
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Key Steps and Catalyst Performance for Conversion of Cellulose to Isosorbide: A Review
State Key Laboratory of Heavy Oil Processing, China University of Petroleum-Beijing, Beijing, 102249, China
* Corresponding Author: Ying Yang. Email:
(This article belongs to the Special Issue: Recent Advances on Renewable Materials)
Journal of Renewable Materials 2025, 13(1), 49-78. https://doi.org/10.32604/jrm.2024.056644
Received 27 July 2024; Accepted 20 September 2024; Issue published 20 January 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Upgrading of abundant cellulosic biomass to isosorbide can reduce the dependence on limited fossil resources and provide a sustainable way to produce isosorbide, utilized for polymers, medicine and health care product synthesis. This review comprehensively examines the key steps and catalytic systems involved in the conversion of cellulose to isosorbide. Initially, the reaction pathway from cellulose to isosorbide is elucidated, emphasizing three critical steps: cellulose hydrolysis, glucose hydrogenation, and the two-step dehydration of sorbitol to produce isosorbide. Additionally, the activation energy and acidic sites during cellulose hydrolysis, the impact of metal particle size and catalyst support on hydrogenation, and the effects of catalyst acidity, pore structure, and reaction conditions on sorbitol dehydration have been thoroughly examined. Finally, the progress made in cellulose conversion to isosorbide is summarized, current challenges are highlighted, and future development trends are projected in this review.Graphic Abstract
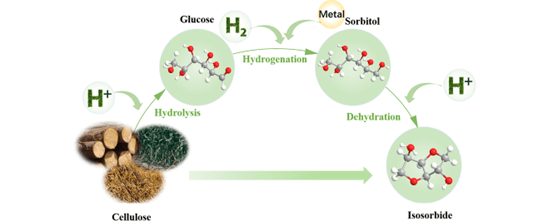
Keywords
The increasing scarcity of fossil resources and the exacerbation of environmental issues have spurred a growing interest in finding renewable alternatives. Biomass resources, due to their abundance and renewability, have emerged as ideal substitutes for fossil resources. Among these, cellulose is the most prevalent and plentiful part of lignocellulosic biomass, found in wood, agricultural residues, and other plant-based waste materials [1] (Fig. 1a). Efficient utilization of cellulose not only helps alleviate resource shortages but also reduces environmental pollution. Consequently, plenty of work has been invested into creating environmentally friendly and productive cellulose conversion techniques to synthesize a range of industrially important chemicals. Isosorbide is a pivotal value-added product from cellulose with significant applications in numerous fields. For instance, it serves as a key component in the synthesis of polymers like polycarbonates, polyurethanes, and epoxy resins. Additionally, it may act as a pharmaceutical intermediate, as a base for the generation of nontoxic organophosphorus flame retardants [2] and an additive to enhance the flexibility of polymers (Fig. 1b), indicating its vast application potential [3].
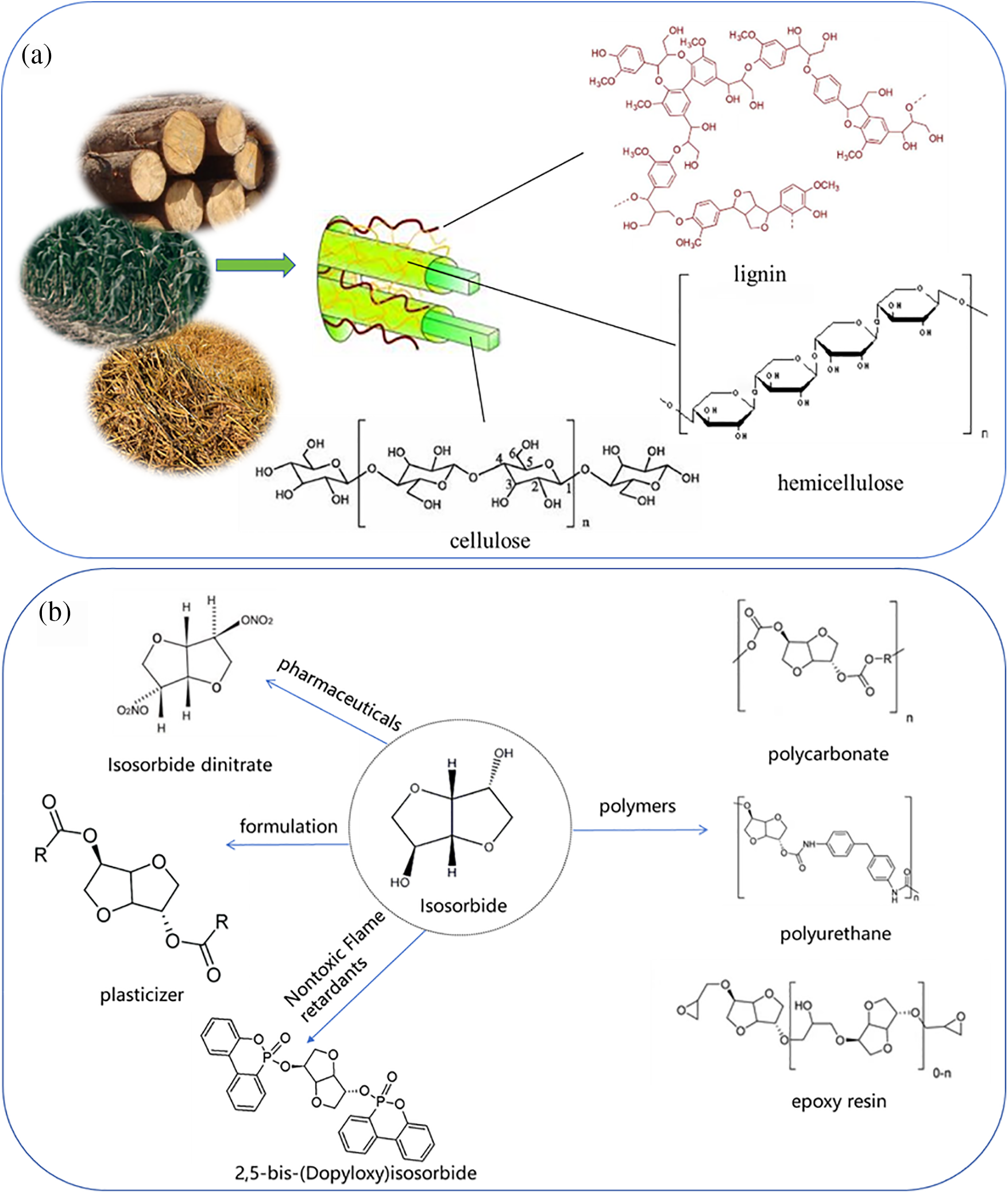
Figure 1: (a) Composition of cellulose and (b) derivatives of isosorbide
Isosorbide, a valuable biobased platform chemical, has gained an extensive amount of attention for its prospective usage in pharmaceuticals, bioplastics, and other high-performance materials. The preparation of isosorbide typically involves the hydrolysis of cellulose-derived materials, hydrogenation of glucose, and subsequent dehydration of sorbitol. Among various methods, acid-catalyzed processes are widely recognized for their efficiency and effectiveness. The initial step in the synthesis of isosorbide involves the disintegration of cellulose’s glycosidic linkages, which is catalyzed by acid to yield glucose. Strong acids including sulfuric acid and phosphoric acid are frequently utilized as catalysts in this process. The resulting glucose is then subjected to hydrogenation over metal catalysts to produce sorbitol. Finally, sorbitol undergoes a two-step acid-catalyzed dehydration to form isosorbide [4]. The acid-catalyzed method is particularly advantageous for large-scale industrial production due to relatively mature technology and high yield of isosorbide produced. The robustness and efficiency of this process make it a promising candidate for the sustainable production of isosorbide, thereby expanding its application potential in various industries.
Considerable advancements have been achieved in the methodology for isosorbide preparation, with in-depth studies conducted on the diversity of raw materials, reaction pathways, synthesis processes, and catalyst optimization. However, comprehensive reviews focusing on the details of key steps and the performance of catalysts in the cellulose-to-isosorbide conversion process are still lacking. A thorough overview of the conversion pathways from cellulose to isosorbide is provided here in. Advancements in three critical steps: cellulose hydrolysis, glucose hydrogenation, and the two-step dehydration of sorbitol are documented. Additionally, the processes, advantages and disadvantages of various catalysts used in these processes along with the existing challenges are analyzed. A valuable reference for future research and industrial applications in the isosorbide synthesis domain is provided.
The transformation process of cellulose to isosorbide is illustrated in Fig. 2. Cellulose, a linear polymer made up of β-1,4-glycosidic linkages connecting D-glucose units, undergoes hydrolysis under acid catalysis, resulting in the splitting apart of these glycosidic linkages and glucose is formed. Subsequently, glucose is hydrogenated in the existence of a metal catalyst to yield sorbitol. Sorbitol then undergoes a two-step high-temperature dehydration process in the presence of a strong acid catalyst, leading to the formation of isosorbide [5]. During this process, glucose can readily isomerize to fructose, which may further undergo dehydration and hydration, producing levulinic acid, formic acid, and 5-hydroxymethylfurfural (HMF) as secondary products.
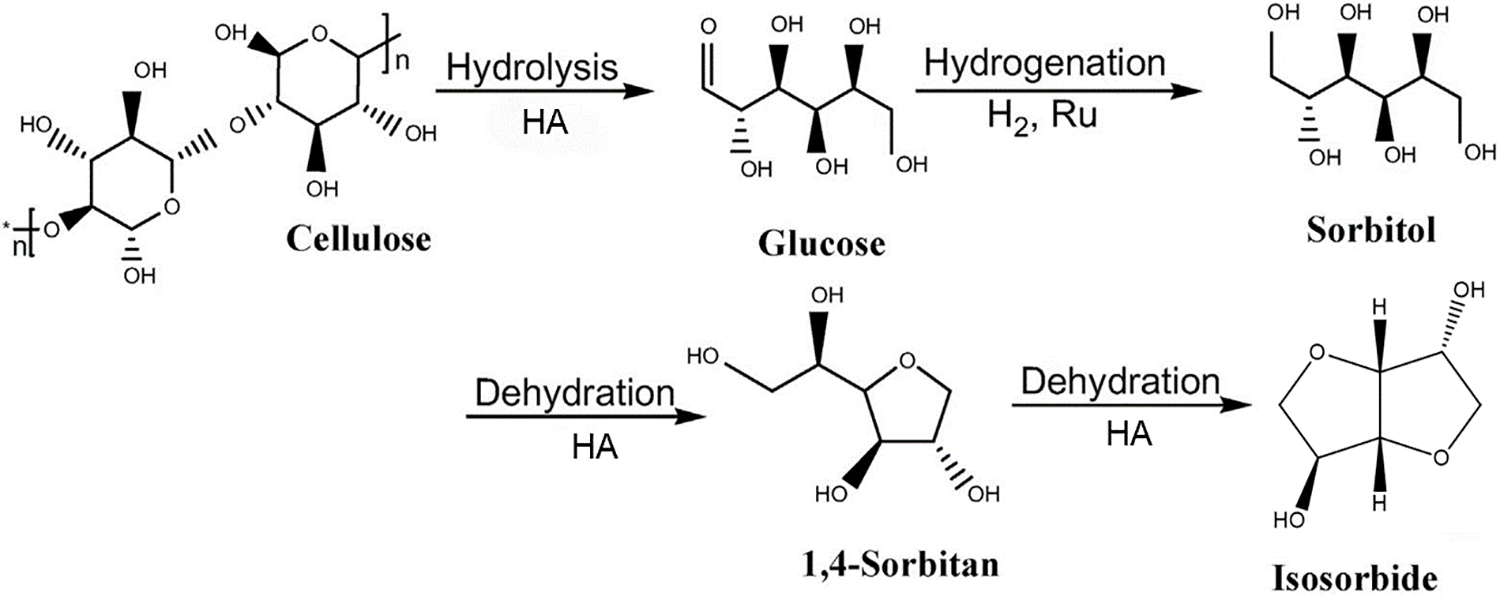
Figure 2: Reaction pathway for the conversion of cellulose to isosorbide. Adapted with permission from Reference [5]. Copyright ©2023, Elsevier
Three crucial processes are involved in converting cellulose to isosorbide: cellulose goes through hydrolysis to glucose, glucose goes through hydrogenation to sorbitol, and sorbitol undergoes dehydration twice to provide isosorbide. The significance of these steps is analyzed as follows:
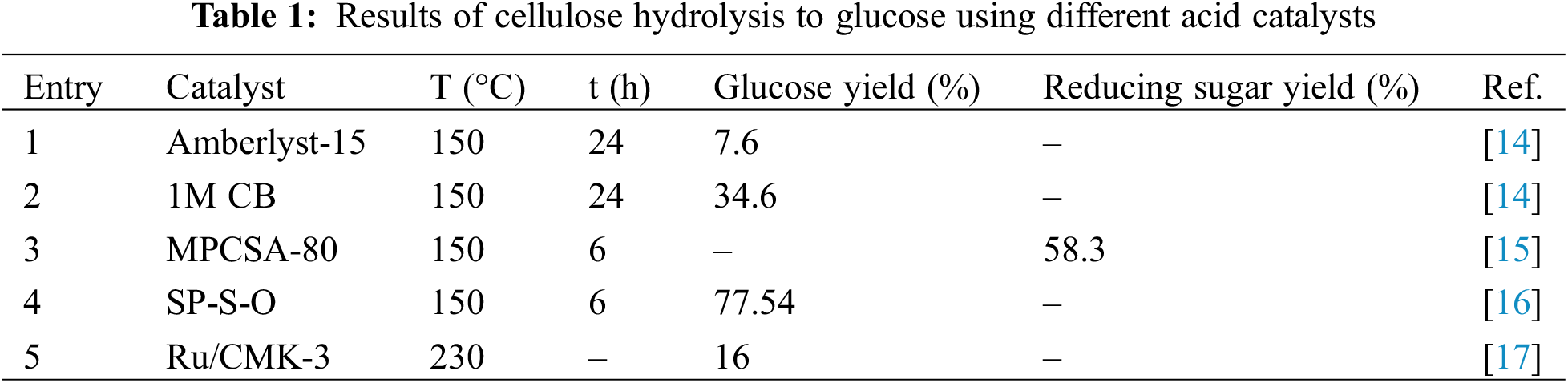
Cellulose, made up of D-pyranoglucose units connected by β-1,4-glycosidic linkages, exhibits a highly ordered crystalline structure. The hydrolysis of cellulose into glucose forms the basis of the entire reaction process, determining the substrate supply and overall reaction efficiency. Kinetic studies have shown that the activation energy for cellulose hydrolysis typically ranges from 120 to 180 kJ/mol. For instance, the characteristics and kinetics for the generation of isosorbide from five types of lignocellulosic biomass-pine sawdust, fern stem, wheat straw, sugarcane bagasse, and jute stick have been investigated using thermogravimetric analysis (TGA) [6]. The distributed activation energy model (DAEM) estimated the apparent average activation energies for these materials to range from 171.04 to 179.54 kJ/mol. Additionally, average activation energies of 194.54 ± 0.76 kJ/mol for hydrolysis of spruce cellulose and 193.17 ± 0.75 kJ/mol for birch cellulose have been reported [7]. Similarly, the hydrolysis of corn straw cellulose exhibits higher activation energies, ranging from 186.9 to 203.6 kJ/mol, compared to those for soybean husk cellulose [8]. The high crystallinity and stable hydrogen-bond network of cellulose necessitate substantial energy for hydrolysis. Therefore, the hydrolysis of cellulose to glucose is considered a crucial step in the overall conversion to isosorbide. Enhancing the efficiency of cellulose hydrolysis is pivotal for improving the yield of isosorbide.
An essential intermediate step in the transformation of cellulose into isosorbide is the hydrogenation of glucose to sorbitol, directly influencing the efficiency of subsequent sorbitol dehydration to isosorbide. The activation energy for this hydrogenation process is typically around 50 kJ/mol. The kinetics of glucose hydrogenation have often been investigated, and optimal reaction processes proposed. The catalytic hydrogenation of a 40 wt% D-glucose aqueous solution in a high-pressure trickling bed reactor has been examined [9]. A kieselgur-supported nickel catalyst (12% NiO, 2% Cr2O3) with a particle diameter of 2–3 mm was utilized to carry out the reaction. Under pressures ranging from 0.5 to 10 MPa, the conditions of operation varied from 388 to 438 K. The reaction showed a hydrogen order of 0.65 and was determined to be initial-order with regard to D-glucose. The range of the apparent activation energy ranged 24–49 kJ/mol.
The catalytic conversion of D-glucose to D-sorbitol using a 5% Ru/C catalyst in a semi-batch slurry high-pressure reactor at 373–403 K and hydrogen pressures of 4.0–7.5 MPa has been examined [10]. The hydrogenation of D-glucose over 5% Ru/C displays near 100% selectivity towards D-sorbitol, with a first-order dependence on hydrogen. The reaction order for D-glucose varies with concentration: initial-order in tiny quantities (approximately 0.3 mol/L) and zero-ordering in greater quantities. To model the kinetic data, three reasonable rate models based on LHHW kinetics have been proposed, considering the surface interaction was rate-determining. Model 1 involves non-competitive adsorption of hydrogen and D-glucose; Model 2 involves competitive adsorption of molecular hydrogen and D-glucose; and for Model 3, competitive adsorption of atomically chemisorbed hydrogen and D-glucose is assumed. Although there is no statistical distinction among these models, Model 3 has been slightly preferred. Model 2 is deemed to be less likely due to its assumption of an interaction with molecularly adsorbed hydrogen. Additionally, only Model 3 predicted the entropy reduction for D-glucose adsorption. The observed activation energy for the reaction was 55 kJ/mol.
An essential step in the synthesis of isosorbide is the dehydration of sorbitol, significantly influencing the yield of the final product. The reaction pathway for the dehydration of sorbitol to isosorbide consists of two steps, as illustrated in Fig. 3. During the first dehydration step, sorbitol can form several intermediates, including 1,4-sorbitan, 3,6-sorbitan, 1,5-sorbitan, and 2,5-sorbitan. However, only 1,4-sorbitan and 3,6-sorbitan can proceed to the second dehydration step to form isosorbide [5]. Consequently, 1,4-sorbitan and 3,6-sorbitan are considered intermediates, while the other types of sorbitan are regarded as stable by-products [11]. To quantify these reactions, the rate constants for the primary and side reactions in the first dehydration step are denoted as k1 and k4, respectively. The second dehydration stage’s both forward and inverse rate constants are denoted as k2 and k3.
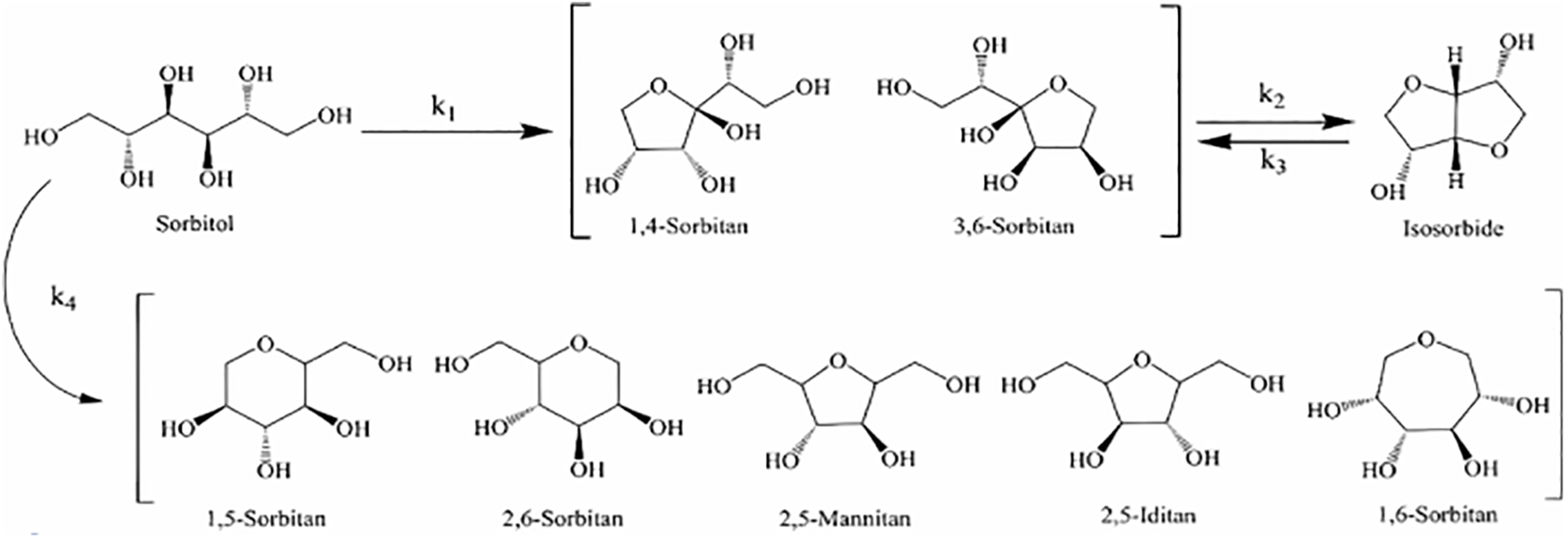
Figure 3: Reaction pathway for the two-step dehydration of sorbitol to isosorbide. Adapted with permission from Reference [12]. Copyright ©2022, American Chemical Society
Wang et al. [12] determined the equilibrium constants for both the initial and subsequent dehydration stages over a temperature range of 473.15 to 533.15 K. The results indicated that the equilibrium constant K1θ for the first dehydration step of sorbitol was on the order of 107, significantly greater than 105, indicating that the initial step involves an irreversible reaction. Conversely, the equilibrium constant K2θ for the dehydration of 1,4-sorbitan was on the order of 100, much less than 105, indicating that the second step is reversible. In a dilute solution using NbOPO4 as a catalyst, the activation energies for the primary and side reactions of the first dehydration step were 117.38 and 139.89 kJ/mol, respectively. The significantly higher activation energy for the side reaction implies that high temperatures would increase the incidence of side reactions, thereby reducing the selectivity for 1,4-sorbitan and 3,6-sorbitan. Therefore, lower reaction temperatures are more favorable for the first dehydration step. For the second dehydration step, the activation energies for k2 and k3 were calculated to be 137.56 and 94.42 kJ/mol, respectively. This indicates that increasing the temperature can accelerate the conversion of intermediates to isosorbide, thereby significantly enhancing the selectivity and yield of the reaction. Thus, a higher reaction temperature is beneficial in the second step to effectively promote the conversion of intermediates and optimize the production of the final product, isosorbide. Furthermore, the reaction rates of the first and second dehydration steps differ markedly. Fig. 4 illustrates the concentration profiles of the dehydration reaction components at different temperatures, showing that the dehydration rate of sorbitol is significantly faster than that of the intermediate, 1,4-sorbitan.
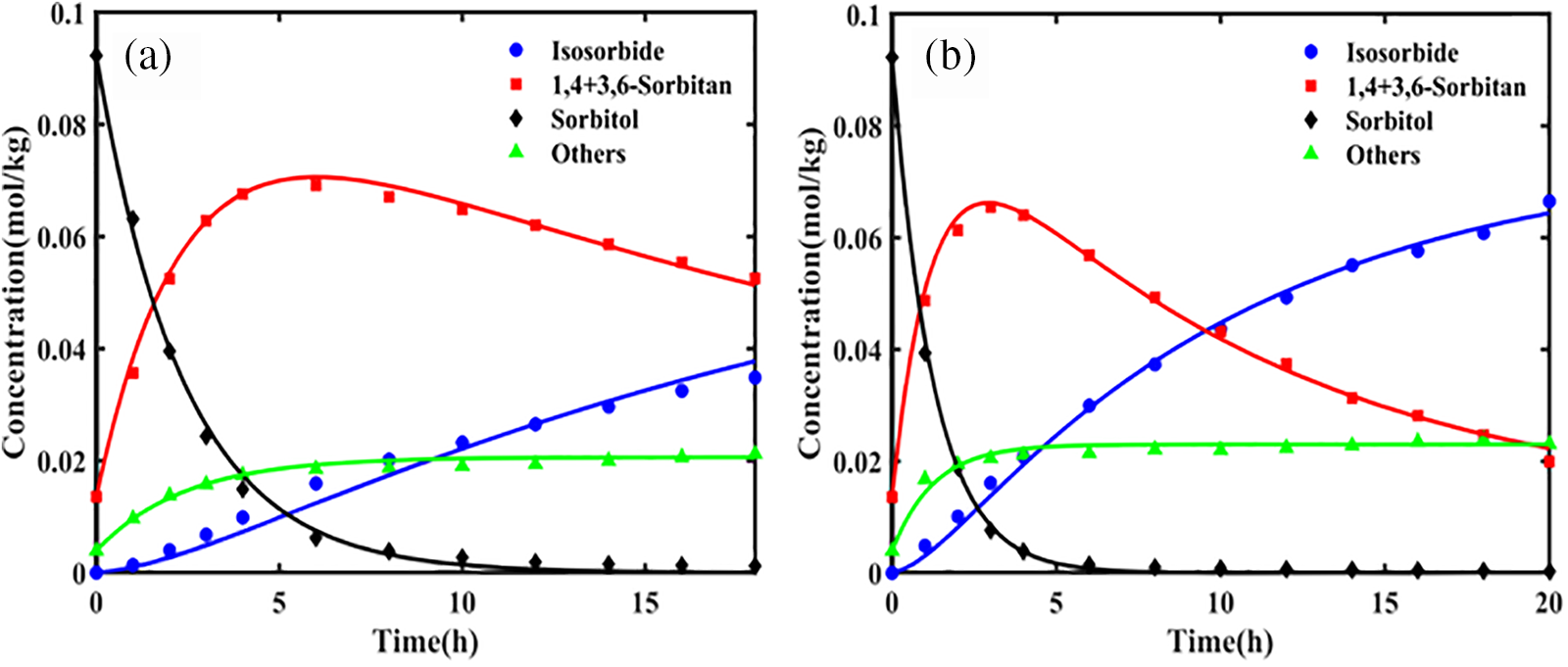
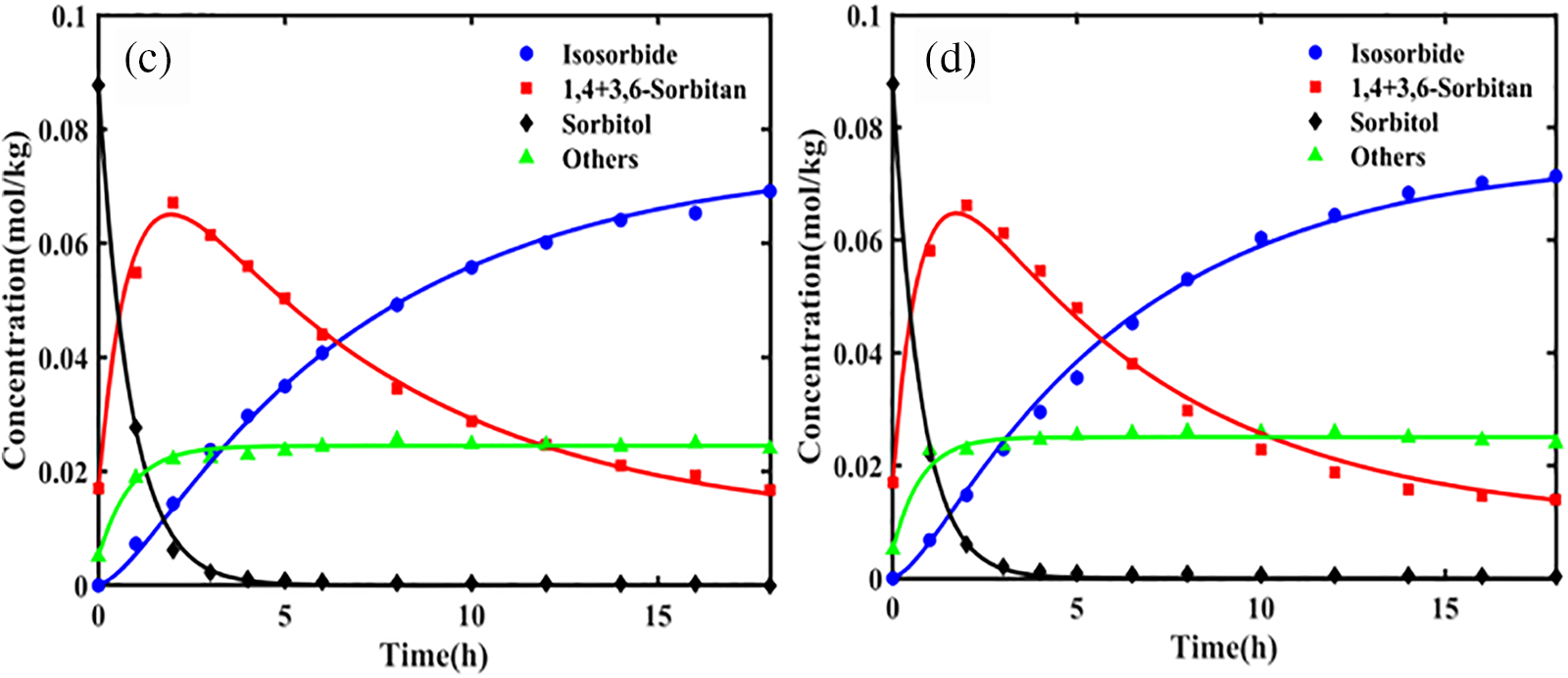
Figure 4: The concentration profiles of the dehydration reaction components at different temperatures are depicted as follows: (a) 493.15, (b) 503.15, (c) 508.15, and (d) 513.15 K. Catalyst amount: 0.5 g. Sorbitol mass: 1.5 g. Water mass: 150 g. Stirring speed: 700 rpm. Nitrogen pressure: 1 MPa. Adapted with permission from Reference [12]. Copyright ©2022, American Chemical Society.
3 Hydrolysis of Cellulose to Glucose
Enzymes or acids can catalyze the dissolution of β-1,4-glycosidic bonds between glucose units, which is the main process in the hydrolysis of cellulose to glucose. Strong hydrogen bonds between equatorial hydroxyl groups and axial stacking of cellulose molecules characterize the crystalline structure of cellulose, providing cellulose significantly resistant to chemical deterioration and insoluble in water and the majority of organic solvents (Fig. 5). The effective conversion of cellulose to glucose is significantly hampered by this structural resilience [13]. It has been demonstrated that the hydrolysis of cellulose can be facilitated by protons (H+) generated by Lewis acids, Brønsted acids, and the ionization of water. These catalytic systems contribute to the breakdown of the intricate hydrogen-bonded network within cellulose, thereby promoting the release of glucose. Table 1 presents an overview of the efficiency of various acid catalysts in the hydrolysis of cellulose.
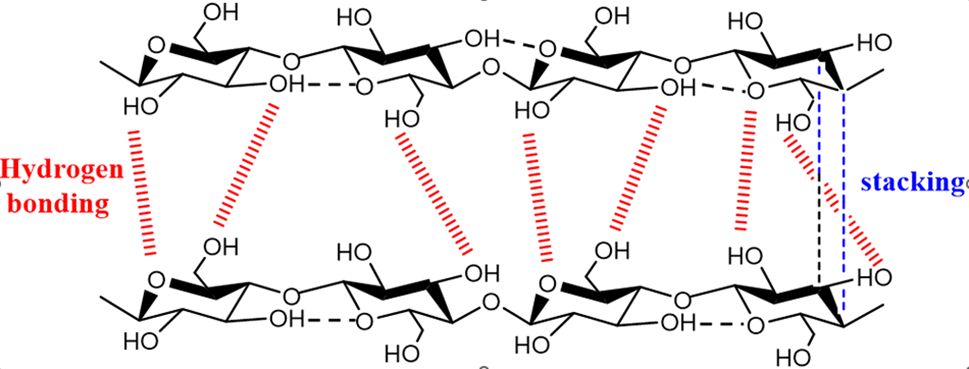
Figure 5: Hydrogen bonding and stacking between cellulose molecules
3.1 Proton Generation from Water Ionization
It is widely recognized that liquid water can produce H+ ions, which can facilitate acid-catalyzed reactions when exposed to high temperatures (above 473 K) (Fig. 6). The protons generated by this in situ acid formation will naturally dissipate at the ambient temperature, making it reversible, thereby completely eliminating issues related to acid recovery and waste disposal.
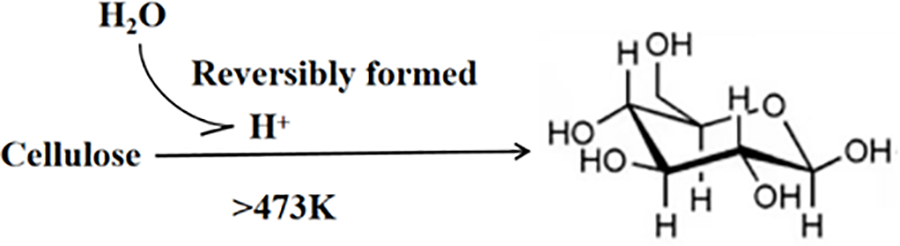
Figure 6: Proton-catalyzed hydrolysis of cellulose provided by water. Adapted with permission from Reference [18]. Copyright ©2007, John Wiley and Sons
By combining in situ generated H+ ions with transient hydrogenation on carbon-supported Ru clusters in hot water. Luo et al. [18] demonstrated an effective approach for converting cellulose to polyols. This method involves the reversible formation of glucose from cellulose degraded by acid catalysis in hot water. In order to validate the in situ emergence of acidic species and their function in the aqueous environment, multiple tests were carried out without the use of water or Ru/C catalyst. Without Ru/C, similar cellulose conversion rates were achieved in water (approximately 38.6% after 5 min and 87.5% after 30 min). Under similar conditions, replacing water with a protic solvent (such as ethanol) or an aprotic solvent (such as dioxane) resulted in no cellulose conversion on Ru/C. However, blending ethanol with water (1:1 volume ratio) under similar conditions resulted in a 10.2% cellulose conversion rate. The reduced conversion rate observed in the mixed solvent system, relative to the pure water system, is attributed to its lower dielectric constant, which correlates with decreased acidity. All of these results point to the necessity of water for the creation of in situ acid and the critical function that acid plays in cellulose hydrolysis, which in turn impacts cellulose conversion efficiency.
Moreover, the hydrolysis process did not alter the crystalline structure of cellulose. Even at a high conversion rate of 85.5% (achieved after 30 min at 518 K), the native cellulose I structure [19] remained intact post-reaction. This result suggests that cellulose hydrolysis takes place on the crystalline surface without inducing swelling or dissolution, processes that would otherwise result in the formation of the cellulose II polymorph. These kinds of changes are usually seen in near-critical or supercritical situations, which ask for high pressures and temperatures (593–673 K and 25 MPa).
Brønsted acid sites provide H+ ions, which facilitate the cleavage and depolymerization of cellulose glycosidic bonds. Increased acidity and higher Brønsted acid site loading will facilitate cellulose depolymerization and related processes more efficiently. Taking cellobiose as an example, its hydrolysis mechanism is illustrated in Fig. 7. When a Brønsted acid gets into interaction and reacts with cellobiose, the procedure of conjugate acid production begins. Thus, it possible for the surface’s Brønsted acid sites to target the β-1,4-glycosidic bond’s oxygen atom. After that, the conjugate acid dissociates and the C-O bond cleaves, releasing glucose and creating an intermediate oxocarbenium ion. A free proton and another glucose molecule are then released as a result of quick hydration. The β-1,4-glycosidic linkages in cellulose are continuously hydrolyzed by the free protons and Brønsted acids present in the hydrolysis solution.
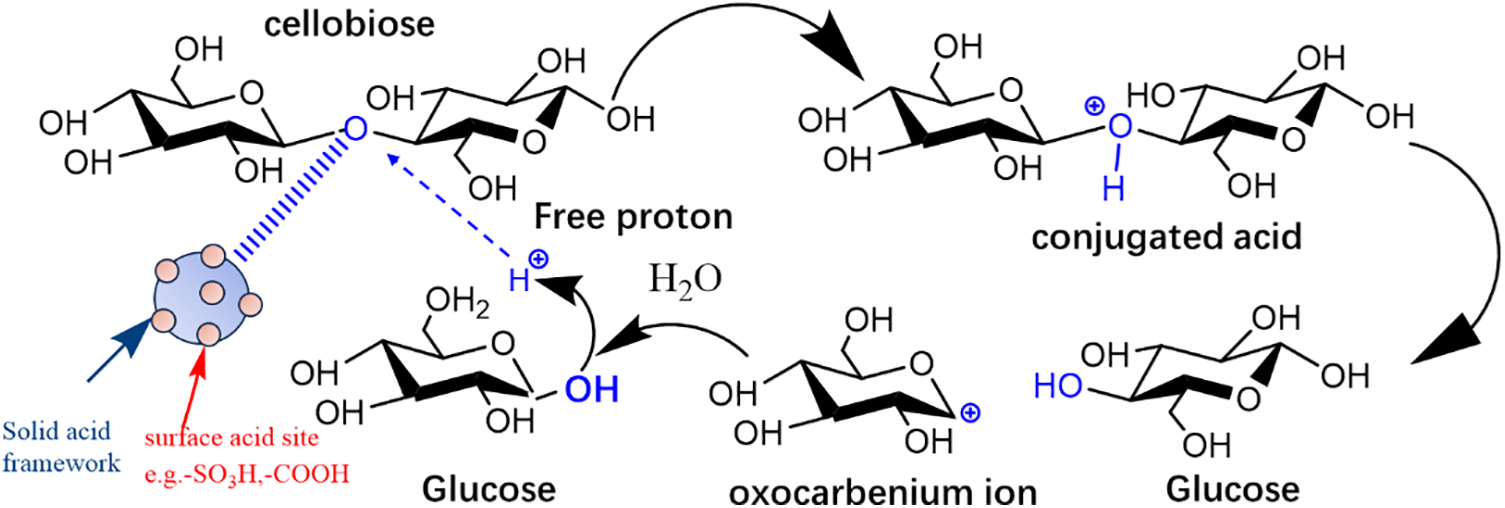
Figure 7: Illustration of cellulose hydrolysis by a solid acid with surface Brønsted acid sites in water
The primary sources of Brønsted acids in catalytic systems include the pyrolysis of water at high temperatures, inorganic acids (such as H2SO4 and HCl), organic acids (containing acidic groups like -SO3H, -PO4H2, and -COOH), hydrolysis of metal species, and solid acids with acidic sites on their surfaces (such as sulfonated resins and zeolites). Homogeneous liquid acids like H2SO4 and HCl exhibit high activity; however, their practical application is limited due to difficulties in recovery and disposal [20]. Therefore, developing new strategies for efficient recycling of acid catalysts is crucial. Since the -SO3H group is a strong acid, it can supply protons needed for the β-1,4-glycosidic linkages in cellulose to hydrolyze. This functional group can be utilized as a strong Brønsted acid site in carbon-based solid acids, polymer-based solid acids, and inorganic solid acids [21]. Pang et al. [22] employed sulfonated ordered mesoporous carbon to catalyze the hydrolysis of cellulose. The acidity of the catalyst was modified by altering the sulfonation temperature. An overall acid density of 2.39 mmol/g and the -SO3H density of 0.63 mmol/g were found in their sulfonated carbon catalyst. This catalyst was capable of hydrolyzing ball-milled cellulose into glucose with an efficiency of 74.5% at 150°C within 24 h.
Additionally to Brønsted acid sites, Lewis acid sites were additionally utilized by researchers to investigate the hydrolysis of cellulose. It is widely believed that the primary catalyst for the hydrolysis of cellulose in water is Brønsted acid sites. However, Lewis acid catalysts can coordinate with cellulose molecules to form coordination compounds, which provide active sites that promote the cleavage of cellulose chains. These cleaved chains are further broken down into smaller carbon units, eventually converting into glucose monomers. Thus, integrating Lewis acid sites within solid acids serves as a method to enhance overall acidity and facilitate effective cellulose hydrolysis. A sustainable method to generate isosorbide from cellulose using a bifunctional ruthenium catalyst supported on mesoporous niobium phosphate was proposed [23]. Under pressured H2 environment, the bifunctional Ru catalyst supported on mesoporous niobium phosphate achieved virtually full cellulose conversion in 1 h and an isosorbide yield of over 50% without requiring additional soluble acids. The authors used phosphate-treated mesoporous (P/mNbO) and bulk (P/NbO) niobium oxide hydrates in addition to mesoporous (mNbO) and bulk (NbO) niobium oxide hydrates to further investigate various niobium oxide supports with differing acidity, pore size, and surface area. They determined the pore size and N2 adsorption-desorption isotherms using density functional theory (DFT) techniques (Fig. 8a–c). The findings showed that the mNbPO isotherms resembled those of type IV and showed hysteresis loops between H2 and H4 types in the 0.4–0.8 relative pressure (P/P0) range, suggesting the presence of sizable mesopores that resembled worms. Fig. 8d depicts NH3-TPD results, showing a significant NH3 desorption peak for mNbPO, which shifted to a higher temperature of approximately 613 K, suggesting stronger acidity, more acid sites, and a larger acid density, leading to better catalytic performance. This bifunctional catalyst maintained its mesoporous structure and showed no signs of Ru leaching in the reaction solution for more than six cycles at a cellulose-to-catalyst mass ratio of 43.3.
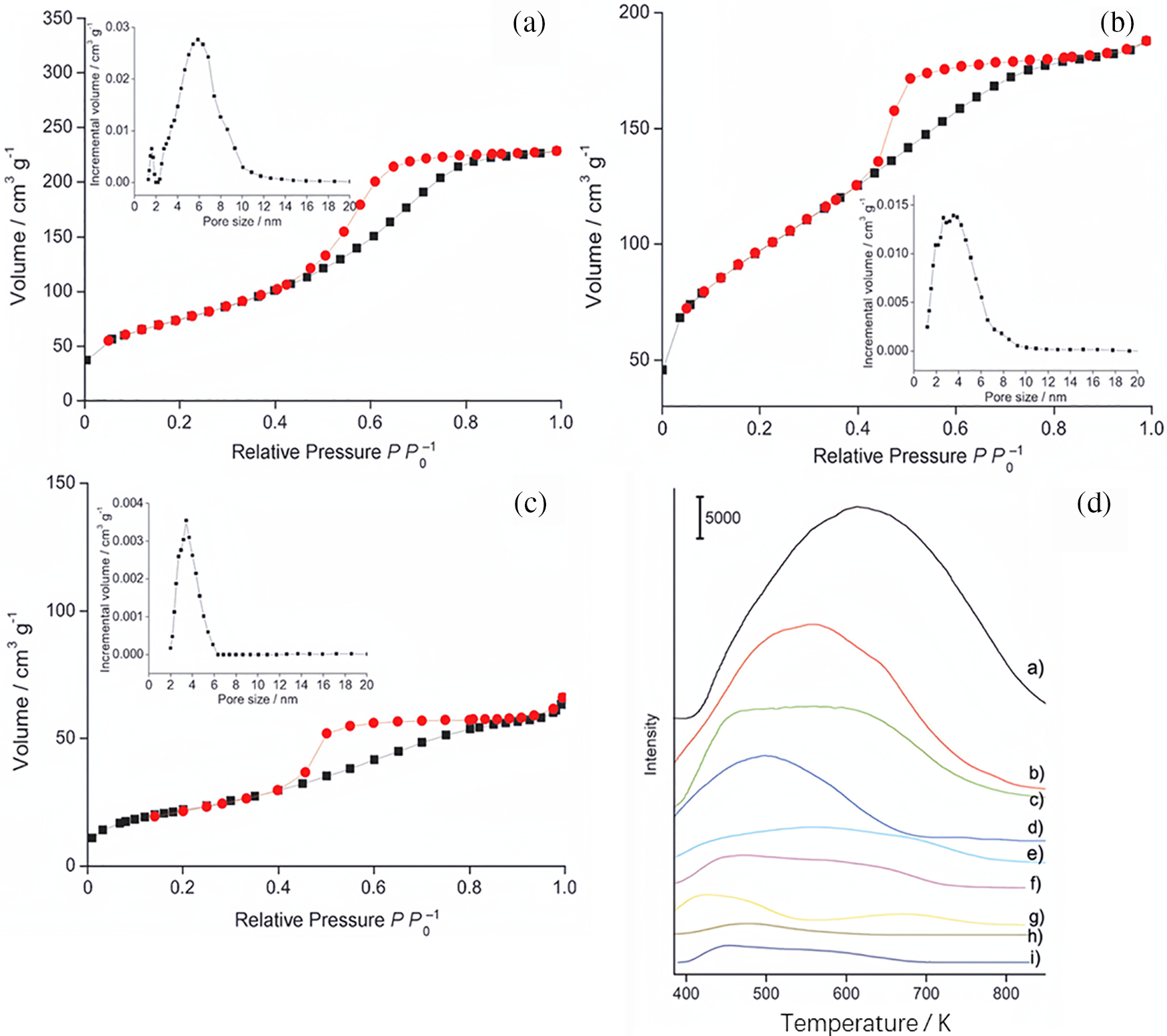
Figure 8: N2 adsorption-desorption isotherms for P/mNbO, mNbO, and mNbPO (a–c). The pore-size distribution curve from the DFT method applied to the adsorption isotherm is displayed in the inset, assuming a model with slit pores. (d) NH3-TPD spectra of the following catalysts: (a) mNbPO, (b) P/mNbO, (c) NbPO, (d) mNbO, (e) P/NbO, (f) NbO, (g) HZSM-5, (h) NaY, and (i) γ-Al2O3. Adapted with permission from Reference [23]. Copyright ©2013, John Wiley and Sons
Komanoya et al. [17] found that Ru catalysts supported on mesoporous carbon CMK-3 exhibited high activity and durability for cellulose hydrolysis into glucose at 503 K. With a combined selectivity of 71% for glucose and oligomers, the CMK-3 support greatly increased the cellulose conversion (54%) and the yields of glucose (16%) and oligomers (22%). The functional groups on CMK-3 that contain acidic oxygen and adsorb β-glucan are responsible for this increase in cellulose hydrolysis [24]. Hydrolysis of cellulose and oligomers was aided in hot compressed water by H3O+ ions that were either formed directly by Ru species on CMK-3 or dissociated from water through direct attack. Ru’s significance in the hydrolysis of cellulose was further highlighted by the authors, who identified the Ru species as RuO2·2H2O. Lewis acidic vacancies were produced by the dissociation of water molecules in concert with Ru. Even in the absence of additional acid, Ru on mesoporous carbon CMK-3 may catalyze the hydrolysis of cellulose into glucose through oligomer intermediates in hot compressed water.
4 Hydrogenation of Glucose to Sorbitol
Several mechanisms for the hydrogenation of glucose have been thoroughly investigated. Crezee et al. [10] and Gallezot et al. [25] developed kinetic models for the hydrogenation of glucose, drawing comparisons with smaller molecules such as acetaldehyde, and concluded that glucose hydrogenation proceeds via a ring-opening process. In contrast, Makkee et al. [26] employed isotope labeling to monitor the chiral isomerization of hydrogenation products, demonstrating that hydrogen may be directly added to the pyranose ring. Additionally, using density functional theory (DFT) simulations, Trinh et al. [27] investigated several glucose adsorption configurations on Pt surfaces, suggesting that glucose adsorbs onto Pt (111) before ring-opening occurs. According to certain research, hydrogenation specifically takes place through a closed-ring formation, while others advocate for an open-chain formation, with the mechanisms of side reactions remaining unclear.
Fang et al. [28] conducted a comprehensive study on investigation into the mechanism of the glucose hydrogenation to sorbitol reaction on a Raney Ni catalyst, combining experimental analysis with computational simulations, including DFT calculations and kinetic modeling (Fig. 9a). Their findings indicate that catalytic hydrogenation necessitate ring-opening. Glucose undergoes ring-opening catalyzed by Raney Ni, followed by desorption into the solution. The exposed aldehyde group of the open-chain glucose re-adsorbs onto the crystal facets of Raney Ni, while hydrogen dissociatively adsorbs onto the catalyst surface, forming a “ring-opened glucose-hydrogen atom-catalyst” co-adsorption state. Subsequently, two hydrogen atoms sequentially add to the aldehyde group of the ring-opened glucose through two transition states (GCHOH* intermediate and GCH2O* intermediate), resulting in the formation of sorbitol. Finally, sorbitol desorbs, completing the hydrogenation process. Within this mechanism, the formation of by-product gluconic acid can affect the catalyst’s stability, potentially due to glucose disproportionation [29,30] or dehydrogenation of cyclic glucose [31,32]. Corroborated by data from HPLC, verified by DFT calculations, and confirmed at different temperatures using glucose hydrogenation, the authors proposed a side reaction mechanism (Fig. 9b). Cyclic glucose adsorbs onto the Ni surface, followed by the sequential adsorption of two hydrogen atoms either pathway B (GCCOH* intermediate) or pathway A (GCCHO* intermediate) to the catalyst. Ultimately, as gluconolactone is generated, it desorbs from the Ni surface and then hydrolyzes to generate gluconic acid.
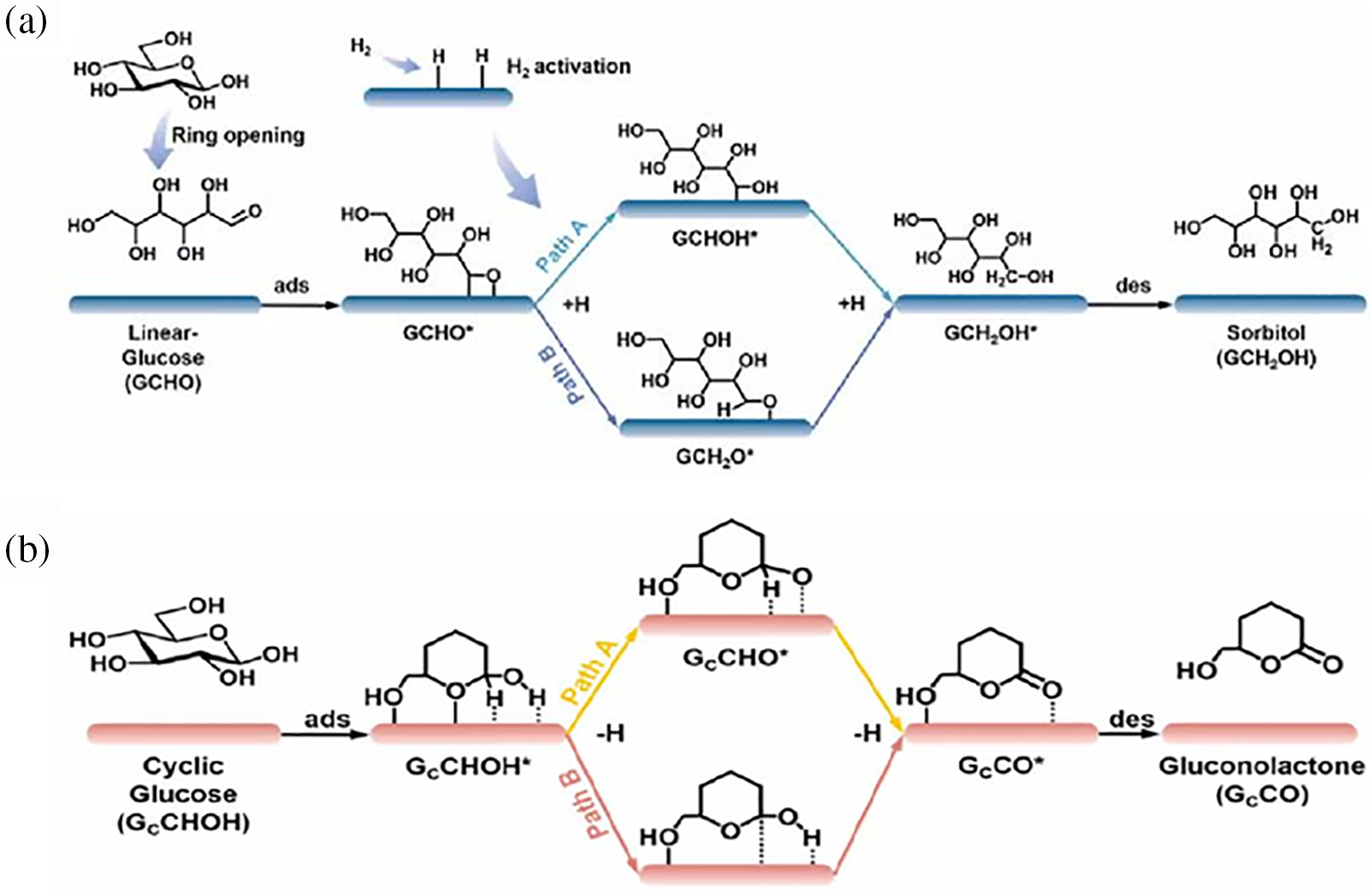
Figure 9: (a) Possible reaction pathways for hydrogenation of glucose to sorbitol on the surface of Raney-Ni; (b) Possible reaction pathways for dehydrogenation of cyclic glucose to gluconolactone on Ni surface. Adapted with permission from Reference [28]. Copyright ©2023, Elsevier
The hydrogenation of glucose is greatly impacted by the selection of metal-based catalysts. Li et al. [33] examined the hydrogenation process of glucose using a Raney Ni-P catalyst, achieving a sorbitol selectivity of 99.5% and a glucose conversion rate of approximately 55.8%. Similarly, Hoffer et al. [34] conducted hydrogenation reactions using Ru supported on carbon and examined various reaction parameters. They found that glucose conversion approached 100% over time, with sorbitol selectivity exceeding 98%. Furthermore, they explored hydrogenation using Raney Ni in combination with Mo and Cr/Fe, observing that the addition of these metals enhanced the selective glucose hydrogenation to produce sorbitol. Thus, the exploration of efficient metal-based catalysts is crucial for improving the hydrogenation efficiency of glucose, thereby facilitating subsequent reactions. Table 2 summarizes the performance of different metal-based catalysts in the hydrogenation of glucose.
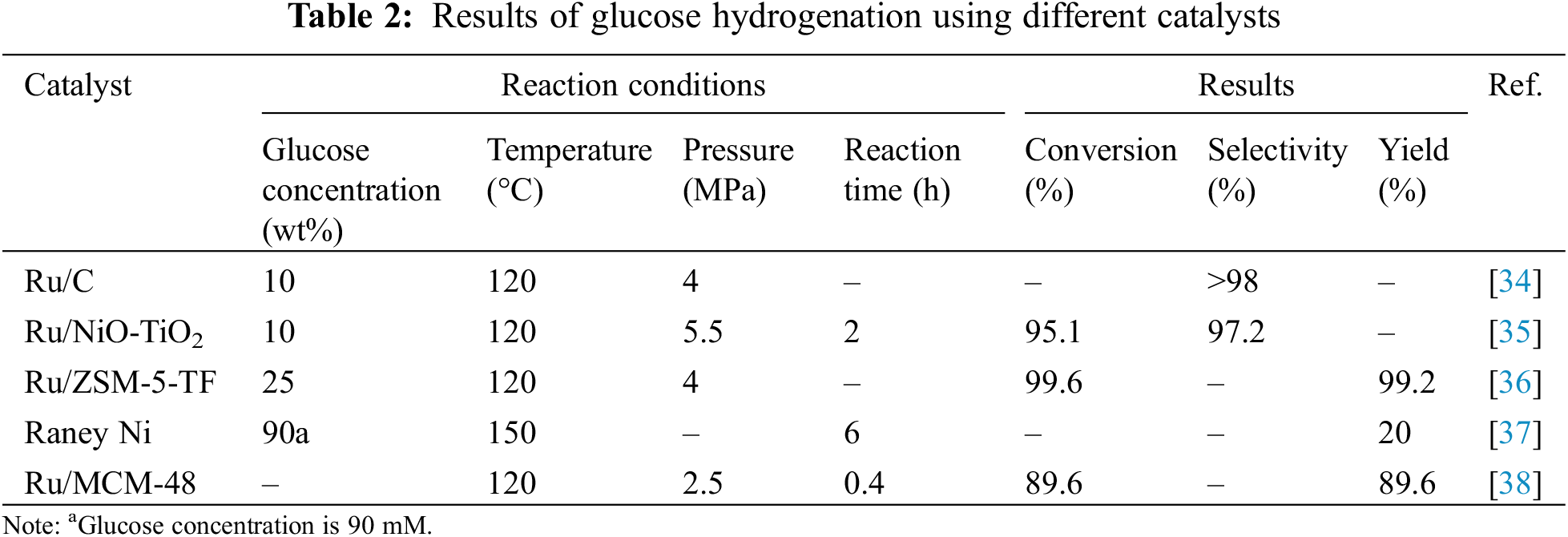
Noble metals like Pt, Rh, and Ru possess unique electronic structures that provide numerous high-activity sites on their surface, facilitating effective adsorption and activation of reactant molecules, thereby accelerating reaction rates. Currently, Ru stands out as the most active catalyst. In recent years, Ru catalysts have garnered significant attention owing to their superior catalytic capabilities in glucose hydrogenation, high activity, stability, and resistance to leaching in aqueous solutions. Hoffer et al. [34] reported that using 2 g of Ru/C catalyst, glucose in aqueous solution containing 5–6.5 weight percent might be transformed into sorbitol within 140 min, achieving a selectivity greater than 98%. Zhao et al. [39] proposed a straightforward synthesis method for core-shell Ru catalysts (Fig. 10a), where γ-Al2O3 particles were impregnated with Ru nanoparticles (NPs) underpinned by a polymer of styrene and maleic acid with amino functionalization (ASMA). High dispersion and solid embedding of Ru NPs on the support surface were guaranteed by the coordination interaction between Ru cations and the hydroxyl- and amino-rich polymer shell, thus enhancing productivity. The resulting catalyst, 5% Ru/γ-Al2O3@ASMA, demonstrated nearly 90% stable sorbitol yield in both batch and trickle-bed reactors during glucose hydrogenation.
Figure 10: (a) Reaction pathway for the hydrogenation of glucose to sorbitol using 5% Ru/γ-Al2O3@ASMA as the catalyst; (b) XPS spectra of Ru 3p for Ru single atoms (SAs) on hollow mesoporous carbon spheres (HMCS-SO3H12); (c) XPS spectra of C 1s for the same catalyst. Adapted with permission from References [5,39]. (a) Open Access. (b) Copyright ©2023, Elsevier
Our research group developed a method to coat HQ-CTS, or 8-hydroxyquinoline-modified chitosan, on the silica sphere surface, followed by pyrolysis and acid treatment to synthesize sulfonic acid-functionalized hollow mesoporous carbon spheres (HMCS-SO3H) via an in-situ templating method. Additionally, we halted aggregated hydroxide formation. by inhibiting the hydrolysis of Ru3+ through N-coordination and acid suppression, achieving a high incorporation of Ru single atoms (SAs). XPS analysis was implemented for investigation into the chemical states of Ru and C in Ru SAs/HMCS-SO3H12. The Ru 3p spectrum exhibited peaks at 462.1 and 464.3 eV for 3p3/2 and at 483.9 and 486.2 eV for 3p1/2 (Fig. 10b), indicating the coexistence of Ru2+ and Ru4+ species. Additionally, Ru 3d photoelectron signals at 285.2 and 280.7 eV for 2d3/2 and 2d5/2 overlapped with the C 1s spectrum (Fig. 10c), suggesting the formation of electron-deficient Ru sites. The anchoring of Ru SAs and the amount of SO3H could be readily altered by varying the adding time of HQ-CTS during the in-situ Stöber templating procedure. The established Ru SAs demonstrated high activity and selectivity for glucose hydrogenation, as was to be predicted. The enhanced synergistic effect with the high concentration of SO3H rendered a high yield of isosorbide while cellulose is converted in a single pot under difficult circumstances [5].
4.1.2 Non-Noble Metal Catalysts
Utilizing non-noble metal catalysts (e.g., Ni, Co, Cu) as opposed to noble metal catalysts can fundamentally reduce costs. Nickel-based catalysts were the first to achieve industrial application in glucose hydrogenation and remain the most widely used catalysts. Numerous catalysts using nickel, include Ni/SiO2, Ni/Al2O3, Ni/TiO2, and Ni-Kieselguhr, have been extensively synthesized and applied in hydrogenation of glucose, conversion of cellulose, and hydrogenolysis of sorbitol and glycerol [40]. Fu et al. [41] explored the Ni particles possessing various morphologies, crystallinities, and compositions: structure-activity relationships in the hydrogenation of glucose. By varying the hydrogen pressure during synthesis, they obtained nanorods and nanospheres, and also synthesized nanobead chains. Comparative analysis revealed that the Ni nanospheres exhibited the lowest activation energy for glucose hydrogenation (32 kJ/mol) as opposed to the (98 kJ/mol) of the nanorods and nanobead chains. Additionally, the Ni nanospheres achieved a constant 100% selectivity for sorbitol at a temperature of 140°C. Our research group developed a range of mesoporous carbons based on nickel that are uniformly dispersed. by neutral-condition self-assembly of chitosan-nickel supramolecular aggregates using P123, followed by pyrolysis. The spherical nanoparticles showed high dispersion and catalytic activity when the pyrolysis temperature was 550°C and the molar ratio of Ni/CTS in the generated gels was 1/2. This was demonstrated by the meticulously controlled production of nickel nanoparticles on the mesoporous carbon that were uniformly spherical. This research may pave the way for the creation of effective non-noble metal catalysts [40]. However, substantial metal leaching occurs during the reaction phase for non-noble metals. By decreasing the concentration of Ni and stabilizing the tiny metal particles with materials like SiO2, zeolites, and activated carbon, the issue can be lessened.
4.1.3 Bimetallic (or Multimetallic) Catalysts
In contrast to the high cost of noble metals and the low selectivity of non-noble metal catalysts for sorbitol, as well as issues related to leaching, active metal sintering, and recyclability, there are advantages to bimetallic (or multimetallic) catalysts in terms of their both cost and performance. The synthesis of bimetallic catalysts (such as Ni-Ru, Fe-Ru, and Ni-Fe) with various metal ratios and compositions through a non-covalent approach has been reported [42]. This method involved introducing bifunctional ligands with pyrene moieties and metal entities onto graphene nanosheets (GNPs) without disrupting their aromatic network. The results of the investigation demonstrated that the catalytic reaction with ruthenium supported on graphene nanosheets (3.03 wt%) had the maximum selectivity, achieving a sorbitol conversion rate of 92% and a selectivity of 87% within 6 h (Fig. 11a). When using Ni-Ru catalysts, an increase in the amount of Ni (up to a 3 Ni:1 Ru ratio) enhanced both the conversion rate and selectivity for sorbitol, achieving a Ru loading of only 1% by weight and a 60% conversion rate (Fig. 11b). Resilient Ru loading and high catalytic activity have been demonstrated by the Fe-Ru/GNP catalyst (1 Fe/3 Ru, Fig. 11c). However, increasing the amount of iron did not further improve the conversion rate. The bimetallic catalyst demonstrated significantly greater selectivity: 40% as opposed to 15% for the monometallic catalyst when sorbitol selectivity was contrasted between bimetallic Ni(II)-Fe/GNP catalysts (containing 3.46–0.66 wt% Ni and Fe, respectively) and monometallic nickel catalysts (7.1 wt% Ni) (Fig. 11d). The bimetallic materials’ catalytic performance exceeded the total results of the separate monometallic catalysts, demonstrating that the two metals function in collaboration.
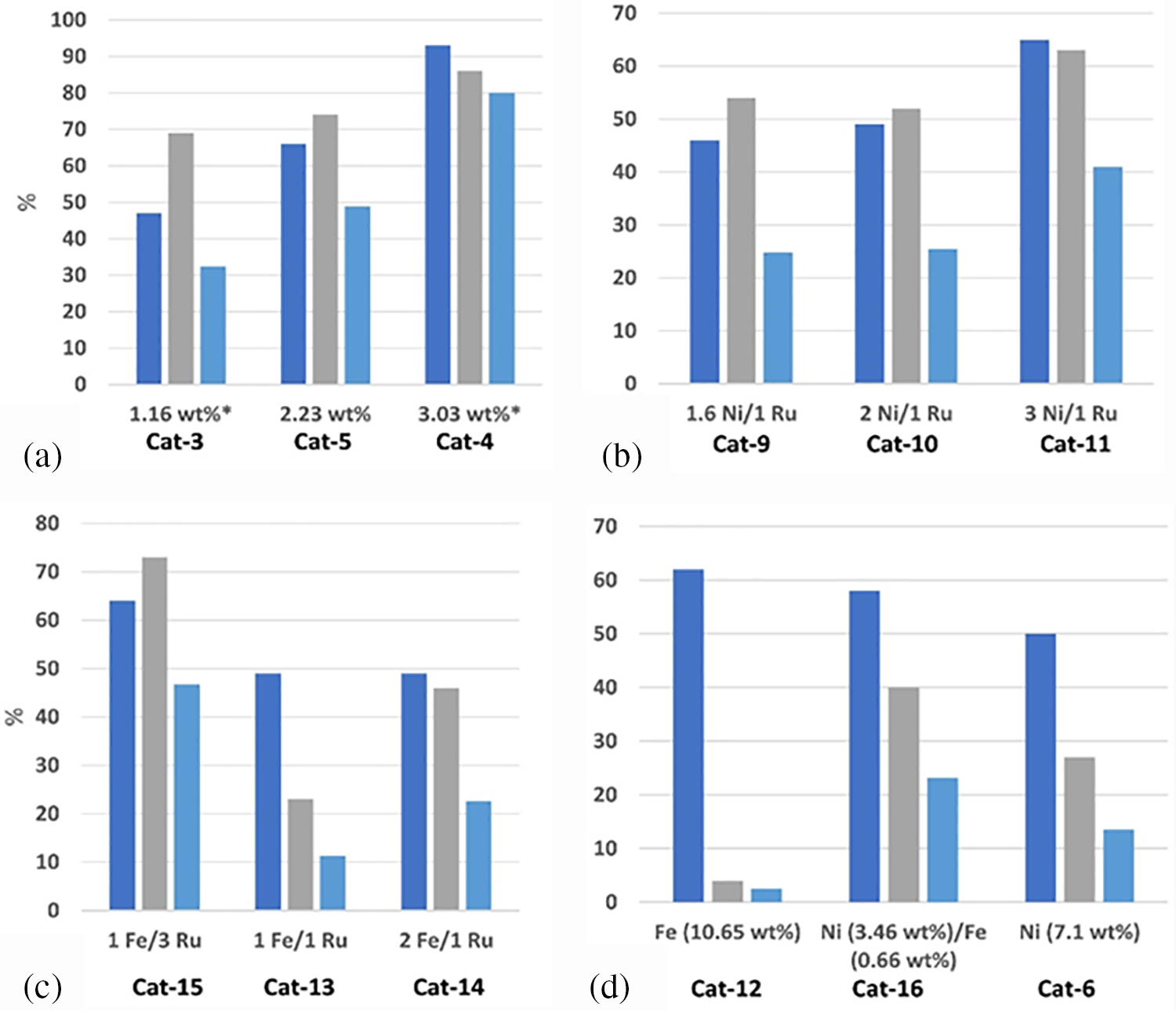
Figure 11: (a) The relationship between conversion rate (blue), sorbitol selectivity (grey), and sorbitol yield (light blue) as a function of Ru loading on GNPs (Catalytic test conditions: 30 bar H2, 140°C, 6 h, 1 g glucose, 30 mg catalyst). (b) Catalytic test results as a function of the Ni:Ru metal weight ratio on GNPs. (c) Catalytic test results as a function of the Fe: Ru metal weight ratio on GNPs. (d) Catalytic test results for non-noble metals, Fe, Ni, and their mixtures. Catalytic test conditions: 30 bar H2, 160°C, 2 h, 0.5 g glucose, 60 mg catalyst. Adapted with permission from Reference [42]. Copyright ©2023, Elsevier
Due to the higher electronegativity of Ru compared to Ni, Ru can be utilized to anchor the less electronegative Ni, thereby facilitating H2 adsorption and reducing kinetic limitations [43]. Furthermore, Ru (101), Ru (100), and Ni (111) planar interaction can improve the functionality of Ni-Ru bimetallic compounds [44]. In an alkaline ethanol-water solvent, formaldehyde and metal salts functioned as cross-linking agents, and pluronic F127 and tannic acid (TA) as an organic ligand and soft template, respectively, to generate coordination spheres [45]. During carbonization and hydrothermal processing, bimetallic Ni-Ru porous carbon spheres (Ni-Ru@PCS) were successfully synthesized (Fig. 12). These Ni-Ru@PCS catalysts possess a rich mesoporous and microporous structure, with uniformly dispersed Ni-Ru bimetallic particles within the carbon framework. Various catalysts were utilized for the catalytic hydrogenation of glucose to sorbitol, including monometallic Ni@PCS, Ru@PCS, and Ni-Rux@PCS that had various Ni: Ru molar ratios. Particularly, the Ni-Ru10:1@PCS and Ni-Ru5:1@PCS catalysts outperformed the monometallic Ru@PCS catalyst in terms of activity and performed similarly to Ni@PCS, achieving a sorbitol yield of 98% with 100% selectivity at 140°C within 120 min.
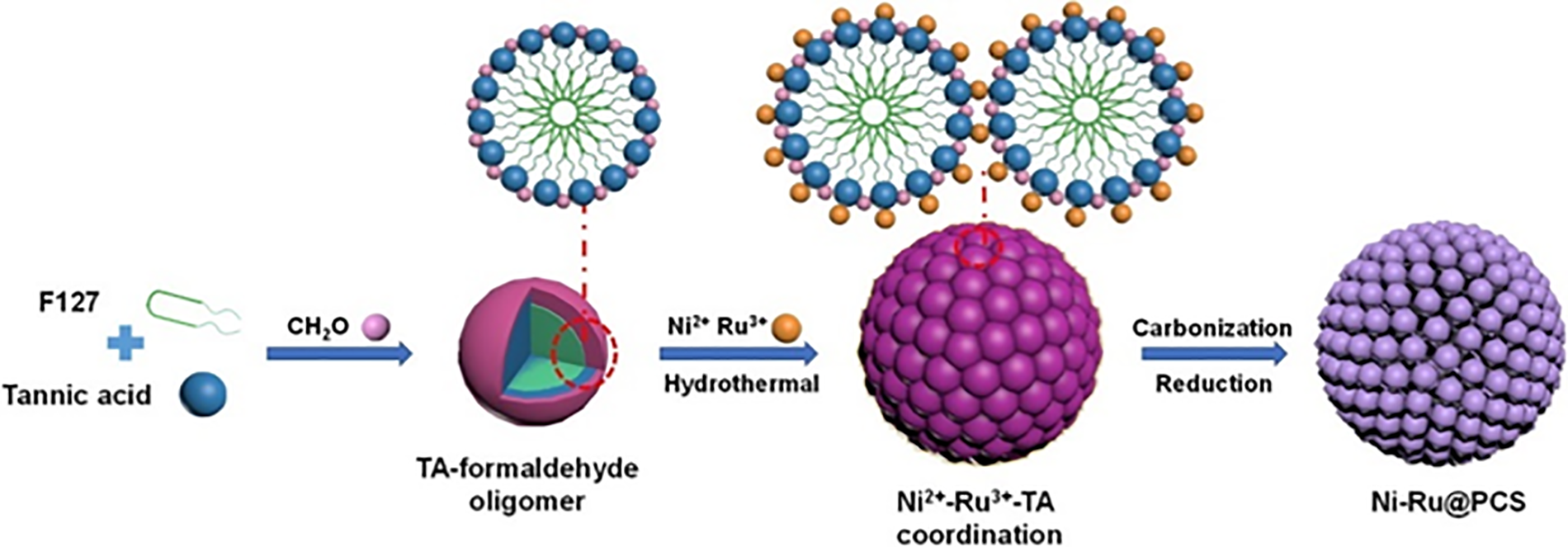
Figure 12: Schematic illustration of the synthesis process of Ni-Ru@PCS materials. Adapted with permission from Reference [45]. Open Access
4.2 Factors Influencing Hydrogenation Activity
The catalyst support is closely tied to catalytic activity, with the specific surface area of the support directly impacting the dispersion of the catalyst. Supports with high specific surface areas, such as carbon nanotubes, activated carbon, SiO2, and Al2O3, can provide more active sites, enhancing metal particle dispersion and thereby improving hydrogenation efficiency. Ali et al. [46] synthesized CNF-doped Y zeolite, which was used as a support for Mo-Ni and Mo-Co catalysts, resulting in the formation of zeolite-CNF/Mo-Ni (Co) (ZFMoNi or ZFMoCo) catalysts. These were compared with catalysts lacking CNF (ZMoNi and ZMoCo). The catalytic performance was evaluated using the hydrodesulfurization (HDS) of dibenzothiophene (DBT), revealing higher activation performance after CNF impregnation. Various characterization techniques were employed to confirm the structure, morphology, and performance of the catalysts. The XRD spectra (Fig. 13a) exhibited diffraction peaks between 5°–35°, characteristic of Y-type zeolite, indicating that all four catalysts retained the Y-type zeolite structure. The introduction of CNF into the catalysts increased the peak intensity at 25.6°, suggesting enhanced dispersion of active phases (Mo, Co, and Ni) on the composite support surface (Z-CNF). Fig. 13b presents the TGA data of the calcined catalysts, demonstrating enhanced thermal stability with the inclusion of CNF. The total weight loss for ZFMoNi was 20.5%, compared to 27.6% for ZMoNi. XPS measurements (Fig. 13c–f) showed that the C 1s peak at 284.5 eV corresponds to the graphitic structure, with MoO3’s oxygen present as O−2. The study found that the introduction of CNF improved the HDS reaction of DBT, ZMoCo’s activity increased from 91.8% to 97.5%, whereas ZFMoNi’s activity increased from 90.1% to 94.6%. Better dispersion of the active phases (Mo, Ni, and Co) on the composite support surface (Z-CNF) was caused by the inclusion of CNF, which enhanced the surface area of the support.
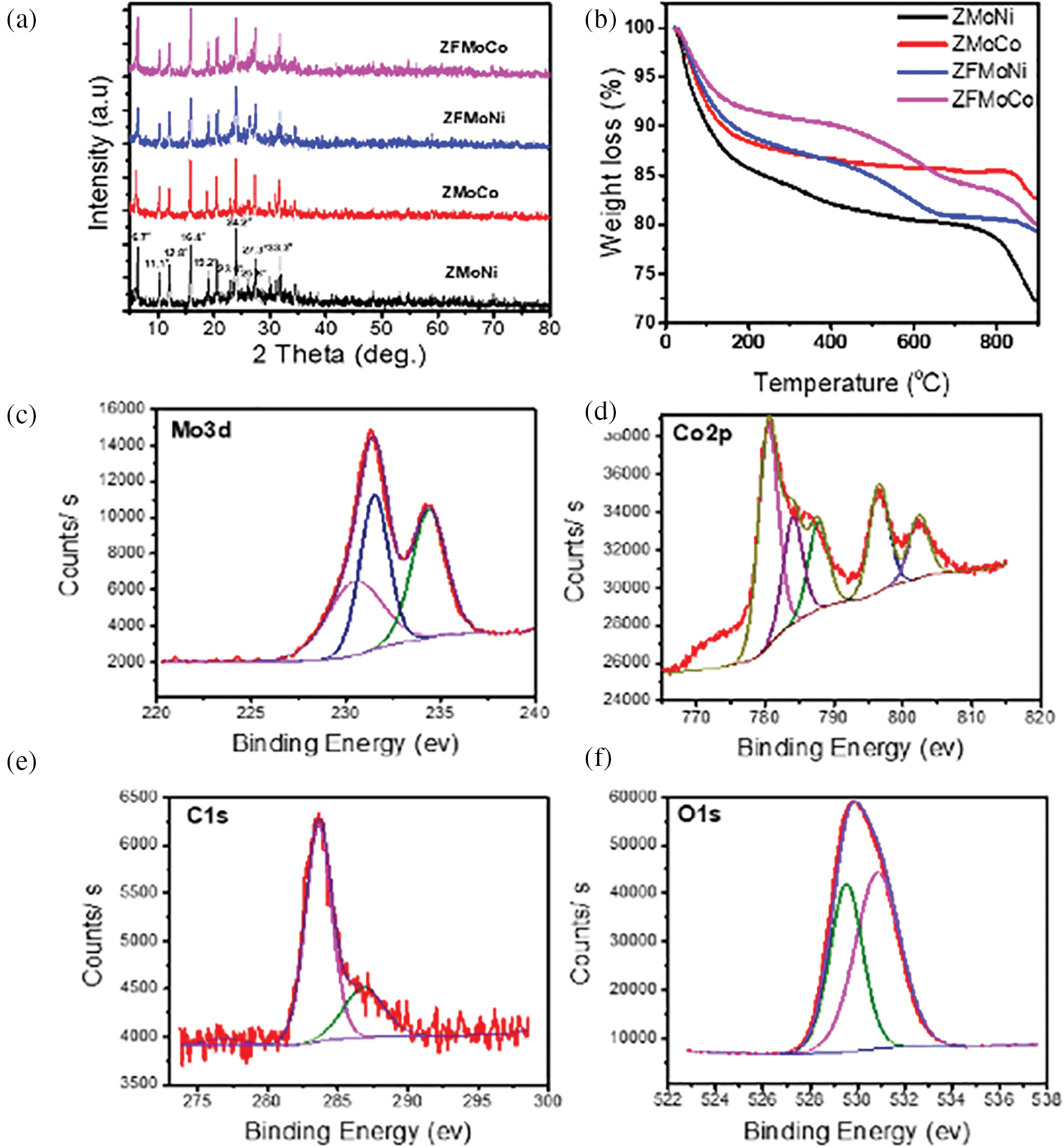
Figure 13: (a) XRD patterns of the catalysts. (b) TGA curves of the four prepared catalysts. (c–f) XPS spectra of ZFMoCo catalyst showing Mo 3d, Co 2p, C 1s, and O 1s signals. Adapted with permission from Reference [46]. Copyright ©2020, Elsevier
The mass transfer of reactants and products is greatly impacted by the pore structure of the support, encompassing pore size, pore volume, and pore distribution. Catalyst pores facilitate mass transfer, with different pore sizes offering varying diffusion rates. The relationship between pore size and the size of reactant or product molecules can induce shape-selective catalysis and spatial confinement effects [11]. Owing to its high surface area, well-dispersed active components, and great chemical and hydrothermal durability, carbon is regarded as one of the most effective catalytic supports [47]. The mechanical durability, high electrical conductivity, large surface area, uniform pore sizes, and adjustable pore structures of porous materials like carbon nanofibers (CNF), activated carbon (AC), and activated carbon nanofibers (ACNF) have attracted a lot of attention in practical research [46]. Among these materials, CNF is particularly noteworthy due to its open morphology, negligible or non-existent micropores, and the significant pore volume created by the gaps between its tubular structures. This results in a remarkably high surface area derived from its outer walls. The excellent activation of metal sites that promotes hydrogen transportation to reactant molecules is accountable for the high activity of CNF-supported catalysts. There are a significant number of active sites available in the reactants due to the low metal-support interaction [48,49].
In addition, the deposition, dispersion, and stability of metal particles are influenced by the surface chemical characteristics of the support, particularly its acidity, basicity, and surface functional groups. For instance, acidic supports can enhance the dispersion of certain metals like platinum and ruthenium. Activated carbon, SiO2, and Al2O3 are frequently used as supporting catalysts in the hydrogenation of glucose to sorbitol. Because of its well-structured pores and large specific surface area, activated carbon can disperse metal particles extensively when utilized as a support, significantly enhancing catalytic efficiency. SiO2 supports can enhance metal activity through their surface acidic sites, although excessive acidity may lead to side reactions. Al2O3 supports, characterized by their high mechanical and thermal stability, maintain excellent catalytic activity and longevity under high-temperature and high-pressure conditions [39].
The characteristics of supported metal catalysts can be tailored by altering the morphology of the metal particles, including adjustments to their size and shape [50]. Given that the electronic structure of metal particles varies with size (ranging from nanoparticles to clusters and single atoms), extensive research has been conducted on the size effects between individual atoms, clusters, and small nanoparticles. Haruta et al. [51] discovered that the turnover frequency for CO oxidation per surface gold atom in Au catalysts supported on TiO2, α-Fe2O3, and Co3O4 was independent of the support used, but dramatically increased when the gold particle diameter was less than 4 nm. Conversely, Pd particles supported on carbon black, centered around 7 nm, exhibited optimal activity in the glucose oxidation reaction [52]. Lykhach et al. [53] combined synchrotron radiation photoelectron spectroscopy, scanning tunneling microscopy, and density functional theory calculations, demonstrating that charge transfer between metal particles and the support is the cause of the size effect, estimating that platinum particles with sizes around 1–1.5 nm exhibit maximum charge transfer. Therefore, most size effects occur in nanoparticles smaller than 10 nm, with catalytic performance stabilizing as particle size increases [54]. Smaller metal particle sizes typically provide a larger specific surface area, enhancing the number of active sites and promoting the hydrogenation of glucose. Additionally, smaller particle sizes usually mean higher dispersion of metal on the support, potentially altering its electronic structure due to quantum size effects. Variations in particle size not only affect catalytic activity but also influence selectivity and stability. Studies have shown that different particle sizes can lead to different reaction pathways in the hydrogenation of glucose, affecting the product distribution. Smaller particles, due to their high surface energy, are more prone to sintering and deactivation, necessitating a balance between particle size and catalyst longevity in practical applications.
However, the size effect can vary depending on the specific reaction and the metal used. Therefore, investigating the size effect of metal nanoparticles larger than 10 nm in glucose hydrogenation is crucial. Fu et al. [55] obtained nickel particles of different sizes by controlling the synthesis temperature using the sol-gel method. They evaluated glucose conversion and sorbitol yield using Ni/CB catalysts and found that nickel particles around 17 nm provided the highest glucose conversion and sorbitol yield. Transmission electron microscopy (TEM) images and corresponding statistical histograms (Fig. 14) revealed that different synthesis temperatures produced nickel nanoparticles with sizes ranging from 6.9 to 23.5 nm. Statistical analysis of 100 particles from TEM images of each sample showed that the average particle size for the CB-400 catalyst was around 6.9 nm. As the synthesis temperature increased, the particle size also increased, with broader size distribution and a standard deviation rising from 1.3 nm at 400°C to 11.6 nm at 650°C. Glucose conversion increased with particle size, peaking at 17.1 ± 6.7 nm, before gradually declining with further size increases, confirming the presence of a size effect for nanoparticles larger than 10 nm.
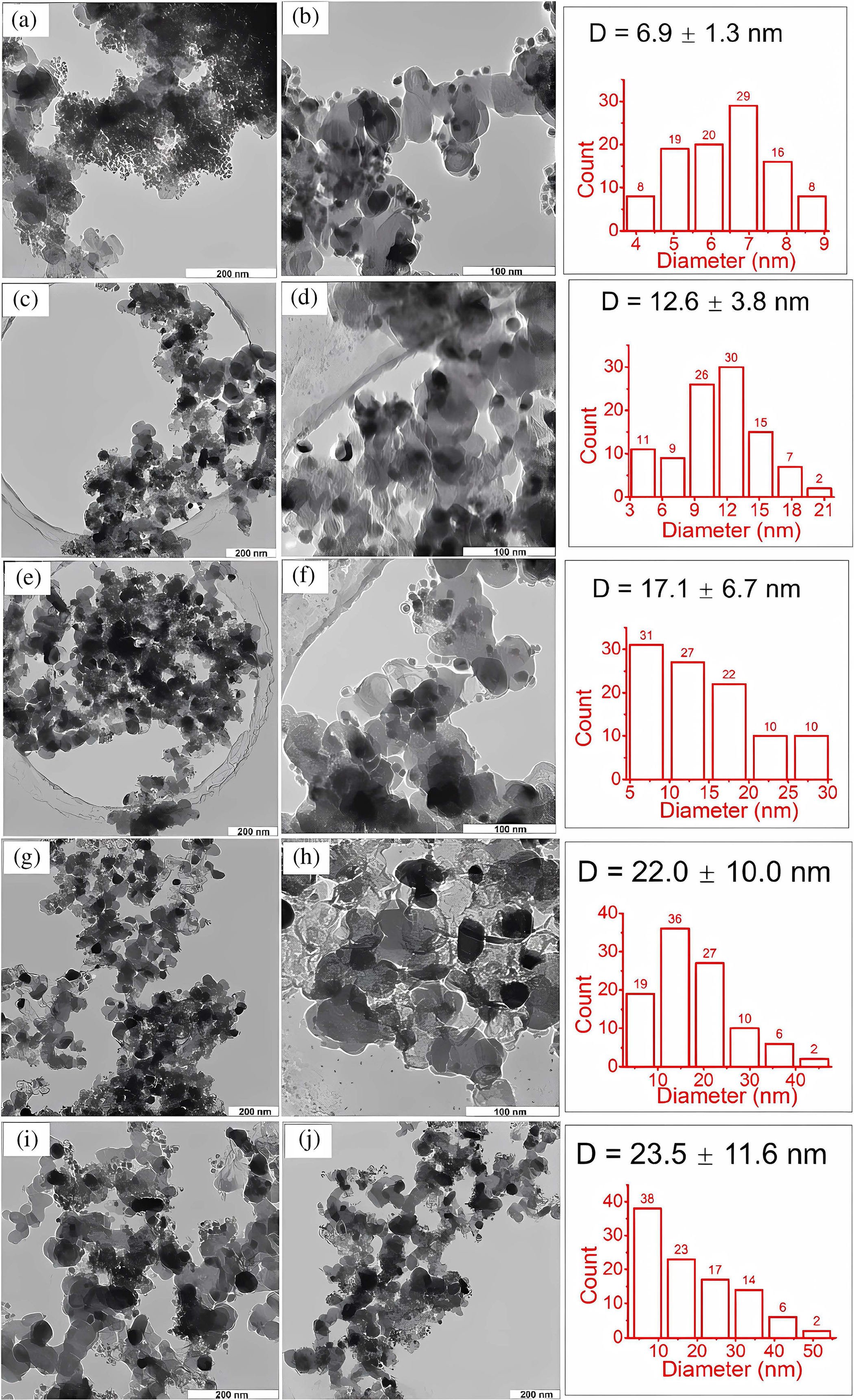
Figure 14: Transmission electron microscopy (TEM) images of samples CB-400 (a, b), CB-500 (c, d), CB-550 (e, f), CB-600 (g, h), and CB-650 (i, j). Corresponding particle size distribution histograms are shown to the right of each pair of images, with the average diameter (D) indicated in the histograms. Adapted with permission from Reference [55]. Copyright ©2022, Elsevier
5 Sorbitol Dehydration to Isosorbide in Two Steps
To produce isosorbide, sorbitol typically needs to be dehydrated twice while a potent acid catalyst is present [56]. The preferred mechanism for hexitol dehydration is SN2 cyclodehydration at C-1 due to reduced steric hindrance, while the SN1 mechanism at C-4, involving a carbocation intermediate, is also possible due to protonation of the hydroxyl group [57]. An improved process for the dehydration of sorbitol to 1,4-sorbitan has been proposed [58]. Researchers found that sorbitol and sulfuric acid often create an adduct, which prevents 1,4-sorbitan from further dehydrating to isosorbide. According to the authors, the hydroxyl group on the secondary carbon (C4) attacks the main carbon (C1) of sorbitol, resulting in an SN2 reaction. However, the production of many by-products frequently limits the reaction yield, a phenomenon primarily linked to difficulties with regioselectivity and chemistry [59]. Shi et al. [60] proposed a potential mechanism for the acid-catalyzed dehydration of sorbitol (Fig. 15). Preferential protonation of the sorbitol hydroxyl group at position C6 or C1 leads to cyclodehydration with the hydroxyl group at position C3 or C4, most frequently creating 1,4-sorbitan (A→C) [61]. Since 3,6-sorbitan production is kinetically unfavorable and occurs in tiny levels, it can be disregarded [62]. Through intramolecular hydrogen bonding, cyclodehydration can also take effect between the C2 and C5 or C1 and C5 locations, generating 1,5-sorbitan (A→D) and 2,5-sorbitan (A→E), respectively; however, these pathways are of low selectivity and can also be disregarded. Subsequently, When the hydroxyl group at position C6 of 1,4-sorbitan is protonated, it turns less complicated to dehydrate the C3 hydroxyl group, which forms isosorbide (C→F).
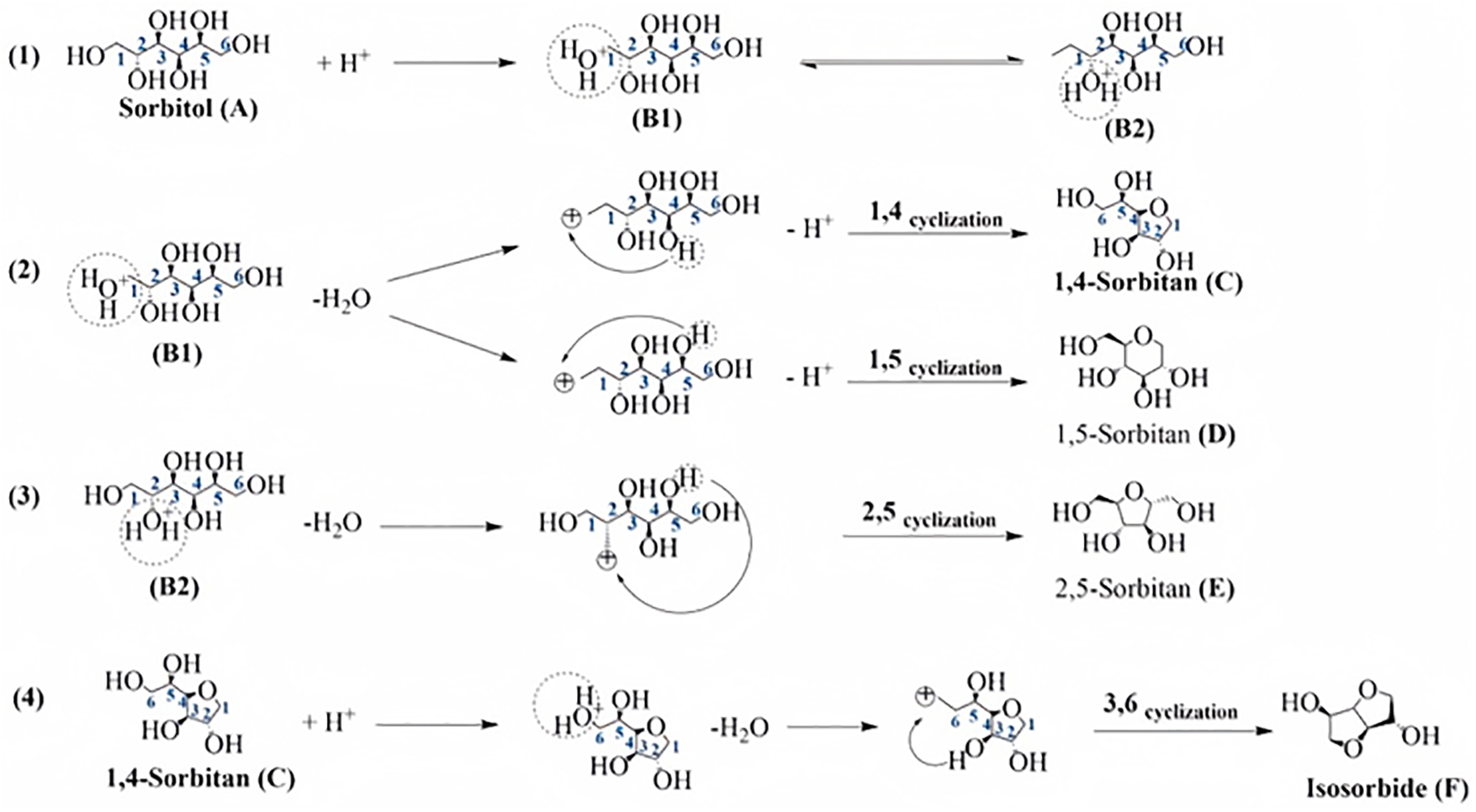
Figure 15: Mechanism of acid-catalyzed dehydration of sorbitol to isosorbide. Adapted with permission from Reference [60]. Copyright ©2024, Elsevier
The final step in industrially converting sorbitol to isosorbide, derived from cellulose, can be effectively carried out using a range of liquid and solid acid catalysts. Various homogeneous catalysts have been reported, such as ionic liquids, Lewis acids (metal chlorides/sulfonates) and Brønsted acids (phosphoric acid, sulfuric acid, and hydrochloric acid) [63]. Acidic resins, zeolites, metal salts, and metal oxides are the main components of heterogeneous catalysts. Table 3 summarizes the catalytic effects of different acid catalysts on the dehydration of sorbitol.
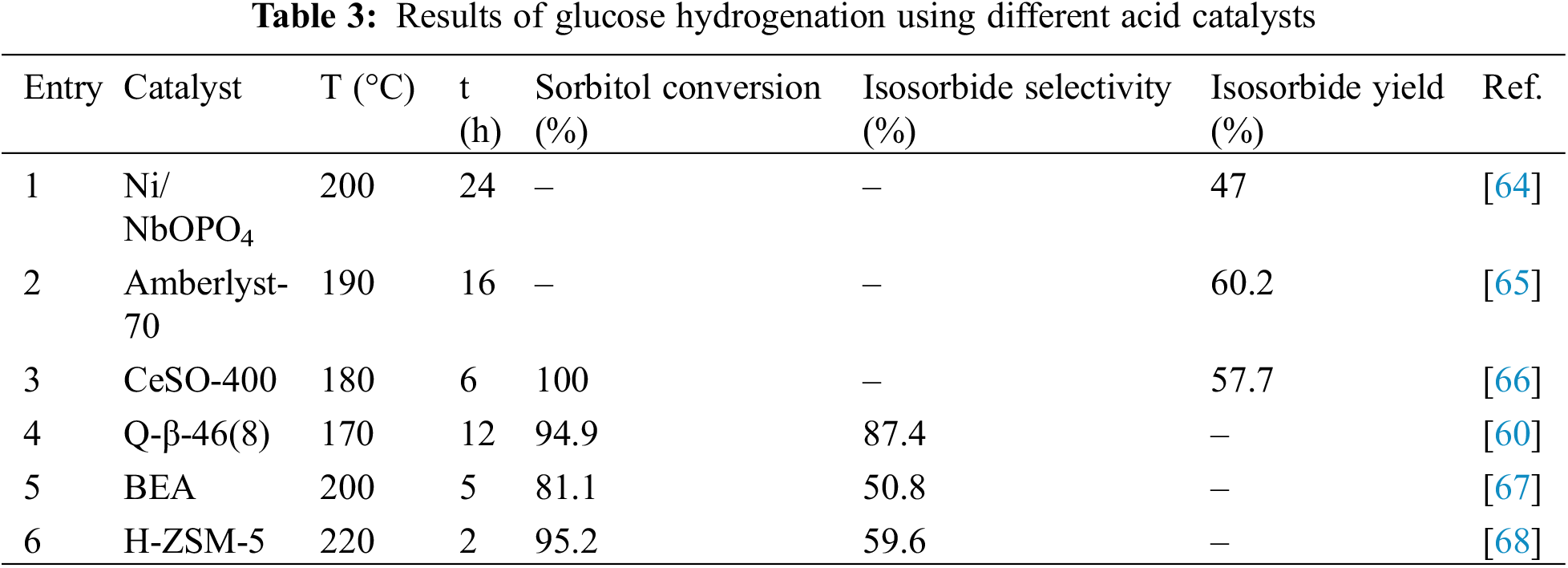
Liquid acid catalysts exhibit high catalytic activity, and sulfuric acid, in particular, is widely used in industry due to its excellent catalytic properties. When sulfuric acid is used at a mass ratio of 1:100 to sorbitol under conditions of 3 kPa and 130°C for 90 min, the yield of isosorbide reaches 80%. When utilizing 3 M sulfuric acid to calculate the chemical reaction rate constants for sorbitol dehydration, Bock et al. [69] found that while the dehydration rate is slower, the generation rate of 1,4-sorbitan is higher than that of 3,6-sorbitan. Sulfuric acid has a larger association constant with sorbitol than it does with 1,4-sorbitan, which accounts for the first dehydration’s overall faster reaction rate than the second [58]. Despite its superior catalytic activity, sulfuric acid poses significant drawbacks, including environmental pollution from post-reaction neutralization, handling risks, high corrosiveness, and stability issues of the product [70]. Ionic liquids (ILs) with cations and anions that have Brønsted substituents added to them can exhibit variable acidity, as demonstrated by Brønsted acidic ionic liquids (BILs), allowing for the functionalization of the organic backbone [62]. Significantly, since BILs are non-volatile and non-corrosive, they are used to replace conventional inorganic acids, which makes catalyst recycling easier [71]. An effective technique for synthesizing isosorbide from sorbitol has been reported with the use of Brønsted acidic ionic liquids as catalysts [72]. Experimental results indicated that different BILs exhibit similar performance during the first dehydration step but vary significantly in the second dehydration step. This discrepancy is attributed to their respective abilities to protonate polyhydroxy substrates. The protonation active centers in acidic ionic liquids primarily originate from active hydrogen, and when the cation remains constant, the activity order of the anions is: BF4− < HSO4− < CF3SO3−.
To address the notable differences in performance of BILs during the second dehydration step, researchers further conducted dehydration reactions using self-prepared BILs and protonated the ionic liquids with various inorganic acids. Fig. 16a compares the yields of isosorbide produced by different BILs and the chemical shifts of unstable protons observed in ANS and 1H NMR. This indicates that, as opposed to intrinsic acidity, each BIL’s reactivity is directly correlated with its AN value. In particular, as the AN value rises, the isosorbide yield first rises before abruptly declining. Therefore, it is necessary to optimize the yield of isosorbide based on an appropriate acid value rather than the inherent acidity. Fig. 16b depicts the conversion of sorbitol and the yield of isosorbide in the continuous dehydration reaction catalyzed by BIL-4. BIL-4 demonstrates excellent catalytic performance in transforming sorbitol into isosorbide. Across five successive runs, the conversion rate of sorbitol and the yield of isosorbide stay relatively consistent at around 99% and 73%, respectively.
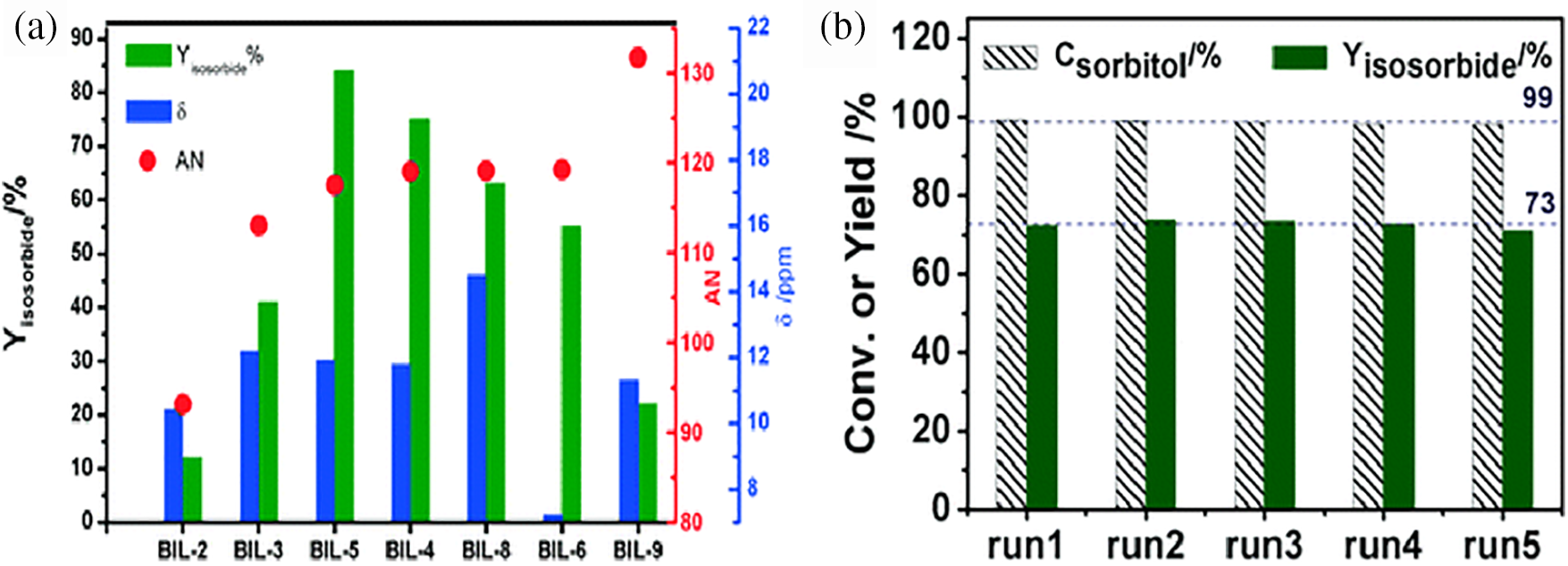
Figure 16: (a) Evaluating the isosorbide ester yield in the catalytic reaction in relation to the protons’ chemical shifts (ppm) and AN values at 600 MHz and 80°C. (b) Sorbitol conversion and isosorbide production from sorbitol’s continuous dehydration, catalyzed by BIL-4 (BIL-4: 0.6 mol%, 130°C, 1.5 h). Adapted with permission from Reference [72]. Copyright ©2017, Royal Society of Chemistry
The corrosive nature of inorganic acids towards equipment and the difficulties associated with subsequent product separation and purification render them unsuitable for meeting green and sustainable requirements. Additionally, the high cost of ionic liquids limits their use in industrial catalysis. Therefore, the development of solid acid catalysts emerges as the optimal solution for achieving continuous industrial production. Solid acid catalysts, known for their environmental friendliness, ease of separation from reaction systems, and reusability, have been employed in various industrial reactions such as alkylation, dehydration, polymerization, condensation, addition, and isomerization [73]. The creation of highly efficient solid acid catalysts, such as metal salts, metal oxides, zeolite molecular sieves, and acidic resins, is the main focus of current research on the dehydration of sorbitol (Fig. 17).
Figure 17: Types of solid acid catalysts
Metal oxide solid acids, including ZrO2, TiO2, SnO2, CaO, MgO, and Al2O3, feature surface acidic sites capable of catalyzing dehydration reactions. By incorporating other modifying substances into these metal oxides, the catalytic activity for sorbitol dehydration can be enhanced. A sulfated catalyst supported on zirconia efficiently converted sorbitol to isosorbide in two hours at a reaction temperature of 210°C. Under these conditions, a 100% sorbitol conversion rate and approximately 61% isosorbide yield were achieved [74]. The impact of acid site density, in addition to the relative activity and selectivity of several zirconia phases (SO42–/ZrO2, SZ), were also investigated in this study. Fig. 18a illustrates the relationship between the specific activity of D-sorbitol and the acid site loading for monoclinic (m-) SZ, SZ, and tetragonal (t-) SZ catalysts. The D-sorbitol conversion rate increased linearly with acid site loading in all cases, with m-SZ showing the strongest correlation with this relationship. For comparable acid site loadings, the performance of pure t-SZ was inferior to that of SZ. Fig. 18b illustrates the selectivity for 1,4-sorbitan and isosorbide at a 10% conversion rate among various catalysts, eliminating mass transfer limitations and minimizing side reactions. The isosorbide selectivity and acid site density were consistent across all catalysts. Previous kinetic studies have demonstrated that the ring dehydration of D-sorbitol to 1,4-sorbitan occurs faster than the ring dehydration of 1,4-sorbitan to isosorbide. Consequently, stronger acids are more effective in catalyzing the second dehydration phase (1,4-sorbitan to isosorbide).
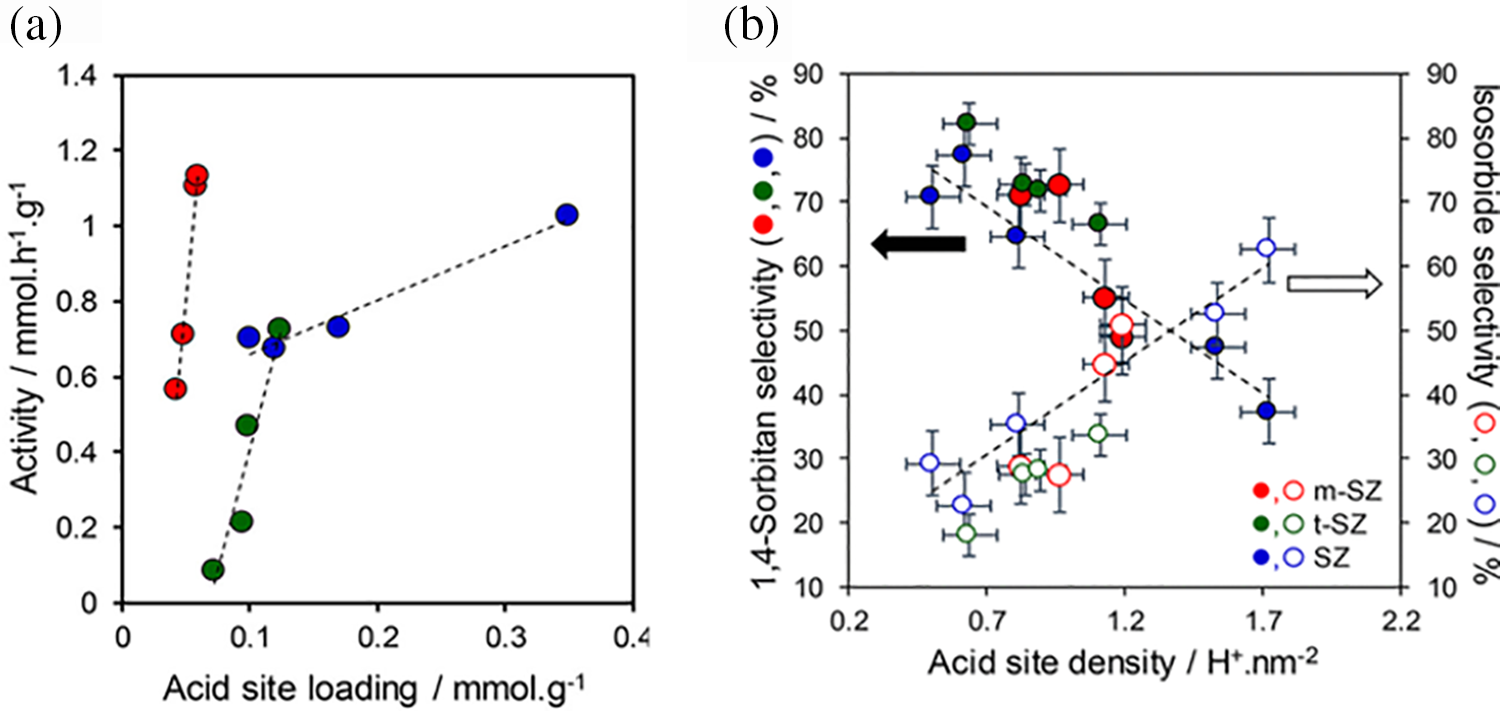
Figure 18: Cyclodehydration over m-SZ, xM-SZ, and t-SZ catalysts of D-sorbitol to isosorbide: (a) D-sorbitol conversion activity; (b) association between catalyst acid site density at 10% D-sorbitol conversion across SZ, m-SZ, and t-SZ catalysts and selectivity for 1,4-sorbitan or isosorbide. Conditions: 40 mL of water, 0.4 g of D-sorbitol, 0.1 mL of DMSO internal standard, 180°C temperature, and 2 h of reaction time. Adapted with permission from Reference [74]. Copyright ©2018, American Chemical Society
The liquid phase conversion of sorbitol to isosorbide using trivalent cations such as B-, Al-, Fe-, Ce-, and La- phosphates as catalysts and tetrapolyphosphate zinc under hydrothermal conditions. A 100% sorbitol conversion rate and a 70.3% selectivity for isosorbide were obtained from the investigation, however, the presence of heterogeneous acidic BP catalysts in a batch reactor caused the selectivity for 1,4-sorbitan to decrease to 6.3% [75]. Using NH3-TPD, the researchers assessed the acidity of metal phosphate catalyst surfaces (Fig. 19a). The findings demonstrated that water molecules and solid acid centers drove the conversion of sorbitol when FeP, ZrP-2, ZrP-1 and CeP were used as catalysts; BP had more acid sites than the other phosphates. The activity of the metal phosphates, in terms of sorbitol conversion, followed this order: AlP < ZrP-1 < ZrP-2 < FeP < LaP < CeP < BP, which was consistent with the selectivity order for isosorbide. Sorbitol dehydration was more favorable at medium and strong acidic sites, while very strong and overly strong acidity was less beneficial. Moreover, researchers observed that BP retained its crystallinity even under high-temperature calcination (Fig. 19b). The BP catalyst’s higher average pore size was primarily responsible for its enhanced catalytic performance.
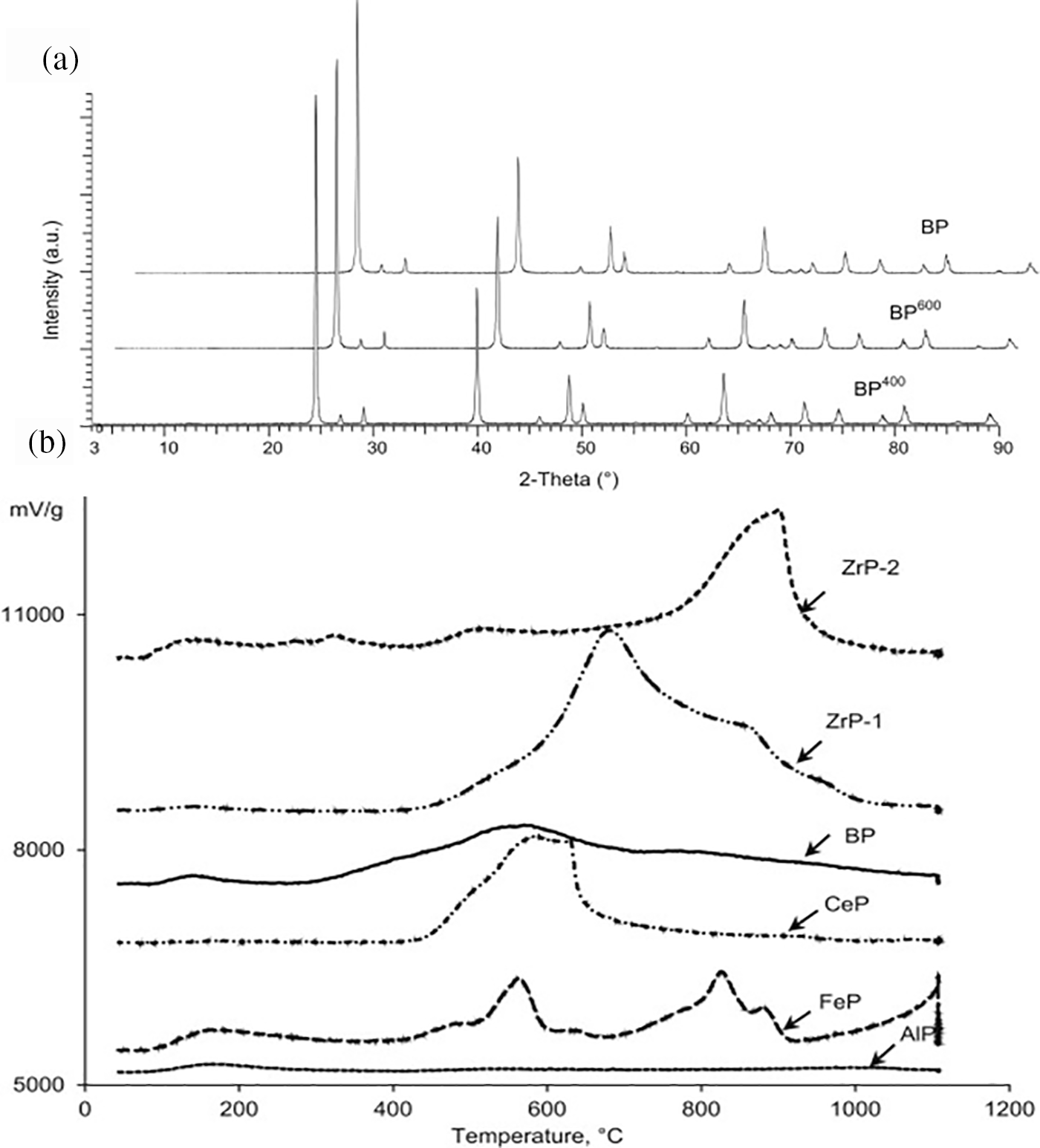
Figure 19: (a) XRD patterns of BP and its calcined forms, BP400 and BP600. (b) NH3-TPD profiles of the metal phosphates. Adapted with permission from Reference [75]. Copyright ©2015, Elsevier
Various metal-modified (Cr, Mn, Fe, Co, Ni, Cu, Al, Bi, Ga) sulfonic acid resins have been discovered to have Brønsted (B) and Lewis (L) acid sites. When applied as catalysts, these resins help byproducts produced at the start of the reaction produce 1,4-sorbitan and isosorbide simultaneously. However, the catalysts lost activity when loaded with metals such as Fe, Al, and Ga, likely due to the deactivation of the acidic sites. Notably, Bi (OTs)3 achieved a 100% sorbitol conversion rate and a 67% selectivity for isosorbide [76]. Using the solid acid ion-exchange resin Amberlyst 36, researchers investigated the effects of temperature, stirring speed, reaction time, and catalyst loading on the dehydration of sorbitol under mild conditions [77]. The data demonstrated that increasing the catalyst loading from 5 to 7 wt% did not significantly alter the isosorbide yield. Extended temperatures facilitated the process, increasing the isosorbide production and sorbitol conversion at the same time. Sorbitol conversion and isosorbide yield both above 99% under ideal circumstances, which included 5 wt% catalyst loading, 300 rpm stirring speed, 423 K, and 4 h of reaction time. When it came to isosorbide yield during sorbitol dehydration, Amberlyst 36 performed better than other resin catalysts that have been described. The pore size of zeolites allows the selective entry of molecules of specific sizes, and their acidic sites facilitate proton transfer, thus accelerating dehydration reactions effectively [40]. H-ZSM-5, a high-silica zeolite with an MFI structure, exhibits excellent thermal stability and acidity. In the sorbitol dehydration process to produce isosorbide, H-ZSM-5 efficiently catalyzes the dehydration reaction while minimizing side reactions, thereby enhancing the yield and purity of isosorbide [78]. At 200°C for 2 h, the BEA-type aluminosilicate zeolite with a Si/Al ratio of 75 generated up to 80% isosorbide. The reaction rate of sorbitol is closely related to temperature; at 180°C for 2 h, the sorbitol conversion rate was 43%, whereas at 220°C, it reached 99%. While increasing the catalyst amount can improve sorbitol conversion, it does not necessarily enhance the isosorbide yield, indicating an increase in by-product formation.
5.3 Factors Influencing Catalytic Performance
While sorbitol dehydration and cellulose hydrolysis can both utilize acid catalysts under specific conditions, there are several key differences between the two processes with respect to catalyst type, strength, and origin. Acid catalysts are generally used to donate protons (H+) to the glycosidic bond oxygen atoms in cellulose molecules during hydrolysis to glucose, increasing the susceptibility of these protonated bonds to nucleophilic attack by water molecules. Cellulose hydrolysis typically occurs at temperatures below 190°C using acid catalysts that effectively break the robust glycosidic bonds without causing excessive side reactions. In contrast, sorbitol dehydration requires temperatures exceeding 220°C and involves a two-step dehydration process to achieve high yields of isosorbide [3]. This process relies on strong acids at high temperatures to facilitate hydroxyl group removal, stabilize reaction intermediates, and overcome the high activation energy barrier, thereby ensuring efficient conversion. In the catalytic dehydration of sorbitol, catalysts initially provide protons to protonate the substrate. Only when the acid strength of the catalyst reaches a certain threshold can the reaction substrate be effectively activated, leading to high reaction activity. Therefore, the acidity of the catalyst is a major factor influencing reaction activity. Researchers compared the dehydration efficiency of sorbitol using Brønsted homogeneous acid catalysts with varying acid strengths (pKa). According to their data, the catalytic activity of Brønsted acids in sorbitol dehydration, as shown in Fig. 20a, increases when the catalyst’s pKa is less than −1.74, resulting in a higher turnover frequency of sorbitol. Conversely, when the pKa exceeds −1.74, the catalytic activity decreases with increasing pKa, reducing both sorbitol conversion and isosorbide yield. This indicates that the acidity of Brønsted acid catalysts significantly impacts sorbitol dehydration, with strong acids (low pKa values) having a high acid dissociation constant (Ka), which fully dissociates in the reaction system, thereby effectively catalyzing the dehydration of the substrate.
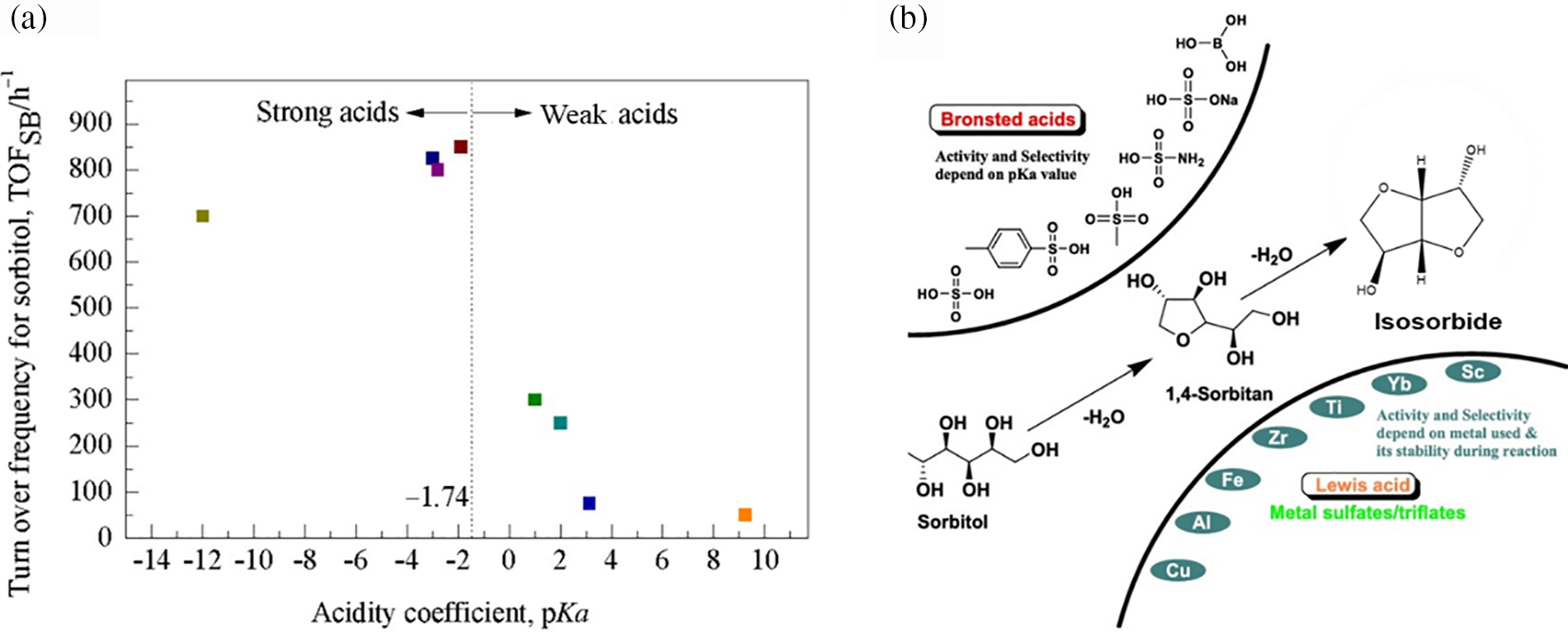
Figure 20: (a) Catalytic activity of Brønsted acids with different acid strengths in the dehydration reaction of sorbitol. (b) Schematic illustration of sorbitol conversion to isosorbide under various Brønsted and Lewis acid catalysts. Adapted with permission from Reference [79]. Copyright ©2015, Elsevier
The transformation of sorbitol into isosorbide using several Brønsted and Lewis acid catalysts is shown in Fig. 20b [79]. After comparing the product selectivity of Lewis and Brønsted acids, researchers discovered that both kinds of acids may catalyze the formation of 1,4-sorbitan by removing the first water molecule from sorbitol. But when it came to taking out the second water molecule from sorbitol, Brønsted acids worked better. The combined action of Lewis and Brønsted acids can produce isosorbide at temperatures above 160°C. Although Brønsted acid sites generally exhibit stronger catalytic activity than Lewis acid sites, isosorbide selectivity is negatively impacted by both excessively high and low acidity. Low acidity results in low catalytic activity, while excessive acidity increases side reactions, thereby reducing isosorbide selectivity.
Solid acid catalysts’ catalytic activity and product selectivity depend significantly on their pore structure, specific surface area, and surface properties [80]. Surface characteristics play a crucial role during the reaction. Hydrophobic surfaces accelerate the reaction by facilitating the removal of water produced, but they also promote coking, leading to catalyst deactivation [34]. Conversely, hydrophilic surfaces can extend the catalyst’s lifespan, although higher acidity is required to maintain catalytic efficiency [11]. The solvent environment also significantly affects the reaction, not only by synergistically activating the substrates with the catalyst and accelerating the reaction rate but also by altering the conformation of sorbitol within the reaction system or directly participating in the reaction, thereby influencing selectivity. The pore structure of catalysts is essential for mass transfer. Different pore sizes exhibit varying diffusion rates, and the relationship between pore size and the molecular size of reactants or products can result in shape-selective catalysis and spatial confinement effects. Studies have shown that microporous Ru/C and Ru/NbOPO4 catalysts yield isosorbide at rates ranging from 20% to 56%, whereas loading Ru onto mesoporous niobium phosphate increases isosorbide yield by 2 to 6 times [23]. This demonstrates that mesoporous structures are more conducive to mass transfer, increasing the beneficial effects of metal and acid sites.
In addition to the factors mentioned above affecting the dehydration rate of sorbitol, the amount of catalyst, temperature, and pressure also influence the reaction rate or equilibrium. Catalyst loading is a significant parameter that influences reaction rate and must be adjusted to reach the desired catalytic efficiency. Increasing catalyst loading can improve the accessibility of active sites to the reactants, leading to adequate catalytic effects [81]. Temperature significantly affects sorbitol dehydration; lower temperatures are insufficient to activate the reactants, while higher temperatures can lead to problems such as metal particle sintering and coking [77]. Therefore, determining the optimal reaction conditions for converting cellulose to isosorbide is essential.
This review has comprehensively examined the reaction mechanisms and catalyst systems involved in the three key steps of converting cellulose to isosorbide. Firstly, we detailed the activation energy and acidic sites involved in the hydrolysis of cellulose to glucose, discussing the types and performances of catalysts used in this process. Secondly, we delved into the mechanism of glucose hydrogenation to sorbitol, analyzing the impact of different metal catalysts on hydrogenation activation, with a focus on factors influencing the catalytic performance of metal catalysts. Finally, we emphasized the critical steps in the two-step dehydration of sorbitol to isosorbide, thoroughly exploring the catalyst systems, catalytic performance, and factors affecting catalytic activity in the sorbitol dehydration process.
Currently, the one-step method for producing isosorbide from cellulose remains in the experimental research stage, presenting significant technical challenges. The catalysts used generally suffer from low activity and poor stability, resulting in slow reaction rates, and the yield and selectivity of isosorbide need improvement.
Future research should, therefore, focus on developing efficient and stable catalysts and optimizing reaction conditions to enhance reaction efficiency and increase the yield of isosorbide. A clear understanding of the pore structure and surface acidic centers of catalysts is crucial for improving isosorbide selectivity and suppressing various side reactions. Consequently, further research is needed in the catalytic synthesis of isosorbide.
Acknowledgement: None.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Dan Wang, Jihuan Song, Qiyuan Wang, Ying Yang. draft manuscript preparation: Dan Wang, Chenmeng Cui. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Wang SY, Cheng AH, Liu FH, Zhang JJ, Xia T, Zeng X, et al. Catalytic conversion network for lignocellulosic biomass valorization: a panoramic view. Ind Chem Mater. 2023;1(2):188–206. doi:10.1039/D2IM00054G. [Google Scholar] [CrossRef]
2. Howell BA. Flame retardants from starch: phosphorus derivatives of isosorbide. Bio-Based Flame-Retardant Technol. 2022;53(1):255–68. doi:10.1016/B978-0-323-90771-2.00015-8. [Google Scholar] [CrossRef]
3. Zhu Y, Durand M, Molinier V, Aubry JM. Isosorbide as a novel polar head derived from renewable resources. Application to the design of short-chain amphiphiles with hydrotropic properties. Green Chem. 2008;10(5):532–40. doi:10.1039/b717203f. [Google Scholar] [CrossRef]
4. Rose M, Palkovits R. Isosorbide as a renewable platform chemical for versatile applications–Quo Vadis? ChemSusChem. 2012;5(1):167–76. doi:10.1002/cssc.201100580. [Google Scholar] [PubMed] [CrossRef]
5. Yang Y, Ding Z, Ren D, Shang C, Li C, Yang F, et al. Dense Ru single-atoms integrated with sulfoacids for cellulose valorization to isosorbide. Mater Today Sustain. 2023;24(24):100494. doi:10.1016/j.mtsust.2023.100494. [Google Scholar] [CrossRef]
6. Chen ZH, Hu M, Zhu XL, Guo DB, Liu SM, Hu ZQ, et al. Characteristics and kinetic study on pyrolysis of five lignocellulosic biomass via thermogravimetric analysis. Bioresour Technol. 2015;192:441–50. doi:10.1016/j.biortech.2015.05.062. [Google Scholar] [PubMed] [CrossRef]
7. Bach QV, Tran KQ, Skreiberg Ø, Trinh TT. Effects of wet torrefaction on pyrolysis of woody biomass fuels. Energy. 2015;88:443–56. doi:10.1016/j.energy.2015.05.062. [Google Scholar] [CrossRef]
8. Miranda MIG, Bica CID, Nachtigall SMB, Rehman N, Rosa SML. Kinetical thermal degradation study of maize straw and soybean hull celluloses by simultaneous DSC-TGA and MDSC techniques. Thermochim Acta. 2013;565:65–71. doi:10.1016/j.tca.2013.04.012. [Google Scholar] [CrossRef]
9. Tukac V. Glucose hydrogenation in a trickle-bed reactor. Collect Czech Chem Commun. 1997;62(9):1423–8. doi:10.1135/cccc19971423. [Google Scholar] [CrossRef]
10. Crezee E, Hoffer BW, Berger RJ, Makkee M, Kapteijn F, Moulijn JA. Three-phase hydrogenation of D-glucose over a carbon-supported ruthenium catalyst-mass transfer and kinetics. Appl Catal A Gen. 2003;251(1):1–17. doi:10.1016/S0926-860X(03)00587-8. [Google Scholar] [CrossRef]
11. Kamimura A, Murata K, Kawamoto T. An efficient and selective conversion of sorbitol in ionic liquids: use of ion exchange resin as a solid acid catalyst. Tetrahedron Lett. 2017;58(37):3616–8. doi:10.1016/j.tetlet.2017.07.105. [Google Scholar] [CrossRef]
12. Wang L, Liu X, Wang Y, Sun W, Zhao L. Thermodynamics and reaction kinetics of the sorbitol dehydration to isosorbide using NbOPO4 as the catalyst. Ind Eng Chem Res. 2022;61(23):7833–41. doi:10.1021/acs.iecr.2c00925. [Google Scholar] [CrossRef]
13. Zeng MJ, Pan XJ. Insights into solid acid catalysts for efficient cellulose hydrolysis to glucose: progress, challenges, and future opportunities. Ind Eng Chem Res. 2020;64(3):445–90. [Google Scholar]
14. Li O, Ikurac RH, Ishizaki T. Hydrolysis of cellulose to glucose over carbon catalysts sulfonated via a plasma process in dilute acids. Green Chem. 2017;19(20):4774–7. doi:10.1039/C7GC02143G. [Google Scholar] [CrossRef]
15. Yuan S, Li T, Wang Y, Cai B, Wen X, Shen S, et al. Double-adsorption functional carbon based solid acids derived from co-pyrolysis of PVC and PE for cellulose hydrolysis. Fuel. 2019;237:895–902. doi:10.1016/j.fuel.2018.10.088. [Google Scholar] [CrossRef]
16. Wu B, Shen S, Guo X, Sun R, Liu Y, Li J, et al. Introducing the sulfone group as a cellulose-binding site to improve the hydrolysis performance of the solid acid. Fuel. 2024;374:132461. doi:10.1016/j.fuel.2024.132461. [Google Scholar] [CrossRef]
17. Komanoya T, Kobayashi H, Hara K, Chun WJ, Fukuoka A. Catalysis and characterization of carbon-supported ruthenium for cellulose hydrolysis. Appl Catal A: Gen. 2011;407(1–2):188–94. doi:10.1016/j.apcata.2011.08.039. [Google Scholar] [CrossRef]
18. Luo C, Wang S, Liu HC. Cellulose conversion to polyols catalyzed by reversibly-formed acids and supported ruthenium clusters in hot water. Angew Chem Int Ed. 2007;46(40):7636–9. doi:10.1002/anie.200702661. [Google Scholar] [PubMed] [CrossRef]
19. Oh SY, Yoo DI, Shin Y, Kim HC, Kim HY, Chung YS, et al. Crystalline structure analysis of cellulose treated with sodium hydroxide and carbon dioxide by means of X-ray diffraction and FTIR spectroscopy. Carbohydr Res. 2005;340(15):2376–81. doi:10.1016/j.carres.2005.08.007. [Google Scholar] [PubMed] [CrossRef]
20. Liu Y, Chen LX, Zhang W, Liu HC. Recyclable Cu salt-derived Brønsted acids for hydrolytic hydrogenation of cellulose on Ru catalyst. CCS Chem. 2021;4(9):3162–9. doi:10.31635/ccschem.021.202101506. [Google Scholar] [CrossRef]
21. Suganuma S, Nakajima K, Kitano M, Yamaguchi D, Kato H, Hayashi S, et al. Hydrolysis of cellulose by amorphous carbon bearing SO3H, COOH, and OH groups. J Am Chem Soc. 2008;130(38):12787–93. doi:10.1021/ja803983h. [Google Scholar] [PubMed] [CrossRef]
22. Pang JF, Wang AQ, Zheng MY, Zhang T. Hydrolysis of cellulose into glucose over carbons sulfonated at elevated temperatures. Chem Commun. 2010;46(37):6935–7. doi:10.1039/c0cc02014a. [Google Scholar] [PubMed] [CrossRef]
23. Sun P, Long XD, He H, Xia CG, Li FW. Conversion of cellulose into isosorbide over bifunctional ruthenium nanoparticles supported on niobium phosphate. ChemSusChem. 2013;6(11):2190–7. doi:10.1002/cssc.v6.11. [Google Scholar] [CrossRef]
24. Kitano M, Yamaguchi D, Suganuma S, Nakajima K, Kato H, Hayashi S, et al. Adsorption-enhanced hydrolysis of β-1,4-glucan on graphene-based amorphous carbon bearing SO3H, COOH, and OH groups. Langmuir. 2009;25(9):5068–75. doi:10.1021/la8040506. [Google Scholar] [PubMed] [CrossRef]
25. Gallezot P, Cerino PJ, Blanc B, Flèche G, Fuertes P. Glucose hydrogenation on promoted Raney-nickel catalysts. J Catal. 1994;146(1):93–102. doi:10.1016/0021-9517(94)90012-4. [Google Scholar] [CrossRef]
26. Michiel M, Antonius PGK, Herman VB. Hydrogenation of D-fructose and D-fructose/D-glucose mixtures. Carbohydr Res. 1985;138(2):225–36. doi:10.1016/0008-6215(85)85106-5. [Google Scholar] [CrossRef]
27. Trinh QT, Chethana BK, Mushrif SH. Adsorption and reactivity of cellulosic aldoses on transition metals. J Phys Chem C. 2015;119(30):17137–45. doi:10.1021/acs.jpcc.5b03534. [Google Scholar] [CrossRef]
28. Fang ZC, Fan HA, Zhao XZ, Lin GB, Li BL, Wang JH, et al. Unveiling the nature of glucose hydrogenation over Raney Ni: DFT and AIMD simulations. Appl Catal A: Gen. 2023;667:119462. doi:10.1016/j.apcata.2023.119462. [Google Scholar] [CrossRef]
29. Weiss J. Mechanism of the Cannizzaro reaction and some allied processes. Trans Faraday Soc. 1941;37:782. doi:10.1039/tf9413700782. [Google Scholar] [CrossRef]
30. Sifontes HVA, Oladele O, Kordás K, Eränen K, Mikkola J-P, Murzin DY, et al. Sugar hydrogenation over a Ru/C catalyst. J Chem Technol Biotechnol. 2011;86(5):658–68. doi:10.1002/jctb.v86.5. [Google Scholar] [CrossRef]
31. Hoffer BW. The role of the active phase of Raney-type Ni catalysts in the selective hydrogenation of D-glucose to D-sorbitol. Appl Catal A: Gen. 2003;253(2):437–52. doi:10.1016/S0926-860X(03)00553-2. [Google Scholar] [CrossRef]
32. Besson M, Gallezot P. Deactivation of metal catalysts in liquid phase organic reactions. Catal Today. 2003;81(4):547–59. doi:10.1016/S0920-5861(03)00153-6. [Google Scholar] [CrossRef]
33. Li HL, Wang WJ, Deng JF. Glucose hydrogenation to sorbitol over a skeletal Ni-P amorphous alloy catalyst (Raney Ni-P). J Catal. 2000;191(1):257–60. doi:10.1006/jcat.1999.2792. [Google Scholar] [CrossRef]
34. Hoffer BW, Crezee E, Mooijman PRM, van Langeveld AD, Kapteijn F, Moulijn JA. Carbon supported Ru catalysts as promising alternative for Raney-type Ni in the selective hydrogenation of D-glucose. Catal Today. 2003;79-80:35–41. doi:10.1016/S0920-5861(03)00040-3. [Google Scholar] [CrossRef]
35. Mishra DK, Lee JM, Chang JS, Hwang JS. Liquid phase hydrogenation of D-glucose to D-sorbitol over the catalyst (Ru/NiO-TiO2) of ruthenium on a NiO-modified TiO2 support. Catal Today. 2012;185(1):104–8. doi:10.1016/j.cattod.2011.11.020. [Google Scholar] [CrossRef]
36. Guo XC, Wang XC, Guan J, Chen XF, Qin ZF, Mu XD, et al. Selective hydrogenation of D-glucose to D-sorbitol over Ru/ZSM-5 catalysts. Chin J Catal. 2014;35(5):733–40. doi:10.1016/S1872-2067(14)60077-2. [Google Scholar] [CrossRef]
37. García B, Moreno J, Iglesias J, Melero JA, Morales G. Transformation of glucose into sorbitol on Raney nickel catalysts in the absence of molecular hydrogen: sugar disproportionation vs catalytic hydrogen transfer. Top Catal. 2019;62:570–8. doi:10.1007/s11244-019-01156-3. [Google Scholar] [CrossRef]
38. Romero A, Alonso E, Sastre Á, Nieto-Márquez A. Conversion of biomass into sorbitol: cellulose hydrolysis on MCM-48 and D-glucose hydrogenation on Ru/MCM-48. Microporous Mesoporous Mater. 2016;224:1–8. doi:10.1016/j.micromeso.2015.11.013. [Google Scholar] [CrossRef]
39. Zhao J, Yang XR, Wang W, Liang JH, Orooji Y, Dai CW, et al. Efficient sorbitol producing process through glucose hydrogenation catalyzed by Ru supported amino poly(styrene-co-maleic) polymer (ASMA) encapsulated on γ-Al2O3. Catalysts. 2020;10(9):1068. doi:10.3390/catal10091068. [Google Scholar] [CrossRef]
40. Yang Y. A simple way to nickel based mesoporous carbons: inexpensive nanocatalysts for the hydrogenolysis of sorbitol. Int J Petrochem Res. 2018;2(3):194–201. doi:10.18689/ijpr. [Google Scholar] [CrossRef]
41. Fu Y, Pichon B, Devred F, Singleton ML, Hermans S. Synthesis of spherical, rod, or chain Ni nanoparticles and their structure-activity relationship in glucose hydrogenation reaction. J Catal. 2022;415:63–76. doi:10.1016/j.jcat.2022.09.028. [Google Scholar] [CrossRef]
42. Kryvutsa N, Eloy P, Hackens B, Hermans S. Synthesis of Ru, Ni and Fe supported graphene nanoplatelets catalysts for hydrogenation of glucose into sorbitol. Mol Catal. 2023;545:113222. doi:10.1016/j.mcat.2023.113222. [Google Scholar] [CrossRef]
43. Ge JM, Zhang DB, Qin Y, Dou T, Jiang MH, Zhang FZ, et al. Dual-metallic single Ru and Ni atoms decoration of MoS2 for high-efficiency hydrogen production. Appl Catal B: Environ. 2021;298:120557. doi:10.1016/j.apcatb.2021.120557. [Google Scholar] [CrossRef]
44. Guo Y, He HR, Liu X, Chen ZF, Rioux RM, Janik MJ, et al. Ring-opening and hydrodenitrogenation of indole under hydrothermal conditions over Ni, Pt, Ru, and Ni-Ru bimetallic catalysts. Chem Eng J. 2021;406:126853. doi:10.1016/j.cej.2020.126853. [Google Scholar] [CrossRef]
45. Xi R, Tang YW, Smith RL, Liu XN, Liu L, Qi XH, et al. Selective hydrogenation of glucose to sorbitol with tannic acid-based porous carbon sphere supported Ni-Ru bimetallic catalysts. Green Energy Environ. 2023;8(6):1719–27. doi:10.1016/j.gee.2022.04.003. [Google Scholar] [CrossRef]
46. Ali I, Al-Arfaj AA, Saleh TA. Carbon nanofiber-doped zeolite as support for molybdenum based catalysts for enhanced hydrodesulfurization of dibenzothiophene. J Mol Liq. 2020;304(17):112376. doi:10.1016/j.molliq.2019.112376. [Google Scholar] [CrossRef]
47. Lee J, Kim J, Hyeon T. Recent progress in the synthesis of porous carbon materials. Adv Mater. 2006;18(16):2073–94. doi:10.1002/adma.200501576. [Google Scholar] [CrossRef]
48. Zhang ZG, Yoshida T. Behaviour of hydrogen transfer in the hydrogenation of anthracene over activated carbon. Energy Fuels. 2001;15(3):708–13. doi:10.1021/ef000253d. [Google Scholar] [CrossRef]
49. Ma YM, Wei XY, Zhou X, Cai KY, Peng YL, Xie RL, et al. Microwave-assisted hydrogen transfer to anthracene and phenanthrene over Pd/C. Energy Fuels. 2009;23(2):638–45. doi:10.1021/ef800808t. [Google Scholar] [CrossRef]
50. Haruta M. Size- and support-dependency in the catalysis of gold. Catal Today. 1997;36(1):153–66. doi:10.1016/S0920-5861(96)00208-8. [Google Scholar] [CrossRef]
51. Haruta M, Tsubota S, Kobayashi T, Kageyama H, Genet MJ, Delmon B. Low-temperature oxidation of CO over gold supported on TiO2, α-Fe2O3, and Co3O4. J Catal. 1993;144(1):175–92. doi:10.1006/jcat.1993.1322. [Google Scholar] [CrossRef]
52. Haynes T, Dubois V, Hermans S. Particle size effect in glucose oxidation with Pd/CB catalysts. Appl Catal A: Gen. 2017;542:47–54. doi:10.1016/j.apcata.2017.05.008. [Google Scholar] [CrossRef]
53. Lykhach Y, Kozlov SM, Skála T, Tovt A, Stetsovych V, Tsud N, et al. Counting electrons on supported nanoparticles. Nat Mater. 2015;15:284–8. [Google Scholar] [PubMed]
54. Bezemer GL, Bitter JH, Kuipers HPC, Oosterbeek H, Holewijn JE, Xu XD, et al. Cobalt particle size effects in the Fischer-Tropsch reaction studied with carbon nanofiber supported catalysts. J Am Chem Soc. 2006;128(12):3956–64. doi:10.1021/ja058282w. [Google Scholar] [PubMed] [CrossRef]
55. Fu Y, Devred F, Eloy P, Haynes T, Singleton ML, Hermans S. The effect of Ni particle size and carbon support on catalytic activity for glucose hydrogenation reaction. Appl Catal A: Gen. 2022;644:118833. doi:10.1016/j.apcata.2022.118833. [Google Scholar] [CrossRef]
56. Hopper JT, Ma RN, Rawlings JB, Ford PC, Abu-Omar MM. Markedly improved catalytic dehydration of sorbitol to isosorbide by sol-gel sulfated zirconia: a quantitative structure-reactivity study. ACS Catal. 2023;13(15):10137–52. doi:10.1021/acscatal.3c00755. [Google Scholar] [PubMed] [CrossRef]
57. Delbecq F, Khodadadi MR, Rodriguez Padron D, Varma R, Isosorbide Len C. Recent advances in catalytic production. Mol Catal. 2020;482:110648. doi:10.1016/j.mcat.2019.110648. [Google Scholar] [CrossRef]
58. Yabushita M, Kobayashi H, Shrotri A, Hara K, Ito S, Fukuoka A. Sulfuric acid-catalyzed dehydration of sorbitol: mechanistic study on preferential formation of 1,4-sorbitan. Bull Chem Soc Jpn. 2015;88(7):996–1002. doi:10.1246/bcsj.20150080. [Google Scholar] [CrossRef]
59. Cheng DC, Ma ZH, Liu ZY, Liu XH, Liu T, Sun WZ, et al. Experiments and kinetic modeling of the sorbitol dehydration to isosorbide catalyzed by sulfuric acid under conditions of non-constant volume. Chin J Chem Eng. 2024;73:281–9. doi:10.1016/j.cjche.2024.05.024. [Google Scholar] [CrossRef]
60. Shi YL, Yang JH, Guo J, Wang SW, Yuan DP, Zhao N, et al. Preparation of isosorbide by dehydration of sorbitol on hierarchical Beta zeolites under solvent-free conditions. Micropor Mesopor Mat. 2024;368:113009. doi:10.1016/j.micromeso.2024.113009. [Google Scholar] [CrossRef]
61. Li JR, Spina A, Moulijn JA, Makkee M. Sorbitol dehydration into isosorbide in a molten salt hydrate medium. Catal Sci Technol. 2013;3(6):1540–6. doi:10.1039/c3cy20809e. [Google Scholar] [CrossRef]
62. Shirai M, Sato O, Hiyoshi N, Yamaguchi A. Enhancement of reaction rates for catalytic benzaldehyde hydrogenation and sorbitol dehydration in water solvent by addition of carbon dioxide. J Chem Sci. 2014;126:395–401. doi:10.1007/s12039-014-0582-3. [Google Scholar] [CrossRef]
63. Dussenne C, Delaunay T, Wiatz V, Wyart H, Suisse I, Sauthier M. Synthesis of isosorbide: an overview of challenging reactions. Green Chem. 2017;19(22):5332–44. doi:10.1039/C7GC01912B. [Google Scholar] [CrossRef]
64. He M, Guo J, Wang X, Song Y, Liu S, Wang H, et al. Direct conversion of cellulose into isosorbide over Ni doped NbOPO4 catalysts in water. New J Chem. 2020;44(25):10292–9. doi:10.1039/D0NJ01403F. [Google Scholar] [CrossRef]
65. Yamaguchi A, Sato O, Mimura N, Shirai M. One-pot conversion of cellulose to isosorbide using supported metal catalysts and ion-exchange resin. Catal Commun. 2015;67(2):59–63. [Google Scholar]
66. Boupan M, Prompang K, Chompunuch A, Boonma P, Neramittagapong A, Theerakulpisut S, et al. Role of calcination temperature on isosorbide production from sorbitol dehydration over the catalyst derived from Ce(IV) sulfate. J Renew Mater. 2023;11(7):2985–3000. doi:10.32604/jrm.2023.026397. [Google Scholar] [CrossRef]
67. Jeong S, Jeon KJ, Park YK, Kim BJ, Chung KH, Jung SC. Catalytic properties of microporous zeolite catalysts in synthesis of isosorbide from sorbitol by dehydration. Catalysts. 2020;10(2):148. doi:10.3390/catal10020148. [Google Scholar] [CrossRef]
68. Deng T, He X, Liu H. Insights into the active acid sites for isosorbide synthesis from renewable sorbitol and cellulose on solid acid catalysts. Chem Res Chin Univ. 2022;38(1):257–64. doi:10.1007/s40242-022-1499-x. [Google Scholar] [CrossRef]
69. Bock K, Pedersen C, Thøgersen H, Krogsgaard-Larsen P, Örn U. Acid catalyzed dehydration of alditols. Part I. D-glucitol and D-mannitol. Acta Chem Scand. 1981;35(6):441–9. [Google Scholar]
70. Chakraborti AK, Roy SR. On catalysis by ionic liquids. J Am Chem Soc. 2009;131(20):6902–3. doi:10.1021/ja900076a. [Google Scholar] [PubMed] [CrossRef]
71. Earle MJ, Esperança JMS, Gilea MA, Lopes JNC, Rebelo LPN, Magee JW, et al. The distillation and volatility of ionic liquids. Nature. 2006;439(7078):831–4. doi:10.1038/nature04451. [Google Scholar] [PubMed] [CrossRef]
72. Deng J, Xu BH, Wang YF, Mo XE, Zhang R, Li Y, et al. Brønsted acidic ionic liquid-catalyzed dehydrative formation of isosorbide from sorbitol: introduction of a continuous process. Catal Sci Technol. 2017;7(10):2065–73. doi:10.1039/C7CY00512A. [Google Scholar] [CrossRef]
73. Nicewicz DA, MacMillan DWC. Merging photoredox catalysis with organocatalysis: the direct asymmetric alkylation of aldehydes. Science. 2008;322(5898):77–80. doi:10.1126/science.1161976. [Google Scholar] [PubMed] [CrossRef]
74. Zhang XG, Rabee AIM, Isaacs M, Lee AF, Wilson K. Sulfated zirconia catalysts for D-sorbitol cascade cyclodehydration to isosorbide: impact of zirconia phase. ACS Sustain Chem Eng. 2018;6(11):14704–12. doi:10.1021/acssuschemeng.8b03268. [Google Scholar] [CrossRef]
75. Rusu OA, Hoelderich WF, Wyart H, Ibert M. Metal phosphate catalyzed dehydration of sorbitol under hydrothermal conditions. Appl Catal B: Environ. 2015;176–177(1):139–49. doi:10.1016/j.apcatb.2015.03.033. [Google Scholar] [CrossRef]
76. Dussenne C, Wyart H, Wiatz V, Suisse I, Sauthier M. Catalytic dehydration of sorbitol to isosorbide in the presence of metal tosylate salts and metallized sulfonic resins. Mol Catal. 2019;463:61–6. doi:10.1016/j.mcat.2018.11.004. [Google Scholar] [CrossRef]
77. Kamaruzaman MR, Jiang XX, Hu XD, Chin SY. High yield of isosorbide production from sorbitol dehydration catalyzed by Amberlyst 36 under mild conditions. Chem Eng J. 2020;388(6):124186. doi:10.1016/j.cej.2020.124186. [Google Scholar] [CrossRef]
78. Otomo R, Yokoi T, Tatsumi T. Synthesis of isosorbide from sorbitol in water over high-silica aluminosilicate zeolites. Appl Catal A: Gen. 2015;505:28–35. doi:10.1016/j.apcata.2015.07.034. [Google Scholar] [CrossRef]
79. Dabbawala AA, Mishra DK, Huber GW, Hwang J-S. Role of acid sites and selectivity correlation in solvent free liquid phase dehydration of sorbitol to isosorbide. Appl Catal A: Gen. 2015;492:252–61. doi:10.1016/j.apcata.2014.12.014. [Google Scholar] [CrossRef]
80. Chen F, Meng XJ, Xiao FS. Mesoporous solid acid catalysts. Catal Surv Asia. 2011;15(1):37–48. doi:10.1007/s10563-010-9107-3. [Google Scholar] [CrossRef]
81. Cao D, Yu B, Zhang SY, Cui L, Zhang JH, Cai WJ. Isosorbide production from sorbitol over porous zirconium phosphate catalyst. Appl Catal A: Gen. 2016;528:59–66. doi:10.1016/j.apcata.2016.09.017. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools