
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

CNPY2 Regulates Macrophage Polarization and Inflammatory Immune Responses via the TLR4/NF-κB Signaling Pathway to Alleviate Osteomyelitis of the Jaw
1 Department of Oral and Maxillofacial Surgery, the First Affiliated Hospital, Fujian Medical University, Fuzhou, China
2 Department of Oral and Maxillofacial Surgery, National Regional Medical Center, Binhai Campus of the First Affiliated Hospital, Fujian Medical University, Fuzhou, China
3 Department of Stomatology, Fujian Health College, Fuzhou, China
4 Department of Stomatology, the First Affiliated Hospital, Fujian MedicalUniversity, Fuzhou, China
5 Department of Stomatology, National Regional Medical Center, Binhai Campus of the First Affiliated Hospital, Fujian Medical University, Fuzhou, China
* Corresponding Author: Li Huang. Email:
# These authors contributed equally to this work as the first author
BIOCELL 2026, 50(5), 8 https://doi.org/10.32604/biocell.2026.075875
Received 10 November 2025; Accepted 22 January 2026; Issue published 13 May 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
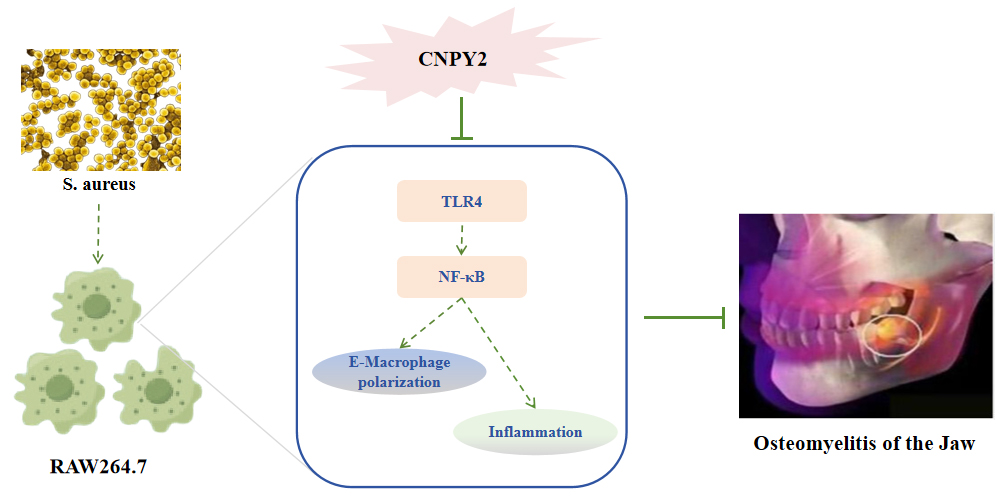
Background: Osteomyelitis of the jaw (OMJ) is a severe infectious bone disease. While Canopy FGF signaling regulator 2 (CNPY2) is known to regulate inflammatory diseases, its role in OMJ remains unclear. The study aimed to investigate the role of CNPY2 in the mandibular joint and its molecular mechanisms. Methods: An in vitro OMJ model was generated by stimulating RAW264.7 macrophages with S. aureus. CNPY2 knockdown and overexpression models were established using siRNA and plasmids. Functional assays assessed cell proliferation, migration, and invasion. Macrophage polarization, cytokine secretion, and osteoclast differentiation were analyzed. The CNPY2-Toll-Like Receptor 4 (TLR4)/Nuclear factor-kappa B (NF-κB) interaction was confirmed by Co-Immunoprecipitation (co-IP) and Western blot. In vivo, an OMJ mouse model was induced by S. aureus jaw injection and treated with si-CNPY2 lentivirus. Therapeutic effects were evaluated through histology and protein analysis. Results: S. aureus stimulation upregulated CNPY2 expression in RAW264.7 cells. Knockdown of CNPY2 inhibited S. aureus-induced cell proliferation, migration, and invasion, promoted macrophage polarization toward the M2 phenotype, suppressed M1 polarization, and reduced the release of pro-inflammatory cytokines. Additionally, CNPY2 knockdown inhibited S. aureus-induced osteoclast differentiation (decreased expression of markers such as Nuclear factor of activated T-cells cytoplasmic 1 [NFATc1] and Cathepsin K [CTSK]). Mechanistically, CNPY2 directly interacts with TLR4, and its knockdown suppresses activation of the TLR4/NF-κB axis. In the OMJ mouse model, CNPY2 knockdown significantly reduced inflammatory infiltration in the jaw, inhibited macrophage M1 polarization, and decreased osteoclastogenesis. Conclusion: CNPY2 exacerbates OMJ by enhancing macrophage M1 polarization, inflammation, and osteoclastogenesis via the TLR4/NF-κB axis. Targeting CNPY2 may offer a therapeutic strategy for S. aureus-induced OMJ.Graphic Abstract
Keywords
Osteomyelitis of the Jaw (OMJ) constitutes a debilitating infectious disease of the maxillofacial skeleton. Its pathological hallmarks include persistent inflammation, ensuing bone necrosis, and a consequent loss of jaw function [1,2]. The pathological process is complex, involving multiple cellular and molecular mechanisms that significantly impact patients’ quality of life. Its challenging treatment and high recurrence rate pose substantial clinical difficulties [3,4]. Recent advances in molecular biology and immunology have increasingly focused on the immune mechanisms of osteomyelitis, particularly the crucial role of inflammatory responses in disease progression [5]. As immune components, macrophages directly regulate the inflammatory response’s intensity and duration through their polarization states [6]. Therefore, investigating macrophage polarization and its regulatory mechanisms is essential for understanding OMJ pathogenesis and identifying novel therapeutic targets.
Macrophages exhibit a functional dichotomy, polarizing into classically activated (M1) or alternatively activated (M2) phenotypes in response to stimuli [7]. The M1 phenotype orchestrates pathogen clearance and inflammatory responses through pro-inflammatory cytokine secretion. In contrast, the M2 phenotype facilitates tissue repair and inflammation resolution by releasing anti-inflammatory factors [8,9]. In OMJ, excessive M1 activation often leads to persistent inflammation and aggravated bone destruction, whereas impaired M2 function hinders effective inflammation resolution and tissue repair [10]. Thus, modulating macrophage polarization to promote M1-to-M2 transition represents a crucial strategy for ameliorating inflammation and enhancing bone regeneration.
The Toll-Like Receptor 4 (TLR4)/Nuclear factor-kappa B (NF-κB) axis plays a critical role in regulating macrophage polarization and inflammatory responses [11]. TLR4 is a key receptor for recognizing bacterial lipopolysaccharides (LPS), but it can also be activated in infections mediated by Gram-positive bacteria such as Staphylococcus aureus by sensing infection-induced endogenous danger-associated molecular patterns (DAMPs) or specific bacterial exotoxins [12,13]. Upon activation, it initiates downstream NF-κB signaling pathways, promoting the expression of numerous inflammatory cytokines and inducing M1 polarization [14,15]. NF-κB regulates the expression of numerous inflammation-related genes. Its dysregulation is a key driver of chronic inflammatory states and subsequent tissue damage [16,17]. Therapeutic inhibition of the upstream TLR4/NF-κB axis has proven effective in reducing inflammatory responses and facilitating a shift toward M2 macrophage polarization [18]. The complexity of this pathway’s regulation, which involves extensive crosstalk and numerous modulators, mandates further exploration to uncover novel regulatory entities.
Canopy FGF Signaling Regulator 2 (CNPY2), a recently identified molecule, demonstrates significant regulatory functions in various diseases [19]. Initially recognized as an endoplasmic reticulum stress modulator [20], emerging evidence suggests CNPY2 influences cell signaling, inflammatory factor expression, and polarization processes, particularly in macrophage regulation [21]. This study systematically investigates CNPY2’s role in OMJ, focusing on its mechanism of regulating macrophage polarization and immune responses via the TLR4/NF-κB axis, and evaluates its impact on OMJ pathology.
RAW264.7 cells (CC-Y2084, Enzyme Research Biotechnology Co., Ltd., Shanghai, China) were cultured in DMEM medium (11965092, Grand Island, NY, USA) supplemented with 10% Fetal Bovine Serum (FBS; SA311.02, Antgene, Wuhan, China) and 1% penicillin-streptomycin solution (C0222, Beyotime, Shanghai, China) at 37°C in a 5% CO2 incubator. Cells were authenticated by Enzyme Research Biotechnology Co., Ltd. using short tandem repeat (STR) profiling. Throughout the entire experimental period, mycoplasma contamination was routinely monitored using a PCR-based mycoplasma detection kit (Solarbio, CA1081, Beijing, China), and all tests yielded negative results.
2.2.1 Staphylococcus aureus (S. aureus) Stimulation Model
The S. aureus strain BAA-2313 (ATCC, Manassas, VA, USA) was grown overnight in tryptic soy broth (TSB) at 37°C with shaking (120 rpm/min; MS310T, Radobio, Shanghai, China). Bacterial cells were harvested (8000 rpm/min, 5 min, 37°C; 5418, Eppendorf, Hamburg, Germany), washed with PBS (pH 7.4, 0.01 M; BL601A, Biosharp, Hefei, China), and resuspended by gentle pipetting in PBS to an OD600 of 0.5 (~1 × 108 CFU/mL). RAW264.7 cells were then stimulated at an multiplicity of stimulation (MOI) of 10 for 1 h [22]. Extracellular bacteria were eliminated by gentamicin (20 μg/mL; YZ-130326, Solarbio) treatment for 30 min. After washing, the cells were incubated in fresh medium with 10% FBS for 24 h prior to subsequent assays.
2.2.2 Gene Knockdown and Overexpression
To generate CNPY2-knockdown or overexpressing cell lines, lentiviruses (si-CNPY2/si-NC) and plasmids (oe-CNPY2/oe-NC) were obtained from GenePharma (Suzhou, China). The targeting si-RNA sequence for CNPY2 knockdown was 5′-CAACAAGATGAAGAGCACCAA-3′. To construct a CNPY2 overexpression plasmid, the cDNA sequence of the CNPY2 gene was amplified using PCR technology and subsequently inserted into the pcDNA3.1 vector. For viral transduction, cells were treated with lentivirus and Polybrene (10 μg/mL, HY-112735, MCE, Monmouth Junction, NJ, USA) for 24 h before replacing the medium. Following an additional 24 h of culture, cells were subjected to selection with puromycin (2 μg/mL, P8230, Solarbio, Beijing, China) for 10–14 days to obtain stable pools.
Plasmid transfections were performed with Lipofectamine 3000 (L3000150, Invitrogen, Carlsbad, CA, USA) using oe-CNPY2 or oe-NC. Complexes were prepared in Opti-MEM (11058021, Gibco, Grand Island, NY, USA), added to cells for 6 h, and then replaced with fresh medium. After 48 h, cells were lysed for Western blot to validate CNPY2 expression.
To investigate the role of CNPY2 in the TLR4/NF-κB signaling pathway, the following procedures were employed. First, cells were transiently transfected with si-CNPY2 or si-NC using Lipofectamine 3000 in Opti-MEM. Briefly, siRNA and Lipofectamine 3000 were separately diluted in Opti-MEM, gently mixed, and incubated at room temperature for 15 min to form a complex. This mixture was then added to the cell culture. After 6 h of incubation, the transfected medium was replaced with fresh complete medium. To validate compensatory signaling pathways in the absence of CNPY2, si-CNPY2-treated cells were further treated with the TLR4 agonist lipopolysaccharide (LPS, 1 μg/mL, L2880, Sigma-Aldrich, St. Louis, MO, USA) for 24 h. The activation of the NF-κB pathway was then assessed. To specifically block TLR4 signaling, cells were pretreated with the small-molecule inhibitor TAK-242 (10 μmol/L, HY-11109, MCE, Monmouth Junction, NJ, USA) for 4 h. TAK-242 is a selective TLR4 signaling inhibitor that covalently binds to the intracellular TIR domain of TLR4, blocking its interaction with adaptor proteins. This mechanism shields downstream signaling mediated by TLR4 without affecting receptor expression [23].
2.3 CCK-8 Cell Proliferation Assay
Following the designated treatments, cell viability was assessed by CCK-8 assay. Briefly, after seeding 5 × 103 cells/well in 96-well plates, 10 μL of CCK-8 reagent (C0037, Beyotime, Shanghai, China) was added to each well and incubated for 1 h (37°C, dark). The absorbance at 450 nm was measured (Benchmark™ Plus microplate reader, Bio-Rad, Hercules, CA, USA), and viability was calculated as:
2.4 EdU Staining for Cell Proliferation
Cell proliferation was evaluated with the BeyoClick™ EdU-594 Kit (C0078S, Beyotime). After CNPY2 modulation and S. aureus stimulation, cells were labeled with EdU (final concentration: 10 μM) for 2 h, fixed in 4% paraformaldehyde (PFA), and permeabilized with 0.5% Triton X-100 (T8200, Solarbio). Subsequently, 0.5 mL Click staining was performed in the dark for 30 min, and nuclei were counterstained with DAPI (1 μg/mL, 5 min; C1002, Beyotime). Images were acquired using a fluorescence microscope (Eclipse Ti2, Nikon, Tokyo, Japan). Quantitative analysis was performed using ImageJ software (version 1.53; NIH, Bethesda, MD, USA). To ensure objectivity, five random non-overlapping fields were captured per well for each of the three independent replicates (n = 3). The proliferation rate was calculated as the percentage of EdU-positive nuclei (red) relative to the total number of DAPI-stained nuclei (blue) in each field.
Cell invasion assays were performed using Matrigel-coated Transwell chambers (8 μm pore, Corning, NY, USA). The upper chamber was seeded with 2 × 104 cells in serum-free medium, while the lower chamber contained medium supplemented with 10% FBS. Following 24 h incubation, non-invading cells were removed, and invaded cells on the lower membrane were stained with 0.1% crystal violet (G1014, Servicebio, Wuhan, China) and quantified microscopically (ECLIPSE Ci-L Plus, Nikon, Tokyo, Japan).
RAW264.7 cells were seeded in 6-well plates at 5 × 105 cells/well. After adherence, uniform scratches were created using a 200 μL pipette tip, followed by washing with PBS (pH 7.4, 0.01 M) to remove detached cells. Serum-free medium was then added. Cell migration was monitored at 0 h and 24 h using microscopy (BX53, Olympus, Tokyo, Japan), with quantitative analysis performed using ImageJ software (version 1.53) to calculate migration rates.
RAW264.7 cells were collected and resuspended in PBS (pH 7.4, 0.01 M) containing 2% FBS to a concentration of 1 × 107 cells/mL. Cells were incubated with CD16-PE (12-0862-85, Invitrogen) and CD206-APC (17-2061-82, Invitrogen) antibodies for 1 h at on ice in the dark. Analysis was performed using a BD FACSCanto II flow cytometer (San Jose, CA, USA), with data processed by FlowJo software (version 7.6.1).
2.8 F-Actin Ring Immunofluorescence
Cellular F-actin was fluorescently labeled by employing phalloidin, a toxin derived from the Amanita phalloides fungus [24]. Cells were fixed (4% PFA, 15 min), permeabilized (0.1% Triton X-100, 10 min), and incubated with Phalloidin-FITC (1:1000, HY-D1817, MCE) for 40 min (25°C, dark). Cells were then stained for nuclei using DAPI (1 μg/mL, 10 min) for 10 min. At least 10 non-overlapping fields were randomly selected per group and examined under a fluorescence microscope (TCS SP8, Leica, Wetzlar, Germany) to count the number of actin rings per cell. To ensure objectivity, image analysis was performed independently by two assessors unaware of the experimental groupings.
2.9 Immunofluorescence Staining
Mouse macrophages were washed three times with PBS, fixed in 4% PFA for 30 min, permeabilized with 0.5% Triton X-100 for 20 min, and blocked with 5% BSA for 30 min. Primary antibodies against TLR4 (1:50, ab22048, Abcam, Cambridge, MA, USA), NF-κB p65 (1:500, ab16502, Abcam), and NFAT1 (1:100, #5861, CST, Danvers, MA, USA) were applied overnight at 4°C. After washing, fluorescent antibodies (1:200, SA00013-1/2, Proteintech, Wuhan, China) were added for 1 h at 37°C. After three PBS washes, nuclei were stained with DAPI (1 μg/mL, room temperature; Ex/Em: 340/488 nm) for 5 min. Finally, observations and photographs were taken using a fluorescence microscope (Axio Imager 2, Zeiss, Oberkochen, Germany). Fluorescence intensity (green) was analyzed using ImageJ software (version 1.53).
Twenty male C57BL/6 mice (10 weeks of age, weighing 25–28 g) were purchased from Speifu Biotechnology Co., Ltd. (Beijing, China). They were maintained under controlled environmental conditions (22 ± 2°C, 55 ± 5% humidity) with a 12-h photoperiod and ad libitum access to food and water. All animal procedures were conducted in accordance with the relevant regulations approved by the Institutional Ethics Committee of The First Affiliated Hospital, Fujian Medical University (Approval No.: IACUC FJMU 2024-0114). Animals were randomly assigned to different experimental groups using a random number table [25].
The mice were randomly divided into four groups, with five animals in each group. For OMJ induction [26], mice were anesthetized with ketamine (100 mg/kg) and xylazine (16 mg/kg). The left mandible was exposed, and a 1 mm diameter hole was drilled. The defect was inoculated with 2.5 μL of fibrin glue containing 2 × 106 CFU S. aureus or PBS (Sham). Concurrently, 5 μL lentivirus suspension (1 × 107 TU, si-NC or si-CNPY2) was injected into the defect site. After 14 days, mandibles were harvested for analysis. All outcome assessments were performed by investigators blinded to the group allocations.
Following fixation in 4% PFA and decalcification in EDTA (0.5M, E1170, Solarbio), mouse mandibles were processed into 4-μm paraffin sections. To perform histological analysis, sections were first deparaffinized in xylene (two changes, 15 min each) and rehydrated through a descending alcohol series (100%, 95%, 85%, and 75% ethanol, 5 min each). The sections were subjected to standard HE staining (G1120, Solarbio) according to the manufacturer’s protocol, including hematoxylin-eosin counterstaining. After mounting with neutral balsam (G8590, Solarbio), samples were examined by light microscopy (BX53, Olympus).
2.12 Tartrate-Resistant Acid Phosphatase (TRAP) Staining
Osteoclast formation was assessed using a TRAP staining kit (387A, Sigma). Cells were seeded in 48-well plates, washed with PBS (pH 7.4, 0.01 M) after treatment, and fixed with 4% paraformaldehyde. The TRAP working solution was applied to both macrophage cultures and mandibular paraffin sections, followed by 40 min incubation at 37°C (dark). After PBS washing, sections were mounted. TRAP-positive osteoclasts were identified as multinucleated (≥3 nuclei) red-stained cells under microscopy (BX53, Olympus) [27].
2.13 Enzyme-Linked Immunosorbent Assay (ELISA)
Mouse mandibles were homogenized in sterile mortars, and the homogenates were centrifuged at 5000× g (10 min, 4°C) to collect supernatants. ELISA kits (MyBioSource, California, USA) were used to quantify IL-6 (MBS2508516), TNF-α (MBS494101), IL-6 (MBS705090), Arg-1 (MBS3805532), CXCL1 (MBS824609), and CXCL10 (MBS824818) levels. After equilibrating all reagents and samples to room temperature, add the standards and cell supernatants to the 96-well plate. Incubate for 90 min, then wash. Next, add the biotin-labeled detection antibody, incubate for 60 min, and wash. Subsequently, add the enzyme conjugate working solution, incubate for 30 min, and wash. Finally, add the substrate solution to develop color, incubate in the dark for 15 min. After adding the stop solution, measure the absorbance at 450 nm using a microplate reader (Benchmark™ Plus, Bio-Rad) and calculate the concentration based on the standard curve.
2.14 Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was extracted from cells and bone homogenates using TRIzol reagent (15596026, Invitrogen) according to the manufacturer’s protocol. RNA concentration and purity were determined using a microvolume spectrophotometer (NanoDrop 2000, Thermo Fisher Scientific, Waltham, MA, USA). Reverse transcribe 1 μg of total RNA into cDNA using the reverse transcription kit (R323-01, Vazyme, Nanjing, China). The 20 μL reverse transcription reaction mixture contained: 4 μL 4× gDNA wiper Mix, 1 μg total RNA, and RNase-free water to a final volume of 16 μL. Incubate at 42°C for 2 min. Add 4 μL 5× HiScript III qRT SuperMix, incubate at 37°C for 15 min, then heat at 85°C for 5 s. Amplification was performed using SYBR Green PCR Master Mix (CN830, TaKaRa, Kyoto, Japan) on a real-time fluorescent quantitative PCR instrument (7500 Fast, Thermo, Waltham, MA, USA). The total qPCR reaction volume was 20 μL, comprising: 10 μL SYBR Premix Ex Taq (2×), 0.4 μL primers (10 μM), 0.4 μL ROX Reference Dye (50×), 2.0 μL cDNA template, and ddH2O. PCR cycling parameters were: 95°C pre-denaturation for 30 s; followed by 40 cycles: 95°C denaturation for 5 s, 60°C annealing/extension for 34 s. Three parallel wells were set up for each sample, and melting curve analysis was performed to verify primer specificity. GAPDH was used as the internal control, and relative gene expression was calculated using the 2−ΔΔCt method. Primer sequences are shown in Table 1.
Table 1: RT-qPCR primer sequences.
| Gene | Forward Primer (5’→3’) | Reverse Primer (5’→3’) |
|---|---|---|
| CNPY2 | AGACCATTCAGATGGGATCTTTC | CTCCTTCATCCGGTCACATATC |
| NFATc1 | AACTTTCTGCAAGACTCCAA | TTATTCTCTGGTTGCGGAAA |
| CTSK | ATGAAATCTCTCGGCGTTTA | GAGAGGCCTCAAGATTATGG |
| Oscar | CTCCAGCTGTCTACTCTCTGTG | TAGGGGCACTGGTGATGTG |
| Trap | TGGATTCATGGGTGGTGCTG | CGTCCTCAAAGGTCTCCTGG |
| GAPDH | CCTCGTCCCGTAGACAAAATG | CCTCGTCCCGTAGACAAAATG |
2.15 Western Blot and Co-Immunoprecipitation (Co-IP)
Mouse jawbone Tissues were pulverized in liquid nitrogen and lysed with RIPA buffer (R0010, Solarbio). RAW264.7 cell proteins were extracted similarly. Protein concentrations were determined using a BCA assay kit (WLA004, Wanleibio, Shenyang, China). Equal protein amounts were separated by SDS-PAGE (WLA013, Wanleibio), transferred to PVDF membranes (E801/E802, Vazyme), blocked with 5% skim milk, and probed overnight at 4°C with primary antibodies against: CNPY2 (1:1000, 14635-1-AP, Proteintech), CD86 (1:1000, 83213-5-RR, Proteintech), iNOS (1:1000, 22226-1-AP, Proteintech), CD206 (1:1000, 83485-1-RR, Proteintech), Arg-1 (1:1000, 16001-1-AP, Proteintech), NFATc1 (1:1000, #8032, CST), MMP9 (1:1000, ab283575, Abcam), c-FOS (1:1000, #2250S, CST), Cathepsin K (CTSK, 1:1000, ab187647, Abcam), TLR4 (1:1000, 19811-1-AP, Proteintech), p-NF-κB p65 (1:1000, #3033S, CST), NF-κB p65 (1:1000, #8242S, CST), and GAPDH (1:5000, ab8245, Abcam). After TBST washing, membranes were incubated with secondary antibody (1:3000, #7074, CST) for 90 min. Signals were developed using ECL substrate (WBULP-100ML, Millipore, Burlington, MA, USA) and captured by a chemiluminescence imaging system (Amersham ImageQuant 800, Cytiva, Marlborough, MA, USA). Band intensities were quantified using ImageJ (version 1.53) with and GAPDH normalization.
For Co-IP, proteins were extracted with NP40 lysis buffer (N274337, Aladdin, Shanghai, China). Equal lysate amounts were incubated overnight at 4°C with Flag (1:1000, b205606, Abcam) or myc (1:1000, b32072, Abcam) antibodies, followed by 2 h incubation with Protein A/G agarose beads (1:1000, A10001, Abmart, Shanghai, China). Beads were washed and eluted in loading buffer for immunoblotting using HRP-conjugated goat anti-rabbit IgG (1:2000, AS014, Abclone, Wuhan, China).
Data were analyzed using GraphPad Prism 8.0 (GraphPad Software, Inc., San Diego, CA, USA) and presented as mean ± standard deviation (SD). Two-group comparisons used t-tests, while multi-group comparisons employed one-way ANOVA with Tukey’s post-hoc test. p < 0.05 was considered statistically significant.
3.1 CNPY2 Regulates S. aureus-Induced RAW264.7 Cell Proliferation, Migration, Invasion, and Apoptosis
S. aureus stimulattion significantly upregulated CNPY2 mRNA and protein expression levels in RAW264.7 cells (Fig. 1A–C). To elucidate CNPY2’s function, we constructed CNPY2 knockdown (si-CNPY2) and overexpression (oe-CNPY2) cell models, with Western blot confirming intervention efficiency (Fig. 1D,E).
CCK-8 and EdU assays demonstrated that S. aureus suppressed RAW264.7 cell proliferation (Fig. 1F–H), while Transwell and wound healing assays indicated enhanced cell invasion and migration capabilities (Fig. 1I–L). Notably, CNPY2 knockdown exacerbated S. aureus-induced proliferation inhibition and attenuated its pro-migration and pro-invasion effects. Conversely, CNPY2 overexpression partially mitigated proliferation suppression while further enhancing cell migration and invasion (Fig. 1F–L).

Figure 1: CNPY2 regulates S. aureus-induced proliferation, migration, invasion, and apoptosis in RAW264.7 cells. (A) RT-qPCR analysis of CNPY2 mRNA expression levels in cells following S. aureus stimulation. ***p < 0.001 vs. Control. (B,C) Western blot analysis of CNPY2 protein expression in cells after S. aureus stimulation. **p < 0.01 vs. Control. (D,E) Western blot validation of CNPY2 protein expression efficiency in RAW264.7 cells after siRNA knockdown and overexpression plasmid transfection. *p < 0.05 vs. si-NC; ***p < 0.001 vs. oe-NC. (F) CCK-8 assay assessing proliferation capacity in different treatment groups (Control, S. aureus-stimulated, S. aureus + si-NC, S. aureus + si-CNPY2, S. aureus + oe-NC, S. aureus + oe-CNPY2). (G,H) EdU staining for proliferative activity in RAW264.7 cells (red fluorescence: proliferating cells; blue fluorescence: nuclei). Scale bar: 50 μm. (I,J) Transwell invasion assay evaluating cell invasiveness over 24 h. Scale bar: 100 μm. (K,L) Wound healing assay measuring cell migration over 24 h. Scale bar: 200 μm. Data are presented as Mean ± SD from three independent experiments (n = 3). ***p < 0.001 vs. Control; ###p < 0.001 vs. S. aureus + si-NC; &&p < 0.01, &&&p < 0.001 vs. S. aureus + oe-NC.
3.2 CNPY2 Modulates S. aureus-Induced RAW264.7 Cell Polarization and Inflammation
This study comprehensively examined CNPY2’s role in regulating S. aureus-induced macrophage polarization and inflammatory responses. S. aureus stimulation significantly increased expression of the M1 macrophage marker CD86 (Fig. 2A,B) while reducing the M2 marker CD206 (Fig. 2C,D). Remarkably, CNPY2 knockdown (si-CNPY2) inhibited M1 polarization but promoted M2 polarization, exhibiting anti-inflammatory effects. In contrast, CNPY2 overexpression (oe-CNPY2) enhanced M1 polarization while suppressing M2 polarization, demonstrating pro-inflammatory activity. ELISA further validated this regulatory pattern. S. aureus stimulation substantially elevated IL-6 and TNF-α while decreasing IL-10 and Arg-1 in macrophages (Fig. 2E–H). CNPY2 knockdown reversed this trend by reducing pro-inflammatory cytokines and elevating anti-inflammatory factors, whereas CNPY2 overexpression exacerbated inflammatory responses. S. aureus stimulation significantly upregulated M1 markers (CD86, iNOS) while downregulating M2 markers (CD206, Arg-1) in macrophages (Fig. 2I–M). CNPY2 knockdown markedly suppressed M1 marker expression but promoted M2 markers, whereas overexpression produced the opposite effect. These results clearly demonstrate that CNPY2 plays a pivotal role in modulating macrophage polarization balance.

Figure 2: CNPY2 regulates S. aureus-induced polarization and inflammation in RAW264.7 cells. (A–D) Flow cytometry analysis of CD86 (M1 marker) and CD206 (M2 marker) expression levels in different treatment groups (Control, S. aureus-stimulated, and CNPY2 genetically modified groups). (E–H) ELISA detection of inflammatory cytokine secretion levels in cell culture supernatants. (I–M) Western blot analysis of macrophage polarization marker protein (CD86, iNOS, CD206 and Arg-1) expression. Data are presented as Mean ± SD from three independent experiments (n = 3). **p < 0.01, ***p < 0.001 vs. Control; ##p < 0.01, ###p < 0.001 vs. S. aureus + si-NC; &&&p < 0.001 vs. S. aureus + oe-NC.
3.3 CNPY2 Regulates S. aureus-Induced Osteoclast Differentiation in RAW264.7 Cells
This study revealed the critical regulatory role of CNPY2 in S. aureus-induced osteoclast differentiation. In vitro culture experiments demonstrated that S. aureus induced RAW264.7 cells to differentiate into multinucleated osteoclasts (Fig. 3A). S. aureus treatment significantly increased the number of TRAP-positive cells (Fig. 3B,C), whereas CNPY2 knockdown (si-CNPY2) inhibited this process, and CNPY2 overexpression (oe-CNPY2) further promoted osteoclast formation.
The actin ring is a key feature of osteoclast functional activation. Phalloidin-FITC staining revealed that S. aureus induced the formation of typical actin ring structures and significantly increased actin polymerization (Fig. 3D,E). CNPY2 knockdown markedly reduced the number of actin rings, while overexpression enhanced their formation (Fig. 3D,E). NFATc1 exhibited significantly increased immunofluorescence intensity in the S. aureus-treated group (Fig. 3F,G), and this effect was also modulated by CNPY2 expression levels. RT-qPCR analysis demonstrated that S. aureus stimulation significantly upregulated mRNA expression of osteoclast differentiation markers (NFATc1, CTSK, Oscar, and Trap) (Fig. 3H). S. aureus treatment markedly increased the protein levels of osteoclast differentiation markers (NFATc1, MMP9, c-FOS, and CTSK) (Fig. 3I,J). CNPY2 knockdown suppressed the expression of these markers, whereas overexpression further enhanced their levels. These findings indicate that CNPY2 modulates S. aureus-induced osteoclast differentiation by regulating the expression of key genes involved in this process.

Figure 3: CNPY2 regulates S. aureus-induced osteoclast differentiation in RAW264.7 cells. (A) Inverted microscopy observation of morphological changes and multinucleated osteoclast formation across treatment groups. Scale bar: 200 μm. (B,C) TRAP staining for osteoclast identification. Scale bar: 50 μm. (D,E) Phalloidin-FITC fluorescence staining to assess characteristic actin ring formation in mature osteoclasts, indicating activation status. Scale bar: 50 μm. (F,G) Immunofluorescence staining detecting expression of NFATc1. Scale bar: 50 μm. (H) RT-qPCR analysis of osteoclast differentiation marker genes. (I,J) Western blot evaluation of osteoclast differentiation-related proteins. Data are presented as Mean ± SD from three independent experiments (n = 3). ***p < 0.001 vs. Control; #p < 0.05, ###p < 0.001 vs. S. aureus + si-NC; &&p < 0.01, &&&p < 0.001 vs. S. aureus + oe-NC.
3.4 CNPY2 Regulates the TLR4/NF-κB Pathway
Co-IP experiments demonstrated that anti-Flag antibody precipitated Flag-CNPY2 protein, while TLR4 was also co-precipitated (Fig. 4A). Similarly, TLR4 was detected in the immunoprecipitates of myc-CNPY2 protein. These results indicate a direct interaction between CNPY2 and TLR4, which was significantly enhanced following S. aureus stimulation. S. aureus stimulation upregulated CNPY2 and TLR4 protein expression in RAW264.7 macrophages and promoted NF-κB p65 phosphorylation (Fig. 4B,C). Notably, CNPY2 knockdown (si-CNPY2) suppressed TLR4 expression and NF-κB activation, whereas CNPY2 overexpression (oe-CNPY2) amplified these effects. Immunofluorescence assays further confirmed this regulatory relationship. S. aureus stimulation increased TLR4-positive expression and facilitated NF-κB p65 nuclear translocation (Fig. 4D,E). CNPY2 knockdown attenuated these changes, while overexpression intensified TLR4 expression and NF-κB activation.
To validate TLR4’s role in CNPY2 regulation, intervention experiments were conducted using LPS (a TLR4 agonist) and the TLR4 inhibitor TAK-242. Western blot results revealed that in si-CNPY2 cells stimulated with S. aureus, LPS treatment significantly upregulated CNPY2 and TLR4 protein expression and NF-κB phosphorylation (Fig. 4F,G), whereas TAK-242 further suppressed these signaling molecules. Immunofluorescence observations demonstrated that compared to the S. aureus + si-CNPY2 group, LPS enhanced TLR4 and NF-κB p65 fluorescence intensity, while TAK-242 counteracted the effects of CNPY2 knockdown (Fig. 4H,I). These findings suggest that CNPY2 activates the downstream NF-κB axis by binding to TLR4.

Figure 4: CNPY2 modulates the TLR4/NF-κB axis. (A) Co-IP assay demonstrating CNPY2-TLR4 interaction. (B,C) Western blot analysis of CNPY2/TLR4/NF-κB axis protein expression patterns. (D,E) Immunofluorescence staining evaluating TLR4 expression and NF-κB p65 nuclear translocation. Scale bar: 50 μm. ***p < 0.001 vs. Control; ##p < 0.01, ###p < 0.001 vs. S. aureus + si-NC; &&&p < 0.001 vs. S. aureus + oe-NC. (F,G) Western blot validation of CNPY2/TLR4/NF-κB pathway components post-genetic intervention. (H,I) Immunofluorescence analysis of TLR4 expression and NF-κB p65 nuclear localization dynamics. Scale bar: 50 μm. Data are presented as Mean ± SD from three independent experiments (n = 3). ***p < 0.001 vs. Control; ###p < 0.001 vs. S. aureus + si-NC; &p < 0.05, &&p < 0.01, &&&p < 0.001 vs. S. aureus + si-CNPY2.
3.5 CNPY2 Knockdown Attenuates Macrophage Polarization, Inflammatory Responses, and Osteoclastogenesis via Suppressing TLR4/NF-κB Signaling
Through intervention experiments using the TLR4 agonist (LPS) and inhibitor (TAK-242), this study confirmed the pivotal role of the CNPY2-TLR4/NF-κB axis. S. aureus stimulation increased the expression of M1 macrophage marker CD86 (Fig. 5A,B) while decreasing M2 marker CD206 (Fig. 5C,D). CNPY2 knockdown (si-CNPY2) reversed this polarization trend, with TLR4 agonist LPS treatment partially counteracting the effects of si-CNPY2, whereas TLR4 inhibitor TAK-242 further enhanced si-CNPY2’s efficacy. ELISA assays demonstrated that si-CNPY2 significantly suppressed S. aureus-induced secretion of pro-inflammatory cytokines (IL-6 and TNF-α) while elevating anti-inflammatory factors (IL-10 and Arg-1) in macrophages (Fig. 5E). LPS treatment attenuated the anti-inflammatory effects of si-CNPY2, while TAK-242 synergistically amplified them (Fig. 5E).
Regarding osteoclast differentiation, TRAP staining showed that si-CNPY2 reduced multinucleated osteoclast formation (Fig. 5F,G), an effect partially reversed by LPS but further enhanced by TAK-242 (Fig. 5F,G). Si-CNPY2 downregulated the expression of osteoclastogenic master regulator NFATc1 and its downstream effectors (MMP9, c-FOS, and CTSK) (Fig. 5H,I), with these changes being similarly modulated by TLR4 activity. LPS treatment weakened si-CNPY2’s inhibitory effect on osteoclast differentiation, while TAK-242 cooperatively strengthened this suppression. These results functionally validate CNPY2’s dual regulatory roles in macrophage polarization and osteoclast differentiation via the TLR4/NF-κB axis.

Figure 5: CNPY2 knockdown ameliorates macrophage polarization, inflammatory response, and osteoclastogenesis by suppressing the TLR4/NF-κB axis. (A–D) Flow cytometric analysis of M1 marker (CD86) and M2 marker (CD206) expression levels. (E) ELISA quantification of secreted inflammatory cytokines and anti-inflammatory factors in cell culture supernatants. (F,G) TRAP staining for osteoclast formation assessment. Scale bar: 50 μm. (H,I) Western blot detection of osteoclast differentiation-related proteins. Data are presented as Mean ± SD from three independent experiments (n = 3). ***p < 0.001 vs. Control; ###p < 0.001 vs. S. aureus + si-NC; &p < 0.05, &&p < 0.01, &&&p < 0.001 vs. S. aureus + si-CNPY2.
3.6 CNPY2 Knockdown Ameliorates S. aureus-Induced OMJ in Mice
This study evaluated the therapeutic effect of CNPY2 knockdown on disease progression using a mouse OMJ model induced by S. aureus. qPCR results demonstrated that the si-CNPY2 lentivirus significantly suppressed local CNPY2 expression in the mouse mandible (Fig. 6A). S. aureus-stimulated mice exhibited significant pathological alterations in jaw tissue, including inflammatory cell infiltration and bone destruction (Fig. 6B). However, CNPY2 knockdown (si-CNPY2) markedly alleviated these pathological features, reducing inflammation and bone damage. TRAP staining analysis revealed that S. aureus stimulation substantially increased TRAP-positive cell numbers in jaw tissue (Fig. 6C,D), which was significantly inhibited by si-CNPY2 treatment. Western blot analysis demonstrated that si-CNPY2 downregulated M1 marker CD86 while upregulating M2 marker CD206 (Fig. 6E,F), indicating that CNPY2 suppression promotes macrophage polarization toward an anti-inflammatory phenotype. ELISA further confirmed that si-CNPY2 treatment significantly reduced concentrations of pro-inflammatory cytokines and chemokines in bone homogenates (Fig. 6G,H), collectively suggesting CNPY2 inhibition exerts anti-inflammatory effects in vivo. Si-CNPY2 suppressed protein expression of osteoclastogenic regulators NFATc1 and its downstream molecules (MMP9, c-FOS, CTSK) (Fig. 6I,J), while downregulating CNPY2 and TLR4 expression and reducing NF-κB p65 phosphorylation (Fig. 6K,L). These in vivo findings strongly corroborate prior in vitro results, further substantiating that CNPY2 knockdown mitigates OMJ pathology by modulating the TLR4/NF-κB signaling pathway.

Figure 6: CNPY2 knockdown alleviates murine OMJ. (A) RT-qPCR detection of CNPY2 knockout efficiency in bone homogenates. (B) HE staining evaluation of histopathological changes in mandibular tissues, analyzing inflammatory infiltration and bone destruction. Scale bar: 100 μm. (C,D) TRAP staining detection of osteoclast formation in mandibular tissues. Scale bar: 100 μm. (E,F) Measurement of macrophage polarization markers (CD86, CD206) in mandibular tissues. (G,H) ELISA quantification of inflammatory cytokines (IL-6, TNF-α, IL-1β) and chemokines (CXCL1, CXCL10) in mandibular tissue homogenates. (I,J) Osteoclast differentiation-related proteins (NFATc1, MMP9, c-FOS, CTSK) in mandibular tissues. (K,L) Western blot detection of key CNPY2/TLR4/NF-κB axis components in mandibular tissues. Data are presented as Mean ± SD (n = 5 mice per group). ***p < 0.001 vs. Sham; ##p < 0.01, ###p < 0.001 vs. S. aureus + si-NC.
CNPY2, as a member of the canopy family, is involved in various physiological and pathological processes, including cell proliferation, migration, apoptosis, and inflammatory responses [28,29]. CNPY2 expression was significantly upregulated in S. aureus-stimulated RAW264.7 macrophages, suggesting CNPY2 may play a crucial role in bacterial stimulation-induced inflammation. Huang et al. reported that CNPY2 promotes vascular endothelial cell injury and inflammation by activating the PERK pathway during atherosclerosis progression [29], further supporting CNPY2’s role as an important inflammatory regulator. Our in vitro experiments demonstrated that CNPY2 knockdown significantly inhibited S. aureus-induced cell migration and invasion while enhancing proliferation, whereas CNPY2 overexpression produced opposite effects. These therapeutic potentials were further validated in a murine model of jaw osteomyelitis, where lentiviral-mediated si-CNPY2 intervention markedly alleviated inflammatory infiltration and tissue damage. Macrophage proliferation, migration, and invasion are critical components of inflammatory responses and tissue injury [30,31], with excessive macrophage activation and infiltration being key drivers of disease progression [32]. Therefore, CNPY2 likely participates in disease pathogenesis by modulating macrophage activity, providing a theoretical foundation for mechanistic studies and therapeutic interventions.
Microenvironmental cues drive macrophage polarization toward pro-inflammatory M1 or anti-inflammatory M2 phenotypes [33]. M1 macrophages promote inflammation and pathogen elimination via pro-inflammatory cytokine secretion, but can also contribute to tissue damage [34]. Conversely, M2 macrophages play pivotal roles in inflammation resolution, tissue repair, immunosuppression and angiogenesis, characterized by anti-inflammatory factors [35]. In chronic inflammatory diseases like osteomyelitis, aberrant macrophage polarization is central to sustained inflammation, exacerbated bone destruction, and impaired tissue repair [36,37]. Our results demonstrate that CNPY2 knockdown significantly suppresses M1 marker expression, while promoting M2 markers, indicating CNPY2 inhibition drives macrophage polarization toward M2 phenotype. ELISA results corroborated this trend, showing reduced pro-inflammatory cytokines and elevated anti-inflammatory factors upon CNPY2 knockdown. Conversely, CNPY2 overexpression markedly upregulated M1 markers and pro-inflammatory cytokine release. These data clearly establish that CNPY2 promotes M1 polarization and pro-inflammatory responses while suppressing M2-mediated anti-inflammatory and reparative processes during S. aureus stimulation. Notably, Zhan et al. found CNPY3 (a CNPY2 homolog) expression correlated with M2 macrophage abundance and negatively regulated macrophage apoptosis in glioma [38], suggesting Canopy family members may broadly influence macrophage polarization across disease contexts.
Macrophage polarization critically determines osteomyelitis progression [39]. Pro-inflammatory cytokines can directly/indirectly stimulate osteoclast precursor proliferation/differentiation and enhance mature osteoclast resorptive activity, while macrophage polarization status directly affects osteoclast fate and bone metabolic balance [40,41]. In the RAW264.7 cell model employed in this study, this complex biological process is vividly demonstrated. As a representative component of the monocyte-macrophage lineage, RAW264.7 cells serve not only as a crucial model for studying inflammatory polarization but are also internationally recognized as an osteoclast precursor (OCP) model, possessing the potential to differentiate into osteoclasts [42,43]. Under S. aureus stimulation, RAW264.7 cells undergo a functional state transition from “pro-inflammatory macrophage activation” to “early osteoclast differentiation”. TRAP, primarily expressed by osteoclasts, increases progressively during osteoclast differentiation [44,45]. Our TRAP staining showed S. aureus stimulation significantly increased multinucleated cell numbers and osteoclast formation, which was markedly suppressed by CNPY2 knockdown. Actin ring formation serves as a hallmark of osteoclast maturation [46], while NFATc1 is the master transcriptional regulator of osteoclastogenesis [47]. CTSK and MMP9 are key enzymes highly expressed in mature osteoclasts, that directly mediate bone matrix degradation [48], and both c-FOS and Oscar can enhance activation of critical transcription factors like NFATc1 [49,50]. Knockdown of CNPY2 significantly inhibited macrophage CNPY2 protein expression, reduced actin ring formation, and downregulated mRNA and protein levels of osteoclast differentiation markers. CNPY2 is essential in regulating macrophage polarization and osteoclast differentiation. Jiang et al. found that miR-146a upregulation mitigated S. aureus-induced osteoblast loss, altered bone remodeling, and suppressed inflammatory cytokine production and osteoclastogenesis, thereby preventing osteomyelitis [51]. Inflammatory environments promote osteoclast differentiation from macrophages, and pro-inflammatory cytokines stimulate osteoclastic bone resorption, leading to extensive bone loss and hindered regeneration [52,53]. These findings confirm that CNPY2 can influence bone metabolic balance and osteomyelitis progression by regulating macrophage polarization.
TLR4 is a key pattern recognition receptor that plays a central role in recognizing PAMPs, particularly LPS [54]. TLR4 activation serves as the first line of defense in initiating host inflammatory responses, activating the downstream NF-κB pathway and inducing the expression of pro-inflammatory cytokines, chemokines, and adhesion molecules to recruit immune cells and eliminate pathogens [55,56]. Our co-IP results revealed an interaction between CNPY2 and TLR4 proteins, consistent with recent reports that CNPY2 directly binds TLR4 and regulates TLR4/NF-κB pathway-dependent cytokine secretion in liver macrophages [57]. Further experimental results suggest that this physical interaction underlies CNPY2’s regulation of TLR4 protein homeostasis. Western blot and immunofluorescence analyses revealed that CNPY2 knockdown significantly reduced both total TLR4 protein levels and its surface abundance, while CNPY2 overexpression produced the opposite effect. Considering the crucial role of Canopy family members (e.g., CNPY3) as molecular chaperones in TLR maturation, folding, and transport to the cell membrane [58], we hypothesize that CNPY2 binding to TLR4 may maintain TLR4 expression levels in macrophages under infectious stress by reducing TLR4 degradation or promoting its processing. Western blot analysis demonstrated that CNPY2 knockdown in S. aureus-stimulated RAW264.7 cells significantly reduced TLR4 protein expression and, more importantly, markedly inhibited NF-κB p65 subunit phosphorylation without affecting total p65 protein levels. Phosphorylation of NF-κB p65 is a critical step for its activation, enabling nuclear translocation to initiate transcription of inflammation-related genes. Thus, CNPY2 knockdown suppressed NF-κB pathway activation. Immunofluorescence results further supported this conclusion: CNPY2 knockdown reduced TLR4 surface expression and significantly inhibited NF-κB p65 nuclear translocation. As nuclear translocation is a prerequisite for NF-κB transcriptional activity, this inhibition suggests suppressed NF-κB-mediated inflammatory gene expression [59]. These findings demonstrate that CNPY2 interacts with TLR4 to influence TLR4 expression and NF-κB pathway activation, thereby regulating macrophage inflammatory responses. Additionally, S. aureus can induce osteoclastogenesis through NF-κB pathway activation [60]. Our results revealed that CNPY2 knockdown suppressed expression of osteoclast differentiation-related proteins and osteoclast formation in OMJ mouse mandible tissues. Collectively, CNPY2 regulates the TLR4/NF-κB axis to influence both macrophage inflammatory responses and osteoclast differentiation/formation, providing new theoretical insights into the pathogenesis of OMJ.
However, this study also has certain limitations that require further refinement in subsequent work. Although we established CNPY2 knockdown and overexpression cell models and conducted experiments using a combination of TLR4 agonists and antagonists, confirming that CNPY2 knockdown suppresses S. aureus-induced TLR4/NF-κB activation and affects macrophage inflammatory responses and osteoclast differentiation. However, it remains unclear whether S. aureus specifically regulates these behaviors through the NF-κB pathway. Furthermore, this study primarily employed in vitro cell models and limited in vivo experiments to elucidate the CNPY2-TLR4/NF-κB axis, lacking long-term, dynamic validation of this pathway in animal models closer to clinical scenarios (e.g., chronic osteomyelitis). Furthermore, we have not yet explored whether CNPY2 exhibits cross-regulation or signaling network integration with other innate immune receptors and downstream signaling pathways (e.g., MAPK, JAK/STAT), which could significantly impact macrophage fate determination and bone remodeling processes. In future studies, we plan to build upon our existing work by conducting functional experiments on macrophage proliferation, migration, and invasion. These will be combined with NF-κB pathway-specific interventions to comprehensively evaluate the regulatory spectrum of S. aureus on macrophage biological behavior. Concurrently, we will establish clinically relevant animal models of stimulatory bone destruction to systematically validate the dynamic role of the CNPY2-TLR4/NF-κB axis in bone destruction progression. We will also explore the interactions between CNPY2 and other inflammatory signaling pathways, along with their combined effects on the immune microenvironment and bone metabolic homeostasis.
This study comprehensively investigated CNPY2’s role in S. aureus-induced macrophage polarization, inflammatory-immune responses, and osteoclast differentiation, revealing its regulatory mechanism through the TLR4/NF-κB signaling and ultimately demonstrating CNPY2’s therapeutic potential for OMJ. Although we confirmed direct CNPY2-TLR4 binding, their precise interaction sites, binding modes, and effects on TLR4 conformation and signal transduction require further elucidation. Moreover, CNPY2’s differential roles in various osteomyelitis types and its functions in other bone-related cells (e.g., osteoblasts and osteocytes) remain unclear. Future research should develop highly effective, low-toxicity CNPY2-specific inhibitors and conduct systematic preclinical and clinical studies to advance CNPY2-targeted therapies for OMJ, offering patients novel treatment options.
Acknowledgement:
Funding Statement: This work was supported by Startup Fund for Scientific Research, Fujian Medical University. (Grant number: 2020QH1026).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Rihui Wang, Wanlu Li; data collection: Canyang Jiang, Jianping Huang; analysis and interpretation of results: Kangwei Zhou, Yan Jiang, Junyang Zhang; draft manuscript preparation: Li Huang. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Ethics Approval: This study was reviewed and approved by the Institutional Ethics Committee of The First Affiliated Hospital, Fujian Medical University. (Approval number: IACUC FJMU 2024-0114).
Conflicts of Interest: The authors declare no conflicts of interest.
Abbreviations
| CNPY2 | Canopy FGF signaling regulator 2 |
| S. aureus | Staphylococcus aureus |
| LPS | lipopolysaccharides |
| TRAP | Tartrate-resistant acid phosphatase |
| NFATc1 | Nuclear factor of activated T-cells cytoplasmic 1 |
| CTSK | Cathepsin K |
| MMP9 | Matrix metalloproteinase 9 |
| c-FOS | Proto-oncogene c-Fos |
| TLR4 | Toll-like receptor 4 |
| OMJ | Osteomyelitis of the Jaw |
| TSB | Tryptic Soy Broth |
| CXCL1 | C-X-C motif chemokine ligand 1 |
| CXCL10 | C-X-C motif chemokine ligand 10 |
| Co-IP | Co-immunoprecipitation |
| PAMPs | Pathogen-associated molecular patterns |
| OCP | osteoclast precursor |
References
1. Justine D , Bhalla AS , Manchanda S , Bhutia O , Roychoudhury A . Osteomyelitis of the jaw bones and its mimics: Resolving the diagnostic enigma. Indian J Radiol Imaging. 2025; 35( 3): 361– 73. doi:10.1055/s-0044-1800972. [Google Scholar] [CrossRef]
2. Lucidarme Q , Lebrun D , Vernet-Garnier V , Le Gall J , Diallo S , Mauprivez C , et al. Chronic osteomyelitis of the jaw: Pivotal role of microbiological investigation and multidisciplinary management—A case report. Antibiotics. 2022; 11( 5): 568. doi:10.3390/antibiotics11050568. [Google Scholar] [CrossRef]
3. Xie S , Yao Z . The correlation between the main pathogenic bacteria and bone metabolism markers in mandibular osteomyelitis. Int J Front Med. 2023; 5( 10): 127– 31. doi:10.25236/IJFM.2023.051020. [Google Scholar] [CrossRef]
4. López-Carriches C , Mateos-Moreno MV , Taheri R , Martínez JL-Q , Madrigal-Martínez-Pereda C . Chronic Osteomyelitis of the jaw. Osteomyelitis. J Clin Exp Dent. 2025; 17( 3): e324. doi:10.4317/jced.62596. [Google Scholar] [CrossRef]
5. Surendar J , Hackenberg RK , Schmitt-Sánchez F , Ossendorff R , Welle K , Stoffel-Wagner B , et al. Osteomyelitis is associated with increased anti-inflammatory response and immune exhaustion. Front Immunol. 2024; 15: 1396592. doi:10.3389/fimmu.2024.1396592. [Google Scholar] [CrossRef]
6. Hu K , Shang Z , Yang X , Zhang Y , Cao L . Macrophage polarization and the regulation of bone immunity in bone homeostasis. J Inflammation Res. 2023: 3563– 80. doi:10.2147/JIR.S423819. [Google Scholar] [CrossRef]
7. Luo M , Zhao F , Cheng H , Su M , Wang Y . Macrophage polarization: An important role in inflammatory diseases. Front Immunol. 2024; 15: 1352946. doi:10.3389/fimmu.2024.1352946. [Google Scholar] [CrossRef]
8. Peng Y , Zhou M , Yang H , Qu R , Qiu Y , Hao J , et al. Regulatory mechanism of M1/M2 macrophage polarization in the development of autoimmune diseases. Mediat Inflamm. 2023; 2023( 1): 8821610. doi:10.1155/2023/8821610. [Google Scholar] [CrossRef]
9. Paschalidi P , Gkouveris I , Soundia A , Kalfarentzos E , Vardas E , Georgaki M , et al. The role of M1 and M2 macrophage polarization in progression of medication-related osteonecrosis of the jaw. Clin Oral Investig. 2021; 25( 5): 2845– 57. doi:10.1007/s00784-020-03602-z. [Google Scholar] [CrossRef]
10. Liu Z , Luo X , Xu R . Interaction between immuno-stem dual lineages in jaw bone formation and injury repair. Front Cell Dev Biol. 2024; 12: 1359295. doi:10.3389/fcell.2024.1359295. [Google Scholar] [CrossRef]
11. Ren S , Zhou R , Tang Z , Song Z , Li N , Shi X , et al. Wuling capsule modulates macrophage polarization by inhibiting the TLR4-NF-κB signaling pathway to relieve liver fibrosis. Int Immunopharmacol. 2024; 129: 111598. doi:10.1016/j.intimp.2024.111598. [Google Scholar] [CrossRef]
12. Brandquist ND , Kielian T . Immune dysfunction during S. aureus biofilm-associated implant infections: Opportunities for novel therapeutic strategies. npj Biofilms and Microbiomes. 2025; 11( 1): 144. doi:10.1038/s41522-025-00782-y. [Google Scholar] [CrossRef]
13. Wood SJ , Goldufsky JW , Seu MY , Dorafshar AH , Shafikhani SH . Pseudomonas aeruginosa cytotoxins: Mechanisms of cytotoxicity and impact on inflammatory responses. Cells. 2023; 12( 1): 195. doi:10.3390/cells12010195. [Google Scholar] [CrossRef]
14. Sawoo R , Dey R , Ghosh R , Bishayi B . TLR4 and TNFR1 blockade dampen M1 macrophage activation and shifts them towards an M2 phenotype. Immunol Res. 2021; 69( 4): 334– 51. doi:10.1007/s12026-021-09209-0. [Google Scholar] [CrossRef]
15. Ciesielska A , Matyjek M , Kwiatkowska K . TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. 2021; 78( 4): 1233– 61. doi:10.1007/s00018-020-03656-y. [Google Scholar] [CrossRef]
16. Sharma KK , Gupta S , Bisen PS . Enhancing Gastrointеstinal (GI) Cancer Therapies with Ganoderma Lucidum: A Review of Mechanisms and Efficacy. J Cancer Biomol Ther. 2025; 2( 1): 15– 44. doi:10.62382/jcbt.v2i1.28. [Google Scholar] [CrossRef]
17. Liu D , Zhong Z , Karin M . NF-κB: A double-edged sword controlling inflammation. Biomedicines. 2022; 10( 6): 1250. doi:10.3390/biomedicines10061250. [Google Scholar] [CrossRef]
18. Pu S , Meng X , Shi Y , Huang N , Zhang C , Pang A , et al. Fermented Cordyceps Powder alleviates silica-induced inflammation and fibrosis by inhibiting M1 macrophage polarization via the HMGB1-TLR4-NF-κB pathway. J Ethnopharmacol. 2025; 345: 119631. doi:10.1016/j.jep.2025.119631. [Google Scholar] [CrossRef]
19. Chen K-q , Zhang Y-q , Wang Z-b , Wang S-z . Progress in Research on CNPY2 in Diseases. Mini Rev Med Chem. 2024; 24( 4): 391– 402. doi:10.2174/1389557523666230601094149. [Google Scholar] [CrossRef]
20. Mullick Chowdhury S , Hong F , Rolfo C , Li Z , He K , Wesolowski R , et al. CNPY2 in Solid Tumors: Mechanisms, Biomarker Potential, and Therapeutic Implications. Biology. 2025; 14( 2): 214. doi:10.3390/biology14020214. [Google Scholar] [CrossRef]
21. Zhang W , Meng L , Zhang X , Li Z , Hong F . CNPY2 drives DSS-induced colitis via the macrophage-ROS axis. Biomed Pharmacother. 2025; 187: 118078. doi:10.1016/j.biopha.2025.118078. [Google Scholar] [CrossRef]
22. Yang B , Shu W , Hu J , Wang Z , Wu J , Su J , et al. Aberrant Expression of SLC7A11 Impairs the Antimicrobial Activities of Macrophages in Staphylococcus Aureus Osteomyelitis in Mice. Int J Biol Sci. 2024; 20( 7): 2555– 75. doi:10.7150/ijbs.93592. [Google Scholar] [CrossRef]
23. Matsunaga N , Tsuchimori N , Matsumoto T , Ii M . TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol. 2011; 79( 1): 34– 41. doi:10.1124/mol.110.068064. [Google Scholar] [CrossRef]
24. Garlick E , Thomas SG , Owen DM . Super-resolution imaging approaches for quantifying F-actin in immune cells. Front Cell Dev Biol. 2021; 9: 676066. doi:10.3389/fcell.2021.676066. [Google Scholar] [CrossRef]
25. Sorzano COS . Statistical experiment design for animal research. OSF Preprints. 2023; 10: 296. doi:10.31219/osf.io/e9s25. [Google Scholar] [CrossRef]
26. Gehrke AE , Mendoza-Bertelli A , Ledo C , Gonzalez CD , Noto Llana M , Blanco C , et al. Neutralization of staphylococcus aureus protein a prevents exacerbated osteoclast activity and bone loss during osteomyelitis. Antimicrob Agents Chemother. 2023; 67( 1): e0114022. doi:10.1128/aac.01140-22. [Google Scholar] [CrossRef]
27. Hulley PA , Knowles HJ . A New Method to Sort differentiating osteoclasts into defined homogeneous subgroups. Cells. 2022; 11( 24): 3973. doi:10.3390/cells11243973. [Google Scholar] [CrossRef]
28. Kakehashi A , Suzuki S , Shiota M , Raymo N , Gi M , Tachibana T , et al. Canopy homolog 2 as a novel molecular target in Hepatocarcinogenesis. Cancers. 2021; 13( 14): 3613. doi:10.3390/cancers13143613. [Google Scholar] [CrossRef]
29. Huang H , Tang N , Li Y , Huo Q , Chen Q , Meng Q . The role of CNPY2 in endothelial injury and inflammation during the progress of atherosclerosis. J Mol Histol. 2023; 54( 3): 195– 205. doi:10.1007/s10735-023-10122-z. [Google Scholar] [CrossRef]
30. Wu Y , Liu W , Xiang Y , Hong L , Zhan Z . DPEP2 deficiency enhances infiltration of macrophages by Akt1-VIM axis. Biochem Biophys Res Commun. 2025: 152427. doi:10.1016/j.bbrc.2025.152427. [Google Scholar] [CrossRef]
31. Yu F , Wang Y , Stetler AR , Leak RK , Hu X , Chen J . Phagocytic microglia and macrophages in brain injury and repair. CNS Neurosci Ther. 2022; 28( 9): 1279– 93. doi:10.1111/cns.13899. [Google Scholar] [CrossRef]
32. Yang S , Zhao M , Jia S . Macrophage: Key player in the pathogenesis of autoimmune diseases. Front Immunol. 2023; 14: 1080310. doi:10.3389/fimmu.2023.1080310. [Google Scholar] [CrossRef]
33. Zhang W , Wang M , Ji C , Liu X , Gu B , Dong T . Macrophage polarization in the tumor microenvironment: Emerging roles and therapeutic potentials. Biomed Pharmacother. 2024; 177: 116930. doi:10.1016/j.biopha.2024.116930. [Google Scholar] [CrossRef]
34. Wang C , Ma C , Gong L , Guo Y , Fu K , Zhang Y , et al. Macrophage polarization and its role in liver disease. Front Immunol. 2021; 12: 803037. doi:10.3389/fimmu.2021.803037. [Google Scholar] [CrossRef]
35. He L , Jhong J-H , Chen Q , Huang K-Y , Strittmatter K , Kreuzer J , et al. Global characterization of macrophage polarization mechanisms and identification of M2-type polarization inhibitors. Cell Rep. 2021; 37( 5): 109955. doi:10.1016/j.celrep.2021.109955. [Google Scholar] [CrossRef]
36. Zhu D , Chen F , Qiang H , Qi H . SPA inhibits hBMSC osteogenic differentiation and M1 macrophage polarization by suppressing SETD2 in acute suppurative osteomyelitis. Sci Rep. 2024; 14( 1): 12728. doi:10.1038/s41598-024-63219-0. [Google Scholar] [CrossRef]
37. Su J , Wu Y , Wang Z , Zhang D , Yang X , Zhao Y , et al. Probiotic biofilm modified scaffolds for facilitating osteomyelitis treatment through sustained release of bacteriophage and regulated macrophage polarization. Materials Today Bio. 2025; 30: 101444. doi:10.1016/j.mtbio.2025.101444. [Google Scholar] [CrossRef]
38. Zhan L , Zeng F , Zheng J , Chen S , Zhang Z , Ju D . Exploring the regulatory role of CNPY3 as a prognostic biomarker on human glioma cell migration, invasion and immune infiltration. Cancer Biomark. 2025; 42( 3): 18758592251328162. doi:10.1177/18758592251328162. [Google Scholar] [CrossRef]
39. Tian L , Tan Z , Yang Y , Liu S , Yang Q , Tu Y , et al. In situ sprayed hydrogels containing resiquimod-loaded liposomes reduce chronic osteomyelitis recurrence by intracellular bacteria clearance. Acta Biomater. 2023; 169: 209– 27. doi:10.1016/j.actbio.2023.07.039. [Google Scholar] [CrossRef]
40. Li J , Zhang L , Peng J , Zhao C , Li W , Yu Y , et al. Mitochondrial metabolic regulation of macrophage polarization in osteomyelitis and other orthopedic disorders: Mechanisms and therapeutic opportunities. Front Cell Dev Biol. 2025; 13: 1604320. doi:10.3389/fcell.2025.1604320. [Google Scholar] [CrossRef]
41. Sun Y , Li J , Xie X , Gu F , Sui Z , Zhang K , et al. Macrophage-osteoclast associations: Origin, polarization, and subgroups. Front Immunol. 2021; 12: 778078. doi:10.3389/fimmu.2021.778078. [Google Scholar] [CrossRef]
42. Cheng Y , Liu H , Li J , Ma Y , Song C , Wang Y , et al. Evaluation of culture conditions for osteoclastogenesis in RAW264. 7 cells. PLoS One. 2022; 17( 11): e0277871. doi:10.1371/journal.pone.0277871. [Google Scholar] [CrossRef]
43. Zhang X , Wang L , Huang N , Zheng Y , Cai L , Ke Q , et al. MicroRNA-455-3p regulates proliferation and osteoclast differentiation of RAW264. 7 cells by targeting PTEN. BMC Musculoskeletal Disord. 2022; 23( 1): 340. doi:10.1186/s12891-025-08576-1. [Google Scholar] [CrossRef]
44. Zhang Y , Yang M , Zhang S , Yang Z , Zhu Y , Wang Y , et al. BHLHE40 promotes osteoclastogenesis and abnormal bone resorption via c-Fos/NFATc1. Cell Biosci. 2022; 12( 1): 70. doi:10.1186/s13578-020-00430-2. [Google Scholar] [CrossRef]
45. Tian H , Chen F , Wang Y , Liu Y , Ma G , Zhao Y , et al. Nur77 prevents osteoporosis by inhibiting the NF-κB signalling pathway and osteoclast differentiation. J Cell Mol Med. 2022; 26( 8): 2163– 76. doi:10.1111/jcmm.17238. [Google Scholar] [CrossRef]
46. Liu X , Wang X , Ma X , Li H , Miao C , Tian Z , et al. Genetic disruption of Ano5 leads to impaired osteoclastogenesis for gnathodiaphyseal dysplasia. Oral Dis. 2024; 30( 3): 1403– 15. doi:10.1111/odi.14562. [Google Scholar] [CrossRef]
47. Bergamin LS , Penolazzi L , Lambertini E , Falzoni S , Sarti AC , Molle CM , et al. Expression and function of the P2X7 receptor in human osteoblasts: The role of NFATc1 transcription factor. J Cell Physiol. 2021; 236( 1): 641– 52. doi:10.1002/jcp.29891. [Google Scholar] [CrossRef]
48. Wu H , Yin G , Pu X , Wang J , Liao X , Huang Z . Coordination of osteoblastogenesis and osteoclastogenesis by the bone marrow mesenchymal stem cell-derived extracellular matrix to promote bone regeneration. ACS applied bio materials. 2022; 5( 6): 2913– 27. doi:10.1021/acsabm.2c00264. [Google Scholar] [CrossRef]
49. Fujii T , Murata K , Mun S-H , Bae S , Lee YJ , Pannellini T , et al. MEF2C regulates osteoclastogenesis and pathologic bone resorption via c-FOS. Bone Res. 2021; 9( 1): 4. doi:10.1038/s41413-020-00120-2. [Google Scholar] [CrossRef]
50. Li F-B , Bao S-Q , Sun X-L , Ma J-X , Ma X-L . Extracellular acidification stimulates OGR1 to modify osteoclast differentiation and activity through the Ca2+-calcineurin-NFATc1 pathway. Exp Ther Med. 2024; 29( 2): 28. doi:10.3892/etm.2024.12778. [Google Scholar] [CrossRef]
51. Jiang C , Lin Y , Shan H , Xia W , Pan C , Wang N , et al. miR-146a protects against Staphylococcus aureus-induced osteomyelitis by regulating inflammation and osteogenesis. ACS Infect Dis. 2022; 8( 5): 918– 27. doi:10.1021/acsinfecdis.1c00459. [Google Scholar] [CrossRef]
52. Granata V , Possetti V , Parente R , Bottazzi B , Inforzato A , Sobacchi C . The osteoblast secretome in Staphylococcus aureus osteomyelitis. Front Immunol. 2022; 13: 1048505. doi:10.3389/fimmu.2022.1048505. [Google Scholar] [CrossRef]
53. Campbell MJ , Bustamante-Gomez C , Fu Q , Beenken KE , Reyes-Pardo H , Smeltzer MS , et al. RANKL-mediated osteoclast formation is required for bone loss in a murine model of Staphylococcus aureus osteomyelitis. Bone. 2024; 187: 117181. doi:10.1016/j.bone.2024.117181. [Google Scholar] [CrossRef]
54. Heine H , Zamyatina A . Therapeutic targeting of TLR4 for inflammation, infection, and cancer: A perspective for disaccharide lipid A mimetics. Pharmaceuticals. 2022; 16( 1): 23. doi:10.3390/ph16010023. [Google Scholar] [CrossRef]
55. Dejban P , Nikravangolsefid N , Chamanara M , Dehpour A , Rashidian A . The role of medicinal products in the treatment of inflammatory bowel diseases (IBD) through inhibition of TLR4/NF-kappaB pathway. Phytother Res. 2021; 35( 2): 835– 45. doi:10.1002/ptr.6866. [Google Scholar] [CrossRef]
56. Dong P , Liu K , Han H . The role of NF-κB in myocardial ischemia/reperfusion injury. Curr Protein Pept Sci. 2022; 23( 8): 535– 47. doi:10.2174/1389203723666220817085941. [Google Scholar] [CrossRef]
57. Kim D , Allen CA , Chung D , Meng L , Zhang X , Zhang W , et al. A novel TLR4 accessory molecule drives hepatic oncogenesis through tumor-associated macrophages. Cancer Lett. 2025; 614: 217543. doi:10.1016/j.canlet.2025.217543. [Google Scholar] [CrossRef]
58. Ghait M , Husain RA , Duduskar SN , Haack TB , Rooney M , Göhrig B , et al. The TLR-chaperone CNPY3 is a critical regulator of NLRP3-inflammasome activation. Eur J Immunol. 2022; 52( 6): 907– 23. doi:10.1002/eji.202149612. [Google Scholar] [CrossRef]
59. Meier-Soelch J , Mayr-Buro C , Juli J , Leib L , Linne U , Dreute J , et al. Monitoring the levels of cellular NF-κB activation states. Cancers. 2021; 13( 21): 5351. doi:10.3390/cancers13215351. [Google Scholar] [CrossRef]
60. Ren LR , Wang ZH , Wang H , He XQ , Song MG , Xu YQ . Staphylococcus Aureus Induces Osteoclastogenesis via the NF-κB Signaling Pathway. Med Sci Monit. 2017; 23: 4579– 90. doi:10.12659/MSM.903371. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools