
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
CASE REPORT

Compound Heterozygous PLD1 Variants in Right-Sided Heart Malformations
1 Ted Rogers Centre for Heart Research, Cardiac Genome Clinic, The Hospital for Sick Children, University of Toronto, Toronto, Canada
2 Department of Paediatrics, Division of Clinical and Metabolic Genetics, The Hospital for Sick Children, University of Toronto, Toronto, Canada
3 MIT Sloan School of Management, Massachusetts Institute of Technology, Cambridge, USA
4 The Prenatal Diagnosis and Medical Genetics Program, Mount Sinai Hospital, University of Toronto, Toronto, Canada
5 Department of Paediatrics, Division of Cardiology, The Hospital for Sick Children, University of Toronto, Toronto, Canada
6 Department of Medical Biophysics, University of Toronto, Toronto, Canada
7 Department of Paediatrics, Translations Medicine Department, SickKids Research Institute, University of Toronto, Toronto, Canada
8 Department of Paediatric Laboratory Medicine, Genome Diagnostics, The Hospital for Sick Children, Toronto, Canada
* Corresponding Author: Rebekah Jobling. Email:
Congenital Heart Disease 2023, 18(2), 213-218. https://doi.org/10.32604/chd.2023.023042
Received 06 August 2022; Accepted 27 December 2022; Issue published 15 March 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
We report a three-year-old male child who presented with congenital valvular defects, right ventricular malformation, and initial developmental delay. Genome sequencing showed rare deleterious biallelic missense variants in PLD1. In his parents’ second pregnancy, echocardiogram at 13 weeks gestation revealed right-sided cardiac malformations resembling the clinical presentation of the family’s first child. Targeted DNA analysis showed that the fetus carried the same biallelic PLD1 variants as their older sibling. This case helps to further delineate the clinical spectrum of PLD1-related defects and highlights the value of both genome sequencing in congenital heart disease and early fetal echocardiography to establish phenotype.Keywords
Nomenclature
| ACMG | American College of Medical Genetics and Genomics |
| ASD | Atrial septal defect |
| CADD | Combined Annotation Dependent Depletion |
| CHD | Congenital heart disease |
| CLIA | Clinical Laboratory Improvement Amendments |
| CVS | Chorionic villi sample |
| GnomAD | The Genome Aggregation Database |
| GS | Genome sequencing |
| NT | Nuchal translucency |
| PDA | Patent ductus arteriosus |
| PolyPhen-2 | Polymorphism Phenotyping v2 |
| PLD1 | Phosphatidylcholine-specific Phospholipase D1 |
| RV | Right ventricle |
| SIFT | Sorting Intolerant from Tolerant |
| TOP | Termination of pregnancy |
| VSD | Ventricular septal defect |
Congenital heart diseases account for almost one-third of all major congenital anomalies, with a prevalence of 7 per 1000 births [1]. While advances in sequencing efforts have enabled the identification of novel causative CHD-gene associations, the molecular etiology for most CHD remains largely unknown, with the majority being multifactorial. Biallelic variants in PLD1 have recently been identified in patients with non-syndromic congenital valve defects and RV malformations [2,3]. To our knowledge, only a few studies on CHD have described the use of genomic investigation to identify a single causative gene in a proband and early fetal echocardiography to facilitate a diagnosis in their fetal sibling. Here we report a family with a three-year-old male child and a fetal sibling, diagnosed at 13 weeks gestation, with compound heterozygous variants in PLD1.
The proband was the product of the first pregnancy of a healthy non-consanguineous couple of European descent. A fetal echocardiogram at 20 weeks 6 days gestation done on the mother showed severe pulmonary valve stenosis, hypoplastic tricuspid valve, large muscular VSD, bilateral hypoplastic main pulmonary arteries and critically hypoplastic and dysmorphic RV (multilobular parts) (26-week fetal echocardiogram, Fig. 1A, and supplementary video S1).
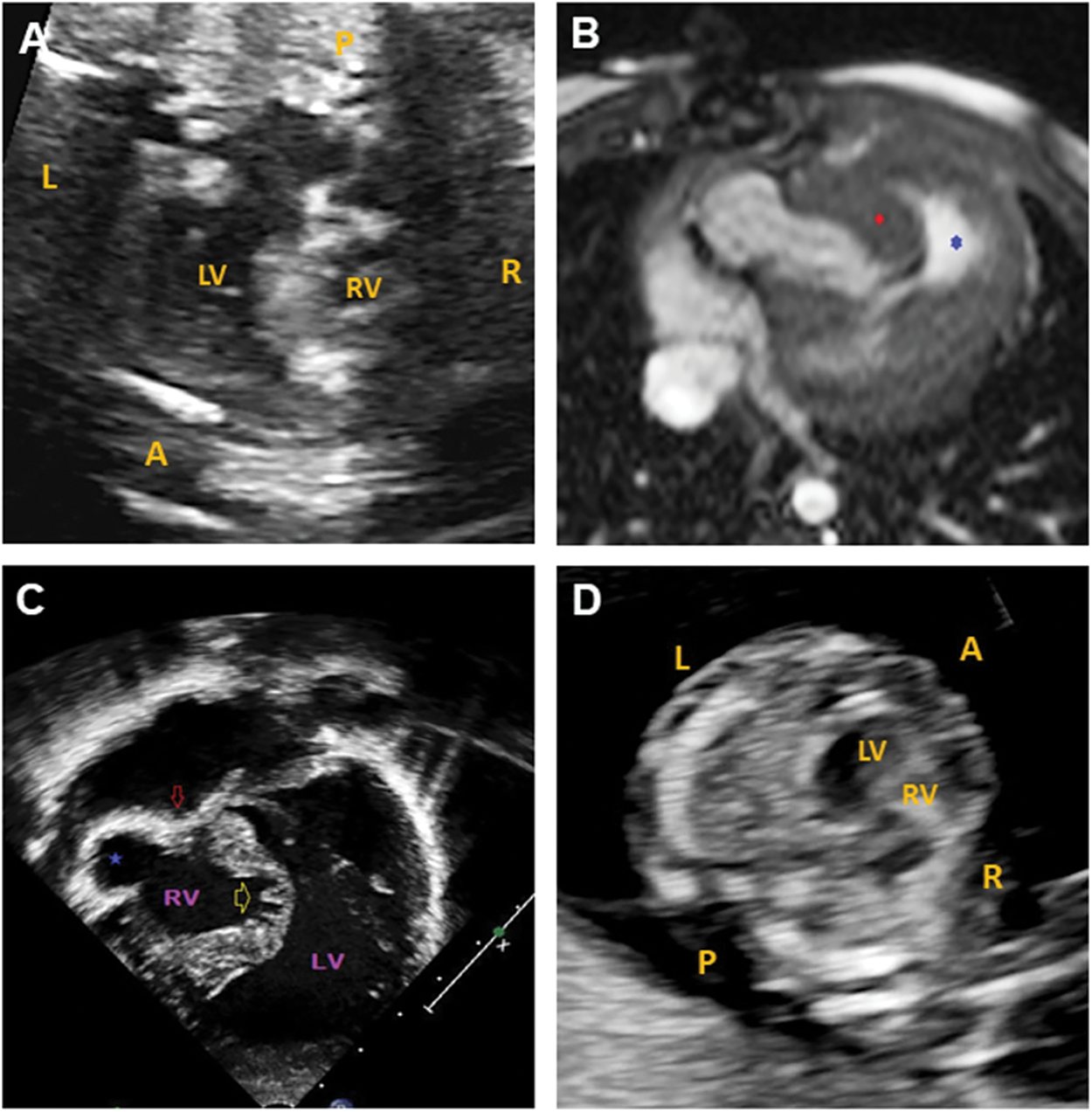
Figure 1: Cardiac imaging for the paediatric case and fetal sibling. (A) 26-week fetal echocardiogram for the paediatric patient showing a hypoplastic RV. (B) Cardiac MRI for the paediatric patient at birth. Axial balanced steady state gradient echo (TRUFISP). Severe tricuspid valve stenosis with RV anterior free wall ballooning (blue star) and hypertrophied moderator band (red diamond). (C) Echocardiography for the paediatric patient at birth. Apical view with severe tricuspid stenosis (red arrow), RV anterior free wall ballooning (blue star), and atrophic and trabecular inter-ventricular septum. (D) 13-week fetal echocardiogram showing a hypoplastic RV in the fetal sibling. L: left, R: right, A: anterior, P: posterior, LV: left ventricle, RV: right ventricle
Delivery was at term and uncomplicated. The patient’s birthweight was 2.85 kg (3–10th centile). Postnatal echocardiography and MRI (Figs. 1B and 1C) confirmed the antenatal diagnosis with the addition of coronary sinusoids (RV) and an aberrant right subclavian artery with left aortic arch.
A univentricular surgical strategy was indicated. At four days of life, the patient underwent a central Blalock-Taussing shunt and main pulmonary artery ligation. At five months, the patient underwent his second stage of repair which included a bidirectional cavopulmonary connection, and takedown of the central shunt. The patient is currently being followed in cardiology and has recently undergone Fontan surgery.
At last assessment at 18 months of age, neurodevelopmental evaluation using the Bayley Scales of Infant and Toddler Development, third edition [4], showed that his receptive language was at the 5th percentile, expressive language was at the 2nd percentile, and his cognitive skills were at the 16th percentile.
Given his complex CHD, the proband was referred for trio GS at the Cardiac Genome Clinic. The analytic workflow was previously described [5]. GS of the proband and his parents showed compound heterozygous missense variants in PLD1 (NM_002662.5): paternally inherited c.2681A>C, p.(Tyr894Ser), and maternally inherited c.1982A>T, p.(Asp661Val). The variants are present in gnomAD [6], with a European minor allele frequency of 0.017% and 0.01%, respectively, and neither variant is seen in the homozygous state. In-silico prediction tools, including CADD [7], PolyPhen-2 [8], SIFT [9], and ClinPred [10], support a deleterious effect for both variants. The CADD scores for p.(Tyr894Ser) and p.(Asp661Val) are 25.9 and 32, respectively. Both variants lie in the catalytic domain of the protein [3].
The paternally inherited missense variant, p.(Tyr894Ser), was previously described in two, unrelated deceased patients from Lahrouchi et al. [3] (Table 1), both of whom harboured the variant in trans with a second variant that severely reduced the catalytic activity of PLD1 on functional assessment: c.2098C>T, p.(Arg700Cys) and c.1062G>A, p.(Trp354Ter), respectively.

In the proband, clinical validation of the variants was performed in a CLIA-approved laboratory using targeted variant testing. GS did not reveal any additional genomic findings thought to contribute to the patient’s phenotype. The family was counselled regarding the 25% recurrence risk for future pregnancies.
The couple’s second pregnancy was naturally conceived and NT ultrasound at 12 weeks gestation was 3.1 mm. Enhanced first trimester screening done subsequently showed an increased risk for Trisomy 21 (1 in 12 at term). Given the history of a child with complex CHD, the family was referred for fetal ultrasound and echocardiography at 13 weeks gestation. The fetal imaging showed hypoplastic, non-apex forming RV and hypoplastic tricuspid valve with minimal, monophasic inflow (Fig. 1D and supplementary videos S2–S4). No VSD was present. Normal left-sided structures were observed.
The right-sided valvular defects with hypoplastic RV had marked overlap with the cardiac features of the family’s three-year-old son. Following counselling, the couple chose to terminate the pregnancy. Targeted DNA analysis for the familial PLD1 gene showed that the fetus had the same compound heterozygous variants as their son. Microarray analysis done on CVS was normal.
Using the ACMG criteria [11], the added segregation data from this family enabled a classification of Likely Pathogenic for the previously reported p.(Tyr894Ser) variant (PM2, PM3, PP3, PP1_Moderate [12]), and a classification of Variant of Uncertain Significance for the p.(Asp661Val) variant (PM2, PP1, PP3).
Phosphatidylcholine-specific Phospholipase D1 (PLD1) catalyzes the hydrolysis of phosphatidylcholine to produce phosphatidic acid and choline. PLD1 has been implicated in numerous cellular pathways, including membrane trafficking, signal transduction, and cytoskeleton organization [13]. Recent research revealed that loss of PLD1 function in a recessive manner results in CHD [2,3]. Cardiac defects caused by loss of PLD1 generally led to abnormal development of the cardiac valves on the right chamber.
The advancement of next-generation sequencing technologies and computational strategies is accelerating the rate of novel gene discovery for CHD, and newly associated rare disorders associated with CHD are continuously identified [14–16]. Given this rapid gene discovery and the rarity of these disorders, panels and targeted testing are impractical, making GS essential for identifying cases with high recurrence risk. In the presented case, GS was critical in providing the family with a clear diagnosis for their three-year-old child and allowed us to appropriately counsel the family on their 25% recurrence risk in future pregnancies.
Evaluation of the heart in the fetal sibling enabled early detection of the cardiac abnormality and genetic diagnosis of the PLD1-related cardiac lesions. The specific clinical manifestation of PLD1-related CHD was identified on echocardiography early in the pregnancy and was beneficial for the family in decision making.
The family’s added segregation data allowed an upgraded classification of Likely Pathogenic for the previously reported p.(Tyr894Ser) variant. Patients reported to have the p.(Tyr894Ser) variant present with a striking phenotypic overlap, consisting of tricuspid valve and pulmonary valve anomalies, and hypoplastic RV [3]. The three-year-old proband is the only reported survivor with the p.(Tyr894Ser) variant.
The proband also presented with initial developmental delay. While developmental concerns are not explicitly described for other PLD1 patients, the sample size of patients who survived past infancy is small. Additional cases are needed to further delineate the clinical spectrum of PLD1-related defects and the possible association of PLD1 with developmental delay.
This family shows the importance of GS in identifying the causative gene in CHD patients and the importance of fetal echocardiography to establish the phenotype. The value of GS for this family extends to patient populations with rare disorders, such as this one, with high recurrence risks. In the context of an accurate recurrence risk, molecular diagnosis through GS can enable preimplantation and prenatal diagnosis, and early fetal echocardiography, at 12–13 weeks gestation, can accurately identify the cardiac abnormalities.
Acknowledgement: We thank the family for their participation in this study.
Funding Statement: This work was funded by the Ted Rogers Centre for Heart Research.
Author Contributions: Conceptualization, C.S., and R.K.J.; Data Acquisition, Analysis, and Interpretation, C.S., J.O., R.M., K.K., Q.D., R.S., D.C., V.T., O.V., and R.K.J.; Writing–original draft, C.S.; Writing–review & editing, K.K., R.M., Q.D., R.S., D.C., V.T., O.V., and R.K.J.; Supervision, R.K.J.
Ethics Approval: The study was approved by the Hospital for Sick Children (REB 1000053844).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Wu, W., He, J., Shao, X. (2020). Incidence and mortality trend of congenital heart disease at the global, regional, and national level, 1990–2017. Medicine, 99(23), e20593. https://doi.org/10.1097/MD.0000000000020593 [Google Scholar] [PubMed] [CrossRef]
2. Ta-Shma, A., Zhang, K., Salimova, E., Zernecke, A., Sieiro-Mosti, D. et al. (2017). Congenital valvular defects associated with deleterious mutations in the PLD1 gene. Journal of Medical Genetics, 54(4), 278–286. https://doi.org/10.1136/jmedgenet-2016-104259 [Google Scholar] [PubMed] [CrossRef]
3. Lahrouchi, N., Postma, A. V., Salazar, C. M., de Laughter, D. M., Tjong, F. et al. (2021). Biallelic loss-of-function variants in PLD1 cause congenital right-sided cardiac valve defects and neonatal cardiomyopathy. Journal of Clinical Investigation, 131(5). https://doi.org/10.1172/JCI142148 [Google Scholar] [PubMed] [CrossRef]
4. Bayley, N. (2006). Bayley scales of infant and toddler development. San Antonio, TX: The Psychological Corporation. [Google Scholar]
5. Reuter, M. S., Chaturvedi, R. R., Liston, E., Manshaei, R., Aul, R. B. et al. (2020). The cardiac genome clinic: Implementing genome sequencing. Genetics in Medicine, 22(6), 1015–1024. https://doi.org/10.1038/s41436-020-0757-x [Google Scholar] [PubMed] [CrossRef]
6. Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J. et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. https://doi.org/10.1038/s41586-020-2308-7 [Google Scholar] [PubMed] [CrossRef]
7. Kircher, M., Witten, D. M., Jain, P., O’Roak, B. J., Cooper, G. M. et al. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46(3), 210–215. https://doi.org/10.1038/ng.2892 [Google Scholar] [PubMed] [CrossRef]
8. Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A. et al. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. https://doi.org/10.1038/nmeth0410-248 [Google Scholar] [PubMed] [CrossRef]
9. Ng, P. C., Henikoff, S. (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Research, 31(13), 3812–3814. https://doi.org/10.1093/nar/gkg509 [Google Scholar] [PubMed] [CrossRef]
10. Alirezaie, N., Kernohan, K. D., Hartley, T., Majewski, J., Hocking, T. D. (2018). ClinPred: Prediction tool to identify disease-relevant nonsynonymous single-nucleotide variants. American Journal of Human Genetics, 103(4), 474–483. https://doi.org/10.1016/j.ajhg.2018.08.005 [Google Scholar] [PubMed] [CrossRef]
11. Richards, S., Aziz, N., Bale, S., Bick, D., Das, S. et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genetics in Medicine, 17(5), 405–424. https://doi.org/10.1038/gim.2015.30 [Google Scholar] [PubMed] [CrossRef]
12. Oza, A. M., DiStefano, M. T., Hemphill, S. E., Cushman, B. J., Grant, A. R. et al. (2018). Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Human Mutation, 39(11), 1593–1613. https://doi.org/10.1002/humu.23630 [Google Scholar] [PubMed] [CrossRef]
13. Jenkins, G. M., Frohman, M. A. (2005). Phospholipase D: A lipid centric review. Cell Molecular and Life Sciences, 62(19–20), 2305–2316. https://doi.org/10.1007/s00018-005-5195-z [Google Scholar] [PubMed] [CrossRef]
14. Homsy, J., Zaidi, S., Shen, Y., Ware, J. S., Samocha, K. E. et al. (2015). De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science, 350(6265), 1262–1266. https://doi.org/10.1126/science.aac9396 [Google Scholar] [PubMed] [CrossRef]
15. Jin, S. C., Homsy, J., Zaidi, S., Lu, Q., Morton, S. et al. (2017). Contribution of rare inherited and de novo variants in 2.871 congenital heart disease probands. Nature Genetics, 49(11), 1593–1601. https://doi.org/10.1038/ng.3970 [Google Scholar] [PubMed] [CrossRef]
16. Bayrak, C. S., Zhang, P., Tristani-Firouzi, M., Gelb, B. D., Itan, Y. (2020). De novo variants in exomes of congenital heart disease patients identify risk genes and pathways. Genome Medicine, 12(1), 9. https://doi.org/10.1186/s13073-019-0709-8 [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools