
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Glucocorticoid reduces the efficacy of afatinib on the head and neck squamous cell carcinoma
State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, Chinese Academy of Medical Sciences Research Unit of Oral Medicine of Carcinogenesis and Management, West China Hospital of Stomatology, Sichuan University, Chengdu, 610041, China
* Corresponding Author: YINGQIANG SHEN. Email:
(This article belongs to the Special Issue: Recent Advancement in Cancer Molecular Signaling)
BIOCELL 2023, 47(2), 329-338. https://doi.org/10.32604/biocell.2023.023489
Received 28 April 2022; Accepted 28 July 2022; Issue published 18 November 2022
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Glucocorticoids (GC) are widely used to counter the adverse events during cancer therapy; nonetheless, previous studies pointed out that GC may reduce the efficacy of chemotherapy on cancer cells, especially in epidermal growth factor receptor (EGFR)-targeted therapy of head and neck squamous cell carcinoma (HNSCC) remaining to be elucidated. The primary aim of the present study was to probe into the GC-induced resistance of EGFR-targeted drug afatinib and the underlying mechanism. HNSCC cell lines (HSC-3, SCC-25, SCC-9, and H-400) and the human oral keratinocyte (HOK) cell lines were assessed for GC receptor (GR) expression. The promoting tumor growth effect of GC was evaluated by the CCK-8 assay and flow cytometry. Levels of signaling pathways participants GR, mTOR, and EGFR were determined by quantitative polymerase chain reaction and western blotting. GC increased the proliferation of HNSCC cells in a GR-dependent manner and promoted AKT/mTOR signaling. But GC failed in counteracting the inhibition of rapamycin in the mTOR signaling pathway. Besides, GC also induced resistance to EGFR-targeted drug afatinib through AKT/mTOR instead of the EGFR/ERK signaling pathway. Thus, GCs reduce the efficacy of afatinib on HNSCC, implicating a cautious use of glucocorticoids in clinical practice.Keywords
The adrenal cortex and peripheral tissues produce the steroid hormone glucocorticoid (GC), which is involved in critical physiological functions such as metabolic control, immunosuppression, anti-inflammatory, and cognitive signaling (Slominski et al., 2013). Cortisol biosynthesis and metabolism also occur in the majority of solid tumors, named the cancer-associated glucocorticoid (CAG) system, while its role in malignancy has not been fully investigated to date (Celentano et al., 2019; Cirillo et al., 2017). In the treatment of solid tumors, GC is used to combat the adverse reactions caused by anti-tumor therapy and control tumor-related complications (Herr and Pfitzenmaier, 2006). According to first-line medication guidelines, GCs have a wide array of use in oral, potentially malignant disorders (Ioannides et al., 2020). Dexamethasone and hydrocortisone (HYD), noted as widely used synthetic GCs are administered before chemotherapy to reduce significant edema, hypersensitivity reactions, and dermatologic toxicities (Dreyfuss et al., 1996). However, the potential unfavorable effects of GC on solid tumors have received increasing attention in recent years (Planey and Litwack, 2000). The landmark study of Obradović et al. (2019) found that GCs promoted breast cancer metastasis through the GC receptor and kinase ROR1. Besides, studies have shown that GCs could promote the progression of cancer and reduce the efficacy of chemotherapy drugs (Azher et al., 2016; Xing et al., 2015), especially in epithelial carcinomas. The effect of GCs on malignant transformation and progression of head and neck squamous cell carcinoma (HNSCC) is unsubstantiated, which demands further studies in view of the CAG system and the wide application of GC in the clinical environment.
Head and neck squamous cell carcinoma (HNSCC) is the sixth highest cause of morbidity due to cancer worldwide. In the past three decades, despite advances in diagnosis and treatment techniques, the survival rate of HNSCC has risen only slightly (Ferlay et al., 2019; Pulte and Brenner, 2010), which ascribes to the recurrence, metastasis, and resistance of HNSCC after treatment (Shen et al., 2018). Cisplatin is one of the preferred chemotherapy drugs for HNSCC, whereas, in those with advanced age patients who cannot tolerate cisplatin or with recurrent/metastatic cancer who are resistant to chemotherapy, immune/targeted therapy is an ideal mode of adjuvant access (Bonner et al., 2006; Johnson et al., 2020). The sole targeted approved therapy for HNSCC is the monoclonal anti-epidermal growth factor receptor (EGFR) antibody cetuximab, which is used to treat HNSCC that has spread to other organs. Afatinib (AFA), a second-generation EGFR tyrosine kinase inhibitor (TKI), is regarded as the perfect medication due to its irreversible activity and the capacity to interact with many receptors (Ferrarotto and Gold, 2014). But it should not be overlooked that salient resistance was acquired in both therapies mentioned above (Hung et al., 2017; Westover et al., 2018). The steroid GC is frequently used to alleviate drug-induced nausea and pain, and its antagonism to the efficacy of chemotherapy drugs on oral squamous epithelial cells has been reported (Celentano et al., 2019). However, it is not clear whether GC might incur resistance to the EGFR-targeted drugs in HNSCC.
GCs bind to the GC receptor (GR, NR3C1) in the cytoplasm to form the GC-GR complex, followed by nuclear translocation for transcriptional regulation (Kleiman and Tuckermann, 2007). GC up-regulates anti-apoptotic genes and inhibits the expression of pro-apoptotic genes in the transcription profile of human keratinocytes (Stojadinovic et al., 2007). In addition, signal molecules such as AKT and ERK, motivated by GC, play vital roles in cell survival and proliferation, for either AKT/mTOR signaling contributing to the occurrence, progression, metastasis, and resistance of HNSCC (Liao et al., 2011; Nguyen et al., 2012), or ERK/MAPK signaling pathway facilitating cancer migration and invasion (Jeong et al., 2016). Given the correlation between the above signaling pathways and EGFR, whether GC adversely affects the efficacy of EGFR-targeting therapies in HNSCC is worth considering (Zhang et al., 2022).
Herein, we investigated the effects of HYD on HNSCC cells with different GR expression levels, and report the GC-induced resistance and the underlying mechanism to EGFR-targeted drug afatinib, to provide a reference for first-line clinical practice for HNSCC.
Five cell lines were used in this study, including HSC3, H400, SCC9, SCC25, and human oral keratinocyte (HOK). SCC9 and SCC25 cell lines were from the American-Type Culture Collection, USA. The H400 cell line was established at Bristol Dental School, the University of Bristol, UK (Prime et al., 1990). HSC3 cell line was gained from the Japanese Collection of Research Bioresource. The HOK cell line was provided by Dr. Xuan Liu, Charles R. Drew University of Medicine and Science.
The complete medium for HSC3 cells consisted of 89% Dulbecco’s Modified Eagle Medium (DMEM; HyClone, SH30243.01B), 10% fetal bovine serum (FBS; HyClone, SH30088.03) and 1% penicillin-streptomycin (Gibco, 15070063). The complete medium for HOK cells was composed of keratinocyte-SFM (Gibco, 10744019) supplemented with 1% penicillin-streptomycin. The complete medium for H400, SCC9 and SCC25 cells consisted of 89% DMEM: F12 1:1(Gibco, A4192002), 10% FBS, 1% penicillin-streptomycin and 1 µg/ mL hydrocortisone (HYD; MCE, HY-N0583). All cell lines were cultured in a cell incubator containing 5% CO2 at 37°C.
The HYD was dissolved in dimethyl sulfoxide (DMSO) to obtain a concentration of 10 mM. Ru486 1 mM; Selleck, S2606) was dissolved in DMSO; Rapamycin (Sigma, V900930) was dissolved in ethanol to obtain a 10 mM solution. Afatinib (MCE, HY-10261S) was dissolved in DMSO to obtain a 100 mM solution. The reagents prepared for tests were stored at −20°C before dilution and treatment. The cells in the experiment were cultured in the HYD-free medium for 24–72 h in advance before the experiment.
The cell densities were planned according to the experimental design. Cells were seeded with HYD-free medium into 96-well plates at 10%–20% confluency per well for the proliferation assay or at 70%–80% confluency per well for the apoptosis assay and incubated overnight. The treatment included HYD (1, 10, and 3 μM), AFA at different concentrations, and Ru486 of 1 μM. After the appropriate treatment time, the medium was replaced with 10 μL CCK-8 (Dojindo Molecular Technologies, DJDB4000X) and 100 μL medium per well, and the 96-well plates were placed back into the incubation for 1 h. The absorbance at 450 nM was measured by Varioskan Flash (Thermo Scientific, Shanghai, China), and the OD values were recorded. Each group was set with at least three replicates.
Quantitative real-time polymerase chain reaction (RT-qPCR)
Total RNA of cells was extracted using a TR250 extraction kit (Tianmobio, Beijing, China) according to instructions. The RNA concentration was measured by a spectrophotometer, and the amount of RNA was controlled at 1 µg for the reverse transcription reaction system, which used Prime-Script RT reagent KIT with gDNA Eraser (Takara, Japan). The reverse transcribed cDNA was stored at −20°C.
The RT-qPCR system in a 20 μL volume was prepared on ice as: cDNA Template 2 μL; 2*SYBR Green qPCR Mix 10 μL, 10 μM forward and reverse primers each 0.4, and 7.2 μL ddH2O. Real-time qPCR System (Thermo QuantStudio 5) was used for the amplification reaction, and Ct values were recorded. Based on the Ct value of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), gene expression levels were calculated by the 2−ΔΔCT method. The primers used in this study included GAPDH: forward primer 5’-CCTGTTCGACAGTCAGCCG-3’ and reverse primer 5’-CGACCAAATCCGTTGACTCC-3’, GR: forward primer 5’-TCCCTTTCTCAACAGCAGGAT-3’ and reverse primer 5’-CAATCATTCCTTCCAGCACAT-3’.
The proportion of apoptotic cells was analyzed using Annexin V/PI Apoptosis Detection Kit (KeyGEN, KGA107). After treatment, apoptotic cells were suspended in the culture medium, and adherent cells were collected and rinsed with PBS. After suspending cells in 500 μL binding buffer, 5 μL Annexin V-FITC was added and mixed in the dark for 10 min. Then 10 μL of PI was added and mixed for 5 min. The proportion of apoptotic cells was analyzed by flow cytometry (Beckman FC500, USA). The data were analyzed and represented using FlowJo V10 software.
After treatment, proteins were extracted from the collected cell pellet using RIPA lysate buffer and separated according to molecular weight by electrophoresis and transferred to polyvinylidene difluoride membranes (EMD Millipore, ISEQ00010). After sealing in 5% skim milk for about 1 h, the membranes were immersed in proportionally diluted primary antibody solutions and incubated overnight at 4°C. The membranes were washed thrice with tris-buffered saline with Tween (TBST) for 10 min on the shaker and incubated with an anti-rabbit antibody (1:4000; 7074, CST) at 37°C for 1 h. The excess antibody was washed with TBST, and the membranes were soaked with the developer. Finally, ECL (EMD Millipore, WBKLS0500) was used to visualize the protein band. Gray values of strips were measured by Image J software and were expressed as the percent relative to GAPDH.
Primary antibodies used in this study included GR (1:2000, AB183127, Abcam), AKT (1:2000, 4691T, CST), P-Akt (1:2000, 4060T, CST), mammalian target of rapamycin (mTOR; 1:2000, AB32028, Abcam), Phospho-S6 Ribosomal Protein (P-S6; 1:2000, 4858T, CST), GAPDH (1:2000, 5174, CST), epidermal growth factor receptor (EGFR; 1:2000, AB52894, Abcam), P-EGFR (1:2000, 3777T, CST), extracellular regulated protein kinases (ERK; 1:2000, AB184699, Abcam), P-ERK (1:2000, AB201015, Abcam).
Data analysis was performed using GraphPad Prism 8.3.0 and Image J software. Shapiro-Wilk normality test was used for normality analysis; one-way ANOVA and independent T-test were adopted for comparison of differences. The data were expressed as mean ± SD. P-value < 0.05 was statistically significant. The data of gene differential expression were obtained from The Cancer Genome Atlas Program (TCGA, USA) and analyzed using R studio.
The differences in GR expression were significant among HNSCC cell lines
To evaluate the effect of GC on HNSCC cells, we examined the level of GR mRNA in different cell lines through qPCR. GR mRNA was significantly lower in H400 and SCC25 cell lines but not in SCC9 and HSC-3 than that in normal squamous epithelial cells, HOK. The level of GR mRNA in SCC9 cells was significantly higher than that in H400 cells (Fig. 1A). Western blotting further confirmed the higher expression of GR protein in SCC9 than that in H400 cells (Fig. 1B). To explore the correlation between the effect of GC and GR expression, HSC-3, SCC9, and H400 were selected for functional validation. Differential gene expression analysis demonstrated that HNSCC tissue samples expressed higher levels of GR than normal tissues (Fig. 1C).
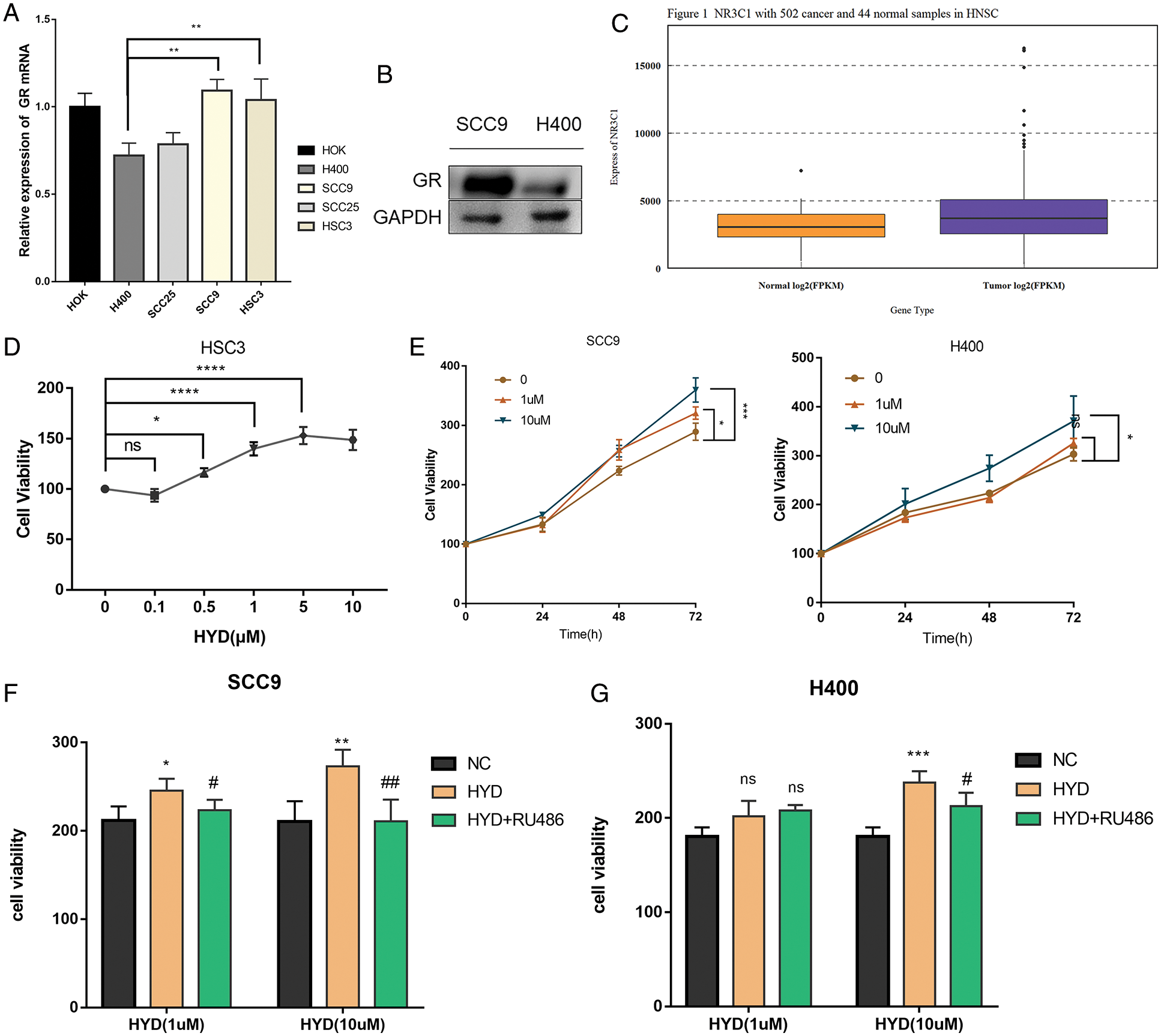
Figure 1: Enhanced proliferation of HNSCC cells in the presence of glucocorticoid, and the rescue by GR inhibitor RU486. (A) Expression of glucocorticoid receptor (GR) mRNA in different cell lines; (B) Difference of GR protein expression between SCC9 and H400 cell lines; (C) Difference of GR protein expression between HNSCC tissue and normal tissue samples. The Promoting proliferative effect of Hydrocortisone (HYD) in HNSCC cell lines: (D) The proliferation activity of HSC-3 cells was increased by increasing concentrations of HYD after 24 h; (E) Effects of different concentrations of HYD on the proliferation of SCC9 and H400 at 48 and 72 h; (F, G) GR inhibitor RU486 inhibited the promoting-proliferation effect of HYD. The cell viability was evaluated by the CCK-8 assay; The data are expressed as the mean ± SD of a minimum of three independent experiments. A P-value of <0.05 was considered to indicate statistical significance. (Negative control-NC; Hydrocortisone treatment-HYD).
Glucocorticoid promoted proliferation of HNSCC cells with the help of endogenous glucocorticoid receptor
Numerous studies have proved that GC is conducive to the survival of tumor cells (He et al., 2019). Data of CCK-8 implied that HYD markedly promoted the proliferation of HSC-3 in a concentration-dependent manner (Fig. 1D). Based on the difference in GR content, dual concentrations of HYD (1, 10 μM) were intervened to SCC9 & H400 cells for analysis at 0, 24, 48, and 72 h. The GR-dependent pro-proliferation effect persisted compared with the parallel control group (Fig. 1E), as low GR expression cell line H400 only responded to a high concentration of HYD, while SCC9 cells were sensitive to either concentration of HYD. These results indicate that HYD-mediated pro-proliferation effects followed a ubiquitous and dose-dependent mode.
To further confirm that HYD promoted the proliferation of HNSCC cells by binding to the GR, cells were treated with HYD combined with the GR inhibitor RU486 to block the GR-induced downstream cascade reactions and corresponding physiological processes. Examination of the cell viability at two concentrations of HYD (1, 10 μM) interfered with or without 1 μM RU486 at 48 h post-dosing. Compared with the HYD treatment group, the cell viability in the combination treatment group was abated (Figs. 1F, 1G), which indicated that GR inhibition by RU486 rescued the pro-proliferation effect of HYD. These results suggest that the effect of HYD on the proliferation of HNSCC was influenced by endogenous GR.
Glucocorticoid-mediated activation of AKT/mTOR signaling pathway in HNSCC cells
Previous studies have confirmed that GC acts on AKT/mTOR to stimulate tumor growth (Alzahrani, 2019; Ediriweera et al., 2019). Treatment with HYD significantly motivated the phosphorylation of both AKT and S6 (Figs. 2A–2E), suggesting that HYD might act as the activator of mTOR in HNSCC. Thus, we tested its antagonistic effect on mTOR inhibition induced by rapamycin. After treating with HYD/rapamycin, p-S6, the downstream target of mTORC1, was evaluated. As expected, treatment with rapamycin offset the HYD-induced phosphorylation of S6 compared with the rapamycin alone group, and HYD failed to counteract the inhibition of rapamycin in the change of p-S6 (Figs. 2F, 2G). The application of HYD or rapamycin alone promoted AKT phosphorylation. In SCC9, combined treatment increased the P-AKT level compared to the rapamycin group (Fig. 2I) but unexpectedly inhibited the expression of AKT in the H400 (Fig. 2H), which might be due to the feedback loop of AKT. Taken together, GC and not rapamycin activated the AKT/mTOR signaling pathway, which means that GC may not induce rapamycin resistance in HNSCC, suggesting the feasibility of rapamycin therapy in combination with GC.
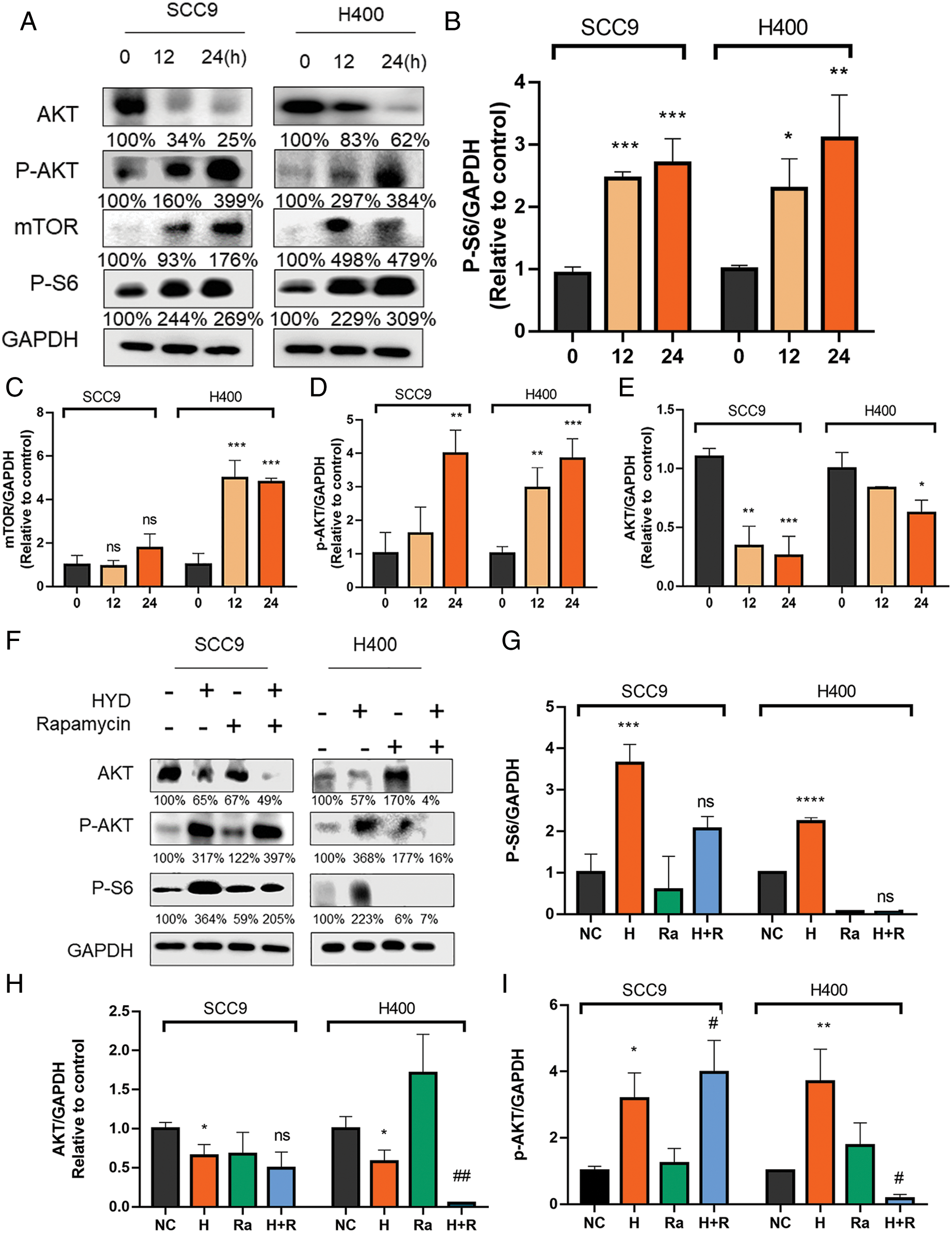
Figure 2: Hydrocortisone activated the AKT/mTOR signaling pathway, which is inhibited by the mTOR inhibitor rapamycin. Western blot analysis and densitometric quantification of pS6 (A, B), AKT (A, E), pAkt (A, D) and Mtor (A, C) in control (vehicle), hydrocortisone (3 μM, 12 or 24 h) treatment groups in the HNSCC cell lines SCC9 and H400. The treatment groups include control (vehicle), hydrocortisone (3 μM), rapamycin (20 nM) and HYD plus rapamycin. Western blot analysis of pS6 (F, G), AKT (F, I) and pAkt (F, H) in the HNSCC cell lines SCC9 and H400. Data are presented as mean ± SEM. n = 3 per group *P < 0.05, **P < 0.01, ***P < 0.001 vs. control. and #P < 0.05, ##P < 0.01, ###P < 0.001 vs. HYD-treated group. Ns, not statistically significant. (Negative control-NC; Hydrocortisone treatment-H; Rapamycin-RA; Hydrocortisone & Rapamycin-H+R).
Reduced efficacy of afatinib on HNSCC due to Glucocorticoid treatment
Next, we investigated the effects of GC on the EGFR signaling pathway in HNSCC. HYD treatment promoted the phosphorylation of EGFR (P-EGFR) and ERK (P-ERK) (Figs. 3A–3E). GCs have been reported to interfere with the ability of the potent EGFR-targeting medication afatinib (AFA) to treat HNSCC in light of the activation of GC on EGFR (Ferrarotto and Gold, 2014; Tagliamento et al., 2018). SCC9 and H400 were treated with AFA at different concentration gradients, with or without HYD, and cell viability was detected by CCK8 assay in 24 h. Compared with the AFA group, the combination treatment group improved the survival rate of both SCC9 and H400 cells (Figs. 4A, 4C). Moreover, the survival-promoting effect of HYD was more apparent in SCC9 than that in H400 cells, consistent with the previous results that SCC9 had a higher expression level of GR and was more sensitive to HYD.
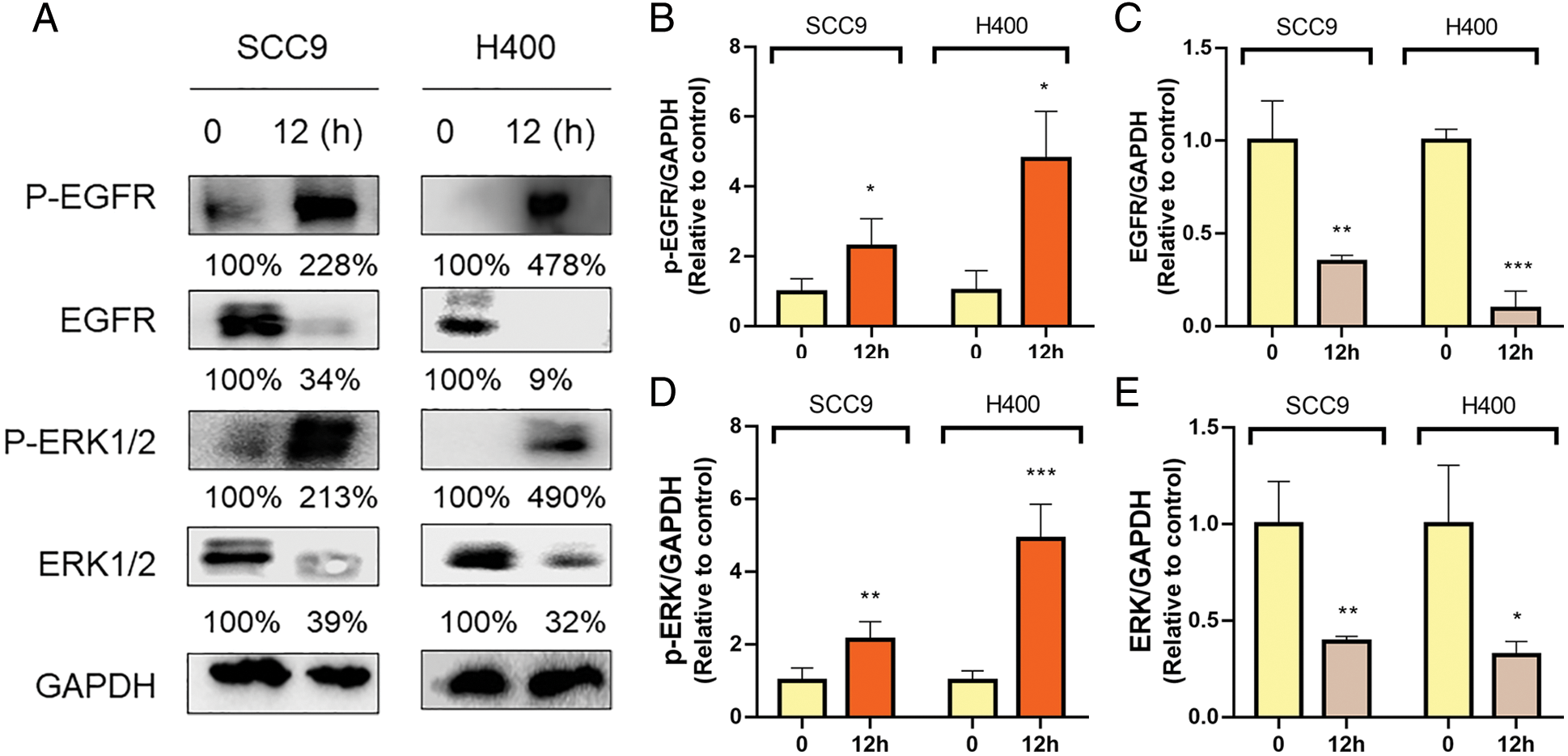
Figure 3: Hydrocortisone treatment modulated the EGFR/ERK signaling pathway. Western blot analysis and densitometric quantification of Pegfr (A, B), EGFR (A, C), pERK1/2 (A, D), and ERK1/2 (A, E) in the HNSCC cell lines SCC9 and H400. Treatment groups include control (vehicle), hydrocortisone (3 μM, 12 h). n = 2 per group. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control. ns means not statistically significant. (epidermal growth factor receptor-EGFR; phosphorylation of EGFR—P-EGFR; extracellular regulated protein kinases-ERK; phosphorylation of ERK—P-ERK).
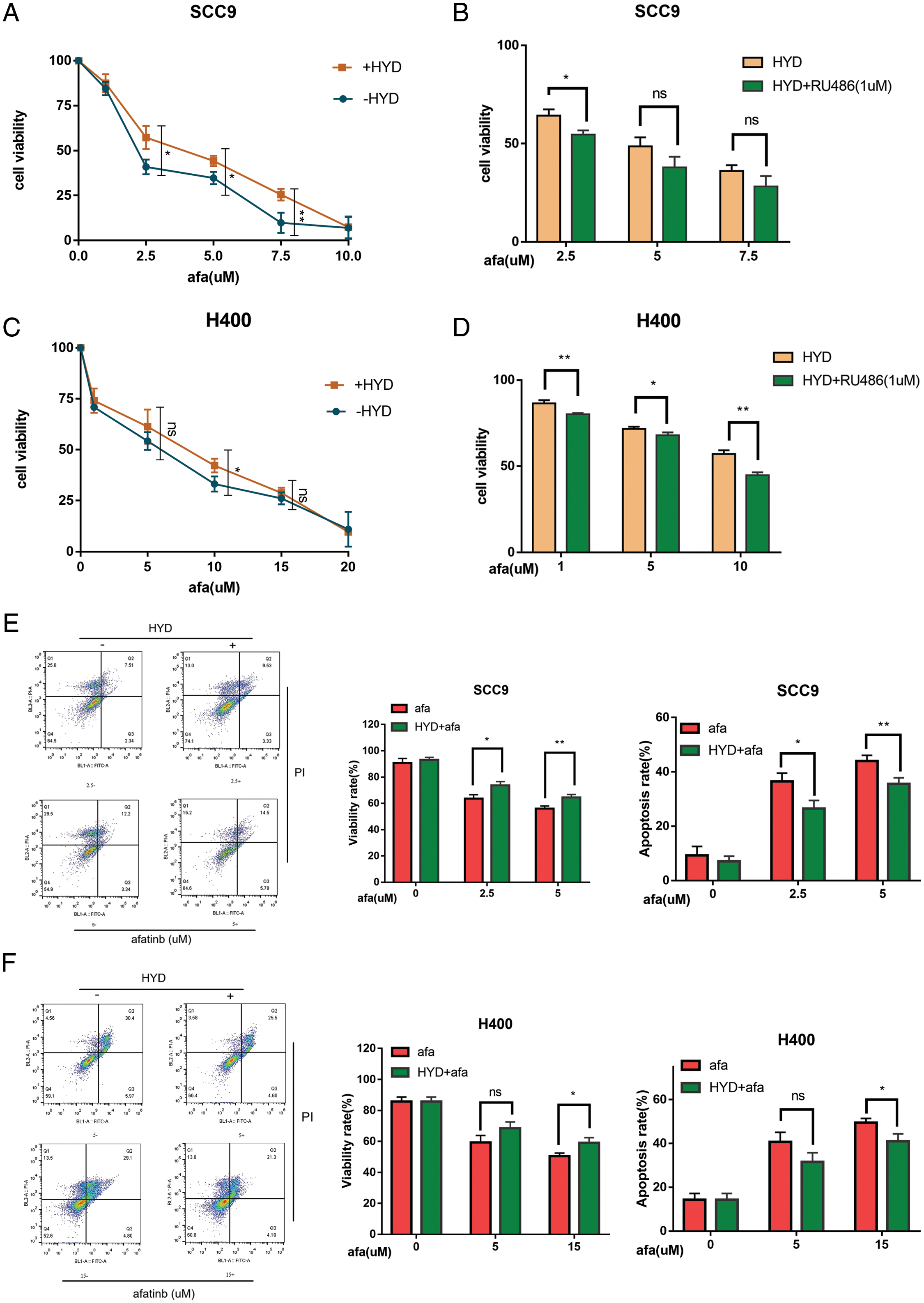
Figure 4: Hydrocortisone (HYD) inhibited the apoptosis-inducing effect of targeted drug afatinib (AFA) on HNSCC cell lines, and this was inhibited by GR inhibitor Ru486. (A, C) SCC9 and H400 cell lines were treated with different concentrations of AFA for 24 h, with or without HYD (3 μM). HYD suppressed the efficacy of AFA and improved IC50; (B, D) GR inhibitor RU496 blocked the effect of HYD on SCC9 and H400 cell lines and decreased the cell survival rate. The cell viability was evaluated by the CCK-8 assay; (E, F) SCC9 and H400 cell lines were treated with different concentrations of AFA for 24 h, with or without HYD (3 μM). The percentage of apoptosis was measured by Flow cytometry. The data are expressed as the mean ± SD of a minimum of three independent experiments. A P-value of <0.05 was considered to indicate statistical significance. ns means not statistically significant.
We proposed that GR dependency also contributes to the enhanced survival effect. To prevent the interaction between GR and HYD, GR inhibitor RU486 was used. Ru486 decreased cell survival and the anti-apoptotic effects of HYD on cells treated with AFA (Figs. 4B, 4D). Additionally, this impact was stronger in H400 cells (Fig. 4D). Furthermore, the results of flow cytometry revealed that combination treatment, when compared to AFA treatment alone, enhanced the survival rate and decreased the apoptotic rate of HNSCC cells (Figs. 4E, 4F). These results prove that GC decreased the effectiveness of AFA in HNSCC through GR.
Glucocorticoids-mediated inhibition of AFA efficacy by mTOR signaling pathway
SCC9 and H400 were treated with HYD to explore the underlying mechanism of GC and AFA interaction. Protein band analysis demonstrated that HYD promoted the phosphorylation of EGFR and ERK, but failed to increase the P-EGFR or P-ERK under the treatment of AFA (Figs. 5A–5C). Given our previous finding by which HYD exerts cancer-promoting effect, we assessed the mTOR signaling pathway. As anticipated, the combined treatment of AFA and HYD increased P-AKT and P-S6 levels compared with AFA treatment alone (Figs. 5D–5E). These findings demonstrate that AFA had no discernible effect on the HYD-induced activation of the mTOR signaling pathway, further suggesting that HYD may reduce the efficiency of AFA by activating the mTOR signaling pathway rather than the EGFR signaling pathway.
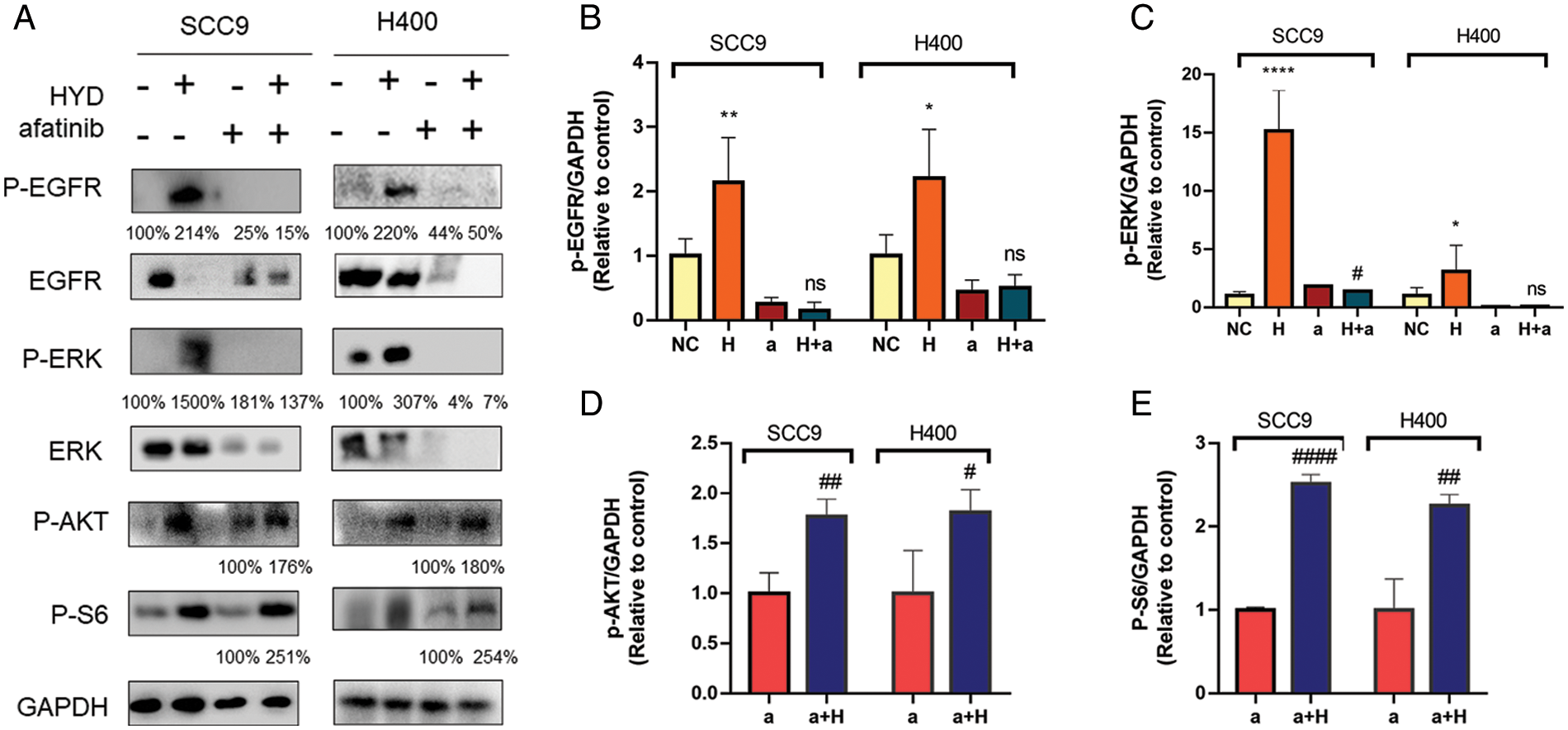
Figure 5: Hydrocortisone (HYD, H) activated mTOR signaling pathway but not EGFR under treatment of afatinib (AFA, A). Western blot analysis and densitometric quantification of p-EGFR (A, B), p-ERK (A, C), p-AKT (A, D), and p-S6 (A, E) in the HNSCC cell lines SCC9 and H400. Treatment groups include control (vehicle), hydrocortisone (3 μM), afatinib (5 μM for SCC9 and 10 μM for H400), and HYD plus AFA. n = 4 per group. Data are presented as mean ± SEM. n = 3 per group. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control, and #P < 0.05, ##P < 0.01, ###P < 0.001 vs. afatinib- treated group. ns means not statistically significant (epidermal growth factor receptor-EGFR; phosphorylation of EGFR—P-EGFR; extracellular regulated protein kinases-ERK; phosphorylation of ERK—P-ERK; Phospho-S6 Ribosomal Protein—P-S6).
Our results demonstrated the importance of GC on HNSCC cell viability and resisting the efficacy of EGFR-targeted drug afatinib. Fig. 6 depicts the schematic representation of the pro-cancerous activity of HYD via a signal network made up of EGFR/ERK and AKT/mTOR in HNSCC. Our findings potentially explain why cancers develop resistance to EGFR-targeted drugs used in clinical treatment and advise caution when using GC.
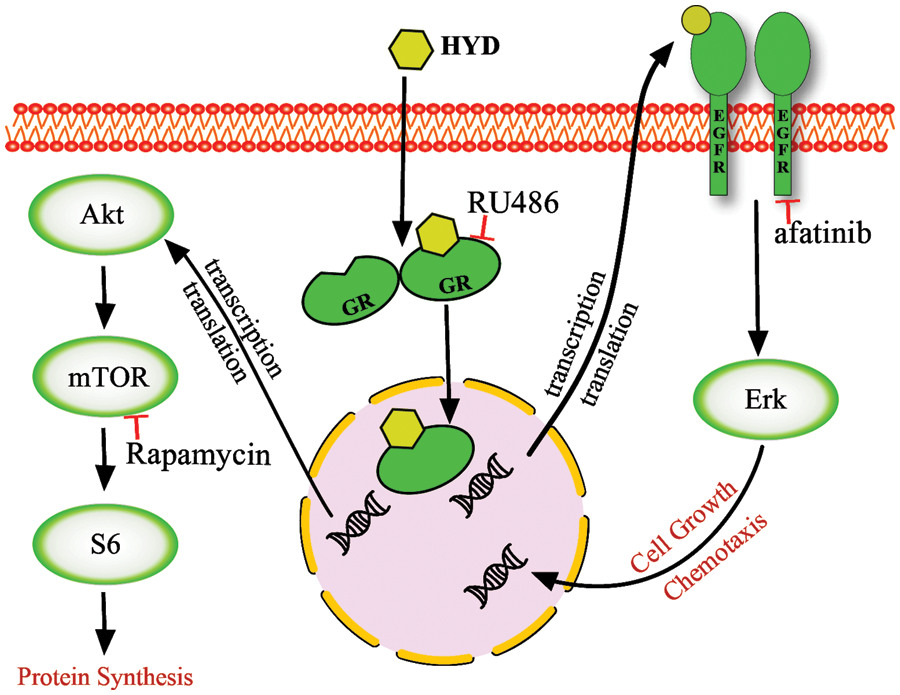
Figure 6: A model of glucocorticoid-mediated cross-signaling leading to tumor progression. Hydrocortisone (HYD) enters the cytoplasm and binds to the glucocorticoid receptor (GR) to form GC–GR homodimers that enter the nucleus, interact with other transcription factors, and bind to glucocorticoid receptor element-containing target genes. This activates the pathway of AKT/mTOR and EGFR/ERK. The downstream target S6 and ERK may converge into the occurrence, progression, and drug resistance of HNSCC. The GR antagonist mifepristone (RU486), the mTOR inhibitor rapamycin, and the EGFR inhibitor afatinib interfere with this crosstalk, but none of the inhibitors on their own can completely block it.
GCs and synthetic derivatives, frequently used to treat solid tumors, work with the GR to generate physiological responses in the transcription of target genes (Timmermans et al., 2019). In the current in-vitro study, HNSCC exhibited differences in susceptibility to the pro-malignant effects of GC in a GR-dependent manner. The increase in GR activation was associated with the proliferation and survival of tumor cells. Although the expression of GR showed no consistent difference between tumor cells and normal epithelial cells, differential analysis of gene expression demonstrated that HNSCC tissue samples expressed higher levels of GR than normal tissues. Together, these findings suggest that GR may contribute to the malignant phenotypes of solid tumors as a component of the cancer-associated GC (CAG) system. Reinforcing our hypothesis, previous studies have reported that GR acts as an oncoprotein and regulates the malignant phenotypes in stem-like cells and lip cancer (de Sena et al., 2018; Wang et al., 2015).
Synergistic with the action of GR, the potential malignant effects of GCs on solid tumors of epithelial origin have often been supported (Azher et al., 2016). We demonstrated that GC promoted HNSCC cell proliferation, which is related to the expression level of GR. A low concentration of GC (1 µm) promoted cell viability of HSC3 and SCC9 with higher GR expression, while the multiplying effect occurred in H400 with a lower GR level in the concentration of 10 µm. The GR inhibitor RU486 declined the proliferative effect of GC and brought cell viability back to or slightly above normal levels. Increased GR activity positively correlated with cell viability, while depletion of GR suppressed the proliferation of carcinoma cells (Tian et al., 2019).
Importantly, we further investigated how GC functions by triggering the AKT/mTOR signaling pathway and encouraging the phosphorylation of downstream protein S6. The GC-induced protein kinase may carry out this process by promoting AKT phosphorylation and activating downstream pathways. The amount of total protein was reduced, including that of AKT, even though changes in total protein were often undetectable. Similar findings for the AKT and EGFR proteins were reported in several studies on oral squamous-cell carcinoma, albeit they were not thoroughly explained (Feng et al., 2021; Li et al., 2018; Yin et al., 2022). As the downstream target of AKT, ribosomal protein S6 promotes protein synthesis and plays an important role in the proliferation of tumors (Yang et al., 2020). The mTOR pathway inhibitor hindered GC-induced S6 phosphorylation, and its presence decreased the proliferative viability of cells (Liao et al., 2011). Apart from the mTOR signaling pathway, we do not rule out the role of GC in tumors promotion through a complex network cascade, such as the activation of EGFR induced by GC shown in our results. Previous research reported that dexamethasone mediates proliferation of pancreatic cancer cells through GR, transforming growth factor-β and c-Jun N-terminal kinases/activator protein-1 (JNK/AP-1) (Liu et al., 2017). While there are reports of the opposite effects through nuclear factor-kB, interleukin-6, and vascular endothelial growth factor (Yao et al., 2020), suggesting complex roles of GC in tumors. In conclusion, we indicated that GC exerts tumor-promoting effects through signaling networks, including the mTOR signaling pathway.
In one study (Celentano et al., 2019), GC was found to reduce the chemotherapeutic sensitivity of HNSCC to doxorubicin and 5-fluoro uracil. In addition to chemotherapeutic drugs, our findings showed that GC has an antagonistic effect on the EGFR-targeted drug afatinib. When SCC9 cells were treated with HYD at varying concentrations of afatinib, its survival rate increased significantly, with a more noticeable decline in its apoptotic rate than that in cell lines with fewer GR. The aforementioned data supported the notion that HNSCC, with increased GR expression, were more vulnerable to the carcinogenic effects of HYD, as demonstrated by our earlier proliferation tests. Unexpectedly, regarding the rescue of GC-induced drug resistance, the same dose of GR inhibitor RU486 displayed a more prominent role in H400 cells. In contrast, RU486 showed no difference in the inhibition of the proliferative effect of GC. In regard to restrictive effect of Ru486 on the malignant effect of GC, inhibition of GR or its downstream pathways may provide an approach to antagonize GC-induced resistance of tumors.
AFA collaborates with EGFR, human epidermal growth factor receptor 2 (HER2), and HER4 as an ERB family blocker to destroy tumor cells (Solca et al., 2012). Protein analysis demonstrated that GC increased the phosphorylation of the mTOR downstream pathway but not that of EGFR, under the treatment of AFA. This result indicated that GC may promote cell viability to reduce the efficacy of AFA by activating the AKT/mTOR signaling pathway instead of the EGFR/ERK signaling pathway, which is completely blocked by AFA. Our theory was supported by an earlier study that showed GC decreased chemotherapeutic effectiveness via a glucose-dependent mechanism controlled by the mTOR signaling pathway (Celentano et al., 2019). Similarly, the resistance of HNSCC to cetuximab was due to the activation of the mTOR signaling pathway through mutations in phosphatidylinositol 3-kinase and RAS (Wang et al., 2014), suggesting the acquisition of resistance of HNSCC to chemotherapeutic agents may have commonalities. Based on the finding that rapamycin inhibited the activation of the mTOR signaling pathway by GC, we predict that rapamycin may provide an approach to rescue the agent resistance. The combined application of cetuximab and rapamycin has shown remarkable anti-tumor activity, especially in HNSCCs resistant to cetuximab (Wang et al., 2014). The limitation and future direction of this study are to investigate the role of rapamycin, mTOR activator, and GR inhibitor in the potential malignant effect of GC.
Our research demonstrated that GC promoted proliferation and inhibited apoptosis of HNSCC cell lines through the activation of the AKT/mTOR signaling pathway after binding with GR. GC also partially decreased the effectiveness of AFA on HNSCC cells, and rapamycin may hold the secret to overcoming agent resistance.
Acknowledgement: The authors acknowledge funding support from Sichuan University, the National Clinical Research Center for Oral Diseases, and West China Hospital of Stomatology, Sichuan University. The authors also acknowledge technical help from Shou-Yi Tang, Zhen Wang, and Lu-Yao Cai.
Availability of Data and Materials: The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Author Contributions: The authors confirm contribution to the paper as follows: study design and data collection: Dong-Yang Wang, Yi Chen; analysis, and interpretation of the results: Dong-Yang Wang, Yi Chen, Jing Huang; drafted manuscript preparation: Jing Huang, You Zhang and Chong-kui Sun; revised the manuscript: Ying-Qiang Shen; All authors reviewed the results and approved the final version of the manuscript.
Ethics Approval: Not applicable.
Funding Statement: This study was supported by the Research Funding (No. RCDWJS 2020-20), Research and Development Program (RD-02-202002) from West China School/Hospital of Stomatology Sichuan University, the Natural Science Foundation of China (81902784 & 81872207), and Sichuan Provincial Fund of China (2022YFSY0058).
Conflicts of Interest: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alzahrani AS (2019). PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Seminars in Cancer Biology 59: 125–132. DOI 10.1016/j.semcancer.2019.07.009. [Google Scholar] [CrossRef]
Azher S, Azami O, Amato C, McCullough M, Celentano A, Cirillo N (2016). The non-conventional effects of glucocorticoids in cancer. Journal of Cellular Physiology 231: 2368–2373. DOI 10.1002/jcp.25408. [Google Scholar] [CrossRef]
Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM et al. (2006). Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. New England Journal of Medicine 354: 567–578. DOI 10.1056/NEJMoa053422. [Google Scholar] [CrossRef]
Celentano A, McCullough M, Cirillo N (2019). Glucocorticoids reduce chemotherapeutic effectiveness on OSCC cells via glucose-dependent mechanisms. Journal of Cellular Physiology 234: 2013–2020. DOI 10.1002/jcp.27227. [Google Scholar] [CrossRef]
Cirillo N, Morgan DJ, Pedicillo MC, Celentano A, Lo Muzio L, McCullough MJ, Prime SS (2017). Characterisation of the cancer-associated glucocorticoid system: Key role of 11β-hydroxysteroid dehydrogenase type 2. British Journal of Cancer 117: 984–993. DOI 10.1038/bjc.2017.243. [Google Scholar] [CrossRef]
de Sena LSB, da Silveira ÉJD, Batista AC, Mendonça EF, Alves PM, Nonaka CFW (2018). Immunoexpression of glucocorticoid receptor alpha (GRα) isoform and apoptotic proteins (Bcl-2 and Bax) in actinic cheilitis and lower lip squamous cell carcinoma. Journal of Oral Pathology and Medicine 47: 788–795. DOI 10.1111/jop.12757. [Google Scholar] [CrossRef]
Dreyfuss AI, Clark JR, Norris CM, Rossi RM, Lucarini JW, Busse PM, Poulin MD, Thornhill L, Costello R, Posner MR (1996). Docetaxel: An active drug for squamous cell carcinoma of the head and neck. Journal of Clinical Oncology 14: 1672–1678. DOI 10.1200/JCO.1996.14.5.1672. [Google Scholar] [CrossRef]
Ediriweera MK, Tennekoon KH, Samarakoon SR (2019). Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Journal of Oral Pathology and Medicine 59: 147–160. DOI 10.1016/j.semcancer.2019.05.012. [Google Scholar] [CrossRef]
Feng Y, Cao X, Zhao B, Song C, Pang B, Hu L, Zhang C, Wang J, He J, Wang S (2021). Nitrate increases cisplatin chemosensitivity of oral squamous cell carcinoma via REDD1/AKT signaling pathway. Science China Life Sciences 64: 1814–1828. DOI 10.1007/s11427-020-1978-4. [Google Scholar] [CrossRef]
Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, Znaor A, Bray F (2019). Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. International Journal of Cancer 144: 1941–1953. DOI 10.1002/ijc.31937. [Google Scholar] [CrossRef]
Ferrarotto R, Gold KA (2014). Afatinib in the treatment of head and neck squamous cell carcinoma. Expert Opinion on Investigational Drugs 23: 135–143. DOI 10.1517/13543784.2014.858696. [Google Scholar] [CrossRef]
He L, Yuan L, Sun Y, Wang P, Zhang H et al. (2019). Glucocorticoid receptor signaling activates TEAD4 to promote breast cancer progression. Cancer Research 79: 4399–4411. DOI 10.1158/0008-5472.CAN-19-0012. [Google Scholar] [CrossRef]
Herr I, Pfitzenmaier J (2006). Glucocorticoid use in prostate cancer and other solid tumours: Implications for effectiveness of cytotoxic treatment and metastases. Lancet Oncology 7: 425–430. DOI 10.1016/S1470-2045(06)70694-5. [Google Scholar] [CrossRef]
Hung CC, Chien CY, Chu PY, Wu YJ, Lin CS et al. (2017). Differential resistance to platinum-based drugs and 5-fluorouracil in p22phox-overexpressing oral squamous cell carcinoma: Implications of alternative treatment strategies. Head and Neck 3: 1621–1630. DOI 10.1002/hed.24803. [Google Scholar] [CrossRef]
Ioannides D, Vakirlis E, Kemeny L, Marinovic B, Massone C et al. (2020). European S1 guidelines on the management of lichen planus: A cooperation of the European Dermatology Forum with the European Academy of Dermatology and Venereology. Journal of the European Academy of Dermatology and Venerology 34: 1403–1414. DOI 10.1111/jdv.16464. [Google Scholar] [CrossRef]
Jeong W, Jung S, Bazer FW, Song G, Kim J (2016). Epidermal growth factor: Porcine uterine luminal epithelial cell migratory signal during the peri-implantation period of pregnancy. Molecular and Cellular Endocrinology 420: 66–74. DOI 10.1016/j.mce.2015.11.023. [Google Scholar] [CrossRef]
Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR (2020). Head and neck squamous cell carcinoma. Nature Reviews Disease Primers 6: 92. DOI 10.1038/s41572-020-00224-3. [Google Scholar] [CrossRef]
Kleiman A, Tuckermann JP (2007). Glucocorticoid receptor action in beneficial and side effects of steroid therapy: Lessons from conditional knockout mice. Molecular and Cellular Endocrinology 275: 98–108. DOI 10.1016/j.mce.2007.05.009. [Google Scholar] [CrossRef]
Li Z, Liu J, Li L, Shao S, Wu J, Bian L, He Y (2018). Epithelial mesenchymal transition induced by the CXCL9/CXCR3 axis through AKT activation promotes invasion and metastasis in tongue squamous cell carcinoma. Oncology Reports 39: 1356–1368. DOI 10.3892/or.2017.6169. [Google Scholar] [CrossRef]
Liao YM, Kim C, Yen Y (2011). Mammalian target of rapamycin and head and neck squamous cell carcinoma. Head and Neck Oncology 3: 22. DOI 10.1186/1758-3284-3-22. [Google Scholar] [CrossRef]
Liu L, Aleksandrowicz E, Schönsiegel F, Gröner D, Bauer N et al. (2017). Dexamethasone mediates pancreatic cancer progression by glucocorticoid receptor, TGFβ and JNK/AP-1. Cell Death and Disease 8: e3064. DOI 10.1038/cddis.2017.455. [Google Scholar] [CrossRef]
Nguyen SA, Walker D, Gillespie MB, Gutkind JS, Day TA (2012). mTOR inhibitors and its role in the treatment of head and neck squamous cell carcinoma. Current Treatment Options in Oncology 13: 71–81. DOI 10.1007/s11864-011-0180-2. [Google Scholar] [CrossRef]
Obradović MMS, Hamelin B, Manevski N, Couto JP, Sethi A et al. (2019). Glucocorticoids promote breast cancer metastasis. Nature 567: 540–544. DOI 10.1038/s41586-019-1019-4. [Google Scholar] [CrossRef]
Planey SL, Litwack G (2000). Glucocorticoid-induced apoptosis in lymphocytes. Biochemical and Biophysical Research Communications 279: 307–312. DOI 10.1006/bbrc.2000.3922. [Google Scholar] [CrossRef]
Prime SS, Nixon SV, Crane IJ, Stone A, Matthews JB, Maitland NJ, Remnant L, Powell SK, Game SM, Scully C (1990). The behaviour of human oral squamous cell carcinoma in cell culture. Journal of Pathology 160: 259–269. DOI 10.1002/path.1711600313. [Google Scholar] [CrossRef]
Pulte D, Brenner H (2010). Changes in survival in head and neck cancers in the late 20th and early 21st century: A period analysis. Oncologist 15: 994–1001. DOI 10.1634/theoncologist.2009-0289. [Google Scholar] [CrossRef]
Shen YQ, Guerra-Librero A, Fernandez-Gil BI, Florido J, García-López S et al. (2018). Combination of melatonin and rapamycin for head and neck cancer therapy: Suppression of AKT/mTOR pathway activation, and activation of mitophagy and apoptosis via mitochondrial function regulation. Journal of Pineal Research 64: e12461. DOI 10.1111/jpi.12461. [Google Scholar] [CrossRef]
Slominski A, Zbytek B, Nikolakis G, Manna PR, Skobowiat C et al. (2013). Steroidogenesis in the skin: Implications for local immune functions. Journal of Steroid Biochemistry and Molecular Biology 137: 107–123. DOI 10.1016/j.jsbmb.2013.02.006. [Google Scholar] [CrossRef]
Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR (2012). Target binding properties and cellular activity of afatinib (BIBW 2992an irreversible ErbB family blocker. Journal of Pharmacology and Experimental Therapeutics 343: 342–350. DOI 10.1124/jpet.112.197756. [Google Scholar] [CrossRef]
Stojadinovic O, Lee B, Vouthounis C, Vukelic S, Pastar I, Blumenberg M, Brem H, Tomic-Canic M (2007). Novel genomic effects of glucocorticoids in epidermal keratinocytes: Inhibition of apoptosis, interferon-gamma pathway, and wound healing along with promotion of terminal differentiation. Journal of Biological Chemistry 282: 4021–4034. DOI 10.1074/jbc.M606262200. [Google Scholar] [CrossRef]
Tagliamento M, Genova C, Rijavec E, Rossi G, Biello F, Dal Bello MG, Alama A, Coco S, Boccardo S, Grossi F (2018). Afatinib and Erlotinib in the treatment of squamous-cell lung cancer. Expert Opinion on Pharmacotherapy 19: 2055–2062. DOI 10.1080/14656566.2018.1540591. [Google Scholar] [CrossRef]
Tian D, Tian M, Han G, Li JL (2019). Increased glucocorticoid receptor activity and proliferation in metastatic colon cancer. Scientific Reports 9: 11257. DOI 10.1038/s41598-019-47696-2. [Google Scholar] [CrossRef]
Timmermans S, Souffriau J, Libert C (2019). A general introduction to glucocorticoid biology. Frontiers In Immunology 10: 1545. DOI 10.3389/fimmu.2019.01545. [Google Scholar] [CrossRef]
Wang Q, Lu PH, Shi ZF, Xu YJ, Xiang J et al. (2015). Glucocorticoid receptor β acts as a co-activator of T-cell factor 4 and enhances glioma cell proliferation. Molecular Neurobiology 52: 1106–1118. DOI 10.1007/s12035-014-8900-9. [Google Scholar] [CrossRef]
Wang Z, Martin D, Molinolo AA, Patel V, Iglesias-Bartolome R, Sol Degese D, Vitale-Cross L, Chen Q, Gutkind JS (2014). mTOR co-targeting in cetuximab resistance in head and neck cancers harboring PIK3CA and RAS mutations. Journal of the National Cancer Institute 106: 3579. DOI 10.1093/jnci/dju215. [Google Scholar] [CrossRef]
Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L (2018). Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Annals of Oncology 29: i10–i19. DOI 10.1093/annonc/mdx703. [Google Scholar] [CrossRef]
Xing K, Gu B, Zhang P, Wu X (2015). Dexamethasone enhances programmed cell death 1 (PD-1) expression during T cell activation: An insight into the optimum application of glucocorticoids in anti-cancer therapy. BMC Immunology 16: 39. DOI 10.1186/s12865-015-0103-2. [Google Scholar] [CrossRef]
Yang X, Xu L, Yang YE, Xiong C, Yu J, Wang Y, Lin Y (2020). Knockdown of ribosomal protein S6 suppresses proliferation, migration, and invasion in epithelial ovarian cancer. Journal of Ovarian Research 13: 100. DOI 10.1186/s13048-020-00707-7. [Google Scholar] [CrossRef]
Yao Y, Yao QY, Xue JS, Tian XY, An QM et al. (2020). Dexamethasone inhibits pancreatic tumor growth in preclinical models: Involvement of activating glucocorticoid receptor. Toxicology and Applied Pharmacology 401: 115118. DOI 10.1016/j.taap.2020.115118. [Google Scholar] [CrossRef]
Yin F, Chen Q, Shi Y, Xu H, Huang J, Qing M, Zhong L, Li J, Xie L, Zeng X (2022). Activation of EGFR-Aurora A induces loss of primary cilia in oral squamous cell carcinoma. Oral Disease 28: 621–630. DOI 10.1111/odi.13791. [Google Scholar] [CrossRef]
Zhang J, Cao R, Peng J, Hu Q, Xia J, Cheng B (2022). In Silico analysis and linking of metabolism-related genes with the immune landscape in head and neck squamous cell carcinoma. BIOCELL 46: 111–125. DOI 10.32604/biocell.2022.016612. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools