
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

MCU-i4, a mitochondrial Ca2+ uniporter modulator, induces breast cancer BT474 cell death by enhancing glycolysis, ATP production and reactive oxygen species (ROS) burst
1 Department of Anesthesia, An Nan Hospital, China Medical University, Tainan, 709, Taiwan
2 Graduate Institute of Medical Sciences, Chang Jung Christian University, Tainan, 711301, Taiwan
3 UNIMED Medical Institute, Hong Kong SAR, China
4 Department of Emergency Medicine, China Medical University Hospital, Taichung, 404327, Taiwan
5 Department of Cosmetic Science, Providence University, Taichung, 43301, Taiwan
6 School of Pharmacy, China Medical University, Taichung, 404328, Taiwan
7 Department of Anatomy, China Medical University, Taichung, 404328, Taiwan
8 Department of Physiology, China Medical University, Taichung, 404328, Taiwan
9 Department of Medicinal Botanicals and Healthcare, Dayeh University, Changhua, 51591, Taiwan
10 Department of Anesthesiology, Kuang Tien General Hospital, Shalu, Taichung, 433, Taiwan
* Corresponding Authors: YUK-MAN LEUNG. Email: ; YI-PING HUANG. Email:
# Contributed equally as first authors
(This article belongs to the Special Issue: Recent Advances in Cancer Pharmacology)
Oncology Research 2025, 33(2), 397-406. https://doi.org/10.32604/or.2024.052743
Received 13 April 2024; Accepted 29 July 2024; Issue published 16 January 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Objectives: Mitochondrial Ca2+ uniporter (MCU) provides a Ca2+ influx pathway from the cytosol into the mitochondrial matrix and a moderate mitochondrial Ca2+ rise stimulates ATP production and cell growth. MCU is highly expressed in various cancer cells including breast cancer cells, thereby increasing the capacity of mitochondrial Ca2+ uptake, ATP production, and cancer cell proliferation. The objective of this study was to examine MCU inhibition as an anti-cancer mechanism. Methods: The effects of MCU-i4, a newly developed MCU inhibitor, on cell viability, apoptosis, cytosolic Ca2+, mitochondrial Ca2+ and potential, glycolytic rate, generation of ATP, and reactive oxygen species, were examined in breast cancer BT474 cells. Results: MCU-i4 caused apoptotic cell death, and it decreased and increased, respectively, mitochondrial and cytosolic Ca2+ concentration. Inhibition of MCU by MCU-i4 revealed that cytosolic Ca2+ elevation resulted from endoplasmic reticulum (ER) Ca2+ release via inositol 1,4,5-trisphosphate receptors (IP3R) and ryanodine receptors (RYR). Unexpectedly, MCU-i4 enhanced glycolysis and ATP production; it also triggered a large production of reactive oxygen species (ROS) and mitochondrial membrane potential collapse. Conclusion: Cytotoxic mechanisms of MCU-i4 in cancer cells involved enhanced glycolysis and heightened formation of ATP and ROS. It is conventionally believed that cancer cell death could be caused by inhibition of glycolysis. Our observations suggest cancer cell death could also be induced by increased glycolytic metabolism.Keywords
Abbreviations
| MCU | Mitochondrial Ca2+ uniporter |
| ER | Endoplasmic reticulum |
| IP3R | Inositol 1,4,5-trisphosphate receptors |
| RYR | Ryanodine receptors |
| ROS | Reactive oxygen species |
Ca2+ ions are important for living cells, and their trafficking and signaling are under control through different kinds of cation channels, uniporters, and receptors [1]. Several studies have shown that regulation of Ca2+ signaling by modulating ion channel gating could induce cell death in cancer cells with different pathways [2–4]. A tight control of mitochondria matrix Ca2+ level is necessary, as a small rise could stimulate tricarboxylic acid (TCA) cycle enzymes, resulting in enhanced ATP production; on the contrary, excessive mitochondrial matrix Ca2+, as a result of cytosolic Ca2+ overload caused by deleterious agents, would result in cell death [5,6]. It has been demonstrated that constitutive endoplasmic reticulum (ER)-mitochondria Ca2+ transfer is necessary for normal mitochondrial functioning. For instance, Ca2+ released via inositol 1,4,5-trisphosphate receptors (IP3R) of ER flows into mitochondria to stimulate oxidative phosphorylation [7–9].
The molecular machinery responsible for the mitochondria to uptake Ca2+ from the cytosol is the mitochondrial calcium uniporter (MCU), a molecular complex residing in the inner mitochondrial membrane [10,11]. This complex comprises the MCU channel itself and accessory regulatory proteins, namely, MCU regulatory subunit β (MCUβ), essential MCU regulator (EMRE), mitochondrial Ca2+ uptake proteins (MICU1, 2, and 3), and mitochondrial Ca2+ uniporter regulator 1 (MCUR1) [12,13]. MCU is positively and negatively regulated by MCUR1 and MICU1, respectively [14].
Remarkably, MCU is highly expressed in various cancer cells including breast cancer cells, thereby increasing the capacity of mitochondrial Ca2+ uptake, ATP production, and cell proliferation [15]. MCU-mediated Ca2+ entry into the mitochondrial matrix stimulates colorectal cancer growth [16]. MCU promotes pancreatic ductal adenocarcinoma metastasis and metabolic stress resistance [17]. There is a high correlation between MCU expression and tumor growth and metastasis of triple-negative breast cancer; down-regulation of MCU attenuates tumor growth and invasiveness [18]. MCU inhibitors are therefore potential anti-cancer drugs [5]. Classical MCU inhibitors such as ruthenium red and ruthenium 360 are direct pore blockers of MCU [19,20]. A new category of MCU-inhibiting drugs was introduced recently: MCU-i4 binds to and stimulates MICU-1, and since the latter negatively regulates MCU, MCU-i4 serves as a negative modulator of MCU and inhibits Ca2+ uptake into the mitochondrial matrix [21]. MCU-i4 fails to inhibit mitochondrial Ca2+ uptake in MICU1-silenced cells or cells expressing a MICU1 mutant lacking an MCU-i4-binding site [21].
In our study, we aim to investigate the cytotoxic actions of MCU-i4 in breast cancer BT474 cells. In particular, we aim to examine how MCU-i4 modulated cytosolic Ca2+ homeostasis, mitochondrial functions and metabolism to cause such cytotoxicity.
Fetal calf serum, Dulbecco’s modified Eagle’s medium (DMEM), and tissue culture reagents were purchased from Invitrogen Corporation (Carlsbad, CA, USA). Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), 2-aminoethoxydiphenyl borate (2-APB) and cyclosporin A were from Sigma-Aldrich chemical Co. (St. Louis, MO, USA). JTV-519 and MCU-i4 were from Tocris BioScience (Bristol, UK). All other chemicals were of reagent grades and were from Sigma-Aldrich. BT474 cells were purchased from American Type Culture Collection (Manassas, VA, USA), and were cultured in RPMI-1640 medium supplemented with L-glutamine (2 mM), 10% fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37°C (98.6°F) with 5% humidified CO2.
Assay of cell viability and apoptosis
BT474 cells were cultured in 96-well plates at a density of 1.5 × 104 and were treated with different agents for 48 h. DMSO was added to the medium as solvent control (final concentration = 0.1%). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenltetrazolium bromide (MTT; final concentration of 0.5 mg/mL) was added to each well and further incubated for 4 h. Culture medium was discarded and DMSO (100 μL) was added to each well for another 15 min with mild shaking to dissolve precipitates. Absorbance at 595 nm was measured by an ELISA reader; absorbance was used to indicate cell viability or metabolic activities. A reduction in MTT absorbance indicates cell death, reduced cell proliferation or metabolic activities. Number of viable cells was quantified by the trypan blue exclusion method: viable cells were unstained by trypan blue and were counted with a hemocytometer. An FITC annexin V apoptosis detection kit (BioLegend, San Diego, CA, USA) and flow cytometer (BD Biosciences, San Jose, CA, USA) were used to quantify apoptosis. Caspase-9 level was measured using an ELISA kit (cat. # E-EL-H0663; Elabscience, Houston, TX, USA) following the instructions in the manufacturer’s manual.
Microfluorimetric measurement of cytosolic Ca2+
Cytosol Ca2+ concentration was measured using fura-2 as a fluorescent probe [22]. Cells were incubated with 5 μM fura-2 AM (Invitrogen) at 37°C (98.6°F) for 1 h; cells were then washed in bath solution (mM): 140 NaCl, 2 CaCl2, 1 MgCl2, 4 KCl, 10 HEPES (pH was adjusted to 7.4 by NaOH). Intracellular Ca2+ release was examined in Ca2+-free bath solution; the latter being the same as the bath solution, except that Ca2+ was removed and EGTA (100 μM) was added. Cells were excited by 340 and 380 nm alternately (switching frequency = 1 Hz) by an optical filter changer (Lambda 10-2, Sutter Instruments, Novato, CA, USA). Emission was collected at 500 nm. and data were captured by a CCD camera (CoolSnap HQ2, Photometrics, Tucson, AZ, USA), which was connected to a Nikon (Tokyo, Japan) TE2000-U microscope. Data were analyzed with an MAG Biosystems Software (Sante Fe, MN, USA). Experiments were conducted at 25°C (77°F). Changes in 340/380 ratio were analyzed at a region of interest of single cells; the same experimental procedures were repeated multiple times to obtain the mean.
Measurement of mitochondrial Ca2+ concentration
Microfluorimetric quantification of Ca2+ concentration within the mitochondrial matrix was performed using a Ca2+-sensitive dye Rhod-2 AM [23]. The cells were incubated with 5 μM Rhod-2 AM (Invitrogen, Carlsbad, CA, USA) at 37°C (98.6°F) for 1 h. Cells were then permeabilized and washed with a digitonin (30 μM)-containing intracellular solution, which contained (mM): 140 KCl, 8 NaCl, 1 CaCl2, 1 MgCl2, 1.85 EGTA, 10 HEPES, and 8 MgATP (KOH was used to adjust pH to 7.25). Free [Ca2+] in this intracellular solution was calculated to be 114 nM. Excitation wavelength was 540 nm and emission wavelength was 605 nm. Images were captured by a CCD camera (CoolSnap HQ2, Photometrics, Tucson, AZ, USA) connected to an inverted microscope (Nikon TE 2000-U). An MAG Biosystems Software (Sante Fe, MN) was used for analysis. All experiments were performed at room temperature (25°C) (77°F).
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential was measured by a mitochondrial membrane potential assay kit (#12664; Cell Signaling, Danvers, MA, USA) as described in a previous report [24]. The cells were plated at a density of 5 × 104 cells per well to settle overnight. The cells were treated with DMSO or other agents for 24 h. JC-1 (2 μM) was added to each well for 30 min. Fluorescent emission was measured by a Varioskan LUX multimode microplate reader (Thermo Fisher Scientific, Waltham, MA, USA). Excitation wavelength was at 485 nm and dual emission wavelengths were at 520 and 590 nm. Mitochondrial membrane potential was calculated as the ratio RFU of red emission (590 nm)/RFU of green emission (520 nm).
Reactive oxygen species (ROS) assay
ROS was measured as described in a previous report [25]. The cells were treated with various agents for 4 h, and were then incubated in serum-free DMEM supplemented with 2,7-dichlorodihydrofluorescein diacetate (DCFH2-DA, 20 μM, Sigma, St. Louis, MO, USA) at 37°C (98.6°F) for 30 min in darkness. Cells were then washed, trypsinized at 37°C for 3 min, and washed again three more times in phosphate-buffered saline using centrifugation. The cells were then dispersed in phosphate-buffered saline, put in polystyrene tubes for FACS (fluorescence-activated cell sorting). The samples (1 × 105 cells/sample) were analyzed by an FACS Canto flow cytometer system (BD Biosciences, San Jose, CA, USA). Data analysis was performed with the aid of a BD FACSDIVA™ software (BD Biosciences).
Cellular ATP content was quantified as described in a previous report [26]. The cells were seeded in 10-cm dishes at a density of 6 × 106 cells per dish and treated with DMSO or 30 μM MCU-i4 for 24 h. The cells were subsequently trypsinized and cell viability was quantified by the trypan blue exclusion method. The cells were sonicated (33 Hz, 90 s) in ice bath, centrifuged (10,000 g, 10 min) and 30 μL supernatant was taken for ATP quantification using an ATP assay kit (catalogue # E-BC-K57-M; Elabscience, Houston, TX, USA). Samples were analyzed by a BioTek Epoch 2 microplate spectrophotometer (Winooski, VT, USA).
Quantification of lactate was used as an indicator of glycolysis [27]. The cells were seeded in 96-well plates at a density of 1 × 104 cells/per well overnight. The cells were then treated with DMSO or 30 μM MCU-i4 for 3 h, and 5 μL supernatant was taken for lactate quantification using a glycolysis assay kit (catalogue # ECGL-100; BioAssay Systems, Hayward, CA, USA), and samples were analyzed by a BioTek Epoch 2 microplate spectrophotometer (Winooski, VT, USA).
Statistical analysis and graphing were performed using Origin8.5 (OriginLab, MA, USA). Results are presented as means ± standard error of mean (S.E.M.). Paired or unpaired Student t-test was employed where appropriate to compare two groups. When multiple groups were analyzed, ANOVA and the Tukey’s HSD post-hoc test were used. Statistical significance was considered to be reached if the p-value is less than 0.05.
MCU-i4 induced apoptotic cell death
Treatment of BT474 cells with MCU-i4 (3–30 μM) for 2 days resulted in a concentration-dependent decrease in cell viability (Fig. 1A). Since the MTT assay shown in Fig. 1A could not discriminate between cell death and reduced cell proliferation, we used the trypan blue exclusion method to quantitate the number of viable cells (Fig. 1B). Cell proliferation was suppressed by 3 μM MCU-i4, while higher concentrations (10–30 μM) concentration-dependently caused cell death. We investigated whether cell death was necrotic or apoptotic. There was a 10-fold increase in annexin-positive/propidium iodide-negative cells (Q4), suggesting early apoptosis had taken place (Fig. 1C). Caspase-9 has been known as a marker of apoptosis [28]. Consistently, MCU-i4 treatment also resulted in a moderate increase in the level of caspase-9 (Fig. 1D).
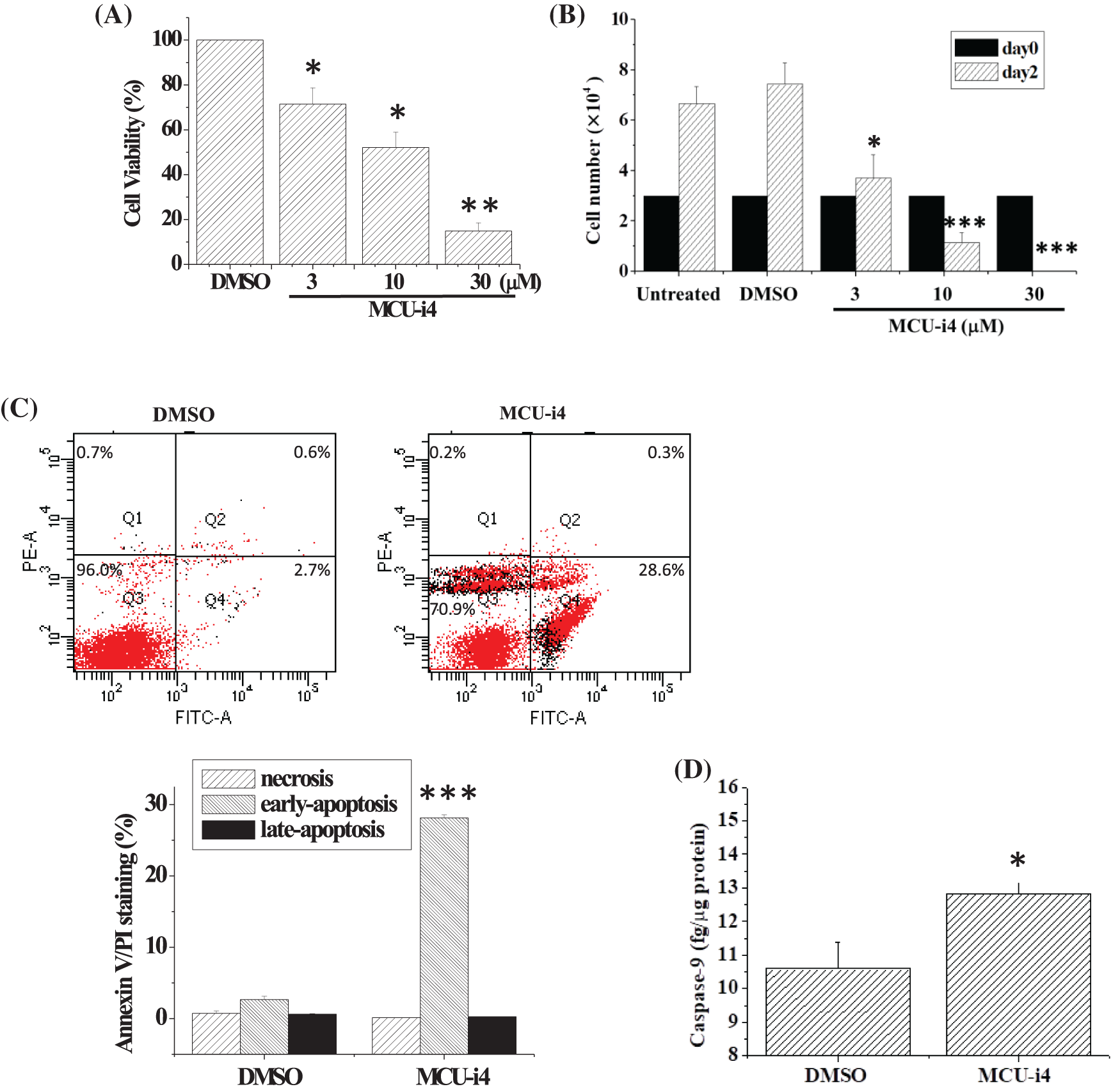
Figure 1: MCU-4 decreased cell viability and caused apoptosis. (A) Cells were treated with different concentrations of MCU-i4 for 2 days before the MTT assay was used to determine cell viability. (B) Cells, initially seeded on 24-well plates at a density of 3 × 104/well, received no vehicle or drug (untreated) or were treated with DMSO (vehicle) or different concentrations of MCU-i4 for 2 days, and viable cells were counted by the trypan blue exclusion method. (C) Cells were treated with DMSO or 30 μM MCU-i4 for 1 day and examined for apoptosis. (D) Cells were cultured on 6-cm wells at a density of 5 × 105/well (in order to have sufficient protein harvest for caspase-9 determination), treated with DMSO or 30 μM MCU-i4 for 2 days, and examined for the level of caspase-9. Results are mean ± SEM from 3–4 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 different from the DMSO control.
Effects of MCU-i4 on cytosolic Ca2+ fluxes
We investigated whether MCU-i4 affected cytosolic Ca2+ levels. MCU-i4 did not cause an immediate elevation in cytosolic Ca2+ concentration [Ca2+]i (Fig. 2A). We then investigated whether a prolonged MCU-i4 pre-treatment (25 min) would affect the Ca2+ level. The cells in Ca2+-containing bath solution were treated with DMSO or MCU-i4 for 25 min (kept in the dark to avoid photobleaching) prior to microfluorimetric measurement. An elevated Ca2+ baseline was observed in the MCU-i4-treated cells (Fig. 2B). Treatment with MCU-i4 for 24 h (followed by fura 2 loading and microfluorimetric measurements) also resulted in an elevated Ca2+ baseline, suggesting prolonged MCU-i4 treatment raised Ca2+ concentration in the cytosol (Fig. 2C). We repeated the above protocol (25 min) in Ca2+-free bath solution (Fig. 2D). An elevated Ca2+ baseline was again observed in the MCU-i4-treated cells, suggesting the raised Ca2+ concentration in the cytosol was in part due to Ca2+ release. Inhibition of inositol 1,4,5-trisphosphate receptors (IP3R) by 2-APB strongly suppressed the elevation of Ca2+ baseline, while inhibition of ryanodine receptors (RYR) by JTV-519 only mildly alleviated it, suggesting the Ca2+ leak was mainly via IP3R and in part via RYR.
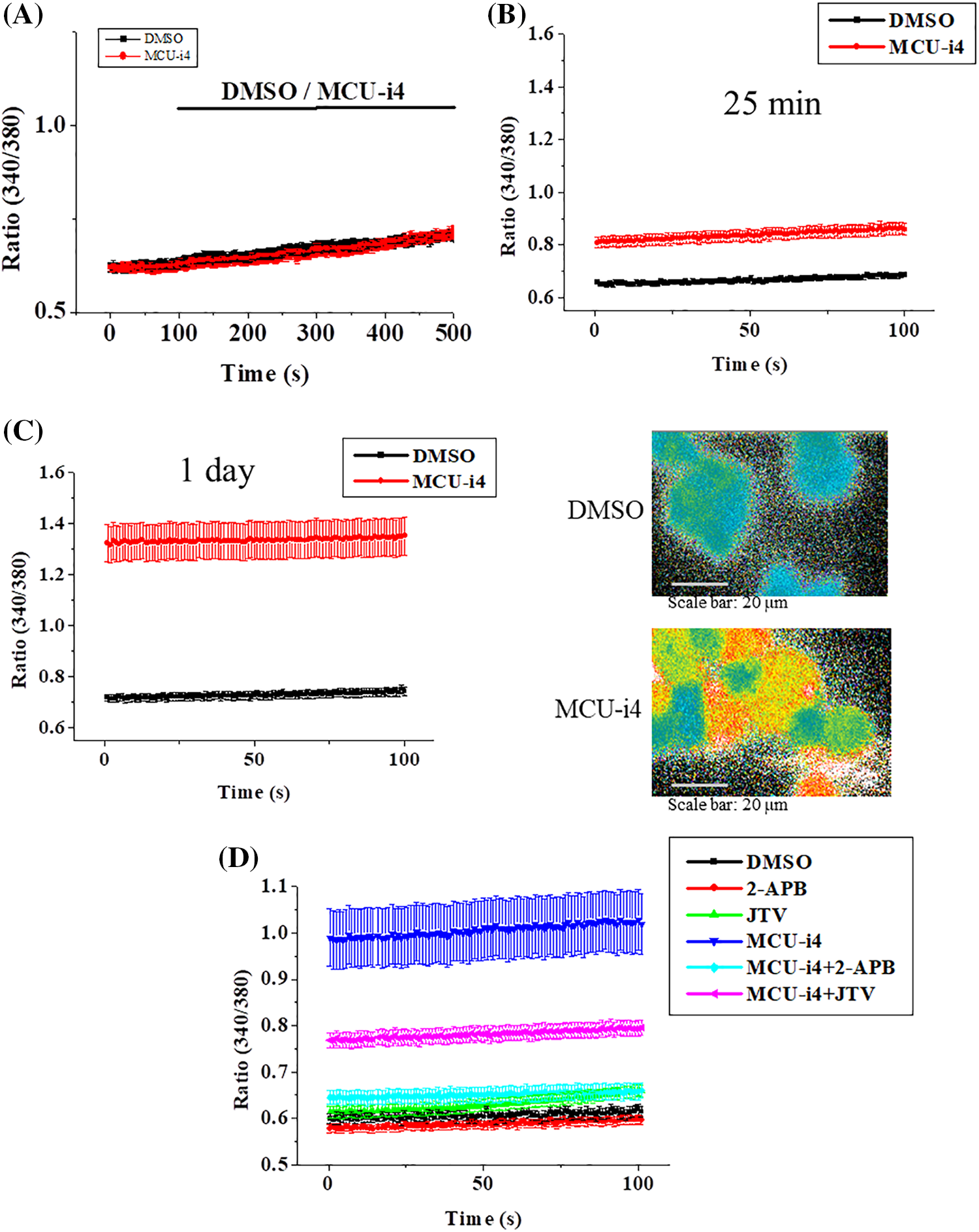
Figure 2: Effects of MCU-i4 (30 μM) on Ca2+ fluxes. (A) Cells in Ca2+-containing solution were exposed to DMSO (vehicle control) or MCU-i4. (B) Cells were pretreated with DMSO or MCU-i4 for 25 min in a Ca2+-containing solution and then subject to microfluorimetric measurement. (C) Cells were cultured in the presence of DMSO or MCU-i4 for 1 day, loaded with fura-2, bathed in Ca2+-containing solution, and then subject to microfluorimetric measurement. The right panels show pseudocolor images (low ratio being blue to high ratio being red) of fluorescent ratio analysis of DMSO-and MCU-i4-treated cells. (D) Cells were pretreated with DMSO or MCU-i4 (in the absence or presence of 30 μM JTV-159 or 30 μM 2-APB) for 25 min in Ca2+-free solution and then subject to microfluorimetric measurement. For (B)–(D), there are significant (p < 0.001) differences between the DMSO and MCU-i4 groups at all time points. For (D), there are significant (p < 0.001) differences between the MCU-i4 group and MCU-i4 plus JTV-159 group or MCU-i4 plus 2-APB group at all time points. Results are mean ± S.E.M.; each group had 14–57 cells from 3 independent experiments.
MCU-i4 reduced mitochondrial matrix Ca2+ level
Ca2+ release from intracellular Ca2+ store caused elevation in cytosolic Ca2+ level under inhibition of mitochondrial Ca2+ uptake by MCU-i4 (Fig. 2). To show that inhibition of mitochondrial Ca2+ uptake by MCU-i4 led to a decrease in mitochondrial Ca2+ concentration, we used Rhod-2, a fluorescent probe for Ca2+ concentration in the mitochondrial matrix (Fig. 3). In the control where DMSO was added, there was a slow decrease in fluorescence which was due to inevitable photobleaching of the fluorescent dye; addition of MCU-i4 caused an immediate and persistent decrease in fluorescence when compared to the control, indicating a decrease in mitochondrial matrix Ca2+ concentration. This result, together with the data in Fig. 2, suggest that upon MCU-i4 inhibition of mitochondrial Ca2+ uptake, Ca2+ released from Ca2+ store failed to enter mitochondria and thus “spilled over” in the cytosol.
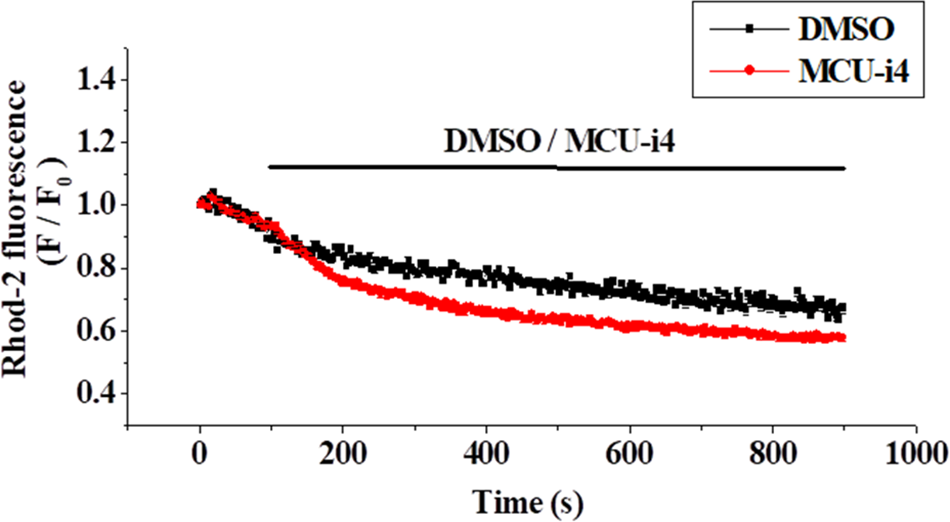
Figure 3: MCU-i4 (30 μM) caused a decrease in mitochondrial Ca2+ level. Cells were loaded with Rhod-2 and permeabilized as described in the methods section. Cells were then treated with DMSO or MCU-i4. Changes in mitochondrial Ca2+ level are quantified as fluorescence/fluorescence at time zero (F/F0). There are significant differences between the DMSO control and the MCU-i4 group after 171 s (p < 0.05). Results are mean ± SEM; each group had 26–31 cells from 3 independent experiments.
MCU-i4 enhanced glycolysis and production of ATP and ROS
We next examined whether MCU-i4-induced lowered mitochondrial matrix Ca2+ level would affect ATP production. MCU-i4 treatment for 24 h resulted in a 51.9 ± 5.8% decrease in viable cell count (trypan blue exclusion test) but only moderately reduced ATP production by 23.9 ± 9.8%. When ATP production was normalized by the number of viable cells, MCU-i4 treatment significantly enhanced ATP production (Fig. 4A). Since lowered mitochondrial matrix Ca2+ level was not compatible with increased mitochondrial ATP production, we examined whether increased ATP production resulted from increased glycolytic activities. As shown in Fig. 4B, MCU-i4 caused a 1.6-fold elevation in secreted lactate concentration, which indicated an increase in glycolysis. Whether MCU-i4 elicited ROS formation was next examined. As shown in Fig. 5, treatment of cells with MCU-i4 for 4 h resulted in large production of ROS.
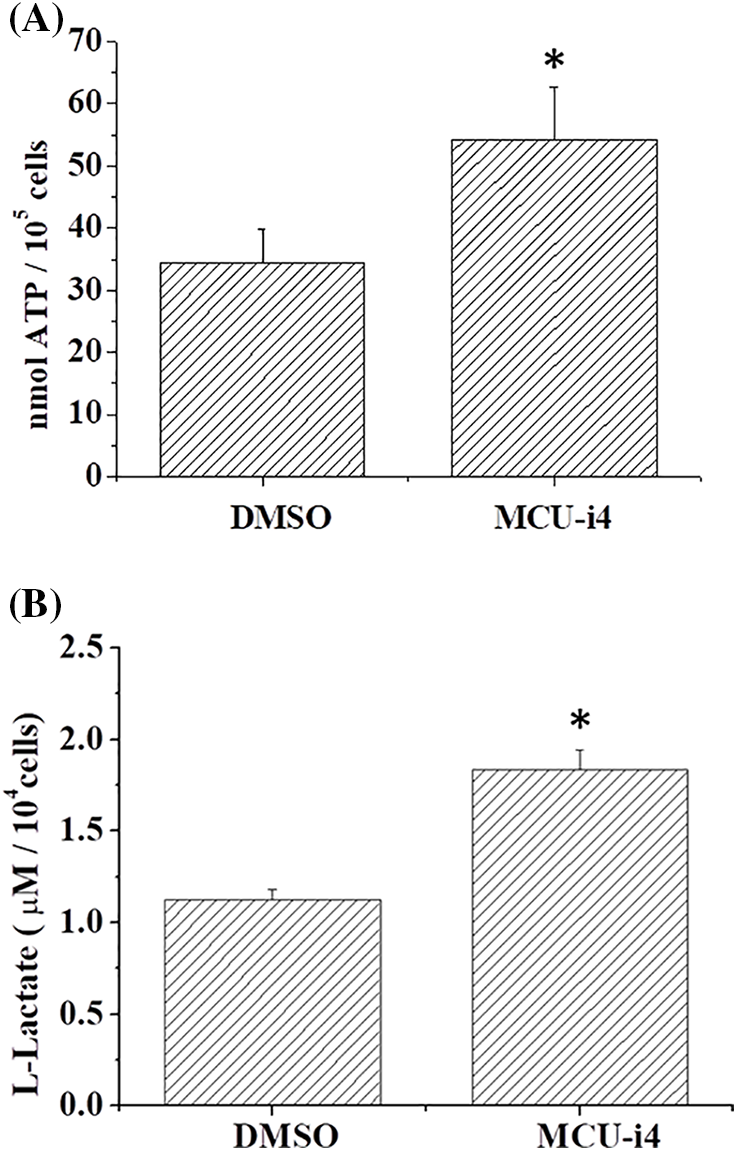
Figure 4: MCU-i4 treatment increased ATP and lactate production. (A) Cells were treated with DMSO or 30 μM MCU-i4 for one day and then subject to ATP quantification. (B) Cells were treated with DMSO or 30 μM MCU-i4 for 3 h and then subject to lactate quantification. Results are mean ± SEM from 4 independent experiments. Significantly different from the DMSO control *p < 0.05.
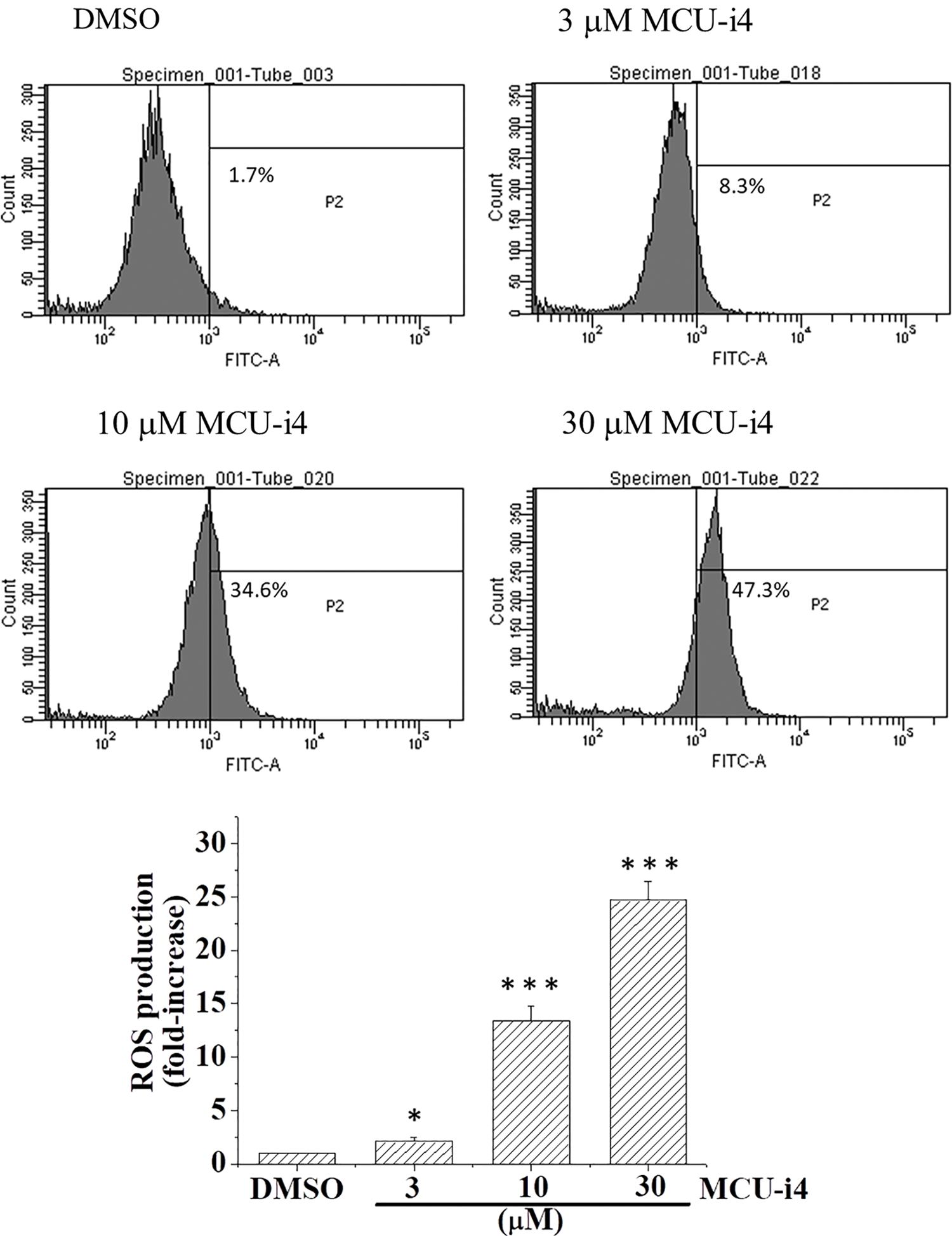
Figure 5: MCU-i4 triggered ROS formation. Cells were treated with DMSO or different concentrations of MCU-i4 for 4 h and then subject to ROS measurement using flow cytometry. Results represent mean ± SEM from 6 independent experiments. *p < 0.05, ***p < 0.001 significantly different from the DMSO control.
Effects of MCU-i4 on mitochondrial membrane potential
Using JC-1 as a fluorescent probe of mitochondrial membrane potential, we examined if MCU-i4 would depolarize mitochondrial potential. MCU-i4 at 10 μM caused marked depolarization whilst at 30 μM it caused collapse of mitochondrial membrane potential to an extent comparable to that caused by FCCP (Fig. 6A). Since mitochondrial membrane potential collapse could be a result of mitochondria permeability transition pore (MPTP) opening and that cyclophilin D is an integral part of the MPTP, we examined if cyclosporin A (an MPTP inhibitor by interacting with cyclophilin D) could reduce MCU-i4 cytotoxicity. However, MCU-i4-inflicted cell death was not prevented by cyclosporin A (Fig. 6B).
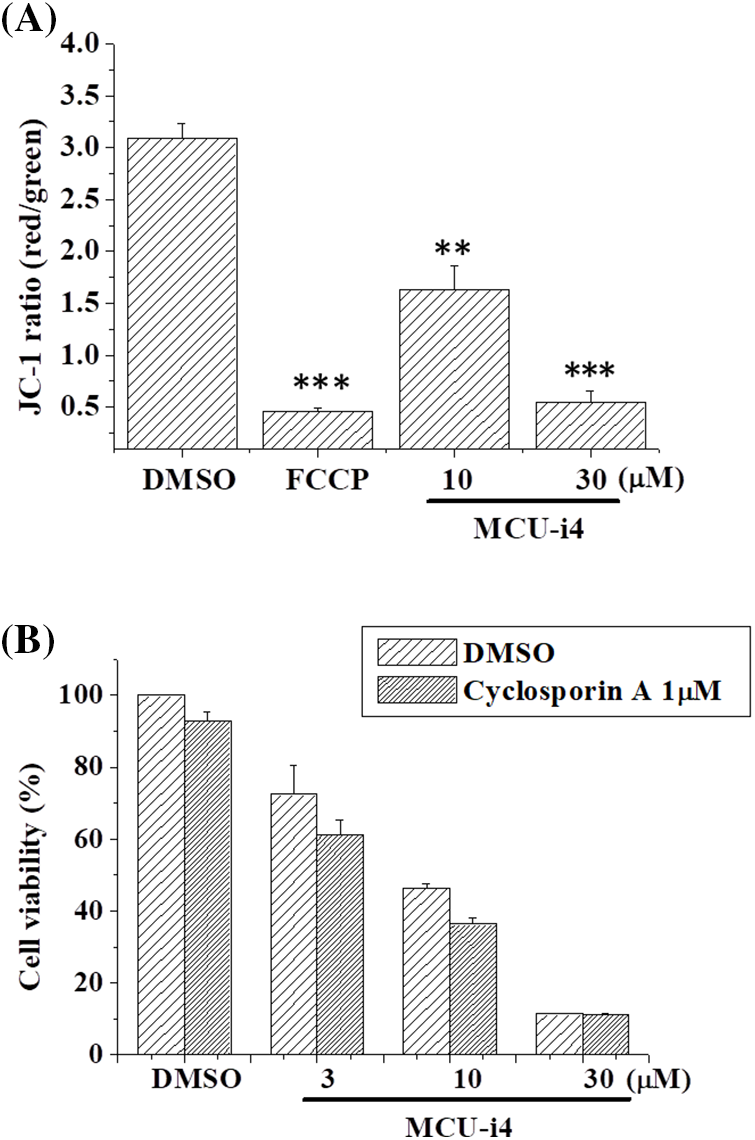
Figure 6: MCU-i4 caused mitochondrial membrane depolarization but decreased viability was not rescued by cyclosporin A. (A) Cells were treated with 10–30 μM MCU-i4 for one day and then subject to mitochondrial membrane potential measurement. FCCP (4-h treatment) was used as a positive control for membrane potential collapse. (B) Cells were treated with different concentrations of MCU-i4 in the absence or presence of 1 μM cyclosporin A for 2 days before MTT assay was employed to quantify cell viability. Results are mean ± SEM from 3 independent experiments. **p < 0.01, ***p < 0.001 significantly different from the DMSO control.
Classical MCU inhibitors (direct pore blockers) such as ruthenium complexes (ruthenium red and Ru360) have been shown to induce apoptosis in kidney tubular cyst cells and colon carcinoma HCT-116 cells [29,30]. By contrast, they inhibit apoptosis in colonocytes, mammary gland adenocarcinoma cells, neuroblastoma cells, and podocytes, by preventing mitochondrial Ca2+ overload [31–34]. The factors governing whether these ruthenium-related compounds prevent or induce apoptosis are unclear. The antibiotics minocycline and doxycycline also cause MCU inhibition, which may lead to their anti-cancer activities [5,35–37]. MCU-i4 differs from the classical MCU pore blockers: it binds to MICU1 and is therefore a negative modulator of MCU [21]. We here provided data to show how MCU-i4 caused apoptotic death in cancer cells.
In our report, we showed MCU-i4 decreased mitochondrial matrix Ca2+ level. This, however, did not lead to the expected reduction of ATP production but instead caused a moderate increase in ATP production. One explanation is that the reduction in mitochondrial matrix Ca2+ level was too mild to cause a significant drop in ATP production, whilst persistent cytosolic Ca2+ rise (Fig. 2) enhanced glycolytic production of ATP. This is supported by our findings that MCU-i4 caused increases in glycolytic activity and ATP production (Fig. 4). Our data are therefore in concordance with previous observations showing that Ca2+ stimulates glycolysis [38,39]. Our study showing that MCU-i4 caused increased ATP production and apoptotic (instead of necrotic) cell death (Fig. 1) is reminiscent of previous reports demonstrating that cells undergoing apoptosis had raised levels of ATP. For instance, cerebellar granule cells undergoing apoptosis had enhanced ATP production derived from both oxidative phosphorylation and glycolysis [40]. HeLa, PC12 and U937 cells had increased cytosolic ATP levels when they underwent apoptosis; inhibition of glycolysis abolished apoptosis [41]. In isolated hypoxic rat cardiac myocytes, cell death shifted from necrosis to apoptosis when cellular ATP level was raised by increasing glucose concentration in the medium [42]. Therefore, lack and abundance of ATP favor, respectively, necrosis and apoptosis [43].
In heart failure, disturbed Ca2+ handling reduces mitochondrial Ca2+ uptake and results in oxidative stress in cardiomyocytes [44]. How ROS formation was raised in MCU-i4-treated BT474 cells was uncertain. It might be partly due to heightened metabolism: increased cytosolic Ca2+ level enhanced glycolysis (see above; Figs. 2 and 4), with pyruvate increasingly fueling the Kreb’s cycle and oxidative phosphorylation. ROS may activate hypoxia-inducible factor 1-α, which further promotes glycolysis [45]. ROS can elicit further ROS formation from proximal mitochondria, a term coined ROS-induced ROS release [46,47]. This may account for the substantial amount of ROS formation in MCU-i4-treated BT474 cells. Thus, ROS formation, together with persistent cytosolic Ca2+ overload (see below), may eventually lead to apoptotic cell death (Fig. 1C). The latter was also evidenced by the increase in caspase-9 level, an indicator of mitochondria-dependent apoptosis (Fig. 1D). Reduced mitochondrial Ca2+ level may sensitize cytotoxicity by apoptotic signals. Remarkably, a reduction in mitochondrial Ca2+ level by chelation or Ru360 potentiates cytotoxicity induced by tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) in apoptosis-resistant tumor cells [48].
One of the lethal causes of MCU-i4 is likely cytosolic Ca2+ overload, as excessive and persistent Ca2+ elevation activates multiple phospholipases, proteases, and caspases [49]. As constitutive ER-mitochondria Ca2+ transfer is necessary for normal mitochondrial functioning such as oxidative phosphorylation [7–9], blockade of Ca2+ influx into the mitochondrial matrix by MCU-i4 resulted in sustained cytosolic Ca2+ elevation. Our data suggest that Ca2+ released from ER was mainly via IP3R and in part RYR. Although MICU1 is the known target of MCU-i4, the possibility that MCU-i4 targets on other protein molecules involved in cell metabolism, and hence inflicts cell death, could not be ruled out. Future work to identify possible off-targets is warranted.
Raised mitochondrial Ca2+ levels and ROS are strongly implicated in mitochondrial permeability transition pore (MPTP) opening and consequent mitochondrial membrane potential collapse [50]. Here we show that although MCU-i4 caused a mild reduction in mitochondrial Ca2+, the drastic ROS production it elicited might have overridden and sufficed to cause mitochondrial membrane potential collapse (which was to the same extent as elicited by FCCP). Ruthenium-related compounds reportedly do not alter mitochondrial membrane potential and thus serve as more selective probes for the MCU [51]. The ability of MCU-i4 to decrease mitochondrial membrane potential limits its selectivity but endows it with anti-cancer activities. Given that cyclophilin D is an integral part of the MPTP, the observation that MCU-i4 caused mitochondrial membrane potential collapse but its cytotoxicity was not alleviated by cyclosporin A (which binds to cyclophilin D and thus inhibits MPTP), appears intriguing. A possible explanation for this is that the MPTP opening caused by MCU-i4 was partly independent of cyclophilin D. To support this notion, evidence comes from observations that cardiomyocytes and embryonic fibroblasts from cyclophilin D knock-out mice still exhibited MPTP opening, albeit to a lesser extent than that of the wild-type counterparts [52,53].
We have presented evidence that MCU-i4 enhanced glycolysis and ATP production (Fig. 4). MCU-i4 also caused mitochondrial dysfunction (Fig. 6A), which might lead to decreased oxygen consumption. Therefore, it is likely that MCU-i4-treated BT474 cells consumed less oxygen when compared to untreated cells. A limitation of our study was the lack of data on oxygen consumption rate, which would warrant further investigation.
MCU-i4 inhibited mitochondrial Ca2+ uptake and thus caused cytosolic Ca2+ overload due to continuous ER Ca2+ release. Increased glycolysis, ATP production, and ROS burst were followed by mitochondrial membrane potential collapse and eventually apoptotic death of BT474 cells. It is conventionally believed that cancer cell death could be caused by inhibition of glycolysis [54]. Our observations suggest cancer cell death could also be induced by increased glycolytic metabolism. The cytotoxic mechanisms of MCU-i4 may shed light on future investigations into the use of anti-cancer drugs that target the MCU.
Acknowledgement: None.
Funding Statement: Yuk-Man Leung, Cheng-Hsun Wu and Chin-Min Chuang would like to thank China Medical University and China Medical University Hospital, Taiwan for providing fundings (CMU111-S-20; CMU112-S-59; DMR-112-067). ECS thanks An Nan Hospital for support (ANHRF112-04).
Author Contributions: Lian-Ru Shiao, Ting-Tsz Ou and Cing Yu Chen did the experimental work and data analysis. Edmund Cheung So, Louis W. C. Chow, Chin-Min Chuang, Cheng-Hsun Wu and Kar-Lok Wong designed research protocol, did data analysis and drafted the manuscript. Yuk-Man Leung and Yi-Ping Huang did the conceptualization of the entire framework, wrote and did the finalization of the paper. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Data that support findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Giorgi, C., Romagnoli, A. C., Pinton, P., Rizzuto, R. (2008). Ca2+ signaling, mitochondria and cell death. Current Molecular Medicine, 8(2), 119–130. https://doi.org/10.2174/156652408783769571. [Google Scholar] [PubMed] [CrossRef]
2. Singh, J., Meena, A., Luqman, S. (2023). New frontiers in the design and discovery of therapeutics that target calcium ion signaling: A novel approach in the fight against cancer. Expert Opinion on Drug Discovery, 18(12), 1379–1392. https://doi.org/10.1080/17460441.2023.2251887. [Google Scholar] [PubMed] [CrossRef]
3. Fernandez Garcia, E., Paudel, U., Noji, MC., Bowman, C. E., Rustgi, A. K. et al. (2023). The mitochondrial Ca2+ channel MCU is critical for tumor growth by supporting cell cycle progression and proliferation. Frontiers in Cell and Developmental Biology, 11, 1082213. https://doi.org/10.3389/fcell.2023.1082213. [Google Scholar] [PubMed] [CrossRef]
4. Zhang, Q., Wang, C., He, L. (2024). ORAI Ca2+ channels in cancers and therapeutic interventions. Biomolecules, 14(4), 417. https://doi.org/10.3390/biom14040417. [Google Scholar] [PubMed] [CrossRef]
5. Cui, C., Yang, J., Fu, L., Wang, M., Wang, X. (2019). Progress in understanding mitochondrial calcium uniporter complex- mediated calcium signalling: A potential target for cancer treatment. British Journal of Pharmacology, 176(9), 1190–1205. https://doi.org/10.1111/bph.v176.9 [Google Scholar] [CrossRef]
6. Dejos, C., Gkika, D., Cantelmo, A. R. (2020). The two-way relationship between calcium and metabolism in cancer. Frontiers in Cell and Developmental Biology, 8, 573747. https://doi.org/10.3389/fcell.2020.573747. [Google Scholar] [PubMed] [CrossRef]
7. CÆrdenas, C., Miller, R. A., Smith, I., Bui, T., Molgó, J. et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell, 142(2), 270–283. https://doi.org/10.1016/j.cell.2010.06.007. [Google Scholar] [PubMed] [CrossRef]
8. Bustos, G., Ahumada-Castro, U., Silva-Pavez, E., Puebla, A., Lovy, A. et al. (2021). The ER-mitochondria Ca2+ signaling in cancer progression: Fueling the monster. International Review of Cell and Molecular Biology, 363, 49–121. https://doi.org/10.1016/bs.ircmb.2021.03.006. [Google Scholar] [PubMed] [CrossRef]
9. Katona, M., Bartók, Á., Nichtova, Z., CsordÆs, G., Berezhnaya, E. et al. (2022). Capture at the ER-mitochondrial contacts licenses IP3 receptors to stimulate local Ca2+ transfer and oxidative metabolism. Nature Communications, 13(1), 6779. https://doi.org/10.1038/s41467-022-34365-8. [Google Scholar] [PubMed] [CrossRef]
10. Murphy, E., Steenbergen, C. (2021). Regulation of mitochondrial Ca2+ uptake. American Physiological Society, 83, 107–126. [Google Scholar]
11. Yoo, J. (2022). Structural basis of Ca2+ uptake by mitochondrial calcium uniporter in mitochondria: A brief review. BMB Reports, 55(11), 528–534. https://doi.org/10.5483/BMBRep.2022.55.11.134. [Google Scholar] [PubMed] [CrossRef]
12. Alevriadou, B. R., Patel, A., Noble, M., Ghosh, S., Gohil, V. M. et al. (2021). Molecular nature and physiological role of the mitochondrial calcium uniporter channel. American Journal of Physiology Cell Physiology, 320(4), C465–82. https://doi.org/10.1152/ajpcell.00502.2020. [Google Scholar] [PubMed] [CrossRef]
13. Tanwar, J., Singh, J. B., Motiani, R. K. (2021). Molecular machinery regulating mitochondrial calcium levels: The nuts and bolts of mitochondrial calcium dynamics. Mitochondrion, 57, 9–22. https://doi.org/10.1016/j.mito.2020.12.001. [Google Scholar] [PubMed] [CrossRef]
14. Kamer, K. J., Mootha, V. K. (2015). The molecular era of the mitochondrial calcium uniporter. Nature Reviews Molecular Cell Biology, 16(9), 545–553. https://doi.org/10.1038/nrm4039. [Google Scholar] [PubMed] [CrossRef]
15. Marchi, S., Giorgi, C., Galluzzi, L., Pinton, P. (2020). Ca2+ fluxes and cancer. Molecular Cell, 78(6), 1055–1069. https://doi.org/10.1016/j.molcel.2020.04.017. [Google Scholar] [PubMed] [CrossRef]
16. Liu, Y., Jin, M., Wang, Y., Zhu, J., Tan, R. et al. (2020). MCU-induced mitochondrial calcium uptake promotes mitochondrial biogenesis and colorectal cancer growth. Signal Transduction and Targeted Therapy, 5(1), 59. https://doi.org/10.1038/s41392-020-0155-5. [Google Scholar] [PubMed] [CrossRef]
17. Wang, X., Li, Y., Li, Z., Lin, S., Wang, H. et al. (2022). Mitochondrial calcium uniporter drives metastasis and confers a targetable cystine dependency in pancreatic cancer. Cancer research, 82(12), 2254–2268. https://doi.org/10.1158/0008-5472.CAN-21-3230. [Google Scholar] [PubMed] [CrossRef]
18. Tosatto, A., Sommaggio, R., Kummerow, C., Bentham, R. B., Blacker, T. S. et al. (2016). The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1α. EMBO Molecular Medicine, 8(5), 569–585. https://doi.org/10.15252/emmm.201606255. [Google Scholar] [PubMed] [CrossRef]
19. Cao, C., Wang, S., Cui, T., Su, X. C., Chou, J. J. (2017). Ion and inhibitor binding of the double-ring ion selectivity filter of the mitochondrial calcium uniporter. Proceedings of the National Academy of Sciences of the United States of America, 114(14), E2846–51. https://doi.org/10.1073/pnas.1620316114. [Google Scholar] [PubMed] [CrossRef]
20. Paillard, M., CsordÆs, G., Huang, K. T., VÆrnai, P., Joseph, S. K. (2018). MICU1 interacts with the D-ring of the MCU pore to control its Ca2+ flux and sensitivity to Ru360. Molecular Cell, 72(4), 778–85.e3. https://doi.org/10.1016/j.molcel.2018.09.008. [Google Scholar] [PubMed] [CrossRef]
21. Di Marco, G., Vallese, F., Jourde, B., Bergsdorf, C., Sturlese, M. et al. (2020). A high-throughput screening identifies MICU1 targeting compounds. Cell Reports, 30(7), 2321–31.e6. https://doi.org/10.1016/j.celrep.2020.01.081. [Google Scholar] [PubMed] [CrossRef]
22. Tsai, T. Y., Leong, I. L., Shiao, L. R., Wong, K. L., Shao, L. et al. (2020). Tannic acid, a vasodilator present in wines and beverages, stimulates Ca2+ influx via TRP channels in bEND.3 endothelial cells. Biochemical and Biophysical Research Communications, 526(1), 117–121. https://doi.org/10.1016/j.bbrc.2020.03.078. [Google Scholar] [PubMed] [CrossRef]
23. Gerasimenko, O., Tepikin, A. (2005). How to measure Ca2+ in cellular organelles? Cell Calcium, 38(3–4), 201–211. https://doi.org/10.1016/j.ceca.2005.06.025. [Google Scholar] [PubMed] [CrossRef]
24. Chen, C. Y., Wu, C. H., Wu, K. C., Shiao, L. R., Chuang, C. M. (2021). A basal level of γ-linolenic acid depletes Ca2+ stores and induces endoplasmic reticulum and oxidative stresses to cause death of breast cancer BT-474 cells. The Chinese Journal of Physiology, 64(4), 202–209. https://doi.org/10.4103/cjp.cjp_30_21. [Google Scholar] [PubMed] [CrossRef]
25. Eruslanov, E., Kusmartsev, S. (2010). Identification of ROS using oxidized DCFDA and flow-cytometry. Methods in Molecular Biology, 594, 57–72. https://doi.org/10.1007/978-1-60761-411-1 [Google Scholar] [CrossRef]
26. Arora, S., Singh, P., Tabassum, G., Dohare, R., Syed, M. A. (2022). miR-495-3p regulates sphingolipid metabolic reprogramming to induce Sphk1/ceramide mediated mitophagy and apoptosis in NSCLC. Free Radical Biology & Medicine, 189, 71–84. https://doi.org/10.1016/j.freeradbiomed.2022.07.001. [Google Scholar] [PubMed] [CrossRef]
27. Schmiedeknecht, K., Kaufmann, A., Bauer, S., Venegas Solis, F. (2022). L-lactate as an indicator for cellular metabolic status: An easy and cost-effective colorimetric L-lactate assay. PLoS One, 17(7), e0271818. https://doi.org/10.1371/journal.pone.0271818. [Google Scholar] [PubMed] [CrossRef]
28. Li, P., Zhou, L., Zhao, T., Liu, X., Zhang, P. et al. (2017). Caspase-9: Structure, mechanisms and clinical application. Oncotarget, 8(14), 23996–24008. https://doi.org/10.18632/oncotarget.v8i14 [Google Scholar] [CrossRef]
29. Yanda, M. K., Tomar, V., Cole, R., Guggino, W. B., Cebotaru, L. (2022). The mitochondrial Ca2+ import complex is altered in ADPKD. Cell Calcium, 101, 102501. https://doi.org/10.1016/j.ceca.2021.102501. [Google Scholar] [PubMed] [CrossRef]
30. Cervinka, J., Gobbo, A., Biancalana, L., Markova, L., Novohradsky, V. et al. (2022). Ruthenium(II)-tris-pyrazolylmethane complexes inhibit cancer cell growth by disrupting mitochondrial calcium homeostasis. Journal of Medicinal Chemistry, 65(15), 10567–10587. https://doi.org/10.1021/acs.jmedchem.2c00722. [Google Scholar] [PubMed] [CrossRef]
31. Kolar, S. S., Barhoumi, R., Lupton, J. R., Chapkin, R. S. (2007). Docosahexaenoic acid and butyrate synergistically induce colonocyte apoptosis by enhancing mitochondrial Ca2+ accumulation. Cancer Research, 67(11), 5561–5568. https://doi.org/10.1158/0008-5472.CAN-06-4716. [Google Scholar] [PubMed] [CrossRef]
32. Ibrahim, A., El-Meligy, A., Lungu, G., Fetaih, H., Dessouki, A. et al. (2011). Curcumin induces apoptosis in a murine mammary gland adenocarcinoma cell line through the mitochondrial pathway. European Journal of Pharmacology, 668(1–2), 127–132. https://doi.org/10.1016/j.ejphar.2011.06.048. [Google Scholar] [PubMed] [CrossRef]
33. Yang, X., Wang, B., Zeng, H., Cai, C., Hu, Q. et al. (2014). Role of the mitochondrial Ca2+ uniporter in Pb2+-induced oxidative stress in human neuroblastoma cells. Brain Research, 1575, 12–21. https://doi.org/10.1016/j.brainres.2014.05.032. [Google Scholar] [PubMed] [CrossRef]
34. Cao, A. Z., Liu, H., Guo, H., Zang, Y., Wang, Y. et al. (2017). Calcium uptake via mitochondrial uniporter contributes to palmitic acid- induced apoptosis in mouse podocytes. Journal of Cellular Biochemistry, 118(9), 2809–2818. https://doi.org/10.1002/jcb.v118.9 [Google Scholar] [CrossRef]
35. Schwartz, J., Holmuhamedov, E., Zhang, X., Lovelace, G. L., Smith, C. D. et al. (2013). Minocycline and doxycycline, but not other tetracycline-derived compounds, protect liver cells from chemical hypoxia and ischemia/reperfusion injury by inhibition of the mitochondrial calcium uniporter. Toxicology and Applied Pharmacology, 273(1), 172–179. https://doi.org/10.1016/j.taap.2013.08.027. [Google Scholar] [PubMed] [CrossRef]
36. Garrido-Mesa, N., Zarzuelo, A., GÆlvez, J. (2013). Minocycline: Far beyond an antibiotic. British Journal of Pharmacology, 169(2), 337–352. https://doi.org/10.1111/bph.2013.169.issue-2 [Google Scholar] [CrossRef]
37. Ali, I., Alfarouk, K. O., Reshkin, S. J., Ibrahim, M. E. (2017). Doxycycline as potential anti-cancer agent. Anti-Cancer Agents in Medicinal Chemistry, 17(12), 1617–1623. https://doi.org/10.2174/1871520617666170213111951. [Google Scholar] [PubMed] [CrossRef]
38. Horvats, A., Muhios, M., Smolios, T., Begi/s, E., Zorecs, R. et al. (2021). Ca2+ as the prime trigger of aerobic glycolysis in astrocytes. Cell Calcium, 95, 102368. https://doi.org/10.1016/j.ceca.2021.102368. [Google Scholar] [PubMed] [CrossRef]
39. PØrez-LiØbana, I., Juaristi, I., GonzÆlez-SÆnchez, P., GonzÆlez- Moreno, L., Rial, E. et al. (2022). A Ca2+-dependent mechanism boosting glycolysis and OXPHOS by activating aralar-malate-aspartate shuttle, upon neuronal stimulation. The Journal of Neuroscience, 42(19), 3879–3895. https://doi.org/10.1523/JNEUROSCI.1463-21.2022. [Google Scholar] [PubMed] [CrossRef]
40. Atlante, A., Giannattasio, S., Bobba, A., Gagliardi, S., Petragallo, V. et al. (2005). An increase in the ATP levels occurs in cerebellar granule cells en route to apoptosis in which ATP derives from both oxidative phosphorylation and anaerobic glycolysis. Biochimica et Biophysica Acta, 1708(1), 50–62. https://doi.org/10.1016/j.bbabio.2005.01.009. [Google Scholar] [PubMed] [CrossRef]
41. Zamaraeva, M. V., Sabirov, R. Z., Maeno, E., Ando-Akatsuka, Y., Bessonova, S. V. et al. (2005). Cells die with increased cytosolic ATP during apoptosis: A bioluminescence study with intracellular luciferase. Cell Death & Differentiation, 12(11), 1390–1397. https://doi.org/10.1038/sj.cdd.4401661. [Google Scholar] [PubMed] [CrossRef]
42. Tatsumi, T., Shiraishi, J., Keira, N., Akashi, K., Mano, A. et al. (2003). Intracellular ATP is required for mitochondrial apoptotic pathways in isolated hypoxic rat cardiac myocytes. Cardiovascular Research, 59(2), 428–440. https://doi.org/10.1016/S0008-6363(03)00391-2. [Google Scholar] [PubMed] [CrossRef]
43. Kushnareva Y., Newmeyer D. D. (2010). Bioenergetics and cell death. Annals of the New York Academy of Sciences, 1201(1), 50–57. https://doi.org/10.1111/j.1749-6632.2010.05633.x. [Google Scholar] [CrossRef]
44. Bertero E., Maack C. (2018). Calcium signaling and reactive oxygen species in mitochondria. Circulation Research, 122(10), 1460–1478. https://doi.org/10.1161/CIRCRESAHA.118.310082. [Google Scholar] [PubMed] [CrossRef]
45. Choudhry H., Harris A. L. (2018). Advances in hypoxia-inducible factor biology. Cell Metabolism, 27(2), 281–298. https://doi.org/10.1016/j.cmet.2017.10.005. [Google Scholar] [PubMed] [CrossRef]
46. Fukai, T., Ushio-Fukai, M. (2020). Cross-talk between NADPH oxidase and mitochondria: Role in ROS signaling and angiogenesis. Cells, 9(8), 1849. https://doi.org/10.3390/cells9081849. [Google Scholar] [PubMed] [CrossRef]
47. Park, J., Lee, J., Choi, C. (2011). Mitochondrial network determines intracellular ROS dynamics and sensitivity to oxidative stress through switching inter-mitochondrial messengers. PLoS One, 6(8), e23211. https://doi.org/10.1371/journal.pone.0023211. [Google Scholar] [PubMed] [CrossRef]
48. Takata, N., Ohshima, Y., Suzuki-Karasaki, M., Yoshida, Y., Tokuhashi, Y. et al. (2017). Mitochondrial Ca2+ removal amplifies TRAIL cytotoxicity toward apoptosis-resistant tumor cells via promotion of multiple cell death modalities. International Journal of Oncology, 51(1), 193–203. https://doi.org/10.3892/ijo.2017.4020. [Google Scholar] [PubMed] [CrossRef]
49. Pinton, P., Giorgi, C., Siviero, R., Zecchini, E., Rizzuto, R. (2008). Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene, 27(50), 6407–6418. https://doi.org/10.1038/onc.2008.308. [Google Scholar] [PubMed] [CrossRef]
50. Leanza, L., Biasutto, L., Manago, A., Gulbins, E., Zoratti, M. (2013). Intracellular ion channels and cancer. Frontiers in Physiology, 4, 227. https://doi.org/10.3389/fphys.2013.00227. [Google Scholar] [PubMed] [CrossRef]
51. Woods, J. J., Nemani, N., Shanmughapriya, S., Kumar, A., Zhang, M. et al. (2019). A selective and cell-permeable mitochondrial calcium uniporter (MCU) inhibitor preserves mitochondrial bioenergetics after hypoxia/reoxygenation injury. ACS Central Science, 5(1), 153–166. https://doi.org/10.1021/acscentsci.8b00773. [Google Scholar] [PubMed] [CrossRef]
52. Nguyen, T. T., Stevens, M. V., Kohr, M., Steenbergen, C., Sack, M. N. et al. (2011). Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. The Journal of Biological Chemistry, 286(46), 40184–40192. https://doi.org/10.1074/jbc.M111.243469. [Google Scholar] [PubMed] [CrossRef]
53. Gordan, R., Fefelova, N., Gwathmey, J. K., Xie, L. H. (2016). Involvement of mitochondrial permeability transition pore (mPTP) in cardiac arrhythmias: Evidence from cyclophilin D knockout mice. Cell Calcium, 60(6), 363–372. https://doi.org/10.1016/j.ceca.2016.09.001. [Google Scholar] [PubMed] [CrossRef]
54. Ganapathy-Kanniappan, S., Geschwind, J. F. (2013). Tumor glycolysis as a target for cancer therapy: Progress and prospects. Molecular Cancer, 12, 152. https://doi.org/10.1186/1476-4598-12-152. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools