
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
Apatinib modulates sorafenib-resistant hepatocellular carcinoma through inhibiting the EGFR/JNK/ERK signaling pathway
1 Department of Hepatobiliary Surgery, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, 250021, China
2 Liver Gall Bladder and Pancreatic Surgery Ward, Qinghai Red Cross Hospital, Xining, 810001, China
3 Department of Hepatobiliary Surgery, Shandong Provincial Hospital, Shandong University, Jinan, 250021, China
4 Departments of Infectious Diseases, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, 250021, China
5 Department of Pediatric Surgery, Shandong Provincial Hospital, Shandong University, Jinan, 250021, China
* Corresponding Authors: ZHENHAI ZHANG. Email: ; JUNLIN GAO. Email:
# These two authors contributed equally to this work
(This article belongs to the Special Issue: Signaling Pathway Crosstalk in Malignant Tumors: Molecular Targets and Combinatorial Therapeutics)
Oncology Research 2025, 33(6), 1459-1472. https://doi.org/10.32604/or.2025.060407
Received 31 October 2024; Accepted 03 January 2025; Issue published 29 May 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Objectives: Apatinib has been reported to be a promising treatment for sorafenib-resistant hepatocellular carcinoma (HCC) patients. However, the underlying mechanism remains ambiguous. The study aimed to explore the efficacy of apatinib in sorafenib-resistant HCC and the underlying mechanism both in vitro and in vivo. Methods: After observing epithelial-mesenchymal transformation (EMT) changes in HepG2 and HepG2/Sorafenib cells, we treated them with varying concentrations of apatinib to assess its impact on sorafenib-resistant HCC. Subsequently, specific inhibitors of c-Jun N-terminal kinase (JNK, SP600125) and extracellular signal-regulated kinase (ERK, PD98059) were introduced to investigate whether apatinib influenced sorafenib-resistant HCC via modulation of the epidermal growth factor receptor (EGFR)/JNK/ERK signaling pathway in vitro and in vivo. Biological behavior changes were assessed through cell counting kit-8 (CCK-8), colony formation, transwell, and immunofluorescence tests. Simultaneously, Western blot analysis was conducted to elucidate the expression of proteins associated with EMT and the EGFR/JNK/ERK signaling pathway. Results: The HepG2/Sorafenib cells exhibited greater resistance to sorafenib compared to HepG2 cells, and sorafenib-resistant HCC was characterized by EMT changes. Apatinib demonstrated concentration-dependent inhibition of biological behaviors in HepG2/Sorafenib cells, with minimal impact on HepG2 cells. Additionally, apatinib had a pronounced effect on the expression of EMT-related proteins in sorafenib-resistant cells similar to that in sorafenib-sensitive cells. Furthermore, there was a dose-dependent reduction in the expression of proteins associated with the EGFR/JNK/ERK pathway in apatinib-treated groups. Notably, SP600125 and PD98059 contributed to the inhibition of EMT and EGFR/JNK/ERK pathway-related proteins by apatinib in sorafenib-resistant HCC. Conclusion: Apatinib potentially hindered the progression of sorafenib-resistant HCC by suppressing both EMT and the EGFR/JNK/ERK pathway.Keywords
Liver cancer, ranked as the fourth most prevalent malignancy, poses a significant threat due to its high mortality rates [1]. Notably, the liver lacks pain receptors (nerve endings), allowing changes within the organ, including the growth of small tumors, to occur without inducing direct pain sensations [2]. The liver’s substantial size and functional reserves enable it to maintain normal function despite partial occupancy by a tumor, leading to inconspicuous symptoms during the early stages [2]. Typically, hepatocellular carcinoma (HCC) remains asymptomatic and is often discovered incidentally during routine check-ups or liver disease monitoring, marking the subclinical phase [3]. However, the onset of HCC symptoms, such as liver pain, fatigue, weight loss, jaundice, and ascites, usually indicates the disease in its middle to late stages [4]. While surgery and radiotherapy are common clinical interventions, the cure rates for advanced stages remain low due to widespread cancer cell dissemination [5]. Recently, multiple studies have indicated that the Royal Marsden Hospital (RMH) score could serve as a readily available prognostic biomarker for cancer patients [6], however, its wide-scale implementation in clinical practice is still insufficient, and its true benefit in clinical decision-making has not been fully realized. Thus, the importance of early detection, diagnosis, and intervention for HCC cannot be overstated.
Sorafenib is a novel multi-targeted oral agent used in the treatment of inoperable or distantly metastatic HCC [7]. Since 2007, sorafenib has been approved to be the first-line therapy for advanced HCC; however, approximately just 30% of HCC patients benefit from treatment with sorafenib and these patients are inclined to develop resistance within 6 months [8]. The mechanisms underlying sorafenib resistance encompass alterations in cell signaling pathways, changes in drug targets, modifications in drug pumps and metabolism, variations in the tumor microenvironment, and evasion of drug feedback inhibition [8–10]. HCC stem cells inherently exhibit resistance to Sorafenib, impeding HCC neovascularization while intensifying local hypoxia and activating hypoxia signaling pathways [11,12]. This fosters epithelial-mesenchymal transition (EMT), and pseudoangiogenesis, ultimately promoting invasion and metastasis [13]. Additionally, multiple studies have reported the key roles of EGFR/JNK/ERK signaling in sorafenib resistance of HCC. For example, EGFR was highly expressed in sorafenib-resistant HCC cells [14]. An ERK inhibitor could enhance the sorafenib efficiency of HCC [15].
The development of resistance in HCC against sorafenib results in suboptimal clinical outcomes, necessitating the exploration of combination therapies involving other targeted drugs to enhance treatment efficacy. Apatinib, an inhibitor of VEGFR-2, has been reported to obviously improve overall survival in patients with advance HCC in a multicentre, double-blind, randomized, and placebo-controlled phase 3 trial [16]. Moreover, a phase II clinical trial showed that apatinib could be an encouraging first-line treatment in patients with advanced HCC [17]. Importantly, Zhang et al. have demonstrated that advanced HCC patients with sorafenib resistance achieved a longer overall survival after apatinib treatment [18]. These results indicated that apatinib could be a promising treatment for HCC patients with sorafenib resistance. However, the underlying mechanism has not been clarified. Therefore, in this study, the effectiveness of apatinib on sorafenib-resistant HCC and the relevant mechanism were investigated in vitro and in vivo.
HepG2 cells were procured from ATCC (Manassas, VA, USA) and maintained in an incubator with a culture medium consisting of 90% DMEM (PM150250, Procell, Wuhan, China), 10% FBS (10099141, GIBCO, Shanghai, China), and 1% penicillin/streptomycin (MA0110, Meilune, Dalian, China). No mycoplasma contamination was detected in the cell line.
Establishment of sorafenib-resistant HepG2 cells
Sorafenib-resistant HepG2 cells were established according to a previous study [19]. The establishment process involved a gradual escalation of sorafenib exposure, starting at a concentration of 1 μM and increasing by increments of 1 μM every fortnight until the cells could tolerate the highest dose of 8 μM. Once the cells reached this tolerance level, they were maintained in a culture medium containing 8 μM sorafenib (HY-10201, MCE, Shanghai, China).
The HepG2 and HepG2/Sorafenib cells were treated with 0.001, 0.01, 0.1, 1 and 10 µM sorafenib for 24 h respectively to investigate the drug resistance. To assess the impacts of apatinib on sorafenib-sensitive and resistant HepG2 cells, the HepG2 and HepG2/Sorafenib cells underwent treatment with varying concentrations (0, 0.5, 1, 2, and 4 μM) of apatinib (S2221, Selleck, Shanghai, China) for 24 h. Subsequently, specific inhibitors of JNK (SP600125, Beyotime, Shanghai, China) and ERK (PD98059, Beyotime, China) were introduced to investigate the potential influence of apatinib on drug resistance in hepatoma cells through modulation of the EGFR/JNK/ERK signaling pathway. The cells were treated with 20 µM SP600125 or 10 µM PD98059 for 24 h.
The resistance of HCC to sorafenib and cell viability was assessed using the CCK-8 assay. Briefly, 5 × 104 cells were seeded in a 96-well plate. The cells in different groups were treated with 0, 0.5, 1, 2, and 4 μmol/L of apatinib for 24 h, with the drug volume not exceeding one-tenth of the cell suspension volume. There were three replicate wells in each group. 10 μL of CCK-8 solution (G4103, Servicebio, Wuhan, China) to each well, and the cells were incubated for an additional 4 h. Finally, the optical density (OD) values at 450 nm were determined using a plate reader (multiskan sky, Thermo Fisher, Bothell, WA, USA).
The HepG2 and HepG2/Sorafenib cells were fixed with 4% paraformaldehyde (G1101, Servicebio, Wuhan, China) and 0.1% TritonX-100 (T8200, Solarbio, Beijing, China), followed by blocking with bovine serum albumin (G5001, Servicebio) for 15 min at room temperature. Subsequently, they were incubated overnight at 4°C with the anti-β-microtubulin antibody (1:400, 10068-1-AP, PTG, Rosemont, IL, USA). Afterward, visualization of microtubules in the HepG2 and HepG2/Sorafenib groups was achieved by incubating with the secondary antibody Multi-rAb CoraLite®® Plus 594-Goat Anti-Rabbit Recombinant Secondary Antibody (H + L) (1:500, RGAR004, PTG, USA) and observing under a fluorescence microscope (Eclipse C1, Nikon, Tokyo, Japan).
After apatinib treatment, a colony formation assay was performed to investigate the effect of apatinib on the proliferation of HepG2 and HepG2/Sorafenib cells. Briefly, 1 × 103 cells were seeded in a 6-well plate and cultured for about two weeks until visible clones were observed by the naked eye. Subsequently, these clones underwent fixation with 4% paraformaldehyde (1 mL/well, XW0130525894011, SinoPharm, Shanghai, China) for 60 min and stained with crystal-free crystal violet staining solution (500 µL/well, CB0331, Sangon Biotech, Shanghai, China) for 20 min. Finally, the clones were counted under an inverted microscope (CKX53, Olympus, Shanghai, China).
Invasion and migration assays of HepG2 and HepG2/Sorafenib cells were performed using a transwell assay kit (3422, Corning, Corning, NY, USA) according to previous studies [20]. Serum-free cell suspensions were prepared using DMEM culture solution without serum. 200 µL cell suspension was added into the upper chambers with a cell density of 1.2 × 105/well. 500 µL DMEM supplemented with 30% FBS was added into the lower chamber. After 48 h, the migrated cells were fixed with 4% paraformaldehyde for 30 min, stained with 0.1% crystal violet (CB0331, Sangon, Shanghai, China) staining solution for 30 min, and observed under an inverted microscope (CKX53, Olympus) to evaluate their migration capability. Invasion experiments were conducted similarly to the migration experiments, with the exception that Matrigel (BD Biocoat 354234, Corning) spreading was applied at the bottom of the Transwell chambers.
The HepG2 and HepG2/Sorafenib cells were lyzed with the RIPA lysis buffer (AR0102, Boster, Wuhan, China) to extract the total proteins. Then the BCA Protein Assay Kit (P0012, Beyotime) was used to measure protein concentration. Proteins were separated and analyzed by SDS-PAGE at 80 V. After transferring to polyvinylidene fluoride (PVDF) membranes (IPVH00010, Millipore, Shanghai, China), proteins were treated with 5% skim milk for blocking and then incubated with primary antibodies against β-Tubulin III (Ab52623, Abcam, 1:5000), keratin (Ab8068, Abcam, 1:250), N-cadherin (Ab52623, Abcam, 1:1000), Vimentin (Ab52623, Abcam, 1:10000), p-EGFR (Ab32430, Abcam, 1:1000), p-JNK (9255, CST, 1:2000), p-ERK (4370, CST, 1:2000), EGFR (Ab52894, Abcam, 1:10000), JNK (9252, CST, 1:1000), ERK (4695, CST, 1:1000), and GAPDH (60004-1-Ig, Proteintech, 1:50000). Subsequent to incubation with HRP-labeled goat anti-mouse or rabbit secondary antibodies (ZB-2305/ZB-2301, 1:3000, ZSGB-Bio, China), protein bands were analyzed using Image Lab Software (Bio-Rad, Boulder, CO, USA). GAPDH was also used as an internal control.
Sixteen four-week-old female Balb/c nude mice were obtained by Charles River (Beijing, China). After two weeks of adaptive feeding, HepG2/sorafenib cells were injected subcutaneously into the mice (1 × 106 cells/mouse). Seven days later, the mice were divided into four groups (n = 4): HepG2/sorafenib (Control), Apatinib, Apatinib + SP600125, and Apatinib + PD98059 groups. For the apatinib group, apatinib was administered orally at a dose of 150 mg/kg once a day by gavage. For the apatinib + SP600125 or PD98059 group, after oral administration of apatinib, the mice were injected with SP600125 or PD98059 at 10 mg/kg every 3 days. The mice were sacrificed after about 4 weeks of treatment and tumors were collected. Subsequently, tumor weight and volume were examined. Tumor volume = 0.52 × length × width2. For the western blot, the tumor tissues were cut into pieces and mixed with RIPA lysis buffer and then homogenized to extract total proteins. The animal research was approved by the Ethics Committee of Shandong First Medical University (No. KY-2021-16).
All the experiments were performed in triplicates. Data processing was conducted using GraphPad Prism (Version 8.3.0, GraphPad, San Diego, CA, USA). The results were presented as mean ± SD and subjected to t-tests for comparisons between the two groups. A significance level of p < 0.05 was considered statistically significant.
The microtubule disease and epithelial-mesenchymal transformation (EMT) changes in sorafenib-resistant HCC
Sorafenib was introduced to both HepG2 and HepG2/Sorafenib cells to assess drug resistance in HCC. As illustrated in Fig. 1A, HepG-2/Sorafenib exhibited greater resistance to sorafenib compared to HepG2. Microtubule structures are associated with sorafenib resistance [21]. Thus, microtubule structures were observed in both cell lines. And there was an obvious change in microtubule structures between HepG2 and HepG2/Sorafenib groups (Fig. 1B and C). Notably, drug-resistant strains demonstrated significantly heightened migration and invasive capabilities compared to normal cancer cells (p < 0.05, respectively, Fig. 1D and 1E).
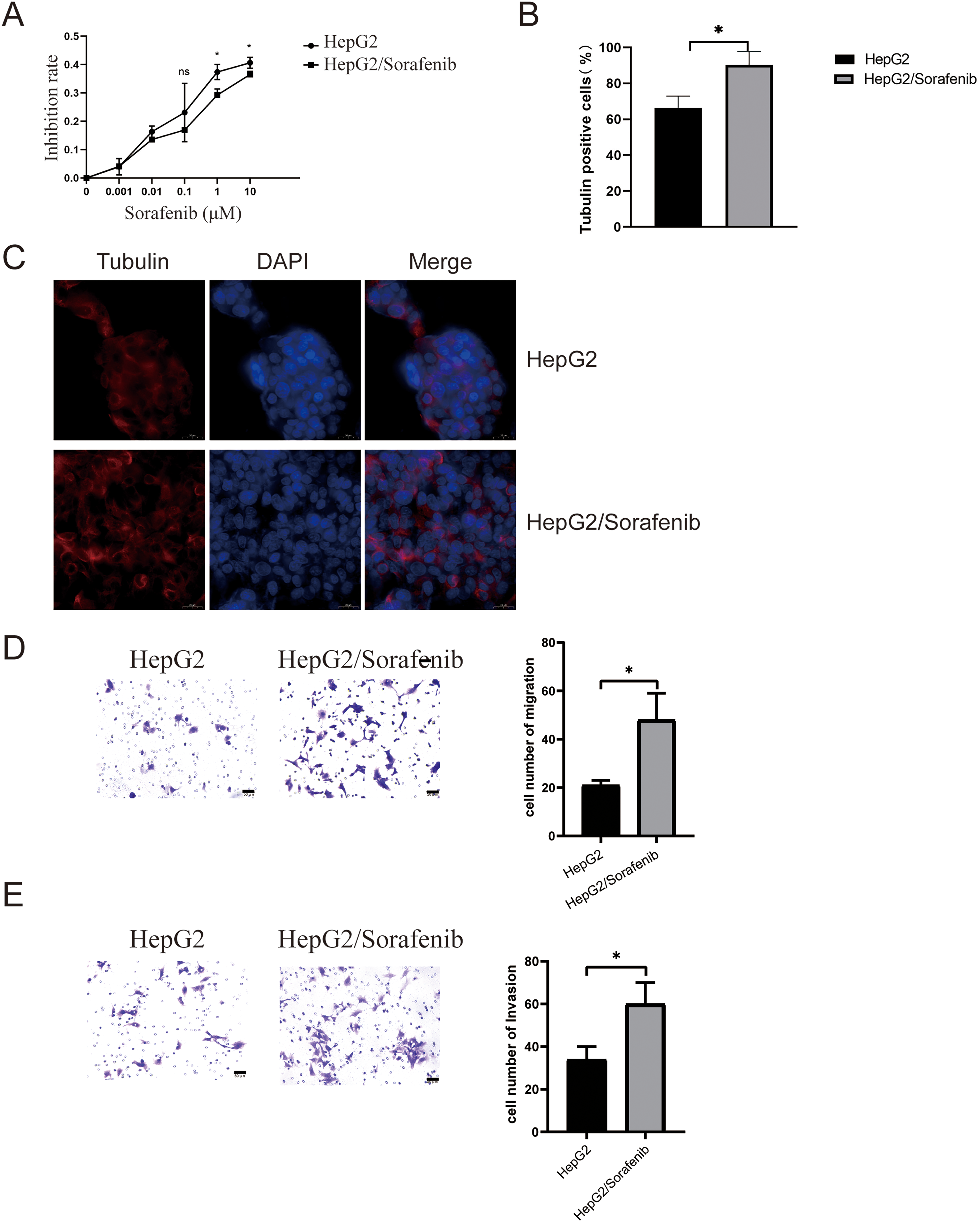
Figure 1: The microtubule disease change in sorafenib-resistant hepatocellular carcinoma cells. (A) CCK-8 assays for drug resistance. (B and C) Immunofluorescence for microtubule structure. Scale bar: 50 μm. (D) Transwell assay for migration. Scale bar: 50 μm. (E) Transwell assay for invasion. Scale bar: 50 μm. Values were shown as mean ± SD. Differences between the two groups were analyzed using a two-tailed t-test. *p < 0.05 vs. HepG2 group.
In consideration of the importance of EMT in sorafenib resistance of HCC, EMT-associated proteins including β-Tubulin III, N-cadherin, and Vimentin were detected. In addition, during the process of EMT in tumor cells, keratin expression would be suppressed and replaced by Vimentin expression [22]. As shown in Fig. 2A, the expression levels of β-Tubulin III, N-cadherin and Vimentin were markedly elevated in HepG2/Sorafenib compared to HepG2, whereas keratin expression was lower in the drug-resistant cells (p < 0.05, respectively). As EGFR/JNK/ERK signaling was highly correlated with sorafenib resistance in HCC, the phosphorylation of EGFR, JNK and ERK were also examined. As Fig. 2B displayed, the phosphorylation of the three proteins were obviously promoted in HepG2/sorafenib cells. These findings implied that sorafenib-resistant HCC was associated with EMT changes and EGFR/JNK/ERK signaling.
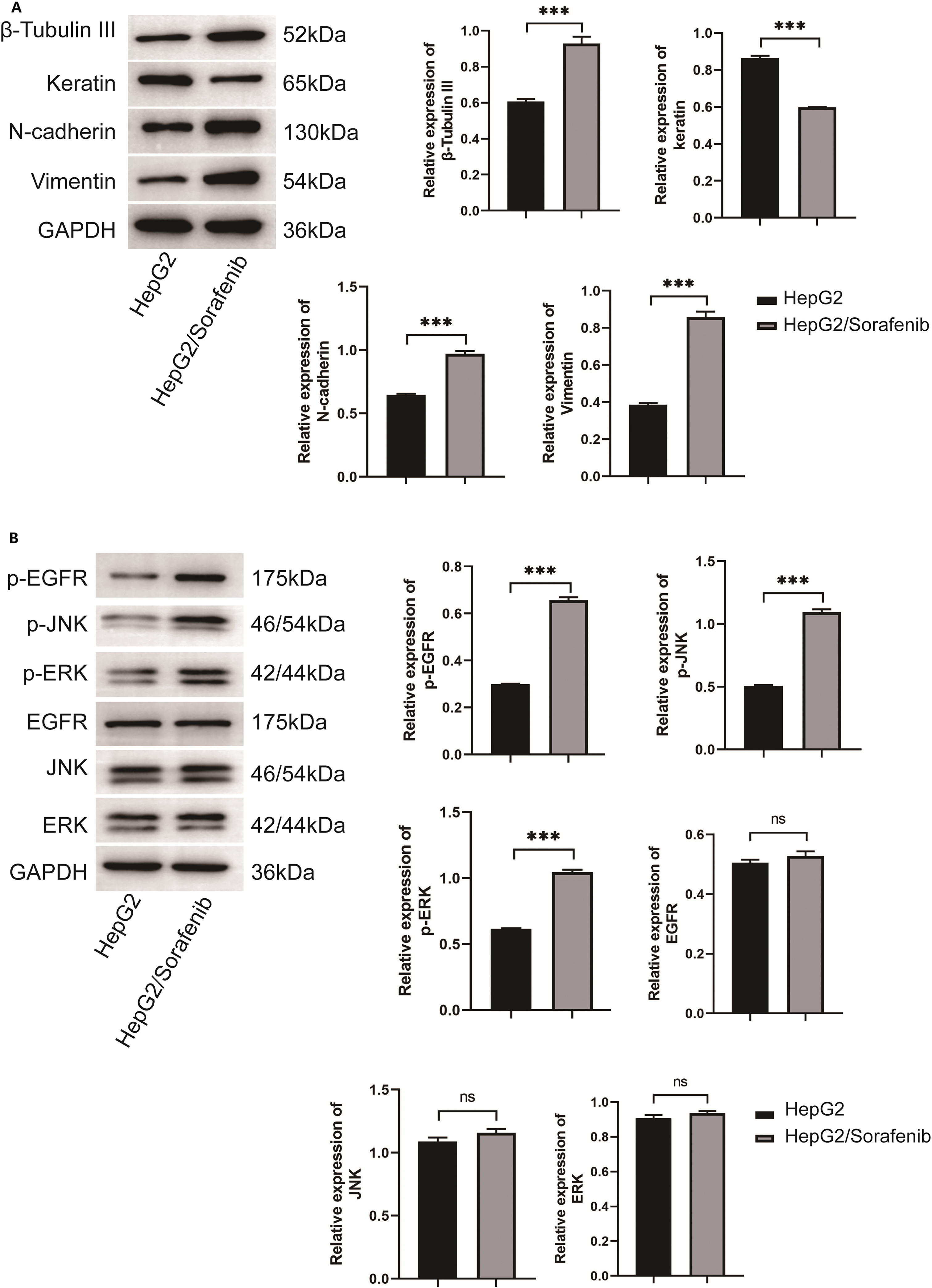
Figure 2: The EMT change in sorafenib-resistant hepatocellular carcinoma cells. (A) Western blot was used to detect the expression of EMT related protein. (B) Western blot was used to detect the expression of EGFR/JNK/ERK signaling pathway related protein. Values were shown as mean ± SD. Differences between two groups were analyzed using a two-tailed t-test. ***p < 0.001 vs. HepG2 group. ns, not significant.
Effect of apatinib on the bioactivity of sorafenib resistant HCC
Next, we examined the effects of apatinib on the malignant phenotypes of sorafenib-resistant HepG2 cells and HepG2 cells. Within HepG2/Sorafenib cells, the groups treated with apatinib exhibited a notable dose-dependent reduction in proliferation, colony formation, migration and invasion capabilities compared to the blank group (Fig. 3A–G and Fig. 4A–D, p < 0.05, respectively). However, the impact of apatinib on HepG2 cell viability and proliferation was minimal (Fig. 3A and E), while the migration and invasion of HepG2 cells were obviously inhibited with apatinib treatment (Fig. 4A–D). These observations collectively suggested that apatinib hindered the bioactivity of HepG2/Sorafenib cells in a concentration-dependent manner, and the inhibitory effects were similar to those of HepG2 cells.
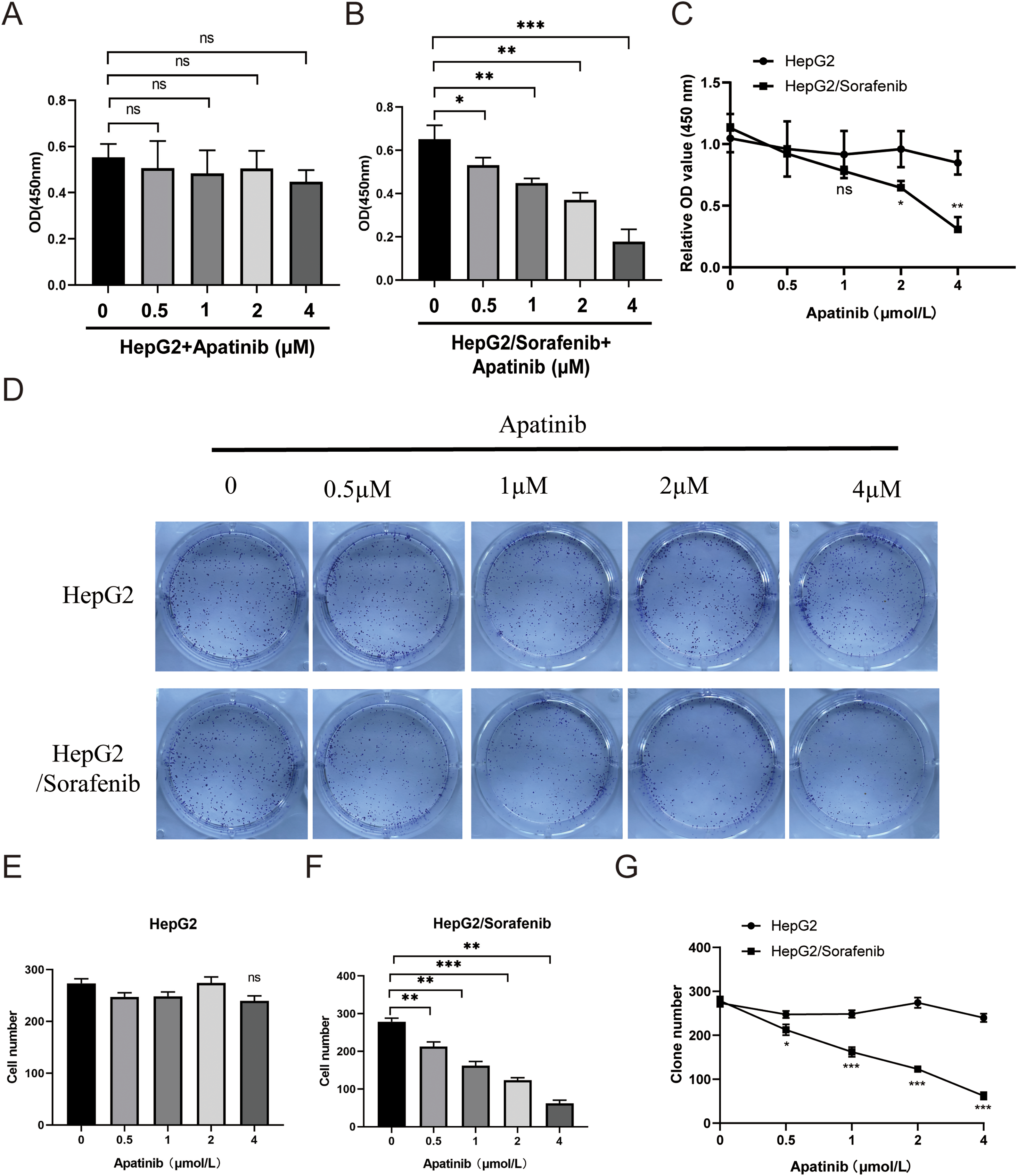
Figure 3: Apatinib inhibited HepG2/Sorafenib cell viability and proliferation in a concentration-dependent manner. (A and B) CCK-8 assay was used to examine the effects of apatinib on proliferation of HepG2 (A) and HepG2/Sorafenib (B) cells; (C) The compasion of proliferation between HepG2 and HepG2/Sorafenib groups; (D) Colony formation assay; (E and F) Quantitative results of colony formation in HepG2 (E) and HepG2/Sorafenib (F) groups; (G) The comparison of colony formation between HepG2 and HepG2/Sorafenib groups. Values were shown as mean ± SD. Differences between two groups were analyzed using a two-tailed t-test. *p < 0.05, **p < 0.01, ***p < 0.001 vs. HepG2/Sorafenib group. ns, not significant.
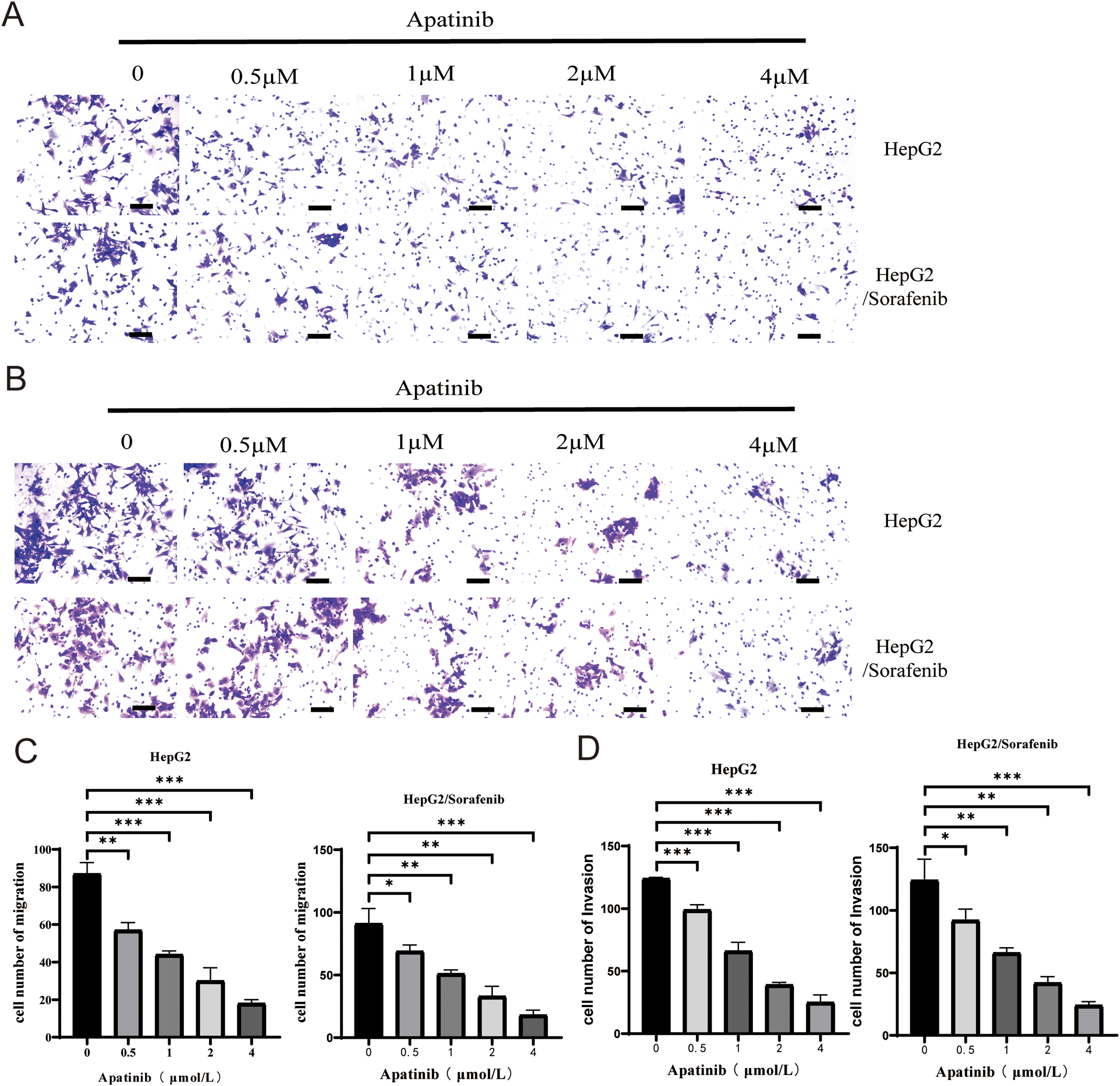
Figure 4: Apatinib inhibited HepG2/Sorafenib migration and invasion in a concentration-dependent manner. (A) Transwell assay for migration. Scale bar: 50 μm; (B) Transwell assay for invasion. Scale bar: 50 μm; (C) Quantitative results of the migration of HepG2 and HepG2/Sorafenib cells; (D) Quantitative results of the invasion of HepG2 and HepG2/Sorafenib cells. Values were shown as mean ± SD. Differences between two groups were analyzed using a two-tailed t-test. *p < 0.05, **p < 0.01, ***p < 0.001 vs. HepG2 group or HepG2/Sorafenib group.
Effect of apatinib on the expression of EMT-related proteins in sorafenib resistant HCC cells
Further, to confirm whether EMT was associated with the effects of apatinib on sorafenib-resistant HepG2 cells, we examined the expression of EMT-associated proteins after apatinib treatment. As depicted in Fig. 5, apatinib demonstrated a concentration-dependent inhibition of β-Tubulin III, N-cadherin, and Vimentin expression while promoting keratin expression in HepG2/Sorafenib and HepG2 cells (p < 0.05, respectively, Fig. 5). Besides, the impact of apatinib on the four EMT-associated proteins in HepG2/Sorafenib cells was similar to that of HepG2 cells (p < 0.05, respectively). These results indicated that apatinib exerted a pronounced effect on the expression of EMT-related proteins in sorafenib-resistant cells similar to that of sorafenib-sensitive HepG2 cells.
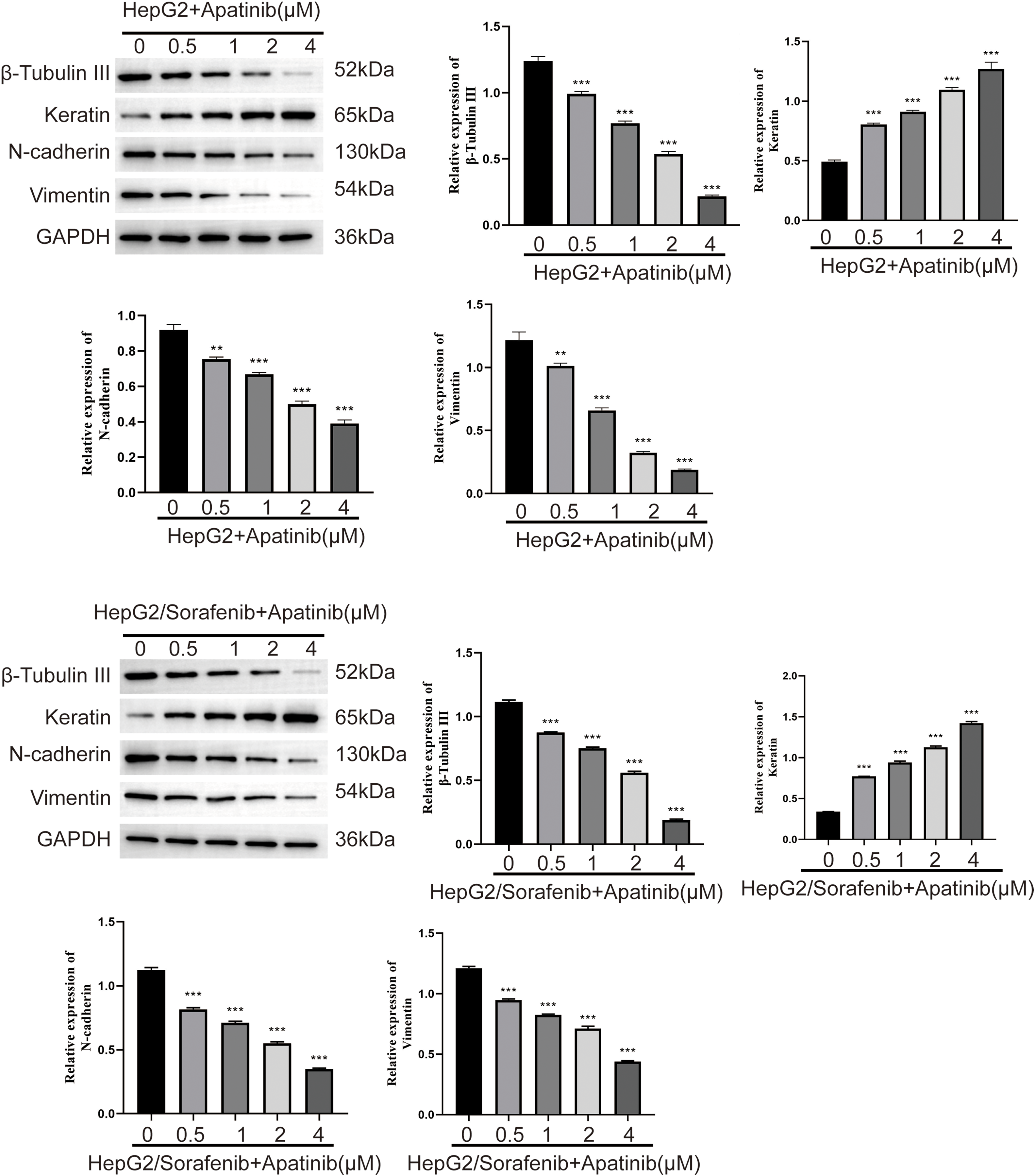
Figure 5: Effect of apatinib on the expression of EMT related proteins in sorafenib-resistant hepatocellular carcinoma cells. Values were shown as mean ± SD. Differences between two groups were analyzed using a two-tailed t-test. **p < 0.01, ***p < 0.001 vs. HepG2 group or HepG2/Sorafenib group.
Effects of Apatinib on EGFR/JNK/ERK signaling pathway in sorafenib-resistant HCC cells
To further elucidate the roles of apatinib in the EGFR/JNK/ERK signaling pathway in sorafenib-resistant HCC, HepG2/Sorafenib cells were exposed to varying apatinib doses, followed by the addition of JNK- and ERK-specific inhibitors, SP600125 and PD98059, for mechanistic exploration. Compared to the HepG2/Sorafenib group, the protein expression of p-EGFR, p-JNK, p-ERK, EGFR, JNK, and ERK significantly decreased in the different apatinib dose groups (p < 0.05, respectively, Fig. 6). Furthermore, SP600125 and PD98059 enhanced apatinib’s inhibition of migration and invasion in resistant strains (p < 0.05, respectively, Fig. 7A and B).
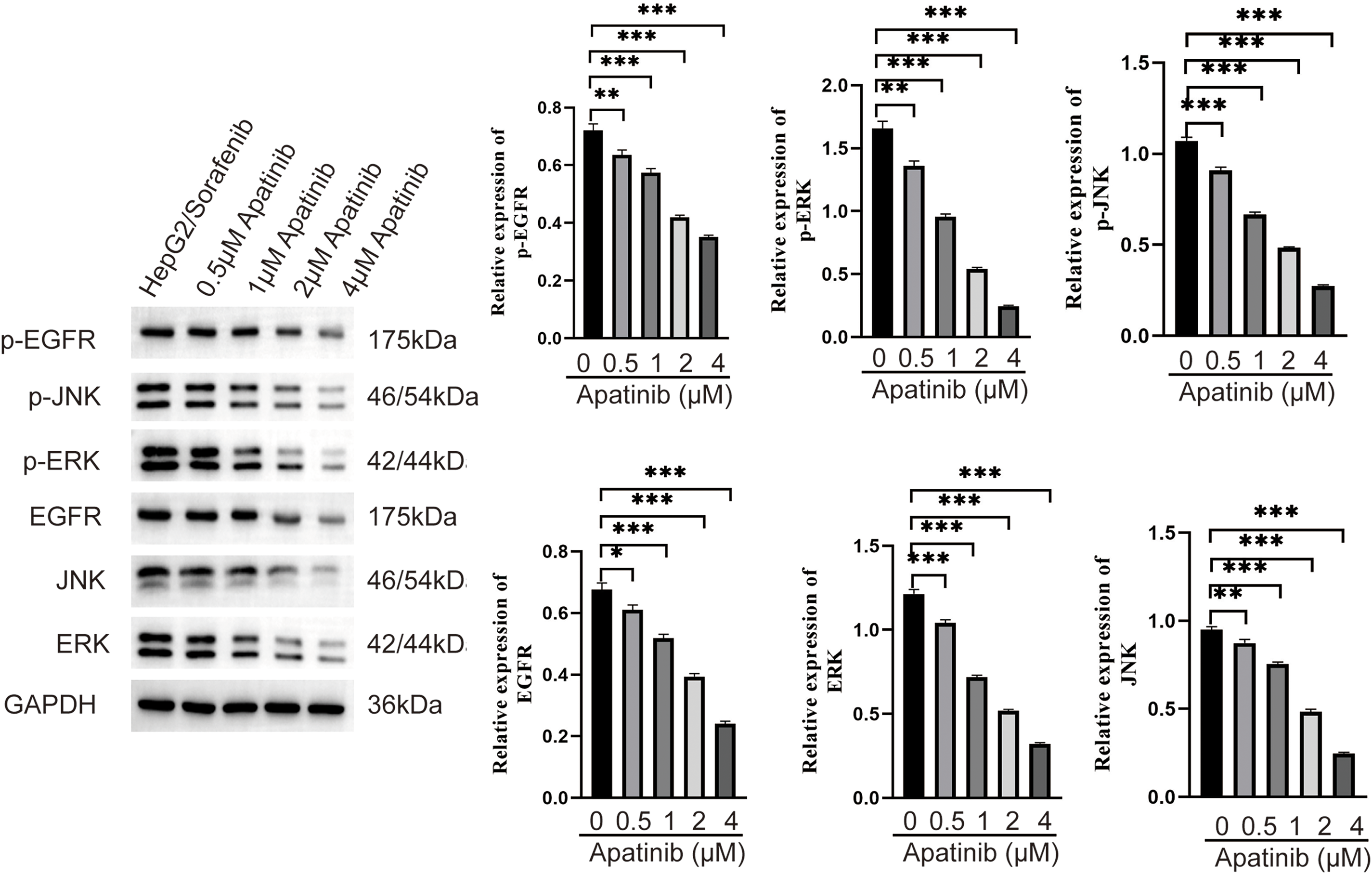
Figure 6: Effect of apatinib on the expression of EGFR/JNK/ERK signaling pathway related proteins in sorafenib-resistant hepatocellular carcinoma cells. Values were shown as mean ± SD. Differences between two groups were analyzed using a two-tailed t-test. *p < 0.05, **p < 0.01, ***p < 0.001 vs. HepG2/Sorafenib group.
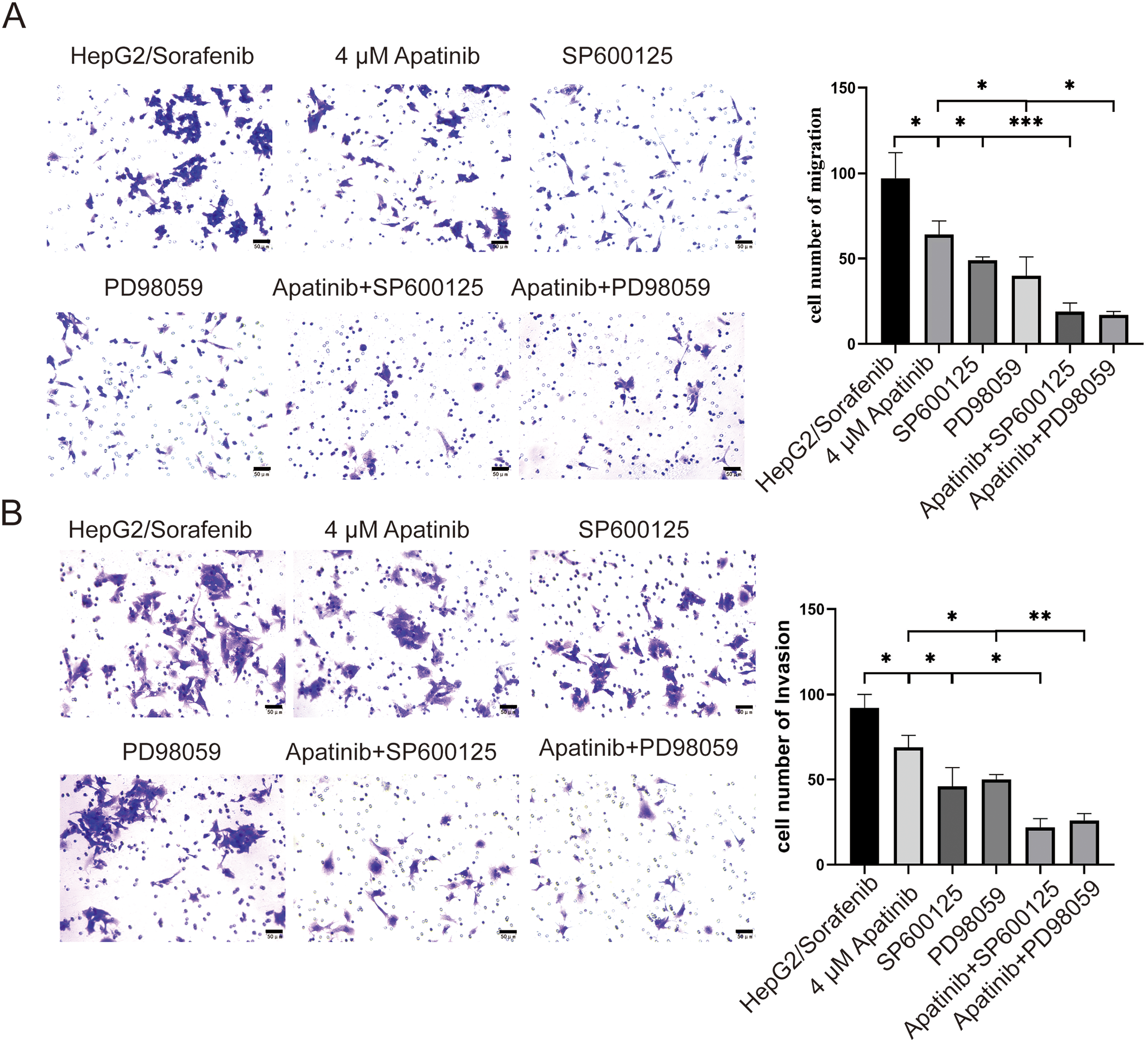
Figure 7: SP600125 and PD98059 promote apatinib inhibition of migration and invasion of HepG2/Sorafenib cells. (A) Transwell assay for migration. (B) Transwell assay for invasion. Values were shown as mean ± SD. Differences between two groups were analyzed using a two-tailed t-test. *p < 0.05, **p < 0.01, ***p < 0.001.
Additionally, SP600125 and PD98059 demonstrated a promoting effect on the inhibition of p-EGFR, p-JNK, p-ERK, EGFR, JNK, ERK, β-Tubulin III, N-cadherin, and Vimentin protein expression by apatinib in HepG2/Sorafenib cells (p < 0.05, respectively, Fig. 8). In summary, these in vitro results indicated that apatinib inhibited the malignancy of sorafenib-resistant HCC by inhibiting EMT and the EGFR/JNK/ERK pathway.
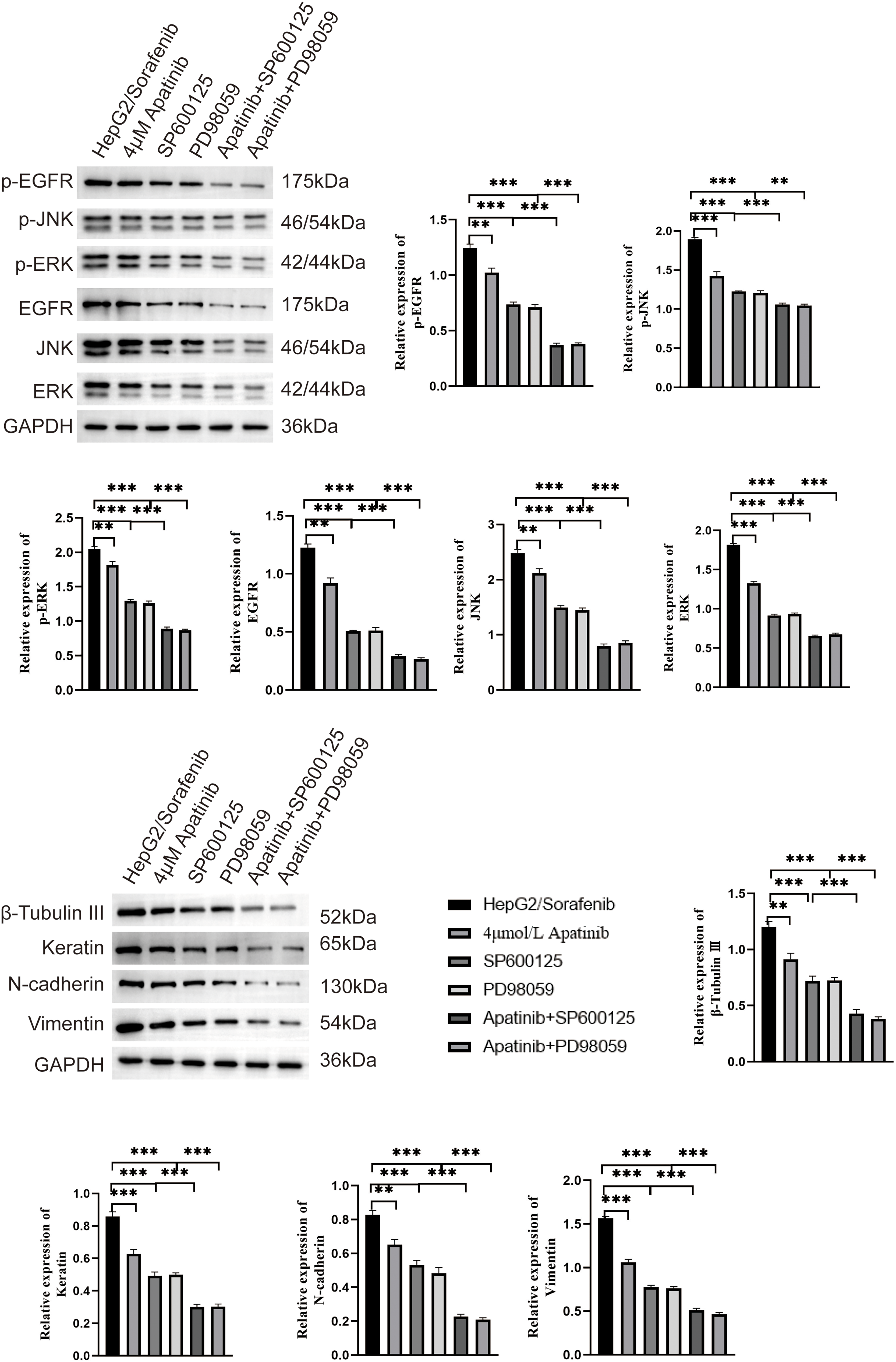
Figure 8: SP600125 and PD98059 contribute to the inhibition of EMT and EGFR/JNK/ERK pathway related protein by apatinib in HepG2/Sorafenib cells. Values were shown as mean ± SD. Differences between two groups were analyzed using a two-tailed t-test. **p < 0.01, ***p < 0.001.
Apatinib affects development of sorafenib-resistant HCC in vivo
To explore the effects of apatinib on sorafenib-resistant HCC in vivo, the HepG2/sorafenib cells were subcutaneously injected into nude mice. As shown in Fig. 9A–D, apatinib treatment inhibited the growth of tumor compared to HepG2/sorafenib group (p < 0.01). And SP600125 and PD98059 enhanced the inhibitory effects of apatinib in tumor growth in HCC (p < 0.01). Similarly, apatinib treatment inhibited the expression of EMT-associated proteins including β-Tubulin III, N-cadherin and Vimentin in vivo (Fig. 9E and F). And SP600125 or PD98059 treatment further reduced the expression of three proteins (Fig. 9E and F). Then similar effects of apatinib on p-EGFR, p-JNK, p-ERK, EGFR, JNK, ERK (Fig. 9G and H). Therefore, the in vivo experiments similarly proved that apatinib alleviated the development of sorafenib-resistant HCC by inhibiting EMT and the EGFR/JNK/ERK pathway.
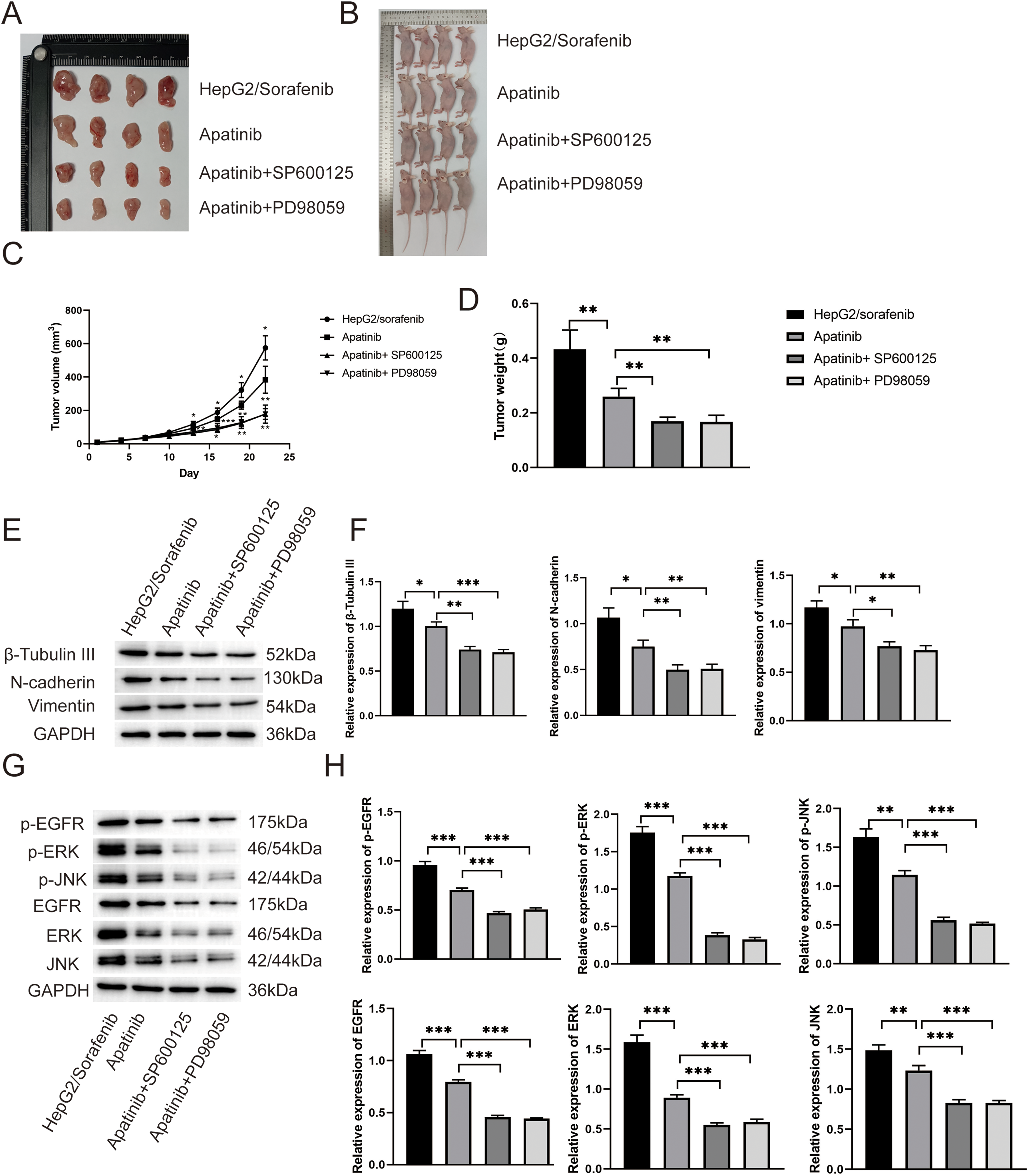
Figure 9: In vivo experiments proved that apatinib alleviated sorafenib resistance of HCC by inhibiting EMT and the EGFR/JNK/ERK pathway. (A and B) The images showed the tumors (A) and mice (B) in HepG2/Sorafenib, apatinib, apatinib + SP600125 and apatinib + PD98059 groups. (C and D) The quantitative results of tumor volume and tumor weight; (E and F) Effect of apatinib on the expression of EMT related proteins, including β-tubulin III, N-cadherin and Vimentin; (G and H) Effect of apatinib on the expression of p-EGFR, p-ERK, p-JNK, EGFR, ERK and JNK. Values were shown as mean ± SD. Differences between two groups were analyzed using a two-tailed t-test. *p < 0.05, **p < 0.01, ***p < 0.001.
Currently, targeted therapy stands out as a predominant approach in liver cancer [23,24], with sorafenib emerging as a leading multikinase inhibitor in this domain. As a novel multi-targeted anti-tumor drug, sorafenib adeptly hinders tumor angiogenesis and restrains tumor cell replication, effectively impeding tumor growth [13]. Despite notable progress in cancer therapeutics that has enhanced clinical outcomes, drug resistance remains a significant challenge [25,26]. Fortunately, in vivo experiments showed that apatinib exhibited antiangiogenic and antitumorigenic effects in HCC and drug safety evaluation indicated that mice treated with sorafenib exhibited diarrhea, skin rash, and reduced body weight, while these adverse effects were not detected in those treated with apatinib [27]. Additionally, apatinib has been reported to improve overall survival of patients with sorafenib-resistant HCC [16,28,29]. Similarly, in our study, apatinib inhibited the growth and movement of HepG2/sorafenib cells. Besides, apatinib exerted an inhibitory effect on the movement of HepG2 cells. Of note, the effects of apatinib on HepG2/sorafenib cells were more obvious than those in HepG2 cells.
In the context of resistance development, EMT serves as a crucial determinant, orchestrating the transformative “switch” of cancer cells from a quiescent to a proliferative state. Approximately 90% of cancer patients eventually suffer from the growth of metastatic tumors beyond the primary site [26,30]. Throughout tumor onset and progression, cancer cells derived from epithelial cells undergo EMT, gaining motility and invasive capabilities and disseminating through the bloodstream or lymphatic system to distant sites [31].
EMT involves a gradual loss of epithelial characteristics in cancer cells originating from epithelial cells, leading them to adopt a more mesenchymal-like state. This transformation enhances cancer cells’ ability to invade surrounding tissues and spread to distant parts of the body [32]. During EMT, increased expression of N-cadherin results in the loss of intercellular adhesion and heightened cell motility, facilitating tumor cell invasion [33]. Notably, β-tubulin III serves as the exclusive marker for resistance to microtubule-targeting drugs [34]. And keratin expression is decreased and replaced by Vimentin expression in EMT [22].
Multiple studies have indicated that sorafenib resistance promoted EMT in HCC cells [35]. Similarly, the present study revealed that the expression levels of β-Tubulin III, N-cadherin, and Vimentin were significantly higher in HepG2/Sorafenib compared to HepG2 cells. Conversely, keratin expression was lower in the drug-resistant cells. The results indicated that EMT process may participate in sorafenib resistance of HCC. After apatinib treatment, the expression levels of EMT-related proteins were obviously reduced in sorafenib-resistant HepG2 cells similar to those in HepG2 cells. Therefore, apatinib exerted inhibitive effects on sorafenib-resistant HCC partly through EMT process.
Given the intricate link between EMT and tumor infiltration and migration, contemporary researchers are committed to unraveling the complexities of cancer dissemination by delving into the underlying molecular mechanisms of EMT. Studies indicate that various signals can trigger the EMT process [36–38]. The EGFR, a glycoprotein in the tyrosine kinase receptor family, resides on the cell membrane surface and becomes activated upon ligand binding. EGFR activates intracellular kinase pathways, such as ERK, by binding with ligands and inducing cell proliferation [39].
Research points to the involvement of the EGFR/JNK/ERK signaling pathway in regulating chemoresistance mechanisms in gastric cancer; however, reports on its role in liver cancer resistance studies are limited [40]. In this study, we observed that the expression levels of p-EGFR, p-JNK, and p-ERK were significantly higher in HepG2/Sorafenib compared to HepG2 cells. This observation suggests that the EGFR/JNK/ERK pathway plays a role in the mechanism of sorafenib resistance in HCC. Apatinib, an anti-angiogenic targeted drug, has demonstrated the ability to reverse drug resistance in cancer cells [41]. Nonetheless, its impact on the EGFR/JNK/ERK signaling pathway in HCC resistance remains unexplored. In light of this, JNK- and ERK-specific inhibitors, SP600125 and PD98059, were employed, and the results suggest that apatinib exerted an inhibitory effect on the progression of sorafenib-resistant HCC by inhibiting both EMT and the EGFR/JNK/ERK pathway.
This study speculated that apatinib could ameliorate sorafenib-resistant HCC by modulating EMT and EGFR/JNK/ERK pathway, which is of great significance. The study provided a new perspective for exploring treatment strategies for sorafenib-resistant HCC. The knowledge gaps include that the proposed mechanism needs further verification through in vitro, in vivo and clinical studies, and other interacting pathways or factors remain to be explored. To address these, large-scale clinical trials and relevant experiments can be conducted. In the next five years, it’s expected that more in-depth preclinical studies will be carried out to clarify the molecular mechanisms using multi-omics technologies. The number of relevant clinical trials will increase, helping to formulate treatment guidelines. Moreover, new drug targets or biomarkers may be discovered, promoting the development of precision medicine and enhancing the understanding and management of sorafenib-resistant HCC.
In conclusion, this study revealed that sorafenib-resistant HCC exhibited both microtubule abnormalities and EMT changes. Considering that apatinib influences sorafenib-resistant HCC by inhibiting the EGFR/JNK/ERK signaling pathway, it is plausible to speculate that apatinib could inhibit the development of sorafenib-resistant HCC through the EMT process and EGFR/JNK/ERK pathway. These findings hold significant implications for comprehending the sorafenib resistance process in HCC and provide insights into the mechanism by which apatinib operates in sorafenib-resistant HCC.
Acknowledgement: None.
Funding Statement: This study was supported by Natural Science Foundation of Shandong Province, No. ZR2021QH186, National Natural Science Foundation of China, No. 81870205, Foundation Research Project of Qinghai Province, 2021-ZJ-719, National Natural Science Foundation of China, No. 82000579, Qinghai Provincial Health System Key Project No. 2021-wjzd-06 and Foundation Research Project of Qinghai Province, 2023-ZJ-786.
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Dexue Fan, Zhenhai Zhang and Junlin Gao; data collection: Dexue Fan, Wei Su, Zhaowen Bi and Xinxing Wang; analysis and interpretation of results: Dexue Fan, Wei Su, Zhaowen Bi, Xinxing Wang, Xianwen Xu, Mingze Ma and Lichao Zhu; draft manuscript preparation: Dexue Fan and Wei Su. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval: The animal research was approved by Ethics Committee of Shandong First Medical University (No. KY-2021-16).
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Salazar J, Le A. The heterogeneity of liver cancer metabolism. Adv Exp Med Biol. 2021;1311:127–36. doi:10.1007/978-3-030-65768-0. [Google Scholar] [CrossRef]
2. Wu D, Yang Y, Hou Y, Zhao Z, Liang N, Yuan P, et al. Increased mitochondrial fission drives the reprogramming of fatty acid metabolism in hepatocellular carcinoma cells through suppression of Sirtuin 1. Cancer Commun. 2022;42(1):37–55. doi:10.1002/cac2.12247. [Google Scholar] [PubMed] [CrossRef]
3. Ricke J, Schinner R, Seidensticker M, Gasbarrini A, van Delden OM, Amthauer H, et al. Liver function after combined selective internal radiation therapy or sorafenib monotherapy in advanced hepatocellular carcinoma. J Hepatol. 2021;75(6):1387–96. doi:10.1016/j.jhep.2021.07.037. [Google Scholar] [PubMed] [CrossRef]
4. Feng J, Dai W, Mao Y, Wu L, Li J, Chen K, et al. Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1α/PPAR-γ/PKM2-mediated glycolysis. J Exper Clin Cancer Res. 2020;39(1):24. doi:10.1186/s13046-020-1528-x. [Google Scholar] [PubMed] [CrossRef]
5. Yildirim HC, Kavgaci G, Chalabiyev E, Dizdar O. Advances in the early detection of hepatobiliary cancers. Cancers. 2023;15(15):3880. doi:10.3390/cancers15153880. [Google Scholar] [PubMed] [CrossRef]
6. Sahin TK, Rizzo A, Aksoy S, Guven DC. Prognostic significance of the royal marsden hospital (RMH) score in patients with cancer: a systematic review and meta-analysis. Cancers. 2024;16(10):1835. [Google Scholar] [PubMed]
7. Qin S, Chan SL, Gu S, Bai Y, Ren Z, Lin X, et al. Camrelizumab plus rivoceranib versus sorafenib as first-line therapy for unresectable hepatocellular carcinoma (CARES-310a randomised, open-label, international phase 3 study. Lancet. 2023;402(10408):1133–46. [Google Scholar] [PubMed]
8. Tang W, Chen Z, Zhang W, Cheng Y, Zhang B, Wu F, et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther. 2020;5(1):87. [Google Scholar] [PubMed]
9. Zhu YJ, Zheng B, Wang HY, Chen L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017;38(5):614–22. [Google Scholar] [PubMed]
10. Chen J, Jin R, Zhao J, Liu J, Ying H, Yan H, et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015;367(1):1–11. [Google Scholar] [PubMed]
11. Shrestha R, Bridle KR, Cao L, Crawford DHG, Jayachandran A. Dual targeting of sorafenib-resistant HCC-derived cancer stem cells. Curr Oncol. 2021;28(3):2150–72. [Google Scholar] [PubMed]
12. Fornari F, Giovannini C, Piscaglia F, Gramantieri L. Elucidating the molecular basis of sorafenib resistance in HCC: current findings and future directions. J Hepatocell Carcinoma. 2021;8:741–57. [Google Scholar] [PubMed]
13. Wang HC, Haung LY, Wang CJ, Chao YJ, Hou YC, Yen CJ, et al. Tumor-associated macrophages promote resistance of hepatocellular carcinoma cells against sorafenib by activating CXCR2 signaling. J Biomed Sci. 2022;29(1):99. [Google Scholar] [PubMed]
14. Pang L, Xu L, Yuan C, Li X, Zhang X, Wang W, et al. Activation of EGFR-KLF4 positive feedback loop results in acquired resistance to sorafenib in hepatocellular carcinoma. Mol Carcinog. 2019;58(11):2118–26. [Google Scholar] [PubMed]
15. Zhang D, Zhou Y, Ma Y, Jiang P, Lv H, Liu S, et al. Ribosomal protein L22-like1 (RPL22L1) mediates sorafenib sensitivity via ERK in hepatocellular carcinoma. Cell Death Discov. 2022;8(1):365. [Google Scholar] [PubMed]
16. Qin S, Li Q, Gu S, Chen X, Lin L, Wang Z, et al. Apatinib as second-line or later therapy in patients with advanced hepatocellular carcinoma (AHELPa multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol. 2021;6(7):559–68. [Google Scholar] [PubMed]
17. Hou Z, Zhu K, Yang X, Chen P, Zhang W, Cui Y, et al. Apatinib as first-line treatment in patients with advanced hepatocellular carcinoma: a phase II clinical trial. Ann Transl Med. 2020;8(17):1047. [Google Scholar] [PubMed]
18. Zhang Y, Fan W, Wang Y, Huang G, Li J. Apatinib for patients with sorafenib-refractory advanced hepatitis B virus related hepatocellular carcinoma: results of a pilot study. Cancer Control. 2019;26(1):1073274819872216. doi:10.1177/1073274819872216. [Google Scholar] [PubMed] [CrossRef]
19. Ma Z, Chen W, Liu Y, Yu L, Mao X, Guo X, et al. Artesunate Sensitizes human hepatocellular carcinoma to sorafenib via exacerbating AFAP1L2-SRC-FUNDC1 axis-dependent mitophagy. Autophagy. 2024;20(3):541–56. [Google Scholar] [PubMed]
20. Munir J, Van Ngu T, Na Ayudthaya PD, Ryu S. Downregulation of glypican-4 facilitates breast cancer progression by inducing cell migration and proliferation. Biochem Biophys Res Commun. 2020;526(1):91–7. [Google Scholar] [PubMed]
21. Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10(3):194–204. [Google Scholar] [PubMed]
22. Fortier AM, Asselin E, Cadrin M. Keratin 8 and 18 loss in epithelial cancer cells increases collective cell migration and cisplatin sensitivity through claudin1 up-regulation. J Biol Chem. 2013;288(16):11555–71. [Google Scholar] [PubMed]
23. Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. [Google Scholar] [PubMed]
24. Liu F, Liao Z, Zhang Z. MYC in liver cancer: mechanisms and targeted therapy opportunities. Oncogene. 2023;42(45):3303–18. [Google Scholar] [PubMed]
25. Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–73. [Google Scholar] [PubMed]
26. Lee MS, Ryoo BY, Hsu CH, Numata K, Stein S, Verret W, et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140an open-label, multicentre, phase 1b study. Lancet Oncol. 2020;21(6):808–20. [Google Scholar] [PubMed]
27. Liang Q, Kong L, Du Y, Zhu X, Tian J. Antitumorigenic and antiangiogenic efficacy of apatinib in liver cancer evaluated by multimodality molecular imaging. Exper Mol Med. 2019;51(7):1–11. [Google Scholar]
28. Zhang Y, Huang G, Miao H, Song Z, Zhang X, Fan W, et al. Apatinib treatment may improve survival outcomes of patients with hepatitis B virus-related sorafenib-resistant hepatocellular carcinoma. Ther Adv Med Oncol. 2020;12:1758835920937422. [Google Scholar] [PubMed]
29. Han Z, He Z, Wang C, Wang Q. The effect of apatinib in the treatment of sorafenib resistant metastatic hepatocellular carcinoma: a case report. Medicine. 2018;97(49):e1338830. [Google Scholar]
30. Ganesh K, Massague J. Targeting metastatic cancer. Nat Med. 2021;27(1):34–44. [Google Scholar] [PubMed]
31. Bakir B, Chiarella AM, Pitarresi JR, Rustgi AK. EMT, MET, plasticity, and tumor metastasis. Trends Cell Biol. 2020;30(10):764–76. [Google Scholar] [PubMed]
32. Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019;29(3):212–26. [Google Scholar] [PubMed]
33. Loh CY, Chai JY, Tang TF, Wong WF, Sethi G, Shanmugam MK, et al. The E-Cadherin and N-Cadherin switch in Epithelial-to-Mesenchymal transition: signaling, therapeutic implications, and challenges. Cells. 2019;8(10):1118. [Google Scholar] [PubMed]
34. Kanakkanthara A, Miller JH. βIII-tubulin overexpression in cancer: causes, consequences, and potential therapies. Biochimica Et Biophysica Acta Reviews on Cancer. 2021;1876(2):188607. [Google Scholar] [PubMed]
35. Kuang Y, Cheng Y, Wang J, Li H, Cao X, Wang Y. KIAA1429 mediates epithelial mesenchymal transition in sorafenib-resistant hepatocellular carcinoma through m6A methylation modification. Cancer Med. 2023;12(6):7222–33. [Google Scholar] [PubMed]
36. Akrida I, Bravou V, Papadaki H. The deadly cross-talk between Hippo pathway and epithelial-mesenchymal transition (EMT) in cancer. Mol Biol Rep. 2022;49(10):10065–76. doi:10.1007/s11033-022-07590-z. [Google Scholar] [PubMed] [CrossRef]
37. Deshmukh AP, Vasaikar SV, Tomczak K, Tripathi S, den Hollander P, Arslan E, et al. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc Natl Acad Sci U S A. 2021;118(19):e2102050118. doi:10.1073/pnas.2102050118. [Google Scholar] [PubMed] [CrossRef]
38. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014;7(344):re8. doi:10.1126/scisignal.2005189. [Google Scholar] [PubMed] [CrossRef]
39. Guo G, Gong K, Wohlfeld B, Hatanpaa KJ, Zhao D, Habib AA. Ligand-independent EGFR signaling. Cancer Res. 2015;75(17):3436–41. doi:10.1158/0008-5472.CAN-15-0989. [Google Scholar] [PubMed] [CrossRef]
40. Cui Y, Li Q, Li H, Wang Y, Wang H, Chen W, et al. Asparaginyl endopeptidase improves the resistance of microtubule-targeting drugs in gastric cancer through IQGAP1 modulating the EGFR/JNK/ERK signaling pathway. Onco Targets Ther. 2017;10:627–43. doi:10.2147/OTT.S125579. [Google Scholar] [PubMed] [CrossRef]
41. Yang QK, Chen T, Wang SQ, Zhang XJ, Yao ZX. Apatinib as targeted therapy for advanced bone and soft tissue sarcoma: a dilemma of reversing multidrug resistance while suffering drug resistance itself. Angiogenesis. 2020;23(3):279–98. [Google Scholar] [PubMed]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools