
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Variants and Molecular Mechanism of NOTCH1 in Congenital Heart Disease
1 College of Life Sciences, Hunan Normal University, Changsha, 410081, China
2 Guangdong Cardiovascular Institute, Guangdong Provincial People’s Hospital (Guangdong Academy of Medical Sciences), Southern Medical University, Guangzhou, 510080, China
3 Guangdong Provincial Key Laboratory of South China Structural Heart Disease, Guangdong Cardiovascular Institute, Guangdong Provincial People’s Hospital (Guangdong Academy of Medical Sciences), Southern Medical University, Guangzhou, 510080, China
4 Department of Ultrasonography, Guangdong Provincial Hospital of Chinese Medicine, Guangzhou, 510120, China
5 Department of Ultrasonography, The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, 510120, China
* Corresponding Author: Yan Shi. Email:
Congenital Heart Disease 2025, 20(2), 245-263. https://doi.org/10.32604/chd.2025.064366
Received 13 February 2025; Accepted 23 April 2025; Issue published 30 April 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Congenital heart disease (CHD) is the most common birth defect, with 34% of cases attributed to genetic variants. NOTCH1, a multi-domain transmembrane protein, regulates heart development by controlling the differentiation and migration of myocardial mesoderm cells, and different variants are present in different types of CHD. In this review, we aim to provide a detailed description of NOTCH1 structural domains and their functions, highlighting NOTCH1 variants in CHD and the molecular mechanisms through which they contribute to CHD occurrence. NOTCH1 has two main domains, the NOTCH extracellular domain (NECD) and the NOTCH intracellular domain (NICD). NECD facilitates ligand binding and NICD formation, while the NICD functions as a transcription factor, forming complexes with co-factors in the nucleus to initiate gene transcription. Among the NOTCH1 variants associated with CHD occurrence, most are loss-of-function variants. Moreover, most of the variants are located in the EGF-like domain. The molecular mechanism behind the NOTCH1 variant-associated CHD occurrence appears to be either due to a loss-of-function or missense variant. In the loss-of-function mutations, NOTCH1 haploinsufficiency is noted and directly reduces the NICD production, causing CHD occurrence. In the less common case of missense variant, only a mild NOTCH1 malfunction is observed, but insufficient to directly lead to CHD occurrence. However, when a missense variant is combined with a risk factor, such as exposure to an environmental toxin, the cumulative effect can lead to CHD. Understanding the genetic and molecular mechanisms linking NOTCH1 variants to CHD is crucial for improving clinical management and patient quality of life.Keywords
Supplementary Material
Supplementary Material FileThe incidence of congenital heart disease (CHD) has increased in recent decades, making it the most common birth defect. While the exact cause of CHD is often unknown, risk factors include contracting rubella, diabetes, alcohol consumption during pregnancy, some medications, smoking and genetics [1,2]. In recent years, early prenatal detection and advances in surgical techniques have improved survival rates among CHD individuals [1,3]. With the widespread application of whole-exome sequencing (WES) technology, numerous CHD-related pathogenic mutations have been identified, including variants in genes such as NKX2.5, GATA4, NOTCH1, TBX1, and TBX20 [1,4]. Notably, of these genes, NOTCH1 mutations account for 6% of CHD cases, highlighting its significance for further exploration.
The NOTCH signaling pathway involves multiple receptor-ligand interactions. The receptors consist of NOTCH1, NOTCH2, NOTCH3, and NOTCH4, while the ligands are divided into two families: the Jagged family (JAG1 and JAG2) and the Delta-like family (Delta1, Delta3, and Delta4). The NOTCH genes are transcribed in the nucleus and translated into precursor NOTCH proteins, such as pre-NOTCH1 (Fig. 1). These precursor proteins are transported to the Golgi apparatus, where they undergo S1 cleavage by a furin-like convertase to form a mature heterodimer transmembrane receptor that is transported to the cell membrane [5]. Upon ligand interaction, the receptor undergoes a conformational change, exposing its S2 site, which is then cleaved by metalloproteases, such as ADAM10 [6,7]. This cleavage releases the NOTCH extracellular domain (NECD) and leaves behind the remaining receptor, NOTCH extracellular truncation (NEXT) (Fig. 1) [8].
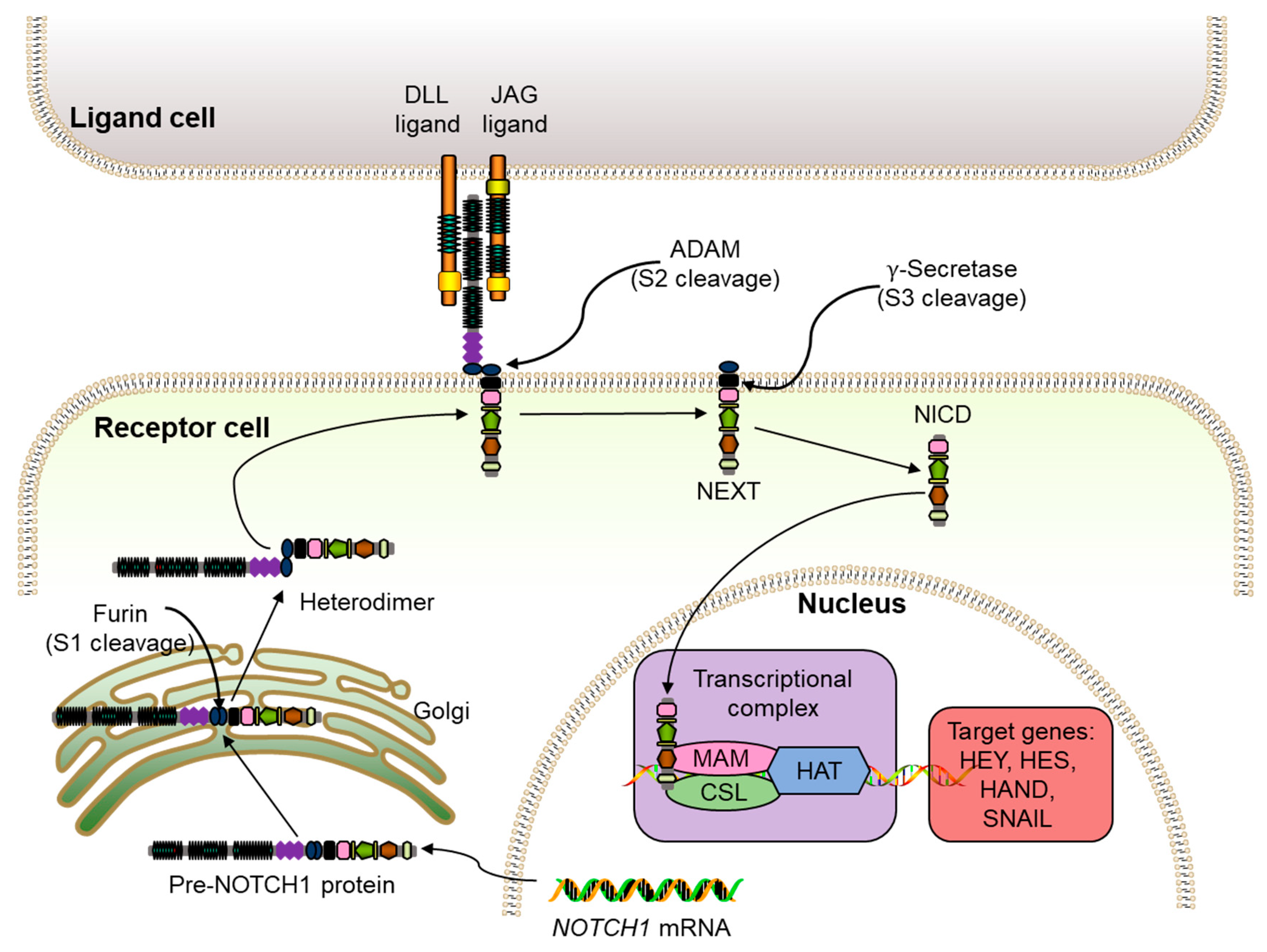
Figure 1: Schematic map of the NOTCH signaling pathway. Pre-NOTCH1 protein, the precursor NOTCH protein; DLL, delta-like ligand; ADAM, ADAM metalloproteases group; NICD, NOTCH intracellular domain; NEXT, NOTCH extracellular truncation; MAM, transcriptional coactivator mastermind; CSL, a nuclear effector (CBF1/RBP-J, Su(H), Lag1); HAT, histone acetyltransferase.
Following S2 cleavage, a γ-secretase cleavage site is exposed [8]. This third cleavage generates the NOTCH1 intracellular domain (NICD) [9], which then translocates to the nucleus to bind nuclear effectors, such as CSL (CBF1/RBP-J, Su(H), Lag1) to transform it from a repressor to a transcriptional activator. This NICD-CSL binding recruits coactivators like Mastermind (MAM)/Lag-3 to form the NICD-CSL-MAM complex, which recruits the ARC-L/MED complexes, a positive transcription regulator [10], histone ubiquitin ligases (Bre1) [11], and histone acetyltransferases (HAT) [12] to promote gene expression (Fig. 1). Overall, the NOTCH1 signaling cascade governs cellular processes such as proliferation, apoptosis, and differentiation, with each linked to specific structural domains of NOTCH1. Therefore, this review will focus on the domains of NOTCH1 and elaborate on the functions of each domain.
During cardiac embryogenesis, NOTCH1 is expressed in the outflow tract (OFT), atrioventricular canal (AVC), trabecular ventricular wall, and epicardium, but is absent in the dense myocardial layers of the atria and ventricles [13]. The NOTCH1 signaling pathway is critical for cardiac development, influencing aortic and pulmonary valve formation and large blood vessel maintenance [13,14,15]. In adults, NOTCH1 plays a role in cardiac repair by limiting myocardial hypertrophy, promoting precursor cell proliferation, improving cardiomyocyte survival, and reducing fibrosis [16]. Furthermore, following myocardial infraction, enhanced NOTCH1 activity has been to increase survival rates, improve cardiac function, and mitigate fibrosis via anti-apoptotic and pro-angiogenic mechanisms [17].
During cardiac development, NOTCH1 plays a role in promoting epithelial-to-mesenchymal transition (EMT), with the targeted deletion of Notch1 or its nuclear partner (RBP-J/CBF1/Su(H)) in mice resulting in EMT impairment, leading to endocardial collapse and the absence of cushion cells [18]. These findings demonstrate the critical role of NOTCH1 in cardiac development and its association with various clinical CHD phenotypes [19,20]. NOTCH1 is highly intolerant to variation, as reflected by its low residual variation intolerance score (0.33%) [21].
NOTCH1 variants are frequently associated with CHD. In 2005, a NOTCH1 variant was linked with an aortic valve malformation for the first time [22]. Furthermore, the NOTCH1 locus is the most frequent site of genetic variants that cause non-syndromic tetralogy of Fallot (TOF), accounting for 4.5% of TOF cases (37/829) [23]. In familial valve diseases and left ventricular outflow tract obstruction (LVOTO), NOTCH1 mutations represent 5.9% of cases, with an additional 16% involving variants of uncertain significance [24,25]. Among CHD patients, NOTCH1 variants are considered the most significant of de novo mutations identified [26]. In addition to CHD, NOTCH1 mutations are implicated in various cancers, including T-cell acute lymphoblastic leukemia (T-ALL), where 62–66% of adults exhibit such mutations [27,28]. However, this review will focus on NOTCH1 variants in CHD.
Herein, a comprehensive view of the mechanisms by which NOTCH1 variants mediate CHD development is explored to provide insight into the role of NOTCH1 in heart development and disease.
2 NOTCH1 Structure and Function
The NOTCH receptor is a type I single channel transmembrane protein consisting of an intracellular ligand-binding domain, a transmembrane domain, and an intracellular domain [13]. In this section, the structures and functions of NECD and NEXT, the membrane bound fragment that is generated following S2 cleavage, are examined.
2.1 NECD Structure and Function
The NECD portion of NOTCH1 receptor is 209 kDa and contains 36 epidermal growth factor-like (EGF) repeats that serve as the ligand-binding domain and a negative regulatory region (NRR) domain (Fig. 2A) [29].
EGF repeats. Each region of the repeated EGF is comprised of a distinctive six cysteine motif consisting of 30–40 amino acid residues from three internal disulfide bridges [30]. These domains interacted with a specific ligand and a conformational change occurs leading to dimerization and subsequent proteolytic cleavages, initiating signal transduction. When binding the delta-like 4 (DLL4) ligand, the EGF11 and EGF12 regions with NECD interacts with the DSL (Delta/Serrate/Lag-2) and MNNL (N-terminus of Notch ligands) ligand domains, respectively [31]. However, when binding JAG, the EGF8 and EGF 11 regions of the NECD interact with the EGF 3 and DSL domains of the ligand, respectively [32].
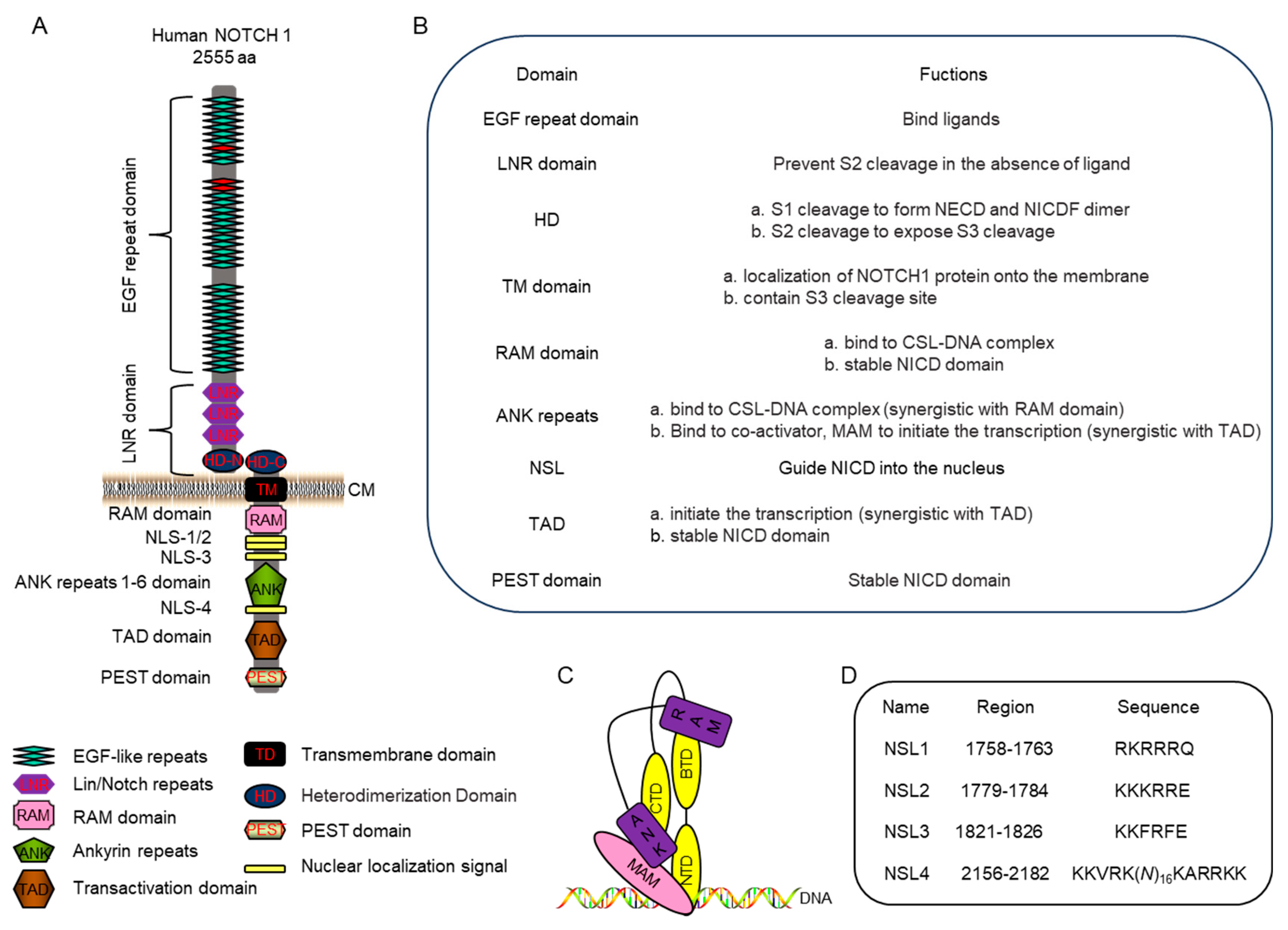
Figure 2: NOTCH1 structure and function. (A): Detailed structure of NOTCH1. The red EGF domain, including EGF8, EGF11 and 12, interact with ligands. CM, cell membrane. (B): The function of each structure listed in 2A. (C): The CSL-NICD-MAM complex with the RAM domain and ANK repeats of NICD (purple) interacting with BTD, NTD and CTD of CSL (yellow) to recruit MAM (pink). MAM, Mastermind; NTD, N-terminal domain; BTD, beta-trefoil domain; and CTD, C-terminal domain. (D): Putative NLS in NOTCH1 protein.
Another important aspect that influences ligand binding is post-translational modifications within the EGF-like domain, specifically O-glycosylation that adds an O-fucose or O-glucose to a serine or threonine residues within a domain. In O-fucosylation, EGF 2/3/5/6/8/9/12/16/18/20/21/23/24/26/27/30/31/32/35/36 have fucose sites, with the specific location of O-fucosylation having different effects, such as a fucose addition at EGF9 affecting receptor trafficking [33,34]. In a patient with a global developmental delay and contained heart development disease, including severe coarctation of the aorta and ventricular septal defect (VSD), a large decrease at EGF9 and the complete absence at EGF12 were observed [35]. When NOTCH1 binds its ligands, its O-fucosylated EGF8 and 12 regions directly interact with the EGF3 and C2 domains of JAG1, while EGF11 and 12 interact with the DSL domain on DLL4 [31,32,36]. Moreover, the addition of O-fucose to residue Thr466 in EGF12 and residue Ser435 in EGF11 has been shown to stabilize the interaction between NOTCH1 and DLL4 [31]. NOTCH signal activation also requires calcium-dependent phospholipid binding in the case of JAG, but in the case of DLL, calcium ions did not directly contribute to activation, but did provide DLL an increased rigidity that potentiated the interaction [31].
Unlike an O-fucose modification that affects ligand binding, an O-glucose modification enhances NOTCH signaling by increasing receptor susceptibility to proteolytic processing [34]. The consensus sequence for O-glucosylation is C-X-S-X-P/A-C, with serine being the only hydroxyamino acid that is modified with O-glucose on an EGF region. These modifications vary across cell types and affect Notch signaling activation differently depending on the modification site [34]. Overall, EGF1–36 plays a role in ligand binding and signaling initiation (Fig. 2B).
NRR domain. Close to the cellular membrane is the NRR, which is comprised of three Lin12/Notch repeats (LNRs) and a heterodimerization domain (HD) that acts as a regulatory switch for Notch signaling (Fig. 2). Each LNR repeat contains 40 residues, with three conserved aspartate (D) or asparagine (N) residues, and three disulfide bonds that pair in a characteristic pattern, cys (I)—cys (V)—cys (II)– cys (IV)—cys (III)–cys (VI) [37]. Without the receptor-ligand interaction, the LNRs maintain the receptor in a resting conformation to prevent S2 cleavage [8], but activate NOTCH in the presence of ligand [29,38]. While adjacent LNR domains can also affect the structural domains, their main function is to modulate cleavage in the presence or absence of ligands (Fig. 2B) [39].
The second portion of the NRR, which is the HD region, contains the S1 cleavage site (cleavage site: 1659-1670-GGRRRR′ELDPMD) that is targeted by a furin-like protease during protein maturation [5]. After cleavage, its N-terminal part (HD-N) terminates the NECD region, while the C-terminal (HD-C) begins the NEXT region and both form a heterodimerization (Fig. 2) [5]. If furin cleavage is disrupted, ligand-dependent signaling through the well-characterized mediator of Notch signal transduction will be abolished [40]. Moreover, the S2 cleavage site (1716-1724-YKIEAVQSE) is located in the HD-C, with cleavage at this site required to expose the S3 site (1705-1720: ALASLG′SLNIPYKIEA) [8]. Thus, the HD plays an important role in heterodimerization formation and modulates S1 and S2 cleavage (Fig. 2B).
In summary, the main function of the sequence of NECD is to bind a specific ligand and ensure that the subsequent cleavage events in the extracellular segment are performed to generate the NEXT intermediate.
2.2 NEXT Structure and Function
The NEXT intermediate of NOTCH1 contains HD-C, which is located at the N-terminal, a transmembrane domain (TD), which anchors the protein to the membrane, and a segment that will become the NICD (Fig. 2). The NICD contains an RBP-J association module (RAM) domain, ankyrin (ANK) repeats, nuclear localization signal (NLS), transcriptional activation domain (TAD) and PEST, a sequence rich in proline (P), glutamic acid (E), serine (S) and threonine (T) [6,8]. The regions on NICD have different functions, with the ANK repeat and TAD domain shown to be required for T-cell leukemogenesis, while the RAM and PEST domain are nonessential [41]. NICD overexpression can lead to complex defects in heart morphology, including the abnormal maturation of myocardial cells due to changes in gene expression [42]. In some cases, NICD can inhibit myoblast differentiation by binding to myocyte enhancer factor 2C (MEF2C), a regulatory factor that is associated with development in various tissues, and inhibit its transcription [43]. Thus, to more fully characterize the function of NICD, we will discuss the function of each domain in NICD.
RAM domain. RAM contains an RBP-J associated sequence and a conserved Trp-Xaa-Pro (WXP) tripeptide that has a high affinity for the β-trefoil domain of CSL [44]. Upon NOTCH signal transduction, NICD is released and enters the nucleus, where it binds CSL, a transcriptional repressor (Fig. 2C) [44]. While NICD, specifically the RAM domain, has been considered crucial in transcriptional activation, one study has showed that deleting the RAM domain only partially reduce NOTCH signaling activity as the ANK region was more pivotal in transcription [45]. Additionally, RAM is essential for NICD stability with ubiquitination of RAM by NEDD4-binding protein 1 (N4BP1) mediating NICD degradation [46]. Therefore, RAM has two functions, one is to interact with RBP-J, and the other is to regulate NICD stability (Fig. 2B).
ANK repeats. ANK repeats, consisting of two or more tandem repeats, are one of the most frequently observed amino acid motifs in protein databases, and form curved solenoid structures that are associated with protein-lipid, protein-sugar, and protein-protein interactions [47]. Each ANK repeat has a highly conserve TPLH (Thr-Pro-Leu-His) tetrapeptide that plays an important role in stability [48]. In NOTCH1, the ANK region contains six repeats. The ANK repeats bind the N-terminal and C-terminal domains of CSL to create a high-affinity binding site for MAM, to form a ternary complex (NICD-CSL-MAM complex) that is necessary for activate transcription [44]. The NICD-CSL-MAM complex formation induces a substantial conformational change within CSL that converts it from a repressor to an activator [49]. A deletion of any of the ANK repeats would completely reduce NOTCH signaling activity [45]. In Caenorhabditis elegans, the ANK repeats were shown to not only be involved in RBP-J interactions, but to also act as an autonomous transactivation domain [50]. Moreover, while the RAM region is the primary determinant of NICD-CSL complex stability, the ANK region largely directs the binding of the co-activator MAM to CSL [51]. Therefore, ANK and RAM act synergistically to promote NOTCH1 transcriptional activity (Fig. 2B).
NSL domain. A NSL domain consists of a short peptide segment, with the sequence K-R/K-X-R/K (X is any amino acid), that guides NICD into the nucleus [52]. NOTCH1 sequencing analysis has identified four putative NLS segments within NICD, the first three NLSs are monopartite, with the first and second closely resembling the RAM domain. The fourth NLS is bipartite given its location between the ANK and TAD domains (Fig. 2A,D) [53,54]. When examining the role of each region, NLS1 and NLS2 were found to be non-essential for NICD nuclear import, while NLS3 and NSL4 were required, with NSL3 showing a clear independence from NLS4 [53]. Furthermore, the deletion of NLS3 and NLS4 has been shown to negatively affect nuclear localization, further confirming the importance of these regions [55]. Additionally, while the specific NLS amino acid sequence is important for nuclear localization, its neighboring amino acids also impact the nuclear localization. In a cancer mouse model, the phosphorylation of serine residue 2152 (2147-2153: K-A-R-K-P-S-T) within the NSL4 region by all three Pim family kinases was shown to be necessary for nuclear localization and signal transduction [56]. In addition to the NSL regions directing nuclear localization, one study examining mice identified S/T-P-S/T domains (2122-2124: T-P-T, 2126-2128: S-P-T) that can also direct nuclear localization of NICD [57]. Overall, the main function of NLS domains within NICD is to guide nuclear entry.
TAD. Located between the ANK repeats and the PEST domains is TAD, which stimulates activation of an associated gene at the promotor (Fig. 2A). After the NICD-CSL-MAM complex is formed, chromosome modifying proteins are recruited, including histone ubiquitin ligase and HAT, to open the chromosome and initiate activate transcription [12]. However, TAD alone has been shown to be insufficient to fully activate NOTCH1 transcriptional activity, and relies on the presence of the ANK repeats to enhance transcriptional activity [41]. Furthermore, TAD also promote the phosphorylation and proteolytic turnover of NICD, which is essential for Notch transcription [10]. At the 3′-terminal of TAD, there is a glutamine-rich OPA sequence that is conserved between drosophila and mammalian [41], but its role has not been researched. Therefore, TAD collaborates with ANK to activate NOTCH1 transcriptional activity and plays a role in maintaining NICD stability.
PEST domain. The PEST domain is a polypeptide sequences that aids in modulating NICD degradation via a ubiquitin-proteasome pathway [58], and modulates by post-transcriptional modification [59]. In one study, a point mutation in the conserved Ser residues within the PEST domain prevented hyperphosphorylation of PEST and stabilized ICD. The PEST domain also interacts with FBW7, an E3 ligase, to ubiquitinate and degrade NICD [60]. In this study, they showed that introducing a T2512A substitution at a potential phosphorylation site abolished the interaction between NICD and FBW7 and enhanced NOTCH1 pathway signaling. Furthermore, when PEST is ubiquitinated, NOTCH signaling is reduced [61]. An overexpression of the NOTCH1 C-terminal, which contains PEST, does not activate NOTCH signaling, and an alteration in PEST can significantly contribute to neoplastic transformation [62]. Therefore, PEST has a lesser role in transactivation, with its primary function being NOTCH1 stability and turnover.
In summary, of the NOTCH1 components, NECD primarily facilitates ligand binding and proper extracellular cleavage, while NEXT contains NICD that undergoes nuclear localization, promotes transcriptional activation, and modulates its stability.
NOTCH1 is highly intolerant of both loss-of-function (LOF) variants, including stop-gain, frameshift, splicing variants, micro-deletion, and missense variation, with a pLI of 1 and a missense z score of 4.48 on the Exome Aggregation Consortium (ExAC) [23]. NOTCH1 variants have been implicated in both sporadic and hereditary CHD cases [24,25,63,64] and pathogenic NOTCH1 variants have been found in approximately 6% of CHD patients, with a higher prevalence in LVOTO and TOF [25]. Currently, a total of 216 NOTCH1 variants have reported related to CHD (Fig. 3 and Supplementary Table).
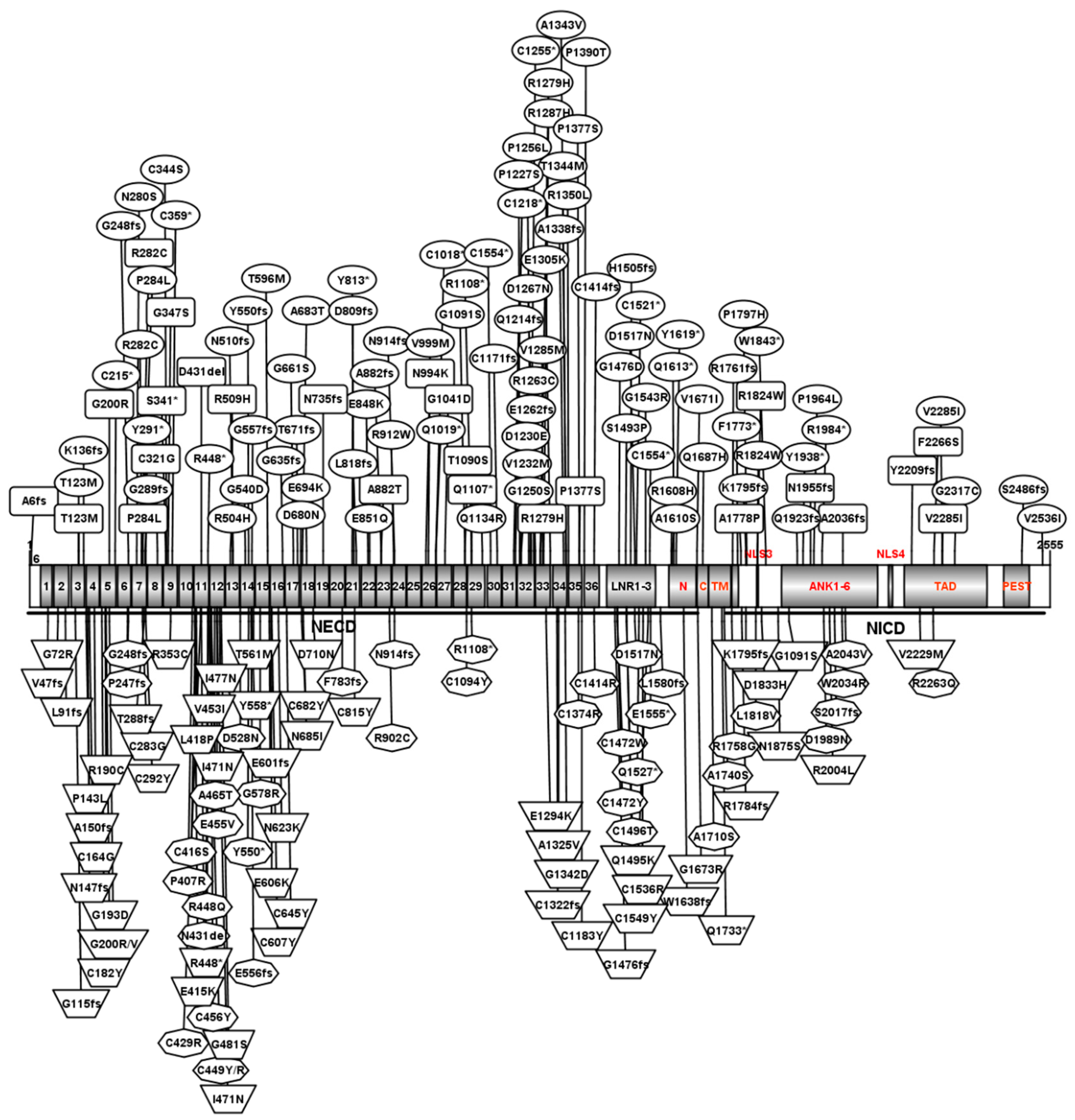
Figure 3: NOTCH1 variants associated with cardiac disease. Variants associated with LVOTO individuals (oval), TOF individuals (trapezoid), AOS individuals (octagon) and other individuals (rounded rectangle) are indicated. C359*, a truncated variant with the cysteine at position 359 changed into a stop codon. N914–1G>A, splicing variant at amino acid N914. D809fs, a frameshift variant at position 809 that contains a frameshift and splice site variant. 1–36, EGF–domain 1–36 repeats; LNR1–3, the LNR1–3 domain; N, the heterodimerization domain located the N-terminal part of the NECD region; C, the heterodimerization domain located the C-terminal begins the NEXT region; TM, transmembrane domain; NLS3 and NLS4, nuclear localization signals 3 and 4, respectively; ANK1–6, the ANK 1–6 domain; TAD, the transcriptional activation domain; PEST, the PEST domain. This figure was prepared using IBS (https://ibs.renlab.org/) based on protein structure.
The LVOTO defect is associated with structural malformations on the left side of the heart, including hypoplastic left heart syndrome (HLHS), aortic coarctation (CoA), aortic stenosis (AS), and bicuspid aortic valve (BAV) [65]. In LVOTO individuals, NOTCH1 mutations represent 5.9% of cases, with an additional 16% involving variants of uncertain significance [25]. Among reported variants, we have searched the NCBI database and found 92 NOTCH1 variants associated with LVOTO patients (Supplementary Table). Among them, 62 variants (out of 92 total variants) are located in the EGF-like domain. EGF-like repeats are short peptides with a distinctive motif of six cysteines with three internal disulfide bridges, and make up the most important domain, accounting for about 60% of the animo acids in NOTCH1 protein (1426/2555) (Fig. 3) [30]. If a mutation site is located at a cysteine residue, disulfide bond formation will be disrupted leading to protein structural instability, S1 clearance failure, and the formation of fewer NICDs, affecting the transmission of NOTCH signals. The missense mutation, c.G1820;p.C607Y, which is located in EGF16, can disrupt conserved disulfide bonds and impair S1 cleavage, thereby reducing NICD formation [23]. Therefore, we classify cysteine variants in the EGF-like domain as LOF mutations that are pathogenic. In LVOTO, a total of 92 NOTCH1 variants were reported (Fig. 3, Supplementary Table), of which 44 were classified as LOF mutations and 62 variants were located in the EGF-like domain (Fig. 3, Table 1).
Table 1: Number of NOTCH1 variants in different type of CHD.
| Disease Type | Total Variants | Loss of Function | All Variants in EGF-Like Repeat | Missense Variants | ||||
|---|---|---|---|---|---|---|---|---|
| Stop-Gain Variants | Frameshifts | Micro-Deletions | C in EGF-Like Repeats | n | ||||
| LVOTO | 92 | 18 | 21 | 4 | 1 | 44/92 | 62/92 | 48 |
| TOF | 57 | 3 | 12 | 2 | 9 | 26/57 | 41/57 | 31 |
| AOS | 42 | 4 | 7 | 3 | 8 | 22/42 | 22/42 | 18 |
| Other CHD | 25 | 2 | 5 | 0 | 1 | 8/25 | 18/25 | 18 |
| Total | 216 | 27 | 45 | 9 | 19 | 100/216 | 143/216 | 115 |
TOF is one of the most common and complex CHD forms, with a prevalence of approximately 1 in every 3000 live births, accounting for 4.5% of newborns with CHD [64]. TOF is considered a malformation of the cardiac outflow tract, which includes four specific structural features: VSD, right ventricular outflow tract stenosis, right ventricular hypertrophy, and aortic override. Genetic factors are the main cause of TOF, with 20% of patients having pathological copy number variants (CNVs) or significant chromosomal abnormalities [66]. In TOF patient, NOTCH1 variants account for at least 4.5% of TOF cases (37/829) [23,67]. Among reported variants, a total of 57 NOTCH1 variants were reported in TOF patients (Fig. 3, Supplementary Table). In TOF individuals, 26 variants (out of 57 total variants) were LOF mutations and 21 variants (out of 57 total variants) were located in the EGF-like domain (Fig. 3, Table 1).
NOTCH1 is an important candidate gene in AOS, a rare disorder featuring congenital scalp defects and terminal limb anomalies, that is often accompanied by CHD, including atrial septal defect (ASD), VSD, aortic valve stenosis, pulmonary valve stenosis and TOF. NOTCH1 variants account for 10% of AOS cases, particularly those with cardiovascular abnormalities [68]. Among reported variants, a total of 42 NOTCH1 variants were reported in AOS patients (Fig. 3, Supplementary Table). In these AOS individuals, 22 variants (total variants were 42) were LOF mutations, and 22 variants (out of 42 total variants) were located within the EGF-like domain (Fig. 3, Table 1).
3.4 NOTCH1 Variants in Other CHDs
NOTCH1 variants have also been associated with other CHDs not mentioned above, including VSD, ASD, outflow tract and coronary artery abnormalities. In ASD and mitral stenosis, a splice variant, c.2207+1G>T:pN735fsX*, was identified [69]; while in VSD and tricuspid atresia, a frameshift variant, c.13_14dupCT:p.A6fs*28, was observed [70]. A frameshift-deletion variant, c.6105Cdel:p.A2036fs3*, was observed in probands with an outflow tract defect, such as double-outlet right ventricle (DORV) and right ventricular dilation [71]. Additionally, a stop-gain variant, c.1023C>A:p.S341*, was found in a patient with coronary artery abnormalities [72]. Up to now, a total of 25 NOTCH1 variants were reported in other CHD patients (Fig. 3, Supplementary Table). Among them, 8 variants (out of 25 total variants) were LOF mutations, and 18 variants (out of 25 total variants) were located in EGF-like domain (Fig. 3, Table 1).
Overall, a total of 216 NOTCH1 variants were reported, and most of the NOTCH1 variants appeared to be predominantly located in the EGF-like domain (143/216), with many being LOF variants (100/216).
4 NOTCH1 Mechanistic Links to CHD
In a computational study performed in 2010, the probability that NOTCH1 function is susceptible to haploinsufficiency was predicted to be 0.957 [73]. Moreover, NOTCH1 and its family members have been found to be downregulated in both NOTCH1 mutant and non-mutant CHD samples [23,74,75]. Therefore, NOTCH1 haploinsufficiency is a significant factor in CHD occurrence.
In induced pluripotent stem cells (iPSCs) that were reprogrammed from fibroblast patients with valve disease, both a stop-gain variant, c.C3322T:p.R1108X*1108, and a frameshift variant, c.C4515del:p.H1505fs74*, can result in NOTCH1 haploinsufficiency [22,75]. In a transgenic mouse model, Notch1-p.V1744G, which is analogous to human p.V1754G, and located within the intramembranous processing site, was shown to significantly reduce NOTCH1 processing [76]. This study also found that homozygous Notch1 knockout mice (Notch1−/−) and homozygous of Notch1-p.V1744G mice displayed that same phenotype, including development retardation, embryonic lethality before e12.5 days, and a distended pericardial sac, but with a beating heart. Meanwhile, our group aimed to investigate the relationship between NOTCH1 variants and TOF. We constructed two variant transgenic mice, both located at the C-amino acid of the EGF-like domain, and generated fewer NICDs in the homozygous and heterozygous mice. The experimental results showed that, homozygous mice with these two mutation sites had the same phenotype as mice with Notch1 knockout (lethality before e12.5 days, with the presence of a pericardial sac and a beating heart). Heterozygotes were born with high permeability CHD, such as VSD, overriding aorta, and right ventricular hypoplasia (data unpublished). Therefore, NOTCH1 variants can lead to a decrease in NICD, affecting NOTCH1 functioning and disrupting normal embryonic development, causing various types of CHD (Fig. 4).
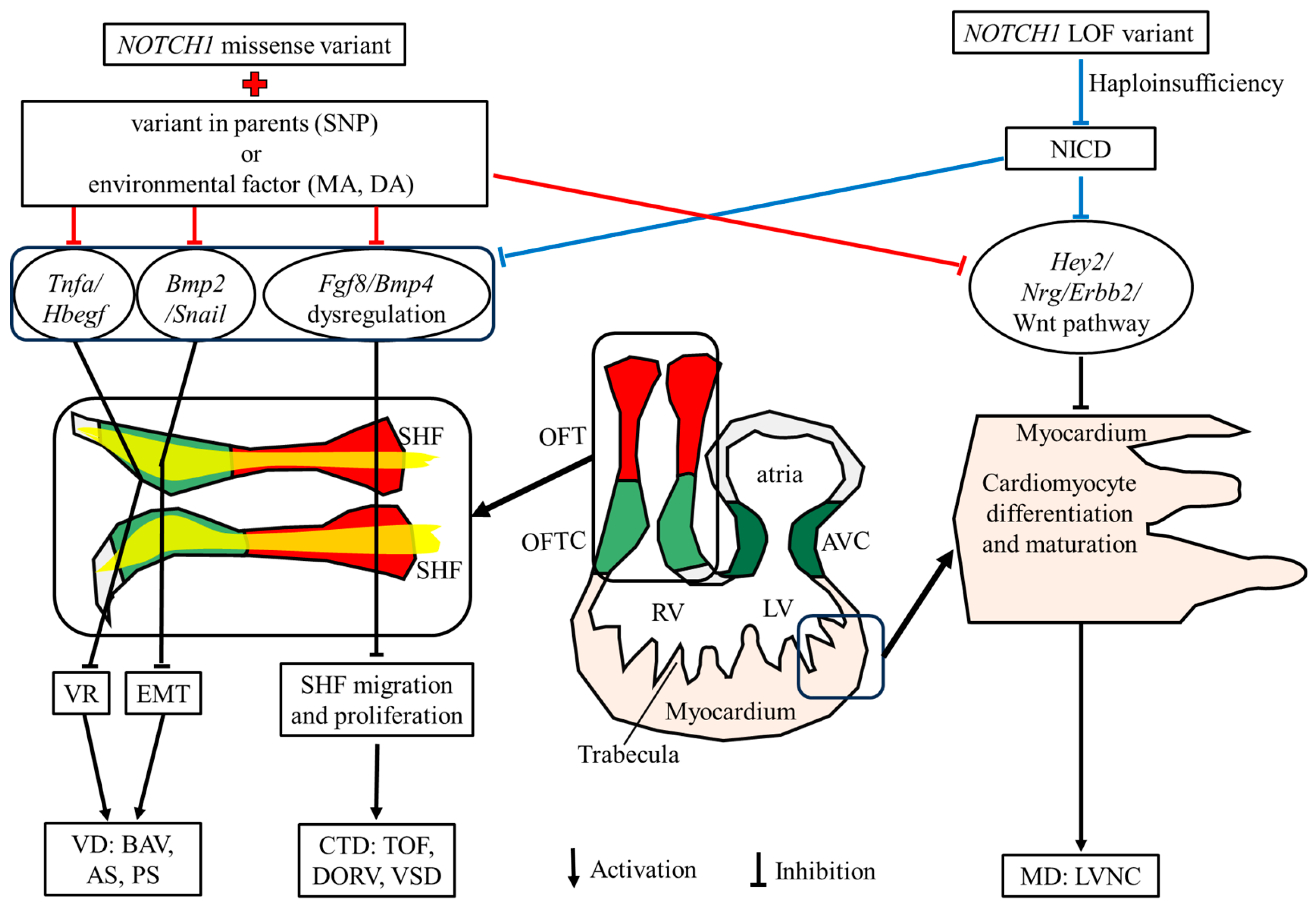
Figure 4: Heart development in embryos at 10.5 days and the two molecular mechanism contributing NOTCH1 variant associated CHD occurrence. OFT, outflow tract; OFTC, OFT cushion; RV, right ventricular; LV, left ventricular; AVC, atrioventricular cushion; CTD, conotruncal defects; MD, myocardial defect; VD, valve defect; NICD, NOTCH intracellular domain, TOF, Tetralogy of Fallot; DORV, double-outlet right ventricle; VSD, ventricular septal defects; AS, aortic stenosis; BAV, bicuspid aortic valve; PS, pulmonary stenosis; LVNC, left ventricular non-compaction; EMT, epithelial-to-mesenchymal transition; LOF, loss-of-function. VR, valve remodeling; MA, maternal alcohol; DA, drug abuse; Red backgrounds indicate OFT; Yellow backgrounds indicate second heart field; and light green and dark green backgrounds indicate OFT and atrial-ventricular cushions, respectively.
NOTCH1 variants associated with LOF variants, including stop-gain, frameshift, splicing, micro-deletion and cysteine variants, have been shown to be commonly located within the EGF-like domain, to reduce NICD function, and can result in haploinsufficiency. In stop-gain and frame shift variants, the conversion of a coding amino acid to a premature stop codon generates a truncation or haploinsufficiency. Splicing variants that are located at the junction of a 5′ or 3′ exon can cause exon skipping or intron-containing transcripts due to aberrant pre-messenger RNA splicing. This aberrant splicing can result in a premature truncation or generation of another unknown protein. In a microdeletion, the NOTCH1 gene, which is located at 9q.34, is completely or partially removed, leading to a haploinsufficiency. A cysteine variant in the EGF-like domain affects the disulfide bonds formation and can cause S1 cleavage failure, reducing NICD production. The mentioned above variants make up 100 (out of 216 total variants) of all CHD associated mutations.
In another transgenic Notch1 mouse model, a substitution in the ANK-repeat domain at the Arg in position 1974 was introduced, and while NICD formation was not affected, sequence-paired site binding was impaired [77]. Moreover, homozygous mice with this substitution had a normal lifespan and did not develop any T-cell abnormalities or CHD, with both defects noted in Notch1 knockout; however, they did show an increased sensitivity to dextran sulfate sodium (DSS), which induced colitis and highly penetrant with severe VSD. This study indicated that a missense NOTCH1 variant could maintain normal target activation functioning, but increase CHD susceptibility when combined with environmental toxins. Taken together, these finding suggest that rare variants found in both unaffected and affected individuals, such as de novo missense variants alone, are, on their own, insufficient to cause CHD. However, when combined with a affected environmental (toxic exposures, such as drug abuse, maternal alcohol, consumption) or inherited variants with unaffected parents (e.g., SNP), the risk of CHD occurrence in the offspring increases (Fig. 4).
NOTCH1 is the one of the High-Confidence Genes in Conotruncal Cardiac Defects by Gene Burden Analyses [78]. Using a fate-mapped mouse model, Notch signal abnormality (Dll4 or Notch1 knockout mouse model) has been found to reduce SHF proliferation via Fgf8 and Bmp4 [79]. Notch1+/− heterozygous mice exhibited an inhibition of Notch signal transmission, and a reduction in second cardiac cell proliferation and migration, through Fgf8 or Bmp4. This can lead to conotruncal defects, such as TOF, DORV and VSD [79]. Furthermore, Notch1 or RBP-J deletion affects outflow tract clockwise rotation and septation [80]. During OFT development, the rotation and alignment of OFT rely on the accurate proliferation and migration of anterior SHF [81]. Therefore, in NOTCH1 variants, NOTCH1 malfunction can lead to the abnormal proliferation and migration of SHF and cause conotruncal defects, such as TOF, DORV, and VSD (Fig. 4).
During early valve development, cardiomyocytes of the conotruncal cushions recruit NCCs into conotruncal cushions, where they promote EMT and play essential roles in aorticopulmonary septation [81]. NOTCH pathways activate the transcription activity of Snail and subsequently repress VE-cadherin expression, which is a prerequisite for the EMT [82]. NOTCH signals also activate Bmp2 expression and promote the proliferation of mesenchymal cells (MCs) during endocardial cushion formation [82]. Additionally, NOTCH1 induces the apoptosis and proliferation of valve interstitial cells via Tnfa and Hbegf, respectively, which controls the cardiac valve remodeling process [82]. NOTCH1 loss-of-function (LOF) mutant embryos severely impaired the EMT and valve remodeling process, and disruption of the NOTCH signaling pathway can lead to valve defects, such as thickened valve leaflets, PS, AS, and BAV (Fig. 4).
The genetic landscape indicated that NOTCH1 is associated with both aorta and aorta valve disease, as well as cardiomyopathy, such as HLHS [83]. In human induced pluripotent stem cells (iPSCs), NOTCH1 is essential for ventricular-like cardiomyocyte differentiation and proliferation through regulating the cell fate of cardiac mesoderms and modulating the cell cycle via the Wnt pathway; notably, insufficient Notch1 functionality results in HLHS [84,85]. In mouse models, Notch1 malfunction can also affect cardiomyocyte differentiation and proliferation during early myocardium formation via Hey1/Nrg/Erbb2 [86]. Furthermore, inhibiting NOTCH signals can reduce the regenerative ability of ventricular myocardia [87]. Therefore, NOTCH1 variants can lead to NOTCH1 malfunctions and cause myocardial defects at the cardiac mesoderm stage, such as left ventricular non-compaction (LVNC) (Fig. 4).
NOTCH1 encodes a protein with two major domains: NECD and NICD. The NECD is comprised of an EGF-like domain that contains 1–36 repeats, and a NRR domain. The EGF-like repeats function in ligand binding and activate the NOTCH signal. The NRR domain harbors the S1 and S2 cleavage sites and negatively regulates NOTCH1 stability to ensure the correct triple cleavage of the extracellular segment to form NICD. NICD, which contains RAM, ANK, TAD, and PEST domains, functions as a transcription factor by entering the nucleus to promote transcription by forming the NICD-CSL-MAM complex, which recruits HAT to form transcription complexes and initiate gene transcription.
After examining NOTCH1 variants associated with CHD, they were found to have a high prevalence in the EGF-like domain. Additionally, LOF variants make up 44 (out of 92 total variants) variants in LVOTO, 26 (out of 57 total variants) in TOF, 22 (out of 42 total variants) in AOS, and 8 (out of 25 total variants) in other types of CHD. The two possible molecular mechanisms underlying NOTCH1-associated CHD occurrence included LOF variants, which directly reduce NICD production, or a missense variant, which causes a mild NOTCH1 mal-function, but is insufficient to CHD occurrence. In the case of a missense variant, other factors, such as toxic exposure, are required to cumulatively promote CHD occurrence.
With the development of larger-scale genetic sequencing platforms, an increasing number of NOTCH1 variants have been discovered in CHD. However, several limitations remain.
- The NOTCH1 variants have not yet been clinically used for improved methods for CHD diagnosis and management.
- To date, there is no validated animal model that accurately mimics the CHD phenotypes caused by NOTCH1 variants.
- Large and more diverse clinical studies are needed to develop and validate the relationship between NOTCH1 variants and CHD, especially TOF.
Future research avenues:
- As sequencing technologies advance and clinical sample sizes increase, it will become possible to develop improved diagnostic and management methods for CHD.
- With current genetic testing, screening can aid in reducing or delaying incidence rates, improve the quality of life of variant carriers.
- A deeper understanding of the functional impact of NOTCH1 mutations may allow their integration into reproductive medicine, potentially reducing the occurrence of severe CHD through the pre-implantation genetic diagnosis and screening of embryos.
Acknowledgement:
Funding Statement: This research was funded by the National Natural Science Foundation of China, Grant Nos. 82100321 and 82370353.
Author Contributions: Conceptualization, Hongqun Xiang, Yan Shi; methodology, Yan Shi; software, Luoning Bao, Jian Zhuang; formal analysis, Hongqun Xiang; investigation, Luoning Bao, Jian Zhuang; resources, Yan Shi; data curation, Yan Shi; writing—original draft preparation, Hongqun Xiang, Yan Shi; writing—review and editing, Jian Zhuang, Yan Shi; visualization, Luoning Bao; supervision, Jian Zhuang; project administration, Yan Shi; funding acquisition, Yan Shi. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: No original data was utilized in the creation of this manuscript.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://www.techscience.com/doi/10.32604/chd.2025.064366/s1.
Abbreviation
| Congenital heart disease | |
| NOTCH extracellular domain | |
| NOTCH intracellular domain | |
| NOTCH extracellular truncation | |
| Whole-exome sequencing | |
| Epithelial-to-mesenchymal transition | |
| Tetralogy of Fallot | |
| Left ventricular outflow tract obstruction | |
| T-cell acute lymphoblastic leukemia | |
| Histone acetyltransferase | |
| Epidermal growth factor-like | |
| Negative regulatory region | |
| Ventricular septal defect | |
| Lin12/Notch repeats | |
| Heterodimerization domain | |
| Transmembrane domain | |
| Nuclear localization signal | |
| RBP-J association module | |
| Ankyrin | |
| Transcriptional activation domain | |
| Myocyte enhancer factor 2C | |
| Loss-of-function | |
| Exome Aggregation Consortium | |
| Hypoplastic left heart syndrome | |
| Aortic coarctation | |
| Aortic stenosis | |
| Bicuspid aortic valve | |
| Atrial septal defect | |
| Copy number variants | |
| Induced pluripotent stem cells | |
| Dextran sulfate sodium | |
| Left ventricular non-compaction | |
| Pulmonary stenosis | |
| Double-outlet right ventricle |
References
1. Zaidi S, Brueckner M. Genetics and genomics of congenital heart disease. Circ Res. 2017;120(6):923–40. doi:10.1161/CIRCRESAHA.116.309140. [Google Scholar] [CrossRef]
2. Zhao L, Chen L, Yang T, Wang T, Zhang S, Chen L, et al. Birth prevalence of congenital heart disease in China, 1980–2019: a systematic review and meta-analysis of 617 studies. Eur J Epidemiol. 2020;35(7):631–42. doi:10.1007/s10654-020-00653-0. [Google Scholar] [CrossRef]
3. Wang Y, Yang X, Ye M, Zhao Y, Chen R, Da M, et al. Machine learning-based intelligent auscultation techniques in congenital heart disease: application and development. Congenit Heart Dis. 2024;19(2):219–31. doi:10.32604/chd.2024.048314. [Google Scholar] [CrossRef]
4. Narayan P, Richter F, Morton S. Genetics and etiology of congenital heart disease. Curr Top Dev Biol. 2024;156(3):297–331. doi:10.1016/bs.ctdb.2024.01.009. [Google Scholar] [CrossRef]
5. Kovall RA, Blacklow SC. Mechanistic insights into notch receptor signaling from structural and biochemical studies. Curr Top Dev Biol. 2010;92:31–71. doi:10.1016/S0070-2153(10)92002-4. [Google Scholar] [CrossRef]
6. Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, et al. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell. 2000;5(2):197–206. doi:10.1016/S1097-2765(00)80416-5. [Google Scholar] [CrossRef]
7. Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, et al. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell. 2000;5(2):207–16. doi:10.1016/S1097-2765(00)80417-7. [Google Scholar] [CrossRef]
8. Sanchez-Irizarry C, Carpenter AC, Weng AP, Pear WS, Aster JC, Blacklow SC. Notch subunit heterodimerization and prevention of ligand-independent proteolytic activation depend, respectively, on a novel domain and the LNR repeats. Mol Cell Biol. 2004;24(21):9265–73. doi:10.1128/MCB.24.21.9265-9273.2004. [Google Scholar] [CrossRef]
9. Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393(6683):382–6. doi:10.1038/30756. [Google Scholar] [CrossRef]
10. Fryer CJ, Lamar E, Turbachova I, Kintner C, Jones KA. Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes Dev. 2002;16(11):1397–411. doi:10.1101/gad.991602. [Google Scholar] [CrossRef]
11. Bray S, Musisi H, Bienz M. Bre1 is required for Notch signaling and histone modification. Dev Cell. 2005;8(2):279–86. doi:10.1016/j.devcel.2004.11.020. [Google Scholar] [CrossRef]
12. Kurooka H, Honjo T. Functional interaction between the mouse notch1 intracellular region and histone acetyltransferases PCAF and GCN5. J Biol Chem. 2000;275(22):17211–20. doi:10.1074/jbc.M000909200. [Google Scholar] [CrossRef]
13. MacGrogan D, Nus M, de la Pompa JL. Notch signaling in cardiac development and disease. Curr Top Dev Biol. 2010;92:333–65. doi:10.1016/S0070-2153(10)92011-5. [Google Scholar] [CrossRef]
14. Garg V. Molecular genetics of aortic valve disease. Curr Opin Cardiol. 2006;21(3):180–4. doi:10.1097/01.hco.0000221578.18254.70. [Google Scholar] [CrossRef]
15. Ackah RL, Yasuhara J, Garg V. Genetics of aortic valve disease. Curr Opin Cardiol. 2023;38(3):169–78. doi:10.1097/HCO.0000000000001028. [Google Scholar] [CrossRef]
16. Yang Y, Duan W, Jin Z, Bi S, Yan J, Jin Y, et al. New role of Notch-mediated signaling pathway in myocardial ischemic preconditioning. Med Hypotheses. 2011;76(3):427–8. doi:10.1016/j.mehy.2010.11.011. [Google Scholar] [CrossRef]
17. Kratsios P, Catela C, Salimova E, Huth M, Berno V, Rosenthal N, et al. Distinct roles for cell-autonomous Notch signaling in cardiomyocytes of the embryonic and adult heart. Circ Res. 2010;106(3):559–72. doi:10.1161/CIRCRESAHA.109.203034. [Google Scholar] [CrossRef]
18. Timmerman LA, Grego-Bessa J, Raya A, Bertrán E, Pérez-Pomares JM, Díez J, et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004;18(1):99–115. doi:10.1101/gad.276304. [Google Scholar] [CrossRef]
19. Kachanova O, Lobov A, Malashicheva A. The role of the notch signaling pathway in recovery of cardiac function after myocardial infarction. Int J Mol Sci. 2022;23(20):12509. doi:10.3390/ijms232012509. [Google Scholar] [CrossRef]
20. Lampada A, Taylor V. Notch signaling as a master regulator of adult neurogenesis. Front Neurosci. 2023;17:1179011. doi:10.3389/fnins.2023.1179011. [Google Scholar] [CrossRef]
21. Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9(8):e1003709. doi:10.1371/journal.pgen.1003709. [Google Scholar] [CrossRef]
22. Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270–4. doi:10.1038/nature03940. [Google Scholar] [CrossRef]
23. Page DJ, Miossec MJ, Williams SG, Monaghan RM, Fotiou E, Cordell HJ, et al. Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of fallot. Circ Res. 2019;124(4):553–63. doi:10.1161/CIRCRESAHA.118.313250. [Google Scholar] [CrossRef]
24. Preuss C, Capredon M, Wünnemann F, Chetaille P, Prince A, Godard B, et al. Family based whole exome sequencing reveals the multifaceted role of notch signaling in congenital heart disease. PLoS Genet. 2016;12(10):e1006335. doi:10.1371/journal.pgen.1006335. [Google Scholar] [CrossRef]
25. Helle E, Córdova-Palomera A, Ojala T, Saha P, Potiny P, Gustafsson S, et al. Loss of function, missense, and intronic variants in NOTCH1 confer different risks for left ventricular outflow tract obstructive heart defects in two European cohorts. Genet Epidemiol. 2019;43(2):215–26. doi:10.1002/gepi.22176. [Google Scholar] [CrossRef]
26. Ji W, Ferdman D, Copel J, Scheinost D, Shabanova V, Brueckner M, et al. De novo damaging variants associated with congenital heart diseases contribute to the connectome. Sci Rep. 2020;10(1):7046. doi:10.1038/s41598-020-63928-2. [Google Scholar] [CrossRef]
27. Aref S, El Agdar M, Salama O, Zeid TA, Sabry M. Significance of NOTCH1 mutations détections in T-acute lymphoblastic leukemia patients. Cancer Biomark Sect A Dis Markers. 2020;27(2):157–62. doi:10.3233/CBM-190967. [Google Scholar] [CrossRef]
28. Asnafi V, Buzyn A, Le Noir S, Baleydier F, Simon A, Beldjord K, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood. 2009;113(17):3918–24. doi:10.1182/blood-2008-10-184069. [Google Scholar] [CrossRef]
29. Rand MD, Grimm LM, Artavanis-Tsakonas S, Patriub V, Blacklow SC, Sklar J, et al. Calcium depletion dissociates and activates heterodimeric notch receptors. Mol Cell Biol. 2000;20(5):1825–35. doi:10.1128/MCB.20.5.1825-1835.2000. [Google Scholar] [CrossRef]
30. Davis CG. The many faces of epidermal growth factor repeats. New Biol. 1990;2(5):410–9. [Google Scholar]
31. Luca VC, Jude KM, Pierce NW, Nachury MV, Fischer S, Garcia KC. Structural biology. Structural basis for Notch1 engagement of Delta-like 4. Science. 2015;347(6224):847–53. doi:10.1126/science.1261093. [Google Scholar] [CrossRef]
32. Luca VC, Kim BC, Ge C, Kakuda S, Wu D, Roein-Peikar M, et al. Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science. 2017;355(6331):1320–4. doi:10.1126/science.aaf9739. [Google Scholar] [CrossRef]
33. Kakuda S, LoPilato RK, Ito A, Haltiwanger RS. Canonical Notch ligands and Fringes have distinct effects on NOTCH1 and NOTCH2. J Biol Chem. 2020;295(43):14710–22. doi:10.1074/jbc.RA120.014407. [Google Scholar] [CrossRef]
34. Rana NA, Haltiwanger RS. Fringe benefits: functional and structural impacts of O-glycosylation on the extracellular domain of Notch receptors. Curr Opin Struct Biol. 2011;21(5):583–9. doi:10.1016/j.sbi.2011.08.008. [Google Scholar] [CrossRef]
35. Matsumoto K, Luther KB, Haltiwanger RS. Analysis of endogenous NOTCH1 from POFUT1 S162L patient fibroblasts reveals the importance of the O-fucose modification on EGF12 in human development. Glycobiology. 2024;34(8):1. doi:10.1093/glycob/cwae047. [Google Scholar] [CrossRef]
36. LoPilato RK, Kroeger H, Mohan SK, Lauderdale JD, Grimsey N, Haltiwanger RS. Two NOTCH1 O-fucose sites have opposing functions in mouse retinal angiogenesis. Glycobiology. 2023;33(8):661–72. doi:10.1093/glycob/cwad048. [Google Scholar] [CrossRef]
37. Aster JC, Simms WB, Zavala-Ruiz Z, Patriub V, North CL, Blacklow SC. The folding and structural integrity of the first LIN-12 module of human Notch1 are calcium-dependent. Biochemistry. 1999;38(15):4736–42. doi:10.1021/bi982713o. [Google Scholar] [CrossRef]
38. Greenwald I, Seydoux G. Analysis of gain-of-function mutations of the lin-12 gene of Caenorhabditis elegans. Nature. 1990;346(6280):197–9. doi:10.1038/346197a0. [Google Scholar] [CrossRef]
39. Vardar D, North CL, Sanchez-Irizarry C, Aster JC, Blacklow SC. Nuclear magnetic resonance structure of a prototype Lin12-Notch repeat module from human Notch1. Biochemistry. 2003;42(23):7061–7. doi:10.1021/bi034156y. [Google Scholar] [CrossRef]
40. Bush G, diSibio G, Miyamoto A, Denault JB, Leduc R, Weinmaster G. Ligand-induced signaling in the absence of furin processing of Notch1. Dev Biol. 2001;229(2):494–502. doi:10.1006/dbio.2000.9992. [Google Scholar] [CrossRef]
41. Kurooka H, Kuroda K, Honjo T. Roles of the ankyrin repeats and C-terminal region of the mouse notch1 intracellular region. Nucleic Acids Res. 1998;26(23):5448–55. doi:10.1093/nar/26.23.5448. [Google Scholar] [CrossRef]
42. Miyamoto M, Andersen P, Sulistio E, Liu X, Murphy S, Kannan S, et al. Noncanonical Notch signals have opposing roles during cardiac development. Biochem Biophys Res Commun. 2021;577:12–6. doi:10.1016/j.bbrc.2021.08.094. [Google Scholar] [CrossRef]
43. Wilson-Rawls J, Molkentin JD, Black BL, Olson EN. Activated notch inhibits myogenic activity of the MADS-Box transcription factor myocyte enhancer factor 2C. Mol Cell Biol. 1999;19(4):2853–62. doi:10.1128/MCB.19.4.2853. [Google Scholar] [CrossRef]
44. Bertagna A, Toptygin D, Brand L, Barrick D. The effects of conformational heterogeneity on the binding of the Notch intracellular domain to effector proteins: a case of biologically tuned disorder. Biochem Soc Trans. 2008;36(Pt 2):157–66. doi:10.1042/BST0360157. [Google Scholar] [CrossRef]
45. Deregowski V, Gazzerro E, Priest L, Rydziel S, Canalis E. Role of the RAM domain and ankyrin repeats on notch signaling and activity in cells of osteoblastic lineage. J Bone Miner Res Off J Am Soc Bone Miner Res. 2006;21(8):1317–26. doi:10.1359/jbmr.060505. [Google Scholar] [CrossRef]
46. Ma Z, Zeng Y, Wang M, Liu W, Zhou J, Wu C, et al. N4BP1 mediates RAM domain-dependent notch signaling turnover during neocortical development. EMBO J. 2023;42(22):e113383. doi:10.15252/embj.2022113383. [Google Scholar] [CrossRef]
47. Islam Z, Nagampalli RSK, Fatima MT, Ashraf GM. New paradigm in ankyrin repeats: beyond protein-protein interaction module. Int J Biol Macromol. 2018;109:1164–73. doi:10.1016/j.ijbiomac.2017.11.101. [Google Scholar] [CrossRef]
48. Mosavi LK, Cammett TJ, Desrosiers DC, Peng ZY. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci Publ Protein Soc. 2004;13(6):1435–48. doi:10.1110/ps.03554604. [Google Scholar] [CrossRef]
49. Wilson JJ, Kovall RA. Crystal structure of the CSL-Notch-Mastermind ternary complex bound to DNA. Cell. 2006;124(5):985–96. doi:10.1016/j.cell.2006.01.035. [Google Scholar] [CrossRef]
50. Roehl H, Bosenberg M, Blelloch R, Kimble J. Roles of the RAM and ANK domains in signaling by the C. elegans GLP-1 receptor. EMBO J. 1996;15(24):7002–12. doi:10.1002/j.1460-2075.1996.tb01092.x. [Google Scholar] [CrossRef]
51. Ramsey KM, Barrick D. Unraveling paralog-specific Notch signaling through thermodynamics of ternary complex formation and transcriptional activation of chimeric receptors. Protein Sci Publ Protein Soc. 2024;33(4):e4947. doi:10.1002/pro.4947. [Google Scholar] [CrossRef]
52. Kim YH, Han ME, Oh SO. The molecular mechanism for nuclear transport and its application. Anat Cell Biol. 2017;50(2):77–85. doi:10.5115/acb.2017.50.2.77. [Google Scholar] [CrossRef]
53. Huenniger K, Krämer A, Soom M, Chang I, Köhler M, Depping R, et al. Notch1 signaling is mediated by importins alpha 3, 4, and 7. Cell Mol Life Sci CMLS. 2010;67(18):3187–96. doi:10.1007/s00018-010-0378-7. [Google Scholar] [CrossRef]
54. Aster JC, Robertson ES, Hasserjian RP, Turner JR, Kieff E, Sklar J. Oncogenic forms of NOTCH1 lacking either the primary binding site for RBP-Jkappa or nuclear localization sequences retain the ability to associate with RBP-Jkappa and activate transcription. J Biol Chem. 1997;272(17):11336–43. doi:10.1074/jbc.272.17.11336. [Google Scholar] [CrossRef]
55. Kopan R, Nye JS, Weintraub H. The intracellular domain of mouse Notch: a constitutively activated repressor of myogenesis directed at the basic helix-loop-helix region of MyoD. Development. 1994;120(9):2385–96. doi:10.1242/dev.120.9.2385. [Google Scholar] [CrossRef]
56. Santio NM, Landor SK, Vahtera L, Ylä-Pelto J, Paloniemi E, Imanishi SY, et al. Phosphorylation of Notch1 by Pim kinases promotes oncogenic signaling in breast and prostate cancer cells. Oncotarget. 2016;7(28):43220–38. doi:10.18632/oncotarget.9215. [Google Scholar] [CrossRef]
57. Han X, Ju JH, Shin I. Glycogen synthase kinase 3-β phosphorylates novel S/T-P-S/T domains in Notch1 intracellular domain and induces its nuclear localization. Biochem Biophys Res Commun. 2012;423(2):282–8. doi:10.1016/j.bbrc.2012.05.111. [Google Scholar] [CrossRef]
58. Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1996;21(7):267–71. doi:10.1016/S0968-0004(96)10031-1. [Google Scholar] [CrossRef]
59. Fryer CJ, White JB, Jones KA. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol Cell. 2004;16(4):509–20. doi:10.1016/j.molcel.2004.10.014. [Google Scholar] [CrossRef]
60. Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med. 2007;204(8):1825–35. doi:10.1084/jem.20070872. [Google Scholar] [CrossRef]
61. Oberg C, Li J, Pauley A, Wolf E, Gurney M, Lendahl U. The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. J Biol Chem. 2001;276(38):35847–53. doi:10.1074/jbc.M103992200. [Google Scholar] [CrossRef]
62. Jeffries S, Capobianco AJ. Neoplastic transformation by Notch requires nuclear localization. Mol Cell Biol. 2000;20(11):3928–41. doi:10.1128/MCB.20.11.3928-3941.2000. [Google Scholar] [CrossRef]
63. Kerstjens-Frederikse WS, van de Laar IM, Vos YJ, Verhagen JM, Berger RM, Lichtenbelt KD, et al. Cardiovascular malformations caused by NOTCH1 mutations do not keep left: data on 428 probands with left-sided CHD and their families. Genet Med Off J Am Coll Med Genet. 2016;18(9):914–23. doi:10.1038/gim.2015.193. [Google Scholar] [CrossRef]
64. Liu Y, Chen S, Zühlke L, Black GC, Choy MK, Li N, et al. Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies. Int J Epidemiol. 2019;48(2):455–63. doi:10.1093/ije/dyz009. [Google Scholar] [CrossRef]
65. Reller MD, Strickland MJ, Riehle-Colarusso T, Mahle WT, Correa A. Prevalence of congenital heart defects in metropolitan Atlanta, 1998–2005. J Pediatr. 2008;153(6):807–13. doi:10.1016/j.jpeds.2008.05.059. [Google Scholar] [CrossRef]
66. Bassett AS, Reuter MS, Malecki S, Silversides C, Oechslin E. Clinically relevant genetic considerations for patients with tetralogy of fallot. CJC Pediatr Congenit Heart Dis. 2023;2(6 Pt A):426–39. doi:10.1016/j.cjcpc.2023.10.002. [Google Scholar] [CrossRef]
67. Manshaei R, Merico D, Reuter MS, Engchuan W, Mojarad BA, Chaturvedi R, et al. Genes and pathways implicated in tetralogy of fallot revealed by ultra-rare variant burden analysis in 231 genome sequences. Front Genet. 2020;11:957. doi:10.3389/fgene.2020.00957. [Google Scholar] [CrossRef]
68. Meester JAN, Sukalo M, Schröder KC, Schanze D, Baynam G, Borck G, et al. Elucidating the genetic architecture of Adams-Oliver syndrome in a large European cohort. Hum Mutat. 2018;39(9):1246–61. doi:10.1002/humu.23567. [Google Scholar] [CrossRef]
69. Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, et al. Contribution of rare inherited and de novo variants in 2871 congenital heart disease probands. Nat Genet. 2017;49(11):1593–601. doi:10.1038/ng.3970. [Google Scholar] [CrossRef]
70. Stanley KJ, Kalbfleisch KJ, Moran OM, Chaturvedi RR, Roifman M, Chen X, et al. Expanding the phenotypic spectrum of NOTCH1 variants: clinical manifestations in families with congenital heart disease. Eur J Hum Genet. 2024;32(7):795–803. doi:10.1038/s41431-024-01629-4. [Google Scholar] [CrossRef]
71. Alankarage D, Ip E, Szot JO, Munro J, Blue GM, Harrison K, et al. Identification of clinically actionable variants from genome sequencing of families with congenital heart disease. Genet Med Off J Am Coll Med Genet. 2019;21(5):1111–20. doi:10.1038/s41436-018-0296-x. [Google Scholar] [CrossRef]
72. Shi X, Liu J, Wu J, Hua Y, Zhou K, Li Y. Hypoplastic coronary arteries in a child with a mutation in Notch1: a case report. Medicine. 2020;99(33):e21355. doi:10.1097/MD.0000000000021355. [Google Scholar] [CrossRef]
73. Huang N, Lee I, Marcotte EM, Hurles ME. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6(10):e1001154. doi:10.1371/journal.pgen.1001154. [Google Scholar] [CrossRef]
74. Bittel DC, Butler MG, Kibiryeva N, Marshall JA, Chen J, Lofland GK, et al. Gene expression in cardiac tissues from infants with idiopathic conotruncal defects. BMC Med Genom. 2011;4:1. doi:10.1186/1755-8794-4-1. [Google Scholar] [CrossRef]
75. Theodoris CV, Li M, White MP, Liu L, He D, Pollard KS, et al. Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell. 2015;160(6):1072–86. doi:10.1016/j.cell.2015.02.035. [Google Scholar] [CrossRef]
76. Huppert SS, Le A, Schroeter EH, Mumm JS, Saxena MT, Milner LA, et al. Embryonic lethality in mice homozygous for a processing-deficient allele of Notch1. Nature. 2000;405(6789):966–70. doi:10.1038/35016111. [Google Scholar] [CrossRef]
77. Kobia FM, Preusse K, Dai Q, Weaver N, Hass MR, Chaturvedi P, et al. Notch dimerization and gene dosage are important for normal heart development, intestinal stem cell maintenance, and splenic marginal zone B-cell homeostasis during mite infestation. PLoS Biol. 2020;18(10):e3000850. doi:10.1371/journal.pbio.3000850. [Google Scholar] [CrossRef]
78. Chui MMC, Mak CCY, Yu MHC, Wong SYY, Lun KS, Yung TC, et al. Evaluating high-confidence genes in conotruncal cardiac defects by gene burden analyses. J Am Heart Assoc. 2023;12(4):e028226. doi:10.1161/JAHA.122.028226. [Google Scholar] [CrossRef]
79. De Zoysa P, Liu J, Toubat O, Choi J, Moon A, Gill PS, et al. Delta-like ligand 4-mediated Notch signaling controls proliferation of second heart field progenitor cells by regulating Fgf8 expression. Development. 2020;147(17):dev185249. doi:10.1242/dev.185249. [Google Scholar] [CrossRef]
80. Miao L, Lu Y, Nusrat A, Abdelnasser HY, Datta S, Zhou B, et al. The spatiotemporal expression of Notch1 and Numb and their functional interaction during cardiac morphogenesis. Cells. 2021;10(9):2192. doi:10.3390/cells10092192. [Google Scholar] [CrossRef]
81. Yamaguchi N, Chang EW, Lin Z, Shekhar A, Bu L, Khodadadi-Jamayran A, et al. An anterior second heart field enhancer regulates the gene regulatory network of the cardiac outflow tract. Circulation. 2023;148(21):1705–22. doi:10.1161/CIRCULATIONAHA.123.065700. [Google Scholar] [CrossRef]
82. Wang Y, Fang Y, Lu P, Wu B, Zhou B. NOTCH signaling in aortic valve development and calcific aortic valve Disease. Front Cardiovasc Med. 2021;8:682298. doi:10.3389/fcvm.2021.682298. [Google Scholar] [CrossRef]
83. Parker LE, Landstrom AP. Genetic etiology of left-sided obstructive heart lesions: a story in development. J Am Heart Assoc. 2021;10(2):e019006. doi:10.1161/JAHA.120.019006. [Google Scholar] [CrossRef]
84. Ye S, Wang C, Xu Z, Lin H, Wan X, Yu Y, et al. Impaired human cardiac cell development due to NOTCH1 deficiency. Circ Res. 2023;132(2):187–204. doi:10.1161/CIRCRESAHA.122.321398. [Google Scholar] [CrossRef]
85. Jaffré F. hiPSCs as a unique platform to model cardiogenesis in NOTCH1-associated HLHS: hiPSCs to model complex congenital heart defects. Circ Res. 2023;132(2):205–7. doi:10.1161/CIRCRESAHA.122.322353. [Google Scholar] [CrossRef]
86. Miao L, Li J, Li J, Tian X, Lu Y, Hu S, et al. Notch signaling regulates Hey2 expression in a spatiotemporal dependent manner during cardiac morphogenesis and trabecular specification. Sci Rep. 2018;8(1):2678. doi:10.1038/s41598-018-20917-w. [Google Scholar] [CrossRef]
87. Wang W, Hu YF, Pang M, Chang N, Yu C, Li Q, et al. BMP and Notch signaling pathways differentially regulate cardiomyocyte proliferation during ventricle regeneration. Int J Biol Sci. 2021;17(9):2157–66. doi:10.7150/ijbs.59648. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools