
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
Factors Affecting the Genetic Diagnostic Rate in Congenital Heart Disease
1
Department of Pediatrics, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
2
3 Billion, Inc., Seoul, Korea
3
Genome Research Center for Birth Defects and Genetic Diseases, Asan Institute for Life Sciences, Asan Medical Center, Seoul,
Korea
4
Division of Cardiology, Department of Pediatrics, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
5
Division of Neonatology, Department of Pediatrics, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
6
Medical Genetics Center, Asan Medical Center Children’s Hospital, University of Ulsan College of Medicine, Seoul, Korea
* Corresponding Author: Beom Hee Lee. Email:
Congenital Heart Disease 2022, 17(6), 653-673. https://doi.org/10.32604/chd.2022.021580
Received 24 February 2022; Accepted 20 June 2022; Issue published 11 October 2022
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Over 400 genes contribute to the development of congenital heart disease (CHD). Additionally, multisystemic manifestations accompanying syndromic CHD pose a higher risk of genetic diseases. This study investigated the diagnostic yield of whole-exome sequencing (WES) in patients with sporadic syndromic CHD and the phenotypic factors affecting the genetic diagnostic rate. Methods: Sixty-four patients with sporadic syndromic CHD aged <18 years underwent WES between May 2018 and December 2020 in a single tertiary center, and the association between genetic testing data and extracardiac phenotypes was analyzed. Results: Extracardiac phenotypes were measured as 3.66 ± 3.05 (standard deviation, interquartile range: 2–5) items per patient. WES detected diagnostic variants in 19 (29.7%) patients: seven (36.8%), seven (36.8%), and five (26.3%) with pathogenic variants, likely pathogenic variants, and variants of unknown significance, respectively. Post-diagnosis surveillance identified the extracardiac phenotype in 54.5% (6/11) of patients. De novo variants accounted for 76.2% (15/19) of variants and autosomal dominant inheritance for 94.7% (18/19). Most diseases were ultra-rare. No significant differences were noted in cardiac and extracardiac phenotypes, single or combined (all P > 0.05), between the groups with and without a diagnostic variant. However, patients with ≥3 extracardiac phenotypes had a significantly higher likelihood of having a diagnostic variant than those with ≤2 (38.3% vs. 5.9%, odds ratio = 9.93, 95% confidence interval = 1.21–81.44, P = 0.013). Conclusions: The number of extracardiac phenotypes is important in predicting the possibility of genetic diagnosis. Physicians will be able to select patients with a high probability of genetic diagnosis and provide appropriate genetic counseling based on the results of this study.Graphic Abstract
Keywords
Congenital heart disease (CHD) develops because of abnormalities in the structure of the heart that arise before birth. It is the most frequently occurring congenital disorder in newborns, with an estimated incidence of 8 per 1000 live births per year (range 3–10) [1]. Despite progress in medical and surgical treatments, CHD remains the leading cause of mortality among all congenital anomalies.
The etiology of CHD has been proposed as both genetic and environmental, with evidence increasingly supporting the role of a genetic contribution [2]. Chromosome disorders, including Down syndrome (OMIM 190685) and Edwards syndrome (OMIM 300484); microdeletion syndrome, including Williams syndrome (OMIM 194050) and 22q11.2 microdeletion syndrome (OMIM 188400, 192430, and 182212); and monogenic diseases, including Noonan syndrome (OMIM 164757, 164760, 164790, etc.) and Kabuki syndrome (OMIM 147920 and 300867), have been revealed to underlie syndromic CHD. To date, approximately 400 genes have been suggested to be involved in CHD development [3].
Karyotyping and chromosome microarray (CMA) can detect chromosome disorders and copy number variants (CNVs). Overall, karyotyping and CNV-related molecular diagnosis can each be made approximately in 10%–25% of patients with CHD [2–4]. However, genomic sequencing, such as whole-exome sequencing (WES) using the next-generation sequencing technique can be effectively applied to diagnose monogenic diseases, considering their genetic heterogeneity [2]. Elucidating the genetic etiology of CHD provides essential information on patient care by surveillance of unpredicted extracardiac manifestations and preemptive prevention of delayed-onset manifestations [5–8]. Furthermore, genetic diagnosis significantly contributes to the provision of appropriate genetic counseling for patients and their family members. Conversely, reckless and over-extensive genetic testing can incur high medical costs and offset its effectiveness [9,10]. Therefore, it is important to determine the clinical features that would enhance the diagnostic yield of the genetic test in terms of cost-effectiveness and understanding the genetic contribution to CHD.
A recent study reported a significant difference between the number of body systems affected and the likelihood of having a pathogenic variant in patients with multiple anomalies [11]. Importantly, extracardiac congenital anomalies are frequently observed in newborns with CHD, with almost twice the prevalence observed in non-CHD individuals [12,13]. Therefore, it is necessary to assess the contribution of these accompanying extracardiac manifestations to the genetic diagnostic rate in patients with CHD. However, there is no research reporting on the association between extracardiac manifestations and the likelihood of detecting the genetic variant in syndromic CHD.
In this study, we performed WES in pediatric patients with sporadic syndromic CHD and assessed the factors affecting diagnostic yield.
2.1 Study Design and Population
We conducted WES in probands aged ≤18 years with sporadic syndromic CHD, which accompanied the diagnosis of at least one extracardiac phenotype at Asan Medical Center Children’s Hospital, Seoul, South Korea, between May 2018 and December 2020. All types of hemodynamically significant CHD were included. Hemodynamically insignificant phenotypes, including mild pulmonary stenosis, spontaneously closure of small atrial septal defect (ASD), left superior vena cava, and so on were excluded. Moreover, patients with cardiomyopathy and isolated transient fetal circulation with spontaneous resolution were excluded; patients with all other physiological abnormalities, including arrhythmia combined with CHD, were included. Patients with positive results in other genetic tests, including chromosome analysis, gene panel, multiplex ligation-dependent probe amplification, and CMA, were excluded. The patients’ detailed demographics and clinical characteristics were reviewed, including age, sex, initial presentation, family history, and cardiac and extracardiac manifestations. Informed consent was obtained from the parents of the patients for genetic testing. The Institutional Review Board for Human Research at Asan Medical Center approved this study (IRB numbers: 2018-0574 and 2018-0180).
2.2 Classification of Manifestations
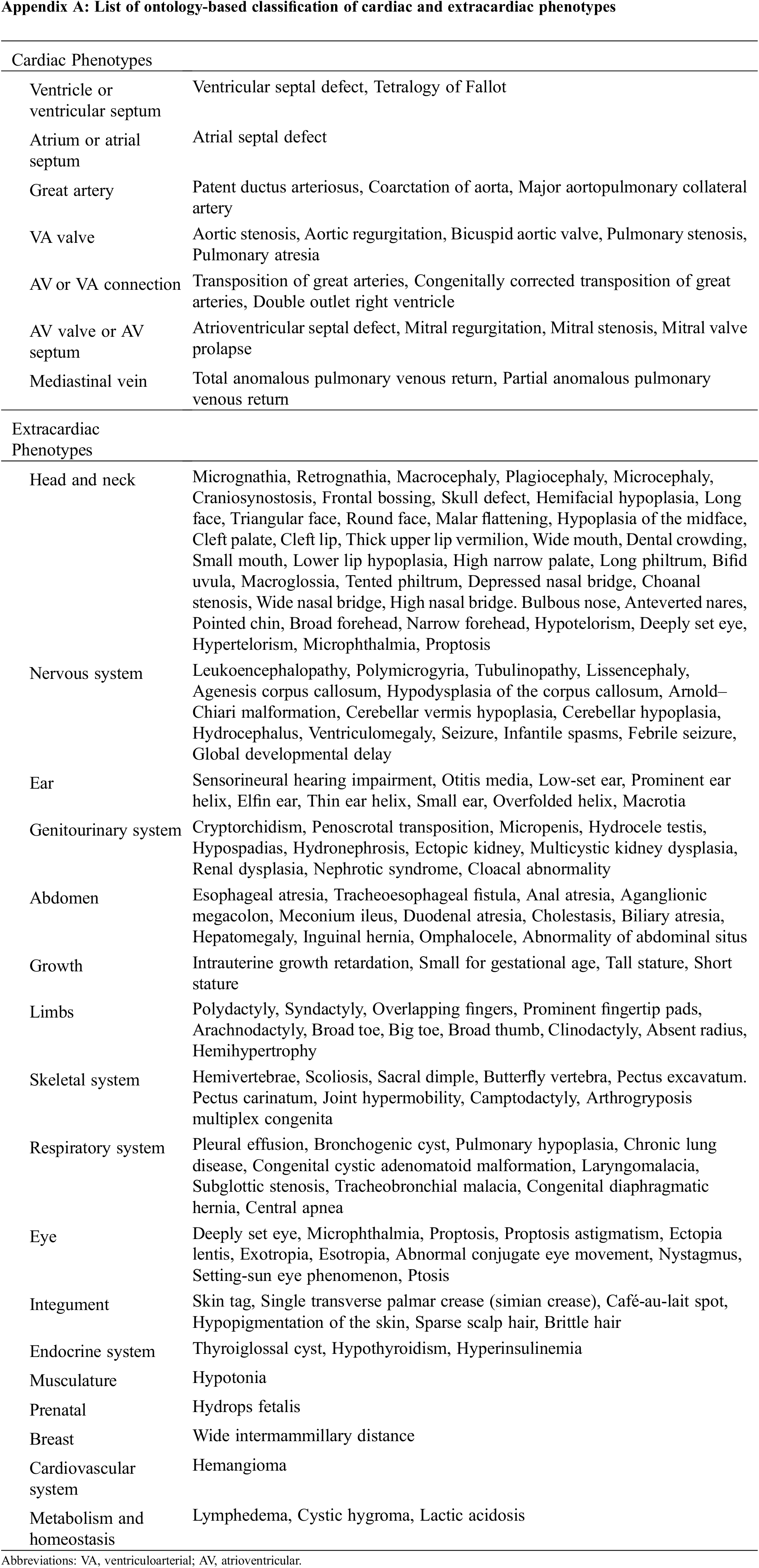
Cardiac phenotypes are classified according to the International Pediatric and Congenital Cardiac Code (IPCCC) and the Eleventh Iteration of the International Classification of Diseases (ICD-11) [14], and extracardiac phenotypes are classified according to the Human Phenotype Ontology (HPO) [15,16]. Cardiac manifestations were assessed using the latest echocardiography or computed tomography or via surgical diagnosis by pediatric cardiologists, pediatric cardiac surgeons, or radiologists. All abnormalities of the morphology and the vasculature, including abnormality of the aorta, coronary artery, vena cava, and pulmonary vasculature, were included (Appendix A).
Patients with CHD underwent routine surveillance, including brain and abdominal ultrasound, tandem mass spectrometry, and complete inspection after birth. Extracardiac manifestations were described by a physician with at least 1 year of experience as a medical geneticist after specializing in pediatrics. Confirmation of findings was provided by a senior geneticist with 10 years of experience. Each phenotype was classified based on the highest level of organ system: head and neck, nervous system, ear, genitourinary system, abdomen, growth, limbs, skeletal system, respiratory system, eye, integument, endocrine system, musculature, prenatal, breast, cardiovascular system, metabolism, and homeostasis. The number of extracardiac phenotypes was counted as the number of systems, not all the individual phenotypes. Multiple phenotypes belonging to the same system were counted as one extracardiac phenotype. For example, the three phenotypes of hypertelorism, high-arched palate, and cryptorchidism were counted as two extracardiac phenotypes: the former two are an abnormality of the head and neck, and the latter is an abnormality of the genitourinary tract.
2.3 Variant Annotation and Interpretation by WES Analysis
We performed WES using genomic DNA isolated from the patient’s whole blood or a buccal swab sample. All exons of all human genes (approximately 22 000) were captured using the Twist Human Core Exome Kit (Twist Bioscience, San Francisco, CA, USA). The captured genomic regions were sequenced using a NovaSeq 6000 platform (Illumina, San Diego, CA, USA). Raw genome sequencing data analysis included alignment to the reference sequence (NCBI genome assembly GRCh37; accessed February 2009). The mean depth of coverage was 100-fold with 99.2% higher coverage than that of 10-fold. Each variant’s pathogenicity was assessed using the automated variant interpretation system EVIDENCE, in line with the American College of Medical Genetics (ACMG) guidelines 12 and relevant patient phenotypes [17]. Candidate variants based on EVIDENCE were reviewed and selected by expert medical geneticists. Sanger sequencing of the variant identified via exome sequencing was performed for patients and their parents. “Diagnostic variant” was defined as the variant that was pathogenic, likely pathogenic, or variant of unknown significance (VUS) noted as a result of “de novo” events or of which the phenotype determined by a gene was fully matched with a patient’s phenotype after complete parental tests.
The primary goal was to investigate the usefulness of WES for identifying diagnostic variants and evaluate the phenotypes after the genetic diagnosis in patients with CHD. The secondary goal was to assess the factors affecting the diagnostic rate of WES in terms of the number and the cardiac and extracardiac phenotypes.
Descriptive data were expressed using a graph and table created using Excel 365 (Microsoft Corp., Redmond, WA, USA). For comparison between the groups, the χ2 test or Fisher’s exact test was performed. These analyses were performed using IBM-SPSS for Windows software, version 21 (SPSS Inc., Chicago, IL, USA). P < 0.05 was considered statistically significant.
WES was conducted for 101 pediatric patients with cardiac abnormalities during the study period. Excluding 19 patients with cardiomyopathy, six with isolated arrhythmia, four with isolated persistent fetal circulation, and eight with CNV in CMA, 64 patients with both syndromic features and cardiac abnormalities were included (Fig. 1).
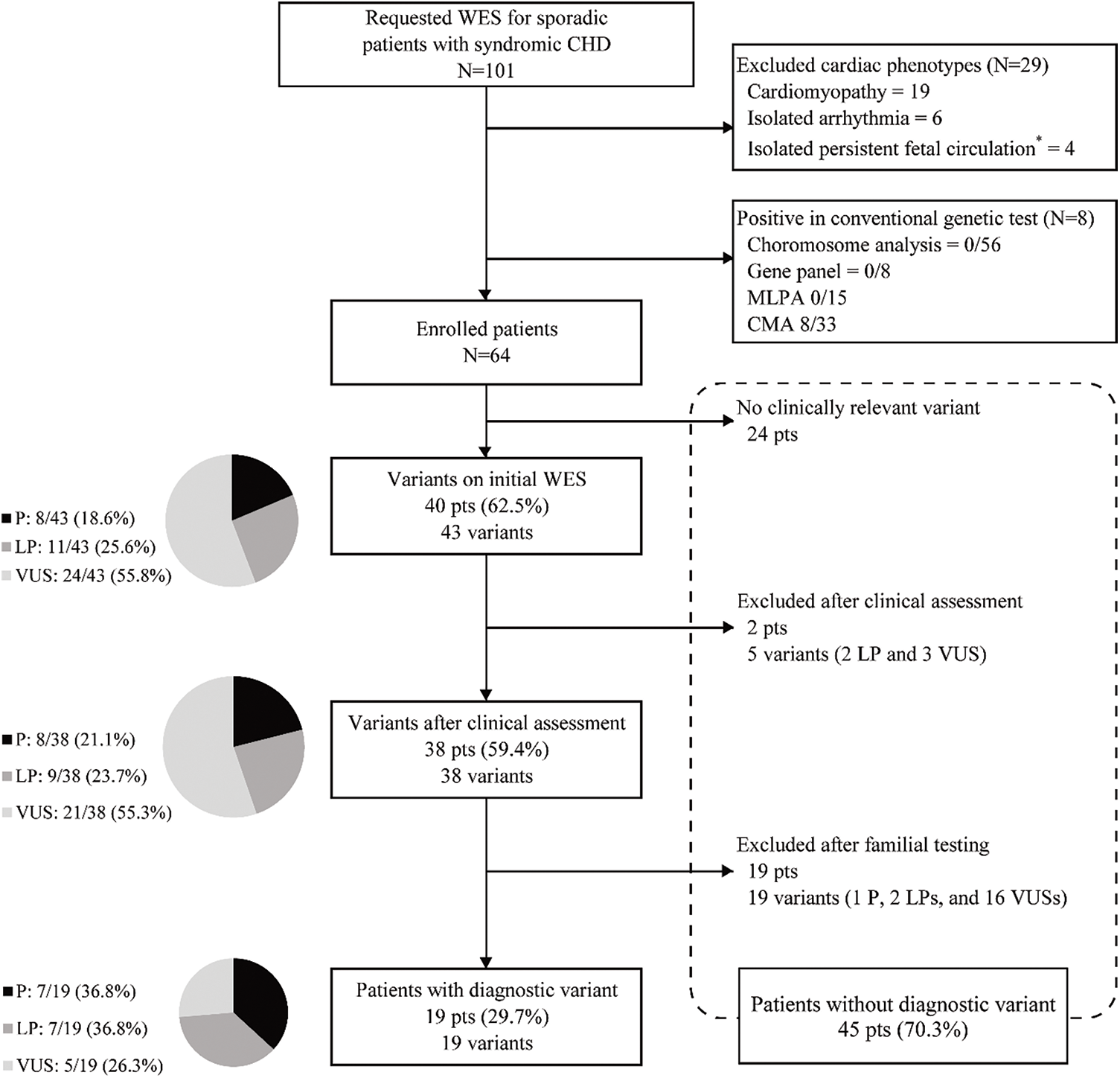
Figure 1: Flow chart of genetic diagnosis of a patient with congenital heart disease using whole-exome sequencing
Abbreviation: WES, whole-exome sequencing; CHD, congenital heart disease; MLPA, multiplex ligation-dependent probe amplification; CMA, chromosome microarray; P, pathogenic; LP, likely pathogenic; VUS, variant of unknown significance.
*Persistent fetal circulations included patent foramen ovale and patent ductus arteriosus.
3.1 Clinical Features of the Patients
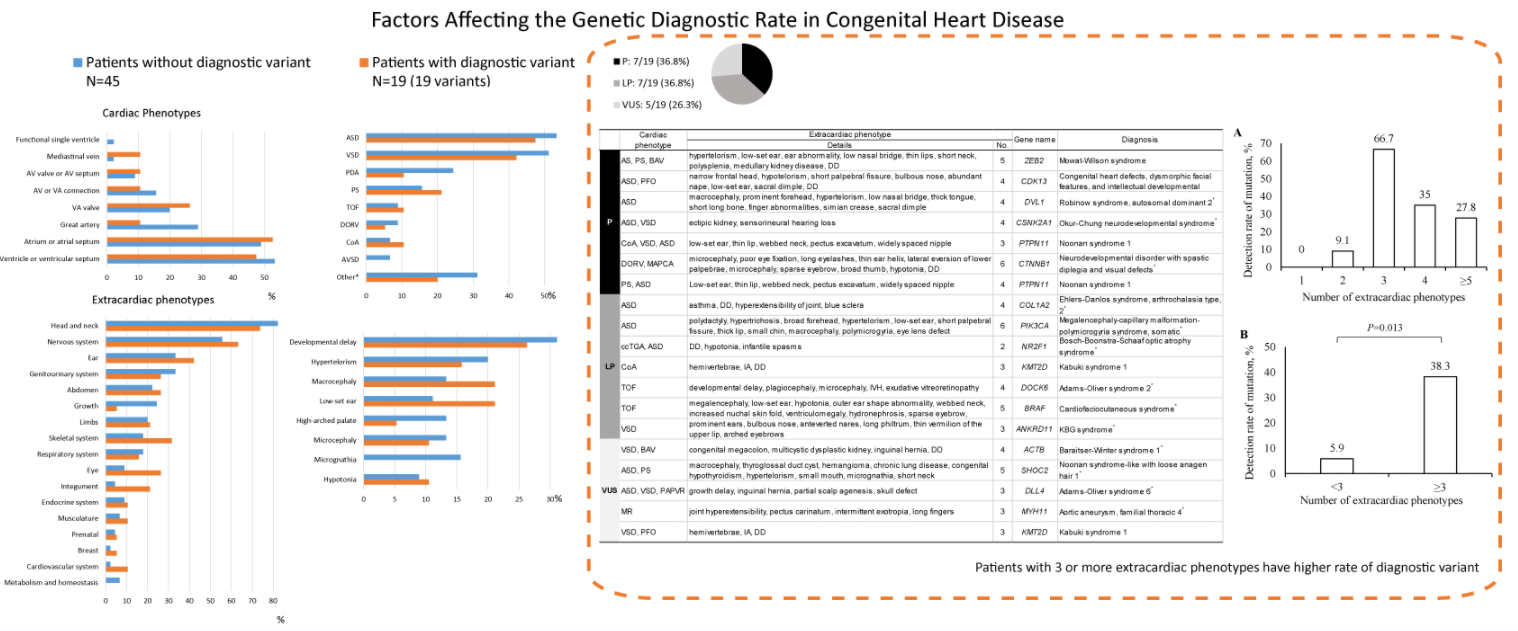
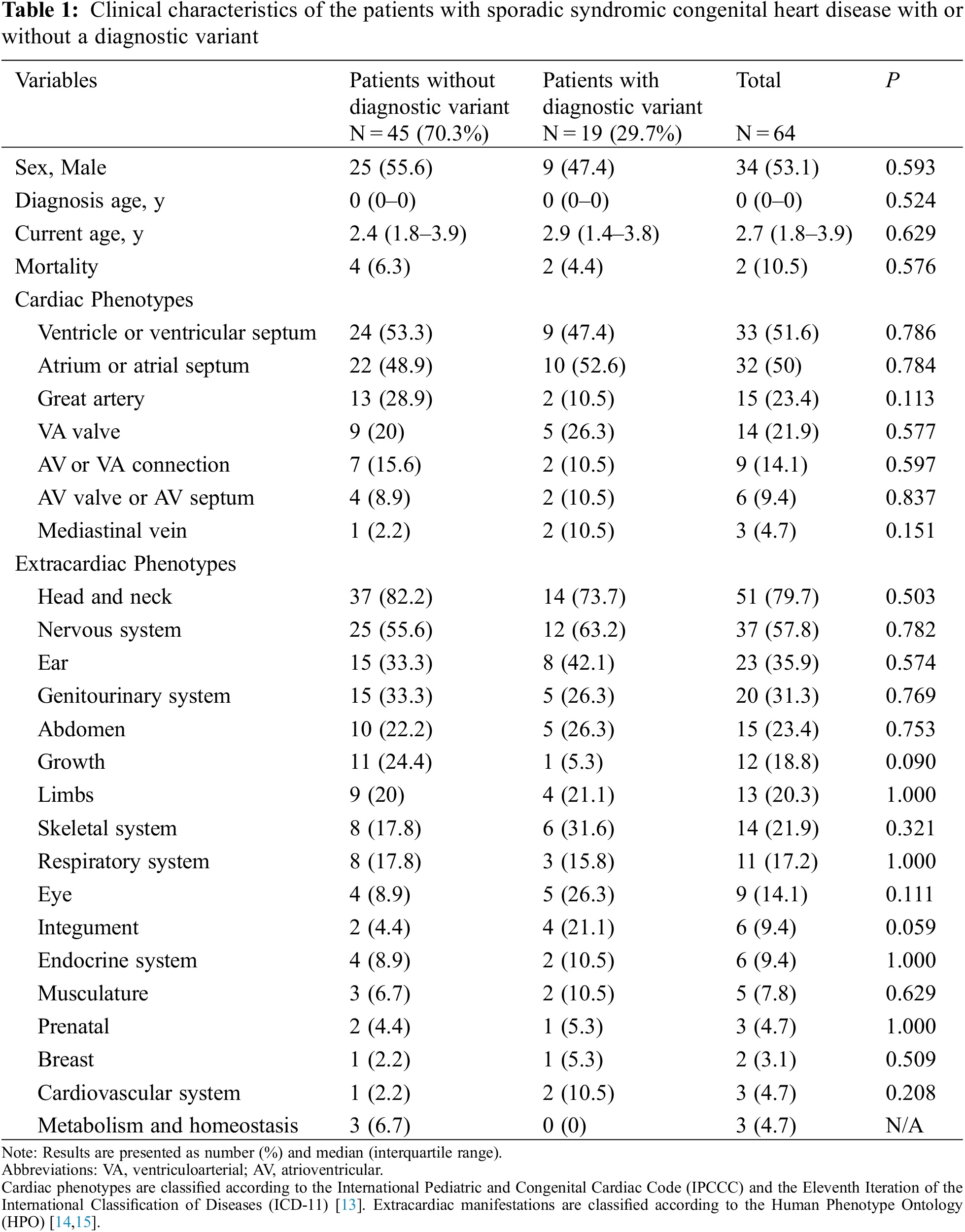
Basic demographics and clinical manifestations of the patients are summarized in Table 1. There were 34 (53.1%) males and 30 (47.9%) females, most of whom were requested for WES in their neonatal periods. Ventricle or ventricular septal defects, atrium or atrial septal defects, and great artery defects were found in 33 (51.6%), 32 (50.0%), and 15 (23.4%) patients, respectively. Among individual phenotypes, ASD (33 patients, 51.6%) and ventricular septal defect (VSD) (31 patients, 48.4%) were the most common cardiac phenotypes (Table 2).
Extracardiac phenotypes were measured as 3.66 ± 3.05 (standard deviation, interquartile range, 2–5) items per patient. The most commonly observed system of phenotypic abnormality was the head and neck (51 patients, 79.7%), followed by the nervous system (37 patients, 57.8%), ear (23 patients, 35.9%), genitourinary system (20 patients, 31.3%), and abdomen (15 patients, 23.4%). Regarding individual phenotypes, developmental delay (19 patients, 29.7%) was the most common, followed by dysmorphic features, such as macrocephaly, microcephaly, hypertelorism, low-set ear, high-arched palate, and micrognathia (Table 2).
The detailed phenotypic information of the individual patients is described in Table 3 (patients with diagnostic variant) and Appendix B (patients without diagnostic variant).
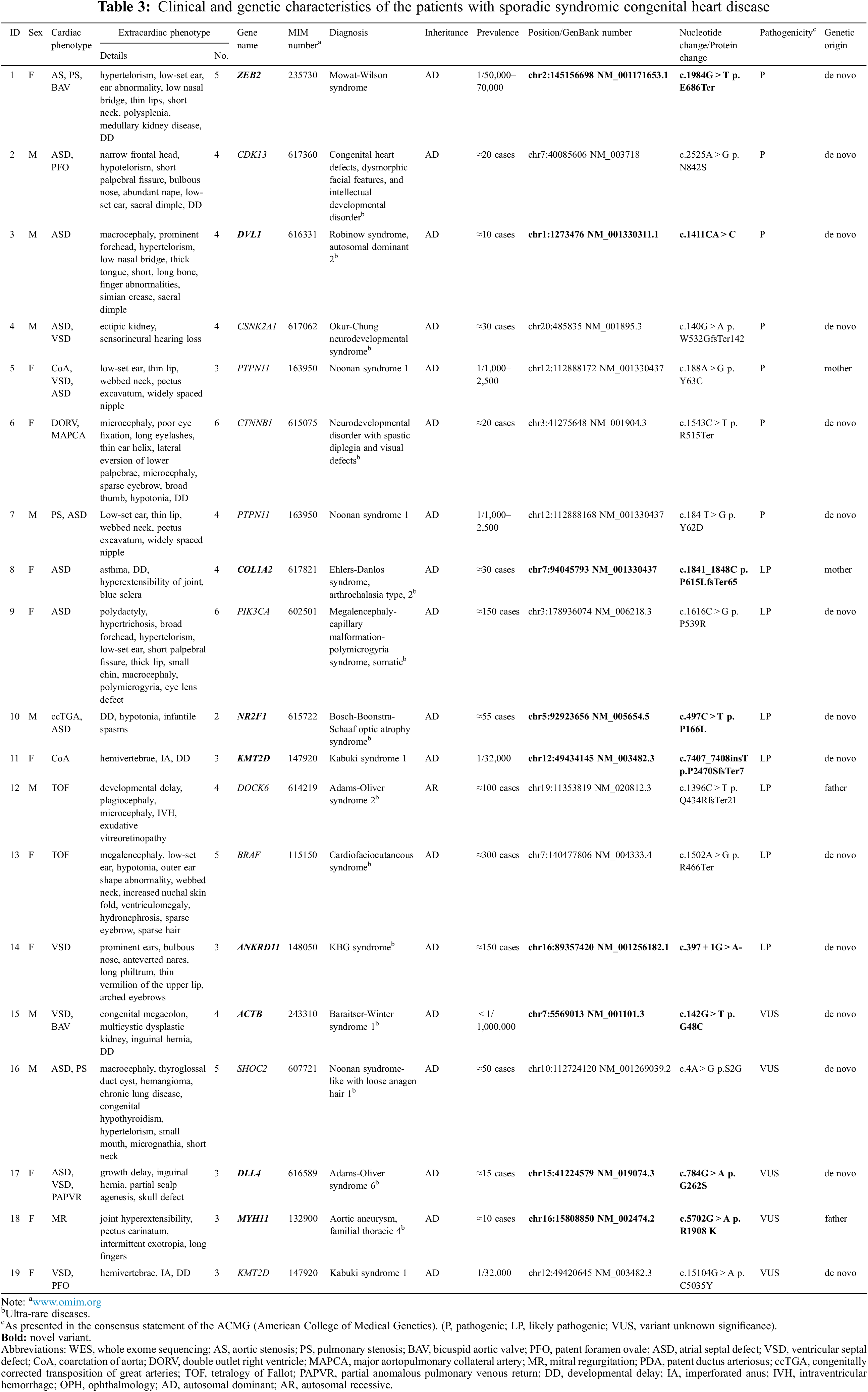
WES identified 43 variants in 40 (63.1%) patients (Fig. 1). According to the ACMG guidelines [18], among the 43 variants, eight (18.6%), 11 (25.6%), and 24 (55.8%) were categorized as pathogenic, likely pathogenic, and VUS, respectively. After clinical reassessment, excluding two variants (RYR2, OMIM 180902; and AP4M1, OMIM 612936) that are highly pathogenic and cause multiple anomalies, but not a cardiac structural abnormality, eight (21.1%), nine (23.7%), and 21 (55.3%) variants were categorized as pathogenic, likely pathogenic, and VUS, respectively. After asymptomatic parental testing, 19 (29.7%) patients were finally genetically confirmed: seven (36.8%), seven (36.8%), and five (26.3%) patients with pathogenic variants, likely pathogenic variants, and VUS, respectively. No patients with more than one diagnostic variant were noted. Detailed information on the variants and phenotypes of each patient are described in Table 3. Among the total number of 19 diagnostic variants found in our study, 15 (78.9%) were de novo variants. With the exception of Patient 12 who had Adams–Oliver syndrome 2 with an autosomal recessive inheritance, all other patients (18/19 patients, 94.7%) had genetic diseases with autosomal dominant inheritance. Nine variants in nine genes were not previously reported (bold text in Table 3, searched by December 24, 2021).
A pathogenic variant of PTPN11, a likely pathogenic variant of COL1A2 or DOCK6, or a VUS in MYH11, was inherited from an asymptomatic parent (4/19, 21.1%, Patients 5, 8, 12, and 18). However, the phenotypes of these variants were highly consistent with associated clinical symptoms and were therefore regarded as a diagnostic variant of probands exhibiting reduced penetrance in the asymptomatic parent. Surveillance for hidden phenotypes was conducted in asymptomatic parents after genetic confirmation, which did not show any affected systems.
3.3 Post-Diagnosis Clinical Evaluation
Post-diagnosis surveillance for the extracardiac phenotypes was conducted in 11 (57.9%) patients. An additional phenotype was identified in six (54.5%) patients: epilepsy in Patient 2 with congenital heart defects, dysmorphic facial features, and intellectual developmental disorder; polymicrogyria in Patient 9 with megalencephaly–capillary malformation–polymicrogyria syndrome, somatic; exudative vitreoretinopathy in Patient 12 with Adams–Oliver syndrome 2; esophoria in Patient 14 with KBG syndrome; multiple hyperechoic foci of the liver and spleen in Patient 17 with Adams–Oliver syndrome 6; and dysphagia in Patient 19 with Kabuki syndrome.
3.4 Assessment of the Factors Affecting the Genetic Diagnostic Rate
No significant difference was noted in the detection rate according to cardiac or extracardiac phenotypes (all P > 0.05, Tables 1 and 2, and Appendix C). Among the various cardiac phenotypes, the genetic diagnostic rate was higher in patients with coarctation of aorta (2/5 patients, 40%), pulmonary stenosis (4/11 patients, 36.4%), and tetralogy of Fallot (2/6 patients, 33.3%). A higher genetic diagnostic rate was observed in patients with extracardiac phenotypes of low-set ear (4/9 patients, 44.4%), macrocephaly (4/10 patients, 40%), and hypotonia (2/6 patients, 33.3%). According to the organ system categorization, a higher genetic diagnostic rate was noted in the eye (5/9 patients, 55.6%), skeletal system (6/14 patients, 42.9%), and ear (8/23 patients, 34.8%).
The correlation of the number of phenotype items according to the extracardiac organ system was analyzed with the genetic diagnostic rate in each patient. Despite no linear correlation of the genetic diagnostic rate with the number of extracardiac phenotypes (Fig. 2A), the genetic diagnostic rate was higher in patients with three or more items in an extracardiac phenotype than in patients with two or fewer (Fig. 2B) (38.3% vs. 5.9%, odds ratio [OR] = 9.93, 95% confidence interval [CI] = 1.21–81.44, P = 0.013). No statistical comparison was noted among the patients with each number of extracardiac phenotypes owing to the small number of patients.
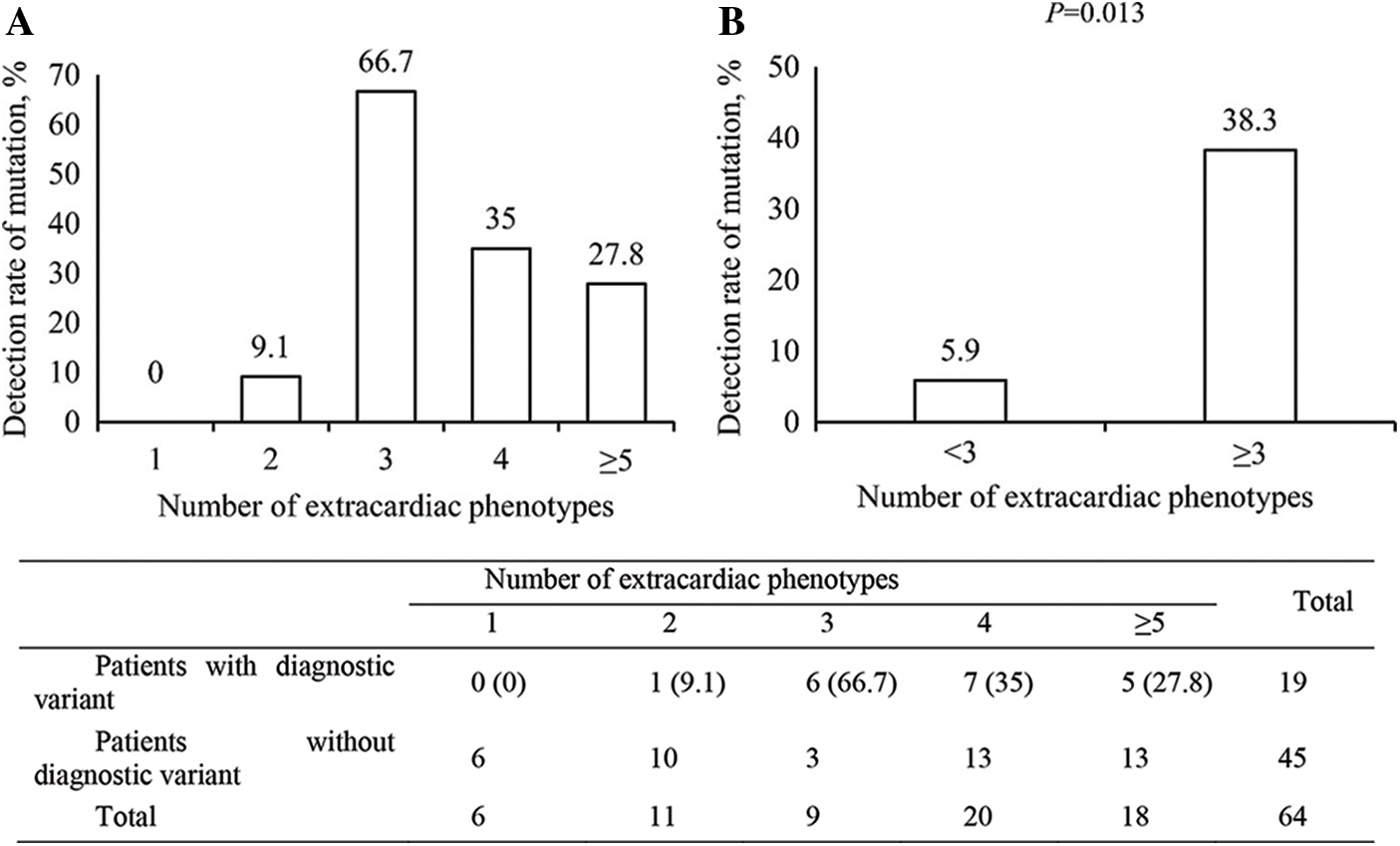
Figure 2: The detection rate of a diagnostic variant according to the number of extracardiac phenotypes
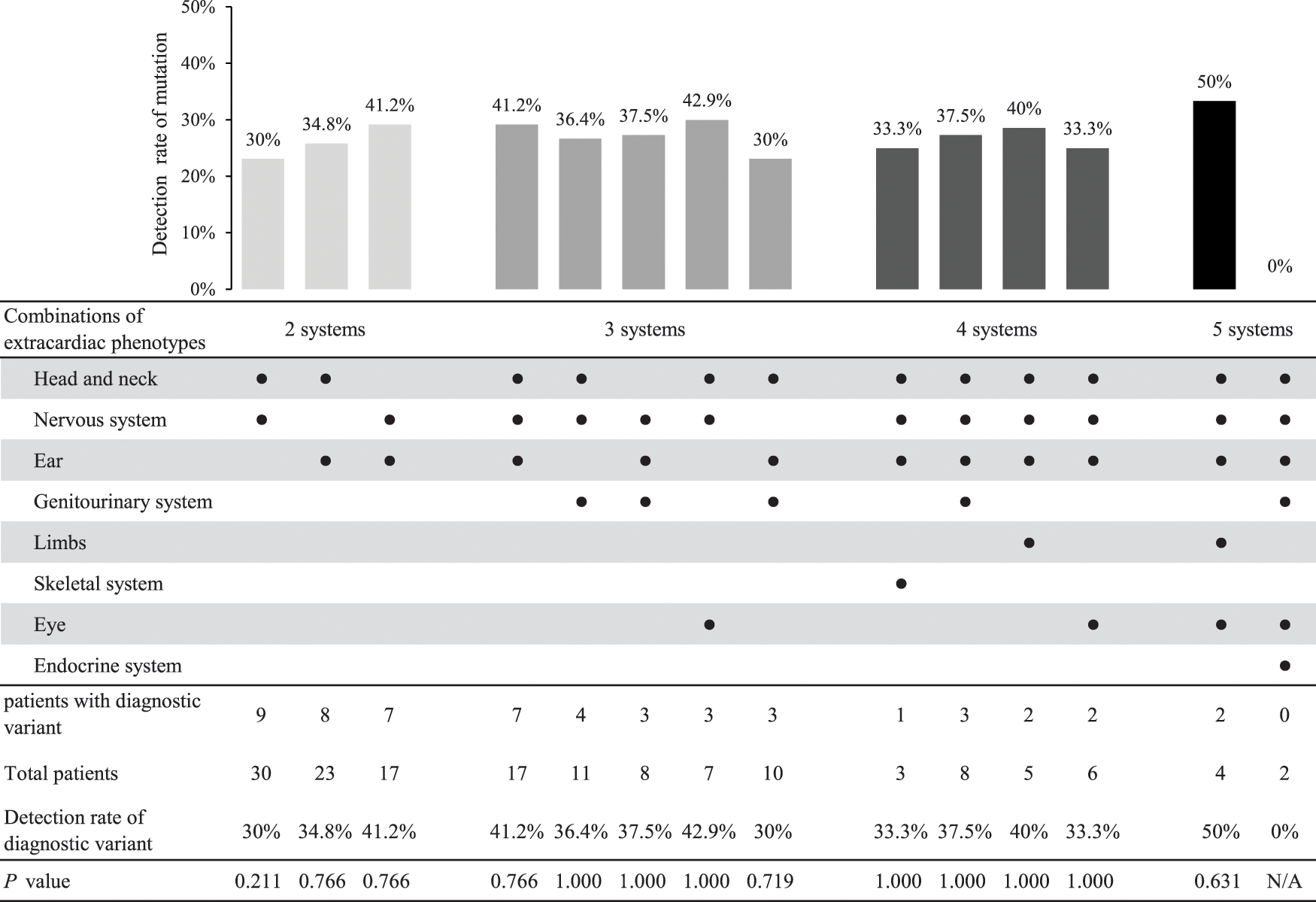
Among those patients with three or more extracardiac phenotypes, common combinations of extracardiac phenotypes and their diagnostic variant detection rates are summarized in Appendix D. Each combination showed a similar detection rate compared with the overall number of patients with three or more extracardiac phenotypes.
The genetic spectra of the patients were diverse, the details of which are described in Table 3. Some genetic diseases were common in the cohort: Noonan syndrome was found in three patients (Patients 5 and 7 with a diagnostic variant of PTPN11; Patient 16 with a diagnostic variant of SHOC2), Kabuki syndrome in two patients (Patients 11 and 19 with a diagnostic variant of KMT2D), and Mowat–Wilson syndrome (Patient 1 with a diagnostic variant of ZEB2); however, no predominant disease phenotype was identified. Meanwhile, most diseases found in the patient cohort were ultra-rare diseases (Table 3).
The current study demonstrated that WES identified a monogenic defect in 29.7% of the patients with sporadic syndromic CHD and normal CMA, which is comparable to that of the previous studies (28%–29%) [12]. Considering that the previous studies included familial cases besides sporadic cases, our similar diagnostic rate in patients with purely sporadic syndromic CHD becomes more meaningful. Furthermore, our study highlights the existence of extracardiac phenotypes and their quantity of items, which will enhance or improve the predictability of genetic diagnosis (patients with three or more extracardiac phenotypes, 38.3%; the others, 5.9%, P = 0.013). These data indicate that genomic sequencing is less likely to detect a genetic alteration in those with isolated CHD, which is consistent with the reports of previous studies [19–21]. However, no study has yet demonstrated the association between the probability of detecting genetic diagnostic variants and the number of extracardiac phenotypes using a specific cut-off value. Thus, our results are important in guiding physicians when discussing the necessity of genomic sequencing and the likelihood of finding a genetic alteration in patients or their guardians.
The results of our study are consistent with several recent reports, indicating that genomic sequencing techniques, such as WES or whole-genome sequencing, would be recommended for the genetic diagnosis of patients with CHD, either familial or sporadic, with normal CMA [22,23]. However, karyotyping and CMA are recommended in general for patients with sporadic CHD as the first-tier test, which reveals genetic alterations in up to 20% of the tested patients [22–24]. Furthermore, although WES can detect CNVs through various algorithms [25], CMA is still an irreplaceable genetic test to detect CNVs. CMA has yielded a diagnostic result in 10%–25% of patients tested [2,4]. In fact, considering there were eight (8/33, 24.2%) patients with positive CMA results who were excluded from our study, WES and CMA exert synergistic effects on each other in terms of the genetic diagnosis of syndromic CHD [26]. Notably, conventional chromosome analysis and gene panel are strongly recommended if the patient shows a typical phenotype, such as Noonan and Down syndrome.
Encouraging genetic elucidation of CHD through WES can help predict and prepare for long-term disease progression. Confirming genetic diagnosis allows the identification of individuals at higher risk of cardiac comorbidities, such as heart failure or arrhythmias, who will benefit from early screening and intervention [27–29]. Preemptive surveillance and intervention for extracardiac manifestations can also promote clinical progress. For example, genetic confirmation of ciliopathy, which decreases mucociliary clearance and causes postoperative respiratory complications [30], can help physicians prepare for augmented respiratory support. Screening and early intervention for neurodevelopmental delay can also contribute to the long-term quality of life for patients [31,32]. Additionally, screening of the families of identified patients for disease is possible [27].
Although most variants were de novo and consistent with those of a previous large cohort study [20], diagnostic variants inherited from asymptomatic parents were detected. These variants are indicative of the confounding genetic inquiry into the genetic diversity of human patients and the heterogeneity associated with CHD [2–5]. In our study, two likely pathogenic, protein-truncating variants were inherited from an asymptomatic parent (Patients 8 and 12). Such reduced penetrance is poorly understood; however, it is hypothetically associated with the genomic context, maternal–fetal environment [33], cardiac biomechanics [34], cell history, microenvironment, cellular states in situ, and other unknown factors impacting the clinical consequences of variants [35–37]. Further additional genomic and clinical data must be accumulated to obtain a more appropriate solution for factors affecting penetrance.
This study had some limitations. The small number of patients caused difficulty in performing a subgroup analysis by phenotype. A previous study has reported that specific phenotypes are associated with a genetic mutation in patients with CHD [21,38]. However, no statistically significant phenotype was noted in our results as detected in those studies. Similarly, despite noting a significantly higher likelihood of diagnostic variants in patients with three or more extracardiac phenotypes, no linear association between the number of extracardiac phenotypes and the detection rate of diagnostic variants was observed. Moreover, structural extracardiac anomalies, such as brain and genitourinary anomalies, would be missed unless appropriate imaging studies have been performed. Phenotypic evaluation was conducted when CHD was detected, and WES was performed; therefore, considering phenotypic progression according to a patient’s age, serial reassessment of a patient’s phenotype is required to improve genetic diagnosis. Finally, despite the possibility of detecting multiple variants of different genes in a single patient [39], only a single or few genetic variants were selected as responsible for the patient’s phenotype in our study. The application of an artificial intelligence-based pipeline to evaluate genomic data may have caused the criteria for the filtration to not detect some lower impact variants.
Authorship: Study conception and design: BHL; data collection: YC, SH, Y-HK, JJY, EA-RK, EJ, BSL, and JSB; analysis and interpretation of results: GHS, MK, H-SD; draft manuscript preparation: JSP. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets generated and analyzed during the current study are not publicly available because the IRB of the Asan Medical Center (IRB Nos. 2018-0574 and 2018-0180) does not allow data to be shared with out-of-hospital facilities due to ethical consideration. However, the datasets are available upon request.
Funding Statement: This work was supported by an Institute for Information and Communications Technology Promotion (IITP) grant funded by the Korean Government (MSIT) (2018-0-00861, Intelligent SW Technology Development for Medical Data Analysis).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. van der Bom, T., Zomer, A. C., Zwinderman, A. H., Meijboom, F. J., Bouma, B. J. et al. (2011). The changing epidemiology of congenital heart disease. Nature Reviews Cardiology, 8(1), 50–60. DOI 10.1038/nrcardio.2010.166. [Google Scholar] [CrossRef]
2. Zaidi, S., Brueckner, M. (2017). Genetics and genomics of congenital heart disease. Circulation Research, 120(6), 923–940. DOI 10.1161/CIRCRESAHA.116.309140. [Google Scholar] [CrossRef]
3. Blue, G. M., Kirk, E. P., Giannoulatou, E., Sholler, G. F., Dunwoodie, S. L. et al. (2017). Advances in the genetics of congenital heart disease: A clinician’s guide. Journal of the American College of Cardiology, 69(7), 859–870. DOI 10.1016/j.jacc.2016.11.060. [Google Scholar] [CrossRef]
4. Costain, G., Silversides, C. K., Bassett, A. S. (2016). The importance of copy number variation in congenital heart disease. NPJ Genomic Medicine, 1, 16031. DOI 10.1038/npjgenmed.2016.31. [Google Scholar] [CrossRef]
5. Williams, K., Carson, J., Lo, C. (2019). Genetics of congenital heart disease. Biomolecules, 9(12), 879. DOI 10.3390/biom9120879. [Google Scholar] [CrossRef]
6. Shabana, N. A., Shahid, S. U., Irfan, U. (2020). Genetic contribution to congenital heart disease (CHD). Pediatric Cardiology, 41(1), 12–23. DOI 10.1007/s00246-019-02271-4. [Google Scholar] [CrossRef]
7. Saliba, A., Figueiredo, A., Baroneza, J. E., Afiune, J. Y., Pic-Taylor, A. et al. (2020). Genetic and genomics in congenital heart disease: A clinical review. Jornal de Pediatria, 96(3), 279–288. DOI 10.1016/j.jped.2019.07.004. [Google Scholar] [CrossRef]
8. Gilboa, S. M., Devine, O. J., Kucik, J. E., Oster, M. E., Riehle-Colarusso, T. et al. (2016). Congenital heart defects in the United States: Estimating the magnitude of the affected population in 2010. Circulation, 134(2), 101–109. DOI 10.1161/CIRCULATIONAHA.115.019307. [Google Scholar] [CrossRef]
9. Landis, B. J., Ware, S. M. (2016). The current landscape of genetic testing in cardiovascular malformations: Opportunities and challenges. Frontiers in Cardiovascular Medicine, 3, 22. DOI 10.3389/fcvm.2016.00022. [Google Scholar] [CrossRef]
10. Schwarze, K., Buchanan, J., Taylor, J. C., Wordsworth, S. (2018). Are whole-exome and whole-genome sequencing approaches cost-effective: A systematic review of the literature. Genetics in Medicine, 20(10), 1122–1130. DOI 10.1038/gim.2017.247. [Google Scholar] [CrossRef]
11. Ziats, M. N., Ahmad, A., Bernat, J. A., Fisher, R., Glassford, M. et al. (2020). Genotype-phenotype analysis of 523 patients by genetics evaluation and clinical exome sequencing. Pediatric Research, 87(4), 735–739. DOI 10.1038/s41390-019-0611-5. [Google Scholar] [CrossRef]
12. Jin, S. C., Homsy, J., Zaidi, S., Lu, Q., Morton, S. et al. (2017). Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nature Genetics, 49(11), 1593–1601. DOI 10.1038/ng.3970. [Google Scholar] [CrossRef]
13. Egbe, A., Lee, S., Ho, D., Uppu, S., Srivastava, S. (2014). Prevalence of congenital anomalies in newborns with congenital heart disease diagnosis. Annals of Pediatric Cardiology, 7(2), 86–91. DOI 10.4103/0974-2069.132474. [Google Scholar] [CrossRef]
14. Franklin, R. C. G., Béland, M. J., Colan, S. D., Walters, H. L., Aiello, V. D. et al. (2017). Nomenclature for congenital and paediatric cardiac disease: The International Paediatric and Congenital Cardiac Code (IPCCC) and the eleventh iteration of the International Classification of Diseases (ICD-11). Cardiology in the Young, 27(10), 1872–1938. DOI 10.1017/S1047951117002244. [Google Scholar] [CrossRef]
15. Köhler, S., Schulz, M. H., Krawitz, P., Bauer, S., Dölken, S. et al. (2009). Clinical diagnostics in human genetics with semantic similarity searches in ontologies. The American Journal of Human Genetics, 85(4), 457–464. DOI 10.1016/j.ajhg.2009.09.003. [Google Scholar] [CrossRef]
16. Köhler, S., Vasilevsky, N. A., Engelstad, M., Foster, E., McMurry, J. et al. (2017). The human phenotype ontology in 2017. Nucleic Acids Research, 45, D865–D876. DOI 10.1093/nar/gkw1039. [Google Scholar] [CrossRef]
17. Seo, G. H., Kim, T., Choi, I. H., Park, J. Y., Lee, J. et al. (2020). Diagnostic yield and clinical utility of whole exome sequencing using an automated variant prioritization system, EVIDENCE. Clinical Genetics, 98(6), 562–570. DOI 10.1111/cge.13848. [Google Scholar] [CrossRef]
18. Richards, S., Aziz, N., Bale, S., Bick, D., Das, S. et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. DOI 10.1038/gim.2015.30. [Google Scholar] [CrossRef]
19. Cowan, J. R., Ware, S. M. (2015). Genetics and genetic testing in congenital heart disease. Clinics in Perinatology, 42(2), 373–393. DOI 10.1016/j.clp.2015.02.009. [Google Scholar] [CrossRef]
20. Sifrim, A., Hitz, M. P., Wilsdon, A., Breckpot, J., Turki, S. H. et al. (2016). Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nature Genetics, 48(9), 1060–1065. DOI 10.1038/ng.3627. [Google Scholar] [CrossRef]
21. Mone, F., Eberhardt, R. Y., Morris, R. K., Hurles, M. E., McMullan, D. J. et al. (2021). Congenital heart disease and the diagnostic yield with exome sequencing (CODE) study: Prospective cohort study and systematic review. Ultrasound in Obstetrics & Gynecology, 57(1), 43–51. [Google Scholar]
22. Qiao, F., Hu, P., Xu, Z. (2019). Application of next-generation sequencing for the diagnosis of fetuses with congenital heart defects. Current Opinion in Obstetrics and Gynecology, 31(2), 132–138. [Google Scholar]
23. Clark, M. M., Stark, Z., Farnaes, L., Tan, T. Y., White, S. M. et al. (2018). Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genomic Medicine, 3, 16. [Google Scholar]
24. Turan, S., Asoglu, M. R., Gabbay-Benziv, R., Doyle, L., Harman, C. et al. (2018). Yield rate of chromosomal microarray analysis in fetuses with congenital heart defects. European Journal of Obstetrics, Gynecology, and Reproductive Biology, 221, 172–176. [Google Scholar]
25. Suzuki, H., Yamada, M., Uehara, T., Takenouchi, T., Kosaki, K. (2020). Parallel detection of single nucleotide variants and copy number variants with exome analysis: Validation in a cohort of 700 undiagnosed patients. American Journal of Medical Genetics Part A, 182(11), 2529–2532. [Google Scholar]
26. Hu, X., Guo, R., Guo, J., Qi, Z., Li, W. et al. (2020). Parallel tests of whole exome sequencing and copy number variant sequencing increase the diagnosis yields of rare pediatric disorders. Frontiers in Genetics, 11, 473. [Google Scholar]
27. de Backer, J., Bondue, A., Budts, W., Evangelista, A., Gallego, P. et al. (2020). Genetic counselling and testing in adults with congenital heart disease: A consensus document of the ESC working group of grown-up congenital heart disease, the ESC working group on aorta and peripheral vascular disease and the european society of human genetics. European Journal of Preventive Cardiology, 27(13), 1423–1435. [Google Scholar]
28. Fahed, A. C., Roberts, A. E., Mital, S., Lakdawala, N. K. (2014). Heart failure in congenital heart disease: A confluence of acquired and congenital. Heart Failure Clinics, 10(1), 219–227. [Google Scholar]
29. Theis, J. L., Zimmermann, M. T., Evans, J. M., Eckloff, B. W., Wieben, E. D. et al. (2015). Recessive MYH6 mutations in hypoplastic left heart with reduced ejection fraction. Circulation: Cardiovascular Genetics, 8(4), 564–571. [Google Scholar]
30. Kennedy, M. P., Omran, H., Leigh, M. W., Dell, S., Morgan, L. et al. (2007). Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation, 115(22), 2814–2821. [Google Scholar]
31. Marino, B. S., Lipkin, P. H., Newburger, J. W., Peacock, G., Gerdes, M. et al. (2012). Neurodevelopmental outcomes in children with congenital heart disease: Evaluation and management: A scientific statement from the American heart association. Circulation, 126(9), 1143–1172. DOI 10.1161/CIR.0b013e318265ee8a. [Google Scholar] [CrossRef]
32. Diamond, A., Barnett, W. S., Thomas, J., Munro, S. (2007). Preschool program improves cognitive control. Science, 318(5855), 1387–1388. DOI 10.1126/science.1151148. [Google Scholar] [CrossRef]
33. Zhu, H., Kartiko, S., Finnell, R. H. (2009). Importance of gene-environment interactions in the etiology of selected birth defects. Clinical Genetics, 75(5), 409–423. DOI 10.1111/j.1399-0004.2009.01174.x. [Google Scholar] [CrossRef]
34. Goenezen, S., Rennie, M. Y., Rugonyi, S. (2012). Biomechanics of early cardiac development. Biomechanics and Modeling in Mechanobiology, 11(8), 1187–1204. DOI 10.1007/s10237-012-0414-7. [Google Scholar] [CrossRef]
35. Fahed, A. C., Gelb, B. D., Seidman, J. G., Seidman, C. E. (2013). Genetics of congenital heart disease: The glass half empty. Circulation Research, 112(4), 707–720. DOI 10.1161/CIRCRESAHA.112.300853. [Google Scholar] [CrossRef]
36. Camp, J. G., Platt, R., Treutlein, B. (2019). Mapping human cell phenotypes to genotypes with single-cell genomics. Science, 365(6460), 1401–1405. DOI 10.1126/science.aax6648. [Google Scholar] [CrossRef]
37. Cooper, D. N., Krawczak, M., Polychronakos, C., Tyler-Smith, C., Kehrer-Sawatzki, H. (2013). Where genotype is not predictive of phenotype: Towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Human Genetics, 132(10), 1077–1130. DOI 10.1007/s00439-013-1331-2. [Google Scholar] [CrossRef]
38. Shikany, A. R., Landis, B. J., Parrott, A., Miller, E. M., Coyan, A. et al. (2020). A comprehensive clinical genetics approach to critical congenital heart disease in infancy. The Journal of Pediatrics, 227, 231–238.e14. DOI 10.1016/j.jpeds.2020.07.065. [Google Scholar] [CrossRef]
39. Bales, N. D., Johnson, N. M., Judge, D. P., Murphy, A. M. (2016). Comprehensive versus targeted genetic testing in children with hypertrophic cardiomyopathy. Pediatric Cardiology, 37(5), 845–851. DOI 10.1007/s00246-016-1358-y. [Google Scholar] [CrossRef]
Appendix A: List of ontology-based classification of cardiac and extracardiac phenotypes
Appendix B: Cardiac and extracardiac phenotypes of a patient without a diagnostic variant

Appendix C: Genetic diagnostic rate according to an individual cardiac or extracardiac phenotype
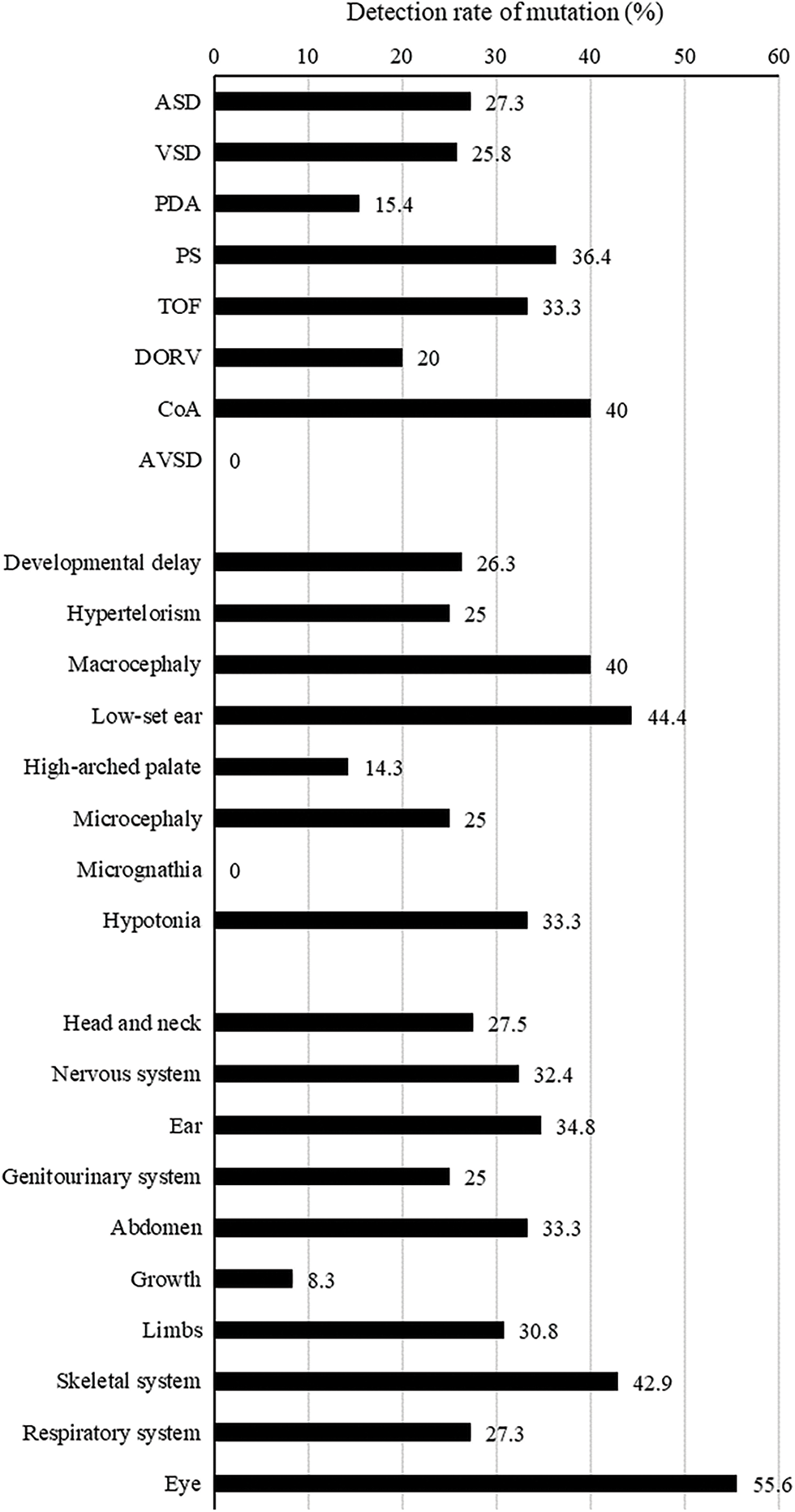
Abbreviations: ASD, atrial septal defect; VSD, ventricular septal defect; PDA, patent ductus arteriosus; PS, pulmonary stenosis; TOF, tetralogy of Fallot; DORV, double outlet right ventricle; CoA, coarctation of aorta; AVSD, atrioventricular septal defect.
Appendix D: The detection rate of a genetic variant according to the combination of extracardiac phenotypes among patients with three or more extracardiac phenotypes
Cite This Article
 Copyright © 2022 The Author(s). Published by Tech Science Press.
Copyright © 2022 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools