
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Pathogenic genes associated with Parkinson’s disease: molecular mechanism overview
1 School of Life Sciences, Institute for Brain Sciences Research, Henan University, Kaifeng, 475004, China
2 Pharmaceutical Department, Peking University Cancer Hospital Inner Mongolia Hospital, Hohhot, 010010, China
* Corresponding Author: LIFENG ZHAO. Email:
# The authors Tingting Liu and Yiwei Hao contributed equally
(This article belongs to the Special Issue: Exploring the Cellular Mechanisms of Neurodegenerative Diseases)
BIOCELL 2024, 48(5), 707-729. https://doi.org/10.32604/biocell.2024.049130
Received 28 December 2023; Accepted 21 February 2024; Issue published 06 May 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Parkinson’s disease (PD) is a common neurodegenerative disease in the elderly, accounting for more than 1% of the population aged 65 years. Monogenic inheritance is relatively rare in PD, accounting for approximately 5% to 10% of PD patients, and there is a growing body of evidence suggesting that multiple genetic risk factors play a significant role in the pathogenesis of PD. Several groups have identified and reported a number of genes carrying mutations associated with affected family members. Mutated genes associated with PD are also candidates for idiopathic PD, and these genes may also carry other mutation sites that increase risk. When multiple genetic risk factors are combined, the risk of PD is increased to a greater extent, and to unravel the pathogenic pathways that lead to different forms of PD. This review focuses on the association of PD genes, such as Parkinson Disease 1–24 (PARK1–24), glucosylceramidase (GBA), GTP cyclohydrolase 1 (GCH1), fibroblast growth factor 20 (FGF20), nuclear receptor-related factor 1 (NURR1), NUS1 dehydrodolichyl diphosphate synthase subunit (NUS1), diacylglycerol Lipase Beta (DAGLB), transmembrane protein (TMEM), ubiquinol-cytochrome c reductase core protein 1 (UQCRC1), glycoprotein non-metastatic melanoma protein B protein (GPNMB), dynactin 1 (DCTN1), LDL receptor related protein 10 (LRP10), monoamine oxidase (MAO), ataxin 2 (ATXN2), microtubule associated protein tau (MAPT), pantothenate kinase 2 (PANK2), spastic parapplegia type 11 (SPG11), polymer gamma (POLG), TATA-box binding protein associated factor 1 (TAF1), dual specificity tyrosine phosphorylation regulated kinase 1A (Dyrk1a), and crystallin alpha A (CRYAA), with the pathogenesis of PD. We introduce what is currently known about the molecular genetics of PD to help explain the molecular mechanisms leading to the neurodegenerative disease.Keywords
Supplementary Material
Supplementary Material FileAbbreviation List
| CREB | AMP response element binding protein |
| MeCP2 | Methyl-CpG-binding protein 2 |
| CSF | Cerebrospinal fluid |
| Ubl | Ubiquitin-like domain |
| SPR | Sepiapterin reductase |
| BH4 | Tetrahydrobiopterin |
| TH | Tyrosine hydroxylase |
| UCH-L1 | Ubiquitin C-terminal hydrolase L1 |
| UPS | Ubiquitin-proteasome system |
| PARL | Presenilin-associated rhomboid-like protein |
| LC3 | Microtubule-associated protein 1 light chain 3 |
| SNpc | Substantia nigra pars compacta |
| Drp1 | Dynamin-relatedprotein 1 |
| iPSC | Induced pluripotent stem cells |
| MAM | Mitochondrial associated membrane |
| COR | C-terminal of ROC |
| mPTP | Mitochondrial permeability transition pore |
| DA | Dopamine |
| Miro | Mitochondrial rho GTPases |
| ATP13A2 | ATPase cation transporting 13A2 |
| RNF11 | RING-finger protein 11 |
| UQCRH | Ubiquinol-cytochrome C reductase hinge protein |
| HIVEP3 | HIVEP zinc finger 3 |
| EIF2B3 | Eukaryotic translation initiation factor 2B subunit gamma |
| USP24 | Ubiquitin specific peptidase 24 |
| ELAVL4 | ELAV like RNA binding protein 4 |
| SNP | Single nucleotide polymorphisms |
| GIGYF2 | GRB10 interacting GYF protein 2 |
| IL-13Rα1 | Interleukin 13 receptor α1 |
| PGK-1 | Phosphoglycerate kinase 1 |
| HTRA2 | High-temperature requirement serine protease A2 |
| PLA2G6 | Phospholipase A2, group VI |
| iPLA2A | Calcium-independent phospholipase A2A |
| FBXO7 | F-box protein 7 |
| CRM1 | Chromosomal region maintenance 1 |
| FBD | F-box domain |
| NF-κB | Nuclear factor kappa-B |
| RPL23 | Ribosomal protein L23 |
| MDM2 | Murine double minute 2 |
| TP53 | Tumor protein P53 |
| SNPs | Single-nucleotide polymorphisms |
| RAB7L1 | RAB7, a member of RAS oncogene family-like 1 |
| SLC41A1 | Solute carrier family 41 member 1 |
| DLP1 | Dynamin-like protein 1 |
| APP | Amyloid precursor protein |
| Lamp2A | Lysosome-associated membrane protein 2A |
| EIF4G1 | Eukaryotic translation initiation factor 4-gamma 1 gene |
| DNAJC6 | DNAJ heat shock protein family (HSP40) member C6 |
| CCVs | Clathrin-coated vesicles |
| SV | Synaptic vesicle |
| LMX1A | LIM homeobox transcription factor 1α |
| SYNJ1 | Synaptojanin1 |
| CHCHD2 | Coiled-coil-helix-coiled-coil-helix domain containing 2 |
| OMA1 | OMA1 zinc metallopeptidase |
| OPA1 | Optic atrophy 1 |
| BCL-XL | B-cell lymphoma-extra-large |
| BAX | BCL2-associated X |
| P62 | Prostacyclin |
| TDP43 | TAR DNA binding protein-43 |
| VPS13C | Vacuolar Protein Sorting 13 Homolog C |
| cGAS | Cyclic GMP-AMP synthase |
| STING | Stimulator of interferon genes |
| PSAP | Prosaposin |
| SAPC | Saposin C |
| GRN | Progranulin |
| PGRN | Progranulin protein |
| GBA | Glucosylceramidase |
| GCase | Glucosidase |
| GCH1 | GTP cyclohydrolase 1 |
| FGF20 | Fibroblast growth factor 20 |
| FGFR-1C | FGF 1C receptor |
| MAPK | Mitochondrial activated protein kinase |
| NURR1 | Nuclear receptor-related factor 1 |
| NUS1 | NUS1 dehydrodolichyl diphosphate synthase subunit |
| ER | Endoplasmic reticulum |
| PERK | Protein kinase r-like ER kinase |
| 2-AG | 2-arachidoniyl-glycerol |
| AEA | N-arachidoniylethanolamine |
| DAGLA | Diacylglycerol lipase α |
| TMEM175 | Transmembrane protein 175 |
| GWAS | Genome-wide association studies |
| UQCRC1 | Ubiquinol-cytochrome c reductase core protein 1 |
| cyt-C | Cytochrome C |
| GPNMB | Glycoprotein non-metastatic melanoma protein B protein |
| TLR2 | Toll like receptor 2 |
| DCTN1 | Dynactin 1 |
| LRP10 | LDL receptor related protein 10 |
| SORL1 | Sortilin 1 |
| MAO | Monoamine oxidase |
| FAD | Flavin adenine dinucleotide |
| ATXN | Protein family ataxin 2 |
| LSm | Like-Sm |
| SCA2 | Spinocerebellar ataxia 2 |
| ALS | Amyotrophic Lateral Sclerosis |
| MAPT | Microtubule associated protein tau |
| PANK2 | Pantothenate kinase 2 |
| PKAN | Pantothenate kinase-associated neurodegeneration |
| DBS | Deep brain stimulation |
| SPG11 | Spastic paraplegia type 11 |
| POLG | Polymer gamma |
| mtDNA | Mitochondrial DNA |
| TAF1 | TATA-box binding protein associated factor 1 |
| TFIID | Transcription factor IID |
| Dyrk | Dual specificity tyrosine phosphorylation regulated kinase |
| CRYAA | Crystallin alpha A |
| ERS | Endoplasmic reticulum stress |
| UPR | Unfolded protein response |
| COMT | Catechol-Omethyl transferase |
| STN | Subthalamic nucleus |
| GPi | Globus pallidus internus |
| CoQ10 | Coenzyme Q10 |
| NFL | Neurofilament light chain |
Parkinson’s disease (PD) is a complex neurodegenerative disorder characterized by the gradual deterioration of autonomic movement control. The clinical syndrome of PD is characterized by delayed movement, static tremors, stiffness, and postural instability. Delayed movement is considered to be a major characteristic of PD and necessary for its diagnosis [1]. Besides these motor symptoms, many patients endure numerous non-motor symptoms, which sometimes take precedence over typical motor disorders, such as hypotension, sleep disturbances, depression, constipation, and other dysautonomic symptoms [2–4]. The genetic risk of PD can be divided into genetic risk related to the high risk of PD and genetic risk that only modestly increases the risk of PD. Mutations associated with an elevated risk of PD account for 5%–10% of PD cases [5]. The pathophysiology of PD is very complex, and various cellular mechanisms are involved in the backdrop of oxidative stress, accompanied by lifestyle, environmental, and genetic factors. Core mechanisms include α-Synuclein (α-Syn) wrong structure and concentration, mitochondrial impaired functioning, impaired protein removal (involving ubiquitin-proteasome and autophagy-lysosomal systems), neuroinflammation, oxidative stress, and immune system disorders. Patients with familial PD (fPD) have gene mutations involved in different biological processes and pathogenic mutations. A hallmark event of fPD is axonal mitochondrial dysfunction leading to impaired axon transport. The far-end axons of the corpus striatum may be the locus of the initiation of PD neurodegeneration. Earliest disease genes were found in an Italian family of autosomal dominant form of PD discovered by Polymeropoulos et al., and found the first with the separation of disease loci, named PARK1 [6,7]. Recent years, it has been shown that mutations are associated with genes related to fPD and candidate genes for idiopathic PD (iPD), as these genes perhaps also bring other mutations that would add hazard. Therefore, understanding the genetic factors that influence the risk, pathogenesis and progression of PD, and the catastrophic ramification of these changes at the molecular and biological standard, will help develop etiologically related therapies. Table 1 displays the genetic inheritance patterns and clinical characteristics of the genes. Additionally, the gene locations on the chromosome are annotated (Fig. 1). The single-cell type clustering analysis of genes is shown in the Suppl. Fig. S1. Genes are expressed in the following cells, including inhibitory neurons, excitatory neurons, astrocytes, oligodendrocytes, and microglia. Different colors represent different cell types. We also used R (version: 4.2.1) software R package: clusterProfiler [4.4.4] to conduct enrichment analysis of gene biological functions (Fig. 2) and signaling pathways (Fig. 3), and the results showed that, the biological processes mainly involved in the regulation of autophagy, regulation of mitochondrion organization, synaptic vesicle transport, synaptic vesicle localization, establishment of organelle localization, regulation of autophagy of mitochondrion, etc. The cellular components mainly involved in the inclusion body, neuronal cell body, neuron projection terminus, lysosomal membrane, lytic vacuole membrane, vacuolar membrane, organelle envelope lumen, mitochondrial outer membrane, organelle outer membrane, etc. The molecular functions mainly involved in the oxidoreductase activity, translation initiation factor binding, protease binding, proteoglycan binding, ubiquitin protein ligase binding, ubiquitin-like protein ligase binding, tau protein binding, etc. The pathway mainly involved in the Parkinson disease, Pathways of neurodegeneration-multiple diseases, Phenylalanine metabolism, Histidine metabolism, Amyotrophic lateral sclerosis, Tyrosine metabolism, Tryptophan metabolism, Cocaine addiction, etc.

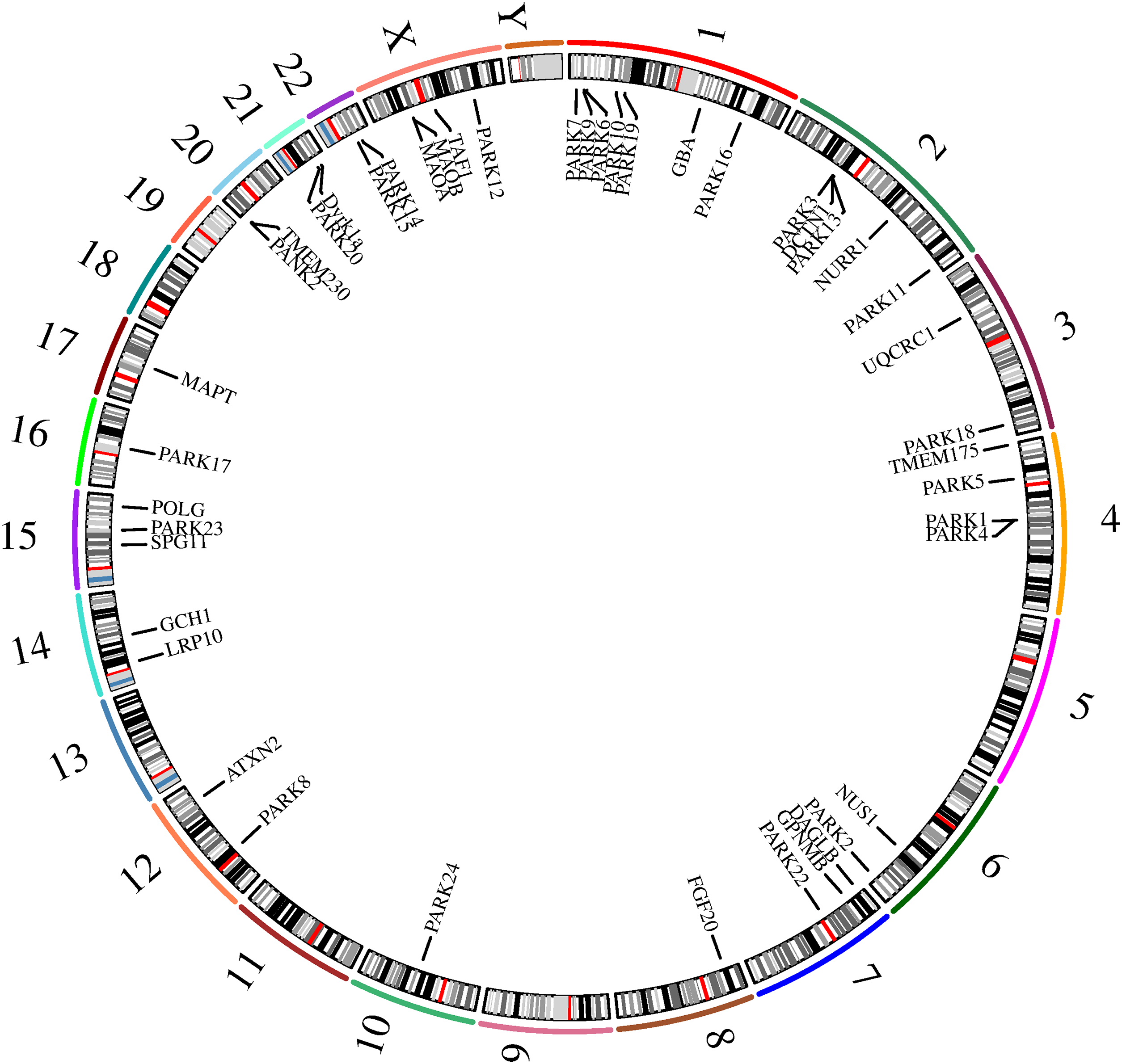
Figure 1: The position of genes in chromosomes.
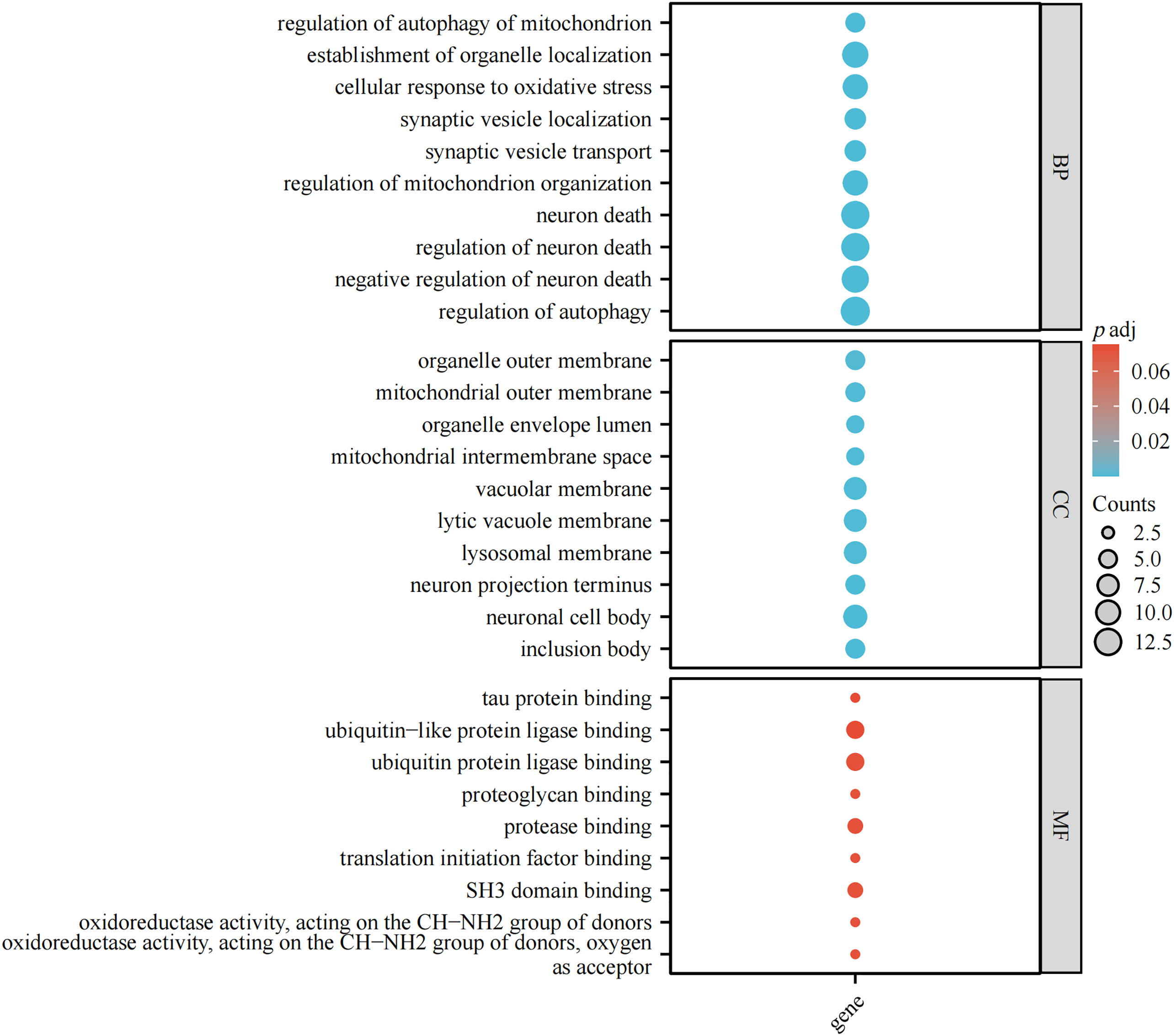
Figure 2: Enrichment analysis of biological functions.
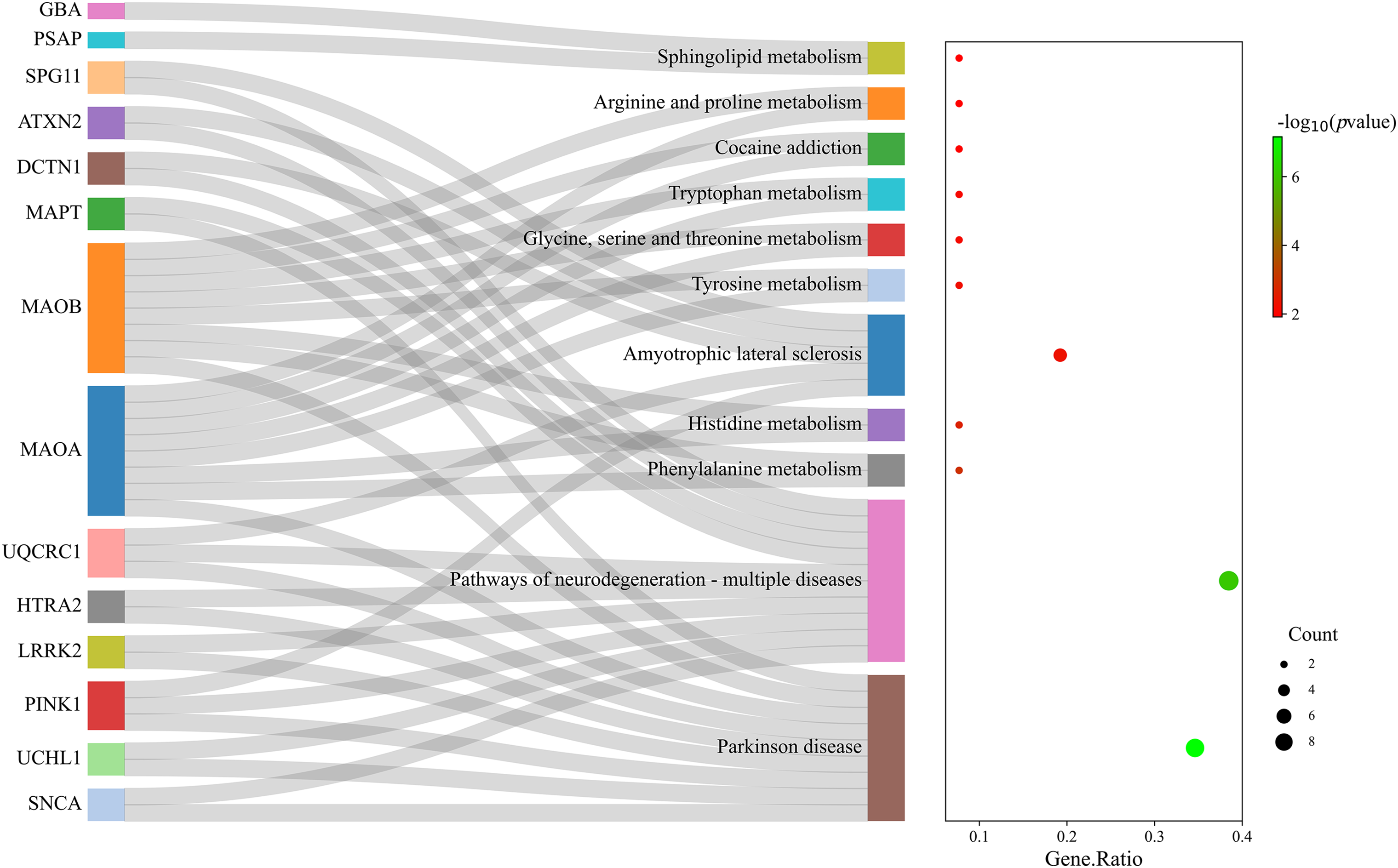
Figure 3: Enrichment analysis of signaling pathways. The size of circles represents the number of enriched genes.
The synuclein clan are composed of α, β and γ subtypes, whose genes are localized to chromosomes 4q21, 5q35 and 10q23 [8,9]. The α and β variants are seen in presynaptic terminals, whereas the γ variant appears in the peripheral nervous system and retina. The α-Syn is the more studied variant due to its link to PD [10]. The α-syn comprises 140 amino acid residues and its structure includes three parts. The amphiphilic N-terminal region (residues 1–60), central non-β-amyloid domain (NAC) (residues 61 to 95), and the acidic C-terminal domain (residues 96 to 140) [11]. α-Syn, mainly expressed in the nervous system and concentrated in the presynaptic terminals of neurons, is an important ingredient of Lewy bodies (LBs) in PD patients. Under physiological conditions, α-Syn is an unfolded, essentially disordered soluble monomer. In addition to the interaction between presynaptic neuron terminals and synaptic vesicles, α-Syn interacts with synaptic proteins and is implicated in regulating neurotransmitter release, synaptic activity and plasticity. It also interacts with the specific inhibitory molecular chaperones heat shock cognate protein 70 (HSC70) and heat shock protein 90β (HSP90β), which cause α-Syn to shift rapidly to mitochondria and eventually aggregate, which is neurotoxic and can cause neuronal dysfunction and death in the nervous system. The coding gene’s missense mutation (A53T) was first discovered in PD family in 1997 [6,7]. Since then, other single-site genetic mutations of α-Syn (A30P, E46K, G51D, H50Q and A53E) are important drivers of fPD [12,13].
These investigations yield new conceptions into the selectivity of α-Syn fiber formation and the pathogenesis of fPD. The α-Syn mainly plays a neurotoxic role through mitochondrial dysfunction, endoplasmic reticulum (ER) dysfunction, protease effect, glial cell inflammation, cell membrane break, lysosome and synaptic impaired functioning. It also induces degeneration of dopaminergic neurons through strangulation of cyclic AMP response element binding protein (CREB) and activation of methyl-CpG-binding protein 2 (MeCP2), leading to down-regulation of brain-derived neurotrophic factor [14]. The Parkin protein is responsible for degrading α-Syn, and mutations cause the loss of Parkin function, which leads to the accumulation of α-Syn. PTEN induced putative kinase 1 (PINK1) protein interacts with α-Syn at the end of the synapse, stabilizing the expression of α-Syn, and PINK1 mutation leads to abnormal aggregation of α-Syn. α-Syn is considered to be one of the key mediators of PD caused by leucine rich repeat kinase 2 (LRRK2) mutation. At present, various methods of preventing and reducing α-Syn toxicity in PD include the use of immunological strategies to clear its pathological aggregates from the brain, inhibit its misfolding and aggregation, reduce its expression levels, enhance the cell clearance mechanism, prevent its transmission in the brain and possibly from peripheral cells. Up to present, phase II clinical trials have been conducted in humans with two anti-α-Syn antibodies: Prasinezumab (PRX002), and Cinpanemab (BIIB054) [15]. In recent years, the methods for α-Syn to alleviate PD have been actively developed and substantial progress has been made. Studies have shown that the presence of elevated levels of synuclein in cerebrospinal fluid (CSF) and other biological samples can indicate the presence of PD. Additionally, the detection of specific forms of synuclein, such as phosphorylated or aggregated forms, may provide further insights into disease severity and progression. However, it is still not confirmed that these therapies are safe and effectively slow down the progression of disability in PD. The complexity of PD pathogenesis stems from its heterogeneous nature, variable gene penetrance, and diverse clinical progression and pathological alterations [16]. In addition, there are currently no available animal models that can realistically reproduce the pathogenesis of the human disease. However, there is still a deprivation of dependable biomarkers to distinguish prodromal PD from progressive PD.
The RBR E3 ubiquitin protein ligase gene (PARKIN), encoded by the PARK2 gene and located on chromosome 6q25.2-q27, which has 465 amino acid [17]. The mechanism involves the recruitment of an N-terminal ubiquitin-like domain (Ubl) linked to the R0RBR module, which consists of four zinc harmonize domains and three zinc finger domains (RING0, RING1, IBR, and RING2). This modular organization enables it to function as an E3 ubiquitin ligase [18]. Parkin protein affects mitochondrial function [19], calcium homeostasis, synaptic function, lysosomal/protease degradation, neuroinflammation, protein folding, apoptosis and oxidative damage [20,21]. Under normal conditions, PARKIN can ubiquitinate CyclinE and other proteins, leading to their protease degradation. However, mutations in protein-coding genes that cause abnormalities in the protein gene can prevent the normal form of ubiquitination, accumulating some cyclins and other functional proteins. It may cause abnormal proliferation in cells that are eligible for mitosis and apoptosis in neurons that cannot undergo mitosis [22]. The PARKIN gene may be due to missense, nonsense, insertion, frameshift mutation (including R402C, T173M, V2441, A82E, P437L, P37P, IVS2 + 248c > t, IVS7 − 60g > c, IVS7 − 58a > g, and IVS8 + 93c > t), premature stop codon, leading to early termination of the translation process or corresponding amino acid sequence change, resulting in a truncated or non-functional protein. As a result, the substrate proteins cannot be ubiquitinated and degraded accumulating in the cell, leading to neuronal death. Ubiquitination, as an E3 ubiquitin ligase, and subsequent autophagy, protein degradation, alteration of protein subcellular localization and protein-protein interactions (PPI) may underlie PARKIN’s protection against PD [23,24]. The study of Parkin-mediated mitochondrial autophagy provides a broad prospect for improving cognitive decline in PD. Still the experimental data in this field is entirely lacking. In conclusion, PARKIN is a multifunctional protein that plays a crucial role in preventing PD. The study on the interaction between PARKIN and PD will facilitate the deeper and earlier diagnosis of PD.
The PARK3 locus was first is targeted at chromosome 2p13 in 1998 in two European families with autosomal dominant PD [25], and no causative mutation and gene not identified. Some carriers of PARK3 mutations present with symptoms of dementia, which manifest clinically similar to sporadic PD (sPD) and involve the formation of LBs. Since the identification of the PARK3 locus, 12 candidate genes have been screened, but no causative gene product has been successfully identified. A haplotype encompassing the sepiapterin reductase (SPR) gene is considered one of the potential candidates for the PARK3 locus. SPR is responsible for the final step in the production of tetrahydrobiopterin (BH4), an essential cofactor for tyrosine hydroxylase (TH) and dopamine (DA) synthesis. Interestingly, PD patients carrying the TT genotype at the rs1876487 locus within the SPR promoter region tend to develop PD approximately 7.5 years earlier compared to those without this genotype [26]. In conclusion, SPR gene DNA polymorphisms seem to be associated with fPD, that is, SPR potentially modulates the onset of or risk for PD.
It was first found in PD patients in the United States and is caused by a triplication events mutation in the SNCA gene. Therefore, most studies believe that PARK4 is actually the SNCA gene [27].
The ubiquitin C-terminal hydrolase L1 (UCH-L1), encoded by the PARK5 gene and located on chromosome 4p13, which is relatively rare. It was first discovered in Germany. The UCH-L1 gene encodes for a deubiquitinating enzyme that plays a key role in the ubiquitin-proteasome system (UPS). It is predominantly expressed in the brain tissue. The UCH-L1 protein is highly conserved across different species and is primarily localized in neurons and neuroendocrine cells. It is estimated to constitute around 5%–10% of the cytoplasmic protein content in these cells [28]. Ile93Met mutation can cause abnormal expression of UCH-L1, abnormal proteolysis pathway of the UPS and accumulation of abnormal proteins in neurons, leading to neuronal degeneration and necrosis, and eventually leading to PD. A common polymorphism (p.S18Y) is associated with a reduced risk for iPD, although controversial [29,30]. It has been discovered that UCH-L1 possesses a unique ligase activity that is dependent on its dimerization. This ligase activity has been shown to promote the aggregation of α-Syn, at least in vitro [31]. Apart from its role as an ubiquitin hydrolase, UCH-L1 has also been suggested to function as an ubiquitin ligase and aid in the stabilization of mono-ubiquitin [32]. Furthermore, UCH-L1 is increasingly being recognized as a potential biomarker for various central nervous system (CNS) injuries, including traumatic brain injury, ischemic and hemorrhagic stroke, pediatric hypoxic-ischemic encephalopathy, spinal cord injury, epileptic seizure, and cardiac arrest. This highlights UCH-L1’s potential as a robust and universal marker target for different types of CNS injuries [33].
The PINK1 encoded by the PARK6 gene and located on chromosome 1p36.12. A polypeptide of 581 amino acids with an N-terminal terminus that serves as a mitochondrial inner membrane transfer termination signal, a serine/threonine domain between 156–509 residues, followed by a C-terminal domain, which serves as a mitochondrial outer membrane retention signal and is involved in mitophagy and mitochondrial function regulation [34]. The PINK1 can inhibit the release of cytochrome C from mitochondria and reduce neuronal apoptosis [35]. Under normal conditions, PINK1 content is extremely low, and PINK1 is constantly transferred to the mitochondrial inner membrane, where it is cleaved by the mitochondrial inner membrane protease presenilin-associated rhomboid-like protein (PARL) to generate an N-terminal degradation motif, which is subsequently cleared. Mutation sites include Ala168Pro, Arg68Pro, Pro296Leu, lle442Thr, Glu476Lys, Asp525Asn, Cys92Phe, Arg464His, R246X, H2710, E417G, L347P, Q239X and R492X. PINK1 regulates PARKIN translocation in response to mitochondrial damage, driving its removal through selective autophagy [36]. In normal circumstances, PINK1 is degraded by proteases in healthy mitochondria. However, when there is a loss of mitochondrial membrane potential, which indicates mitochondrial dysfunction, the import of PINK1 into the inner mitochondrial membrane is disrupted. As a result, PINK1 accumulates on the outer mitochondrial membrane. The accumulated PINK1 on the outer mitochondrial membrane recruits PARKIN, an E3 ubiquitin ligase enzyme, to the damaged mitochondria. PARKIN then promotes the ubiquitination of various proteins on the outer mitochondrial membrane, marking the damaged mitochondria for degradation. While Yang et al. demonstrated degenerated neurons in the brain of adult PINK1 knockdown monkeys with increased lysosomes and phagocytic vacuoles, but no clear evidence of mitochondrial abnormalities [37]. An important process in PD is mitochondrial autophagy, which selectively removes damaged mitochondria through autophagy. Mitochondrial autophagy ensures the clearance of dysfunctional mitochondria and helps maintain cellular homeostasis. This leads to the recruitment of autophagy mechanisms, such as the protein microtubule-associated protein 1 light chain 3 (LC3), which engulfs damaged mitochondria and forms autophagosomes. The autophagosome then fuses with the lysosome, resulting in the degradation of damaged mitochondria and the recycling of components. Mutations in the PARKIN or PINK1 genes, which are associated with PD, can impair the process of mitophagy. Mitophagy is the selective removal and clearance of damaged or dysfunctional mitochondria. Loss-of-function mutations in PARKIN or PINK1 disrupt the normal pathway of mitophagy, leading to defective clearance of damaged mitochondria. As a result, these dysfunctional mitochondria accumulate within cells, particularly in dopaminergic neurons of the substantia nigra pars compacta (SNpc). The accumulation of dysfunctional mitochondria can cause an imbalance in cellular redox status, leading to increased levels of oxidative stress. This oxidative stress, coupled with impaired mitochondrial function, can eventually contribute to the degeneration and death of dopaminergic neurons in the brain, which is a characteristic feature of PD [38]. Therefore, understanding the mechanism and regulation of mitochondrial autophagy mediated by PARKIN-PINK1 is crucial for elucidating the pathogenesis of PD. PINK1 protects neurons by phosphorylating key neural proteins, rather than mitochondrial autophagy. Phosphorylation of dynamin-related protein 1 (Drp1) at S616 promotes fission during mitosis and oxidative stress [39]. PINK1 knockdown leads to substantial reduction in the phosphorylation of many proteins. PINK1 drug development is mainly based on the protection of mitochondrial function. Protecting mitochondrial function and protein phosphorylation are two completely different methods for drug target design, and after clarifying the phosphorylation role of PINK1, drug development ideas will be more toward protein phosphorylation. This is of great significance for screening drug targets and discovering new biomarkers. Targeted drug research and development, along with the investigation of induced pluripotent stem cells (iPSC)-derived dopamine cell replacement therapy [40], are considered promising treatment approaches. These methods aim to utilize gene therapy to intervene early and correct gene mutations, with the ultimate goal of offering a potential cure for PD.
DJ-1, encoded by the PARK7 gene and located on chromosome 1p36, a member of the peptidase C56 protein family. DJ-1 can be used as an androgen receptor-mediated transcriptional regulator, and also as an oxidative stress sensor and redox-sensitive molecular chaperone, which can protect nerve cells from oxidative stress damage and resist cell death. Defects in this gene cause autosomal recessive early-onset PD. The PARK7 gene was first discovered as an oncogene by Nagakubo et al. in 1997 [41]. The PARK7 gene widely exists in brain, liver, kidney, skeletal muscle and reproductive organs, and participates in gene transcription regulation, signal transduction pathways, oxidative stress, mitophagy pathway, molecular chaperones and other biological effects. These include direct antioxidant activity by scavenging reactive oxygen species (ROS) and activation of endogenous antioxidant effect by DJ-1/NFE2 like bZIP transcription factor 2 (Nrf2) signaling pathway [42]. It has been reported that DJ-1 is mutated, including M26I, D149A and L166P, resulting in structural disruption and loss of function. DJ-1 is highly susceptible to oxidation, especially the cysteine residue (C106), which determines the activity of DJ-1 and can act as a scavenger of ROS [43]. The presence of the E64D mutation in the DJ-1 gene has been found to have detrimental effects on mitochondrial respiration and the generation of ROS. This mutation specifically impairs mitochondrial respiration and enhances the production of ROS, resulting in elevated levels of mitochondrial oxidative stress within dopaminergic neurons located in the SNpc. Consequently, this oxidative stress leads to lysosomal dysfunction and the accumulation of α-Syn protein. Furthermore, the deficiency of DJ-1 is also associated with a disorder in the formation of membrane proteins within the mitochondria-associated membrane (MAM). DJ-1 mutation can lead to ER calcium release disorder and mitochondrial calcium uptake disorder, followed by loss of antioxidant activity and increase of cellular oxidative stress, leading to neuronal death. Ibrahimet et al. discovered a splicing defect that relies on U1, which leads to a significant decrease in DJ-1 protein content, leading to dysfunction of mitochondrial function [44]. U1 splice site mutations are concentrated in sPD patients [45]. Further exploration of DJ-1 targets will become a breakthrough for the precise treatment of PD.
The LRRK2 encoded by the PARK8 gene and located on chromosome 12q12 and is a large molecular protein with multiple enzymatic activities, including the ROC (Ras of complex proteins, belonging to the small G protein family) with gtpase activity and Kinase domain with kinase activity. And C-terminal of ROC (COR), WD40, and other domains associated with protein interactions [46]. PARK8 gene is the most common autosomal dominant PD related gene with high penetrance. Although the physiological functions of LRRK2 in cells are currently largely unexplored, accumulating evidence points to a role for LRRK2 in autophagy regulation, microtubule dynamics, and mitochondrial function [47]. LRRK2 gene mutations (including G2019S, R1441C, R1441G, R1441H, I2020T, N1437H, Y1699C, S1761R, G2385R, R1628P) are the most common genetic risk factors for PD [48], mitochondrial effects of expression of a PD-associated pathogenic mutant of the LRRK2 gene include: increasing mitochondrial fragmentation and reduced membrane potential, producing more ROS and less adenosine triphosphate (ATP), cause mitochondrial Ca2+ to be squeezed through the Na+/Ca2+/Li+ exchanger, reducing the mitochondrial permeability transition pore (mPTP) threshold and increasing cell death, leading to increased vulnerability to stressors. Recent findings show that LRRK2 can modulate TH-DA pathway, which is related to LRRK2 mutations induced DA neurodegeneration [49]. There are protective variants of LRRK2 in Asian population. New LRRK2 substrates have been reported, including ribosome proteins. In addition, PINK1 and PARKIN can cooperate with LRRK2 to degrade mitochondrial rho GTPases (Miro) and affect the ubiquitination process to trigger fPD, which may be a common feature of PD pathogenesis [50,51]. At present, there are three LRRK2 inhibitors in clinical practice, namely DNL201, DNL151 and BIIB094 [52]. Inhibition of LRRK2 activity and consequent improvement of membrane transport function and lysosomal function is an up-and-coming pristine approach for the remedy of PD.
The ATPase cation transporting 13A2 (ATP13A2), encoded by the PARK9 gene and located on chromosome 1p36.13. It has 29 exons and is translated into at least three isoforms by alternative splicing. It belongs to the P5 subfamily of P-type ATPase transporters. ATP13A2 is a late lysosomal transporter that transports polyamines into cells through lysosomes, and its gene mutation disrupts this transport process, resulting in the accumulation of polyamines in lysosomes, which eventually swelter and rupture, leading to cell death [53]. ATP13A2 gene mutations included F182L, G504R, G877R, T12M, G533R and A746T [54]. ATP13A2 mutation leads to changes in lysosomal function in fibroblasts from PD patients, including incomplete maturation of lysosomal proteases and cathepsins B and D; The increase of intracellular pH in lysosomes; and an apparent defect in the lysosome-mediated protein degradation pathway. The lysosome itself also showed a perinuclear aggregation and a larger shape. In addition, ATP13A2 protein may carry some cofactors localized on the lysosomal membrane, which increase the activity of lysosomes, stabilize the function of lysosomes, and make cells more susceptible to handle adverse environmental stimuli such as oxidative stress, thereby reducing the occurrence of PD. The experiments have shown that ATP13A2 mutation can lead to the decrease of lysosomal function, and the lysosomal function can be restored after repair. Regulating the expression of ATP13A2 to regulate the function of nerve cell lysosomes may provide a pristine way for the treatment of neurodegenerative diseases.
The PARK10 locus is associated with iPD and located on chromosome 1q32, but it remains to be tested. The experimental verification of fPD and sPD in clinical and animal models practice shows that the components of UPS are closely related to the pathogenesis of PD. The candidate genes attracted by PARK10 were RING-finger protein 11 (RNF11), ubiquinol-cytochrome C reductase hinge protein (UQCRH), HIVEP zinc finger 3 (HIVEP3), eukaryotic translation initiation factor 2B subunit gamma (EIF2B3), ubiquitin specific peptidase 24 (USP24) and ELAV like RNA binding protein 4 (ELAVL4), which may affect the susceptibility or age of onset of PD [55]. RNF11 is expressed only in neurons, including the SN, and in addition has been detected in LBs in the brains of iPD patients [56]. Non-synonymous single nucleotide polymorphisms (SNP) in the coding region of the USP24 gene affecting susceptibility to PD in different populations may be extended in regulating the DNA damage response in cancer cells, in part by regulating the protein stability of p53 [57]. There is no association between PARK10 polymorphism and PD. Most importantly, we need to investigate the association between PARK10 polymorphism and the age of PD patients, and to clarify whether rare mutations in this locus affect the susceptibility of PD patients.
The GRB10 interacting GYF protein 2 (GIGYF2), encoded by the PARK11 gene and located on chromosome 2q36-q37, has a causal role in fPD in Italian and French population, but subsequent studies in Portuguese and US cohorts found no evidence of an association with PD [58]. Many other studies, including different Italian cohorts and multiple other ethnic groups, have also failed to determine the connection of GIGYF2 variants with PD [59–61]. Therefore, it is impossible that GIGYF2 is integrated with PD. However, research has shown that overexpression of GIGYF2 is not only associated with increased neuronal apoptosis but also linked to cognitive impairment, resulting in insulin dysregulation [62,63]. Insulin dysregulation has been implicated in the late-life susceptibility to neurodegenerative diseases. As a result, carriers of GIGYF2 mutations may not exhibit complete PD symptoms until advanced age [64]. The exact role of GIGYF2 in PD is still to be elucidated.
The PARK12 gene is located on chromosome Xq21-25. Studies have shown that the PARK12 gene regulates the expression of interleukin 13 receptor α1 (IL-13Rα1) and phosphoglycerate kinase 1 (PGK-1), and affects the susceptibility to PD [65,66]. The expression of IL-13 and IL-4 can reduce the inflammatory response, and they act by promoting the activation of M2 microglia and leading to the death of M1 microglia [67]. The expression of IL-13Rα1 leads to the damage of dopaminergic neurons and accelerates the progression of PD. PGK-1 is known to regulate the production of ATP in the SN, and its deficiency leads to insufficient ATP regeneration and lack of energy supply [65,66]. Therefore, the study of PARK12 contributes to our deeper understanding of the pathogenesis of PD.
The high-temperature requirement serine protease A2 (HTRA2), encoded by the PARK13 gene and located on chromosome 2p13.1. PARK13 can directly degrade PD proteins SNCA and DJ-1 without the involvement of autophagy. HTRA2 accumulation led to concomitant accumulation of PINK1, suggesting a correlation between the two proteins. In addition, HTRA2 and PINK1 levels were increased in H2O2 and L-levodopa treated cells and gradually migrated to the cytoskeleton under the influence of H2O2. This H2O2-mediated response was abolished when PARK13 was overexpressed. The deficiency of HTRA2 protein can lead to apoptosis disorder and cause disease [68]. HTRA2/Omi/PARK13 is a key regulator of mitochondrial molecular mass control but is released into the cytoplasm under stress conditions to promote cell death through various pathways, including caspase-dependent pathways and ER stress-mediated apoptosis [69]. Point mutations in HTRA2 are a susceptibility factor for PD, leading to mitochondrial dysfunction, autophagy dysfunction, and neuronal death [70].
The phospholipase A2, group VI (PLA2G6), encoded by the PARK14 gene and located on chromosome 22q13. PARK14 gene mainly encodes two proteins with phospholipase activity: calcium-independent phospholipase A2A (iPLA2A) and iPLA2β. They are involved in the maintenance of cell membrane homeostasis, mitochondrial function, calcium signal transduction, phospholipid remodeling, apoptosis, and prostaglandin and leukotriene synthesis and play an anti-apoptosis and neuroprotection role in SN dopaminergic cells [71–73]. PARK14 mutants included D331Y, G517C, T572I, R632W, N659S, S519A, and R741Q. Its mutation leads to the loss of function of PLA2G6, the reduction of protease activity, and the abnormal intracellular iron metabolism. Moreover, it has defects in preventing mitochondrial dysfunction, ROS production and activation of mitochondrial apoptosis pathways, the occurrence of brain and cerebellum atrophy, the accumulation of iron in basal ganglia, and the decline of cognitive ability, leading to neuroaxonal degeneration, and eventually leading to the occurrence of PD [71,74].
The F-box protein 7 (FBXO7), encoded by the PARK15 gene and located on chromosome 22q12.3. FBXO7 activity in the nucleus is essential for the maintenance of brain neurons. A leucine-rich nuclear export sequence (NES) is present in FBXO7, and NES is responsible for binding to chromosomal region maintenance 1 (CRM1) and the F-box domain (FBD) binds to S-phase kinase-associated protein 1 [75]. FBXO7 is a stress response protein that can be influenced by stress challenges. Under stress conditions, FBXO7 can undergo transfer from the nucleus to the mitochondria, and this process can even lead to the formation of detrimental protein aggregates within the mitochondria. FBXO7 plays a crucial role in maintaining mitochondrial health through its direct interactions with PINK1 and PARKIN. It is involved in PARKIN-mediated mitochondrial autophagy, which helps eliminate damaged mitochondria [76]. However, when FBXO7 is not functioning properly, it can lead to the activation of nuclear factor kappa-B (NF-κB) and the production of pro-inflammatory cytokines. This, in turn, can contribute to the aggravation of neuronal degenerative changes seen in various neurodegenerative diseases. Thus, inhibition of NF-κB activity by FBXO7 is expected to be a therapeutic strategy to protect dopaminergic neurons. Mutations within FBXO7 have been found to cause an early-onset PD (including T22M, Y52C, 187T, M115I, D328R, R378G, R481C, Arg498Stop, Thr22M and R498X). The mutant FBXO7 can form protein aggregations in mitochondria, which is relevant to FBXO7 mutation induced DA neurodegeneration. In FBXO7 knockout mice, reduced proteasome activity and early-onset motor defects as well as premature death were found, with dopaminergic degeneration via the ribosomal protein L23 (RPL23)-murine double minute 2 (MDM2)-tumor protein P53 (TP53) pathway [77]. FBXO7 mutant aggravated protein aggregation in mitochondria and inhibited mitophagy. In addition, LBs-like aggregates positive for p62 and α-Syn were found in neurons.
The PARK16 located on chromosome 1q32 and distributed in the pituitary and brain. The five single-nucleotide polymorphisms (SNPs) (rs823083, rs708723, rs4951261, rs823076 and rs16856110) at the PARK16 locus with PD [78]. Deletion of RAB7, a member of RAS oncogene family-like 1 (RAB7L1) in the PARK16 locus gene and LRRK2-related defects lead to defects in sorting lysosomal and Golgi apparatus and in the vacuolar protein sorting-associated protein 35 (VPS35) component of the retro body complex [79]. The genome-wide association study showed that compared with non-carriers, patients with PARK16 mutations showed greater motor dysfunction after 5 years of disease, which may affect the disease over time. At the same time, RAB7L1 (p.K157R) and solute carrier family 41 member 1 (SLC41A1) (p.A350V) mutations have also been found in PD patients, which need further study.
The VPS35 encoded by the PARK17 gene and located on chromosome 16q13. VPS35 is a component of the retro complex that controls retrograde transport of proteins from the endosomal to the Golgi network, promotes synaptic vesicle trafficking and recycling, regulates mitochondrial function [80], removes Tau and α-Syn, and prevents their aggregation. The expression of D620N VPS35 leads to a significant degeneration of dopaminergic neurons in the SN, along with the development of axonal pathology. Mutations in VPS35 (including Asp620Asn, p.P316S, p.Y507F and p.E787K) have been identified in PD patients that induce mitochondrial fragmentation and autophagy defects and increase dopaminergic neuronal death, they can also interact with dynamin-like protein 1 (DLP1), enhance proteolytic turnover of the DLP1 complex from mitochondria to lysosome, and increase mitochondrial fission and organelle dysfunction [81]. The PARK17 mutant (D620N) impairs the activity of Wnt/β-catenin signaling pathway and leads to neurodegeneration of in the SN DA cells [82]. VPS35 is positioned upstream of LRRK2, and when VPS35 is knocked down, it leads to a notable elevation in LRRK2-mediated phosphorylation of numerous Rab proteins. The VPS35 D620N mutant demonstrates hyperphosphorylation and interaction with amyloid precursor protein (APP) at Thr668. By inhibiting the function of APP, the detrimental effects caused by VPS35 D620N on neurogenesis, neural migration, and maturation can be rescued [83]. There is evidence to suggest that lysosome-associated membrane protein 2A (Lamp2A) is a cargo protein of VPS35/retromer in dopaminergic neurons. This finding highlights the importance of the VPS35-Lamp2A-α-Syn pathway in preventing the development of PD [84]. Given the significant overlap in the molecular mechanisms underlying neurodegeneration in different types of PD, it is worthwhile to investigate whether neuroprotective drugs can be tested using the VPS35 model. Additionally, it is important to conduct efficacy, safety, and reliability validation studies and clinical trials in other more common PD syndromes [85].
The eukaryotic translation initiation factor 4-gamma 1 gene (EIF4G1), encoded by the PARK18 gene and located on chromosome 3q27.1. EIF4G1 is a component of the translation initiation complex eukaryotic Initiation factor 4F, which is widely expressed in different tissues and interacts with a variety of initiation factor as a scaffold protein. The EIF4G1 mutants included p.R1205H, p.A502V, p.G686C, p.S1164R, p.R1197W, p.T318I, p.V541G, p.G698A, and p.R1205H. EIF4G1 is involved in protecting mitochondria from cellular stress. Both mutations at the p.A502V and p.R1205H sites can damage cell resistance to oxidative stress and affect the protein turnover process, which is very important for cell survival [86]. Development of individual targeted therapies against EIF4G1.
DNAJ heat shock protein family (HSP40) member C6, also known as DNAJC6, is encoded by the PARK19 gene and can be found on chromosome 1p31. Several DNAJC proteins associated with PD (Auxilin/DNAJC6, GAK/DNAJC26, RME-8/DNAJC13, DNAJC10/ERdj5, DNAJC12) play crucial roles in regulating clathrin trafficking and dynamics [87]. The DNAJC family members belong to the HSP family and DNAJC6 specifically assists in the uncoating of clathrin-coated vesicles (CCVs), thereby promoting the regeneration of synaptic vesicles (SV) at presynaptic sites. In addition, LRRK2 mutation is a common cause of PD, and DNAJC6 may play a role in the pathogenesis. Loss of DNAJC6 function leads to defective autophagy that impairs Wnt-LIM homeobox transcription factor 1α (LMX1A) signaling, and reduced LMX1A expression leads to the generation of vulnerable DA neurons with pathological manifestations. The α-Syn is accumulated due to impaired autophagy-lysosomal degradation in neuronal cells caused by DNAJC6 mutation. Moreover, mutations in the DNAJC6 gene (variant: Q734X, AK4p.S61C) and DNAJC13 (variant: p.E1740Q, p.R1516H, p.N855S, p.N855S, p.L2170W, p.P336A, p.V722L, p.N855S, p.R1266Q) result in disrupted autophagy-lysosomal degradation in neurons. This disruption leads to the accumulation of α-Syn and alterations in synaptic function, which can ultimately result in synaptic loss, axonal degeneration, and eventual death of neuronal cells. These processes are associated with the development of early-onset PD [88]. It is imperative to gain a more comprehensive understanding of the molecular genetics of the DNAJC gene and extensively investigate the mechanisms underlying parkinsonism across different spectrums.
Synaptojanin1 (SYNJ1), encoded by the PARK20 gene and located on chromosome 21q22. SYNJ1, another key gene in clathrin trafficking, is mainly enriched in neurons and centered at synapses. Previous studies have shown that an inactivating mutation (R258Q) of SYNJ1 in the inositol phosphatase domain of Sac can cause early-onset PD. SYNJ1 heterozygous knockout and LRRK2 G2019S transgenic mice show reduced synaptic density and CCVs accumulation, particularly in dopaminergic neurons, leading to dystrophies and selective degeneration of axons in these neurons. The Sac domain of SYNJ1 plays an important role in clathrin coating dynamics. Studies have shown that in all brain regions of PD mice and cultured neurons, abnormal metabolism of clathrin coated intermediates (mainly CCVs) in synapses, including inhibitory synapses, results in accumulation [89].
PARK21, is related to non-syndromic X-linked intellectual disability and combined oxidative phosphorylation deficiency. Late PD, which is characterized by often chromosomal dominants, the average age of the disease is 67 years [90,91]. Affiliated tissues include brain and subthalamic nucleus, and related phenotypes are tremor and rigidity.
The coiled-coil-helix-coiled-coil-helix domain containing 2 (CHCHD2), encoded by the PARK22 gene and located on chromosome 7p11.2. CHCHD2 mutation site include p.Thr61Ile, p.Ala32Thr, p.Pro34Leu, p.Arg145Gln, 300 + 5G > A, and p.Ile80Val. The CHCHD family is highly conserved in function, and CHCHD mutations or deletions will result in targeted and location-related loss of mitochondrial function [92]. The biological functions of CHCHD2 and CHCHD10 include regulating mitochondrial dynamics, participating in oxidative phosphorylation regulation, inhibiting apoptosis, regulating synaptic plasticity, stimulating cell migration, and regulating lipid metabolism. In addition, the ability of these proteins to function depends on their interactions with many other proteins, including optic atrophy 1 (OPA1), OMA1 zinc metallopeptidase (OMA1), B-cell lymphoma-extra-large (BCL-XL), BCL2-associated X (BAX), PINK, Prostacyclin (P62) and TAR DNA binding protein-43 (TDP43), which lead to mitochondrial dysfunction, cognitive impairment and Motor neuron damage [93–96]. If the therapeutic effects of CHCHD2 and CHCHD10 are further confirmed, it will be necessary to conduct additional and more comprehensive experimental studies to elucidate their potential applications in clinical practice [96].
The vacuolar Protein Sorting 13 Homolog C (VPS13C), encoded by the PARK23 gene and located on chromosome 15q22 [97]. VPS13C encodes a lipid transfer protein, which is located in the contact area between the ER membrane and the late endothelium/fox body. It is partially located in the outer mitochondrial membrane, and its silencer is related to the reduction of mitochondrial membrane potential, mitochondrial fragmentation, increased respiratory rate, increased PINK1 protein, PANK2 dependent mitochondria, and the improvement of PANK2 transcriptional regulation after mitochondrial damage [98]. The VPS13C p.D620N mutation weakens the classification function of reverse transcriptase complex, destroys the maturation and automation of endoscope, and destroys the circulation of membrane receptor and the formation of mitochondrial capsule. Deletion of VPS13C resulted in mitochondrial dysfunction and lysosomal homeostasis disorder (downregulation of lysophosphatidylcholine (lysoPC) and lysoPE, regulation of ceramide, hexose ceramide and cholesterol ester) [99,100]. It also activates the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway, which has a strong association with PD [101].
Prosaposin (PSAP), encoded by the PARK24 gene, is located on chromosome 10q22.1. Occasional lysosomal dysfunction in PD is at least partially attributed to changes in saposin C (SAPC) caused by a decrease in PSAP levels. If a stove occurs, it will result in a significant decrease in β-GCase activity, leading to the accumulation of α-Syn. In a case-control association study, two variants (rs4747203 and rs885828) located in the intronic regions of the PSAP saposin D domain were found to have significantly higher allele frequencies in individuals with sPD in a combined cohort from Japan and Taiwan [102]. Researchers also observed abnormal accumulation of autophagic vacuoles, impaired autophagic flux, altered intracellular localization of prosaposin, and aggregation of α-Syn in patient-derived skin fibroblasts or induced pluripotent stem cell-derived dopaminergic neurons. Mutations in PSAP have been linked to progressive motor decline and dopaminergic neurodegeneration. These findings suggest that PSAP plays a role in the pathogenesis of PD and may serve as a potential therapeutic target for the disease [102]. The loss of progranulin (GRN) mutation function leading to progranulin protein (PGRN) haploid deficiency is a common genetic cause of frontal lobe dementia. PSAP knockdown results in increased PGRN monomers, while PSAP overexpression leads to increased PGRN oligomers, partially through a protein-protein interaction (PPI). The PSAP-induced alterations in PGRN levels and oligomerization are observed in human fibroblasts derived from a carrier of the GRN mutation, further suggesting PSAP as a potential therapeutic target related to PGRN. Future studies should center on elucidating the significance and cellular mechanisms through which PGRN oligomeric forms offer neuroprotection [103]. Absolutely, the observation of changes in PGRN levels and oligomerization induced by PSAP in human fibroblast derivatives obtained from GRN mutation vectors reinforces the notion that PSAP could be a promising therapeutic target for PGRN-related conditions. To advance our understanding, future research should prioritize investigating the correlation and cellular mechanisms by which PGRN polyhedra contribute to neuroprotection. This information could provide valuable insights for the development of potential treatments targeting PGRN and related disorders [104].
It is worth noting that the significant decrease in SAPC protein levels directly affects glucosylceramidase (GBA) function and Grave Sin clearance. The SAPC encoded by PSAP is the GBA encoded on chromosome 1q21, which is necessary for the activity of glucosidase (GCase) in GBA [105]. Mutations in the GBA gene (including N370S, L444P, 84 Gg, IVS+1, V394L, and R496H) may lead to loss of GCase activity and lissom dysfunction, thereby impairing α-Syn metabolism. GCase lacks a model to demonstrate dysfunction of the automatic enzyme fox body pathway and subsequent α-Syn invasion aggregation. This dysfunction can also result in abnormal lipid metabolism, characterized by the buildup of glycosides and glucosylceramide [106]. Certain mutations hinder the proper functioning of GCase and cause it to accumulate in the ER, activating stress responses that can lead to neurodegeneration, including prolonged protein responses. Besides these mechanisms, GCase deficiency is also associated with mitochondrial dysfunction and neuroinflammation, both of which contribute to the pathogenesis of PD [107]. Mutations in the GBA gene represent the most significant genetic risk factor for PD in the general population, increasing the risk of developing PD by over fivefold.
Analysis of Rare Variants in PD
The gene GTP cyclohydrolase 1 (GCH1) encoding GTP ring hydrolase 1 belongs to the GTP ring hydrolase family, which is known to be involved in BH4 biosynthesis. BH4 deficiency plays a crucial role in mitochondrial oxidation and homeostasis. BH4 is a cofactor of TH, which limits the enzyme rate of dopamine biosynthesis. GCH1 weakens homology, activates innate immune mechanisms in the brain, leading to microglial activation and phagocytic activity. The accumulation of roses caused by GCH1 mutation damages proteins, lipids, and DNA, thereby disrupting mitochondrial ATP synthesis and mitochondrial metabolism, leading to the death of dopaminergic neurons. Therefore, variants in the GCH1 gene may lead to dysregulation of dopa response [108].
A protein encoded as fibroblast growth factor 20 (FGF20), located on chromosomes 8p21.3-p22, is a neurotrophic substance, best manifested as the nigra pars compacta subtype. FGF20 can significantly improve the survival rate of dopaminergic neurons by activating the FGF 1C receptor (FGFR-1C) and activating mitochondrial activated protein kinase (MAPK). FGF20 is significantly associated with degenerative brain diseases and is expected to become an important new drug for the treatment of PD with recombinant proteins [109].
Nuclear receptor-related factor 1 (NURR1), located on chromosome 2q22-q23, is one of the transcription factors required for differentiation, maturation and maintenance of dopaminergic neuron development. The NURR1 gene is mainly expressed in the CNS and plays an important role in the development and maintenance of dopaminergic neuron function in the midbrain, including DA metabolism, neurotransmission, axon development, mitochondrial function, activation of TH transcription, and cell survival [110–112]. In microglia and astrocytes, NURR1 expression has been found to suppress the expression of proinflammatory mediators. Thus, it protects against inflammation-mediated death of DA neurons. NURR1 interacts with α-Syn to regulate energy metabolism, glucose metabolism, and lipid metabolism, and its expression is reduced in elderly and PD post-mortem brain [113]. Although the value of NURR1 in PD treatment has not been carefully evaluated, determining its molecular mechanisms in dopaminergic neuronal development and cellular metabolic function may ultimately help develop personalized treatment methods to restore functional integrity in brain pathology. Another promising strategy is to identify selective and safe NURR1 agonists to support dopaminergic neural function and reduce neuroinflammatory activity [114].
NUS1 dehydrodolichyl diphosphate synthase subunit (NUS1) encodes the NOGOB receptor (NGBR), which mainly locates the ER membrane. Functional studies on Drosophila have demonstrated that the absence of NUS1 results in a decrease in elevation, DA levels, and the quantity of dopaminergic neurons, ultimately leading to the occurrence of apoptosis in the Drosophila brain [115]. NUS1 deficiency notably triggers stress by activating the protein kinase r-like ER kinase (PERK) pathway. This activation subsequently induces the expression of ER stress-related proteins and promotes neuronal apoptosis in the mouse model [116].
The cell bodies and axons of SN DA neurons can release endogenous cannabinoids such as 2-arachidoniyl-glycerol (2-AG) and N-arachidoniylethanolamine (AEA). The diacylglycerol lipase α (DAGLA) and DAGLB regulate 2-AG synthesis in the brain. DAGLB was significantly more expressed than DAGLA in human and mouse SN dopaminergic neurons. The biosynthesis of DGLB mediated agent 2-AG is involved in regulating the normal physiological function of dopaminergic neurons, which may help explain how DAGLB deficiency leads to the occurrence of PD-related motor symptoms. Therefore, increasing the production of 2-AG may be a potential mechanical therapy intervention to increase the release of DA in Niger and maintain sufficient neural activity in patients with DA neurons in Niger [117].
Transmembrane protein 175 (TMEM175) is a potassium channel in the lysosomal membrane, genome-wide association studies (GWAS) have revealed that mutations in some genes closely related to lysosomal function are highly associated with the risk of PD, and about 20% of PD patients have TMEM175 mutations, which is considered to be a lysosomal H+ channel activated by H+ [118]. The common TMEM175 variant rs34311866 (M393T) can increase the risk of PD, reduce the channel current, and the mutant channel is more vulnerable to nerve injury, and accelerate the accumulation of pathological α-Syn, leading to the loss of dopaminergic neurons and the impairment of motor function. In contrast, another common variant, rs3488217 (Q65P), is associated with a reduced risk of PD, a stronger channel current during cell starvation, and a neuroprotective mechanism that enables neurons to resist injury [119]. TMEM230 is a ubiquitously expressed transmembrane protein and the mutants include R141L, Y92C, *184Wext5, p.Arg141Leu, p.Tyr92Cys, p.*184Trpext*5 and p.*184Pro Glyext*5. It is a transport protein that secretes and recycling vesicles, including neuronal synaptic vesicles, which store neurotransmitters, proteins involved in packaging the neurotransmitter DA in neurons. The expression of mutant TMEM230 has been shown to lead to an increase in α-Syn levels. The loss of TMEM230 function disrupts the secret automata, gold sac secretions, and retrospective circulation [120].
Ubiquinol-cytochrome c reductase core protein 1 (UQCRC1) is the main component of complex III in the respiratory chain. UQCRC1 is highly expressed in neurons and astroglial cells. It may also play a role in maintaining normal brain ischemia tolerance, learning, and memory by forming complex III [121]. Flies with reduced neuronal expression of UQCRC1 display age-related defects resembling PD, such as a decrease in dopaminergic neurons and impaired movement. However, these defects can be improved by introducing UQCRC1 expression. Additionally, the lethality observed in flies lacking UQCRC1 can be rescued by specifically expressing UQCRC1 in neurons, but not by expressing the disease-causing mutation, enabling us to investigate its pathogenicity [122]. Furthermore, UQCRC1 is found to interact with cytochrome C (cyt-C), a trigger for programmed cell death. Deficiency of UQCRC1 leads to an accumulation of cyt-C in the cytoplasmic fraction, activating the caspase cascade associated with cell death. However, reducing cyt-C levels or expressing the anti-apoptotic protein p35 can alleviate the neurodegeneration caused by UQCRC1 deficiency. These findings highlight the involvement of UQCRC1 in the regulation of cyt-C-induced apoptosis [122]. The impact of the p.Tyr314Ser mutation in the UQCRC1 gene, which encodes a subunit of mitochondrial respiratory chain complex III. The age-dependent locomotor defects observed in these models suggest a progressive impairment in motor function over time. Furthermore, the loss of dopaminergic neurons is a characteristic feature of PD, indicating that this model may provide insights into the molecular mechanisms underlying neurodegeneration in this disorder [122,123]. Therefore, UQCRC1 may become a novel target for the treatment of PD by regulating mitochondrial function and oxidative stress to alleviate symptoms or delay disease progression. However, further research is still needed to determine the specific role and underlying mechanisms of UQCRC1 in the treatment of PD.
The glycoprotein non-metastatic melanoma protein B protein (GPNMB) located on chromosome 7. The role of GPNMB in the regulation of systemic immune responses. It includes suppression of t-lymphocyte activation in macrophages and microglymes and reduction of inflammatory reactions to lipopolysaccharides (LPS). It can also perform its neuroprotective role by reducing neuropathy mediated by astrocytes based on CD44. It has been shown that CD44 interacts with toll like receptor 2 (TLR2) and reduces NF-κB signal transmission to suppress inflammation. The loss of GPNMB results in a loss of ability to internalize α-Syn-proton fibres and develop an α-Syn pathology that is necessary and sufficient to absorb α-Syn-fibres in neurons. At the same time, the role of GPNMB and CD44 in astrocytes provides a powerful model of the mechanism and suggests that GPNMB may be a viable treatment method, opening up new ways to treat PD [124,125].
Dynactin 1 (DCTN1) affects neurons, mitochondrial autophagy, protein stability, and mitochondrial dynamics disorders are observed in most neurodegenerative diseases. Mutation types include G59S, G71R, G71E, G71A, T72P and Q74P. Neurological degenerative diseases caused by manganese (Mn) disrupt the transport of mitochondrium when exposed to manganese, reducing DCTN1, kinetic protein-1 (KIF5B) and increasing cell skeleton instability at protein and genetic levels. Genetic mutations caused by atypical Parkinson’s syndrome, apathy or depression, respiratory failure and weight loss in middle age [126,127].
The LDL receptor related protein 10 (LRP10) is mainly expressed in astrocytes and the nervous vascular system, but is not detected in neurons, and its main function is to remove proteins that easily accumulate. LRP10 is also localized and interacts in part with the receptor associated with sortilin 1 (SORL1). The study of the hyperexpression model links LRP10 to bubble transport, demonstrating its localization in plasma membranes, internal culverts, trans-galactic networks and partial overlap with a complex of retrovirus enzymes. LRP10 has been detected in non-neuronal cells with the highest expression in astrocytes cells and neurovascular units of the adult brain [128,129]. Although our current understanding of the relationship between the LRP10 and PD is still limited, extensive research in this field is underway, offering hope for the development of new therapeutic approaches in the future.
The monoamine oxidase (MAO) gene is located on chromosome Xp11.23. MAO is an enzyme combined with mitochondria, which plays an important role in DA metabolism. It can be divided into two types of homogeneous enzymes: MAO-A and MAO-B. Both MAO-A and MAO-B have been extensively studied in terms of their molecular and genetic characteristics. Although they are composed of different proteins, they do share 70% identical amino acid sequences. Additionally, both MAO-A and MAO-B bind to the same coenzyme, flavin adenine dinucleotide (FAD), which forms a covalent thioester linkage with a cysteine residue within a pentapeptide sequence (Ser-Gly-Gly-Cys-Tyr) [130]. Both MAO-A and MAO-B are expressed in the brain and most peripheral tissues, and they are localized on the outer membrane of mitochondria. In the brain, MAO-A is predominantly found in catecholaminergic neurons, while MAO-B is present in serotonergic and histaminergic neurons as well as astrocytes. These enzymes play important roles in the metabolism of neurotransmitters, and alterations in their activity have been implicated in various neurological and psychiatric disorders [131]. The oxygen radicals generated in DA can cause oxidative damage to neurons, while MAO-B can activate the formation of neurotoxic substance 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), thereby increasing oxygen free radicals and active neurotoxic substances [132]. Therefore, inhibiting the activity of MAO-B is considered a therapeutic strategy for treating PD. Some drugs, such as levodopa and rasagiline (a selective MAO-B inhibitor), work by suppressing the activity of MAO-B to increase DA levels and alleviate motor symptoms in PD patients [133]. In summary, both MAO-A and MAO-B may play a significant role in the development of PD, and modulating the activity of these enzymes can help increase DA levels, thereby alleviating related symptoms.
The extension of the repeated CAG sequence in the Like-Sm (LSm) protein family ataxin 2 (ATXN) demonstrates typical symptoms of PD. PARK2 mutates in Parkinson pathogen and has been found to interact with ATXN2, a protein that intensifies polyglutamine amplication, causing the maturation of the hormonal muscle of Spinocerebellar ataxia 2 (SCA2) or increases the risk of levodopa-responsive PD and Amyotrophic Lateral Sclerosis (ALS) of motor neuron disease [134].
Associated with microtubule associated protein tau (MAPT), located on the 17th chromosome, is an internally disturbed protein, expressed at the highest level in the neurons of the entire CNS. MAPT mutations (including R5H, R5L, G55R, A152T, K257T, I260V, L266V, G272V, N279K, Δ280K, S285R, Δ296N, N296H, K298E, N410H) leads to Tau dysfunction and aggregation with neuronal loss and atrophy. Leading to neurodegenerative diseases such as PD [135].
Pantothenate kinase 2 (PANK2) is located on the chromosome 20p13, and pantothenate kinase-associated neurodegeneration (PKAN) is caused by mutation PANK2. Enzyme PANK2, localized in mitochondria, is crucial for the biosynthesis coenzyme a, which in turn is a key factor in the synthesis of ATP, fatty acids and metabolism of neurotransmitters. Loss or anomaly of the PANK2 function can lead to iron accumulation in certain areas of the brain, causing excessive ROS production, which leads to neurodegenerative changes in iron accumulation in the brain [136]. In typical PKAN symptoms usually occur before the age of 6 years, and most patients need a wheelchair in adolescence. Atypical PKAN usually becomes apparent only after 10 years with lighter symptoms and progresses more slowly than classic PKAN [137]. It is believed that the classic PKAN was caused by a deficiency of PANK2, while the atypical disease was caused by a serious deficiency of the enzyme PANK2. It is reported that deep brain stimulation (DBS) can effectively alleviate symptoms.
Spastic paraplegia type 11 (SPG11) is the most common non-conspicuous genetic spasmatic paralysis of the common chromosome, accompanied by depletion of the callous body. Spatacsin is a protein encoded by the genome SPG11 and associated with autophagy [138]. In normal operation SPG11 is important for the development of nerves and neurodegenerative changes due to its role in the process of removal of lipids and functional lysosomes. In patients with hereditary convulsions caused by SPG11 mutation, DA degeneration, DA transmitter densities, anomalies or defects in the transit of lysis were observed, leading to lysosomal dysfunction, DNA recovery defects, cell division anomalies, and autophagy, which eventually led to PD in adolescents. These results, as well as the data that determine the structural lysosomal anomaly SPG11, may expand the new link between lysosomal dysfunction and PD [139,140].
Polymer gamma (POLG) is an enzyme responsible for replicating and restoring mitochondrial DNA. POLG mutations (including G737R, R853W, R722H, Y831C, Q1236H, S1230F, P587L, W748S, etc.) destroy mitochondrial DNA (mtDNA), causing respiratory circuit dysfunction in complex I and depletion of mtDNA. Therefore, alterations in POLG genes can impact the development of different genetic neurodegenerative disorders, including monogenic forms of PD [141].
The TATA-box binding protein associated factor 1 (TAF1) coding the transcription factor IID (TFIID). This facilitates the connection TFIID to the initiating subsystem and plays an important role at the beginning of the transcription. TFIID is a complex of TAFs related factors required for the transcription of RNA polymerase II [142]. It is a transcription of many protein encoding genes in eucaryotes. In addition, TFIID is also involved in cell cycle, cell growth, embryonic development and neural development. Variants in many components of TFIID, such as TBP, TAF2, TAF6, and TAF13, have been reported to be associated with neurodevelopmental disorders. TAF1 is a pathogenic gene for diseases such as X-linked dystonic-Parkinsonism and X-linked mental retardation. Because of its important role in the transcription process, TAF1 has emerged as a topic of interest in the research of the pathogenesis and development mechanisms of various diseases [143].
Members of the dual specificity tyrosine phosphorylation regulated kinase (Dyrk) participate in the management of key signal routes that control post-embryonic neurogenesis, development, survival, differentiation and death of cells. Dyrk1a, located on chromosome 21q22.2, plays a crucial role in processes like nerve multiplication, neurogenesis, and survival of dopaminergic neurons in the developing brain. This genomic region, also known as the critical area of the down syndrome chromosome region (DSCR), is susceptible to mixed mutations that can lead to microcephaly and neurological disorders [144]. Development of new fluorinated polyphenol as Dyrk1a/b kinase selective inhibitors for the treatment of neurological diseases, including PD.
Mutations in the gene crystallin alpha A (CRYAA) lead to protein aggregate ER stress (ERS), causing a reaction of unfolded protein response (UPR), which leads to cell death. In mice with CRYAA-Y118D mutation, long-term UPR activation and severe stress responses resulted in cell deaths caused by protein toxicity and ERS. This could be related to the pathogenesis of PD [145].
The drug treatment of PD includes compound levodopa (Madopar, Sinemet), DA receptor agonists (pramipexole, Piribedil sustained-release tablets, Robiniro), catechol-Omethyl transferase (COMT) inhibitors (Entacapun), MAO-B inhibitors (Slegilan, Rasagiline), amantadine, Benzhexol, etc. [146]. PD patients should also receive training and guidance on language, eating, walking, and various daily activities to enhance their quality of life. For patients with significantly reduced long-term drug treatment efficacy, surgical treatment can also be considered. In addition to immunological and genetic approaches, there are many other strategies to target the pathogenesis, such as disease modification therapy aimed at correcting autophagy lysosome and mitochondrial dysfunction. In terms of surgical treatment, the subthalamic nucleus (STN) or globus pallidus internus (GPi) are the most commonly targeted brain regions for DBS in PD.commonly targeted brain regions for DBS in PD patients [147]. Although DBS is an effective treatment, its therapeutic efficacy is still controversial. Currently, improvements in DBS technology, including the utilization of adaptive stimulation, improved connectivity, directed stimulation, and the investigation of novel targets are still in progress. In recent years, the research of PD biomarkers has made remarkable progress, but there are still many challenges. Transcranial ultrasound and olfaction detection, gene detection, specific diagnostic biomarkers (α-Syn, coenzyme Q10 (CoQ10)), metabolism enzyme cytochrome P450, Tau, neurofilament light chain (NFL), etc.) [148–150], biochemical markers, molecular imaging, digital symptom monitoring and other multi-dimensional evaluation methods obtained from cerebrospinal fluid, blood, peripheral tissues, etc., are all effective ways to diagnose PD and evaluate prognosis. In addition, microRNA (miRNA) also serves as a biomarker for diagnosing PD, these small RNA molecules regulate gene expression by binding to the messenger RNA (mRNA) transcripts, which can subsequently lead to the inhibition of protein synthesis or degradation of mRNA. They have been implicated in various biological processes and diseases, including PD. Studies have identified dysregulation of specific miRNAs in PD, suggesting their involvement in the pathogenesis of the disease. The altered expression of miRNAs in PD may contribute to the dysfunction of cellular processes, such as protein aggregation, oxidative stress, and neuroinflammation [151,152]. Moreover, miRNAs have the potential to serve as biomarkers for PD. The levels of miRNAs in biological fluids, such as blood and cerebrospinal fluid, have been found to be altered in PD patients compared to healthy individuals [153,154]. These changes in miRNA expression profiles could potentially serve as diagnostic or prognostic biomarkers for PD, aiding in early detection and monitoring of the disease. Additionally, miRNAs are being explored as a potential therapeutic strategy for PD. Modulating the expression levels of miRNAs through various approaches, such as miRNA replacement or miRNA inhibition, may offer a novel avenue for the development of targeted therapies. By correcting or attenuating the miRNA dysregulation observed in PD, it is possible to manipulate the expression of specific genes and restore cellular homeostasis [155]. The research on miRNAs in PD is rapidly evolving and holds significant promise for better understanding the disease and developing effective strategies for diagnosis and treatment. With the development of many technologies such as genetics, molecular imaging, proteomics and artificial intelligence, we expect that more reliable biomarkers can be applied to the clinic as early as possible to solve the current problems of early diagnosis and individualized treatment of PD.
There are numerous known pathogenic genes related to PD, and the interactions between genes and the environment have an impact on the occurrence, development, and prognosis of PD. Traditional PD drugs and surgical treatments can provide relief from symptoms, they do not offer a cure for PD, while gene therapy has brought us new treatment methods to further understand the pathogenic mechanisms of these genes and find breakthroughs. At the same time, it is necessary to better understand the relationship between synaptic, mitochondrial and lysosomal dysfunctions and the selective vulnerability of neurons with SN DA in patients with PD, which is expected to open up new prospects for the treatment of PD. Mitochondria and lysosomes are the two main organelles in the pathogenesis of PD. Mitochondria are responsible for the production of cellular energy and play a vital role in maintaining the health of neurons. Mitochondrial dysfunction leads to oxidative stress, insufficient energy, and impaired protein processing, all of which have been linked to PD. Lysosomes, on the other hand, are involved in the degradation and recycling of cellular waste, including damaged proteins and organelles. Impaired lysosomal function can lead to the accumulation of toxic substances, leading to the neurodegeneration observed in PD. Research has shown that in the pathogenesis of PD, SNCA may regulate the phosphorylation of LRRK2 and inhibit the phosphorylation activity of PINK1 [156,157]. When the PARKIN protein is functionally deficient or mutated, SNCA can aggregate [157]. Mutations in the GBA gene can lead to abnormal metabolism of glucosylceramide, resulting in abnormal accumulation of α-syn in the CNS, leading to cellular toxicity and neuronal damage [158]. PINK1 regulates its kinase activity by interacting with LRRK2, thereby affecting mitochondrial function. Mutations in LRRK2 may disrupt its interaction with PINK1, leading to mitochondrial quality control imbalance and promoting the development of PD. The presence of DJ-1 can promote the phosphorylation and activation of PINK1, thereby enhancing PINK1’s selective autophagy towards damaged mitochondria [159]. Accumulated PINK1 can phosphorylate (activate) PARKIN and facilitate the localization of PARKIN to damaged mitochondria [36]. There is also an interaction between FBOX7 and PINK1, research has shown that FBOX7 can form a complex with PINK1 and regulate the stability and activity of PINK1. By binding to PINK1, FBOX7 inhibits its degradation, thereby increasing the expression level and function of PINK1 [160]. This interaction is crucial for maintaining mitochondrial quality control and the occurrence and development of PD. Understanding the interaction of PD genes with organelles and proteins such as mitochondria and lysosomes, α-syn-LRRK2, LRRK2-PINK1, FBOX7-PINK1-PARKIN, GBA-α-syn provides valuable insights into the pathogenesis of PD. Additionally, it would be beneficial to clarify that while mutations in some of the mentioned genes may be infrequent in PD. The gene products of these mutations are hypothesized to contribute to the development of PD. These gene products participate in diverse cellular processes including intracellular trafficking, oxidative stress, mitochondria, phospholipid membranes, and the UPS, all of which have been implicated in the pathogenesis of PD [161]. Further research in this area may reveal potential therapeutic targets to slow the progression of neurodegenerative disease.
Acknowledgement: None.
Funding Statement: This work was supported partly by Henan University graduate “Talent Program” of Henan Province (SYLYC2023092).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: TL; data collection: YH; analysis and interpretation of results: TL, LZ; draft manuscript preparation: TL, YH. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All data and material generated or analyzed during this study are included in this published article. The datasets generated and/or analysed during the current study are available in the Human Protein Atlas database (https://www.proteinatlas.org/).
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no competing financial or non-financial interests.
Supplementary Materials: The supplementary material is available online at https://doi.org/10.32604/biocell.2024.049130 (https://www.jianguoyun.com/p/DYWHsLkQ1sCuDBid1cEFIAA).
References
1. Leite Silva ABR, Gonçalves de Oliveira RW, Diógenes GP, de Castro Aguiar MF, Sallem CC, Lima MPP, et al. Premotor, nonmotor and motor symptoms of Parkinson’s disease: a new clinical state of the art. Ageing Res Rev. 2023;84:101834. [Google Scholar] [PubMed]
2. Verhoeff MC, Eikenboom D, Koutris M, de Vries R, Berendse HW, van Dijk KD, et al. Parkinson’s disease and oral health: a systematic review. Arch Oral Biol. 2023;151:105712. [Google Scholar] [PubMed]
3. Blesa J, Foffani G, Dehay B, Bezard E, Obeso JA. Motor and non-motor circuit disturbances in early Parkinson disease: which happens first? Nat Rev Neurosci. 2022;23(2):115–28. [Google Scholar] [PubMed]
4. Meles SK, Oertel WH, Leenders KL. Circuit imaging biomarkers in preclinical and prodromal Parkinson’s disease. Mol Med. 2021;27(1):111. [Google Scholar] [PubMed]
5. Guadagnolo D, Piane M, Torrisi MR, Pizzuti A, Petrucci S. Genotype-phenotype correlations in monogenic Parkinson disease: a review on clinical and molecular findings. Front Neurol. 2021;12:648588. [Google Scholar] [PubMed]
6. Polymeropoulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, di Iorio G, et al. Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Sci. 1996;274(5290):1197–9. [Google Scholar]
7. Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7(4):306–18. [Google Scholar] [PubMed]
8. Spillantini MG, Divane A, Goedert M. Assignment of human alpha-synuclein (SNCA) and beta-synuclein (SNCB) genes to chromosomes 4q21 and 5q35. Genom. 1995;27(2):379–81. [Google Scholar]
9. Lavedan C, Leroy E, Dehejia A, Buchholtz S, Dutra A, Nussbaum RL, et al. Identification, localization and characterization of the human gamma-synuclein gene. Hum Genet. 1998;103(1):106–12. [Google Scholar] [PubMed]
10. Ali R, Alam A, Rajput SK, Ahmad R. Pharmacotherapeutics and molecular docking studies of alpha-synuclein modulators as promising therapeutics for Parkinson’s disease. BIOCELL. 2022;46(12):2681–94. doi:10.32604/biocell.2022.021224. [Google Scholar] [CrossRef]
11. Yin J, Han J, Zhang C, Ma QL, Li X, Cheng F, et al. C-terminal part of α-synuclein mediates its activity in promoting proliferation of dopaminergic cells. J Neural Transm. 2011;118(8):1155–64. [Google Scholar] [PubMed]
12. Sun Y, Long H, Xia W, Wang K, Zhang X, Sun B, et al. The hereditary mutation G51D unlocks a distinct fibril strain transmissible to wild-type α-synuclein. Nat Commun. 2021;12(1):6252. [Google Scholar] [PubMed]
13. Dorszewska J, Kowalska M, Prendecki M, Piekut T, Kozłowska J, Kozubski W. Oxidative stress factors in Parkinson’s disease. Neural Regen Res. 2021;16(7):1383–91. [Google Scholar] [PubMed]
14. Cao Q, Luo S, Yao W, Qu Y, Wang N, Hong J, et al. Suppression of abnormal α-synuclein expression by activation of BDNF transcription ameliorates Parkinson’s disease-like pathology. Mol Ther Nucleic Acids. 2022;29:1–15. [Google Scholar] [PubMed]
15. Teng JS, Ooi YY, Chye SM, Ling APK, Koh RY. Immunotherapies for Parkinson’s disease: progression of clinical development. CNS Neurol Disord Drug Targets. 2021;20(9):802–13. [Google Scholar] [PubMed]
16. Morris HR, Spillantini MG, Sue CM, Williams-Gray CH. The pathogenesis of Parkinson’s disease. Lancet. 2024;403(10423):293–304. [Google Scholar] [PubMed]
17. Cesari R, Martin ES, Calin GA, Pentimalli F, Bichi R, McAdams H, et al. Parkin, a gene implicated in autosomal recessive juvenile Parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc Natl Acad Sci USA. 2003;100(10):5956–61. [Google Scholar] [PubMed]
18. Fakih R, Sauvé V, Gehring K. Structure of the second phosphoubiquitin-binding site in parkin. J Biol Chem. 2022;298(7):102114. [Google Scholar] [PubMed]
19. Trinh J, Hicks AA, König IR, Delcambre S, Lüth T, Schaake S, et al. Mitochondrial DNA heteroplasmy distinguishes disease manifestation in PINK1/PRKN-linked Parkinson’s disease. Brain. 2023;146(7):2753–65. [Google Scholar] [PubMed]
20. Grossmann D, Malburg N, Glaß H, Weeren V, Sondermann V, Pfeiffer JF, et al. Mitochondria-endoplasmic reticulum contact sites dynamics and calcium homeostasis are differentially disrupted in PINK1-PD or PRKN-PD neurons. Mov Disord. 2023;38(10):1822–36. [Google Scholar] [PubMed]
21. Chakrabarti S, Bisaglia M. Oxidative stress and neuroinflammation in Parkinson’s disease: the role of dopamine oxidation products. Antioxidants. 2023;12(4):955. [Google Scholar] [PubMed]
22. Veeriah S, Morris L, Solit D, Chan TA. The familial Parkinson disease gene PARK2 is a multisite tumor suppressor on chromosome 6q25.2-27 that regulates cyclin E. Cell Cycle. 2010;9(8):1451–2. [Google Scholar] [PubMed]
23. Le Guerroué F, Youle RJ. Ubiquitin signaling in neurodegenerative diseases: an autophagy and proteasome perspective. Cell Death Differ. 2021;28(2):439–54. [Google Scholar] [PubMed]
24. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nat. 2015;524(7565):309–14. [Google Scholar]
25. Gasser T, Müller-Myhsok B, Wszolek ZK, Oehlmann R, Calne DB, Bonifati V, et al. A susceptibility locus for Parkinson’s disease maps to chromosome 2p13. Nat Genet. 1998;18(3):262–5. [Google Scholar] [PubMed]
26. Sharma M, Mueller JC, Zimprich A, Lichtner P, Hofer A, Leitner P, et al. The sepiapterin reductase gene region reveals association in the PARK3 locus: analysis of familial and sporadic Parkinson’s disease in European populations. J Med Genet. 2006;43(7):557–62. [Google Scholar] [PubMed]
27. Fukusumi H, Togo K, Sumida M, Nakamori M, Obika S, Baba K, et al. Alpha-synuclein dynamics in induced pluripotent stem cell-derived dopaminergic neurons from a Parkinson’s disease patient (PARK4) with SNCA triplication. FEBS Open Bio. 2021;11(2):354–66. [Google Scholar] [PubMed]
28. Day IN, Thompson RJ. UCHL1 (PGP 9.5neuronal biomarker and ubiquitin system protein. Prog Neurobiol. 2010;90(3):327–62. [Google Scholar] [PubMed]
29. Lee YC, Hsu SD. Familial mutations and post-translational modifications of UCH-L1 in Parkinson’s disease and neurodegenerative disorders. Curr Protein Pept Sci. 2017;18(7):733–45. [Google Scholar] [PubMed]
30. Liu Y, Chen YY, Liu H, Yao CJ, Zhu XX, Chen DJ, et al. Association between ubiquitin carboxy-terminal hydrolase-L1 S18Y variant and risk of Parkinson’s disease: the impact of ethnicity and onset age. Neurol Sci. 2015;36(2):179–88. [Google Scholar] [PubMed]
31. Pukaß K, Richter-Landsberg C. Inhibition of UCH-L1 in oligodendroglial cells results in microtubule stabilization and prevents α-synuclein aggregate formation by activating the autophagic pathway: implications for multiple system atrophy. Front Cell Neurosci. 2015;9:163. [Google Scholar]
32. Zhang M, Chen MY, Wang SL, Ding XM, Yang R, Li J, et al. Association of ubiquitin C-terminal hydrolase-L1 (Uch-L1) serum levels with cognition and brain energy metabolism. Eur Rev Med Pharmacol Sci. 2022;26(10):3656–63. [Google Scholar] [PubMed]
33. Wang KK, Yang Z, Sarkis G, Torres I, Raghavan V. Ubiquitin C-terminal hydrolase-L1 (UCH-L1) as a therapeutic and diagnostic target in neurodegeneration, neurotrauma and neuro-injuries. Expert Opin Ther Targets. 2017;21(6):627–38. [Google Scholar] [PubMed]
34. Guo Y, Jiang H, Wang M, Ma Y, Zhang J, Jing L. Metformin alleviates cerebral ischemia/reperfusion injury aggravated by hyperglycemia via regulating AMPK/ULK1/PINK1/Parkin pathway-mediated mitophagy and apoptosis. Chem Biol Interact. 2023;384:110723. [Google Scholar] [PubMed]
35. Li J, Yang D, Li Z, Zhao M, Wang D, Sun Z, et al. PINK1/Parkin-mediated mitophagy in neurodegenerative diseases. Ageing Res Rev. 2023;84:101817. [Google Scholar] [PubMed]
36. Han R, Liu Y, Li S, Li XJ, Yang W. PINK1-PRKN mediated mitophagy: differences between in vitro and in vivo models. Autophagy. 2023;19(5):1396–405. [Google Scholar] [PubMed]
37. Yang W, Li S, Li XJ. A CRISPR monkey model unravels a unique function of PINK1 in primate brains. Mol Neurodegener. 2019;14(1):17. [Google Scholar] [PubMed]
38. Quinn PMJ, Moreira PI, Ambrósio AF, Alves CH. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol Commun. 2020;8(1):189. [Google Scholar] [PubMed]
39. Han H, Tan J, Wang R, Wan H, He Y, Yan X, et al. PINK1 phosphorylates Drp1S616 to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020;21(8):e48686. [Google Scholar] [PubMed]
40. Schweitzer JS, Song B, Herrington TM, Park TY, Lee N, Ko S, et al. Personalized iPSC-derived dopamine progenitor cells for Parkinson’s disease. N Engl J Med. 2020;382(20):1926–32. [Google Scholar] [PubMed]
41. Nagakubo D, Taira T, Kitaura H, Ikeda M, Tamai K, Iguchi-Ariga SM, et al. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem Biophys Res Commun. 1997;231(2):509–13. [Google Scholar] [PubMed]
42. Heremans IP, Caligiore F, Gerin I, Bury M, Lutz M, Graff J, et al. Parkinson’s disease protein PARK7 prevents metabolite and protein damage caused by a glycolytic metabolite. Proc Natl Acad Sci USA. 2022;119(4):e2111338119. [Google Scholar] [PubMed]
43. Hijioka M, Inden M, Yanagisawa D, Kitamura Y. DJ-1/PARK7: a new therapeutic target for neurodegenerative disorders. Biol Pharm Bull. 2017;40(5):548–52. [Google Scholar] [PubMed]
44. Imberechts D, Kinnart I, Wauters F, Terbeek J, Manders L, Wierda K, et al. DJ-1 is an essential downstream mediator in PINK1/parkin-dependent mitophagy. Brain. 2022;145(12):4368–84. [Google Scholar] [PubMed]
45. Boussaad I, Obermaier CD, Hanss Z, Bobbili DR, Bolognin S, Glaab E, et al. A patient-based model of RNA mis-splicing uncovers treatment targets in Parkinson’s disease. Sci Transl Med. 2020;12(560):eaau3960. [Google Scholar] [PubMed]
46. Pathak P, Alexander KK, Helton LG, Kentros M, LeClair TJ, Zhang X, et al. Doubly constrained C-terminal of roc (COR) domain-derived peptides inhibit leucine-rich repeat kinase 2 (LRRK2) dimerization. ACS Chem Neurosci. 2023;14(11):1971–80. [Google Scholar] [PubMed]
47. Quintero-Espinosa DA, Velez-Pardo C, Jimenez-Del-Rio M. High yield of functional dopamine-like neurons obtained in NeuroForsk 2.0 medium to study acute and chronic rotenone effects on oxidative stress, autophagy, and apoptosis. Int J Mol Sci. 2023;24(21):15744. [Google Scholar] [PubMed]
48. Simpson C, Vinikoor-Imler L, Nassan FL, Shirvan J, Lally C, Dam T, et al. Prevalence of ten LRRK2 variants in Parkinson’s disease: a comprehensive review. Parkinsonism Relat Disord. 2022;98:103–13. [Google Scholar] [PubMed]
49. Zhou ZD, Saw WT, Ho PGH, Zhang ZW, Zeng L, Chang YY, et al. The role of tyrosine hydroxylase-dopamine pathway in Parkinson’s disease pathogenesis. Cell Mol Life Sci. 2022;79(12):599. [Google Scholar] [PubMed]
50. Siddique MM, Tan EK. Neurochemistry changes associated with mutations in familial Parkinson’s disease. Curr Med Chem. 2010;17(35):4378–91. [Google Scholar] [PubMed]
51. Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St Lawrence E, et al. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell. 2016;19(6):709–24. [Google Scholar] [PubMed]
52. Ding X, Ren F. Leucine-rich repeat kinase 2 inhibitors: a patent review (2014–present). Expert Opin Ther Pat. 2020;30(4):275–86. [Google Scholar] [PubMed]
53. van Veen S, Martin S, van den Haute C, Benoy V, Lyons J, Vanhoutte R, et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature. 2020;578(7795):419–24. [Google Scholar] [PubMed]
54. Podhajska A, Musso A, Trancikova A, Stafa K, Moser R, Sonnay S, et al. Common pathogenic effects of missense mutations in the P-type ATPase ATP13A2 (PARK9) associated with early-onset parkinsonism. PLoS One. 2012;7(6):e39942. [Google Scholar] [PubMed]
55. Wan JY, Edwards KL, Hutter CM, Mata IF, Samii A, Roberts JW, et al. Association mapping of the PARK10 region for Parkinson’s disease susceptibility genes. Parkinsonism Relat Disord. 2014;20(1):93–8. [Google Scholar] [PubMed]
56. Anderson LR, Betarbet R, Gearing M, Gulcher J, Hicks AA, Stefánsson K, et al. PARK10 candidate RNF11 is expressed by vulnerable neurons and localizes to lewy bodies in Parkinson disease brain. J Neuropathol Exp Neurol. 2007;66(10):955–64. [Google Scholar] [PubMed]
57. Thayer JA, Awad O, Hegdekar N, Sarkar C, Tesfay H, Burt C, et al. The PARK10 gene USP24 is a negative regulator of autophagy and ULK1 protein stability. Autophagy. 2020;16(1):140–53. [Google Scholar] [PubMed]
58. Bras J, Simón-Sánchez J, Federoff M, Morgadinho A, Januario C, Ribeiro M, et al. Lack of replication of association between GIGYF2 variants and Parkinson disease. Hum Mol Genet. 2009;18(2):341–6. [Google Scholar] [PubMed]
59. Zheng R, Jin CY, Chen Y, Ruan Y, Gao T, Lin ZH, et al. Analysis of rare variants of autosomal-dominant genes in a Chinese population with sporadic Parkinson’s disease. Mol Genet Genomic Med. 2020;8(10):e1449. [Google Scholar] [PubMed]
60. Bonetti M, Ferraris A, Petracca M, Bentivoglio AR, Dallapiccola B, Valente EM. GIGYF2 variants are not associated with Parkinson’s disease in Italy. Mov Disord. 2009;24(12):1868–9. [Google Scholar]
61. Zimprich A, Schulte C, Reinthaler E, Haubenberger D, Balzar J, Lichtner P, et al. PARK11 gene (GIGYF2) variants Asn56Ser and Asn457Thr are not pathogenic for Parkinson’s disease. Parkinsonism Relat Disord. 2009;15(7):532–4. [Google Scholar] [PubMed]
62. Saini P, Rudakou U, Yu E, Ruskey JA, Asayesh F, Laurent SB, et al. Association study of DNAJC13, UCHL1, HTRA2, GIGYF2, and EIF4G1 with Parkinson’s disease. Neurobiol Aging. 2021;100(3):119.e7–e13. [Google Scholar] [PubMed]
63. Xie J, Wei Q, Deng H, Li G, Ma L, Zeng H. Negative regulation of Grb10 interacting GYF Protein 2 on insulin-like growth factor-1 receptor signaling pathway caused diabetic mice cognitive impairment. PLoS One. 2014;9(9):e108559. [Google Scholar] [PubMed]
64. Sousa L, Guarda M, Meneses MJ, Macedo MP, Vicente Miranda H. Insulin-degrading enzyme: an ally against metabolic and neurodegenerative diseases. J Pathol. 2021;255(4):346–61. [Google Scholar] [PubMed]
65. Mori S, Maher P, Conti B. Neuroimmunology of the interleukins 13 and 4. Brain Sci. 2016;6(2):18. [Google Scholar] [PubMed]
66. Sakaue S, Kasai T, Mizuta I, Suematsu M, Osone S, Azuma Y, et al. Early-onset parkinsonism in a pedigree with phosphoglycerate kinase deficiency and a heterozygous carrier: do PGK-1 mutations contribute to vulnerability to parkinsonism? NPJ Parkinsons Dis. 2017;3:13. [Google Scholar] [PubMed]
67. Carvalho NZM, Chiarotto GB, Bernardes D, Kempe PRG, Oliveira ALR. Neuroprotection by dimethyl fumarate following ventral root crush in C57BL/6J mice. Brain Res Bull. 2020;164:184–97. [Google Scholar] [PubMed]
68. Patil KS, Basak I, Lee S, Abdullah R, Larsen JP, Møller SG. PARK13 regulates PINK1 and subcellular relocation patterns under oxidative stress in neurons. J Neurosci Res. 2014;92(9):1167–77. [Google Scholar] [PubMed]
69. Chung HJ, Jamal MAHM, Hong ST. The function of bacterial HtrA is evolutionally conserved in mammalian HtrA2/Omi. Sci Rep. 2020;10(1):5284. [Google Scholar] [PubMed]
70. Chen C, McDonald D, Blain A, Mossman E, Atkin K, Marusich MF, et al. Parkinson’s disease neurons exhibit alterations in mitochondrial quality control proteins. NPJ Parkinsons Dis. 2023;9(1):120. [Google Scholar] [PubMed]
71. Wong HY, Langlotz M, Gan-Schreier H, Xu W, Staffer S, Tuma-Kellner S, et al. Constitutive oxidants from hepatocytes of male iPLA2β-null mice increases the externalization of phosphatidylethanolamine on plasma membrane. Free Radic Res. 2021;55(6):625–33. [Google Scholar] [PubMed]
72. Nadalin S, Zatković L, Peitl V, Karlović D, Vidrih B, Puljić A, et al. Association between PLA2 gene polymorphisms and treatment response to antipsychotic medications: a study of antipsychotic-naïve first-episode psychosis patients and nonadherent chronic psychosis patients. Prostaglandins Leukot Essent Fat Acids. 2023;194:102578. [Google Scholar]
73. Sumi-Akamaru H, Beck G, Kato S, Mochizuki H. Neuroaxonal dystrophy in PLA2G6 knockout mice. Neuropathol. 2015;35(3):289–302. [Google Scholar]
74. Beck G, Sugiura Y, Shinzawa K, Kato S, Setou M, Tsujimoto Y, et al. Neuroaxonal dystrophy in calcium-independent phospholipase A2β deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J Neurosci. 2011;31(31):11411–20. [Google Scholar] [PubMed]
75. Wang J, Joseph S, Vingill S, Dere E, Tatenhorst L, Ronnenberg A, et al. Loss of the parkinsonism-associated protein FBXO7 in glutamatergic forebrain neurons in mice leads to abnormal motor behavior and synaptic defects. J Neurochem. 2023;167(2):296–317. [Google Scholar] [PubMed]
76. Kraus F, Goodall EA, Smith IR, Jiang Y, Paoli JC, Adolf F, et al. PARK15/FBXO7 is dispensable for PINK1/Parkin mitophagy in iNeurons and HeLa cell systems. EMBO Rep. 2023;24(8):e56399. [Google Scholar] [PubMed]
77. Stott SR, Randle SJ, Al Rawi S, Rowicka PA, Harris R, Mason B, et al. Loss of FBXO7 results in a Parkinson’s-like dopaminergic degeneration via an RPL23-MDM2-TP53 pathway. J Pathol. 2019;249(2):241–54. doi:10.1002/path.5312. [Google Scholar] [PubMed] [CrossRef]
78. Cui HG, Tian XF, Luo XG, Li FR, Zhu LH, Zhou YS, et al. Association of five SNPs at the PARK16 locus as a susceptibility locus with Parkinson’s disease for forensic application. Fa Yi Xue Za Zhi. 2013;29(3):185–9. [Google Scholar] [PubMed]
79. Tan X, Wei Y, Cao J, Wu D, Lai N, Deng R, et al. RAB7L1 participates in secondary brain injury induced by experimental intracerebral hemorrhage in rats. J Mol Neurosci. 2021;71(1):9–18. [Google Scholar] [PubMed]
80. Sen A, Kallabis S, Gaedke F, Jüngst C, Boix J, Nüchel J, et al. Mitochondrial membrane proteins and VPS35 orchestrate selective removal of mtDNA. Nat Commun. 2022;13(1):6704. [Google Scholar] [PubMed]
81. Wang W, Wang X, Fujioka H, Hoppel C, Whone AL, Caldwell MA, et al. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat Med. 2016;22(1):54–63. [Google Scholar] [PubMed]
82. Niu M, Zhao F, Bondelid K, Siedlak SL, Torres S, Fujioka H, et al. VPS35 D620N knockin mice recapitulate cardinal features of Parkinson’s disease. Aging Cell. 2021;20(5):e13347. [Google Scholar] [PubMed]
83. Jiang M, Tu HT, Zhang K, Zhang W, Yu WP, Xu J, et al. Impaired neurogenesis in the hippocampus of an adult VPS35 mutant mouse model of Parkinson’s disease through interaction with APP. Neurobiol Dis. 2021;153:105313. [Google Scholar] [PubMed]
84. Tang FL, Erion JR, Tian Y, Liu W, Yin DM, Ye J, et al. VPS35 in dopamine neurons is required for endosome-to-golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for α-synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J Neurosci. 2015;35(29):10613–28. [Google Scholar] [PubMed]
85. Sassone J, Reale C, Dati G, Regoni M, Pellecchia MT, Garavaglia B. The role of VPS35 in the pathobiology of Parkinson’s disease. Cell Mol Neurobiol. 2021;41(2):199–227. doi:10.1007/s10571-020-00849-8. [Google Scholar] [PubMed] [CrossRef]
86. Dhungel N, Eleuteri S, Li LB, Kramer NJ, Chartron JW, Spencer B, et al. Parkinson’s disease genes VPS35 and EIF4G1 interact genetically and converge on α-synuclein. Neuron. 2023;111(1):138. [Google Scholar] [PubMed]
87. Li C, Ou R, Chen Y, Gu X, Wei Q, Cao B, et al. Mutation analysis of DNAJC family for early-onset Parkinson’s disease in a Chinese cohort. Mov Disord. 2020;35(11):2068–76. [Google Scholar] [PubMed]
88. Wulansari N, Darsono WHW, Woo HJ, Chang MY, Kim J, Bae EJ, et al. Neurodevelopmental defects and neurodegenerative phenotypes in human brain organoids carrying Parkinson’s disease-linked DNAJC6 mutations. Sci Adv. 2021;7(8):eabb1540. [Google Scholar] [PubMed]
89. Cao M, Wu Y, Ashrafi G, McCartney AJ, Wheeler H, Bushong EA, et al. Parkinson Sac domain mutation in synaptojanin 1 impairs clathrin uncoating at synapses and triggers dystrophic changes in dopaminergic axons. Neuron. 2017;93(4):882–96.e5. [Google Scholar] [PubMed]
90. Appel-Cresswell S, Rajput AH, Sossi V, Thompson C, Silva V, McKenzie J, et al. Clinical, positron emission tomography, and pathological studies of DNAJC13 p.N855S Parkinsonism. Mov Disord. 2014;29(13):1684–7. [Google Scholar] [PubMed]
91. Vilariño-Güell C, Rajput A, Milnerwood AJ, Shah B, Szu-Tu C, Trinh J, et al. DNAJC13 mutations in Parkinson disease. Hum Mol Genet. 2014;23(7):1794–801. [Google Scholar]
92. Ren Y, Wang X, Guo J, Wang D, Li X, Cheng X, et al. CHCHD2 regulates mitochondrial function and apoptosis of ectopic endometrial stromal cells in the pathogenesis of endometriosis. Reprod Sci. 2022;29(8):2152–64. [Google Scholar] [PubMed]
93. Liu Y, Clegg HV, Leslie PL, Di J, Tollini LA, He Y, et al. CHCHD2 inhibits apoptosis by interacting with Bcl-x L to regulate bax activation. Cell Death Differ. 2015;22(6):1035–46. [Google Scholar] [PubMed]
94. Ruan Y, Hu J, Che Y, Liu Y, Luo Z, Cheng J, et al. CHCHD2 and CHCHD10 regulate mitochondrial dynamics and integrated stress response. Cell Death Dis. 2022;13(2):156. [Google Scholar] [PubMed]
95. Sato S, Noda S, Torii S, Amo T, Ikeda A, Funayama M, et al. Homeostatic p62 levels and inclusion body formation in CHCHD2 knockout mice. Hum Mol Genet. 2021;30(6):443–53. [Google Scholar] [PubMed]
96. Jiang T, Wang Y, Wang X, Xu J. CHCHD2 and CHCHD10: future therapeutic targets in cognitive disorder and motor neuron disorder. Front Neurosci. 2022;16:988265. [Google Scholar] [PubMed]
97. Dall’Armellina F, Stagi M, Swan LE. In silico modeling human VPS13 proteins associated with donor and target membranes suggests lipid transfer mechanisms. Proteins. 2023;91(4):439–55. [Google Scholar] [PubMed]
98. Li W, Fu Y, Halliday GM, Sue CM. PARK genes link mitochondrial dysfunction and alpha-synuclein pathology in sporadic Parkinson’s disease. Front Cell Dev Biol. 2021;9:612476. [Google Scholar] [PubMed]
99. Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/parkin-dependent mitophagy. Am J Hum Genet. 2016;98(3):500–13. [Google Scholar] [PubMed]
100. Kumar N, Leonzino M, Hancock-Cerutti W, Horenkamp FA, Li P, Lees JA, et al. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J Cell Biol. 2018;217(10):3625–39. [Google Scholar] [PubMed]
101. Hancock-Cerutti W, Wu Z, Xu P, Yadavalli N, Leonzino M, Tharkeshwar AK, et al. ER-lysosome lipid transfer protein VPS13C/PARK23 prevents aberrant mtDNA-dependent STING signaling. J Cell Biol. 2022;221(7):e202106046. [Google Scholar] [PubMed]
102. Oji Y, Hatano T, Ueno SI, Funayama M, Ishikawa KI, Okuzumi A, et al. Variants in saposin D domain of prosaposin gene linked to Parkinson’s disease. Brain. 2020;143(4):1190–205. [Google Scholar] [PubMed]
103. Nicholson AM, Finch NA, Almeida M, Perkerson RB, van Blitterswijk M, Wojtas A, et al. Prosaposin is a regulator of progranulin levels and oligomerization. Nat Commun. 2016;7:11992. [Google Scholar] [PubMed]
104. Ruz C, Barrero FJ, Pelegrina J, Bandrés-Ciga S, Vives F, Duran R, et al. Key regulator in the alpha-synuclein degradation mediated by lysosome. Int J Mol Sci. 2022;23(19):12004. [Google Scholar] [PubMed]
105. Smith L, Schapira AHV. GBA variants and Parkinson disease: mechanisms and treatments. Cells. 2022;11(8):1261. [Google Scholar] [PubMed]
106. Kinghorn KJ, Asghari AM, Castillo-Quan JI. The emerging role of autophagic-lysosomal dysfunction in Gaucher disease and Parkinson’s disease. Neural Regen Res. 2017;12(3):380–4. [Google Scholar] [PubMed]
107. Sosero YL, Yu E, Estiar MA, Krohn L, Mufti K, Rudakou U, et al. Rare PSAP variants and possible interaction with GBA in REM sleep behavior disorder. J Parkinsons Dis. 2022;12(1):333–40. [Google Scholar] [PubMed]
108. Larbalestier H, Keatinge M, Watson L, White E, Gowda S, Wei W, et al. GCH1 deficiency activates brain innate immune response and impairs tyrosine hydroxylase homeostasis. J Neurosci. 2022;42(4):702–16. [Google Scholar] [PubMed]
109. Van Greenen JD, Hockman D. FGF20. Differ. 2023;7:100737. [Google Scholar]
110. Zhang Z, Yu J. Nurr1 exacerbates cerebral ischemia-reperfusion injury via modulating YAP-INF2-mitochondrial fission pathways. Int J Biochem Cell Biol. 2018;104:149–60. [Google Scholar] [PubMed]
111. Chen S, Luo GR, Li T, Liu TX, Le W. Correlation of Nr4a2 expression with the neuron progenitors in adult zebrafish brain. J Mol Neurosci. 2013;51(3):719–23. [Google Scholar] [PubMed]
112. Taraskina AE, Grunina MN, Zabotina AM, Nasyrova RF, Ivanov MV, Krupitsky EM, et al. The key proteins of dopaminergic neurotransmission of human peripheral blood lymphocytes: changed mRNA level in alcohol dependence syndrome. Bull Exp Biol Med. 2015;160(2):271–4. [Google Scholar] [PubMed]
113. Jakaria M, Haque ME, Cho DY, Azam S, Kim IS, Choi DK. Molecular insights into NR4A2(Nurr1an emerging target for neuroprotective therapy against neuroinflammation and neuronal cell death. Mol Neurobiol. 2019;56(8):5799–814. [Google Scholar] [PubMed]
114. Al-Nusaif M, Yang Y, Li S, Cheng C, Le W. The role of NURR1 in metabolic abnormalities of Parkinson’s disease. Mol Neurodegener. 2022;17(1):46. [Google Scholar] [PubMed]
115. Xue J, Zhu Y, Wei L, Huang H, Li G, Huang W, et al. Loss of drosophila NUS1 results in cholesterol accumulation and Parkinson’s disease-related neurodegeneration. FASEB J. 2022;36(7):e22411. [Google Scholar] [PubMed]
116. Ma J, Zeng P, Liu L, Zhu M, Zheng J, Wang C, et al. Peroxisome proliferator-activated receptor-gamma reduces ER stress and inflammation via targeting NGBR expression. Front Pharmacol. 2022;12:817784. [Google Scholar] [PubMed]
117. Liu Z, Yang N, Dong J, Tian W, Chang L, Ma J, et al. Deficiency in endocannabinoid synthase DAGLB contributes to early onset Parkinsonism and murine nigral dopaminergic neuron dysfunction. Nat Commun. 2022;13(1):3490. [Google Scholar] [PubMed]
118. Hu M, Li P, Wang C, Feng X, Geng Q, Chen W, et al. Parkinson’s disease-risk protein TMEM175 is a proton-activated proton channel in lysosomes. Cell. 2022;185(13):2292–308.e20. [Google Scholar] [PubMed]
119. Wie J, Liu Z, Song H, Tropea TF, Yang L, Wang H, et al. A growth-factor-activated lysosomal K+ channel regulates Parkinson’s pathology. Cah Rev The. 2021;591(7850):431–7. [Google Scholar]
120. Kim MJ, Deng HX, Wong YC, Siddique T, Krainc D. The Parkinson’s disease-linked protein TMEM230 is required for Rab8a-mediated secretory vesicle trafficking and retromer trafficking. Hum Mol Genet. 2017;26(4):729–41. [Google Scholar] [PubMed]
121. Shan W, Li J, Xu W, Li H, Zuo Z. Critical role of UQCRC1 in embryo survival, brain ischemic tolerance and normal cognition in mice. Cell Mol Life Sci. 2019;76(7):1381–96. [Google Scholar] [PubMed]
122. Hung YC, Huang KL, Chen PL, Li JL, Lu SH, Chang JC, et al. UQCRC1 engages cytochrome c for neuronal apoptotic cell death. Cell Rep. 2021;36(12):109729. [Google Scholar] [PubMed]
123. Lin CH, Tsai PI, Lin HY, Hattori N, Funayama M, Jeon B, et al. Mitochondrial UQCRC1 mutations cause autosomal dominant parkinsonism with polyneuropathy. Brain. 2020;143(11):3352–73. [Google Scholar] [PubMed]
124. Diaz-Ortiz ME, Seo Y, Posavi M, Carceles Cordon M, Clark E, Jain N, et al. GPNMB confers risk for Parkinson’s disease through interaction with α-synuclein. Sci. 2022;377(6608):eabk0637. [Google Scholar]
125. Neal ML, Boyle AM, Budge KM, Safadi FF, Richardson JR. The glycoprotein GPNMB attenuates astrocyte inflammatory responses through the CD44 receptor. J Neuroinflammation. 2018;15(1):73. [Google Scholar] [PubMed]
126. Dulski J, Konno T, Wszolek Z. DCTN1-related neurodegeneration. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al., editors. GeneReviews®. University of Washington: Seattle (WASeattle; 2010. [Google Scholar]
127. Morcillo P, Cordero H, Ijomone OM, Ayodele A, Bornhorst J, Gunther L, et al. Defective mitochondrial dynamics underlie manganese-induced neurotoxicity. Mol Neurobiol. 2021;58(7):3270–89. [Google Scholar] [PubMed]
128. Grochowska MM, Carreras Mascaro A, Boumeester V, Natale D, Breedveld GJ, Geut H, et al. LRP10 interacts with SORL1 in the intracellular vesicle trafficking pathway in non-neuronal brain cells and localises to lewy bodies in Parkinson’s disease and dementia with lewy bodies. Acta Neuropathol. 2021;142(1):117–37. [Google Scholar] [PubMed]
129. Manini A, Straniero L, Monfrini E, Percetti M, Vizziello M, Franco G, et al. Screening of LRP10 mutations in Parkinson’s disease patients from Italy. Parkinsonism Relat Disord. 2021;89:17–21. [Google Scholar] [PubMed]
130. Naoi M, Riederer P, Maruyama W. Modulation of monoamine oxidase (MAO) expression in neuropsychiatric disorders: genetic and environmental factors involved in type A MAO expression. J Neural Transm. 2016;123(2):91–106. [Google Scholar] [PubMed]
131. Tong J, Meyer JH, Furukawa Y, Boileau I, Chang LJ, Wilson AA, et al. Distribution of monoamine oxidase proteins in human brain: implications for brain imaging studies. J Cereb Blood Flow Metab. 2013;33(6):863–71. [Google Scholar] [PubMed]
132. Mentis AA, Dardiotis E, Katsouni E, Chrousos GP. From warrior genes to translational solutions: novel insights into monoamine oxidases (MAOs) and aggression. Transl Psychiatry. 2021;11(1):130. [Google Scholar] [PubMed]
133. Tan YY, Jenner P, Chen SD. Monoamine oxidase-B inhibitors for the treatment of Parkinson’s disease: past, present, and future. J Parkinsons Dis. 2022;12(2):477–93. [Google Scholar] [PubMed]
134. Casse F, Courtin T, Tesson C, Ferrien M, Noël S, Fauret-Amsellem AL, et al. Detection of ATXN2 expansions in an exome dataset: an underdiagnosed cause of parkinsonism. Mov Disord Clin Pract. 2023;10(4):664–9. [Google Scholar] [PubMed]
135. Habekost M, Qvist P, Denham M, Holm IE, Jørgensen AL. Directly reprogrammed neurons express MAPT and APP splice variants pertinent to ageing and neurodegeneration. Mol Neurobiol. 2021;58(5):2075–87. [Google Scholar] [PubMed]
136. Munshi MI, Yao SJ, Ben Mamoun C. Redesigning therapies for pantothenate kinase-associated neurodegeneration. J Biol Chem. 2022;298(3):101577. [Google Scholar] [PubMed]
137. Huang Y, Wan Z, Tang Y, Xu J, Laboret B, Nallamothu S, et al. Pantothenate kinase 2 interacts with PINK1 to regulate mitochondrial quality control via acetyl-CoA metabolism. Nat Commun. 2022;13(1):2412. [Google Scholar] [PubMed]
138. Mori S, Honda H, Hamasaki H, Sasagasako N, Suzuki SO, Furuya H, et al. Transactivation response DNA-binding protein of 43 kDa proteinopathy and lysosomal abnormalities in spastic paraplegia type 11. Neuropathol. 2021;41(4):253–65. [Google Scholar]
139. Faber I, Martinez ARM, Martins CRJr, Maia ML, Souza JP, Lourenço CM, et al. SPG11-related parkinsonism: clinical profile, molecular imaging and l-dopa response. Mov Disord. 2018;33(10):1650–6. [Google Scholar] [PubMed]
140. Renvoisé B, Chang J, Singh R, Yonekawa S, FitzGibbon EJ, Mankodi A, et al. Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann Clin Transl Neurol. 2014;1(6):379–89. [Google Scholar] [PubMed]
141. Hsieh PC, Wang CC, Tsai CL, Yeh YM, Lee YS, Wu YR. POLG R964C and GBA L444P mutations in familial Parkinson’s disease: case report and literature review. Brain Behav. 2019;9(5):e01281. [Google Scholar] [PubMed]
142. Antonova SV, Boeren J, Timmers HTM, Snel B. Epigenetics and transcription regulation during eukaryotic diversification: the saga of TFIID. Genes Dev. 2019;33(15–16):888–902. [Google Scholar] [PubMed]
143. Al Ali J, Vaine CA, Shah S, Campion L, Hakoum A, Supnet ML, et al. TAF1 transcripts and neurofilament light chain as biomarkers for X-linked dystonia-parkinsonism. Mov Disord. 2021;36(1):206–15. [Google Scholar] [PubMed]
144. Demuro S, di Martino RMC, Ortega JA, Cavalli A. FYN, and DYRK1A: master regulators in neurodegenerative pathways. Int J Mol Sci. 2021;22(16):9098. [Google Scholar] [PubMed]
145. Jia ZK, Fu CX, Wang AL, Yao K, Chen XJ. Cataract-causing allele in CRYAA (Y118D) proceeds through endoplasmic reticulum stress in mouse model. Zool Res. 2021;42(3):300–9. [Google Scholar] [PubMed]
146. Armstrong MJ, Okun MS. Diagnosis and treatment of Parkinson disease: a review. JAMA. 2020;323(6):548–60. [Google Scholar] [PubMed]
147. Wang F, He L, Zhao H, Guo X. Efficacy of deep brain stimulation for camptocormia in Parkinson’s disease: a systematic review and meta-analysis. J Integr Neurosci. 2023;22(1):11. [Google Scholar] [PubMed]
148. Xiang C, Cong S, Tan X, Ma S, Liu Y, Wang H, et al. A meta-analysis of the diagnostic utility of biomarkers in cerebrospinal fluid in Parkinson’s disease. NPJ Parkinsons Dis. 2022;8(1):165. [Google Scholar] [PubMed]
149. Sarparast M, Dattmore D, Alan J, Lee KSS. Cytochrome P450 metabolism of polyunsaturated fatty acids and neurodegeneration. Nutr. 2020;12(11):3523. [Google Scholar]
150. Rauchová H. Coenzyme Q10 effects in neurological diseases. Physiol Res. 2021;70(Suppl4):S683–714. [Google Scholar] [PubMed]
151. Selvakumar SC, Preethi KA, Tusubira D, Sekar D. MicroRNAs in the epigenetic regulation of disease progression in Parkinson’s disease. Front Cell Neurosci. 2022;16:995997. [Google Scholar] [PubMed]
152. Pinnell JR, Cui M, Tieu K. Exosomes in Parkinson disease. J Neurochem. 2021;157(3):413–28. [Google Scholar] [PubMed]
153. Joo HS, Jeon HY, Hong EB, Kim HY, Lee JM. Exosomes for the diagnosis and treatment of dementia. Curr Opin Psychiatry. 2023;36(2):119–25. [Google Scholar] [PubMed]
154. Zimmermann M, Brockmann K. Blood and cerebrospinal fluid biomarkers of inflammation in Parkinson’s disease. J Parkinsons Dis. 2022;12(s1):S183–200. [Google Scholar] [PubMed]
155. Paccosi E, Proietti-De-Santis L. Parkinson’s disease: from genetics and epigenetics to treatment, a miRNA-based strategy. Int J Mol Sci. 2023;24(11):9547. [Google Scholar] [PubMed]
156. Xu E, Boddu R, Abdelmotilib HA, Sokratian A, Kelly K, Liu Z, et al. Pathological α-synuclein recruits LRRK2 expressing pro-inflammatory monocytes to the brain. Mol Neurodegener. 2022;17(1):7. [Google Scholar] [PubMed]
157. Panicker N, Ge P, Dawson VL, Dawson TM. The cell biology of Parkinson’s disease. J Cell Biol. 2021;220(4):e202012095. [Google Scholar] [PubMed]
158. Galvagnion C, Marlet FR, Cerri S, Schapira AHV, Blandini F, di Monte DA. Sphingolipid changes in Parkinson L444P GBA mutation fibroblasts promote α-synuclein aggregation. Brain. 2022;145(3):1038–51. [Google Scholar] [PubMed]
159. Li JL, Lin TY, Chen PL, Guo TN, Huang SY, Chen CH, et al. Mitochondrial function and Parkinson’s disease: from the perspective of the electron transport chain. Front Mol Neurosci. 2021;14:797833. [Google Scholar] [PubMed]
160. Cardona F, Perez-Tur J. Other proteins involved in Parkinson’s disease and related disorders. Curr Protein Pept Sci. 2017;18(7):765–78. [Google Scholar] [PubMed]
161. Funayama M, Nishioka K, Li Y, Hattori N. Molecular genetics of Parkinson’s disease: contributions and global trends. J Hum Genet. 2023;68(3):125–30. [Google Scholar] [PubMed]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools