 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

A Comprehensive Insight to the Physical Properties of NaCuZrS3
1 College of Electronic and Information Engineering, Yangtze Normal University, Fuling, Chongqing, China
2 School of Physics, Electronics and Intelligent Manufacturing, Huaihua University, Huaihua, China
3 School of Materials Engineering, Yancheng Institute of Technology, Yancheng, China
* Corresponding Author: Hongli Guo. Email:
Chalcogenide Letters 2026, 23(2), 3 https://doi.org/10.32604/cl.2026.076290
Received 18 November 2025; Accepted 19 January 2026; Issue published 28 February 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
To gain deeper insights into the characteristics of orthorhombic NaCuZrS3, this study investigates its mechanical, electronic and thermal properties using a theoretical approach grounded in density functional theory (DFT). The computed structural parameters align closely with experimental data. Additionally, its elastic constants and moduli show an increasing trend as pressure increases. It confirms that this material is brittle at 0 GPa, but as the pressure increases, it transforms into a ductile material after 10 GPa. Through computational analysis, the electronic properties for NaCuZrS3 are determined. The material is an indirect bandgap semiconductor with a bandgap of 0.61 eV. The elastic constants were used to determine mechanical stability. Furthermore, thermodynamic properties were calculated and analyzed the physical and chemical nature of NaCuZrS3.Keywords
Generally, copper-containing chalcogenides have recently attracted considerable interest due to their promising thermoelectric applications, owing to their low thermal conductivity. They are also applicable in areas such as gas sensing, solar cell design and photocatalysis [1,2,3]. Now, lithium-ion batteries have seen increasingly widespread application. However, safety concerns are drawing more attention owing to the use of combustible organic liquid electrolytes. All-solid-state batteries (ASSBs) have become the most competitive alternative due to their improved safety, greater energy density, extended cycle life, and superior capacity [4,5]. Compared to lithium-ion ASSBs, sodium-ion ASSBs have lower costs, making them more suitable for large-scale applications. NaCuZrS3 is an excellent sodium battery material, composed of abundant, earth-rich, and environmentally friendly elements. This compound adopts the orthorhombic KCuZrS3-type crystal structure and is assigned to the Cmcm space group [6,7]. The performance of this material is excellent, but its mechanical and thermodynamic properties are not yet fully developed. The orthorhombic structures with Cmcm was not discussed. Unfortunately, the influence of pressure on the structural, mechanical and thermodynamic properties of orthorhombic NaCuZrS3 is unknown. To explore the high pressure behavior, in this paper, we report the results of calculations of NaCuZrS3 based on DFT. The correctness of first-principles calculations has been verified [8,9,10,11,12,13,14]. The findings presented in this paper will offer fundamental information for the future utilization of NaCuZrS3. The purpose of this paper is to fill in this gap. This is accomplished by calculating the structural, electronic and elastic properties of the NaCuZrS3 using a first-principle method. The general trends in crystal structures, elastic and thermodynamic properties of NaCuZrS3 was analyzed in detail.
We used CASTEP within the Material Studio package [15,16]. The exchange-correlation functional employed was the Generalized Gradient Approximation (GGA-PBE), and the pseudopotential used was the OTFG ultrasoft pseudopotential [17,18,19]. For structural relaxation calculations, the plane-wave energy cutoff in reciprocal k-space was set to 600 eV, and the Brillouin zone k-points were set to a 5 × 5 × 5 mesh. During the calculations, the tolerances of the self-consistent field (SCF), energy, maximum forces per atom and maximum displacement were 5 × 10−7 eV/atom, 5 × 10−6 eV, 0.01 eV/Å and 5 × 10−4 Å, respectively.
NaCuZrS3 (Fig. 1) is a chalcogenide with a structure similar to post-perovskite, featuring an orthorhombic crystal structure belonging to the Cmcm space group. According to x-ray diffraction data, the lattice parameters are: a = 3.69 Å, b = 12.84 Å, c = 9.73 Å [9]. This study uses theoretical values a = 3.66 Å, b = 12.94 Å, c = 9.68 Å [20]. The atomic coordinates are: Na (0, 0.7482, 0.25), Cu (0, 0.46692, 0.25), Zr (0, 0, 0), S1 (0, 0.36743, 0.05630), S2 (0, 0.07682, 0.25).
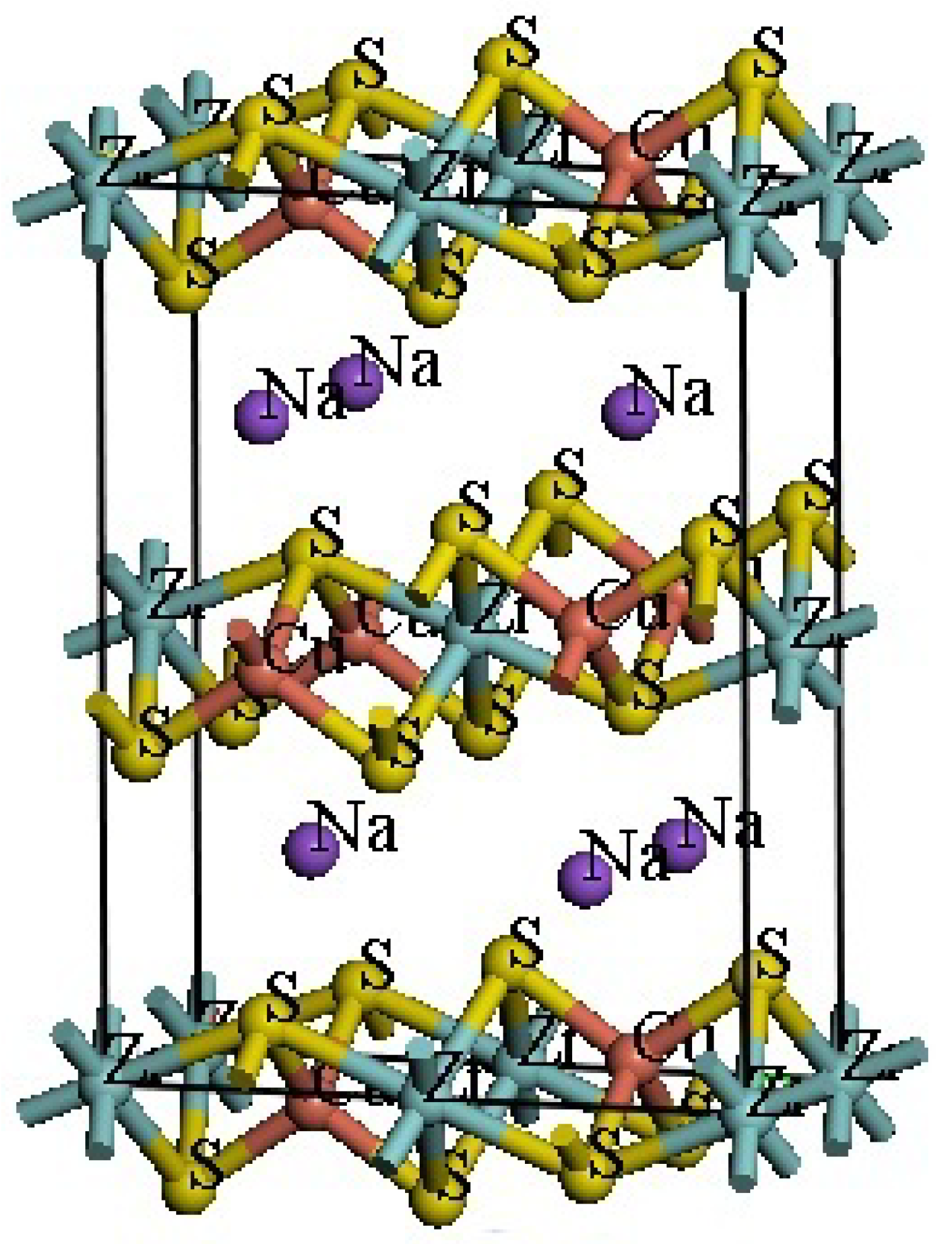
Figure 1: Unit cell of NaCuZrS3.
The calculated lattice parameters show small errors compared to experimentally and theoretical data, as listed in Table 1 for lattice parameters under different pressures. Furthermore, according to Refs. [21,22,23], the formation energy (−811.95 eV/atom), cohesive energy (890.39 eV/atom), binding energy (−822.47 eV/atom), tolerance factor (0.89), and octahedral factor (0.40) is calculated. The negative formation energy observed for the material confirms structural stability.
The electronic structure is very important for semiconductor materials. The band structure diagram of NaCuZrS3 at 0 GPa, obtained after optimization using the GGA-PBE method, is shown below. From Fig. 2, it can be seen that this material exhibits an indirect bandgap valence. The calculated bandgap size using the PBE method is approximately 0.61 eV.
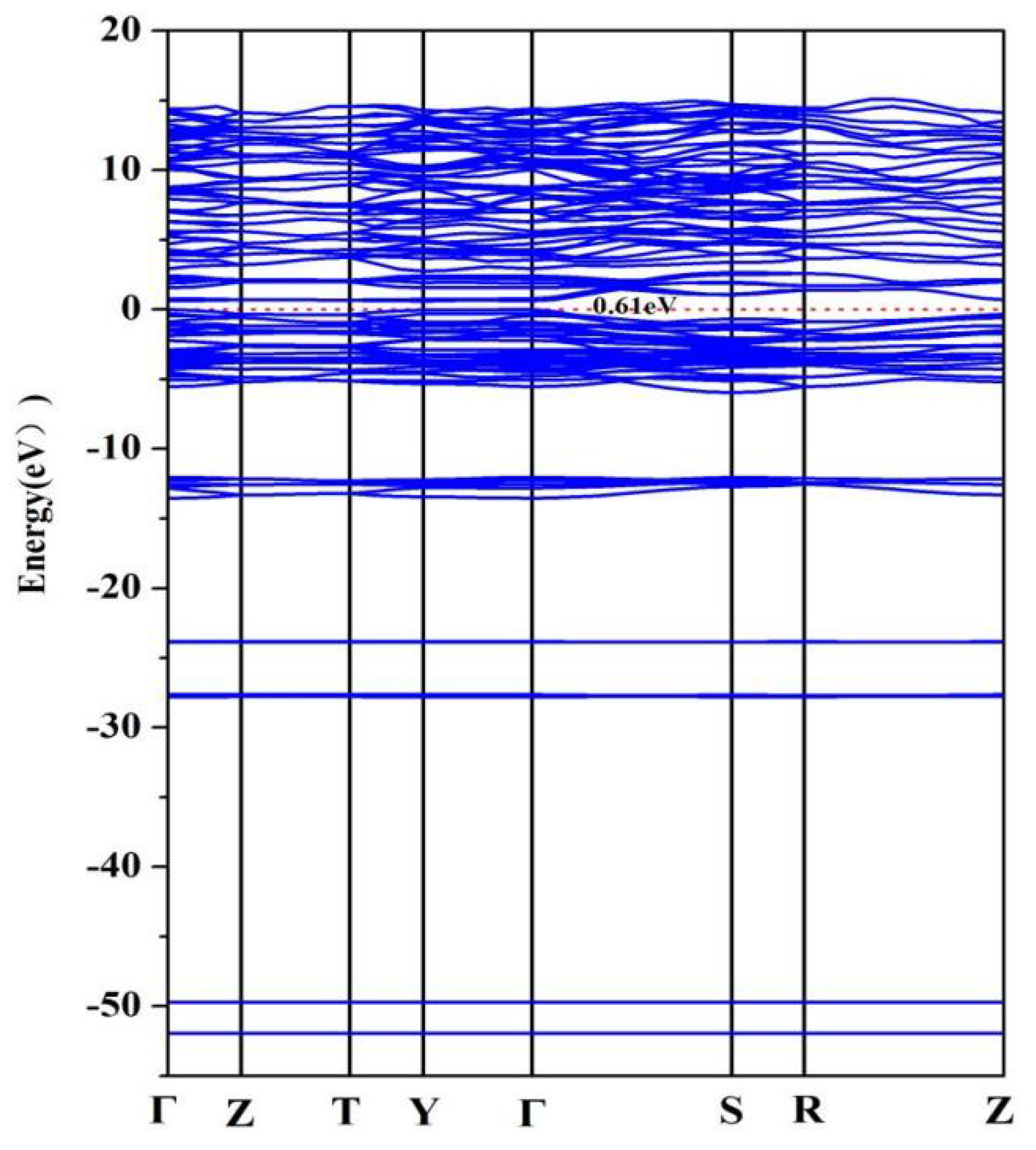
Figure 2: Band structure of NaCuZrS3.
Analysis of the density of States (DOS) plots in Fig. 3 shows that the valence band near the Fermi level spans from −60 eV to 0 eV. Based on the electronic structure analysis, the conduction band minimum is primarily contributed by the Zr d-orbitals, whereas the valence band maximum is mainly dominated by the Na p-orbitals, Cu d-orbitals, Zr p-orbitals and S p-orbitals.
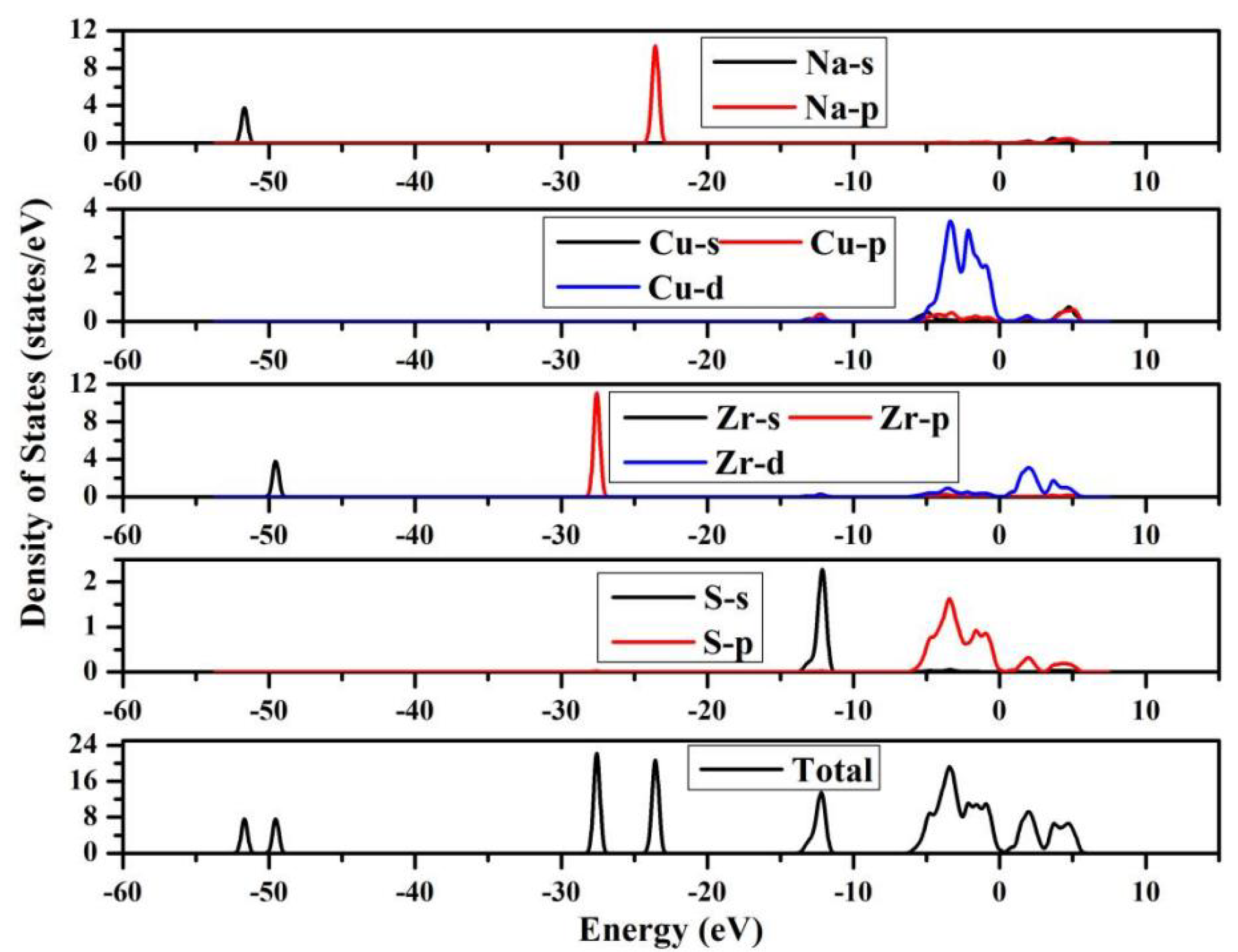
Figure 3: Density of States of NaCuZrS3.
3.3 Elastic Properties of NaCuZrS3
The calculated elastic constants Cij (GPa) for the NaCuZrS3 crystal are presented in Table 2. NaCuZrS3 has an orthorhombic crystal structure with nine independent elastic components.
Table 2: Elastic constants Cij (GPa) of NaCuZrS3 under different pressures.
| Pressure (GPa) | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 133.58 | 16.03 | 16.301 | 46.86 | 12.36 | 135.25 | 11.30 | 35.26 | 20.28 |
| 0 Ref. [20] | 208.59 | 23.87 | 30.04 | 122.25 | 25.94 | 226.50 | 35.42 | 71.30 | 51.59 |
| 10 | 184.35 | 59.38 | 44.58 | 117.62 | 51.12 | 213.83 | 15.02 | 51.30 | 47.11 |
| 20 | 233.85 | 105.10 | 73.10 | 176.24 | 79.12 | 259.39 | 26.02 | 66.37 | 73.84 |
| 30 | 277.67 | 147.37 | 101.28 | 231.89 | 108.98 | 307.11 | 24.99 | 80.88 | 97.67 |
| 40 | 316.45 | 188.96 | 121.49 | 294.75 | 145.40 | 357.28 | 40.45 | 94.28 | 118.11 |
| 50 | 353.14 | 224.51 | 148.26 | 328.60 | 164.73 | 391.05 | 41.60 | 107.69 | 133.82 |
Fig. 4 clearly demonstrates that the elastic constants Cij rise as pressure increases, but the growth rates of each Cij are different. The uniaxial compression stiffness C33 increases the fastest, indicating that the covalent bonding between atomic layers along this axis is more sensitive to pressure regulation. In contrast, the growth rate of C44 is the slowest compared to others, because the lattice slip barrier in the shear deformation mode is less affected by pressure, resulting in shear stiffness being less sensitive to pressure changes.
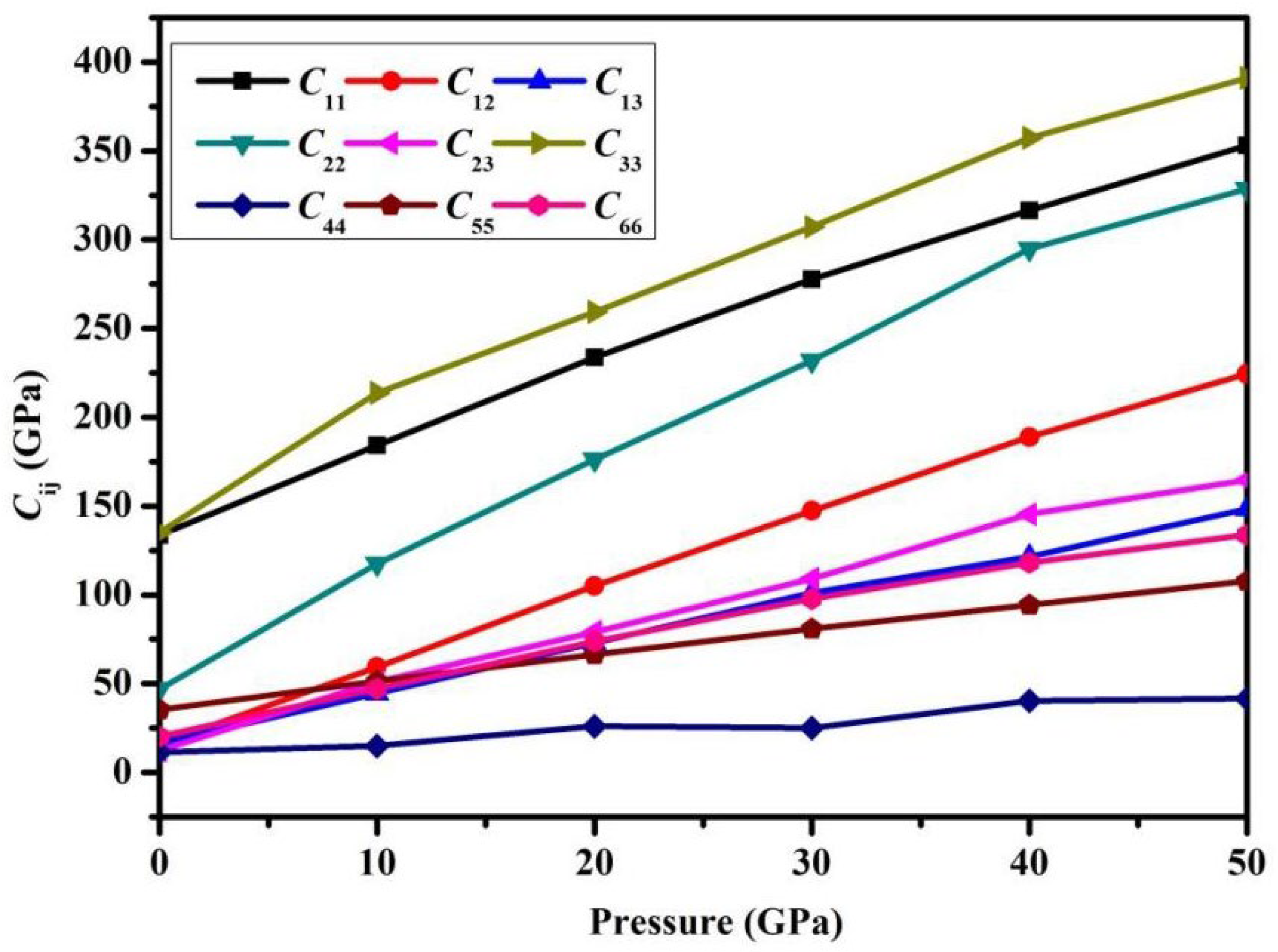
Figure 4: Relationship between pressure and elastic constants.
To evaluate its mechanical stability, the Cij of the orthorhombic NaCuZrS3 structure were inserted into Eqs. (1) and (2) [24]. The results (Table 2 and Table 3) fulfilled all necessary stability criteria, confirming that the material is mechanically stable.
Table 3: Mechanical stability criteria for NaCuZrS3.
| Pressure (GPa) | ||||||
|---|---|---|---|---|---|---|
| 0 | 133.58 | 1885.18 | 11.30 | 35.26 | 20.28 | 785,502.78 |
| 10 | 184.35 | 7421.04 | 15.02 | 51.30 | 47.11 | 3,437,892.26 |
| 20 | 233.85 | 13,532.12 | 26.02 | 66.37 | 73.84 | 6,635,552.03 |
| 30 | 277.67 | 19,202.46 | 24.99 | 80.88 | 97.67 | 10,682,142.89 |
| 40 | 316.45 | 24,090.98 | 40.45 | 94.28 | 118.11 | 16,204,149.55 |
| 50 | 353.14 | 28,878.89 | 41.60 | 107.69 | 133.82 | 19,828,770.12 |
The values of the bulk modulus, shear modulus, Young’s modulus, and Poisson’s ratio were calculated from the Cij using the Hill average method [25,26,27], as shown in Table 4. According to Pugh, the ratio of the B to the G can also determine whether a material is brittle (ductile), with a B/G value less than (greater than) the critical value of 1.75 [28]. Typically, a value of 1.496, as determined in Table 4, confirms that this material is brittle, but as the pressure increases, it transforms into a ductile material after 10 GPa.
Table 4: Elastic moduli under different pressures.
| Pressure (GPa) | Bv | BR | B | Gv | GR | G | E | v | B/G |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 45.01 | 35.48 | 40.24 | 31.44 | 22.35 | 26.90 | 65.99 | 0.23 | 1.50 |
| 10 | 91.78 | 87.43 | 89.60 | 46.74 | 34.46 | 40.60 | 105.81 | 0.30 | 2.21 |
| 20 | 131.57 | 129.58 | 130.58 | 60.73 | 50.23 | 55.48 | 145.79 | 0.31 | 2.35 |
| 30 | 170.22 | 169.38 | 169.8 | 71.313 | 54.45 | 62.88 | 167.92 | 0.34 | 2.70 |
| 40 | 208.91 | 208.91 | 208.91 | 84.747 | 71.20 | 77.97 | 208.03 | 0.33 | 2.68 |
| 50 | 238.65 | 238.53 | 238.59 | 92.313 | 75.13 | 83.72 | 224.86 | 0.34 | 2.85 |
The thermodynamic properties of NaCuZrS3 were investigated using energy-volume (E-V) (Fig. 5) data in conjunction with the quasi-harmonic Debye model approach [29,30].
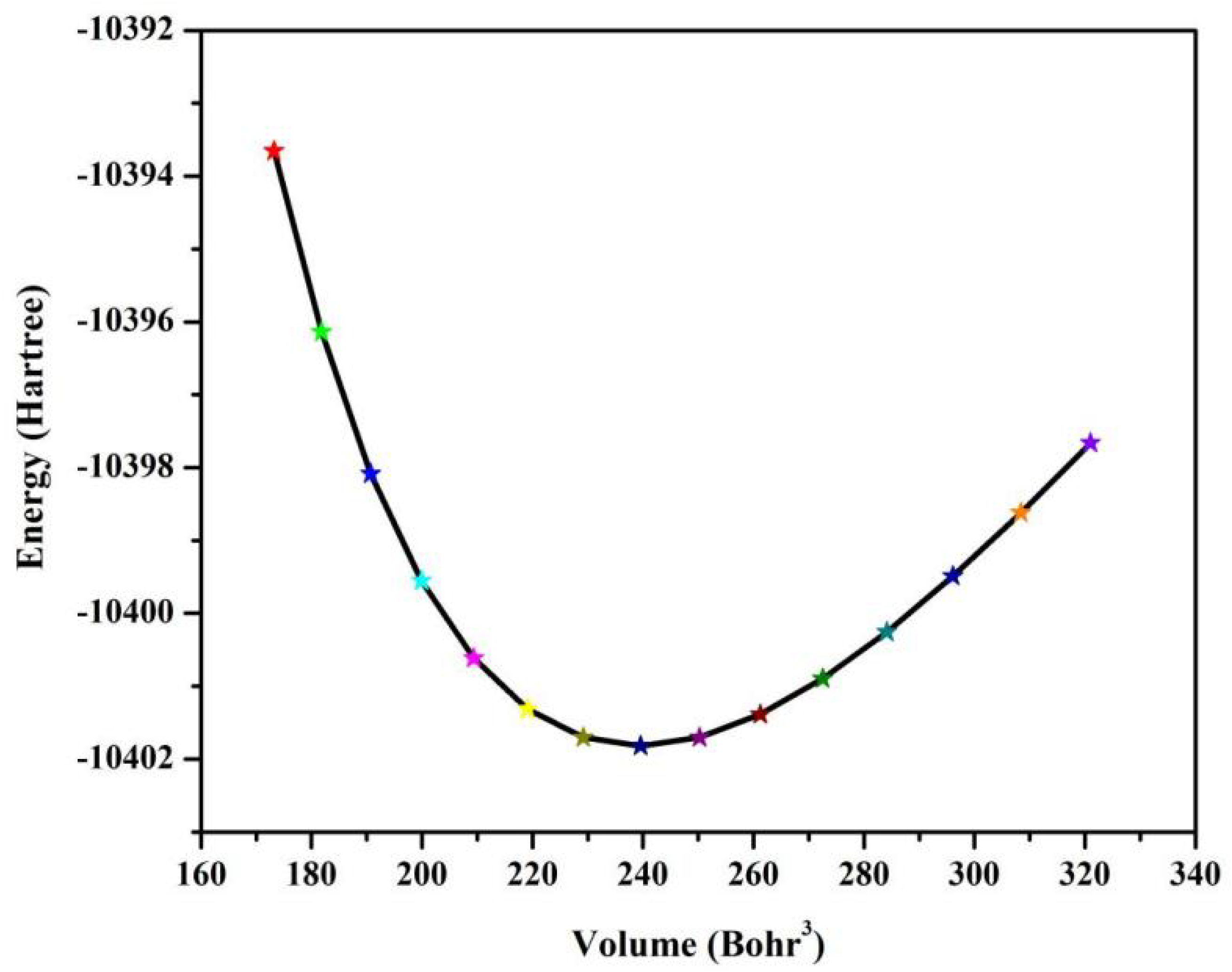
Figure 5: Relationship between total energy E and volume V.
The trend in Fig. 6 and Fig. 7 is as follows: at a fixed temperature, B is positively correlated with P. Under constant pressure conditions, as T increases, B decreases. The co-design of pressure/temperature is more instructive for material development.
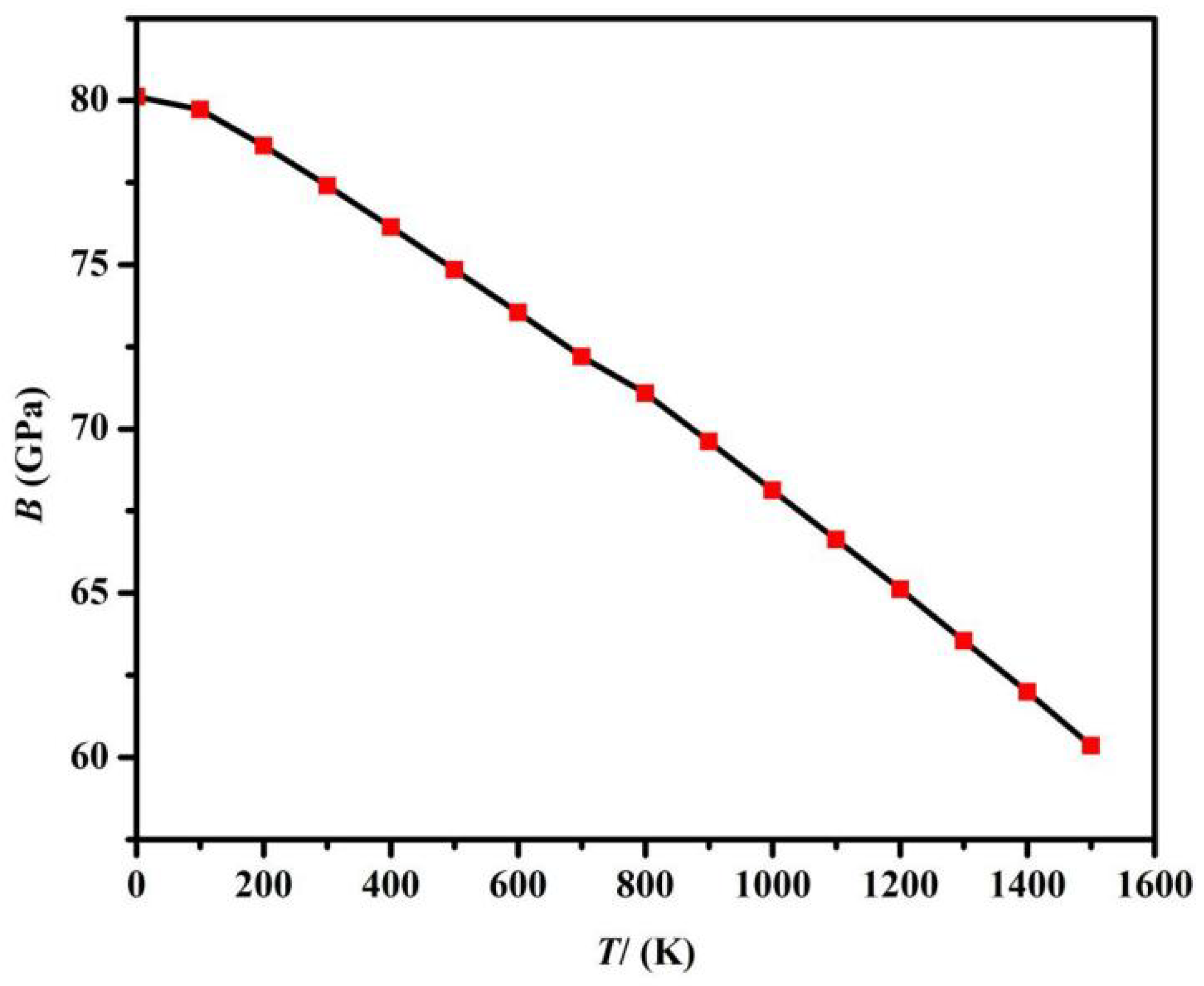
Figure 6: Relationship between B and T.
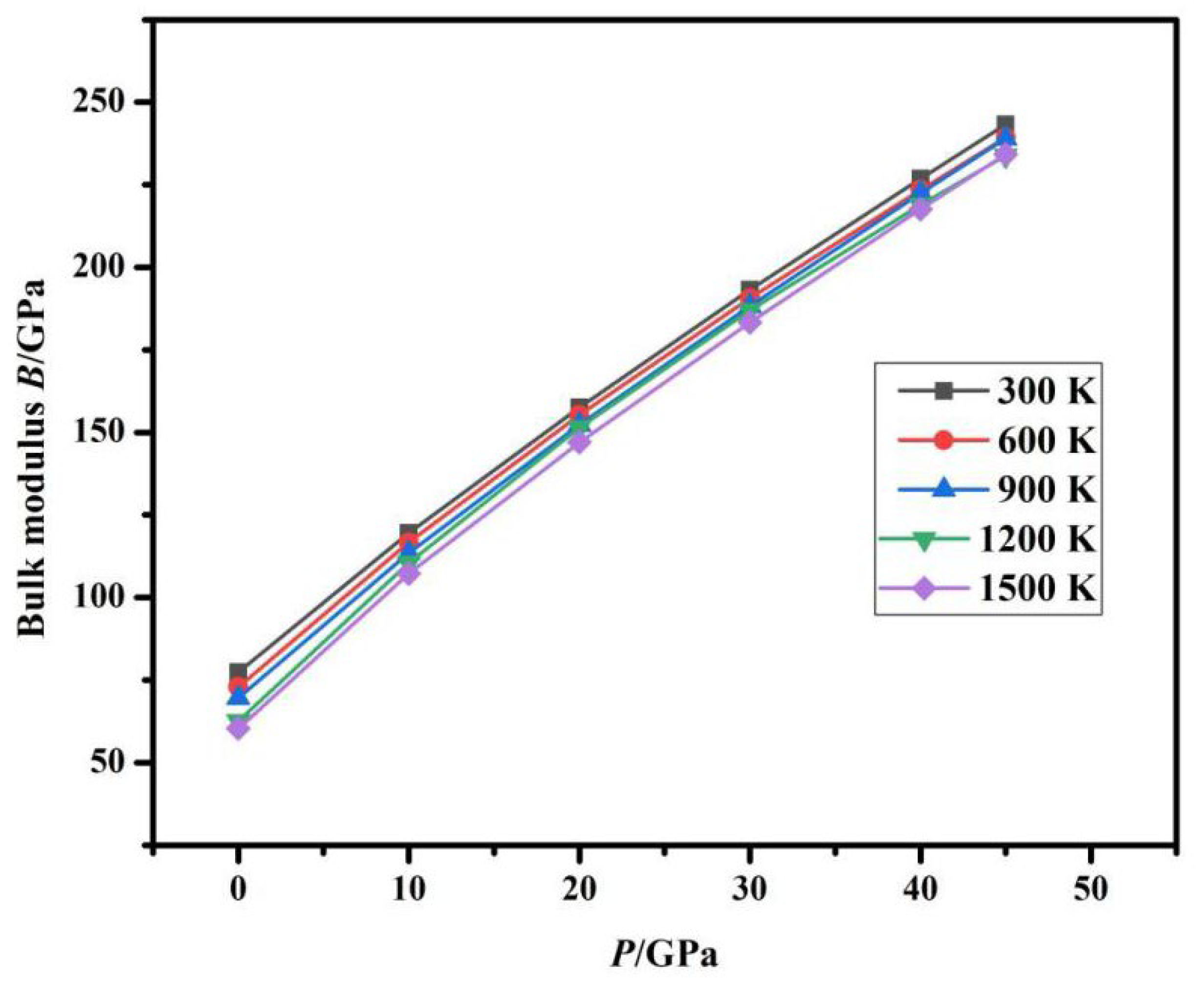
Figure 7: Relationship between B and P.
The thermodynamic properties are given in Table 5. As the P rises from 0 GPa to 50 GPa, in the low-temperature range of 300 K to 600 K, the entropy S decreases by 40.43%, the Debye temperature ΘD increases by 68.79%, and the Grüneisen constant γ decreases by 41.32%. In the mid-temperature range of 600 K to 900 K, the entropy S decreases by 28.85% and 23.86% respectively; the Debye temperature ΘD increases by 71.72% and 76.15% respectively; the Grüneisen constant γ decreases by 42.11% and 42.85% respectively. In the mid-to-high temperature range of 900 K to 1500 K, the entropy S decreases by 22.68% and 21.59% respectively, the Debye temperature ΘD increases by 79.14% and 83.33% respectively, and the Grüneisen constant γ decreases by 44.02% and 45.17% respectively. It can be concluded that with increasing pressure, S decreases slowly due to the suppression of low-frequency phonons by high pressure, while ΘD exhibits minimal variation between high and low temperatures, γ decreases somewhat more rapidly at elevated temperatures compared to lower ones.
Table 5: Entropy S/J·mol−1·K−1, Debye temperature ΘD/K, and Grüneisen constant γ for NaCuZrS3 at temperatures (300–1500 K) and pressures (0–50 GPa).
| T/K | P/GPa | 0 | 10 | 20 | 30 | 40 | 50 |
|---|---|---|---|---|---|---|---|
| 300 | S | 168.56 | 140.17 | 123.63 | 112.30 | 103.90 | 100.40 |
| ΘD | 383.97 | 473.49 | 537.51 | 587.96 | 629.55 | 648.11 | |
| γ | 2.24 | 1.86 | 1.65 | 1.50 | 1.37 | 1.32 | |
| 600 | S | 270.80 | 238.68 | 219.81 | 206.70 | 196.82 | 192.67 |
| ΘD | 376.36 | 468.98 | 534.34 | 585.54 | 627.62 | 646.32 | |
| γ | 2.28 | 1.88 | 1.66 | 1.50 | 1.38 | 1.32 | |
| 900 | S | 350.70 | 315.73 | 295.67 | 281.80 | 271.37 | 267.01 |
| ΘD | 365.44 | 462.55 | 529.77 | 582.04 | 624.84 | 643.73 | |
| γ | 2.31 | 1.88 | 1.66 | 1.50 | 1.38 | 1.32 | |
| 1200 | S | 380.43 | 343.85 | 323.15 | 309.17 | 298.56 | 294.13 |
| ΘD | 359.13 | 459.2 | 527.96 | 580.23 | 623.38 | 642.37 | |
| γ | 2.38 | 1.91 | 1.68 | 1.52 | 1.39 | 1.33 | |
| 1500 | S | 417.85 | 378.74 | 357.36 | 342.96 | 332.12 | 327.61 |
| ΘD | 349.3 | 454.03 | 524.14 | 577.48 | 621.21 | 640.39 | |
| γ | 2.44 | 1.93 | 1.68 | 1.53 | 1.39 | 1.34 |
Fig. 8 and Fig. 9 is based on calculations using the quasi-harmonic approximation method, where T and P represent two key parameters influencing Cv and Cp. In the temperature range of 0–300 K, the heat capacity increases exponentially, primarily due to the gradual excitation of lattice vibration modes (phonons). In this stage, the Debye model dominates, and the Cv ∝ T3 characteristic is significant. In the mid-temperature range of 300–700 K, the rate of heat capacity increase begins to slow down, transitioning to a linear growth region, corresponding to the activation of high-frequency modes in the phonon density of states and the gradual emergence of electronic thermal excitation contributions. When T is greater than 700 K (high temperature), Cv hardly changes anymore and begins to saturate, as the thermodynamic degrees of freedom reach their maximum. At a constant temperature T, as the pressure P increases, the heat capacity Cv exhibits a generally decreasing trend: in the low-temperature region of 0~300 K, Cv decreases rapidly, as high pressure significantly suppresses phonon vibrational entropy, leading to a rapid reduction in Cv; in the mid-temperature region of 300 K~700 K, the decrease in Cv slows down; at high temperatures greater than 700 K, Cv is almost unaffected by pressure. As T and P continuously increase, the heat capacity Cv is entirely determined by the Dulong-Petit limit value. According to Fig. 9, at low temperatures, the change in CP is similar to the change in CV.
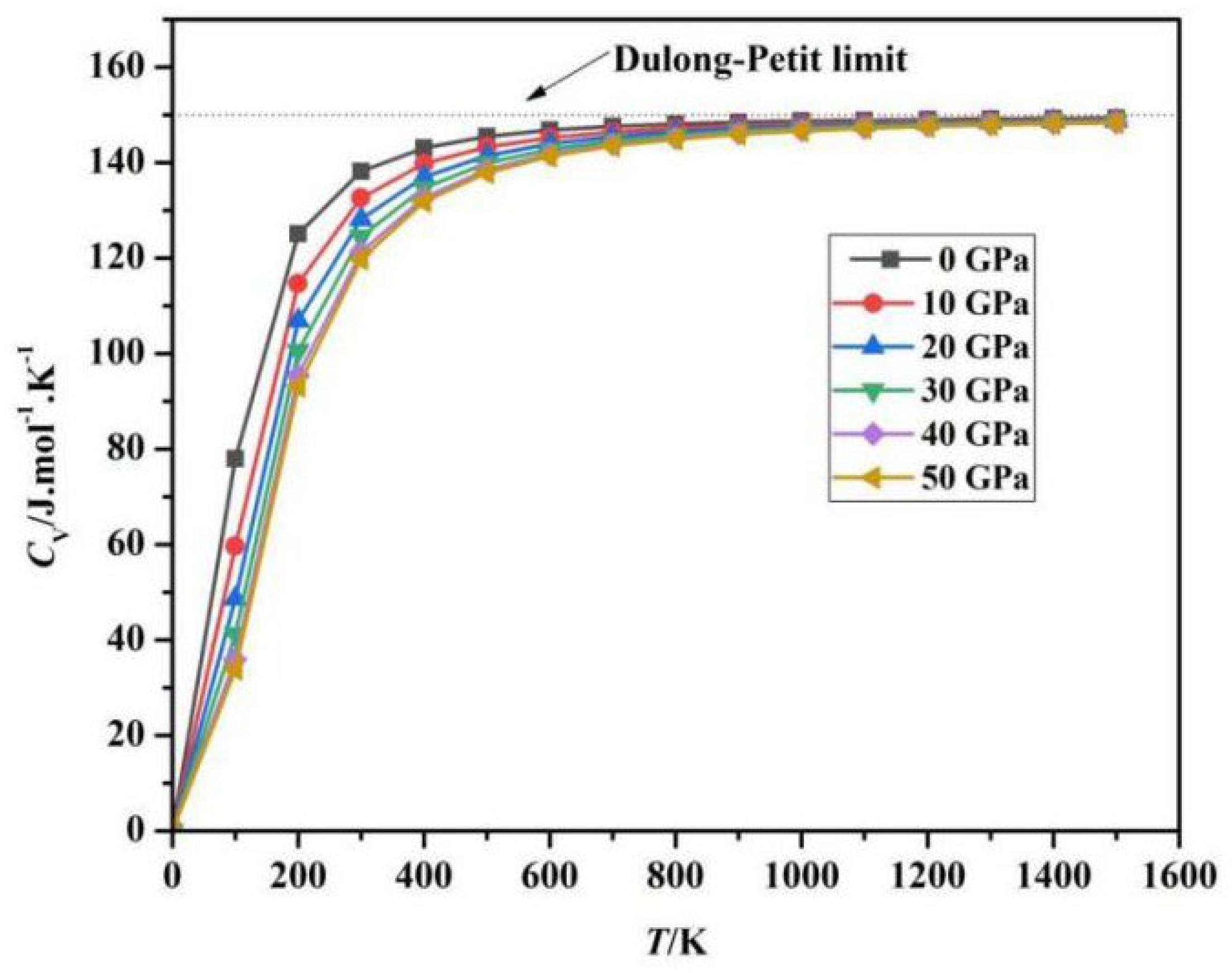
Figure 8: Relationship between Cv and T under different pressures.
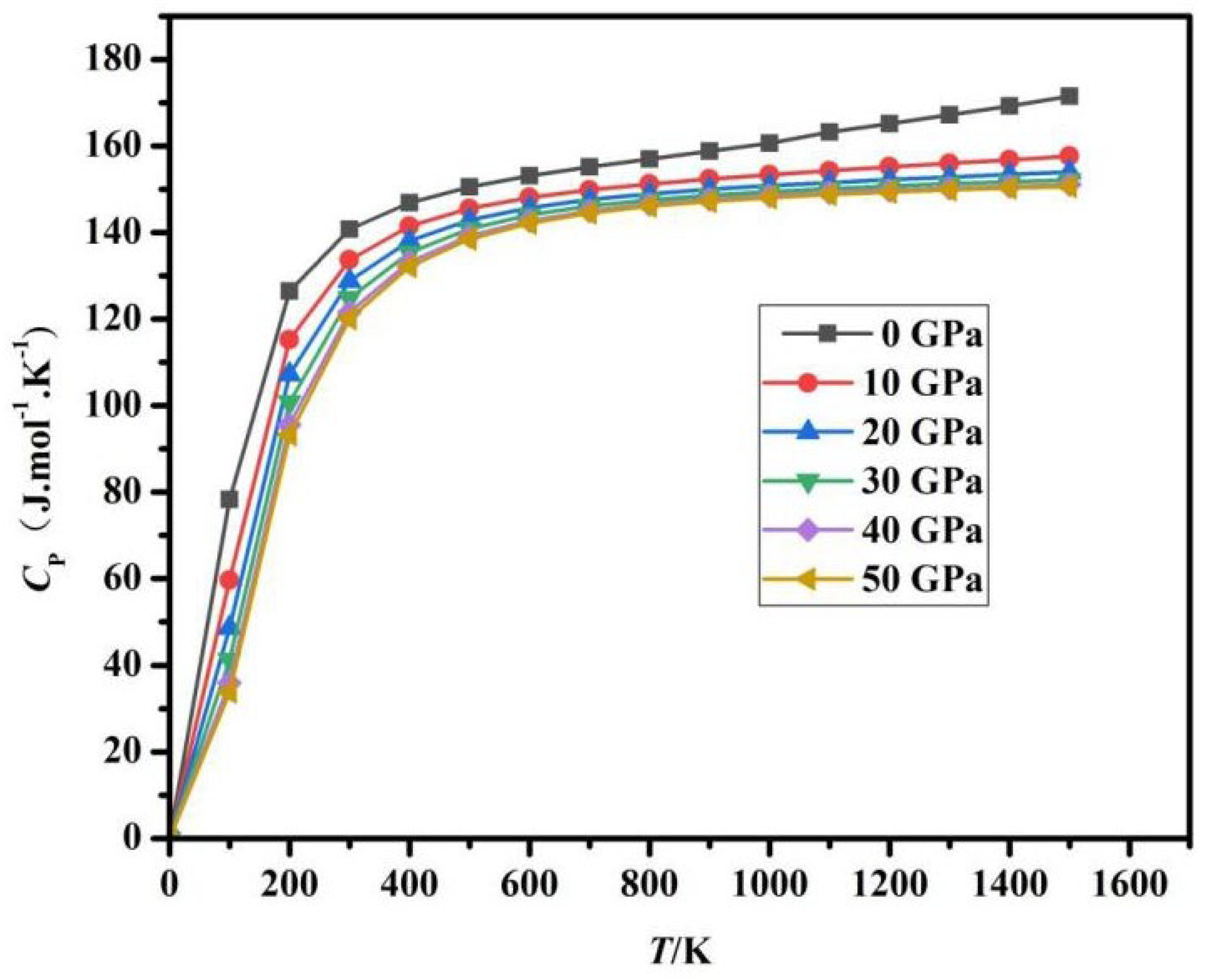
Figure 9: Relationship between Cp and T under different pressures.
Fig. 10 presents the calculated dependence of the thermal expansion coefficient α on temperature T under various pressure conditions. Under low pressure (0 GPa and 10 GPa) and low temperature (less than 300 K), α increases super-linearly; in the mid-temperature range (greater than 300 K), the growth rate of α tends to flatten. Especially under high-temperature conditions, α remains almost constant. Under high-pressure environments, the anharmonic vibrations of high-energy optical phonons are suppressed, and their thermal excitation contribution to volume change tends to be isotropic, leading to the disappearance of temperature sensitivity in α. Fig. 11 illustrates the calculated variation of the α with pressure P at various temperatures. At a constant temperature, as the pressure gradually increases, α shows a decreasing trend. Under high pressure (exceeding 15 GPa), pressure suppresses the anisotropy of thermal expansion, and the expansion coefficient gradually levels off, with α being almost unaffected by T and P.
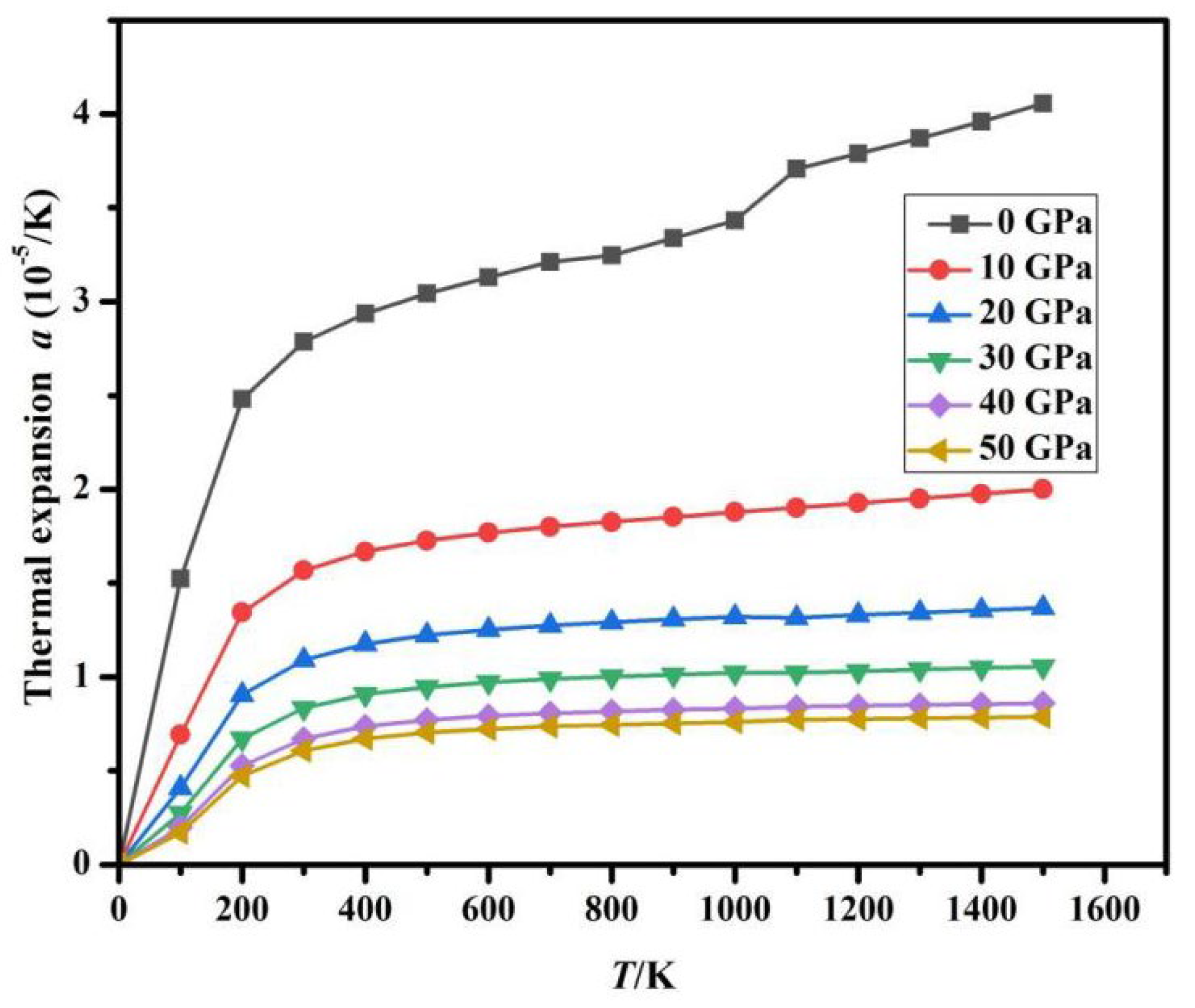
Figure 10: Relationship between α and T under pressure gradients.
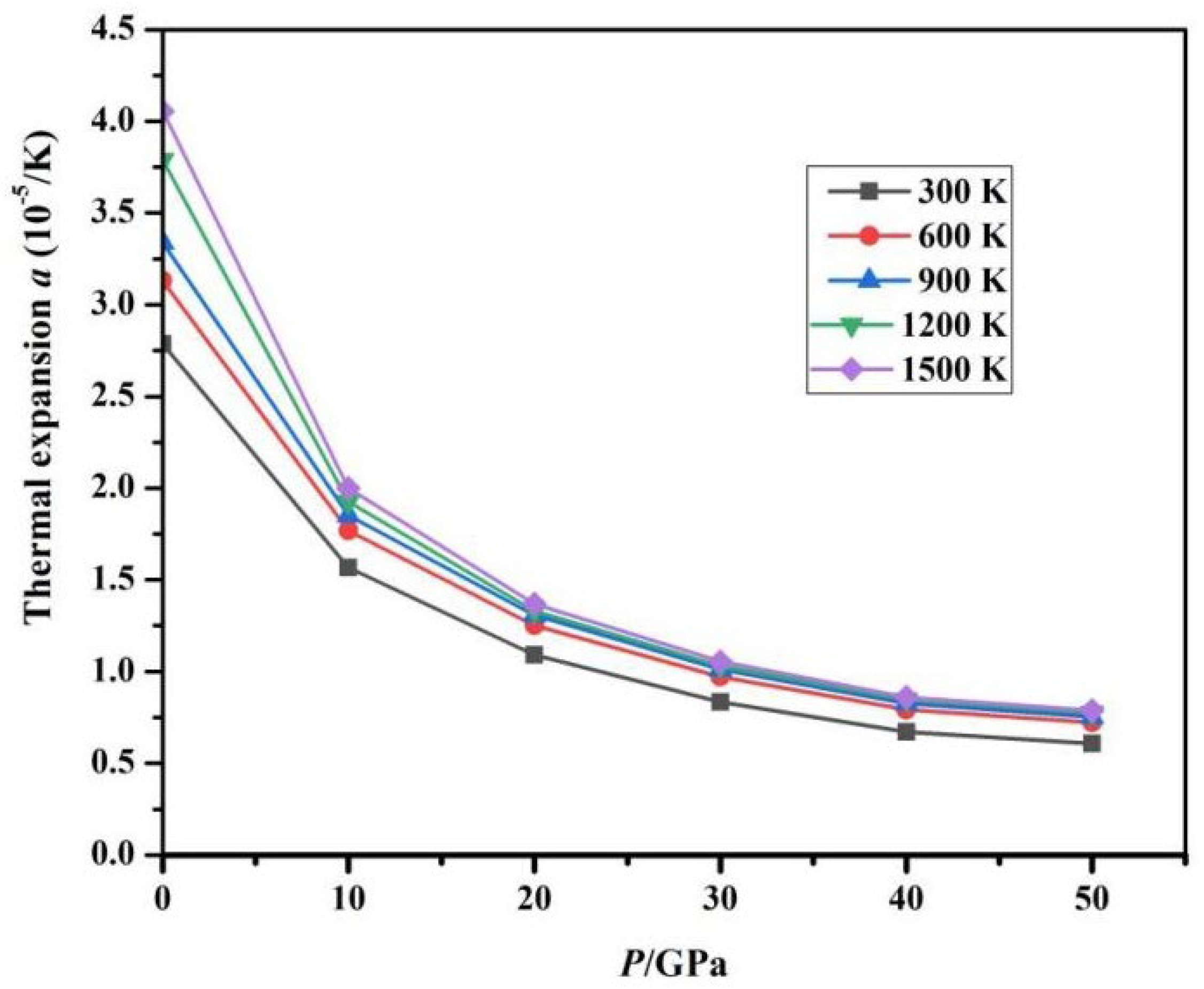
Figure 11: Relationship between α and P at different temperatures.
In this work, we employed a first-principles methodology to investigate the structural, elastic, electronic, and thermodynamic properties of orthorhombic NaCuZrS3. First, structural optimization was performed, and then the physical properties of NaCuZrS3 were calculated. Optimization under different pressures found that the lattice parameters of NaCuZrS3 continuously decrease. Through band structure calculations, the bandgap at 0 GPa is 0.61 eV, which matches the theoretical calculated value. Calculation of elastic moduli yielded the elastic constants Cij and physical parameters of mechanical properties for NaCuZrS3. In the thermodynamic property calculations, we found that entropy decreases significantly at low temperatures, γ changes little with temperature, and the graphs of Cv vs. T and Cp vs. T under different pressures show that the Cv eventually approaches the Dulong-Petit limit value. The α tends to level off with increasing temperature or pressure.
Acknowledgement:
Funding Statement: Key Science and Technology Project of Chongqing Municipal Education Commission (No. KJZD-K202501409).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Hongli Guo; data collection: Hongli Guo; analysis and interpretation of results: Huanyin Yang; draft manuscript preparation: Hongli Guo, Huanyin Yang, Suihu Dang, Shunru Zhang, Haijun Hou. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest.
References
1. Grigoriev MV , Ruseikina AV , Chernyshev VA , Oreshonkov AS , Garmonov AA , Molokeev MS , et al. Single crystals of EuScCuSe3: Synthesis, experimental and DFT investigations. Materials. 2023; 16( 4): 1555. doi:10.3390/ma16041555. [Google Scholar] [CrossRef]
2. Lu YJ , Ibers JA . Synthesis and characterization of the new quaternary two-dimensional materials KCu2NbQ4 (Q = Se, S). J Solid State Chem. 1991; 94( 2): 381– 5. doi:10.1016/0022-4596(91)90204-U. [Google Scholar] [CrossRef]
3. Ishtiyak M , Jana S , Karthikeyan R , Ramesh M , Tripath B , Malladi SK , et al. Syntheses of five new layered quaternary chalcogenides SrScCuSe3, SrScCuTe3, BaScCuSe3, BaScCuTe3, and BaScAgTe3: Crystal structures, thermoelectric properties, and electronic structures. Inorg Chem Front. 2021; 8: 4086. doi:10.1039/D1QI00717C. [Google Scholar] [CrossRef]
4. Janek J , Zeier WG . A solid future for battery development. Nat Energy. 2016; 1: 16141. doi:10.1038/nenergy.2016.141. [Google Scholar] [CrossRef]
5. Kato Y , Hori S , Saito T , Suzuki K , Hirayama M , Mitsui A , et al. High-power all-solid-state batteries using sulfide superionic conductors. Nat Energy. 2016; 1( 4): 16030. doi:10.1038/nenergy.2016.30. [Google Scholar] [CrossRef]
6. Koscielski LA , Ibers JA . The structural chemistry of quaternary chalcogenides of the type AMM’Q3. Zeitschrift Anorg Allge Chemie. 2012; 638( 15): 2585– 93. doi:10.1002/zaac.201200301. [Google Scholar] [CrossRef]
7. Pal K , Xia Y , He J , Wolverton C . Intrinsically low lattice thermal conductivity derived from rattler cations in an AMM′Q3 family of chalcogenides. Chem Mater. 2019; 31( 21): 8734– 41. doi:10.1021/acs.chemmater.9b02484. [Google Scholar] [CrossRef]
8. Pan Y . Exploring the structural, physical properties and hydrogen storage properties of LiBHx (x = 1 and 4) lithium borohydrides. Ceram Int. 2024; 50: 3837– 42. doi:10.1016/j.ceramint.2023.11.140. [Google Scholar] [CrossRef]
9. Biswas M , Ghosh S , Chowdhury J , Ali MA , Hossain MM , Naqib SH , et al. An inclusive study of lead-free perovskite CsMI3 materials for photovoltaic and optoelectronic appliance explored by a first principles study. Mater Today Commun. 2024; 40: 109422 doi:10.1016/j.mtcomm.2024.109422. [Google Scholar] [CrossRef]
10. Rana MR , Islam S , Hoque K , Mahmud S , Ali MA . Newly synthesized Pb-based 312 MAX phases M3PbC2 (M = Zr and Hf): A First-principles study, Diam. Rela. Mater. 2024; 146: 111245. doi:10.1016/j.diamond.2024.111245. [Google Scholar] [CrossRef]
11. Guo H , Yang H , Dang S , Zhang S , Hou H . Structural, elastic, electronic, dynamic, and thermal properties of SrAl2O4 with an orthorhombic structure under pressure. Molecules. 2024; 29( 21): 5192. doi:10.3390/molecules29215192. [Google Scholar] [CrossRef]
12. Wang Y , Lin A , Chai J , Ming C , Sun YY . First-principles design of Na-ion superionic conductors: Interstitial-based Na diffusion in NaCuZrS3. Chem. 2022; 28( 32): e202200234. doi:10.1002/chem.202200234. [Google Scholar] [CrossRef]
13. Guan H , Hou HJ , Zhang SR , Xie LH . Theoretical study on the elastic and thermodynamic properties of CdS. Chalcogenide Lett. 2024; 21( 1): 39– 51. doi:10.15251/CL.2024.211.39. [Google Scholar] [CrossRef]
14. Alay-e-Abbas SM , Shaukat A . FP-LAPW calculations of structural, electronic, and optical properties of alkali metal tellurides: M2Te [M: Li, Na, K and Rb]. J Mater Sci. 2011; 46( 4): 1027– 37. doi:10.1007/s10853-010-4868-7. [Google Scholar] [CrossRef]
15. Clark SJ , Segall MD , Pickard CJ , Kristallogr Z . First principles methods using CASTEP. J Crystallogr Cryst Mater. 2005; 220: 567. doi:10.1524/zkri.220.5.567.65075. [Google Scholar] [CrossRef]
16. Hussain S , Rehman JU . First-principles calculation to investigate structural, electronic, optical, and thermodynamics properties of perovskite KXO3 (KTa and Zn) alloys for photovoltaic and smart window applications. Phys B Condens Matter. 2024; 687: 416116. doi:10.1016/j.physb.2024.416116. [Google Scholar] [CrossRef]
17. Vanderbilt D . Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys Rev B. 1990; 41( 11): 7892– 5. doi:10.1103/PhysRevB.41.7892. [Google Scholar] [CrossRef]
18. Perdew JP , Burke K , Ernzerhof M . Generalized gradient approximation made simple. Phys Rev Lett. 1996; 77( 18): 3865– 8. doi:10.1103/PhysRevLett.77.3865. [Google Scholar] [CrossRef]
19. Li TJ , Yue Y , Qu LP , Hou HJ , Fan SH , Guo HL . Electronic structure, optical properties, and elastic properties of solar material CuInSe2 from theoretical simulation. Chalcogenide Lett. 2025; 22( 6): 529– 40. doi:10.15251/CL.2025.226.529. [Google Scholar] [CrossRef]
20. Rugut E . Theoretical study on mechanical and optoelectronic properties of NaCuZrS3 chalcogenide. Phys B Condens Matter. 2025; 707: 417159. doi:10.1016/j.physb.2025.417159. [Google Scholar] [CrossRef]
21. Qureshi MW , Ma XX , Tang GZ , Paude R . Ab initio predictions of structure and physical properties of the Zr2GaC and Hf2GaC MAX phases under pressure. Sci Rep. 2021; 11: 3260. doi:10.1038/s41598-021-82402-1. [Google Scholar] [CrossRef]
22. Travis W , Glover ENK , Bronstein H , Scanlon DO , Palgrave RG . On the application of the tolerance factor to inorganic and hybrid halide perovskites: A revised system. Chem Sci. 2016; 7( 7): 4548– 56. doi:10.1039/C5SC04845A. [Google Scholar] [CrossRef]
23. Ahmed B , Tahir MB , Ali A , Sagir M . DFT insights on structural, electronic, optical and mechanical properties of double perovskites X2FeH6 (X = Ca and Sr) for hydrogen-storage applications. Int J Hydrogen Energy. 2024; 50: 316– 23. doi:10.1016/j.ijhydene.2023.10.237. [Google Scholar] [CrossRef]
24. Mouhat F , Coudert FX . Necessary and sufficient elastic stability conditions in various crystal systems. Phys Rev B. 2014; 90( 22): 224104. doi:10.1103/PhysRevB.90.224104. [Google Scholar] [CrossRef]
25. Voigt W . Lehrbuchder kristallphysik teubner leipzig. Leipzig, Germany: Teubner Verlag; 1928. [Google Scholar]
26. Reuss A . Berechnung der fließgrenze von mischkristallen auf grund der plastizitätsbedingung für einkristalle. Z Angew Math Mech. 1929; 9( 1): 49– 58. doi:10.1002/zamm.19290090104. [Google Scholar] [CrossRef]
27. Hill R . The elastic behaviour of a crystalline aggregate. Proc Phys Soc Sect A. 1952; 65( 5): 349. doi:10.1088/0370-1298/65/5/307. [Google Scholar] [CrossRef]
28. Pugh SF . XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond Edinb Dublin Philos Mag J Sci. 1954; 45( 367): 823– 43. doi:10.1080/14786440808520496. [Google Scholar] [CrossRef]
29. Blanco MA , Francisco E , Luaña V . GIBBS: Isothermal-isobaric thermodynamics of solids from energy curves using a quasi-harmonic Debye model. Comput Phys Commun. 2004; 158( 1): 57– 72. doi:10.1016/j.comphy.2003.12.001. [Google Scholar] [CrossRef]
30. Hou HJ , Fan S , Wang HY , Chen WX , Lu XW , Zhang SR , et al. Study of electronic structure, elastic and thermodynamic properties of Cu2MgSnS4 under different pressures. Chalcogenide Lett. 2024; 21( 2): 189– 200. doi:10.15251/CL.2024.212.189. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools