
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
Amitriptyline inhibits NLRP3 inflammasome activation via the ASM/CE pathway in a cell model of NAFLD
1 Shengli Clinical Medical College, Fujian Medical University, Fuzhou, 350001, China
2 Department of Gastroenterology, Nanjing Lishui People’s Hospital, Zhongda Hospital Lishui Branch, Southeast University, Nanjing, 211200, China
3 Department of Gastroenterology, Yancheng Third People’s Hospital (The Yancheng School of Clinical Medicine of Nanjing Medical University), Yancheng, 224000, China
4 Administrative Department of Teaching and Scientific Research, Nanjing Lishui People’s Hospital, Zhongda Hospital Lishui Branch, Southeast University, Nanjing, 211200, China
* Corresponding Author: CHUNYAN NIU. Email:
# Contributed equally
BIOCELL 2024, 48(5), 759-769. https://doi.org/10.32604/biocell.2024.048551
Received 11 December 2023; Accepted 07 February 2024; Issue published 06 May 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Nonalcoholic fatty liver disease (NAFLD) is a global health concern with the acid sphingomyelinase (ASM)/ceramide (CE) pathway and the NOD-like receptor family, pyrin domain-containing protein 3 (NLRP3) inflammasome identified as pivotal players in lipid disorders and inflammation. This study explores the interaction mechanism between the ASM/CE pathway and NLRP3 in NAFLD cell models, aiming to understand the impact of amitriptyline (Ami), an ASM inhibitor, on lipid deposition and hepatocyte injury by regulating the ASM/CE-NLRP3 pathway. Methods: HepG2 and HL-7702 cells were exposed to free fatty acids (FFAs) to establish the NAFLD model. The cells were divided into 5 groups: control group, model group, Ami group, tumor necrosis factor-alpha (TNF-α) group, and Ami + TNF-α group. Intracellular lipid droplets were visualized using Oil Red O staining, and Western blot analysis quantified ASM, NLRP3, and caspase 1 protein expression. Enzyme linked immunosorbent assay (ELISA) was measured CE and ASM levels, while qRT-PCR assessed mRNA expression. The apoptotic rate was evaluated by flow cytometry (FCM). Results: Following FFAs incubation, significant increases in ASM and CE levels were observed in HepG2 and HL-7702 cells, accompanied by elevated expression of NLRP3, and caspase 1, and IL-1β. TNF-α treatment further amplified these indicators. Ami demonstrated a reduction in lipid deposition, suppressed ASM/CE pathway activation, downregulated NLRP3 and caspase 1 expression, and improved apoptosis. Additionally, MCC950, a selective inhibitor of the NLRP3, mitigated NLRP3, caspase 1, and IL-1β expression, alleviating lipid deposition and apoptosis in the NAFLD cell model. Conclusion: The ASM/CE-NLRP3 pathway in NAFLD cells promotes hepatocyte steatosis, inflammation, and cell damage. Ami emerges as a promising therapeutic agent by inhibiting the ASM/CE-NLRP3 pathway, underscoring its potential as a key target for NAFLD treatment.Keywords
Nonalcoholic fatty liver disease (NAFLD) stands as the prevalent chronic liver disease worldwide, affecting approximately 25% of the global population [1]. Nonalcoholic steatohepatitis (NASH), an advanced stage of NAFLD, poses a risk of evolving into cirrhosis and hepatocellular carcinoma (HCC) [2]. The exact pathogenesis of NAFLD remains elusive. The latest “multiple parallel hits” [3] hypothesis suggests that insulin resistance (IR), lipotoxicity, inflammation, genetic factors, intestinal microecology, and some other factors [4] jointly participate in the occurrence of NAFLD and its sphingolipid mediators, such as ceramide (CE), with lipotoxicity and the NLRP3 inflammasome with proinflammatory effects are vital drivers of lipid metabolism disorder and inflammation, and the ASM/CE pathway plays a critical role in the pathophysiological mechanism of liver disease. Therefore, elucidating the relationship between the ASM/CE pathway and NLRP3 inflammasome is of great significance to further clarify the pathogenesis of NAFLD.
CE belongs to the sphingolipid family. As the second messenger of the sphingomyelin signaling pathway, CE can induce lipotoxicity and further mediate IR, the inflammatory response, and endoplasmic reticulum stress (ERS) [5] as well as regulate cell migration, apoptosis, autophagy, and other cellular responses [6]. Therefore, CE has been described as the most important harmful pathway in lipotoxicity events [7]. Studies have shown that CE synthesis in liver tissue is significantly increased in NASH patients [8,9], and plasma CE levels are closely related to adolescent hepatic steatosis (HS) and may be a novel biomarker of HS independent of obesity [10]. Acid sphingomyelinase (ASM) is a hydrolase that acts on sphingomyelin and is a key enzyme regulating the synthesis and secretion of CE. ASM can be activated by the TNF-α inflammatory factor [11] and inhibited by amitriptyline (Ami). In NAFLD conditions, the TNF-α level is elevated; subsequently, activated ASM can further promote the sensitivity of liver cells to the cytotoxic effects of TNF-α [12], and this interaction eventually accelerates the progression of liver disease. Ami, a tricyclic antidepressant, is a functional inhibitor of ASM that can lead to ASM dissociation, protein degradation, and inactivation [13], further inhibiting the generation of CE and its subsequent toxic biological effects. Ami has been shown to protect high-fat diet (HFD)-fed ASM+/+ mice from HS, ERS, fibrosis, liver damage, and early NASH [14], and it significantly reduces liver steatosis, inflammation, and IR in LDLR−/− mice [15] and Mat1a−/−mice [16].
The NLRP3 inflammasome is a multiprotein complex that senses and assembles danger signals from damaged cells and pathogens, and it mediates the activation of caspase 1 and release of the IL-1β cytokine, ultimately leading to metabolic inflammation [17]. Serving as the regulatory center of the inflammatory cascade, the NLRP3 inflammasome plays a pivotal role in the progression of NAFLD to NASH, fibrosis, and HCC [18,19]. Both animal models and patients with NASH exhibit significantly elevated levels of caspase 1 activity and serum IL-1β expression [20]. MCC950 is one of the most potent and selective inhibitors of the NLRP3 inflammasome, and it reduces caspase 1 activity and IL-1β production [21], inhibiting NLRP3 with MCC950 significantly improves metabolic disorders and protects hepatocytes from injury and fibrosis [22].
In previous studies, only a few animal experiments have shown that the ASM/CE pathway promotes steatosis in NAFLD, but no relevant studies have been reported in vitro to date. Moreover, the activation effect of the ASM/CE pathway on the NLRP3 inflammasome has not been verified in a NAFLD cell model. Given our previous results that the levels of serum CE and ASM increase in patients with NAFLD and continuously increase with the aggravation of hepatic steatosis [23,24], we aimed to examine the role of Ami in lipid deposition and its effect on NLRP3 activation from the perspective of the ASM/CE pathway in a cell model of NAFLD, and further demonstrate whether the ASM/CE-NLRP3 pathway plays a central role in lipid metabolism, inflammation and cell damage in NAFLD cell models.
HepG2 cells (procell, CL-0103) and HL-7702 cells (Wuhan biofavor biotech service Co., Ltd., Wuhan, China) were cultured in RPMI 1640 (GIBCO, 11875-093) +10% fetal bovine serum (FBS) (GIBCO, 26170035) +1% Penicillin‒Streptomycin (GIBCO, 15070-063) Solution at 37°C under a 5% CO2 atmosphere. The control group underwent standard culturing procedures, while the NAFLD model group was exposed to 1 mmol/L FFA [oleic acid (Sigma, O1383): palmitic acid (Sigma, P5585) = 2:1] for 24 h. The Ami group received 100 μM amitriptyline (Aladdin, A129730, hereinafter referred to as Ami), and the TNF-α group was exposed to 20 ng/mL recombinant human TNF-α (PeproTech, 96-300-01A). The Ami+TNF-α group received both 100 μM Ami and 20 ng/mL TNF-α. The Ami group, TNF-α group and Ami+TNF-α group were pretreated for 1 h before modeling, and the cell medium remained unchanged during modeling.
Cell viability was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) colorimetry. Cells in the logarithmic growth phase were seeded in 96-well plates and cultured at 37°C for 24 h. Subsequently, 10 μL of MTT (BIOSHARP, 0793) was added to each well, and the plates were then incubated at 37°C for 4 h. After discarding the medium, 150 μL of dimethyl sulfoxide (DMSO) was introduced. The plate was shaken for 10 min before measuring the absorbance values at 568 nm using a microplate reader.
Cells of each group were seeded on slides in 24-well plates and incubated for 48 h. The cells underwent fixation with 4% paraformaldehyde (Beyotime, 142287) for 15 min, followed by washing with distilled water, incubation in 60% isopropyl alcohol, staining with Oil Red O (Sigma, O0625) for 10 min, subsequent incubation with 60% isopropyl alcohol to achieve a colorless background, and final washing. Cells were then restained with hematoxylin for several minutes, washed, and mounted with Fluoromount-G (southernbiotech, 0100-01). The presence of lipid droplets within the cells was observed under a microscope (OLYMPUS, BX53).
Protein extraction was performed using RIPA buffer (Beyotime, P0013B) supplemented with PMSF (Aladdin, P105539) and phosphatase inhibitors. The protein concentration was quantified using a BCA kit (Beyotime, P0010). Isolated proteins were separated by SDS‒PAGE (10% gels) and then transferred onto PVDF membranes (Millipore, IPVH00010). The membranes were blocked with 5% skimmed milk powder in TBST (Tris-Buffered Saline with 0.1% Tween 20 detergent) at room temperature for 2 h, and the membranes were then incubated overnight at 4°C with the following primary antibodies: anti-ASM (affinity, DF13384) (1:1000), anti-NLRP3 (affinity, DF7438) (1:1000), and anti-caspase 1 (affinity, AF5418) (1:1000). Subsequently, the membranes were incubated with the secondary antibody (BOSTER, BA1054) (1:50000) at 37°C on a shaker for 2 h. Following three washes with TBST, protein bands were visualized using an enhanced ECL kit (Thermo Scientific, 34580).
Total RNA extraction from each cell group was conducted using Trizol reagent (Aidlab, Lot:252250AX). RNA purity and concentration were determined by ultraviolet spectrophotometry. HiScript Reverse Transcriptase (VAZYME, R101-01/02) was used to reverse transcribe RNA into cDNA. PCR amplification was carried out using AceQ qPCR SYBR Green Master Mix (VAZYME, Q111-02) on a real-time PCR instrument (ABI, QuantStudio 6). GAPDH served as the internal control for mRNA expression analysis. Relative expression levels of ASM and IL-1β mRNA were calculated by the 2−∆∆Ct method. Primer sequences are detailed in Table 1.
The cell culture medium from each group underwent centrifugation at 1000 rpm (Labnet, C2500-R-230V) for 20 min, after which the supernatant was harvested and stored at −20°C for future utilization. The total ASM, CE, and IL-1β contents were detected using the Human Ceramide ELISA Kit (JONLN, JL19781), Human ASM ELISA Kit (Elabscience, E-EL-H0511c) and Human IL-1β ELISA Kit (Elabscience, E-EL-H0149c), respectively, following the manufacturer’s instructions.
Determination of biochemical indices
The intracellular triglyceride (TG), total cholesterol (TC), alanine aminotransferase (ALT), and aspartate aminotransferase (AST) contents were detected by an automatic biochemical analyzer. The contents of non-esterified fatty acid (NEFA) were determined separately using commercial kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according the manufacturer’s instructions and then normalized to the total protein content.
Cell apoptosis rates of cells were assessed through flow cytometry (FCM) analysis utilizing an Annexin V-FITC/PI Apoptosis Kit (KeyGEN Biotech, KGA108, Nanjing, China). Cells were plated in 6-well plates for 24 h, resuspended in 500 μL of binding buffer, and stained with a mixture of 5 μL of Annexin V-FITC and 5 μL of propidium iodide (PI). Following a 15-min incubation period, the samples were analyzed by FCM. The results are expressed as the percentage of cells undergoing apoptosis.
Data analysis was conducted using SPSS 25.0 and GraphPad 8.0. One-way ANOVA was used to compare the measurement data between multiple groups, followed by an additional pairwise comparisons using Tukey’s test. Statistical significance was defined as p < 0.05. Data are presented as the mean ± standard deviation. All experiments were independently repeated at least three times.
Effects of different concentrations of Ami on lipid accumulation in a cell model of NAFLD
Cells were treated with 1 mmol/L FFA to induce steatosis. As shown in the Oil Red O staining pictures, HepG2 and HL-7702 cells in the control group were spindle-shaped or oval with no obvious red staining in the cytoplasm. However, the cytoplasm of the NAFLD model group was extensively stained, and the accumulation of red lipid droplets was significantly increased (Fig. 1A). Compared to the control group, the intracellular triglyceride (TG), total cholesterol (TC), and nonesterified fatty acid (NEFA) contents of the model group were significantly increased (Figs. 1C–1E), which indicated that a cell model of NAFLD was successfully established.
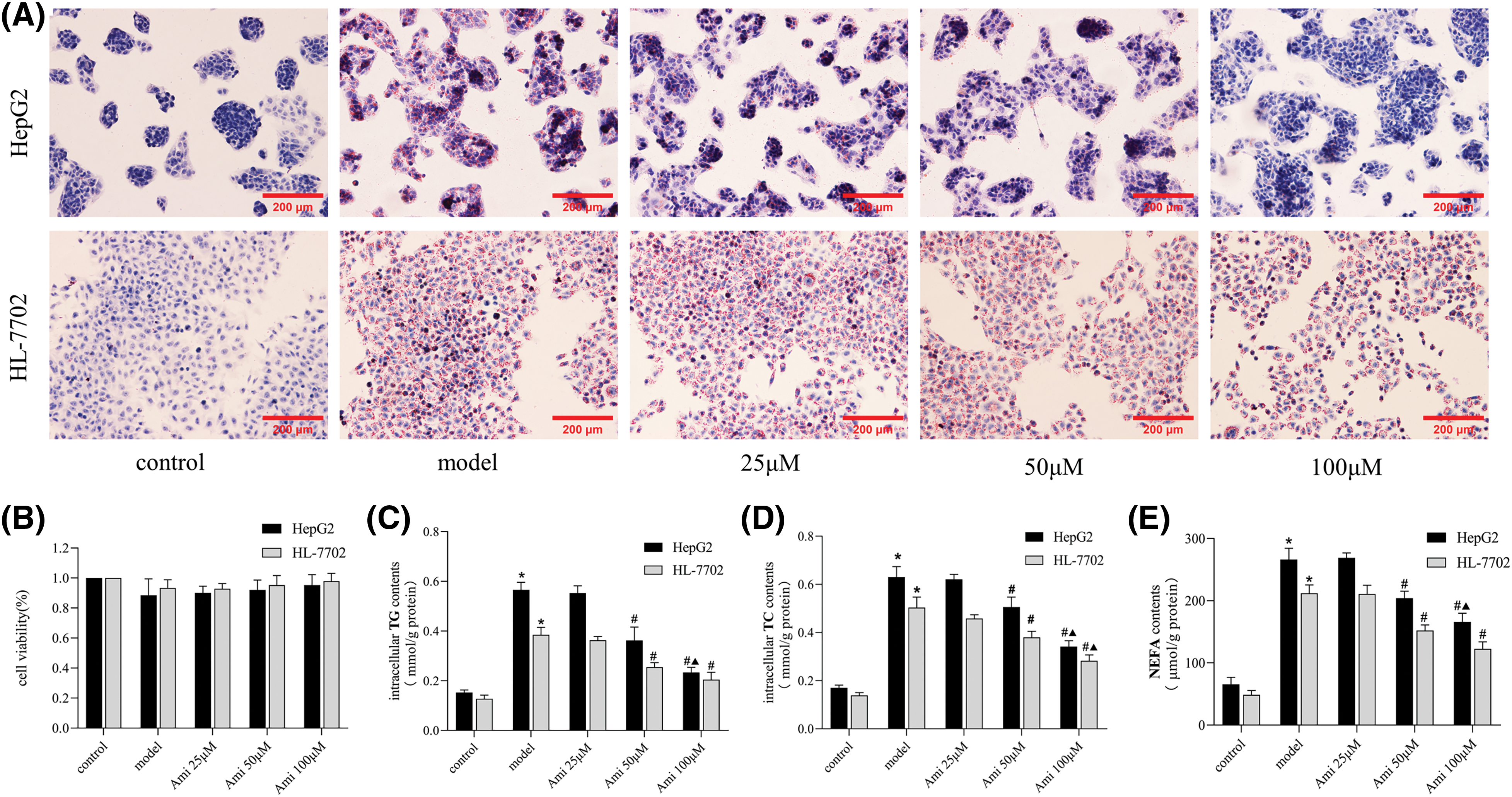
Figure 1: The inhibitory effect of Ami on hepatocyte lipid deposition is dose-dependent. (A): Oil Red O staining results (×200). (B): Cell viability was evaluated by a MTT assay. (C–E): Intracellular TG, TC, and NEFA contents in the five groups. *p < 0.05 vs. control group; #p < 0.05 vs. model group; ▴p < 0.05 vs. Ami 50 μM group, n = 3 experiments. Ami: amitriptyline; TG: total triglyceride; TC: total cholesterol; NEFA: nonesterified fatty acid.
Cells in the control, model, 25, 50 and 100 μM Ami groups all grew well, and there were no statistically significant differences in cell proliferation rates among the groups (Fig. 1B). Ami attenuated FFA-induced intracellular lipid droplet accumulation in a dose-dependent manner (Fig. 1A). Compared to the model group, the intracellular TG, TC, and NEFA contents in the Ami-treated groups decreased with increasing Ami concentration, especially in the Ami 50 μM and Ami 100 μM groups (Figs. 1C–1E). Therefore, Ami at a concentration of 100 μM, which had the strongest inhibitory effect, was selected for further study.
Effects of Ami and TNF-α on the ASM/CE pathway
To verify Ami’s inhibitory impact on ASM and TNF-α’s activating influence on ASM, the ASM protein and mRNA expression levels were examined. As anticipated, preincubation with Ami eliminated the TNF-α-induced activation of ASM (Figs. 2A–2C). ELISA was used to detect the intracellular total CE and ASM levels, which were notably elevated in the model group and further increased in the TNF-α group. However, treatment with Ami nearly eliminated each of these responses (Figs. 2D and 2E), indicating Ami’s ability to mitigate the alterations in ASM and CE expression induced by TNF-α.
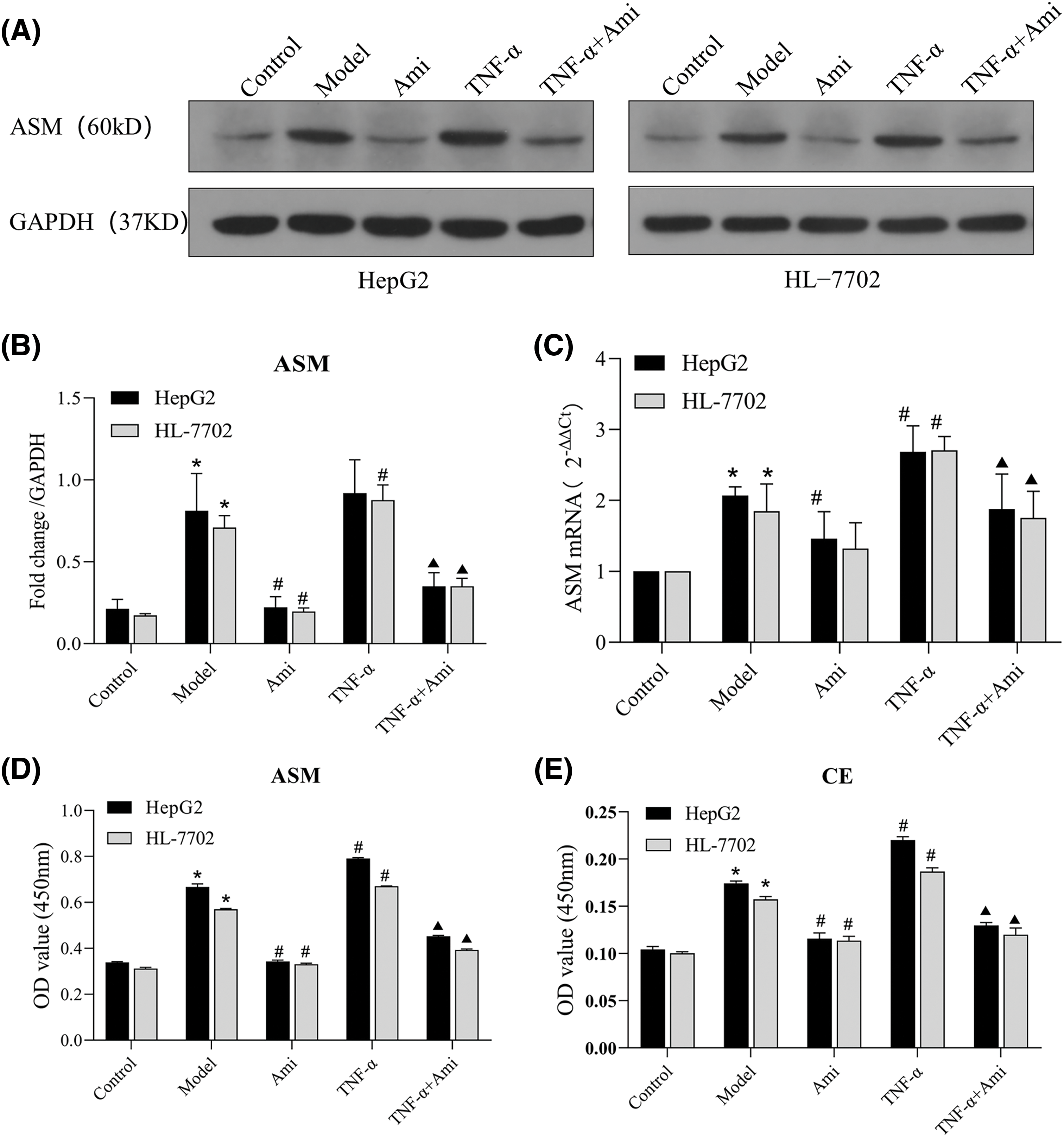
Figure 2: TNF-α activates the ASM/CE pathway and Ami inhibits the ASM/CE pathway. (A and B): Protein expression of ASM was detected by Western blot analysis, and densitometric analysis was performed. (C): mRNA expression of ASM was determined using qRT-PCR. (D and E): The total CE and ASM levels were detected by ELISA. *p < 0.05 vs. control group; #p < 0.05 vs. model group; ▴p < 0.05 vs. TNF-α group, n = 3 experiments. ASM: acid sphingomyelinase; Ami: amitriptyline; TNF-α: tumor necrosis factor-alpha.
Effects of the ASM/CE pathway on lipid deposition, hepatocyte injury, and apoptosis in NAFLD cell models
Having clarified the effects of Ami and TNF-α on the ASM/CE pathway, we proceeded to investigate the involvement of this pathway in hepatic steatosis. As shown by Oil Red O staining, Ami attenuated TNF-α-induced intracellular lipid droplets accumulation (Fig. 3A). TNF-α-induced activation of the ASM/CE pathway led to elevated intracellular levels of TG, TC, and NEFA, all of which were significantly attenuated by Ami (Figs. 3C–3E). Activation of the ASM/CE pathway by TNF-α markedly upregulated the levels of ALT and AST, while Ami attenuated the TNF-α-induced hepatocyte injury with a marked decrease in ALT and AST levels (Figs. 3F and 3G). These findings indicated that Ami alleviates lipid deposition and hepatocyte injury by inhibiting the ASM/CE pathway in a cell model of NAFLD.
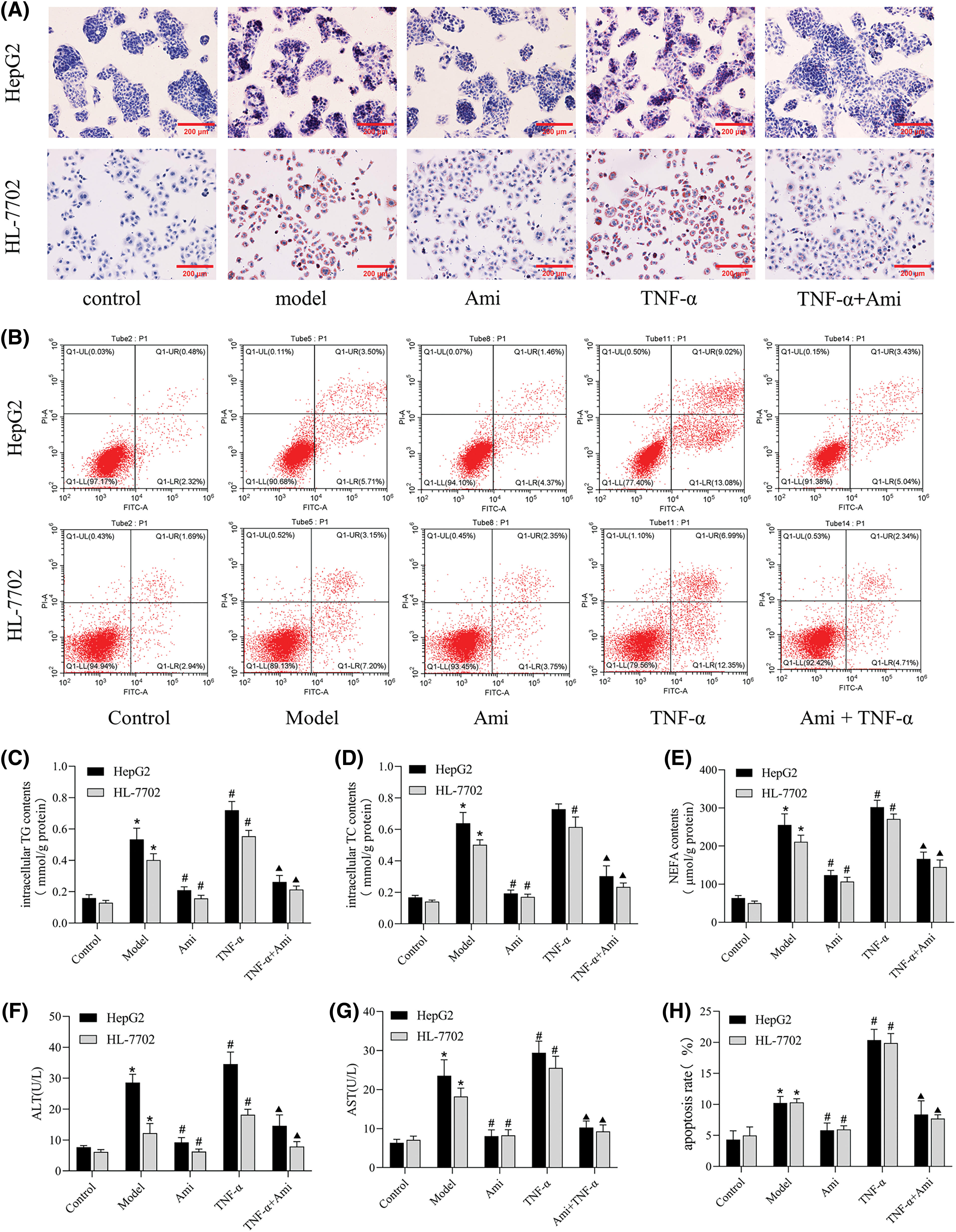
Figure 3: Ami improves lipid deposition, metabolism, and apoptosis in NAFLD cell models by inhibiting the ASM/CE pathway. (A): Oil Red O staining results (×200). (B): Annexin V-FITC/PI staining was used to detect apoptosis in the five groups. (C–E): Intracellular TG, TC, and NEFA contents in the five groups. (F and G): ALT and AST expression levels in cell supernatants; (H): The apoptosis rate was analyzed by FCM. *p < 0.05 vs. control group; #p < 0.05 vs. model group; ▴p < 0.05 vs. TNF-α group, n = 3 experiments. Ami: amitriptyline; TNF-α: tumor necrosis factor-alpha; TG: total triglyceride; TC: total cholesterol; NEFA: nonesterified fatty acid; ALT: alanine aminotransferase; AST: aspartate aminotransferase.
Subsequently, we conducted an Annexin V/PI assay (Figs. 3B and 3H) to explore the effect of the ASM/CE pathway on apoptosis. Cells in the control group were mainly distributed in the lower left region, and the number of dead cells caused by mechanical operation and other reasons was low. Compared to the control group, apoptotic cells (lower right and upper right regions) were increased in the NAFLD model group. Compared to the model group, apoptotic cells were further increased in the TNF-α group but significantly decreased in the Ami group. Compared to the TNF-α group, the apoptotic cells in the Ami+TNF-α group were reduced.
The ASM/CE pathway activates the NLRP3 inflammasome in NAFLD cell models
In order to explore the potential activation of the NLRP3 inflammasome by CE in a cell model of NAFLD, we next determined the expression of NLRP3 and its downstream components. Western blot analysis revealed a significant upregulation in the protein expression levels of NLRP3 and caspase 1 in the NAFLD model group were significantly increased compared to the control group. Interestingly, treatment with Ami led to a notable decrease in the protein expression of NLRP3 and caspase 1 (Figs. 4A–4C). In comparison to the control group, both IL-1β mRNA levels and supernatant IL-1β contents in the model group were significantly elevated. Compared to the model group and TNF-α group, treatment with Ami significantly reduced the mRNA level of IL-1β and effectively suppressed the release of intracellular IL-1β (Figs. 4D and 4E).
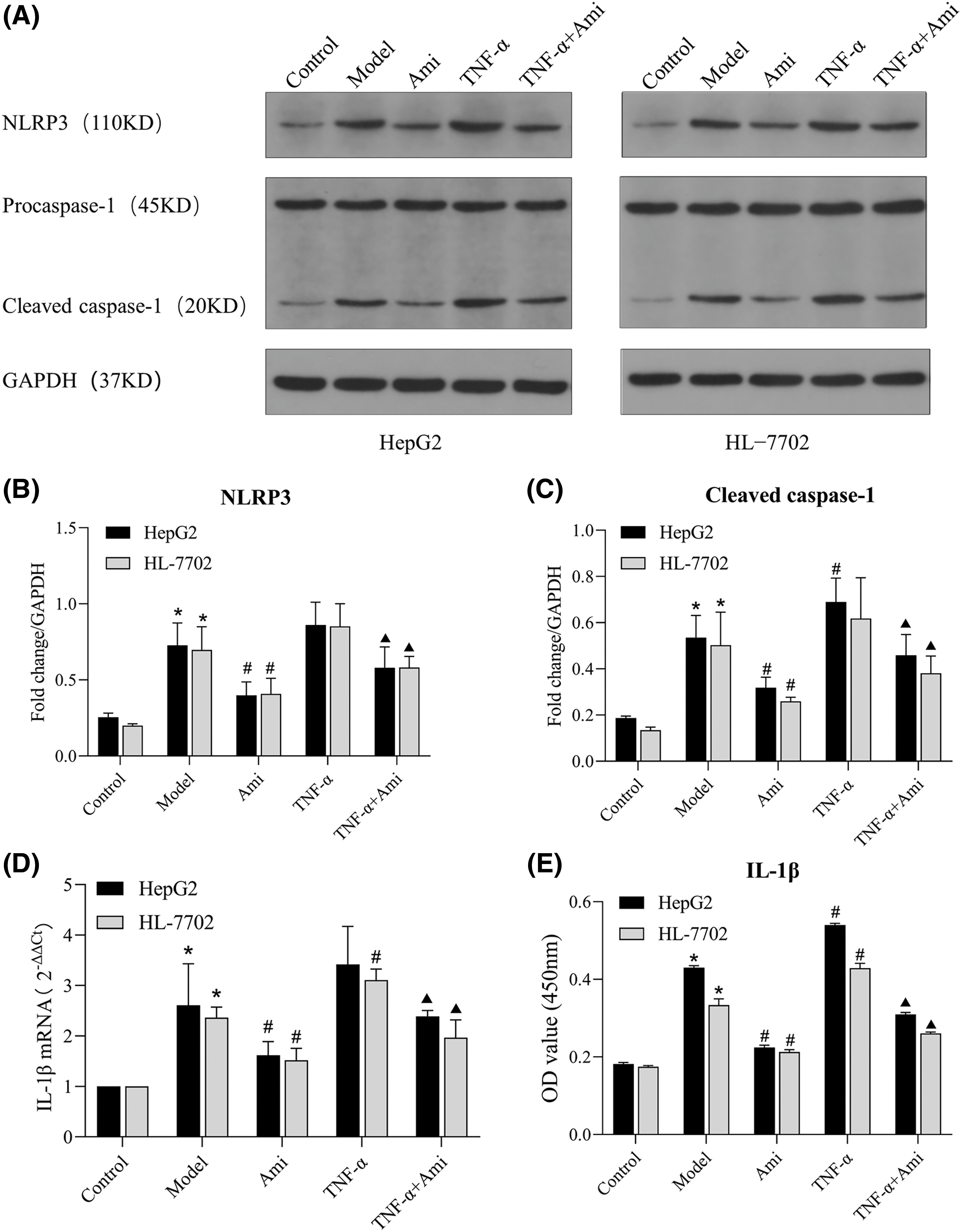
Figure 4: Ami inhibits NLRP3 inflammasome activation through the ASM/CE pathway. (A–C): The protein expression of NLRP3 and caspase 1 was detected by Western blot analysis, and densitometric analysis was performed; (D): The mRNA expression of IL-1β was determined by RT-PCR. (E): IL-1β content in the cell supernatant was detected by ELISA. *p < 0.05 vs. control group; #p < 0.05 vs. model group; ▴p < 0.05 vs. TNF-α group, n = 3 experiments. NLRP3: NOD-like receptor family, pyrin domain-containing protein 3; IL-1β: interleukin-1 beta; Ami: amitriptyline; TNF-α: tumor necrosis factor-alpha.
MCC950 downregulates the expression of NLRP3, caspase 1, and IL-1β as well as inhibits apoptosis in NAFLD cell models
To further explore whether the effect of the ASM/CE pathway in NAFLD cell models is related to NLRP3 activation, we administered MCC950, a specific inhibitor of NLRP3, to the NAFLD cell models. Results from the Annexin V/PI assay revealed a notable increase in the number of apoptotic cells in the NAFLD model group compared to the control group. While in the MCC950 group,the number of apoptotic cells decreased significantly in comparison to the model group (Figs. 5A and 5C). Western blot analysis (Fig. 5B) revealed significant inhibition in the protein expression of NLRP3 and caspase 1 in the MCC950 group compared to the model group (Figs. 5D and 5E). Additionally, the mRNA expression of IL-1β in the MCC950 group notably reduced compared to the model group (Fig. 5F). Thus, the activation of the NLRP3 inflammasome by the CE lipid mediator leads to the further induction of apoptosis.
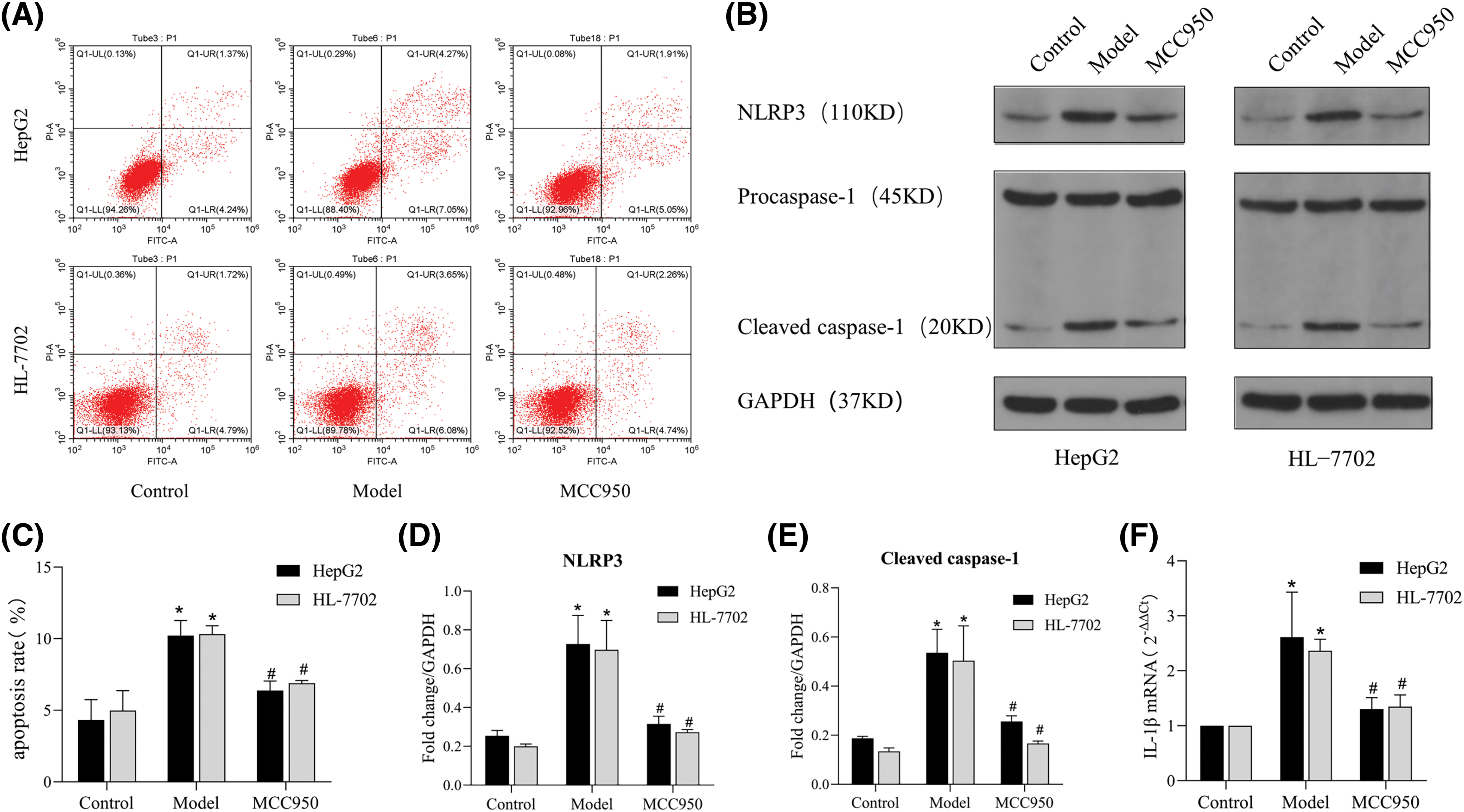
Figure 5: MCC950 downregulates the expression of NLRP3, caspase 1, and IL-1β as well as inhibits apoptosis in NAFLD cell models. (A): Apoptosis was detected by Annexin/PI flow cytometry. (B): The protein expression of NLRP3 and caspase 1 was detected by Western blot analysis. (C): The apoptosis rate was determined by FCM. (D and E): Quantitative analysis of NLRP3 and caspase 1 protein expression. (F): The expression of IL-1β was detected by qRT‒PCR. *p < 0.05 vs. control group; #p < 0.05 vs. model group, n = 3 experiments. NLRP3: NOD-like receptor family, pyrin domain-containing protein 3; IL-1β: interleukin-1 beta.
Inhibition of NLRP3 improves lipid deposition and metabolic damage
We proceeded to explore the impact of NLRP3 on lipid accumulation and metabolic alterations in NAFLD model cells. Notably, the MCC950 group exhibited significant reductions in intracellular lipid droplets accumulation (Fig. 6A) and the levels of intracellular TG, TC and NEFA compared to the model group (Figs. 6D–6F). Moreover, the MCC950 group showed decreased intracellular CE level (Fig. 6K), along with reductions in the protein expression levels of ASM, a key upstream signaling molecule of CE (Figs. 6G–6J). In addition, MCC950 markedly lowered ALT levels, AST levels, and mitigated hepatocyte injury (Figs. 6B and 6C). Collectively, these results suggest that inhibition of NLRP3-induced apoptosis also inhibits the release of lipid mediators, such as CE, thereby ameliorating metabolic injury in liver cells.
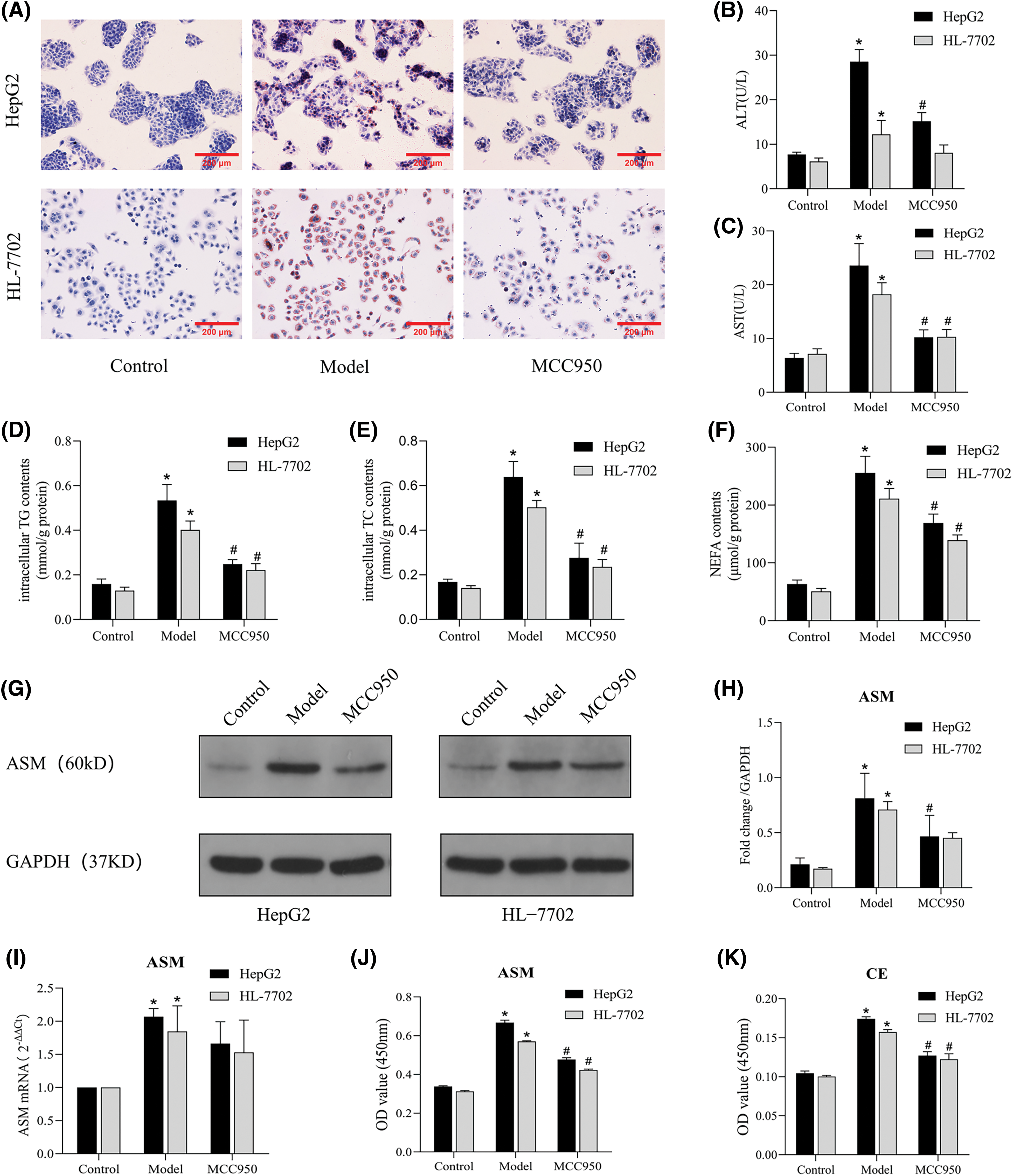
Figure 6: MCC950 improves lipid deposition and metabolic damage by inhibiting the NLRP3 inflammasome. (A): Oil Red O staining results (×200). (B and C): Expression levels of ALT and AST in the cell supernatants. (D–F): Intracellular TG, TC, and NEFA contents in three groups. (G): The protein expression of ASM was detected by Western blot analysis. (H): Quantitative analysis of ASM protein expression. (I): The mRNA expression of ASM was determined by qRT-PCR. (J and K): The total ASM and CE levels were detected by ELISA. *p < 0.05 vs. control group; #p < 0.05 vs. model group, n = 3 experiments. ASM: acid sphingomyelinase; CE: ceramide; TG: total triglyceride; TC: total cholesterol; NEFA: nonesterified fatty acid; ALT: alanine aminotransferase; AST: aspartate aminotransferase.
The present in vitro study demonstrated that the ASM/CE pathway is activated in a cell model of NAFLD, resulting in an imbalance in lipid homeostasis. Ami inhibits the activation of NLRP3 inflammasomes by regulating the ASM/CE pathway and exerts anti-lipid deposition and anti-inflammatory effects. In addition, the ASM/CE-NLRP3 pathway activates its downstream molecules, namely, caspase 1 and IL-1β, which induces inflammation and apoptosis. MCC950, a specific inhibitor of NLRP3, improves lipid metabolism and inflammatory damage in NAFLD cell models. To the best of our knowledge, the results of this study confirmed for the first time that Ami exerts an anti-inflammatory effect via the NLRP3 inflammasome pathway in vitro, further revealing the key role of the ASM/CE-NLRP3 pathway in hepatocyte lipid metabolism, inflammation, and apoptosis (Fig. 7). Thus, these findings suggested that targeted inhibition of the ASM/CE-NLRP3 pathway may be a potential NAFLD treatment.
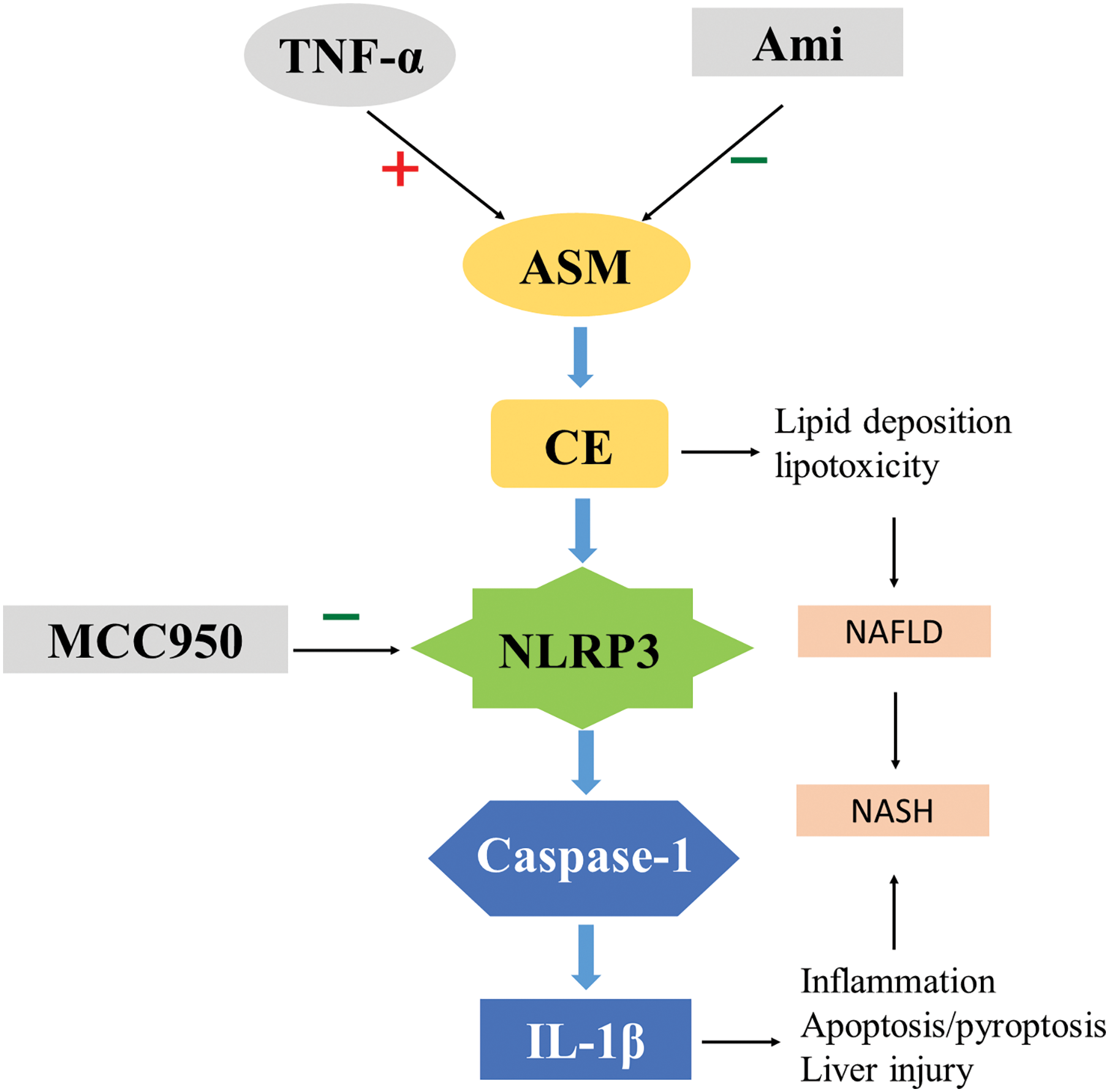
Figure 7: Role of ASM/CE-NLRP3 inflammasome pathway in NAFLD disease progression. Ami: amitriptyline; TNF-α: Tumor necrosis factor-alpha; ASM: Acid sphingomyelinase; CE: Ceramide; NLRP3: NOD-like receptor family, pyrin domain-containing protein 3; IL-1β: interleukin-1 beta; NAFLD: Non-alcoholic fatty liver disease; NASH: Non-alcoholic steatohepatitis.
We have previously reported the serum ASM and CE levels in NAFLD patients are increased and that there is a positive correlation with the severity of disease [23,24], which agreed with the findings of Maldonado-Hernández et al. [10]. Based on this evidence, we intend to further explore the specific mechanism of ASM and CE in the occurrence and development of NAFLD. It has been established that hepatic ceramide, steatosis, and fibrosis escalate in liver tissue from rats with NAFLD [25]. In animal models of alcoholic hepatitis (ASH) and NASH, ASM activation triggers lipotoxicity, IR, steatosis, and fibrosis [14,26]. Imipramine, an ASM inhibitor, has been found to alleviate IR and hepatic inflammation, and improve NAFLD in HFD-fed mice [27]. Mice deficient in ASM (Smpd1−/−) are protected against adipocyte hypertrophy and diet-induced HS [28]. These findings underline the significance of the ASM/CE pathway as a pivotal target regulating multiple pathways in NASH [29]. In the present study, we observed significantly elevated ASM and CE levels in the model group compared to the control group, indicating abnormal ASM/CE pathway activation in NAFLD. The elevated expression of ASM and CE in the TNF-α group verified the activation effect of TNF-α on the ASM/CE pathway. Ami reduced the intracellular lipid droplet contents, suggesting that Ami improves lipid deposition in the NAFLD cell model. Compared to the model group, treatment with Ami significantly reduced the lipid contents, metabolic enzyme levels, ASM protein levels, ASM mRNA levels, and CE contents in cells. These indices were also decreased in the TNF-α+Ami group than in the TNF-α group. These results indicated that the ASM/CE pathway promotes lipid accumulation and steatosis. By inhibiting this pathway, Ami improves the lipid deposition and liver function indicators in NAFLD liver cells, corroborating previous animal experimental findings.
The activation of the NLRP3 inflammasome is a well-established factor in various inflammation-related diseases [30], indicating the importance of understanding its molecular mechanism for devising new diagnostic and therapeutic approaches in inflammatory conditions. Emerging evidence suggests that the ASM/CE pathway initiates the assembly and activation of NLRP3 inflammasomes under different pathological conditions, although its role remains unverified in a NAFLD model. Increased CE levels lead to activation of NLRP3 inflammasomes in adipose tissue macrophages, peritoneal macrophages, and microglia [31–33]. Podocyte pretreatment with Ami reduces CE production and downregulates inflammasome components (caspase 1 and IL-1β) [34]. Animal experiments have also demonstrated that traumatic brain injury activates ASM and increases CE production, which activates NLRP3 [35]. In our investigation, we observed a positive association between the ASM/CE pathway and the activation and formation of NLRP3 and its downstream products in the NAFLD cell model. TNF-α-induced ASM/CE pathway activation promoted NLRP3 inflammasomes assembly and IL-1β release, whereas Ami treatment reduced expression of NLRP3, caspase 1 and IL-1β. These findings underscore the role of the ASM/CE pathway and NLRP3 inflammasomes in regulating lipid metabolism and inflammation in hepatocytes. Although it is not fully understood whether Ami suppresses NLRP3 inflammasomes by regulating the ASM/CE pathway, based on the findings that the ASM/CE pathway activates NLRP3 and that Ami inhibits ASM/CE, the present study suggested a potential mechanism by which Ami inhibits the ASM/CE-NLRP3 pathway from the source to alleviate NASH. However, further research is needed to confirm this hypothesis.
The NLRP3/caspase 1/IL-1β pathway is a crucial pathway leading to liver inflammation, particularly in the progression of NASH, where diverse lipid toxicants may activate caspase 1 and its downstream inflammatory factor, IL-1β, exacerbating inflammation, tissue damage and HSCs activation, consequently fostering fibrosis [36]. In addition, the NLRP3/caspase 1/IL-1β pathway is also an important pathway involved in inflammasome-mediated cell death, triggering not only apoptosis but also pyroptosis [37]. Recent studies have found that the lipid mediator, CE, promotes the assembly of NLRP3 inflammasomes; in contrast, abnormal activation of NLRP3 escalates lipid mediators release post-pyroptosis, potentially inducing systemic metabolic damage [30]. In the present study, we delineate how MCC950 and Ami target distinct nodes of the ASM/CE-NLRP3 pathway, directly and indirectly curbing NLRP3-mediated effects. Additionally, we showed that MCC950 and Ami mitigate lipid mediator release, ameliorating hepatocyte lipid deposition and damage. Moreover, we hypothesize that the downregulation of caspase 1 and IL-1β via ASM/CE-NLRP3 pathway inhibition may ameliorate pyroptosis in NAFLD cells, though this conjecture warrants further exploration.
The present study has several limitations. While focusing on the ASM/CE-NLRP3 pathway and its downstream NLRP3/caspase 1/IL-1β pathway, we acknowledge that the NLRP3 inflammasome may not be the only target protein regulated by the ASM/CE pathway. Furthermore, elucidating the precise alterations in NLRP3 downstream metabolites or signaling molecules and their interaction mechanisms in inflammation, fibrosis, and pyroptosis necessitates further investigation. Treatments for NAFLD encompass various drugs like hepatoprotective, anti-inflammatory, and insulin sensitizers [38]. While Ami exhibits anti-inflammatory properties in our investigation, it also poses a risk of drug-induced liver injury [39]. Therefore, to prevent an adverse reaction, the exposure period and dosage for Ami must be carefully selected during clinical application [40], warranting further exploration.
Taken together, the present study provides in vitro evidence supporting the activation of the ASM/CE-NLRP3 pathway in NAFLD pathogenesis. As an inhibitor of ASM, Ami attenuates NLRP3 activation by modulating the ASM/CE pathway, ameliorating lipid deposition and inflammation progression. Meanwhile, MCC950, a specific NLRP3 inhibitor, downregulates the expression of NLRP3/caspase 1/IL-1β and mitigates cell apoptosis. These findings underscore the potential of ASM and CE as predictive markers for NAFLD diagnosis and highlight the therapeutic promise targeting the ASM/CE-NLRP3 pathway in NAFLD treatment.
Acknowledgement: None.
Funding Statement: This work was supported by the Initial Scientific Research Fund of the Talents Introduced in Nanjing Lishui People’s Hospital (Project 2021YJ02).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Chunyan Niu and Qin Liu; references collection: Yue Chen and Yongqiang Shi; performing the experiments: Qin Liu and Qiang Zhang; analysis and interpretation of results: Qin Liu; draft manuscript preparation: Qin Liu and Chunyan Niu; putting forward suggestions on experimental scheme: Shiqing Sun. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2021;18(4):223–38. [Google Scholar] [PubMed]
2. Llovet JM, Willoughby CE, Singal AG, Greten TF, Heikenwälder M, El-Serag HB, et al. Nonalcoholic steatohepatitis-related hepatocellular carcinoma: pathogenesis and treatment. Nat Rev Gastroenterol Hepatol. 2023;20(8):487–503. [Google Scholar] [PubMed]
3. Tilg H, Adolph TE, Moschen AR. Multiple parallel hits hypothesis in nonalcoholic fatty liver disease: revisited after a decade. Hepatol. 2021;73(2):833–42. [Google Scholar]
4. Karkucinska-Wieckowska A, Simoes ICM, Kalinowski P, Lebiedzinska-Arciszewska M, Zieniewicz K, Milkiewicz P, et al. Mitochondria, oxidative stress and nonalcoholic fatty liver disease: a complex relationship. Eur J Clin Invest. 2022;52(3):e13622. [Google Scholar] [PubMed]
5. Yu XD, Wang JW. Ceramide de novo synthesis in non-alcoholic fatty liver disease: pathogenic mechanisms and therapeutic perspectives. Biochem Pharmacol. 2022;202:115157. [Google Scholar] [PubMed]
6. Zhu C, Huai Q, Zhang X, Dai H, Li X, Wang H. Insights into the roles and pathomechanisms of ceramide and sphigosine-1-phosphate in nonalcoholic fatty liver disease. Int J Biol Sci. 2023;19(1):311–30. [Google Scholar] [PubMed]
7. Unger RH. Lipotoxic diseases. Annu Rev Med. 2002;53:319–36. [Google Scholar] [PubMed]
8. Apostolopoulou M, Gordillo R, Koliaki C, Gancheva S, Jelenik T, de Filippo E, et al. Specific hepatic sphingolipids relate to insulin resistance, oxidative stress, and inflammation in nonalcoholic steatohepatitis. Diabetes Care. 2018;41(6):1235–43. [Google Scholar] [PubMed]
9. Luukkonen PK, Zhou Y, Sädevirta S, Leivonen M, Arola J, Orešič M, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol. 2016;64(5):1167–75. [Google Scholar] [PubMed]
10. Maldonado-Hernández J, Saldaña-Dávila GE, Piña-Aguero MI, Núñez-García BA, López-Alarcón MG. Association between plasmatic ceramides profile and AST/ALT ratio: C14:0 ceramide as predictor of hepatic steatosis in adolescents independently of obesity. Can J Gastroenterol Hepatol. 2017;2017:3689375. [Google Scholar]
11. Beckmann N, Sharma D, Gulbins E, Becker KA, Edelmann B. Inhibition of acid sphingomyelinase by tricyclic antidepressants and analogons. Front Physiol. 2014;5:331. doi:10.3389/fphys.2014.00331. [Google Scholar] [PubMed] [CrossRef]
12. Mühle C, Weinland C, Gulbins E, Lenz B, Kornhuber J. Peripheral acid sphingomyelinase activity is associated with biomarkers and phenotypes of alcohol use and dependence in patients and healthy controls. Int J Mol Sci. 2018;19(12):4028. [Google Scholar]
13. Guan Y, Li X, Umetani M, Boini KM, Li PL, Zhang Y. Tricyclic antidepressant amitriptyline inhibits autophagic flux and prevents tube formation in vascular endothelial cells. Basic Clin Pharmacol Toxicol. 2019;124(4):370–84. [Google Scholar] [PubMed]
14. Fucho R, Martínez L, Baulies A, Torres S, Tarrats N, Fernandez A, et al. ASMase regulates autophagy and lysosomal membrane permeabilization and its inhibition prevents early stage non-alcoholic steatohepatitis. J Hepatol. 2014;61(5):1126–34. [Google Scholar] [PubMed]
15. Lu Z, Li Y, Syn WK, Wang Z, Lopes-Virella MF, Lyons TJ, et al. Amitriptyline inhibits nonalcoholic steatohepatitis and atherosclerosis induced by high-fat diet and LPS through modulation of sphingolipid metabolism. Am J Physiol Endocrinol Metab. 2020;318(2):E131–44. doi:10.1152/ajpendo.00181.2019. [Google Scholar] [PubMed] [CrossRef]
16. Alarcón-Vila C, Insausti-Urkia N, Torres S, Segalés-Rovira P, Conde de la Rosa L, Nuñez S, et al. Dietary and genetic disruption of hepatic methionine metabolism induce acid sphingomyelinase to promote steatohepatitis. Redox Biol. 2023;59:102596. [Google Scholar]
17. Fu J, Wu H. Structural mechanisms of NLRP3 inflammasome assembly and activation. Annu Rev Immunol. 2023;41:301–16. [Google Scholar] [PubMed]
18. Kumar R, Goh BG, Kam JW, Chang PE, Tan CK. Comparisons between non-alcoholic steatohepatitis and alcohol-related hepatocellular carcinoma. Clin Mol Hepatol. 2020;26(2):196–208. [Google Scholar] [PubMed]
19. Horn P, Newsome PN. Emerging therapeutic targets for NASH: key innovations at the preclinical level. Expert Opin Ther Targets. 2020;24(3):175–86. [Google Scholar] [PubMed]
20. Farrell GC, Haczeyni F, Chitturi S. Pathogenesis of NASH: how metabolic complications of overnutrition favour lipotoxicity and pro-inflammatory fatty liver disease. Adv Exp Med Biol. 2018;1061:19–44. [Google Scholar] [PubMed]
21. Unamuno X, Gómez-Ambrosi J, Ramírez B, Rodríguez A, Becerril S, Valentí V, et al. NLRP3 inflammasome blockade reduces adipose tissue inflammation and extracellular matrix remodeling. Cell Mol Immunol. 2021;18(4):1045–57. [Google Scholar] [PubMed]
22. Zeng W, Wu D, Sun Y, Suo Y, Yu Q, Zeng M, et al. The selective NLRP3 inhibitor MCC950 hinders atherosclerosis development by attenuating inflammation and pyroptosis in macrophages. Sci Rep. 2021;11(1):19305. [Google Scholar] [PubMed]
23. Qi X. Study on serum ASMase expression and its significance in patients with nonalcoholic fatty liver disease (Master Thesis). Xi’an Medical College: China; 2018. [Google Scholar]
24. Han HJ. Diagnostic value of ceramide in nonalcoholic fatty liver disease (Master Thesis). Xi’an Medical College: China; 2019. [Google Scholar]
25. Jiang M, Li C, Liu Q, Wang A, Lei M. Inhibiting ceramide synthesis attenuates hepatic steatosis and fibrosis in rats with non-alcoholic fatty liver disease. Front Endocrinol. 2019;10:665. [Google Scholar]
26. Fernandez A, Matias N, Fucho R, Ribas V, Von Montfort C, Nuño N, et al. ASMase is required for chronic alcohol induced hepatic endoplasmic reticulum stress and mitochondrial cholesterol loading. J Hepatol. 2013;59(4):805–13. [Google Scholar] [PubMed]
27. Lu Z, Li Y, Chowdhury N, Yu H, Syn WK, Lopes-Virella M, et al. The presence of periodontitis exacerbates non-alcoholic fatty liver disease via sphingolipid metabolism-associated insulin resistance and hepatic inflammation in mice with metabolic syndrome. Int J Mol Sci. 2023;24(9):8322. [Google Scholar] [PubMed]
28. Sydor S, Sowa JP, Megger DA, Schlattjan M, Jafoui S, Wingerter L, et al. Acid sphingomyelinase deficiency in Western diet-fed mice protects against adipocyte hypertrophy and diet-induced liver steatosis. Mol Metab. 2017;6(5):416–27. [Google Scholar] [PubMed]
29. Garcia-Ruiz C, Mato JM, Vance D, Kaplowitz N, Fernández-Checa JC. Acid sphingomyelinase-ceramide system in steatohepatitis: a novel target regulating multiple pathways. J Hepatol. 2015;62(1):219–33. [Google Scholar] [PubMed]
30. Fusco R, Siracusa R, Genovese T, Cuzzocrea S, di Paola R. Focus on the role of NLRP3 inflammasome in diseases. Int J Mol Sci. 2020;21(12):4223. [Google Scholar] [PubMed]
31. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–88. [Google Scholar] [PubMed]
32. Sanches JM, Branco LM, Duarte GHB, Oliani SM, Bortoluci KR, Moreira V, et al. Annexin A1 regulates NLRP3 inflammasome activation and modifies lipid release profile in isolated peritoneal macrophages. Cells. 2020;9(4):926. [Google Scholar] [PubMed]
33. Scheiblich H, Schlütter A, Golenbock DT, Latz E, Martinez-Martinez P, Heneka MT. Activation of the NLRP3 inflammasome in microglia: the role of ceramide. J Neurochem. 2017;143(5):534–50. [Google Scholar] [PubMed]
34. Hong J, Bhat OM, Li G, Dempsey SK, Zhang Q, Ritter JK, et al. Lysosomal regulation of extracellular vesicle excretion during d-ribose-induced NLRP3 inflammasome activation in podocytes. Biochim Biophys Acta Mol Cell Res. 2019;1866(5):849–60. [Google Scholar] [PubMed]
35. Novgorodov SA, Voltin JR, Wang W, Tomlinson S, Riley CL, Gudz TI. Acid sphingomyelinase deficiency protects mitochondria and improves function recovery after brain injury. J Lipid Res. 2019;60(3):609–23. [Google Scholar] [PubMed]
36. Ramos-Tovar E, Muriel P. NLRP3 inflammasome in hepatic diseases: a pharmacological target. Biochem Pharmacol. 2023;217:115861. [Google Scholar] [PubMed]
37. Gaul S, Leszczynska A, Alegre F, Kaufmann B, Johnson CD, Adams LA, et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol. 2021;74(1):156–67. [Google Scholar] [PubMed]
38. Jiang G, Sun C, Wang X, Mei J, Li C, Zhan H, et al. Hepatoprotective mechanism of Silybum marianum on nonalcoholic fatty liver disease based on network pharmacology and experimental verification. Bioengineered. 2022;13(3):5216–35. [Google Scholar] [PubMed]
39. Mu W, Xu G, Wang Z, Li Q, Sun S, Qin Q, et al. Tricyclic antidepressants induce liver inflammation by targeting NLRP3 inflammasome activation. Cell Commun Signal. 2023;21(1):123. [Google Scholar] [PubMed]
40. Mir IH, Thirunavukkarasu C. The relevance of acid sphingomyelinase as a potential target for therapeutic intervention in hepatic disorders: current scenario and anticipated trends. Arch Toxicol. 2023;97(8):2069–87. [Google Scholar] [PubMed]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools