
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Targeting myeloid-derived suppressor cells in the tumor microenvironment: potential therapeutic approaches for osteosarcoma
Research Institute of Women’s Health, Sookmyung Women’s University, Seoul, 04312, Republic of Korea
* Corresponding Author: YOUNG YANG. Email:
# These authors contributed equally to this work
Oncology Research 2025, 33(3), 519-531. https://doi.org/10.32604/or.2024.056860
Received 01 August 2024; Accepted 14 October 2024; Issue published 28 February 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Osteosarcoma is a bone malignancy characterized by strong invasiveness and rapid disease progression. The tumor microenvironment of osteosarcoma contains various types of immune cells, including myeloid-derived suppressor cells, macrophages, T cells, and B cells. Imbalances of these immune cells can promote the proliferation and metastasis of osteosarcoma. Recent studies have indicated a substantial increase in the levels of myeloid-derived suppressor cells, an immune cell associated with immunosuppressive and pro-cancer effects, in the peripheral blood of patients with osteosarcoma. Moreover, the levels of the pro-inflammatory cytokine interleukin 18 are positively correlated with those of myeloid-derived suppressor cells in the peripheral blood of animal models of osteosarcoma. In this review, we explore the function of myeloid-derived suppressor cells in osteosarcoma based on the clinical diagnoses of patients with osteosarcoma and discuss future therapeutic approaches for targeting osteosarcoma. Targeting myeloid-derived suppressor cells represents a promising approach to improving the prognosis and survival rates of patients with osteosarcoma.Keywords
Abbreviations
| 5-FU | 5-fluorouracil |
| ACT | Adoptive cell transfer |
| ARG1 | Arginase 1 |
| ATRA | All-trans retinoic acid |
| C/EBP | CCAAT-enhancer-binding protein |
| CAR | Chimeric antigen receptor |
| CCL | C-C motif chemokine ligand |
| CCR | C-C chemokine receptor |
| CTL | Cytotoxic T cell |
| CXCR | C-X-C chemokine receptor |
| DC | Dendritic cell |
| G-CSF | Granulocyte colony-stimulating factor |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| HK2 | Hexokinase 2 |
| ICI | Immune checkpoint inhibitor |
| IDO | Indoleamine 2,3-dioxygenase |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| IRF8 | Interferon regulatory factor 8 |
| JAK | Janus kinase 2 |
| M-CSF | Macrophage colony-stimulating factor |
| M-MDSC | Monocytic myeloid-derived suppressor cell |
| MCT | Monocarboxylate transporter |
| MDSC | Myeloid-derived suppressor cell |
| MOF | Metal-organic framework |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | Natural killer |
| NO | Nitric oxide |
| NOX2 | Nicotinamide adenine dinucleotide phosphate hydrogen oxidase 2 |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed death ligand 1 |
| PDE | Phosphodiesterase |
| PKM2 | Pyruvate kinase M2 |
| PMN-MDSC | Polymorphonuclear myeloid-derived suppressor cell |
| ROS | Reactive oxygen species |
| STAT3 | Signal transducer and activator of the transcription 3 |
| TAM | Tumor-associated macrophage |
| TCR | T-cell receptor |
| TME | Tumor microenvironment |
| VEGF | Vascular endothelial growth factor |
Myeloid-derived suppressor cells in osteosarcoma
Myeloid-derived suppressor cells (MDSCs) are classified based on the presence of cytoplasmic granules into two types: granulocytic/polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs) [1]. PMN-MDSCs, the largest proportion of immune cells in all types of cancer, resemble neutrophils, whereas M-MDSCs are similar to monocytes. In mice, MDSCs are identified based on the expression of the markers CD11b and Gr1. PMN-MDSCs show high Ly6G and low Ly6C levels, whereas M-MDSCs express low Ly6G and high Ly6C levels [2–4]. In humans, MDSCs are distinguished based on the expression of markers such as CD33, CD14, CD15, and low HLA-DR levels [4]. Specifically, human PMN-MDSCs are identified as Lin−HLA-DR−/lowCD33+ or Lin−HLA-DR−/lowCD11b+CD14−CD15+CD33+, whereas human M-MDSCs are characterized as CD14+HLA-DR−/low or Lin−HLA-DR−/lowCD11b+CD14+CD15− [3]. A newly identified type, early-stage MDSCs, expresses both granulocytic and monocytic markers [5]. MDSCs are a diverse group of immature myeloid cells that accumulate during cancer proliferation and other pathological conditions [6,7]. They are potent immune suppressors and facilitate tumor growth and metastasis through various mechanisms, such as suppression of T-cell activation and promotion of tumor immune evasion in various cancers including melanoma, non-small cell lung cancer, and osteosarcoma [8]. Conversely, MDSCs perform distinct functions in autoimmune diseases such as rheumatoid arthritis [9–12] having protective pro-inflammatory effects. Thus, dual mechanisms and functions of MDSCs have been identified across different disease contexts.
Osteosarcoma is a highly aggressive bone malignancy characterized by rapid progression and a high potential for metastasis, particularly affecting children, adolescents, and young adults [13]. The tumor microenvironment (TME) in osteosarcoma is a complex and dynamic network comprising cancer cells along with various stromal and immune cells, including MDSCs, macrophages, T cells, and B cells [14]. These components interact to create an environment that supports tumor growth, immune evasion, and metastasis. An imbalance among these immune cells can lead to an immunosuppressive TME which favors tumor progression and therapeutic resistance. Recent research has highlighted the important role of MDSCs in the TME of osteosarcoma, contributing to immunosuppression and tumor progression [15]. The number of MDSCs is significantly increased in patients with osteosarcoma, indicating their critical role in the TME of osteosarcoma. A review of MDSCs in musculoskeletal diseases reported that MDSCs, particularly PMN-MDSCs, accumulate in the TME and inhibit T cell-mediated immune responses, induced by high expression levels of interleukin (IL)-18 and C-X-C motif chemokine ligand 12 [16]. Blocking these factors improved the efficacy of anti-programmed cell death protein 1 (PD-1) treatment in mice. Using an osteosarcoma tumor model, Guan et al. found that the levels of MDSCs are positively correlated with those of IL-18, suggesting that IL-18 may attract MDSCs into the tumor parenchyma. A combination therapy with IL-18 inhibition and anti-PD-1 decreased tumor burden and increased T-cell infiltration [17]. Long et al. reported that osteosarcoma-induced MDSCs are associated with poor chimeric antigen receptor (CAR) T-cell efficacy against xenografts [18]. Osteosarcomas significantly induced the proliferation of CD11b+Ly6G+ and CD11b+Ly6C+ myeloid cell populations compared to nontumor-bearing controls in a murine model. Clinically, proliferating monocytic and granulocytic MDSCs adversely affected the efficiency of GD2-CAR T cells generated from apheresis products of patients with osteosarcoma [19].
The multifaceted immunosuppressive roles of MDSCs in the TME of osteosarcoma: Crosstalk with other immune cells
Osteosarcoma tissue forms a heterogeneous and complex immune microenvironment with extensive and varied immune cell infiltration [20]. This TME accelerates cancer cell growth within the bone by establishing an immunosuppressive milieu that supports cancer cell survival and proliferation [20,21]. Guan et al. reported significant increases in MDSC populations in the peripheral blood of patients with osteosarcoma [17]. These cells heavily infiltrate the osteosarcoma TME, promoting cancer progression by inhibiting the proliferation of anti-cancer cytotoxic T cells (CTLs) and preventing CTL-associated anti-PD-1 cancer therapy [22]. By interacting with various immune cells, including T cells, dendritic cells (DCs), and natural killer (NK) cells, MDSCs create an immunosuppressive environment. They also modulate immunosuppressive functions through humoral interactions with osteoblasts, osteoclasts, chondrocytes, and other stromal cells in the osteosarcoma TME [20,23]. Additionally, MDSCs downregulate the anti-cancer immune response by further differentiating into cancer-favoring and immunosuppressive DCs, tumor-associated macrophages (TAMs), and tumor-associated neutrophils [20,24]. However, this review focuses on how MDSCs contribute to the immunosuppressive TME by interacting with immune cells rather than their differentiation into other immunosuppressive cell types.
T cell-targeted immunotherapy is a promising strategy for treating osteosarcoma [17,25–27]. Considering that MDSCs are potent inhibitors of T cell-mediated anti-cancer immunity, understanding how MDSCs dysregulate T cells is crucial for optimizing T cell-mediated anti-osteosarcoma therapies. MDSCs interact closely with T cells, suppressing their antitumor activity through humoral mechanisms and direct cell–cell interactions. MDSCs express and secrete high levels of arginase 1 (ARG1) into the TME, thus depleting L-arginine, an essential amino acid for T-cell metabolism [28]. This depletion leads to T-cell cycle arrest before entering the G1 phase by suppressing cyclin D3 expression [28]. MDSC-derived ARG1 also downregulates the CD3ζ transmembrane component of the T-cell receptor (TCR), thereby disrupting T-cell activation via TCR engagement [28]. Additionally, MDSCs deplete cysteine, another critical amino acid for T-cell proliferation and activation. Cysteine is the rate-limiting substrate for the synthesis of the tripeptide glutathione [29]. T cells acquire cysteine by interacting with antigen-presenting cells via a neutral amino acid transporter. MDSCs compete with antigen-presenting cells, including macrophages and DCs, thereby interrupting the cysteine uptake by T cells [28,30]. Furthermore, MDSC-derived indoleamine 2,3-dioxygenase (IDO) depletes tryptophan, leading to T-cell cycle arrest in the mid-G1 phase and inhibition of T-cell proliferation [28,31].
Reactive oxygen species (ROS) and nitric oxide (NO), major humoral factors produced by MDSCs, disrupt T-cell functions. PMN-MDSCs primarily produce ROS, whereas M-MDSCs are characterized by enhanced NO production. TME-derived IL-6 activates the Janus kinase 2 (JAK)/signal transducer and activator of the transcription 3 (STAT3) signaling pathways, thereby increasing the expression of nicotinamide adenine dinucleotide phosphate hydrogen oxidase 2 (NOX2), which mediates ROS production in PMN-MDSCs [32,33]. M-MDSCs express high levels of inducible nitric oxide synthase (iNOS), leading to significantly increased NO production. Hypoxic conditions within solid tumors also upregulate iNOS and ARG1 expression in MDSCs [34]. ROS and NO produce the byproduct peroxynitrite, which disrupts the conjugation of the TCR component CD8 with major histocompatibility complex molecules on cancer cell surfaces, thereby reducing the cytotoxic activity of CD8+ CTLs [28,35]. Additionally, MDSCs inactivate the T cell-activating IL-2 signaling pathway and destabilize IL-2 mRNA, thus decreasing IL-2 production and secretion, which suppresses T-cell activity [36,37]. This multifaceted suppression orchestrated by MDSC-derived ROS and NO underscores the critical role of MDSCs in modulating anti-cancer responses within the TME.
Programmed death ligand 1 (PD-L1), expressed on the surfaces of PMN-MDSCs and M-MDSCs directly binds PD-1 on CD8+ T cells. This interaction inhibits T-cell functions through the PD-1–PD-L1 signaling pathway. The hypoxic conditions within the TME increase PD-L1 expression in infiltrating MDSCs by enhancing the expression and stability of hypoxia-inducible factor 1α, which promotes PD-L1 transcription in MDSCs [38,39]. Anti-PD-1/anti-PD-L1 therapy is a promising strategy for treating various types of cancers, as cancer cells express high levels of PD-L1 to evade anti-cancer T-cell activity [40,41]. However, patients with osteosarcoma are resistant to PD-1/PD-L1 inhibitors, and anti-PD-1 monotherapy has been ineffective against osteosarcoma in a murine model of K7M2 osteosarcoma grafts [22]. Interestingly, these authors found that combining a C-X-C chemokine receptor (CXCR)4 antagonist with an anti-PD-1 monoclonal antibody can overcome the limitations of anti-PD-1 therapy in their osteosarcoma model. They reported that continuous substantial MDSC infiltration into the TME undermines the efficacy of anti-PD-1 therapy. The CXCR4 antagonist reduced MDSC chemotaxis into the osteosarcoma TME, thereby enhancing the antitumor effects of anti-PD-1 therapy [22]. However, the impact of MDSC suppression, the identification of effective strategies to inhibit MDSC infiltration into the tumor site, and the implications for anti-osteosarcoma therapy need further preclinical and clinical evaluation.
Although the major immunosuppressive and anti-cancer strategies of MDSCs are primarily mediated through their crosstalk with T cells, MDSCs also interact with other immune cells, such as NK cells, by altering the expression levels of natural cytotoxicity receptors on the surfaces of NK cells. For instance, MDSCs downregulate CD247 [42], natural cytotoxicity triggering receptor 3 [43], and natural killer group 2D, impairing the anti-cancer function of interferon-gamma production by NK cells [44]. The role of MDSC–NK cell crosstalk in the TME of osteosarcoma remains largely unknown, particularly regarding the pathophysiology and metastasis of osteosarcoma, highlighting the need for further investigation. These MDSC interactions in osteosarcoma collectively create a complex immunosuppressive and oncogenic network within the TME, effectively inhibiting multiple arms of the immune system, including both innate (NK cells, DCs) and adaptive (T cells) responses.
MDSCs can interact not only with immune cells but also directly with cancer cells within the osteosarcoma TME. Taylor et al. demonstrated that MDSCs expressing high levels of IL-1β bind directly to osteosarcoma cells through their interaction with the IL1R1 receptor, which is highly expressed in osteosarcoma. This binding activates downstream nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling [45]. NF-κB is recognized as a critical oncogene in osteosarcoma, promoting cell proliferation, survival, migration, and invasion, while inhibiting apoptosis in 143B and MG63 human osteosarcoma cells [46].
In summary, MDSCs play crucial roles in shaping the immunosuppressive TME of osteosarcoma. They suppress T-cell functions, promote the expansion of regulatory T cells, reduce NK cell-mediated antitumor immunity, and impair DC-mediated antigen presentation. By interacting with macrophages and differentiating into immunosuppressive cell types, MDSCs further reinforce the immunosuppressive network within the TME. Understanding these multifaceted interactions and mechanisms is essential for developing effective therapeutic strategies to counteract MDSC-mediated immunosuppression and enhance antitumor immune responses in osteosarcoma.
The role of MDSCs in osteosarcoma metastasis
MDSCs also play a considerable role in promoting osteosarcoma metastasis through several mechanisms. They contribute to forming a pre-metastatic niche, creating a favorable environment for tumor cells to establish metastases by secreting factors that alter the extracellular matrix, promoting angiogenesis, and recruiting other immunosuppressive cells [47]. MDSCs also enhance tumor cell invasion by producing factors that facilitate the spread of osteosarcoma cells to distant sites [47]. They create an immunosuppressive microenvironment that allows tumor cells to evade immune detection and destruction by suppressing T-cell function through ROS production, depleting L-arginine, inhibiting NK-cell activity through phagocytosis, and promoting Treg proliferation [33]. Tumors recruit MDSCs through various chemokine pathways, including C-C motif chemokine ligand (CCL)2/CCL12–C-C chemokine receptor (CCR)2, CCL3/4/5–CCR5, and CXCL8–CXCR1/CXCR2, which subsequently promote metastasis through similar chemokine axes [33]. These MDSCs also secrete pro-metastatic factors such as transforming growth factor beta, S100A8/A9, vascular endothelial growth factor (VEGF), and exosomes, which interact with various cells in the TME, making it more conducive to metastasis [8,33,48]. Additionally, MDSCs enhance metastasis by promoting β-adrenergic signaling and the IL-6–STAT3 pathway and can differentiate into osteoclasts in bone metastatic cancers, thereby contributing to bone destruction and promoting tumor growth [49]. In bone metastatic cancers, MDSC-derived NO mediates immunosuppression and promotes osteoclastogenesis, further facilitating metastasis. Moreover, the presence of MDSCs in the TME can impair the efficacy of immunotherapeutic drugs, such as PD-1 inhibitors, by suppressing T-cell responses [50].
Targeting MDSCs in the TME: Potential Approaches for Osteosarcoma Therapy
To improve the survival rates of patients with osteosarcoma, various treatment approaches are employed, including surgery [51,52], chemotherapy [53], immunotherapy [54,55], and targeted therapy [56–59]. Each method plays a crucial role in the comprehensive management of the disease, aiming to enhance patient outcomes and increase the likelihood of long-term survival. Recent research has focused on targeting MDSCs within the TME as a promising therapeutic strategy for cancer, including osteosarcoma (Table A1, Fig. 1). However, research on cancer therapies targeting MDSCs specifically in osteosarcoma is limited. Therefore, we aim to describe tumor therapies that target MDSCs in the TME and discuss the potential application of these approaches in the treatment of osteosarcoma.
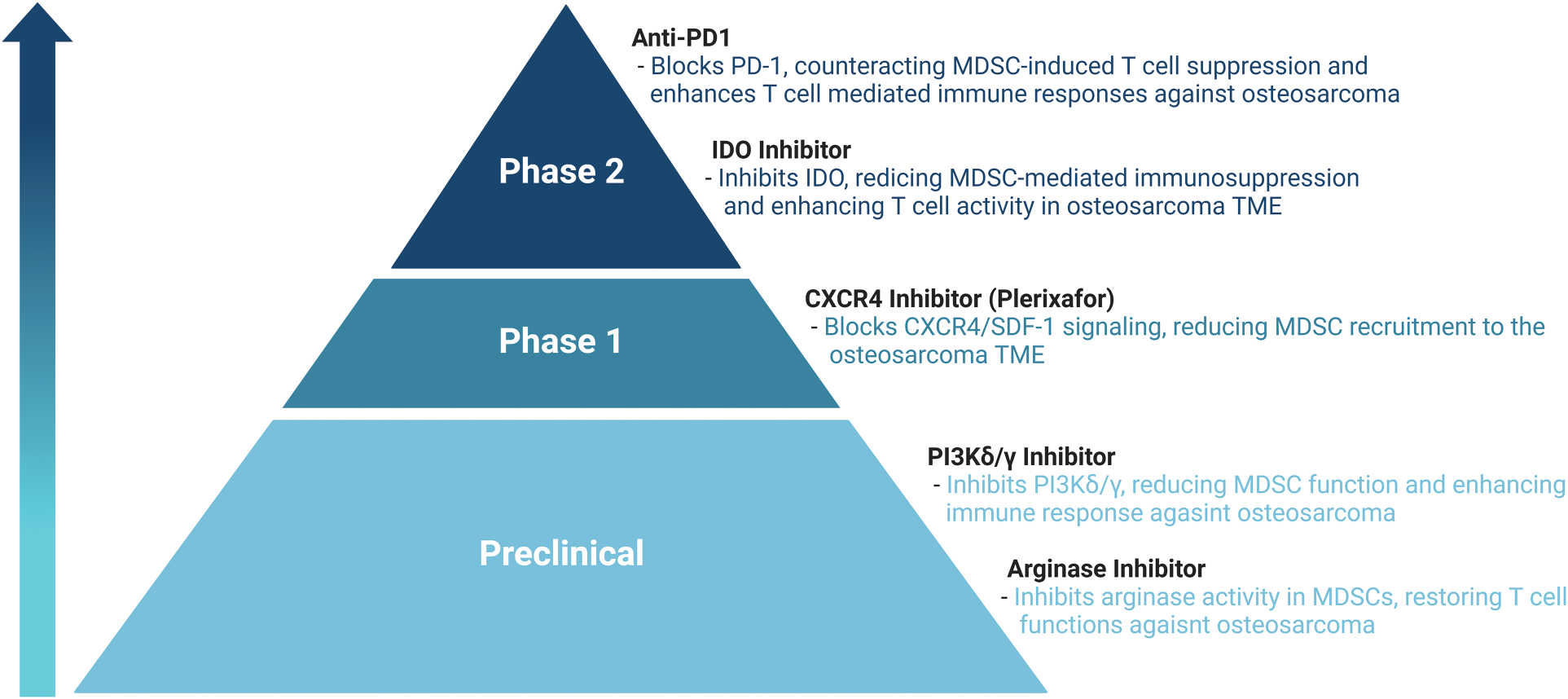
Figure 1: Therapeutic approaches targeting MDSCs in TME.
Blocking the activation of MDSCs is a critical therapeutic strategy to mitigate their immunosuppressive effects in the TME. This approach involves targeting key signaling pathways and transcription factors essential for MDSC activation. Several signaling pathways, including the STAT3, CCAAT-enhancer-binding protein (C/EBP) β, and interferon regulatory factor 8 (IRF8) pathways, are linked to MDSC proliferation and activation. Inhibiting the STAT3 pathway can reduce MDSC proliferation and their immunosuppressive activities [60,61]. STAT3 downregulation, caused by activated CD45 phosphatase, has been observed in M-MDSCs, leading to a unique differentiation into TAMs [62]. Preclinically, STAT3 inhibitors have shown promising results by reducing MDSC numbers and enhancing the efficacy of other cancer therapies. Inhibitors of the JAK–STAT3 pathway, such as JSI-124, significantly reduce MDSC numbers and suppress their activity, leading to enhanced antitumor immune responses [63]. STAT3 plays major roles in the progression and metastasis of osteosarcoma. Inhibition of STAT3 blocks protein synthesis and reduces tumor metastasis in osteosarcoma cells. This has led to preclinical studies exploring STAT3 inhibitors as a potential therapeutic approach in osteosarcoma [64,65]. The C/EBPβ pathway is regulated by IL-6 and controls the expression of immune-suppressive genes (such as those encoding ARG1, iNOS, and NOX2), thereby regulating the differentiation and function of MDSCs. C/EBPβ promotes MDSC generation in the bone marrow or spleen and is closely associated with the expression of granulocyte-macrophage colony-stimulating factor (GM-CSF) and granulocyte colony-stimulating factor (G-CSF) in myeloid cells. Targeting C/EBPβ in osteosarcoma treatment is a less common strategy. However, it has been reported that M-MDSCs are reduced in the spleen of C/EBPβ-deficient mice [66–68], and this transcription factor is known to regulate immunosuppressive genes and participate in the TME in various cancers [69]. Therefore, there is potential for its application in combination therapy for osteosarcoma. IRF8 pathways are also involved in MDSC differentiation and function. Downregulation of IRF8 induces the production of PMN-MDSCs. In vivo, IRF8 overexpression inhibits MDSC proliferation and enhances antitumor efficacy through the STAT3 and STAT5 signaling pathways. IRF8 overexpression selectively induces the expansion of granulocytes and PMN-MDSCs while not significantly affecting monocytes or M-MDSCs. Other downstream effectors in MDSC activation include S100A9 and NOX2, which are involved in ROS production. Inhibitors targeting these molecules can effectively impair MDSC function and enhance antitumor immune responses [70,71]. Research directly targeting IRF8 in osteosarcoma is not well established. However, its role in modulating immune responses suggests it might be a future target for therapies aimed at enhancing antitumor immunity in patients with osteosarcoma [65]. A study by Li et al. demonstrated that disruption of the COX-2/PGE2 pathway and the use of phosphodiesterase (PDE) 5 inhibitors, such as sildenafil, vardenafil, and tadalafil, neutralize MDSC immunosuppressive capacities [7]. Sildenafil reduces ARG1 and iNOS expression in MDSCs, thereby inhibiting their immunosuppressive functions [72]. Furthermore, sildenafil prolongs the survival of melanoma-bearing mice by reducing MDSC levels and activity, leading to restored CD8+ T-cell infiltration and function in the TME [73]. PDE inhibitors can reactivate antitumor immunity by enhancing T- and NK-cell functions. In preclinical murine models, the administration of sildenafil and tadalafil significantly reduces MDSC levels and their suppressive activity, leading to improved T-cell proliferation and tumor cell apoptosis. These agents not only destroy tumor cells but also diminish the suppressive effects of MDSCs on the immune system, thereby restoring the antitumor immune response. Several studies focus on the pleiotropic effects of PDE5 inhibitors, which include reducing tumor growth by enhancing NO signaling and inhibiting tumor angiogenesis. These mechanisms are being explored in preclinical models of osteosarcoma. For instance, the results of some studies suggest that by modulating blood flow and reducing hypoxia, PDE5 inhibitors may reduce the aggressive spread of cancer cells and improve the efficacy of other treatments like chemotherapy [74,75]. This potential can also be seen in research on osteosarcoma using the phosphodiesterase family member PDE2. In a study investigating the role of PDE2 in human osteosarcoma cells, PDE2 inhibition regulated cell proliferation and migration. This suggests potential therapeutic implications for targeting PDE pathways in osteosarcoma, particularly in manipulating cAMP and cGMP signaling in osteosarcoma cells [76].
MDSCs are recruited to the TME through various tumor-derived growth factors such as GM-CSF, G-CSF, VEGF, macrophage colony-stimulating factor (M-CSF), and IL-6. Targeting these pathways can reduce MDSC recruitment. GM-CSF drives the proliferation and immunosuppressive activity of MDSCs. Inhibiting GM-CSF can reduce MDSC proliferation and subdue their immunosuppressive functions [77]. G-CSF promotes MDSC proliferation, and targeting G-CSF can help limit the accumulation of these cells in the TME [78]. VEGF is involved in MDSC recruitment and impairs DC maturation, thereby promoting an immunosuppressive TME. Anti-VEGF therapies can reduce MDSC levels and improve antitumor immune responses [79]. IL-6 positively correlates with MDSC levels in various cancers. Targeting IL-6 can disrupt MDSC recruitment and support the anti-cancer ability of the immune system [80]. GM-CSF, G-CSF, VEGF, M-CSF, and IL-6 are all key cytokines that can be linked to osteosarcoma therapy due to their roles in immune regulation, inflammation, and tumor growth. GM-CSF and G-CSF have been used to stimulate immune responses, whereas VEGF is often targeted to inhibit tumor angiogenesis. M-CSF and IL-6 also play roles in promoting macrophage activity and inflammation, which can influence tumor progression. Emerging therapies explore how modulating these cytokines can enhance treatment outcomes in osteosarcoma [81–84].
According to chemokine studies examining MDSCs, the CCL2–CCR2 axis is crucial for recruiting MDSCs to tumor sites. Inhibiting this axis can reduce MDSC accumulation in the TME, thereby alleviating immunosuppression. Blocking CCR2 reduces MDSC migration to tumors and enhances antitumor T-cell responses. This strategy helps in reprogramming the TME to support antitumor immunity [85,86]. CXCR2 plays a considerable role in the trafficking of PMN-MDSCs. Inhibiting CXCR2 disrupts the recruitment of these cells to the tumor site, thereby reducing their immunosuppressive effects. CXCR2 inhibitors, such as SX-682, impair PMN-MDSC trafficking, which subsequently enhances the efficacy of T cell- and NK cell-based immunotherapies by allowing better immune cell infiltration and activity within the tumor [85,87]. In osteosarcoma, crucial roles of CCL2 and its receptor CCR2 have been identified. The CCL2–CCR2 pathway contributes to the migration of immune cells, such as TAMs, as well as to tumor growth and metastasis within the osteosarcoma microenvironment. Study findings suggest that inhibition of the CCL2–CCR2 pathway can suppress the progression of osteosarcoma and prevent bone destruction [88–90]. Other studies have demonstrated that the SDF-1–CXCR4 signaling axis plays a crucial role in the accumulation of MDSCs within the TME. Notably, the CXCR4 antagonist AMD3100 showed a synergistic effect when used in combination with anti-PD-1 antibodies in murine models of osteosarcoma. These findings suggest that co-administration of CXCR4 antagonists with PD-1 inhibitors might be a promising therapeutic strategy for improving treatment outcomes in patients with osteosarcoma [22]. This research highlights that inhibiting the SDF-1-CXCR4 axis, a key signaling pathway that regulates the accumulation and function of immune cells in osteosarcoma, can suppress tumor growth and enhance immune responses. In particular, the combination of CXCR4 antagonists with immune checkpoint inhibitors (ICIs, such as anti-PD-1 drugs) has the potential to maximize therapeutic efficacy [91].
Reprogramming MDSCs within the TME aims to transform these immunosuppressive cells into non-suppressive or even antitumor immune cells. This approach seeks to enhance the body’s natural immune response against cancer by modifying the behavior and function of MDSCs [92]. One strategy involves inducing the differentiation of MDSCs into mature DCs or macrophages that do not support tumor growth. This differentiation can be promoted using cytokines or specific drugs. All-trans retinoic acid (ATRA) supports antitumor therapy by reprogramming MDSCs within the TME [93,94]. Tobin et al. reported that ATRA reduces the expression of immunosuppressive genes, including those encoding PD-L1, IL-10, and IDO, in MDSCs [95]. The use of ATRA promotes the differentiation of MDSCs into mature cells, thereby diminishing their suppressive activity [96–98]. In an osteosarcoma study, ATRA decreased osteosarcoma metastases by reprogramming MDSCs in the TME. This provides insights into how such strategies can convert immune-resistant “cold” tumors into “hot” tumors, making them more responsive to immunotherapies [99].
Targeting MDSC metabolic pathways
Targeting the metabolic pathways of MDSCs is a promising therapeutic strategy to overcome immunosuppressive TME. These cells express high levels of ARG1, which depletes arginine, and consequently inhibit T-cell proliferation and function. Inhibitors of ARG1 can restore T-cell activity and enhance antitumor immunity [100–102]. Additionally, MDSCs produce NO through iNOS, thereby suppressing T-cell function and promoting tumor growth. iNOS competes with ARG1 for the same substrate and metabolizes L-arginine into citrulline and NO in tumor progression and T-cell activation [103]. Therefore, iNOS inhibitors may reduce MDSC-mediated immunosuppression and improve the effectiveness of other cancer therapies [104–106]. Targeting ARG1 and iNOS involved in the L-arginine pathway that can induce the inhibition of L-arginine metabolism. It is an essential strategy to activate T-cell function and decrease the activity of immune-suppressive cells. Rodriguez et al. found that L-arginine starvation arrests the cell cycle from the G1 to the S phase with impairment of cyclin D expression [102]. The suppressive effects of MDSCs are attributed to L-arginine metabolism. The Warburg effect is a hallmark of cancer characterized by metabolic reprogramming in tumors [107]. Excess lactate is transported across membranes by the monocarboxylate transporters (MCT) 1 and MCT4, which are crucial for tumor aggressiveness [108,109]. Lymphoid-derived myeloid-derived suppressor cells often upregulate glycolytic pathways to meet their energy demands. Inhibiting key glycolytic enzymes such as hexokinase 2 (HK2) or pyruvate kinase M2 (PKM2) can reduce MDSC survival and function [110–112]. Targeting these pathways may shift the TME toward a more immune-permissive state. HK2 and PKM2 impair glucose metabolism, thereby affecting the survival and function of both MDSCs and tumor cells. This intervention disrupts glycolysis, which is crucial for the energy supply and function of both MDSCs and tumor cells, potentially enhancing antitumor immunity [113,114]. Thus, glycolytic inhibition could be a viable strategy to modulate the immune environment in favor of antitumor responses, making the TME less supportive of cancer progression. Lactate is an immunosuppressive metabolite that promotes tumor expansion, induces angiogenesis, stimulates amino acid metabolism, inhibits CTLs, NK cells, and DCs, and further impedes the antitumor response [109]. Additionally, lactate promotes cancer growth by inducing protumor abilities, such as MDSC differentiation [115]. Lactate-induced MDSCs directly inhibit the antitumor response of immune cells. Moreover, the levels of immunosuppressive MDSCs increase when cultured with high concentrations of lactate [116]. Research on osteosarcoma therapies targeting L-arginine, lactate, and HK2 primarily aims to inhibit cancer cell metabolism and enhance immune responses. Therapeutic strategies that target the glycolytic pathway can be pivotal in promoting antitumor immune responses and inhibiting tumor cell growth. For instance, using HK2 inhibitors can block glycolysis in cancer cells, disrupting their energy production. Additionally, by reducing lactate production, these treatments can help improve the immunosuppressive environment, thereby enhancing immune function and limiting tumor progression [117]. Moreover, MDSCs exhibit altered lipid metabolism, characterized by increased fatty acid uptake and oxidation. Inhibiting fatty acid transporters or enzymes involved in fatty acid oxidation can impair MDSC function. This approach can potentially reduce MDSC numbers and their immunosuppressive activity, thereby enhancing the antitumor immune response [118,119]. PMN-MDSCs suppress immune responses by increasing the expression of CPT1 and fatty acid uptake to promote fatty acid oxidation in tumors [118]. Overexpression of fatty acid transport protein 2 in PMN-MDSCs contributes to tumor growth through PGE2 synthesis [107]. Additionally, lectin-type oxidized LDL receptor 1, which is highly expressed in PMN-MDSCs, is associated with endoplasmic reticulum stress and lipid metabolism in tumors [120]. Research on therapies targeting lipid metabolism in osteosarcoma highlights this critical aspect of energy metabolism in cancer cells. Osteosarcoma cells utilize lipid metabolism to obtain the energy necessary for tumor growth and metastasis. By regulating lipid metabolism, potential therapeutic strategies to inhibit osteosarcoma growth have been proposed [121]. Additionally, a model that predicts the prognosis of patients with osteosarcoma based on the relationship between lipid metabolism and macrophages has been suggested [122]. Fatty acid amide hydrolase has also been emphasized as an important marker for therapies targeting lipid metabolism in osteosarcoma [123]. Therefore, inhibiting lipid metabolism might be an effective strategy for suppressing cancer cell growth.
Nanomedicine-based targeting of MDSCs
Cancer treatment strategies using nanoengineering employ nanomaterials to regulate MDSCs in the TME and enhance immune responses. By targeting and inhibiting MDSCs through these techniques, researchers aim to improve the effectiveness of cancer therapies, such as treatment with ICIs or chemotherapy. Targeted delivery using nanoparticles can selectively transport drugs to the TME. These nanoparticles can inhibit the accumulation of MDSCs and block the immunosuppressive signals they release. For example, metal-organic frameworks (MOFs) can be engineered at the nanoscale to deliver therapeutic agents directly to MDSCs, simultaneously promoting tumor suppression and immune activation. IDO, an enzyme expressed by MDSCs, suppresses immune cell activity. Nanoengineering can be used to deliver IDO inhibitors through nanoparticles, preventing MDSCs from releasing immunosuppressive signals and enabling immune cells to attack tumor cells more effectively. Fan et al. developed MOF-based nanomaterials to modulate both IDO and MDSCs, enhancing chemo-immunotherapy for osteosarcoma. By inhibiting the accumulation of MDSCs, the study improved the tumor’s immunosuppressive environment and demonstrated a synergistic effect between chemotherapy and immunotherapy [25]. These approaches aim to reduce or eliminate the immunosuppressive factors in the TME, allowing immune cells to attack the tumor more effectively, thus offering new possibilities for cancer treatment.
Combination therapies targeting MDSCs
ICIs such as pembrolizumab (anti-PD-1) have shown limited efficacy in some tumors due to the presence of immunosuppressive cells like MDSCs. Combining ICIs with agents that deplete or inhibit MDSCs can enhance the overall antitumor immune responses. Blocking MDSCs can improve the efficacy of ICIs by reducing immunosuppression and promoting effective T-cell functions [94,124]. Combining Adoptive cell transfer (ACT) with MDSC-targeting strategies can improve the persistence and function of the transferred T cells. For example, reducing MDSC levels can decrease the immunosuppressive environment, thus enhancing the efficacy of ACT in eradicating tumors [125]. Although the combination of MDSC inhibition and ACT has the potential to enhance T-cell function and persistence, this has primarily been demonstrated in experimental models, and the clinical efficacy remains uncertain. Combining these strategies with cancer vaccines aims to stimulate the patient’s immune system to recognize and attack tumor cells. Notably, MDSCs can inhibit the efficacy of these vaccines. Combining cancer vaccines with MDSC inhibitors can enhance the vaccine-induced immune response by reducing MDSC-mediated suppression. The study by Iclozan et al., conducted in animal models of small cell lung cancer, showed that combining an MDSC inhibitor with a cancer vaccine enhanced immune responses [126]. Another study highlighted the potential of combining cancer vaccines with ICIs, which can further enhance the antitumor response by reducing MDSC-mediated suppression and blocking other inhibitory pathways used by tumors to evade the immune system [94–96]. Traditional osteosarcoma therapies, such as chemotherapy and radiation therapy, can sometimes paradoxically increase MDSC levels, leading to immune suppression. Combining these therapies with MDSC-targeting agents might mitigate this effect and enhance the overall therapeutic outcome. Gemcitabine and 5-fluorouracil (5-FU) reduce both the number and function of MDSC. These drugs not only diminish tumor growth but also extend survival in mouse models. Clinical observations have further corroborated these findings, demonstrating that gemcitabine and 5-FU decrease MDSC populations and enhance CD8+ T-cell responses, which are crucial for effective antitumor immunity [127,128]. Studies in humans and animal models have demonstrated that chemotherapeutic agents, such as platinum-based drugs, cyclophosphamide, gemcitabine, and 5-FU, can reduce MDSC levels, which can help restore the antitumor immune response, making traditional treatments more effective. For example, combining low-dose chemotherapy with fatty acid oxidation inhibitors in animal models delayed tumor growth and reduced MDSC-mediated immunosuppressive activity, thereby enhancing antitumor immunity [129–131]. However, clinical validation remains needed.
Mechanistic insights into the reversibility of epigenetic modifications through small-molecule inhibitors have raised the prospect of targeting specific epigenetic pathways to reprogram the MDSC population into an immunostimulatory phenotype. The histone deacetylase inhibitor entinostat blocks the formation of the pre-metastatic niche by promoting MDSC differentiation into pro-inflammatory macrophages. However, the therapeutic applicability of entinostat in clinical trials has shown limited efficacy [132]. Small-molecule inhibitors, such as STAT3 or CSF1R inhibitors, targeting MDSC pathways have been used in combination with ICIs to improve treatment outcomes. These inhibitors can reduce the number and suppressive function of MDSC, thereby enhancing the antitumor immune response initiated by ICIs [129]. Some studies reported on the combination of MDSC inhibition with ICIs or chemotherapy in osteosarcoma treatment. Inhibition of IL-18-mediated MDSC accumulation improves the efficacy of ICIs, such as anti-PD-1 drugs, in treating osteosarcoma. One study demonstrated a significant reduction in MDSC levels, which enhanced the tumor’s response to immune checkpoint blockade [17]. Another study on tumoral immune infiltrates, PD-L1 expression, and the role of CD8/TIA-1 lymphocytes in patients with localized osteosarcoma examined how MDSCs and ICIs influence tumor progression and how combining these therapies can increase treatment efficacy [133]. The SDF-1–CXCR4 axis facilitates the accumulation of MDCs in the osteosarcoma microenvironment, which blunts the response to anti-PD-1 therapy [22]. Additionally, a study on nanoengineering an MFO for osteosarcoma chemo-immunotherapy shows how modulation of IDO and MDSCs can enhance the effectiveness of chemo-immunotherapy [25]. These studies present promising approaches to improving osteosarcoma treatments by combining MDSC inhibition with ICIs and chemotherapy.
Future Directions and Conclusion
MDSCs in osteosarcoma have been increasingly recognized as a critical factor in the progression and immune evasion of this aggressive cancer. Thus, targeting MDSCs represents a promising approach to improving the prognosis and survival of patients with osteosarcoma. Elucidating the mechanisms underlying MDSC proliferation and function, as well as their interactions with other immune cells in the TME, is essential for developing effective therapeutic strategies.
However, there are limitations to our review. The preclinical findings need to be validated in clinical settings to assess the efficacy and safety of MDSC-targeting therapies in patients with osteosarcoma. Additionally, while we have focused on the immunosuppressive roles of MDSCs in the TME, further research is necessary to understand the full scope of MDSC interactions with other stromal and cancer-associated cells. Recent studies have demonstrated that MDSCs play dual roles, exerting both immunosuppressive and pro-inflammatory functions. Therefore, the detailed mechanisms and consequences of the pro-inflammatory roles of MDSCs in immune cells in osteosarcoma TME need to be elucidated. In addition, future research should focus on identifying novel targets for modulating MDSC activity and evaluating the efficacy of combination therapies that target multiple components of the TME.
Acknowledgement: None.
Funding Statement: This work was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2021R1A2C3003414, 2021R1A6A1A03038890, 2022R1I1A1A01053839, RS-2023-00246963), the Korea government (MSIT) (RS-2024-00344635), and the Korea Basic Science Institute (National Research Facilities and Equipment Center, No. 2021R1A6C101A564).
Author Contributions: The authors confirm their contribution to the paper as follows: study concept: Young Yang; draft manuscript preparation: Hye In Ka, Sora Han, Se Hwan Mun; review and editing: Hye In Ka, Sora Han, Se Hwan Mun, Young Yang. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19(2):108–19. doi:10.1038/s41590-017-0022-x. [Google Scholar] [PubMed] [CrossRef]
2. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32(1):19–25. doi:10.1016/j.it.2010.10.002. [Google Scholar] [PubMed] [CrossRef]
3. Umansky V, Blattner C, Gebhardt C, Utikal J. The role of myeloid-derived suppressor cells (MDSC) in cancer progression. Vaccines. 2016;4(4):36. doi:10.3390/vaccines4040036. [Google Scholar] [PubMed] [CrossRef]
4. Cassetta L, Baekkevold ES, Brandau S, Bujko A, Cassatella MA, Dorhoi A, et al. Deciphering myeloid-derived suppressor cells: isolation and markers in humans, mice and non-human primates. Cancer Immunol Immunother. 2019;68(4):687–97. doi:10.1007/s00262-019-02302-2. [Google Scholar] [PubMed] [CrossRef]
5. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7(1):12150. doi:10.1038/ncomms12150. [Google Scholar] [PubMed] [CrossRef]
6. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013;13(10):739–52. doi:10.1038/nrc3581. [Google Scholar] [PubMed] [CrossRef]
7. Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther. 2021;6(1):362. doi:10.1038/s41392-021-00670-9. [Google Scholar] [PubMed] [CrossRef]
8. Wang Y, Ding Y, Guo N, Wang S. MDSCs: key criminals of tumor pre-metastatic niche formation. Front Immunol. 2019;10:172. doi:10.3389/fimmu.2019.00172. [Google Scholar] [PubMed] [CrossRef]
9. Rajabinejad M, Salari F, Gorgin Karaji A, Rezaiemanesh A. The role of myeloid-derived suppressor cells in the pathogenesis of rheumatoid arthritis; anti- or pro-inflammatory cells? Artif Cells Nanomed Biotechnol. 2019;47(1):4149–58. doi:10.1080/21691401.2019.1687504. [Google Scholar] [PubMed] [CrossRef]
10. Bekic M, Tomic S. Myeloid-derived suppressor cells in the therapy of autoimmune diseases. Eur J Immunol. 2023;53(12):e2250345. doi:10.1002/eji.v53.12. [Google Scholar] [CrossRef]
11. Nepal MR, Shah S, Kang KT. Dual roles of myeloid-derived suppressor cells in various diseases: a review. Arch Pharm Res. 2024;47(7):597–616. doi:10.1007/s12272-024-01504-2. [Google Scholar] [PubMed] [CrossRef]
12. Xiong X, Zhang Y, Wen Y. Diverse functions of myeloid-derived suppressor cells in autoimmune diseases. Immunol Res. 2024;72(1):34–49. doi:10.1007/s12026-023-09421-0. [Google Scholar] [PubMed] [CrossRef]
13. Mirabello L, Troisi RJ, Savage SA. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int J Cancer. 2009;125(1):229–34. doi:10.1002/ijc.v125:1. [Google Scholar] [CrossRef]
14. Zhu T, Han J, Yang L, Cai Z, Sun W, Hua Y, et al. Immune microenvironment in osteosarcoma: components, therapeutic strategies and clinical applications. Front Immunol. 2022;13:907550. doi:10.3389/fimmu.2022.907550. [Google Scholar] [PubMed] [CrossRef]
15. Pilavaki P, Gahanbani Ardakani A, Gikas P, Constantinidou A. Osteosarcoma: current concepts and evolutions in management principles. J Clin Med. 2023;12(8):2785. doi:10.3390/jcm12082785. [Google Scholar] [PubMed] [CrossRef]
16. Ren Y, Backer H, Muller M, Kienzle A. The role of myeloid derived suppressor cells in musculoskeletal disorders. Front Immunol. 2023;14:1139683. doi:10.3389/fimmu.2023.1139683. [Google Scholar] [PubMed] [CrossRef]
17. Guan Y, Zhang R, Peng Z, Dong D, Wei G, Wang Y. Inhibition of IL-18-mediated myeloid derived suppressor cell accumulation enhances anti-PD1 efficacy against osteosarcoma cancer. J Bone Oncol. 2017;9:59–64. doi:10.1016/j.jbo.2017.10.002. [Google Scholar] [PubMed] [CrossRef]
18. Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol Res. 2016;4(10):869–80. doi:10.1158/2326-6066.CIR-15-0230. [Google Scholar] [PubMed] [CrossRef]
19. Stroncek DF, Ren J, Lee DW, Tran M, Frodigh SE, Sabatino M, et al. Myeloid cells in peripheral blood mononuclear cell concentrates inhibit the expansion of chimeric antigen receptor T cells. Cytotherapy. 2016;18(7):893–901. doi:10.1016/j.jcyt.2016.04.003. [Google Scholar] [PubMed] [CrossRef]
20. Tian H, Cao J, Li B, Nice EC, Mao H, Zhang Y, et al. Managing the immune microenvironment of osteosarcoma: the outlook for osteosarcoma treatment. Bone Res. 2023;11(1):11. doi:10.1038/s41413-023-00246-z. [Google Scholar] [PubMed] [CrossRef]
21. Wang H, Liu H, Wang L, Xu S, Pi H, Cheng Z. Subtype classification and prognosis signature construction of osteosarcoma based on cellular senescence-related genes. J Oncol. 2022;2022:4421952. doi:10.1155/2022/4421952. [Google Scholar] [PubMed] [CrossRef]
22. Jiang K, Li J, Zhang J, Wang L, Zhang Q, Ge J, et al. SDF-1/CXCR4 axis facilitates myeloid-derived suppressor cells accumulation in osteosarcoma microenvironment and blunts the response to anti-PD-1 therapy. Int Immunopharmacol. 2019;75(27):105818. doi:10.1016/j.intimp.2019.105818. [Google Scholar] [PubMed] [CrossRef]
23. Ling Z, Yang C, Tan J, Dou C, Chen Y. Beyond immunosuppressive effects: dual roles of myeloid-derived suppressor cells in bone-related diseases. Cell Mol Life Sci. 2021;78(23):7161–83. doi:10.1007/s00018-021-03966-9. [Google Scholar] [PubMed] [CrossRef]
24. De Vlaeminck Y, Gonzalez-Rascon A, Goyvaerts C, Breckpot K. Cancer-associated myeloid regulatory cells. Front Immunol. 2016;7(10):113. doi:10.3389/fimmu.2016.00113. [Google Scholar] [PubMed] [CrossRef]
25. Fan Q, Zuo J, Tian H, Huang C, Nice EC, Shi Z, et al. Nanoengineering a metal-organic framework for osteosarcoma chemo-immunotherapy by modulating indoleamine-2,3-dioxygenase and myeloid-derived suppressor cells. J Exp Clin Cancer Res. 2022;41(1):162. doi:10.1186/s13046-022-02372-8. [Google Scholar] [PubMed] [CrossRef]
26. Park JA, Cheung NV. Promise and challenges of T cell immunotherapy for osteosarcoma. Int J Mol Sci. 2023;24(15):12520. doi:10.3390/ijms241512520. [Google Scholar] [PubMed] [CrossRef]
27. Cheng S, Wang H, Kang X, Zhang H. Immunotherapy innovations in the fight against osteosarcoma: emerging strategies and promising progress. Pharmaceutics. 2024;16(2):251. doi:10.3390/pharmaceutics16020251. [Google Scholar] [PubMed] [CrossRef]
28. He ZN, Zhang CY, Zhao YW, He SL, Li Y, Shi BL, et al. Regulation of T cells by myeloid-derived suppressor cells: emerging immunosuppressor in lung cancer. Discov Oncol. 2023;14(1):185. doi:10.1007/s12672-023-00793-1. [Google Scholar] [PubMed] [CrossRef]
29. Castellano F, Molinier-Frenkel V. Control of T-cell activation and signaling by amino-acid catabolizing enzymes. Front Cell Dev Biol. 2020;8:613416. doi:10.3389/fcell.2020.613416. [Google Scholar] [PubMed] [CrossRef]
30. Ping Y, Shen C, Huang B, Zhang Y. Reprogramming T-cell metabolism for better anti-tumor immunity. Cells. 2022;11(19):3103. doi:10.3390/cells11193103. [Google Scholar] [PubMed] [CrossRef]
31. Lee GK, Park HJ, Macleod M, Chandler P, Munn DH, Mellor AL. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology. 2002;107(4):452–60. doi:10.1046/j.1365-2567.2002.01526.x. [Google Scholar] [PubMed] [CrossRef]
32. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182(9):5693–701. doi:10.4049/jimmunol.0900092. [Google Scholar] [PubMed] [CrossRef]
33. Chen S, Lei J, Mou H, Zhang W, Jin L, Lu S, et al. Multiple influence of immune cells in the bone metastatic cancer microenvironment on tumors. Front Immunol. 2024;15:1335366. doi:10.3389/fimmu.2024.1335366. [Google Scholar] [PubMed] [CrossRef]
34. Tumino N, Fiore PF, Pelosi A, Moretta L, Vacca P. Myeloid derived suppressor cells in tumor microenvironment: interaction with innate lymphoid cells. Semin Immunol. 2022;61–4:101668. doi:10.1016/j.smim.2022.101668. [Google Scholar] [PubMed] [CrossRef]
35. Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, et al. Monocytic CCR2+ myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012;72(4):876–86. doi:10.1158/0008-5472.CAN-11-1792. [Google Scholar] [PubMed] [CrossRef]
36. Macphail SE, Gibney CA, Brooks BM, Booth CG, Flanagan BF, Coleman JW. Nitric oxide regulation of human peripheral blood mononuclear cells: critical time dependence and selectivity for cytokine versus chemokine expression. J Immunol. 2003;171(9):4809–15. doi:10.4049/jimmunol.171.9.4809. [Google Scholar] [PubMed] [CrossRef]
37. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumors. Nat Rev Immunol. 2012;12(4):253–68. doi:10.1038/nri3175. [Google Scholar] [PubMed] [CrossRef]
38. Lu C, Redd PS, Lee JR, Savage N, Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology. 2016;5(12):e1247135. doi:10.1080/2162402X.2016.1247135. [Google Scholar] [PubMed] [CrossRef]
39. Jachetti E, Sangaletti S, Chiodoni C, Ferrara R, Colombo MP. Modulation of PD-1/PD-L1 axis in myeloid-derived suppressor cells by anti-cancer treatments. Cell Immunol. 2021;362(3):104301. doi:10.1016/j.cellimm.2021.104301. [Google Scholar] [PubMed] [CrossRef]
40. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. 2020;10(3):727–42. [Google Scholar] [PubMed]
41. Parvez A, Choudhary F, Mudgal P, Khan R, Qureshi KA, Farooqi H, et al. PD-1 and PD-L1: architects of immune symphony and immunotherapy breakthroughs in cancer treatment. Front Immunol. 2023;14:1296341. doi:10.3389/fimmu.2023.1296341. [Google Scholar] [PubMed] [CrossRef]
42. Vaknin I, Blinder L, Wang L, Gazit R, Shapira E, Genina O, et al. A common pathway mediated through Toll-like receptors leads to T- and natural killer-cell immunosuppression. Blood. 2008;111(3):1437–47. doi:10.1182/blood-2007-07-100404. [Google Scholar] [PubMed] [CrossRef]
43. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50(3):799–807. doi:10.1002/hep.23054. [Google Scholar] [PubMed] [CrossRef]
44. Nausch N, Galani IE, Schlecker E, Cerwenka A. Mononuclear myeloid-derived suppressor cells express RAE-1 and activate natural killer cells. Blood. 2008;112(10):4080–9. doi:10.1182/blood-2008-03-143776. [Google Scholar] [PubMed] [CrossRef]
45. Taylor AM, Sheng J, Shing Ng PK, Harder JM, Kumar P, Ahn JY, et al. Immunosuppressive tumor microenvironment of osteosarcoma. bioRxiv. 2024. doi:10.1101/2023.11.01.565008. [Google Scholar] [CrossRef]
46. Tan B, Yuan Z, Zhang Q, Xiqiang X, Dong J. The NF-κB pathway is critically implicated in the oncogenic phenotype of human osteosarcoma cells. J Appl Biomed. 2021;19(4):190–201. doi:10.32725/jab.2021.021. [Google Scholar] [PubMed] [CrossRef]
47. Nirala BK, Yamamichi T, Petrescu DI, Shafin TN, Yustein JT. Decoding the impact of tumor microenvironment in osteosarcoma progression and metastasis. Cancers. 2023;15(20):5108. doi:10.3390/cancers15205108. [Google Scholar] [PubMed] [CrossRef]
48. Cole K, Al-Kadhimi Z, Talmadge JE. Role of myeloid-derived suppressor cells in tumor recurrence. Cancer Metastasis Rev. 2023;42(1):113–42. doi:10.1007/s10555-023-10079-1. [Google Scholar] [PubMed] [CrossRef]
49. An J, Feng L, Ren J, Li Y, Li G, Liu C, et al. Chronic stress promotes breast carcinoma metastasis by accumulating myeloid-derived suppressor cells through activating beta-adrenergic signaling. Oncoimmunology. 2021;10(1):2004659. doi:10.1080/2162402X.2021.2004659. [Google Scholar] [PubMed] [CrossRef]
50. Sawant A, Ponnazhagan S. Myeloid-derived suppressor cells as osteoclast progenitors: a novel target for controlling osteolytic bone metastasis. Cancer Res. 2013;73(15):4606–10. doi:10.1158/0008-5472.CAN-13-0305. [Google Scholar] [PubMed] [CrossRef]
51. Iwata S, Ishii T, Kawai A, Hiruma T, Yonemoto T, Kamoda H, et al. Prognostic factors in elderly osteosarcoma patients: a multi-institutional retrospective study of 86 cases. Ann Surg Oncol. 2014;21(1):263–8. doi:10.1245/s10434-013-3210-4. [Google Scholar] [PubMed] [CrossRef]
52. Bertrand TE, Cruz A, Binitie O, Cheong D, Letson DG. Do surgical margins affect local recurrence and survival in extremity, nonmetastatic, high-grade osteosarcoma? Clin Orthop Relat Res. 2016;474(3):684–6. doi:10.1007/s11999-015-4416-5. [Google Scholar] [PubMed] [CrossRef]
53. Anninga JK, Gelderblom H, Fiocco M, Kroep JR, Taminiau AH, Hogendoorn PC, et al. Chemotherapeutic adjuvant treatment for osteosarcoma: where do we stand? Eur J Cancer. 2011;47(16):2431–45. doi:10.1016/j.ejca.2011.05.030. [Google Scholar] [PubMed] [CrossRef]
54. Corrales L, Matson V, Flood B, Spranger S, Gajewski TF. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017;27(1):96–108. doi:10.1038/cr.2016.149. [Google Scholar] [PubMed] [CrossRef]
55. Demaria O, Cornen S, Daeron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature. 2019;574(7776):45–56. doi:10.1038/s41586-019-1593-5. [Google Scholar] [PubMed] [CrossRef]
56. Joseph CG, Hwang H, Jiao Y, Wood LD, Kinde I, Wu J, et al. Exomic analysis of myxoid liposarcomas, synovial sarcomas, and osteosarcomas. Genes Chromosomes Cancer. 2014;53(1):15–24. doi:10.1002/gcc.22114. [Google Scholar] [PubMed] [CrossRef]
57. Perry JA, Kiezun A, Tonzi P, Van Allen EM, Carter SL, Baca SC, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A. 2014;111(51):E5564–73. doi:10.1073/pnas.1419260111. [Google Scholar] [PubMed] [CrossRef]
58. Behjati S, Tarpey PS, Haase K, Ye H, Young MD, Alexandrov LB, et al. Recurrent mutation of IGF signaling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat Commun. 2017;8(1):15936. doi:10.1038/ncomms15936. [Google Scholar] [PubMed] [CrossRef]
59. Matsuoka K, Bakiri L, Wolff LI, Linder M, Mikels-Vigdal A, Patino-Garcia A, et al. Wnt signaling and Loxl2 promote aggressive osteosarcoma. Cell Res. 2020;30(10):885–901. doi:10.1038/s41422-020-0370-1. [Google Scholar] [PubMed] [CrossRef]
60. Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, et al. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol. 2004;172(1):464–74. doi:10.4049/jimmunol.172.1.464. [Google Scholar] [PubMed] [CrossRef]
61. Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69(6):2506–13. doi:10.1158/0008-5472.CAN-08-4323. [Google Scholar] [PubMed] [CrossRef]
62. Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity. 2016;44(2):303–15. doi:10.1016/j.immuni.2016.01.014. [Google Scholar] [PubMed] [CrossRef]
63. Nefedova Y, Nagaraj S, Rosenbauer A, Muro-Cacho C, Sebti SM, Gabrilovich DI. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 2005;65(20):9525–35. doi:10.1158/0008-5472.CAN-05-0529. [Google Scholar] [PubMed] [CrossRef]
64. Zuo D, Shogren KL, Zang J, Jewison DE, Waletzki BE, Miller AL, et al. Inhibition of STAT3 blocks protein synthesis and tumor metastasis in osteosarcoma cells. J Exp Clin Cancer Res. 2018;37(1):244. doi:10.1186/s13046-018-0914-0. [Google Scholar] [PubMed] [CrossRef]
65. Li S, Zhang H, Liu J, Shang G. Targeted therapy for osteosarcoma: a review. J Cancer Res Clin Oncol. 2023;149(9):6785–97. doi:10.1007/s00432-023-04614-4. [Google Scholar] [PubMed] [CrossRef]
66. Akagi T, Saitoh T, O’Kelly J, Akira S, Gombart AF, Koeffler HP. Impaired response to GM-CSF and G-CSF, and enhanced apoptosis in C/EBPβ-deficient hematopoietic cells. Blood. 2008;111(6):2999–3004. doi:10.1182/blood-2007-04-087213. [Google Scholar] [PubMed] [CrossRef]
67. Gao Y, Sun W, Shang W, Li Y, Zhang D, Wang T, et al. Lnc-C/EBPβ negatively regulates the suppressive function of myeloid-derived suppressor cells. Cancer Immunol Res. 2018;6(11):1352–63. doi:10.1158/2326-6066.CIR-18-0108. [Google Scholar] [PubMed] [CrossRef]
68. Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab. 2018;28(1):87–103. doi:10.1016/j.cmet.2018.04.022. [Google Scholar] [PubMed] [CrossRef]
69. Xu G, Ma Z, Yang F, Bai Y, Li J, Luo W, et al. TRIM59 promotes osteosarcoma progression via activation of STAT3. Hum Cell. 2022;35(1):250–9. doi:10.1007/s13577-021-00615-y. [Google Scholar] [PubMed] [CrossRef]
70. Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205(10):2235–49. doi:10.1084/jem.20080132. [Google Scholar] [PubMed] [CrossRef]
71. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181(7):4666–75. doi:10.4049/jimmunol.181.7.4666. [Google Scholar] [PubMed] [CrossRef]
72. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203(12):2691–702. doi:10.1084/jem.20061104. [Google Scholar] [PubMed] [CrossRef]
73. Meyer C, Sevko A, Ramacher M, Bazhin AV, Falk CS, Osen W, et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc Natl Acad Sci U S A. 2011;108(41):17111–6. doi:10.1073/pnas.1108121108. [Google Scholar] [PubMed] [CrossRef]
74. Lim HX, Kim TS, Poh CL. Understanding the differentiation, expansion, recruitment and suppressive activities of myeloid-derived suppressor cells in cancers. Int J Mol Sci. 2020;21(10):3599. doi:10.3390/ijms21103599. [Google Scholar] [PubMed] [CrossRef]
75. Tomaselli D, Lucidi A, Rotili D, Mai A. Epigenetic polypharmacology: a new frontier for epi-drug discovery. Med Res Rev. 2020;40(1):190–244. doi:10.1002/med.v40.1. [Google Scholar] [CrossRef]
76. Murata T, Shimizu K, Kurohara K, Tomeoku A, Koizumi G, Arai N. Role of phosphodiesterase2A in proliferation and migration of human osteosarcoma cells. Anticancer Res. 2019;39(11):6057–62. doi:10.21873/anticanres.13812. [Google Scholar] [PubMed] [CrossRef]
77. Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. 2010;40(1):22–35. doi:10.1002/eji.200939903. [Google Scholar] [PubMed] [CrossRef]
78. Casbon AJ, Reynaud D, Park C, Khuc E, Gan DD, Schepers K, et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci U S A. 2015;112(6):E566–75. doi:10.1073/pnas.1424927112. [Google Scholar] [PubMed] [CrossRef]
79. Melani C, Chiodoni C, Forni G, Colombo MP. Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood. 2003;102(6):2138–45. doi:10.1182/blood-2003-01-0190. [Google Scholar] [PubMed] [CrossRef]
80. Almand B, Resser JR, Lindman B, Nadaf S, Clark JI, Kwon ED, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6(5):1755–66. [Google Scholar] [PubMed]
81. Rutkowski P, Kaminska J, Kowalska M, Ruka W, Steffen J. Cytokine and cytokine receptor serum levels in adult bone sarcoma patients: correlations with local tumor extent and prognosis. J Surg Oncol. 2003;84(3):151–9. doi:10.1002/jso.10305. [Google Scholar] [PubMed] [CrossRef]
82. Guihard P, Danger Y, Brounais B, David E, Brion R, Delecrin J, et al. Induction of osteogenesis in mesenchymal stem cells by activated monocytes/macrophages depends on oncostatin M signaling. Stem Cells. 2012;30(4):762–72. doi:10.1002/stem.1040. [Google Scholar] [PubMed] [CrossRef]
83. Chimal-Ramirez GK, Espinoza-Sanchez NA, Fuentes-Panana EM. Protumor activities of the immune response: insights in the mechanisms of immunological shift, oncotraining, and oncopromotion. J Oncol. 2013;2013:835956. doi:10.1155/2013/835956. [Google Scholar] [PubMed] [CrossRef]
84. Wu MY, Li CJ, Yiang GT, Cheng YL, Tsai AP, Hou YT, et al. Molecular regulation of bone metastasis pathogenesis. Cell Physiol Biochem. 2018;46(4):1423–38. doi:10.1159/000489184. [Google Scholar] [PubMed] [CrossRef]
85. Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, et al. Inhibition of MDSC trafficking with SX-682, a CXCR1/2 inhibitor, enhances NK-cell immunotherapy in head and neck cancer models. Clin Cancer Res. 2020;26(6):1420–31. doi:10.1158/1078-0432.CCR-19-2625. [Google Scholar] [PubMed] [CrossRef]
86. Tan Z, Chiu MS, Yue M, Kwok HY, Tse MH, Wen Y, et al. Enhancing the efficacy of vaccinia-based oncolytic virotherapy by inhibiting CXCR2-mediated MDSC trafficking. J Leukoc Biol. 2024;115(4):633–46. doi:10.1093/jleuko/qiad150. [Google Scholar] [PubMed] [CrossRef]
87. Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight. 2019;4(7):e126853. doi:10.1172/jci.insight.126853. [Google Scholar] [PubMed] [CrossRef]
88. Pevida M, Gonzalez-Rodriguez S, Lastra A, Garcia-Suarez O, Hidalgo A, Menendez L, et al. Involvement of spinal chemokine CCL2 in the hyperalgesia evoked by bone cancer in mice: a role for astroglia and microglia. Cell Mol Neurobiol. 2014;34(1):143–56. doi:10.1007/s10571-013-9995-7. [Google Scholar] [PubMed] [CrossRef]
89. Xu M, Wang Y, Xia R, Wei Y, Wei X. Role of the CCL2-CCR2 signaling axis in cancer: mechanisms and therapeutic targeting. Cell Prolif. 2021;54(10):e13115. doi:10.1111/cpr.v54.10. [Google Scholar] [CrossRef]
90. Tatsuno R, Komohara Y, Pan C, Kawasaki T, Enomoto A, Jubashi T, et al. Surface markers and chemokines/cytokines of tumor-associated macrophages in osteosarcoma and other carcinoma microenviornments-contradictions and comparisons. Cancers. 2024;16(16):2801. doi:10.3390/cancers16162801. [Google Scholar] [PubMed] [CrossRef]
91. D’Oronzo S, Coleman R, Brown J, Silvestris F. Metastatic bone disease: pathogenesis and therapeutic options: up-date on bone metastasis management. J Bone Oncol. 2019;15:4. [Google Scholar]
92. Nie SC, Jing YH, Lu L, Ren SS, Ji G, Xu HC. Mechanisms of myeloid-derived suppressor cell-mediated immunosuppression in colorectal cancer and related therapies. World J Gastrointest Oncol. 2024;16(5):1690–704. doi:10.4251/wjgo.v16.i5.1690. [Google Scholar] [PubMed] [CrossRef]
93. Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells. 2020;9(3):561. doi:10.3390/cells9030561. [Google Scholar] [PubMed] [CrossRef]
94. Zhao Y, Du J, Shen X. Targeting myeloid-derived suppressor cells in tumor immunotherapy: current, future and beyond. Front Immunol. 2023;14:1157537. doi:10.3389/fimmu.2023.1157537. [Google Scholar] [PubMed] [CrossRef]
95. Tobin RP, Jordan KR, Robinson WA, Davis D, Borges VF, Gonzalez R, et al. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int Immunopharmacol. 2018;63:282–91. doi:10.1016/j.intimp.2018.08.007. [Google Scholar] [PubMed] [CrossRef]
96. Bauer R, Udonta F, Wroblewski M, Ben-Batalla I, Santos IM, Taverna F, et al. Blockade of myeloid-derived suppressor cell expansion with all-trans retinoic acid increases the efficacy of antiangiogenic therapy. Cancer Res. 2018;78(12):3220–32. doi:10.1158/0008-5472.CAN-17-3415. [Google Scholar] [PubMed] [CrossRef]
97. Liu Y, Wei G, Cheng WA, Dong Z, Sun H, Lee VY, et al. Targeting myeloid-derived suppressor cells for cancer immunotherapy. Cancer Immunol Immunother. 2018;67(8):1181–95. doi:10.1007/s00262-018-2175-3. [Google Scholar] [PubMed] [CrossRef]
98. Tobin RP, Cogswell DT, Cates VM, Davis DM, Borgers JSW, Van Gulick RJ, et al. Targeting MDSC differentiation using ATRA: a Phase I/II clinical trial combining pembrolizumab and all-trans retinoic acid for metastatic melanoma. Clin Cancer Res. 2023;29(7):1209–19. doi:10.1158/1078-0432.CCR-22-2495. [Google Scholar] [PubMed] [CrossRef]
99. Rytlewski J, Milhem MM, Monga V. Turning ‘Cold’ tumors ‘Hot’: immunotherapies in sarcoma. Ann Transl Med. 2021;9(12):1039. doi:10.21037/atm. [Google Scholar] [CrossRef]
100. Sinha P, Clements VK, Ostrand-Rosenberg S. Interleukin-13-regulated M2 macrophages in combination with myeloid suppressor cells block immune surveillance against metastasis. Cancer Res. 2005;65(24):11743–51. doi:10.1158/0008-5472.CAN-05-0045. [Google Scholar] [PubMed] [CrossRef]
101. Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65(8):3044–8. doi:10.1158/0008-5472.CAN-04-4505. [Google Scholar] [PubMed] [CrossRef]
102. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. 2007;109(4):1568–73. doi:10.1182/blood-2006-06-031856. [Google Scholar] [PubMed] [CrossRef]
103. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5(8):641–54. doi:10.1038/nri1668. [Google Scholar] [PubMed] [CrossRef]
104. Huang B, Lei Z, Zhao J, Gong W, Liu J, Chen Z, et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 2007;252(1):86–92. doi:10.1016/j.canlet.2006.12.012. [Google Scholar] [PubMed] [CrossRef]
105. Gehad AE, Lichtman MK, Schmults CD, Teague JE, Calarese AW, Jiang Y, et al. Nitric oxide-producing myeloid-derived suppressor cells inhibit vascular E-selectin expression in human squamous cell carcinomas. J Invest Dermatol. 2012;132(11):2642–51. doi:10.1038/jid.2012.190. [Google Scholar] [PubMed] [CrossRef]
106. Vannini F, Kashfi K, Nath N. The dual role of iNOS in cancer. Redox Biol. 2015;6:334–43. doi:10.1016/j.redox.2015.08.009. [Google Scholar] [PubMed] [CrossRef]
107. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019;569(7754):73–8. doi:10.1038/s41586-019-1118-2. [Google Scholar] [PubMed] [CrossRef]
108. Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118(12):3930–42. doi:10.1172/JCI36843. [Google Scholar] [PubMed] [CrossRef]
109. Payen VL, Mina E, Van Hee VF, Porporato PE, Sonveaux P. Monocarboxylate transporters in cancer. Mol Metab. 2020;33:48–66. doi:10.1016/j.molmet.2019.07.006. [Google Scholar] [PubMed] [CrossRef]
110. Li J, Bolyard C, Xin G, Li Z. Targeting metabolic pathways of myeloid cells improves cancer immunotherapy. Front Cell Dev Biol. 2021;9:747863. doi:10.3389/fcell.2021.747863. [Google Scholar] [PubMed] [CrossRef]
111. Zhang Z, Zheng Y, Chen Y, Yin Y, Chen Y, Chen Q, et al. Gut fungi enhances immunosuppressive function of myeloid-derived suppressor cells by activating PKM2-dependent glycolysis to promote colorectal tumorigenesis. Exp Hematol Oncol. 2022;11(1):88. doi:10.1186/s40164-022-00334-6. [Google Scholar] [PubMed] [CrossRef]
112. Kim J, Choi JY, Min H, Hwang KW. Exploring the potential of glycolytic modulation in myeloid-derived suppressor cells for immunotherapy and disease management. Immune Netw. 2024;24(3):e26. doi:10.4110/in.2024.24.e26. [Google Scholar] [PubMed] [CrossRef]
113. Miao G, Han J, Zhang J, Wu Y, Tong G. Targeting pyruvate kinase M2 and hexokinase II, pachymic acid impairs glucose metabolism and induces mitochondrial apoptosis. Biol Pharm Bull. 2019;42(1):123–9. doi:10.1248/bpb.b18-00730. [Google Scholar] [PubMed] [CrossRef]
114. Huang Y, Ouyang F, Yang F, Zhang N, Zhao W, Xu H, et al. The expression of hexokinase 2 and its hub genes are correlated with the prognosis in glioma. BMC Cancer. 2022;22(1):900. doi:10.1186/s12885-022-10001-y. [Google Scholar] [PubMed] [CrossRef]
115. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016;24(5):657–71. doi:10.1016/j.cmet.2016.08.011. [Google Scholar] [PubMed] [CrossRef]
116. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. 2013;191(3):1486–95. doi:10.4049/jimmunol.1202702. [Google Scholar] [PubMed] [CrossRef]
117. Paunovic V, Kosic M, Misirkic-Marjanovic M, Trajkovic V, Harhaji-Trajkovic L. Dual targeting of tumor cell energy metabolism and lysosomes as an anticancer strategy. Biochim Biophys Acta Mol Cell Res. 2021;1868(4):118944. doi:10.1016/j.bbamcr.2020.118944. [Google Scholar] [PubMed] [CrossRef]
118. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. 2015;3(11):1236–47. doi:10.1158/2326-6066.CIR-15-0036. [Google Scholar] [PubMed] [CrossRef]
119. Wang Y, Jia A, Bi Y, Wang Y, Liu G. Metabolic regulation of myeloid-derived suppressor cell function in cancer. Cells. 2020;9(4):1011. doi:10.3390/cells9041011. [Google Scholar] [PubMed] [CrossRef]
120. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. 2016;1(2):aaf8943. doi:10.1126/sciimmunol.aaf8943. [Google Scholar] [PubMed] [CrossRef]
121. Ding X, Zhang Y, Liang J, Li Q, Hu H, Zhou Y, et al. Dihydroartemisinin potentiates VEGFR-TKIs antitumorigenic effect on osteosarcoma by regulating Loxl2/VEGFA expression and lipid metabolism pathway. J Cancer. 2023;14(5):809–20. doi:10.7150/jca.81623. [Google Scholar] [PubMed] [CrossRef]
122. Lin Z, Wu Z, Luo W. Bulk and single-cell sequencing identified a prognostic model based on the macrophage and lipid metabolism related signatures for osteosarcoma patients. Heliyon. 2024;10(4):e26091. doi:10.1016/j.heliyon.2024.e26091. [Google Scholar] [PubMed] [CrossRef]
123. Shi Y, Wu S, Zhang X, Cao Y, Zhang L. Lipid metabolism-derived FAAH is a sensitive marker for the prognosis and immunotherapy of osteosarcoma patients. Heliyon. 2024;10(1):e23499. doi:10.1016/j.heliyon.2023.e23499. [Google Scholar] [PubMed] [CrossRef]
124. Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol. 2020;38(1):1–10. doi:10.1200/JCO.19.02105. [Google Scholar] [PubMed] [CrossRef]
125. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–8. doi:10.1126/science.aaa4967. [Google Scholar] [PubMed] [CrossRef]
126. Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother. 2013;62(5):909–18. doi:10.1007/s00262-013-1396-8. [Google Scholar] [PubMed] [CrossRef]
127. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70(8):3052–61. doi:10.1158/0008-5472.CAN-09-3690. [Google Scholar] [PubMed] [CrossRef]
128. Naiditch H, Shurin MR, Shurin GV. Targeting myeloid regulatory cells in cancer by chemotherapeutic agents. Immunol Res. 2011;50(2–3):276–85. [Google Scholar] [PubMed]
129. Grover A, Sanseviero E, Timosenko E, Gabrilovich DI. Myeloid-derived suppressor cells: a propitious road to clinic. Cancer Discov. 2021;11(11):2693–706. doi:10.1158/2159-8290.CD-21-0764. [Google Scholar] [PubMed] [CrossRef]
130. Li JY, Chen YP, Li YQ, Liu N, Ma J. Chemotherapeutic and targeted agents can modulate the tumor microenvironment and increase the efficacy of immune checkpoint blockades. Mol Cancer. 2021;20(1):27. doi:10.1186/s12943-021-01317-7. [Google Scholar] [PubMed] [CrossRef]
131. Zhu S, Zhang T, Zheng L, Liu H, Song W, Liu D, et al. Combination strategies to maximize the benefits of cancer immunotherapy. J Hematol Oncol. 2021;14(1):156. doi:10.1186/s13045-021-01164-5. [Google Scholar] [PubMed] [CrossRef]
132. Lu Z, Zou J, Li S, Topper MJ, Tao Y, Zhang H, et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature. 2020;579(7798):284–90. doi:10.1038/s41586-020-2054-x. [Google Scholar] [PubMed] [CrossRef]
133. Palmerini E, Agostinelli C, Picci P, Pileri S, Marafioti T, Lollini PL, et al. Tumoral immune-infiltrate (IFPD-L1 expression and role of CD8/TIA-1 lymphocytes in localized osteosarcoma patients treated within protocol ISG-OS1. Oncotarget. 2017;8(67):111836–46. doi:10.18632/oncotarget.22912. [Google Scholar] [PubMed] [CrossRef]
134. Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer. 2010;10(1):464. doi:10.1186/1471-2407-10-464. [Google Scholar] [PubMed] [CrossRef]
135. Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–57. doi:10.1158/1078-0432.CCR-08-1332. [Google Scholar] [PubMed] [CrossRef]
136. Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol. 2009;9(7–8):900–9. doi:10.1016/j.intimp.2009.03.015. [Google Scholar] [PubMed] [CrossRef]
137. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539(7629):437–42. doi:10.1038/nature19834. [Google Scholar] [PubMed] [CrossRef]
138. He X, Lin H, Yuan L, Li B. Combination therapy with L-arginine and α-PD-L1 antibody boosts immune response against osteosarcoma in immunocompetent mice. Cancer Biol Ther. 2017;18(2):94–100. doi:10.1080/15384047.2016.1276136. [Google Scholar] [PubMed] [CrossRef]
139. Fujiwara T, Yakoub MA, Chandler A, Christ AB, Yang G, Ouerfelli O, et al. CSF1/CSF1R signaling inhibitor pexidartinib (PLX3397) reprograms tumor-associated macrophages and stimulates T-cell infiltration in the sarcoma microenvironment. Mol Cancer Ther. 2021;20(8):1388–99. doi:10.1158/1535-7163.MCT-20-0591. [Google Scholar] [PubMed] [CrossRef]
140. Rose P, van den Engel NK, Kovacs JR, Hatz RA, Boon L, Winter H. Anti-Gr-1 antibody provides short-term depletion of MDSC in lymphodepleted mice with active-specific melanoma therapy. Vaccines. 2022;10(4):560. doi:10.3390/vaccines10040560. [Google Scholar] [PubMed] [CrossRef]
141. Deyhle MR, Callaway CS, Neyroud D, D’Lugos AC, Judge SM, Judge AR. Depleting Ly6G positive myeloid cells reduces pancreatic cancer-induced skeletal muscle atrophy. Cells. 2022;11(12):1893. doi:10.3390/cells11121893. [Google Scholar] [PubMed] [CrossRef]
142. Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, et al. MDSC targeting with Gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. eBioMedicine. 2019;47(10):235–46. doi:10.1016/j.ebiom.2019.08.025. [Google Scholar] [PubMed] [CrossRef]
143. Qiao J, Liu Z, Dong C, Luan Y, Zhang A, Moore C, et al. Targeting tumors with IL-10 prevents dendritic cell-mediated CD8+ T cell apoptosis. Cancer Cell. 2019;35(6):901–15. doi:10.1016/j.ccell.2019.05.005. [Google Scholar] [PubMed] [CrossRef]
144. Li J, Srivastava RM, Ettyreddy A, Ferris RL. Cetuximab ameliorates suppressive phenotypes of myeloid antigen presenting cells in head and neck cancer patients. J Immunother Cancer. 2015;3(1):54. doi:10.1186/s40425-015-0097-6. [Google Scholar] [PubMed] [CrossRef]
145. Koinis F, Vetsika EK, Aggouraki D, Skalidaki E, Koutoulaki A, Gkioulmpasani M, et al. Effect of first-line treatment on myeloid-derived suppressor cells’ subpopulations in the peripheral blood of patients with non-small cell lung cancer. J Thorac Oncol. 2016;11(8):1263–72. doi:10.1016/j.jtho.2016.04.026. [Google Scholar] [PubMed] [CrossRef]
Appendix A
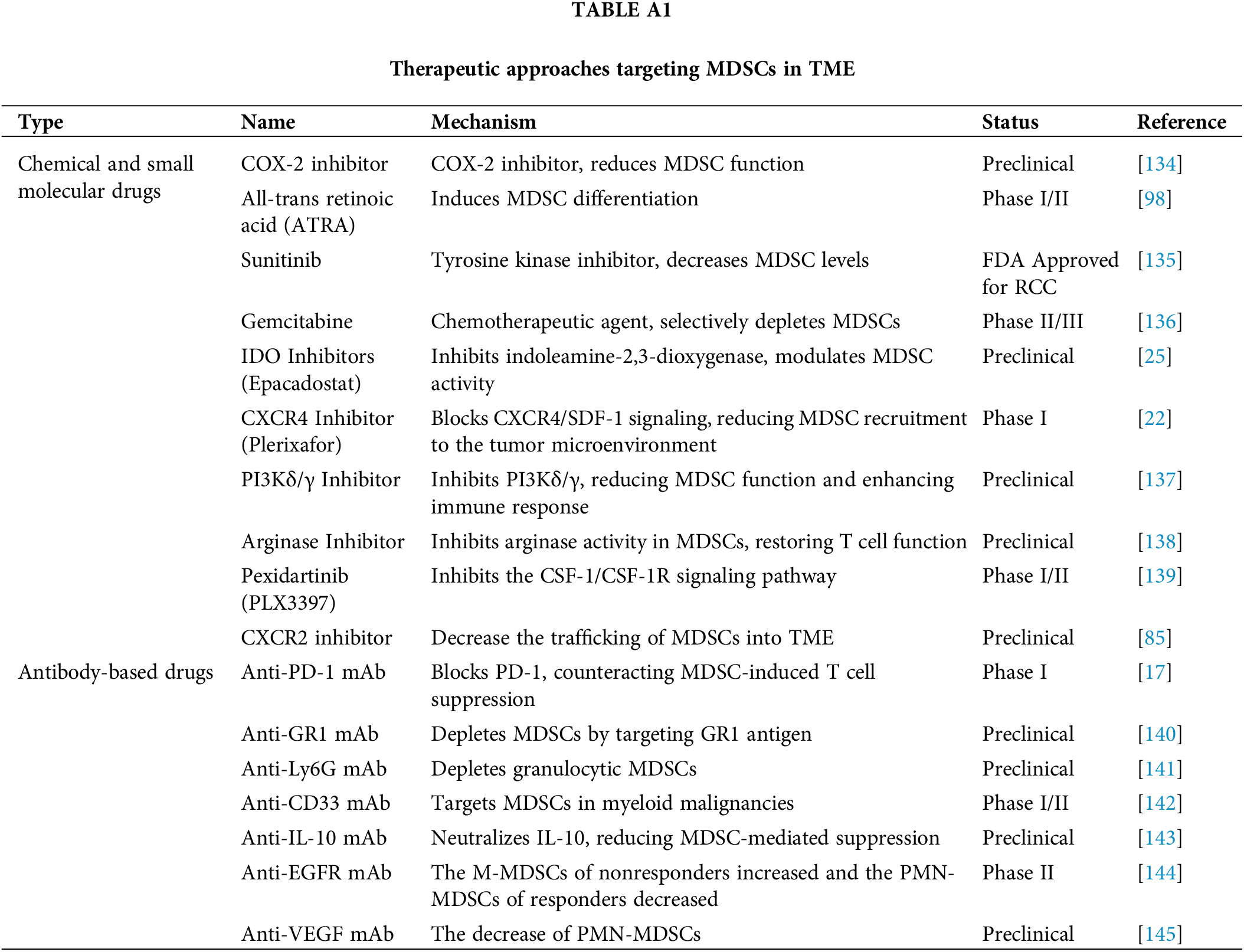
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools