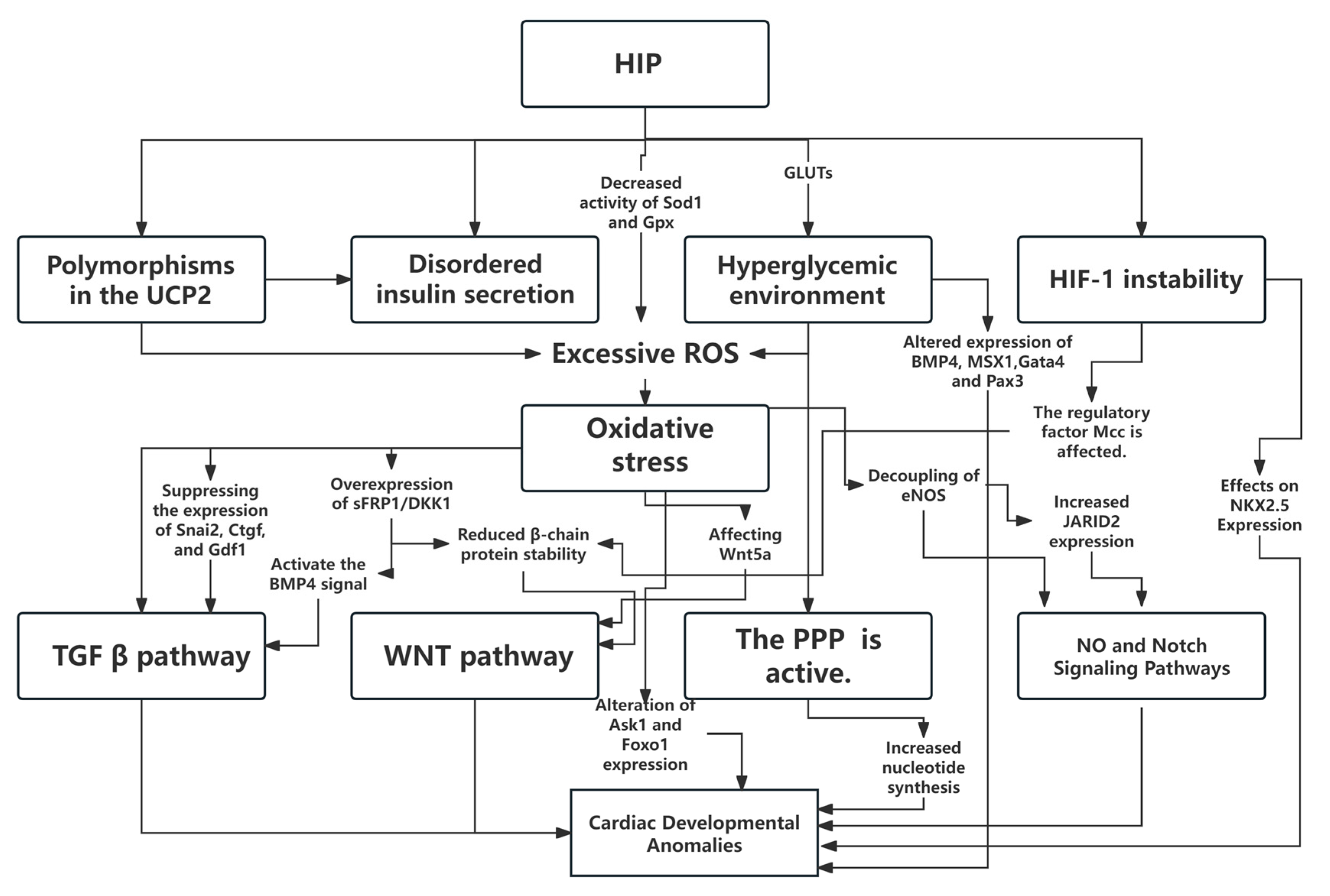
Figure 1: Schematic Diagram of Mechanisms by Which Gestational Hyperglycemia Affects Cardiac Development in Offspring. HIP, hyperglycemia in pregnancy; Sod1, superoxide dismutase; Gpx, glutathione peroxidase; GLUTS, glucose transporters; MCC, a regulator of the cell cycle and the wnt/β-catenin signaling pathway; ROS, reactive oxygen species; eNOS, endothelial nitric oxide synthase; PPP, pentose phosphate pathway; HIF-1, hypoxia-inducible factor 1.
4.1 The Core Role of Oxidative Stress and Its Related Signaling Pathways
Oxygen is essential for normal cellular metabolism and the maintenance of life processes. During metabolism, cells naturally produce small amounts of reactive oxygen species (ROS), including hydroxyl radicals (•OH) and hydrogen peroxide (H2O2). Glucose can undergo auto-oxidation to generate •OH; therefore, under hyperglycemic conditions, ROS production increases. The generated ROS play crucial roles in cellular signaling and physiological processes, such as proliferation, differentiation, and migration. Excessive ROS can lead to protein oxidation, lipid peroxidation, and DNA damage, causing cellular injury and death, which in turn impairs normal cardiac development. Oxidative stress (OS) occurs when ROS production exceeds the capacity of the antioxidant defense system to process it [28,36].
Multiple studies have demonstrated that hyperglycemia can induce excessive ROS production and reduce intracellular antioxidant capacity. During maternal diabetes mellitus (matDM), the expression and activity of key ROS scavenging enzymes, such as superoxide dismutase (Sod1) and glutathione peroxidase (Gpx), decline, exacerbating ROS production. Excessive ROS-induced oxidative stress appears to be an initial response to maternal hyperglycemia, subsequently modulating multiple molecular pathways essential for cardiac development and playing a central role in disrupting normal cardiac developmental mechanisms [24]. For example, during cardiac development, the expression and function of uncoupling protein UCP2 play crucial roles. UCP2 is widely distributed across various tissues and cell types in the body, where it regulates free fatty acid metabolism and transport, reduces ROS production, and modulates insulin secretion. In the presence of maternal diabetes, hyperglycemia, UCP2 gene polymorphisms, and disrupted insulin secretion pathways lead to OS, causing alterations in the intrauterine environment that are associated with the development of CHD in offspring [37]. Furthermore, OS can disrupt cellular homeostasis by altering key molecular pathways essential for normal cardiac development, such as transforming growth factor-β (TGFβ), Wnt signaling, nitric oxide, and Notch signaling, and by inducing excessive apoptosis, thereby increasing the risk of CHD in the offspring of diabetic mothers [5,28].
4.2 Abnormalities in Key Signaling Pathways
Nitric oxide (NO) and Notch signaling pathways play crucial roles in cardiac development. NO is a vital cardiovascular signaling molecule. Endothelial nitric oxide synthase (eNOS) participates in vascular endothelial relaxation by synthesizing NO, thereby regulating hemodynamic changes, and promoting angiogenesis, which is essential for embryonic heart development. Animal studies have revealed that hyperglycemia in pregnancy alters the expression of eNOS and VEGF genes in embryos of matDM mice, both of which play roles in regulating cell growth and angiogenesis [24]. Furthermore, eNOS deficiency leads to the emergence of most CHD phenotypes in mice. Tetrahydrobiquinoline (BH4) is a key factor in maintaining eNOS stability. As an antioxidant and eNOS cofactor, it enhances eNOS stability and activity, thereby improving vascular endothelial function During hyperglycemia-induced OS, NOS3 expression decreases while NOS2 expression increases [38,39]. This leads to substantial depletion of BH4 and decoupling of eNOS, resulting in excessive production of peroxides rather than NO. This further exacerbates oxidative stress and disrupts normal cardiac development processes [5].
The Notch signaling pathway involves multiple receptor-ligand interactions, with receptors including Notch1–4, among which Notch1 is the key receptor. Notch1 signaling cascades regulate cellular processes such as proliferation, apoptosis, and differentiation, with each process associated with specific domains of Notch1. During fetal cardiac development, Notch1 is expressed in the outflow tract (OFT), atrioventricular canal (AVC), interventricular septum, and epicardium, influencing aortic and pulmonary valve formation and the maintenance of the great vessels. Studies indicate that global eNOS deficiency in mice leads to the development of CHD, and eNOS−/−; Notch1+/− compound mutant mice exhibit a series of conotruncal and semilunar valve malformations [24,40]. Beyond the genetic interaction between NO and Notch1, a hyperglycemic intrauterine environment during early embryonic development (embryonic days E6–7.5) can disrupt the Notch signaling pathway, affecting the establishment of the left-right axis in mouse embryos. Additionally, oxidative stress-induced decoupling of eNOS leads to reduced NO production, which in turn increases the expression of the transcription repressor JARID2. Overexpression of JARID2 inhibits Notch1 and the transcription factor Hey2, whereas eNOS deficiency may further diminish Notch1 signaling through NO loss, resulting in a high risk of CHD in affected children [5].
The Wnt signaling pathway is a multifunctional signaling system that is extensively involved in embryonic development, cell proliferation, differentiation, maintenance of tissue homeostasis, and disease onset. Targeted deletion of key components in both canonical and non-canonical Wnt pathways may induce CHD, exhibiting phenotypes similar to those observed in human diabetic pregnancies [24]. Research indicates that in the canonical pathway, OS induced by a maternal hyperglycemic environment leads to increased expression of two Wnt antagonists (sFRP1 and DKK1), resulting in decreased phosphorylation of Wnt ligand-binding transmembrane receptor proteins Frizzleds and Dishevelled (Dvl) proteins. By reducing phosphorylated GSK3β levels, it enhances GSK3β activity, decreases overall β-catenin stability, and consequently blocks typical Wnt signaling in the developing heart [41,42]. Furthermore, elevated FRP1 expression can activate BMP4 signaling, leading to dysregulation of the TGFβ signaling pathway and adversely affecting cardiac development [43]. On the other hand, the emergence of OS has impacted the activation of the Ca2+/calmodulin signaling pathway by the atypical Wnt ligand Wnt5a, a pathway critical for normal outflow tract development in the heart [42,44].
Another signaling pathway suppressed in embryonic hearts exposed to maternal hyperglycemia is the TGFβ pathway, which plays a crucial role in early cardiac development. During normal cardiac development, TGFβ signaling plays a critical role in the migration, homing, and maturation of neural crest cells (NCCs). Mutations in one or more components of this pathway are known to cause outflow tract (OFT) and septal defects [24]. Research indicates that when mothers have diabetes, the high-glucose environment induces OS, downregulates its ligands (Tgfβ1 and Tgfβ3), leading to reduced phosphorylation levels of downstream effectors (TβRII, Smad2/3) and suppression of Tgfβ target gene expression (Snai2, Ctgf, and Gdf1). This inhibits Tgfβ signaling in embryonic hearts, resulting in the development of cardiac malformations [45].
Hypoxia plays a crucial physiological role in cardiac development, particularly during early embryonic development and angiogenesis; however, excessive, or prolonged hypoxia may lead to cardiac developmental abnormalities, including cardiac malformations, impaired angiogenesis, and myocardial cell damage. The primary regulator of the hypoxic response is hypoxia-inducible factor 1 (HIF-1), which is composed of two subunits: HIF-1α (oxygen-sensitive subunit) and HIF-1β (constantly expressed subunit). Recent studies have identified 36 downregulated and 32 upregulated protein-coding gene transcripts in Hif1aCKO neurons through differential expression analysis during cardiac development. Increased expression of Nkx2.5, Tbx5, Nppa, Cx43, and Mef2c was observed [46,47,48,49,50]. Additionally, Gene Ontology (GO) categories associated with proliferation, cell cycle, and mitosis were significantly enriched, such as Mcc, a regulator of the cell cycle and Wnt/β-catenin signaling pathways [46]. Transcription factors such as E2f1 and cyclin B1 (Ccnb1) that influence cell cycle progression and apoptosis [47,48]. HIF-1α is crucial for normal embryonic development, while a hyperglycemic intrauterine environment leads to HIF-1α instability, causing fetal cells to lose their ability to adapt to hypoxia. In mouse experiments, homozygous Hif1a knockout mice died due to cardiac malformations and vascular defects. Concurrently, research indicates that offspring from diabetic pregnancies with Hif1α+/− status exhibit left ventricular dysfunction at 12 weeks of age, manifested as reduced shortening fraction and myocardial structural remodeling. This may reflect the embryo’s response to the maternal diabetic intrauterine hyperglycemic environment, resulting from increased OS and hypoxia [49,50,51].
4.3 Other Potential Mechanisms
Beyond the aforementioned OS and related pathways, CHD development also involves multiple glucose-mediated developmental pathways: during normal cardiac development, the energy supply pathways for cardiac cell growth and development undergo profound shifts, transitioning from an early stage of high dependence on glucose-dependent glycolysis and the pentose phosphate pathway (PPP) to increasingly efficient oxidative phosphorylation [52]. Under conditions of high blood sugar, large amounts of glucose are directed toward the PPP pathway to generate two primary products: reduced NADPH and pentose sugars that provide the backbone for nucleotide biosynthesis. This enables rapid cell division, but compared to cells exposed to normal glucose levels, these cells fail to mature properly This phenomenon has been observed both in vitro in human embryonic stem cell-derived cardiomyocytes (hESC-CMs) and in vivo in mouse models of diabetes. Additionally, hyperglycemia inhibits the differentiation of embryonic stem cells (ESCs) into cardiomyocytes by suppressing the potassium channels HCN1 and KCN1-essential for myocardial cell contraction, and by reducing the maturation markers TNNT2 and MEF2C [25,53]. Additionally, mitochondria in immature cardiomyocytes appear small and round or oval in shape, with a relatively low number. This may further lead to a significant increase in ROS levels, ultimately resulting in oxidative stress-induced cell death [54,55,56].
Another study indicated that endothelial progenitor cells (EPCs) possess the potential to differentiate into endothelial cells, participate in vascular endothelial repair, and angiogenesis, and play a crucial role in ensuring normal vascular supply during cardiac development. Exposure to an intrauterine hyperglycemic environment impairs fetal EPC function and induces gene expression alterations, thereby increasing the risk of CHD in the offspring [57].
4.4 Epigenetic Regulation and Alterations in Gene Expression
During embryonic and fetal development, epigenetic mechanisms precisely regulate the normal formation and differentiation of the heart through multiple pathways, including DNA methylation, histone modifications, and the expression of non-coding RNAs such as miRNAs and lncRNAs. These epigenetic processes are tightly regulated in utero. However, maternal hyperglycemia can significantly alter the metabolic and biochemical microenvironment within the uterus, thereby disrupting the aforementioned epigenetic regulatory networks. This leads to abnormal expression patterns of genes associated with cardiac development, ultimately affecting the normal structure and function of the fetal heart [58,59,60].
Specifically, during pregnancy in diabetic mothers, glucose exceeding physiological requirements is transported across the placenta to the fetus, leading to intracellular glucose overload. This not only results in excessive ROS and ATP production, inducing OS, but also alters the concentration of intermediates such as S-adenosylmethionine (SAM). Consequently, this affects DNA methyltransferase activity and disrupts DNA methylation [61]. Simultaneously, under hyperglycemic conditions, abnormal activation of glycolysis and the PPP drives increased acetyl-CoA production, potentially leading to enhanced histone modification remodeling. For instance, high glucose may induce inflammation and OS via MLL1-dependent H3K4me3 modification, significantly impairing endothelial cell function and precipitating early onset of adverse cardiovascular phenotypes [60]. Additionally, the expression of non-coding RNAs is also influenced by high glucose levels during cardiac development. Research indicates that alterations in glucose concentration lead to the upregulation of miR-1 and miR-206 expression, which act in myocardial cell apoptosis by targeting HSP60. Additionally, these changes influence the regulation of GLUT4 expression by miR-133 and 195-5p, potentially contributing to cardiac hypertrophy and other outcomes [58,62]. Another study using a mouse model of pregnancy-induced diabetes demonstrated that fetal cardiac tissue exhibited upregulation of let-7e-5p, miR-139-5p, and miR-195-5p, accompanied by increased cardiac wall thickness [63]. Other animal studies also support this view. For instance, offspring born to pregnant baboons exposed to a diabetic environment exhibit altered expression of certain miRNAs in their hearts, potentially increasing their future risk of cardiac hypertrophy, myocardial infarction, or even cardiomyopathy [64]. These epigenetic alterations may heighten the offspring’s risk of developing metabolic disorders and cardiovascular diseases later in life, suggesting a mechanism for transgenerational transmission of metabolic risk [65].
4.5 Interactions between Oxidative Stress and Epigenetic Modifications
The mechanism by which HIP influences cardiac development—whether through direct alteration of gene expression or indirect effects via associated OS and hypoxia processes—remains controversial. For example, hyperglycemia during early pregnancy affects the expression of regulatory genes in the embryo (including Bmp4, Msx1, Gata4 and Pax3), leading to cardiac neural crest cell death and increased risk of CHD [21]; Other studies, however, have demonstrated differing perspectives; for instance, the apoptosis signal-regulated kinase 1 (Ask1) gene, activated by oxidative stress, exerts teratogenic effects by inducing endoplasmic reticulum stress and apoptosis while impairing essential cardiotrophic factors [28].
Research on STZ-induced pre-gestational diabetic rat models indicates that maternal hyperglycemia increases Forkhead Box O1 (Foxo1) activity, affecting the expression of its downstream effector genes in embryonic heart grafts. These genes possess pro-oxidative and pro-inflammatory properties, leading to OS, inflammation, and a high risk of cardiac defects [66].
Numerous studies on epigenetic mediators have examined the association between intrauterine GDM exposure and DNA methylation in the placenta, offspring umbilical cord, and infant blood. ROS-induced OS was found to act as a catalyst for DNA methylation, inducing hypermethylation at specific genomic sites by either upregulating Dnmts expression or generating novel Dnmt content complexes. Notably, pretreatment of offspring with the ROS inhibitor N-acetylcysteine (NAC) reversed GDM-mediated Dnmt3A overexpression and hypermethylation of DNA in cardiac tissue, suggesting a potential causal relationship between GDM-induced ROS and DNA methylation [67,68,69,70] These findings underscore the necessity of rigorous monitoring and management of maternal blood glucose levels during pregnancy to mitigate the potential risk of CHD in offspring. Further research is needed to fully elucidate these mechanisms and explore potential intervention and prevention targets.
5 Long-Term Effects of Hyperglycemia in Pregnancy on Cardiac Structure and Function
5.1 Cardiac Changes and Variations in Cardiac Markers during the Neonatal Period
Prolonged fetal exposure to a hyperglycemic intrauterine environment may subject offspring to both short- and long-term adverse effects [71]. For example, newborns of mothers with GDM may exhibit impaired cardiac function, which manifests as impaired ventricular filling, tachycardia, outflow tract obstruction, and reduced ventricular compliance. These alterations lead to abnormalities in myocardial systolic and diastolic functions, with particularly pronounced changes in the right ventricle [72].
Possible mechanisms for right ventricular dysfunction in the offspring of diabetic mothers include insulin resistance, activation of the sympathetic nervous system, and activation of the renin-angiotensin-aldosterone system. These pathways share the common features of myocardial collagen accumulation and the development of myocardial fibrosis, which may be associated with prolonged hyperglycemia, advanced glycation end products, and excessive production of oxygen free radicals. These changes ultimately lead to loss of elasticity and increased ventricular stiffness, initially causing diastolic dysfunction, followed by myocardial hypertrophy, and eventually progressing to systolic dysfunction [73].
Relevant studies indicate that HIP triggers a cascade of molecular events by promoting hyperglycemia, hyperinsulinemia, and increased insulin receptor expression during cardiac development. This disrupts the delicate balance between myocardial cell proliferation, cell cycle arrest, and terminal differentiation. First, maternal hyperglycemia transmitted through the placenta elevates fetal blood glucose levels, directly damaging fetal cardiomyocytes and inducing excessive cardiomyocyte apoptosis [74]. Concurrently, hyperglycemic conditions may cause fetal hypoxia, leading to increased ROS release in both the fetus and placenta [75]. This subsequently induces membrane lipid oxidation damage and mitochondrial DNA deterioration, resulting in mitochondrial dysfunction within cardiomyocytes and disrupting normal developmental cycles [76,77]. Second, fetal hyperinsulinemia induced by HIP promotes fat and protein synthesis in cardiomyocytes, leading to lipid accumulation within these cells and increasing dependence on fatty acids for energy. Initially, the heart can compensate to maintain cardiac output. However, if the fetal heart remains continuously exposed to this metabolic environment, it will ultimately lead to cell apoptosis, fibrosis, and contractile dysfunction [73]. Additionally, elevated insulin levels can cause dysregulation of insulin-like growth factor I (IGF-1) signaling, leading to myocardial proliferation and hypertrophy, resulting in increased myocardial mass. Some neonates may develop interventricular septal dysfunction, most commonly observed during diastole. Such interventricular septal hypertrophy is typically transient and often asymptomatic [73,78]. On the other hand, large amounts of glucose are diverted to the PPP pathway for nucleotide biosynthesis, enabling rapid cell division but hindering proper differentiation and maturation compared to cells exposed to normal glucose levels [55]. In summary, the pathophysiological mechanisms by which HIP affects myocardial development and function are multifactorial and multi-layered, and remain incompletely understood.
Furthermore, the study revealed that newborns exposed to an intrauterine hyperglycemic environment exhibit persistent alterations in left ventricular geometry, characterized by thickened ventricular walls and shortened and narrowed ventricular cavities. Concurrently, increased right ventricular end-diastolic dimensions and reduced right ventricular length lead to a persistent increase in the right ventricular sphericity index. Using M-mode ultrasound, pulsed-wave tissue Doppler imaging, and speckle tracking to assess cardiac function in newborns with GDM revealed significantly reduced longitudinal contractility in both ventricles, indicating impaired myocardial function and suggesting possible subendocardial microvascular ischemic changes. Persistent alterations in ventricular geometry and function on the first day of life may indicate long-term cardiovascular disease susceptibility in offspring of mothers with gestational diabetes mellitus [79].
Cardiac function assessment indicators in the neonatal period include left ventricular ejection fraction (LVEF), right ventricular function (RVEF), left ventricular end-diastolic volume (LVEDV), and left ventricular end-systolic volume (LVESV) for evaluating neonatal cardiac function. Myocardial injury can be detected using specific cardiac biomarkers such as troponin I (TnI), B-type natriuretic peptide (BNP), and its precursor N-terminal pro-B-type natriuretic peptide (NT-proBNP). Although TnI levels in the umbilical cord blood of healthy infants are extremely low compared to maternal levels, elevated levels have been reported in infants with CHD. Recent studies indicate that certain biochemical markers of cardiac dysfunction are elevated in infants born to diabetic mothers, particularly those with cardiomyopathy or adverse perinatal outcomes [80]. Furthermore, epigenetic markers can integrate genetic and non-genetic factors in a biologically stable and technically reproducible manner, indicating their high potential as biomarkers [81]. For example, researchers have identified specific genes associated with CHD whose promoter methylation status may hold predictive value, including RUNX3, MYLK, GALNT2, TRAPPC9, PRDM16, and NR2F2 [82,83,84]. Additionally, microRNAs participate in key pathways of cardiac development. Large-scale clinical studies have revealed significantly altered levels of these miRNAs in the serum of pregnant women with CHD, with detectable differences as early as the first trimester, demonstrating potential for early prediction [13].
5.2 Cardiovascular and Metabolic Risks from Childhood to Adulthood
A 40-year follow-up study revealed that elevated systolic blood pressure and mean arterial pressure in children and adolescents are associated with maternal diabetes, and that offspring of mothers with diabetes exhibit a relatively higher incidence of early onset cardiovascular disease [85]. This suggests that gestational hyperglycemia may influence offspring’s long-term cardiovascular health through a “developmental programming effect.”
The developmental origin of health and disease (DOHaD) hypothesis has been validated in multiple studies: For example, children aged 3–5 years born to mothers with GDM exhibited increased BMI, skinfold thickness, body fat, blood pressure, lipid profile, and glucose metabolism indicators. Children aged 10–14 years who were exposed to maternal GDM in utero developed impaired glucose tolerance [71]. Concurrently, offspring of diabetic mothers may experience lifelong impairment in cardiac function even without manifesting structural heart abnormalities. Studies indicate that newborns of diabetic mothers exhibit lower birth weight, cardiomyopathy, and reduced systolic and diastolic function. Although cardiac function improves postnatally and into adulthood, it reemerges in older offspring, particularly males [86,87]. Furthermore, offspring exposed to high blood glucose levels in utero also exhibit reduced tolerance to myocardial ischemia [88]. This indicates that early-life exposure to hyperglycemia profoundly impacts offspring cardiac health, with risks persisting not only during fetal development but potentially extending into adolescence and adulthood [89,90]. According to DOHaD theory, maternal hyperglycemia induces structural remodeling of the offspring heart (such as abnormal myocardial arrangement) that is largely irreversible in adulthood. However, the resulting functional and metabolic “setpoint” abnormalities (such as predisposition to hypertrophy and fibrosis) retain plasticity. Numerous animal studies have demonstrated that antioxidants effectively scavenge excess ROS, reverse certain epigenetic reprogramming events such as DNA hypermethylation, thereby significantly mitigating cardiac structural defects and enhancing functional compensation capacity in adulthood. For example, adding the antioxidant NAC to the drinking water of diabetic C57BL/6 mice resulted in significant rescue of ventricular septal defects (VSD) in offspring from diabetic dams, compared to the untreated diabetic group. However, this effect was not replicated in another group [26,38]. Additionally, zinc possesses the ability to reduce levels of lipid peroxidation, peroxides, and oxidized glutathione in developing hearts. In experiments with diabetic mice receiving zinc supplementation, OS decreased in embryonic hearts exposed to matDM, and the incidence of CHD in offspring was reduced [91]. Oral administration of sapropterin (a stable, orally active synthetic form of BH4) to pregnant C57BL/6 mice from E0.5 to E18.5 successfully reduced fetal coronary artery malformations from 50.0% to 20.6%, likely achieved by increasing eNOS activity and reducing OS [92]. These findings strongly suggest that ROS reduction represents a promising therapeutic approach for improving cardiac health in offspring of diabetic mothers [87].
6 Clinical Intervention and Management Strategies
6.1 Early Screening and Glycemic Control
Given the significant role of HIP in the development of CHD, early clinical intervention and gestational blood glucose management are crucial. First, it is recommended to conduct screening during early pregnancy or at the start of prenatal checkups to provide an opportunity window for subsequent dietary and lifestyle interventions. There is currently no consensus on early screening tools for existing diabetes: fasting blood glucose, random blood glucose, glycated hemoglobin, and the 75 g 2-h OGTT are all recommended as screening tools in one or more national guidelines Additionally, DNA methylation and maternal circulating miRNAs may serve as potential biomarkers, offering an additional layer of screening to enhance consensus. This would enable the detection of adverse maternal and fetal outcomes during early pregnancy, thereby providing more time to implement intervention strategies [87].
Second, managing gestational blood glucose levels to reduce the risk of CHD in offspring is the primary intervention strategy currently employed, with lifestyle interventions typically recommended as the first-line approach for mothers with diabetes, including dietary adjustments based on medical nutrition therapy, appropriate physical exercise, and blood glucose monitoring [6]. Research indicates that 70% to 85% of women diagnosed according to the American Diabetes Association (ADA) criteria can achieve glycemic targets through lifestyle interventions [93].
6.2 Selection of Pharmacological Therapy
When lifestyle interventions prove ineffective, intensified blood glucose monitoring should be implemented alongside dietary management and pharmacological therapy (insulin and/or oral hypoglycemic agents such as metformin and glibenclamide) to achieve glycemic normalization [6]. The American College of Obstetricians and Gynecologists and the ADA recommend insulin as the first-line medication due to its placental barrier, relative safety for the fetus, and reliable hypoglycemic efficacy. Metformin is indicated for patients who cannot use or refuse insulin [94]. The reason lies in the fact that metformin is effective in maintaining maternal blood glucose control and may help limit weight gain during pregnancy. Compared to insulin, its advantages lie in greater convenience of use and reduced insulin burden. However, metformin crosses the placenta and is present in clinically relevant concentrations in fetal and placental tissues (50%–100% of maternal concentrations). Simultaneously, exposure to metformin may affect the developing placental unit through direct or indirect pathways, and long-term data on offspring remain unknown. Concerns persist in several areas—such as small-for-gestational-age infants, body composition changes, and potential increased risk of certain types of fetal CHD—prompting hesitation [95,96,97]. Several recent meta-analyses have demonstrated that metformin offers more favorable pregnancy outcomes and the fastest rate of glycemic control, particularly among obese patients with GDM. However, it exhibits the lowest average glycemic control rate. Concurrently, exposure to metformin may influence anthropometric measurements in offspring; however, evidence regarding the risk of metabolic dysfunction remains inconclusive, and there is no evidence suggesting an impact on adverse cardiovascular outcomes or neurodevelopmental disorders later in life [96,97,98]. In summary, clinicians must weigh the potential benefits of using metformin during pregnancy against the uncertainty of its long-term effects on offspring health when managing blood glucose levels.
Additional studies have indicated that insulin analog therapy during early pregnancy significantly reduces the risk of congenital heart defects to 7.6‰, compared to 40.2‰ with human insulin alone and 35.5‰ with combined insulin therapy. These findings were observed in the offspring of mothers with GDM. Notably, insulin analog therapy is associated with a higher incidence of spontaneous abortion (1.5% vs. 1.0% for human insulin) [5]. Therefore, whether insulin analogs (both short- and long-acting) are safe alternatives to human insulin during pregnancy requires further investigation.
6.3 Integrated Management and Other Measures
Beyond early screening and blood glucose management, fundamental improvements in cardiovascular health “programming” of offspring must be achieved through known multiple synergistic mechanisms and signaling pathways to enable early prevention of CHD. First, improve the intrauterine metabolic environment and nutrient supply. Hyperglycemia causes excessive glucose, free fatty acids, and other substances to flood into the fetal circulation via the placenta, forcing the fetus to develop hyperinsulinemia and trigger abnormal metabolism. Early intervention, through precise management of blood glucose and lipids, restores nutrient supply to physiological ranges. This directly alleviates excessive strain on fetal metabolic organs—such as the pancreas, liver, and adipose tissue—laying the foundation for establishing normal energy metabolism balance. Research indicates that breastfeeding helps lower blood pressure and improve skeletal muscle function, thereby preventing insulin resistance and type 2 diabetes, while reducing children’s risk of noncommunicable diseases. This is associated with the long-chain polyunsaturated fatty acids in breast milk [99]. Secondly, reducing OS and chronic low-grade inflammation is crucial, as hyperglycemia is a potent OS inducer. It triggers excessive production of reactive ROS and pro-inflammatory cytokines (such as TNF-α and IL-6) at the maternal-fetal interface. These harmful substances can directly damage developing fetal vascular endothelial cells, impairing their function. Early supplementation with antioxidants and multivitamins holds potential for reducing the incidence of CHD and improving cardiac health in offspring [100]. Ultimately, the key lies in regulating epigenetic programming. Adverse prenatal environments can persistently alter the expression patterns of fetal genes associated with metabolic regulation, inflammatory responses, and vascular function—such as those involved in the TGFβ pathway, Wnt pathway, and insulin signaling pathway—through epigenetic mechanisms including DNA methylation and histone modifications. In the future, by leveraging advanced technologies such as single-cell scRNA-seq and organoid models, early identification and intervention may partially prevent or reverse these harmful “epigenetic memories.” This approach could optimize the developmental trajectory of cardiovascular systems in offspring at its source.
This review acknowledges several limitations. The precise mechanisms linking gestational hyperglycemia to CHD remain incompletely understood, with complex signaling pathways yet to be fully elucidated. Additionally, the lack of validated biomarkers for early risk prediction and limited data on novel interventions highlight significant research gaps. Furthermore, insufficient long-term follow-up studies on offspring exposed to gestational diabetes restrict our understanding of their cardiovascular health trajectory, underscoring the need for further investigation.
In summary, the management of hyperglycemia during pregnancy is a dynamic process requiring multidisciplinary collaboration among obstetrics, endocrinology, nutrition, and neonatology departments. Public awareness regarding the association between gestational hyperglycemia and offspring cardiac development should be enhanced. Women planning pregnancy should control their blood glucose levels prior to conception and optimize prenatal care through multidisciplinary collaboration to minimize the risk of pregnancy complications (Fig. 2). Furthermore, animal model research serves as a vital tool for understanding and exploring novel therapeutic approaches and should be more widely applied.
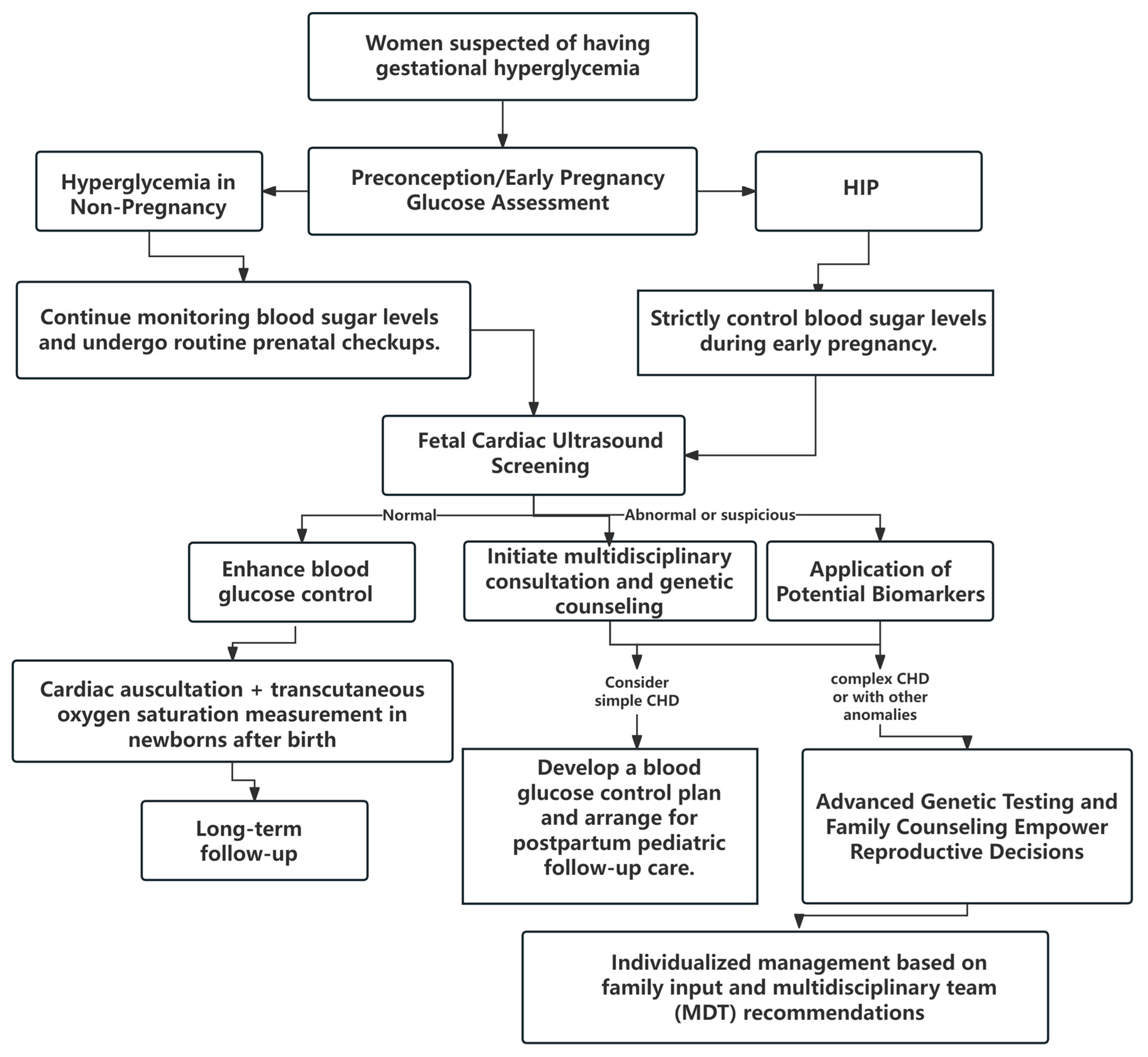
Figure 2: Clinical management flowchart. HIP, hyperglycemia in pregnancy; CHD, congenital heart disease.
7 Conclusion and Future Perspective
This review systematically summarizes the close association between HIP and CHD in offspring. This review elucidates the mechanisms by which maternal hyperglycemia affects fetal cardiac development, involving the interaction of multiple signaling pathways, including nitric oxide and Notch signaling, oxidative stress, TGFβ, and Wnt pathways, as well as the regulation of relevant genetic factors. Cardiac development requires a complex sequence of biological signals, cell-cell interactions, niche specification of cardiac progenitor cells, and the circulation of the heart tube.
Although many underlying mechanisms linking gestational hyperglycemia to CHD remain incompletely understood, existing research indicates that a hyperglycemic environment during pregnancy exerts long-term effects on offspring’s cardiovascular function. These include cardiac dysfunctions, such as impaired ventricular filling, increased heart rate, outflow tract obstruction, and reduced ventricular compliance, along with persistent alterations in cardiac geometry. Such conditions may lead to the premature onset of cardiovascular diseases in offspring.
Therefore, in the foreseeable future, we need to focus on the following research directions: (1) elucidating the interaction networks among signaling pathways and continuously exploring the underlying mechanisms of action; (2) identifying biomarkers capable of predicting CHD risk at an early stage; (3) exploring the safety and efficacy of novel intervention strategies, such as antioxidant therapy; and (4) strengthening long-term follow-up of offspring with GDM to clarify their cardiovascular health trajectory. Through the close integration of basic research and clinical practice, it is hoped that the adverse effects of gestational hyperglycemia on offspring heart health can ultimately be mitigated.
Acknowledgement:
Funding Statement: This study was kindly supported by the Hainan Provincial Health Science and Technology Innovation Joint Project Fund (No.: WSJK2024QN054), the Hainan Provincial Clinical Medical Center (No.: QWYH202175), and the Excellent Talent Team of Hainan Province (No.: QRCBT202121).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Jiafei He, Renwei Chen. Data collection: Jiafei He, Ailixiati Alifu. Analysis and interpretation of results: Jiafei He, Haifan Wang. Draft manuscript preparation: Jiafei He, Renwei Chen, Haifan Wang, Ailixiati Alifu. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: The authors confirm that the data supporting the findings of this study are available in this article.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest.
References
1. Liu Y , Chen S , Zühlke L , Black GC , Choy MK , Li N , et al. Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies. Int J Epidemiol. 2019; 48( 2): 455– 63. doi:10.1093/ije/dyz009. [Google Scholar] [CrossRef]
2. Zhang S , Zhang B , Wu J , Luo J , Shi H , Qi J , et al. The prevalence of congenital heart disease among school-age children in China: a meta-analysis and systematic review. Congenit Heart Dis. 2023; 18( 2): 127– 50. doi:10.32604/chd.2023.025616. [Google Scholar] [CrossRef]
3. Wahab RJ , Jaddoe VWV , Roest AAW , Toemen L , Gaillard R . Associations of maternal glycemia in the first half of pregnancy with alterations in cardiac structure and function in childhood. Diabetes Care. 2020; 43( 9): 2272– 80. doi:10.2337/dc19-2580. [Google Scholar] [CrossRef]
4. Nagasawa M , Ikehara S , Aochi Y , Tanigawa K , Kitamura T , Sobue T , et al. Maternal diabetes and risk of offspring congenital heart diseases: the Japan Environment and Children’s Study. Environ Health Prev Med. 2024; 29: 23. doi:10.1265/ehpm.23-00358. [Google Scholar] [CrossRef]
5. Chen ZY , Mao SF , Guo LH , Qin J , Yang LX , Liu Y . Effect of maternal pregestational diabetes mellitus on congenital heart diseases. World J Pediatr. 2023; 19( 4): 303– 14. doi:10.1007/s12519-022-00582-w. [Google Scholar] [CrossRef]
6. Tocantins C , Diniz MS , Grilo LF , Pereira SP . The birth of cardiac disease: mechanisms linking gestational diabetes mellitus and early onset of cardiovascular disease in offspring. WIREs Mech Dis. 2022; 14( 4): e1555. doi:10.1002/wsbm.1555. [Google Scholar] [CrossRef]
7. Saeedi P , Petersohn I , Salpea P , Malanda B , Karuranga S , Unwin N , et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. 2019; 157: 107843. doi:10.1016/j.diabres.2019.107843. [Google Scholar] [CrossRef]
8. Choudhury AA , Devi Rajeswari V . Gestational diabetes mellitus—A metabolic and reproductive disorder. Biomed Pharmacother. 2021; 143: 112183. doi:10.1016/j.biopha.2021.112183. [Google Scholar] [CrossRef]
9. Wang M , Zhang TH , Li Y , Chen X , Zhang Q , Zheng Y , et al. Atractylenolide-I alleviates hyperglycemia-induced heart developmental malformations through direct and indirect modulation of the STAT3 pathway. Phytomedicine. 2024; 129: 155698. doi:10.1016/j.phymed.2024.155698. [Google Scholar] [CrossRef]
10. Cheng Y , Gu WR . Metabolic changes and hyperglycemia in pregnancy. Chin J Pract Gynecol Obstet. 2025; 41( 4): 392– 6. (In Chinese). [Google Scholar]
11. Juan J , Shu XY , Yang HX . Changes in the cognition of hyperglycemia in pregnancy. Chin J Pract Gynecol Obstet. 2025; 41( 4): 385– 8. (In Chinese). doi:10.19538/j.fk2025040101. [Google Scholar] [CrossRef]
12. Antar SA , Ashour NA , Sharaky M , Khattab M , Ashour NA , Zaid RT , et al. Diabetes mellitus: classification, mediators, and complications; A gate to identify potential targets for the development of new effective treatments. Biomed Pharmacother. 2023; 168: 115734. doi:10.1016/j.biopha.2023.115734. [Google Scholar] [CrossRef]
13. Owen MD , Kennedy MG , Quilang RC , Scott EM , Forbes K . The role of microRNAs in pregnancies complicated by maternal diabetes. Clin Sci. 2024; 138( 18): 1179– 207. doi:10.1042/CS20230681. [Google Scholar] [CrossRef]
14. Brito Nunes C , Borges MC , Freathy RM , Lawlor DA , Qvigstad E , Evans DM , et al. Understanding the genetic landscape of gestational diabetes: insights into the causes and consequences of elevated glucose levels in pregnancy. Metabolites. 2024; 14( 9): 508. doi:10.3390/metabo14090508. [Google Scholar] [CrossRef]
15. Galluzzo RN , Da Correggio KS , von Wangenheim A , Werner H , Bravo-Valenzuela NJ , Júnior EA , et al. Maternal diabetes mellitus and congenital heart diseases: systematic review. Congenit Heart Dis. 2025; 20( 1): 89– 101. doi:10.32604/chd.2025.063014. [Google Scholar] [CrossRef]
16. Choudhury TZ , Majumdar U , Basu M , Garg V . Impact of maternal hyperglycemia on cardiac development: insights from animal models. Genesis. 2021; 59( 11): e23449. doi:10.1002/dvg.23449. [Google Scholar] [CrossRef]
17. Papazoglou AS , Moysidis DV , Panagopoulos P , Kaklamanos EG , Tsagkaris C , Vouloagkas I , et al. Maternal diabetes mellitus and its impact on the risk of delivering a child with congenital heart disease: a systematic review and meta-analysis. J Matern Fetal Neonatal Med. 2022; 35( 25): 7685– 94. doi:10.1080/14767058.2021.1960968. [Google Scholar] [CrossRef]
18. Maduro C , de Castro LF , Moleiro ML , Guedes-Martins L . Pregestational diabetes and congenital heart defects. Rev Bras Ginecol Obstet. 2022; 44( 10): 953– 61. doi:10.1055/s-0042-1755458. [Google Scholar] [CrossRef]
19. Morgan SC , Relaix F , Sandell LL , Loeken MR . Oxidative stress during diabetic pregnancy disrupts cardiac neural crest migration and causes outflow tract defects. Birth Defects Res A Clin Mol Teratol. 2008; 82( 6): 453– 63. doi:10.1002/bdra.20457. [Google Scholar] [CrossRef]
20. Zhao M , Diao J , Huang P , Li J , Li Y , Yang Y , et al. Association of maternal diabetes mellitus and polymorphisms of the NKX2.5 gene in children with congenital heart disease: a single centre-based case-control study. J Diabetes Res. 2020; 2020: 3854630. doi:10.1155/2020/3854630. [Google Scholar] [CrossRef]
21. Asoglu MR , Gabbay-Benziv R , Turan OM , Turan S . Exposure of the developing heart to diabetic environment and early cardiac assessment: a review. Echocardiography. 2018; 35( 2): 244– 57. doi:10.1111/echo.13811. [Google Scholar] [CrossRef]
22. Jin YM , Zhao SZ , Zhang ZL , Chen Y , Cheng X , Chuai M , et al. High glucose level induces cardiovascular dysplasia during early embryo development. Exp Clin Endocrinol Diabetes. 2019; 127( 9): 590– 7. doi:10.1055/s-0043-109696. [Google Scholar] [CrossRef]
23. Rousseau-Ralliard D , Couturier-Tarrade A , Thieme R , Brat R , Rolland A , Boileau P , et al. A short periconceptional exposure to maternal type-1 diabetes is sufficient to disrupt the feto-placental phenotype in a rabbit model. Mol Cell Endocrinol. 2019; 480: 42– 53. doi:10.1016/j.mce.2018.10.010. [Google Scholar] [CrossRef]
24. Basu M , Garg V . Maternal hyperglycemia and fetal cardiac development: clinical impact and underlying mechanisms. Birth Defects Res. 2018; 110( 20): 1504– 16. doi:10.1002/bdr2.1435. [Google Scholar] [CrossRef]
25. Nakano H , Minami I , Braas D , Pappoe H , Wu X , Sagadevan A , et al. Glucose inhibits cardiac muscle maturation through nucleotide biosynthesis. eLife. 2017; 6: e29330. doi:10.7554/eLife.29330. [Google Scholar] [CrossRef]
26. Moazzen H , Lu X , Ma NL , Velenosi TJ , Urquhart BL , Wisse LJ , et al. N-Acetylcysteine prevents congenital heart defects induced by pregestational diabetes. Cardiovasc Diabetol. 2014; 13: 46. doi:10.1186/1475-2840-13-46. [Google Scholar] [CrossRef]
27. Øyen N , Diaz LJ , Leirgul E , Boyd HA , Priest J , Mathiesen ER , et al. Prepregnancy diabetes and offspring risk of congenital heart disease: a nationwide cohort study. Circulation. 2016; 133( 23): 2243– 53. doi:10.1161/circulationaha.115.017465. [Google Scholar] [CrossRef]
28. Ibrahim S , Gaborit B , Lenoir M , Collod-Beroud G , Stefanovic S . Maternal pre-existing diabetes: a non-inherited risk factor for congenital cardiopathies. Int J Mol Sci. 2023; 24( 22): 16258. doi:10.3390/ijms242216258. [Google Scholar] [CrossRef]
29. Liu S , Joseph KS , Lisonkova S , Rouleau J , Van den Hof M , Sauve R , et al. Association between maternal chronic conditions and congenital heart defects: a population-based cohort study. Circulation. 2013; 128( 6): 583– 9. doi:10.1161/CIRCULATIONAHA.112.001054. [Google Scholar] [CrossRef]
30. Srivastava D . Making or breaking the heart: from lineage determination to morphogenesis. Cell. 2006; 126( 6): 1037– 48. doi:10.1016/j.cell.2006.09.003. [Google Scholar] [CrossRef]
31. Nishino T , Ranade SS , Pelonero A , van Soldt BJ , Ye L , Alexanian M , et al. Single cell multimodal analyses reveal epigenomic and transcriptomic basis for birth defects in maternal diabetes. Nat Cardiovasc Res. 2023; 2( 12): 1190– 203. doi:10.1038/s44161-023-00367-y. [Google Scholar] [CrossRef]
32. Thompson E , Hensley J , Taylor RS . Effect of high glucose on embryological development of zebrafish, brachyodanio, rerio through Wnt pathway. Int J Mol Sci. 2024; 25( 17): 9443. doi:10.3390/ijms25179443. [Google Scholar] [CrossRef]
33. Little MH , Combes AN . Kidney organoids: accurate models or fortunate accidents. Genes Dev. 2019; 33( 19–20): 1319– 45. doi:10.1101/gad.329573.119. [Google Scholar] [CrossRef]
34. Hofbauer P , Jahnel SM , Papai N , Giesshammer M , Deyett A , Schmidt C , et al. Cardioids reveal self-organizing principles of human cardiogenesis. Cell. 2021; 184( 12): 3299– 317.e22. doi:10.1016/j.cell.2021.04.034. [Google Scholar] [CrossRef]
35. Nees SN , Chung WK . Genetic basis of human congenital heart disease. Cold Spring Harb Perspect Biol. 2020; 12( 9): a036749. doi:10.1101/cshperspect.a036749. [Google Scholar] [CrossRef]
36. Suda-Całus M , Dąbrowska K , Gulczyńska E . Infant of a diabetic mother: clinical presentation, diagnosis and treatment. Pediatr Endocrinol Diabetes Metab. 2024; 30( 1): 36– 41. doi:10.5114/pedm.2024.137891. [Google Scholar] [CrossRef]
37. Luo L , Huang P , Wang TT , Zhao LJ , Ye ZW , Zhang SM , et al. Association of maternal diabetes mellitus and UCP2 gene polymorphisms with congenital heart disease in offspring: a case-control study. Chin J Contemp Pediatr. 2020; 22( 10): 1092– 9. doi:10.7499/j.issn.1008-8830.2004011. [Google Scholar] [CrossRef]
38. Basu M , Zhu JY , LaHaye S , Majumdar U , Jiao K , Han Z , et al. Epigenetic mechanisms underlying maternal diabetes-associated risk of congenital heart disease. JCI Insight. 2017; 2( 20): e95085. doi:10.1172/jci.insight.95085. [Google Scholar] [CrossRef]
39. Weng H , Li X , Reece EA , Yang P . SOD1 suppresses maternal hyperglycemia-increased iNOS expression and consequent nitrosative stress in diabetic embryopathy. Am J Obstet Gynecol. 2012; 206( 5): 448.e1– 7. doi:10.1016/j.ajog.2012.02.011. [Google Scholar] [CrossRef]
40. Xiang H , Zhuang J , Bao L , Shi Y . Variants and molecular mechanism of NOTCH1 in congenital heart disease. Congenit Heart Dis. 2025; 20( 2): 245– 63. doi:10.32604/chd.2025.064366. [Google Scholar] [CrossRef]
41. Gessert S , Kühl M . The multiple phases and faces of Wnt signaling during cardiac differentiation and development. Circ Res. 2010; 107( 2): 186– 99. doi:10.1161/CIRCRESAHA.110.221531. [Google Scholar] [CrossRef]
42. Wang F , Fisher SA , Zhong J , Wu Y , Yang P . Superoxide dismutase 1 in vivo ameliorates maternal diabetes mellitus-induced apoptosis and heart defects through restoration of impaired Wnt signaling. Circ Cardiovasc Genet. 2015; 8( 5): 665– 76. doi:10.1161/CIRCGENETICS.115.001138. [Google Scholar] [CrossRef]
43. Misra K , Matise MP . A critical role for sFRP proteins in maintaining caudal neural tube closure in mice via inhibition of BMP signaling. Dev Biol. 2010; 337( 1): 74– 83. doi:10.1016/j.ydbio.2009.10.015. [Google Scholar] [CrossRef]
44. Schleiffarth JR , Person AD , Martinsen BJ , Sukovich DJ , Neumann A , Baker CVH , et al. Wnt5a is required for cardiac outflow tract septation in mice. Pediatr Res. 2007; 61( 4): 386– 91. doi:10.1203/pdr.0b013e3180323810. [Google Scholar] [CrossRef]
45. Wang F , Reece EA , Yang P . Oxidative stress is responsible for maternal diabetes-impaired transforming growth factor beta signaling in the developing mouse heart. Am J Obstet Gynecol. 2015; 212( 5): 650.e1– 11. doi:10.1016/j.ajog.2015.01.014. [Google Scholar] [CrossRef]
46. Song H , Zhuang L , Xu X , Shi J , Hu W , Liu Z , et al. MCC regulator of WNT signaling pathway (MCC) is a podocyte essential gene. Front Med. 2021; 8: 777563. doi:10.3389/fmed.2021.777563. [Google Scholar] [CrossRef]
47. Kolesova H , Hrabalova P , Bohuslavova R , Abaffy P , Fabriciova V , Sedmera D , et al. Reprogramming of the developing heart by Hif1a-deficient sympathetic system and maternal diabetes exposure. Front Endocrinol. 2024; 15: 1344074. doi:10.3389/fendo.2024.1344074. [Google Scholar] [CrossRef]
48. Hagey DW , Topcic D , Kee N , Reynaud F , Bergsland M , Perlmann T , et al. CYCLIN-B1/2 and-D1 act in opposition to coordinate cortical progenitor self-renewal and lineage commitment. Nat Commun. 2020; 11( 1): 2898. doi:10.1038/s41467-020-16597-8. [Google Scholar] [CrossRef]
49. Bohuslavova R , Skvorova L , Sedmera D , Semenza GL , Pavlinkova G . Increased susceptibility of HIF-1α heterozygous-null mice to cardiovascular malformations associated with maternal diabetes. J Mol Cell Cardiol. 2013; 60: 129– 41. doi:10.1016/j.yjmcc.2013.04.015. [Google Scholar] [CrossRef]
50. Han SS , Wang G , Jin Y , Ma ZL , Jia WJ , Wu X , et al. Investigating the mechanism of hyperglycemia-induced fetal cardiac hypertrophy. PLoS One. 2015; 10( 9): e0139141. doi:10.1371/journal.pone.0139141. [Google Scholar] [CrossRef]
51. Cerychova R , Bohuslavova R , Papousek F , Sedmera D , Abaffy P , Benes V , et al. Adverse effects of Hif1a mutation and maternal diabetes on the offspring heart. Cardiovasc Diabetol. 2018; 17( 1): 68. doi:10.1186/s12933-018-0713-0. [Google Scholar] [CrossRef]
52. Schraps N , Tirre M , Pyschny S , Reis A , Schlierbach H , Seidl M , et al. Cardiomyocyte maturation alters molecular stress response capacities and determines cell survival upon mitochondrial dysfunction. Free Radic Biol Med. 2024; 213: 248– 65. doi:10.1016/j.freeradbiomed.2024.01.034. [Google Scholar] [CrossRef]
53. Yang P , Chen X , Kaushal S , Reece EA , Yang P . High glucose suppresses embryonic stem cell differentiation into cardiomyocytes: high glucose inhibits ES cell cardiogenesis. Stem Cell Res Ther. 2016; 7( 1): 187. doi:10.1186/s13287-016-0446-5. [Google Scholar] [CrossRef]
54. Ramaccini D , Montoya-Uribe V , Aan FJ , Modesti L , Potes Y , Wieckowski MR , et al. Mitochondrial function and dysfunction in dilated cardiomyopathy. Front Cell Dev Biol. 2021; 8: 624216. doi:10.3389/fcell.2020.624216. [Google Scholar] [CrossRef]
55. Zhou B , Tian R . Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Investig. 2018; 128( 9): 3716– 26. doi:10.1172/JCI120849. [Google Scholar] [CrossRef]
56. Murphy E , Liu JC . Mitochondrial calcium and reactive oxygen species in cardiovascular disease. Cardiovasc Res. 2023; 119( 5): 1105– 16. doi:10.1093/cvr/cvac134. [Google Scholar] [CrossRef]
57. Kwon H , Jung YJ , Lee Y , Son GH , Kim HO , Maeng YS , et al. Impaired angiogenic function of fetal endothelial progenitor cells via PCDH10 in gestational diabetes mellitus. Int J Mol Sci. 2023; 24( 22): 16082. doi:10.3390/ijms242216082. [Google Scholar] [CrossRef]
58. Lock MC , Botting KJ , Tellam RL , Brooks D , Morrison JL . Adverse intrauterine environment and cardiac miRNA expression. Int J Mol Sci. 2017; 18( 12): 2628. doi:10.3390/ijms18122628. [Google Scholar] [CrossRef]
59. Lizárraga D , Gómez-Gil B , García-Gasca T , Ávalos-Soriano A , Casarini L , Salazar-Oroz A , et al. Gestational diabetes mellitus: genetic factors, epigenetic alterations, and microbial composition. Acta Diabetol. 2024; 61( 1): 1– 17. doi:10.1007/s00592-023-02176-y. [Google Scholar] [CrossRef]
60. Di Pietrantonio N , Sánchez-Ceinos J , Shumliakivska M , Rakow A , Mandatori D , Di Tomo P , et al. The inflammatory and oxidative phenotype of gestational diabetes is epigenetically transmitted to the offspring: role of methyltransferase MLL1-induced H3K4me3. Eur Heart J. 2024; 45( 48): 5171– 85. doi:10.1093/eurheartj/ehae688. [Google Scholar] [CrossRef]
61. Xiao Y , Su X , Huang W , Zhang J , Peng C , Huang H , et al. Role of S-adenosylhomocysteine in cardiovascular disease and its potential epigenetic mechanism. Int J Biochem Cell Biol. 2015; 67: 158– 66. doi:10.1016/j.biocel.2015.06.015. [Google Scholar] [CrossRef]
62. Shan ZX , Lin QX , Deng CY , Zhu JN , Mai LP , Liu JL , et al. miR-1/miR-206 regulate Hsp60 expression contributing to glucose-mediated apoptosis in cardiomyocytes. FEBS Lett. 2010; 584( 16): 3592– 600. doi:10.1016/j.febslet.2010.07.027. [Google Scholar] [CrossRef]
63. He L , Wang X , Jin Y , Xu W , Guan Y , Wu J , et al. Identification and validation of the miRNA-mRNA regulatory network in fetoplacental arterial endothelial cells of gestational diabetes mellitus. Bioengineered. 2021; 12( 1): 3503– 15. doi:10.1080/21655979.2021.1950279. [Google Scholar] [CrossRef]
64. Maloyan A , Muralimanoharan S , Huffman S , Cox LA , Nathanielsz PW , Myatt L , et al. Identification and comparative analyses of myocardial miRNAs involved in the fetal response to maternal obesity. Physiol Genomics. 2013; 45( 19): 889– 900. doi:10.1152/physiolgenomics.00050.2013. [Google Scholar] [CrossRef]
65. Howe CG , Cox B , Fore R , Jungius J , Kvist T , Lent S , et al. Maternal gestational diabetes mellitus and newborn DNA methylation: findings from the pregnancy and childhood epigenetics consortium. Diabetes Care. 2020; 43( 1): 98– 105. doi:10.2337/dc19-0524. [Google Scholar] [CrossRef]
66. Sato H , Leonardi ML , Roberti SL , Jawerbaum A , Higa R . Maternal diabetes increases FOXO1 activation during embryonic cardiac development. Mol Cell Endocrinol. 2023; 575: 111999. doi:10.1016/j.mce.2023.111999. [Google Scholar] [CrossRef]
67. Chu AHY , Godfrey KM . Gestational diabetes mellitus and developmental programming. Ann Nutr Metab. 2020; 76( Suppl 3): 4– 15. doi:10.1159/000509902. [Google Scholar] [CrossRef]
68. Wu Q , Ni X . ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr Drug Targets. 2015; 16( 1): 13– 9. doi:10.2174/1389450116666150113121054. [Google Scholar] [CrossRef]
69. Pillai VB , Sundaresan NR , Gupta MP . Regulation of Akt signaling by sirtuins: its implication in cardiac hypertrophy and aging. Circ Res. 2014; 114( 2): 368– 78. doi:10.1161/CIRCRESAHA.113.300536. [Google Scholar] [CrossRef]
70. Chen Z , Gong L , Zhang P , Li Y , Liu B , Zhang L , et al. Epigenetic down-regulation of sirt 1 via DNA methylation and oxidative stress signaling contributes to the gestational diabetes mellitus-induced fetal programming of heart ischemia-sensitive phenotype in late life. Int J Biol Sci. 2019; 15( 6): 1240– 51. doi:10.7150/ijbs.33044. [Google Scholar] [CrossRef]
71. Hromadnikova I , Kotlabova K , Dvorakova L , Krofta L , Sirc J . Substantially altered expression profile of diabetes/cardiovascular/cerebrovascular disease associated microRNAs in children descending from pregnancy complicated by gestational diabetes mellitus—one of several possible reasons for an increased cardiovascular risk. Cells. 2020; 9( 6): 1557. doi:10.3390/cells9061557. [Google Scholar] [CrossRef]
72. Hou Q , Yan F , Li X , Liu H , Yang X , Dong X . ATP5me alleviates high glucose-induced myocardial cell injury. Int Immunopharmacol. 2024; 129: 111626. doi:10.1016/j.intimp.2024.111626. [Google Scholar] [CrossRef]
73. Cade WT , Levy PT , Tinius RA , Patel MD , Choudhry S , Holland MR , et al. Markers of maternal and infant metabolism are associated with ventricular dysfunction in infants of obese women with type 2 diabetes. Pediatr Res. 2017; 82( 5): 768– 75. doi:10.1038/pr.2017.140. [Google Scholar] [CrossRef]
74. Chen A , Tan B , Du R , Chong YS , Zhang C , Koh AS , et al. Gestational diabetes mellitus and development of intergenerational overall and subtypes of cardiovascular diseases: a systematic review and meta-analysis. Cardiovasc Diabetol. 2024; 23( 1): 320. doi:10.1186/s12933-024-02416-7. [Google Scholar] [CrossRef]
75. Kolac UK , Kurek Eken M , Ünübol M , Donmez Yalcin G , Yalcin A . The effect of gestational diabetes on the expression of mitochondrial fusion proteins in placental tissue. Placenta. 2021; 115: 106– 14. doi:10.1016/j.placenta.2021.09.015. [Google Scholar] [CrossRef]
76. Zhao Q , Sun Q , Zhou L , Liu K , Jiao K . Complex regulation of mitochondrial function during cardiac development. J Am Heart Assoc. 2019; 8( 13): e012731. doi:10.1161/JAHA.119.012731. [Google Scholar] [CrossRef]
77. Kulkarni A , Li L , Craft M , Nanda M , Lorenzo JMM , Danford D , et al. Fetal myocardial deformation in maternal diabetes mellitus and obesity. Ultrasound Obstet Gynecol. 2017; 49( 5): 630– 6. doi:10.1002/uog.15971. [Google Scholar] [CrossRef]
78. Dearden L , Bouret SG , Ozanne SE . Nutritional and developmental programming effects of insulin. J Neuroendocrinol. 2021; 33( 4): e12933. doi:10.1111/jne.12933. [Google Scholar] [CrossRef]
79. Patey O , Carvalho JS , Thilaganathan B . Perinatal changes in fetal cardiac geometry and function in diabetic pregnancy at term. Ultrasound Obstet Gynecol. 2019; 54( 5): 634– 42. doi:10.1002/uog.20187. [Google Scholar] [CrossRef]
80. Lee-Tannock A , Hay K , Kumar S . Differences in biomarkers of cardiac dysfunction in cord blood between normal pregnancies and pregnancies complicated by maternal diabetes. Aust N Z J Obstet Gynaecol. 2022; 62( 1): 79– 85. doi:10.1111/ajo.13415. [Google Scholar] [CrossRef]
81. Mirzakhani H . From womb to wellness: early environmental exposures, cord blood DNA methylation and disease origins. Epigenomics. 2024; 16( 17): 1175– 83. doi:10.1080/17501911.2024.2390823. [Google Scholar] [CrossRef]
82. Hoff K , Lemme M , Kahlert AK , Runde K , Audain E , Schuster D , et al. DNA methylation profiling allows for characterization of atrial and ventricular cardiac tissues and hiPSC-CMs. Clin Epigenetics. 2019; 11( 1): 89. doi:10.1186/s13148-019-0679-0. [Google Scholar] [CrossRef]
83. Zhu MJ , Ma XY , Ding PC , Tang HF , Peng R , Lu L , et al. Novel mutations of AXIN2 identified in a Chinese congenital heart disease cohort. J Hum Genet. 2019; 64( 5): 427– 35. doi:10.1038/s10038-019-0572-x. [Google Scholar] [CrossRef]
84. Krolevets M , Ten Cate V , Prochaska JH , Schulz A , Rapp S , Tenzer S , et al. DNA methylation and cardiovascular disease in humans: a systematic review and database of known CpG methylation sites. Clin Epigenetics. 2023; 15( 1): 56. doi:10.1186/s13148-023-01468-y. [Google Scholar] [CrossRef]
85. Huluta I , Wright A , Cosma LM , Hamed K , Nicolaides KH , Charakida M . Fetal cardiac function at midgestation in women who subsequently develop gestational diabetes. JAMA Pediatr. 2023; 177( 7): 718– 25. doi:10.1001/jamapediatrics.2023.1174. [Google Scholar] [CrossRef]
86. Louwagie EJ , Larsen TD , Wachal AL , Gandy TCT , Eclov JA , Rideout TC , et al. Age and sex influence mitochondria and cardiac health in offspring exposed to maternal glucolipotoxicity. iScience. 2020; 23( 11): 101746. doi:10.1016/j.isci.2020.101746. [Google Scholar] [CrossRef]
87. Ren Z , Luo S , Cui J , Tang Y , Huang H , Ding G . Research progress of maternal metabolism on cardiac development and function in offspring. Nutrients. 2023; 15( 15): 3388. doi:10.3390/nu15153388. [Google Scholar] [CrossRef]
88. Gao L , Zhao YC , Liang Y , Lin XH , Tan YJ , Wu DD , et al. The impaired myocardial ischemic tolerance in adult offspring of diabetic pregnancy is restored by maternal melatonin treatment. J Pineal Res. 2016; 61( 3): 340– 52. doi:10.1111/jpi.12351. [Google Scholar] [CrossRef]
89. Yu Y , Arah OA , Liew Z , Cnattingius S , Olsen J , Sørensen HT , et al. Maternal diabetes during pregnancy and early onset of cardiovascular disease in offspring: population based cohort study with 40 years of follow-up. BMJ. 2019; 367: l6398. doi:10.1136/bmj.l6398. [Google Scholar] [CrossRef]
90. Aguilera J , Semmler J , Anzoategui S , Zhang H , Nicolaides KH , Charakida M . Cardiac function in gestational diabetes mellitus: a longitudinal study from fetal life to infancy. BJOG. 2021; 128( 2): 272– 9. doi:10.1111/1471-0528.16434. [Google Scholar] [CrossRef]
91. Kumar SD , Vijaya M , Samy RP , Dheen ST , Ren M , Watt F , et al. Zinc supplementation prevents cardiomyocyte apoptosis and congenital heart defects in embryos of diabetic mice. Free Radic Biol Med. 2012; 53( 8): 1595– 606. doi:10.1016/j.freeradbiomed.2012.07.008. [Google Scholar] [CrossRef]
92. Engineer A , Lim YJ , Lu X , Kim MY , Norozi K , Feng Q . Sapropterin reduces coronary artery malformation in offspring of pregestational diabetes mice. Nitric Oxide. 2020; 94: 9– 18. doi:10.1016/j.niox.2019.10.002. [Google Scholar] [CrossRef]
93. Johns EC , Denison FC , Norman JE , Reynolds RM . Gestational diabetes mellitus: mechanisms, treatment, and complications. Trends Endocrinol Metab. 2018; 29( 11): 743– 54. doi:10.1016/j.tem.2018.09.004. [Google Scholar] [CrossRef]
94. Bolte E , Dean T , Garcia B , Seferovic MD , Sauter K , Hummel G , et al. Initiation of metformin in early pregnancy results in fetal bioaccumulation, growth restriction, and renal dysmorphology in a primate model. Am J Obstet Gynecol. 2024; 231( 3): 352.e1– 352.e16. doi:10.1016/j.ajog.2024.06.002. [Google Scholar] [CrossRef]
95. Helle E , Priest JR . Maternal obesity and diabetes mellitus as risk factors for congenital heart disease in the offspring. J Am Heart Assoc. 2020; 9( 8): e011541. doi:10.1161/JAHA.119.011541. [Google Scholar] [CrossRef]
96. Malek R , Davis SN . Is metformin safe in pregnancy: a focus on offspring outcomes. Expert Opin Drug Saf. 2025; 24( 1): 5– 8. doi:10.1080/14740338.2024.2424410. [Google Scholar] [CrossRef]
97. Tarry-Adkins JL , Aiken CE , Ozanne SE . Neonatal, infant, and childhood growth following metformin versus insulin treatment for gestational diabetes: a systematic review and meta-analysis. PLoS Med. 2019; 16( 8): e1002848. doi:10.1371/journal.pmed.1002848. [Google Scholar] [CrossRef]
98. Mayadunne T , Saadati S , Asmelash D , Mason T , Vanky E , Teede H , et al. Long-term effects of metformin on offspring health: a review of current evidence and future directions. Diabetes Obes Metab. 2025; 27( Suppl 3): 48– 63. doi:10.1111/dom.16418. [Google Scholar] [CrossRef]
99. Pathirana MM , Ali A , Lassi ZS , Arstall MA , Roberts CT , Andraweera PH . Protective influence of breastfeeding on cardiovascular risk factors in women with previous gestational diabetes mellitus and their children: a systematic review and meta-analysis. J Hum Lact. 2022; 38( 3): 501– 12. doi:10.1177/08903344211034779. [Google Scholar] [CrossRef]
100. Lin X , Yang P , Reece EA , Yang P . Pregestational type 2 diabetes mellitus induces cardiac hypertrophy in the murine embryo through cardiac remodeling and fibrosis. Am J Obstet Gynecol. 2017; 217( 2): 216.e1– 216.e13. doi:10.1016/j.ajog.2017.04.008. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools