
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Histone deacetylase inhibitors as a novel therapeutic approach for pheochromocytomas and paragangliomas
1 Endocrine Unit, Second Department of Internal Medicine Propaedeutic & Research Institute, Medical School, National and Kapodistrian University of Athens,
Attikon University Hospital, Athens, 12461, Greece
2 First Department of Internal Medicine, Medical School, National and Kapodistrian University of Athens, Laikon General Hospital, Athens, 11527, Greece
3 Department of Obstetrics & Gynecology, General Hospital of Elefsina Thriassio, Elefsina, 19600, Greece
* Corresponding Author: Dimitrios C. Ziogas,
Oncology Research 2022, 30(5), 211-219. https://doi.org/10.32604/or.2022.026913
Received 03 October 2022; Accepted 11 January 2023; Issue published 03 February 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Epigenetic mechanisms, such as DNA methylation and histone modifications (e.g., acetylation and deacetylation), are strongly implicated in the carcinogenesis of various malignancies. During transcription, the expression and functionality of coding gene products are altered following the histone acetylation and deacetylation. These processes are regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. HDAC inhibitors (HDACis) have been developed as promising therapeutic agents, to limit exposure to traditional and toxic chemotherapies and offer more alternatives for some specific malignant diseases with limited options. Mechanistically, these agents affect many intracellular pathways, including cell cycle arrest, apoptosis and differentiation, and their mechanism of action mainly depends on the type of cancer. Currently, five HDACis have been approved for the treatment of several hematological malignancies (e.g., T-cell lymphoma subtypes and multiple myeloma); while, many of them are tested for further therapeutic indications in solid tumors (e.g., colorectal, thyroid, breast, lung and pancreatic cancer). Herein, we review the literature and gather all available evidence, from in vitro and in vivo data to clinical trial results, that recognizes the antitumor activity of HDACis on pheochromocytomas and paragangliomas; and supports their clinical implementation in the treatment of these rare neuroendocrine tumors at metastatic setting.Keywords
Pheochromocytomas (PCCs) and paragangliomas (PGLs) are rare neuroendocrine tumors that arise from chromaffin cells and frequently secrete one or more catecholamines. Pheochromocytomas arise from the adrenal medulla, whereas PGLs originate from extra-adrenal sympathetic or parasympathetic ganglia [1]. The diagnosis may be disregarded during life and be discovered in 0.05%–0.1% of autopsies [1]. Usually, PCCs are detected by chance (21.1%–57.6%) and constitute approximately 4%–8% of all adrenal incidentalomas [2,3]. Updating the previous epidemiological data where 10% of PCCs/PGLs were identified as malignant [4], all PCCs/PGLs are now considered potentially metastatic [5], and all patients should be advised for genetic counseling [1]. Currently, 25%–30% or more of these tumors are attributed to genetic background [6]; at least 15 PCC/PGL-related genes have been recognized, and 12 syndromes have been described [7].
The diagnosis of PCC/PGL is based on detecting urinary metanephrines [8]. Following the biochemical diagnosis, CT scanning should be performed [1]. At the same time, functional imaging should also be used in suspicion of metastatic disease, including positron emission tomography (PET)/CT with various radiotracers [9] and 123I-metaiodobenzylguanidine (123I-MIBG) scintigraphy, especially to recognize those patients that could also be treated with 131I-MIBG [10]. After a multidisciplinary team consideration, most PCCs and PGLs can be treated surgically [1]. Still, for some unfit cases, therapy with 131I-MIBG could also be a reasonable option, if 123I-MIBG scintigraphy is positive [11,12]. For metastatic PCCs/PGLs, different combinations of conventional chemotherapy, mainly the regimen cyclophosphamide, vincristine and dacarbazine, have been used for many years [13–15], but recently, novel agents, including tyrosine kinase inhibitors [16], somatostatin analogs [17], hypoxia-inducible factor (HIF) inhibitors, mTOR inhibitors, histone deacetylase inhibitors (HDACis), DNA-alkylating agents and immune checkpoint inhibitors [18] are under testing for the treatment of metastatic/unresectable setting of these rare neuroendocrine tumors.
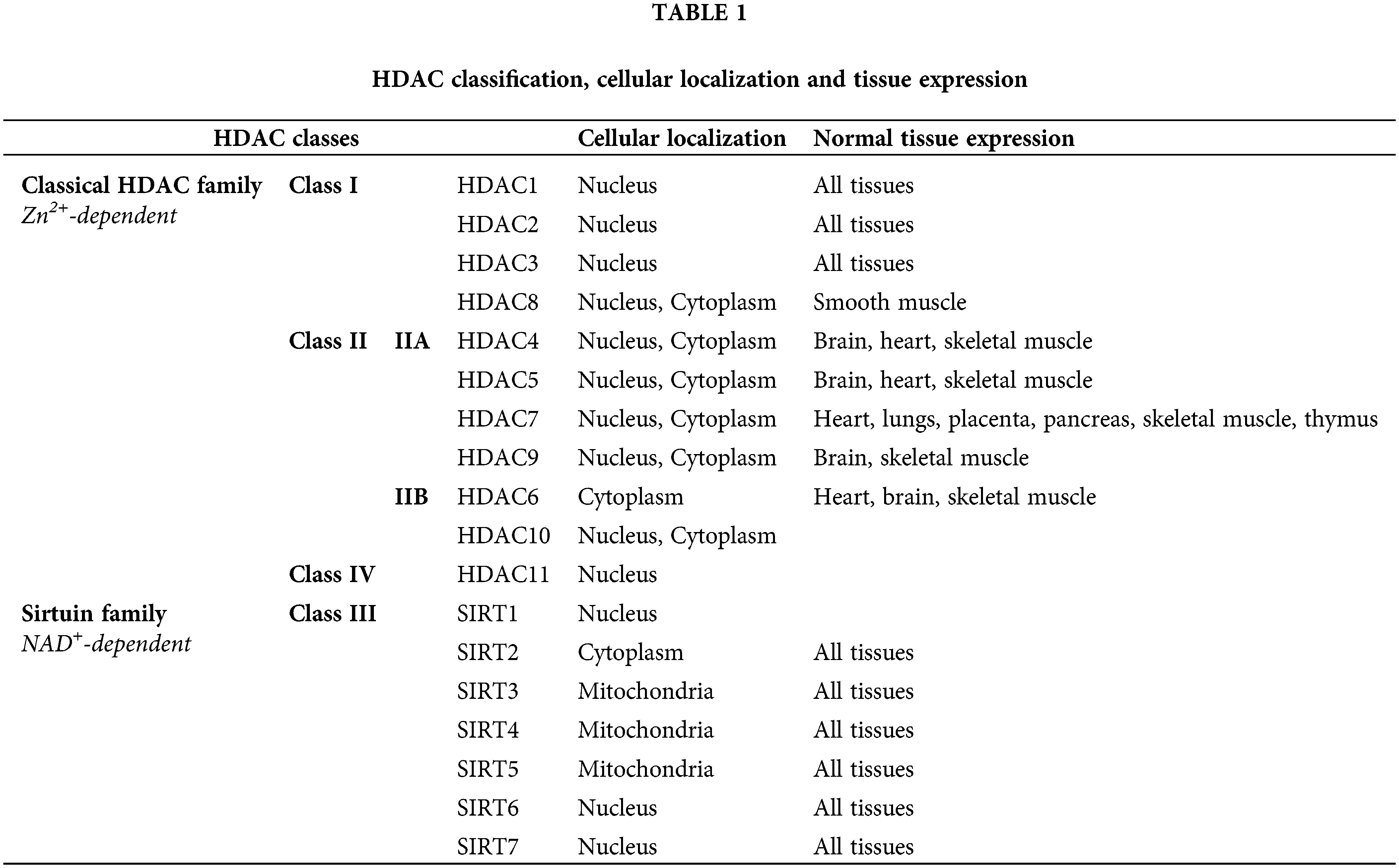
Among those approaches, the inhibition of histone deacetylation via HDACis has been entered into the focus of this study. The imbalance between histone acetylation and deacetylation can epigenetically change the expression of tumor suppressor genes and/or proto-oncogenes [19–21], that control cancer evolution and progression [22,23]. So far, 18 human HDACs have been identified into two families according to the implicated co-factor [24–26]. Different classes of HDACs are located in different cellular compartments [23,27]. An overview of the classification of human HDACs, their cellular localization and their tissue expression is presented in Table 1 [25–28].
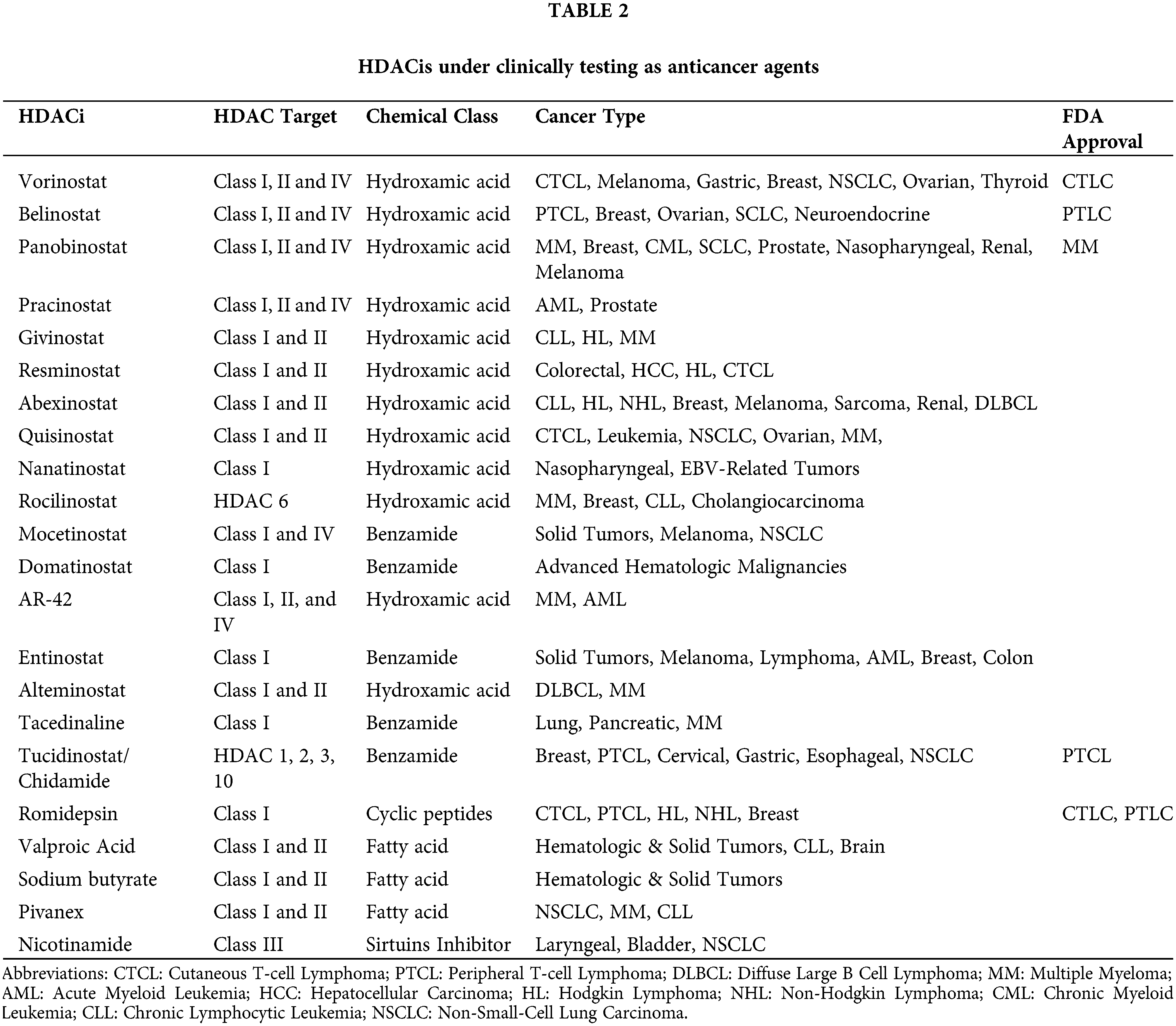
HDACs are overexpressed in hematologic and solid malignancies, and their inhibition became a promising anti-cancer theory. HDACis include both natural and synthetic compounds. Some HDACis selectively inhibit specific HDAC classes while others are pan-HDAC inhibitors [29]. HDACis increase acetylation of both histone and non-histone proteins to a significant degree, resulting in cell cycle arrest, cell differentiation, induction of cell death/apoptosis (e.g., oxidative stress generation, disruption of mitosis and mitotic cell death, autophagy, etc.), as well as blocking of angiogenesis [30]. Fig. 1 depicts the multiple mechanisms of action of HDACis [29]. The main HDACis under clinical testing, their targeted HDACs, their chemical nature and their approved indications are presented in Table 2 [23,29,31,32]. The antitumor activity of these agents depends on the specific type and stage of cancer, the characteristics of each patient and the administered dose [23,33].
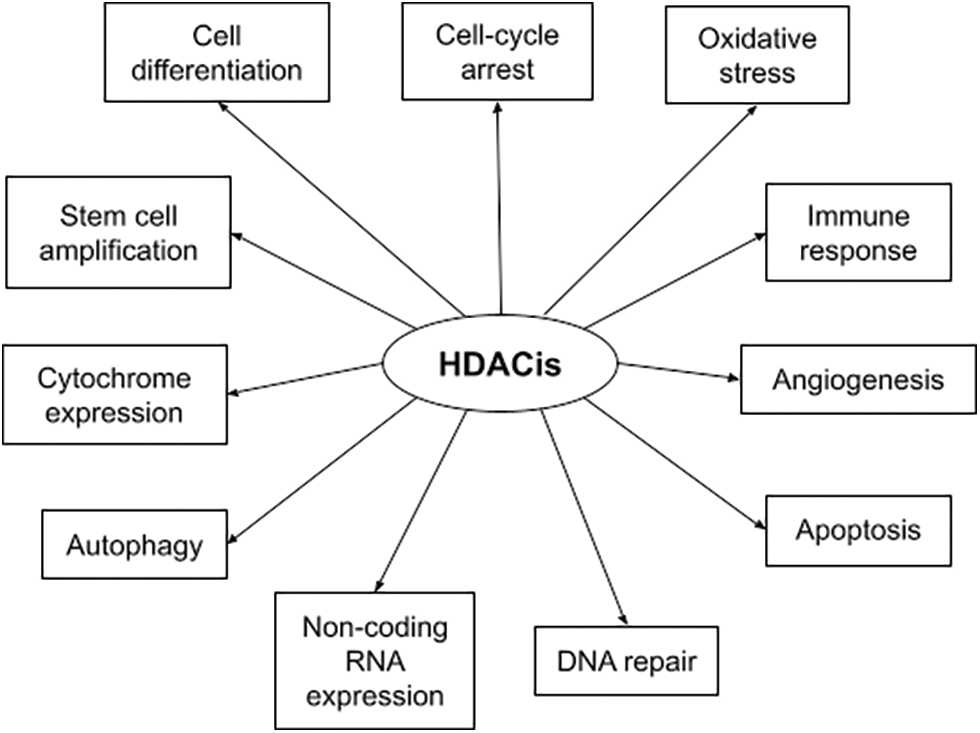
FIGURE 1: Summary of HDACis’ mechanisms of action in cancer.
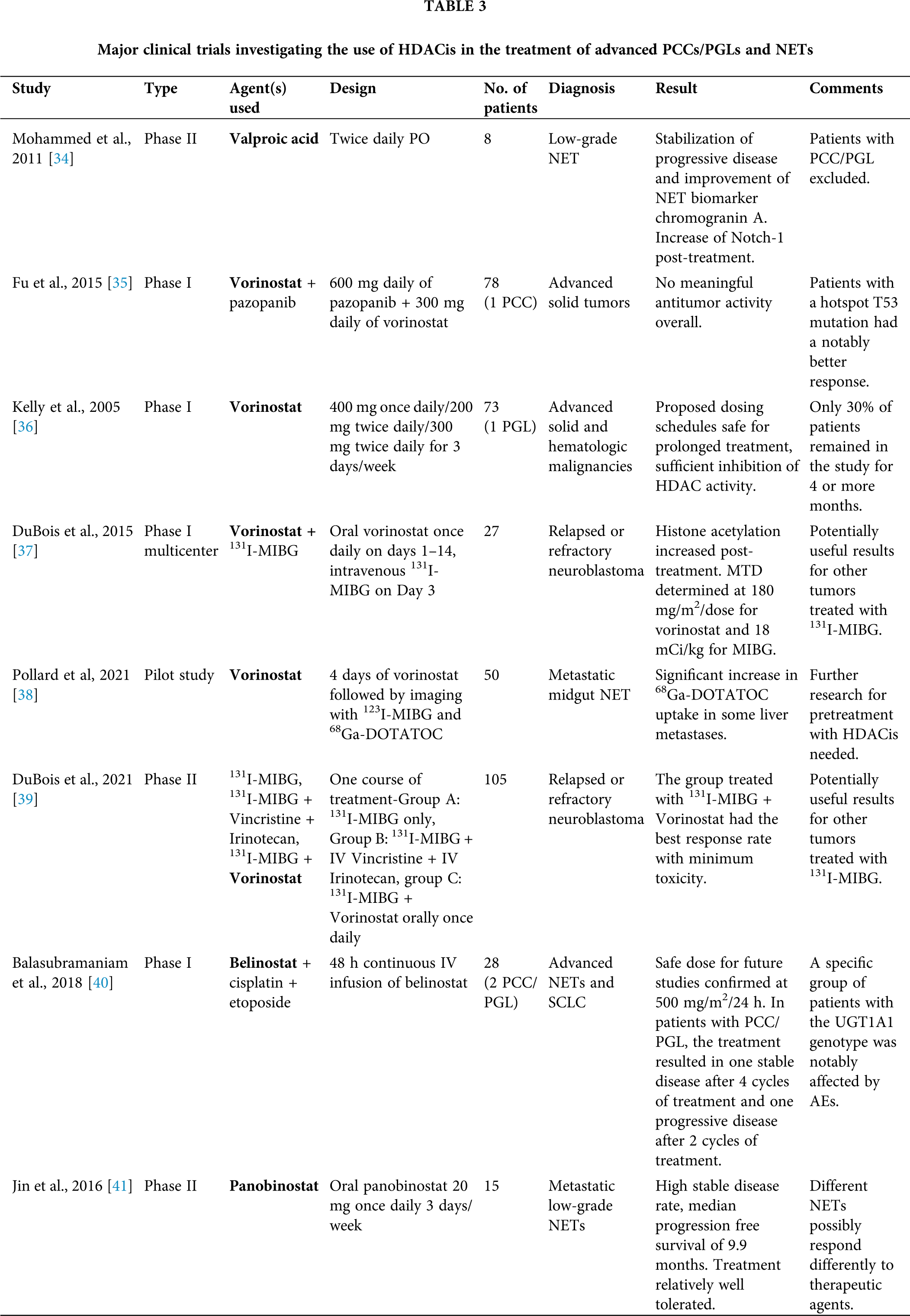
In this review, we summarize the research background and the development status of currently tested HDACis for treating metastatic/unresectable PCCs/PGLs and gather all published evidence from in vitro and in vivo studies up to clinical trials, supporting their implementation in oncological practice. The most important clinical trials investigating the use of HDACis in the treatment of PCCs/PGLs and NETs are presented in Table 3.
HDACis in Pheochromocytomas & Paragangliomas
Valproic acid is a branched short-chained fatty acid that was synthesized in 1882 and was approved for treating epilepsy in 1967 [42]. Since then, it has been used as an effective anticonvulsant medication in many neurological indications. After discovering that VPA can inhibit both HDAC classes I and IIA, many in vitro studies and clinical trials examined its use in different tumor types, including ovarian, breast, lung, pancreatic and thyroid cancer [43,44]. Adler et al. observed that rat PCC cells treated with increasing doses of VPA reduced their neuroendocrine tumor (NET)-related biomarkers, achaete-scute complex-like1 (ASCL1) and chromogranin A (CgA), and subsequently decreased their hormone secretion. The authors concluded that VPA-treated cells suppressed their growth rate by nearly 70% compared to controls in a dose-dependent manner by activating apoptotic pathways, mainly Notch-1 signaling [45]. Notch-1 is inactive in NETs, and its activation is associated with tumor growth inhibition and analogical decrease in NET-related biomarkers [46]. In another experiment in rat PCC cells, treatment with VPA for 48 h showed a similar dose-dependent effect on HDAC-mediated cancer cell growth [47]. In the clinical setting, an interesting phase II study including patients with low-grade neuroendocrine neoplasms was published in 2011 (Table 3). Although patients with PCC/PGL were excluded from this study, treatment with VPA resulted in stable disease in most of included patients, improving the levels of chromogranin A. It is worth noting that post-treatment Notch-1 was upregulated by ten times on average, in the VPA-treated population [34].
Vorinostat (Suberanilohydroxamic acid–SAHA)
Vorinostat is an antitumor agent with inhibitory effects on both classes of HDACs (I and II). At doses that have little or no toxicity on normal cells, it was found that it can cause growth arrest and cell death [48] and subsequently, its therapeutic efficacy was examined as a monotherapy or in combined regimens with promising results [48,49]. Since October 2006, vorinostat has received approval from FDA for patients with cutaneous T-cell lymphoma (CTCL) who have progressive, persistent or recurrent disease on or following two prior systemic therapies [50]. Repositioning some new therapeutic options to the metastatic PCC/PGL, Giubellino et al. suggested that the synergistic combination of the HDAC inhibitor SAHA and the topoisomerase inhibitor epirubicin could represent an example of possible successful treatment [51]. In patients with PCC/PGL, a mutation in the mitochondrial complex II subunit (succinate dehydrogenase subunit B, SDHB) is associated with more aggressive and extensive disease [52,53]. Yang et al. showed that SDHB expression was reduced in SDHB-mutated tumors, and the administration of vorinostat could prevent further degradation and restore the quantity of functional SDHB, blocking the proliferative signaling [54]. The effect of vorinostat on advanced solid and hematologic malignancies has been examined by 3 phase I and 1 phase II clinical trials as well as by a pilot study on imaging of metastatic midgut NET (Table 3). The dosing schedules were safe with sufficient inhibition of HDAC activity either as vorinostat monotherapy or in a combination with other agents, including pazopanib and [131I]-MIBG. Overall, the efficacy results from these trials were modest [35,38], and only Kelly et al. [36] achieved keeping 30% of patients with advanced solid and hematologic malignancies in the study for more than four months [47–50]. Recently, a randomized phase II clinical trial compared 131I-MIBG plus vorinostat vs. 131I-MIBG alone and vs. 131I-MIBG plus vincristine plus irinotecan in 105 patients with neuroblastoma. The combination of 131I-MIBG plus vorinostat offered the highest response rate [39].
Belinostat is a hydroxamic acid that was approved for treating peripheral T-cell lymphoma (PTCL) in 2014. It is currently under investigation for the treatment of both hematologic and solid malignancies, as monotherapy or in a combination with other anticancer agents [23,29]. It is considered a pan-HDACi, blocking Class I, II and IV HDACs [22]. Recently, a prodrug of belinostat, ZL277, was reported to be superior in bioavailability and efficacy. Because of its superior biocompatibility and intratumoral penetration, ZL277 was found to be more effective than belinostat in vivo, not only preventing tumor development but also dramatically lowering tumor sizes in an MCF-7 xenograft tumor model [55]. However, these results are preliminary enough, and more studies are needed to support its potential use in humans [52]. A phase I clinical trial tried to determine the maximum tolerated dose of the combination of belinostat (in a 48-h continuous IV infusion on days 1–2, reached 500 mg/m2/24 h) with cisplatin (a 1-h IV infusion of 60 mg/m2 on day 2), and etoposide (a 1-h IV infusion of 80 mg/m2 on days 2, 3, and 4) in 28 patients with advanced small cell lung cancer (SCLC) (Table 3). The combination was safe and active in SCLC and other neuroendocrine cancers. Objective responses were observed in 11 (39%) of 28 patients and seven (47%) of 15 patients with neuroendocrine tumors. In the 2 included patients with PCC/PGL, the combination resulted in one stable disease after four cycles, and one progressive disease after two cycles of treatment Patients carrying more than three copies of variant UGT1A1 (*28 and *60) had higher serum levels of belinostat because of slower clearance. Future phase II studies incorporating the genotyping information for UGT1A1*28 and UGT1A1*60 are needed to identify candidates for this combination [40].
Sodium Butyrate is a short-chain fatty acid and one of the oldest identified HDACis [56,57]. It is typically produced in the gastrointestinal tract through anaerobic bacterial fermentation of dietary fibers. Among the fatty acids, NaB displays the most remarkable efficacy in inhibiting HDAC activity, including most HDAC classes. Many studies have examined its use in different types of cancer. Primarily due to its production in the colon, NaB may have a protective role in the development of colon cancer [58,59]. The impact of NaB in rat PCC cells was initially described in 1987, when Byrd et al. observed that treatment with NaB ceased cell division and altered the cellular “malignant” phenotype [56]. In rat PCC cells, NaB reduced cell growth and the levels of NET biomarkers ASCL1 and CgA in a dose-dependent manner, activating the Notch-1 pathway and subsequent carcinogenesis, as mentioned above. The tumor growth was inhibited due to the concurrent arrest of the cell cycle and the induction of apoptosis [60].
Trichostatin A is a metabolic compound isolated from the strains of Streptomyces hygroscopicus in 1975 primarily used for its antifungal activity [61]. TSA displays a similar inhibitory activity in Classes I and II HDAC, keeping some slight differences among particular HDAC counterparts, but remains ineffective in Class III HDAC [62,63]. It has shown anti-proliferative properties, inducing cell cycle arrest, differentiation and apoptosis [64]. Treatment with TSA inhibited the proliferation of mouse pheochromocytoma cells (MPC) in a dose- and time-dependent manner, while increased specific [3H]-norepinephrine and 123I-MIBG uptake. In vivo experiments showed that TSA-treated tumor-bearing mice presented an increased uptake of 123I-MIBG and 18F-fluorodopamine in their metastatic liver lesions. Although TSA may enhance the response to 131I-MIBG treatment and make more effective the123I-MIBG-mediated diagnosis of metastatic disease, its poor in vivo availability will never permit its use in clinical trials in patients [65].
Romidepsin (FR901228 or FK228)
Romidepsin is a bicyclic depsipeptide that was isolated from Chromobacterium violaceum cultures, first reported in the literature in 1994 [66]. In 1998, Nakajima et al. showed that romidepsin could inhibit intracellular HDAC. Its mechanism of action is similar to TSA, despite their chemical and structural differences [67]. Romidepsin was found to inhibit the growth of tumor cell lines, but its anticancer efficacy varied against different tumor tissues [68]. Currently, romidepsin has been approved for treating patients with CTCL or PTCL who have received at least one prior line of therapy [25]. Martiniova et al. found that in vitro exposure of MPC cells to romidepsin increases the uptake of 3H-norepinephrine, 18F-fluorodopamine and 123I-MIBG and causes a dose- and time-dependent decrease of cell proliferation. Further in vivo studies revealed that mice with metastatic PCC treated with romidepsin had increased uptake of 123I-MIBG and 18F-fluorodopamine in their liver lesions. Taken together these results, we could suggest that romidepsin may work as a useful diagnostic and therapeutic tool, improving the accuracy of 123I-MIBG scintigraphy or 18F-fluorodopamine PET, and in parallel, increasing the response of 131I-MIBG treatment in patients with PCC [65].
Suberoyl bis-hydroxamic acid (SBHA)
Suberoyl bis-hydroxamic acid is a close analog of SAHA that acts as a HDACi and has been tested in treating NETs. It activates Notch-1 signaling, suppresses the secretion of NET-related biomarkers and hormones and inhibits cell proliferation by inducing cell cycle arrest or apoptosis in various cancer cell lines, including gastrointestinal and pulmonary carcinoid cells and medullary thyroid cancer cells [69–71]. Adler et al. treated rat PCC cells with gradually increasing doses of HDACis. Treatment of PCC cells with SBHA had similar results as treatment with VPA. The NET biomarkers ASCL1 and CgA were decreased. SBHA in a dose of 40μM suppressed tumor growth in more than 70% of PCC cells after six days of treatment and activated the apoptotic pathway. More specifically, the Notch-1 signaling pathway was upregulated 3-fold upon treatment with 40 μM of SBHA [45].
(−)-Epigallocatechin-3-gallate (EGCG)
(−)-Epigallocatechin-3-gallate (EGCG) is one of the most substantial polyphenolic extracts in green tea. It has been reported that EGCG inhibits DNA methyltransferase and reactivates methylation-silenced genes in various cancer cell lines (e.g., human colon cancer HT-29 cells, esophageal cancer KYSE 150 cells, and prostate cancer PC3 cells) [72]. This induced inhibition of DNA methylation by a commonly consumed dietary constituent suggested a potential use of EGCG for the reversal of related gene silencing in the prevention of carcinogenesis. EGCG acts in a concentration-dependent manner affecting class I HDACs, a especially HDAC1 [73,74]. Hu et al. studied the effect of EGCG on PCC-xenografted mice. Treatment with EGCG affected both tumor growth and apoptosis via activating the caspases 3 & 7 and decreasing amyloid precursor protein (APP) levels [75]. This specific APP protein seems to play a significant role in various diseases, including Alzheimer’s disease. It has been studied as a diagnostic tumor biomarker or as a targetable molecule in distinct cancer types, including pancreatic adenocarcinoma or colon carcinoma. Except EGCG, studies have shown that also other HDACis (such as VPA) could downregulate the levels of APP, leading to decreased tumor growth, invasion and angiogenesis [76].
Panobinostat is a pan-HDAC inhibitor with high efficacy that has shown substantial antineoplastic activity in various cancer cell lines and is currently being clinically tested against hematologic and solid malignancies [77]. In 2015, it was approved for treating multiple myeloma in patients who have received at least two previous treatments [78]. A Phase II clinical trial showed that treatment with panobinostat resulted in a high rate of stable disease and a median progression-free survival (PFS) of 9.9 months with tolerable toxicity in patients with metastatic low-grade neuroendocrine tumors. Still, no data is available regarding PCCs/PGLs (Table 3) [41].
PCCs and PGLs are rare neuroendocrine tumors with complicated genetic backgrounds and an unmet need for a more individualized approach, especially in the metastatic or unresectable setting. The benefits from conventional chemotherapy are limited, and the prognosis remains poor. Based on some promising preclinical results, HDACis grew the expectations of being a considerable alternative treatment for these tumors with the limited options in the metastatic context. The preliminary clinical studies confirmed that HDACis could inhibit tumor growth, activate specific molecular pathways and act synergistically with other already approved treatments, such as 131I-MIBG. However, more data are required to prove the benefit of their use, to determine the specific HDACis compound that should be used in the rare indication of metastatic PCCs/PGLs, and to detect the optimal dosing and the way of administration (as monotherapy or in combination with other agents), following in parallel their safety profile.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: Aspasia Manta: Conceptualization, Writing, Reviewing and Editing. Spyridon Kazanas: Data collection and curation, Writing-original draft preparation. Stefanos Karamaroudis: Data collection and curation, Writing-original draft preparation. Helen Gogas: Reviewing and Editing. Dimitrios C. Ziogas: Supervision, Conceptualization, Reviewing and Editing. All authors approved the final version of the manuscript.
Availability of Data and Materials: Data supporting this article are included within the reference list. Please contact corresponding author for any further information.
Ethics Approval: Not applicable
Conflicts of Interest: Helen Gogas has received grants and personal fees by Roche, BMS, MSD, Novartis and personal fees by Amgen and Pierre Fabre, outside the submitted work. All other authors declare no conflict of interest.
References
1. Lenders, J. W. M., Duh, Q. Y., Eisenhofer, G., Gimenez-Roqueplo, A. P., Grebe, S. K. G. et al. (2014). Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. The Journal of Clinical Endocrinology & Metabolism, 99(6), 1915–1942. DOI 10.1210/jc.2014-1498. [Google Scholar] [CrossRef]
2. Fassnacht, M., Arlt, W., Bancos, I., Dralle, H., Newell-Price, J. et al. (2016). Management of adrenal incidentalomas: European society of endocrinology clinical practice guideline in collaboration with the European network for the study of adrenal tumors. European Journal of Endocrinology, 175(2), G1–G34. DOI 10.1530/EJE-16-0467. [Google Scholar] [CrossRef]
3. Patel, D., Phay, J. E., Yen, T. W. F., Dickson, P. V, Wang, T. S. et al. (2020). Update on pheochromocytoma and paraganglioma from the SSO endocrine and head and neck disease site working group, part 2 of 2: Perioperative management and outcomes of pheochromocytoma and paraganglioma. Annals of Surgical Oncology, 27(5), 1338–1347. DOI 10.1245/s10434-020-08221-2. [Google Scholar] [CrossRef]
4. Plouin, P. F., Amar, L., Dekkers, O. M., Fassnacht, M., Gimenez-Roqueplo, A. P. et al. (2016). European society of endocrinology clinical practice guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. European Journal of Endocrinology, 174(5), G1–G10. DOI 10.1530/EJE-16-0033. [Google Scholar] [CrossRef]
5. Lam, A. K. (2017). Update on adrenal tumours in 2017 world health organization (WHO) of endocrine tumours. Endocrine Pathology, 28(3), 213–227. DOI 10.1007/s12022-017-9484-5. [Google Scholar] [CrossRef]
6. Gimenez-Roqueplo, A. P., Dahia, P., Robledo, M. (2012). An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Hormone and Metabolic Research, 44(5), 328–333. DOI 10.1055/s-0031-1301302. [Google Scholar] [CrossRef]
7. Katabathina, V. S., Rajebi, H., Chen, M., Restrepo, C. S., Salman, U. et al. (2020). Genetics and imaging of pheochromocytomas and paragangliomas: Current update. Abdominal Radiology, 45(4), 928–944. DOI 10.1007/s00261-019-02044-w. [Google Scholar] [CrossRef]
8. Manu, P., Runge, L. A. (1984). Biochemical screening for pheochromocytoma. Superiority of urinary metanephrines measurements. American Journal of Epidemiology, 120(5), 788–790. DOI 10.1093/oxfordjournals.aje.a113947. [Google Scholar] [CrossRef]
9. Timmers, H. J. L. M., Chen, C. C., Carrasquillo, J. A., Whatley, M., Ling, A. et al. (2009). Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. The Journal of Clinical Endocrinology & Metabolism, 94(12), 4757–4767. DOI 10.1210/jc.2009-1248. [Google Scholar] [CrossRef]
10. Čtvrtlík, F., Koranda, P., Schovánek, J., Škarda, J., Hartmann, I. et al. (2018). Current diagnostic imaging of pheochromocytomas and implications for therapeutic strategy (Review). Experimental and Therapeutic Medicine, 15(4), 3151–3160. DOI 10.3892/etm.2018.5871. [Google Scholar] [CrossRef]
11. van Hulsteijn, L. T., Niemeijer, N. D., Dekkers, O. M., Corssmit, E. P. M. (2014). (131)I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: Systematic review and meta-analysis. Clinical Endocrinology, 80(4), 487–501. DOI 10.1111/cen.12341. [Google Scholar] [CrossRef]
12. Pryma, D. A., Chin, B. B., Noto, R. B., Dillon, J. S., Perkins, S. et al. (2019). Efficacy and safety of high-specific-activity 131I-MIBG therapy in patients with advanced pheochromocytoma or paraganglioma. Journal of Nuclear Medicine, 60(5), 623–630. DOI 10.2967/jnumed.118.217463. [Google Scholar] [CrossRef]
13. Corssmit, E. P. M., Snel, M., Kapiteijn, E. (2020). Malignant pheochromocytoma and paraganglioma: Management options. Current Opinion in Oncology, 32(1), 20–26. DOI 10.1097/CCO.0000000000000589. [Google Scholar] [CrossRef]
14. Averbuch, S. D. (1988). Malignant pheochromocytoma: Effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Annals of Internal Medicine, 109(4), 267. DOI 10.7326/0003-4819-109-4-267. [Google Scholar] [CrossRef]
15. Tanabe, A., Naruse, M., Nomura, K., Tsuiki, M., Tsumagari, A. et al. (2013). Combination chemotherapy with cyclophosphamide, vincristine, and dacarbazine in patients with malignant pheochromocytoma and paraganglioma. Hormones and Cancer, 4(2), 103–110. DOI 10.1007/s12672-013-0133-2. [Google Scholar] [CrossRef]
16. Ayala-Ramirez, M., Chougnet, C. N., Habra, M. A., Palmer, J. L., Leboulleux, S. et al. (2012). Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. The Journal of Clinical Endocrinology & Metabolism, 97(11), 4040–4050. DOI 10.1210/jc.2012-2356. [Google Scholar] [CrossRef]
17. Nastos, K., Cheung, V. T. F., Toumpanakis, C., Navalkissoor, S., Quigley, A. M. et al. (2017). Peptide receptor radionuclide treatment and (131)I-MIBG in the management of patients with metastatic/progressive phaeochromocytomas and paragangliomas. Journal of Surgical Oncology, 115(4), 425–434. DOI 10.1002/jso.24553. [Google Scholar] [CrossRef]
18. Pang, Y., Liu, Y., Pacak, K., Yang, C. (2019). Pheochromocytomas and paragangliomas: From genetic diversity to targeted therapies. Cancers, 11(4), 436. DOI 10.3390/cancers11040436. [Google Scholar] [CrossRef]
19. Verdone, L. (2006). Histone acetylation in gene regulation. Briefings in Functional Genomics and Proteomics, 5(3), 209–221. DOI 10.1093/bfgp/ell028. [Google Scholar] [CrossRef]
20. Bottomley, M. J. (2004). Structures of protein domains that create or recognize histone modifications. EMBO Reports, 5(5), 464–469. DOI 10.1038/sj.embor.7400146. [Google Scholar] [CrossRef]
21. Haberland, M., Montgomery, R. L., Olson, E. N. (2009). The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nature Reviews Genetics, 10(1), 32–42. DOI 10.1038/nrg2485. [Google Scholar] [CrossRef]
22. Shah, R. R. (2019). Safety and tolerability of histone deacetylase (HDAC) inhibitors in oncology. Drug Safety, 42(2), 235–245. DOI 10.1007/s40264-018-0773-9. [Google Scholar] [CrossRef]
23. Singh, A., Bishayee, A., Pandey, A. (2018). Targeting histone deacetylases with natural and synthetic agents: An emerging anticancer strategy. Nutrients, 10(6), 731. DOI 10.3390/nu10060731. [Google Scholar] [CrossRef]
24. Kim, H. J., Bae, S. C. (2011). Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. American Journal of Translational Research, 3(2), 166–179. [Google Scholar]
25. Bondarev, A. D., Attwood, M. M., Jonsson, J., Chubarev, V. N., Tarasov, V. V. et al. (2021). Recent developments of HDAC inhibitors: Emerging indications and novel molecules. British Journal of Clinical Pharmacology, 87(12), 4577–4597. DOI 10.1111/bcp.14889. [Google Scholar] [CrossRef]
26. de Ruijter, A. J. M., van Gennip, A. H., Caron, H. N., Kemp, S., van Kuilenburg, A. B. P. (2003). Histone deacetylases (HDACsCharacterization of the classical HDAC family. Biochemical Journal, 370(3), 737–749. DOI 10.1042/bj20021321. [Google Scholar] [CrossRef]
27. Bassett, S., Barnett, M. (2014). The role of dietary histone deacetylases (HDACs) inhibitors in health and disease. Nutrients, 6(10), 4273–4301. DOI 10.3390/nu6104273. [Google Scholar] [CrossRef]
28. Poniewierska-Baran, A., Warias, P., Zgutka, K. (2022). Sirtuins (SIRTs) as a novel target in gastric cancer. International Journal of Molecular Sciences, 23(23), 15119. DOI 10.3390/ijms232315119. [Google Scholar] [CrossRef]
29. Eckschlager, T., Plch, J., Stiborova, M., Hrabeta, J. (2017). Histone deacetylase inhibitors as anticancer drugs. International Journal of Molecular Sciences, 18(7), 1414. DOI 10.3390/ijms18071414. [Google Scholar] [CrossRef]
30. Bao, L., Diao, H., Dong, N., Su, X., Wang, B. et al. (2016). Histone deacetylase inhibitor induces cell apoptosis and cycle arrest in lung cancer cells via mitochondrial injury and p53 up-acetylation. Cell Biology and Toxicology, 32(6), 469–482. DOI 10.1007/s10565-016-9347-8. [Google Scholar] [CrossRef]
31. ClinicalTrials.gov. NIH U.S. National Library of Medicine. [Google Scholar]
32. Ceccacci, E., Minucci, S. (2016). Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. British Journal of Cancer, 114(6), 605–611. DOI 10.1038/bjc.2016.36. [Google Scholar] [CrossRef]
33. Kretsovali, A., Hadjimichael, C., Charmpilas, N. (2012). Histone deacetylase inhibitors in cell pluripotency, differentiation, and reprogramming. Stem Cells International, 2012, 184154. DOI 10.1155/2012/184154. [Google Scholar] [CrossRef]
34. Mohammed, T. A., Holen, K. D., Jaskula-Sztul, R., Mulkerin, D., Lubner, S. J. et al. (2011). A pilot phase II study of valproic acid for treatment of low-grade neuroendocrine carcinoma. The Oncologist, 16(6), 835–843. DOI 10.1634/theoncologist.2011-0031. [Google Scholar] [CrossRef]
35. Fu, S., Hou, M. M., Naing, A., Janku, F., Hess, K. et al. (2015). Phase I study of pazopanib and vorinostat: a therapeutic approach for inhibiting mutant p53-mediated angiogenesis and facilitating mutant p53 degradation. Annals of Oncology, 26(5), 1012–1018. DOI 10.1093/ANNONC/MDV066. [Google Scholar] [CrossRef]
36. Kelly, W. K., O’Connor, O. A., Krug, L. M., Chiao, J. H., Heaney, M. et al. (2005). Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. Journal of Clinical Oncology, 23(17), 3923–3931. DOI 10.1200/JCO.2005.14.167. [Google Scholar] [CrossRef]
37. DuBois, S. G., Groshen, S., Park, J. R., Haas-Kogan, D. A., Yang, X. et al. (2015). Phase I study of vorinostat as a radiation sensitizer with 131I-metaiodobenzylguanidine (131I-MIBG) for patients with relapsed or refractory neuroblastoma. Clinical Cancer Research, 21(12), 2715–2721. DOI 10.1158/1078-0432.CCR-14-3240. [Google Scholar] [CrossRef]
38. Pollard, J. H., Menda, Y., Zamba, K. D., Madsen, M., O’Dorisio, M. S. et al. (2021). Potential for increasing uptake of radiolabeled 68Ga-DOTATOC and 123I-MIBG in patients with midgut neuroendocrine tumors using a histone deacetylase inhibitor vorinostat. Cancer Biotherapy & Radiopharmaceuticals, 36(8), 632–641. DOI 10.1089/CBR.2020.4633. [Google Scholar] [CrossRef]
39. DuBois, S. G., Meaghan Granger, M., Groshen, S., Tsao-Wei, D., Ji, L. et al. (2021). Randomized phase II trial of MIBG versus MIBG, vincristine, and irinotecan versus MIBG and vorinostat for patients with relapsed or refractory neuroblastoma: A report from NANT consortium. Journal of Clinical Oncology, 39(31), 3506–3514. DOI 10.1200/JCO.21.00703. [Google Scholar] [CrossRef]
40. Balasubramaniam, S., Redon, C. E., Peer, C. J., Bryla, C., Lee, M. J. et al. (2018). Phase I trial of belinostat with cisplatin and etoposide in advanced solid tumors, with a focus on neuroendocrine and small cell cancers of the lung. Anti-Cancer Drugs, 29(5), 457–465. DOI 10.1097/CAD.0000000000000596. [Google Scholar] [CrossRef]
41. Jin, N., Lubner, S. J., Mulkerin, D. L., Rajguru, S., Carmichael, L. (2016). A phase II trial of a histone deacetylase inhibitor panobinostat in patients with low-grade neuroendocrine tumors. The Oncologist, 21(7), 785–786. DOI 10.1634/theoncologist.2016-0060. [Google Scholar] [CrossRef]
42. Perucca, E. (2002). Pharmacological and therapeutic properties of valproate: A summary after 35 years of clinical experience. CNS Drugs, 16(10), 695–714. DOI 10.2165/00023210-200216100-00004. [Google Scholar] [CrossRef]
43. Lipska, K., Gumieniczek, A., Filip, A. A. (2020). Anticonvulsant valproic acid and other short-chain fatty acids as novel anticancer therapeutics: Possibilities and challenges. Acta Pharmaceutica, 70(3), 291–301. DOI 10.2478/acph-2020-0021. [Google Scholar] [CrossRef]
44. Duenas-Gonzalez, A., Candelaria, M., Perez-Plascencia, C., Perez-Cardenas, E., de la Cruz-Hernandez, E. et al. (2008). Valproic acid as epigenetic cancer drug: Preclinical, clinical and transcriptional effects on solid tumors. Cancer Treatment Reviews, 34(3), 206–222. DOI 10.1016/j.ctrv.2007.11.003. [Google Scholar] [CrossRef]
45. Adler, J. T., Hottinger, D. G., Kunnimalaiyaan, M., Chen, H. (2008). Histone deacetylase inhibitors upregulate Notch-1 and inhibit growth in pheochromocytoma cells. Surgery, 144(6), 956–961. DOI 10.1016/j.surg.2008.08.027 discussion 961-2. [Google Scholar] [CrossRef]
46. Egloff, A. M., Grandis, J. R. (2012). Molecular pathways: Context-dependent approaches to Notch targeting as cancer therapy. Clinical Cancer Research, 18(19), 5188–5195. DOI 10.1158/1078-0432.CCR-11-2258. [Google Scholar] [CrossRef]
47. Gotfryd, K., Skladchikova, G., Lepekhin, E. A., Berezin, V., Bock, E. et al. (2010). Cell type-specific anti-cancer properties of valproic acid: independent effects on HDAC activity and Erk1/2 phosphorylation. BMC Cancer, 10, 383. DOI 10.1186/1471-2407-10-383. [Google Scholar] [CrossRef]
48. Marks, P. A., Breslow, R. (2007). Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nature Biotechnology, 25(1), 84–90. DOI 10.1038/NBT1272. [Google Scholar] [CrossRef]
49. Richon, V. M. (2006). Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acida novel histone deacetylase inhibitor. British Journal of Cancer, 95, S2–S6. DOI 10.1038/sj.bjc.6603463. [Google Scholar] [CrossRef]
50. Mann, B. S., Johnson, J. R., Cohen, M. H., Justice, R., Pazdur, R. (2007). FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. The Oncologist, 12(10), 1247–1252. DOI 10.1634/theoncologist.12-10-1247. [Google Scholar] [CrossRef]
51. Giubellino, A., Shankavaram, U., Bullova, P., Schovanek, J., Zhang, Y. et al. (2014). High-throughput screening for the identification of new therapeutic options for metastatic pheochromocytoma and paraganglioma. PLoS One, 9(4), e90458. DOI 10.1371/JOURNAL.PONE.0090458. [Google Scholar] [CrossRef]
52. Neumann, H. P. H., Bausch, B., McWhinney, S. R., Bender, B. U., Gimm, O. et al. (2002). Germ-line mutations in nonsyndromic pheochromocytoma. The New England Journal of Medicine, 346(19), 1459–1466. DOI 10.1056/NEJMOA020152. [Google Scholar] [CrossRef]
53. Amar, L., Bertherat, J., Baudin, E., Ajzenberg, C., Bressac-De Paillerets, B. et al. (2005). Genetic testing in pheochromocytoma or functional paraganglioma. Journal of Clinical Oncology,23(34), 8812–8818. DOI 10.1200/JCO.2005.03.1484. [Google Scholar] [CrossRef]
54. Yang, C., Matro, J. C., Huntoon, K. M., Ye, D. Y., Huynh, T. T. et al. (2012). Missense mutations in the human SDHB gene increase protein degradation without altering intrinsic enzymatic function. FASEB Journal, 26(11), 4506–4516. DOI 10.1096/FJ.12-210146. [Google Scholar] [CrossRef]
55. Zhang, C., Guo, S., Zhong, Q., Zhang, Q., Hossain, A. et al. (2019). Metabolism and pharmacokinetic study of the boron-containing prodrug of belinostat (ZL277a pan HDAC inhibitor with enhanced bioavailability. Pharmaceuticals, 12(4), 180. DOI 10.3390/PH12040180. [Google Scholar] [CrossRef]
56. Byrd, J. C., Alho, H. (1987). Differentiation of PC12 pheochromocytoma cells by sodium butyrate. Brain Research, 428(1), 151–155. DOI 10.1016/0165-3806(87)90096-4. [Google Scholar] [CrossRef]
57. Sealy, L., Chalkley, R. (1978). The effect of sodium butyrate on histone modification. Cell, 14(1), 115–121. DOI 10.1016/0092-8674(78)90306-9. [Google Scholar] [CrossRef]
58. Jiang, W., Guo, Q., Wu, J., Guo, B., Wang, Y. et al. (2012). Dual effects of sodium butyrate on hepatocellular carcinoma cells. Molecular Biology Reports, 39(5), 6235–6242. DOI 10.1007/S11033-011-1443-5. [Google Scholar] [CrossRef]
59. Davie, J. R. (2003). Inhibition of histone deacetylase activity by butyrate. The Journal of Nutrition, 133(7 Suppl), 2485S–2493S. DOI 10.1093/JN/133.7.2485S. [Google Scholar] [CrossRef]
60. Cayo, M. A., Cayo, A. K., Jarjour, S. M., Chen, H. (2009). Sodium butyrate activates Notch1 signaling, reduces tumor markers, and induces cell cycle arrest and apoptosis in pheochromocytoma. American Journal of Translational Research, 1(2), 178–183. [Google Scholar]
61. Tsuji, N., Kobayashi, M., Nagashima, K., Wakisaka, Y., Koizumi, K. (1976). A new antifungal antibiotic, trichostatin. The Journal of Antibiotics, 29(1), 1–6. DOI 10.7164/ANTIBIOTICS.29.1. [Google Scholar] [CrossRef]
62. Furumai, R., Komatsu, Y., Nishino, N., Khochbin, S., Yoshida, M. et al. (2001). Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proceedings of the National Academy of Sciences of the United States of America, 98(1), 87–92. DOI 10.1073/pnas.98.1.87. [Google Scholar] [CrossRef]
63. Yoshida, M., Kijima, M., Akita, M., Beppu, T. (1990). Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. The Journal of Biological Chemistry, 265(28), 17174–17179. [Google Scholar]
64. Vanhaecke, T., Papeleu, P., Elaut, G., Rogiers, V. (2004). Trichostatin A-like hydroxamate histone deacetylase inhibitors as therapeutic agents: Toxicological point of view. Current Medicinal Chemistry, 11(12), 1629–1643. DOI 10.2174/0929867043365099. [Google Scholar] [CrossRef]
65. Martiniova, L., Perera, S. M., Brouwers, F. M., Alesci, S., Abu-Asab, M. et al. (2011). Increased uptake of [123I]meta-iodobenzylguanidine, [18F]fluorodopamine, and [3H]norepinephrine in mouse pheochromocytoma cells and tumors after treatment with the histone deacetylase inhibitors. Endocrine-Related Cancer, 18(1), 143–157. DOI 10.1677/ERC-10-0090. [Google Scholar] [CrossRef]
66. Ueda, H., Nakajima, H., Hori, Y., Fujita, T., Nishimura, M. et al. (1994). FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. The Journal of Antibiotics, 47(3), 301–310. DOI 10.7164/ANTIBIOTICS.47.301. [Google Scholar] [CrossRef]
67. Nakajima, H., Kim, Y. B., Terano, H., Yoshida, M., Horinouchi, S. (1998). FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Experimental Cell Research, 241(1), 126–133. DOI 10.1006/EXCR.1998.4027. [Google Scholar] [CrossRef]
68. Sasakawa, Y., Naoe, Y., Inoue, T., Sasakawa, T., Matsuo, M. et al. (2003). Effects of FK228, a novel histone deacetylase inhibitor, on tumor growth and expression of p21 and c-myc genes in vivo. Cancer Letters, 195(2), 161–168. DOI 10.1016/S0304-3835(03)00184-8. [Google Scholar] [CrossRef]
69. Greenblatt, D. Y., Cayo, M., Ning, L., Jaskula-Sztul, R., Haymart, M. et al. (2007). Suberoyl bishydroxamic acid inhibits cellular proliferation by inducing cell cycle arrest in carcinoid cancer cells. Journal of Gastrointestinal Surgery, 11(11), 1515–1520. DOI 10.1007/S11605-007-0249-1. [Google Scholar] [CrossRef]
70. Ning, L., Greenblatt, D. Y., Kunnimalaiyaan, M., Chen, H. (2008). Suberoyl bis-hydroxamic acid activates Notch-1 signaling and induces apoptosis in medullary thyroid carcinoma cells. The Oncologist, 13(2), 98–104. DOI 10.1634/THEONCOLOGIST.2007-0190. [Google Scholar] [CrossRef]
71. Adler, J. T., Hottinger, D. G., Kunnimalaiyaan, M., Chen, H. (2009). Combination therapy with histone deacetylase inhibitors and lithium chloride: A novel treatment for carcinoid tumors. Annals of Surgical Oncology, 16(2), 481–486. DOI 10.1245/S10434-008-0194-6. [Google Scholar] [CrossRef]
72. Fang, M. Z., Wang, Y., Ai, N., Hou, Z., Sun, Y. et al. (2003). Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Research, 63(22), 7563–7570. [Google Scholar]
73. Groh, I. A. M., Chen, C., Lüske, C., Cartus, A. T., Esselen, M. (2013). Plant polyphenols and oxidative metabolites of the herbal alkenylbenzene methyleugenol suppress histone deacetylase activity in human colon carcinoma cells. Journal of Nutrition and Metabolism, 2013, 821082. DOI 10.1155/2013/821082. [Google Scholar] [CrossRef]
74. Thakur, V. S., Gupta, K., Gupta, S. (2012). Green tea polyphenols increase p53 transcriptional activity and acetylation by suppressing class I histone deacetylases. International Journal of Oncology, 41(1), 353–361. DOI 10.3892/IJO.2012.1449. [Google Scholar] [CrossRef]
75. Hu, Q., Chang, X., Yan, R., Rong, C., Yang, C. et al. (2015). (-)-Epigallocatechin-3-gallate induces cancer cell apoptosis via acetylation of amyloid precursor protein. Medical Oncology, 32(1), 390. DOI 10.1007/s12032-014-0390-0. [Google Scholar] [CrossRef]
76. Venkataramani, V., Rossner, C., Iffland, L., Schweyer, S., Tamboli, I. Y. et al. (2010). Histone deacetylase inhibitor valproic acid inhibits cancer cell proliferation via down-regulation of the alzheimer amyloid precursor protein. The Journal of Biological Chemistry, 285(14), 10678–10689. DOI 10.1074/JBC.M109.057836. [Google Scholar] [CrossRef]
77. Atadja, P. (2009). Development of the pan-DAC inhibitor panobinostat (LBH589Successes and challenges. Cancer Letters, 280(2), 233–241. DOI 10.1016/j.canlet.2009.02.019. [Google Scholar] [CrossRef]
78. Eleutherakis-Papaiakovou, E., Kanellias, N., Kastritis, E., Gavriatopoulou, M., Terpos, E. et al. (2020). Efficacy of panobinostat for the treatment of multiple myeloma. Journal of Oncology, 2020, 1–11. DOI 10.1155/2020/7131802. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2022 The Author(s). Published by Tech Science Press.
Copyright © 2022 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools