
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Understanding Endoplasmic Reticulum Stress as a Central Driver of Atherosclerosis
1 Unit for Study of Aortic, Valvular and Coronary Pathologies, Centro Cardiologico Monzino IRCCS, via Carlo Parea 4, Milan, 20138, Italy
2 School of Medicine and Health Sciences, The George Washington University, Washington, DC 20037, USA
3 R&D Lab, Institute for Atherosclerosis Research, Osennyaya Street 4-1-207, Moscow, 121609, Russia
* Corresponding Author: Anastasia V. Poznyak. Email:
(This article belongs to the Special Issue: Molecular Basis for the Involvement of Inflammation and Lipids in Pathologies)
BIOCELL 2026, 50(3), 6 https://doi.org/10.32604/biocell.2025.074266
Received 07 October 2025; Accepted 21 November 2025; Issue published 23 March 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Atherosclerosis (AS) remains a major contributor to cardiovascular disease (CVD) mortality worldwide. Its development involves dysregulated lipid handling, persistent vascular inflammation, and endothelial cell (EC) dysfunction, influenced by genetic, environmental, and lifestyle factors. Increasing evidence highlights a pivotal role of endoplasmic reticulum (ER) stress as a molecular link between lipid dysregulation and inflammatory signaling in AS pathogenesis. ER stress is triggered by modified LDL, oxidized lipids, hyperhomocysteinemia, oxidative stress (OS), and disrupted calcium (Ca2+) homeostasis, leading to activation of the unfolded protein response (UPR). Core UPR mediators—inositol-requiring enzyme 1 (IRE1), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6)—initially act to restore ER homeostasis but, when persistently activated, may drive pro-inflammatory cytokine production, apoptosis, and plaque destabilization. The aim of this review is to critically synthesize primary research evidence on ER stress as a mediator of lipid-driven inflammation in ECs, macrophages, and vascular smooth muscle cells (VSMCs), emphasizing disease-stage–specific effects. Current debates include whether macrophage ER stress promotes necrotic core expansion vs. apoptosis-mediated clearance, and whether ER stress in ECs is initially protective or primarily pathogenic. Emerging therapeutic strategies targeting ER stress are summarized, including chemical chaperones, AMPK activators, and natural compounds. We highlight the importance of lipid- and inflammation-specific ER stress modulation, noting limitations such as off-target effects and poor bioavailability that hinder translation. Our goal is to achieve a deeper understanding of the lipid–ER stress–inflammation axis to facilitate the design of therapies that may slow AS progression.Keywords
Atherosclerosis (AS) is a major problem in modern medicine. As the leading cause of cardiovascular disease (CVD) progression and related mortality, its prevention and treatment remain a global priority. AS progression is an intricate process influenced by risk factors such as abnormal lipid metabolism, genetic predisposition, diet, smoking, and physical inactivity [1]. Lesions arise from lipid deposition, thickening of the vessel walls, and persistent vascular inflammation. Impaired endothelial function and increased permeability to lipids and proteins further accelerate plaque progression [2–4].
Low-density lipoproteins (LDL) are the major source of cholesterol deposition. Modified LDL particles—including oxidized, desialylated, and glycated forms—play a particularly pathogenic role by stimulating inflammatory responses. Elevated levels of such modified LDL in the bloodstream constitute a major risk factor for AS. Lesion development is closely linked to inflammatory processes and the recruitment of immune cells (ICs) [5,6]. Resident vascular cells (e.g., vascular smooth muscle cells (VSMCs)) and infiltrating macrophages also contribute to the pathogenesis.
Trials in young subjects have shown that AS development can begin early in life and remain clinically silent for decades. With ageing, atheromatous lesions may evolve into vulnerable plaques [7–9]. Such plaques can rupture and trigger thrombosis, leading to acute cardiovascular events. Therefore, anti-AS therapies must focus not only on late-stage interventions but also on early diagnosis and prevention, which in turn requires elucidation of the cellular and molecular mechanisms driving AS [10,11].
Emerging evidence indicates a major role of endoplasmic reticulum (ER) stress in all phases of AS. The ER is a large membranous organelle responsible for protein folding and maturation. It is also central to intracellular signaling, serving as the primary Ca2+ reservoir and maintaining Ca2+ homeostasis [12–14]. Disruption of ER function—for example, by excess lipid accumulation, oxidative stress, Ca2+ dysregulation, defective protein folding diseases (DPFDs), or increased protein synthesis—can lead to ER stress and activation of the unfolded protein response (UPR) [15,16].
Importantly, whether ER stress acts as an adaptive response that restores vascular homeostasis or as a maladaptive process that accelerates AS progression remains controversial. This duality underpins both the scientific interest and therapeutic challenges of the field, forming the basis of this review. Therefore, the aim of the present study is to critically evaluate the context-dependent roles of ER stress in atherosclerosis across different cell types and disease stages, and to highlight unresolved controversies with implications for targeted therapeutic strategies.
Proteins that fail to fold correctly in the ER are typically degraded through endoplasmic-reticulum-associated protein degradation (ERAD) in the cytosol. Because protein quality control in the ER is tightly regulated, accumulation of misfolded proteins activates theUPR [17,18].
Three canonical UPR sensors—inositol-requiring enzyme 1 (IRE1), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6)—monitor ER protein-folding status. Their luminal domains normally bind glucose-regulated protein 78 (GRP78/BiP), which keeps them inactive. During ER stress, GRP78 dissociates, thereby activating these sensors and initiating signaling cascades [19,20]. An overview of ER stress and the canonical UPR pathways is presented in Fig. 1.
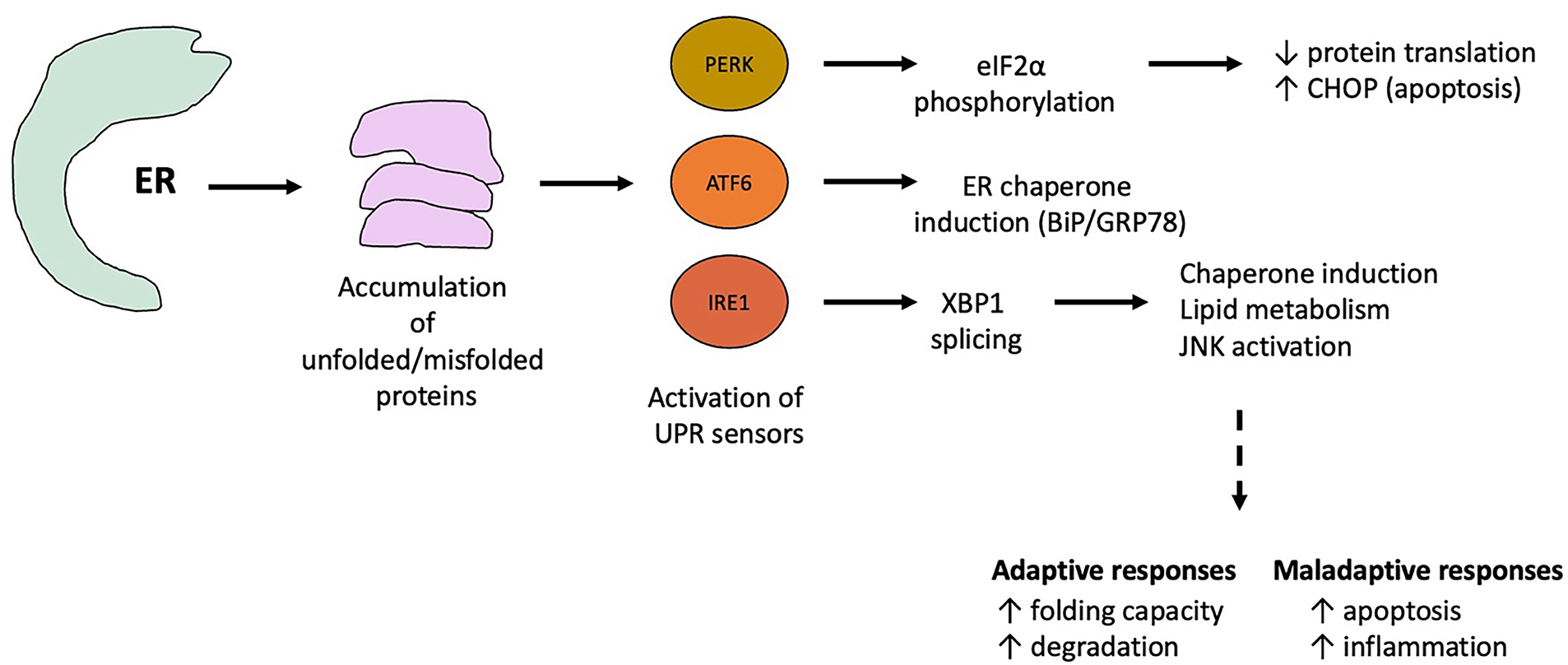
Figure 1: Overview of endoplasmic reticulum (ER) stress and the canonical unfolded protein response (UPR) pathways relevant to atherosclerosis. Under physiological conditions, the ER maintains protein folding, lipid synthesis, and Ca2+ homeostasis. Accumulation of misfolded or unfolded proteins—triggered by oxidative stress, lipid overload, inflammation, or Ca2+ dysregulation—activates ER stress and induces the UPR. The three principal UPR sensors—inositol-requiring enzyme 1 (IRE1), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6)—remain inactive through binding to the ER chaperone BiP/GRP78. Under stress, BiP dissociates, leading to sensor activation. eIF2α: Eukaryotic translation initiation factor 2α; XBP1: X-box binding protein 1; BiP: Binding Immunoglobulin Protein; CHOP: C/EBP Homologous Protein; GRP78: of glucose-regulated protein 78; JNK: c-Jun N-terminal kinase
The UPR employs adaptive mechanisms such as upregulating chaperones to enhance folding, attenuating translation to reduce ER load, and expanding ER biogenesis [21–23]. If stress remains unresolved, however, the UPR shifts from adaptive to pro-apoptotic signaling, contributing to cell loss and tissue damage [24–27].
IRE1 is the most evolutionarily conserved ER stress sensor. Upon stress, its dissociation from GRP78 and subsequent autophosphorylation activate endoribonuclease activity, which splices X-box binding protein 1 (XBP1) mRNA, producing the active transcription factor XBP1s [28–32]. XBP1s upregulates molecular chaperones and ERAD components, facilitating recovery. Additionally, IRE1-mediated mRNA degradation reduces ER protein load [33,34].
However, prolonged IRE1 activation recruits TNF Receptor-associated Factor 2 (TRAF2) and caspase-12, linking ER stress to apoptosis and inflammation through Mitogen-Activated Protein Kinase (MAPK) and Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling [35–39]. This dual role is particularly relevant in atherosclerosis, where IRE1 may initially enhance survival but later promote chronic inflammation.
PERK, a serine/threonine kinase, is activated through a mechanism similar to IRE1 [40–42]. Once active, PERK phosphorylates eukaryotic initiation factor 2α (eIF2α), suppressing general protein synthesis while selectively allowing translation of activating transcription factor 4 (ATF4) [43,44]. ATF4 coordinates stress adaptation but also induces growth arrest and DNA damage-inducible protein (GADD)34, which restores translation via eIF2α dephosphorylation [45,46].
Critically, ATF4 also induces GADD153/C/EBP Homologous Protein (CHOP), a pro-apoptotic transcription factor that upregulates Endoplasmic Reticulum Oxidoreductin 1 Alpha (ERO1α), increases reactive oxygen species (ROS), promotes Ca2+ release from the ER, and suppresses anti-apoptotic proteins such as B-cell lymphoma-2 (Bcl-2) [47–50]. This signaling cascade links prolonged PERK activation to oxidative stress, mitochondrial dysfunction, and apoptosis—mechanisms strongly implicated in plaque instability [51–54].
ATF6 is a transmembrane ER protein that, under stress, translocates to the Golgi where it is cleaved by site-1 and site-2 proteases (S1P/S2P). The cytosolic fragment then acts as a transcription factor, upregulating GRP78, XBP1, and ERAD components [55,56]. ATF6 also promotes Derlin-3 expression, enhancing ERAD efficiency [57–59].
Unlike IRE1 and PERK, ATF6 signaling is primarily adaptive. However, its activity may indirectly contribute to apoptosis by enhancing protein degradation and metabolic demand. The relative importance of ATF6 in vascular pathology remains debated, as most studies emphasize PERK- and IRE1-driven pathways.
Together, IRE1, PERK, and ATF6 illustrate the double-edged nature of the UPR: initially protective but ultimately pathogenic when stress is sustained. In atherosclerosis, these pathways are not equally implicated—PERK/CHOP-driven apoptosis and IRE1-mediated inflammation have been most strongly linked to plaque progression, whereas ATF6 may act more as a compensatory regulator. Future research should clarify whether selectively targeting one branch of the UPR can provide therapeutic benefit without disrupting adaptive responses.
A conceptual integration of UPR branches is necessary: IRE1–TRAF2–NF-κB links primarily to inflammation, PERK–eIF2α–ATF4–CHOP drives apoptosis, and ATF6 enhances adaptive folding but indirectly increases metabolic demand. In atherosclerosis, these signals converge on macrophage apoptosis, VSMC cap thinning, and endothelial dysfunction, collectively destabilizing plaques.
3 Endoplasmic Reticulum Stress in Endothelial Cells (ECs)
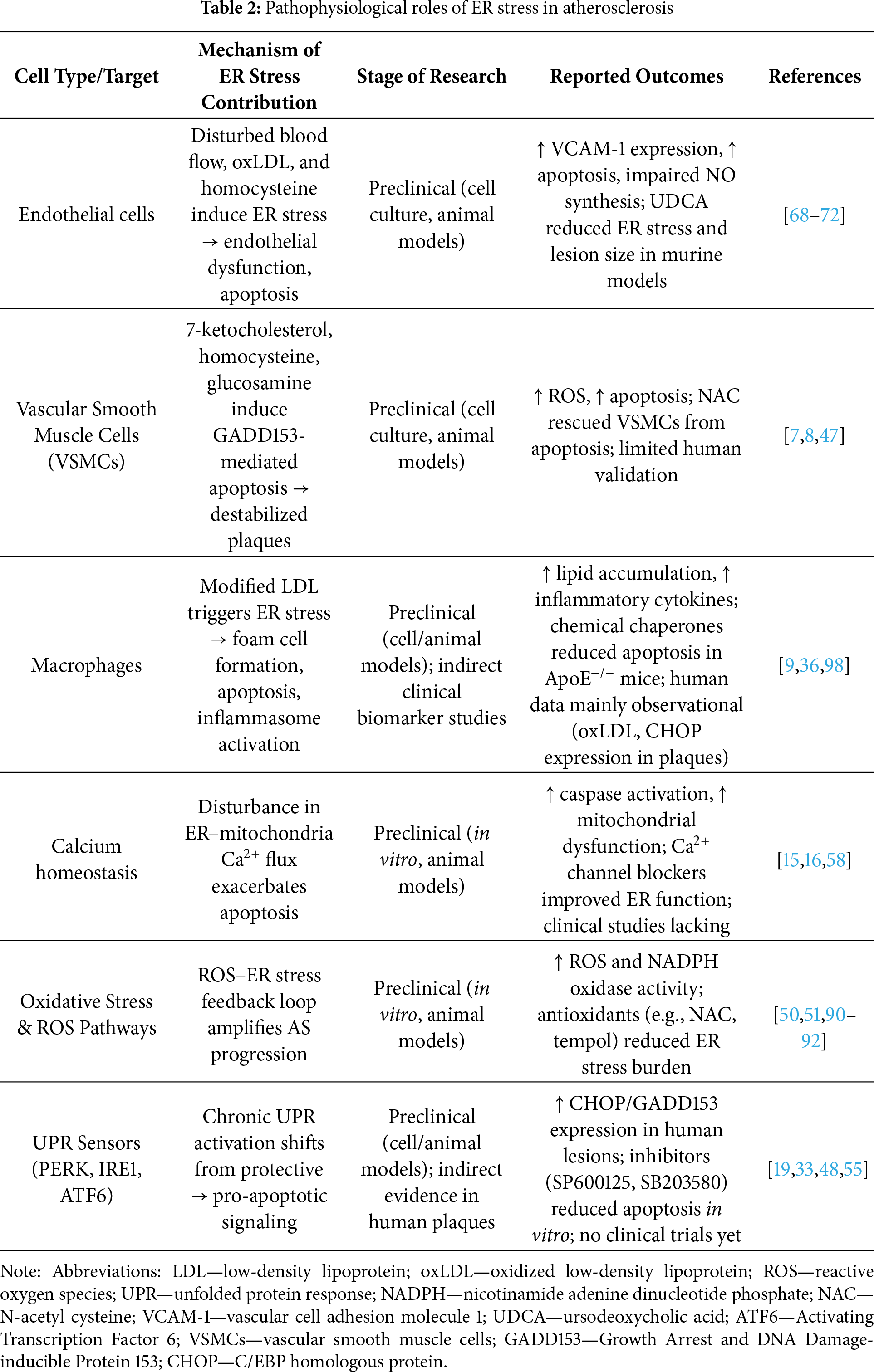
The hypothesis of vascular endothelial cell (VEC) damage response is an important pathophysiological model of AS. Impaired function of ECs facilitates AS progression. Atheromatous lesions mostly appear in sites of disturbed blood flow, e.g., where vessels bend or fork. Endothelial cells are permanently exposed to hemodynamic forces and are therefore especially vulnerable in such sites [60,61].
3.1 Shear Stress and ER Stress in ECs
In non-atherosclerotic pigs, ER stress biomarkers—IRE1, XBP1, and ATF6—undergo activation in ECs located in AS-prone regions of the aorta. Recent studies demonstrated that turbulent blood flow with lower shear stress (SS), a key factor in endothelial dysfunction, stimulates ER stress in ECs, thereby contributing to AS development [62].
In vitro studies on cultured ECs showed that SS enhanced GRP78 expression via a p38 mitogen-activated protein kinase (p38 MAPK)- and α2β1-integrin-dependent process prior to lesion development. These findings suggest a possible anti-atherogenic response to ER stress. However, other reports emphasize that SS can also induce a pro-inflammatory endothelial phenotype through ER stress activation [63,64]. This contradiction highlights that ER stress responses may be context-dependent and not uniformly detrimental.
Bailey et al. reported that SS controls the inflammatory response of aortic ECs via activation of XBP1. During this process, a temporary SS-driven increase in p38 phosphorylation promoted XBP1 nuclear translocation and upregulated vascular cell adhesion molecule-1 (VCAM1) expression [65,66]. Thus, SS increases EC sensitivity to ER stress induced by cytokines, thereby regulating pro-inflammatory processes that accelerate AS. Additionally, SS promotes apoptosis in aortic ECs through ER stress-mediated interleukin-1 receptor-associated kinase 2 (IRAK2)/GADD153 signaling [67]. Interestingly, ursodeoxycholic acid (UDCA) has been shown to mitigate these effects: in a murine model of turbulent-flow-induced AS, UDCA reduced ER stress markers (XBP1 and GADD153) in ECs and suppressed inflammation and apoptosis, ultimately inhibiting AS lesion development [68].
3.2 Homocysteine-Induced ER Stress
Homocysteine (Hcy) is another potent inducer of ER stress in ECs. Experimental data have shown that ER stress caused by Hcy promotes gene expression changes and cell death in human umbilical vein endothelial cells (HUVECs) [69,70]. In hyperhomocysteinemia, ER stress contributes to vascular inflammation and endothelial dysfunction. Mechanistically, a reactive thiol group in Hcy can alter protein function by exchanging disulfide bonds with cysteine residues in key proteins [71–73]. These alterations affect not only ER-resident proteins but also membrane-bound and secreted proteins. Consistent with this, reticulon protein (RTP) expression was increased in HUVECs under ER stress conditions [74,75]. Importantly, Hcy-induced ER stress exerts atherogenic effects not only in ECs but also in other vascular cell types [76].
In AS, LDL particles modified by oxidative or enzymatic processes impair ER Ca2+ metabolism, stimulating oxidative stress (OS) and endothelial unfolded protein response, while suppressing sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) activity. Studies have demonstrated that phospholipolyzed LDL triggers inflammatory responses in ECs via ER stress [77–79]. Moreover, oxidized LDL (oxLDL) promotes inflammatory changes in ECs, activating inflammasome signaling through apoptosis signal-regulating kinase 1 (ASK1) and NOD-like receptor family pyrin domain containing 3 (NLRP3) in an ER stress–dependent manner. OxLDL also induces EC apoptosis via the PERK/ eIF2α/GADD153 pathway [80,81]. Notably, simvastatin suppresses oxLDL-induced ER stress and apoptosis, suggesting therapeutic potential. Collectively, altered LDL is a major driver of EC dysfunction, inflammation, and apoptosis through ER stress mechanisms [82–84].
Although ER stress is fundamentally a protective response that enables cells to cope with adverse stimuli, failure of the UPR to restore ER homeostasis results in prolonged stress, activation of apoptotic pathways, and progression of AS and cardiovascular disease [85–87]. Prolonged ER stress induces mitochondrial (Mt)-mediated apoptosis in ECs. Studies have shown that GADD153 disrupts the balance of the Bcl-2 family, promoting pro-apoptotic protein activation at the mitochondrial membrane. This leads to cytochrome c (Cyt C) release and subsequent caspase-dependent apoptosis [88,89].
3.4 Calcium Dysregulation and Mitochondrial Dysfunction
Furthermore, disturbed Ca2+ homeostasis exacerbates mitochondrial dysfunction and increases production of ROS and nicotinamide adenine dinucleotide phosphate (NADPH), both of which impair endothelial nitric oxide synthase (eNOS) activity and reduce nitric oxide (NO) bioavailability, thereby aggravating OS and endothelial dysfunction [90–92]. Cytoplasmic Ca2+ overload during ER stress also activates the proenzyme procaspase-12, which is cleaved into active caspase-12 in the ER membrane, subsequently activating caspase-3 and caspase-9 via calpain-dependent mechanisms [93,94]. In apolipoprotein E knockout (ApoE−/−) mice with AS, endothelial anti-apoptotic Bcl-2 expression was markedly reduced, while caspase-3 was elevated. Similarly, in HUVECs, silica nanoparticles (SiNPs) enhanced ER stress by activating the IRE1/c-Jun N-terminal kinase (JNK) pathway, increasing GADD153 and caspase-12 expression, and shifting the Bcl-2/Bcl-2 Associated X-protein (BAX) ratio toward apoptosis, with elevated Cyt C, caspase-3, and caspase-9 levels [95–97].
4 Macrophage Endoplasmic Reticulum Stress and AS
Macrophages are essential for pathogen defense and tissue homeostasis. However, chronic macrophage activation can contribute to tissue injury, particularly in AS and metabolic disorders such as diabetes mellitus (DM) [98,99]. Understanding the pathways leading to macrophage activation may reveal novel therapeutic opportunities.
Beyond phagocytosis of cellular debris, macrophages handle toxic lipid cargo, including altered LDL and saturated fatty acids (FAs). Macrophages are especially vulnerable to lipotoxicity in hyperlipidemia or DM, and exposure to these toxic environments can induce ER stress and apoptosis [100]. The fate of lipids, particularly cholesterol, determines whether macrophage activity is protective or deleterious: efficient cholesterol efflux promotes plaque stability, whereas lipid accumulation triggers cellular stress and pro-inflammatory signaling [101–103]. In adiposity, uptake of FAs and necrotic lipocytes initially supports clearance but ultimately impairs macrophage function, amplifying stress and cell death [104–106].
In advanced AS, macrophage apoptosis contributes to plaque instability and rupture, raising questions about whether macrophage death is protective (through removal of dysfunctional, lipid-laden cells and reduced inflammatory signaling) or harmful (due to impaired clearance of accumulated cholesterol and necrotic core expansion). Determining the stage-specific roles of macrophages is therefore critical for therapeutic targeting [107–109].
4.1 Lipotoxicity and ER Stress Pathways
The mechanisms by which toxic lipids induce macrophage apoptosis remain incompletely understood. Emerging evidence suggests that certain ER stress pathways are selectively engaged by lipids, rather than representing a nonspecific cellular decline [110,111]. Genes involved in ER stress and associated signaling appear to link metabolic dysfunction, inflammation, and apoptotic processes, highlighting ER stress as a potential target in obesity and type 2 diabetes mellitus (T2DM) [112–114].
Two critical observations underscore the importance of ER stress in macrophages: (1) ER stress pathways are activated in lipid-loaded macrophages in human and mouse AS plaques; (2) cholesterol loading in vitro induces ER stress and subsequent apoptosis [115–117].
4.2 Stage-Specific Roles of Macrophage ER Stress
The contribution of macrophage ER stress to AS is complex and likely stage-dependent. While most studies focus on advanced AS, ER stress-induced macrophage apoptosis may be particularly relevant for plaque rupture and sudden cardiac death (SCD) [118,119]. ER stress is partially linked to insulin resistance via IRE1-mediated JNK activation, which may exacerbate macrophage apoptosis in AS [120,121].
A useful framework is to distinguish upstream ER stress sensing and protein folding capacity from downstream apoptotic signaling, as these processes may exert different effects at various AS stages [122–124]. Some evidence suggests a double-edged effect of macrophage apoptosis: promoting early lesion formation but potentially limiting lesion expansion in advanced AS [125,126].
5 Regulators and Therapeutic Implications
Key signaling modulators—including JNK-2, GADD153, STAT-1, p38 MAPK, and Glycogen Synthase Kinase 3 (GSK3)—can influence AS outcomes following ER stress or macrophage apoptosis, impacting plaque composition and stability [127,128]. Targeting ER folding capacity or upstream stress pathways may therefore represent a therapeutic strategy, particularly when considering systemic metabolic effects, such as hepatic lipid handling [129,130].
5.1 Pharmacological Modulation of ER Stress
Chemical chaperones and ER stress modulators (e.g., phenylbutyric acid, aP2 suppression) have shown promising effects in reducing macrophage ER stress, lipid-induced apoptosis, and atherosclerotic lesion formation in murine models [131–134]. These findings highlight the translational potential of targeting ER stress in macrophages, while also raising questions about specificity, stage-dependence, and long-term outcomes [135–137].
5.2 Lipid-Specific ER Stress Responses
Emerging data suggest that distinct lipids can selectively modulate ER stress pathways, influencing lipid metabolism, desaturation products, and membrane composition. For example, aP2-deficient macrophages exhibit increased monounsaturated fatty acids (MUFAs), alleviating ER stress and improving cellular resilience to saturated fats [138–140]. These observations suggest that ER stress modulation may extend beyond apoptosis regulation, affecting broader aspects of macrophage lipid handling and metabolic signaling [141–145].
5.3 Research Gaps and Future Directions
Further studies are needed to:
• Identify lipid-specific triggers of ER stress and UPR branches.
• Clarify whether UPR mediators act solely via ER function or through ER-independent pathways.
• Determine how ER stress in macrophages versus endothelial or smooth muscle cells contributes to AS progression.
• Explore systemic metabolic influences, including hepatic ER function, on AS development [146–149].
Overall, macrophage ER stress is a multifaceted contributor to AS, with both protective and detrimental effects depending on context, lipid load, and disease stage. Understanding these complexities may enable precision-targeted therapies to mitigate AS progression and improve cardiovascular outcomes [150,151].
6 Endoplasmic Reticulum Stress in Vascular Smooth Muscle Cells
VSMCs play a pivotal role in maintaining plaque stability, and their apoptosis can compromise the fibrous cap, increasing the risk of rupture. ER stress-mediated apoptosis in VSMCs is less studied than in endothelial cells or macrophages, representing a critical gap in understanding AS progression [152–154].
6.1 ER Stress-Induced Apoptotic Pathways
Multiple ER stressors, including 7-ketocholesterol (7KC), Hcy, glucosamine, and free cholesterol, promote GADD153-dependent apoptosis in VSMCs. This process is associated with increased ROS generation, highlighting the interplay between ER stress and oxidative stress. Notably, antioxidants such as N-acetylcysteine (NAC) can mitigate ROS-mediated apoptosis in cultured VSMCs, suggesting potential therapeutic avenues [155,156].
6.2 Homocysteine and Lipid-Induced ER Stress
Hyperhomocysteinemia (HHcy) is linked to elevated AS risk and appears to trigger ER stress by disrupting Ca2+ homeostasis and upregulating Sterol Regulatory Element-binding Protein 2 (SREBP2), which enhances lipid accumulation in VSMCs. Similarly, glucosamine accumulation in diabetic vessel cells stimulates ER stress through increased GRP78 expression, reflecting metabolic vulnerability in DM [157].
6.3 Critical Gaps and Translational Perspectives
Despite evidence of ER stress in VSMCs, mechanistic studies remain limited. Notably, VSMCs contribute critically to plaque stability through collagen and extracellular matrix production. Their apoptosis, often CHOP-dependent, weakens the fibrous cap and predisposes lesions to rupture. In addition to 7-ketocholesterol and homocysteine, mechanical stress and inflammatory cytokines can also trigger ER stress in VSMCs, further linking systemic metabolic and vascular factors. Compared with endothelial cells and macrophages, VSMC ER stress pathways are underexplored, and it remains unclear whether interventions targeting VSMC ER stress would meaningfully alter AS outcomes in vivo [158,159].
Emerging data suggest that stage-specific roles of VSMC ER stress may exist: in early AS, mild ER stress may contribute to adaptive remodeling, whereas in advanced lesions, prolonged stress may promote cap thinning and plaque vulnerability. Targeting these pathways—either via chemical chaperones, antioxidant therapy, or lipid-modulating strategies—represents a promising but largely untested therapeutic frontier.
7 Therapeutic Potential of Endoplasmic Reticulum Stress Modulators in Atherosclerosis
Targeting ER stress and the UPR represents a promising therapeutic strategy in disorders where ER stress contributes to pathophysiology, such as AS. Chemical chaperones such as phenylbutyrate (PBA) and tauroursodeoxycholic acid (TUDCA) facilitate nonselective protein folding and trafficking, thereby reducing misfolded protein accumulation in the ER [160]. PBA, approved for urea cycle disorders, ameliorated hyperglycemia and ER stress in murine DM models, and in apolipoprotein E knockout AS models, reduced ER stress markers (GRP78, Cluster of Differentiation 36 [CD36], IRE1 phosphorylation) in macrophage-rich lesions [161–164]. However, while promising, the translational potential remains uncertain, particularly regarding optimal dosing, off-target effects, and long-term outcomes in humans. Key therapeutic classes, mechanisms of action, and reported outcomes are summarized in Table 1.
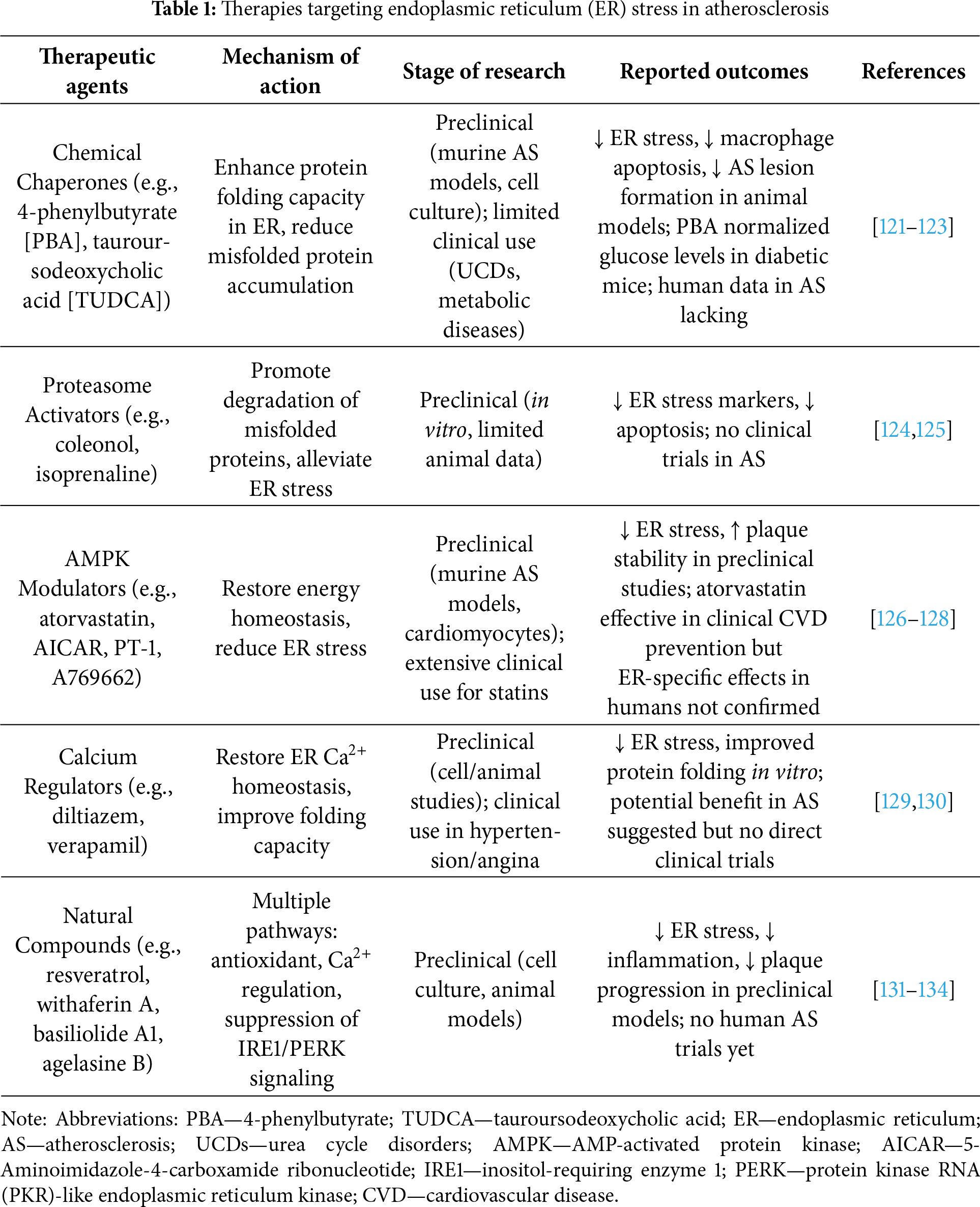
TUDCA similarly decreased ER stress and attenuated AS lesion development in LDL receptor-deficient mice, and in vitro studies confirmed reduced ER stress in macrophages exposed to oxidized LDL [165,166]. Despite encouraging preclinical results, clinical validation is lacking, and the specificity of these agents for different cell types within lesions requires further investigation.
While chemical chaperones such as PBA and TUDCA show strong efficacy in animal models, their pharmacokinetics in humans (rapid clearance, poor oral bioavailability) and off-target actions (hepatic, pancreatic, and neurological effects) raise major translational barriers. For example, PBA requires gram-scale dosing for metabolic disorders, limiting feasibility in CVD patients. Moreover, very few randomized controlled trials (RCTs) have evaluated ER stress modulators in AS, and no ER stress–targeted drug has yet advanced beyond Phase II trials.
7.1 Proteasome and Translational Control
ER stress can also be alleviated by enhancing proteasome function to reduce misfolded protein accumulation. Agents such as coleonol or isoprenaline have shown protective effects by activating proteasomal degradation, while suppression of protein synthesis via eIF2α signaling may reduce ER load [167–169]. Salubrinal, an eIF2α phosphatase inhibitor, protects cardiomyocytes from ER stress-induced apoptosis but paradoxically induces severe ER stress in pancreatic β-cells, highlighting cell-specific responses and the need for precision therapy [170,171].
7.2 Targeting ER Stress Signaling Pathways
Pharmacological modulation of stress-induced signaling represents another therapeutic approach. For instance, TNF-α inhibition, pravastatin therapy, or GADD153 suppression via p38 MAPK or JNK inhibitors can mitigate ER stress-mediated apoptosis [172–174]. Valproate upregulates the ER chaperone BiP and inhibits GADD153/caspase-12 activation, showing preclinical efficacy in AS models, yet human applicability and safety profiles remain to be determined.
Specific inhibitors of ERO1α (EN-460, QM-295) and AMPK activators (AICAR, PT-1, atorvastatin, A769662) have been shown to reduce ER stress and protect cardiomyocytes, while AMPK inhibition exacerbates ER stress and AS [175–177]. However, AMPK agonists are mainly approved for metabolic syndromes, and their effectiveness specifically in vascular lesions requires further clinical evaluation.
Restoration of ER Ca2+ homeostasis (via agents such as diltiazem or verapamil) may improve protein folding capacity and mitigate apoptosis, with potential crosstalk to mitochondrial Ca2+ regulation. Nevertheless, robust preclinical and clinical studies in AS are still limited, and off-target cardiovascular effects must be carefully assessed [178,179].
Several natural products exhibit ER stress-modulating activity, including proteasome inhibitors (tunicamycin, lactacystin, Brefeldin A [BFA]), Ca2+ regulators (basiliolide A1, agelasine B, thapsigargin), and IRE1/PERK pathway modulators (withaferin A, resveratrol) [180]. While these agents demonstrate mechanistic potential, their in vivo efficacy, bioavailability, and safety profiles remain largely unexplored, highlighting the need for rigorous translational studies. The cell-specific roles and mechanisms of ER stress in atherosclerosis are summarized in Table 2.
8 Discussion: Interplay of Lipids, ER Stress, and Inflammation in Atherosclerosis
A central theme of atherosclerosis research is the dynamic interplay between lipid metabolism and vascular inflammation. Growing evidence suggests that lipid dysregulation is not only a metabolic disorder but also a key trigger of inflammation, acting through ER stress-dependent mechanisms in various vascular cell types. This relationship is bidirectional: lipid accumulation induces inflammation through activation of ER stress, while inflammatory signaling further exacerbates lipid imbalance, creating a self-perpetuating cycle that accelerates atherogenesis.
Modified LDL particles, particularly oxLDL, electronegative LDL, and enzymatically modified LDL, exert potent proinflammatory effects on endothelial cells (ECs). Upon internalization, modified LDL disrupts calcium homeostasis in the ER and generates reactive oxygen species (ROS), promoting activation of the UPR and downstream proinflammatory pathways such as IRE1–TRAF2–NF-κB and PERK–CHOP. This leads to increased expression of adhesion molecules (VCAM-1, ICAM-1) and cytokine secretion (IL-6, IL-1β, TNF-α), thereby promoting monocyte recruitment and early plaque formation. Thus, lipids act as molecular triggers that convert endothelial cells to an inflammatory phenotype via ER stress signaling.
Macrophages are the primary cells that process lipids in plaques and play a key role in amplifying inflammation. Excess lipids in macrophages cause ER stress through the accumulation of non-esterified cholesterol and saturated fatty acids, activating the IRE1-JNK and PERK-ATF4-CHOP axes. Activation of these pathways stimulates inflammasome signaling, apoptotic cell death, and defective efferocytosis, promoting expansion of the necrotic core in advanced lesions. This demonstrates that lipid-induced ER stress not only triggers innate immune activation but also impairs inflammation resolution, highlighting its role in plaque progression and instability.
In vascular smooth muscle cells (SMCs), modified lipids induce a maladaptive phenotypic switch and apoptosis via ER stress-mediated mechanisms. While SMCs normally stabilize plaques by producing extracellular matrix components, lipid-induced ER stress shifts SMCs toward a synthetic, inflammatory, and ultimately apoptotic profile, compromising the integrity of the fibrous cap. The combined effects on endothelial cells (ECs), macrophages, and SMCs highlight that lipids mediate ER stress as a unified mechanistic pathway linking metabolic stress to chronic inflammation and plaque vulnerability.
Importantly, not all lipids exert the same negative effects. Certain lipid types, including omega-3 polyunsaturated fatty acids and lipid mediators such as resolvins, can attenuate ER stress and promote inflammation resolution. These findings highlight the need to distinguish between lipid subtypes and their context-dependent roles in modulating vascular inflammation. Therapies that selectively target lipid-induced ER stress pathways, rather than globally suppressing the UPR, may provide more effective vascular protection with fewer side effects.
Taken together, these mechanistic concepts provide a more comprehensive understanding of the pathogenesis of atherosclerosis, positioning ER stress as a mechanistic bridge between lipid dysregulation and chronic vascular inflammation. This conceptual framework not only aligns with the theme of this special issue but also identifies critical intervention points for future therapeutic approaches.
9 Conclusion and Future Perspective
Overall, while ER stress-targeted therapies hold promise, most data derive from preclinical models. Cell-specific, stage-specific, and dose-dependent effects remain poorly understood, limiting immediate clinical application. Future research should focus on defining which ER stress pathways are most pathogenic at specific AS stages, evaluating combination therapies, and establishing safety and efficacy in humans.
AS represents a multifaceted pathological condition driven by a complex interplay of metabolic, inflammatory, and cellular processes. Central to the progression of AS is ER stress, which emerges as a pivotal player influencing endothelial dysfunction, smooth muscle cell apoptosis, and macrophage responses. The intricate network of the UPR underscores the importance of cellular homeostasis in mitigating AS progression.
While the UPR can instigate protective mechanisms, its chronic activation often leads to detrimental outcomes, such as pro-inflammatory signaling and cell death, thereby exacerbating AS. Future investigations should clarify the stage-specific and cell-type–specific roles of ER stress in AS, since responses in endothelial cells, smooth muscle cells, and macrophages appear to differ significantly. Defining these nuances is essential to avoid therapeutic strategies that inadvertently worsen disease progression in particular contexts.
Targeting the pathways associated with ER stress presents a promising therapeutic strategy for managing AS and its associated cardiovascular risks. However, translation into clinical practice remains limited. Biomarkers such as GRP78, CHOP, and spliced XBP1 show potential for monitoring ER stress activity in patients, while imaging modalities assessing plaque vulnerability could further link molecular stress responses with clinical outcomes. Although human trials directly targeting ER stress are scarce, early studies using chemical chaperones or metabolic modulators provide proof-of-concept that these pathways can be manipulated in vivo. Greater integration of molecular research with clinical studies will be critical for validating therapeutic targets.
Future studies are vital to unravel the specific molecular mechanisms through which ER stress contributes to AS at various stages and to identify potential pharmacological agents that can effectively restore ER function without provoking adverse effects. Promising areas include the exploration of ER–mitochondria crosstalk, identification of non-invasive ER stress biomarkers, and repurposing of clinically approved drugs with ER-modulating properties. Further, large-scale clinical trials are needed to determine whether ER stress modulators can complement existing lipid-lowering or anti-inflammatory therapies in reducing cardiovascular events.
Acknowledgement: None.
Funding Statement: This research was funded by Russian Science Foundation, grant number 25-15-00080.
Author Contributions: Writing—original draft preparation, Alessio L. Ravani; writing—review and editing, Anastasia V. Poznyak, Michael I. Bukrinsky. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
Abbreviations
| ABs | Antibodies |
| AICAR | 5-Aminoimidazole-4-carboxamide ribonucleotide |
| AIS | Acute Ischemic Stroke |
| AMPK | AMP-activated protein kinase |
| AP-1 | Activator Protein-1 |
| aP2 | Adipocyte Protein 2 (also known as Fatty Acid Binding Protein 4, FABP4) |
| AS | Atherosclerosis |
| ASK | Apoptosis Signal-regulating Kinase |
| ATF4 | Activating Transcription Factor 4 |
| ATF6 | Activating Transcription Factor 6 |
| Bcl2 | B-cell lymphoma 2 |
| BIM | Bcl2-interacting mediator of cell death |
| Bnip3 | Bcl2/E1B 19 kDa-interacting protein 3 |
| BFA | Brefeldin A |
| Ca2+ | Calcium ion |
| C/PL ratio | Cholesterol/Phospholipid ratio |
| CD36 | Cluster of Differentiation 36 (scavenger receptor) |
| CVD | Cardiovascular Disease |
| Cyt C | Cytochrome c |
| DM | Diabetes Mellitus |
| DPFDs | Diseases with Protein Folding Defects |
| ECs | Endothelial Cells |
| eIF2α | Eukaryotic Initiation Factor 2 Alpha |
| ENOX | Endoplasmic Reticulum Oxidoreductin-like protein (if you meant ERO1, see below) |
| ERO1α | Endoplasmic Reticulum Oxidoreductin 1 Alpha |
| ER | Endoplasmic Reticulum |
| FA | Fatty Acid |
| FLD | Fatty Liver Disease |
| GADD153 | Growth Arrest and DNA Damage-inducible Protein 153 (also known as CHOP) |
| GSK3 | Glycogen Synthase Kinase 3 |
| HHcy | Hyperhomocysteinemia |
| Hcy | Homocysteine |
| HUVEC | Human Umbilical Vein Endothelial Cells |
| ICs | Immune Cells |
| IKK | IκB kinase |
| IL | Interleukin |
| IP3R1 | Inositol 1,4,5-triphosphate receptor type 1 |
| IRE1 | Inositol-Requiring Enzyme 1 |
| IR | Insulin Resistance |
| JNK | c-Jun N-terminal Kinase |
| KO | Knockout |
| LDL | Low-Density Lipoprotein |
| Lp | Lipoprotein |
| LRR | Leucine-Rich Repeat |
| LXRα | Liver X Receptor Alpha |
| MAPK | Mitogen-Activated Protein Kinase |
| Mt | Mitochondria |
| MUFA | Monounsaturated Fatty Acid |
| NAC | N-Acetylcysteine |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate (reduced form) |
| NF-κB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | NOD-, LRR- and Pyrin Domain-containing Protein 3 (inflammasome) |
| NO | Nitric Oxide |
| NR | Nuclear Receptor |
| OS | Oxidative Stress |
| oxLDL | Oxidized Low-Density Lipoprotein |
| PBA | Phenylbutyric Acid |
| PERK | Protein kinase RNA-like Endoplasmic Reticulum Kinase |
| PKA | Protein Kinase A |
| PL | Phospholipid |
| PT-1 | Phosphotyrosyl phosphatase activator-1 |
| RTP | Response to Protein (context suggests ER stress-responsive protein, please confirm exact gene/protein) |
| ROS | Reactive Oxygen Species |
| S1P/S2P | Site-1 Protease / Site-2 Protease |
| SCD | Sudden Cardiac Death |
| SERCA | Sarco/Endoplasmic Reticulum Ca2+-ATPase |
| SiNPs | Silica Nanoparticles |
| SP600125 | JNK-specific inhibitor compound |
| SREBP2 | Sterol Regulatory Element-binding Protein 2 |
| SS | Shear Stress |
| STAT1 | Signal Transducer and Activator of Transcription 1 |
| T2DM | Type 2 Diabetes Mellitus |
| TNF-α | Tumor Necrosis Factor Alpha |
| TRAF2 | TNF Receptor-associated Factor 2 |
| TUDCA | Tauroursodeoxycholic Acid |
| UCDs | Urea Cycle Disorders |
| UDCA | Ursodeoxycholic Acid |
| UPR | Unfolded Protein Response |
| VCAM1 | Vascular Cell Adhesion Molecule 1 |
| VEC | Vascular Endothelial Cell |
| VSMCs | Vascular Smooth Muscle Cells |
| XBP1 | X-box Binding Protein 1 |
| 7KC | 7-Ketocholesterol |
References
1. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors 1990–2019: update from the gbd 2019 study. J Am Coll Cardiol. 2020;76(25):2982–3021. [Google Scholar] [PubMed]
2. Gimbrone MAJr, Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118(4):620–36. [Google Scholar] [PubMed]
3. Borén J, Packard CJ, Binder CJ. Apolipoprotein B-containing lipoproteins in atherogenesis. Nat Rev Cardiol. 2025;22(6):399–413. doi:10.1038/s41569-024-01111-0. [Google Scholar] [PubMed] [CrossRef]
4. Malekmohammad K, Bezsonov EE, Rafieian-Kopaei M. Role of lipid accumulation and inflammation in atherosclerosis. Front Cardiovasc Med. 2021;8:707529. doi:10.3389/fcvm.2021.707529. [Google Scholar] [PubMed] [CrossRef]
5. Boren J, Chapman MJ, Krauss RM, Packard CJ, Bentzon JF, Binder CJ, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. Eur Heart J. 2020;41(24):2313–30. [Google Scholar] [PubMed]
6. Jin X, Yang S, Lu J, Wu M. Small dense low-density lipoprotein-cholesterol and atherosclerosis. Front Cardiovasc Med. 2022;8:804214. [Google Scholar] [PubMed]
7. Uryga AK, Bennett MR. Ageing induced vascular smooth muscle cell senescence in atherosclerosis. J Physiol. 2016;594(8):2115–24. doi:10.1113/jp270923. [Google Scholar] [PubMed] [CrossRef]
8. Sorokin V, Vickneson K, Kofidis T, Woo CC, Lin XY, Foo R, et al. Role of vascular smooth muscle cell plasticity and interactions. Front Immunol. 2020;11:599415. doi:10.3389/fimmu.2020.599415. [Google Scholar] [PubMed] [CrossRef]
9. Soehnlein O, Libby P. Targeting inflammation in atherosclerosis. Nat Rev Drug Discov. 2021;20:589–610. [Google Scholar] [PubMed]
10. Olie RH, Van der Meijden PEJ, Ten Cate H. The coagulation system in atherothrombosis. Res Pract Thromb Haemost. 2018;2(2):188–98. [Google Scholar] [PubMed]
11. Asada Y, Yamashita A, Sato Y, Hatakeyama K. Pathophysiology of atherothrombosis. Pathol Int. 2020;70(6):309–22. [Google Scholar] [PubMed]
12. Chen X, Shi C, He M, Xiong S, Xia X. Endoplasmic reticulum stress: molecular mechanism and therapeutic targets. Signal Transduct Target Ther. 2023;8:352. doi:10.1038/s41392-023-01570-w. [Google Scholar] [PubMed] [CrossRef]
13. Spencer BG, Finnie JW. Role of endoplasmic reticulum stress in cell survival and death. J Comp Pathol. 2020;181:86–91. doi:10.1016/j.jcpa.2020.10.006. [Google Scholar] [PubMed] [CrossRef]
14. Kettel P, Karagoz GE. Endoplasmic reticulum: monitoring and maintaining protein homeostasis. Int J Biochem Cell Biol. 2024;172:106598. [Google Scholar] [PubMed]
15. Carreras-Sureda A, Pihan P, Hetz C. Calcium signaling at the endoplasmic reticulum. Cell Calcium. 2018;70:24–31. doi:10.1016/j.ceca.2017.08.004. [Google Scholar] [PubMed] [CrossRef]
16. Wang CH, Wei YH. Role of mitochondrial dysfunction and Ca2+ dysregulation in insulin resistance. J Biomed Sci. 2017;24:70. [Google Scholar] [PubMed]
17. Guerriero CJ, Brodsky JL. Secreted protein folding and ERAD balance. Physiol Rev. 2012;92(2):537–76. [Google Scholar] [PubMed]
18. Chipurupalli S, Samavedam U, Robinson N. Crosstalk between ER stress autophagy and inflammation. Front Med. 2021;8:758311. doi:10.3389/fmed.2021.758311. [Google Scholar] [PubMed] [CrossRef]
19. Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. ER stress sensing in the UPR. Cold Spring Harb Perspect Biol. 2013;5(3):a013169. [Google Scholar] [PubMed]
20. Adams CJ, Kopp MC, Larburu N, Nowak PR, Ali MMU. ER stress signaling mechanism of IRE1. Front Mol Biosci. 2019;6:11. [Google Scholar] [PubMed]
21. Dauer P, Sharma NS, Gupta VK, Durden B, Hadad R, Banerjee S, et al. GRP78 regulates redox status in pancreatic cancer. Cell Death Dis. 2019;10(2):132. [Google Scholar] [PubMed]
22. Akinyemi AO, Simpson KE, Oyelere SF, Nur M, Ngule CM, Owoyemi BCD, et al. Dark side of GRP78 in cancers and pathology. Mol Med. 2023;29(1):112. [Google Scholar] [PubMed]
23. Diaz-Villanueva JF, Diaz-Molina R, Garcia-Gonzalez V. Protein folding and proteostasis. Int J Mol Sci. 2015;16(8):17193–230. [Google Scholar] [PubMed]
24. Needham PG, Guerriero CJ, Brodsky JL. Chaperoning ERAD in protein diseases. Cold Spring Harb Perspect Biol. 2019;11(8):a033928. [Google Scholar] [PubMed]
25. Craig EA. Hsp70 at the membrane. BMC Biol. 2018;16:11. [Google Scholar] [PubMed]
26. Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B, et al. ER stress signalling. FEBS J. 2019;286(2):241–78. [Google Scholar] [PubMed]
27. Bhattarai KR, Riaz TA, Kim HR, Chae HJ. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp Mol Med. 2021;53:151–67. doi:10.1038/s12276-021-00560-8. [Google Scholar] [PubMed] [CrossRef]
28. Riaz TA, Junjappa RP, Handigund M, Ferdous J, Kim HR, Chae HJ. Role of IRE1α in physiology and inflammation. Cells. 2020;9(5):1160. [Google Scholar] [PubMed]
29. Huang S, Xing Y, Liu Y. Roles for IRE1α in metabolic regulation. J Biol Chem. 2019;294(49):18726–41. [Google Scholar] [PubMed]
30. Kriegermeier A, Hyon A, LeCuyer B, Hubchak S, Liu X, Green RM. IRE1/XBP1 pathway in pediatric cholestasis. PLoS One. 2022;17(12):e0279016. [Google Scholar] [PubMed]
31. Tavernier SJ, Osorio F, Vandersarren L, Vetters J, Vanlangenakker N, Van Isterdael G, et al. Regulated IRE1-dependent mRNA decay sets the threshold for dendritic cell survival. Nat Cell Biol. 2017;19(6):698–710. doi:10.1038/ncb3518. [Google Scholar] [PubMed] [CrossRef]
32. Poznyak AV, Sukhorukov VN, Popov MA, Chegodaev YS, Postnov AY, Orekhov AN. Mechanisms of the wnt pathways as a potential target pathway in atherosclerosis. J Lipid Atheroscler. 2023;12(3):223–36. doi:10.12997/jla.2023.12.3.223. [Google Scholar] [PubMed] [CrossRef]
33. Wang T, Zhou J, Zhang X, Wu Y, Jin K, Wang Y, et al. X-box binding protein 1: an adaptor in the pathogenesis of atherosclerosis. Aging Dis. 2023;14(2):350–69. doi:10.14336/ad.2022.0824. [Google Scholar] [PubMed] [CrossRef]
34. Gebert M, Sobolewska A, Bartoszewska S, Cabaj A, Crossman DK, Króliczewski J, et al. Genome-wide mRNA profiling identifies X-box-binding protein 1 (XBP1) as an IRE1 and PUMA repressor. Cell Mol Life Sci. 2021;78(21–22):7061–80. doi:10.21203/rs.3.rs-527235/v1. [Google Scholar] [CrossRef]
35. Siwecka N, Rozpędek-Kamińska W, Wawrzynkiewicz A, Pytel D, Diehl JA, Majsterek I. The structure, activation and signaling of IRE1 and its role in determining cell fate. Biomedicines. 2021;9(2):156. doi:10.3390/biomedicines9020156. [Google Scholar] [PubMed] [CrossRef]
36. Sukhorukov VN, Khotina VA, Bagheri Ekta M, Ivanova EA, Sobenin IA, Orekhov AN. Endoplasmic reticulum stress in macrophages: the vicious circle of lipid accumulation and pro-inflammatory response. Biomedicines. 2020;8(7):210. doi:10.3390/biomedicines8070210. [Google Scholar] [PubMed] [CrossRef]
37. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. 2020;5:209. doi:10.1038/s41392-020-00312-6. [Google Scholar] [PubMed] [CrossRef]
38. Prescott JA, Cook SJ. Targeting IKKβ in cancer: challenges and opportunities for the therapeutic utilisation of IKKβ inhibitors. Cells. 2018;7(9):115. doi:10.3390/cells7090115. [Google Scholar] [PubMed] [CrossRef]
39. Poznyak AV, Orekhova VA, Sukhorukov VN, Khotina VA, Popov MA, Orekhov AN. Atheroprotective aspects of heat shock proteins. Int J Mol Sci. 2023;24(14):11750. doi:10.3390/ijms241411750. [Google Scholar] [PubMed] [CrossRef]
40. Junjappa RP, Patil P, Bhattarai KR, Kim HR, Chae HJ. IRE1α implications in endoplasmic reticulum stress-mediated development and pathogenesis of autoimmune diseases. Front Immunol. 2018;9:1289. doi:10.3389/fimmu.2018.01289. [Google Scholar] [PubMed] [CrossRef]
41. Zhou Z, Wang Q, Michalak M. Inositol Requiring Enzyme (IREa multiplayer in sensing endoplasmic reticulum stress. Anim Cells Syst. 2021;25(6):347–57. doi:10.1080/19768354.2021.2020901. [Google Scholar] [PubMed] [CrossRef]
42. Yang Y, Lu D, Wang M, Liu G, Feng Y, Ren Y, et al. Endoplasmic reticulum stress and the unfolded protein response: emerging regulators in progression of traumatic brain injury. Cell Death Dis. 2024;15:156. doi:10.1038/s41419-024-06515-x. [Google Scholar] [PubMed] [CrossRef]
43. Rios-Fuller TJ, Mahe M, Walters B, Abbadi D, Pérez-Baos S, Gadi A, et al. Translation regulation by eIF2α phosphorylation and mTORC1 signaling pathways in non-communicable diseases. Int J Mol Sci. 2020;21(15):5301. doi:10.3390/ijms21155301. [Google Scholar] [PubMed] [CrossRef]
44. Liu Y, Wang M, Cheng A, Yang Q, Wu Y, Jia R, et al. The role of host eIF2α in viral infection. Virol J. 2020;17:112. [Google Scholar] [PubMed]
45. Wortel IMN, Van der Meer LT, Kilberg MS, Van Leeuwen FN. Surviving stress: modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol Metab. 2017;28(11):794–806. doi:10.1016/j.tem.2017.07.003. [Google Scholar] [PubMed] [CrossRef]
46. Neill G, Masson GR. A stay of execution: ATF4 regulation and potential outcomes for the integrated stress response. Front Mol Neurosci. 2023;16:1112253. doi:10.3389/fnmol.2023.1112253. [Google Scholar] [PubMed] [CrossRef]
47. Chistiakov DA, Sobenin IA, Orekhov AN, Bobryshev YV. Role of endoplasmic reticulum stress in atherosclerosis and diabetic macrovascular complications. Biomed Res Int. 2014;2014:610140. doi:10.1155/2014/610140. [Google Scholar] [PubMed] [CrossRef]
48. Hu H, Tian M, Ding C, Yu S. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front Immunol. 2018;9:3083. doi:10.3389/fimmu.2018.03083. [Google Scholar] [PubMed] [CrossRef]
49. Blagov AV, Sukhorukov VN, Guo S, Zhang D, Popov MA, Orekhov AN. Impaired mitochondrial function in T-lymphocytes as a result of exposure to HIV and ART. Cells. 2023;12(7):1072. doi:10.3390/cells12071072. [Google Scholar] [PubMed] [CrossRef]
50. Shergalis AG, Hu S, Bankhead AIII, Neamati N. Role of the ERO1-PDI interaction in oxidative protein folding and disease. Pharmacol Ther. 2020;210:107525. doi:10.1016/j.pharmthera.2020.107525. [Google Scholar] [PubMed] [CrossRef]
51. Zito E. ERO1: a protein disulfide oxidase and H2O2 producer. Free Radic Biol Med. 2015;83:299–304. doi:10.1016/j.freeradbiomed.2015.01.011. [Google Scholar] [PubMed] [CrossRef]
52. Chen P, Sharma A, Weiher H, Schmidt-Wolf IG. Biological mechanisms and clinical significance of endoplasmic reticulum oxidoreductase 1 alpha (ERO1α) in human cancer. J Exp Clin Cancer Res. 2024;43:71. doi:10.1186/s13046-024-02990-4. [Google Scholar] [PubMed] [CrossRef]
53. Mo G, Liu X, Zhong Y, Mo J, Li Z, Li D, et al. IP3R1 regulates Ca2+ transport and pyroptosis through the NLRP3/Caspase-1 pathway in myocardial ischemia/reperfusion injury. Cell Death Discov. 2021;7(1):31. doi:10.1038/s41420-021-00404-4. [Google Scholar] [PubMed] [CrossRef]
54. Moon DO. Calcium’s role in orchestrating cancer apoptosis: mitochondrial-centric perspective. Int J Mol Sci. 2023;24(10):8982. doi:10.3390/ijms24108982. [Google Scholar] [PubMed] [CrossRef]
55. Park SM, Kang TI, So JS. Roles of XBP1s in transcriptional regulation of target genes. Biomedicines. 2021;9(7):791. doi:10.3390/biomedicines9070791. [Google Scholar] [PubMed] [CrossRef]
56. Corazzari M, Gagliardi M, Fimia GM, Piacentini M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front Oncol. 2017;7:78. doi:10.3389/fonc.2017.00078. [Google Scholar] [PubMed] [CrossRef]
57. Kadowaki H, Nishitoh H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes. 2013;4(3):306–33. doi:10.3390/genes4030306. [Google Scholar] [PubMed] [CrossRef]
58. Xu L, Wang ZH, Xu D, Lin G, Li DR, Wan T, et al. Expression of Derlin-1 and its effect on expression of autophagy marker genes under endoplasmic reticulum stress in lung cancer cells. Cancer Cell Int. 2014;14:50. doi:10.1186/1475-2867-14-50. [Google Scholar] [PubMed] [CrossRef]
59. Badawi S, Mohamed FE, Varghese DS, Ali BR. Genetic disruption of mammalian endoplasmic reticulum-associated protein degradation: human phenotypes and animal and cellular disease models. Traffic. 2023;24(8):312–33. doi:10.1111/tra.12902. [Google Scholar] [PubMed] [CrossRef]
60. Feng S, Chen JW, Shu XY, Aihemaiti M, Quan JW, Lu L, et al. Endothelial microparticles: a mechanosensitive regulator of vascular homeostasis and injury under shear stress. Front Cell Dev Biol. 2022;10:980112. doi:10.3389/fcell.2022.980112. [Google Scholar] [PubMed] [CrossRef]
61. Nikiforov NG, Kirichenko TV, Kubekina MV, Chegodaev YS, Zhuravlev AD, Ilchuk LA, et al. Macrophages derived from LPS-stimulated monocytes from individuals with subclinical atherosclerosis were characterized by increased pro-inflammatory activity. Cytokine. 2023;172:156411. doi:10.1016/j.cyto.2023.156411. [Google Scholar] [PubMed] [CrossRef]
62. Lindholm D, Korhonen L, Eriksson O, Kõks S. Recent insights into the role of unfolded protein response in ER stress in health and disease. Front Cell Dev Biol. 2017;5:48. doi:10.3389/fcell.2017.00048. [Google Scholar] [PubMed] [CrossRef]
63. Visioli F, Wang Y, Alam GN, Ning Y, Rados PV, Nör JE, et al. Glucose-regulated protein 78 (Grp78) confers chemoresistance to tumor endothelial cells under acidic stress. PLoS One. 2014;9(6):e101053. doi:10.1371/journal.pone.0101053. [Google Scholar] [PubMed] [CrossRef]
64. Vorotnikov AV, Khapchaev AY, Nickashin AV, Shirinsky VP. In vitro modeling of diabetes impact on vascular endothelium: are essentials engaged to tune metabolism? Biomedicines. 2022;10(12):3181. doi:10.3390/biomedicines10123181. [Google Scholar] [PubMed] [CrossRef]
65. Bailey KA, Moreno E, Haj FG, Simon SI, Passerini AG. Mechanoregulation of p38 activity enhances endoplasmic reticulum stress-mediated inflammation by arterial endothelium. FASEB J. 2019;33(11):12888–99. doi:10.1096/fj.201900236r. [Google Scholar] [PubMed] [CrossRef]
66. Bailey KA, Haj FG, Simon SI, Passerini AG. Atherosusceptible shear stress activates endoplasmic reticulum stress to promote endothelial inflammation. Sci Rep. 2017;7:8196. doi:10.1038/s41598-017-08417-9. [Google Scholar] [PubMed] [CrossRef]
67. Hauger PC, Hordijk PL. Shear stress-induced AMP-activated protein kinase modulation in endothelial cells: its role in metabolic adaptions and cardiovascular disease. Int J Mol Sci. 2024;25(11):6047. doi:10.3390/ijms25116047. [Google Scholar] [PubMed] [CrossRef]
68. Chung J, Kim KH, Lee SC, An SH, Kwon K. Ursodeoxycholic acid exerts anti-atherogenic effects by inhibiting endoplasmic reticulum stress induced by disturbed flow. Mol Cells. 2015;38(10):851–8. doi:10.14348/molcells.2015.0094. [Google Scholar] [PubMed] [CrossRef]
69. Zhang S, Lv Y, Luo X, Weng X, Qi J, Bai X, et al. Homocysteine promotes atherosclerosis through macrophage pyroptosis via endoplasmic reticulum stress and calcium disorder. Mol Med. 2023;29:73. doi:10.1186/s10020-023-00656-z. [Google Scholar] [PubMed] [CrossRef]
70. Nasoni MG, Crinelli R, Iuliano L, Luchetti F. When nitrosative stress hits the endoplasmic reticulum: possible implications in oxLDL/oxysterols-induced endothelial dysfunction. Free Radic Biol Med. 2023;208:178–85. doi:10.1016/j.freeradbiomed.2023.08.008. [Google Scholar] [PubMed] [CrossRef]
71. Wu X, Zhang L, Miao Y, Yang J, Wang X, Wang CC, et al. Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 2019;20:46–59. doi:10.1016/j.redox.2018.09.021. [Google Scholar] [PubMed] [CrossRef]
72. Yuan D, Chu J, Lin H, Zhu G, Qian J, Yu Y, et al. Mechanism of homocysteine-mediated endothelial injury and its consequences for atherosclerosis. Front Cardiovasc Med. 2023;9:1109445. doi:10.3389/fcvm.2022.1109445. [Google Scholar] [PubMed] [CrossRef]
73. Esse R, Barroso M, Tavares de Almeida I, Castro R. The contribution of homocysteine metabolism disruption to endothelial dysfunction: state-of-the-art. Int J Mol Sci. 2019;20(4):867. doi:10.3390/ijms20040867. [Google Scholar] [PubMed] [CrossRef]
74. Chatterjee B, Fatima F, Seth S, Sinha Roy S. Moderate elevation of homocysteine induces endothelial dysfunction through adaptive UPR activation and metabolic rewiring. Cells. 2024;13(3):214. doi:10.3390/cells13030214. [Google Scholar] [PubMed] [CrossRef]
75. Poznyak AV, Kashirskikh DA, Postnov AY, Popov MA, Sukhorukov VN, Orekhov AN. Sialic acid as the potential link between lipid metabolism and inflammation in the pathogenesis of atherosclerosis. Braz J Med Biol Res. 2023;56:e12972. doi:10.1590/1414-431x2023e12972. [Google Scholar] [PubMed] [CrossRef]
76. Osman A, El-Gamal H, Pasha M, Zeidan A, Korashy HM, Abdelsalam SS, et al. Endoplasmic reticulum stress-generated extracellular vesicles self-perpetuate ER stress and mediate endothelial cell dysfunction independently of cell survival. Front Cardiovasc Med. 2020;7:584791. doi:10.3389/fcvm.2020.584791. [Google Scholar] [PubMed] [CrossRef]
77. Maamoun H, Abdelsalam SS, Zeidan A, Korashy HM, Agouni A. Endoplasmic reticulum stress: a critical molecular driver of endothelial dysfunction and cardiovascular disturbances associated with diabetes. Int J Mol Sci. 2019;20(7):1658. doi:10.3390/ijms20071658. [Google Scholar] [PubMed] [CrossRef]
78. Ni L, Yang L, Lin Y. Recent progress of endoplasmic reticulum stress in the mechanism of atherosclerosis. Front Cardiovasc Med. 2024;11:1413441. doi:10.3389/fcvm.2024.1413441. [Google Scholar] [PubMed] [CrossRef]
79. Shariatzadeh M, Nagtzaam NMA, Van Vark-van der Zee L, Van Holten-Neelen C, Verhoeven AJM, Dehairs J, et al. Altered functionality of lipoprotein(a) impacts on angiogenesis in diabetic retinopathy. Investig Ophthalmol Vis Sci. 2023;64(5):8. doi:10.1167/iovs.64.5.8. [Google Scholar] [PubMed] [CrossRef]
80. Hang L, Peng Y, Xiang R, Li X, Li Z. Ox-LDL causes endothelial cell injury through ASK1/NLRP3-mediated inflammasome activation via endoplasmic reticulum stress. Drug Des Devel Ther. 2020;14:731–44. doi:10.2147/dddt.s231916. [Google Scholar] [PubMed] [CrossRef]
81. Jiang H, Zhou Y, Nabavi SM, Sahebkar A, Little PJ, Xu S, et al. Mechanisms of oxidized LDL-mediated endothelial dysfunction and its consequences for the development of atherosclerosis. Front Cardiovasc Med. 2022;9:925923. doi:10.3389/fcvm.2022.925923. [Google Scholar] [PubMed] [CrossRef]
82. Zhang GQ, Tao YK, Bai YP, Yan ST, Zhao SP. Inhibitory effects of simvastatin on oxidized low-density lipoprotein-induced endoplasmic reticulum stress and apoptosis in vascular endothelial cells. Chin Med J. 2018;131(8):950–5. doi:10.4103/0366-6999.229891. [Google Scholar] [PubMed] [CrossRef]
83. Wu ZH, Chen YQ, Zhao SP. Simvastatin inhibits ox-LDL-induced inflammatory adipokine secretion via amelioration of ER stress in 3T3-L1 adipocyte. Biochem Biophys Res Commun. 2013;432(2):365–9. doi:10.1016/j.bbrc.2013.01.094. [Google Scholar] [PubMed] [CrossRef]
84. Khan S, Huda B, Bhurka F, Patnaik R, Banerjee Y. Molecular and immunomodulatory mechanisms of statins in inflammation and cancer therapeutics with emphasis on the NF-κB, NLRP3 inflammasome, and cytokine regulatory axes. Int J Mol Sci. 2025;26(17):8429. doi:10.3390/ijms26178429. [Google Scholar] [PubMed] [CrossRef]
85. Schonthal AH. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica. 2012;2012:857516. doi:10.6064/2012/857516. [Google Scholar] [PubMed] [CrossRef]
86. Li T, Zhao H, Guo G, Xia S, Wang L. VMP1 affects endoplasmic reticulum stress sensitivity via differential modulation of the three unfolded protein response arms. Cell Rep. 2023;42(3):112209. doi:10.1016/j.celrep.2023.112209. [Google Scholar] [PubMed] [CrossRef]
87. Lam M, Marsters SA, Ashkenazi A, Walter P. Misfolded proteins bind and activate death receptor 5 to trigger apoptosis during unresolved endoplasmic reticulum stress. eLife. 2020;9:e52291. doi:10.7554/elife.52291. [Google Scholar] [PubMed] [CrossRef]
88. Yang S, Wu M, Li X, Zhao R, Zhao Y, Liu L, et al. Role of endoplasmic reticulum stress in atherosclerosis and its potential as a therapeutic target. Oxid Med Cell Longev. 2020;2020:9270107. doi:10.1155/2020/9270107. [Google Scholar] [PubMed] [CrossRef]
89. Zhang W, Shi Y, Oyang L, Cui S, Li S, Li J, et al. Endoplasmic reticulum stress—a key guardian in cancer. Cell Death Discov. 2024;10:343. doi:10.1038/s41420-024-02110-3. [Google Scholar] [PubMed] [CrossRef]
90. Negri S, Faris P, Moccia F. Reactive oxygen species and endothelial Ca2+ signaling: brothers in arms or partners in crime? Int J Mol Sci. 2021;22(18):9821. doi:10.3390/ijms22189821. [Google Scholar] [PubMed] [CrossRef]
91. Albano GD, Gagliardo RP, Montalbano AM, Profita M. Overview of the mechanisms of oxidative stress: impact in inflammation of the airway diseases. Antioxidants. 2022;11(11):2237. doi:10.3390/antiox11112237. [Google Scholar] [PubMed] [CrossRef]
92. Nita M, Grzybowski A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxid Med Cell Longev. 2016;2016:3164734. doi:10.1155/2016/3164734. [Google Scholar] [PubMed] [CrossRef]
93. Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis: cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277(37):34287–94. doi:10.1074/jbc.M204973200. [Google Scholar] [PubMed] [CrossRef]
94. Orekhov AN, Summerhill VI, Khotina VA, Popov MA, Uzokov JK, Sukhorukov VN. Role of mitochondria in the chronification of inflammation: focus on dysfunctional mitophagy and mitochondrial DNA mutations. Gene Expr. 2023;22(4):329–44. doi:10.14218/GE.2023.00061. [Google Scholar] [CrossRef]
95. Dissanayaka WL, Han Y, Zhang L, Zou T, Zhang C. Bcl-2 overexpression and hypoxia synergistically enhance angiogenic properties of dental pulp stem cells. Int J Mol Sci. 2020;21(17):6159. doi:10.3390/ijms21176159. [Google Scholar] [PubMed] [CrossRef]
96. Yang D, He L, Ma S, Li S, Zhang Y, Hu C, et al. Pharmacological targeting of Bcl-2 induces caspase-3-mediated cleavage of HDAC6 and regulates autophagy in colorectal cancer. Int J Mol Sci. 2023;24(7):6662. doi:10.3390/ijms24076662. [Google Scholar] [PubMed] [CrossRef]
97. Liang Z, Chen Y, Gu R, Guo Q, Nie X. Asiaticoside prevents oxidative stress and apoptosis in endothelial cells by activating ROS-dependent p53/Bcl-2/caspase-3 signaling pathway. Curr Mol Med. 2023;23(10):1116–29. doi:10.2174/1566524023666221024120825. [Google Scholar] [PubMed] [CrossRef]
98. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–37. doi:10.1038/nri3073. [Google Scholar] [PubMed] [CrossRef]
99. Mass E, Nimmerjahn F, Kierdorf K, Kierdorf K, Schlitzer A. Tissue-specific macrophages: how they develop and choreograph tissue biology. Nat Rev Immunol. 2023;23:563–79. doi:10.1038/s41577-023-00848-y. [Google Scholar] [PubMed] [CrossRef]
100. Hill AA, Kim M, Zegarra-Ruiz DF, Chang LC, Norwood K, Assié A, et al. Acute high-fat diet impairs macrophage-supported intestinal damage resolution. JCI Insight. 2023;8(3):e164489. doi:10.1172/jci.insight.164489. [Google Scholar] [PubMed] [CrossRef]
101. Linton MRF, Yancey PG, Davies SS, Jerome WG, Linton EF, Song WL, et al. The role of lipids and lipoproteins in atherosclerosis. In: Feingold KR, Ahmed SF, Anawalt B, Blackman MR, Boyce A, Chrousos G, et al., editors. Endotext [Internet]. South Dartmouth, MA, USA: MDText.com, Inc.; 2000 [cited 2025 Nov 21]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK343489/. [Google Scholar]
102. Susser LI, Rayner KJ. Through the layers: how macrophages drive atherosclerosis across the vessel wall. J Clin Investig. 2022;132(9):e157011. doi:10.1172/JCI157011. [Google Scholar] [PubMed] [CrossRef]
103. Soppert J, Lehrke M, Marx N, Jankowski J, Noels H. Lipoproteins and lipids in cardiovascular disease: from mechanistic insights to therapeutic targeting. Adv Drug Deliv Rev. 2020;159:4–33. doi:10.1016/j.addr.2020.07.019. [Google Scholar] [PubMed] [CrossRef]
104. Dahik VD, Frisdal E, Le Goff W. Rewiring of lipid metabolism in adipose tissue macrophages in obesity: impact on insulin resistance and type 2 diabetes. Int J Mol Sci. 2020;21(15):5505. doi:10.3390/ijms21155505. [Google Scholar] [PubMed] [CrossRef]
105. Li X, Ren Y, Chang K, Wu W, Griffiths HR, Lu S, et al. Adipose tissue macrophages as potential targets for obesity and metabolic diseases. Front Immunol. 2023;14:1153915. doi:10.3389/fimmu.2023.1153915. [Google Scholar] [PubMed] [CrossRef]
106. Stansbury CM, Dotson GA, Pugh H, Rehemtulla A, Rajapakse I, Muir LA. A lipid-associated macrophage lineage rewires the spatial landscape of adipose tissue in early obesity. JCI Insight. 2023;8(19):e171701. doi:10.1172/jci.insight.171701. [Google Scholar] [PubMed] [CrossRef]
107. Guha Ray A, Odum OP, Wiseman D, Weinstock A. The diverse roles of macrophages in metabolic inflammation and its resolution. Front Cell Dev Biol. 2023;11:1147434. doi:10.3389/fcell.2023.1147434. [Google Scholar] [PubMed] [CrossRef]
108. Martinet W, Coornaert I, Puylaert P, De Meyer GRY. Macrophage death as a pharmacological target in atherosclerosis. Front Pharmacol. 2019;10:306. doi:10.3389/fphar.2019.00306. [Google Scholar] [PubMed] [CrossRef]
109. Sansonetti M, Al Soodi B, Thum T, Jung M. Macrophage-based therapeutic approaches for cardiovascular diseases. Basic Res Cardiol. 2024;119(1):1–33. doi:10.1007/s00395-023-01027-9. [Google Scholar] [PubMed] [CrossRef]
110. Wieder N, Fried JC, Kim C, Sidhom EH, Brown MR, Marshall JL, et al. FALCON systematically interrogates free fatty acid biology and identifies a novel mediator of lipotoxicity. Cell Metab. 2023;35(5):887–905.e11. doi:10.1016/j.cmet.2023.03.018. [Google Scholar] [PubMed] [CrossRef]
111. Zhang Z, Yang Z, Wang S, Wang X, Mao J. Decoding ferroptosis: revealing the hidden assassin behind cardiovascular diseases. Biomed Pharmacother. 2024;176:116761. doi:10.1016/j.biopha.2024.116761. [Google Scholar] [PubMed] [CrossRef]
112. Ajoolabady A, Liu S, Klionsky DJ, Lip GYH, Tuomilehto J, Kavalakatt S, et al. ER stress in obesity pathogenesis and management. Trends Pharmacol Sci. 2022;43(2):97–109. doi:10.1016/j.tips.2021.11.011. [Google Scholar] [PubMed] [CrossRef]
113. Li A, Song NJ, Riesenberg BP, Li Z. Emerging roles of endoplasmic reticulum stress in balancing immunity and tolerance in health and diseases: mechanisms and opportunities. Front Immunol. 2019;10:3154. doi:10.3389/fimmu.2019.03154. [Google Scholar] [PubMed] [CrossRef]
114. Dong H, Sun Y, Nie L, Cui A, Zhao P, Leung WK, et al. Metabolic memory: mechanisms and diseases. Signal Transduct Target Ther. 2024;9:38. doi:10.1038/s41392-024-01755-x. [Google Scholar] [PubMed] [CrossRef]
115. Horn P, Tacke F. Unboxing the cell-type specific contribution of endoplasmic reticulum stress to NASH pathophysiology: myeloid X-box-binding protein 1 as a driver of steatohepatitis and fibrosis. Transl Gastroenterol Hepatol. 2022;8:14. doi:10.21037/tgh-22-65. [Google Scholar] [PubMed] [CrossRef]
116. Díaz-Bulnes P, Saiz ML, López-Larrea C, Rodríguez RM. Crosstalk between hypoxia and ER stress response: a key regulator of macrophage polarization. Front Immunol. 2019;10:2951. doi:10.3389/fimmu.2019.02951. [Google Scholar] [PubMed] [CrossRef]
117. Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med. 2009;15(12):1383–91. doi:10.1038/nm.2067. [Google Scholar] [PubMed] [CrossRef]
118. Shan R, Liu N, Yan Y, Liu B. Apoptosis, autophagy and atherosclerosis: relationships and the role of Hsp27. Pharmacol Res. 2021;166:105169. doi:10.1016/j.phrs.2020.105169. [Google Scholar] [PubMed] [CrossRef]
119. Liu C, Jiang Z, Pan Z, Yang L. Function, regulation and mechanism of programmed cell death of macrophages in atherosclerosis. Front Cell Dev Biol. 2021;9:809516. doi:10.3389/fcell.2021.809516. [Google Scholar] [PubMed] [CrossRef]
120. Amen OM, Sarker SD, Ghildyal R, Arya A. Endoplasmic reticulum stress activates unfolded protein response signaling and mediates inflammation, obesity and cardiac dysfunction: therapeutic and molecular approach. Front Pharmacol. 2019;10:977. doi:10.3389/fphar.2019.00977. [Google Scholar] [PubMed] [CrossRef]
121. Herlea-Pana O, Eeda V, Undi RB, Lim HY, Wang W. Pharmacological inhibition of inositol-requiring enzyme 1α RNase activity protects pancreatic beta cell and improves diabetic condition in insulin mutation-induced diabetes. Front Endocrinol. 2021;12:749879. doi:10.3389/fendo.2021.749879. [Google Scholar] [PubMed] [CrossRef]
122. Yuan S, She D, Jiang S, Deng N, Peng J, Ma L. Endoplasmic reticulum stress and therapeutic strategies in metabolic, neurodegenerative diseases and cancer. Mol Med. 2024;30(1):40. doi:10.1186/s10020-024-00808-9. [Google Scholar] [PubMed] [CrossRef]
123. Wu D, Huang LF, Chen XC, Huang XR, Li HY, An N, et al. Research progress on endoplasmic reticulum homeostasis in kidney diseases. Cell Death Dis. 2023;14:473. doi:10.1038/s41419-023-05905-x. [Google Scholar] [PubMed] [CrossRef]
124. Kuzu OF, Granerud LJT, Saatcioglu F. Navigating the landscape of protein folding and proteostasis: from molecular chaperones to therapeutic innovations. Signal Transduct Target Ther. 2025;10:358. doi:10.1038/s41392-025-02439-w. [Google Scholar] [PubMed] [CrossRef]
125. Linton MF, Babaev VR, Huang J, Linton EF, Tao H, Yancey PG. Macrophage apoptosis and efferocytosis in the pathogenesis of atherosclerosis. Circ J. 2016;80(11):2259–68. doi:10.1253/circj.CJ-16-0924. [Google Scholar] [PubMed] [CrossRef]
126. Simion V, Zhou H, Haemmig S, Pierce JB, Mendes S, Tesmenitsky Y, et al. A macrophage-specific lncRNA regulates apoptosis and atherosclerosis by tethering HuR in the nucleus. Nat Commun. 2020;11:6135. doi:10.1038/s41467-020-19664-2. [Google Scholar] [PubMed] [CrossRef]
127. Sprenkle NT, Sims SG, Sánchez CL, Meares GP. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol Neurodegener. 2017;12:42. doi:10.1186/s13024-017-0183-y. [Google Scholar] [PubMed] [CrossRef]
128. Gao S, Brown J, Wang H, Feng X. The role of glycogen synthase kinase 3-β in immunity and cell cycle: implications in esophageal cancer. Arch Immunol Ther Exp. 2014;62(2):131–44. doi:10.1007/s00005-013-0263-9. [Google Scholar] [PubMed] [CrossRef]
129. Merighi A, Lossi L. Endoplasmic reticulum stress signaling and neuronal cell death. Int J Mol Sci. 2022;23(23):15186. doi:10.3390/ijms232315186. [Google Scholar] [PubMed] [CrossRef]
130. Kapuy O. Mechanism of decision making between autophagy and apoptosis induction upon endoplasmic reticulum stress. Int J Mol Sci. 2024;25(8):4368. doi:10.3390/ijms25084368. [Google Scholar] [PubMed] [CrossRef]
131. Zhu W, Niu X, Wang M, Li Z, Jiang HK, Li C, et al. Endoplasmic reticulum stress may be involved in insulin resistance and lipid metabolism disorders of the white adipose tissues induced by high-fat diet containing industrial trans-fatty acids. Diabetes Metab Syndr Obes. 2019;12:1625–38. doi:10.2147/DMSO.S218336. [Google Scholar] [PubMed] [CrossRef]
132. Lemmer IL, Willemsen N, Hilal N, Bartelt A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol Metab. 2021;47:101169. doi:10.1016/j.molmet.2021.101169. [Google Scholar] [PubMed] [CrossRef]
133. Wang T, Zhao Y, You Z, Li X, Xiong M, Li H, et al. Endoplasmic reticulum stress affects cholesterol homeostasis by inhibiting LXRα expression in hepatocytes and macrophages. Nutrients. 2020;12(10):3088. doi:10.3390/nu12103088. [Google Scholar] [PubMed] [CrossRef]
134. Zhu G, Gao H, Li Y, Li X, Yang X, Wang C, et al. Suppression of endoplasmic reticulum stress by 4-PBA enhanced atherosclerotic plaque stability via up-regulating CLOCK expression. Pathol Res Pract. 2024;253:154969. doi:10.1016/j.prp.2023.154969. [Google Scholar] [PubMed] [CrossRef]
135. Liang Y, Kaushal D, Wilson RB. Cellular senescence and extracellular vesicles in the pathogenesis and treatment of obesity—a narrative review. Int J Mol Sci. 2024;25(14):7943. doi:10.3390/ijms25147943. [Google Scholar] [PubMed] [CrossRef]
136. Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107(7):839–50. doi:10.1161/CIRCRESAHA.110.224766. [Google Scholar] [PubMed] [CrossRef]
137. Zhao Y, Jiang Y, Chen L, Zheng X, Zhu J, Song X, et al. Inhibition of the endoplasmic reticulum (ER) stress-associated IRE-1/XBP-1 pathway alleviates acute lung injury via modulation of macrophage activation. J Thorac Dis. 2020;12(3):284–95. doi:10.21037/jtd.2020.01.45. [Google Scholar] [PubMed] [CrossRef]
138. Ertunc ME, Hotamisligil GS. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J Lipid Res. 2016;57(12):2099–114. doi:10.1194/jlr.R066514. [Google Scholar] [PubMed] [CrossRef]
139. Lipke K, Kubis-Kubiak A, Piwowar A. Molecular mechanism of lipotoxicity as an interesting aspect in the development of pathological states—current view of knowledge. Cells. 2022;11(5):844. doi:10.3390/cells11050844. [Google Scholar] [PubMed] [CrossRef]
140. Jalil A, Bourgeois T, Ménégaut L, Lagrost L, Thomas C, Masson D. Revisiting the role of LXRs in PUFA metabolism and phospholipid homeostasis. Int J Mol Sci. 2019;20(15):3787. doi:10.3390/ijms20153787. [Google Scholar] [PubMed] [CrossRef]
141. Batista-Gonzalez A, Vidal R, Criollo A, Carreño LJ. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front Immunol. 2020;10:2993. doi:10.3389/fimmu.2019.02993. [Google Scholar] [PubMed] [CrossRef]
142. Rosas-Ballina M, Guan XL, Schmidt A, Bumann D. Classical activation of macrophages leads to lipid droplet formation without de novo fatty acid synthesis. Front Immunol. 2020;11:131. doi:10.3389/fimmu.2020.00131. [Google Scholar] [PubMed] [CrossRef]
143. Ménégaut L, Jalil A, Thomas C, Masson D. Macrophage fatty acid metabolism and atherosclerosis: the rise of PUFAs. Atherosclerosis. 2019;291:52–61. doi:10.1016/j.atherosclerosis.2019.10.002. [Google Scholar] [PubMed] [CrossRef]
144. Moreews M, Karlsson MCI. Endoplasmic reticulum stress: a key player in immune cell regulation and autoimmune disorders. Semin Immunol. 2025;78:101954. doi:10.1016/j.smim.2025.101954. [Google Scholar] [PubMed] [CrossRef]
145. Baccouch R, Shi Y, Vernay E, Mathelié-Guinlet M, Taib-Maamar N, Villette S, et al. The impact of lipid polyunsaturation on the physical and mechanical properties of lipid membranes. Biochim Biophys Acta Biomembr. 2023;1865(2):184084. doi:10.1016/j.bbamem.2022.184084. [Google Scholar] [PubMed] [CrossRef]
146. Keylani K, Arbab Mojeni F, Khalaji A, Rasouli A, Aminzade D, Karimi MA, et al. Endoplasmic reticulum as a target in cardiovascular diseases: is there a role for flavonoids? Front Pharmacol. 2023;13:1027633. doi:10.3389/fphar.2022.1027633. [Google Scholar] [PubMed] [CrossRef]
147. Wojtasińska A, Frąk W, Lisińska W, Sapeda N, Młynarska E, Rysz J, et al. Novel insights into the molecular mechanisms of atherosclerosis. Int J Mol Sci. 2023;24(17):13434. doi:10.3390/ijms241713434. [Google Scholar] [PubMed] [CrossRef]
148. Ghemrawi R, Battaglia-Hsu SF, Arnold C. Endoplasmic reticulum stress in metabolic disorders. Cells. 2018;7(6):63. doi:10.3390/cells7060063. [Google Scholar] [PubMed] [CrossRef]
149. Cheng H, Zhong W, Wang L, Zhang Q, Ma X, Wang Y, et al. Effects of shear stress on vascular endothelial functions in atherosclerosis and potential therapeutic approaches. Biomed Pharmacother. 2023;158:114198. doi:10.1016/j.biopha.2022.114198. [Google Scholar] [PubMed] [CrossRef]
150. Deshmukh K, Apte U. The role of endoplasmic reticulum stress response in liver regeneration. Semin Liver Dis. 2023;43(3):279–92. doi:10.1055/a-2129-8977. [Google Scholar] [PubMed] [CrossRef]
151. Venkatesan N, Doskey LC, Malhi H. The role of endoplasmic reticulum in lipotoxicity during metabolic dysfunction-associated steatotic liver disease (MASLD) pathogenesis. Am J Pathol. 2023;193(12):1887–99. doi:10.1016/j.ajpath.2023.08.007. [Google Scholar] [PubMed] [CrossRef]
152. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118(4):692–702. doi:10.1161/CIRCRESAHA.115.306361. [Google Scholar] [PubMed] [CrossRef]
153. Qin HL, Bao JH, Tang JJ, Xu DY, Shen L. Arterial remodeling: the role of mitochondrial metabolism in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2023;324(1):C183–92. doi:10.1152/ajpcell.00074.2022. [Google Scholar] [PubMed] [CrossRef]
154. Jiang Y, Qian HY. Transcription factors: key regulatory targets of vascular smooth muscle cell in atherosclerosis. Mol Med. 2023;29:2. doi:10.1186/s10020-022-00586-2. [Google Scholar] [PubMed] [CrossRef]
155. Sciaccotta R, Gangemi S, Penna G, Giordano L, Pioggia G, Allegra A. Potential new therapies “ROS-based” in CLL: an innovative paradigm in the induction of tumor cell apoptosis. Antioxidants. 2024;13(4):475. doi:10.3390/antiox13040475. [Google Scholar] [PubMed] [CrossRef]
156. Li X, Cao D, Sun S, Wang Y. Anticancer therapeutic effect of ginsenosides through mediating reactive oxygen species. Front Pharmacol. 2023;14:1215020. doi:10.3389/fphar.2023.1215020. [Google Scholar] [PubMed] [CrossRef]
157. Fu Y, Wang X, Kong W. Hyperhomocysteinaemia and vascular injury: advances in mechanisms and drug targets. Br J Pharmacol. 2018;175(8):1173–89. doi:10.1111/bph.13988. [Google Scholar] [PubMed] [CrossRef]
158. Furmanik M, Van Gorp R, Whitehead M, Ahmad S, Bordoloi J, Kapustin A, et al. Endoplasmic reticulum stress mediates vascular smooth muscle cell calcification via increased release of Grp78 (glucose-regulated protein, 78 kDa)-loaded extracellular vesicles. Arterioscler Thromb Vasc Biol. 2021;41(2):898–914. doi:10.1161/ATVBAHA.120.315506. [Google Scholar] [PubMed] [CrossRef]
159. Cicalese S, Okuno K, Elliott KJ, Kawai T, Scalia R, Rizzo V, et al. 78 kDa glucose-regulated protein attenuates protein aggregation and monocyte adhesion induced by angiotensin II in vascular cells. Int J Mol Sci. 2020;21(14):4980. doi:10.3390/ijms21144980. [Google Scholar] [PubMed] [CrossRef]
160. Ghemrawi R, Khair M. Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. Int J Mol Sci. 2020;21(17):6127. doi:10.3390/ijms21176127. [Google Scholar] [PubMed] [CrossRef]
161. Rellmann Y, Gronau I, Hansen U, Dreier R. 4-Phenylbutyric acid reduces endoplasmic reticulum stress in chondrocytes that is caused by loss of the protein disulfide isomerase ERp57. Oxid Med Cell Longev. 2019;2019:6404035. doi:10.1155/2019/6404035. [Google Scholar] [PubMed] [CrossRef]
162. Pao HP, Liao WI, Tang SE, Wu SY, Huang KL, Chu SJ. Suppression of endoplasmic reticulum stress by 4-PBA protects against hyperoxia-induced acute lung injury via up-regulating claudin-4 expression. Front Immunol. 2021;12:674316. doi:10.3389/fimmu.2021.674316. [Google Scholar] [PubMed] [CrossRef]
163. Tang W, Cai D, Fu Y, Zheng Z, Huang X, Khouzam RN, et al. 4-Phenylbutyric acid re-trafficking hERG/G572R channel protein by modulating the endoplasmic reticulum stress-associated chaperones and endoplasmic reticulum-associated degradation gene. J Thorac Dis. 2023;15(8):4472–85. doi:10.21037/jtd-23-1252. [Google Scholar] [PubMed] [CrossRef]
164. Kawanaka R, Jin H, Aoe T. Unraveling the connection: pain and endoplasmic reticulum stress. Int J Mol. 2024;25(9):4995. doi:10.3390/ijms25094995. [Google Scholar] [PubMed] [CrossRef]
165. Yao S, Miao C, Tian H, Sang H, Yang N, Jiao P, et al. Endoplasmic reticulum stress promotes macrophage-derived foam cell formation by up-regulating cluster of differentiation 36 (CD36) expression. J Biol Chem. 2014;289(7):4032–42. doi:10.1074/jbc.M113.524512. [Google Scholar] [PubMed] [CrossRef]
166. Bucci T, Menichelli D, Palumbo IM, Pastori D, Ames PRJ, Lip GYH, et al. Statins as an adjunctive antithrombotic agent in thrombotic antiphospholipid syndrome: mechanisms and clinical implications. Cells. 2025;14(5):353. doi:10.3390/cells14050353. [Google Scholar] [PubMed] [CrossRef]
167. Qu J, Zou T, Lin Z. The roles of the ubiquitin-proteasome system in the endoplasmic reticulum stress pathway. Int J Mol Sci. 2021;22(4):1526. doi:10.3390/ijms22041526. [Google Scholar] [PubMed] [CrossRef]
168. Lozada Ortiz J, Betancor M, Pérez Lázaro S, Bolea R, Badiola JJ, Otero A. Endoplasmic reticulum stress and ubiquitin-proteasome system impairment in natural scrapie. Front Mol Neurosci. 2023;16:1175364. doi:10.3389/fnmol.2023.1175364. [Google Scholar] [PubMed] [CrossRef]
169. Zhu Y, Ju H, Lu H, Tang W, Lu J, Wang Q. The function role of ubiquitin proteasome pathway in the ER stress-induced AECII apoptosis during hyperoxia exposure. BMC Pulm Med. 2021;21:379. doi:10.1186/s12890-021-01751-9. [Google Scholar] [PubMed] [CrossRef]
170. Matsuoka M, Komoike Y. Experimental evidence shows salubrinal, an eIF2α dephosphorylation inhibitor, reduces xenotoxicant-induced cellular damage. Int J Mol Sci. 2015;16(7):16275–87. doi:10.3390/ijms160716275. [Google Scholar] [PubMed] [CrossRef]
171. Lu W, Ni K, Li Z, Xiao L, Li Y, Jiang Y, et al. Salubrinal protects against cisplatin-induced cochlear hair cell endoplasmic reticulum stress by regulating eukaryotic translation initiation factor 2α signalling. Front Mol Neurosci. 2022;15:916458. doi:10.3389/fnmol.2022.916458. [Google Scholar] [PubMed] [CrossRef]
172. Akhter N, Wilson A, Arefanian H, Thomas R, Kochumon S, Al-Rashed F, et al. Endoplasmic reticulum stress promotes the expression of TNF-α in THP-1 cells by mechanisms involving ROS/CHOP/HIF-1α and MAPK/NF-κB pathways. Int J Mol Sci. 2023;24(20):15186. doi:10.3390/ijms242015186. [Google Scholar] [PubMed] [CrossRef]
173. Yang R, Ma S, Zhuo R, Xu L, Jia S, Yang P, et al. Suppression of endoplasmic reticulum stress-dependent autophagy enhances cynaropicrin-induced apoptosis via attenuation of the P62/Keap1/Nrf2 pathways in neuroblastoma. Front Pharmacol. 2022;13:977622. doi:10.3389/fphar.2022.977622. [Google Scholar] [PubMed] [CrossRef]
174. Falcicchia C, Tozzi F, Arancio O, Watterson DM, Origlia N. Involvement of p38 MAPK in synaptic function and dysfunction. Int J Mol Sci. 2020;21(16):5624. doi:10.3390/ijms21165624. [Google Scholar] [PubMed] [CrossRef]
175. Ramirez MU, Hernandez SR, Soto-Pantoja DR, Cook KL. Endoplasmic reticulum stress pathway, the unfolded protein response, modulates immune function in the tumor microenvironment to impact tumor progression and therapeutic response. Int J Mol Sci. 2019;21(1):169. doi:10.3390/ijms21010169. [Google Scholar] [PubMed] [CrossRef]
176. Huang Y, Smith CA, Chen G, Sharma B, Miner AS, Barbee RW, et al. The AMP-dependent protein kinase (AMPK) activator A-769662 causes arterial relaxation by reducing cytosolic free calcium independently of an increase in AMPK phosphorylation. Front Pharmacol. 2017;8:756. doi:10.3389/fphar.2017.00756. [Google Scholar] [PubMed] [CrossRef]
177. Timm KN, Tyler DJ. The role of AMPK activation for cardioprotection in doxorubicin-induced cardiotoxicity. Cardiovasc Drugs Ther. 2020;34(2):255–69. doi:10.1007/s10557-020-06941-x. [Google Scholar] [PubMed] [CrossRef]
178. Bahar E, Kim H, Yoon H. ER stress-mediated signaling: action potential and Ca2+ as key players. Int J Mol Sci. 2016;17(9):1558. doi:10.3390/ijms17091558. [Google Scholar] [PubMed] [CrossRef]
179. Lizák B, Birk J, Zana M, Kosztyi G, Kratschmar DV, Odermatt A, et al. Ca2+ mobilization-dependent reduction of the endoplasmic reticulum lumen is due to influx of cytosolic glutathione. BMC Biol. 2020;18:19. doi:10.1186/s12915-020-0749-y. [Google Scholar] [PubMed] [CrossRef]
180. Martucciello S, Masullo M, Cerulli A, Piacente S. Natural products targeting ER stress, and the functional link to mitochondria. Int J Mol Sci. 2020;21(6):1905. doi:10.3390/ijms21061905. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools