
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

E3 ubiquitin ligase CBL-B suppresses vascular endothelial cell pyroptosis and injury in intracranial aneurysm by facilitating NLRP3 degradation
1 Department of Neurosurgery, Jingzhou Hospital of Traditional Chinese Medicine, Jingzhou, 434000, China
2 Department of Critical Care Medicine, Jingzhou Hospital of Traditional Chinese Medicine, Jingzhou, 434000, China
* Corresponding Author: CHENG LIU. Email:
BIOCELL 2024, 48(2), 293-301. https://doi.org/10.32604/biocell.2023.044211
Received 24 July 2023; Accepted 24 October 2023; Issue published 23 February 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Objective: Intracranial aneurysm (IA) represents a devastating disease with high rates of disability and mortality, which is initiated by dysfunction of endothelial cells (ECs). Evidence suggests the dysregulation of the E3 ubiquitin ligase family during EC injury. In this work, the role of an E3 ubiquitin ligase, casitas B lymphoma-B (CBL-B), was explored in human brain microvascular EC (HBMEC) function through the NLRP3 pathway. Methods: In vitro IA model was induced by treating HBMECs with oxidized low-density lipoprotein (ox-LDL). The levels of CBL-B and pyroptosis-related proteins NLRP3, ASC, cleaved caspase-1, and GSDME-N were determined by real-time-quantitative polymerase chain reaction and western blotting. HBMEC apoptosis, proliferation, and migration were evaluated by flow cytometry, MTT, and scratch tests. An enzyme-linked immunosorbent assay was conducted to measure interleukin (IL)-1β and IL-18 levels in cell culture supernatant. The ubiquitination of NLRP3 was detected by co-immunoprecipitation. Results: NLRP3 was highly expressed while CBL-B was weakly expressed in ox-LDL-treated HBMECs. Cell apoptosis was enhanced, proliferation and migration were weakened, NLRP3 inflammasome was activated, and levels of gasdermin E-N, IL-1β, and IL-18 were increased in ox-LDL-induced HBMECs. The above tendencies were counteracted by NLRP3 knockdown or CBL-B overexpression. Additionally, CBL-B overexpression increased NLRP3 ubiquitination, suggesting that CBL-B hampered the activation of NLRP3 inflammasome by ubiquitinating NLRP3. Conclusion: E3 ubiquitin ligase CBL-B accelerates NLRP3 degradation to impede HBMEC pyroptosis, thus palliating vascular EC injury in IA.Keywords
Intracranial aneurysm (IA) is conceived as an outward bulging of arterial walls, featured primarily by chronic inflammation and weakening of arterial walls [1]. IA afflicts approximately 3% of the general population, with a markedly high prevalence in women [2]. Although most of them remain stable and are asymptomatic; once the IA ruptures, the consequent subarachnoid hemorrhage (SAH) usually yields high rates of mortality, disability, and morbidity, as well as heavy socioeconomic burden [3,4]. Survival from aneurysmal SAH has risen by approximately 17% in recent few decades, presumably due to better diagnosis, prescription of nimodipine, and early aneurysm repair, as well as advanced intensive care support [5]. Patients with aneurysmal SAH may have better outcomes when cared for in the specialized neurologic intensive care unit with a multi-/inter-disciplinary clinical team [6]. Currently, microsurgical clipping and endovascular modalities have made profound advancements in the treatment of IA [7]. However, therapeutic efficacy remains unsatisfactory due to high recurrence rates and complications, highlighting a need to elucidate its complicated molecular mechanisms [8]. Intrinsically, endothelial cells (ECs) are a dominant type of cells composing the intracranial arteries, which are essential in maintaining cerebrovascular integrity [9]. EC dysfunction is widely-acknowledged as a contributor to IA pathogenesis, with damage or injury to the EC layer as the first event in IA formation [10]. Consequently, a deeper understanding of the complex mechanisms of vascular EC function is urgently needed to treat IA effectively.
Current evidence has indicated the involvement of endothelium pyroptosis in multiple vascular diseases, including cerebrovascular diseases [11]. More specifically, EC pyroptosis occurs after intracerebral hemorrhage, and can be repressed by directly targeting nucleotide-binding oligomerization domain-like receptor pyrin domain-containing 3 (NLRP3) or its upstream molecules, thereby improving the prognosis of intracerebral hemorrhage [12]. The NLRP3 inflammasome is considered an intracellular protein complex composed of a sensor NLRP3, an effector caspase-1, and an adaptor apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), which is vital in mediating inflammation in the context of diverse pathologies [13,14]. The assembly of NLRP3 inflammasome contributes to the caspase 1-dependent generation of pro-inflammatory cytokines interleukin (IL)-1β and IL-18, in addition to gasdermin D (GSDMD)-regulated pyroptosis [15]. NLRP3 inflammasome-mediated pyroptosis and inflammation are pivotal function in endothelial dysfunction [16]. Importantly, pharmacologic manipulation of NLRP3 ubiquitination can hinder NLRP3 inflammasome assembly and activation and protect against atherosclerosis and vascular inflammation [17].
Ubiquitination is a crucial mechanism related to post-translational modification, and casitas B lymphoma-B (CBL-B) emerges as a known RING finger E3 ubiquitin ligase to mediate ubiquitination modification of proteins [18]. CBL-B is shown to control NLRP3 inflammasome by targeting NLRP3 for K48-linked ubiquitination [19]. More importantly, heterozygous variants in the CBL gene are extremely related to cerebral arteriopathy [20]. CBL-B expression in human plaques decreases during atherosclerosis progression [21]. In particular, CBL is implicated in angiogenesis in part by mediating EC growth [22]. Moreover, the suppression of tyrosine phosphorylation of phospholipase Cγ1 by CBL is imperative in vascular endothelial growth factor-induced cellular responses in ECs [23]. We, therefore, dissected the possible impact and mechanism of CBL-B and NLRP3 on the function of vascular ECs.
Cell culture and construction of in vitro intracranial aneurism model
Human brain microvascular ECs (HBMECs; ScienCell, Carlsbad, CA, USA) were cultured in the extracellular matrix medium (ScienceCell) at 37°C in a 5% CO2 incubator. Once reaching 70% confluence, HBMECs were treated with 100 mg/L oxidized low-density lipoprotein (ox-LDL; Solarbio, Beijing, China) for 24 h [24].
Cell transfection and grouping
HBMECs were placed in 96-well plates prior to transfection. Small interfering RNA for NLRP3 (si-NLRP3), overexpression vectors for CBL-B and NLRP3 (oe-CBL-B and oe-NLRP3), and their negative controls (si-NC and oe-NC) were ordered from GenePharma (Shanghai, China). When cells reached 70%–80% confluence, transfection (48 h) was executed strictly in the presence of Lipofectamine 2000 transfection following the kit instructions.
Cells were treated with trypsin (0.25%), centrifuged for 5 min at 800 rpm, and suspended in binding buffer (1×) to adjust the cell density to 1 × 107 cells/mL. Next, the cell suspension (100 μL) was incubated for 20 min with 5 μL propidium iodide (20 μg/mL) and Annexin V-fluorescein isothiocyanate and subsequently mixed with 400 μL binding buffer (1×). Within 1 h, cell apoptosis was evaluated with the help of a flow cytometer (BD FACSArial I cell sorter, BD Biosciences, San Jose, CA, USA).
Assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
Cell suspension was prepared by detaching cells with trypsin. The cell concentration was adjusted to 5 × 104 cells/mL and cultured in 96-well plates. Following 48 h, cells were incubated with 20 μL MTT for 4 h. MTT solution in each well was dissolved in 150 μL dimethyl sulfoxide. The optical density (OD) value of cells was measured at 570 nm, and then the viability rate of HBMECs was calculated.
Cells were inoculated in 24-well plates at a density of 2 × 105 cells/well. On reaching 90% confluence, a 1 mL pipette tip was used to create a straight scratch, after which the floating cells were removed. Following 24-h culture, the scratch spacing was observed under a microscope. National Instrument Vision Assistant 8.6 software was utilized to calculate the wound healing area. Cell mobility rate = healing area of scratch wound/initial scratched area × 100%.
Enzyme-linked immunosorbent assay (ELISA)
The collected HBMECs were centrifuged at 300×g for 10 min, and the sediment was removed. Thereafter, levels of IL-1β and IL-18 in cell culture supernatant were determined using the corresponding kits (EK101B; EK218, Multisciences (Lianke) Biotech, Hangzhou, Zhejiang, China). Finally, the OD value was measured, and a standard curve was plotted.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
The extraction of total RNA from cells was conducted by means of RNAsimple Total RNA Extraction Kits (DP419, TIANGEN Biotech, Beijing, China), followed by determination of RNA concentration and purity. The reverse transcription into cDNA was performed utilizing PrimeScript RT reagent Kits (Takara, Tokyo, Japan). RT-qPCR was implemented with the help of SYBR GreenPCR Master Mix (Takara) and Real-Time PCR System (Roche, Basel, Switzerland). Primers were synthesized by (Sangon Biotech, Shanghai, China) and exhibited in Table 1. The Ct value of each well was recorded, and the relative expression of the product was calculated using the 2−ΔΔCt method, with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) selected as an internal control.
Cells were rinsed with pre-cooled phosphate-buffered saline (PBS) and lysed with protein extraction lysis buffer. Protein samples were quantified using bicinchoninic acid kits. Samples were mixed with 1/4 volume of 5×sample buffer, followed by 5-min heating in a boiling water bath. Following electrophoresis using 10% separation gels and 5% concentration gels and transferring to membranes, the membranes were sealed with 5% dry skim milk for 60 min and then probed overnight with following primary antibodies at 4°C: NLRP3 (1:1000, ab263899, Abcam, Cambridge, UK), CBL-B (1:200, sc-8006, Santa Cruz Biotechnology, Santa Cruz, CA, USA), ASC (1:1000, ab283684, Abcam), cleaved caspase-1 (1:1000, #4199, CST, Beverly, MA, USA), GSDME-N (1:1000, ab222408, Abcam), and GAPDH (1:500, ab8245, Abcam). Thereafter, membranes were incubated with secondary antibody IgG for 60 min. The membranes were immersed in an electrochemiluminescence reaction solution (Beyotime) for 1 min, and after the removal of the liquid, the membranes were covered. After the dropwise addition of the developing solution, membranes were detected using a chemiluminescence imaging system (Tanon, Shanghai, China).
Co-immunoprecipitation (Co-IP)
NLRP3 ubiquitination was detected by immunoblotting after NLRP3 was isolated using IP assay. Cells were subjected to transfection for 48 h, and the next treatment with MG132 (HY-13259, MedChemExpress, Shanghai, China) for 4–6 h. Subsequently, cells were rinsed with PBS and lysed utilizing protein extraction kits (KeyGen Biotechnology, Nanjing, Jiangsu, China). Following centrifugation of lysates, the obtained cytoplasmic proteins were incubated overnight with NLRP3 antibody and then added with 100 μL protein A/G agarose beads to capture the antigen-antibody complex. The complex was subsequently incubated for 4 h on a shaker at 4°C, rinsed thrice with lysis buffer, and boiled in 2×sodium dodecyl sulfate loading buffer to release the proteins, which were then incubated with anti-ubiquitin antibody (1:2000, ab134953, Abcam) for immunoblotting.
Statistical analysis was processed by means of GraphPad Prism 7 software, and data were described as mean ± SD. For normally-distributed samples, two-group comparisons were conducted through the t-test, and multiple-group comparisons were made using ANOVA, followed by Tukey’s test. For samples not conforming to the normal distribution, the analysis of two independent samples was carried out by the Mann-Whitney U test, and for multiple independent samples, Kruskal-Wallis was performed. Statistical significance was declared when p < 0.05.
Mitigation of human brain microvascular endothelial-cell injury due to NLRP3 silencing
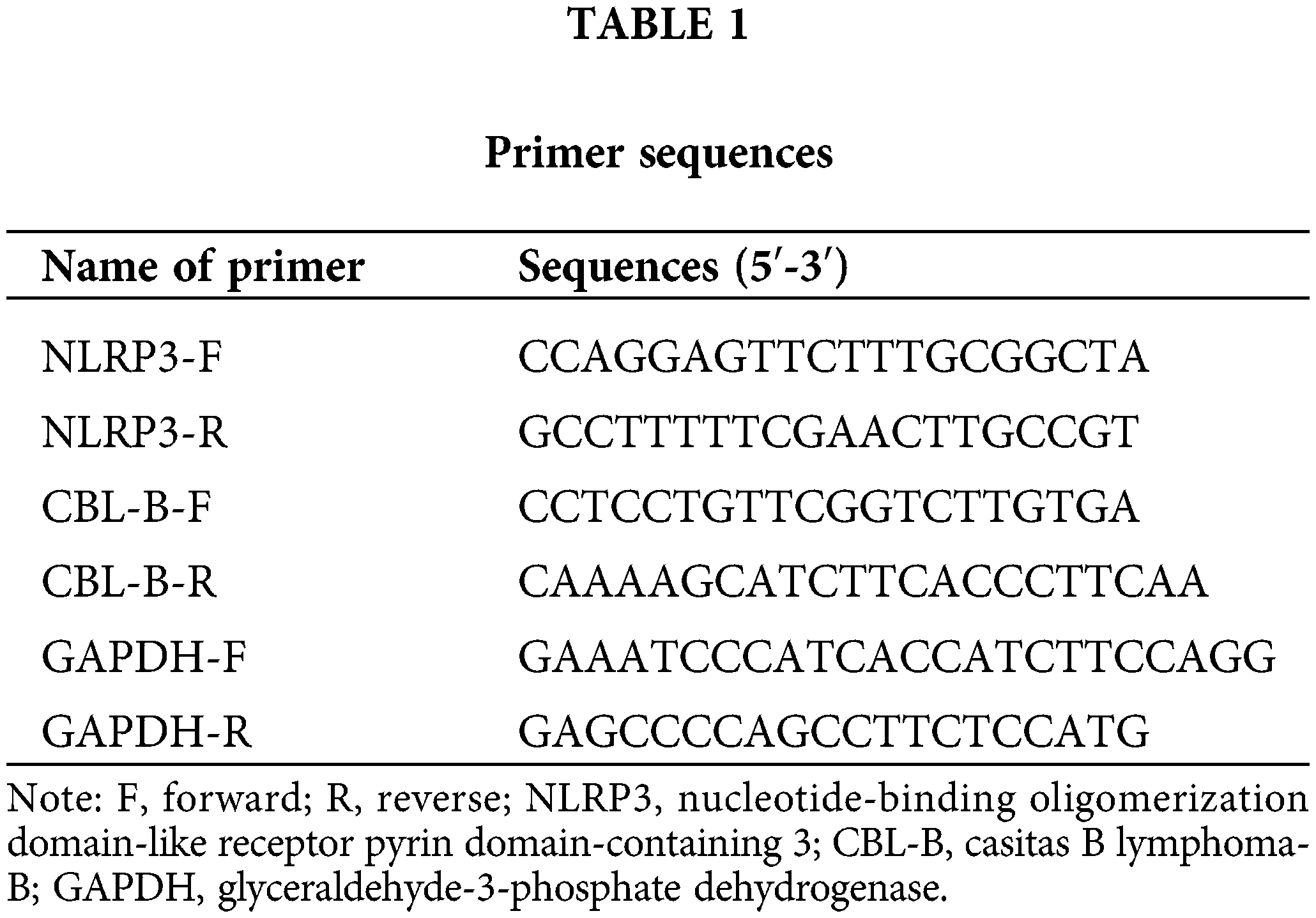
We first constructed an in vitro model of IA by inducing HBMECs with ox-LDL. RT-qPCR and Western blot revealed that NLRP3 levels were prominently increased in the ox-LDL group compared with the control group (Figs. 1A and 1B). To dissect the exact effect of NLRP3 on the function of HBMECs, si-NLRP3 or its NC was transfected into ox-LDL-induced HBMECs. RT-qPCR and Western blot evinced the successful transfection (Figs. 1A and 1B). Flow cytometry suggested that the ox-LDL group exhibited a higher apoptosis rate than the control group, and the ox-LDL + si-NLRP3 group showed a lower rate of apoptosis than the ox-LDL + si-NC group (Fig. 1C). The proliferative and migratory capacities of HBMECs were assessed by MTT (Fig. 1D) and scratch tests (Fig. 1E), which unveiled that cell viability and mobility were weakened in the ox-LDL group vs. the control group, and enhanced in the ox-LDL + si-NLRP3 group vs. the ox-LDL + si-NC group. Additionally, Western blot suggested that the expression levels of ASC, cleaved caspase-1, and GSDME-N were markedly increased in the ox-LDL group, whereas diminished in the ox-LDL + si-NLRP3 group (Fig. 1F). As indicated by ELISA, levels of inflammatory factors IL-1β and IL-18 in the cell supernatant were higher in the ox-LDL group than in the control group, which were lower in the ox-LDL + si-NLRP3 group than in the ox-LDL + si-NC group (Figs. 1G and 1H). Taken together, NLRP3 silencing contributed to the alleviation of HBMEC injury.
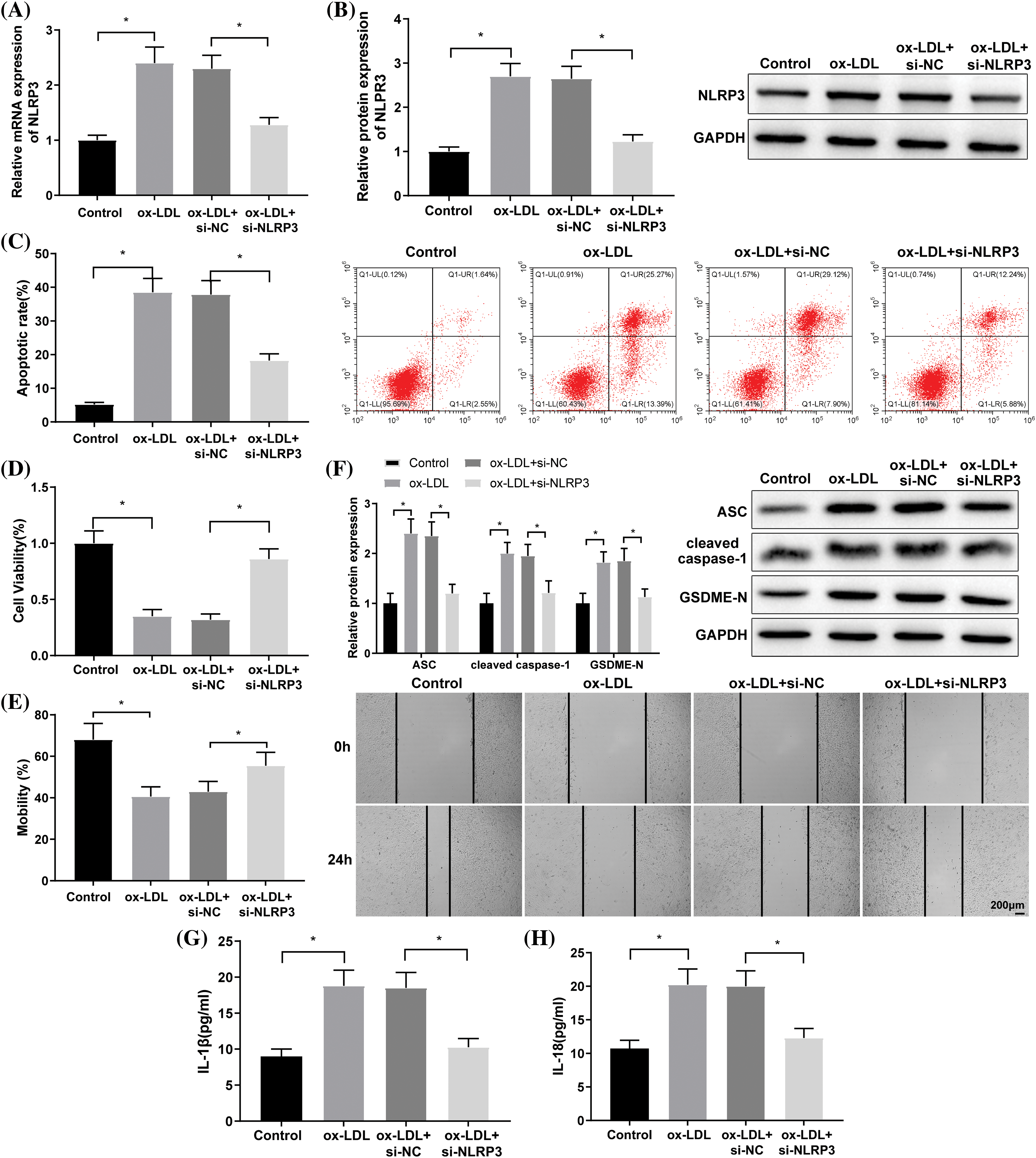
Figure 1: NLRP3 downregulation alleviates HBMEC injury. The expression of NLRP3 was determined through (A) RT-qPCR and (B) western blotting; (C) cell apoptosis was assessed by flow cytometry; (D) cell viability was assessed by MTT assay; (E) cell mobility was evaluated by a scratch test; (F) Western blotting was adopted to measure the protein levels of ASC, cleaved caspase-1, and GSDME-N; (G and H) ELISA was employed to determine IL-1β and IL-18 levels. *p < 0.05 compared with the control or ox-LDL + si-NC group. Experimentation was repeated thrice. NLRP3: nucleotide-binding oligomerization domain-like receptor pyrin domain-containing 3; RT-qPCR: real-time quantitative polymerase chain reaction; ASC: a caspase recruitment domain; GSDME: gasdermin E; ELISA: enzyme linked immunosorbent assay; IL-1β: interleukin-1β; ox-LDL: oxidized low-density lipoprotein; NC: negative control.
Inhibition of NLRP3 expression through ubiquitination regulation by casitas B lymphoma-B
The prediction from the UbiBrowser database revealed that CBL-B acted as an E3 ubiquitin ligase of NLRP3 (Fig. 2A). RT-qPCR and Western blot suggested the low expression of CBL-B in ox-LDL-induced HBMECs (Figs. 2B and 2C). Subsequently, oe-CBL-B or oe-NC was introduced into ox-LDL-treated HBMECs. Compared with the ox-LDL + oe-NC group, CBL-B expression was significantly increased, NLRP3 protein level was evidently reduced, but NLRP3 mRNA level was not distinctly altered in the ox-LDL + oe-CBL-B group (Figs. 2D and 2E). As reflected by the Co-IP assay, ubiquitination of NLRP3 was remarkably enhanced after CBL-B overexpression (Fig. 2F). Collectively, the aforementioned results evinced that CBL-B suppressed the expression of NLRP3 by mediating ubiquitination.
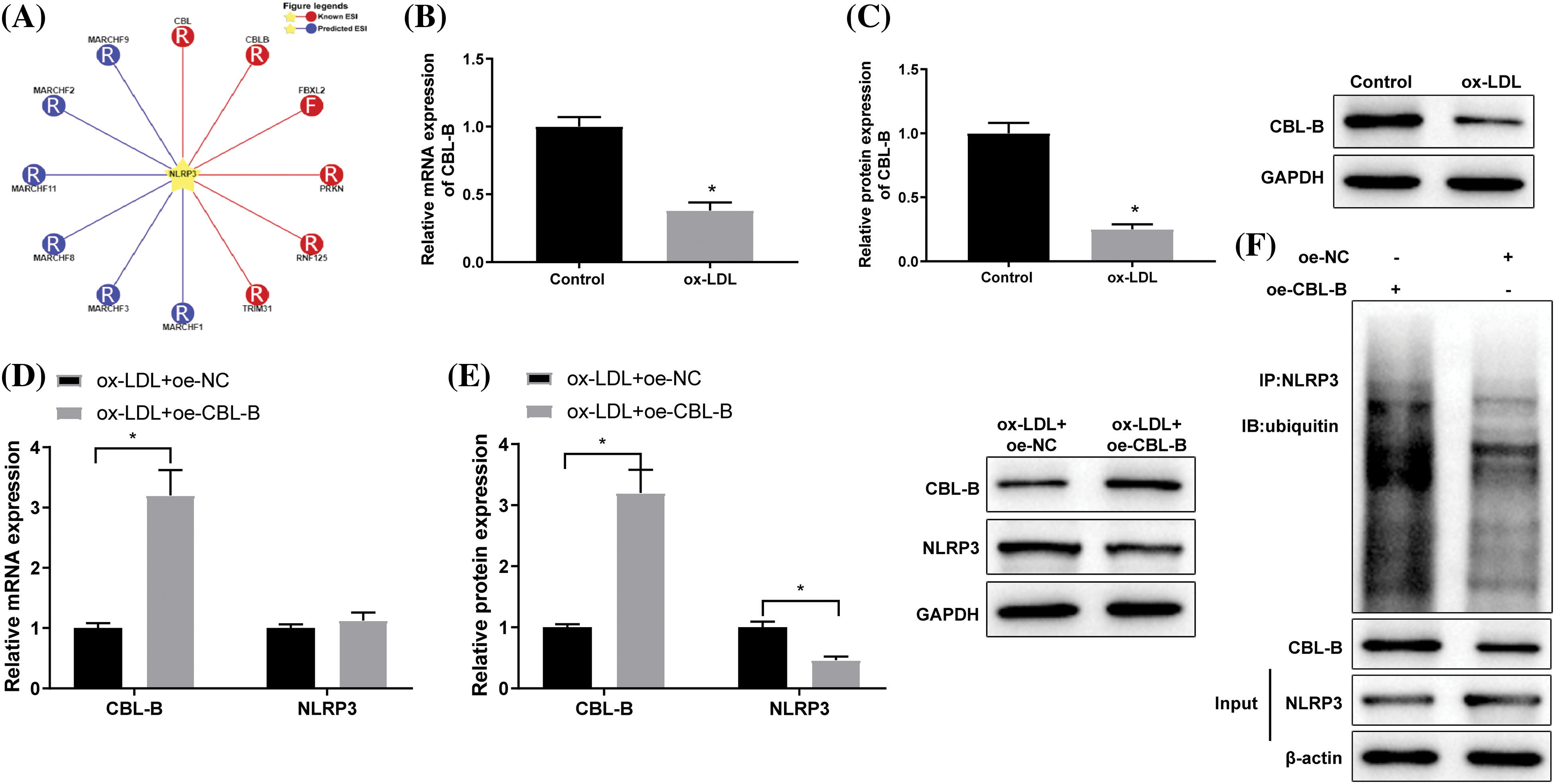
Figure 2: CBL-B regulates ubiquitination to repress NLRP3 expression. (A) The UbiBrowser database was employed to predict the E3 ubiquitin ligases of NLRP3, F: F-box family; R: RING family; CBL-B expression was determined through (B) RT-qPCR and (C) western blotting. The changes in CBL-B and NLPR3 levels after transfection with oe-CBL-B were determined by (D) RT-qPCR and (E) western blotting. NLRP3 ubiquitination after treatment with MG132 was determined by (F) Co-IP was implemented to examine. *p < 0.05 compared with the control or ox-LDL + oe-NC group. The experiment was performed thrice in duplicate. CBL-B: casitas B lymphoma-B; NLRP3: nucleotide-binding oligomerization domain-like receptor pyrin domain-containing 3; RT-qPCR: real-time quantitative polymerase chain reaction; Co-IP: co-immunoprecipitation; IL-1β: interleukin-1β; ox-LDL: oxidized low-density lipoprotein; oe-NC: overexpressed negative control.
Alleviation of human brain microvascular endothelial-cell injury by casitas B lymphoma-B treatment
Subsequently, we probed into the specific role of CBL-B in the biological behaviors of HBMECs. As reflected by flow cytometry (Fig. 3A), MTT (Fig. 3B), and scratch tests (Fig. 3C), HBMEC apoptosis was evidently weakened, cell viability was enhanced, and cell mobility was strengthened in the ox-LDL + oe-CBL-B group compared with the ox-LDL + oe-NC group. Additionally, western blot manifested that the ox-LDL + oe-CBL-B group presented lower ASC, cleaved caspase-1, and GSDME-N levels than the ox-LDL + oe-NC group (Fig. 3D). ELISA demonstrated a decline of IL-1β and IL-18 levels in the ox-LDL + oe-CBL-B group (Figs. 3E and 3F). In summary, overexpression of CBL-B impeded HBMEC pyroptosis and palliated cell injury.
Figure 3: CBL-B is shown to extenuate HBMEC injury. (A) HBMEC apoptosis was evaluated using flow cytometry; (B) HBMEC viability was estimated by MTT; (C) HBMEC mobility was assessed by scratch test; (D) The protein levels of ASC, cleaved caspase-1, and GSDME-N were determined by means of western blotting; (E and F) The levels of IL-1β and IL-18 were measured using ELISA. *p < 0.05 compared with the ox-LDL + oe-NC group. The experimentation was performed thrice in duplicate. CBL-B: casitas B lymphoma-B; HBMEC: human brain microvascular endothelial cells; ASC: a caspase recruitment domain; GSDME-N: gasdermin N; ELISA: enzyme linked immunosorbent assay; Co-IP: co-immunoprecipitation; IL-1β: interleukin-1β; ox-LDL: oxidized low-density lipoprotein; oe-NC: overexpressed negative control.
Casitas B lymphoma-B attenuated vascular endothelial cell injury in intracranial aneurism by promoting NLRP3 degradation
The ox-LDL-induced HBMECs were co-transfected with oe-CBL-B and oe-NLRP3 or oe-NC. Next, RT-qPCR and western blot indicated a successful co-transfection (Figs. 4A and 4B). A series of experiments, including flow cytometry (Fig. 4C), MTT (Fig. 4D), and scratch tests (Fig. 4E), unraveled that the oe-CBL-B + oe-NLRP3 group exhibited higher HBMEC apoptosis and lower cell viability and mobility capabilities than the oe-CBL-B + oe-NC group. Moreover, western blot elucidated the increase of ASC, cleaved caspase-1, and GSDME-N levels in the oe-CBL-B + oe-NLRP3 group in contrast to the oe-CBL-B + oe-NC group (Fig. 4F). ELISA demonstrated that IL-1β and IL-18 levels were predominantly raised in the oe-CBL-B + oe-NLRP3 group (Figs. 4G and 4H). The result from co-IP illustrated that there was no obvious change in NLRP3 ubiquitination between the oe-CBL-B + oe-NC group and the oe-CBL-B + oe-NLRP3 group (Fig. 4I). Altogether, overexpression of NLRP3 nullified the mitigative effect of CBL-B upregulation on HBMEC injury, suggesting that CBL-B unleashed an alleviating effect on vascular EC injury in IA by facilitating degradation of NLRP3.
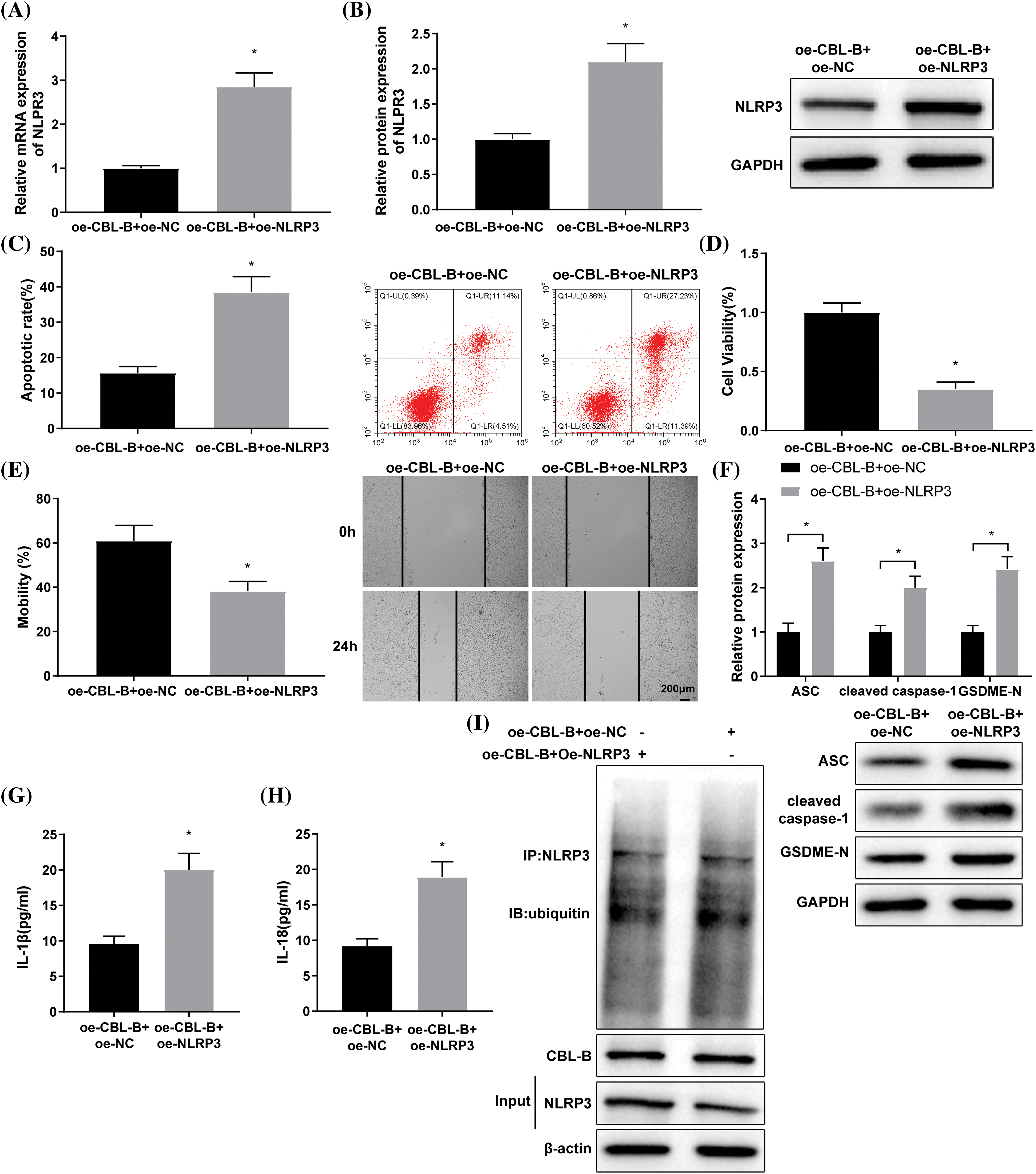
Figure 4: CBL-B facilitates NLRP3 degradation to mitigate vascular EC injury in IA. NLRP3 expression was measured by (A) RT-qPCR and (B) western blotting. (C) Cell apoptosis was determined by flow cytometry. (D) cell viability was assessed by MTT assay. (E) Cell mobility was determined by a scratch test. (F) Levels of ASC, cleaved caspase-1, and GSDME-N were determined by western blotting. (G and H) IL-1β and IL-18 levels were determined by ELISA. (I) NLRP3 ubiquitination was assessed by Co-IP. *p < 0.05 compared with the oe-CBL-B + oe-NC group. The experiment was performed thrice in duplicates. CBL-B: casitas B lymphoma-B; NLRP3: nucleotide-binding oligomerization domain-like receptor pyrin domain-containing 3; EC: endothelial cells; ASC: a caspase recruitment domain; ELISA: enzyme linked immunosorbent assay; GSDME-N: gasdermin N; RT-qPCR: real-time quantitative polymerase chain reaction; Co-IP: co-immunoprecipitation; IL-1β: interleukin-1β; ox-LDL: oxidized low-density lipoprotein; oe-NC: overexpressed negative control.
IA refers to a pathological and irreversible dilatation of cerebral arteries, the rupture of which is a considerably destructive event leading to SAH [25]. In terms of etiology, IA is initiated mainly by EC disruption and dysfunction driven by aberrant physiological wall shear stress [26]. Therefore, maintenance of EC function is essential for preventing and delaying IA progression. In this study, HBMECs induced by ox-LDL was used as in vitro model to investigate the intrinsic mechanisms associated with EC injury. Our results elucidated that CBL-B curtailed HBMEC pyroptosis and injury by promoting NLRP3 degradation, thus palliating vascular EC injury in IA (Fig. 5).
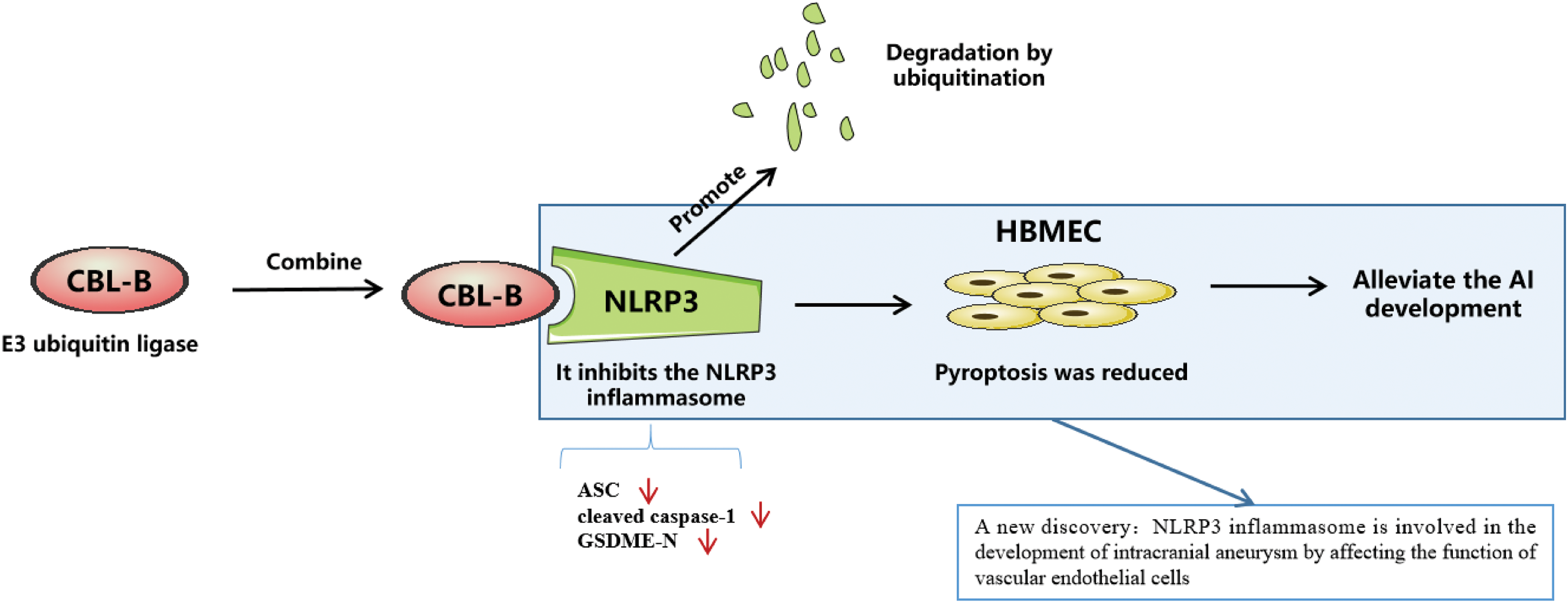
Figure 5: E3 ubiquitin ligase CBL-B accelerates degradation of NLRP3 to inhibit the expression of NLRP3, and low expression of NLRP3 impedes endothelial cell pyroptosis, thus palliating vascular endothelial cell injury in intracranial aneurysm. CBL-B: casitas B lymphoma-B; NLRP3: nucleotide-binding oligomerization domain-like receptor pyrin domain-containing 3.
As an extensively explored inflammasome, NLRP3 inflammasome is linked with IL-1β and IL-18 secretion, which aggravates inflammation responses following SAH [27]. NLRP3 inflammasome is expressed in the wall of human IAs and is more abundant in ruptured IAs than in unruptured ones [28]. Inhibition of NLRP3 function can palliate early brain injury as well as delayed cerebral vasospasm following SAH [29]. Of note, NLRP3 inflammasome activation in ECs in the context of pathophysiological conditions appears to exacerbate endothelial dysfunction, thereby resulting in multiple disorders [16]. Similar to these findings, our results also evinced that NLRP3, ASC, and cleaved caspase-1 were highly expressed in ox-LDL-induced HBMECs, confirming the activation of NLRP3 inflammasome. Compelling evidence has indicated that NLRP3 inflammasome activation exacerbates cerebrovascular disorders by manipulating pyroptosis and apoptosis [30]. Intrinsically, GSDME can be cleaved by caspase-3 to release the GSDME-N domain, which facilitates pyroptosis by forming membrane pores, and GSDME knockdown weakens pyroptosis and inflammation in TNF-α-induced human umbilical vein ECs [31]. In our assays, the knockdown of NLRP3 hindered cell apoptosis, enhanced cell proliferative and migratory capabilities, impeded NLRP3 inflammasome activation, and downregulated the expression of GSDME-N, IL-1β, and IL-18. Likewise, in human aortic ECs, NLRP3 inflammasome activation contributes to impaired aorta relaxation and pyroptosis [32], whereas NLRP3 silencing efficiently halts IL-18 and IL-1β generation and pyroptosis [33]. Additionally, lysophosphatidylcholine triggers inflammatory damage and apoptosis in HBMECs through GPR4-modulated NLRP3 inflammasome activation [34]. Equally importantly, silenced NLRP3 can palliate cardiac microvascular EC injury, evidenced by promotion roles in cell proliferation and cell cycle progression, as well as inhibition impact on cell apoptosis [35]. Collectively, the aforementioned evidence and findings underscored the mitigation effect of NLRP3 silencing on HBMEC injury.
It is noteworthy that the manipulation of NLRP3 ubiquitination and deubiquitylation emerges as a potential and promising therapeutic target for NLRP3 inflammasome-related inflammatory lesions [36]. Ubiquitin-regulated protein degradation is considered the most ubiquitous mechanism for controlling protein availability and activity, and specifically, downregulating NLRP3 ubiquitination is implicated in facilitating NLRP3 inflammasome activation [37]. Using the UbiBrowser database, we could screen CBL-B as a potential E3 ubiquitin ligase of NLRP3. Mechanistically, CBL-B binds to the NLRP3 leucine-rich repeat domain and targets NLRP3 at the K496 site for K48-linked ubiquitination, leading to proteasome-mediated degradation [19]. Consistently, our study unearthed that CBL-B overexpression apparently diminished NLRP3 protein level and enhanced NLRP3 ubiquitination. Subsequent RT-qPCR and western blotting also revealed downregulated CBL-B in ox-LDL-induced HBMECs. Loss of CBL-B elevates the expression levels of chemokine receptors on monocytes, such as CCR1/2/7, thus facilitating vascular inflammation [38]. Notably, targeting CBL could extensively induce neutrophil adhesion and endothelial inflammatory injury, indicating the potential role of CBL agonist as a promising therapeutic modality for vascular inflammatory injury [39]. More importantly, CBL exerts a pivotal role in repressing NLRP3 inflammasome activation via the inhibition of Pyk2-mediated ASC oligomerization [40]. Furthermore, upregulation of CBL-B expression conferred a definitive alleviating effect on HBMEC injury and NLRP3 inflammasome-mediated pyroptosis. However, the above-mentioned effects were nullified by NLRP3 overexpression.
In conclusion, the present study showed that CBL-B mitigated HBMEC pyroptosis and injury by accelerating NLRP3 degradation, providing prominent insight into the progress of novel therapeutic targets for IA. Nevertheless, this work has several limitations. First, there is a lack of animal studies for in vivo validation of the mechanism. Second, the downstream pathways implicated in NLRP3-mediated vascular EC function warrant further investigations. Third, exploring other E3 ubiquitin ligases of NLRP3 is also worthwhile. Therefore, future experiments need to be designed to address the above shortcomings. Moreover, other mechanisms, such as reactive oxygen species (ROS), may also possibly regulate vascular injury. In one previous study, the effect of ROS on autophagy and centrocyte neutrophil extracellular traps, suggested that ROS may regulate trabecular neovascularization [41]. Whether ROS affect vascular injury for this study is also a worthy direction to explore, and we will investigate this in subsequent studies.
Acknowledgement: None.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: ZW and LC conceived the ideas. ZW designed and performed the experiments, analyzed the data, provided critical materials, and wrote the manuscript. LC supervised the study. Both authors have read and approved the final version for publication.
Availability of Data and Materials: The datasets used or analyzed during the current study are available from the corresponding author upon reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare there is no conflict of interest.
References
1. Strange F, Gruter BE, Fandino J, Marbacher S. Preclinical intracranial aneurysm models: a systematic review. Brain Sci. 2020;10(3):134. doi:10.3390/brainsci10030134. [Google Scholar] [PubMed] [CrossRef]
2. Freneau M, Baron-Menguy C, Vion AC, Loirand G. Why are women predisposed to intracranial aneurysm? Front Cardiovasc Med. 2022;9:815668. doi:10.3389/fcvm.2022.815668. [Google Scholar] [PubMed] [CrossRef]
3. Morel S, Bijlenga P, Kwak BR. Intracranial aneurysm wall (in) stability-current state of knowledge and clinical perspectives. Neurosurg Rev. 2022;45:1233–53. doi:10.1007/s10143-021-01672-5. [Google Scholar] [PubMed] [CrossRef]
4. Morga R, Pera J. Transcriptomic studies on intracranial aneurysms. Genes. 2023;14:613. doi:10.3390/genes14030613. [Google Scholar] [PubMed] [CrossRef]
5. Macdonald RL, Schweizer TA. Spontaneous subarachnoid haemorrhage. Lancet. 2017;389:655–66. doi:10.1016/S0140-6736(16)30668-7. [Google Scholar] [PubMed] [CrossRef]
6. Neifert SN, Chapman EK, Martini ML, Shuman WH, Schupper AJ, Oermann EK, et al. Aneurysmal subarachnoid hemorrhage: the last decade. Transl Stroke Res. 2021;12:428–46. doi:10.1007/s12975-020-00867-0. [Google Scholar] [PubMed] [CrossRef]
7. Lee KS, Zhang JJY, Nguyen V, Han J, Johnson JN, Kirollos R, et al. The evolution of intracranial aneurysm treatment techniques and future directions. Neurosurg Rev. 2022;45(1):1–25. doi:10.1007/s10143-021-01543-z. [Google Scholar] [PubMed] [CrossRef]
8. Tang H, Luo Y, Zuo Q, Wang C, Huang Q, Zhao R, et al. Current understanding of the molecular mechanism between hemodynamic-induced intracranial aneurysm and inflammation. Curr Protein Pept Sci. 2019b;20(8):789–98. doi:10.2174/1389203720666190507101506. [Google Scholar] [PubMed] [CrossRef]
9. Xu Z, Rui YN, Hagan JP, Kim DH. Intracranial aneurysms: pathology, genetics, and molecular mechanisms. Neuromol Med. 2019;21:325–43. doi:10.1007/s12017-019-08537-7. [Google Scholar] [PubMed] [CrossRef]
10. Sheinberg DL, McCarthy DJ, Elwardany O, Bryant JP, Luther E, Chen SH, et al. Endothelial dysfunction in cerebral aneurysms. Neurosurg Focus. 2019;47(1):E3. doi:10.3171/2019.4.FOCUS19221. [Google Scholar] [PubMed] [CrossRef]
11. Ju J, Liu Y, Liang H, Yang B. The role of pyroptosis in endothelial dysfunction induced by diseases. Front Immunol. 2022;13:1093985. doi:10.3389/fimmu.2022.1093985. [Google Scholar] [PubMed] [CrossRef]
12. Song D, Yeh CT, Wang J, Guo F. Perspectives on the mechanism of pyroptosis after intracerebral hemorrhage. Front Immunol. 2022;13:989503. doi:10.3389/fimmu.2022.989503. [Google Scholar] [PubMed] [CrossRef]
13. Coll RC, Schroder K, Pelegrin P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci. 2022;43:653–68. doi:10.1016/j.tips.2022.04.003. [Google Scholar] [PubMed] [CrossRef]
14. Duez H, Pourcet B. Nuclear receptors in the control of the NLRP3 inflammasome pathway. Front Endocrinol. 2021;12:630536. doi:10.3389/fendo.2021.630536. [Google Scholar] [PubMed] [CrossRef]
15. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477–89. doi:10.1038/s41577-019-0165-0. [Google Scholar] [PubMed] [CrossRef]
16. Bai B, Yang Y, Wang Q, Li M, Tian C, Liu Y, et al. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020;11(9):776. doi:10.1038/s41419-020-02985-x. [Google Scholar] [PubMed] [CrossRef]
17. Chen S, Wang Y, Pan Y, Liu Y, Zheng S, Ding K, et al. Novel role for tranilast in regulating NLRP3 ubiquitination, vascular inflammation, and atherosclerosis. J Am Heart Assoc. 2020a;9(12):e015513. doi:10.1161/JAHA.119.015513. [Google Scholar] [PubMed] [CrossRef]
18. Tang R, Langdon WY, Zhang J. Regulation of immune responses by E3 ubiquitin ligase Cbl-b. Cell Immunol. 2019a;340(3):103878. doi:10.1016/j.cellimm.2018.11.002. [Google Scholar] [PubMed] [CrossRef]
19. Tang J, Tu S, Lin G, Guo H, Yan C, Liu Q, et al. Sequential ubiquitination of NLRP3 by RNF125 and Cbl-b limits inflammasome activation and endotoxemia. J Exp Med. 2020;217(4):e20182091. doi:10.1084/jem.20182091. [Google Scholar] [PubMed] [CrossRef]
20. Hong Y, Keylock A, Jensen B, Jacques TS, Ogunbiyi O, Omoyinmi E, et al. Cerebral arteriopathy associated with heterozygous variants in the casitas B-lineage lymphoma gene. Neurol Genet. 2020;6(4):e448. doi:10.1212/NXG.0000000000000448. [Google Scholar] [PubMed] [CrossRef]
21. Seijkens TTP, Poels K, Meiler S, van Tiel CM, Kusters PJH, Reiche M, et al. Deficiency of the T cell regulator Casitas B-cell lymphoma-B aggravates atherosclerosis by inducing CD8+ T cell-mediated macrophage death. Eur Heart J. 2019;40(4):372–82. doi:10.1093/eurheartj/ehy714. [Google Scholar] [PubMed] [CrossRef]
22. Lyle CL, Belghasem M, Chitalia VC. c-Cbl: an important regulator and a target in angiogenesis and tumorigenesis. Cells. 2019;8(5):498. doi:10.3390/cells8050498. [Google Scholar] [PubMed] [CrossRef]
23. Husain D, Meyer RD, Mehta M, Pfeifer WM, Chou E, Navruzbekov G, et al. Role of c-Cbl-dependent regulation of phospholipase Cγ1 activation in experimental choroidal neovascularization. Invest Ophth Vis Sci. 2010;51:6803–9. doi:10.1167/iovs.10-5255. [Google Scholar] [PubMed] [CrossRef]
24. Yu H, Pan Y, Dai M, Xu H, Li J. Circ_0003423 alleviates ox-LDL- induced human brain microvascular endothelial cell injury via the miR-589-5p/TET2 network. Neurochem Res. 2021;46(11):2885–96. doi:10.1007/s11064-021-03387-x. [Google Scholar] [PubMed] [CrossRef]
25. Chen S, Yang D, Liu B, Wang L, Chen Y, Ye W, et al. Identification and validation of key genes mediating intracranial aneurysm rupture by weighted correlation network analysis. Ann Transl Med. 2020;8(21):1407. doi:10.21037/atm-20-4083. [Google Scholar] [PubMed] [CrossRef]
26. Liu Z, Ajimu K, Yalikun N, Zheng Y, Xu F. Potential therapeutic strategies for intracranial aneurysms targeting aneurysm pathogenesis. Front Neurosci. 2019;13:1238. doi:10.3389/fnins.2019.01238. [Google Scholar] [PubMed] [CrossRef]
27. Jin J, Duan J, Du L, Xing W, Peng X, Zhao Q. Inflammation and immune cell abnormalities in intracranial aneurysm subarachnoid hemorrhage (SAHrelevant signaling pathways and therapeutic strategies. Front Immunol. 2022;13:1027756. doi:10.3389/fimmu.2022.1027756. [Google Scholar] [PubMed] [CrossRef]
28. Zhang D, Yan H, Hu Y, Zhuang Z, Yu Z, Hang C. Increased expression of NLRP3 inflammasome in wall of ruptured and unruptured human cerebral aneurysms: preliminary results. J Stroke Cerebrovasc Dis. 2015;24(5):972–9. doi:10.1016/j.jstrokecerebrovasdis.2014.12.019. [Google Scholar] [PubMed] [CrossRef]
29. Dodd WS, Noda I, Martinez M, Hosaka K, Hoh BL. NLRP3 inhibition attenuates early brain injury and delayed cerebral vasospasm after subarachnoid hemorrhage. J Neuroinflamm. 2021;18:163. doi:10.1186/s12974-021-02207-x. [Google Scholar] [PubMed] [CrossRef]
30. Bai R, Lang Y, Shao J, Deng Y, Refuhati R, Cui L. The role of NLRP3 inflammasome in cerebrovascular diseases pathology and possible therapeutic targets. ASN Neuro. 2021;13:17590914211018100. doi:10.1177/175909142110. [Google Scholar] [CrossRef]
31. Yao F, Jin Z, Zheng Z, Lv X, Ren L, Yang J, et al. HDAC11 promotes both NLRP3/caspase-1/GSDMD and caspase-3/GSDME pathways causing pyroptosis via ERG in vascular endothelial cells. Cell Death Discov. 2022;8(1):112. doi:10.1038/s41420-022-00906-9. [Google Scholar] [PubMed] [CrossRef]
32. Liao K, Lv DY, Yu HL, Chen H, Luo SX. iNOS regulates activation of the NLRP3 inflammasome through the sGC/ cGMP/PKG/TACE/TNF-α axis in response to cigarette smoke resulting in aortic endothelial pyroptosis and vascular dysfunction. Int Immunopharmacol. 2021;101(10):108334. doi:10.1016/j.intimp.2021.108334. [Google Scholar] [PubMed] [CrossRef]
33. Wu X, Zhang H, Qi W, Zhang Y, Li J, Li Z, et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018;9(2):171. doi:10.1038/s41419-017-0257-3. [Google Scholar] [PubMed] [CrossRef]
34. Liu T, Wang X, Guo F, Sun X, Yuan K, Wang Q, et al. Lysophosphatidylcholine induces apoptosis and inflammatory damage in brain microvascular endothelial cells via GPR4-mediated NLRP3 inflammasome activation. Toxicol In Vitro. 2021;77(5):105227. doi:10.1016/j.tiv.2021.105227. [Google Scholar] [PubMed] [CrossRef]
35. Zhou T, Xiang DK, Li SN, Yang LH, Gao LF, Feng C. MicroRNA-495 ameliorates cardiac microvascular endothelial cell injury and inflammatory reaction by suppressing the NLRP3 inflammasome signaling pathway. Cell Physiol Biochem. 2018;49(2):798–815. doi:10.1159/000493042. [Google Scholar] [PubMed] [CrossRef]
36. Akther M, Haque ME, Park J, Kang TB, Lee KH. NLRP3 ubiquitination—a new approach to target NLRP3 inflammasome activation. Int J Mol Sci. 2021;22(16):8780. doi:10.3390/ijms22168780. [Google Scholar] [PubMed] [CrossRef]
37. Xia X, Shi Q, Song X, Fu J, Liu Z, Wang Y, et al. Tetrachlorobenzoquinone stimulates NLRP3 inflammasome-mediated post-translational activation and secretion of IL-1β in the HUVEC endothelial cell line. Chem Res Toxicol. 2016;29(3):421–9. doi:10.1021/acs.chemrestox.6b00021. [Google Scholar] [PubMed] [CrossRef]
38. Poels K, Vos WG, Lutgens E, Seijkens TTP. E3 ubiquitin ligases as immunotherapeutic target in atherosclerotic cardiovascular disease. Front Cardiovasc Med. 2020;7:106. doi:10.3389/fcvm.2020.00106. [Google Scholar] [PubMed] [CrossRef]
39. Zhu Y, Liu L, Chu L, Lan J, Wei J, Li W, et al. Microscopic polyangiitis plasma-derived exosomal miR-1287-5p induces endothelial inflammatory injury and neutrophil adhesion by targeting CBL. PeerJ. 2023;11(16):e14579. doi:10.7717/peerj.14579. [Google Scholar] [PubMed] [CrossRef]
40. Lin HC, Chen YJ, Wei YH, Chuang YT, Hsieh SH, Hsieh JY, et al. Cbl negatively regulates NLRP3 inflammasome activation through GLUT1-dependent glycolysis inhibition. Int J Mol Sci. 2020;21(14):5104. doi:10.3390/ijms21145104. [Google Scholar] [PubMed] [CrossRef]
41. Migliario M, Tonello S, Rocchetti V, Rizzi M, Reno F. Near infrared laser irradiation induces NETosis via oxidative stress and autophagy. Laser Med Sci. 2018;33(9):1919. doi:10.1007/s10103-018-2556-z. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools