
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

MAD2L2 overexpression attenuates the effects of TNF-α-induced migration and invasion capabilities in colorectal cancer cells
1 School of Basic Medicine, Medical Genetics and Cell Biology, Ningxia Medical University, Yinchuan, 750004, China
2 Key Laboratory of Reproduction and Genetics, Ningxia Medical University, Yinchuan, 750004, China
* Corresponding Author: FANG XU. Email:
# These authors contributed equally to this work
(This article belongs to the Special Issue: New Perspectives on Inflammatory Cancer Transformation)
BIOCELL 2024, 48(9), 1311-1322. https://doi.org/10.32604/biocell.2024.052451
Received 25 February 2024; Accepted 11 June 2024; Issue published 04 September 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Colorectal cancer is a major global health concern, exacerbated by tumor necrosis factor-alpha(TNF-α) and its role in inflammation, with the effects of Mitotic Arrest Deficient 2 Like 2 (MAD2L2) in this context still unclear. Methods: The colorectal carcinoma cell lines HCT116 and SW620 were exposed to TNF-α for a period of 24 h to instigate an inflammatory response. Subsequent assessments were conducted to measure the expression of inflammatory cytokines, the activity within the p38 mitogen-activated protein kinase (p38 MAPK) and Phosphoinositide 3-Kinase/AKT Serine/Threonine Kinase pathway (PI3K/AKT) signaling cascades. Transcriptome sequencing and subsequent integrative analysis with the Cancer Genome Atlas (TCGA) program database revealed a significant downregulation of the key factor MAD2L2. Enhancement of MAD2L2 expression was facilitated via lentiviral vector-mediated transfection. The influence of this overexpression on TNF-α-prompted inflammation, intracellular signaling pathways, and the migratory and invasive behaviors of the colorectal cancer cells was then scrutinized. Results: TNF-α treatment significantly increased the expression of Interleukin-1 beta (IL-1β) and Interleukin-6 (IL-6), activated the MAPK p38 and PI3K/AKT signaling pathways, and enhanced cell migration and invasion. A decrease in MAD2L2 expression was observed following TNF-α treatment. However, overexpression of MAD2L2 reversed the effects of TNF-α, reducing IL-1β and IL-6 levels, attenuating PI3K/AKT pathway activation, and inhibiting cell migration and invasion. Conclusions: Overexpression of MAD2L2 attenuates the pro-inflammatory effects of TNF-α, suggesting that MAD2L2 plays a protective role against TNF-α-induced migration and invasion of colorectal carcinoma cells. Therefore, MAD2L2 holds potential as a therapeutic target in the treatment of colorectal cancer.Keywords
Abbreviations
| CRC | Colorectal cancer |
| CAC | Colitis-associated cancer |
| STR | Short tandem repeat |
| TMB | 3,3′,5,5′-Tetramethylbenzidine |
| TCGA | The Cancer Genome Atlas Program |
| DMEM | Dulbecco’s Modified Eagle Medium |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| EMT | Epithelial-mesenchymal transition |
| CCK-8 | Cell Counting Kit-8 |
| ELISA | The enzyme-linked immunosorbent assay |
| TNF-α | Tumor necrosis factor-alpha |
| MAD2L2 | Mitotic Arrest Deficient 2 Like 2 |
| p38 MAPK | P38 mitogen-activated protein kinase |
| NF-κB | Nuclear factor kappa-B |
| IL-1β | Interleukin-1 beta |
| IL-6 | Interleukin 6 |
| NCOA3 | Nuclear receptor coactivator 3 |
Colorectal cancer (CRC) is a highly prevalent malignant tumor, ranking second in cancer-related deaths [1]. Its pathogenesis is a complex, multistep process involving various factors such as genetics, environment, diet, and medication [2]. Among these factors, inflammation has been reported to participate in the onset and progression of CRC [3]. The inflammatory response is a complex biological reaction of tissues to harmful stimuli, with macrophages playing a central role in innate immunity [4]. Numerous studies have confirmed that the inflammatory tumor microenvironment is closely related to tumor cell proliferation, invasion, and metastasis [5,6]. Particularly during CRC treatment, surgery, radiotherapy, and chemotherapy are accompanied by inflammatory responses, which have been demonstrated to be associated with CRC recurrence [7].
Furthermore, tumor necrosis factor-alpha (TNF-α) induces CRC cell proliferation and reduces apoptosis by activating STAT3 [8], while anti-TNF-α monoclonal antibody inhibits the growth of colorectal cancer in an orthotopic transplantation mouse model [9]. In the bioinformatics analysis of multiple transcriptomic datasets of acute colitis and colitis-associated cancer (CAC) mouse colon tissues, genes such as complement component 3 (C3) [10], TYRO protein tyrosine kinase binding protein (Tyrobp) [11], matrix metallopeptidase 3 (Mmp3) [12], tissue inhibitor of metalloproteinases 1 (Timp1) [13], and A disintegrin and metalloproteinase domain 8 (Adam8) have been found to be associated with inflammation and malignant lesions of colon tissues [11].
Therefore, understanding the role of inflammatory regulation in CRC progression is crucial for revealing the pathogenesis of CRC and developing effective therapeutic strategies [14]. MAD2L2, an integral component of the translesion synthesis (TLS) pathway, is implicated in the regulation of diverse biological processes, including cellular cycle progression and the repair of DNA damage [15]. Research studies have demonstrated that MAD2L2 play a vital role in various types of cancer [16,17]. Specifically, overexpression of MAD2L2 fosters the tumorigenic phenotype in MDA-MB-157 cells through facilitation of dysregulated mitotic processes. Conversely, the silencing of MAD2L2 is conducive to the apoptotic demise of human breast cancer cell lines subsequent to DNA damage induced by cisplatin treatment [18]. According to our previous study, we discovered CRC tissues have a lower expression of MAD2L2 compared to adjacent tissues, making it a reliable diagnostic molecule for CRC [19]. MAD2L2 is crucial in regulating inflammation by inhibiting excessive inflammatory response and preventing tissue damage and disease occurrence [20–22]. However, the significance of MAD2L2 overexpression in CRC requires further investigation.
This study aims to explore the regulatory role of MAD2L2 in modulating the inflammatory environment associated with colorectal cancer, with a specific focus on how MAD2L2 expression affects the behavior of CRC cells under TNF-α stimulation. Given the potential of MAD2L2 to influence key pathways involved in cancer progression, such as the PI3K/AKT signaling axis, our research specifically investigates whether enhancing MAD2L2 expression can mitigate the migration and invasion of CRC cells and reduce the oncogenic effects of inflammation. This could provide valuable insights into the potential of MAD2L2 as a therapeutic target for limiting cancer progression in the inflammatory context of colorectal cancer.
The human colorectal carcinoma cell lines HCT116 (CL-0096) and SW620 (CL-0225B), procured from ATCC (Manassas, VA, USA). Cellular short tandem repeat (STR) characterization was performed by Wuhan Procell Life Science and Technology Co, Ltd., Wuhan, China. Underwent cultivation in high glucose Dulbecco’s Modified Eagle Medium (DMEM, Cytiva, SH30022.01, Shanghai, China) enriched with 10% fetal bovine serum (Vivacell, C04001-500HI, Shanghai, China), and penicillin-streptomycin at a concentration of 10,000 U/mL (Beyotime, C0223, Shanghai, China). The culture environment was maintained at 37°C with a 5% CO2 atmosphere in a cell culture incubator (Thermo Fisher, Thermo317, Shanghai, China). Sterility assessments confirmed the absence of bacteria, yeast, and mycoplasma within these cells. Authentication of the cell lines was accomplished through STR profiling. Cell harvesting was conducted using a 0.25% trypsin/EDTA solution (Beyotime, C0223, Shanghai, China). Cells (2 × 103/mL) were seeded into 96-well plates, upon achieving over 70% confluence, the cells were treated with different concentration of the cytokine TNF-α (Peprotech, AF-300-01A, Suzhou, China) for a duration of 24 h. Cell death was measured by a Cell Counting Kit-8 (CCK-8) assay (40203ES60, YEASEN, Shanghai, China) according to the manufacturer’s protocol. The samples were then analyzed by measuring the absorbance of 450 nm using FLx800 Fluorescence Microplate Reader (cat. 9022898, Biotek, USA).
Transcriptome sequencing was conducted on cell specimens prepared under standard cell culture conditions. HCT116 cells were cultured until they reached 80% confluence and were in the logarithmic phase of growth. At this juncture, a concentration of 30 ng/mL TNF-α cytokine was administered to stimulate cellular proliferation. Post 24-h exposure to TNF-α, the cells were trypsinized and subsequently centrifuged at 1000 revolutions per minute for a duration of 5 min (Eppendorf, 5804/R, Hamburg, Germany), followed by discarding of the supernatant. The cell pellets were then rapidly frozen using liquid nitrogen and maintained on dry ice for 2 h prior to dispatching the samples. The collection comprised three aliquots each of untreated HCT116 cells, TNF-α-treated HCT116 cells, and HCT116 cells post TNF-α induction. Transcriptomic analysis of these samples was performed by Beijing BioMac Biotechnology Co., Beijing, China.
Differential expression analysis
To pinpoint genes with notable changes in expression between various experimental scenarios, we apply differential expression analysis. This method employs a statistical approach, typically the negative binomial model, which is applied to normalized counts via DESeq2. We define differentially expressed genes (DEGs) as those with an absolute log2 fold change greater than 0.25 coupled with an adjusted p-value below 0.05.
The enzyme-linked immunosorbent assay (ELISA) for expression of inflammatory factors IL-6, IL-1β
HCT116 cells treated with 30 ng/mL of TNF-α cytokine and SW620 cells treated with 10 ng/mL of TNF-α cytokine were utilized in the study. Employing an ELISA kit (Dayou, IL-6-1310602, IL-1β-1310122, Yichun, Jiangxi, China), antibodies were diluted to a concentration of 5 μg/mL using a carbonate coating buffer. Subsequently, 100 μL of this antibody solution was dispensed into each well of a polystyrene enzyme-linked immunosorbent assay plate, followed by an overnight incubation at 4°C. After a period of 24 h, the contents of the wells were discarded and the wells were subjected to a trio of washes, each lasting 3 min, utilizing a washing buffer. Thereafter, 200 μL of a blocking reagent was introduced to each well, which was then incubated at 37°C for 2 h. Following the removal of the blocking solution, the wells were filled with 300 μL of washing solution for a 2-min immersion. This washing step was repeated once more with an additional 300 μL of washing buffer. Subsequently, 100 μL of the analytical specimen was added to the antibody-precoated wells and sealed with adhesive film for a 2-h incubation at 37°C. After discarding the sample solution, the wells were again filled with 300 μL of washing solution for a 2-min immersion. The next phase involved the addition of 100 μL of a diluted biotinylated detection antibody solution to each well, followed by the introduction of 100 μL of 3,3′,5,5′-Tetramethylbenzidine (TMB) substrate solution, with the plate then incubated for 30 min at 37°C in the absence of light. The optical density at 450 nm (OD) value for each well was subsequently determined after calibrating the plate reader (BioTeK, Epoch #800TS, Montpelier, VT, USA).
MAD2L2 overexpression via lentiviral transfection
Cells in 60 mm dishes were ready for transfection once they reached 60%–70% confluence and exhibited stable cell line morphology under high magnification microscopy (Leica Microsystems, DM2700M, Wetzlar, Germany). Lentiviruses (LV3-NC, LV5-REV7-homo) were added (Genepharma, Suzhou, China) and fluorescence expression was observed using fluorescence microscopy (Leica Microsystems, DM2700M, Wetzlar, Germany) 72 h after infection.
Total RNA isolation, cDNA synthesis, and quantitative real-time PCR
Following a 72-h period post-transfection, cells were collected and lysed with the addition of 1 mL of RNA extract (Servicebio, G3013, Wuhan, China). Subsequently, 250 μL of chloroform was introduced, and the tube was inverted for 15 s and then allowed to stand for 3 min. Centrifugation of the sample ensued at 12,000 rpm (Eppendorf, 5804/R, Hamburg, Germany) for 10 min at a temperature of 4°C. Thereafter, 400 μL of the supernatant was carefully transferred into a fresh tube and combined with 0.8 volumes of isopropanol, followed by inversion to mix. The RNA-containing mixture was subjected to another centrifugation at 12,000 rpm for 10 min at 4°C. The resultant white RNA pellet at the tube’s base was then aspirated and washed with 1.5 mL of 75% ethanol. After a further centrifugation at 12,000 rpm for 10 min at 4°C, the supernatant was discarded. For RNA resuspension, 15 μL of nuclease-free water was added, and the solution was incubated at 55°C for 5 min to facilitate dissolution.
RNase-free PCR tubes were prepared with a volume of 200 μL, and reverse transcription was conducted in accordance with the protocol provided by the TaKaRa SYBR Premix Ex TaqII (TliRNaseH Plus) (TaKaRa, RR820A, Japan) kit. The procedure entailed an initial reverse transcription reaction at 37°C for 15 min, followed by inactivation of the reverse transcriptase at 85°C for 5 s, and then the samples were maintained at 4°C to stabilize the temperature. This process yielded cDNA, which was subsequently stored at −20°C. For primer preparation, gene-specific primers were combined with an appropriate volume of buffer as per synthesis guidelines to generate a 100 μM stock solution. Further, 1 μL of this stock solution was diluted to achieve a working concentration of 500 nM, suitable for experimental applications. Citing species genera as human. The primer sequences are detailed in Table 1.
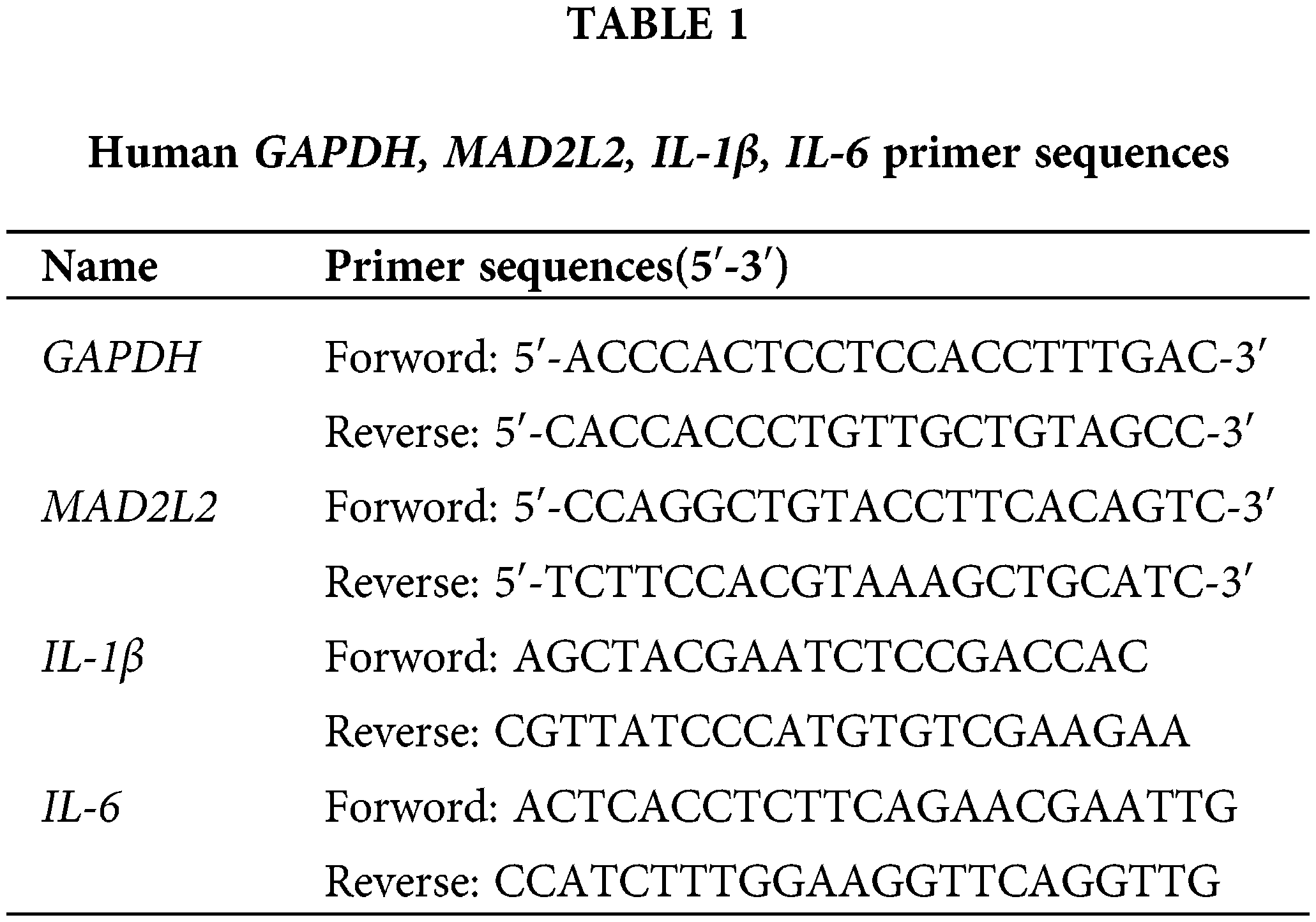
The reaction system was prepared according to the Thermo Fisher PowerUp™ SYBR™ Green Master Mix Real-Time PCR (Thermo Fisher, DRR081A, New York, USA) kit system, using 10 μL per tube, with three replicates for each sample. After all samples were added, they were mixed, centrifuged, and assayed on a 7500 FAST fluorescence PCR instrument (Thermo Fisher Scientific, ABI 7500 Fast, Boston, MA, USA). After the completion of the tests, the data were organized and recorded, ensuring that the entire experiment was repeated three times. The data from the three experiments were subjected to statistical processing, and the CT values were analyzed for relative quantification using the 2−ΔΔCt formula.
Western blot analysis was utilized for protein isolation and identification. Proteins were extracted using RIPA lysis buffer (Beyotime, P0013B, Shanghai, China), and their concentrations were quantified via a BCA assay (KeyGEN BioTECH, KGP902, Nanjing, Jiangsu, China). Samples containing 10–20 µg of protein per well were resolved by 10% SDS-PAGE at 80 V and subsequently transferred to 0.45 µm Polyvinylidene Difluoride (PVDF) membranes (Millipore, Darmstadt, Germany) under wet transfer conditions at 200 mA. The membranes were then blocked with 5% non-fat milk for 1 h at ambient temperature. Primary antibodies, diluted in an appropriate antibody diluent, were applied and incubated for 3 h at room temperature, followed by a 1-h incubation with secondary antibodies. Chemiluminescent detection was performed using the Pierce™ ECL Western Blotting Substrate (Servicebio, G2161-200ML, Wuhan, China), and signal analysis was facilitated by Image Lab software (version 5.2.1, Bio-Rad Laboratories, Inc.). The primary antibodies utilized included: MAD2L2 (cat. ab180579, 1:1,000, Abcam, Cambridge, UK), β-actin (GB150003, 1:1,000, Servicebio, Wuhan, China), p38 (GB114685, 1:1,000, Servicebio, Wuhan, China), phosphorylated p38 (GB113380, 1:1,000, Servicebio, Wuhan, China), AKT (4691, 1:1,000, Cell Signaling Technology, Boston, MA, USA), phosphorylated AKT (4060, 1:1,000, Cell Signaling Technology, USA), PI3K (4249, 1:1,000, Cell Signaling Technology, USA), and phosphorylated PI3K (17366, 1:1,000, Cell Signaling Technology, USA). The horseradish peroxidase-conjugated secondary antibody employed was goat anti-rabbit IgG H&L (HRP) (cat. ZB-2306; 1:10,000, Zsbio, Beijing, China).
Transwell assays were employed to evaluate the invasive and migratory capacities of cells. During the logarithmic growth phase, 1 × 104 cells were collected and enzymatically dissociated with trypsin. A 24-well plate was arranged by introducing 600 µL of complete culture medium into the bottom chamber and inserting a Transwell apparatus. Post a 48-h culture period, cells were fixed using 4% paraformaldehyde (absin, abs9179, Shanghai, China) for 20 min, rinsed thrice with PBS, and stained with crystal violet (Yuanye Bio-Technology, B26890, Shanghai, China) for 15 min. Examination and quantification were performed under a microscope at 200× magnification. The above steps are migration experiments, and serum-free DMEM basic medium (Gibco, C11995500BT, MA, USA) was used for the upper chamberlets. For invasion assessment, a matrix gel barrier was established prior to adding 600 µL of a chemotactic agent into the lower compartment of a 24-well plate. The Transwell inserts, coated with the matrix gel, were placed into the wells, and 100–200 µL of cell suspension was combined and deposited into the upper chamber. Following a 48-h incubation, the cells underwent fixation and staining similar to the migration assay. Matrigel and non-invasive cells from the upper chamber were delicately expunged using cotton swabs. After PBS washes, the cells were visualized and counted under the microscope at 200× magnification. The principal materials used included: Transwell inserts (Corning Inc., 3422, USA) and Matrigel (Becton Dickinson and Company, 354248, NY, USA). Microscope (Olympus, CKX41, Tokyo, Japan).
To ensure the reliability and consistency of the experimental outcomes, each assay was replicated three times. Advanced analysis and processing of the experimental data were conducted using sophisticated statistical software packages, IBM SPSS Statistics 26.0 version (IBM Corporation, Armonk, NY, USA) and data and graphic processing software Prism 6.0 (GraphPad Software, San Diego, CA, USA). For statistical comparisons, the t-test was employed when comparing two groups. In contrast, comparisons among multiple groups were evaluated using one-way ANOVA, with Tukey’s HSD (Honestly Significant Difference) Test applied to determine significant differences between all possible paired combinations of groups. A p-value below 0.05 was recognized as the threshold for statistical significance.
TNF-α induces the production of inflammatory cytokines in colorectal cancer cells
Upon evaluating the dose-response effect of TNF-α on the colorectal cancer cell lines HCT116 and SW620, a differential optimal concentration for cell viability was discerned: 30 ng/mL for HCT116 and 10 ng/mL for SW620 after a 24-h treatment period (Fig. 1A,B, p < 0.05). This heightened viability, however, was not maintained with higher concentrations of TNF-α or beyond the initial 24 h, suggesting a nuanced profile of concentration- and time-dependent cytotoxicity. HCT116 and SW620 cell lines underwent a 24-h TNF-α treatment to emulate an inflammatory milieu. Post-treatment, quantitative analysis of Interleukin-1 beta (IL-1β) and Interleukin 6 (IL-6) at the mRNA and protein levels was conducted via qRT-PCR and enzyme-linked immunosorbent assays (ELISA), respectively. The findings demonstrated a significant elevation in IL-1β and IL-6 within the carcinoma cells upon TNF-α administration at defined concentrations (Fig. 1C–F, p < 0.05), implying that TNF-α elicits an inflammatory response in colorectal cancer cells.
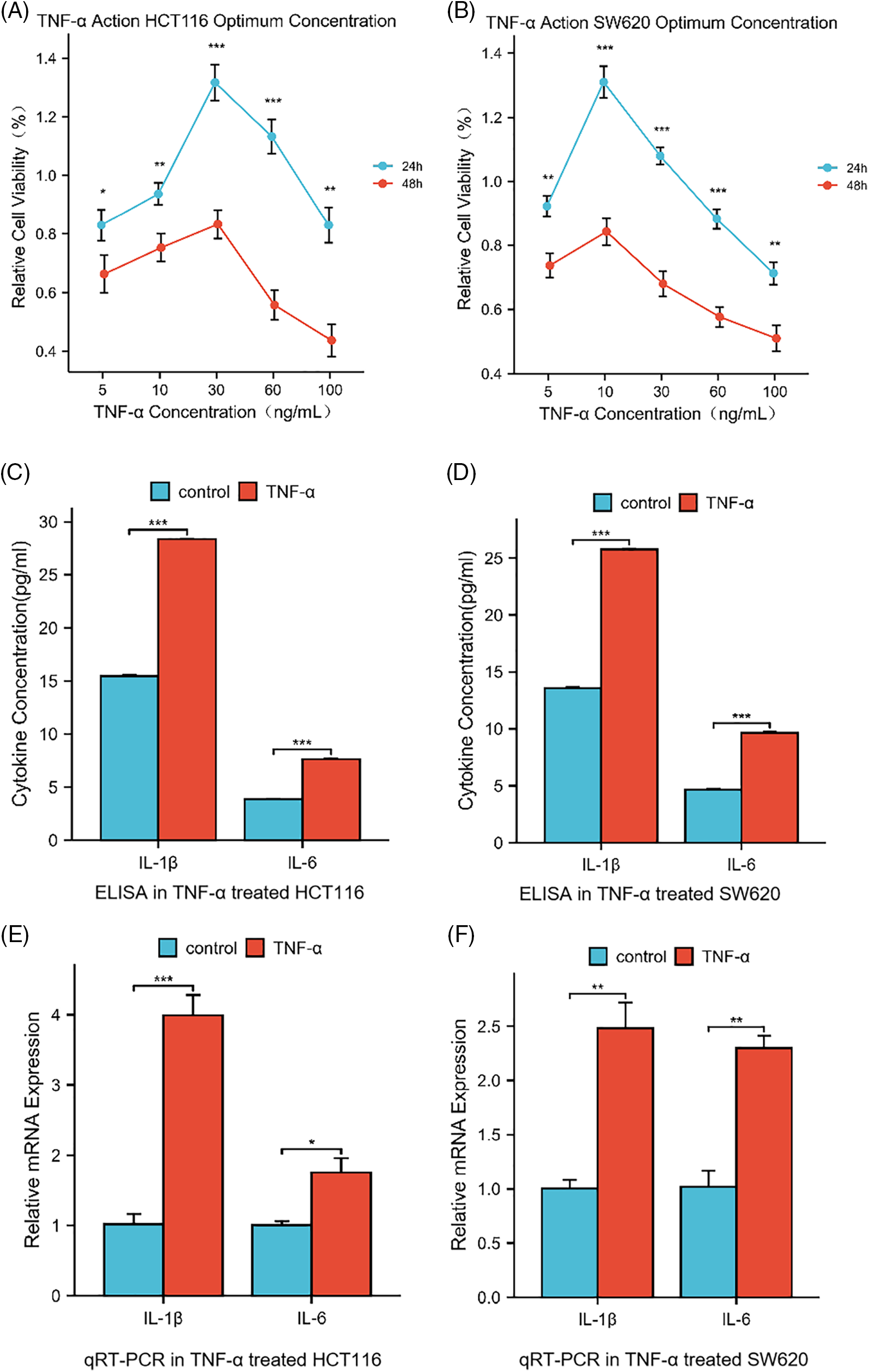
Figure 1: Cytokine levels in HCT116 (30 ng/mL) and SW620 (10 ng/mL) cells after 24 h of treatment with tumor necrosis factor-alpha (TNF-α). (A, B) Dose- and Time-Dependent Cytotoxicity of TNF-α on HCT116 and SW620 Cell Viability. Relative cell viability percentages are plotted against varying concentrations of TNF-α (5, 10, 30, 60, 100 ng/mL) for 24 and 48 h. (C, D) ELISA determination of IL-1β and IL-6 levels secreted by HCT116 and SW620 cells. (E, F) qRT-PCR determination of the relative mRNA expression of IL-1β and IL-6, two inflammatory cytokines, in HCT116 and SW620 cells. Data are presented as mean ± SEM, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001.
TNF-α promotes the invasion and migration of colorectal cancer cells
Subsequent investigations were conducted on the influence of TNF-α on the invasive and migratory properties of HCT116 and SW620 cells via Transwell assays. The outcomes of these assays underscored a considerable enhancement in the invasive and migratory capabilities of both cell lines post TNF-α exposure, relative to the untreated control group (Fig. 2A–D, p < 0.001). These results imply that the inflammatory response provoked by TNF-α may augment the invasion and migration potential of colorectal cancer cells.
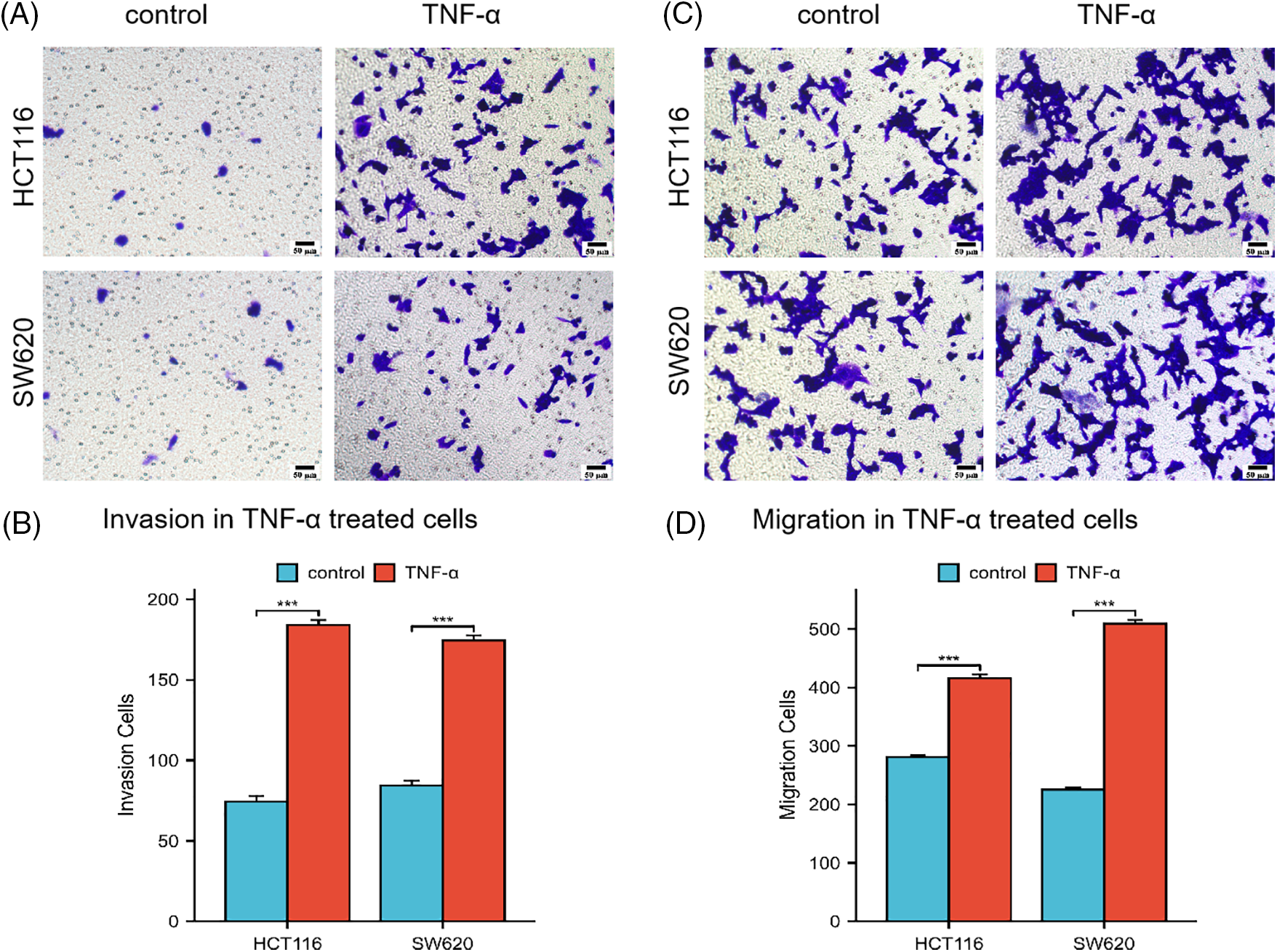
Figure 2: Transwell assay assessing the invasion and migration abilities of HCT116 and SW620 cells. Scale bar = 50 µm. (A, B) Invasion abilities of HCT116 and SW620 cells. (C, D) Migration abilities of HCT116 and SW620 cells. Data are presented as mean ± SEM, n = 3, ***p < 0.001.
TNF-α activates the MAPK p38 and PI3K/AKT signaling pathways in colorectal cancer cells
To elucidate the molecular underpinnings of TNF-α-induced activation in colorectal cancer cells, HCT116 cells were exposed to TNF-α (at 30 ng/mL specified concentrations) for a duration of 24 h, followed by total RNA extraction for transcriptome sequencing. Through our transcriptomic analysis of HCT116 cells treated with TNF-α, we identified 284 down-regulated and 425 up-regulated differentially expressed genes. Notable among the down-regulated genes are LHPP, MACN1, and MAD2L2, whereas genes such as IL1B, IL6, CD79B, BMP4, and LEP were observed to be up-regulated (Fig. 3A). The sequencing data underwent Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses, which identified the MAPK and PI3K/AKT pathways as highly ranked in activity (Fig. 3B). To further assess the impact on these pathways at the protein level, TNF-α-stimulated HCT116 and SW620 cells were examined, revealing a notable association between MAPK p38 pathway activation and inflammatory response. Western blotting demonstrated a significant upregulation of phosphorylated p38, PI3K, and AKT proteins in the TNF-α-treated groups of both cell lines compared with control groups not receiving treatment (Fig. 3C,D, p < 0.001), indicating that TNF-α incites the activation of MAPK p38 and PI3K/AKT pathways, which are integral to inflammation in colorectal cancer cells. Additionally, we queried the transcriptome data of CRC patients from the TCGA database, generating an expression matrix of high-frequency differential genes across 808 samples. We observed that common differential genes, such as ETV5, KLF4, MAD2L2, TCEA1, etc., exhibit highly similar variations among different patients (Fig. 3E).
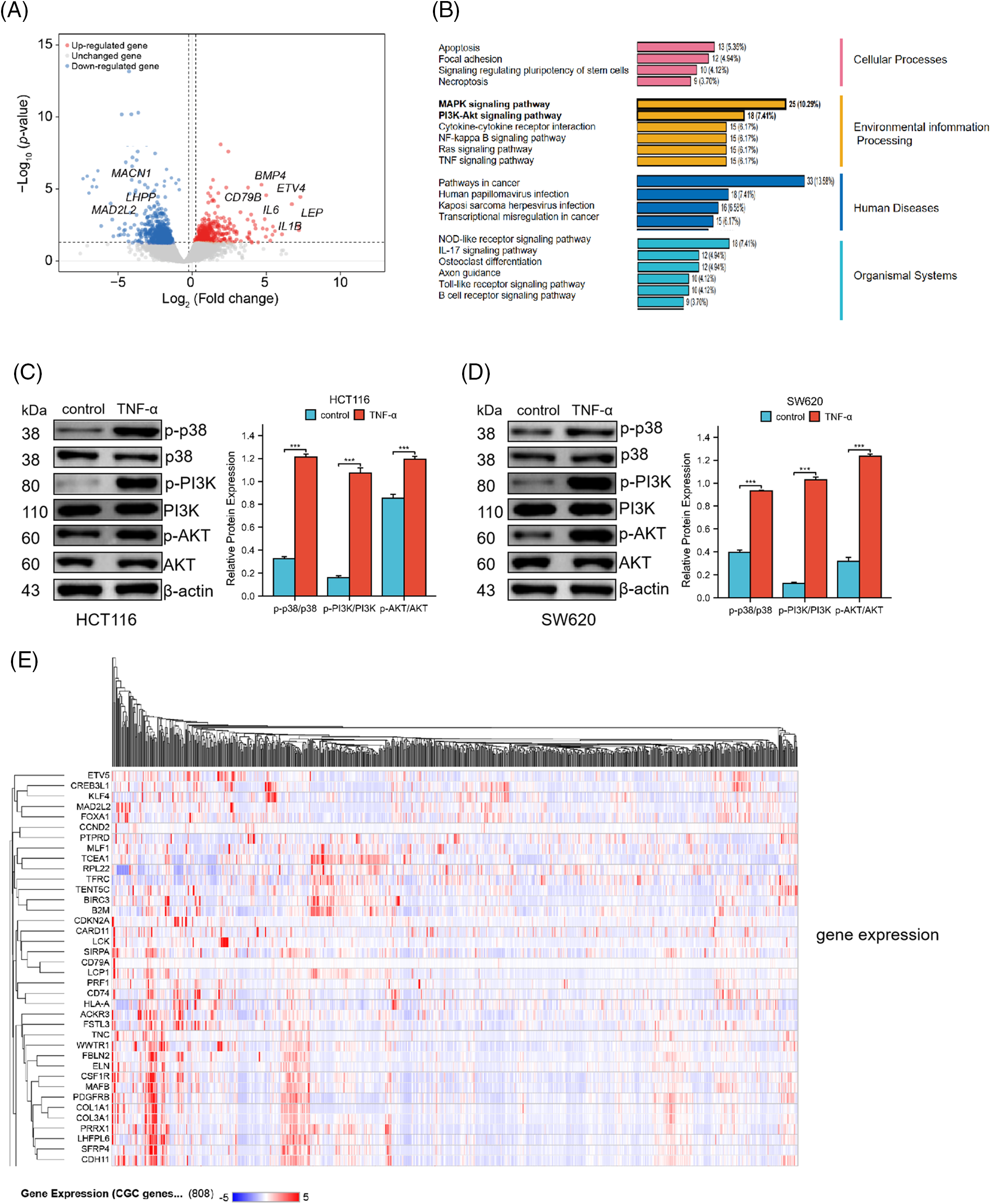
Figure 3: TNF-α triggers the activation of the MAPK p38 and PI3K/AKT signaling pathways in HCT116 (30 ng/mL) and SW620 (10 ng/mL) cells. (A) Volcano plot represents the differential gene expression profile in HCT116 cells following treatment with TNF-α. (B) Schematic representation of the biological responses involved in HCT116 cell transcriptome sequencing. (C, D) Western blot analysis shows the phosphorylation levels of p38, PI3K, and AKT protein expression in HCT116 and SW620 cells. (E) The heatmap displays the expression matrix of high-frequency differential genes in CRC patients within the TCGA database. Data are presented as mean ± SEM, n = 3, ***p < 0.001.
TNF-α treatment reduces MAD2L2 expression levels in colorectal cancer cells
Based on the differential gene expression data from HCT116 cells and high-frequency variant genes in CRC patient data, we conducted a joint analysis to identify common regulated genes. We identified several genes consistently upregulated across both datasets, including KLF4, Myc, and IL6, among others. Additionally, our analysis revealed common downregulated genes, such as MAD2L2, FOXA1, and TFRC (Fig. 4A). These findings suggest a shared pattern of gene expression alterations that may play crucial roles in the pathogenesis and progression of CRC, offering potential targets for therapeutic intervention and biomarkers for disease diagnosis. Notably, MAD2L2 is integral to inflammation regulation, curbing overactive inflammatory responses and thus safeguarding against tissue damage and disease by modulating the NF-κB and JAK/STAT signaling pathways. In pursuit of understanding the role of MAD2L2 within the inflammatory response of cancer cells, the expression dynamics of MAD2L2 were scrutinized in TNF-α-stimulated colorectal cancer cells. qRT-PCR and Western blot analyses revealed a reduction in MAD2L2 expression in HCT116 (30 ng/mL) and SW620 (10 ng/mL) cells following treatment with TNF-α. To corroborate the association between MAD2L2 expression and the inflammatory state of colorectal cancer cells, lentiviral vectors were employed to enhance MAD2L2 expression. This intervention successfully reinstated the expression of MAD2L2 under inflammatory conditions, observable at both mRNA and protein levels (Fig. 4B–E, p < 0.05).
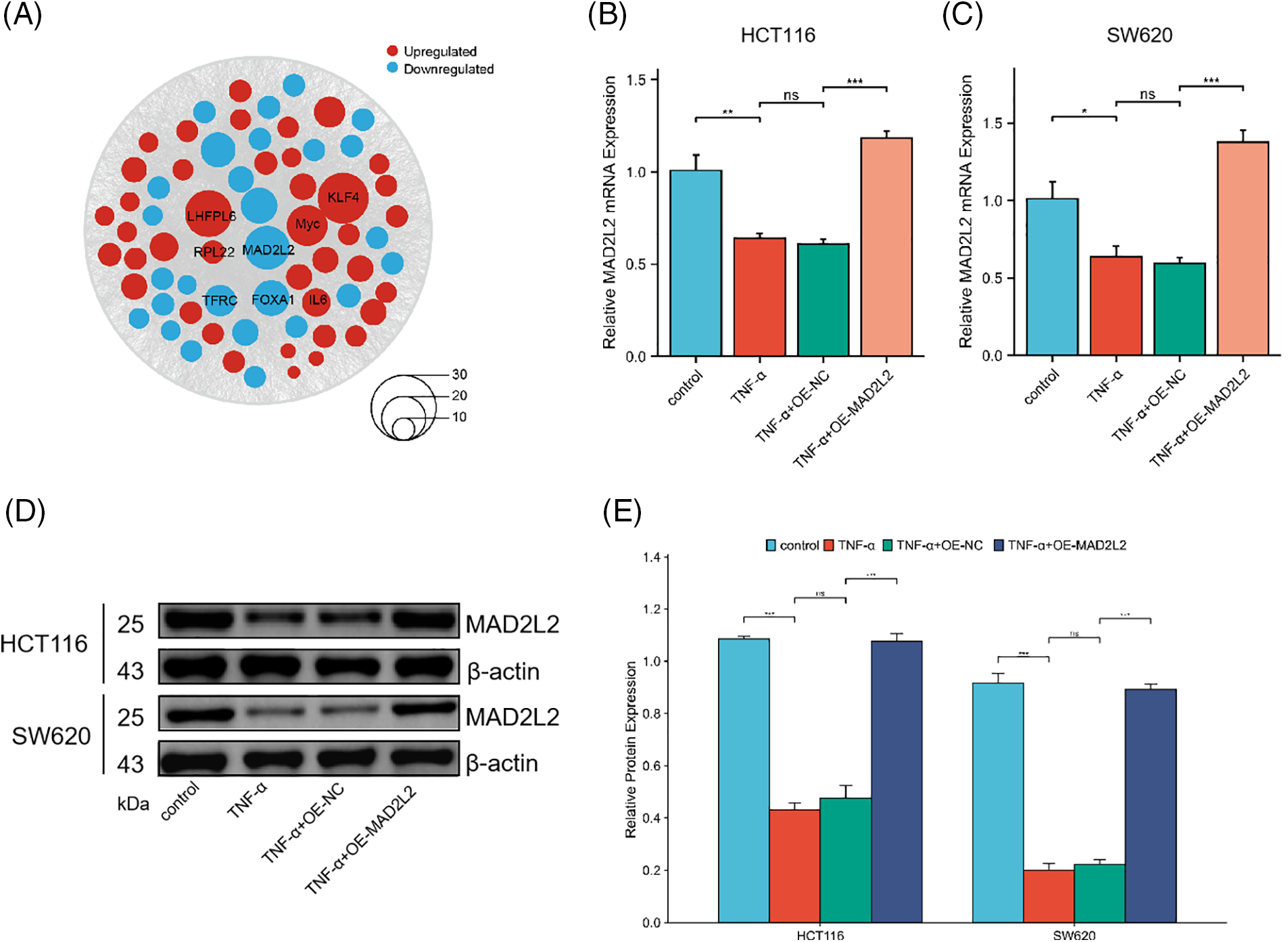
Figure 4: qRT-PCR and Western blot assays assessing Mitotic Arrest Deficient 2 Like 2 (MAD2L2) expression in HCT116 (30 ng/mL) and SW620 (10 ng/mL) cells following TNF-α treatment. (A) Bubble plot illustrating the joint analysis of differential gene expression in CRC patient samples from the TCGA database and HCT116 transcriptomic data. Genes found to be upregulated are depicted in red, while those that are downregulated are shown in blue, with the size of each node corresponding to the magnitude of expression change. (B, C) qRT-PCR analysis of MAD2L2 expression in HCT116 and SW620 cells. (D, E) Western blot analysis of MAD2L2 expression in HCT116 and SW620 cells. Data are presented as mean ± SEM, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, ns: Not Significant. OE: Overexpression, NC: Negative Control.
MAD2L2 overexpression attenuates TNF-α-induced inflammatory response in colorectal cancer cells
To investigate the role of MAD2L2 in the inflammatory response of colorectal cancer cells, we analyzed the production of inflammatory cytokines in HCT116 and SW620 cell lines that overexpressed MAD2L2 following TNF-α treatment. Our results, obtained through qRT-PCR and ELISA, showed that HCT116 and SW620 cells transfected with OE-NC exhibited elevated IL-1β and IL-6 levels after TNF-α stimulation, However, the overexpression of MAD2L2 significantly suppressed their expression. These findings suggest that MAD2L2 overexpression can alleviate the inflammatory response of colorectal cancer cells under TNF-α stimulation (Fig. 5A–D, p < 0.05).
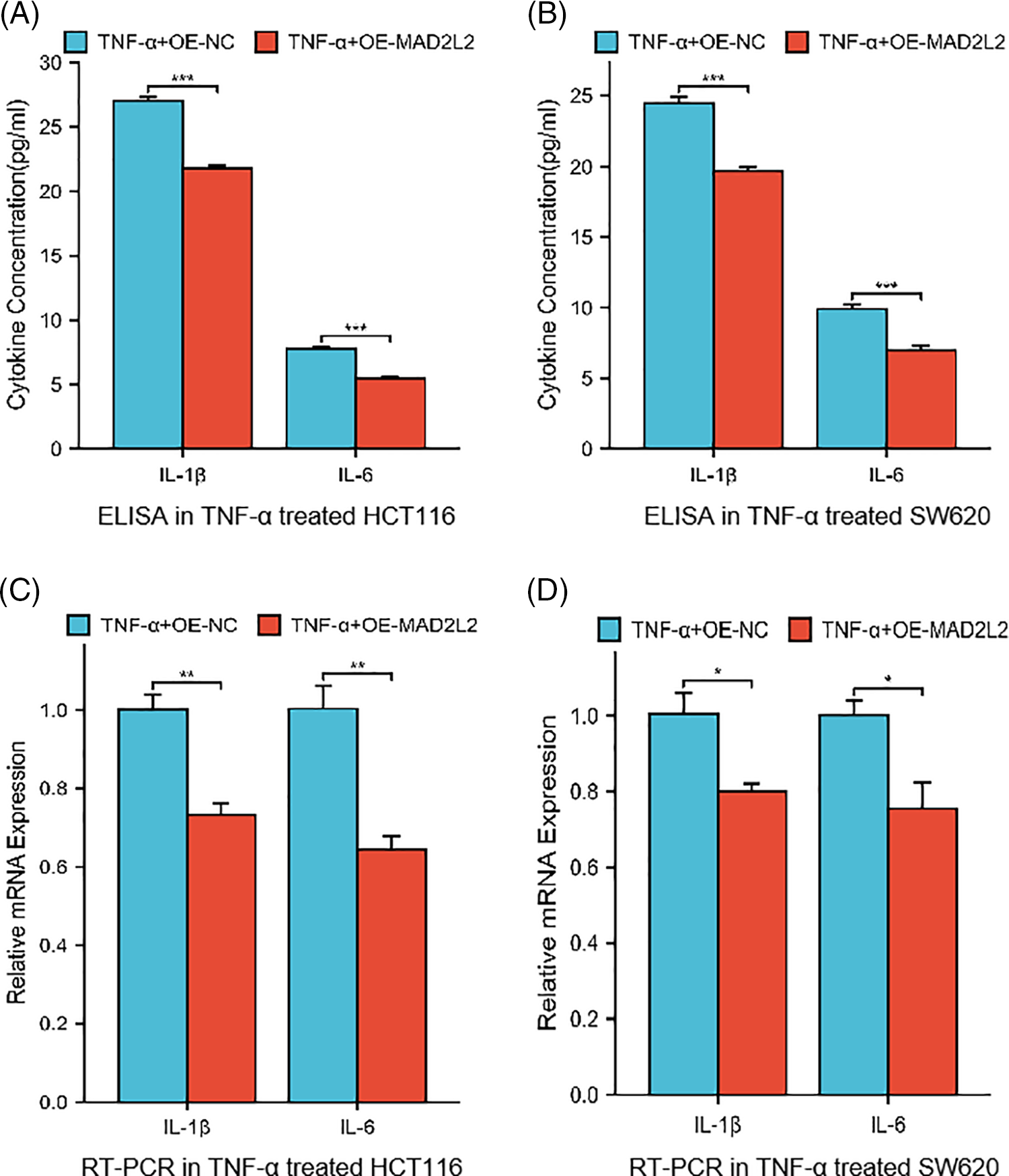
Figure 5: Cytokine levels in HCT116 (30 ng/mL) and SW620 (10 ng/mL) cells stably overexpressed MAD2L2 and treated with TNF-α for 24 h. (A, B) ELISA measuring secreted IL-1β and IL-6 levels in HCT116 and SW620 cells. (C, D) qRT-PCR measuring relative mRNA expression of IL-1β and IL-6 inflammatory cytokines in HCT116 and SW620 cells. Data are presented as mean ± SEM, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001. OE: Overexpression, NC: Negative Control.
MAD2L2 overexpression blocks the PI3K/AKT pathway in colorectal cancer cells
Our research suggests that MAD2L2 overexpression could alleviate changes in inflammatory factors in cancer cells after inflammatory stimulation. We hypothesized that this could be achieved through either the MAPK or PI3K pathway. Therefore, to test this hypothesis, we subjected stably transfected MAD2L2-overexpressed HCT116 and SW620 cells to TNF-α treatment for 24 h and conducted a Western blot analysis. The results showed that compared to the NC group of stable cells treated with TNF-α, the MAD2L2 overexpression group exhibited decreased PI3K and AKT phosphorylation levels and increased p38 phosphorylation levels (Fig. 6A,B, p < 0.001). Overall, the Western blot analysis results suggest that the overexpression of MAD2L2 in colorectal cancer cells can inhibit the TNF-α-activated PI3K/AKT pathway.
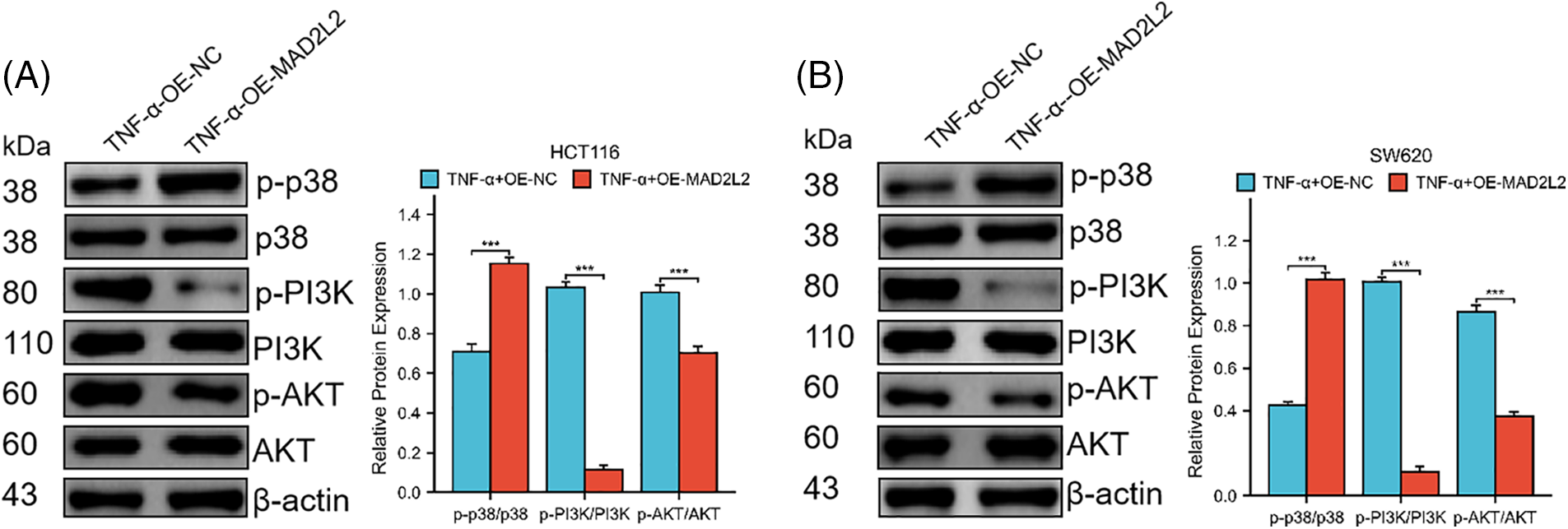
Figure 6: Western blotting was employed to investigate the expression of MAPK p38 and PI3K/AKT pathway proteins in HCT116 (30 ng/mL) and SW620 (10 ng/mL) cells following their induction with TNF-α, both of which stably overexpress MAD2L2. (A, B) Western blot showing the phosphorylation levels of p38, PI3K, and AKT protein expression in HCT116 and SW620 cells. Data are presented as mean ± SEM, n = 3, ***p < 0.001. OE: Overexpression, NC: Negative Control.
Effect of MAD2L2 overexpression on TNF-α-induced migration and invasion of colorectal cancer cells
To delineate the contribution of MAD2L2 to the migratory and invasive behaviors of colorectal cancer cells post TNF-α treatment, Transwell invasion, and migration assays were conducted on TNF-α-treated HCT116 and SW620 cells with MAD2L2 overexpression. The assays revealed diminished migration and invasion in the MAD2L2-overexpressing cohorts of both HCT116 and SW620 cells, compared to the negative control (NC) group (Fig. 7A–D, p < 0.001). These findings indicate that the upregulation of MAD2L2 may mitigate the pro-migratory and invasive effects induced by TNF-α in these colorectal cancer cell lines.
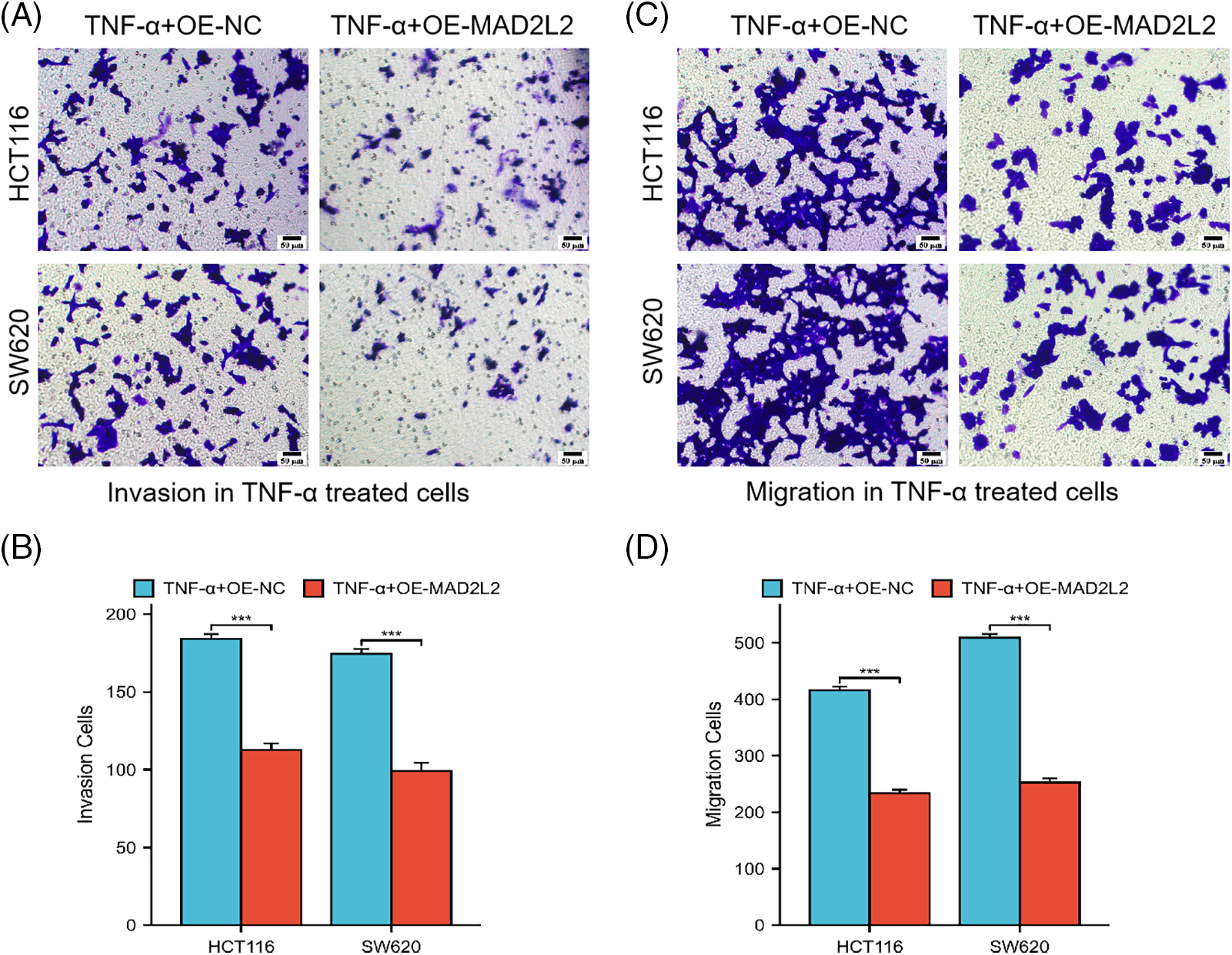
Figure 7: Transwell assay was conducted to evaluate the invasion and migration capabilities of HCT116 cells treated with 30 ng/mL TNF-α and SW620 cells treated with 10 ng/mL TNF-α, both of which stably overexpress MAD2L2. Scale bar = 50 μM. (A, B) Invasion ability of HCT116 and SW620 cells. (C, D) Migration ability of HCT116 and SW620 cells. Data are presented as mean ± SEM, n = 3, ***p < 0.001. OE: Overexpression, NC: Negative Control.
Inflammatory colorectal cancer has a recurrence and metastasis rate of over 30% [23]. Despite advancements, the pivotal mechanisms driving metastasis in colorectal cancer remain incompletely elucidated. The invasiveness and metastatic progression of neoplastic cells are critical determinants of the reduced patient survival rates observed. Research has substantiated that chemokine and cytokine within the tumor microenvironment act as stimulants for tumoral invasion and metastasis, with TNF-α identified as a significant contributor to this process [24]. The persistent release of TNF-α by macrophages and lymphocytes within the tumor microenvironment is a critical element contributing to tumor progression [25]. TNF-α is an inflammatory trigger and a significant inflammatory mediator that promotes cancer stem cell-like phenotypes, metastasis, and proliferation [26]. However, the mechanisms by which chronic inflammation leads to tumor occurrence and the development of colorectal cancer cells are not yet fully understood.
MAD2L2, alternatively designated as MAD2B or REV7, pertains to the mitotic arrest deficient-like 2 family of proteins. Research has demonstrated its expression across diverse cancer cell types, where it functions as a crucial regulator of the proliferation of human malignant neoplasms [20]. Liu et al. documented that augmented levels of REV7 enhance migratory, invasive, and epithelial-mesenchymal transition (EMT) activities in breast cancer cells [27]. Concurrently, research by Hoshino A indicates that a reduction in REV7 within malignant melanoma cell lines MEWO and G361 curtails their proliferation, migration, and invasion [28]. This depletion of REV7 additionally influences the expression of intracellular signaling proteins AKT and extracellular signal-regulated kinases (ERK) in MEWO cells, culminating in the diminished activation of the ERK pathway. MAD2L2 is reported to be associated with epithelial-mesenchymal transition (EMT) and metastasis of lung cancer [29] and mediates the tumor microenvironment, promoting the growth of oral squamous cell carcinoma [30].
Our study shows that there is a significant correlation between MAD2L2 overexpression and the reduction of inflammatory factors, which can alleviate TNF-α-induced colorectal cancer invasion and metastasis. To study the potential molecular mechanisms of TNF-α-induced cell invasion and migration, we used transcriptome sequencing to analyze the possible biological response processes involved in TNF-α-induced colorectal cancer cells HCT116. Transcriptome sequencing and subsequent integrative analysis with the TCGA database revealed a significant downregulation of the key factor MAD2L2. Our findings indicate that under TNF-α induction, the expression level of MAD2L2 decreases. Meanwhile, the overexpression of MAD2L2 using lentivirus reduced the levels of IL-1β and IL-6 in tumor cells and significantly inhibited TNF-α-induced invasion and migration of colorectal cancer cells. These findings reveal that TNF-α treatment may promote the progression of colorectal cancer. However, we discovered that upregulating MAD2L2 leads to the suppression of cellular invasion and migration. While MAD2L2 alone may not constitute an autonomous prognostic indicator for patients with colorectal cancer of an inflammatory nature, the results underscore the pivotal role of MAD2L2 in the pathology of inflammatory colorectal cancer.
This study explores the impact of the PI3K/AKT signaling pathway on cellular behaviors associated with epithelial-to-mesenchymal transition characteristics in colorectal cancer, such as migration and invasion. It was observed that enhancing the expression of MAD2L2 in TNF-α-stimulated colorectal cancer cells reduces the phosphorylation of PI3K and AKT, leading to decreased cellular invasion and motility. This is consistent with the findings of Li et al., who indicated that increased MAD2L2 expression inhibits colorectal cancer cell proliferation, motility, and colony formation by promoting the degradation of nuclear receptor coactivator 3 (NCOA3) [31]. Moreover, this study revealed that the activation of p38 phosphorylation by TNF-α and MAD2L2 involves different pathways and mechanisms. It is well known that the activation of the p38 MAPK signaling pathway by TNF-α is part of the inflammatory response, typically through the involvement of the TNF receptor, triggering a cascade of phosphorylation events that activate p38 MAPK [32]. This process is crucial for the regulation of cytokine production and apoptosis. Conversely, MAD2L2 has been found to play a role in the activation of p38, necessary for the phosphorylation and subsequent degradation of specific substrates such as NCOA3. This finding suggests that MAD2L2 can modulate the activity of p38, highlighting its potential roles in cellular processes like protein degradation regulation and tumor suppression in colorectal cancer. However, in the context of TNF-α acting on colon cancer, a reduction in MAD2L2 expression was noted, raising questions about their interaction and the pathways involved in activating the p38 pathway, which is a key area for further exploration in this study. Therefore, increased expression of MAD2L2 can reduce the invasiveness and metastasis of colorectal cancer cells induced by TNF-α.
To further elucidate the translational potential of these laboratory findings for practical therapeutic applications, assessing the feasibility and challenges associated with targeting MAD2L2 in clinical settings is imperative. This necessitates a comprehensive investigation into the mechanisms by which MAD2L2 modulation can be safely and effectively implemented in human subjects, taking into account the likelihood of off-target effects and resistance development. Moreover, designing drug delivery systems capable of precisely targeting MAD2L2 within the tumor microenvironment, while preserving the integrity of normal tissues, poses a formidable challenge that must be addressed. Subsequent research should prioritize the identification of biomarkers predictive of patient responses to MAD2L2-targeted therapies, thereby facilitating the adoption of personalized treatment approaches. Ultimately, bridging the divide between these encouraging cell-level findings and their application in clinical practice demands a concerted multidisciplinary effort, integrating molecular biology, pharmacology, and clinical research, to affirm the safety, efficacy, and specificity of MAD2L2 as a therapeutic target in CRC management. While this study has unveiled certain insights, it is important to acknowledge the inherent limitations associated with our predominantly in vitro methodology, which may not fully capture the intricate dynamics of tumor microenvironments in living systems. Our findings, though indicative of potential therapeutic pathways, underscore the necessity for subsequent in vivo validation to ascertain the efficacy and practical relevance of overexpressing MAD2L2 in counteracting TNF-α-induced invasion and metastasis in colorectal cancer. Furthermore, our exploration into the molecular interplay between MAD2L2 expression and the PI3K/AKT and p38 MAPK signaling pathways, while informative, highlights the need for more comprehensive studies to fully delineate their roles in colorectal cancer progression. Additionally, focusing exclusively on TNF-α as an inflammatory mediator may not do justice to the complex and multifaceted nature of the tumor microenvironment, which is shaped by a variety of critical factors. Consequently, future research endeavors should aim to expand the investigation to include a broader spectrum of inflammatory mediators and their interactions with MAD2L2, striving for a more thorough understanding of its therapeutic potential.
To encapsulate, the findings of this study propose a linkage where a decrease in MAD2L2 expression correlates with an increase in the inflammatory mediators IL-1β and IL-6, which may play a role in the facilitation of cancer cell invasion and migration following TNF-α induction. At the same time, we have discovered a novel mechanism by which TNF-α-induced colorectal cancer cells can inhibit cell invasion and migration by upregulating MAD2L2 and activating the PI3K/AKT pathway. These findings suggest that MAD2L2 plays a crucial inhibitory role in regulating inflammation during the progression of colorectal cancer, offering potential therapeutic value. Our study provides new insights into the pathogenesis of colorectal cancer and offers theoretical support for future investigations related to the development of effective treatment strategies.
Acknowledgement: None.
Funding Statement: The present study was supported by the Ningxia Hui Autonomous Region key research and development programs (Grant No. 2021BEG03084) and the National Natural Science Foundation of China (Grant No. 31660336).
Author Contributions: The authors confirm their contribution to the paper as follows: study conception and design: Haotong Sun and Heying Wang; data collection: Haotong Sun and Yanjie Hao; analysis and interpretation of results: Xin Li, Jun Ling and Huan Wang; draft manuscript preparation: Fang Xu. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets used and/or analyzed in the current study are available from the corresponding author upon reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Baidoun F, Elshiwy K, Elkeraie Y, Merjaneh Z, Khoudari G, Sarmini MT, et al. Colorectal cancer epidemiology: recent trends and impact on outcomes. Curr Drug Targets. 2021;22(9):998–1009. doi:10.2174/18735592MTEx9NTk2y. [Google Scholar] [CrossRef]
2. Migliore L, Coppede F. Genetic and environmental factors in cancer and neurodegenerative diseases. Mutat Res. 2002;512(2–3):135–53. [Google Scholar] [PubMed]
3. Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. 2021;6(1):263. doi:10.1038/s41392-021-00658-5. [Google Scholar] [PubMed] [CrossRef]
4. Chen Y, Hu M, Wang L, Chen W. Macrophage M1/M2 polarization. Eur J Pharmacol. 2020;877:173090. doi:10.1016/j.ejphar.2020.173090. [Google Scholar] [PubMed] [CrossRef]
5. Tauriello DVF, Sancho E, Batlle E. Overcoming TGFβ-mediated immune evasion in cancer. Nat Rev Cancer. 2022;22(1):25–44. doi:10.1038/s41568-021-00413-6. [Google Scholar] [PubMed] [CrossRef]
6. Shigematsu Y, Tanaka K, Amori G, Kanda H, Takahashi Y, Takazawa Y, et al. Potential involvement of oncostatin M in the immunosuppressive tumor-immune microenvironment in hepatocellular carcinoma with vessels encapsulating tumor clusters. Hepatol Res. 2023;54(4):368–81. [Google Scholar] [PubMed]
7. Long J, He Q, Yin Y, Lei X, Li Z, Zhu W. The effect of miRNA and autophagy on colorectal cancer. Cell Prolif. 2020;53(10):e12900. doi:10.1111/cpr.v53.10. [Google Scholar] [CrossRef]
8. Wei W, Wang J, Huang P, Gou S, Yu D, Zong L. Tumor necrosis factor-α induces proliferation and reduces apoptosis of colorectal cancer cells through STAT3 activation. Immunogenetics. 2023;75(2):161–9. doi:10.1007/s00251-023-01302-y. [Google Scholar] [PubMed] [CrossRef]
9. Takasago T, Hayashi R, Ueno Y, Ariyoshi M, Onishi K, Yamashita K, et al. Anti-tumor necrosis factor-α monoclonal antibody suppresses colorectal cancer growth in an orthotopic transplant mouse model. PLoS One. 2023;18(3):e0283822. doi:10.1371/journal.pone.0283822. [Google Scholar] [PubMed] [CrossRef]
10. Lin Y, Wang D, Zhao H, Li D, Li X, Lin L. Pou3f1 mediates the effect of Nfatc3 on ulcerative colitis-associated colorectal cancer by regulating inflammation. Cell Mol Biol Lett. 2022;27(1):75. doi:10.1186/s11658-022-00374-0. [Google Scholar] [PubMed] [CrossRef]
11. Markov AV, Savin IA, Zenkova MA, Sen’kova AV. Identification of novel core genes involved in malignant transformation of inflamed colon tissue using a computational biology approach and verification in murine models. Int J Mol Sci. 2023;24(5):4311. doi:10.3390/ijms24054311. [Google Scholar] [PubMed] [CrossRef]
12. He L, Kang Q, Chan KI, Zhang Y, Zhong Z, Tan W. The immunomodulatory role of matrix metalloproteinases in colitis-associated cancer. Front Immunol. 2022;13:1093990. [Google Scholar] [PubMed]
13. Pan Z, Lin H, Fu Y, Zeng F, Gu F, Niu G, et al. Identification of gene signatures associated with ulcerative colitis and the association with immune infiltrates in colon cancer. Front Immunol. 2023;14:1086898. doi:10.3389/fimmu.2023.1086898. [Google Scholar] [PubMed] [CrossRef]
14. Xu H, Liu L, Li W, Zou D, Yu J, Wang L, et al. Transcription factors in colorectal cancer: molecular mechanism and therapeutic implications. Oncogene. 2021;40(9):1555–69. doi:10.1038/s41388-020-01587-3. [Google Scholar] [PubMed] [CrossRef]
15. Marima R, Hull R, Penny C, Dlamini Z. Mitotic syndicates Aurora Kinase B (AURKB) and mitotic arrest deficient 2 like 2 (MAD2L2) in cohorts of DNA damage response (DDR) and tumorigenesis. Mutat Res Rev Mutat Res. 2021;787:108376. doi:10.1016/j.mrrev.2021.108376. [Google Scholar] [PubMed] [CrossRef]
16. Kim JH, Patel R. Mad2B forms a complex with Cdc20, Cdc27, Rev3 and Rev1 in response to cisplatin-induced DNA damage. Korean J Physiol Pharmacol. 2023;27(5):427–36. doi:10.4196/kjpp.2023.27.5.427. [Google Scholar] [PubMed] [CrossRef]
17. Shimada Y, Kato T, Sakurai Y, Watanabe H, Nonaka M, Nanaura N, et al. Identification of the promoter region regulating the transcription of the REV7 gene. Biochem Biophys Res Commun. 2023;662:8–17. doi:10.1016/j.bbrc.2023.04.056. [Google Scholar] [PubMed] [CrossRef]
18. Pernicone N, Peretz L, Grinshpon S, Listovsky T. MDA-MB-157 cell line presents high levels of MAD2L2 and dysregulated mitosis. Anticancer Res. 2020;40(10):5471–80. doi:10.21873/anticanres.14558. [Google Scholar] [PubMed] [CrossRef]
19. Sun H, Wang H, Li X, Hao Y, Ling J, Wang H, et al. Increased MAD2L2 expression predicts poor clinical outcome in Colon Adenocarcinoma. BIOCELL. 2023;47(3):607–18. doi:10.32604/biocell.2023.026445. [Google Scholar] [CrossRef]
20. Xu K, Zheng X, Shi H, Ou J, Ding H. MAD2L2, a key regulator in ovarian cancer and promoting tumor progression. Sci Rep. 2024;14(1):130. doi:10.1038/s41598-023-50744-7. [Google Scholar] [PubMed] [CrossRef]
21. Iwai H, Kim M, Yoshikawa Y, Ashida H, Ogawa M, Fujita Y, et al. A bacterial effector targets Mad2L2, an APC inhibitor, to modulate host cell cycling. Cell. 2007;130(4):611–23. doi:10.1016/j.cell.2007.06.043. [Google Scholar] [PubMed] [CrossRef]
22. Liu Z, Wang S, Yu K, Chen K, Zhao L, Zhang J, et al. The promoting effect and mechanism of MAD2L2 on stemness maintenance and malignant progression in glioma. J Transl Med. 2023;21(1):863. doi:10.1186/s12967-023-04740-0. [Google Scholar] [PubMed] [CrossRef]
23. Miyoshi J, Toden S, Yoshida K, Toiyama Y, Alberts SR, Kusunoki M, et al. MiR-139-5p as a novel serum biomarker for recurrence and metastasis in colorectal cancer. Sci Rep. 2017;7:43393. doi:10.1038/srep43393. [Google Scholar] [PubMed] [CrossRef]
24. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20(1):131. doi:10.1186/s12943-021-01428-1. [Google Scholar] [PubMed] [CrossRef]
25. Kasprzak A. The role of tumor microenvironment cells in colorectal cancer (CRC) cachexia. Int J Mol Sci. 2021;22(4):1565. doi:10.3390/ijms22041565. [Google Scholar] [PubMed] [CrossRef]
26. Cruceriu D, Baldasici O, Balacescu O, Berindan-Neagoe I. The dual role of tumor necrosis factor-alpha (TNF-α) in breast cancer: molecular insights and therapeutic approaches. Cell Oncol. 2020;43(1):1–18. doi:10.1007/s13402-019-00489-1. [Google Scholar] [PubMed] [CrossRef]
27. Liu F, Wang W, Zhang H, Han YT, Wang CB. Knockdown of REV7 inhibits breast cancer cell migration and invasion. Oncol Res. 2016;24(5):315–25. doi:10.3727/096504016X14666990347590. [Google Scholar] [PubMed] [CrossRef]
28. Hoshino A, Nakayama C, Jiang SX, Sakurai Y, Kato T, Numata Y, et al. Upregulation of REV7 correlates with progression of malignant melanoma. Pathol Int. 2022;72(1):14–24. doi:10.1111/pin.v72.1. [Google Scholar] [CrossRef]
29. Zhang H, He X, Yu W, Yue B, Yu Z, Qin Y. Mitotic arrest-deficient protein 2B overexpressed in lung cancer promotes proliferation, EMT, and metastasis. Oncol Res. 2019;27(8):859–69. doi:10.3727/096504017X15049209129277. [Google Scholar] [PubMed] [CrossRef]
30. Diniz MG, de Fatima Correia Silva J, de Souza FT, Pereira NB, Gomes CC, Gomez RS. Association between cell cycle gene transcription and tumor size in oral squamous cell carcinoma. Tumour Biol. 2015;36(12):9717–22. doi:10.1007/s13277-015-3735-1. [Google Scholar] [PubMed] [CrossRef]
31. Li Y, Li L, Chen M, Yu X, Gu Z, Qiu H, et al. MAD2L2 inhibits colorectal cancer growth by promoting NCOA3 ubiquitination and degradation. Mol Oncol. 2018;12(3):391–405. doi:10.1002/1878-0261.12173. [Google Scholar] [PubMed] [CrossRef]
32. Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol. 2014;26(3):237–45. doi:10.1016/j.smim.2014.02.009. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools