
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Bioinformatics and In-Silico Findings Reveal Candidate Genes for Tetralogy of Fallot via Integrative Multi-Omics Data
1 Department of Ultrasound Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430022, China
2 Clinical Research Center for Medical Imaging in Hubei Province, Huazhong University of Science and Technology, Wuhan, 430022, China
3 Hubei Province Key Laboratory of Molecular Imaging, Huazhong University of Science and Technology, Wuhan, 430022, China
* Corresponding Authors: Haiyan Cao. Email: ; Jing Wang. Email:
# These authors contributed equally to this work
Congenital Heart Disease 2025, 20(2), 213-229. https://doi.org/10.32604/chd.2025.064950
Received 27 February 2025; Accepted 17 April 2025; Issue published 30 April 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Tetralogy of Fallot (TOF), the predominant cyanotic congenital heart defect, arises from multifactorial gene-environment interactions disrupting cardiac developmental networks. This study investigated TOF-specific transcriptional alterations and identified high-confidence candidate genes. Methods: Based on GSE36761 transcriptome data, a weighted gene co-expression network analysis (WGCNA) and protein-protein interaction (PPI) network were conducted to identify TOF-related sub-network and Hub genes. The potential biological functions among these genes were revealed by enrichment analysis. Genetic, epigenetic and transcriptional alteration in the Hub genes were analyzed with leveraged public resources: a methylation dataset (GSE62629) and two single-cell datasets (EGAS00001003996 and GSE126128). Results: Eight Hub genes were identified using the WGCNA network and PPI network, and functional enrichment analysis revealed that GJA1, RUNX2, PTK7, PRICKLE1, and SFRP1 were involved in the morphogenesis of an epithelium, and dysregulation of the signaling were also found in the other two TOF datasets. Furthermore, the study found that the promoters of GJA1, RUNX2, PTK7, and PRICKLE1 genes were hypermethylated and that GJA1 and SFRP1 are highly expressed in mouse second heart field cells and neural crest cells, and the latter is expressed in human embryonic outflow tract cells. Since RUNX2 was not expressed in human and mouse embryonic hearts, GJA1, PTK7, PRICKLE1, and SFRP1 were ultimately identified as TOF candidate genes. Conclusion: Based on the WGCNA network and various bioinformatics analysis approaches, we screened 4 TOF candidate pathogenic genes, and found that the signaling pathways related to the morphogenesis of an epithelium may be involved in the pathogenesis of TOF.Keywords
Supplementary Material
Supplementary Material FileTetralogy of Fallot (TOF), the most common cyanotic congenital heart disease, is characterized by four cardinal anatomical abnormalities: ventricular septal defects, pulmonary artery stenosis, aortic cross, and right ventricular hypertrophy [1,2]. Epidemiological data have shown that TOF occurs in 3–5 per 10,000 newborns and the family recurrence risk of TOF approximating 3% [3,4,5]. Advances in congenital cardiac surgical techniques have significantly reduced early mortality rates from 25% to less than 2% in recent decades [6]. However, postoperative morbidity and mortality remain substantial for patients with complex congenital heart disease, particularly among infants with TOF [7,8].
The multifactorial etiology of TOF may contribute to disorders of transcription regulation networks [9,10]. TOF exhibiting genetic heterogeneity, including various forms such as single-nucleotide variants, copy number variations, transcriptional dysregulation, abnormal epigenetic modifications, and their complex interactions, all of which ultimately lead to gene network dysfunction [10,11,12,13]. Marcel and colleagues showed that deleterious private and rare mutations are in genes essential for the development of right ventricle and outflow tract in TOF patients, and that the interaction of these mutate genes form a similar abnormal gene regulatory network [12]. Similarly, Reuter et al. found that affected genes in TOF patients form an interactive network in which NOTCH1 plays a core role [13]. Weighted gene co-expression network analysis (WGCNA), a systems biology analysis method, has been widely used to reveal the gene modules and networks underlying abnormalities in various diseases [14,15,16]. Therefore, it is necessary to exploit systems biology analysis to synthesize the interactions between these genetic factors and their effects on TOF.
The maturity of the outflow tract (OFT) development that is closely related to TOF mainly depends on the neural crest cells (NCC) migrating towards the heart and the anterior second heart field cells (aSHFC) from the pharyngeal mesoderm, as well as involving various cell-cell interactions [17,18]. Profiling spatiotemporal gene expression of the NCC and aSHFC provides a new perspective for understanding the embryonic origin of TOF. Thus, the present study will further uncover the single-cell expression profile of Hub genes screened by WGCNA in the embryonic heart.
2.1 Data Availability and Preprocessing
All publicly available sequencing datasets related to TOF were retrieved from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/). Four datasets were ultimately included: two microarray expression profiles (accession numbers GSE26125 [19] and GSE35776 [20]), one transcriptomic dataset (GSE36761 [12]), and one methylation dataset (GSE62629 [21]). The preprocessed matrix data were downloaded from the GSE26125 and GSE35776, the original gene count expression matrix was downloaded from the GSE36761, and the original CpG score data were downloaded from the GSE62629. The analytical workflow is summarized in Fig. 1.
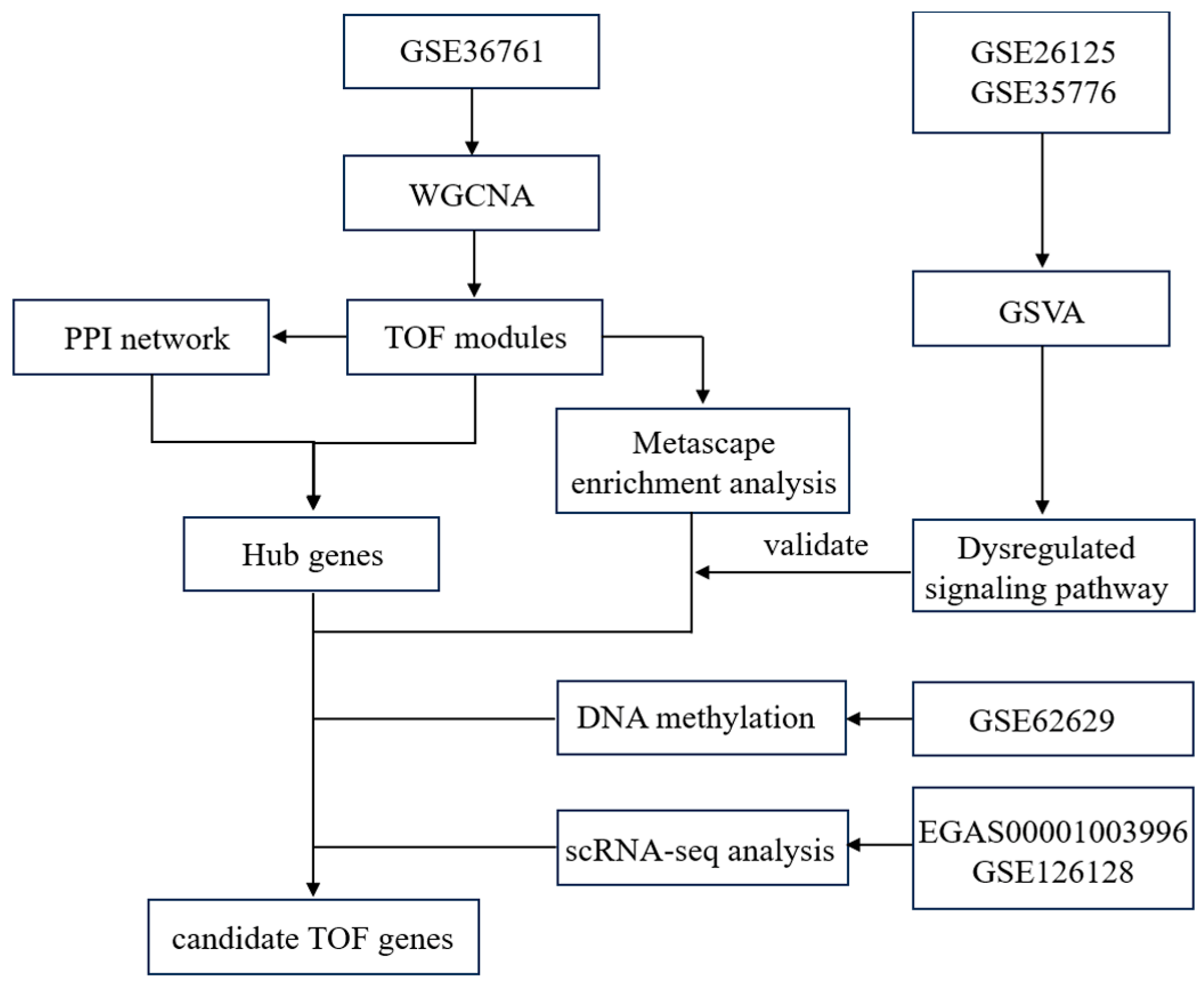
Figure 1: Flowchart of the data collection and analysis.
The GSE26215 data were normalized, and the GSE35776 expression matrix was subjected to log2 logarithmic transformation. The original expression matrix of GSE36761 was normalized by the vst function in the DESeq2 package (version 1.34.0; https://bioconductor.org/packages/release/bioc/html/DESeq2.html, accessed on 01 April 2025). Both GSE36761 and the methylation data were annotated using the GRCh37/hg19 genome assembly. To reduce technical noise, only genes exhibiting the top 60% intergroup variance between TOF and control samples in the GSE36761 expression matrix were retained for subsequent co-expression network construction.
A scale-free WGCNA network was constructed for the GSE36761 dataset using the WGCNA package (version 1.63) in R 4.0 (https://www.r-project.org, accessed on 01 April 2025), with the scale-free topology fit index exceeding 0.7. Detailed methodological protocols for WGCNA implementation can be found in our previous publication [22]. First, pairwise gene comparisons were performed to calculate absolute Pearson correlation coefficients. A soft threshold power β was determined as the minimal integer enabling scale-free network conformity. The adjacency matrix was then computed using the formula
We assessed module-clinical trait associations by computing Pearson correlations between ME and clinical parameters (including age, gender, oxygen partial pressure, and disease status) to identify key modules of interest. The WGCNA package implements module significance (MS) as a metric quantifying module-trait relationships, calculated as the mean gene significance (GS) across all module genes. In our study, the module with maximal MS value for TOF status was designated as the TOF-associated module. Statistical significance was defined as p < 0.01 for MS values.
Pathway enrichment analysis was conducted using Metascape (v3.5.20220101; https://metascape.org, accessed on 01 April 2025) to annotate biological functions, incorporating Gene Ontology Biological Processes (GOBP), KEGG, Reactome, and WikiPathways databases. Gene Set Variation Analysis (GSVA) was performed with the GSVA package (v1.42.0) in R under default parameters. The GOBP gene sets were retrieved from the Molecular Signatures Database (MSigDB; http://www.gsea-msigdb.org/gsea/downloads.jsp, accessed on 01 April 2025). Differential pathway analysis between TOF and control groups was executed using the limma package (v3.50.1), with false discovery rate (FDR) correction applied for multiple testing. The simplifyEnrichment package (v1.4.0) subsequently clustered significant GOBP terms based on semantic similarity, generating condensed visualizations to facilitate rapid interpretation of predominant biological processes.
2.5 Identification of Hub Genes
To robustly identify Hub genes, we integrated protein-protein interaction (PPI) networks with WGCNA analysis through a dual-filter strategy. These filtered genes were subsequently used to construct a PPI network via STRING database (v11.5; https://string-db.org, accessed on 01 April 2025) with medium confidence threshold (combined score ≥ 0.4). Hub genes were defined as nodes with ≥5 interaction partners in the PPI network according to previous reports [23]. All network visualizations were generated using Cytoscape 3.9 (https://cytoscape.org, accessed on 01 April 2025), with node degree represented as topological centrality measure.
2.6 DNA Methylation and Single-Cell Transcriptomic Analysis
Differentially methylated CpGs analysis was performed using the R package methylKit (v1.20.0). Spatial scRNA-seq analysis was performed via the online database (EGAS00001003996) [24], which used to display the spatiotemporal expression information of the Hub genes in the embryonic heart. A murine single-cell dataset (GSE126128) encompassing E7.75-E9.5 cardiac development was analyzed to further validate Hub gene expression dynamics [25].
2.7 Data and Code Availability
All datasets analyzed in this study were obtained from the aforementioned repositories. The WGCNA network construction workflow was implemented using code adapted from the official WGCNA tutorial (Version 1.72-1; https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/, accessed on 01 April 2025), with full reproducibility ensured through open access to all analytical scripts.
3.1 WGCNA Network Construction
After hierarchical clustering analysis, TOF-18 was identified as an outlier sample subsequently excluded to mitigate confounding effects. As shown in Fig. 2, the reclustered samples demonstrated distinct segregation between the case and control groups. The adjacency matrix transformation was performed on the 10,503 genes of the 28 samples, and then the transformation into a topological matrix was continued (Fig. 3A–C), confirming the resultant network exhibited scale-free topology characteristics. In addition, module detection was implemented using dynamic tree cutting with a minimum module size of 100 genes, followed by merging of highly correlated modules (similarity threshold > 0.75). This process ultimately yielded eight distinct co-expression modules (Fig. 3D).
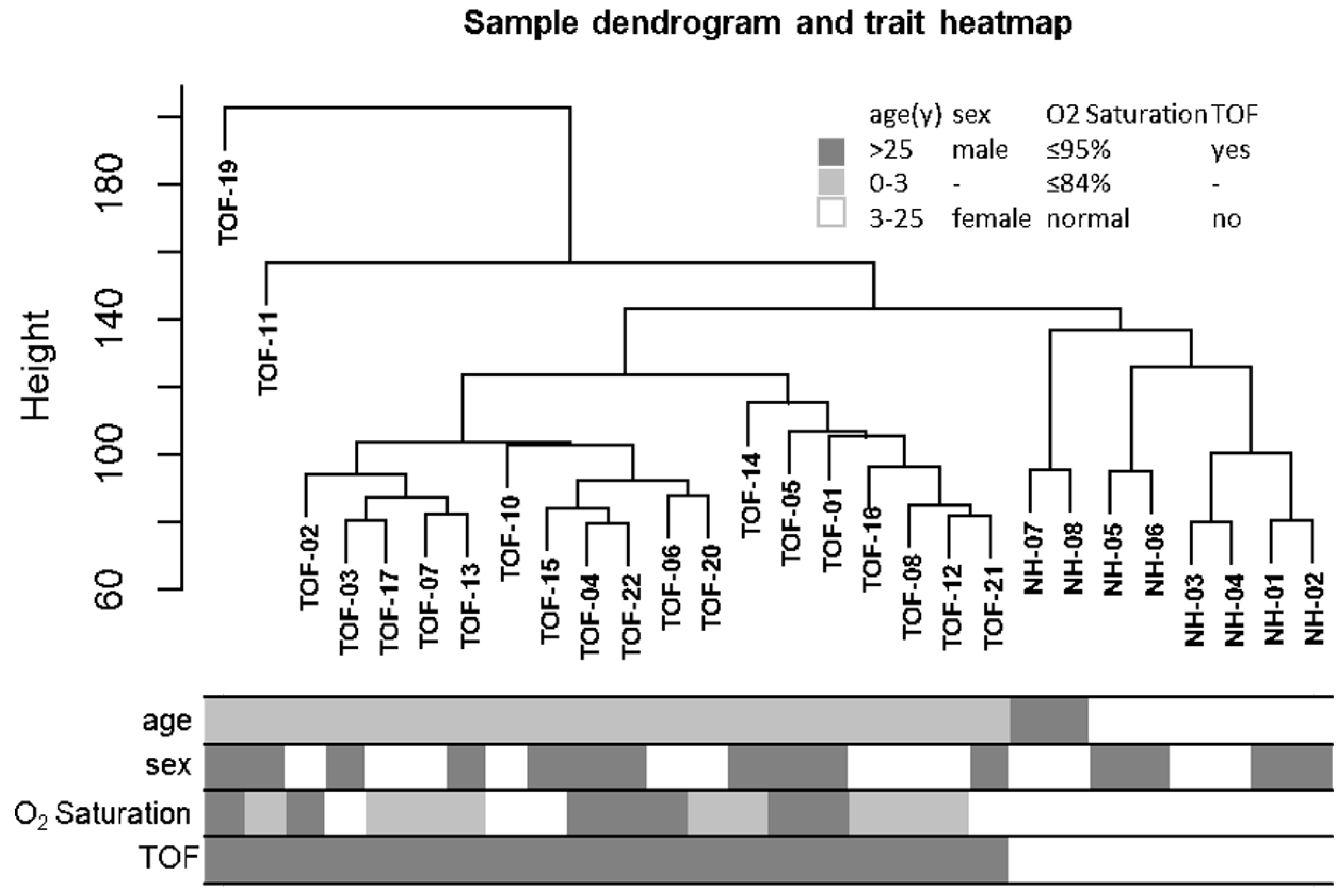
Figure 2: Sample clustering tree and clinical information heatmap. The branches in the clustering tree correspond to the samples in the squares in the heatmap below.
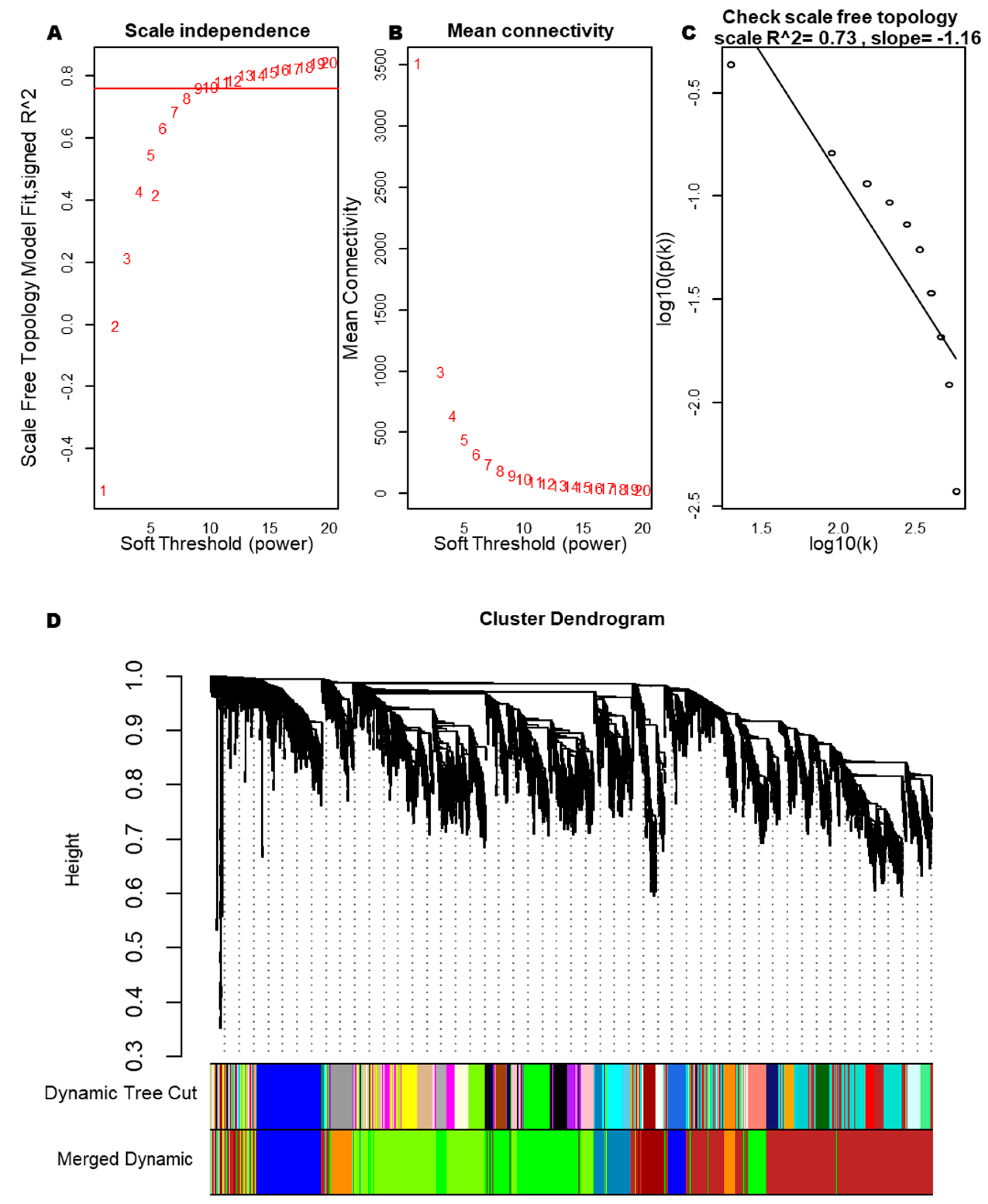
Figure 3: Soft threshold and clustering tree diagram of the gene module. (A) Relationship analysis between different soft thresholds (weighting coefficients) and network topology. (B) Relationships between different soft thresholds and the average connectivity in the network. (C) Correlation scatter plot of log10(k) and log10(p(k)). (D) Clustering tree diagram of all genes.
3.2 Module-Trait Association Analysis
We systematically evaluated the associations of eight co-expression modules with clinical characteristics (age, sex, oxygen saturation, and disease status) to identify clinically relevant modules. Notably, the ME of the darkorange and blue modules demonstrated highly significant correlations with TOF, with correlation coefficients of 0.84 and 0.78, respectively (Fig. 4A). In addition, the darkorange module also presented the highest MS value (Fig. 4B). Although the blue module ranked second in MS and showed statistically significant TOF correlation, its intramodular connectivity metrics within the WGCNA framework were suboptimal. Consequently, only the darkorange module was retained as the clinically meaningful, TOF-associated module for downstream analyses.
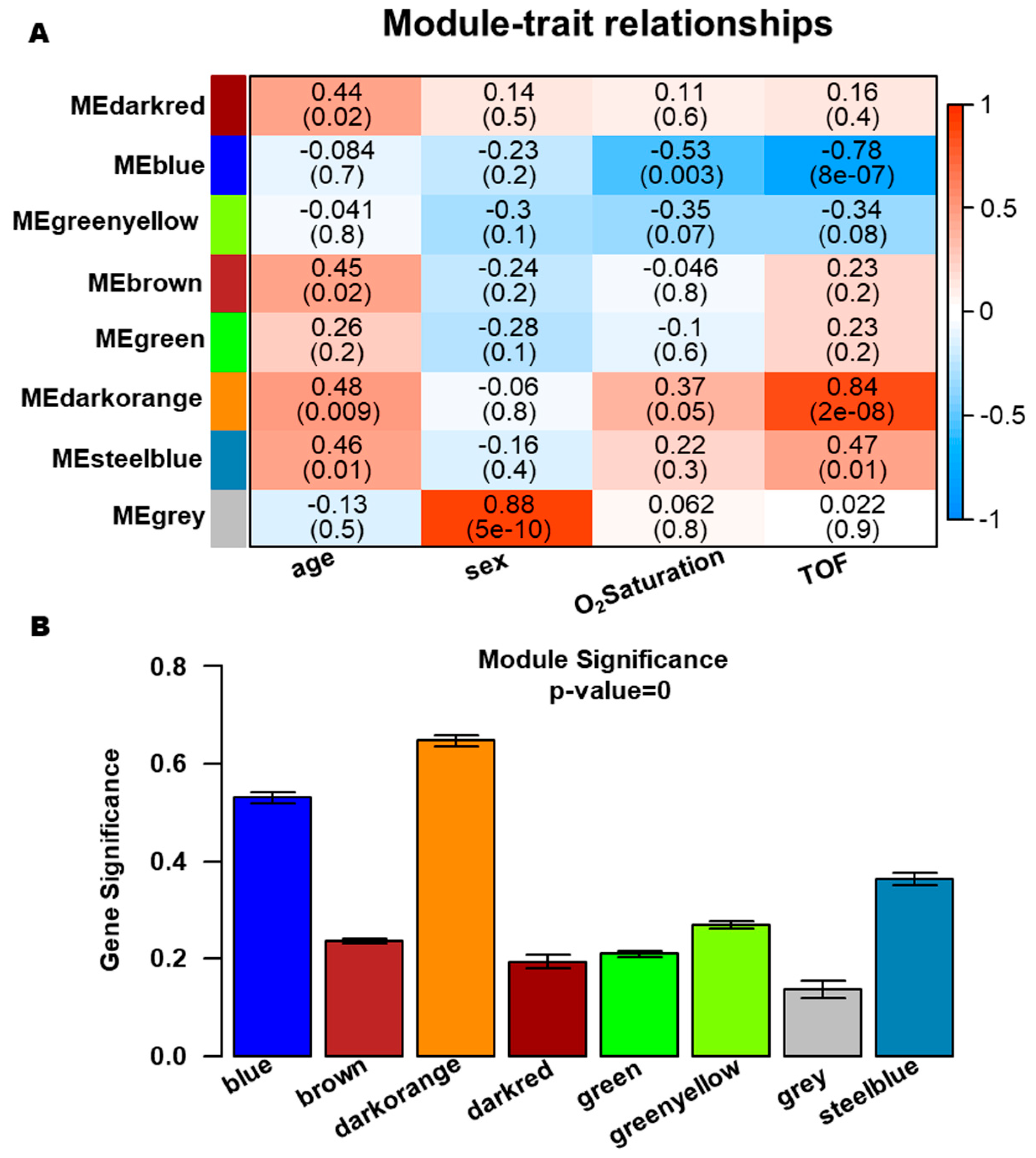
Figure 4: Identification of clinically significant gene modules. (A) Module-trait association heatmap demonstrating correlations between clinical features (x-axis) and module eigengenes (ME) values (y-axis). (B) Module significance histogram indicating the average gene significance (GS) values across eight modules.
3.3 Identification of Hub Genes in the Darkorange Module
Hub gene identification within the darkorange module was performed by analyzing intramodular connectivity metrics. For the 481 genes assigned to this key module, a robust correlation was observed between GS and module membership (MM), reinforcing the biological relevance of this module to TOF pathogenesis (Fig. 5A). Functional enrichment analysis via Metascape revealed predominant associations with organogenesis (e.g., cardiac, renal, and cranial development; epithelial morphogenesis; cell junction assembly) and WNT signaling-mediated cardiac development, alongside system-level processes (cardiac contraction and circulatory regulation) (Table 1). PPI network analysis using the STRING database identified eight Hub genes (GJA1, PTK7, PRICKLE1, SFRP1, RUNX2, FBN2, PIK3R1, and KCNJ2), each exhibiting ≥5 direct interactions within the network (Fig. 5B).
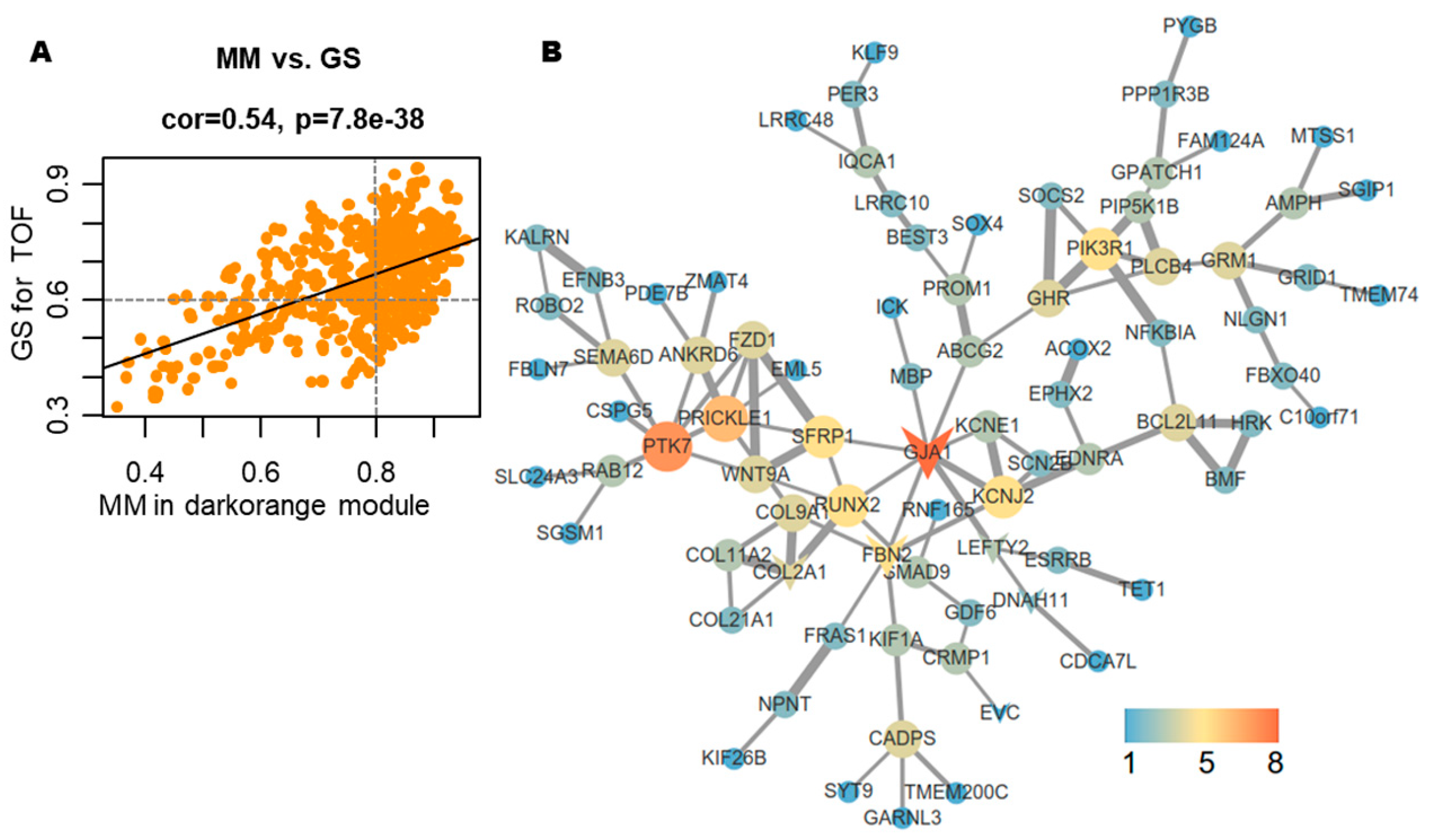
Figure 5: Identification of Hub genes. (A) Scatter plot demonstrating the correlation between module membership (MM) and gene significance (GS) values. (B) Protein-protein interaction (PPI) network of 185 WGCNA-identified Hub genes analyzed via STRING database.
Table 1: Representative enrichment analysis of the Darkorange module genes.
| Pathway Term Name | Log(q) | Names of Genes that Were Enriched in the Pathway |
|---|---|---|
| GO:0044057, regulation of system process | −5.16 | ADRB1, ADRB2, AGTR2, APOA1, CRHBP, ACE, EDNRA, FGF13, GJA1, GRM1, HRH2, INHA, KCNE1, KCNJ2, KCNJ3, MYOG, PTGER3, RGS4, SCN2B, SCT, TH, TNNT3, BVES, NLGN1, TMEM98, CACNG4, EHD3, CELF4, TMEM38A, KCNH6, NPNT, NCMAP, GABRB1, GABRP, GRIA1, GRID1, MPP2, P2RX7, PIEZO2, MYH1, MYOM1, NMUR1, UTS2, CNTNAP2, KCNA4, TSC1, SLC24A3, LRRC55, SYNE2, PTK7, RAC3, MICAL2, MTSS1, USH1C, ABI2, AVIL, TNIK, EVL, ANTXR1, CGNL1 |
| GO:0001822, kidney development | −3.48 | AGTR2, ACE, EDNRA, HMGCS2, PTK7, ROBO2, SFRP1, SOX4, TSC1, PROM1, MTSS1, BCL2L11, ADAMTS6, KIF26B, FRAS1, AMER1, NPNT, FREM2, GDF6 |
| GO:0044089, positive regulation of cellular component biogenesis | −3.34 | APOA1, P2RX7, PIK3R1, SFRP1, TSC1, ST8SIA2, HRK, MAP4K4, MTSS1, BCL2L11, ABI2, AVIL, NLGN1, FLRT3, FLRT2, CNTNAP2, RGCC, CALY, EVL, ATF7IP, LRRN1, BHLHB9, AJUBA, NAV3, BMF, CGNL1, FGF13, KCNJ2, MDK, NTF3, RAC3, RGS4, ADRB2, SLC25A5, SYT9, LDB1 |
| GO:0000902, cell morphogenesis | −3.32 | CDH8, CRMP1, EDNRA, EFNB3, MDK, NTF3, P2RX7, PIK3R1, ROBO2, ST8SIA2, KALRN, MAP4K4, ULK2, ABI2, DPYSL4, IGF2BP1, CSPG5, BVES, LAMB4, NTNG1, NLGN1, TNIK, FLRT3, FLRT2, CNTNAP2, SEMA6D, ANTXR1, LHX4, NTN5, RNF165, GPR37, NRTN, RAC3, NREP, USH1C, CTHRC1, TNNT3, SOX30, NCMAP, APOA1, CTSG, CX3CR1, CCL8, GPNMB |
| GO:0007507, heart development | −3.20 | COL2A1, ACE, EDNRA, FZD2, GJA1, PTK7, ROBO2, SNAI2, SOX4, TH, TSC1, FZD1, DNAH11, DHRS3, MICAL2, BVES, ADAMTS6, FLRT3, FLRT2, SMYD4, PRICKLE1, C1orf127, RBM24, RBM20, FREM2, LRRC10, ZC4H2 |
| GO:1901888, Regulation of cell junction assembly | −3.16 | ACE, ROBO2, SFRP1, SNAI2, TSC1, ST8SIA2, FZD1, LDB1, MAP4K4, NLGN1, FLRT3, FLRT2, CNTNAP2, LRRN1, BHLHB9, KIF1A, CDH8, FRMPD4, ABI2, CPEB3, FOXO6 |
| GO:0002009, morphogenesis of an epithelium | −2.78 | EDNRA, FZD2, GJA1, PGR, PTK7, SFRP1, SOX4, TSC1, FZD1, MICAL2, MTSS1, FLRT3, SOSTDC1, KIF26B, HHIP, FRAS1, ARHGAP24, AJUBA, CTHRC1, PRICKLE1, NPNT, FREM2, ACTG2, ROBO2, SNAI2, CCND1, RAC3, WNT9A, LDB1, TNIK, AMER1, NOTUM, MPP2, P2RX7, ANKRD6, AGTR2, RUNX2, MDK, SOX30, ACE, CCL8, FGF18, BCL2L11, ESM1, E2F8, COL2A1, MSH2, SOX15, CUL4A, ARNT2, FBXW8, CELF4, TET1, MCM2 |
| GO:0016055, WNT signaling pathway | −2.28 | CCND1, EDNRA, FZD2, PTK7, RAC3, SFRP1, WNT9A, FZD1, LDB1, TNIK, SOSTDC1, CTHRC1, AMER1, PRICKLE1, NOTUM |
| GO:0090175, Regulation of establishment of planar polarity | −2.06 | FZD2, PTK7, SFRP1, FZD1, ANKRD6, CTHRC1, PRICKLE1 |
3.4 Dysregulated Cell Polarity-Related Signaling Pathway in TOF
After the identification of the Hub genes, we observed that five Hub genes were significantly enriched in pathways governing epithelial morphogenesis, in which SFRP1, PRICKLE1 and PTK7 were mainly involved in the establishment of planar cell polarity in epithelial morphogenesis (Table 1). To further validate these results, we used two TOF transcriptomic datasets (GSE26125 and GSE35776) for GSVA. The results suggest that epithelial polarization-related pathways (embryonic epithelial morphogenesis pathway and anterior-posterior axis specification in embryos pathway) and their upstream noncanonical WNT signaling are also inactivated, whereas the canonical WNT signaling are activated (Fig. 6A). Therefore, the next analysis focused on five genes involved in the morphological and polarity development of embryonic epithelial cells: GJA1, PTK7, PRICKLE1, SFRP1 and RUNX2. Clustering of 1357 dysregulated biological processes identified through GSVA revealed predominant involvement in metabolic regulation, organogenesis, signal transduction, macromolecular assembly in organelles, and cell cycle/proliferation (Fig. 6B). These findings align with established pathobiological mechanisms underlying congenital cardiac malformations, reinforcing the hypothesis that disrupted epithelial polarity and WNT signaling dynamics contribute to TOF pathogenesis.
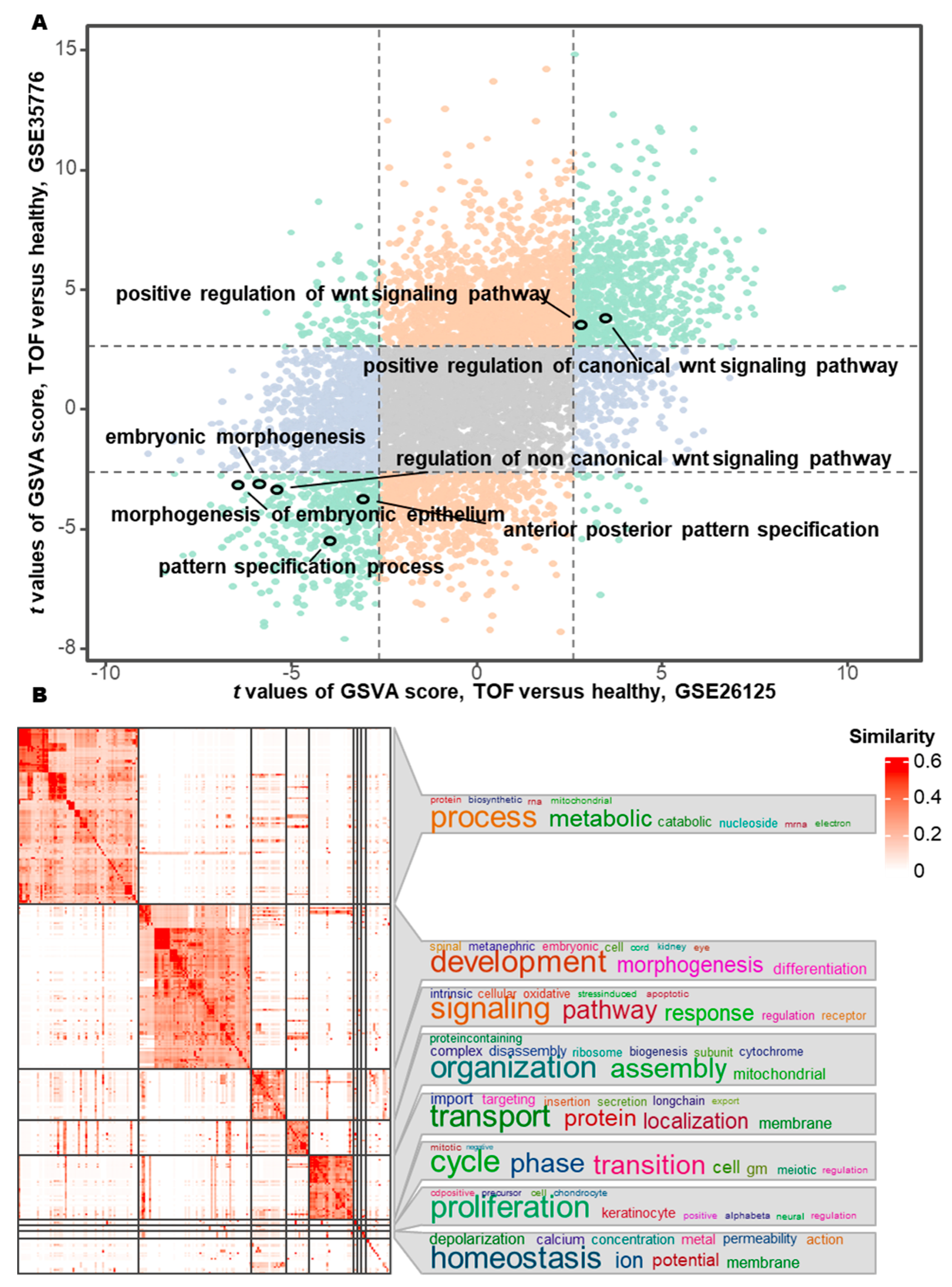
Figure 6: Differential pathway activity and functional clustering between TOF and healthy control. (A) Differential pathway activity scores between TOF and controls quantified by Gene Set Variation Analysis (GSVA) across GSE26125 and GSE35776 datasets. (B) Functional similarity clustering of differentially active pathways from GSVA.
3.5 Methylation Modifications on GJA1, PRICKLE1, PTK7, and RUNX2 in TOF
To investigate the potential causes of epithelial morphogenesis pathway disruption, we analyzed the mutational landscapes of GJA1, PTK7, PRICKLE1, SFRP1 and RUNX2 in two whole-exome sequencing cohorts (the US CHD cohort [26] and the UK TOF cohort [27]). In the US CHD cohort, two novel de novo variants were identified in conotruncal defect patients: a missense mutation (GJA1:c.121768138C>G) and a synonymous variant (PTK7:c.43106663C>T). Additionally, a loss-of-function mutation in PTK7 and SFRP1 was detected in control subjects. The UK cohort revealed two rare RUNX2 mutations (Table 2). Given the rarity of coding sequence alterations, we hypothesized alternative regulatory mechanisms and interrogated methylation profiles using the GSE62629 dataset. Strikingly, promoter hypermethylation was consistently observed in TOF cases for GJA1, PRICKLE1, RUNX2, and PTK7, suggesting transcriptional repression of these polarity regulators (Table 3).
Table 2: Hub gene mutations in the CHD cohort.
| Gene Name | UK Cohort (n = 829 TOF) | US Cohort (n = 2871 CHD) | ||
|---|---|---|---|---|
| De Novo Mutations (Case/Control) | LoF Mutations (Case/Control)* | Homozygous/Compound Heterozygous Mutations (Case/Control) | ||
| SFRP1 | 0/0 | 0/0 | 0/1 | 0/0 |
| PRICKLE1 | 0/0 | 0/0 | 0/0 | 0/0 |
| GJA1 | 0/0 | 1/0 | 0/0 | 0/0 |
| PTK7 | 0/0 | 1/0 | 0/1 | 0/0 |
| RUNX2 | 2/0 | 0/0 | 0/0 | 0/0 |
Table 3: Differential methylation analysis of Hub gene promoter regions.
| Gene Name | Transcript | Genomic Coordinates | SLIM-Corrected p Value | Differential Methylation Value (%) |
|---|---|---|---|---|
| GJA1 | NM_000165 | chr6:121756559 | 8.9E−94 | 43.1 [35.0–51.2] |
| PRICKLE1 | NM_001144883 | chr12:42878912 | 5.0E−103 | 41.4 [35.2–47.6] |
| RUNX2 | NM_001015051 | chr6: 45295333 | 1.5E−48 | 33.5 [26.9–40.1] |
| RUNX2 | NM_001015051 | chr6: 45295367 | 2.4E−69 | 38.7 [24.8–52.6] |
| PTK7 | NM_152881 | chr6: 43042751 | 7.4E−51 | 29.3 [24.1–34.5] |
3.6 Expression of GJA1 and SFRP1 in Embryonic OFT-Derived Cells
Systematic analysis of the candidate genes in TOF bulk RNA-seq data (GSE36761, 22 TOF cases vs 8 controls) revealed: All candidate genes (GJA1, PTK7, PRICKLE1, SFRP1 and RUNX2) showed significant upregulation in TOF myocardial tissues, as visualized in Supplementary Fig. S1.
We used two single-cell datasets to analyze the expression of these genes in different locations and at different times in the embryo. The results revealed the selective expression of GJA1 and SFRP1 in human embryonic hearts at 4.5–9 gestational weeks (Carnegie CS13-CS23) (Table 4) [24]. GJA1 was expressed in trabecular ventricular myocardium, whereas SFPR1 was expressed in OFT/large vessels, mediastinal mesenchyme and vessels (Table 4). Similar expression patterns were also found in mouse hearts at the embryonic age of E7.75–9.25 days (Carnegie CS9-CS13) [25]. GJA1 demonstrated ubiquitous expression across critical cardiac lineages, including multipotent progenitors, NCC, endocardial cells, lateral plate mesoderm (LPM), and paraxial mesoderm (PM) cells (Fig. 7). SFRP1 exhibited stage-specific enrichment, predominantly in multipotent progenitors, mesodermal structural cells, and NCC, LPM and epicardial cells at E8.25 and E9.25 (Fig. 6). PTK7 was only slightly expressed in the LPM and PM, RUNX2 was virtually absent in the heart, and PRICKLE1 was expressed in trace amounts in the heart (Fig. 7).
Table 4: Cell type-specific expression profiles of Hub genes in human embryonic heart.
| Gene Name | Source of Cell Tissue | Fold Change§ | p Value |
|---|---|---|---|
| GJA1 | Trabecular ventricular myocardium | 0.28 | 3.75E−23 |
| GJA1 | Atrial myocardium | −0.46 | 1.28E−27 |
| GJA1 | Outflow tract/large vessels | −0.28 | 3.13E−05 |
| GJA1 | Atrioventricular mesenchyme & vessels | −0.35 | 2.25E−06 |
| SFRP1 | Compact ventricular myocardium | −0.35 | 7.56E−10 |
| SFRP1 | Trabecular ventricular myocardium | −0.45 | 1.36E−10 |
| SFRP1 | Outflow tract/large vessels | 0.79 | 4.79E−54 |
| SFRP1 | Mediastinal mesenchyme & vessels | 1.06 | 1.02E−41 |
Given the essential roles of NCC and aSHFC in the development of the OFT, we observed the expression patterns of five Hub genes. Our analysis revealed that GJA1 was significantly expressed in the aSHFC-derived multipotent progenitors, posterior second heart field (pSHF) cells, and branchiomeric muscle progenitors, with progressive downregulation culminating in near-complete silencing at E9.5 (Fig. 7). In contrast, SFRP1 maintained stable expression in aSHF and pSHF, and both SFRP1 and GJA1 began to be expressed in large quantities in NCC at E8.25 (Fig. 7). PTK7 and PRICKLE1 were weakly expressed in these three types of cells, whereas RUNX2 was not expressed (Fig. 8). Thus, the GJA1, SFRP1, PRICKLE1 and PTK7 genes were ultimately identified as candidate pathogenic genes of TOF.
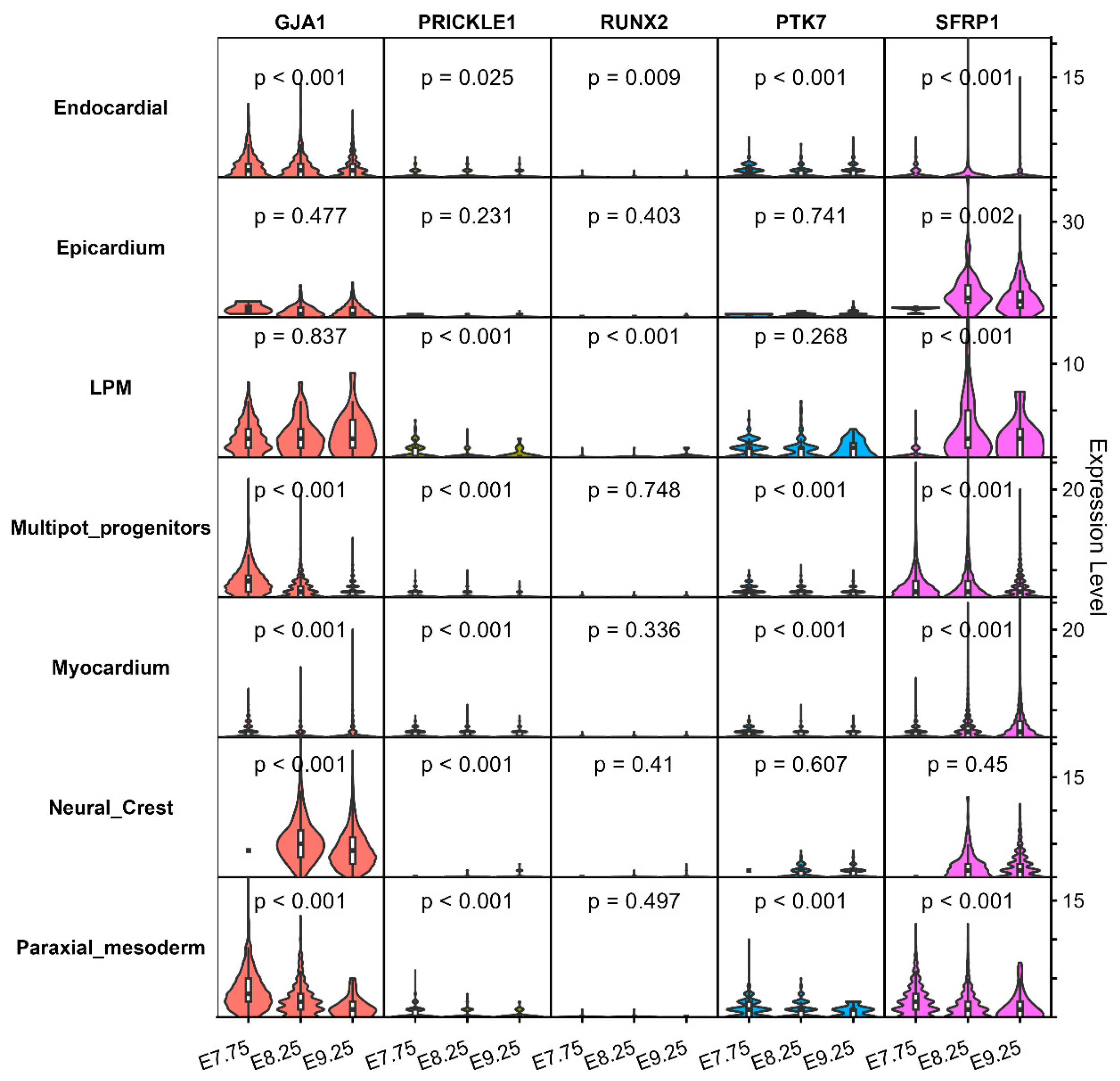
Figure 7: Expression profiles of Hub genes in the hearts of mice at E7.57, E8.25, and E9.25.
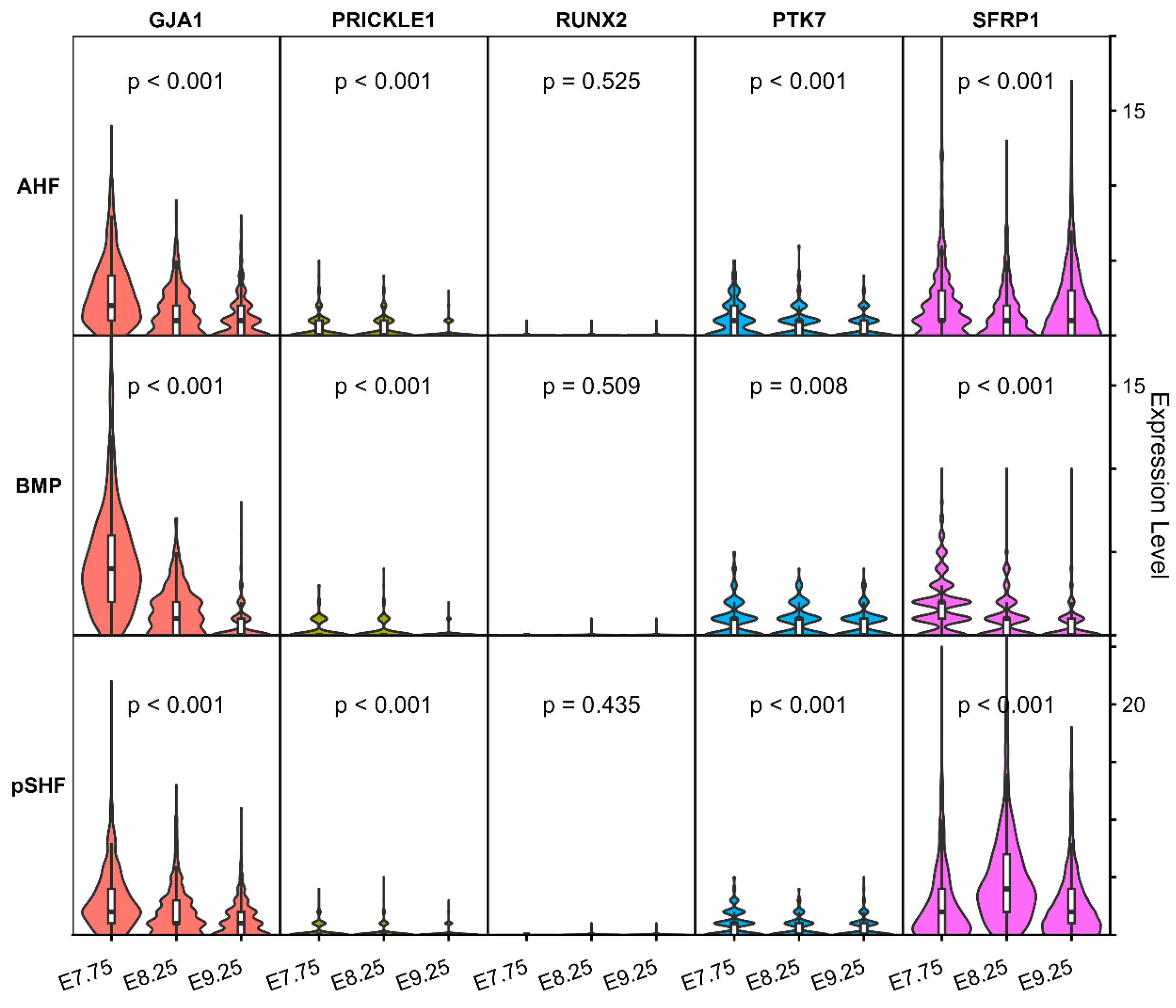
Figure 8: Expression profiles of Hub genes in mouse hearts at embryonic ages E7.57, E8.25, and E9.25.
In this study, we constructed WGCNA network to identify disease-associated modules, subsequently prioritizing eight Hub genes through PPI network analysis. Functional enrichment revealed five genes (GJA1, PTK7, PRICKLE1, SFRP1 and RUNX2) converging on epithelial morphogenesis pathways, particularly planar cell polarity (PCP) signaling. Epigenetic profiling demonstrated promoter hypermethylation in GJA1, PRICKLE1, PTK7 and RUNX2, aligning with observed PCP pathway inactivation. In addition, developmental validation across NCC and aSHFC lineages confirmed the GJA1 and SFRP1 expression, while RUNX2 showed no detectable expression in human or murine cardiac progenitors. Thus, the GJA1, SFRP1, PRICKLE1 and PTK7 genes were ultimately identified as candidate pathogenic genes of TOF. Our findings implicate epithelial polarity pathways as central mechanistic drivers of TOF, highlighting their therapeutic potential for congenital heart disease.
Cardiac development initiates from a linear epithelial tube with arteriovenous polarity, requiring precisely orchestrated epithelial morphogenesis involving cellular polarity establishment. Cell polarity is a basic attribute of various cells and manifests as asymmetry in morphology, macromolecule distribution and function, including apicobasal polarity (ABP) and PCP, which play important roles in embryonic organogenesis and development [28,29,30,31]. Notably, aSHFC exhibit hallmarks of an atypical ABP epithelium, with a single cilium at the apex and basally enriched dynamic actin-rich filopodia [32,33]. Disruption of these polarized features triggers aSHFC proliferative arrest and ectopic differentiation, ultimately impairing heart tube elongation and OFT morphogenesis [32,34,35,36,37]. In addition, epithelial cell polarity dysregulation has been shown to induce embryonic OFT truncation, perturbing ventriculoarterial alignment and culminating in the cardinal pathological features of TOF [38,39]. Therefore, the dysregulation of epithelial cell polarity and related pathways is a potential mechanism of the embryopathogenesis of TOF.
GJA1 encodes connexin 43 (Cx43), which is a critical component of myocardial gap junctions that mediates intercellular communication [40]. Online Mendelian inheritance in humans catalogs pathogenic information on GJA1 (MIM* 121014), which associated with ventricular septal defects and hypoplastic left heart syndrome. A cohort study of 152 TOF patients identified GJA1 point mutations in only 5.3% of cases (8/152). however, murine models showed neither congenital heart defects nor reduced viability upon monoallelic or biallelic Gja1 knockout [41]. Paradoxically, Rhee et al. reported that Gja1 knockout mice exhibit conus malformation and coronary artery abnormalities, with mechanistic analyses revealing Cx43-microtubule interactions as essential regulators of cellular polarity [42]. Another study has demonstrated that cardiomyocytes in children with TOF exhibit reduced Cx43 expression and an abnormally diffuse distribution across the cell surface, resembling patterns seen in immature tissues [43]. Thus, these observations suggest potential genetic mechanisms underlying GJA1 dysregulation that extend beyond conventional genetic sequence variations, possibly involving DNA methylation patterns identified in our investigation or alternative epigenetic regulatory pathways.
SFRP1, a soluble member of the SFRP family, functions as a canonical WNT signaling antagonist by competitively binding to WNT ligands through its conserved frizzled-like cysteine-rich domain [44,45]. During murine cardiac development (between E8.5 and E12.5), SFRP1 and WNT8 are localized specifically in cardiomyocytes, but neither detected in the pericardium nor the endocardium [46]. This localization pattern aligns with our observations in human and mouse embryonic hearts within the cardiogenic zone and OFT. Co-immunoprecipitation assays confirmed direct binding between SFRP1 and WNT8, establishing their functional interplay during cardiogenesis [46]. Gibb et al. reported that differentiating cardiomyocytes robustly upregulate SFRP1 expression to antagonize canonical WNT signaling, thereby facilitating OFT morphogenesis [47]. These findings suggest a critical role for SFRP1 in OFT development and its potential involvement in TOF pathogenesis.
The PRICKLE1 protein, a core component of the PCP pathway, is essential for establishing both PCP and ABP in embryonic cells [48]. PRICKLE1 knockout mice exhibit failures in distal visceral endoderm migration and mesoderm formation, leading to early embryonic lethality [48]. Additional studies demonstrate that embryonic loss of PRICKLE1 disrupts cellular polarity, causing disorganized myocardial fiber alignment and OFT malformations [49,50]. Chiapparo et al. further associated PRICKLE1 deficiency with OFT defects, identifying it as a downstream target of MESP1 that regulates migration velocity, polarity, and directional movement of cardiovascular progenitor cells [51].
PTK7, a member of the protein tyrosine kinase (PTK) family, is a transmembrane glycoprotein critical for embryonic development and tissue homeostasis [52,53]. Structurally, it comprises seven extracellular immunoglobulin-like domains, a transmembrane region, and an intracellular pseudokinase domain lacking catalytic activity, enabling its role as a co-receptor and signaling scaffold [52]. As a core PCP regulator, PTK7 coordinates tissue morphogenesis via non-canonical WNT signaling [54]. Studies in mice reveal that Ptk7 mutations can induce severe neural tube defects alongside multisystem anomalies, including cardiac malformations such as double outlet right ventricle and ventricular septal defects [55,56]. However, the role of PTK7 in OFT development and TOF pathogenesis requires further investigation.
This study has several limitations. First, as a retrospective investigation, its conclusions are inherently constrained by the sample size limitations of the database analyzed; future prospective studies with larger, multi-center cohorts are warranted to validate these findings. Second, while DNA methylation is a well-studied epigenetic modification, other mechanisms such as histone modifications, non-coding RNA regulation, and chromatin accessibility also play critical roles in gene regulation. Thus, it is necessary to explore these additional layers of epigenetic regulation in future studies. Finally, while partial molecular mechanisms of GJA1 and PRICKLE1 have been documented in prior research, comprehensive validation through integrated in vitro and in vivo experimental models remains essential to elucidate the detailed pathogenic mechanisms and functional interplay among all four candidate genes identified in this work.
In summary, our study systematically dissects dysregulated signaling networks in in TOF. We identified GJA1, SFRP1, PRICKLE1, and PTK7 as critical modulators of epithelial cell polarity in TOF. These coordinated epigenetic, transcriptional, and functional perturbations collectively define a pathogenic cascade that translates cellular polarity defects into macroscopic cardiac structural malformations. Our findings implicate epithelial polarity pathways as central mechanistic drivers of TOF, highlighting their therapeutic potential for congenital heart disease.
Acknowledgement:
Funding Statement: This work was supported by the National Natural Science Foundation of China (No. 82300451 for Zhen Wang, 82302230 for Jiawei Shi, 82202194 for Jing Wang and 82171961 for Haiyan Cao).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Jiawei Shi, Zhen Wang; data collection: Ying Bai, Shiying Li, Xin Zhang, Tianshu Liu, Liu Hong, Li Cui, Yi Zhang, Jing Ma, Juanjuan Liu, Jing Zhang; analysis and interpretation of results: Jiawei Shi, Zhen Wang; draft manuscript preparation: Jiawei Shi, Zhen Wang, Haiyan Cao, Jing Wang. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets generated and/or analyzed during the current study are available in the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/, accessed on 01 April 2025).
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://www.techscience.com/doi/10.32604/chd.2025.064950/s1.
References
1. Crea F. Hot topics in congenital heart disease: tetralogy of fallot, Ross operation, immunodeficiency, cardiac arrest, and end-stage heart failure. Eur Heart J. 2023;44(34):3201–4. doi:10.1093/eurheartj/ehad549. [Google Scholar] [CrossRef]
2. Edo B, Eli C, Herra J, Amna Q, Louis JS, Rajab TK. Tetralogy of fallot: anatomy, physiology, and outcomes. Congenit Heart Dis. 2024;19(6):541–62. doi:10.32604/chd.2025.059788. [Google Scholar] [CrossRef]
3. Liu Y, Chen S, Zühlke L, Black GC, Choy MK, Li N, et al. Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies. Int J Epidemiol. 2019;48(2):455–63. doi:10.1093/ije/dyz009. [Google Scholar] [CrossRef]
4. Offen S, Puranik R, Baker D, Cordina R, Chard R, Celermajer DS. Prevalence and determinants of tricuspid regurgitation after repair of tetralogy of Fallot. Int J Cardiol. 2023;372:55–9. doi:10.1016/j.ijcard.2022.11.039. [Google Scholar] [CrossRef]
5. Shi JW, Cao H, Hong L, Ma J, Cui L, Zhang Y, et al. Diagnostic yield of whole exome data in fetuses aborted for conotruncal malformations. Prenat Diagn. 2022;42(7):852–61. doi:10.1002/pd.6147. [Google Scholar] [CrossRef]
6. Forman J, Beech R, Slugantz L, Donnellan A. A review of tetralogy of fallot and postoperative management. Crit Care Nurs Clin N Am. 2019;31(3):315–28. doi:10.1016/j.cnc.2019.05.003. [Google Scholar] [CrossRef]
7. Love BA. Management of infants with tetralogy of fallot: questioning conventional wisdom. J Am Coll Cardiol. 2023;82(7):628–30. doi:10.1016/j.jacc.2023.06.008. [Google Scholar] [CrossRef]
8. Miller JR, Stephens EH, Goldstone AB, Glatz AC, Kane L, Van Arsdell GS, et al. The American association for thoracic surgery (AATS) 2022 expert consensus document: management of infants and neonates with tetralogy of fallot. J Thorac Cardiovasc Surg. 2023;165(1):221–50. doi:10.1016/j.jtcvs.2022.07.025. [Google Scholar] [CrossRef]
9. Zulibiya A, Wen J, Yu H, Chen X, Xu L, Ma X, et al. Single-cell RNA sequencing reveals potential for endothelial-to-mesenchymal transition in tetralogy of fallot. Congenit Heart Dis. 2023;18(6):611–25. doi:10.32604/chd.2023.047689. [Google Scholar] [CrossRef]
10. Gou Z, Zhou Y, Jia H, Yang Z, Zhang Q, Yan X. Prenatal diagnosis and mRNA profiles of fetal tetralogy of Fallot. BMC Pregnancy Childbirth. 2022;22(1):853. doi:10.1186/s12884-022-05190-0. [Google Scholar] [CrossRef]
11. Lahm H, Schön P, Doppler S, Dreßen M, Cleuziou J, Deutsch MA, et al. Tetralogy of fallot and hypoplastic left heart syndrome—complex clinical phenotypes meet complex genetic networks. Curr Genomics. 2015;16(3):141–58. doi:10.2174/1389202916666150303232520. [Google Scholar] [CrossRef]
12. Grunert M, Dorn C, Schueler M, Dunkel I, Schlesinger J, Mebus S, et al. Rare and private variations in neural crest, apoptosis and sarcomere genes define the polygenic background of isolated Tetralogy of Fallot. Hum Mol Genet. 2014;23(12):3115–28. doi:10.1093/hmg/ddu021. [Google Scholar] [CrossRef]
13. Reuter MS, Chaturvedi RR, Jobling RK, Pellecchia G, Hamdan O, Sung WWL, et al. Clinical genetic risk variants inform a functional protein interaction network for tetralogy of fallot. Circ Genom Precis Med. 2021;14(4):e003410. doi:10.1161/CIRCGEN.121.003410. [Google Scholar] [CrossRef]
14. Maertens A, Tran V, Kleensang A, Hartung T. Weighted gene correlation network analysis (WGCNA) reveals novel transcription factors associated with bisphenol a dose-response. Front Genet. 2018;9:508. doi:10.3389/fgene.2018.00508. [Google Scholar] [CrossRef]
15. Zhang T, Wong G. Gene expression data analysis using Hellinger correlation in weighted gene co-expression networks (WGCNA). Comput Struct Biotechnol J. 2022;20:3851–63. doi:10.1016/j.csbj.2022.07.018. [Google Scholar] [CrossRef]
16. Wang Z, Chen X, Li C, Tang W. Application of weighted gene co-expression network analysis to identify novel key genes in diabetic nephropathy. J Diabetes Investig. 2022;13(1):112–24. doi:10.1111/jdi.13628. [Google Scholar] [CrossRef]
17. Schussler O, Gharibeh L, Mootoosamy P, Murith N, Tien V, Rougemont AL, et al. Cardiac neural crest cells: their rhombomeric specification, migration, and association with heart and great vessel anomalies. Cell Mol Neurobiol. 2021;41(3):403–29. doi:10.1007/s10571-020-00863-w. [Google Scholar] [CrossRef]
18. Dominguez MH, Krup AL, Muncie JM, Bruneau BG. Graded mesoderm assembly governs cell fate and morphogenesis of the early mammalian heart. Cell. 2023;186(3):479–96.e23.doi:10.1016/j.cell.2023.01.001. [Google Scholar] [CrossRef]
19. Bittel DC, Butler MG, Kibiryeva N, Marshall JA, Chen J, Lofland GK, et al. Gene expression in cardiac tissues from infants with idiopathic conotruncal defects. BMC Med Genom. 2011;4:1. doi:10.1186/1755-8794-4-1. [Google Scholar] [CrossRef]
20. O’Brien JE Jr, Kibiryeva N, Zhou XG, Marshall JA, Lofland GK, Artman M, et al. Noncoding RNA expression in myocardium from infants with tetralogy of Fallot. Circ Cardiovasc Genet. 2012;5(3):279–86. doi:10.1161/CIRCGENETICS.111.961474. [Google Scholar] [CrossRef]
21. Grunert M, Dorn C, Cui H, Dunkel I, Schulz K, Schoenhals S, et al. Comparative DNA methylation and gene expression analysis identifies novel genes for structural congenital heart diseases. Cardiovasc Res. 2016;112(1):464–77. doi:10.1093/cvr/cvw195. [Google Scholar] [CrossRef]
22. Shi J, Zhang P, Liu L, Min X, Xiao Y. Weighted gene coexpression network analysis identifies a new biomarker of CENPF for prediction disease prognosis and progression in nonmuscle invasive bladder cancer. Mol Genet Genomic Med. 2019;7(11):e982. doi:10.1002/mgg3.982. [Google Scholar] [CrossRef]
23. Li Y, Du J, Liu B, She Q. Identifying key genes and related molecules as potential biomarkers in human dilated cardiomyopathy by comprehensive bioinformatics analysis. Cardiovasc Innov Appl. 2023;8(1):996. doi:10.15212/cvia.2023.0018. [Google Scholar] [CrossRef]
24. Asp M, Giacomello S, Larsson L, Wu C, Fürth D, Qian X, et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell. 2019;179(7):1647–60.e19. doi:10.1016/j.cell.2019.11.025. [Google Scholar] [CrossRef]
25. de Soysa TY, Ranade SS, Okawa S, Ravichandran S, Huang Y, Salunga HT, et al. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature. 2019;572(7767):120–4. doi:10.1038/s41586-019-1414-x. [Google Scholar] [CrossRef]
26. Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, et al. Contribution of rare inherited and de novo variants in 2, 871 congenital heart disease probands. Nat Genet. 2017;49(11):1593–601. doi:10.1038/ng.3970. [Google Scholar] [CrossRef]
27. Page DJ, Miossec MJ, Williams SG, Monaghan RM, Fotiou E, Cordell HJ, et al. Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of fallot. Circ Res. 2019;124(4):553–63. doi:10.1161/CIRCRESAHA.118.313250. [Google Scholar] [CrossRef]
28. Karner C, Wharton KA Jr, Carroll TJ. Planar cell polarity and vertebrate organogenesis. Semin Cell Dev Biol. 2006;17(2):194–203. doi:10.1016/j.semcdb.2006.05.003. [Google Scholar] [CrossRef]
29. Shi DL. Planar cell polarity regulators in asymmetric organogenesis during development and disease. J Genet Genomics. 2023;50(2):63–76. doi:10.1016/j.jgg.2022.06.007. [Google Scholar] [CrossRef]
30. Shi DL. Wnt/planar cell polarity signaling controls morphogenetic movements of gastrulation and neural tube closure. Cell Mol Life Sci. 2022;79(12):586. doi:10.1007/s00018-022-04620-8. [Google Scholar] [CrossRef]
31. Mlodzik M. Planar cell polarity: moving from single cells to tissue-scale biology. Development. 2020;147(24):dev186346. doi:10.1242/dev.186346. [Google Scholar] [CrossRef]
32. Francou A, Saint-Michel E, Mesbah K, Kelly RG. TBX1 regulates epithelial polarity and dynamic basal filopodia in the second heart field. Development. 2014;141(22):4320–31. doi:10.1242/dev.115022. [Google Scholar] [CrossRef]
33. Yang D, Gomez-Garcia J, Funakoshi S, Tran T, Fernandes I, Bader GD, et al. Modeling human multi-lineage heart field development with pluripotent stem cells. Cell Stem Cell. 2022;29(9):1382–401.e8. doi:10.1016/j.stem.2022.08.007. [Google Scholar] [CrossRef]
34. Cortes C, Francou A, De Bono C, Kelly RG. Epithelial properties of the second heart field. Circ Res. 2018;122(1):142–54. doi:10.1161/CIRCRESAHA.117.310838. [Google Scholar] [CrossRef]
35. Leung C, Liu Y, Lu X, Kim M, Drysdale TA, Feng Q. Rac1 signaling is required for anterior second heart field cellular organization and cardiac outflow tract development. J Am Heart Assoc. 2015;5(1):e002508. doi:10.1161/JAHA.115.002508. [Google Scholar] [CrossRef]
36. Schmidt C, Deyett A, Ilmer T, Haendeler S, Torres Caballero A, Novatchkova M, et al. Multi-chamber cardioids unravel human heart development and cardiac defects. Cell. 2023;186(25):5587–605.e27. doi:10.1016/j.cell.2023.10.030. [Google Scholar] [CrossRef]
37. Yamaguchi N, Chang EW, Lin Z, Shekhar A, Bu L, Khodadadi-Jamayran A, et al. An anterior second heart field enhancer regulates the gene regulatory network of the cardiac outflow tract. Circulation. 2023;148(21):1705–22. doi:10.1161/CIRCULATIONAHA.123.065700. [Google Scholar] [CrossRef]
38. Zhao Y, Kang X, Gao F, Guzman A, Lau RP, Biniwale R, et al. Gene-environment regulatory circuits of right ventricular pathology in tetralogy of fallot. J Mol Med. 2019;97(12):1711–22. doi:10.1007/s00109-019-01857-y. [Google Scholar] [CrossRef]
39. Tambi R, Zehra B, Nandkishore S, Sharafat S, Kader F, Nassir N, et al. Single-cell reconstruction and mutation enrichment analysis identifies dysregulated cardiomyocyte and endothelial cells in congenital heart disease. Physiol Genomics. 2023;55(12):634–46. doi:10.1152/physiolgenomics.00070.2023. [Google Scholar] [CrossRef]
40. Palatinus JA, Valdez S, Taylor L, Whisenant C, Selzman CH, Drakos SG, et al. GJA1-20k rescues Cx43 localization and arrhythmias in arrhythmogenic cardiomyopathy. Circ Res. 2023;132(6):744–6. doi:10.1161/circresaha.122.322294. [Google Scholar] [CrossRef]
41. Huang GY, Xie LJ, Linask KL, Zhang C, Zhao XQ, Yang Y, et al. Evaluating the role of connexin43 in congenital heart disease: screening for mutations in patients with outflow tract anomalies and the analysis of knock-in mouse models. J Cardiovasc Dis Res. 2011;2(4):206–12. doi:10.4103/0975-3583.89804. [Google Scholar] [CrossRef]
42. Rhee DY, Zhao XQ, Francis RJB, Huang GY, Mably JD, Lo CW. Connexin 43 regulates epicardial cell polarity and migration in coronary vascular development. Development. 2009;136(18):3185–93. doi:10.1242/dev.032334. [Google Scholar] [CrossRef]
43. Kołcz J, Drukała J, Bzowska M, Rajwa B, Korohoda W, Malec E. The expression of connexin 43 in children with tetralogy of fallot. Cell Mol Biol Lett. 2005;10(2):287–303. [Google Scholar]
44. Baharudin R, Tieng FYF, Lee LH, Ab Mutalib NS. Epigenetics of SFRP1: the dual roles in human cancers. Cancers. 2020;12(2):445. doi:10.3390/cancers12020445. [Google Scholar] [CrossRef]
45. Yamamoto T, Kambayashi Y, Otsuka Y, Afouda BA, Giuraniuc C, Michiue T, et al. Positive feedback regulation of frizzled-7 expression robustly shapes a steep Wnt gradient in Xenopus heart development, together with sFRP1 and heparan sulfate. eLife. 2022;11:e73818. doi:10.7554/eLife.73818. [Google Scholar] [CrossRef]
46. Jaspard B, Couffinhal T, Dufourcq P, Moreau C, Duplàa C. Expression pattern of mouse sFRP-1 and mWnt-8 gene during heart morphogenesis. Mech Dev. 2000;90(2):263–7. doi:10.1016/s0925-4773(99)00236-1. [Google Scholar] [CrossRef]
47. Gibb N, Lavery DL, Hoppler S. sfrp1 promotes cardiomyocyte differentiation in Xenopus via negative-feedback regulation of Wnt signalling. Development. 2013;140(7):1537–49. doi:10.1242/dev.088047. [Google Scholar] [CrossRef]
48. Tao H, Suzuki M, Kiyonari H, Abe T, Sasaoka T, Ueno N. Mouse prickle1, the homolog of a PCP gene, is essential for epiblast apical-basal polarity. Proc Natl Acad Sci U S A. 2009;106(34):14426–31. doi:10.1073/pnas.0901332106. [Google Scholar] [CrossRef]
49. Liu C, Lin C, Gao C, May-Simera H, Swaroop A, Li T. Null and hypomorph Prickle1 alleles in mice phenocopy human Robinow syndrome and disrupt signaling downstream of Wnt5a. Biol Open. 2014;3(9):861–70. doi:10.1242/bio.20148375. [Google Scholar] [CrossRef]
50. Gibbs BC, Damerla RR, Vladar EK, Chatterjee B, Wan Y, Liu X, et al. Prickle1 mutation causes planar cell polarity and directional cell migration defects associated with cardiac outflow tract anomalies and other structural birth defects. Biol Open. 2016;5(3):323–35. doi:10.1242/bio.015750. [Google Scholar] [CrossRef]
51. Chiapparo G, Lin X, Lescroart F, Chabab S, Paulissen C, Pitisci L, et al. Mesp1 controls the speed, polarity, and directionality of cardiovascular progenitor migration. J Cell Biol. 2016;213(4):463–77. doi:10.1083/jcb.201505082. [Google Scholar] [CrossRef]
52. Berger H, Wodarz A, Borchers A. PTK7 faces the Wnt in development and disease. Front Cell Dev Biol. 2017;5:31. doi:10.3389/fcell.2017.00031. [Google Scholar] [CrossRef]
53. Jin Z, Guo T, Zhang X, Wang X, Liu Y. PTK7: an underestimated contributor to human cancer. Front Oncol. 2024;14:1448695. doi:10.3389/fonc.2024.1448695. [Google Scholar] [CrossRef]
54. Berger H, Breuer M, Peradziryi H, Podleschny M, Jacob R, Borchers A. PTK7 localization and protein stability is affected by canonical Wnt ligands. J Cell Sci. 2017;130(11):1890–903. doi:10.1242/jcs.198580. [Google Scholar] [CrossRef]
55. Paudyal A, Damrau C, Patterson VL, Ermakov A, Formstone C, Lalanne Z, et al. The novel mouse mutant, chuzhoi, has disruption of Ptk7 protein and exhibits defects in neural tube, heart and lung development and abnormal planar cell polarity in the ear. BMC Dev Biol. 2010;10:87. doi:10.1186/1471-213X-10-87. [Google Scholar] [CrossRef]
56. Golubkov VS, Aleshin AE, Strongin AY. Potential relation of aberrant proteolysis of human protein tyrosine kinase 7 (PTK7) chuzhoi by membrane type 1 matrix metalloproteinase (MT1-MMP) to congenital defects. J Biol Chem. 2011;286(23):20970–6. doi:10.1074/jbc.M111.237669. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools