
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Dexmedetomidine Protects Intestinal Mucosal Barrier via Activating the α7-nAChR-GDNF in Enteric Glial Cells
Department of Anaesthesiology, First Affiliated Hospital of Kunming Medical University, Kunming, China
* Corresponding Author: Kun Yang. Email:
# These authors contributed equally to this paper
§ The two authors named “Kun Yang” are different individuals
BIOCELL 2026, 50(3), 11 https://doi.org/10.32604/biocell.2026.075138
Received 25 October 2025; Accepted 27 December 2025; Issue published 23 March 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Objective: Intestinal barrier disruption is a critical event in sepsis and ischemia–reperfusion (I/R) injury. Enteric glial cells (EGCs) maintain barrier integrity by secreting glial cell line–derived neurotrophic factor (GDNF). This study aimed to determine whether Dexmedetomidine (Dex) protects the intestinal barrier via α7-nicotinic acetylcholine receptor (α7-nAChR) signaling in EGCs. Methods: An in vitro EGC-intestinal epithelial cell (IEC) co-culture system and a murine intestinal I/R model were established. EGCs were selectively ablated in vivo using benzalkonium chloride (BAC). Barrier integrity was evaluated by transmembrane electrical resistance (TEER) and plasma FITC-dextran permeability. Enzyme-Linked Immunosorbent Assay (ELISA) and Western blotting quantified levels of GDNF and Occludin. The α7-nAChR antagonist methyllycaconitine (MLA) was applied for mechanistic validation. Results: In vitro, Dex (40–100 μm) dose-dependently increased GDNF expression in EGCs (p < 0.05) and enhanced IEC TEER. These protective effects were abolished by MLA pre-treatment (p < 0.05). In vivo, Dex significantly reduced I/R-induced mucosal injury and decreased plasma FITC-dextran concentrations compared to the untreated I/R group (0.30 ± 0.01 vs. 0.43 ± 0.02 mg/mL, p < 0.05). Notably, in EGC-ablated mice, Dex failed to restore Occludin levels or reduce permeability (p > 0.05), confirming EGC-dependence. Conclusion: Dexmedetomidine protects the intestinal mucosal barrier via an EGC-dependent mechanism involving α7-nAChR activation and GDNF-mediated tight junction reinforcement. These findings highlight EGCs as key effectors of Dex-induced intestinal protection and potential therapeutic targets for barrier dysfunction in critical illness.Keywords
Sepsis is a common and life-threatening complication following severe trauma, hemorrhagic shock, and major surgery. It is one of the leading causes of death among critically ill surgical patients, with mortality rates ranging from 30% to 66% [1–5]. Damage to the intestinal mucosal barrier plays a key role in the pathogenesis of intestinal-derived sepsis, often serving as the ‘motor’ of systemic inflammation [6,7]. Emerging evidence further indicates that preserving the integrity of the gut–vascular barrier is crucial for improving survival outcomes in patients with sepsis [8]. Barrier integrity relies on intestinal epithelial cells (IECs) and intercellular tight junctions, specialized structures regulating permeability by restricting harmful substances while permitting nutrient passage [9,10]. Tight junction proteins, including zonula occludens-1(ZO-1) and Occludin, seal the paracellular space [11,12]. Occludin, the first identified tight junction protein, reflects barrier functional status; its loss significantly increases gastrointestinal permeability [13,14]. Enteric glial cells (EGCs), a major component of the enteric nervous system marked by glial fibrillary acidic protein (GFAP), are crucial for maintaining intestinal epithelial barrier integrity [15–17]. Evidence indicates EGCs support barrier function by secreting glial-derived neurotrophic factor (GDNF), which upregulates the tight junction protein Occludin [15,18,19].
Dexmedetomidine (Dex), a highly selective α2-adrenergic receptor agonist, provides perioperative sedation and analgesia by activating locus coeruleus α2 receptors to suppress norepinephrine release [20–22]. EGCs express functional α7 nicotinic acetylcholine receptors (α7-nAChRs), activated by vagal nerve signaling [23–25]. Dex’s sympathoinhibitory effects may potentiate vagal tone, and preclinical studies show it enhances α7-nAChR expression in the hippocampus [26]. We therefore hypothesized that Dex preserves intestinal barrier integrity via α7-nAChR-dependent EGC activation, leading to GDNF-mediated Occludin upregulation.
Therefore, the specific objective of this study was to determine the cellular and molecular mechanisms by which Dex modulates barrier function. We established an EGC-IEC co-culture system to investigate Dex-induced alterations in Occludin expression and transmembrane electrical resistance (TEER), providing cellular and molecular insights into barrier function modulation. In vivo, EGCs-depletion models with intestinal ischemia-reperfusion(I/R) injury were utilized to further assess the expression of intestinal Occludin. Meanwhile, serum FITC-dextran assay systematically confirmed the EGC-dependence of Dex’s protective effects on mucosal barrier integrity.
This study was approved by the Animal Care and Use Committee of Kunming Medical University (Approval No.: KMMU20211240). All animal procedures adhered to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, USA), minimizing animal discomfort and numbers used.
A total of 48 male C57BL/6 mice (6–8 weeks, 18–22 g) were purchased from the Animal Facility of Kunming Medical University. Mice were acclimatized for 1 week in a specific pathogen-free facility (22 ± 2°C, 50% ± 10% humidity, 12-h light/dark cycle) with ad libitum access to standard chow and water. Mice were randomly assigned to 6 experimental groups (n = 8 per group): (1) EGC(+) Sham; (2) EGC(−) Sham; (3) EGC(+) I/R; (4) EGC(−) I/R; (5) EGC(+) Dex(+) I/R; (6) EGC(−) Dex(+) I/R.
Rat EGCs (CRL-2690, catalog #FH1201, Fuheng Biology, Shanghai, China) and rat small intestinal epithelial cells (IEC-6, catalog #iCell-h642, Cellverse, Shanghai, China) were used. Cells were verified to be free of mycoplasma contamination using a PCR-based mycoplasma detection kit. Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS; #FH100-900, Fuheng Biology) and 1% penicillin/streptomycin (#FH-K001TM, Fuheng Biology) in a humidified incubator at 37°C with 5% CO2.
Dexmedetomidine (TYZ24L01, Jiangsu Enhua Pharmaceutical, Jiangsu, China), Benzalkonium Chloride (catalog #B6295-100 g, Sigma, MO, USA), Fluorescein isothiocyanate–dextran (catalog # 100 MG, Sigma), Methyllycaconitine citrate (MLA) (catalog #ab120072, Abcam, Cambridge, UK), DAPI Staining Solution (#C1006-10 mL; Beyotime, Shanghai, China). The following antibodies were used in this study: Anti-Occludin (1:1000 for WB, 1:100 for IF; EPR20992, #ab216327; Abcam), anti-GFAP (1:1000; #ab7260; Abcam), anti-GAPDH (1:5000; EPR16891, #ab181602; Abcam), anti-GDNF (1:1000; EPR2714-N, #ab176564; Abcam), Anti-NeuN (1:200; EPR12763, #ab177487; Abcam), Anti-S100β (1:200; SP127, #ab115803; Abcam), FITC-labeled Goat Anti-Rabbit IgG (1:500; #A0562; Beyotime), Recombinant Rat GDNF (Catalog # 512-GF, R&D Systems, Minneapolis, MN, USA), and Cy3-labeled Goat Anti-Mouse IgG (1:500; Catlog #A0521, Beyotime).
Immunocytochemistry was performed to visualize protein expression. Briefly, EGCs were washed with PBS, fixed (4% PFA, 10 min, RT), and washed three times with PBS. Cells were then permeabilized (0.1% Triton X-100, 10 min) and blocked 1% BSA (#ST023, Beyotime, Shanghai, China) in PBS for 30 min at RT. Primary antibodies (diluted in blocking buffer as listed in Section 2.4) were applied (4°C, 12 h). After washing, secondary antibodies (diluted in blocking buffer as listed in Section 2.4) were applied (30 min, RT, dark). Following washes, samples were mounted and visualized by fluorescence microscopy (IX71, Olympus, Tokyo, Japan).
2.6 Cell Counting Kit-8 (CCK-8) Assay
EGCs were seeded (5 × 103 cells/well, 200 μL) in 96-well plates. After adhesion, the medium was replaced with 100 μL of a 1:9 (v/v) mixture of CCK-8 solution (#C0038, Beyotime) and serum-free DMEM (#FH-D02; Fuheng Biology). Plates were incubated (37°C, dark, 2 h). Absorbance was measured at 450 nm using a microplate reader (MULTISKAN FC, Thermo Fisher Scientific, WA, USA). Five replicate wells per group were used.
2.7 Real-Time Quantitative Polymerase Chain Reaction (qPCR)
Total RNA (1 μg) was extracted from EGCs using TRIzol reagent (#15596026, Invitrogen, Carlsbad, CA, USA), including a genomic DNA elimination step (DNase I treatment), and reverse-transcribed into cDNA using a PrimeScript™ RT Reagent Kit (#RR037A, Takara, Dalian, China). qPCR was performed using SYBR® Green Premix Ex Taq™ (#RR420A, Takara) on a Real-Time PCR System (Model 7500; Applied Biosystems). Relative mRNA expression was calculated using the 2−∆∆Ct method, normalized to GAPDH. Primer sequences were as follows: GDNF (Forward: 5′-GCGCTGACCAGTGACTCCAAT-3′, Reverse: 5′-GTGCCGCCGCTTGTTTATC-3′); GAPDH (Forward: 5′-AGTGCCAGCCTCGTCTCATA-3′, Reverse: 5′-ATGAAGGGGTCGTTGATGGC-3′). The total reaction volume was 20 μL. The cycling conditions were: 95°C for 30 s, followed by 40 cycles of 95°C for 5 s and 60°C for 34 s. Experiments were performed with 3 biological replicates.
2.8 Hematoxylin-Eosin (H&E) Staining and Immunofluorescence (IFC)
Mice were anesthetized and transcardially perfused with PBS followed by 4% PFA in PBS. Ileal segments were dissected and immersed in the same fixative overnight. Following fixation, tissues were dehydrated through an ethanol gradient (70%, 80%, 90%, 95%, 100%), embedded in Paraplast® (Leica, Nussloch, Germany), and sectioned at 5 μm using a rotary microtome (Leica RM2235, Leica). For H&E staining, sections were deparaffinized in xylene, rehydrated through graded ethanol solutions, and stained with hematoxylin and eosin (#G1120, Solarbio, Beijing, China) or immunohistochemistry.
IFC sections underwent antigen retrieval in citrate buffer (pH 6.0) via microwave heating. Sections were blocked with 5% BSA (#ST023, Beyotime) for 1 h at RT, then incubated with primary antibodies overnight at 4°C including Anti-Occludin (1:100; EPR20992, #ab216327; Abcam), anti-GFAP (1:1000; #ab7260; Abcam), Anti-NeuN (1:200; EPR12763, #ab177487; Abcam) and Anti-S100β (1:200; SP127, #ab115803; Abcam). After washing, sections were incubated with fluorescent secondary antibodies FITC-labeled Goat Anti-Rabbit IgG (1:500; #A0562; Beyotime) and Cy3-labeled Goat Anti-Mouse IgG (1:500; #A0521, Beyotime) for 1 h at RT. Images were acquired using the Olympus IX71 inverted microscope (Olympus).
Mice were fasted for 6 h before surgery. Under isoflurane anesthesia (2% induction, 1.5% maintenance), mice were placed on a heating pad to maintain body temperature at 37°C. A midline laparotomy exposed the superior mesenteric artery (SMA). The SMA was occluded at its origin for 30 min (confirmed by pulsation loss and mucosal pallor), followed by clip removal and 2 h reperfusion (confirmed by mucosal re-coloration). Both EGC(+) Sham and EGC(−) Sham mice underwent dissection without occlusion. Mice were euthanized by cervical dislocation under deep anesthesia six hours post-closure. Ileal tissues and portal vein blood were collected for subsequent analysis.
2.10 Enzyme-Linked Immunosorbent Assay (ELISA)
For in vitro analysis, cell culture supernatants were collected from the EGC-IEC co-culture wells after 48 h of treatment. For in vivo analysis, ileal tissue segments were harvested, weighed, and immediately homogenized in ice-cold RIPA lysis buffer containing protease inhibitors. Homogenates were centrifuged at 12,000× g for 15 min at 4°C, and the supernatant was collected. Total protein was quantified using a BCA Protein Assay Kit (#P0012, Beyotime). GDNF concentrations were quantified using species-specific GDNF ELISA kit (Mouse: E-El-M3028; Rat: E-EL-R0420; Elabscience, Wuhan, China) according to the manufacturer’s manual. In vivo GDNF levels were normalized to the total protein and expressed as pg/mg tissue; in vitro levels were expressed as pg/mL.
2.11 Benzalkonium Chloride (BAC) Induced EGCs Ablation
Mouse EGCs ablation was performed as previously described [25,27]. Mice were secured on a heating pad under isoflurane anesthesia. A midline laparotomy exposed the small intestine, and a 5-cm ileal segment was isolated. Sterile gauze soaked in 0.005% BAC/saline or saline alone was applied around the segment and mesentery for 15 min. After rinsing with saline, the bowel was returned, and the abdomen was closed. Mice recovered on a heating pad. Six hours post-BAC application, animals received food/water ad libitum and were monitored daily.
2.12 Intestinal Permeability Assay
Six hours post—Benzalkonium chloride (BAC) application (0.005% concentration, topical application via gauze, 15-min duration), mice were anesthetized, and a ~5 cm ileal segment was isolated. Both ends of this segment were ligated with silk suture to create a closed intestinal pouch. Then, 200 μL of phosphate-buffered saline (PBS) containing FITC-labeled 4 kDa dextran (FD-4) was injected into the pouch lumen. The abdomen was closed, and the mice were allowed to recover for 30 min. Subsequently, mice were euthanized, and portal vein blood was collected into heparinized tubes. Blood samples were centrifuged (3000× g, 10 min, 4°C) to obtain plasma. A 200 μL aliquot of plasma supernatant was analyzed for FD-4 concentration using a fluorescence spectrophotometer (MULTISKAN FC, Thermo Fisher) at excitation/emission wavelengths of 490/520 nm. To quantify FD-4 levels, a standard curve ranging from 0 to 100 μg/mL was generated using known concentrations of FD-4 dissolved in plasma obtained from untreated control mice.
Data analysis was performed using SPSS 20.0 (IBM Corp., Armonk, NY, USA). Normally distributed continuous data are presented as mean ± SEM and were compared using the independent samples t-test (for pairwise comparisons) or one-way analysis of variance (ANOVA) (for comparisons across multiple groups). Non-normally distributed continuous data are presented as median and interquartile range (IQR) and were compared using the Mann-Whitney U test. p value < 0.05 was considered statistically significant.
3.1 Dex Activates Cultured EGCs in a Dose-Dependent Manner
CCK-8 assays showed Dex concentrations ≤ 100 μm were non-cytotoxic to rat EGCs, with mean viability exceeding 90% (Fig. 1A). Consequently, Dex concentrations ≤ 100 μm were used for subsequent experiments. GFAP immunofluorescence revealed no significant changes in morphology or GFAP fluorescent signal in EGCs treated with 60 μm Dex for 24 h (Fig. 1B,C). GDNF mRNA expression showed no significant change at 20 μm Dex (p > 0.05 vs. control) but was significantly increased at 40, 60, 80, and 100 μm Dex (p < 0.05), exhibiting dose-dependence within this range (Fig. 1D). Western blot analysis confirmed gradually increasing GDNF protein expression from 40 to 100 μm Dex (Fig. 1E). The GDNF/GAPDH ratio was unchanged at 20 μm Dex (p > 0.05) but significantly increased at concentrations ≥40 μm (p < 0.01) (Fig. 1F). These results indicate that Dex dose-dependently increases GDNF expression in EGCs at concentrations between 40 and 100 μm.
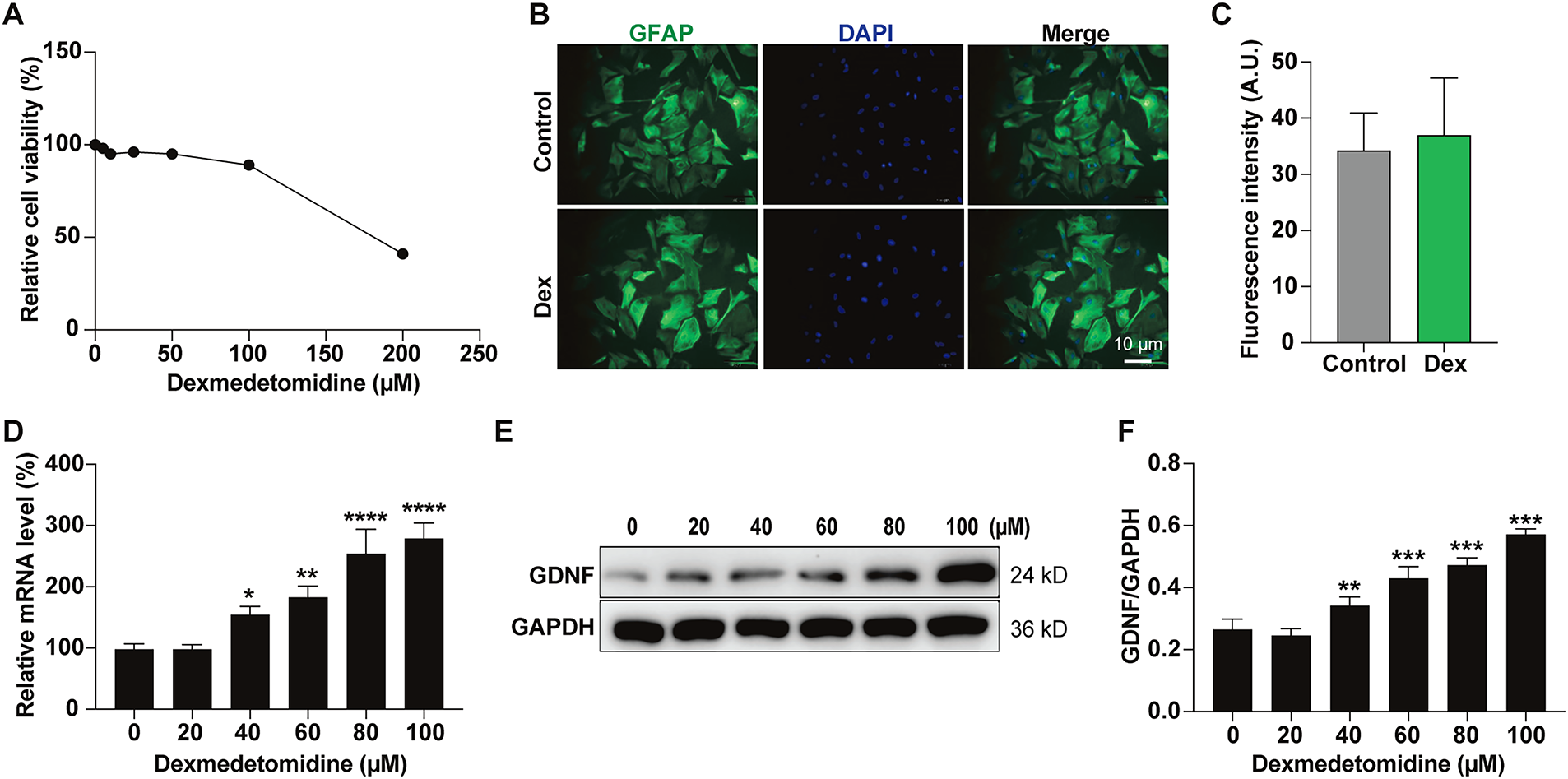
Figure 1: Dexmedetomidine (Dex) dose-dependently activates enteric glial cells (EGCs) and increases glial cell line-derived neurotrophic factor (GDNF) expression. (A) EGC proliferation was determined by a Cell Counting Kit-8 (CCK-8) cell viability assay. (B) Representative immunofluorescence images of EGCs using glial fibrillary acidic (GFAP) antibody, scale bar: 10 µm, accompanied by (C) quantification (n = 6, p > 0.05). (D) EGCs were analyzed by qPCR for changes in mRNA levels of GDNF after treatment with Dex (n = 3). (E) EGCs were analyzed by Western blotting for changes in protein levels of GDNF after treatment with Dex. (F) Quantification and statistical analysis, n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. One-way ANOVA followed by Tukey’s test.
3.2 Dex Enhances IEC Barrier Integrity via the α7-nAChR-EGC-GDNF Axis In Vitro
To elucidate the mechanism of Dex-mediated protection, we used an EGC-IEC co-culture system (Fig. 2A). First, we measured GDNF protein secretion into the culture medium via ELISA (Fig. 2B). Dex treatment significantly increased GDNF release in the IEC + EGC + Dex group compared to controls. This effect was completely blocked by the addition of MLA, an α7-nAChR antagonist. Next, we assessed IEC barrier integrity by measuring TEER (Fig. 2C). After 48 h, Dex alone (IEC + Dex) did not affect TEER (p > 0.05). However, TEER was significantly increased in groups where EGCs were present (IEC + EGC + Dex), EGC supernatant was used (IEC + EGC-Sup + Dex), or recombinant GDNF (100 ng/mL) was added directly (IEC + GDNF) (p < 0.05 vs. IEC). Critically, the protective effect of Dex was abolished in the IEC + EGC + Dex + MLA group, confirming the role of α7-nAChR activation. Finally, Western blot analysis (Fig. 2D) showed that Occludin expression was unchanged in the IEC + Dex group, but was significantly elevated in the IEC + GDNF, IEC + EGC + Dex, and IEC + EGC-sup/Dex groups (p < 0.05). These findings demonstrate that Dex enhances tight junction function by activating the α7-nAChR on EGCs, which in turn stimulates GDNF secretion and leads to the upregulation of Occludin.
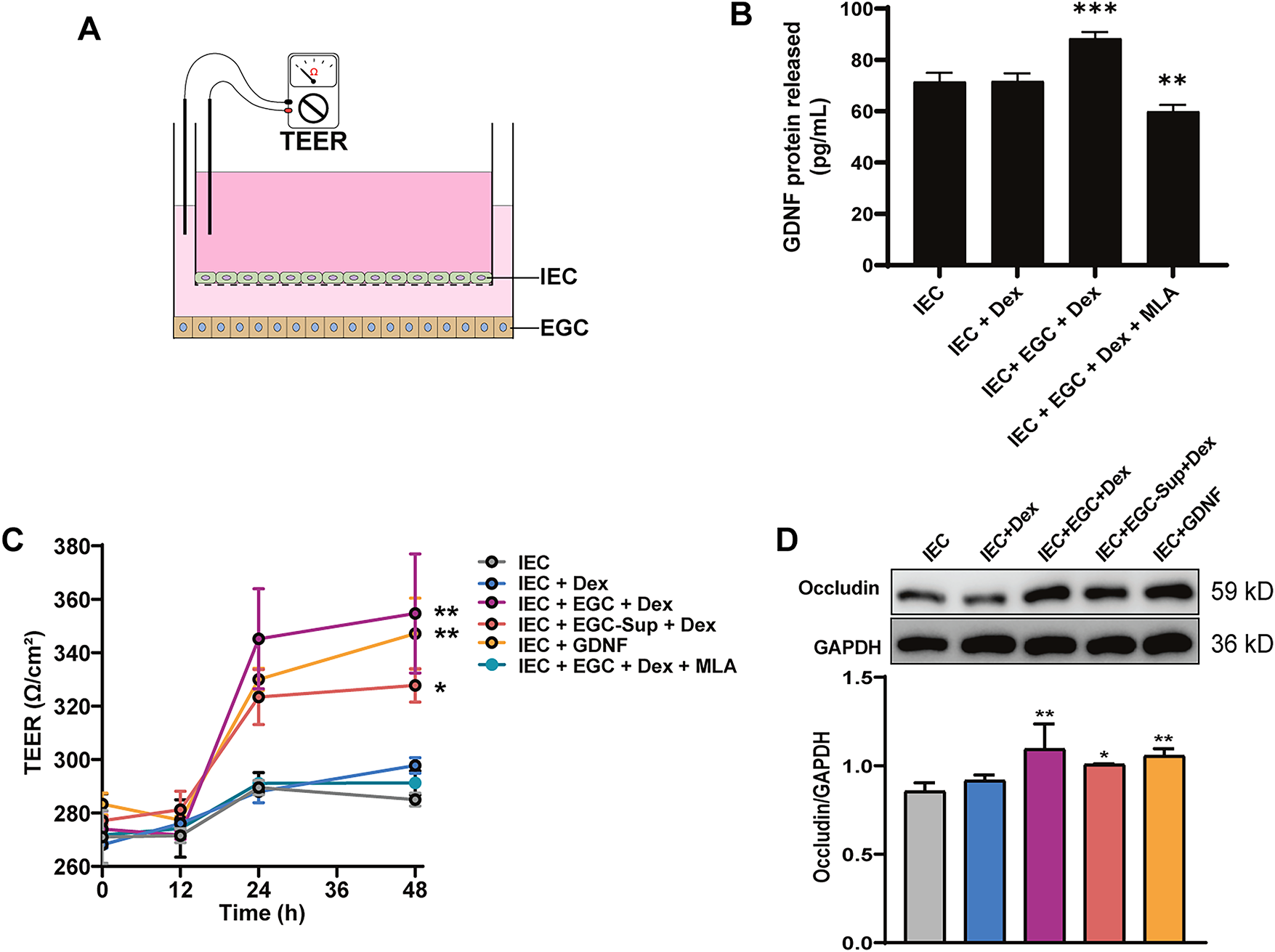
Figure 2: Dex enhances Occludin expression and tight junction integrity via α7-nAChR-EGC-GDNF axis in vitro. (A) Schematic of the EGC-IEC co-culture system, with an IEC monolayer on the apical side of the membrane, and EGC positions on the underside of the membrane and in the basolateral chamber. IEC monolayer permeability was evaluated by measuring transmembrane electrical resistance (TEER) between electrodes located in each compartment. (B) GDNF protein levels in the co-culture supernatant after 48 h, measured by Enzyme-Linked Immunosorbent Assay (ELISA). (C) TEER values measured at 0, 12, 24, and 48 h. (D) Representative Western blots and quantification for Occludin expression in IECs after 48 h (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. One-way ANOVA followed by Tukey’s test.
3.3 BAC Successfully Depletes EGCs without Neuronal Damage
As a critical component of the intestinal epithelial barrier, enteric glial cells (EGCs) play a pivotal role in maintaining epithelial barrier integrity [15,16]. To investigate the EGC-dependence of Dex’s effects on mucosal barrier integrity in vivo, ileal EGCs were selectively depleted by topical application of 0.005% benzalkonium chloride (BAC) [25,27]. H&E staining of ileum sections showed intact intestinal layers, villus structure, and no inflammation, hemorrhage, or glandular abnormalities in saline-treated Controls. BAC-treated animals retained mucosal and villus integrity, with only mild lymphocyte and neutrophil infiltration in the lamina propria and normal glands (Fig. 3A). Chiu’s scores showed no significant difference (p > 0.05) (Fig. 3B). Immunofluorescence confirmed a significant reduction in S100β protein fluorescence (an EGC marker) in the BAC group vs. Controls (p < 0.01) (Fig. 3C,D), indicating EGC depletion. NeuN fluorescence (a neuronal marker) showed no significant intergroup variation (p > 0.05) (Fig. 3C,E), confirming selective EGC loss without neuronal damage.
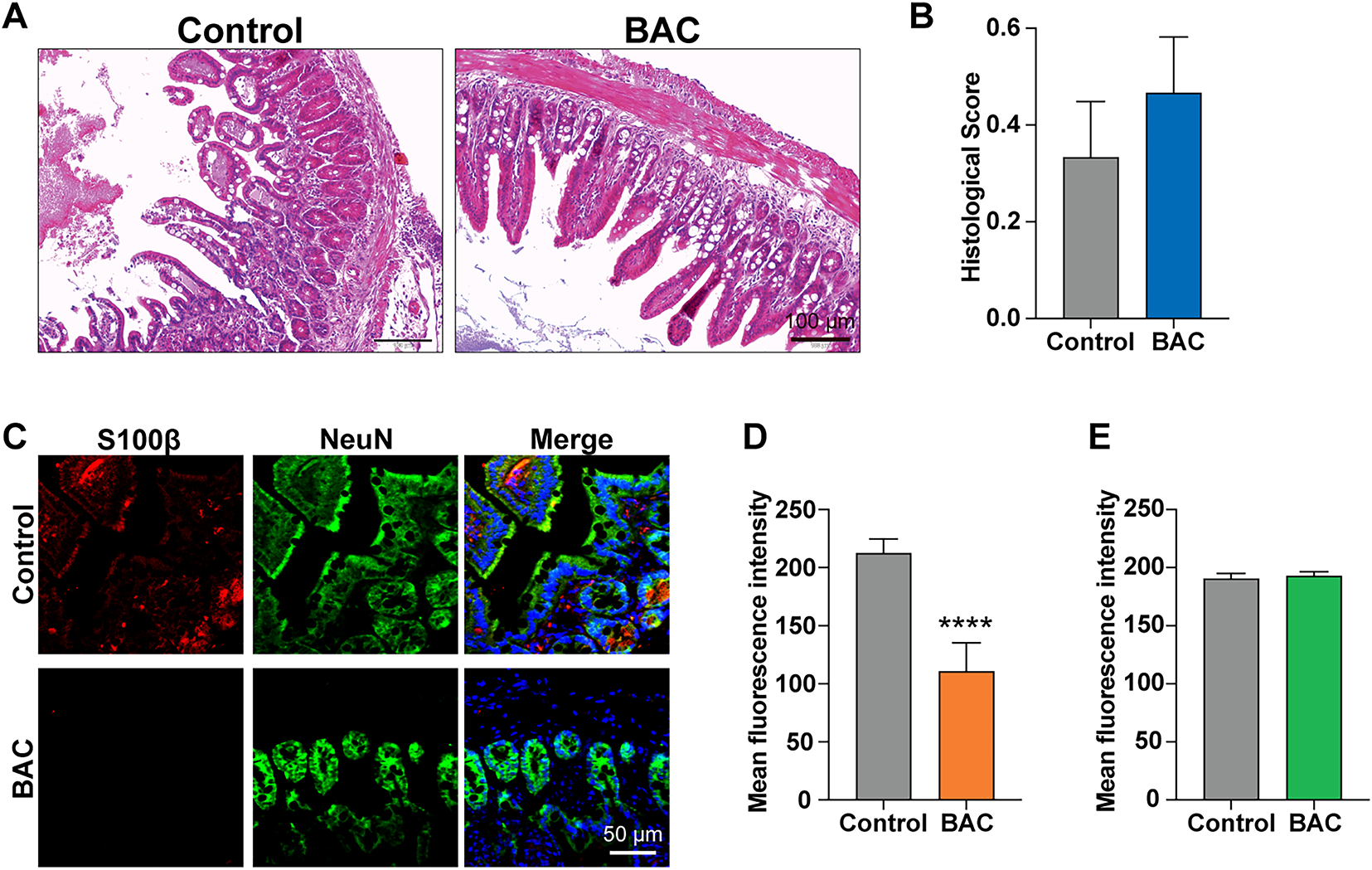
Figure 3: BAC selectively induces EGC loss without neuronal damage. (A) Representative image of H&E staining on the cross-section of ileum, scale bar: 100 µm, and (B) statistical analysis of the histological score (n = 3, p > 0.05). (C) Representative image of immunohistology for S100β and NeuN on the section of ileum, Scale bar: 50 µm, accompanied with (D) quantification and statistical analysis of S100β and (E) NeuN fluorescence intensity (n = 9 fields per animal, from 3 animals, ****p < 0.0001, t-test).
3.4 Dex Protects against I/R Injury via Activating EGCs In Vivo
Intestinal ischemia-reperfusion (I/R) injury impairs the epithelial barrier. Chiu’s scores showed no difference between EGC-intact sham and EGC-deficient sham groups (p > 0.05). Both EGC-intact I/R and EGC-deficient I/R groups exhibited significantly higher scores vs. their respective sham controls (p < 0.01). The EGC-deficient I/R group had a significantly higher Chiu’s score than the EGC-intact I/R group (p < 0.05), indicating exacerbated damage upon EGC depletion. In EGC-intact mice, Dex treatment (EGC-intact Dex-I/R) significantly reduced Chiu’s scores vs. the EGC-intact I/R group (p < 0.05). In contrast, Dex treatment (EGC-deficient Dex-I/R) showed no significant reduction vs. the EGC-deficient I/R group (p > 0.05) (Fig. 4A,B).
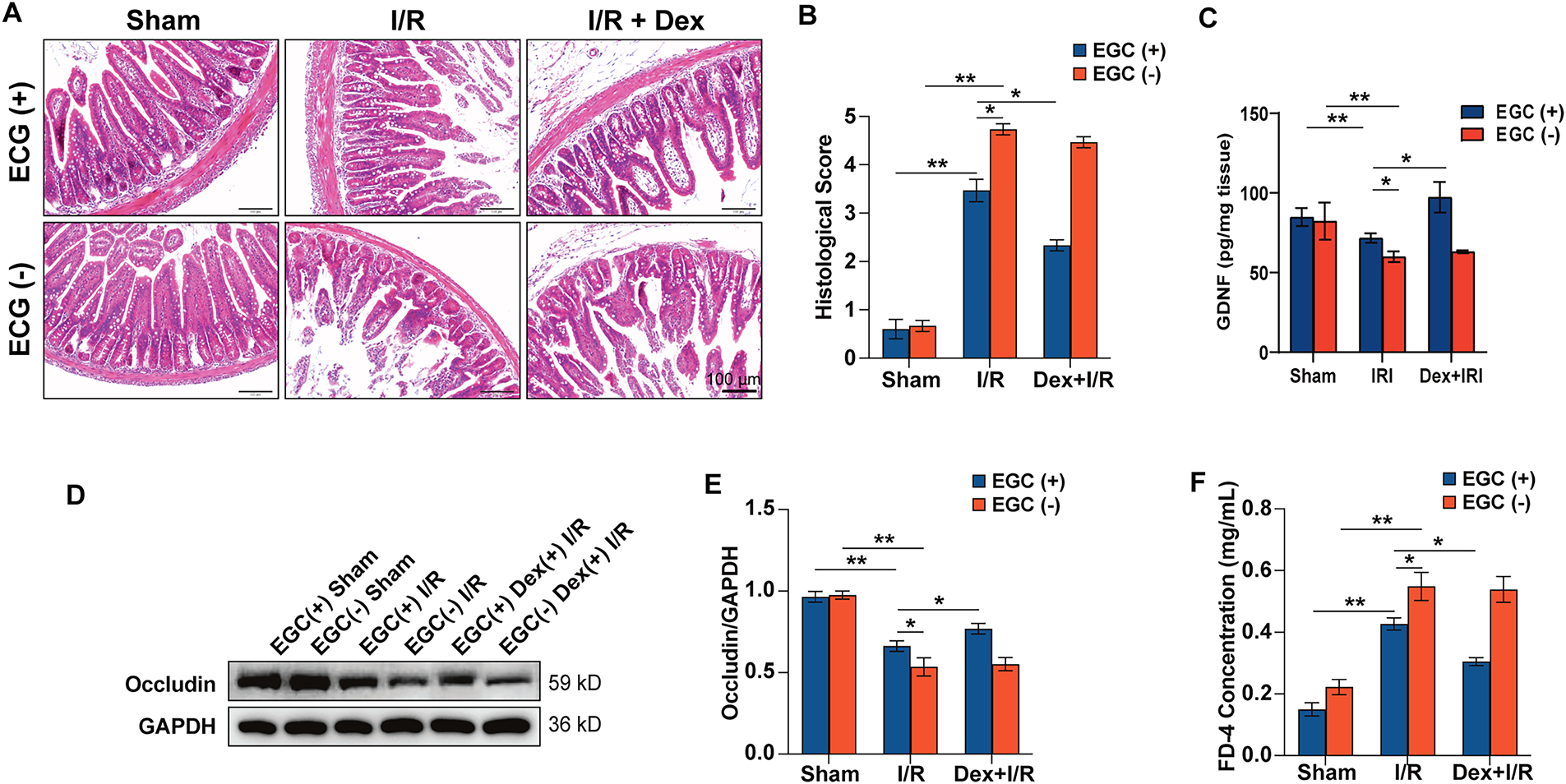
Figure 4: Dex protects against I/R injury via activating EGCs in vivo. (A,B) Representative images of H&E staining and statistical analysis of the histological score, scale bar: 100 µm, n = 3. (C) GDNF protein levels in ileal tissue homogenates, measured by ELISA. (D) Representative Western blot of Occludin expression in ileal tissue. (E) Quantification and statistical analysis of the Occludin/GAPDH ratio. (F) Statistical analysis of plasma FITC-dextran (FD-4) concentration. (n = 3). *p < 0.05, **p < 0.01, one-way ANOVA followed by Tukey’s test or t-test.
To confirm this mechanism in vivo, GDNF protein levels were measured in ileal tissue (Fig. 4C). I/R injury significantly reduced GDNF levels compared to sham controls in both EGC-intact and EGC-deficient mice (p < 0.01). In EGC-intact mice, Dex treatment (EGC(+) Dex(+) I/R) significantly restored GDNF levels (p < 0.05 vs. EGC(+) I/R). In contrast, Dex did not affect GDNF levels in EGC-deficient mice (EGC(−) Dex(+) I/R), demonstrating that EGCs are the essential source of Dex-induced GDNF in vivo.
Occludin expression did not differ between EGC-intact sham and EGC-deficient sham groups (p > 0.05). Both I/R groups showed significantly reduced Occludin expression vs. their sham controls (p < 0.01). Occludin expression was significantly lower in the EGC-deficient I/R group than in the EGC-intact I/R group (p < 0.05). In EGC-intact mice, Dex treatment (EGC-intact Dex-I/R) significantly upregulated Occludin expression vs. the EGC-intact I/R group (p < 0.05). No significant difference was observed between the EGC-deficient Dex-I/R and EGC-deficient I/R groups (p > 0.05) (Fig. 4D,E), indicating Dex improves Occludin expression only when EGCs are present.
Plasma FITC-dextran (FD-4) concentration (Table 1) showed no significant difference between sham groups (p > 0.05). Both I/R groups exhibited significantly elevated FD-4 concentrations vs. their sham controls (p < 0.01). The EGC-deficient I/R group had a significantly higher FD-4 level than the EGC-intact I/R group (p < 0.05). In EGC-intact mice, Dex treatment (EGC-intact Dex-I/R) significantly reduced FD-4 concentration vs. the EGC-intact I/R group (p < 0.05). No significant difference was observed between the EGC-deficient Dex-I/R and EGC-deficient I/R groups (p > 0.05) (Fig. 4F). This demonstrates Dex effectively ameliorates I/R-induced intestinal hyperpermeability only when EGCs are preserved.

This study identifies a novel mechanism for dexmedetomidine (Dex)-mediated protection of intestinal mucosal barrier integrity: activation of enteric glial cells (EGCs) to upregulate the tight junction protein Occludin. In vitro experiments demonstrated that Dex (40–100 μm) promoted EGCs to secrete glial-derived neurotrophic factor (GDNF) in a dose-dependent manner. GDNF, as a key effector of EGCs, directly upregulates Occludin expression and enhances trans-epithelial electrical resistance (TEER) in cultured intestinal epithelial cells. This highlights the central role of the EGCs-GDNF-Occludin pathway in barrier homeostasis [15,16]. In the mouse models, Dex improves intestinal Occludin expression and ameliorates I/R-induced intestinal hyperpermeability only when EGCs are intact. These findings align with studies showing reduced Occludin expression and barrier defects in transgenic mice with glial disruption (GFAP-HA), which are rescued by exogenous GDNF [28].
Regarding signaling pathways, Dex, as a selective α2-adrenergic receptor agonist, likely acts via the cholinergic anti-inflammatory pathway. Dex inhibits sympathetic activity, potentially enhancing vagal tone. The vagus nerve activates EGCs via α7-nicotinic acetylcholine receptors (α7-nAChRs). Dex upregulates α7-nAChR expression in the hippocampus [26]—a mechanism potentially conserved in the intestine. Supporting this, Dex-mediated EGC activation, subsequent GDNF secretion, and the resulting enhancement of epithelial barrier function were all blocked by the α7-nAChR antagonist MLA. Crucially, Dex alone did not affect Occludin in IEC monoculture, confirming EGCs are essential mediators.
This study challenges the traditional view of Dex’s intestinal protective mechanisms, which focused primarily on antioxidant and anti-inflammatory effects [29,30]. However, emerging evidence indicates that the gut-protective functions of Dex extend far beyond classical immunosuppression. Dex has been shown to upregulate the intestinal chloride transporter SLC26A3, thereby enhancing epithelial ion transport and contributing to mucosal homeostasis independently of anti-inflammatory pathways [31]. In addition, Dex and its analogue Dexmedetomidine have been shown to promote epithelial and gut-vascular barrier repair by activating receptor-dependent signaling cascades [32]. These findings expand the mechanistic landscape of Dex, suggesting that its therapeutic effects involve not only suppression of oxidative and inflammatory injury but also direct reinforcement of epithelial transport capacity, cytoskeletal stability, and barrier structural remodeling. To our knowledge, we demonstrate the pivotal role of EGCs in mediating these protective effects. In vivo, Dex improved histopathology and reduced intestinal permeability (FITC-dextran) in I/R mice, but only when EGCs were intact. Selective EGC depletion using 0.005% BAC abolished Dex’s protection, providing direct evidence that EGCs are prerequisite for Dex-mediated intestinal barrier protection.
However, the study has limitations: 1. Translational Discrepancy in Concentration: The effective in vitro Dex concentrations (40–100 μm) that showed efficacy in our EGC model significantly exceed typical clinical serum levels (nmol/L range) [33,34]. Although our CCK-8 assay showed no cytotoxicity at these levels (Fig. 1A), this discrepancy is a known translational challenge [35]. The observed tolerance might be attributable to several factors, including cell-type-specific resistance (as EGCs may be more robust than other cell types), the specific in vitro exposure duration (24–48 h), or protective components in the culture medium [36,37]. Future dose-effect optimization studies are necessary to bridge this gap between in vitro findings and potential in vivo applications. 2. Species Mismatch: A species mismatch exists between our experimental models. Our in vitro studies utilized rat-derived cell lines (EGCs and IEC-6), whereas our in vivo intestinal I/R model was established in mice. While these pathways are generally conserved, potential species-specific differences in EGC biology or the α7-nAChR-GDNF axis cannot be ruled out. Therefore, caution is warranted when directly translating mechanistic findings from the in vitro rat co-culture system to the in vivo mouse model. 3. Ablation Model Selectivity: While BAC is used to selectively ablate EGCs, its potential off-target effects on other enteric neuroglial subsets were not definitively ruled out. Validation using more specific genetic knockout models is required to confirm these findings. 4. Mechanistic Gaps: Our mechanistic claims are based primarily on in vitro data. While we demonstrated the role of α7-nAChRs in vitro (Fig. 2), direct in vivo evidence linking the cholinergic pathway to EGC activation in the I/R model is lacking. The absence of in vivo antagonist (e.g., MLA) administration means we cannot definitively conclude that Dex protects against I/R injury via this specific mechanism in vivo. This remains a critical point for future investigation. Furthermore, even in vitro, our study did not confirm that MLA directly blocks the upregulation of Occludin protein (Fig. 2D), only its effect on the functional outcome (TEER) and GDNF secretion, which warrants further investigation.
A further limitation is the species mismatch between our experimental models. Our in vitro studies utilized rat-derived cell lines (EGCs and IEC-6), whereas our in vivo intestinal I/R model was established in mice. Although these pathways are generally conserved, potential species-specific differences in enteric glial cell biology or the signaling pathways investigated, such as the α7-nAChR-GDNF axis, may exist. Therefore, caution is warranted when directly translating the specific mechanistic findings from our in vitro rat co-culture system to the in vivo mouse model. Finally, we acknowledge the presence of background signal in the immunofluorescence images. We recognize that this is likely attributable to the inherent limitations of antibody specificity within the complex tissue architecture. Although image processing was applied to minimize this noise, some non-specific staining remained. However, the quantitative analysis supports the efficacy of the EGC ablation models as the difference in S100β fluorescence intensity between the control and BAC groups despite the background.
From a translational perspective, this study identifies EGCs as a novel target for perioperative intestinal barrier protection. Given EGC dysfunction in inflammatory bowel disease and intestinal ischemia [28,38], targeting EGCs with Dex may offer a new strategy for preventing and treating intestinal mucosal barrier dysfunction. Future studies should integrate clinical research to explore optimal Dex regimens in patients and identify EGC-related biomarkers, facilitating the translation of these findings.
In conclusion, this study demonstrates that dexmedetomidine (Dex) protects the intestinal mucosal barrier through a novel, EGC-dependent mechanism. We have provided both in vitro and in vivo evidence that Dex activates the α7-nAChR on EGCs, stimulating the secretion of GDNF. This EGC-derived GDNF, in turn, enhances intestinal epithelial integrity by upregulating the tight junction protein Occludin. Our findings using a selective EGC-ablation model definitively show that the protective effects of Dex against I/R-induced intestinal injury, including the restoration of Occludin and the reduction of gut permeability, are lost in the absence of EGCs. Collectively, these findings identify the EGC-GDNF-Occludin axis as a critical signaling pathway for Dex-induced intestinal protection, highlighting EGCs as a promising therapeutic target for managing barrier dysfunction in critical illness.
Acknowledgement: Not applicable.
Funding Statement: This research was supported by Yunnan Provincial Science and Technology Basic Research Project (202201AY070001-075), Yunnan Health Training Project of High-Level Talents (L-2024008), and “Xingdian Talents” Support Project of Yunnan Province.
Author Contributions: The authors confirm contribution to the paper as follows: Conceptualization, Yuanhong Mao, Yunlan Yang, and Kun Yang (the 2nd one); methodology, Yunlan Yang; formal analysis, Yongqiang Sun; investigation, Yuanhong Mao, Yunlan Yang, Kun Yang (the 1st one), Yongqiang Sun and Kun Yang (the 2nd one); data curation, Yunlan Yang; writing—original draft preparation, Yuanhong Mao; writing—review and editing, Kun Yang (the 2nd one); visualization, Yongqiang Sun; funding acquisition, Kun Yang (the 2nd one). All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: The authors confirm that the data supporting the findings of this study are available within the article.
Ethics Approval: This study was approved by the Animal Care and Use Committee of Kunming Medical University (Approval No.: KMMU20211240). All animal procedures adhered to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, USA).
Conflicts of Interest: The authors declare no conflicts of interest.
References
1. Hatfield KM, Dantes RB, Baggs J, Sapiano MRP, Fiore AE, Jernigan JA, et al. Assessing variability in hospital-level mortality among U.S. medicare beneficiaries with hospitalizations for severe sepsis and septic shock. Crit Care Med. 2018;46(11):1753–60. doi:10.1097/ccm.0000000000003324. [Google Scholar] [PubMed] [CrossRef]
2. Rhee C, Wang R, Zhang Z, Fram D, Kadri SS, Klompas M. Epidemiology of hospital-onset vs. community-onset sepsis in U.S. hospitals and association with mortality: a retrospective analysis using electronic clinical data. Crit Care Med. 2019;47(9):1169–76. doi:10.1097/ccm.0000000000003817. [Google Scholar] [PubMed] [CrossRef]
3. Weng L, Zeng XY, Yin P, Wang LJ, Wang CY, Jiang W, et al. Sepsis-related mortality in China: a descriptive analysis. Intensive Care Med. 2018;44(7):1071–80. doi:10.1007/s00134-018-5203-z. [Google Scholar] [PubMed] [CrossRef]
4. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395(10219):200–11. doi:10.1016/s0140-6736(19)32989-7. [Google Scholar] [PubMed] [CrossRef]
5. Fleischmann-Struzek C, Rudd K. Challenges of assessing the burden of sepsis. Med Klin Intensiv Notfmed. 2023;118(Suppl 2):68–74. doi:10.1007/s00063-023-01088-7. [Google Scholar] [PubMed] [CrossRef]
6. Tao YL, Wang JR, Liu M, Liu YN, Zhang JQ, Zhou YJ, et al. Progress in the study of the correlation between sepsis and intestinal microecology. Front Cell Infect Microbiol. 2024;14:1357178. doi:10.3389/fcimb.2024.1357178. [Google Scholar] [PubMed] [CrossRef]
7. di Tommaso N, Santopaolo F, Gasbarrini A, Ponziani FR. The gut–vascular barrier as a new protagonist in intestinal and extraintestinal diseases. Int J Mol Sci. 2023;24(2):1470. doi:10.3390/ijms24021470. [Google Scholar] [PubMed] [CrossRef]
8. McMullan RR, McAuley DF, O’Kane CM, Silversides JA. Vascular leak in sepsis: physiological basis and potential therapeutic advances. Crit Care. 2024;28(1):97. doi:10.1186/s13054-024-04875-6. [Google Scholar] [PubMed] [CrossRef]
9. Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med. 2018;50(8):126. doi:10.1038/s12276-018-0126-x. [Google Scholar] [PubMed] [CrossRef]
10. Ou J, Guan X, Wang J, Wang T, Zhang B, Li R, et al. Epithelial NELF guards intestinal barrier function to ameliorate colitis by maintaining junctional integrity. Mucosal Immunol. 2022;15(2):279–88. doi:10.1038/s41385-021-00465-9. [Google Scholar] [PubMed] [CrossRef]
11. Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol. 2009;124(1):3–20; quiz 1-2. doi:10.1016/j.jaci.2009.05.038. [Google Scholar] [PubMed] [CrossRef]
12. Kuo WT, Odenwald MA, Turner JR, Zuo L. Tight junction proteins occludin and ZO-1 as regulators of epithelial proliferation and survival. Ann N Y Acad Sci. 2022;1514(1):21–33. doi:10.1111/nyas.14798. [Google Scholar] [PubMed] [CrossRef]
13. Shen L. Tight junctions on the move: molecular mechanisms for epithelial barrier regulation. Ann N Y Acad Sci. 2012;1258(1):9–18. doi:10.1111/j.1749-6632.2012.06613.x. [Google Scholar] [PubMed] [CrossRef]
14. Yu ASL, McCarthy KM, Francis SA, McCormack JM, Lai J, Rogers RA, et al. Knockdown of occludin expression leads to diverse phenotypic alterations in epithelial cells. Am J Physiol Cell Physiol. 2005;288(6):C1231–41. doi:10.1152/ajpcell.00581.2004. [Google Scholar] [PubMed] [CrossRef]
15. Neunlist M, Rolli-Derkinderen M, Latorre R, van Landeghem L, Coron E, Derkinderen P, et al. Enteric glial cells: recent developments and future directions. Gastroenterology. 2014;147(6):1230–7. doi:10.1053/j.gastro.2014.09.040. [Google Scholar] [PubMed] [CrossRef]
16. Ochoa-Cortes F, Turco F, Linan-Rico A, Soghomonyan S, Whitaker E, Wehner S, et al. Enteric glial cells: a new frontier in neurogastroenterology and clinical target for inflammatory bowel diseases. Inflamm Bowel Dis. 2016;22(2):433–49. doi:10.1097/mib.0000000000000667. [Google Scholar] [PubMed] [CrossRef]
17. Osman I, Wang L, Hu G, Zheng Z, Zhou J. GFAP (glial fibrillary acidic protein)-positive progenitor cells contribute to the development of vascular smooth muscle cells and endothelial cells—brief report. Arterioscler Thromb Vasc Biol. 2020;40(5):1231–8. doi:10.1161/atvbaha.120.314078. [Google Scholar] [PubMed] [CrossRef]
18. Bauman BD, Meng J, Zhang L, Louiselle A, Zheng E, Banerjee S, et al. Enteric glial–mediated enhancement of intestinal barrier integrity is compromised by morphine. J Surg Res. 2017;219:214–21. doi:10.1016/j.jss.2017.05.099. [Google Scholar] [PubMed] [CrossRef]
19. Meir M, Kannapin F, Diefenbacher M, Ghoreishi Y, Kollmann C, Flemming S, et al. Intestinal epithelial barrier maturation by enteric glial cells is GDNF-dependent. Int J Mol Sci. 2021;22(4):1887. doi:10.3390/ijms22041887. [Google Scholar] [PubMed] [CrossRef]
20. Rajan S, Hutcherson MT, Sessler DI, Kurz A, Yang D, Ghobrial M, et al. The effects of dexmedetomidine and remifentanil on hemodynamic stability and analgesic requirement after craniotomy: a randomized controlled trial. J Neurosurg Anesth. 2016;28(4):282–90. doi:10.1097/ana.0000000000000221. [Google Scholar] [PubMed] [CrossRef]
21. Fukuda M, Vazquez AL, Zong X, Kim SG. Effects of the α2-adrenergic receptor agonist dexmedetomidine on neural, vascular and BOLD fMRI responses in the somatosensory cortex. Eur J Neurosci. 2013;37(1):80–95. doi:10.1111/ejn.12024. [Google Scholar] [PubMed] [CrossRef]
22. Bao N, Tang B. Organ-protective effects and the underlying mechanism of dexmedetomidine. Mediat Inflamm. 2020;2020:6136105. doi:10.1155/2020/6136105. [Google Scholar] [PubMed] [CrossRef]
23. Costantini TW, Krzyzaniak M, Cheadle GA, Putnam JG, Hageny AM, Lopez N, et al. Targeting α-7 nicotinic acetylcholine receptor in the enteric nervous system: a cholinergic agonist prevents gut barrier failure after severe burn injury. Am J Pathol. 2012;181(2):478–86. doi:10.1016/j.ajpath.2012.04.005. [Google Scholar] [PubMed] [CrossRef]
24. Cheadle GA, Costantini TW, Bansal V, Eliceiri BP, Coimbra R. Cholinergic signaling in the gut: a novel mechanism of barrier protection through activation of enteric glia cells. Surg Infect. 2014;15(4):387–93. doi:10.1089/sur.2013.103. [Google Scholar] [PubMed] [CrossRef]
25. Langness S, Kojima M, Coimbra R, Eliceiri BP, Costantini TW. Enteric glia cells are critical to limiting the intestinal inflammatory response after injury. Am J Physiol Gastrointest Liver Physiol. 2017;312(3):G274–82. doi:10.1152/ajpgi.00371.2016. [Google Scholar] [PubMed] [CrossRef]
26. Xu KL, Liu XQ, Yao YL, Ye MR, Han YG, Zhang T, et al. Effect of dexmedetomidine on rats with convulsive status epilepticus and association with activation of cholinergic anti-inflammatory pathway. Biochem Biophys Res Commun. 2018;495(1):421–6. doi:10.1016/j.bbrc.2017.10.124. [Google Scholar] [PubMed] [CrossRef]
27. Yoneda A, Shima H, Nemeth L, Oue T, Puri P. Selective chemical ablation of the enteric plexus in mice. Pediatr Surg Int. 2002;18(4):234–7. doi:10.1007/s003830100681. [Google Scholar] [PubMed] [CrossRef]
28. Aube AC, Cabarrocas J, Bauer J, Philippe D, Aubert P, Doulay F, et al. Changes in enteric neurone phenotype and intestinal functions in a transgenic mouse model of enteric glia disruption. Gut. 2006;55(5):630–7. doi:10.1136/gut.2005.067595. [Google Scholar] [PubMed] [CrossRef]
29. Cong D, Yu Y, Meng Y, Qi X. Dexmedetomidine (Dex) exerts protective effects on rat neuronal cells injured by cerebral ischemia/reperfusion via regulating the Sphk1/S1P signaling pathway. J Stroke Cerebrovasc Dis. 2023;32(1):106896. doi:10.1016/j.jstrokecerebrovasdis.2022.106896. [Google Scholar] [PubMed] [CrossRef]
30. Zhao B, Li D, Zhang S, He L, Ai Y. Dexmedetomidine attenuates cerebral ischemia-reperfusion injury in rats by inhibiting the JNK pathway. Ann Palliat Med. 2021;10(6):6768–78. doi:10.21037/apm-21-1218. [Google Scholar] [PubMed] [CrossRef]
31. Kumar A, Husain N, Anbazhagan AN, Jayawardena D, Priyamvada S, Singhal M, et al. Dexamethasone upregulates the expression of the human SLC26A3 (DRA Down-Regulated in Adenoma) transporter (an IBD susceptibility gene) in intestinal epithelial cells and attenuates gut inflammation. Inflamm Bowel Dis. 2025;31(3):625–35. doi:10.1093/ibd/izae271. [Google Scholar] [PubMed] [CrossRef]
32. Zhou X, Yang Y, Su Z, Luo Z. Dexmedetomidine protects the brain: exploring the α2AR/FAK pathway in post-stroke intestinal barrier repair. Front Biosci. 2025;30(2):27159. doi:10.31083/fbl27159. [Google Scholar] [PubMed] [CrossRef]
33. Wen J, McCann S, Balevic SJ, Muller WJ, Hornik CD, Autmizguine J, et al. Pharmacokinetics of dexamethasone in children and adolescents with obesity. J Clin Pharmacol. 2024;64(12):1491–504. doi:10.1002/jcph.6108. [Google Scholar] [PubMed] [CrossRef]
34. Czock D, Keller F, Rasche FM, Häussler U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin Pharmacokinet. 2005;44(1):61–98. doi:10.2165/00003088-200544010-00003. [Google Scholar] [PubMed] [CrossRef]
35. Jiao T, Yao X, Zhao Y, Zhou Y, Gao Y, Fan S, et al. Dexamethasone-induced liver enlargement is related to PXR/YAP activation and lipid accumulation but not hepatocyte proliferation. Drug Metab Dispos. 2020;48(9):830–9. doi:10.1124/dmd.120.000061. [Google Scholar] [PubMed] [CrossRef]
36. Krishna G, Urda G, Tefera W, Lalwani ND, Theiss J. Simultaneous evaluation of dexamethasone-induced apoptosis and micronuclei in rat primary spleen cell cultures. Mutat Res. 1995;332(1–2):0027510795000753. doi:10.1016/0027-5107(95)00075-3. [Google Scholar] [PubMed] [CrossRef]
37. Bailly-Maitre B, de Sousa G, Boulukos K, Gugenheim J, Rahmani R. Dexamethasone inhibits spontaneous apoptosis in primary cultures of human and rat hepatocytes via Bcl-2 and Bcl-xL induction. Cell Death Differ. 2001;8(3):279–88. doi:10.1038/sj.cdd.4400815. [Google Scholar] [PubMed] [CrossRef]
38. Cornet A, Savidge TC, Cabarrocas J, Deng WL, Colombel JF, Lassmann H, et al. Enterocolitis induced by autoimmune targeting of enteric glial cells: a possible mechanism in Crohn’s disease? Proc Natl Acad Sci U S A. 2001;98(23):13306–11. doi:10.1073/pnas.231474098. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools