
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

The Role of Linker Histone Mutation in Oncogenesis: Molecular Mechanism and Structural Impact
Institute of Biophysics and Department of Physics, Central China Normal University, Wuhan, 430079, China
* Corresponding Author: Yunhui Peng. Email:
# These authors contributed equally to this work
(This article belongs to the Special Issue: Genetic Biomarkers of Cancer: Insights into Molecular and Cellular Mechanisms)
BIOCELL 2025, 49(4), 519-538. https://doi.org/10.32604/biocell.2025.061470
Received 25 November 2024; Accepted 14 February 2025; Issue published 30 April 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Nucleosomes play a vital role in chromatin organization and gene regulation, acting as key hubs that interact with various chromatin-associated factors through diverse binding mechanisms. Recent research has highlighted the prevalence of mutations in linker histones across different types of cancer, emphasizing their critical involvement in cancer progression. These cancer-associated mutations in linker histones have been shown to disrupt nucleosome stacking and the formation of higher-order chromatin structures, which in turn significantly affect epigenetic regulatory processes. In this review, we provide a comprehensive analysis of how cancer-associated linker histone mutations alter their physicochemical properties, influencing their binding to nucleosomes, and overall chromatin architecture. Additionally, we explore the significant impact of mutations near post-translational modification sites, which further modulate chromatin dynamics and regulatory functions, offering insights into their role in oncogenesis and potential therapeutic targets.Keywords
In eukaryotes, chromatin provides the foundation for epigenetic regulation and plays a crucial role in numerous biological processes, including gene regulation, the repair of DNA damage, its replication process, as well as the structuring and separation of chromosomes [1–4]. Nucleosomes, the fundamental units of chromatin compaction, consist of approximately 147 base pairs of DNA coiled around a histone octamer, which is made up of two copies of each core histone: H2A, H2B, H3, and H4 [5]. The linker histone (LH) H1 interacts with nucleosomes by binding to linker DNA at the entry and exit points, forming chromatin structures [6]. The LH is essential for chromatin organization, facilitating the compaction of nucleosome arrays and assisting in the assembly of higher-order chromatin structures [7,8]. By dynamically interacting with chromatin, histone H1 affects the nucleosome repeat length and is vital for preserving the stability of higher-order chromatin structure [7,9,10].
Histone H1 is among the most variable histone families, consisting of up to 11 subtypes [11,12]. All H1 proteins consist of three distinct structural domains: a disordered N-terminal domain (NTD), a central globular domain (GD), and an unstructured C-terminal domain (CTD) [12]. Similar to other core histones, histone H1 undergoes various post-translational modifications (PTMs). As a key epigenetic regulator, linker histones are involved in several cellular processes, such as gene expression, mitotic chromosome structure and segregation, heterochromatin function, and the heterogeneity of cancer cells [11,13].
In recent years, an increasing number of studies have highlighted the essential roles of LH H1 in human diseases including cancer [14–17]. The deletion or mutation-induced loss of H1 results in notable alterations to chromatin structure and function, affecting nucleosome stacking and the global chromatin architecture [18,19]. Although cancer-related mutations significantly impact LH structure and function, their molecular mechanisms in oncogenesis have been less studied than those of core histones. Recent experimental and computational research has made notable advances in understanding the impact of H1 cancer mutations on chromatin structure and function, providing novel perspectives on the epigenetic mechanisms driving tumorigenesis [20–23]. This review offers a comprehensive summary of recent research on LH mutations in cancers, with a focus on how these mutations potentially drive oncogenic development by altering chromatin structure and interactions. We also explore how mutations affect the post-translational modification sites of LH and their potential influence on higher-order chromatin structures.
2 The Function of Linker Histones and Their Association with Diseases across Different Variants
Similar to core histone proteins, the LH family consists of various variants [24]. Recent studies suggest that different LH variants and their binding modes can lead to diverse chromatin fiber structures, which, in turn, influence gene expression and chromatin function [9,25]. In human, there are a total of 11 LH variants. Seven, including H1.0, H1.1–H1.5, and H1.10, are expressed in somatic cells, while four others (H1.6–H1.9) are expressed in germ cells [12]. These variants in somatic cells can be classified into DNA replication-dependent variants (H1.1–H1.5) and replication-independent variants (H1.10 and H1.0) (Fig. 1a) [26].

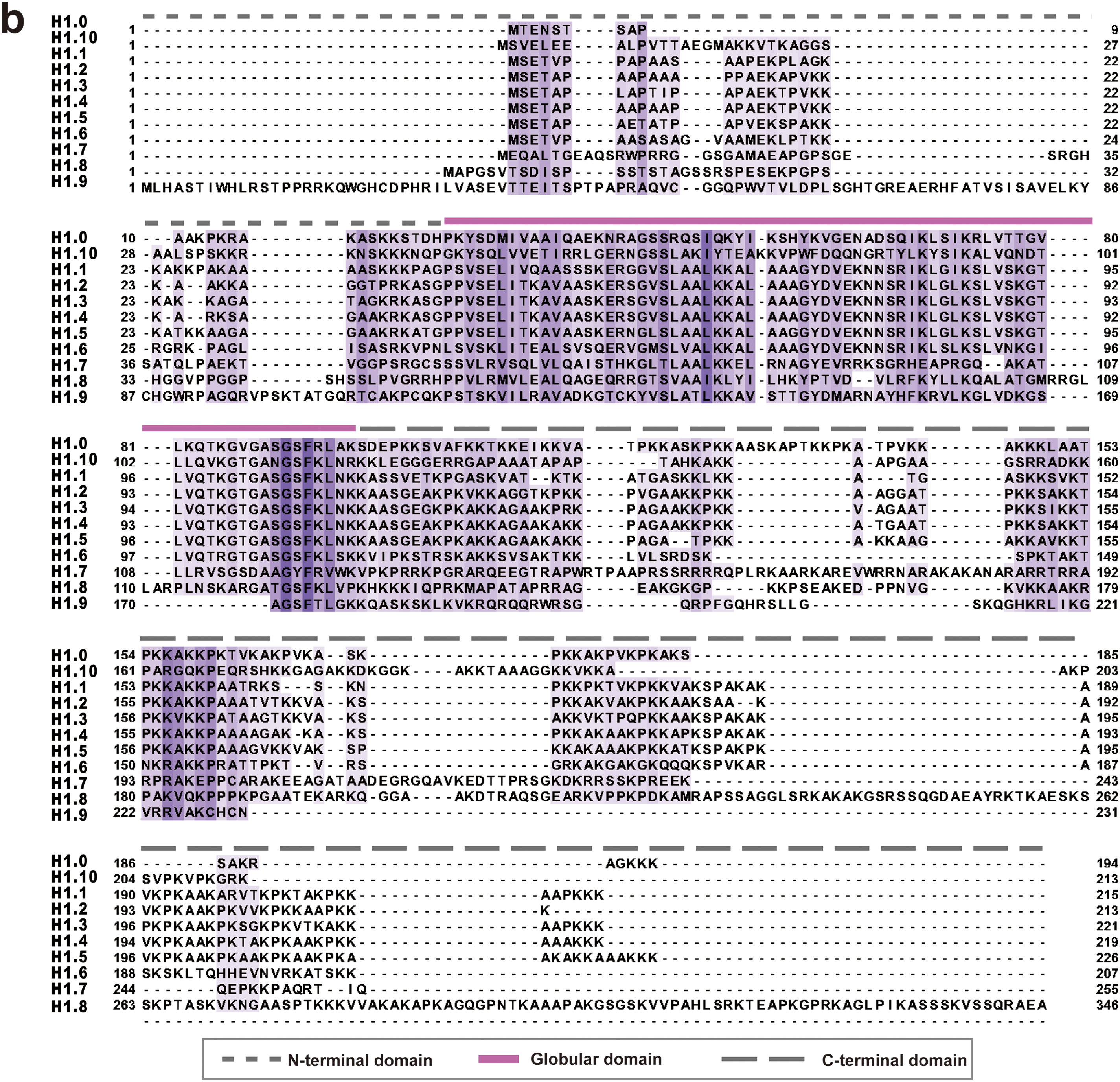
Figure 1: Human linker histones subtypes and their sequences. (a) The different types of H1 histones found in somatic cells and germ cells are displayed in this figure. H1 histones in somatic cells are categorized into replication-dependent and replication-independent types. Additionally, testis-specific and oocyte-specific H1 histones in germ cells are listed. The UniProt IDs and gene names are provided for each subtype. (b) A multiple sequence alignment of LH family members, color-coded by sequence conservation. Darker colors represent more conserved sequences, highlighting the evolutionary conservation of specific regions within the LH family. The alignment was performed using Clustal Omega (v1.2.4) [29]. The visualization was generated using Jalview (version 2.11.4.1) [30]
In higher eukaryotes, linker histones have a three-part structure: a central GD consisting of approximately 80 amino acids, with intrinsically disordered tails at both the N- and C-termini. The NTD is approximately 25–35 amino acids in length, while the CTD is longer, spanning around 100 amino acids [27,28]. We performed a sequence alignment of 11 human linker histones, and as shown in Fig. 1b, the GD of H1 variants is generally more conserved than the unstructured tail regions. The H1 tails are highly basic and lysine-rich but are notably lacking in acidic and aromatic residues. Recent studies have revealed that these long, less conserved tails may be crucial in regulating chromatin structure and function by interacting specifically with chromatin-binding factors and undergoing PTMs [12].
Different H1 variants exhibit varying binding affinities and interaction modes with nucleosomes [10,31,32]. High-affinity binding of H1 to nucleosomes, both in vitro and in vivo, relies on its CTD, which is also critical for chromatin condensation [33,34]. Interestingly, a potential correlation has been observed between the binding affinity of H1 to nucleosomes and the length of its CTD. Among somatic H1 variants, H1.4 and H1.5 exhibit the highest binding affinities, while H1.0, H1.2, and H1.3 display intermediate affinities [35,36]. The binding affinity of H1 variants primarily depends on the length of the C-terminal region [35]. Additionally, the presence of DNA-binding S/TPKK motifs and an increase in positively charged residues also contribute to the enhancement of binding affinity [35]. H1.4 and H1.5 have the highest binding affinity, mainly due to their longer C-terminal regions [35]. H1.10 has the weakest binding affinity and is the most divergent and least conserved of the somatic variants [10,33,37]. Despite its lower chromatin-binding affinity, H1.10 effectively promotes chromatin compaction [33]. Interestingly, studies have found that H1.10 is also associated with more open and transcriptionally active chromatin regions [37]. The differences in H1.10’s chromatin compaction ability may be closely related to its cellular localization [10,37,38]. For example, in the nucleolus, H1.10 inhibits gene expression by promoting chromatin condensation, whereas in the nucleoplasm, it is primarily associated with transcriptionally active regions [10,37,38]. H1.0, the most conserved somatic variant, predominates in adult cells and is essential for forming highly compacted chromatin. Overexpression of H1.0 in proliferating cells inhibits both DNA replication and transcription. In contrast, H1.1, which has a low chromatin-binding affinity and limited chromatin compaction ability, leads to a more open chromatin structure, particularly in undifferentiated cells [39,40]. This variant also exhibits rapid exchange between chromatin regions, likely due to its shorter CTD [41]. In addition to the CTD, the NTD of H1 is crucial for determining both the specificity and strength of its interactions with nucleosomes [42]. Moreover, the N-terminus appears to play a critical role in ensuring the proper localization of H1 at its specific binding site within the nucleosome [43,44]. Supporting this, studies have demonstrated that H1 variants lacking their N-terminal regions are unable to localize to the nucleosome in vitro. This proper localization is essential not only for H1’s binding properties but also for the subsequent formation of chromatin structure [43,44].
The chromatosome, a fundamental structural unit of chromatin, consists of a nucleosome and an LH that binds to the linker DNA connecting adjacent nucleosomes. Recent research highlights that the proximity between the two linker DNA segments within the chromatosome is governed not only by the net positive charge in the C-terminal tails of H1 subtypes but also by the presence of distinct T/SPKK motifs [9]. For instance, H1.0 contains two T/SPKK motifs, while H1.4 possesses three. Notably, a recent study has shown that mutating threonine or serine residues to glutamic acid (T or S to E) within the T/SPKK motifs of H1.4’s CTD can disrupt the interaction between H1.4 tail and linker DNA [9]. These mutations may alter the binding strength or the mode of interaction between H1.4 and the DNA. Therefore, the number of T/SPKK motifs in the C-terminal tail of the LH likely plays an important role in regulating the openness of the linker DNA [9,45]. Notably, frameshift mutations, such as Thr146HisfsTer50, in the C-terminal tail of H1.4 result in the loss of T/SPKK motifs and positively charged residues, a change that has been linked to autism and progeria [15]. However, so far, there have been no germline mutations in the H1.0 gene associated with Rahman syndrome (RMNS). To date, over 20 frameshift mutations have been identified in H1.4 variants, most of which led to premature truncation of the CTD. These germline mutations often create pathogenic variants with abnormal functions, including those associated with RMNS [14]. In addition to frameshift mutations, missense mutations in LH genes, particularly within the H1.2–H1.5 gene family, are frequently associated with follicular lymphoma [46]. These mutations, which are predominantly located in the C-terminal region, are somatically acquired in follicular lymphoma [46]. Furthermore, mutations in the H1.5 gene are commonly observed in colorectal cancer, underscoring the involvement of histone H1 variants in various diseases [47].
The interactions of H1 with the nucleosome are highly dynamic and involve different binding modes [9,48,49]. Typically, the GD of H1 attaches to the nucleosome at the dyad, forming connections with both linker DNA strands through the α2 and α3 helices, as well as the W1 “wing” and L1 loop. At the same time, the disordered CTD is asymmetrically associated with a single strand of linker DNA [50]. The interaction of H1 with both the nucleosome and linker DNA leads to more compact and rigid conformations, facilitating chromatin compaction [50,51]. Recent experiments have also revealed “off-dyad” binding of H1 in chromatosomes, where the GD associates with the nucleosome near the dyad axis and binds to 10 or 20 base pairs of linker DNA [25,48,52] (Fig. 2). In the “off-dyad” binding modes, H1 establishes close contact with both the C-terminal tail of H2A and the N-terminal tail of H3. The α3 helix within its GD extends toward the nucleosome core, where key residues K91 and K95 engage with nucleosomal DNA near the dyad axis. Simultaneously, residues K58, K102, K107, and K116 form interactions with a nearby 10-base pair segment of linker DNA [48].
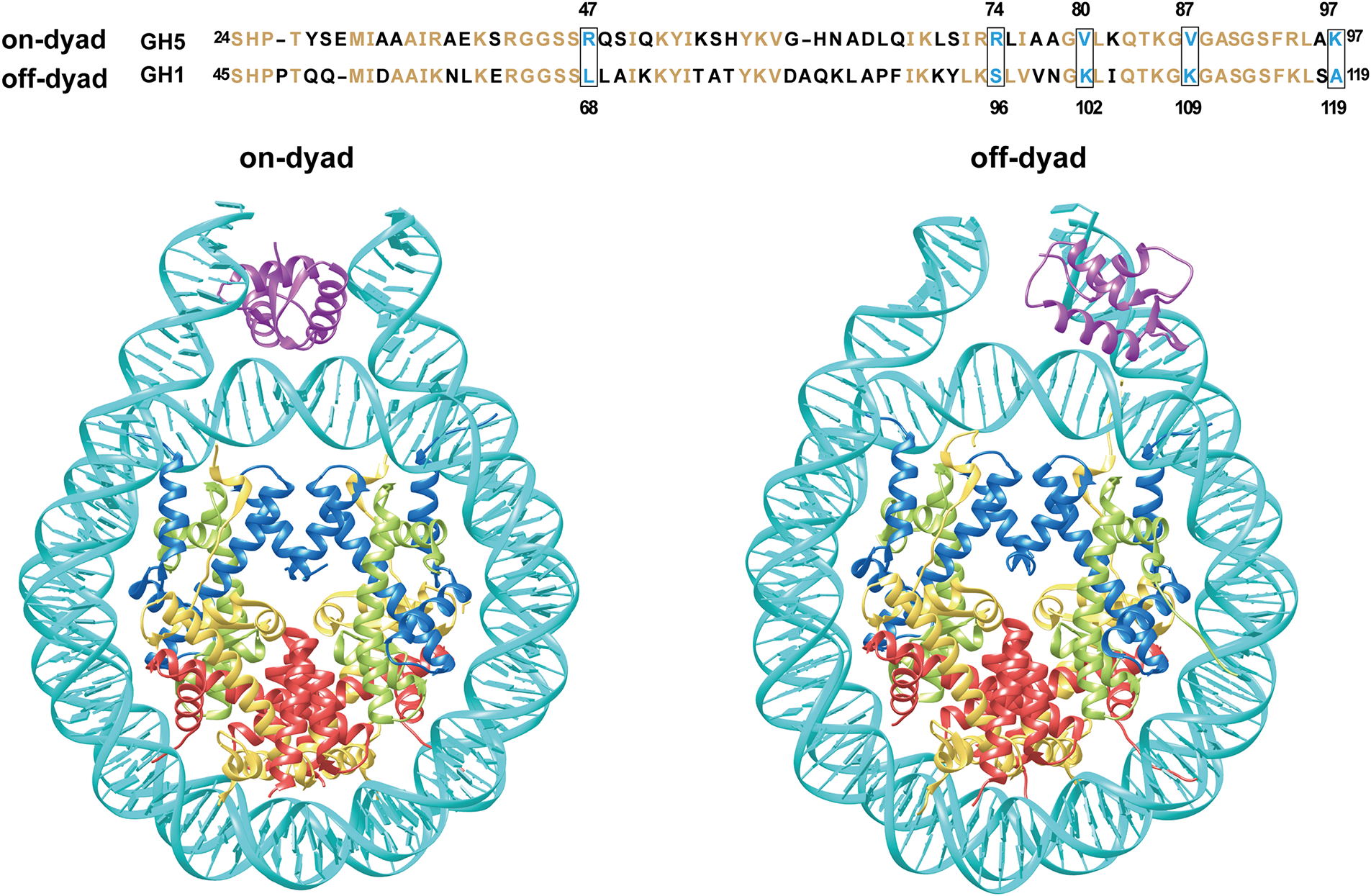
Figure 2: Different H1 binding modes in the chromatosome structure. The top panel presents a sequence comparison of GH5 and GH1, with conserved residues highlighted in orange. Five positively charged residues, critical for the binding of the globular domains of Drosophila H1 or chicken H5, are highlighted within boxes. The bottom panel illustrates schematic representations of two H1 binding modes: on-dyad and off-dyad. The on-dyad structure is modeled based on PDB 4QLC, while the off-dyad structure is derived from HADDOCK docking using 1HST and 1ZBB [49,53,54]. Given the flexibility of histone tails and lack of structural information, the figure emphasizes the interaction between the H1 GD and DNA
The GD primarily dictates the binding mode of the LH, and modifications to several key residues within this domain can regulate on- or off-dyad binding [49,55]. For example, the study of chicken H5 and Drosophila H1 linker histones has shown distinct binding preferences [49]. Replacement of the five residues within the GD of H5/H1.0 with the corresponding residues from Drosophila H1 causes the LH to associate with the nucleosome at a position off the dyad. Importantly, these residues are highly conserved across multiple species, including humans [9]. In chicken H5, the on-dyad binding mode is stabilized by three positively charged residues—R47, R74, and K97—which are replaced by neutral residues (L68, S96, and A119) in Drosophila H1 (Fig. 2). Conversely, in the off-dyad mode, Drosophila H1 depends on two positively charged residues, K102 and K109, to interact with linker DNA and nucleosomal DNA, respectively. These residues correspond to neutral residues (V80 and V87) in chicken H5 [49]. Mutations in critical residues of the GD can disrupt H1’s interaction with the nucleosome, thereby affecting nucleosome stability and higher-order chromatin organization. Notably, introducing specific mutations in chicken H5 (R47L, R74S, K97A, V80K, V87K) shifts its binding mode, transitioning from on-dyad to off-dyad binding (Fig. 2). This demonstrates how individual residues regulate H1’s binding dynamics and influence chromatin structure [49,56,57]. R47, R74, and K97 are highly conserved in human H1 variants H1.0, H1.4, and H1.10, but whether they play determinant roles in the human H1 binding mode remains to be further validated [9].
3 The Role of Linker Histones in Cancer
The expression patterns of histone H1 variants, particularly their expression levels, are closely associated with cancer grading and invasiveness. For instance, two independent immunohistochemical studies on prostate adenocarcinoma biopsy samples have shown that H1.5 is highly expressed in high-grade tumors but significantly reduced in low-grade tumors [58,59]. This makes H1.5 a potential biomarker for assessing prostate cancer progression and aggressiveness [58,60]. Similarly, increased expression of H1.2 in pancreatic cancer has been associated with a worse prognosis, further reinforcing the notion that H1 variants could be potential markers of cancer severity [61]. In contrast, the expression of various H1 variants, including H1.0, H1.1, H1.4, and H1.10, is notably reduced in breast and ovarian cancers, suggesting that the loss of these variants may contribute to malignant transformation and progression [62,63]. H1.0, typically found in fully differentiated cells, is significantly reduced in breast cancer tissue, regardless of the tumor’s differentiation status. This reduction in H1.0 expression is associated with tumor malignancy [62]. Notably, H1.0 is an effective immunohistochemical marker for evaluating cancer cell proliferative activity, underscoring its relevance in cancer diagnostics and prognostics [64–66]. These findings collectively highlight the critical role of H1 variant expression in cancer biology. Alterations in the expression of specific H1 variants not only correlate with tumor grade and differentiation but also offer insights into the invasive potential and prognosis of various cancers. Consequently, H1 variants hold significant promise as indicators for cancer detection, progression monitoring, and potential therapeutic targets [67].
Histone mutations are frequently observed across various cancer types and significantly impact chromatin structure and gene expression [21–23,68]. While previous research has primarily focused on mutations in core histones, particularly at positions K36, K27, and G34 of H3 [69–71]. Recent studies have also begun to explore the molecular mechanisms underlying cancer mutations in linker histones [22]. Mutations in linker histones have been frequently identified across various cancers, such as skin cancer, prostate cancer, endometrial cancer, and mature B-cell tumors [18,22,23]. Furthermore, multiple histone mutation databases have identified H1.2, H1.4, and H1.5 as the most commonly mutated histone variants [18,22,23]. Hotspot mutation sites are primarily located at A64 and A163 in both H1.4 and H1.2, while in H1.1, the hotspot mutation site is at A67 [23].
Large-scale analyses using pan-cancer atlas data from The Cancer Genome Atlas (TCGA) project have shown that B-cell lymphoma has the highest occurrence of mutant alleles in H1.1–H1.5 variants across all cancer types [18]. Interestingly, in B-cell lymphoma, most H1 mutations (97%) are missense variants that predominantly target the globular and C-terminal domains of H1 histones. Among the different isoforms, H1.2 and H1.4 are the most frequently affected [18]. The study also highlighted a significant co-occurrence between mutations in H1.2 and H1.4 as well as in other H1 alleles. Furthermore, 85% of mutations in the globular regions of H1.2–H1.5 affect key conserved residues at interaction sites, including the ASGS motif that directly binds DNA [18]. Another recent study identified heterozygous sequence variants in H1.4 associated with RMNS through clinical exome sequencing, with these mutations occurring in germline cells [15]. Most H1.4 variants were concentrated in the c.360–c.450 region of the CTD, resulting in frameshift mutations [15]. The predicted mutant protein exhibited an increased negative charge in comparison with the wild-type H1.4, implying that deleterious mutations in the CTD of H1.4 may impair H1-DNA interactions by reducing the positive charge in the tail regions [15].
In 2023, a large-scale analysis of publicly available whole-genome and exome data found that mutations in H1 are not limited to lymphomas, but also occur in a range of both pediatric and adult cancers, highlighting the widespread involvement of H1 mutations in cancer [22]. Notably, a mutation hotspot, K22 in the H1.4, was identified as one of the most frequently mutated histone residues. Additionally, H1 mutations were found to be clonal and repeatedly present in a minimum of two patients, with mutations such as H1.4 L42V, H1.5 K187N, and H1.2 P146S [22]. In the same year, Bennett and colleagues analyzed 205 unique cancer studies from cBioPortal, encompassing 65,489 patient samples [23]. Their analysis revealed that genetic changes, including point mutations, structural rearrangements, and variations in copy number, were present in H1 genes in approximately 7% of cancer patients. Further experimental work using embryonic stem cells from H1 knockout mice has demonstrated that the H1.2 S101F mutation acts as a loss-of-function mutation, significantly decreasing the binding affinity of H1 for chromatin [72]. Moreover, recent analyses suggest that mutations at G102A, S101F, and S103F in H1.2 may disrupt the DNA-binding region of the protein, impairing interactions between H1 and the nucleosome [23].
In our recent study, we investigated how cancer-related LH mutations impact nucleosome structure and interactions by analyzing whole-exome and whole-genome sequencing data from cBioPortal and our recently developed histone interaction network [21,73,74]. Our analysis revealed that although cancer-associated mutations in H1 are not the most prevalent among histone types, they still represent a relatively substantial proportion, accounting for 18.1% (Fig. 3a). Among the H1 subtypes, H1.4 exhibited the highest mutation frequency, contributing to 22.1% of all H1-related cancer mutations. In contrast, the mutation frequencies of H1.0 and H1.1 were relatively lower, at 3.4% and 3.1%, respectively (Fig. 3b). Notably, H1 mutations occur across a wide range of cancers, with the highest mutation frequencies observed in mature B-cell tumors, melanoma, and colorectal cancer (Fig. 3c). Our findings also suggest that cancer mutations in linker histones often decrease the number of positively charged residues at the binding interface while increasing negatively charged residues. This change could weaken the interaction between linker histones and negatively charged DNA, ultimately affecting chromatosome stability (Fig. 3d). The most commonly mutated sites in linker histones include G91D/A/C, G92V, and G94D/S (Fig. 3e). Overall, these findings highlight that LH mutations are significantly associated with various cancers and may disrupt chromatosome stability and higher-order chromatin structure by altering the physicochemical properties of linker histones.
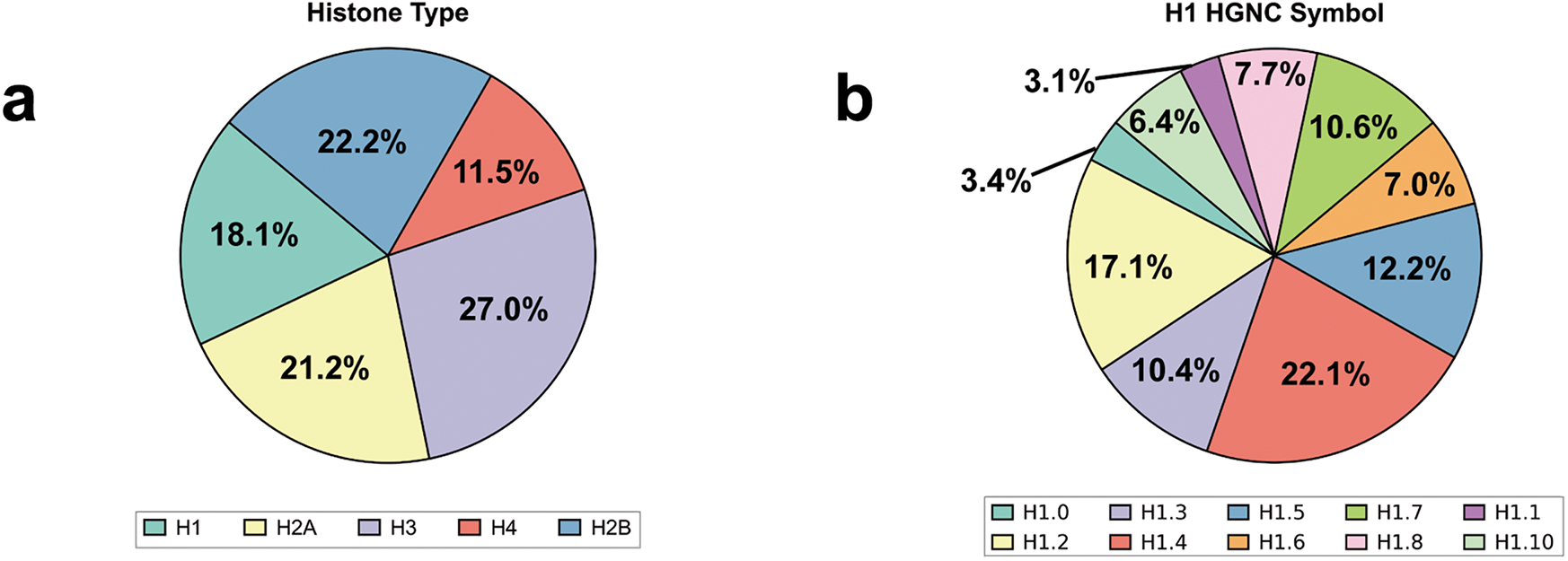
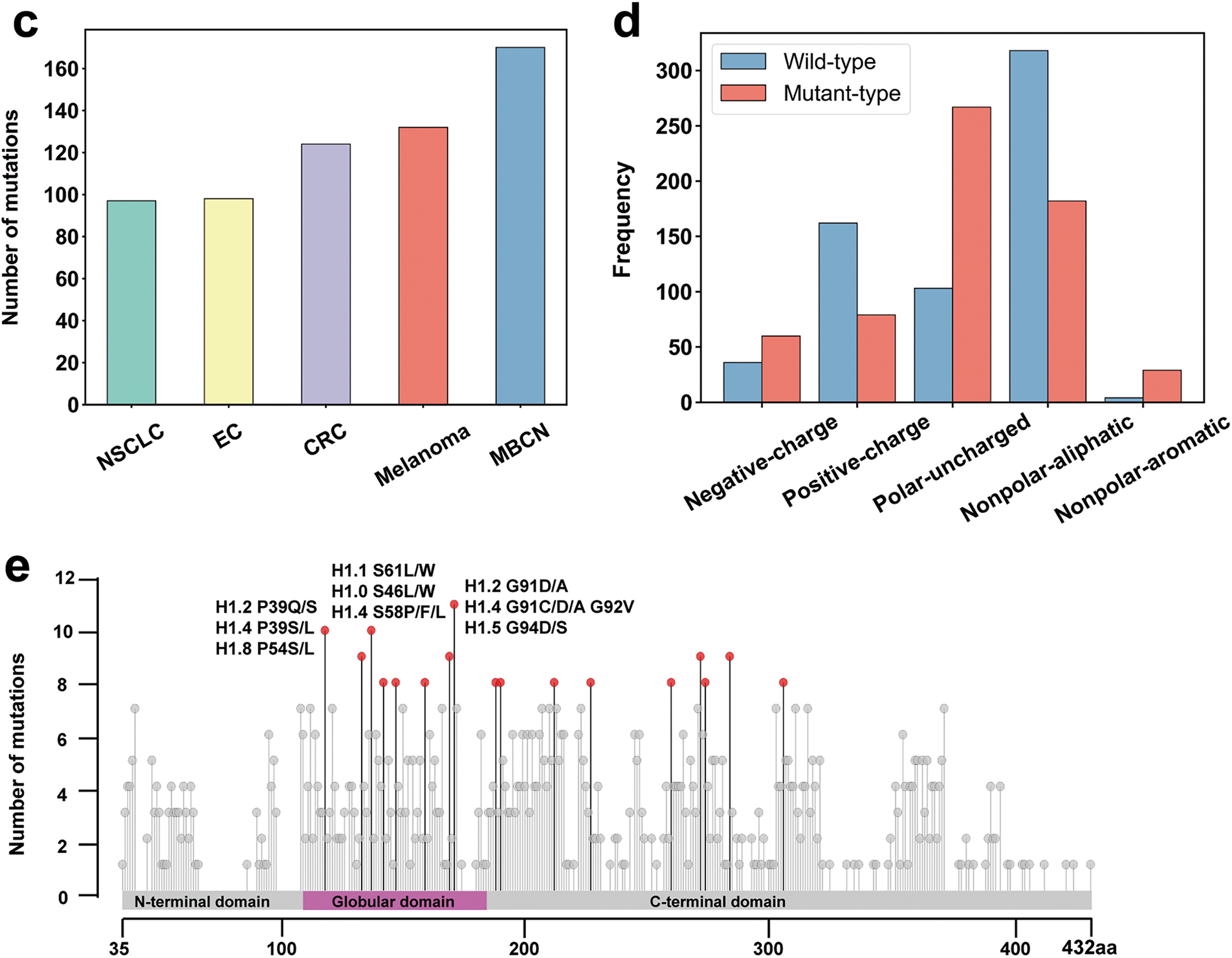
Figure 3: Statistical analysis of cancer-associated mutations in LH using the data from cBioPortal: (a) The proportion of cancer-associated mutations across different histone types in the mutation data; (b) The number of LH cancer-associated mutations across LH genes; (c) Number of LH cancer-associated mutations across the top five cancer types. NSCLC: Non-Small Cell Lung Cancer; EC: Endometrial Cancer; CRC: Colorectal Cancer; MBCN: Mature B-cell Neoplasms; (d) Number of wild-type and mutant residues in H1 mutation data categorized by physicochemical properties. The blue bars represent the quantity of each residue type in the wild-type H1 before mutation, while the red bars represent the quantity of each residue type in the mutant H1 following mutation. The differences between the blue and red bars for each residue type reflect the changes in residue numbers before and after mutation; (e) Mutations are mapped onto the consensus sequence of H1 from multiple sequence alignment. The x-axis shows the coordinates of the aligned consensus sequence (Fig. 1), while the mutation sites are annotated based on the coordinates of the original sequence. Residues with no less than eight mutations are marked with red dots, and the top three sites are labeled with their specific mutation types. The alignment was performed using Clustal Omega (v1.2.4) [29] and the data visualization was conducted using RStudio [75]
Recent computational studies have offered in-depth insights into how mutations affect the interactions between the globular domains of histone H1 subtypes (H1.2, H1.4 and H1.5) and nucleosomes [76]. By mapping the mutations within the H1.2 GD and its interaction sites with nucleosomal DNA onto its sequence, it was observed that certain mutations occur directly at the interaction interface (Fig. 4a). Notably, mutations S58F and S104F in H1.2 were found to significantly alter the binding free energy of H1 to nucleosomes [76]. The serine at position 104 of H1.2 interacts with DNA through hydrogen bonds, and the mutations S104F may disrupt the formation of these hydrogen bonds, thereby affecting the binding free energy of H1 to the nucleosome, potentially leading to decreased stability of H1 binding and subsequently impacting the compaction of chromatin structure (Fig. 4b). In the mutation dataset obtained from our recent study, we also found that the S104F mutation in H1.2 was observed in various cancers, including glioma, skin cancer, non-melanoma, and mature B-cell tumors [21]. Additionally, mutations in H1.2, including D72N, E74Q, I80M, S86I, and K109N, as well as mutations in H1.4, such as E42D and K75N, have been shown to alter the H1-nucleosome binding free energies, suggesting potential disruptions in H1’s ability to interact with the nucleosome [76].
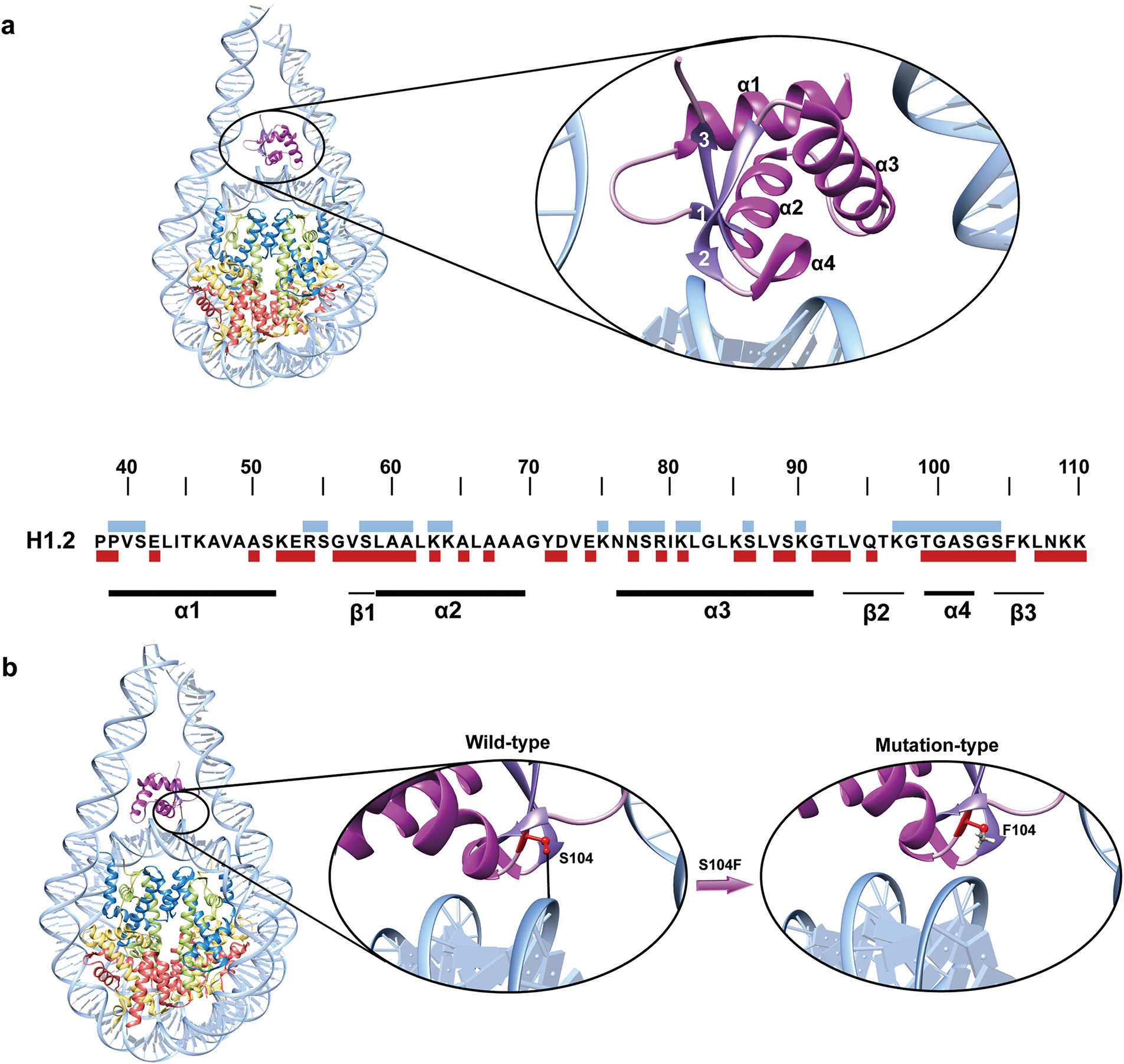
Figure 4: Cancer-associated mutations disrupt DNA-H1 interaction: (a) Mapping of cancer-associated mutations onto the DNA binding interfaces of H1.2. The sequence of the GD structure of H1.2 is shown, with blue blocks above the sequence representing sites that interact with DNA. The dark red blocks below the sequence correspond to mutation sites identified in our study. Secondary structures, including alpha helices and beta strands, are indicated by black lines beneath the sequence; (b) The S104F mutation in H1.2 disrupts hydrogen bonds, impairing the interaction between H1.2 and the nucleosome. Hydrogen bonds are depicted by black lines, mutation sites are highlighted in red, and histone H1.2 is shown in purple. The structural representation is generated using PDB entry 8HOV
4 Linker Histone Cancer Mutations Disrupt the Higher-Order Chromatin Structure
Histone H1 is crucial for regulating chromatin states and is believed to influence cellular transformation by facilitating higher-order chromatin structure formation [11]. Recent research reveals that mutations in H1 variants can impair its function, leading to significant structural effects on chromatin organization [46,72]. Specifically, these mutations can lead to the loss of H1 function, triggering both global and local transitions from a tightly packed to a more relaxed chromatin state, ultimately resulting in extensive structural remodeling of the genome [18,19,76].
In a recent study, transmission electron microscopy was used to investigate chromatin structure in H1-deficient lymphocytes, revealing notable changes in nuclear morphology, including nuclear enlargement and decreased signal density within heterochromatin. These findings indicate that H1 deficiency promotes chromatin decompaction [18]. A recent study on follicular lymphoma progression identified recurrent somatic mutations, including the S102F alteration in H1.2 [72]. This mutation significantly weakens the interaction between H1.2 and chromatin compared to the wild-type protein. The reduced binding of H1.2 to chromatin results in chromatin decompaction, which may disrupt gene expression and PTMs [72]. However, the precise molecular mechanisms by which these changes affect the expression of specific genes remain incompletely understood. Moreover, a study employing Mg2+ precipitation of 12-mer nucleosome arrays combined with atomic force microscopy revealed that chromatin compaction was markedly impaired in H1.2 variants harboring C-terminal mutations, as compared to the wild-type protein [18]. Additionally, H1 deficiency has been associated with a decrease in nucleosome repeat length (NRL), a critical factor in determining nucleosome array compaction [18,77]. Studies have demonstrated that shorter NRLs enhance the formation of highly ordered nucleosome-nucleosome stacks, which are less affected by LH binding [78]. This leads to less compact chromatin fibers with diameters of approximately 21 nm. In contrast, longer NRLs, in conjunction with LH binding, facilitate the packing of nucleosome arrays into tightly folded, regular fibers with diameters from 33 to 35 nm [78].
Recent studies have further underscored the crosstalk of LH H1 with various proteins associated with cancer progression [17,73,79,80]. For instance, H1 interacts with PTEN, a key tumor suppressor that is often inactivated in many cancers. Evidence suggests that H1 and PTEN cooperate to silence cancer-promoting genes [17,79]. Specifically, PTEN directly interacts with the C-terminal region of H1, stabilizing the association of the histone H1 and heterochromatin protein 1α (H1-HP1α) complex with chromatin and thereby regulating H1’s chromatin binding. In the absence of PTEN, the H1-HP1α complex dissociates from chromatin, resulting in reduced H1 binding [79]. PTEN physically interacts with multiple H1 variants, enhancing their chromatin binding, and H1 may contribute to mediating PTEN’s tumor-suppressive functions within the nucleus [79]. The absence of PTEN causes H1 to detach from chromatin, resulting in global chromatin decondensation and a reduction in chromatin stability. Moreover, the metastasis-associated protein 1 (MTA1), which plays a role in enhancing malignant traits such as invasion and metastasis, displaces H1 from chromatin when overexpressed [80]. It competes with H1 for binding to chromatin in a concentration-dependent manner, weakening the H1-chromatin interactions and promoting cancer-related phenotypes [17,81]. Notably, MTA1 is overexpressed in various cancers, including breast, ovarian, and gastric cancer [17,81]. This overexpression is closely linked to malignant progression, metastasis, and poor prognosis in cancer patients. Additionally, MTA1 overexpression can disrupt the interaction between H1 and chromatin, further contributing to tumorigenesis [80]. In addition, histone H1 interacts with the tumor suppressor p53, with this binding mediated by chromodomain helicase DNA-binding protein 8 (CHD8). Under genotoxic stress conditions, the interaction between H1 and p53 inhibits the transcriptional activation typically driven by p53, modulating the cellular response to stress [17,82]. However, abnormal expression of CHD8 in cancer can disrupt this interaction, impairing the tumor-suppressive functions of p53. Interestingly, CHD8 also inhibits the Wnt/β-catenin signaling pathway by recruiting histone H1 to Wnt-responsive elements, thereby suppressing the transcriptional activation of β-catenin. While the exact role of H1 in regulating β-catenin activity remains unclear, possibly involving inhibition of T-cell factor/Lymphoid enhancer-binding factor (TCF/LEF)-bound gene expression, this pathway is known to play a pivotal role in cancer development and progression. In this context, the CHD8-H1 interaction may have potential anti-cancer effects [17,83].
In germinal center B cells of B-cell lymphoma, the loss of H1 induces extensive chromatin decompaction and significant alterations in the distribution of PTMs of core histones [18,84]. The hallmark of chromatin decompaction is the global transition from B to A compartments. Compartment B, characterized by dense, transcriptionally silent chromatin, shifts to a more open configuration resembling compartment A, where chromatin is transcriptionally poised or active [18]. This is reflected by an increase in H3K36 monomethylation and dimethylation, while H3K27 dimethylation and trimethylation show a significant decrease [7,18]. Notably, the increase in H3K36me2 aligns more closely with the transition from compartment B to compartment A compared to the reduction of H3K27me3 [18]. In compartment B of chromatin, both marks are generally absent, with H3K27me3 detected only in the least compact regions [18]. In compartment A of chromatin, H3K36me2 progressively increases, while H3K27me3 is significantly reduced in highly decompacted regions [18]. These dynamic changes suggest that the distribution of H3K36me2 and H3K27me3 marks effectively reflects the degree of chromatin decompaction [18]. H3K27me modification serves as a pivotal marker of transcriptional repression in facultative heterochromatin, essential for gene silencing throughout development [85]. Moreover, chromatin decompaction is accompanied by the activation of genes that are typically silenced by polycomb repressive complex 2 (PRC2) during hematopoietic differentiation [7,18]. Although the precise mechanisms by which the loss of H1 modulates PRC2 activity in vivo remain unclear, dysregulation of PRC2 is associated with a wide range of cancers, suggesting its potential involvement in lymphoma [86]. A recent study found that PRC2 mutations in follicular lymphoma result in significant downregulation of histone genes, especially H1.2, but their effects on H1’s chromatin compaction ability remains unclear [87]. These findings highlight the critical role of H1 in chromatin compaction and suggest that its loss leads to chromatin decompaction. This alteration not only changes the physical structure of chromatin but may also have significant effects on gene regulation and expression. The loss of H1 affects a specific subset of genes, primarily those associated with stem cell characteristics, immune response, and chromatin regulation [18,88–90]. However, the changes in gene expression are not uniform, as not all genes are upregulated; for example, enhancers of zeste homolog 2 (EZH2) target genes remain unaffected [18]. Moreover, the loss of H1 also disrupts signaling pathways related to immune response and histone modifications, underscoring the critical role of H1 in these biological processes [18].
5 Cancer Mutations Disrupt Post-Translational Modifications in Linker Histone
A growing body of research highlights the crucial role of PTMs of linker histones in regulating chromatin compaction during the cell cycle, responding to DNA damage, and influencing cell differentiation [91,92]. Among the most common PTMs in human linker histones are phosphorylation, methylation, and acetylation [14,93,94]. The interaction between H1 and chromatin is primarily driven by basic amino acids, while phosphorylation reduces H1’s positive charge, potentially increasing its dissociation constant and enhancing chromatin accessibility [93,95]. In contrast, methylation of H1 is commonly linked to transcriptional repression [93]. A study using mass spectrometry analysis identified methylation modifications at the K34/35, K64/65, and K75 sites in different H1 variants [93]. Lysine modifications in the GD of H1 histone are commonly located near its DNA-binding sites, including K34, K52, K64, K85, and K97 [96,97]. Methylation at these sites can protect the ε-amino group of lysine and increase the affinity of the histone for DNA, potentially facilitating the transition of chromatin into a locally repressed state [93]. The K26 site in H1.4 is particularly noteworthy as it can be both acetylated and methylated (Fig. 5). Methylation at this site facilitates the binding of Heterochromatin Protein 1 (HP1) to H1.4, contributing to heterochromatin formation and transcriptional repression [93]. Conversely, acetylation at this site may prevent methylation, inhibiting HP1 binding to H1.4, which promotes chromatin opening and transcriptional activation [93].
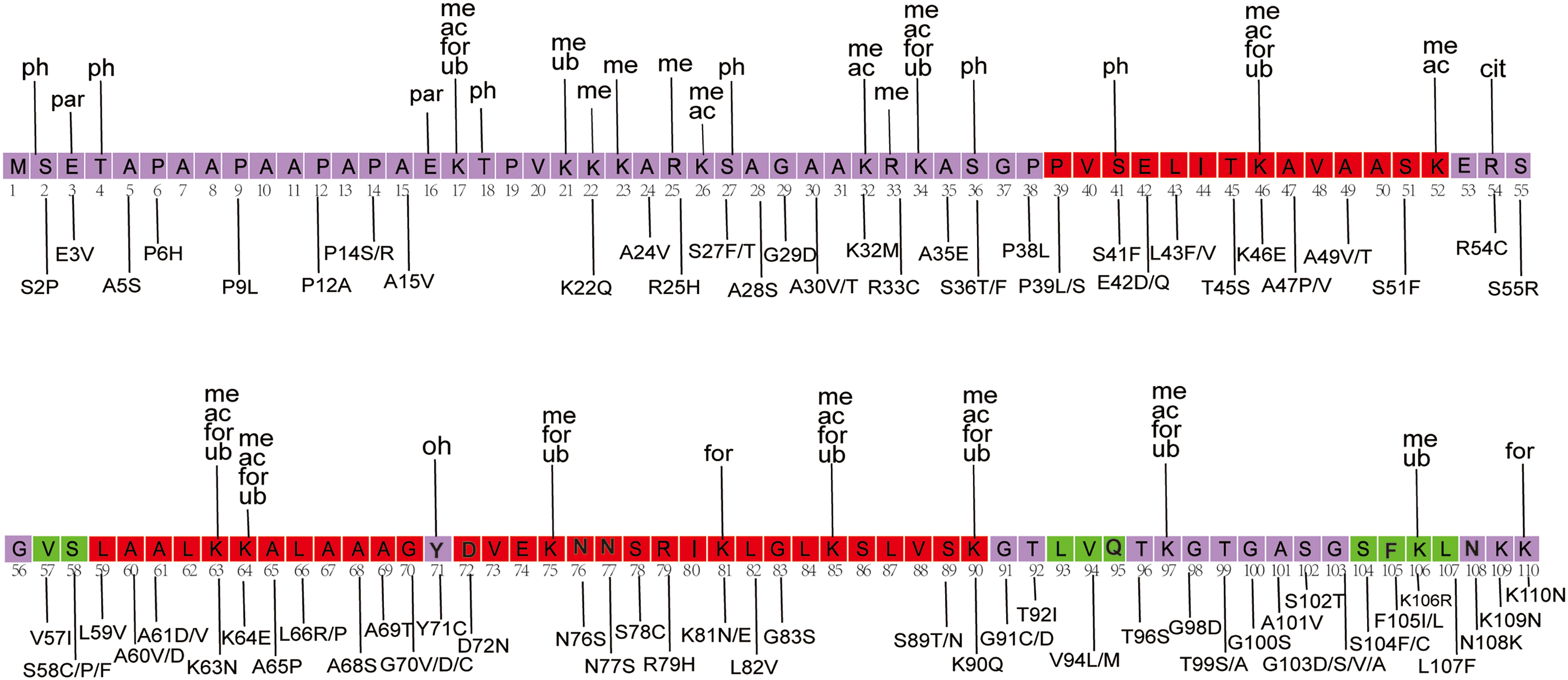
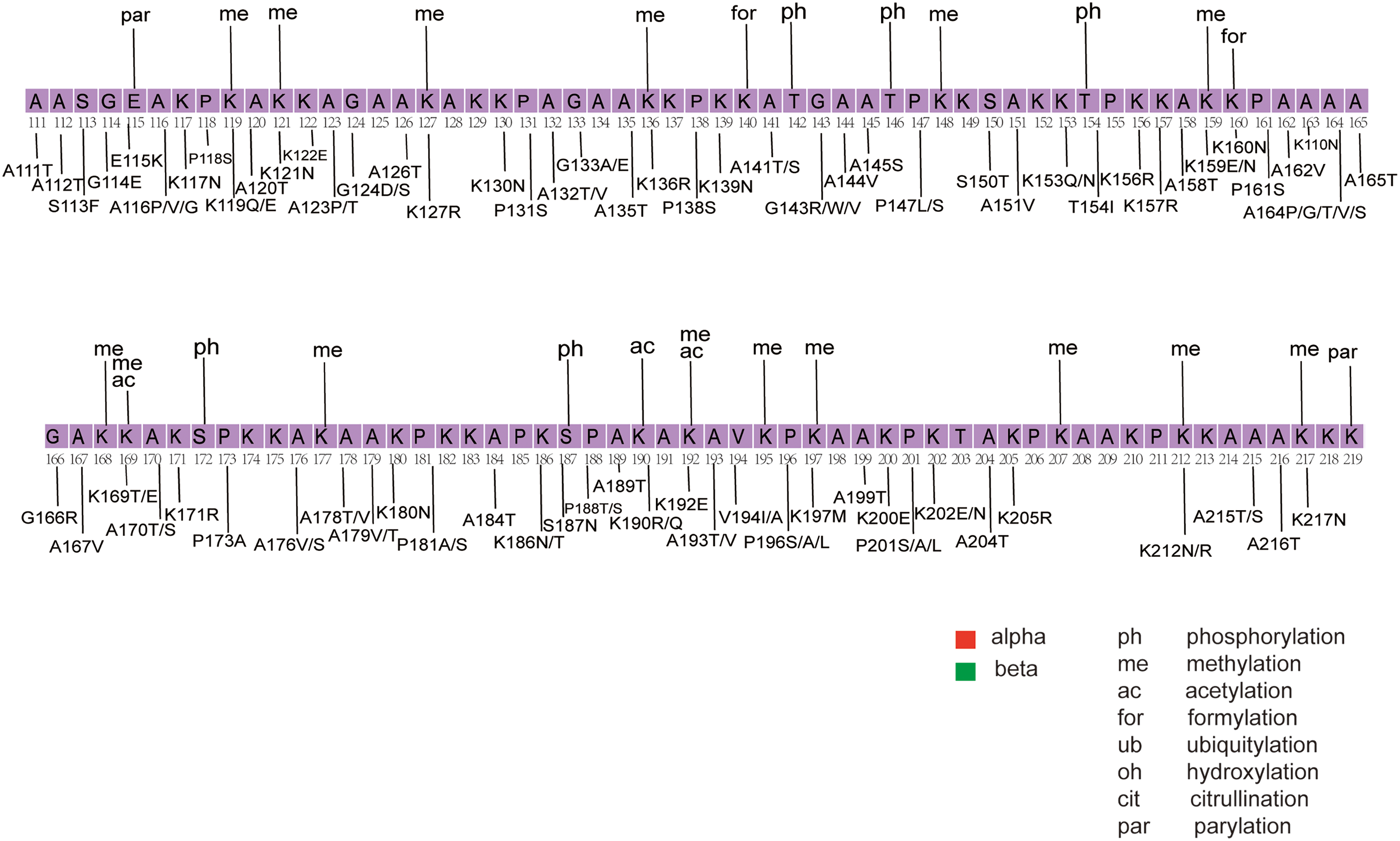
Figure 5: Mapping of PTMs and cancer-associated mutations onto the H1.4 sequence. The figure was created using Adobe Illustrator [98]. The structural sequence of H1.4 is obtained from the PDB structural database. The main structural sequence is highlighted within the purple box, with red and green indicating alpha helices and beta strands, respectively. PTM types are labeled above the sequence, with data sourced from existing literature [14]. Mutation types are labeled below the sequence, with mutation data derived from our dataset and previous studies [21]
PTMs of linker histones are also closely linked to cancers. For example, a study analyzing changes in histone modifications during bladder cancer progression found significant variations in the phosphorylation levels of histone H1 among normal cells, non-invasive cancer cells, and invasive cancer cells [99]. When comparing non-invasive low-grade and invasive high-grade bladder cancer cell lines, it was observed that the phosphorylation level of H1 at the T146 site gradually increased from low-grade to high-grade cancer cells [99]. Additionally, research has suggested that cyclin dependent kinase (CDK)-dependent phosphorylation may serve as an important marker for cell proliferation and tumor formation, further supporting the key role of phosphorylation modifications in the development of bladder cancer [99]. Recent studies have also revealed that MTA1 inhibits H1.2 phosphorylation at T145 by promoting the breakdown of DNA-PK, a mechanism linked to the initiation and metastatic progression of hepatocellular carcinoma (HCC) [100]. In addition, wolf-hirschhorn syndrome candidate 1 (WHSC1)-mediated mono-methylation of H1.4K85 in squamous cell carcinoma of the head and neck may contribute to the enhancement of stemness traits in cancer cells [101].
Recent studies have highlighted the intricate crosstalk between H1 PTMs and core histone PTMs, emphasizing their roles in cancer progression [102–104]. For example, studies have demonstrated that G9a histone methyltransferase (HMT), which typically mediates dimethylation of histone H3 at lysine 9 (H3K9me2), can also methylate histone H1.4 at lysine K26 in vivo. This modification is crucial for chromatin condensation and heterochromatin formation [103]. More importantly, studies have shown that in the context of histone H1, phosphorylation at H1.4S27 (H1.4S27ph) inhibits the interaction between HP1 and H1.4K26me2. Additionally, H1.4K26 competes with H3K9me3 for binding HP1, affecting its association with heterochromatin [105]. These nearby H1 phosphorylation events oppose the effects of histone lysine methylation. Additionally, recent research shows that the distribution of phosphorylated histone H1 and H3K9me3 in the nucleus remains consistent throughout the G1, S, and G2 phases of the cell cycle [106].
Recently, a comprehensive analysis using two-dimensional TAU/SDS electrophoresis combined with mass spectrometry was conducted to explore histone PTMs in cell lines derived from breast cancer [102]. In comparison to normal cells, cancer cells exhibited significant dysregulation in specific histone markers. Specifically, the levels of H4K16ac and H4K20me3 were reduced, while H3K9me2-3 levels were elevated, suggesting their potential involvement in the epigenetic regulation of cancer cells [102,107]. The study also focused on histone H1, revealing significant increases in tyrosine phosphorylation, particularly at Y74 of H1.5, Y70 of H1.2, and Y71 of H1.3 [102]. This modification was closely linked to cell proliferation and was regulated by Focal Adhesion Kinase (FAK), a tyrosine kinase with nuclear localization capability [108,109]. FAK was found to directly interact with H1 and catalyze its tyrosine phosphorylation [102]. FAK is often overexpressed in HCC [110]. Silencing FAK significantly suppressed EZH2’s activity in tri-methylating histone H3 lysine 27 (H3K27me3) [110]. These findings suggest that tyrosine phosphorylation of H1 may indirectly influence H3K27me3 levels by regulating EZH2 activity. In human HCC, the expression levels of FAK, EZH2, and H3K27me3were significantly higher than those in non-tumor liver tissues, further validating the interplay between H1 and H3 PTMs and their critical role in cancer progression [110].
It is also important to note that numerous cancer-related mutations in linker histones can interfere with the processes of reading, writing, and erasing histone modifications. For example, a recent study used protein sequence-based prediction tools to explore the impact of mutations on common PTMs in the histone H1 sequence, such as acetylation, methylation, phosphorylation, and ubiquitination. The study found that the stability of proteins with mutations at modification sites was significantly reduced, indicating that these mutations may disrupt the normal function of these PTMs [111]. The c.435dupC mutation is a germline mutation and is relatively common in RMNS. This mutation introduces a premature stop codon, resulting in a protein that is 25 amino acids shorter than the normal protein, thereby causing the loss of the phosphorylation site at position 146 of H1 [112]. T146 is a phosphorylation site in the human LH H1.4 subtype and is also present across other somatic subtypes (H1.2–H1.5) [99,100]. In bladder cancer, phosphorylation of the T146 site in H1.2-H1.5 has been proposed as a potential biomarker, suggesting that disruption of this critical PTM may impair the normal function of the protein [99].
By mapping the cancer mutations and PTMs sites onto the H1.4 sequences, we show that several mutations occur at known PTM sites (Fig. 5) [14,21]. The results showed that approximately 50% of the PTM sites overlapped with mutations. Several of these sites undergo multiple PTMs, including K17, K34, K46, K63, K64, K85, K90, and K97. Notably, mutations such as K46E and K63N were identified in endometrial cancer, while K64E was detected in melanoma [21]. In addition, we observed that many mutations in mature B-cell neoplasms overlapped with or were located near PTM sites. Some of these overlapping mutations included R33C, K81E, K90Q, K110N, E115K, K119E, K136R, T154I, K160N, K169T, and S187N. Previous studies have suggested that H1 mutations may contribute to the pathogenesis of B-cell lymphoma [18]. Of particular interest is the phosphorylation of the H1.4 S187 site by CDK9, a crucial early step in gene activation that facilitates RNA polymerase release to initiate transcription elongation [104]. Mutations at this site could disrupt the normal PTM process, altering the physicochemical properties and molecular functions of H1, and potentially contributing to disease development.
Mutations in linker histones have emerged as significant contributors to various cancers, highlighting their potential as key players in tumorigenesis. These mutations compromise chromatin structural integrity by modifying the interactions between linker histones and nucleosomes, which in turn affects chromatin compaction and higher-order organization. Furthermore, PTMs of linker histones, including methylation, acetylation, and phosphorylation, are crucial in regulating chromatin accessibility and gene expression and are also closely linked to cancer development [101,111]. Recent studies have uncovered the specific mutations in linker histones that disrupt these processes, providing valuable insights into the molecular mechanisms underlying cancer progression [18].
The emerging significance of LH mutations in cancer biology opens new opportunities for future research. Although substantial progress has been achieved in elucidating the structural and functional impacts of these mutations, there remains a need for deeper exploration of the molecular mechanisms by which linker histones influence the gene expression landscape and oncogenesis. Advancements in high-throughput sequencing technologies and computational models are likely to facilitate the identification of additional mutations and the analysis of their functional consequences. Moreover, studying the impact of LH mutations on their interactions with other chromatin-associated proteins, as well as their influence on the 3D organization of the genome, could provide critical insights into the epigenetic regulation of gene expression in cancer. These efforts could also facilitate the development of targeted therapies designed to modulate LH function, providing new treatment strategies for cancers driven by these mutations.
Acknowledgement: None.
Funding Statement: This work was supported by the National Natural Science Foundation of China (No. 12205112) and financially supported by self-determined research funds of CCNU from the colleges’ basic research and operation of MOE (CCNU24JC012) and supported by Natural Science Foundation of Wuhan (No. 2024040801020302).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Yunhui Peng; data collection: Gege Liu and Houfang Zhang; analysis and interpretation of results: Yunhui Peng, Gege Liu and Houfang Zhang; draft manuscript preparation: Yunhui Peng, Gege Liu and Houfang Zhang. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article. Additional data that support the findings of this study are available on request from the corresponding author.
Ethics Approval: This review did not involve human participants or animal experiments, therefore ethics approval and consent to participate were not required.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Stewart-Morgan KR, Petryk N, Groth A. Chromatin replication and epigenetic cell memory. Nat Cell Biol. 2020;22(4):361–71. doi:10.1038/s41556-020-0487-y. [Google Scholar] [PubMed] [CrossRef]
2. Mohan C, Das C, Tyler J. Histone and chromatin dynamics facilitating DNA repair. DNA Repair. 2021;107:103183. doi:10.1016/j.dnarep.2021.103183. [Google Scholar] [PubMed] [CrossRef]
3. Ren M, Greenberg MM, Zhou C. Participation of histones in DNA damage and repair within nucleosome core particles: mechanism and applications. Acc Chem Res. 2022;55(7):1059–73. doi:10.1021/acs.accounts.2c00041. [Google Scholar] [PubMed] [CrossRef]
4. Zaib S, Rana N, Khan I. Histone modifications and their role in epigenetics of cancer. Curr Med Chem. 2022;29(14):2399–411. doi:10.2174/0929867328666211108105214. [Google Scholar] [PubMed] [CrossRef]
5. McGinty RK, Tan S. Nucleosome structure and function. Chem Rev. 2015;115(6):2255–73. doi:10.1021/cr500373h. [Google Scholar] [PubMed] [CrossRef]
6. Li W, Hu J, Song F, Yu J, Peng X, Zhang S, et al. Structural basis for linker histone H5-nucleosome binding and chromatin fiber compaction. Cell Res. 2024;34(10):707–24. doi:10.1038/s41422-024-01009-z. [Google Scholar] [PubMed] [CrossRef]
7. Willcockson MA, Healton SE, Weiss CN, Bartholdy BA, Botbol Y, Mishra LN, et al. H1 histones control the epigenetic landscape by local chromatin compaction. Nature. 2021;589(7841):293–8. doi:10.1038/s41586-020-3032-z. [Google Scholar] [PubMed] [CrossRef]
8. Chen P, Li W, Li G. Structures and functions of chromatin fibers. Annu Rev Biophys. 2021;50(1):95–116. doi:10.1146/annurev-biophys-062920-063639. [Google Scholar] [PubMed] [CrossRef]
9. Zhou B-R, Feng H, Kale S, Fox T, Khant H, de Val N, et al. Distinct structures and dynamics of chromatosomes with different human linker histone isoforms. Mol Cell. 2021;81(1):166–82. e6. doi:10.1016/j.molcel.2020.10.038. [Google Scholar] [PubMed] [CrossRef]
10. Prendergast L, Reinberg D. The missing linker: emerging trends for H1 variant-specific functions. Genes Dev. 2021;35(1–2):40–58. doi:10.1101/gad.344531.120. [Google Scholar] [PubMed] [CrossRef]
11. Fyodorov DV, Zhou B-R, Skoultchi AI, Bai Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol. 2018;19(3):192–206. doi:10.1038/nrm.2017.94. [Google Scholar] [PubMed] [CrossRef]
12. Sokolova V, Sarkar S, Tan D. Histone variants and chromatin structure, update of advances. Comput Struct Biotechnol J. 2023;21:299–311. doi:10.1016/j.csbj.2022.12.002. [Google Scholar] [PubMed] [CrossRef]
13. Saha A, Dalal Y. A glitch in the snitch: the role of linker histone H1 in shaping the epigenome in normal and diseased cells. Open Biol. 2021;11(8):210124. doi:10.1098/rsob.210124. [Google Scholar] [PubMed] [CrossRef]
14. Kumar A, Maurya P, Hayes JJ. Post-translation modifications and mutations of human linker histone subtypes: their manifestation in disease. Int J Mol Sci. 2023;24(2):1463. doi:10.3390/ijms24021463. [Google Scholar] [PubMed] [CrossRef]
15. Tremblay MW, Green MV, Goldstein BM, Aldridge AI, Rosenfeld JA, Streff H, et al. Mutations of the histone linker H1-4 in neurodevelopmental disorders and functional characterization of neurons expressing C-terminus frameshift mutant H1.4. Hum Mol Genet. 2021;31(9):1430–42. doi:10.1093/hmg/ddab321. [Google Scholar] [PubMed] [CrossRef]
16. Soshnev AA, Allis CD, Cesarman E, Melnick AM. Histone H1 mutations in lymphoma: a Link(er) between chromatin organization, developmental reprogramming, and cancer. Cancer Res. 2021;81(24):6061–70. doi:10.1158/0008-5472.CAN-21-2619. [Google Scholar] [PubMed] [CrossRef]
17. Scaffidi P. Histone H1 alterations in cancer. BBA-Gene Regul Mech. 2016;1859(3):533–9. doi:10.1016/j.bbagrm.2015.09.008. [Google Scholar] [PubMed] [CrossRef]
18. Yusufova N, Kloetgen A, Teater M, Osunsade A, Camarillo JM, Chin CR, et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature. 2021;589(7841):299–305. doi:10.1038/s41586-020-3017-y. [Google Scholar] [PubMed] [CrossRef]
19. Gokey NG, Ward JM, Milliman EJ, Deterding LJ, Trotter KW, Archer TK. The loss of the H1.4 linker histone impacts nascent transcription and chromatin accessibility. bioRxiv. 2023;7:382. doi:10.1101/2023.05.14.540702. [Google Scholar] [CrossRef]
20. Chen Q, Yang M, Duan X, Zhang J, Shi F, Chen R, et al. Linker Histone H1.4 inhibits the growth, migration and EMT process of non-small cell lung cancer by regulating ERK1/2 expression. Biochem Genet. 2024;63:1–16. doi:10.1007/s10528-024-10760-2. [Google Scholar] [PubMed] [CrossRef]
21. Xu W, Zhang H, Guo W, Jiang L, Zhao Y, Peng Y. Deciphering principles of nucleosome interactions and impact of cancer-associated mutations from comprehensive interaction network analysis. Brief Bioinform. 2024;25(2):bbad532. doi:10.1093/bib/bbad532. [Google Scholar] [PubMed] [CrossRef]
22. Bonner ER, Dawood A, Gordish-Dressman H, Eze A, Bhattacharya S, Yadavilli S, et al. Pan-cancer atlas of somatic core and linker histone mutations. npj Genomic Med. 2023;8(1):23. doi:10.1038/s41525-023-00367-8. [Google Scholar] [PubMed] [CrossRef]
23. Espinoza Pereira KN, Shan J, Licht JD, Bennett RL. Histone mutations in cancer. Biochem Soc Trans. 2023;51(5):1749–63. doi:10.1042/BST20210567. [Google Scholar] [PubMed] [CrossRef]
24. Kasinsky HE, Lewis JD, Dacks JB, Ausló J. Origin of H1 linker histones. FASEB J. 2001;15(1):34–42. doi:10.1096/fj.00-0237rev. [Google Scholar] [PubMed] [CrossRef]
25. Zhou B-R, Jiang J, Feng H, Ghirlando R, Xiao TS, Bai Y. Structural mechanisms of nucleosome recognition by linker histones. Mol Cell. 2015;59(4):628–38. doi:10.1016/j.molcel.2015.06.025. [Google Scholar] [PubMed] [CrossRef]
26. Ponte I, Andrés M, Jordan A, Roque A. Towards understanding the regulation of histone H1 somatic subtypes with OMICs. J Mol Biol. 2021;433(2):166734. doi:10.1016/j.jmb.2020.166734. [Google Scholar] [PubMed] [CrossRef]
27. Talbert PB, Henikoff S. Histone variants at a glance. J Cell Sci. 2021;134(6):jcs244749. doi:10.1242/jcs.244749. [Google Scholar] [PubMed] [CrossRef]
28. Luzhetskaya OP, Sedykh SE, Nevinsky GA. How human H1 histone recognizes DNA. Molecules. 2020;25(19):4556. doi:10.3390/molecules25194556. [Google Scholar] [PubMed] [CrossRef]
29. Madeira F, Madhusoodanan N, Lee J, Eusebi A, Niewielska A, Tivey AR, et al. The EMBL-EBI Job Dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 2024;52:gkae241. doi:10.1093/nar/gkae241. [Google Scholar] [PubMed] [CrossRef]
30. Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–91. doi:10.1093/bioinformatics/btp033. [Google Scholar] [PubMed] [CrossRef]
31. Woods DC, Wereszczynski J. Elucidating the influence of linker histone variants on chromatosome dynamics and energetics. Nucleic Acids Res. 2020;48(7):3591–604. doi:10.1093/nar/gkaa121. [Google Scholar] [PubMed] [CrossRef]
32. Leicher R, Osunsade A, Chua GN, Faulkner SC, Latham AP, Watters JW, et al. Single-stranded nucleic acid binding and coacervation by linker histone H1. Nat Struct Mol Biol. 2022;29(5):463–71. doi:10.1038/s41594-022-00760-4. [Google Scholar] [PubMed] [CrossRef]
33. Hendzel MJ, Lever MA, Crawford E, Th’ng JP. The C-terminal domain is the primary determinant of histone H1 binding to chromatin in vivo. J Biol Chem. 2004;279(19):20028–34. doi:10.1074/jbc.M400070200. [Google Scholar] [PubMed] [CrossRef]
34. Rudnizky S, Khamis H, Ginosar Y, Goren E, Melamed P, Kaplan A. Extended and dynamic linker histone-DNA Interactions control chromatosome compaction. Mol Cell. 2021;81(16):3410–21.e4. doi:10.1016/j.molcel.2021.06.006. [Google Scholar] [PubMed] [CrossRef]
35. Th’ng JPH, Sung R, Ye M, Hendzel MJ. H1 family histones in the nucleus: control of binding and localization by the C-terminal domain. J Biol Chem. 2005;280(30):27809–14. doi:10.1074/jbc.M501627200. [Google Scholar] [PubMed] [CrossRef]
36. Vyas P, Brown DT. N-and C-terminal domains determine differential nucleosomal binding geometry and affinity of linker histone isotypes H10 and H1c. J Biol Chem. 2012;287(15):11778–87. doi:10.1074/jbc.M111.312819. [Google Scholar] [PubMed] [CrossRef]
37. Pan C, Fan Y. Role of H1 linker histones in mammalian development and stem cell differentiation. BBA-Gene Regul Mech. 2016;1859(3):496–509. doi:10.1016/j.bbagrm.2015.12.002. [Google Scholar] [PubMed] [CrossRef]
38. Burge NL. Investigations of the mechanisms of transcription regulation by histone H1. USA: The Ohio State University; 2022. [Google Scholar]
39. Clausell J, Happel N, Hale TK, Doenecke D, Beato M. Histone H1 subtypes differentially modulate chromatin condensation without preventing ATP-dependent remodeling by SWI/SNF or NURF. PLoS One. 2009;4(10):e0007243. doi:10.1371/journal.pone.0007243. [Google Scholar] [PubMed] [CrossRef]
40. Bieluszewski T, Prakash S, Roulé T, Wagner D. The role and activity of SWI/SNF chromatin remodelers. Annu Rev Plant Biol. 2023;74(1):139–63. doi:10.1146/annurev-arplant-102820-093218. [Google Scholar] [PubMed] [CrossRef]
41. Lever MA, Th’ng JP, Sun X, Hendzel MJ. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature. 2000;408(6814):873–6. doi:10.1038/35048603. [Google Scholar] [PubMed] [CrossRef]
42. Peng Y, Li S, Landsman D, Panchenko AR. Histone tails as signaling antennas of chromatin. Curr Opin Struct Biol. 2021;67:153–60. doi:10.1016/j.sbi.2020.10.018. [Google Scholar] [PubMed] [CrossRef]
43. Öberg C, Belikov S. The N-terminal domain determines the affinity and specificity of H1 binding to chromatin. Biochem Biophys Res Commun. 2012;420(2):321–4. doi:10.1016/j.bbrc.2012.02.157. [Google Scholar] [PubMed] [CrossRef]
44. Sridhar A, Orozco M, Collepardo-Guevara R. Protein disorder-to-order transition enhances the nucleosome-binding affinity of H1. Nucleic Acids Res. 2020;48(10):5318–31. doi:10.1093/nar/gkaa285. [Google Scholar] [PubMed] [CrossRef]
45. Turner AL, Watson M, Wilkins OG, Cato L, Travers A, Thomas JO, et al. Highly disordered histone H1−DNA model complexes and their condensates. Proc Nat Acad Sci. 2018;115(47):11964–9. doi:10.1073/pnas.1805943115. [Google Scholar] [PubMed] [CrossRef]
46. Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones S, et al. Mutations in linker histone genes HIST1H1, OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood. 2014;123(10):1487–98. doi:10.1182/blood-2013-05-500264. [Google Scholar] [PubMed] [CrossRef]
47. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–74. doi:10.1126/science.1133427. [Google Scholar] [PubMed] [CrossRef]
48. Zhou B-R, Feng H, Kato H, Dai L, Yang Y, Zhou Y, et al. Structural insights into the histone H1-nucleosome complex. Proc Nat Acad Sci. 2013;110(48):19390–5. doi:10.1073/pnas.1314905110. [Google Scholar] [PubMed] [CrossRef]
49. Zhou B-R, Feng H, Ghirlando R, Li S, Schwieters CD, Bai Y. A small number of residues can determine if linker histones are bound on or off dyad in the chromatosome. J Mol Biol. 2016;428(20):3948–59. doi:10.1016/j.jmb.2016.08.016. [Google Scholar] [PubMed] [CrossRef]
50. Bednar J, Garcia-Saez I, Boopathi R, Cutter AR, Papai G, Reymer A, et al. Structure and dynamics of a 197 bp nucleosome in complex with linker histone H1. Mol Cell. 2017;66(3):384–97.e8. doi:10.1016/j.molcel.2017.04.012. [Google Scholar] [PubMed] [CrossRef]
51. Hao F, Kale S, Dimitrov S, Hayes JJ. Unraveling linker histone interactions in nucleosomes. Curr Opin Struct Biol. 2021;71:87–93. doi:10.1016/j.sbi.2021.06.001. [Google Scholar] [PubMed] [CrossRef]
52. Pachov GV, Gabdoulline RR, Wade RC. On the structure and dynamics of the complex of the nucleosome and the linker histone. Nucleic Acids Res. 2011;39(12):5255–63. doi:10.1093/nar/gkr101. [Google Scholar] [PubMed] [CrossRef]
53. Honorato RV, Trellet ME, Jiménez-García B, Schaarschmidt JJ, Giulini M, Reys V, et al. The HADDOCK2.4 web server for integrative modeling of biomolecular complexes. Nat Protoc. 2024;19:1–23. doi:10.1038/s41596-024-01011-0. [Google Scholar] [PubMed] [CrossRef]
54. Honorato RV, Koukos PI, Jiménez-García B, Tsaregorodtsev A, Verlato M, Giachetti A, et al. Structural biology in the clouds: the WeNMR-EOSC ecosystem. Front Mol Biosci. 2021;8:729513. doi:10.3389/fmolb.2021.729513. [Google Scholar] [PubMed] [CrossRef]
55. Chikhirzhina E, Starkova TY, Polyanichko A. The role of linker histones in chromatin structural organization. 2. Interaction with DNA and Nuclear proteins. Biophysics. 2020;65:202–12. doi:10.1134/S0006350920020049. [Google Scholar] [CrossRef]
56. Wu H, Dalal Y, Papoian GA. Binding dynamics of disordered linker histone H1 with a nucleosomal particle. J Mol Biol. 2021;433(6):166881. doi:10.1016/j.jmb.2021.166881. [Google Scholar] [PubMed] [CrossRef]
57. Melters DP, Dalal Y. Nano-surveillance: tracking individual molecules in a sea of chromatin. J Mol Biol. 2021;433(6):166720. doi:10.1016/j.jmb.2020.11.019. [Google Scholar] [PubMed] [CrossRef]
58. Behrends M, Engmann O. Linker histone H1.5 is an underestimated factor in differentiation and carcinogenesis. Environ Epigenet. 2020;6(1):dvaa013. doi:10.1093/eep/dvaa013. [Google Scholar] [PubMed] [CrossRef]
59. El-Rashidy MA, Bedeer AE, Kabel AM. Histone H1.5 expression in prostatic carcinoma: an immunohistochemical study. J Cancer Res Treat. 2016;4:21–5. doi:10.1016/j.humpath.2014.06.015. [Google Scholar] [PubMed] [CrossRef]
60. Hechtman JF, Beasley MB, Kinoshita Y, Ko HM, Hao K, Burstein DE. Promyelocytic leukemia zinc finger and histone H1.5 differentially stain low-and high-grade pulmonary neuroendocrine tumors: a pilot immunohistochemical study. Hum Pathol. 2013;44(7):1400–5. doi:10.1016/j.humpath.2012.11.014. [Google Scholar] [PubMed] [CrossRef]
61. Zhou S, Yan Y, Chen X, Zeng S, Wei J, Wang X, et al. A two-gene-based prognostic signature for pancreatic cancer. Aging. 2020;12(18):18322. doi:10.18632/aging.103698. [Google Scholar] [PubMed] [CrossRef]
62. Medrzycki M, Zhang Y, McDonald JF, Fan Y. Profiling of linker histone variants in ovarian cancer. Front Biosci. 2012;17:396. doi:10.2741/3934. [Google Scholar] [PubMed] [CrossRef]
63. Noberini R, Morales Torres C, Savoia EO, Brandini S, Jodice MG, Bertalot G, et al. Label-free mass spectrometry-based quantification of linker histone H1 variants in clinical samples. Int J Mol Sci. 2020;21(19):7330. doi:10.3390/ijms21197330. [Google Scholar] [PubMed] [CrossRef]
64. Lashen AG, Almalki N, Toss M, Mirza S, Malki MI, Rutland CS, et al. The characteristics and prognostic significance of histone H1 expression in breast cancer. Pathology. 2024;56:826–33. doi:10.1016/j.pathol.2024.03.012. [Google Scholar] [PubMed] [CrossRef]
65. Dhahri H, Saintilnord WN, Chandler D, Fondufe-Mittendorf YN. Beyond the usual suspects: examining the role of understudied histone variants in breast cancer. Int J Mol Sci. 2024;25(12):6788. doi:10.3390/ijms25126788. [Google Scholar] [PubMed] [CrossRef]
66. Morales Torres C, Wu MY, Hobor S, Wainwright EN, Martin MJ, Patel H, et al. Selective inhibition of cancer cell self-renewal through a Quisinostat-histone H1.0 axis. Nat Commun. 2020;11(1):1792. doi:10.1038/s41467-020-15615-z. [Google Scholar] [PubMed] [CrossRef]
67. Lyubitelev A, Kirpichnikov M, Studitsky V. The role of linker histones in carcinogenesis. Russ J Bioorg Chem. 2021;47:278–87. doi:10.1134/S1068162021010143. [Google Scholar] [CrossRef]
68. Espiritu D, Gribkova AK, Gupta S, Shaytan AK, Panchenko AR. Molecular mechanisms of oncogenesis through the lens of nucleosomes and histones. J Phys Chem B. 2021;125(16):3963–76. doi:10.1021/acs.jpcb.1c00694. [Google Scholar] [PubMed] [CrossRef]
69. Mitchener MM, Muir TW. Oncohistones: exposing the nuances and vulnerabilities of epigenetic regulation. Mol Cell. 2022;82(16):2925–38. doi:10.1016/j.molcel.2022.07.008. [Google Scholar] [PubMed] [CrossRef]
70. Ahmad K, Henikoff S. The H3. 3K27M oncohistone antagonizes reprogramming in Drosophila. PLoS Genet. 2021;17(7):e1009225. doi:10.1371/journal.pgen.1009225. [Google Scholar] [PubMed] [CrossRef]
71. Bryant L, Li D, Cox SG, Marchione D, Joiner EF, Wilson K, et al. Histone H3.3 beyond cancer: germline mutations in Histone 3 family 3A and 3B cause a previously unidentified neurodegenerative disorder in 46 patients. Sci Adv. 2020;6(49):eabc9207. doi:10.1126/sciadv.abc9207. [Google Scholar] [PubMed] [CrossRef]
72. Okosun J, Bödör C, Wang J, Araf S, Yang C-Y, Pan C, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46(2):176–81. doi:10.1038/ng.2856. [Google Scholar] [PubMed] [CrossRef]
73. Peng Y, Markov Y, Goncearenco A, Landsman D, Panchenko AR. Human histone interaction networks: an old concept, new trends. J Mol Biol. 2021;433(6):166684. doi:10.1016/j.jmb.2020.10.018. [Google Scholar] [PubMed] [CrossRef]
74. Zhang H, Guo W, Xu W, Li A, Jiang L, Li L, et al. Electrostatic interactions in nucleosome and higher-order structures are regulated by protonation state of histone ionizable residue; 2024. doi: 10.7554/eLife.100738.1. [Google Scholar] [CrossRef]
75. Kronthaler F, Zöllner S. Data analysis with RStudio. Data Anal RStudio. 2021. doi:10.1007/978-3-662-62518-7. [Google Scholar] [CrossRef]
76. Bass M, Armeev G, Shaitan K, Shaytan A. The effect of oncomutations and posttranslational modifications of histone H1 on chromatosome structure and stability. Moscow Univ Biol Sci Bull. 2019;74:121–6. doi:10.3103/S0096392519030015. [Google Scholar] [CrossRef]
77. Dombrowski M, Engeholm M, Dienemann C, Dodonova S, Cramer P. Histone H1 binding to nucleosome arrays depends on linker DNA length and trajectory. Nat Struct Mol Biol. 2022;29(5):493–501. doi:10.1038/s41594-022-00768-w. [Google Scholar] [PubMed] [CrossRef]
78. Routh A, Sandin S, Rhodes D. Nucleosome repeat length and linker histone stoichiometry determine chromatin fiber structure. Proc Nat Acad Sci. 2008;105(26):8872–7. doi:10.1073/pnas.0802336105. [Google Scholar] [PubMed] [CrossRef]
79. Chen ZH, Zhu M, Yang J, Liang H, He J, He S, et al. PTEN interacts with histone H1 and controls chromatin condensation. Cell Rep. 2014;8(6):2003–14. doi:10.1016/j.celrep.2014.08.008. [Google Scholar] [PubMed] [CrossRef]
80. Toh Y, Nicolson GL. The role of the MTA family and their encoded proteins in human cancers: molecular functions and clinical implications. Clin Exp Metastasis. 2009;26:215–27. doi:10.1007/s10585-008-9233-8. [Google Scholar] [PubMed] [CrossRef]
81. Liu J, Wang H, Ma F, Xu D, Chang Y, Zhang J, et al. MTA1 regulates higher-order chromatin structure and histone H1-chromatin interaction in-vivo. Mol Oncol. 2015;9(1):218–35. doi:10.1016/j.molonc.2014.08.007. [Google Scholar] [PubMed] [CrossRef]
82. Nishiyama M, Oshikawa K, Tsukada Y-I, Nakagawa T, Iemura S-I, Natsume T, et al. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat Cell Biol. 2009;11(2):172–82. doi:10.1038/ncb1831. [Google Scholar] [PubMed] [CrossRef]
83. Nishiyama M, Skoultchi AI, Nakayama KI. Histone H1 recruitment by CHD8 is essential for suppression of the Wnt–β-catenin signaling pathway. Mol Cell Biol. 2012;32(2):501–12. doi:10.1128/MCB.06409-11. [Google Scholar] [PubMed] [CrossRef]
84. Bakhshi TJ, Georgel PT. Genetic and epigenetic determinants of diffuse large B-cell lymphoma. Blood Cancer J. 2020;10(12):123. doi:10.1038/s41408-020-00389-w. [Google Scholar] [PubMed] [CrossRef]
85. Poepsel S, Kasinath V, Nogales E. Cryo-EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. Nat Struct Mol Biol. 2018;25(2):154–62. doi:10.1038/s41594-018-0023-y. [Google Scholar] [PubMed] [CrossRef]
86. Nacev BA, Dabas Y, Paul MR, Pacheco C, Mitchener M, Perez Y, et al. Cancer-associated Histone H3 N-terminal arginine mutations disrupt PRC2 activity and impair differentiation. Nat Commun. 2024;15(1):5155. doi:10.1038/s41467-024-49486-5. [Google Scholar] [PubMed] [CrossRef]
87. Liu X, Liu X. PRC2, chromatin regulation, and human disease: insights from molecular structure and function. Front Oncol. 2022;12:894585. doi:10.3389/fonc.2022.894585. [Google Scholar] [PubMed] [CrossRef]
88. Serna-Pujol N, Salinas-Pena M, Mugianesi F, Le Dily F, Marti-Renom MA, Jordan A. Coordinated changes in gene expression, H1 variant distribution and genome 3D conformation in response to H1 depletion. Nucleic Acids Res. 2022;50(7):3892–910. doi:10.1093/nar/gkac226. [Google Scholar] [PubMed] [CrossRef]
89. Sheikh AH, Nawaz K, Tabassum N, Almeida-Trapp M, Mariappan KG, Alhoraibi H, et al. Linker histone H1 modulates defense priming and immunity in plants. Nucleic Acids Res. 2023;51(9):4252–65. doi:10.1093/nar/gkad106. [Google Scholar] [PubMed] [CrossRef]
90. Mahadevan IA, Kumar S, Rao MRS. Linker histone variant H1t is closely associated with repressed repeat-element chromatin domains in pachytene spermatocytes. Epigenet Chromatin. 2020;13:1–19. doi:10.1186/s13072-020-00335-x. [Google Scholar] [PubMed] [CrossRef]
91. Chu C-S, Hsu P-H, Lo P-W, Scheer E, Tora L, Tsai H-J, et al. Protein kinase A-mediated serine 35 phosphorylation dissociates histone H1.4 from mitotic chromosome. J Biol Chem. 2011;286(41):35843–51. doi:10.1074/jbc.M111.228064. [Google Scholar] [PubMed] [CrossRef]
92. Höllmüller E, Geigges S, Niedermeier ML, Kammer K-M, Kienle SM, Rösner D, et al. Site-specific ubiquitylation acts as a regulator of linker histone H1. Nat Commun. 2021;12(1):3497. doi:10.1038/s41467-021-23636-5. [Google Scholar] [PubMed] [CrossRef]
93. Starkova TY, Polyanichko A, Artamonova T, Khodorkovskii M, Kostyleva E, Chikhirzhina E, et al. Post-translational modifications of linker histone H1 variants in mammals. Phys Biol. 2017;14(1):016005. doi:10.1088/1478-3975/aa551a. [Google Scholar] [PubMed] [CrossRef]
94. Andrés M, García-Gomis D, Ponte I, Suau P, Roque A. Histone H1 post-translational modifications: update and future perspectives. Int J Mol Sci. 2020;21(16):5941. doi:10.3390/ijms21165941. [Google Scholar] [PubMed] [CrossRef]
95. Zheng Y, John S, Pesavento JJ, Schultz-Norton JR, Schiltz RL, Baek S, et al. Histone H1 phosphorylation is associated with transcription by RNA polymerases I and II. J Cell Biol. 2010;189(3):407–15. doi:10.1083/jcb.201001148. [Google Scholar] [PubMed] [CrossRef]
96. Izzo A, Schneider R. The role of linker histone H1 modifications in the regulation of gene expression and chromatin dynamics. Biochim Biophys Acta, Gene Regul Mech. 2016;1859(3):486–95. doi:10.1016/j.bbagrm.2015.09.003. [Google Scholar] [PubMed] [CrossRef]
97. Lai S, Jia J, Cao X, Zhou P-K, Gao S. Molecular and cellular functions of the linker histone H1.2. Front Cell Dev Biol. 2022;9:773195. doi:10.3389/fcell.2021.773195. [Google Scholar] [PubMed] [CrossRef]
98. Hoppe J. Adobe illustrator. USA: Rocky Nook, Inc.; 2020. [Google Scholar]
99. Telu KH, Abbaoui B, Thomas-Ahner JM, Zynger DL, Clinton SK, Freitas MA, et al. Alterations of histone H1 phosphorylation during bladder carcinogenesis. J Proteome Res. 2013;12(7):3317–26. doi:10.1021/pr400143x. [Google Scholar] [PubMed] [CrossRef]
100. Li Y-H, Zhong M, Zang H-L, Tian X-F. MTA1 promotes hepatocellular carcinoma progression by downregulation of DNA-PK-mediated H1. 2T146 phosphorylation. Front Oncol. 2020;10:567. doi:10.3389/fonc.2020.00567. [Google Scholar] [PubMed] [CrossRef]
101. Saloura V, Vougiouklakis T, Bao R, Kim S, Baek S, Zewde M, et al. WHSC1 monomethylates histone H1 and induces stem-cell like features in squamous cell carcinoma of the head and neck. Neoplasia. 2020;22(8):283–93. doi:10.1016/j.neo.2020.05.002. [Google Scholar] [PubMed] [CrossRef]
102. Perri AM, Agosti V, Olivo E, Concolino A, De Angelis M, Tammè L, et al. Histone proteomics reveals novel post-translational modifications in breast cancer. Aging. 2019;11(23):11722. doi:10.18632/aging.102577. [Google Scholar] [PubMed] [CrossRef]
103. Kuzmichev A, Jenuwein T, Tempst P, Reinberg D. Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol Cell. 2004;14(2):183–93. doi:10.1016/S1097-2765(04)00185-6. [Google Scholar] [PubMed] [CrossRef]
104. Saha A, Seward CH, Stubbs L, Mizzen CA. Site-specific phosphorylation of histone H1.4 is associated with transcription activation. Int J Mol Sci. 2020;21(22):8861. doi:10.3390/ijms21228861. [Google Scholar] [PubMed] [CrossRef]
105. Healton SE, Pinto HD, Mishra LN, Hamilton GA, Wheat JC, Swist-Rosowska K, et al. H1 linker histones silence repetitive elements by promoting both histone H3K9 methylation and chromatin compaction. Proc Nat Acad Sci. 2020;117(25):14251–8. doi:10.1073/pnas.1920725117. [Google Scholar] [PubMed] [CrossRef]
106. Legartová S, Lochmanová G, Bártová E. The highest density of phosphorylated histone H1 appeared in prophase and prometaphase in parallel with reduced H3K9me3, and HDAC1 depletion increased H1.2/H1.3 and H1.4 Serine 38 Phosphorylation. Life. 2022;12(6):798. doi:10.3390/life12060798. [Google Scholar] [PubMed] [CrossRef]
107. Yokoyama Y, Matsumoto A, Hieda M, Shinchi Y, Ogihara E, Hamada M, et al. Loss of histone H4K20 trimethylation predicts poor prognosis in breast cancer and is associated with invasive activity. Breast Cancer Res. 2014;16:1–13. doi:10.1186/bcr3681. [Google Scholar] [PubMed] [CrossRef]
108. Chatzizacharias NA, Kouraklis GP, Theocharis SE. Clinical significance of FAK expression in human neoplasia. Histol Histopathol. 2008. doi: 10.14670/HH-23.629. [Google Scholar] [PubMed] [CrossRef]
109. Kleinschmidt EG, Schlaepfer DD. Focal adhesion kinase signaling in unexpected places. Curr Opin Cell Biol. 2017;45:24–30. doi:10.1016/j.ceb.2017.01.003. [Google Scholar] [PubMed] [CrossRef]
110. Gnani D, Romito I, Artuso S, Chierici M, De Stefanis C, Panera N, et al. Focal adhesion kinase depletion reduces human hepatocellular carcinoma growth by repressing enhancer of zeste homolog 2. Cell Death Differ. 2017;24(5):889–902. doi:10.1038/cdd.2017.34. [Google Scholar] [PubMed] [CrossRef]
111. Kowalski A. Sequence-based prediction of the effects of histones H1 post-translational modifications: impact on the features related to the function. J Biomol Struct Dyn. 2024;1–10. doi:10.1080/07391102.2024.2316773. [Google Scholar] [PubMed] [CrossRef]
112. Duffney LJ, Valdez P, Tremblay MW, Cao X, Montgomery S, McConkie–Rosell A, et al. Epigenetics and autism spectrum disorder: a report of an autism case with mutation in H1 linker histone HIST1H1E and literature review. Am J Med Genet Part B: Neuropsychiatr Genet. 2018;177(4):426–33. doi:10.1002/ajmg.b.32631. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools