
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Re-evaluating cytokine storm syndromes: dysregulated host defense or contextual immune adaptation?
1 Department of Pharmacy Practice, ISF College of Pharmacy, Moga-142001, Punjab, India
2 School of Pharmaceutical Sciences, CT University, Ferozepur Rd, Sidhwan Khurd, Ludhiana-142024, Punjab, India
* Corresponding Author: Hardik Kumar. Email:
European Cytokine Network 2026, 37(1), 13-24. https://doi.org/10.32604/ecn.2026.078458
Received 31 December 2025; Accepted 25 March 2026; Issue published 13 April 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Cytokine storm syndromes have become a much-invoked concept to describe severe immunopathology in infectious, inflammatory, and iatrogenic diseases, but the concept is poorly defined and often mechanistically imprecise. High levels of systemic cytokines have often been viewed as indicators of immune dysfunction, and based on this notion, therapeutic interventions focused on general cytokine inhibition are proposed. Nevertheless, a growing number of clinical and experimental data dispute the notion that hypercytokinemia is necessarily pathological. The present paper reconsiders cytokine storm syndromes in the light of an evolutionary, systems-immunology model, and suggests that most cytokine amplification conditions are context-specific host defence programmes, however, not uniform maladaptations of immune regulation. Its major objective is to redefine the cytokine storm syndromes, which are no longer understood as a single, uniformly pathological situation, but as a spectrum of context-specific immune programmes. The paper reviews disease-specific phenotypes of cytokine storms in viral infections, bacterial sepsis, autoimmune diseases, and immune-based therapies, noting considerable variability in cytokine composition, kinetics of production, cytokine-producing cell frequency, and localization in tissues. It also explains how the temporal dynamics, host-specific conditions, and failure of counter-regulatory mechanisms define the difference between cytokine amplification as an adaptation or as a pathological condition. In addition, it considers the clinical consequences of such a refined framework and argues that exploring precision strategies need to consider immune context, disease stage, and signalling thresholds, as opposed to a general use of blocking cytokines. In conclusion, this review attempts to draw a line between adaptive cytokine amplification and maladaptive immunopathology by integrating the insights gained in evolutionary biology and systems immunology.Keywords
Cytokine storm is a name that has become predominant in explaining severe immunopathology in a wide variety of clinical settings, such as in viral diseases, bacterial sepsis, autoimmune disease, and immunotherapy with immune cells [1]. The term was originally used to refer to excessive inflammatory reactions with adverse effects, but has since been extended to situations in which cytokines are raised in the bloodstream. This terminology has simplified clinical communication and hypothesis generation, but has also created an artificially simplified and mechanistically ambiguous perspective on immune-mediated pathology.
The focal point of the cytokine storm paradigm is that hyperproduction of cytokines is an indication of immune regulation failure, and a major contributor to tissue injury and death [2]. This conjecture has, subsequently, influenced therapeutic approaches that focus on general cytokine inhibition, which often results in inconsistent or unsatisfactory clinical outcomes. Importantly, the same levels of hypercytokinemia can be linked to a divergent outcome in different diseases and even among individuals with the same disease, which poses basic questions regarding the causal role of cytokines in immunopathology [3]. Additionally, the dependence on systemic cytokine analyses has obscured vital aspects of immune signalling, such as temporal dynamics, tissue compartmentalization, and cellular source specificity.
Recent findings in the areas of systems immunology, evolutionary biology, and clinical immunology have cast doubt on the idea that the amplification of cytokines is inherently maladaptive. Cytokines evolved as fast and scalable communication information used to coordinate host defence in situations of acute threat [4]. In this sense, strong cytokine generation may reflect an adaptive response to control pathogens, protect tissues, or coordinate immunity, and pathology only occurs when regulatory limits are exceeded or when situational factors alter the balance between collateral damage [5]. Binary division of responses to cytokines as normal or storm-like, thus, poses a danger of overlooking the context-dependent, graded nature of immune signalling. These considerations support a continuum-based view of cytokine storms as context-dependent immune programs rather than discrete pathological entities (figure 1).
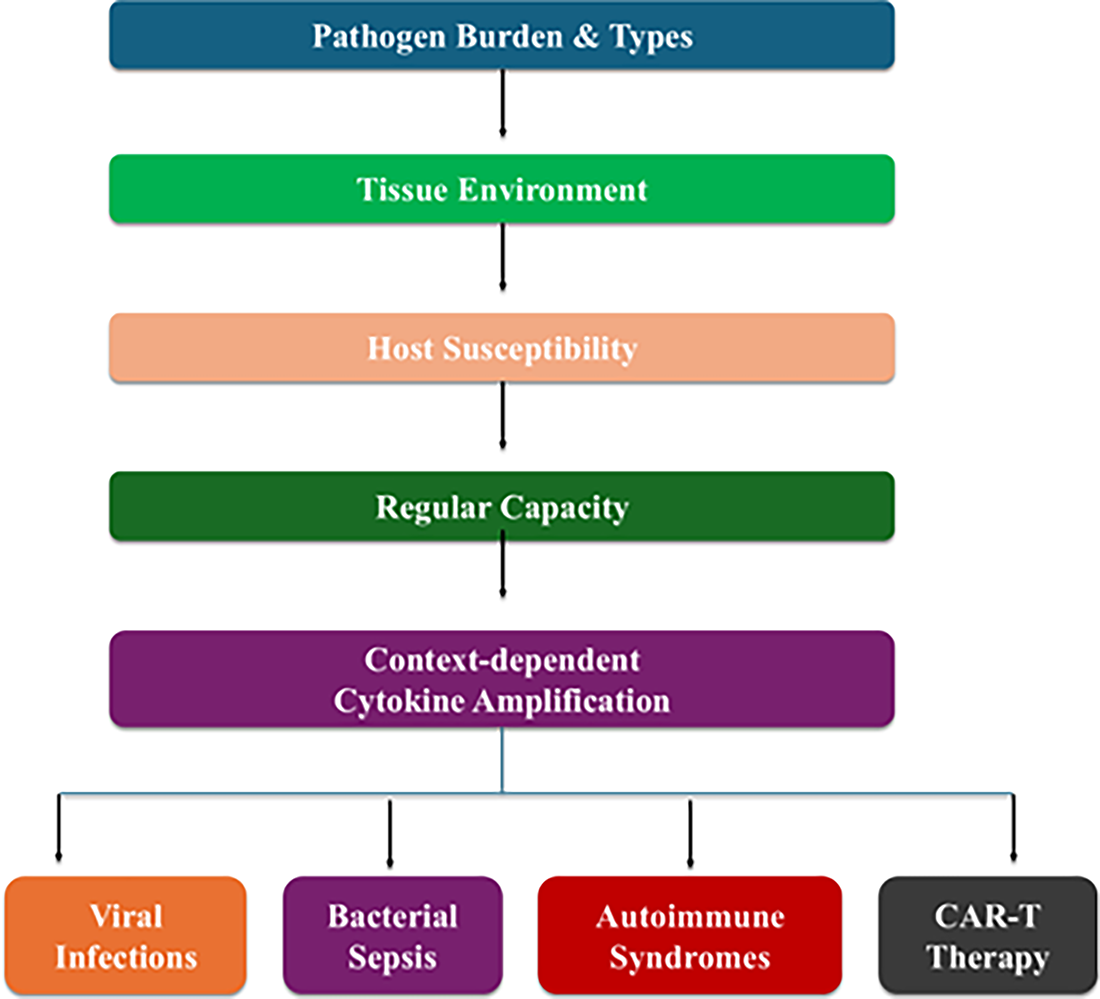
Figure 1: Conceptual continuum of cytokine amplification from adaptive host defense to pathological cytokine storm. CAR-T: Chimeric Antigen Receptor T cells.
This article re-examines cytokine storm syndromes as heterogeneous, condition-specific immune programmes but not a singular pathological entity. The incorporation of evolutionary logic, pathogen-specific phenotypes and time, it suggests an updated conceptual model that separates the adaptive cytokine amplification and maladaptive immunopathology. This has significant implications for the interpretation of biomarkers, clinical stratification, and the development of precision immunomodulatory therapies.
2 Cytokine Storm Syndromes: Diagnostic and Conceptual Ambiguities
Cytokine storm is a term that describes a wide group of clinical and lab findings, but its definition is not universal or standardised. Historically, cytokine storm syndromes are defined as hyperinflammatory systemic responses, which involve excessive secretion of pro-inflammatory cytokines, including interleukin-6 (IL-6), tumour necrosis factor-alpha (TNF-α), and IL-1β, and are associated with the dysfunction of multiple organs and mortality [6]. Initial names were coined based on graft-versus-host disease and hemophagocytic lymphohistiocytosis, although the term is now applied to various situations such as sepsis, viral infections and toxicities associated with immunotherapy. Although widely used, even with such a broad application, a discussion exists on quantitative thresholds of cytokine elevations that reliably indicate a storm state and not an appropriate inflammatory response, with multi-organ dysfunction and high mortality [7]. Initial nomenclatures were the graft-versus-host disease and hemophagocytic lymphohistiocytosis, but the term has since been applied to a broader range of situations, such as sepsis and viral infection.
Additionally, Systematic reviews indicate that severe COVID-19 is associated with modest increases in circulating IL-6 and IL-8 compared with non-severe disease, with concentrations generally remaining in the tens of pg/mL range, substantially lower than those observed in classical cytokine release syndromes [8]. This numerical difference questions the standard categorization of severe COVID-19 as a paradigm cytokine storm and indicates that comparative, and not absolute, cytokine alteration might be more instructive in particular illness conditions. Notably, small systemic cytokines may exist in the absence of significant immunopathology, suggesting that other determinants of clinical severity exist besides circulating cytokine levels, including localization, sensitivity of receptors and competence of downstream signalling [9].
The latter observations also point to a major limitation of using serum cytokine measurements as a primary diagnostic criterion. The levels of circulating cytokines are an integrated and temporally averaged signal that might not be a good indicator of local foci of inflammatory activity in the involved organs, especially the lung and vascular endothelium [10]. These investigations using tissue transcriptomics and single-cell technologies have revealed significant cytokine and chemokine expression in pulmonary and myeloid compartments in severe COVID-19, despite a relative containment of systemic levels. This type of compartmentalised inflammation can be the cause of organ dysfunction without showing up in extreme hypercytotoxinemia in peripheral blood [11].
Altogether, these statistics highlight the intellectual ambiguity inherent in the construct of a cytokine storm. Cytokine profiles can have similarities in separate clinical syndromes with different etiologies and outcomes, and also, strikingly different quantitative cytokine levels can exist with identical clinical phenotypes [12]. The lack of standardised thresholds, and the use of the term disease-agnostically, run the risk of confusion of mechanistically different inflammatory programmes within a single term. A fine definition of cytokine storm syndromes thus requires that one go beyond absolute cytokine concentrations to complex frameworks that embrace context-dependent benchmarks, time dynamics, compartmentalization of tissues, and host susceptibility variables [13].
To further complicate the matter, the diagnostic criteria based on hemophagocytic lymphohistiocytosis and macrophage activation syndrome are not typically effective to apply to infections such as COVID-19, as the criteria did not undergo calibration to reflect pathogen-induced systemic inflammation [14]. This has led to redundant and even conflicting definitions within clinical practise with a paucity of consensus on what combinations of biomarkers or clinical phenotypes should constitute a storm state. Collectively, this information supports the notion of conceptual and diagnostic vagueness in the cytokine storm construct, which necessitates streamlined criteria that include disease-specific context, quantitative thresholds, and mechanistic understanding.
3 Evolutionary and Physiological Rationale for Hypercytokinemia
Evolutionarily, the ability to amplify cytokine signalling quickly has been a conserved property of vertebrate immunity, and this indicates that there has been strong selection pressure to provide decisive responses to acute, life-threatening insults. Cytokines are high-affinity communication molecules, which allow a rapid coordination between immune cells, stromal compartments, and distant organs [15]. In this regard, hypercytokinemia cannot be considered pathological, but as an extreme of a physiological range aimed at optimising host survival in the case of severe infection or tissue injury.
The pro-inflammatory cytokines implicated in cytokine storm syndromes, including TNF-α, IL-1β, IL-6, and IFN-γ, all have a role to play in early host defence [16]. TNF-α and IL-1β stimulate endothelial activation, vascular permeability, and leukocyte recruitment, promoting rapid immune cell access to infected tissues. IL-6 bridges between local inflammation and systemic metabolic and acute-phase responses and makes an energy redistribution in favour of immune activity [17]. IFN-γ improves the antimicrobial effect of macrophages and induces the differentiation of cytotoxic lymphocytes [18]. Experimental models show that genetic deletion or early neutralisation of these cytokines typically leads to poor clearing of pathogens and elevated mortality rates, highlighting their protective roles in the early stages of infection [19].
Hypercytokinemia can thus be an evolutionarily adaptive emergency form of immune response, which is most efficient at short-term survival and least efficient at long-term tissue maintenance [20]. This trade-off is consistent with the evolutionary fitness principle, where survival to reproductive age is more important than risk of collateral tissue damage or chronic disease. In pre-modern settings where medical treatment was minimal and pathogen load was high, the inability to develop adequately robust inflammatory reactions would have most probably resulted in a more severe relative disadvantage than short-term immunopathology.
At the physiological level, the cytokine amplification is tightly controlled by the feedback and counter-regulatory systems, such as anti-inflammatory cytokines like IL-10 and TGF-β, anti-inflammatory proteins like SOCS, and immune checkpoints [21]. The damaging nature of hypercytokinemia is mainly seen when these regulatory loops are surpassed, delayed or made useless by host conditions like ageing, metabolic impairment, genetic susceptibility, or massive tissue damage. Notably, the shift to adaptive or maladaptive inflammation is seldom sudden; instead, it is an active imbalance between the pro-inflammatory force and restraint represented by regulatory mechanisms [22].
The experimental infection models have clearly shown that early pro-inflammatory cytokinesignalling plays a critical role in host survival. The deletion of tumour necrosis factor-alpha or interleukin 1 beta, or the pharmacological blocking of these cytokines, in murine systems exposed to bacterial and intracellular pathogens, alters the clearance of the pathogen, the recruitment of the myeloid cells and increases mortality, hence stressing the critical role of early cytokine amplification in effective host defence [23].
These studies validate that cytokines historically linked with the so-called cytotoxic storm pathways are evolutionarily preserved mediators of protective immunity and that the indiscriminate inhibition of such pathways can undermine survival in acute infection [24,25].
This physiological and evolutionary framing disputes the assumed intrinsic abnormality of cytokine storms. Rather, they can be imagined as context-specific immune programmes whose pathological implications can only be realised when the scale, duration and localization of cytokine signalling surpass the ability of the host to regulate and repair. The acknowledgement of hypercytokinemia as a conditional response is a more subtle basis to the explanation of the fact that similar cytokine reactions can prove useful in certain situations and disastrous in others, and it highlights the significance of timing and context in therapy.
4 Context Matters: Disease-Specific Cytokine Storm Phenotypes
Despite the common deployment of cytokine storm as a unifying term to describe extreme forms of inflammatory conditions, growing evidence suggests that cytokine amplification is more of a phenotype per disease, not a pathological process [26], as shown in table 1. Superficially analogous rises in circulating cytokines may be a consequence of profoundly disparate immunological structures, cellular stimuli and tissue settings. To understand clinical data, and develop effective immunomodulatory strategies, it is crucial to appreciate this heterogeneity.
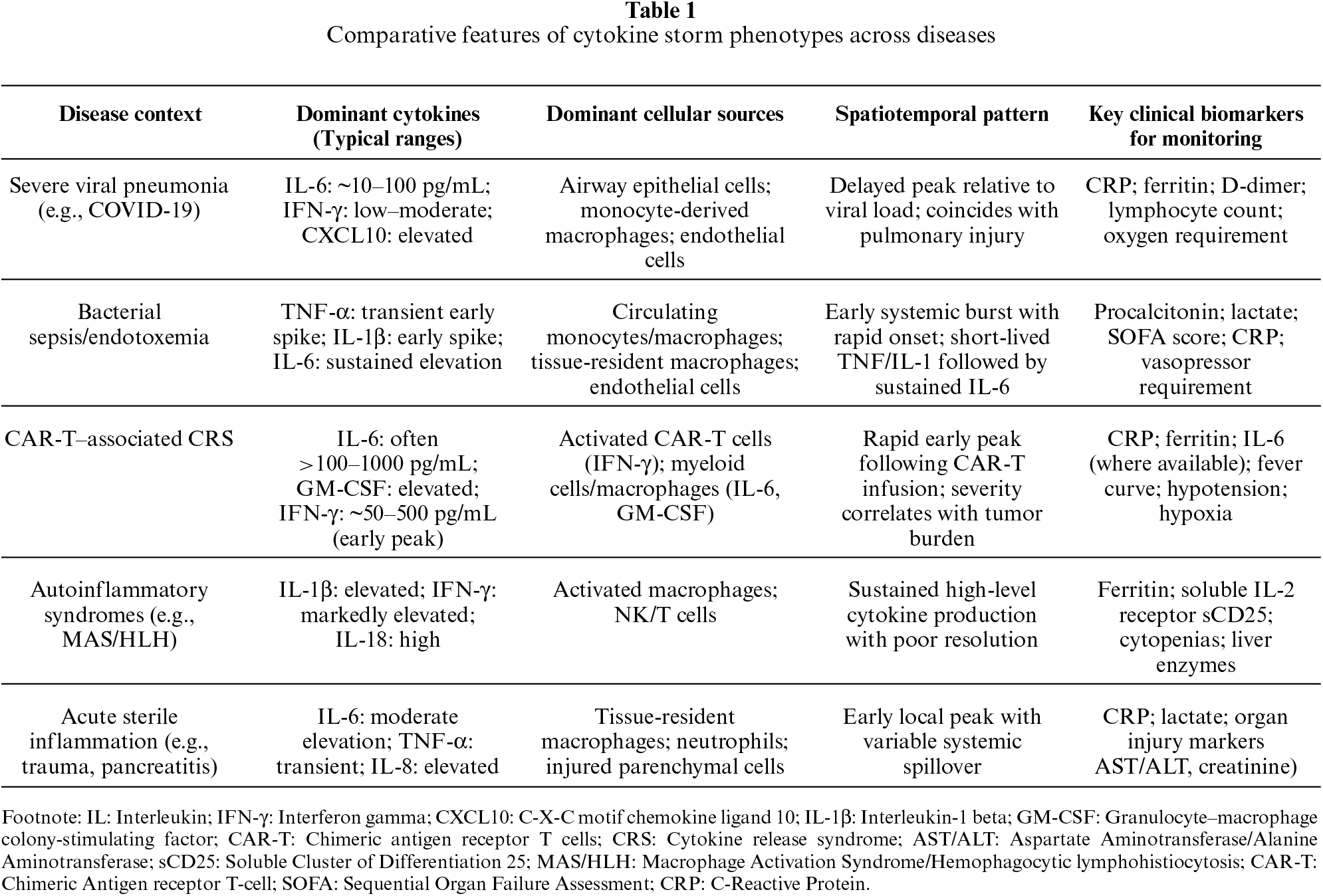
The dominant cytokine patterns are suggestive of the etiological character of the inflammatory stimulus and the particular pattern-recognition pathways activated. Viral recognition by intracellular means selectively triggers interferon-mediated transcriptional programmes; endotoxin-mediated responses by the innate immune system predominantly result in TNF or IL-1 mediated responses [27]. Increased IL-1 family cytokine response in autoinflammatory diseases is linked to dysregulated inflammasome activity, and concerted T-cell activation is linked to IL-6-dominant cytokine responses in cytokine release syndromes after chimeric antigen receptor T-cell therapy [28].
Not only has evolutionary selection shaped the magnitude of inflammatory responses, but it has also limited the architecture of cytokine programs in a trigger-specific fashion, producing predictable disease-associated inflammatory phenotypes. Intracellularly replicating pathogens selectively activate nucleic acid-sensing pathways, and interferon-dominated antiviral restriction and cytotoxic effector activation immune programs. By contrast, extracellular bacterial products and circulating pattern-recognition receptor-binding pathogen-associated molecular patterns selectively activate surface and endosomal pattern-recognition receptors, thereby causing an immediate tumor necrosis factor and IL-1-driven program leading to endothelial activation, increase vascular permeability, and recruitment of myeloid cells [29]. Such conserved response architectures produce stereotyped patterns of cytokine dominance across disease categories and make inflammatory phenotypes partially predictable according to pathogen localization and the mode of immune sensing [30]. Pathological cytokine storm phenotypes are therefore not the consequence of haphazard dysregulation, but of the hyper-activation of the modules of response that are evolutionarily shaped to operate within an adaptive range of cytokine programs.
Cytokine amplification is an adaptive immunity-innate antiviral signalling interaction in viral diseases like influenza and SARS-CoV-2 infection [31]. Notable ones include type I and type II interferons, IL-6, IL-8 and chemokines, including CXCL10, with major sources being the myeloid cells and infected epithelial cells. Notably, the production of cytokines in such environments tends to be spatially confined in infected tissues, especially in the lung, where local signalling coordinates immune cell recruitment and antiviral defences. Systemic cytokine levels are modest in severe COVID-19 in comparison with classical cytokine release syndromes, but lung-specific inflammation and endothelial activation are eminent [32]. This trend indicates that tissue-focal cytokine action, instead of systemic hypercytokinemia, might be the major pathogenic force in most viral diseases.
4.2 Bacterial Sepsis and Endotoxemia
In comparison, the rapid cytokine release typical of bacterial sepsis occurs as a systemic response to pathogen-associated molecular patterns, including lipopolysaccharide. TNF-α, IL-1β, and IL-6 are early and high in concentration and may be released long before the organ dysfunction becomes apparent [32]. Here, cytokine amplification is closely linked with systemic vascular reactions, such as vasodilation, capillary leakage, and coagulopathy. In contrast to viral infections, in which cytokine signalling can be confined to a compartment, sepsis-related hypercytokinemia is often associated with global endothelial and stromal activation, which is part of distributive shock and multi-organ failure [33]. Importantly, there is a high rate of failure of clinical trials of single cytokines in sepsis, indicating the redundancy and strength of these inflammatory networks.
4.3 Autoimmune and Autoinflammatory Disorders
In autoimmune and autoinflammatory diseases, a cytokine storm, as a consequence of immune dysregulation, occurs due to non-exogenous pathogens. Diseases like systemic juvenile idiopathic arthritis and adult-onset Still’s disease involve repeated episodes of cytokine amplification mediated by IL-1β, IL-6, and IL-18, which frequently occur with macrophage activation syndrome [34]. In this case, hypercytokinemia indicates the inability of immune regulation and tolerance, and the formation of self-perpetuating inflammatory processes [35]. In contrast to infection-related storms, these syndromes tend to be very sensitive to specific cytokine blockage, and the role of disease aetiology in shaping therapeutic susceptibility is essential.
4.4 Iatrogenic Cytokine Storms
Chimeric antigen receptor T-cell therapy-related iatrogenic cytokine storms are an exclusive immunological entity [36]. In this context, engineered T cells can activate in vast numbers and simultaneously on antigen presentation to produce supraphysiologic amounts of IL-6, IFN-γ and GM-CSF, thereby triggering cytokine release syndrome. Cytokine release in CAR-T therapy can be greater in magnitude and kinetics than that found in infectious diseases, but the syndrome can be reversed with prompt treatment [37]. Such reversibility is an indication of the temporally transient character of the provoking stimulus, and the comparative maintenance of regulatory pathways after cytokine amplification is inhibited.
4.5 Implications of Phenotypic Heterogeneity
Combined, these disease-specific patterns evinced that cytokine storms can be viewed as the convergent outcomes of a wide variety of upstream processes but not as a single mechanism [2]. There can be similar cytokines, but the sources, timing, spatial changes, and downstream effects vary greatly. The inconsistent use of the cytokine storm label thus runs the risk of obscuring any biological differences and may lead to poor treatment results [38]. The accurate diagnosis, prognostication and choice of treatment require a context-sensitive model which separates pathogen-mediated, autoimmune, and iatrogenic cytokine amplification.
5 Temporal Dynamics and Threshold Effects in Cytokine Amplification
The process of cytokine signalling dynamism, its biological outcomes depend not only on the magnitude but also on the time, duration, and sequence [39]. Classical definitions of cytokine storms tend to be based on fixed measurements of circulating cytokines, but increasingly growing data are showing that time dependence and threshold effects are essential elements to define whether cytokine responses are adaptive or rendered immunopathological [9]. The lack of consideration of these dimensions has been one of the contributing factors to inconsistent interpretations of cytokine data and variable therapeutic effects.
Initial production of cytokines is often protective, mediating the activation of antimicrobial defences, immune cell recruitment, and tissue-resident repair programmes. Rapid induction of TNF-α, IL-1β and type I interferons is transiently associated with the successful control of pathogens in both experimental models of infection and in clinical environments [40]. On the other hand, slow or delayed cytokine signalling is associated more with tissue injury and organ dysfunction. To illustrate, prolonged existence of IL-6 and IFN-γ further than the initial inflammatory window upgrades endothelial activation, metabolic deviation, and immune cell fatigue, especially in profound viral illnesses and sepsis [41].
The interpretation of cytokine amplification is also complicated by threshold effects. Cytokines tend to have a nonlinear dose response because at a certain threshold, a further increase in dose causes a qualitatively new cellular programme to be activated [42]. At these lower limits, cytokine signalling can lead to increased host protection with minimal collateral tissue damage; at higher limits, the pathways can trigger feed-forward inflammatory responses, vascular exudation, and coagulopathy [43]. Significantly, these thresholds are not absolute but depend on tissue context, receptor expression, and intracellular signalling competence. Older age, chronic inflammation and metabolic disease can reduce inflammatory thresholds, making hosts more vulnerable to cytokine-mediated damage at cytokine levels that would be tolerated by younger or healthier hosts.
Immune outcomes are also determined by the temporal sequencing of cytokine signals. The characteristic features of an effective immune resolution are coordinated responses in cytokine production, including early TNF-α and IL-1β followed by IL-6 mediated acute-phase reactions and subsequent induction of regulatory mediators [44]. Alteration of this cascade, either by inhibition of counter-regulatory cytokines, such as IL-10, or the untimely depletion of immune cells, pre-empts unregulated amplification. In this respect, cytokine storms might not necessarily indicate excessive inflammation, but a defect in the switch to resolution phases.
The threshold effects of cytokine amplification at the molecular level are supported by self-amplifying inflammatory loops that couple cytokine signalling to regulated cell death and secondary danger signalling. PANoptosis, a collective term including pyroptosis, apoptosis, and necroptosis, is triggered by innate sensors, including ZBP1 and NLRP3, in response to viral nucleic acids, microbial products, and cytokines. Upon its activation, PANoptotic cell death facilitates the loss of damage-associated molecular patterns that then activate innate immune signalling, resulting in positive-feedback loops enhancing cytokine synthesis [45].
Simultaneously, the synergistic interaction of tumour necrosis factor and interferon-7 enhances inflammatory transcriptional programmes through the JAK-STAT1-IRF1-iNOS axis, and reduces the threshold of effective endothelial triggering, metabolic re-programming of tissue injury. These molecular feedbacks give a mechanistic account of non-linear dose-response behaviour, in which small increases in cytokine signalling may trigger qualitatively different inflammatory settings when regulatory buffering capacity is crossed [46].
Such sources of knowledge are significant to clinical intervention. Interventions to treat disease conditions without taking into consideration the temporal context will suppress positive initial inflammation or act too late to reverse tissue damage [47]. The low potency of blanket cytokine inhibition under various conditions, including sepsis and severe viral infection, is probably partly due to the late intervention compared to the overall inflammatory progression. An elegant comprehension of cytokine storm syndromes will thus necessitate incorporation of time dynamics and threshold effects in both diagnostic and therapeutic decisions [48]. Regarding cytokine amplification as a context-dependent and time-sensitive process offers a better perspective for defining an adaptive immune escalation versus maladaptive immunopathology.
Molecular feedback mechanisms further contribute to nonlinear cytokine amplification. Processes such as PANoptosis and cytokine-driven transcriptional activation can generate positive inflammatory feedback loops that intensify cytokine production once regulatory thresholds are crossed. These mechanisms link cytokine signalling to regulated cell death pathways and secondary danger signalling, amplifying inflammatory cascades during severe immune activation. The detailed molecular mechanisms underlying these processes are discussed in Section 6.
6 When Does Adaptation Become Pathology? Determinants of Harmful Cytokine Storms
The shift between adaptive cytokine amplification and pathological cytokine storm is not marked by a single, solitary molecular event but is instead a product of a combination of host-, tissue-, and context-specific determinants that combine to overwhelm regulatory capacity [49] as shown in table 2. Although the proper maintenance of cytokine signalling is a critical requirement of the host defence system, its pathophysiological implications are manifested when the magnitude, duration, or localization of the inflammatory responses surpass the tolerance and resolution capacity of the tissues to immune-mediated stress [50].

Host intrinsic factors are at the centre of determining vulnerability to adverse cytokine storms. Cytokine receptors, signalling intermediates and regulatory pathways can vary genetically, and inflammatory thresholds can be altered, making some individuals more susceptible to hyper-response [51]. Severe inflammatory phenotypes in infectious and autoimmune diseases have been linked to polymorphisms of either IL-6 signalling, interferon pathways, or inflammasome components. Another essential predictor is age; immunosenescence and inflammaging are defined by baseline increases in pro-inflammatory substances and the inability to resolve the situation, which reduces the threshold at which the amplification of cytokines turns pathogenic [52]. Similarly, metabolic comorbidities, including obesity and diabetes, increase inflammatory signalling via increased production of adipokines, mitochondrial malfunction, and chronic low-grade inflammation.
Tissue-specific vulnerability is also the factor that can define whether cytokine amplification leads to reversible adaptation or irreversible damage. TNF-α, IL-1, and IL-6, the endothelial cells are particularly sensitive to prolonged exposure, which leads to increased permeability, procoagulant activity and leukocyte adhesions [53]. Prolonged cytokine signalling disrupts gas exchange, alveolar-capillary barriers, and causes impairment in the lung, and acute-phase responses in the liver that may contribute to system-wide inflammation [54]. Notably, tissue damage per se is a form of feedback that enhances the generation of cytokines by the release of damage-associated molecular patterns, which generate self-perpetuating loops of inflammatory response [55].
The overlapping of cytokine-induced signalling and cell death-induced amplification loops combine to switch between adaptive cytokine amplification and pathological cytokine storm. The inflammatory cytokine exposure is linked to massive immunogenic cell death by dysregulated activation of PANoptosis triggered by sensors like ZBP1 and NLRP3, therefore, enhancing the local load of damage-associated molecular patterns and sustaining the innate immune activation [56]. Meanwhile, prolonged joint presence of tumor necrosis factor and interferon-gamma supports an inflammatory feed-forward by the JAK-STAT1-IRF1-iNOS pathway, which induces endothelial dysfunction, nitric oxide-mediated metabolic derangement and barrier destabilization [57]. Stimulated by delayed or inadequate anti-inflammatory cytokines and intracellular negative regulators, these self-reinforcing loops allow cytokine signalling to decouple with pathogen burden, leading to threshold-crossing pathology with progressive tissue damage and multi-organ failure. Such a mechanistic interplay of cytokine amplification with controlled cell death is a molecular account of how initially adaptive immune amplification may shift to self-perpetuating immunopathology [58].
The breakdown of the counter-regulatory mechanisms is a leading edge in the development of pathology. Inflammatory quieting agents that include anti-inflammatory cytokines (IL-10 and TGF-b), negative regulators like suppressor of cytokine signalling proteins and immune checkpoint pathways usually control the inflammatory escalation process and resolution [59]. In the case of the delayed or insufficient, or functionally inadequate functioning of these systems, such as in severe sepsis, macrophage activation syndrome, and severe viral infections, the amplification of cytokines is decoupled from regulatory control. This environment allows inflammation to continue despite pathogen burden reduction, converting a protective response into tissue damage [60]. The convergence of host susceptibility, tissue context, and impaired regulatory feedback determines the transition from adaptive cytokine amplification to immunopathology (figure 2).
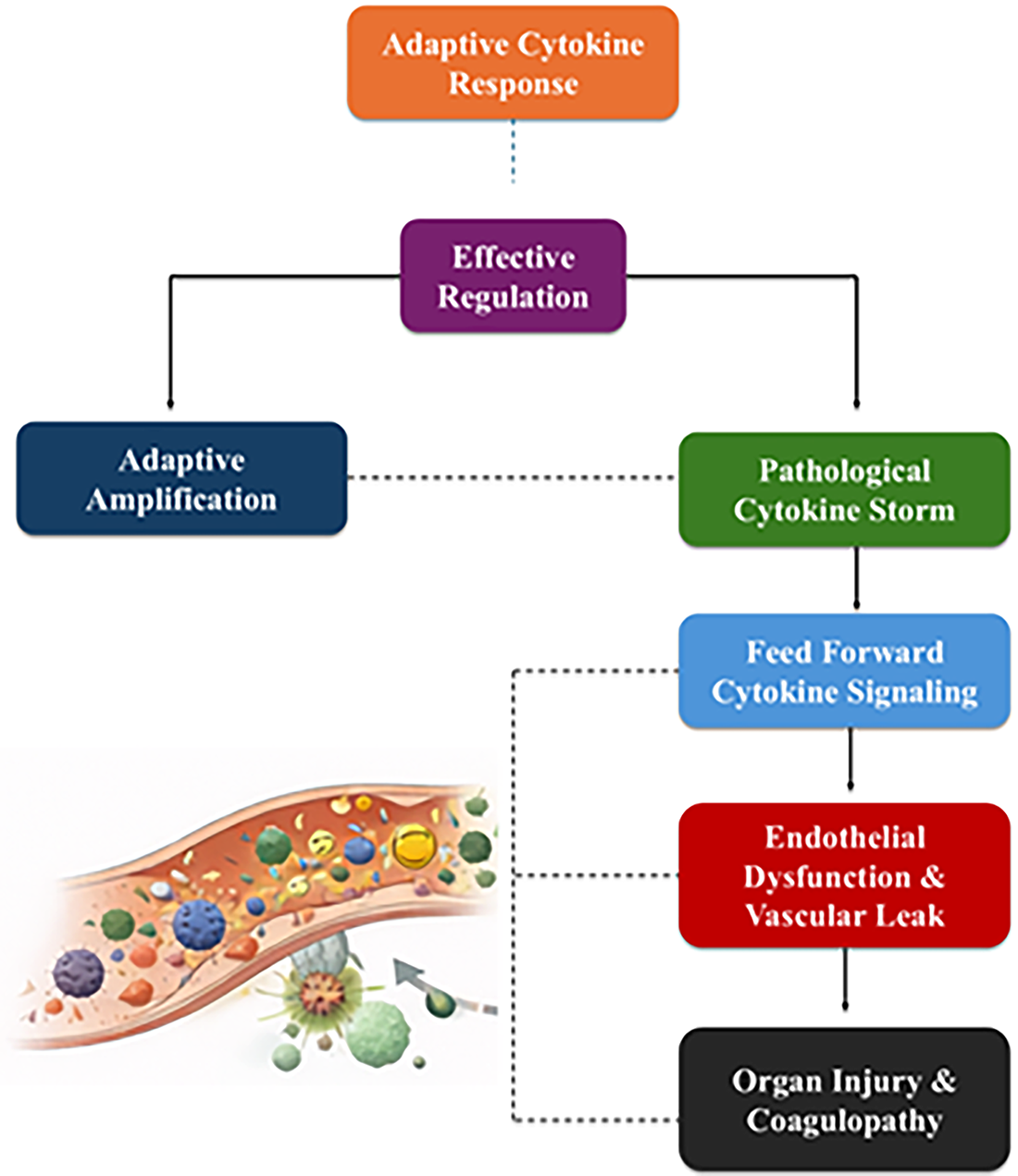
Figure 2: Determinants and progression to pathological cytokine storms.
The shift between adaptive and pathological amplification of cytokines provides a biologically stable construct in clinical decision-making. Risk stratification must identify those with persistent systemic inflammation, endothelial damage, organ dysfunction, and as having proven unable to maintain regulatory mechanisms since such individuals have the highest probability of responding to specific cytokine modulation. In contrast, patients with an early, focal, and strictly controlled cytokine reaction can experience the negative effects of early cytokine inhibition, due to impaired clearance of pathogens. The therapeutic window is thus time-limited; protection often is provided by early signals of cytokines, but in the long term, sustained signals of cytokines are thought to mediate immunopathology [58,61].
Lastly, this transition can be affected by outside sources like the timing of therapeutic intervention and iatrogenic immune modulation. Excessive immunosuppressive therapy can impair host defence, but late treatment can do nothing to correct the inflammatory damage that has already occurred [62]. Collectively, these determinants demonstrate that harmful cytokine storms are not due to excessive production of cytokines, but to the failure of coherent regulatory architecture. Such a multifactorial transition is critical to differentiate adaptive immune escalation and maladaptive immunopathology and to address patients most prone to respond to a targeted intervention.
7 Therapeutic Implications: Rethinking Anti-Cytokine Strategies
The predominant perception of cytokine storms as a pathological process has driven the development of therapies towards general inhibition of inflammatory mediators. Nevertheless, the small and frequently contradictory efficacy of anti-cytokine interventions in a wide range of inflammatory diseases highlights the necessity to reevaluate the approach [63]. Single cytokine trials, including TNF-α, IL-1β, or IL-6 in sepsis, have failed to enhance survival, although they were able to reduce the levels of circulating cytokines. These consequences indicate that alone cytokine amplification is an incomplete therapeutic target and that indiscriminate blockade could interfere with protective immune activities needed to control pathogens and repair tissue [64].
The timing has become an important determinant of therapeutic efficacy. Host defence is often mediated by early cytokine signalling, with later or chronic signalling playing a more direct role in tissue injury. Interventions that fail to consider the stage of disease risk either suppress good early inflammation or, in contrast, are administered late during disease progression and are no longer able to modify existing immunopathology [47]. This principle can be demonstrated by the context-dependent effectiveness of corticosteroids in severe COVID-19: any benefits were found in patients with more severe respiratory failure, whereas early treatment in the case of mild disease did not benefit and may harm. These results highlight that anti-cytokine strategies should be timely in correspondence with the underlying inflammatory course.
The heterogeneity of patients also makes it difficult to target therapy. Cytokine storm syndromes are a diverse group of immunological phenotypes whose upstream and host factors, as well as tissue contexts, are different [2]. Homogenous treatment strategies used in diverse groups of patients water down what might otherwise be beneficial and blunt appreciable responses in characterised subsets. Strict stratification according to inflammatory profiles, aetiology of the disease, and immune regulation biomarkers is thus a necessity to select patients who are most likely to respond to cytokine-directed therapies.
The development of new therapeutic paradigms is focused on managing but not suppressing cytokine signalling. Inhibition of selective pathways, partial agonism, and context-sensitive modulation promise the potential to reduce harmful inflammation without compromising key immune functions [65]. Downstream signalling node or endothelial dysfunction, or metabolic outcome of inflammation approaches can be of broader benefit than upstream cytokine blockade alone. Furthermore, approaches to modulate endogenous regulatory processes, including the amplification of IL-10 signalling or reinstatement of immune checkpoint functions, are promising alternatives to direct cytokine inhibition [66].
Therapeutic approaches must shift to biomarker-directed, context-sensitive immunomodulation, rather than generalised cytokine blockade. Dynamic inflammatory patterns, signs of regulatory malfunction, and early signs of endothelial damage and organ dysfunction can help to identify patients who are most susceptible to targeted intervention and avoid premature cytokine-mediated inhibition of protective immune reactions. The clinical experience of immune-effector-cell-related toxicities and stage-specific immunomodulation in severe viral pneumonia shows that precision strategies, when administered at the right stages of inflammation, can mitigate immunopathology without damaging vital host responses [67,68].
Taken together, these observations suggest the necessity to stop using one-size-fits-all anti-cytokine interventions and instead move towards contextualised therapeutic approaches. The reconsideration of cytokine storms as conditional immune programmes and not homogeneous pathologies offers a more logical basis to formulate interventions with better correlation to disease stage, host vulnerability and immunological context, which leads to better clinical outcomes.
8 Toward a Revised Framework: Cytokine Storms as Contextual Immune Programs
The accumulated data discussed in this paper question the current view of cytokine storms as homogenous obliterations of immune regulation. Rather, cytokine amplification ought to be conceptualized as a continuum of context-specific immune programs that are the result of interactions of pathogen weight, tissue microenvironment, host vulnerability, and regulatory efficiency [69]. An updated paradigm should thus step out of the stagnant measures of circulating cytokines and into a multidimensional approach that can reflect the dynamic and circumstantial character of the inflammatory signalling.
The most critical aspect of this framework is the understanding that cytokine storms are characterized not only by their magnitude, but also by qualitative characteristics of the signalling processes, such as the source of cells, localization, kinetics, and engagement with downstream pathways [2]. The same cytokine pattern in peripheral blood can be indicative of entirely different biological processes when signalling is confined to damaged tissues versus being distributed throughout the system, is temporary or persistent, and is checked or unchecked by regulation [70]. These parameters can be included to discriminate adaptive immune escalation against maladaptive inflammatory pathology.
We hypothesize the idea of defining cytokine storm syndromes as a continuum with two extremes. On one end, there are adaptive amplification states, where healthy cytokine signalling amplifies pathogen clearance, or tissue repair, and is solved via intact counter-regulatory states. On the opposite end are the pathological amplification states, which is a state of prolonged, or spatially imbalanced signalling, endothelial injury, metabolic derangement, and resolution failure [71]. The direction of the movement along this scale is determined by the age, genetic background, comorbidities of a host, disease-specific provoking factors, and therapeutic interventions.
There are practical implications of this framework to both research and clinical care. The biomarker strategies should emphasize dynamic and compartment-specific measurements rather than static systemic thresholds, with temporal sampling and relevant tissue readouts where possible [72]. Clinically, it is possible to stratify patients based on where they are placed on the adaptive-pathological continuum and inform the timing, intensity, and targets of immunomodulatory therapy. Notably, this methodology embraces heterogeneity within and between disease entities and minimises dependence on disease-agnostic nomenclatures that blur mechanistic differences.
In opposition to traditional cytokine storm models, which rely on predetermined systemic cytokine thresholds and see hypercytokinemia as an a priori pathological process, this model views cytokine amplification as a dynamic, context-specific immune programme determined by temporal dynamics, localisation, cellular origins, regulatory capacity and host vulnerability. This view redefines cytokine storms as conditional immune states and not monolithic regulatory failures, which offers a more accurate conceptual basis of biomarker-guided, stage-specific immunomodulation.
This framework offers a consistent basis for further inquiry by replacing the current cytokine storms with the rebranded contextual immune programs, instead of sole regulatory malfunctions. It highlights the necessity of integrative, systems-level studies and the formation of precision immunomodulatory approaches based on immune context, but not only on the amount of cytokines.
This framework is mostly conceptual, and thus it is based on the synthesis of heterogeneous literature as opposed to prospective validation. The scarcity of tissue-specific and longitudinal immunoprofiling datasets restricts the possibility of establishing causal relationships related to cytokine kinetics and dysregulated immune control. The heterogeneity of the disease and variability among individuals may hinder direct clinical application of the findings. In turn, the proposed clinical implications are hypothesis-generating in nature and should be validated using prospective, biomarker-directed interventional research.
9 Future Directions and Conclusion
To translate this framework into clinical practise, methodological improvements will be needed that go beyond the measurement of static systemic cytokines to more integrative, tissue-resolved, and temporally dynamic immune profiling. The longitudinal monitoring of inflammatory patterns, the use of compartment-specific biomarkers that represent tissue-specific immune action, and the combination of cellular and molecular markers of regulatory breakdown should be a priority in future research. Recent technological advances, such as single-cell and spatially resolved immune profiling, have the potential to map patient-specific inflammatory programmes and to determine actionable thresholds between adaptive immune amplification and pathological amplification. Such strategies can guide the creation of precision immunomodulatory strategies that optimise therapeutic targets and timing to the individual immune context and therefore reduce interference of protective host defence, in addition to selectively suppressing damaging inflammatory pathways. Finally, future biomarker-directed interventional research will be necessary to understand that context-dependent immunomodulation enhances clinical outcomes in different phenotypes of cytokine storms.
Cytokine storm syndromes are often described in the context of homogeneous approaches towards immune dysregulation due to excessive cytokine release. It is in opposition to this reductionist perspective and in favour of a re-conceptualization of cytokine amplification as a spectrum of context-dependent immune programmes facilitated by disease aetiology, tissue microenvironment, host susceptibility, time dynamics, and regulatory capacity. Instead of being a pathologically pathological entity, hypercytokinemia is often an evolutionarily conserved and physiologically significant part of host defence and pathological effects manifest where containment of space, time or counter-regulatory responses are not maintained. This view offers a consistent explanation of the pronounced heterogeneity of infectious,autoimmune/autoinflammatory and iatrogenic inflammatory syndromes, and of the variable clinical efficacy of non-selective cytokine blockade.
Hypercytokinemia has often been perceived as a discrete pathological condition, but evidence indicates that it should be viewed as a spectrum of immune activation that can be adaptive or maladaptive depending on the circumstances. These findings and the determination of exact thresholds and dynamic immune landscapes will enable clinicians to maximise the timing and specificity of treatment interventions by weighing between the necessity to control pathogens and the danger of increasing tissue damage.
Future research should prioritize several key questions: (1) what quantitative cytokine thresholds reliably distinguish adaptive immune amplification from pathological inflammation across disease contexts; (2) how tissue-specific cytokine signaling differs from systemic measurements in predicting clinical outcomes; (3) whether longitudinal cytokine profiling can identify early transition points from protective to pathological inflammation; and (4) how host genetic or metabolic factors modulate cytokine signaling thresholds in infectious and inflammatory diseases.
This viewpoint presents a holistic approach to the variability of the cytokine storm phenotypes, where patients have unique trajectories that require individualised treatment. It highlights the significance of considering the dynamic interplay of cytokine signatures, cellular infiltrates, and tissue-specific responses to facilitate more subtle clinical decision-making.
Notably, the redefinition of cytokine storms as conditional immune states highlights the primacy of immune context and time of day in defining whether cytokine amplification is adaptive or maladaptive and opens the way to the development of efficacious and safe precision-guided immunomodulatory therapies.
Acknowledgement: We thank the authors for permitting the use of their details and acknowledge the support of ISF College of Pharmacy, Moga, and the School of Pharmaceutical Sciences, CT University, Ludhiana.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: Hardik Kumar and Kamaljeet contributed towards the conception or design of the manuscript and manuscript writing. Kamaljeet is the first and main author of the manuscript, writing and collecting data for the present study. Kamaljeet, Abhishek Vijukumar, Sourabh Kosey and Shilpa Debnath are contributors to the manuscript for collecting and analyzing data. Hardik Kumar reviewed the final manuscript and gave suggestions. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: No approval is required.
Conflicts of Interest: The authors declare no conflicts of interest.
References
1. Jarczak D, Nierhaus A. Cytokine storm—definition, causes, and implications. Int J Mol Sci 2022;23(19):11740. doi:10.3390/ijms231911740. [Google Scholar] [PubMed] [CrossRef]
2. Karki R, Kanneganti TD. The ‘Cytokine storm’: molecular mechanisms and therapeutic prospects. Trends Immunol 2021;42(8):681–705. doi:10.1016/j.it.2021.06.001. [Google Scholar] [PubMed] [CrossRef]
3. Henrie R, Cherniawsky H, Marcon K et al. Inflammatory diseases in hematology: a review. Am J Physiol Cell Physiol 2022;323(4):C1121–36. doi:10.1152/ajpcell.00356.2021. [Google Scholar] [PubMed] [CrossRef]
4. Navarro Quiroz R, Villarreal Camacho J, Zarate Peñata E et al. Multiscale information processing in the immune system. Front Immunol 2025;16:1563992. doi:10.3389/fimmu.2025.1563992. [Google Scholar] [PubMed] [CrossRef]
5. Cumming E, Peters C. Immune response to infection. Anaesth Intensive Care Med 2024;25(6):439–43. doi:10.1016/j.mpaic.2024.04.003. [Google Scholar] [CrossRef]
6. Roshanravan N, Seif F, Ostadrahimi A, Pouraghaei M, Ghaffari S. Targeting cytokine storm to manage patients with COVID-19: a mini-review. Arch Med Res 2020;51(7):608–12. doi:10.1016/j.arcmed.2020.06.012. [Google Scholar] [PubMed] [CrossRef]
7. Carcillo JA, Shakoory B. Cytokine storm and sepsis-induced multiple organ dysfunction syndrome. In: Cytokine Storm Syndrome. Cham, Switzerland: Springer International Publishing; 2019:451–64. doi:10.1007/978-3-030-22094-5_27 [Google Scholar] [CrossRef]
8. Del Valle DM, Kim-Schulze S, Huang HH et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med 2020;26(10):1636–43. doi:10.1038/s41591-020-1051-9. [Google Scholar] [PubMed] [CrossRef]
9. Liu C, Chu D, Kalantar-Zadeh K, George J, Young HA, Liu G. Cytokines: from clinical significance to quantification. Adv Sci 2021;8(15):2004433. doi:10.1002/advs.202004433. [Google Scholar] [PubMed] [CrossRef]
10. Sabioni L, De Lorenzo A, Lamas C et al. Systemic microvascular endothelial dysfunction and disease severity in COVID-19 patients: evaluation by laser doppler perfusion monitoring and cytokine/chemokine analysis. Microvasc Res 2021;134(8):104119. doi:10.1016/j.mvr.2020.104119. [Google Scholar] [PubMed] [CrossRef]
11. Xu G, Qi F, Li H et al. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discov 2020;6(1):73. doi:10.1038/s41421-020-00225-2. [Google Scholar] [PubMed] [CrossRef]
12. Masselli E, Pozzi G, Gobbi G et al. Cytokine profiling in myeloproliferative neoplasms: overview on phenotype correlation, outcome prediction, and role of genetic variants. Cells 2020;9(9):2136. doi:10.3390/cells9092136. [Google Scholar] [PubMed] [CrossRef]
13. Adekola P. Multi-scale modeling in cancer systems biology: linking transcriptomic landscapes to clinical decision-making. Clin Transl Rep 2025;2(2):39–51. [Google Scholar]
14. Mehta P, Fajgenbaum DC. Is severe COVID-19 a cytokine storm syndrome: a hyperinflammatory debate. Curr Opin Rheumatol 2021;33(5):419–30. doi:10.1097/bor.0000000000000822. [Google Scholar] [PubMed] [CrossRef]
15. West NR. Coordination of immune-stroma crosstalk by IL-6 family cytokines. Front Immunol 2019;10:1093. doi:10.3389/fimmu.2019.01093. [Google Scholar] [PubMed] [CrossRef]
16. Moradian N, Gouravani M, Salehi MA et al. Cytokine release syndrome: inhibition of pro-inflammatory cytokines as a solution for reducing COVID-19 mortality. Eur Cytokine Netw 2020;31(3):81–93. doi:10.1684/ecn.2020.0451. [Google Scholar] [PubMed] [CrossRef]
17. Kistner TM, Pedersen BK, Lieberman DE. Interleukin 6 as an energy allocator in muscle tissue. Nat Metab 2022;4(2):170–9. doi:10.1038/s42255-022-00538-4. [Google Scholar] [PubMed] [CrossRef]
18. Meftahi GH, Jangravi Z, Sahraei H, Bahari Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: the contribution of “inflame-aging”. Inflamm Res 2020;69(9):825–39. doi:10.1007/s00011-020-01372-8. [Google Scholar] [PubMed] [CrossRef]
19. Torrado E, Cooper AM. Cytokines in the balance of protection and pathology during mycobacterial infections. Adv Exp Med Biol 2013;783(8):121–40. doi:10.1007/978-1-4614-6111-1_7. [Google Scholar] [PubMed] [CrossRef]
20. Bektas A, Schurman SH, Franceschi C, Ferrucci L. A public health perspective of aging: do hyper-inflammatory syndromes such as COVID-19, SARS, ARDS, cytokine storm syndrome, and post-ICU syndrome accelerate short- and long-term inflammaging? Immun Ageing 2020;17(1):23. doi:10.1186/s12979-020-00196-8. [Google Scholar] [PubMed] [CrossRef]
21. Al Razee A. Pharmacologic Neuromodulation Targeting Neuroinflammation as a Novel Therapeutic Strategy for Experimental Pulmonary Hypertension. Dissertation. Los Angeles, CA, USA: University of California; 2023. [Google Scholar]
22. Halper-Stromberg A, Jabri B. Maladaptive consequences of inflammatory events shape individual immune identity. Nat Immunol 2022;23(12):1675–86. doi:10.1038/s41590-022-01342-8. [Google Scholar] [PubMed] [CrossRef]
23. Pfeffer K, Matsuyama T, Kündig TM et al. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 1993;73(3):457–67. doi:10.1016/0092-8674(93)90134-c. [Google Scholar] [PubMed] [CrossRef]
24. Raupach B, Kaufmann SHE. Bacterial virulence, proinflammatory cytokines and host immunity: how to choose the appropriate vaccine strain? Microbes Infect 2001;3(14–15):1261–9. doi:10.1016/s1286-4579(01)01486-1. [Google Scholar] [PubMed] [CrossRef]
25. Mahdavi J, Royer PJ, Sjölinder HS et al. Pro-inflammatory cytokines can act as intracellular modulators of commensal bacterial virulence. Open Biol 2013;3(10):130048. doi:10.1098/rsob.130048. [Google Scholar] [PubMed] [CrossRef]
26. Saboowala HK. What is “Cytokine Storm”? Understanding its role in the pathology of clinical & infectious disease at molecular level. 2020 [cited 2026 Jan 1]. https://www.news-medical.net/health/What-is-a-Cytokine-Storm.aspx [Google Scholar]
27. Arora H. ABCF1 is a Novel E2 Ubiquitin-Conjugating Enzyme That Controls Toll-Like Receptor-Mediated Innate Immune Responses and Cytokine Storm During Sepsis. Vancouver, BC, Canada: University of British Columbia; 2018. [Google Scholar]
28. Broderick L, De Nardo D, Franklin BS, Hoffman HM, Latz E. The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol Mech Dis 2015;10(1):395–424. doi:10.1146/annurev-pathol-012414-040431. [Google Scholar] [PubMed] [CrossRef]
29. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010;140(6):805–20. doi:10.1016/j.cell.2010.01.022. [Google Scholar] [PubMed] [CrossRef]
30. Karki R, Netea MG, Diorio C, Kanneganti TD. Cytokine storm. Nat Rev Dis Primers 2026;12(1):1. doi:10.1038/s41572-025-00677-4. [Google Scholar] [PubMed] [CrossRef]
31. Schiuma G, Beltrami S, Bortolotti D, Rizzo S, Rizzo R. Innate immune response in SARS-CoV-2 infection. Microorganisms 2022;10(3):501. doi:10.3390/microorganisms10030501. [Google Scholar] [PubMed] [CrossRef]
32. McGonagle D, Ramanan AV, Bridgewood C. Immune cartography of macrophage activation syndrome in the COVID-19 era. Nat Rev Rheumatol 2021;17(3):145–57. doi:10.1038/s41584-020-00571-1. [Google Scholar] [PubMed] [CrossRef]
33. Makic MBF, Morata LT. Shock, sepsis, and multiple organ dysfunction syndrome. In: Sole’s Introduction to Critical Care Nursing-E-Book: Sole’s Introduction to Critical Care Nursing-E-Book. Amsterdam, The Netherlands: Elsevier; 2024. p. 258. [Google Scholar]
34. Hausmann JS. Targeting cytokines to treat autoinflammatory diseases. Clin Immunol 2019;206:23–32. doi:10.1016/j.clim.2018.10.016. [Google Scholar] [PubMed] [CrossRef]
35. Yasmeen F, Pirzada RH, Ahmad B, Choi B, Choi S. Understanding autoimmunity: mechanisms, predisposing factors, and cytokine therapies. Int J Mol Sci 2024;25(14):7666. doi:10.3390/ijms25147666. [Google Scholar] [PubMed] [CrossRef]
36. Cron RQ, Goyal G, Chatham WW. Cytokine storm syndrome. Annu Rev Med 2023;74(1):321–37. doi:10.1146/annurev-med-042921-112837. [Google Scholar] [PubMed] [CrossRef]
37. Santurio DS, Barros LRC, Glauche I, Fassoni AC. Mathematical modeling unveils the timeline of CAR-T cell therapy and macrophage-mediated cytokine release syndrome. PLoS Comput Biol 2025;21(4):e1012908. doi:10.1371/journal.pcbi.1012908. [Google Scholar] [PubMed] [CrossRef]
38. Panoskaltsis N. Are all cytokine storms the same? Cancer Immunol Immunother 2021;70(4):887–92. doi:10.1007/s00262-020-02822-2. [Google Scholar] [PubMed] [CrossRef]
39. Callard R, George AJT, Stark J. Cytokines, chaos, and complexity. Immunity 1999;11(5):507–13. doi:10.1016/s1074-7613(00)80125-9. [Google Scholar] [PubMed] [CrossRef]
40. Laczko R, Chang A, Watanabe L et al. Anti-inflammatory activities of Waltheria indica extracts by modulating expression of IL-1β, TNF-α, TNFRII and NF-κB in human macrophages. Inflammopharmacology 2020;28(2):525–40. doi:10.1007/s10787-019-00658-6. [Google Scholar] [PubMed] [CrossRef]
41. Kunkl M, Sambucci M, Ruggeri S et al. CD28 and associated class 1A P13K regulates the glycolytic metabolic program associated to pro-inflammatory T cell responses in multiple sclerosis. Eur J Immunol 2019;49(suppl. 1):188–9. [Google Scholar]
42. Guéguen Y, Bontemps A, Ebrahimian TG. Adaptive responses to low doses of radiation or chemicals: their cellular and molecular mechanisms. Cell Mol Life Sci 2019;76(7):1255–73. doi:10.1007/s00018-018-2987-5. [Google Scholar] [PubMed] [CrossRef]
43. Thimmappa PY, Vasishta S, Ganesh K, Nair AS, Joshi MB. Neutrophil (dys)function due to altered immuno-metabolic axis in type 2 diabetes: implications in combating infections. Hum Cell 2023;36(4):1265–82. doi:10.1007/s13577-023-00905-7. [Google Scholar] [PubMed] [CrossRef]
44. Guo Y, Hu K, Li Y et al. Targeting TNF-α for COVID-19: recent advanced and controversies. Front Public Health 2022;10:833967. doi:10.3389/fpubh.2022.833967. [Google Scholar] [PubMed] [CrossRef]
45. Karki R, Sundaram B, Sharma BR et al. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep 2021;37(3):109858. doi:10.1016/j.celrep.2021.109858. [Google Scholar] [PubMed] [CrossRef]
46. Karki R, Sharma BR, Tuladhar S et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 2021;184(1):149–68.e17. doi:10.1016/j.cell.2020.11.025. [Google Scholar] [PubMed] [CrossRef]
47. Rossi JF, Lu ZY, Massart C, Levon K. Dynamic immune/inflammation precision medicine: the good and the bad inflammation in infection and cancer. Front Immunol 2021;12:595722. doi:10.3389/fimmu.2021.595722. [Google Scholar] [PubMed] [CrossRef]
48. Nie J, Zhou L, Tian W et al. Deep insight into cytokine storm: from pathogenesis to treatment. Sig Transduct Target Ther 2025;10(1):112. doi:10.1038/s41392-025-02178-y. [Google Scholar] [PubMed] [CrossRef]
49. Kumar NR, Balraj TA, Kempegowda SN, Prashant A. Multidrug-resistant sepsis: a critical healthcare challenge. Antibiotics 2024;13(1):46. doi:10.3390/antibiotics13010046. [Google Scholar] [PubMed] [CrossRef]
50. Ahmad HI, Jabbar A, Mushtaq N et al. Immune tolerance vs. immune resistance: the interaction between host and pathogens in infectious diseases. Front Vet Sci 2022;9:827407. doi:10.3389/fvets.2022.827407. [Google Scholar] [PubMed] [CrossRef]
51. Webb CE, Vautrinot J, Hers I. IL-6 as a mediator of platelet hyper-responsiveness. Cells 2025;14(11):766. doi:10.3390/cells14110766. [Google Scholar] [PubMed] [CrossRef]
52. Teissier T, Boulanger E, Cox LS. Interconnections between inflammageing and immunosenescence during ageing. Cells 2022;11(3):359. doi:10.3390/cells11030359. [Google Scholar] [PubMed] [CrossRef]
53. Wilhelm G, Mertowska P, Mertowski S et al. The crossroads of the coagulation system and the immune system: interactions and connections. Int J Mol Sci 2023;24(16):12563. doi:10.3390/ijms241612563. [Google Scholar] [PubMed] [CrossRef]
54. Gigliotti S, Guerriero G, Mazza G et al. Perioperative blood biomarkers of infectious and non-infectious postoperative pulmonary complications: a narrative review. J Clin Med 2026;15(2):699. doi:10.3390/jcm15020699. [Google Scholar] [PubMed] [CrossRef]
55. Siametis A, Garinis GA. From genome to geroscience: how DNA damage shapes systemic decline. BioEssays 2025;47(10):e70051. doi:10.1002/bies.70051. [Google Scholar] [PubMed] [CrossRef]
56. Pandeya A, Kanneganti TD. Therapeutic potential of PANoptosis: innate sensors, inflammasomes, and RIPKs in PANoptosomes. Trends Mol Med 2024;30(1):74–88. doi:10.1016/j.molmed.2023.10.001. [Google Scholar] [PubMed] [CrossRef]
57. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-γ: an overview of signals, mechanisms and functions. J Leukoc Biol 2004;75(2):163–89. doi:10.1189/jlb.0603252. [Google Scholar] [PubMed] [CrossRef]
58. Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev 2018;281(1):8–27. doi:10.1111/imr.12621. [Google Scholar] [PubMed] [CrossRef]
59. Hadžimusić N, Hadžijunuzović-Alagić D. Mediators of the acute phase response: the role of cytokines. Int J Agric Environ Res 2025;11(2):337–50. doi:10.51193/ijaer.2025.11203. [Google Scholar] [CrossRef]
60. Crayne CB, Albeituni S, Nichols KE, Cron RQ. The immunology of macrophage activation syndrome. Front Immunol 2019;10:119. doi:10.3389/fimmu.2019.00119. [Google Scholar] [PubMed] [CrossRef]
61. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med 2020;383(23):2255–73. doi:10.1056/nejmra2026131. [Google Scholar] [PubMed] [CrossRef]
62. Beaugerie L, Kirchgesner J. Balancing benefit vs risk of immunosuppressive therapy for individual patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol 2019;17(3):370–9. doi:10.1016/j.cgh.2018.07.013. [Google Scholar] [PubMed] [CrossRef]
63. Sharma Y, Bala K. Multifarious aspect of cytokines as an immuno-therapeutic for various diseases. J Interf Cytokine Res 2024;44(11):477–85. doi:10.1089/jir.2024.0090. [Google Scholar] [PubMed] [CrossRef]
64. Strzelec M, Detka J, Mieszczak P, Sobocińska MK, Majka M. Immunomodulation—a general review of the current state-of-the-art and new therapeutic strategies for targeting the immune system. Front Immunol 2023;14:1127704. doi:10.3389/fimmu.2023.1127704. [Google Scholar] [PubMed] [CrossRef]
65. Maziz MNH, Chakravarthi S, Aung T et al. Microglia-mediated neuroinflammation through phosphatidylinositol 3-kinase signaling causes cognitive dysfunction. Int J Mol Sci 2025;26(15):7212. doi:10.3390/ijms26157212. [Google Scholar] [PubMed] [CrossRef]
66. Rallis KS, Corrigan AE, Dadah H et al. IL-10 in cancer: an essential thermostatic regulator between homeostatic immunity and inflammation—a comprehensive review. Future Oncol 2022;18(29):3349–65. doi:10.2217/fon-2022-0063. [Google Scholar] [PubMed] [CrossRef]
67. Varadarajan I, Kindwall-Keller TL, Lee DW. Management of cytokine release syndrome. In: Chimeric Antigen Receptor T-Cell Therapies for Cancer E-Book: A Practical Guide. Amsterdam, The Netherlands: Elsevier; 2020. p. 45–64. doi:10.1016/b978-0-323-66181-2.00005-6 [Google Scholar] [CrossRef]
68. Yang C, Zhao H. Tocilizumab in COVID-19 therapy: who benefits, and how? Lancet 2021;398(10297):299. doi:10.1016/s0140-6736(21)01380-5. [Google Scholar] [PubMed] [CrossRef]
69. Mallick S, Duttaroy AK, Bose B. A snapshot of cytokine dynamics: a fine balance between health and disease. J Cell Biochem 2025;126(1):e30680. doi:10.1002/jcb.30680. [Google Scholar] [PubMed] [CrossRef]
70. Serrano MA, Gomes AMC, Fernandes SM. Monitoring of the forgotten immune system during critical illness—a narrative review. Medicina 2023;59(1):61. doi:10.3390/medicina59010061. [Google Scholar] [PubMed] [CrossRef]
71. Pacinella G, Ciaccio AM, Tuttolomondo A. Endothelial dysfunction and chronic inflammation: the cornerstones of vascular alterations in age-related diseases. Int J Mol Sci 2022;23(24):15722. doi:10.3390/ijms232415722. [Google Scholar] [PubMed] [CrossRef]
72. Adaku KC. Exploring the relationship between body fluid compartments and health outcomes in patients with sickle cell disease among sub-Saharan African: a systematic mini-review. Int J Eng Res Technol 2025;12(10):TIJER2510063. doi:10.55248/gengpi.4.723.49251. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools