
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Targeting the “undruggable” cancer driver genes: Ras, myc, and tp53
State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, Research Unit of Oral Carcinogenesis and Management, Chinese Academy of Medical Sciences, West China Hospital of Stomatology, Sichuan University, Chengdu, 610041, China
* Corresponding Author: YINGQIANG SHEN. Email:
(This article belongs to the Special Issue: Recent Advancement in Cancer Molecular Signaling)
BIOCELL 2023, 47(7), 1459-1472. https://doi.org/10.32604/biocell.2023.028790
Received 07 January 2023; Accepted 20 March 2023; Issue published 21 June 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
The term “undruggable” is to describe molecules that are not targetable or at least hard to target pharmacologically. Unfortunately, some targets with potent oncogenic activity fall into this category, and currently little is known about how to solve this problem, which largely hampered drug research on human cancers. Ras, as one of the most common oncogenes, was previously considered “undruggable”, but in recent years, a few small molecules like Sotorasib (AMG-510) have emerged and proved their targeted anti-cancer effects. Further, myc, as one of the most studied oncogenes, and tp53, being the most common tumor suppressor genes, are both considered “undruggable”. Many attempts have been made to target these “undruggable” targets, but little progress has been made yet. This article summarizes the current progress of direct and indirect targeting approaches for ras, myc, two oncogenes, and tp53, a tumor suppressor gene. These are potential therapeutic targets but are considered “undruggable”. We conclude with some emerging research approaches like proteolysis targeting chimeras (PROTACs), cancer vaccines, and artificial intelligence (AI)-based drug discovery, which might provide new cues for cancer intervention. Therefore, this review sets out to clarify the current status of targeted anti-cancer drug research, and the insights gained from this review may be of assistance to learn from experience and find new ideas in developing new chemicals that directly target such “undruggable” molecules.Keywords
Targeted therapy is one of the major modalities of medical treatment for cancer, which blocks the growth of cancer cells by interfering with specific target molecules.
Multiple targeting drugs are available for several exhaustively studied oncogenes. For example, for epidermal growth factor receptor (egfr) gene-activating mutations that occur in about 15% of lung adenocarcinomas, at least 3 generations of the US Food and Drug Administration (FDA) approved EGFR inhibitors are available with several more undergoing clinical trials (Cooper et al., 2022).
However, currently approved treatments are available for only a small fraction of approximately 700 identified cancer genes (Sondka et al., 2018). As a consequence, we still lack valid treatments for the vast majority of cancer-causing genes, including some of those that are most frequently altered.
Ras, myc, and tp53 are the most frequently altered cancer-driving genes as highly attractive targets for cancer treatment (Leroy et al., 2014; Simanshu et al., 2017; Priestley et al., 2019). However, the protein products of each of these genes are difficult to target. Therefore, these highly studied but poorly targeted cancer-driver genes have traditionally been referred to as “undruggable” (Dang et al., 2017). RAS, MYC, and P53 are difficult to be targeted by drugs for the following four reasons. First, all these proteins lack a deep protein binding pocket, which is a cavity on the surface or inside of a protein with ligand-binding properties, whose shape and position in the protein and the sequence of the surrounding amino acid residues determine its function (Stank et al., 2016). Second, protein-protein interactions (PPIs) of these proteins are only through large and flat interfaces (Dang et al., 2017; Tsubamoto et al., 2019). Targeting the biological functions of these genes is rather difficult because PPIs are indispensable for most of their genetic functions (Athanasios et al., 2017). Third, all three proteins are located intracellularly, that is, while RAS is present on the inner layer of the cell membrane, both MYC and P53 are in the nucleus. Consequently, the size of the drug molecule is strictly restrained. Finally, except for RAS, both MYC, and P53 lack enzymatic activity. Thus, their targeting cannot be performed by catalytic inhibitors.
The ras family of genes consists of three members, k-ras, n-ras, and h-ras. RAS proteins play the role of “binary switch” in cellular signaling pathways by cycling between an active state and an inactive state (Vigil et al., 2010). The guanylate exchange cycle plays a decisive role in the activation of RAS proteins. GTP-bound RAS protein is activated whereas GDP-bound RAS protein is inactivated (Pylayeva-Gupta et al., 2011; Stephen et al., 2014). RAS proteins have GTPase activity, which is enhanced by RAS GTPase activating protein (RAS GAP), turning the RAS protein from the activated (GTP-bound) form back to the inactivated state (GDP-bound) (Lito et al., 2016). RAS guanine nucleotide exchange factor (RAS GEF) plays a role in catalyzing the ejection of GTPase-bound GDP and the loading of GTP, thereby meditating the activation of RAS proteins (Rossman et al., 2005). For membrane localization of RAS proteins, post-translational modifications (PTMs) are necessary. PTMs are catalyzed by farnesyl transferase (FTase), geranylgeranyl transferase (GGTase), RAS-converting enzyme (Rce1), and isoprenylcysteine carboxyl methyltransferase (ICMT) (Moore et al., 2020). After being processed by PTMs, RAS proteins undergo oligomerization. To elaborate, RAS proteins assemble on the plasma membrane as nanoclusters or dimers, which act as recruitment and activation sites for downstream RAS effector molecules (Šolman et al., 2015; Zhou and Hancock, 2015).
About 30% of human cancers have mutations in the ras family (k-ras, n-ras, and h-ras), which can cause persistent activity of RAS proteins (Cox et al., 2014; Simanshu et al., 2017). Besides, a large proportion of drug-resistant tumors activate the RAS pathway to overcome the effects of kinase inhibitors (Massarelli et al., 2007). Among all 3 forms of ras, k-ras is the most frequently mutated in human cancers (85%), followed by n-ras (12%) and h-ras (3%) (Simanshu et al., 2017). Frequencies of mutations in ras isoforms vary in different tissues. For example, k-ras is most frequently mutated in the pancreas (91%), colon (42%), and lung (33%) while n-ras is most frequently mutated in the skin, especially in melanoma (27%), bone marrow (8%) and thyroid (8%) and h-ras is most frequently mutated in adrenal glands (10%), thymus (8%) and bladder (5%) (Simanshu et al., 2017). Most ras mutations are found in codons 12, 13, or 61, corresponding to glycine 12 (G12), glycine 13 (G13), or glutamine 61 (Q61), respectively (Simanshu et al., 2017; Asimgil et al., 2022).
The myc family consists of 3 members, c-myc, l-myc, and n-myc. All three MYC proteins function as transcriptional regulators, which can integrate multiple cellular signals and regulate the expression of genes involved in multiple biological processes that are essential to normal as well as neoplastic cells, such as cell proliferation, metabolism, differentiation, DNA repair, immune response and apoptosis (Carroll et al., 2018; Casey et al., 2018; Singh et al., 2019). To regulate gene expression programs like growth, proliferation, differentiation, apoptosis, and metabolism, the MYC protein employs its basic-helix-loop-helix-zipper (bHLHZ) domain to form the heterodimer “MYC-MAX” with the bHLHZ domains of the MAX protein. Similarly, MAX also forms heterodimers with the MXD family (as well as MNT and MGA proteins) via the bHLHZ domains to form what is known as “MXD-MAX”. Both MYC-MAX and MXD-MAX directly bind to DNA regulatory sites, especially to canonical consensus 5′-CACGTG--3′ sequences located in E-box regions to perform their functions (Carroll et al., 2018).
Myc and its deregulation in cancer
Myc genes are rarely mutated in contrast to ras. However, the myc family (c-myc, l-myc, n-myc) is one of the most commonly deregulated oncogenes in human cancers, which is estimated to be aberrantly expressed in up to 70% of human cancers (Llombart and Mansour, 2022). Some tumors activate myc expression, mediating resistance to targeted therapies (Dang, 2012; Elbadawy et al., 2019). Among all 3 forms of myc genes, c-myc is the most frequently amplified gene (21%) across all cancer types. In contrast, n-myc and l-myc genes amplified less commonly (7%) at the pan-cancer level (Schaub et al., 2018). Further, c-myc is most frequently amplified in ovarian serous cystadenocarcinoma (64.8%), esophageal carcinoma (45.3%), and lung squamous cell carcinoma (37.2%) (Schaub et al., 2018). While n-myc and l-myc are less frequently amplified, n-myc amplification, however, has been documented in 20%–30% of neuroblastoma patients (Otte et al., 2021).
In contrast to ras and myc, tp53 is a tumor suppressor gene, which is frequently referred to as the “guardian of the genome” and functions to suppress cancer formation (Nguyen et al., 2017; Binayke et al., 2019; Janani et al., 2021). Although discovered decades ago, the exact mechanism by which the tp53 gene prevents cancer formation is still unclear. Wild type (WT) P53 acts as a transcription factor to regulate the expression of target genes that are involved in many distinct biological processes like cell cycle arrest and apoptosis (Kastan et al., 1995; Fridman and Lowe, 2003). In normal unstressed cells, P53 protein levels are kept low and the biochemical activity of P53 is restrained. This is mainly attributed to the proteasomal degradation of P53 controlled by mouse double minute 2 (MDM2), which is an E3 ubiquitin ligase (Hassin and Oren, 2023).
The tp53 gene family is one of the most frequently mutated genes in human cancers with its mutation generally associated with poor prognosis (Leroy et al., 2014; Kastenhuber and Lowe, 2017). For instance, across all human cancers, the overall mutational rate of tp53 is about 50% (Levine, 2019). The frequency of tp53 mutations varies in different cancers. For example, tp53 mutations are more likely to be found in malignancies such as ovarian cancers, lung cancers, and breast cancers (Cancer Genome Atlas Research Network, 2011; Cancer Genome Atlas Research Network, 2012; Cancer Genome Atlas Network, 2012; George et al., 2015). Most tp53 mutations are missense mutations, followed by nonsense mutations (Cai et al., 2022).
We conclude this review with the current approaches and possible challenges of targeting ras, myc, tp53 that are considered “undruggable”. This review set out to clarify the current status of targeted anti-cancer drug research, and the insights gained from this review may be of assistance to learn from experience and find new ideas in developing new chemicals that target those “undruggable” molecules directly.
The Current Progress in Targeting ras, myc, and tp53
In 2021, the FDA approved the first K-RAS inhibitor Sotorasib (AMG-510) (Mullard, 2022), which showed an overall response rate (ORR) (n = 124) of 36% [95% confidence interval (CI), 28–45] in patients with K-RAS-G12C-mutated non-small-cell lung cancer (NSCLC) (Nakajima et al., 2022). Behind this achievement, tremendous efforts have been made by scientific workers on targeting RAS directly. The work of targeting K-RAS-G12C was started by Shokat and his colleagues, who identified a binding pocket Switch-II Pocket (S-IIP) in the mutated K-RAS-G12C protein (Ostrem et al., 2013), which brought a turnaround in targeting K-RAS that was once thought to be undruggable. Based on this, many covalent inhibitors targeting K-RAS-G12C have entered clinical trials and even been approved for marketing, including Sotorasib (also AMG-510), Adagrasib (also MRTX849), JNJ-74699157 (also ARS-3248), LY3499446 and numerous other K-RAS-G12C inhibitors (Nagasaka et al., 2020; Sidaway, 2021; Brazel et al., 2022). Most of the current K-RAS-G12C inhibitors are only applicable for GDP-bound K-RAS-G12C proteins, which are RAS proteins in the inactivated state, whereas some recently developed K-RAS-G12C inhibitors, like RM-018, are valid for GTP-bound K-RAS-G12C proteins, which are RAS proteins in the activated state (Tanaka et al., 2021). Additionally, non-specific RAS inhibitors are also being developed. While some of the RAS mutant proteins are specifically inhibited, it is currently difficult to identify effective treatments for each mutated RAS protein. Direct targeting of conserved ligand binding sites on all kinds of RAS proteins may provide a single therapeutic pathway to inhibit RAS across many mutations and tumor types. For example, compound 3144 targets the switch-I pocket, whereas compound Abd series, compound Ch series, and BI-2852 target the 4,6-dichloro-2-methyl-3-aminoethyl-indole (DCAI) pocket (Welsch et al., 2017; Quevedo et al., 2018; Cruz-Migoni et al., 2019; Kessler et al., 2019). However, the problem with this approach is that non-specific binding is highly toxic to healthy tissue cells as the RAS pathway is essential in cell signaling.
To target ras indirectly, past studies have mainly focused on the inhibition of RAS PTMs, oligomerization, nucleotide exchange, and downstream binding. Altering any of these basic steps could be a chance to inhibit ras indirectly.
Several enzymes catalyzing RAS PTMs are necessary for RAS membrane localization. These include FTase, GGTase, Rce1, and ICMT (Moore et al., 2020). Inhibitors against each of these enzymes have the potential to be effective agents targeting RAS indirectly. In 2020, the FDA approved the first farnesyltransferase inhibitor, lonafarnib, for use as a treatment for Hutchinson-Gilford progeria syndrome and some progeroid laminopathies (Mullard, 2021). In addition, several more farnesyltransferase inhibitors are in clinical trials, such astipifarnib, L778123, BMS 214662, FTI-277, alpha-hydroxy farnesyl phosphonic acid, and manumycin-A (Bolick et al., 2003; Johnson and Heymach, 2004; Ryan et al., 2004; Mesa, 2006; Costa et al., 2012; Tsuda et al., 2013). There are also a few studies on GGTase inhibitors, such as GGTI-2418 and GGTI-298 (Liu et al., 2018; Karasic et al., 2019). Recently, some potential ICMT inhibitors have also emerged, such as UCM-1336 and C75 (Marín-Ramos et al., 2019; Chen et al., 2021). Finally, recent efforts to find new Rce1 inhibitors have focused on the development of non-peptide-based small molecule compounds. Such compounds are expected to have better cell permeability and be easier to synthesize compared to peptide and natural compound-based inhibitors. However, no effective and specific Rce1 inhibitors have been identified yet (Mohammed et al., 2016; Hampton et al., 2018).
The α4-β6-α5 interface (α4-β6-α5 interface) was found to be important for RAS dimerization or nanocluster formation on the plasma membrane. NS1, a drug that is currently being investigated, binds to the α4-β6-α5 region of RAS, disrupting RAS oligomerization to block its normal function (Spencer-Smith et al., 2017).
The RAS guanylate exchange cycle is mediated by RAS guanine nucleotide exchange factor (RAS GEF) and RAS GTPase activating protein (RAS GAP). RAS GEF catalyzes the conversion of GDP to GTP, turning RAS from an inactive to an active state. RAS GAP activates GTPase, turning the GTP-bound RAS back to GDP-bound RAS (Lito et al., 2016). RAS GEFs include son of sevenless (SOS), Src homology-2-containing protein tyrosine phosphatase 2 (SHP2), RAS guanine nucleotide releasing factor (rasGRF), to name a few. While inhibitors of SOS and SHP2 have been studied more deeply, some inhibitors being currently studied include BAY-293, SHP-099, RMC-4550, etc. (Hillig et al., 2019; Wang et al., 2020a; Song et al., 2021). According to a report, the selective suppression of at least one of the RAS GAPs (RASAL1, DAB2IP, or NF1) results in unrestrained activation of wild-type RAS in human hepatocellular carcinomas (HCC) (Calvisi et al., 2011). A similar situation was found in breast cancer, resulting in the suppression of RASAL2 (Shen et al., 2013). This indicates the potential of targeting RAS GAPs in certain cancers.
Active RAS proteins interact with many downstream effector molecules through RAS binding domains (RBDs). A novel pan-RAS inhibitor called Pen-cRaf-v1 with desirable target specificity has been reported by searching a combinatorial library consisting of RBDs and cell-permeable peptides (CPPs). This provides a promising scaffold for further development of RAS inhibitors (Nomura et al., 2021). In another approach, RAS simulants could bind RBDs, but no downstream effector molecule was activated. For example, Rigosertib, a small molecule RAS simulant, binds to RBDs of multiple RAS effectors and blocked their interaction with RAS proteins (Wang et al., 2019). In summary, ras can now be directly or indirectly targeted, as shown in Fig. 1.
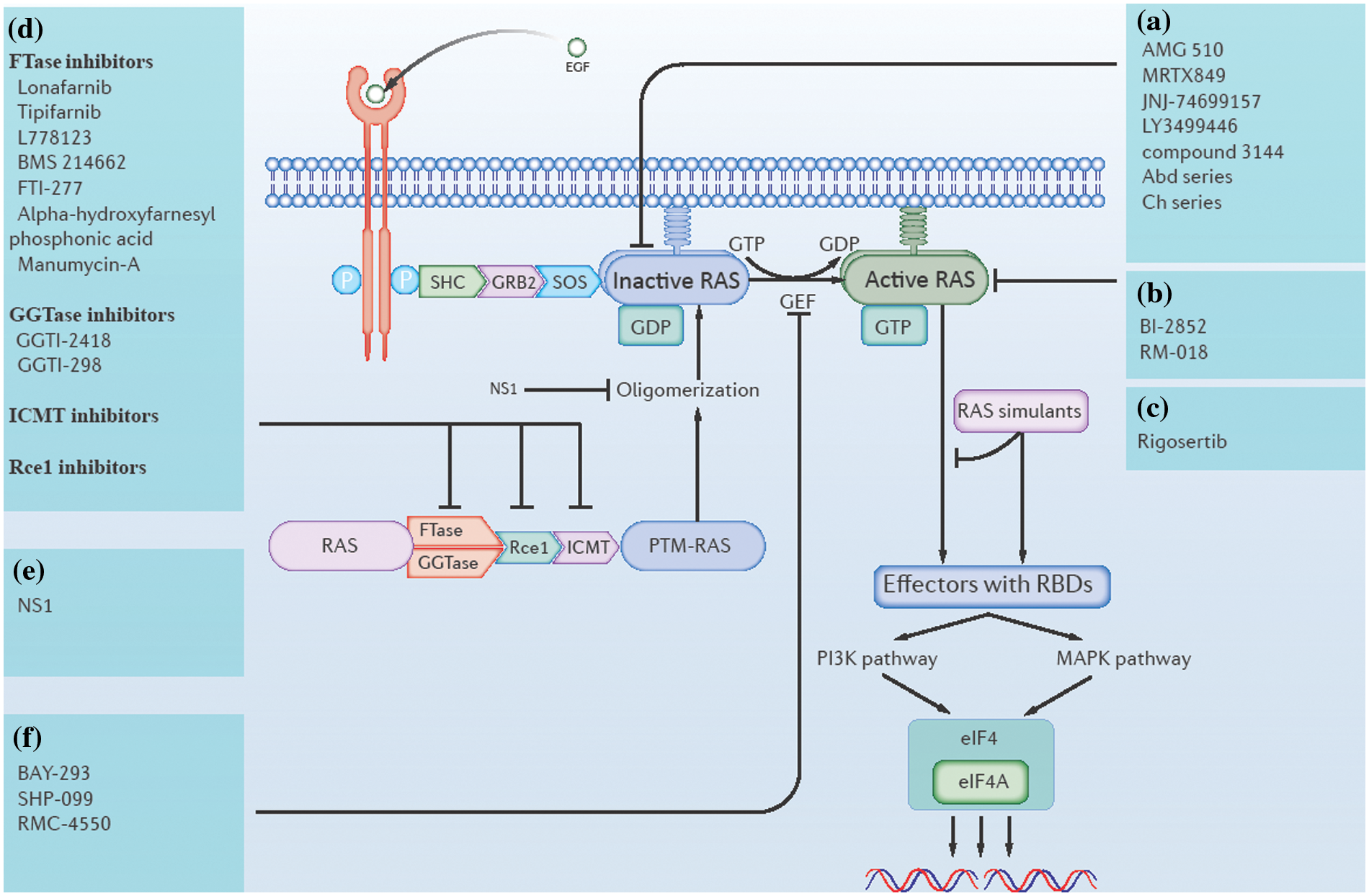
Figure 1: Direct and indirect approaches of ras inhibition. (a) Inhibitors that directly target GDP-bound (inactive state) RAS. (b) Inhibitors that directly target GTP-bound (active state) RAS. (c) RAS simulants block downstream RAS activation. (d) RAS post-translational modification (PTM) inhibitors, including farnesyl transferase (FTase) inhibitors, geranylgeranyl transferase (GGTase) inhibitors, isoprenylcysteine carboxyl methyltransferase (ICMT) inhibitors, and RAS-converting enzyme (Rce1) inhibitors. (e) Inhibitors of RAS oligomerization. (f) RAS guanine nucleotide exchange factor (RAS GEF) inhibitors.
Previous studies of myc pathway inhibition mainly focused on the transcription, translation, stabilization, and activation of myc, as shown in Fig. 2.
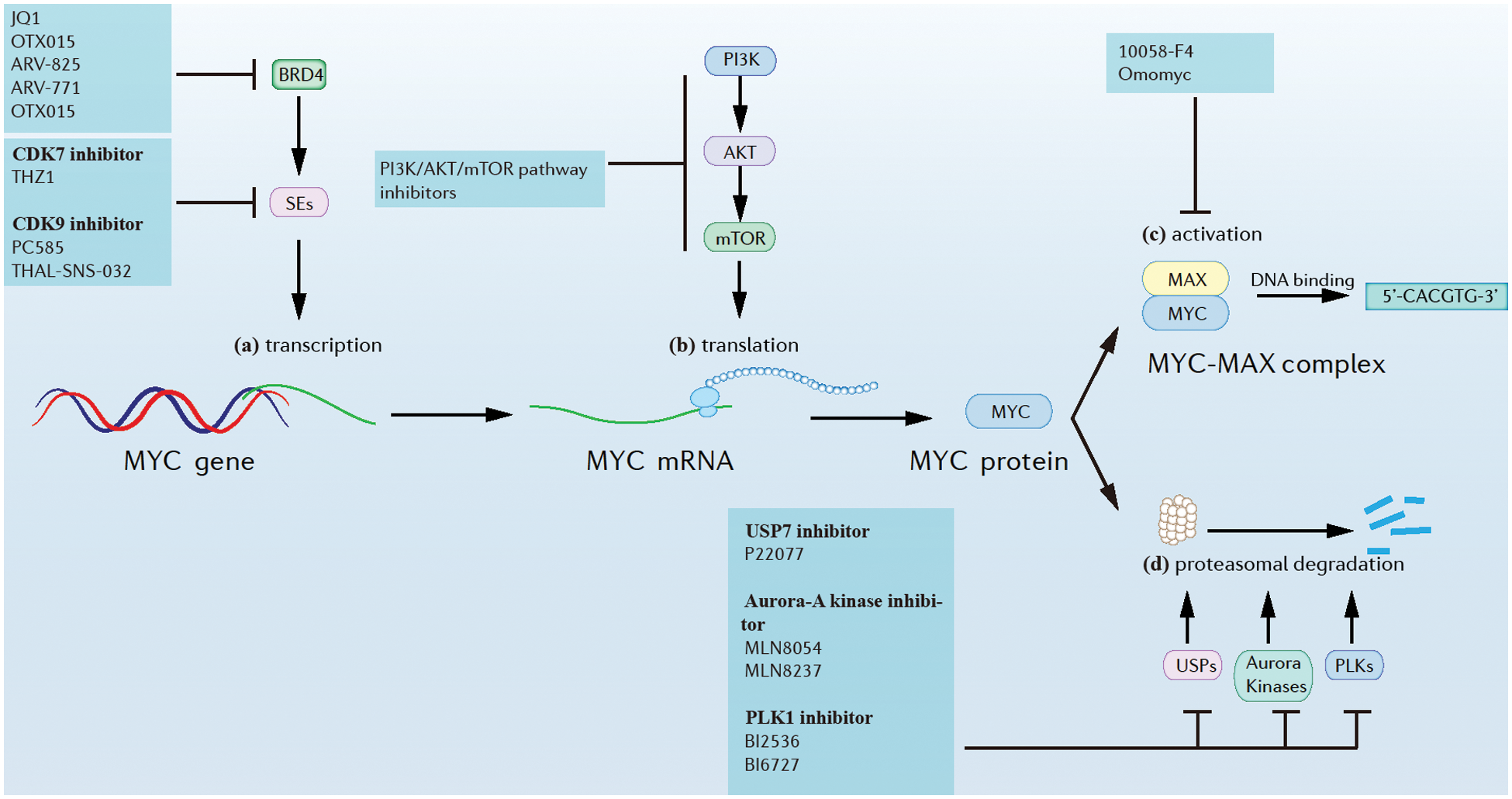
Figure 2: Approaches of myc inhibition at different levels. (a) Myc transcription. (b) Myc translation. (c) MYC activation. (d) MYC proteasomal degradation.
A general feature of myc dysregulation in tumor cells is that it is transcriptionally enhanced by Super-Enhancers (SEs), implying that inhibition of SEs might suppress myc transcription. For example, the inhibition of Cyclin-dependent kinase (CDK) 7 and/or CDK9, which are SEs, was found greatly reduce MYC expression, accompanied by extensive transcriptional downregulation of myc target genes. Additionally, CDK7 inhibitors like THZ1 and CDK9 inhibitors like PC585 were found to have potent antitumor effects against MYC in many myc-driven cancers (Chen et al., 2018; Thandapani, 2019). Further, CDK9 could also be rapidly degraded induced by a selective CDK9 degrader, THAL-SNS-032, consisting of a CDK-binding SNS-032 ligand linked to a thalidomide derivative that binds the E3 ubiquitin ligase Cereblon (CRBN) (Olson et al., 2018). Bromodomain-containing protein 4 (BRD4) is a member of the bromodomain and extra-terminal domain (BET) family of proteins and is widely recognized for its role in the organization of super-enhancers and the regulation of oncogene expression (Donati et al., 2018). BRD4 could be inhibited by small molecules such as JQ1 and OTX015 (Delmore et al., 2011; Boi et al., 2015), and could be also degraded by PROTACs (Otto et al., 2019). In one study, the novel BET-PROTACs (ARV-825 and ARV-771) that degrade BRD4 were compared with the BET bromodomain inhibitor (BETi) OTX015. At equimolar concentrations, ARV-825 and ARV-771 were more potent than the BETi OTX015 in inducing apoptosis of cultured and primary mantle cell lymphoma (MCL) cells (Sun et al., 2018).
A possible biochemical crosstalk between myc and mechanistic target of rapamycin (mTOR) pathways was found in liver cancer. mTORC1 functions by regulating the downstream cascades that are necessary for c-myc-induced carcinogenesis (Liu et al., 2017). Interestingly, the pharmacological inhibition of the phosphoinositide 3-kinase (PI3K)/AKT/mTOR pathway significantly reduced myc translational levels and showed significant therapeutic effects in many myc-driven cancers (Chen et al., 2018).
Though the stability of MYC is tightly controlled, suppressing Ubiquitin-specific proteases (USPs), Aurora kinases, and Polo-like kinases (PLKs) was found to destabilize MYC and suppress MYC-driven tumors (Chen et al., 2018; Tavana et al., 2016). For example, USP7, known as a regulator of N-MYC function in neuroblastoma could be suppressed by small molecule inhibitor P22077, which caused tumor suppression (Tavana et al., 2016). Further, aurora-A kinase, a kinase that protects N-MYC from proteasomal degradation, could be inhibited by several small molecules like MLN8054 and MLN8237. This resulted in N-MYC degradation and tumor suppression in neuroblastoma (Brockmann et al., 2013). PLK1, a kinase that forms a positive feedforward activation loop with MYC could be inhibited by inhibitors like BI2536 and BI6727, resulting in the potent apoptosis of myc-overexpressing tumor cells (Xiao et al., 2016).
The MYC-MAX complex is required for MYC binding to DNA and subsequent activation of target gene transcription. In one report, the inhibitor 10058-F4 targeted and disrupted the MYC-MAX complex in HL60 cells, while Omomyc preferentially bound MAX over MYC and inhibited MYC-mediated transcription by replacing MYC/MAX heterodimers with Omomyc/max heterodimers (Chen et al., 2018; Demma et al., 2019).
To summarize, substantial progress has been made in the study of targeting myc pathways. However, targeting MYC proteins directly still seems to be challenging, since MYC proteins lack appropriate pockets or grooves that can serve as inhibitor binding sites (Wang et al., 2019).
On one hand, as the wild-type tp53 gene is considered one of the tumor suppressor genes, previous studies have focused on preventing tp53 from losing its function as a tumor suppressor gene. On the other hand, mutant tp53 genes are considered therapeutic targets as oncogenes. Such previously researched ideas mainly included restoring wild type function of P53 mutants by altering their protein structures, altering the proteasomal degradation of P53 proteins, tp53 gene editing, and inducing synthetic lethality in tp53-mutated cancer cells, as shown in Fig. 3.
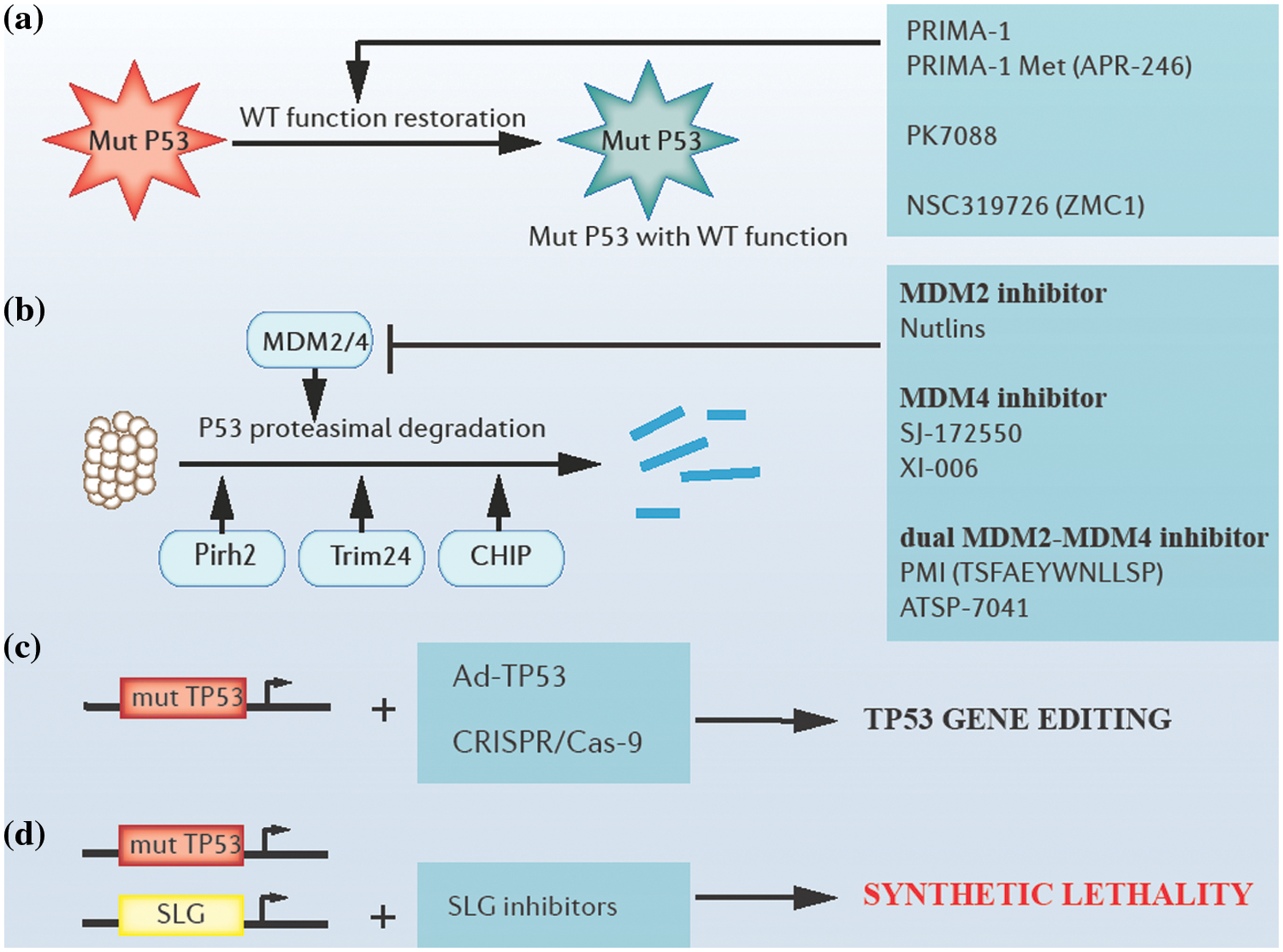
Figure 3: Approaches of targeting tp53. (a) Restoring the wild-type function of P53 mutants by altering their protein structures. (b) Blocking proteasomal degradation of P53 by targeting E3 ubiquitin ligases. (c) Tp53 adenoviral vectors and clustered regulatory interspaced short palindromic repeats (CRISPR)/CRISPR-associated system (Cas9)-based gene editing. (d) Synthetic lethality-based p53 targeting. Mut P53: mutant P53 proteins; WT: wild type; mut TP53: mutant tp53 genes; Ad-TP53: tp53 adenoviral vectors; SLG: synthetic lethality gene.
To restore the wild-type function of P53 mutants, several attempts have been tried and reported. For example, PRIMA-1 (P53 reactivation and induction of massive apoptosis-1) and its structural analog PRIMA-1Met (APR-246) played a role as cysteine-binding compounds in refolding mutant P53 (Peng et al., 2013). Additionally, compounds like PK7088 bound to a crevice in p.Y220C mutant P53 proteins, increasing the fraction of correctly folded p.Y220C mutant P53 in cells (Liu et al., 2013). Further, compounds like NSC319726 (ZMC1) could reactivate several P53 mutants with impaired zinc binding through their zinc ion-chelating properties (Blanden et al., 2015; Soussi and Wiman, 2015; Bykov et al., 2018).
The inhibition of MDM2 protein, a P53-specific E3 ubiquitin ligase, and (or) MDM4, a protein with structural homology to MDM2 has been explored as potential approaches to block the proteasomal degradation of P53. Several MDM2/4 antagonists can block the degradation of P53 (Duffy et al., 2014). For example, MDM2 antagonists, such as Nutlins could inhibit MDM2-P53 interactions, resulting in the apoptosis of malignant cells (Demicco, 2019). Additionally, efforts have been made in searching for MDM4 antagonists, for example, compounds such as SJ-172550 and XI-006 could inhibit MDM4 effectively (Reed et al., 2010; Pishas et al., 2015). Moreover, several dual MDM2-MDM4 antagonists, such as PMI (TSFAEYWNLLSP) and ATSP-7041 presented a stronger affinity with MDM2 /4 than the affinity between P53 and MDM2/4 (Pazgier et al., 2009; Tiwari and Verma, 2017). However, a possible side effect of treatment with MDM2/4 antagonists is that WT P53 levels may accumulate in normal cells and induce inappropriate apoptosis and cell death, which refrains us from jumping to optimistic conclusions (Duffy et al., 2014). Apart from MDM2, many other ubiquitin ligases that participate in the degradation of P53 have been identified recently, including Pirh2, Trim24, Carboxyl terminus of HSP70-interacting protein (CHIP), etc. (Jain and Barton, 2009; Wang et al., 2011; Soussi and Wiman, 2015; Wang et al., 2020b).
Tp53 gene therapy is also a current hot topic. Firstly, efficacy in tp53 gene replacement employing some viral vectors like tp53 adenoviral vectors was reported in prostate cancer in clinical trials (Tamura et al., 2018). Secondly, clustered regulatory interspaced short palindromic repeats (CRISPR)/ CRISPR-associated system (Cas9)-mediated genome editing has a prospect in editing the tp53 gene, and relevant tests are in progress (Mirgayazova et al., 2020).
Moreover, an interesting concept has recently achieved brilliant clinical success, that is, synthetic lethality. This includes mutations in genes that lead to cell or organism death when occurring in combination with mutations in one or more other genes. One of the most classic examples is that of tumors lacking DNA repair function due to the loss of expression of functional breast cancer 1 (BRCA1) or breast cancer 2 (BRCA2) proteins that are extremely sensitive to poly (ADP-ribose) polymerase (PARP) enzyme inhibition (Farmer et al., 2005). Similar mechanisms can be applied to the targeting of tp53, and valuable progress has been made in the synthetic lethality between the tp53 gene and ataxia telangiectasia and Rad3-related protein (ATR)/Chk1, ataxia telangiectasia mutated (ATM)/Chk2 or p38/MAPK-activated protein kinase 2 (MK2), and other signaling pathways (Kawasumi et al., 2014; Morandell and Yaffe, 2012; Pabla et al., 2008).
Emerging Research Ideas for Targeting the “Undruggable” Cancer Driver Genes
Protein-protein interaction (PPI) interfaces may be large and flat between “undruggable” proteins and other proteins, which makes it tricky to design drugs against them. However, a question comes as to whether we use the PPIs of the human body to help us target these proteins. In the process of protein degradation, E3 ubiquitin ligase specifically recognizes the substrate through PPI, which allows ubiquitin transfer to the substrate protein and directs the substrate to degrade at the proteasome. PROTACs technology aims to develop a linker molecule that binds to the E3 ubiquitin ligase and the target protein, simultaneously, promoting the ubiquitination and subsequent hydrolysis of the target protein, as shown in Fig. 4. Since PROTACs work with E3 ubiquitin ligases, they only need to bind briefly and loosely to the target protein without continuously occupying the active site of the target protein to block the function (Neklesa et al., 2017; Schapira et al., 2019).
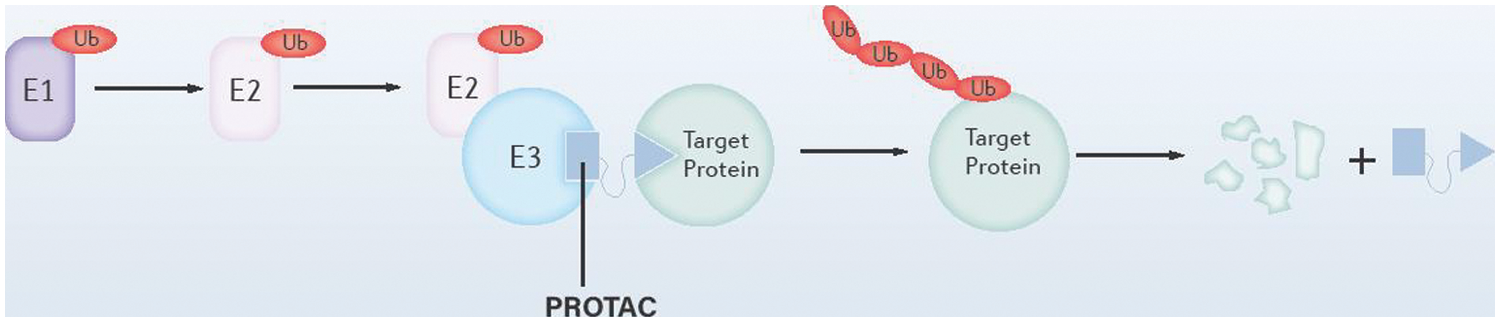
Figure 4: Schematic diagram of the structure of the proteolysis targeting chimera (PROTAC) system. As illustrated, PROTAC binds to both E3 ubiquitin ligase and the target proteins, which promotes the ubiquitination and degradation of the target proteins.
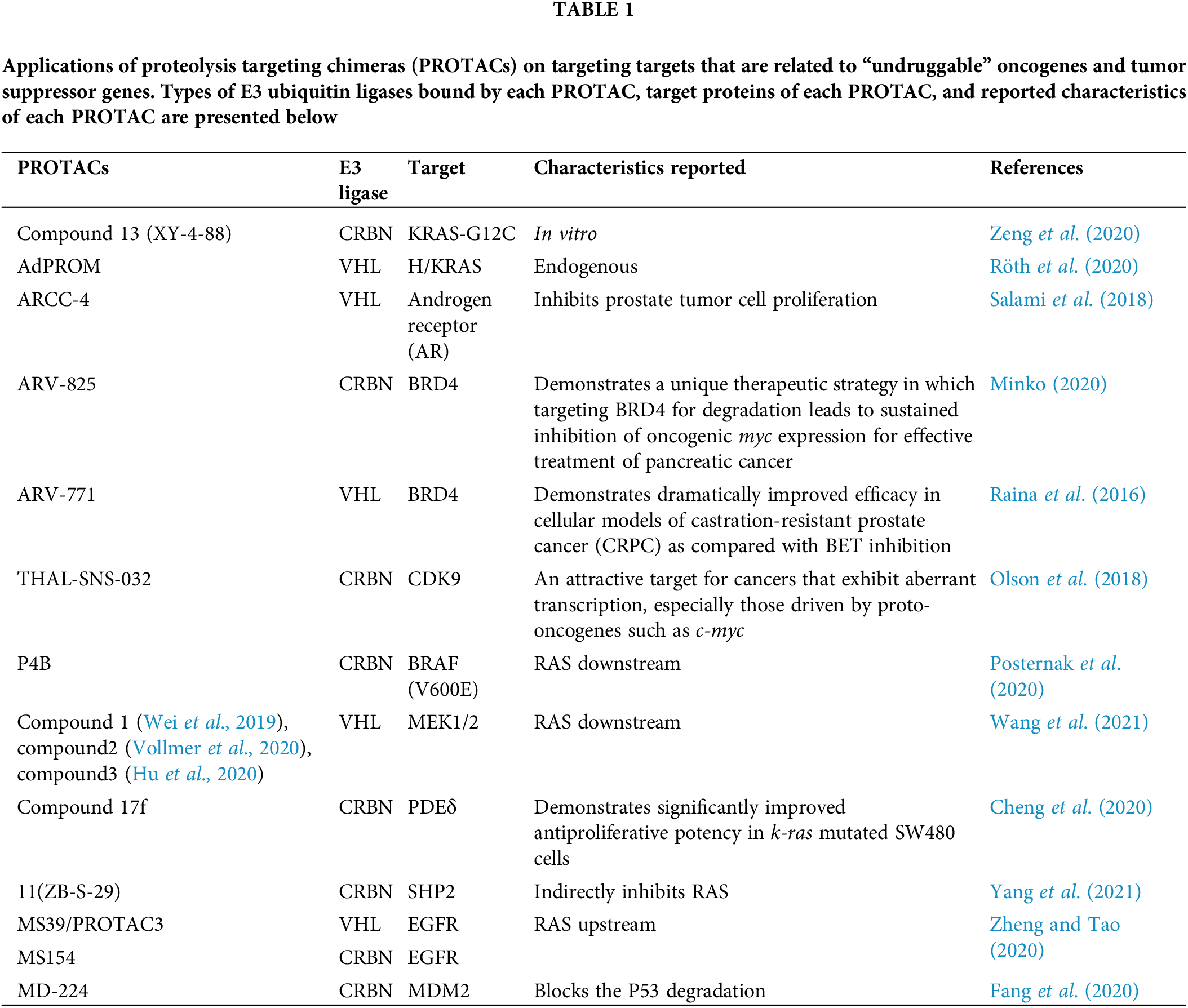
The potential of PROTACs in tackling the “undruggable” molecules was shown in some experiments, as shown in Table 1. For example, K-RAS-G12C could be degraded ubiquitously in vitro by PROTACs, whereas endogenous H/K-RAS proteins could be degraded by the affinity-directed protein missile (AdPROM) system (Röth et al., 2020; Simpson et al., 2020; Zeng et al., 2020). As for transcription factors, especially nuclear receptors (NRs), which are traditionally considered “undruggable”, the application of PROTACs technology has progressed significantly. For example, ARCC-4, a low-nanomolar androgen receptor PROTAC derivative could degrade cellular androgen receptors (Salami et al., 2018; Flanagan and Neklesa, 2019). Additionally, indirect ubiquitination and degradation of transcriptional regulator BRD4 could be induced by the novel BET-PROTACs like ARV-825 and ARV-771 to cause MYC inhibition and antitumor activity in a few cancer cell lines including colorectal cancer (CRC) cells (Sun et al., 2018; Otto et al., 2019). Further, selective degradation of CDK9 could also be induced by PROTAC molecules like THAL-SNS-032 (Olson et al., 2018). Downstream of the RAS pathway, mutants of BRAF (V600E), MEK1/2, could be ubiquitinated and degraded by PROTACs (Posternak et al., 2020; Wang et al., 2021). PDEδ (PDE6 delta subunit) has a role in promoting the diffusion and proper localization of RAS proteins on the cell membrane. The normal function of RAS proteins was inhibited by ubiquitination and degradation by the PROTACs technology (Jiang et al., 2017; Cheng et al., 2020). It is also possible to indirectly inhibit RAS by degrading SHP2 using PROTACs (Yang et al., 2021). Several PROTACs have been developed to target the degradation of EGFR (Zheng and Tao, 2020), which could indirectly inhibit downstream pathways such as RAS and the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway. For targeting TP53, MDM2 could be degraded by PROTACs technology, which inhibited the degradation of P53 by MDM2 and activated P53 (Fang et al., 2020; Demir et al., 2021).
Immunotherapy plays a critical role in cancer treatment in recent years. Many such novel methods may be effective in overcoming undruggable targets. In the following section, bispecific antibodies and cancer vaccines are mainly introduced.
Bispecific antibodies (bsAb) are antibodies that specifically target two antigens at the same time. Here is an example to illustrate the principle. Recently, a bispecific antibody was reported that targets the neoantigen R175H generated by tp53 mutation on the one hand and the T cell receptor (TCR) on the other hand, causing T cells to release cytotoxic proteins that kill tumor cells (Hsiue et al., 2021). In theory, this approach would only bind T cells to the vicinity of tumor cells. It was thus demonstrated that T cells could be effectively activated by such bispecific antibodies to kill tumor cells despite the low antigen density on their surface. Theoretically, this approach could be used to treat cancers containing other mutations that are difficult to target by conventional means.
Cancer vaccines induce and amplify antigen-specific immune responses and have long been considered potentially valuable tools for the treatment of cancer. Protein cancer vaccines and mRNA cancer vaccines are discussed herewith. Protein cancer vaccines are based on the principle of activating dendritic cells and triggering T-cell responses to target proteins by injecting them with target proteins and some cytokines. Examples include the intradermal injection of mutated RAS protein peptides and granulocyte-macrophage colony-stimulating factor (GM-CSF) (Moore et al., 2020). In contrast to protein cancer vaccines, mRNA vaccines are based on the principle of injecting lipid-soluble nanoparticles containing the target protein mRNA. The mRNA nanoparticles are then taken up by antigen-presenting cells, translated into proteins, and presented to the cell surface, which triggers the T-cell response to the target protein. Examples include the intramuscular injection of mRNA nanoparticles containing mRNAs encoding common K-RAS mutations (G12C, G12D, G12V, and G13D) elicited T-cell responses (Moore et al., 2020). Overall, cancer vaccines are a smart alternative for the genes that are difficult to target by conventional approaches.
Although tremendous effort has been made on such “undruggable” molecules, little progress has been made yet. However, graphics processing unit (GPU) computing and deep learning now accelerate drug discovery, including the discovery of drugs targeting these “undruggable” molecules. Procedures like molecular dynamics simulations and protein structure determination can be accelerated with the help of deep learning models and GPU parallel computing (Pandey et al., 2022). Traditionally, evaluating the druggability of potential targets requires considerable time and financial cost in the analysis of three-dimensional (3D) protein structures. However, the situation is about to change thanks to the development of network-based and machine learning (ML)-based artificial intelligence algorithms (You et al., 2022).
The application prospects of AI in the pocket druggability prediction are promising. For example, PockDrug-Server is a web server based on the protein-protein interaction network for pocket druggability prediction, which presents efficiency in both estimated pockets guided by the ligand proximity or solely based on protein structure information (Hussein et al., 2015). Since approaches like PockDrug heavily depend on accurate 3D protein structures, recent ML-based AI algorithms which can predict the 3D structure of a protein from its genetic sequence could be very helpful. Another example is AlphaFold2, one of the state-of-the-art machine learning methods developed by the DeepMind company in 2021. It is claimed to be able to predict structures of almost the entire human proteome (Tunyasuvunakool et al., 2021). Moreover, other protein structure-predicting ML methods like the trRosetta algorithm also showed promising potential in the accurate prediction of protein structure (Yang et al., 2020).
In addition to pocket druggability prediction, the prediction of interactions between drug and target or drug and the drug is critical for new drug discovery. For processes like drug screening and drug repurposing, network-based and ML-based artificial intelligence algorithms can be employed as powerful tools (You et al., 2022). AI-based drug-target interactions (DTIs) predictions are mainly divided into 2 categories, one is “top-down”, which starts from observable characteristics to predict the interaction, whereas the other is “bottom-up”, which starts from molecular features to predict interactions (You et al., 2022).
Last but not least, AI is also widely used in the prediction of drug properties, comprising ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) prediction and predicting properties applied in clinical trials for seeking optimal treatment plans (You et al., 2022).
More specifically, artificial intelligence can be employed as the right helper to accelerate the development of some aforementioned emerging approaches of targeting “undruggable” molecules. Several computational modeling tools are now available to accelerate PROTAC design, which may minimize the spending cost and time (Drummond and Williams, 2019; Drummond et al., 2020). As with cancer immunotherapies, similar advantages of AI are demonstrated in this regard. With the use of high-throughput sequencing data, several in silico methods or tools which facilitate the identification of tumor neoantigens may accelerate the development of cancer immunotherapies (Zhou et al., 2019). Preclinical trials of cancer immunotherapies like chimeric antigen receptor (CAR)--T cell therapy and cancer vaccines can be reduced by applying mathematical models in silico (Barros et al., 2021; Lőrincz et al., 2021; Valle et al., 2021).
In summary, we consider that with the development of AI, an increasing number of AI-based applications like potential target druggability prediction, new drug discovery, and the prediction of drug properties would provide better help in developing potent drugs, including those targeting currently “undruggable” molecules. With the application of AI, it may be easier for scientific workers to remove the barriers when developing drugs for those targets that are presently “undruggable”.
The Possible Challenges of Targeting the “Undruggable” Cancer Driver Genes
The possible challenges of targeting the “undruggable” oncogenes and tumor suppressor genes can be mainly summarized in three parts, that is, technical challenges, financial challenges, and ideological challenges.
Firstly, in addition to the difficulties of targeting summarized in the introduction section, toxicity could be another challenge for targeting the “undruggable” molecules. This is mainly because most of the “undruggable” oncogenes and tumor suppressor genes, including ras, myc, and tp53, all play crucial roles in normal cells (Simanshu et al., 2017; Levine, 2020; Duffy et al., 2021). Further, drug resistance is a great challenge besides effectiveness and toxicity. For example, resistance to some KRAS-G12C inhibitors could be caused by the increased level of RAS dimerization or the GTP-bound RAS proteins (Moore et al., 2020). Tumor cells can suppress or evade the immune system by expressing several immune checkpoints, leading to downregulation of the immune response in human cancer and hindrance to immunotherapy. Elevated levels of immune checkpoints like programmed cell death protein ligand 1 (PDL1) in NSCLC tumors with k-ras mutations and increased cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) expression in liver cancer tumors with tp53 mutations can be an obstacle to applying immunotherapy to the “undruggable” oncogenes and tumor suppressor genes (Ninomiya and Hotta, 2018; Long et al., 2019).
Secondly, the investment in this research with technical challenges is high and risky at the moment. Therefore, persuading stakeholder commitment to resources to invest may be a challenge as big as technical challenges. Thirdly, we need to break away from dogmatism and avoid conventional thinking. Targeting these “undruggable” molecules has been done for many years, but the current progress is few, so we need to reflect on what is wrong with our current research methods and ideas, and how to improve them.
To summarize, how to overcome these challenges is the current question we need to think about.
While direct targeting of “undruggable” molecules has always been the goal of the research, indirect targeting is a temporary alternative measure. Ras is a critical oncogene in cancer development and its associated proteins are now gradually becoming directly targetable by several RAS inhibitors represented by Sotorasib. MYC and P53 proteins encoded by myc and tp53, however, are both unable to be currently directly targeted, but several alternative approaches have documented results in indirectly targeting them. In the quest to target “undruggable” molecules, some emerging approaches like PROTACs, bispecific antibodies, and cancer vaccines all have great potential. In addition, AI can play a significant role in accelerating drug discovery. This article has summarized the conventional and some novel approaches that target ras, myc, tp53, and a few other molecules that are considered “undruggable”. This contributes to our understanding of the current status of targeted anti-cancer drug development. Looking ahead, there are challenges not only from drug effectiveness, toxicity, and drug resistance but also challenges from economical and ideological aspects. The ways in which we can tackle those barriers is the question raised by this article. Drug discovery of the “undruggable” molecules previously seemed to be an impossible mission, but now with the rapid development of technology, this would be a fruitful area for future work.
Funding Statement: This work was supported by Grants from the National Natural Science Foundation of China (81902784), the CAMS Innovation Fund for Medical Sciences (CIFMS, 2019-I2M-5-004), the Fund of Sichuan Provincial Department of Science and Technology (2022YFSY0058), the Research Funding (RCDWJS 2020-20), and the Research and Development Program (RD-02-202002) from West China School/Hospital of Stomatology Sichuan University.
Author Contributions: The authors confirm their contribution to the paper as follows: Xingbo Wu & Yingqiang Shen: Conceptualization; Xingbo Wu: Methodology, Software; Xingbo Wu & Shouyi Tang: Data curation, Writing—original draft preparation. Dan Pan: Visualization, Investigation. Yingqiang Shen: Supervision. Xingbo Wu & Yingqiang Shen: Writing—reviewing, Editing, and Yingqiang Shen: Funding acquisition. All authors reviewed the results and approved the final version of the manuscript.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
Asimgil H, Ertetik U, Çevik NC, Ekizce M, Doğruöz A et al. (2022). Targeting the undruggable oncogenic KRAS: The dawn of hope. JCI Insight 7. [Google Scholar]
Athanasios A, Charalampos V, Vasileios T (2017). Protein-protein interaction (PPI) network: Recent advances in drug discovery. Current Drug Metabolism 18: 5–10. [Google Scholar] [PubMed]
Barros LR, Paixão EA, Valli AM, Naozuka GT, Fassoni AC, Almeida RC (2021). CARTmath–a mathematical model of CAR-T immunotherapy in preclinical studies of hematological cancers. Cancers 13: 2941. [Google Scholar] [PubMed]
Binayke A, Mishra S, Suman P, Das S, Chander H (2019). Awakening the “guardian of genome”: Reactivation of mutant p53. Cancer Chemotherapy and Pharmacology 83: 1–15. [Google Scholar] [PubMed]
Blanden AR, Yu X, Wolfe AJ, Gilleran JA, Augeri DJ et al. (2015). Synthetic metallochaperone ZMC1 rescues mutant p53 conformation by transporting zinc into cells as an ionophore. Molecular Pharmacology 87: 825–831. [Google Scholar] [PubMed]
Boi M, Gaudio E, Bonetti P, Kwee I, Bernasconi E et al. (2015). The BET bromodomain inhibitor OTX015 affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clinical Cancer Research 21: 1628–1638. [Google Scholar] [PubMed]
Bolick SC, Landowski TH, Boulware D, Oshiro MM, Ohkanda J, Hamilton AD, Sebti SM, Dalton WS (2003). The farnesyl transferase inhibitor, FTI-277, inhibits growth and induces apoptosis in drug-resistant myeloma tumor cells. Leukemia 17: 451–457. https://doi.org/10.1038/sj.leu.2402832 [Google Scholar] [PubMed] [CrossRef]
Brazel D, Arter Z, Nagasaka M (2022). A long overdue targeted treatment for KRAS mutations in NSCLC: Spotlight on adagrasib. Lung Cancer 13: 75–80. [Google Scholar] [PubMed]
Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE et al. (2013). Small molecule inhibitors of aurora–a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell 24: 75–89. https://doi.org/10.1016/j.ccr.2013.05.005 [Google Scholar] [PubMed] [CrossRef]
Bykov VJ, Eriksson SE, Bianchi J, Wiman KG (2018). Targeting mutant p53 for efficient cancer therapy. Nature Reviews Cancer 18: 89–102. https://doi.org/10.1038/nrc.2017.109 [Google Scholar] [PubMed] [CrossRef]
Cai BH, Hsu YC, Yeh FY, Lin YR, Lu RY et al. (2022). P63 and P73 activation in cancers with p53 mutationbell. Biomedicines 10: 1490. https://doi.org/10.3390/biomedicines10071490 [Google Scholar] [PubMed] [CrossRef]
Calvisi DF, Ladu S, Conner EA, Seo D, Hsieh JT, Factor VM, Thorgeirsson SS (2011). Inactivation of Ras GTPase-activating proteins promotes unrestrained activity of wild-type Ras in human liver cancer. Journal of Hepatology 54: 311–319. https://doi.org/10.1016/j.jhep.2010.06.036 [Google Scholar] [PubMed] [CrossRef]
Cancer Genome Atlas Network (2012). Comprehensive molecular portraits of human breast tumours. Nature 490: 61–70. https://doi.org/10.1038/nature11412 [Google Scholar] [PubMed] [CrossRef]
Cancer Genome Atlas Research Network (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474: 609–615. https://doi.org/10.1038/nature10166 [Google Scholar] [PubMed] [CrossRef]
Cancer Genome Atlas Research Network (2012). Comprehensive genomic characterization of squamous cell lung cancers. Nature 489: 519–525. https://doi.org/10.1038/nature11404 [Google Scholar] [PubMed] [CrossRef]
Carroll PA, Freie BW, Mathsyaraja H, Eisenman RN (2018). The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Frontiers of Medicine 12: 412–425. https://doi.org/10.1007/s11684-018-0650-z [Google Scholar] [PubMed] [CrossRef]
Casey SC, Baylot V, Felsher DW (2018). The MYC oncogene is a global regulator of the immune response. Blood 131: 2007–2015. https://doi.org/10.1182/blood-2017-11-742577 [Google Scholar] [PubMed] [CrossRef]
Chen H, Liu H, Qing G (2018). Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduction and Targeted Therapy 3: 635. https://doi.org/10.1038/s41392-018-0008-7 [Google Scholar] [PubMed] [CrossRef]
Chen X, Yao H, Kashif M, Revêchon G, Eriksson M et al. (2021). A small-molecule ICMT inhibitor delays senescence of Hutchinson-Gilford progeria syndrome cells. eLife 10: e63284. [Google Scholar]
Cheng J, Li Y, Wang X, Dong G, Sheng C (2020). Discovery of novel PDEδ degraders for the treatment of KRAS mutant colorectal cancer. Journal of Medicinal Chemistry 63: 7892–7905. https://doi.org/10.1021/acs.jmedchem.0c00929 [Google Scholar] [PubMed] [CrossRef]
Cooper AJ, Sequist LV, Lin JJ (2022). Third-generation EGFR and ALK inhibitors: Mechanisms of resistance and management. Nature Reviews Clinical Oncology 19: 499–514. https://doi.org/10.1038/s41571-022-00639-9 [Google Scholar] [PubMed] [CrossRef]
Costa CB, Casalta-Lopes J, Andrade C, Moreira D, Oliveira A et al. (2012). Farnesyltransferase inhibitors: Molecular evidence of therapeutic efficacy in acute lymphoblastic leukemia through cyclin D1 inhibition. Anticancer Research 32: 831–838. [Google Scholar] [PubMed]
Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ (2014). Drugging the undruggable RAS: Mission possible? Nature Reviews Drug Discovery 13: 828–851. https://doi.org/10.1038/nrd4389 [Google Scholar] [PubMed] [CrossRef]
Cruz-Migoni A, Canning P, Quevedo CE, Bataille CJ, Bery N, Miller A, Russell AJ, Phillips SE, Carr SB, Rabbitts TH (2019). Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds. Proceedings of the National Academy of Sciences of the United States of America 116: 2545–2550. [Google Scholar] [PubMed]
Dang CV (2012). MYC on the path to cancer. Cell 149: 22–35. [Google Scholar] [PubMed]
Dang CV, Reddy EP, Shokat KM, Soucek L (2017). Drugging the ‘undruggable’ cancer targets. Nature Reviews Cancer 17: 502–508. [Google Scholar] [PubMed]
Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146: 904–917. [Google Scholar] [PubMed]
Demicco EG (2019). Molecular updates in adipocytic neoplasms⋆. Seminars in Diagnostic Pathology 36: 85–94. [Google Scholar] [PubMed]
Demir Ö, Barros EP, Offutt TL, Rosenfeld M, Amaro RE (2021). An integrated view of p53 dynamics, function, and reactivation. Current Opinion in Structural Biology 67: 187–194. [Google Scholar] [PubMed]
Demma MJ, Mapelli C, Sun A, Bodea S, Ruprecht B et al. (2019). Omomyc reveals new mechanisms to inhibit the MYC oncogene. Molecular and Cellular Biology 39. [Google Scholar]
Donati B, Lorenzini E, Ciarrocchi A (2018). BRD4 and cancer: Going beyond transcriptional regulation. Molecular Cancer 17. [Google Scholar]
Drummond ML, Henry A, Li H, Williams CI (2020). Improved accuracy for modeling PROTAC-mediated ternary complex formation and targeted protein degradation via new in silico methodologies. Journal of Chemical Information and Modeling 60: 5234–5254. [Google Scholar] [PubMed]
Drummond ML, Williams CI (2019). In silico modeling of PROTAC-mediated ternary complexes: Validation and application. Journal of Chemical Information and Modeling 59: 1634–1644. [Google Scholar] [PubMed]
Duffy MJ, O’Grady S, Tang M, Crown J (2021). MYC as a target for cancer treatment. Cancer Treatment Reviews 94. [Google Scholar]
Duffy MJ, Synnott NC, McGowan PM, Crown J, O’Connor D, Gallagher WM (2014). p53 as a target for the treatment of cancer. Cancer Treatment Reviews 40: 1153–1160. https://doi.org/10.1016/j.ctrv.2014.10.004 [Google Scholar] [PubMed] [CrossRef]
Elbadawy M, Usui T, Yamawaki H, Sasaki K (2019). Emerging roles of c-Myc in cancer stem cell-related signaling and resistance to cancer chemotherapy: A potential therapeutic target against colorectal cancer. International Journal of Molecular Sciences 20: 2340. https://doi.org/10.3390/ijms20092340 [Google Scholar] [PubMed] [CrossRef]
Fang Y, Liao G, Yu B (2020). Small-molecule MDM2/X inhibitors and PROTAC degraders for cancer therapy: Advances and perspectives. Acta Pharmaceutica Sinica B 10: 1253–1278. https://doi.org/10.1016/j.apsb.2020.01.003 [Google Scholar] [PubMed] [CrossRef]
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921. https://doi.org/10.1038/nature03445 [Google Scholar] [PubMed] [CrossRef]
Flanagan JJ, Neklesa TK (2019). Targeting nuclear receptors with PROTAC degraders. Molecular and Cellular Endocrinology 493: 110452. https://doi.org/10.1016/j.mce.2019.110452 [Google Scholar] [PubMed] [CrossRef]
Fridman JS, Lowe SW (2003). Control of apoptosis by p53. Oncogene 22: 9030–9040. https://doi.org/10.1038/sj.onc.1207116 [Google Scholar] [PubMed] [CrossRef]
George J, Lim JS, Jang SJ, Cun Y, Ozretić L et al. (2015). Comprehensive genomic profiles of small cell lung cancer. Nature 524: 47–53. https://doi.org/10.1038/nature14664 [Google Scholar] [PubMed] [CrossRef]
Hampton SE, Dore TM, Schmidt WK (2018). Rce1: Mechanism and inhibition. Critical Reviews in Biochemistry and Molecular Biology 53: 157–174. https://doi.org/10.1080/10409238.2018.1431606 [Google Scholar] [PubMed] [CrossRef]
Hassin O, Oren M (2023). Drugging p53 in cancer: One protein, many targets. Nature Reviews Drug Discovery 22: 127–144. https://doi.org/10.1038/s41573-022-00571-8 [Google Scholar] [PubMed] [CrossRef]
Hillig RC, Sautier B, Schroeder J, Moosmayer D, Hilpmann A et al. (2019). Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proceedings of the National Academy of Sciences of the United States of America 116: 2551–2560. https://doi.org/10.1073/pnas.1812963116 [Google Scholar] [PubMed] [CrossRef]
Hsiue EH, Wright KM, Douglass J, Hwang MS, Mog BJ et al. (2021). Targeting a neoantigen derived from a common TP53 mutation. Science 371. [Google Scholar]
Hu J, Wei J, Yim H, Wang L, Xie L et al. (2020). Potent and selective mitogen-activated protein kinase kinase 1/2 (MEK1/2) heterobifunctional small-molecule degraders. Journal of Medicinal Chemistry 63: 15883–15905. [Google Scholar] [PubMed]
Hussein HA, Borrel A, Geneix C, Petitjean M, Regad L, Camproux AC (2015). PockDrug-Server: A new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Research 43: W436–W442. [Google Scholar] [PubMed]
Jain AK, Barton MC (2009). Regulation of p53: TRIM24 enters the RING. Cell Cycle 8: 3668–3674. [Google Scholar] [PubMed]
Janani SK, Dhanabal SP, Sureshkumar R, Upadhyayula N, Chenmala K (2021). Guardian of genome on the tract: Wild type p53-mdm2 complex inhibition in healing the breast cancer. Gene 786: 145616. https://doi.org/10.1016/j.gene.2021.145616 [Google Scholar] [PubMed] [CrossRef]
Jiang Y, Zhuang C, Chen L, Lu J, Dong G, Miao Z, Zhang W, Li J, Sheng C (2017). Structural biology-inspired discovery of novel KRAS-PDEδ inhibitors. Journal of Medicinal Chemistry 60: 9400–9406. [Google Scholar] [PubMed]
Johnson BE, Heymach JV (2004). Farnesyl transferase inhibitors for patients with lung cancer. Clinical Cancer Research 10: 4254s–4257s. https://doi.org/10.1158/1078-0432.CCR-040016 [Google Scholar] [PubMed] [CrossRef]
Karasic TB, Chiorean EG, Sebti SM, O’Dwyer PJ (2019). A phase I study of GGTI-2418 (geranylgeranyl transferase I inhibitor) in patients with advanced solid tumors. Targeted Oncology 14: 613–618. https://doi.org/10.1007/s11523-019-00661-5 [Google Scholar] [PubMed] [CrossRef]
Kastan MB, Canman CE, Leonard CJ (1995). P53, cell cycle control and apoptosis: implications for cancer. Cancer Metastasis Reviews 14: 3–15. https://doi.org/10.1007/BF00690207 [Google Scholar] [PubMed] [CrossRef]
Kastenhuber ER, Lowe SW (2017). Putting p53 in context. Cell 170: 1062–1078. https://doi.org/10.1016/j.cell.2017.08.028 [Google Scholar] [PubMed] [CrossRef]
Kawasumi M, Bradner JE, Tolliday N, Thibodeau R, Sloan H, Brummond KM, Nghiem P (2014). Identification of ATR-Chk1 pathway inhibitors that selectively target p53-deficient cells without directly suppressing ATR catalytic activity. Cancer Research 74: 7534–7545. https://doi.org/10.1158/0008-5472.CAN-14-2650 [Google Scholar] [PubMed] [CrossRef]
Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A et al. (2019). Drugging an undruggable pocket on KRAS. Proceedings of the National Academy of Sciences of the United States of America 116: 15823–15829. https://doi.org/10.1073/pnas.1904529116 [Google Scholar] [PubMed] [CrossRef]
Leroy B, Anderson M, Soussi T (2014). TP53 mutations in human cancer: database reassessment and prospects for the next decade. Human Mutation 35: 672–688. https://doi.org/10.1002/humu.22552 [Google Scholar] [PubMed] [CrossRef]
Levine AJ (2019). Targeting therapies for the p53 protein in cancer treatments. Annual Review of Cancer Biology 3: 21–34. https://doi.org/10.1146/annurev-cancerbio-030518-055455 [Google Scholar] [CrossRef]
Levine AJ (2020). p53: 800 million years of evolution and 40 years of discovery. Nature Reviews Cancer 20: 471–480. https://doi.org/10.1038/s41568-020-0262-1 [Google Scholar] [PubMed] [CrossRef]
Lito P, Solomon M, Li LS, Hansen R, Rosen N (2016). Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351: 604–608. https://doi.org/10.1126/science.aad6204 [Google Scholar] [PubMed] [CrossRef]
Liu BS, Dai XY, Xia HW, Xu HJ, Tang QL, Gong QY, Nie YZ, Bi F (2018). Geranylgeranyl transferase 1 inhibitor GGTI-298 enhances the anticancer effect of gefitinib. Molecular Medicine Reports 18: 4023–4029. https://doi.org/10.3892/mmr.2018.9371 [Google Scholar] [PubMed] [CrossRef]
Liu P, Ge M, Hu J, Li X, Che L et al. (2017). A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology 66: 167–181. https://doi.org/10.1002/hep.29183 [Google Scholar] [PubMed] [CrossRef]
Liu X, Wilcken R, Joerger AC, Chuckowree IS, Amin J, Spencer J, Fersht AR (2013). Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Research 41: 6034–6044. https://doi.org/10.1093/nar/gkt305 [Google Scholar] [PubMed] [CrossRef]
Llombart V, Mansour MR (2022). Therapeutic targeting of “undruggable” MYC. eBioMedicine 75: 103756. https://doi.org/10.1016/j.ebiom.2021.103756 [Google Scholar] [PubMed] [CrossRef]
Long J, Wang A, Bai Y, Lin J, Yang X, Wang D, Yang X, Jiang Y, Zhao H (2019). Development and validation of a TP53-associated immune prognostic model for hepatocellular carcinoma. EBioMedicine 42: 363–374. https://doi.org/10.1016/j.ebiom.2019.03.022 [Google Scholar] [PubMed] [CrossRef]
Lőrincz O, Tóth J, Molnár L, Miklós I, Pántya K, Megyesi M, Somogyi E, Csiszovszki Z, Tőke ER (2021). In silico model estimates the clinical trial outcome of cancer vaccines. Cells 10. [Google Scholar]
Marín-Ramos NI, Balabasquer M, Ortega-Nogales FJ, Torrecillas IR, Gil-Ordóñez A et al. (2019). A potent isoprenylcysteine carboxylmethyltransferase (ICMT) inhibitor improves survival in Ras-driven acute myeloid leukemia. Journal of Medicinal Chemistry 62: 6035–6046. https://doi.org/10.1021/acs.jmedchem.9b00145 [Google Scholar] [PubMed] [CrossRef]
Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, Liu DD, Bekele BN, Herbst RS, Wistuba II (2007). KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clinical Cancer Research 13: 2890–2896. https://doi.org/10.1158/1078-0432.CCR-06-3043 [Google Scholar] [PubMed] [CrossRef]
Mesa RA (2006). Tipifarnib: Farnesyl transferase inhibition at a crossroads. Expert Review of Anticancer Therapy 6: 313–319. https://doi.org/10.1586/14737140.6.3.313 [Google Scholar] [PubMed] [CrossRef]
Minko T (2020). Nanoformulation of BRD4-degrading PROTAC: Improving druggability to target the ‘undruggable’ MYC in pancreatic cancer. Trends in Pharmacological Sciences 41: 684–686. [Google Scholar] [PubMed]
Mirgayazova R, Khadiullina R, Chasov V, Mingaleeva R, Miftakhova R, Rizvanov A, Bulatov E (2020). Therapeutic editing of the TP53 gene: Is CRISPR/Cas9 an option? Genes 11: 704. [Google Scholar] [PubMed]
Mohammed I, Hampton SE, Ashall L, Hildebrandt ER, Kutlik RA et al. (2016). 8-Hydroxyquinoline-based inhibitors of the Rce1 protease disrupt Ras membrane localization in human cells. Bioorganic & Medicinal Chemistry 24: 160–178. [Google Scholar]
Moore AR, Rosenberg SC, McCormick F, Malek S (2020). RAS-targeted therapies: Is the undruggable drugged? Nature Reviews Drug Discovery 19: 533–552. [Google Scholar] [PubMed]
Morandell S, Yaffe MB (2012). Exploiting synthetic lethal interactions between DNA damage signaling, checkpoint control, and p53 for targeted cancer therapy. Progress in Molecular Biology and Translational Science 110: 289–314. [Google Scholar] [PubMed]
Mullard A (2021). The FDA approves a first farnesyltransferase inhibitor. Nature Reviews Drug Discovery 20: 806. https://doi.org/10.1038/d41573-021-00168-7 [Google Scholar] [PubMed] [CrossRef]
Mullard A (2022). 2021 FDA approvals. Nature Reviews Drug Discovery 21: 83–88. https://doi.org/10.1038/d41573-022-00001-9 [Google Scholar] [PubMed] [CrossRef]
Nagasaka M, Li Y, Sukari A, Ou SI, Al-Hallak MN, Azmi AS (2020). KRAS G12C game of thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treatment Reviews 84. [Google Scholar]
Nakajima EC, Drezner N, Li X, Mishra-Kalyani PS, Liu Y et al. (2022). FDA approval summary: Sotorasib for KRAS G12C-mutated metastatic NSCLC. Clinical Cancer Research 28: 1482–1486. https://doi.org/10.1158/1078-0432.CCR-21-3074 [Google Scholar] [PubMed] [CrossRef]
Neklesa TK, Winkler JD, Crews CM (2017). Targeted protein degradation by PROTACs. Pharmacology & Therapeutics 174: 138–144. https://doi.org/10.1016/j.pharmthera.2017.02.027 [Google Scholar] [PubMed] [CrossRef]
Nguyen D, Liao W, Zeng SX, Lu H (2017). Reviving the guardian of the genome: Small molecule activators of p53. Pharmacology & Therapeutics 178: 92–108. https://doi.org/10.1016/j.pharmthera.2017.03.013 [Google Scholar] [PubMed] [CrossRef]
Ninomiya K, Hotta K (2018). Pembrolizumab for the first-line treatment of non-small cell lung cancer. Expert Opinion on Biological Therapy 18: 1015–1021. https://doi.org/10.1080/14712598.2018.1522300 [Google Scholar] [PubMed] [CrossRef]
Nomura TK, Heishima K, Sugito N, Sugawara R, Ueda H, Yukihiro A, Honda R (2021). Specific inhibition of oncogenic RAS using cell-permeable RAS-binding domains. Cell Chemical Biology 28: 1581–1589. https://doi.org/10.1016/j.chembiol.2021.04.013 [Google Scholar] [PubMed] [CrossRef]
Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM et al. (2018). Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nature Chemical Biology 14: 163–170. https://doi.org/10.1038/nchembio.2538 [Google Scholar] [PubMed] [CrossRef]
Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503: 548–551. https://doi.org/10.1038/nature12796 [Google Scholar] [PubMed] [CrossRef]
Otte J, Dyberg C, Pepich A, Johnsen JI (2021). MYCN function in neuroblastoma development. Frontiers in Oncology 10: 22. https://doi.org/10.3389/fonc.2020.624079 [Google Scholar] [PubMed] [CrossRef]
Otto C, Schmidt S, Kastner C, Denk S, Kettler J, Müller N, Germer CT, Wolf E, Gallant P, Wiegering A (2019). Targeting bromodomain-containing protein 4 (BRD4) inhibits MYC expression in colorectal cancer cells. Neoplasia 21: 1110–1120. https://doi.org/10.1016/j.neo.2019.10.003 [Google Scholar] [PubMed] [CrossRef]
Pabla N, Huang S, Mi QS, Daniel R, Dong Z (2008). ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. The Journal of Biological Chemistry 283: 6572–6583. https://doi.org/10.1074/jbc.M707568200 [Google Scholar] [PubMed] [CrossRef]
Pandey M, Fernandez M, Gentile F, Isayev O, Tropsha A, Stern AC, Cherkasov A (2022). The transformational role of GPU computing and deep learning in drug discovery. Nature Machine Intelligence 4: 211–221. https://doi.org/10.1038/s42256-022-00463-x [Google Scholar] [CrossRef]
Pazgier M, Liu M, Zou G, Yuan W, Li C et al. (2009). Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proceedings of the National Academy of Sciences of the United States of America 106: 4665–4670. https://doi.org/10.1073/pnas.0900947106 [Google Scholar] [PubMed] [CrossRef]
Peng X, Zhang MQ, Conserva F, Hosny G, Selivanova G, Bykov VJ, Arnér ES, Wiman KG (2013). APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death & Disease 4: e881. https://doi.org/10.1038/cddis.2013.417 [Google Scholar] [PubMed] [CrossRef]
Pishas KI, Adwal A, Neuhaus SJ, Clayer MT, Farshid G, Staudacher AH, Callen DF (2015). XI-006 induces potent p53-independent apoptosis in Ewing sarcoma. Scientific Reports 5. [Google Scholar]
Posternak G, Tang X, Maisonneuve P, Jin T, Lavoie H et al. (2020). Functional characterization of a PROTAC directed against BRAF mutant V600E. Nature Chemical Biology 16: 1170–1178. https://doi.org/10.1038/s41589-020-0609-7 [Google Scholar] [PubMed] [CrossRef]
Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E et al. (2019). Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575: 210–216. https://doi.org/10.1038/s41586-019-1689-y [Google Scholar] [PubMed] [CrossRef]
Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D (2011). RAS oncogenes: Weaving a tumorigenic web. Nature Reviews Cancer 11: 761–774. https://doi.org/10.1038/nrc3106 [Google Scholar] [PubMed] [CrossRef]
Quevedo CE, Cruz-Migoni A, Bery N, Miller A, Tanaka T et al. (2018). Small molecule inhibitors of RAS-effector protein interactions derived using an intracellular antibody fragment. Nature Communications 9: 3169. https://doi.org/10.1038/s41467-018-05707-2 [Google Scholar] [PubMed] [CrossRef]
Raina K, Lu J, Qian Y, Altieri M, Gordon D et al. (2016). PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proceedings of the National Academy of Sciences of the United States of America 113: 7124–7129. https://doi.org/10.1073/pnas.1521738113 [Google Scholar] [PubMed] [CrossRef]
Reed D, Shen Y, Shelat AA, Arnold LA, Ferreira AM et al. (2010). Identification and characterization of the first small molecule inhibitor of MDMX. The Journal of Biological Chemistry 285: 10786–10796. https://doi.org/10.1074/jbc.M109.056747 [Google Scholar] [PubMed] [CrossRef]
Rossman KL, Der CJ, Sondek J (2005). GEF means go: Turning on RHO GTPases with guanine nucleotide-exchange factors. Nature Reviews Molecular Cell Biology 6: 167–180. https://doi.org/10.1038/nrm1587 [Google Scholar] [PubMed] [CrossRef]
Ryan DP, Eder JPJr, Puchlaski T, Seiden MV, Lynch TJ, Fuchs CS, Amrein PC, Sonnichsen D, Supko JG, Clark JW (2004). Phase I clinical trial of the farnesyltransferase inhibitor BMS-214662 given as a 1-hour intravenous infusion in patients with advanced solid tumors. Clinical Cancer Research 10: 2222–2230. https://doi.org/10.1158/1078-0432.CCR-0980-3 [Google Scholar] [PubMed] [CrossRef]
Röth S, Macartney TJ, Konopacka A, Chan KH, Zhou H, Queisser MA, Sapkota GP (2020). Targeting endogenous K-RAS for degradation through the affinity-directed protein missile system. Cell Chemical Biology 27: 1151–1163. https://doi.org/10.1016/j.chembiol.2020.06.012 [Google Scholar] [PubMed] [CrossRef]
Salami J, Alabi S, Willard RR, Vitale NJ, Wang J et al. (2018). Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Communications Biology 1: 100. https://doi.org/10.1038/s42003-018-0105-8 [Google Scholar] [PubMed] [CrossRef]
Schapira M, Calabrese MF, Bullock AN, Crews CM (2019). Targeted protein degradation: expanding the toolbox. Nature Reviews Drug Discovery 18: 949–963. https://doi.org/10.1038/s41573-019-0047-y [Google Scholar] [PubMed] [CrossRef]
Schaub FX, Dhankani V, Berger AC, Trivedi M, Richardson AB et al. (2018). Pan-cancer alterations of the MYC oncogene and its proximal network across the cancer genome atlas. Cell Systems 6: 282–300. https://doi.org/10.1016/j.cels.2018.03.003 [Google Scholar] [PubMed] [CrossRef]
Shen J, Wang Y, Hung MC (2013). RASAL2: Wrestling in the combat of Ras activation. Cancer Cell 24: 277–279. https://doi.org/10.1016/j.ccr.2013.08.024 [Google Scholar] [PubMed] [CrossRef]
Sidaway P (2021). Sotorasib effective in KRAS-mutant NSCLC. Nature Reviews Clinical Oncology 18: 748. https://doi.org/10.1038/s41571-021-00569-y [Google Scholar] [CrossRef]
Simanshu DK, Nissley DV, McCormick F (2017). RAS proteins and their regulators in human disease. Cell 170: 17–33. https://doi.org/10.1016/j.cell.2017.06.009 [Google Scholar] [PubMed] [CrossRef]
Simpson LM, Macartney TJ, Nardin A, Fulcher LJ, Röth S, Testa A, Maniaci C, Ciulli A, Ganley IG, Sapkota GP (2020). Inducible degradation of target proteins through a tractable affinity-directed protein missile system. Cell Chemical Biology 27: 1164–1180. https://doi.org/10.1016/j.chembiol.2020.06.013 [Google Scholar] [PubMed] [CrossRef]
Singh K, Lin J, Zhong Y, Burčul A, Mohan P et al. (2019). c-MYC regulates mRNA translation efficiency and start-site selection in lymphoma. The Journal of Experimental Medicine 216: 1509–1524. https://doi.org/10.1084/jem.20181726 [Google Scholar] [PubMed] [CrossRef]
Šolman M, Ligabue A, Blaževitš O, Jaiswal A, Zhou Y et al. (2015). Specific cancer-associated mutations in the switch III region of Ras increase tumorigenicity by nanocluster augmentation. eLife 4. [Google Scholar]
Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, Forbes SA (2018). The COSMIC cancer gene census: Describing genetic dysfunction across all human cancers. Nature Reviews Cancer 18: 696–705. https://doi.org/10.1038/s41568-018-0060-1 [Google Scholar] [PubMed] [CrossRef]
Song Y, Zhao M, Wu Y, Yu B, Liu HM (2021). A multifunctional cross-validation high-throughput screening protocol enabling the discovery of new SHP2 inhibitors. Acta Pharmaceutica Sinica B 11: 750–762. https://doi.org/10.1016/j.apsb.2020.10.021 [Google Scholar] [PubMed] [CrossRef]
Soussi T, Wiman KG (2015). TP53: An oncogene in disguise. Cell Death and Differentiation 22: 1239–1249. https://doi.org/10.1038/cdd.2015.53 [Google Scholar] [PubMed] [CrossRef]
Spencer-Smith R, Koide A, Zhou Y, Eguchi RR, Sha F et al. (2017). Inhibition of RAS function through targeting an allosteric regulatory site. Nature Chemical Biology 13: 62–68. https://doi.org/10.1038/nchembio.2231 [Google Scholar] [PubMed] [CrossRef]
Stank A, Kokh DB, Fuller JC, Wade RC (2016). Protein binding pocket dynamics. Accounts of Chemical Research 49: 809–815. https://doi.org/10.1021/acs.accounts.5b00516 [Google Scholar] [PubMed] [CrossRef]
Stephen AG, Esposito D, Bagni RK, McCormick F (2014). Dragging Ras back in the ring. Cancer Cell 25: 272–281. https://doi.org/10.1016/j.ccr.2014.02.017 [Google Scholar] [PubMed] [CrossRef]
Sun B, Fiskus W, Qian Y, Rajapakshe K, Raina K et al. (2018). BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia 32: 343–352. https://doi.org/10.1038/leu.2017.207 [Google Scholar] [PubMed] [CrossRef]
Tamura RE, de Luna IV, Lana MG, Strauss BE (2018). Improving adenoviral vectors and strategies for prostate cancer gene therapy. Clinics 73: e476s. https://doi.org/10.6061/clinics/2018/e476s [Google Scholar] [PubMed] [CrossRef]
Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J et al. (2021). Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discovery 11: 1913–1922. [Google Scholar] [PubMed]
Tavana O, Li D, Dai C, Lopez G, Banerjee D et al. (2016). HAUSP deubiquitinates and stabilizes N-Myc in neuroblastoma. Nature Medicine 22: 1180–1186. https://doi.org/10.1038/nm.4180 [Google Scholar] [PubMed] [CrossRef]
Thandapani P (2019). Super-enhancers in cancer. Pharmacology & Therapeutics 199: 129–138. https://doi.org/10.1016/j.pharmthera.2019.02.014 [Google Scholar] [PubMed] [CrossRef]
Tiwari G, Verma CS (2017). Toward understanding the molecular recognition of albumin by p53-activating stapled peptide ATSP-7041. The Journal of Physical Chemistry B 121: 657–670. https://doi.org/10.1021/acs.jpcb.6b09900 [Google Scholar] [PubMed] [CrossRef]
Tsubamoto M, Le TK, Li M, Watanabe T, Matsumi C, Parvatkar P, Fujii H, Kato N, Sun J, Ohkanda J (2019). A guanidyl-based bivalent peptidomimetic inhibits K-Ras prenylation and association with c-Raf. Chemistry 25: 13531–13536. https://doi.org/10.1002/chem.201903129 [Google Scholar] [PubMed] [CrossRef]
Tsuda M, Okamoto K, Muguruma N, Sannomiya K, Nakagawa T et al. (2013). Suppressive effect of RAS inhibitor manumycin A on aberrant crypt foci formation in the azoxymethane-induced rat colorectal carcinogenesis model. Journal of Gastroenterology and Hepatology 28: 1616–1623. https://doi.org/10.1111/jgh.12287 [Google Scholar] [PubMed] [CrossRef]
Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M et al. (2021). Highly accurate protein structure prediction for the human proteome. Nature 596: 590–596. https://doi.org/10.1038/s41586-021-03828-1 [Google Scholar] [PubMed] [CrossRef]
Valle PA, Coria LN, Plata C, Salazar Y (2021). CAR-T cell therapy for the treatment of ALL: Eradication conditions and in silico experimentation. Hemato 2: 441–462. https://doi.org/10.3390/hemato2030028 [Google Scholar] [CrossRef]
Vigil D, Cherfils J, Rossman KL, Der CJ (2010). Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nature Reviews Cancer 10: 842–857. https://doi.org/10.1038/nrc2960 [Google Scholar] [PubMed] [CrossRef]
Vollmer S, Cunoosamy D, Lv H, Feng H, Li X, Nan Z, Yang W, Perry MWD (2020). Design, synthesis, and biological evaluation of MEK PROTACs. Journal of Medicinal Chemistry 63: 157–162. https://doi.org/10.1021/acs.jmedchem.9b00810 [Google Scholar] [PubMed] [CrossRef]
Wang RR, Liu WS, Zhou L, Ma Y, Wang RL (2020a). Probing the acting mode and advantages of RMC-4550 as an Src-homology 2 domain-containing protein tyrosine phosphatase (SHP2) inhibitor at molecular level through molecular docking and molecular dynamics. Journal of Biomolecular Structure & Dynamics 38: 1525–1538. https://doi.org/10.1080/07391102.2019.1613266 [Google Scholar] [PubMed] [CrossRef]
Wang XN, Su XX, Cheng SQ, Sun ZY, Huang ZS, Ou TM (2019). MYC modulators in cancer: A patent review. Expert Opinion on Therapeutic Patents 29: 353–367. https://doi.org/10.1080/13543776.2019.1612878 [Google Scholar] [PubMed] [CrossRef]
Wang T, Wang W, Wang Q, Xie R, Landay A, Chen D (2020b). The E3 ubiquitin ligase CHIP in normal cell function and in disease conditions. Annals of the New York Academy of Sciences 1460: 3–10. https://doi.org/10.1111/nyas.14206 [Google Scholar] [PubMed] [CrossRef]
Wang C, Wang H, Zheng C, Liu Z, Gao X, Xu F, Niu Y, Zhang L, Xu P (2021). Research progress of MEK1/2 inhibitors and degraders in the treatment of cancer. European Journal of Medicinal Chemistry 218: 113386. https://doi.org/10.1016/j.ejmech.2021.113386 [Google Scholar] [PubMed] [CrossRef]
Wang Z, Yang B, Dong L, Peng B, He X, Liu W (2011). A novel oncoprotein Pirh2: Rising from the shadow of MDM2. Cancer Science 102: 909–917. https://doi.org/10.1111/j.1349-7006.2011.01899.x [Google Scholar] [PubMed] [CrossRef]
Wei J, Hu J, Wang L, Xie L, Jin MS, Chen X, Liu J, Jin J (2019). Discovery of a first-in-class mitogen-activated protein kinase kinase 1/2 degrader. Journal of Medicinal Chemistry 62: 10897–10911. https://doi.org/10.1021/acs.jmedchem.9b01528 [Google Scholar] [PubMed] [CrossRef]
Welsch ME, Kaplan A, Chambers JM, Stokes ME, Bos PH et al. (2017). Multivalent small-molecule Pan-RAS inhibitors. Cell 168: 878–889. https://doi.org/10.1016/j.cell.2017.02.006 [Google Scholar] [PubMed] [CrossRef]
Xiao D, Yue M, Su H, Ren P, Jiang J, Li F, Hu Y, Du H, Liu H, Qing G (2016). Polo-like kinase-1 regulates Myc stabilization and activates a feedforward circuit promoting tumor cell survival. Molecular Cell 64: 493–506. https://doi.org/10.1016/j.molcel.2016.09.016 [Google Scholar] [PubMed] [CrossRef]
Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D (2020). Improved protein structure prediction using predicted interresidue orientations. Proceedings of the National Academy of Sciences of the United States of America 117: 1496–1503. https://doi.org/10.1073/pnas.1914677117 [Google Scholar] [PubMed] [CrossRef]
Yang X, Wang Z, Pei Y, Song N, Xu L et al. (2021). Discovery of thalidomide-based PROTAC small molecules as the highly efficient SHP2 degraders. European Journal of Medicinal Chemistry 218. [Google Scholar]
You Y, Lai X, Pan Y, Zheng H, Vera J, Liu S, Deng S, Zhang L (2022). Artificial intelligence in cancer target identification and drug discovery. Signal Transduction and Targeted Therapy 7. [Google Scholar]
Zeng M, Xiong Y, Safaee N, Nowak RP, Donovan KA et al. (2020). Exploring targeted degradation strategy for oncogenic KRASG12C. Cell Chemical Biology 27: 19–31. [Google Scholar] [PubMed]
Zheng S, Tao W (2020). Targeting Cullin-RING E3 ligases for radiosensitization: From NEDDylation inhibition to PROTACs. Frontiers in Oncology 10. [Google Scholar]
Zhou Y, Hancock JF (2015). Ras nanoclusters: Versatile lipid-based signaling platforms. Biochimica et Biophysica Acta 1853: 841–849. [Google Scholar] [PubMed]
Zhou C, Zhu C, Liu Q (2019). Toward in silico identification of tumor neoantigens in immunotherapy. Trends in Molecular Medicine 25: 980–992. [Google Scholar] [PubMed]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools