
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
Prognostic Significance of DNA Repair Gene mRNA Expression in Early-Stage Breast Cancer: Insights into Clinical Relevance
1 Department of Obstetrics and Gynecology, University Medical Center, Johannes Gutenberg-University Mainz, Mainz, 55131, Germany
2 Department of Gynaecology and Obstetrics, Johann Wolfgang Goethe University, Frankfurt am Main, 60596, Germany
* Corresponding Author: Ina Shehaj. Email:
# These authors contributed equally to this work
Oncology Research 2026, 34(3), 11 https://doi.org/10.32604/or.2025.072222
Received 22 August 2025; Accepted 19 December 2025; Issue published 24 February 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
This article has no abstract.Keywords
Supplementary Material
Supplementary Material FileAdvances in research using microarray-based analysis enhance our ability to predict better, improve patients’ outcomes, and personalize their treatment. Knowledge about gene expression profiling has led to the classification of breast cancer into four molecular subtypes with different disease outcomes Haibe-Kains et al. [1]. Further studies have investigated the prognostic impact of messenger ribonucleic acid (mRNA) expression of several genes on patients with early-stage breast cancer (eBC) [2–4]. However, research on deoxyribonucleic acid (DNA) repair gene expression, such as non-Breast Cancer (BRCA) genes and their impact on the prognosis of eBC patients, remains limited.
Building on our previous findings on the mRNA expression of BRCA1, BRCA2, and Partner and localizer of BRCA2 (PALB2), we further examined the impact of 20 non-BRCA genes on survival in a cohort of 461 eBC patients, using Kaplan-Meier curves and Cox regression analyses [5]. In the present study, we investigated the predictive significance of Ataxia-telangiectasia mutated (ATM), Bloom helicase gene (BLM), and WRN RecQ-Like Helicase (WRN) mRNA expression.
ATM is a tumor suppressor gene that plays a key role in cell cycle control, apoptosis, and oxidative stress [6]. Recently published studies have shown that certain variants of ATM are linked to a higher risk of developing breast cancer and poor prognosis [7,8]. Further studies have investigated the impact of mRNA expression of ATM on the survival of patients with eBC [9–11].
BLM and WRN belong to the RecQ DNA helicase family, which plays a critical role in protecting and stabilizing the genome [12]. Since their prognostic role in eBC remains unknown, a few studies aimed to investigate the prognostic impact of mRNA expression of the RecQ family [13]. BLM plays an important role in replication and proliferation, and it is not surprising that BLM mRNA overexpression is more commonly observed in eBC with a worse prognosis [13]. WRN, a known tumor suppressor, plays an important role in DNA repair, replication, recombination, and maintenance of nuclear telomeres. Reduced WRN expression leads to genomic instability [14,15]. Preclinical studies have also shown that expression of WRN in eBC cell lines inhibits tumor growth in mouse xenograft models [16].
Microarray analysis presents a modality to measure gene expression signatures, which can then be used as a component of genomic-based prognostic tests. Furthermore, expression profiling of DNA repair genes could help stratify patients for future clinical trials investigating targeted therapies. Thus, we provide crucial insights regarding the prognostic significance and the potential clinical relevance of mRNA expression of non-BRCA genes such as ATM, BLM, and WRN in patients with eBC. Such tests can help identify subgroups of eBC patients with worse prognosis and integrate their role in modern diagnostic and prognostic models.
This study was conducted in a consecutive cohort of 461 patients with eBC and long-term follow-up, treated at the Department of Obstetrics and Gynecology, University Medical Center Mainz, between 1986 and 2000. eBC was defined as tumors classified as T1–T3 with N0–N1 disease, as well as high-risk eBC cases (T4 or N2). The cohort comprised all patients with eBC who showed no evidence of distant metastasis at diagnosis. Patients presenting with metastatic disease or lacking adequate clinical information and/or mRNA expression data were excluded. Written informed consent was obtained from all participants, and all procedures adhered to ethical and legal standards consistent with the Declaration of Helsinki. The study protocol was approved by the Institutional Review Board of the Ethics Committee of Rheinland-Pfalz, Germany (No. 837.139.05 [4797], 20 October 2005).
Clinicopathological information was retrieved from pathology records and the institutional breast cancer database. Collected data included age at diagnosis, histological grade, tumor size, nodal status, estrogen receptor (ER) and progesterone receptor (PR) status, human epidermal growth factor receptor 2 (HER2) status, Ki-67 index, protocols of administered treatments, and follow-up outcomes. Of the full cohort, 200 node-negative patients did not receive any adjuvant therapy following surgery. Adjuvant tamoxifen was administered to 165 patients, whereas 96 patients received adjuvant chemotherapy. As all treatments were administered postoperatively and Affymetrix microarray analyses were performed on fresh-frozen primary tumor tissue, these therapies did not influence the measured mRNA expression profiles. The clinical characteristics of this cohort have been described previously [5].
Fresh-frozen breast tumor samples were collected from the Department of Obstetrics and Gynecology, University Medical Center Mainz and analyzed using HG-U133A microarrays (Affymetrix, Santa Clara, CA, USA) to assessrelative transcript levels in breast cancer tissue, as described in previous results [3,5,17].
Tumor samples were snap-frozen and stored at −80°C withtumor cell content exceeding 40% in all samples. Approximately 50 mg of frozen breast tumor tissue was crushed in liquid nitrogen. RLT buffer was added to the tissue, and the homogenate was processed through a QIAshredder column (Qiagen, Hilden, Germany). Total RNA was isolated with the RNeasy Kit (Qiagen, Hilden, Germany, Qiagen Kit: Cat no./ID. 74106), following the manufacturer’s instructions.
RNA yield was quantified by UV absorbance, and RNA quality was assessed via rRNA band integrity using an Agilent 2100 Bioanalyzer with a RNA 6000 LabChip kit (Agilent Technologies, Santa Clara, CA, USA). From 5 μg of total RNA, labeled cRNA was synthesized using Roche Microarray cDNA Synthesis, Microarray RNA Target Synthesis (T7), and Microarray Target Purification kits (Roche Applied Science, Mannheim, Germany), according to the manufacturer’s instructions.
Microarray Data Processing and Normalization
Raw expression data (CEL files) were normalized using frozen robust multiarray analysis (fRMA), with global scaling applied to achieve a mean target intensity of 500. Samples exhibiting suboptimal signal intensities (scaling factors > 25) or high glyceraldehyde-3-phosphate dehydrogenase (GAPDH) 3′/5′ ratios (>5) were relabeled and rehybridized on new arrays.
For the gene expression analysis, data derived from fresh frozen tissue and measured using HG-U133A arrays are reported using TGT500 scaling. The complete dataset of 461 samples, along with updated follow-up information, has been deposited in the NCBI GEO database under accession number GSE158309 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158309).
The following single genes were considered for this gene expression analysis (Table 1).
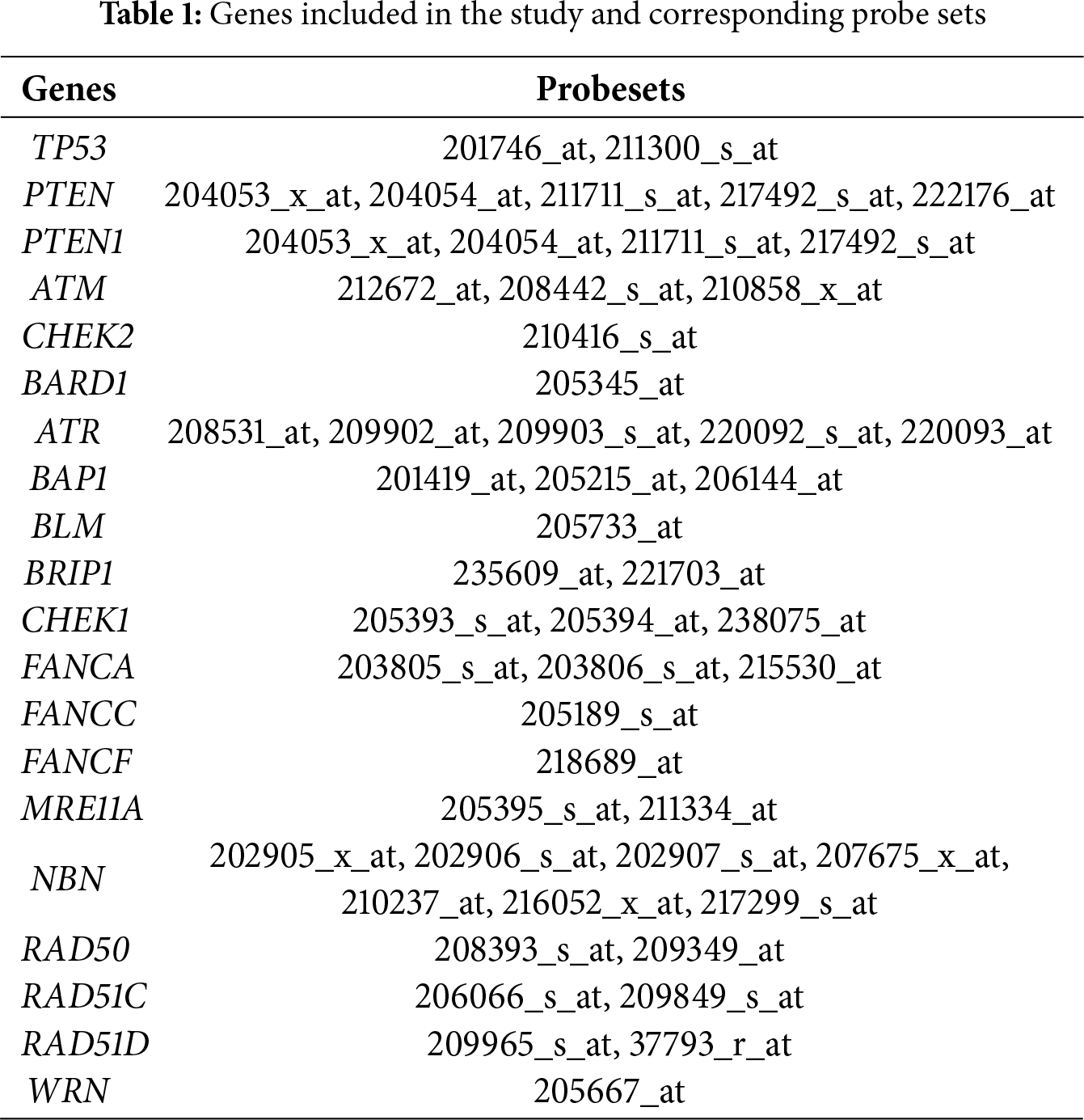
The metagenes of tumor protein p53 (TP53), phosphatase and tensin homolog (PTEN), Ataxia Telangiectasia Mutated (ATM), checkpoint kinase 2 (CHEK2), BRCA1-Associated Ring Domain protein 1 (BARD1), ATR serine/threonine kinase (ATR), BRCA1 associated protein 1 (BAP1), Bloom helicase (BLM), BRCA1 Interacting Helicase 1 (BRIP1), checkpoint kinase 1 (CHEK1), Fanconi anemia complementation group A (FANCA), NIBRIN (NBN), Fanconi anemia complementation group C (FANCC), Fanconi anemia complementation group F (FANCF), Meiotic recombination 11A (MRE11A), RAD50 Double Strand Break Repair Protein (RAD50), RAD51 paralog C (RAD51C), RAD51 paralog D (RAD51D), WRN RecQ Like Helicase (WRN) were calculated for each sample as the average expression of all genes in the gene cluster. As mentioned earlier, the median mRNA expression was used as the cut-off value. All patients included in this study were subdivided into two groups based on low and high mRNA expression levels.
Statistical analyses were conducted using SPSS software, version 27.0 (SPSS Inc., Chicago, IL, USA). We performed Kaplan–Meier survival analysis as well as univariable and multivariable Cox proportional hazards regression models to evaluate the prognostic relevance of mRNA expression of the above-mentioned genes with respect to metastasis-free survival (MFS) in the overall cohort. MFS was defined as the interval between initial diagnosis and the occurrence of distant metastasis.
Subgroup analyses were performed to assess differences in mRNA expression levels across molecular subtypes. The multivariable Cox models were adjusted for tumor stage, histological grade, lymph node status, and Ki-67 index. Survival distributions were compared using the log-rank test, and statistical significance was defined as a two-sided p-value < 0.05. Post hoc Dunn’s tests with Bonferroni correction were applied to identify specific pairwise differences in gene expression. In addition, survival analyses were repeated in the subgroup of patients who did not receive any systemic therapy (N = 200).
The primary endpoint of the study was the association between mRNA expression levels and MFS. Secondary endpoints included the relationships between median mRNA expression levels of ATM, BLM, and WRN and both patient-related and tumor-related characteristics.
2.4 Validation of mRNA Expression of ATM, BLM, and WRN in Independent Cohorts
We utilized the Kaplan-Meier Plotter database (http://kmplot.com/analysis) to validate our results on a larger, independent cohort among all samples of eBC and within various intrinsic subtypes and clinicopathological characteristics. The prognostic significance of ATM (p = 0.022, Log Rank), was successfully validated in previously published datasets. In contrast, the prognostic impact of BLM and WRN could not be validated in the same datasets (Appendix A, Figs. A1–A3) [18].
Intrinsic subtypes were determined according to the classification proposed by Haibe-Kains et al., which is based on the expression of ESR1, HER2, and AURKA [1].
Survival analyses were subsequently conducted within the major molecular breast cancer subgroups. Tumors classified as Luminal A–like were characterized by ESR1 positivity, absence of HER2 overexpression, and low proliferative activity (low AURKA expression). Luminal B–like tumors were also ESR1-positive and HER2-negative but showed high proliferation (high AURKA expression). The HER2-positive group consisted of tumors demonstrating HER2 amplification or overexpression. The Basal-like subtype was defined by the lack of both ESR1 and HER2 expression.
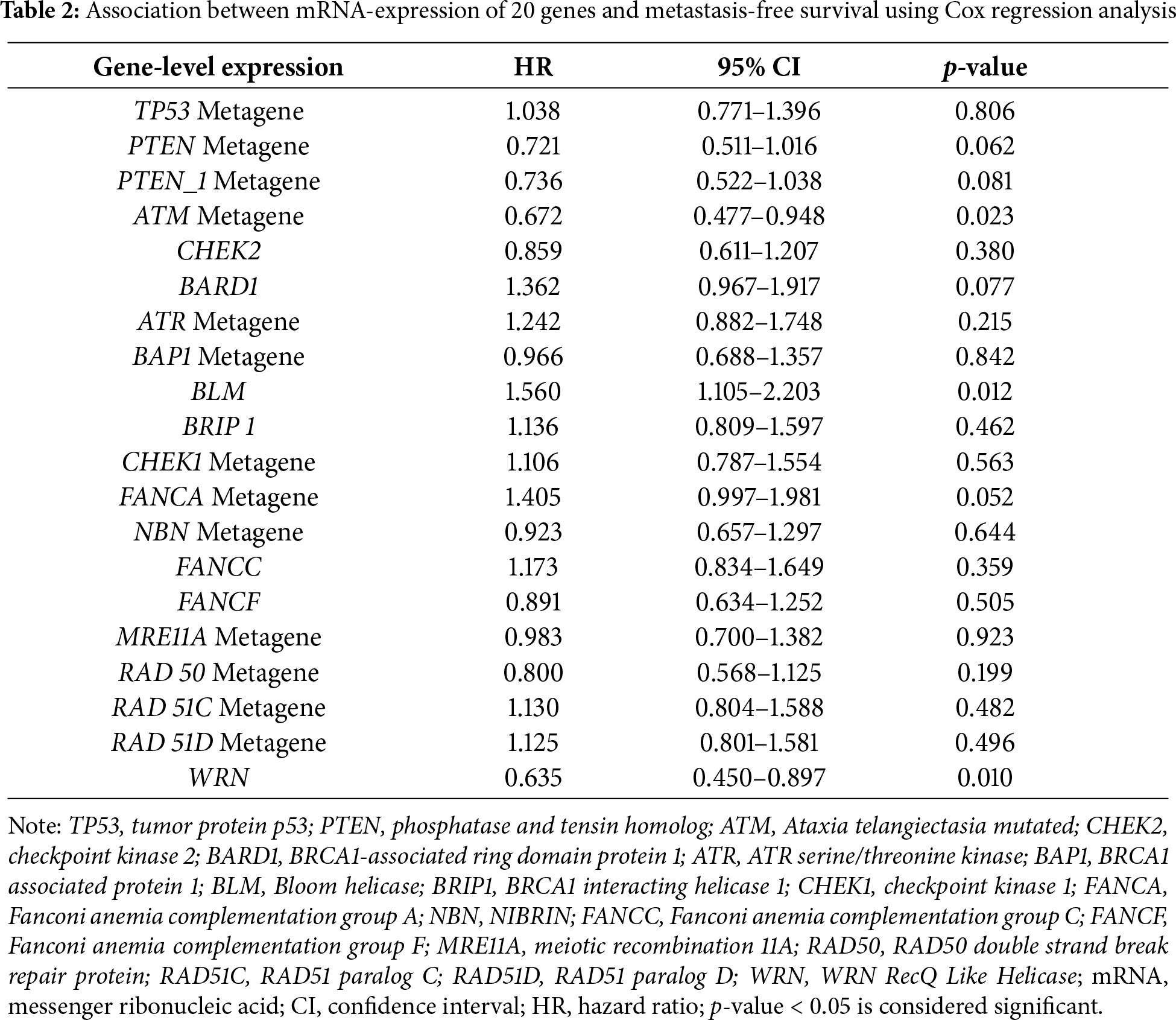
The effect of mRNA expression levels of 20 non-BRCA susceptibility genes on patient survival was evaluated in a cohort of eBC patients with long-term follow-up (median 147 months, ranging from 1 to 306 months). Only three genes (ATM, BLM, WRN) were identified in our analyses for association with MFS. The identified associations remained significant after Bonferroni correction.
3.1 The Prognostic Effect of mRNA Expression of 20 Genes in Patients with eBC
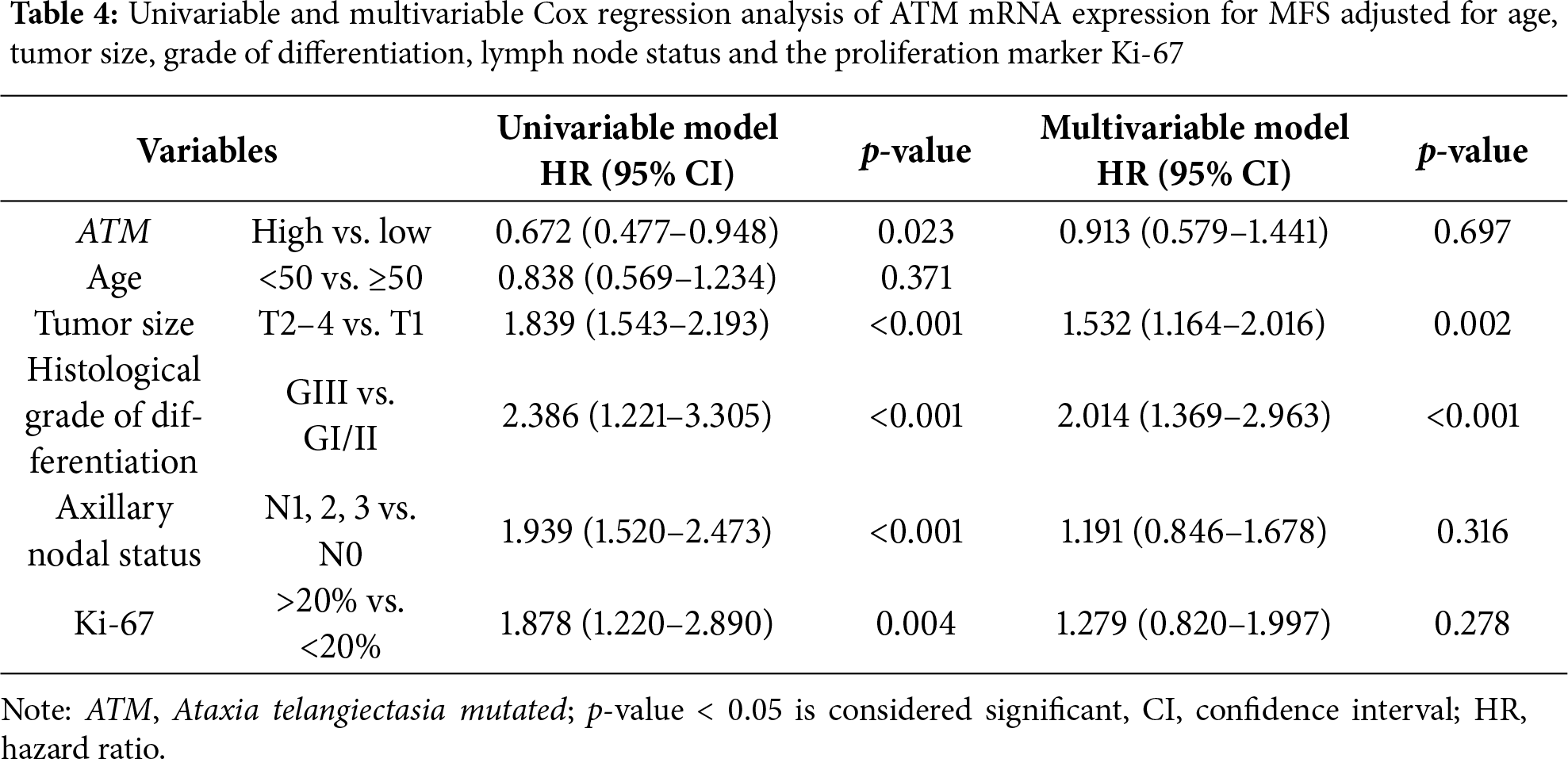
As shown in Table 1, the prognostic significance of non-BRCA susceptibility genes’ mRNA expression was assessed using the available data. ATM mRNA expression levels were significantly associated with MFS (Hazard Ratio, HR 0.672; 95% Confidence Interval, CI 0.477–0.948; p = 0.023). Interestingly, even mRNA expression of BLM and WRN, known as RecQ deoxyribonucleic acid (DNA) helicase family members, demonstrated a significant impact on MFS in the whole cohort (p < 0.05). In contrast, mRNA expression of the remaining genes showed no significant association with MFS (all p > 0.05; Table 2).
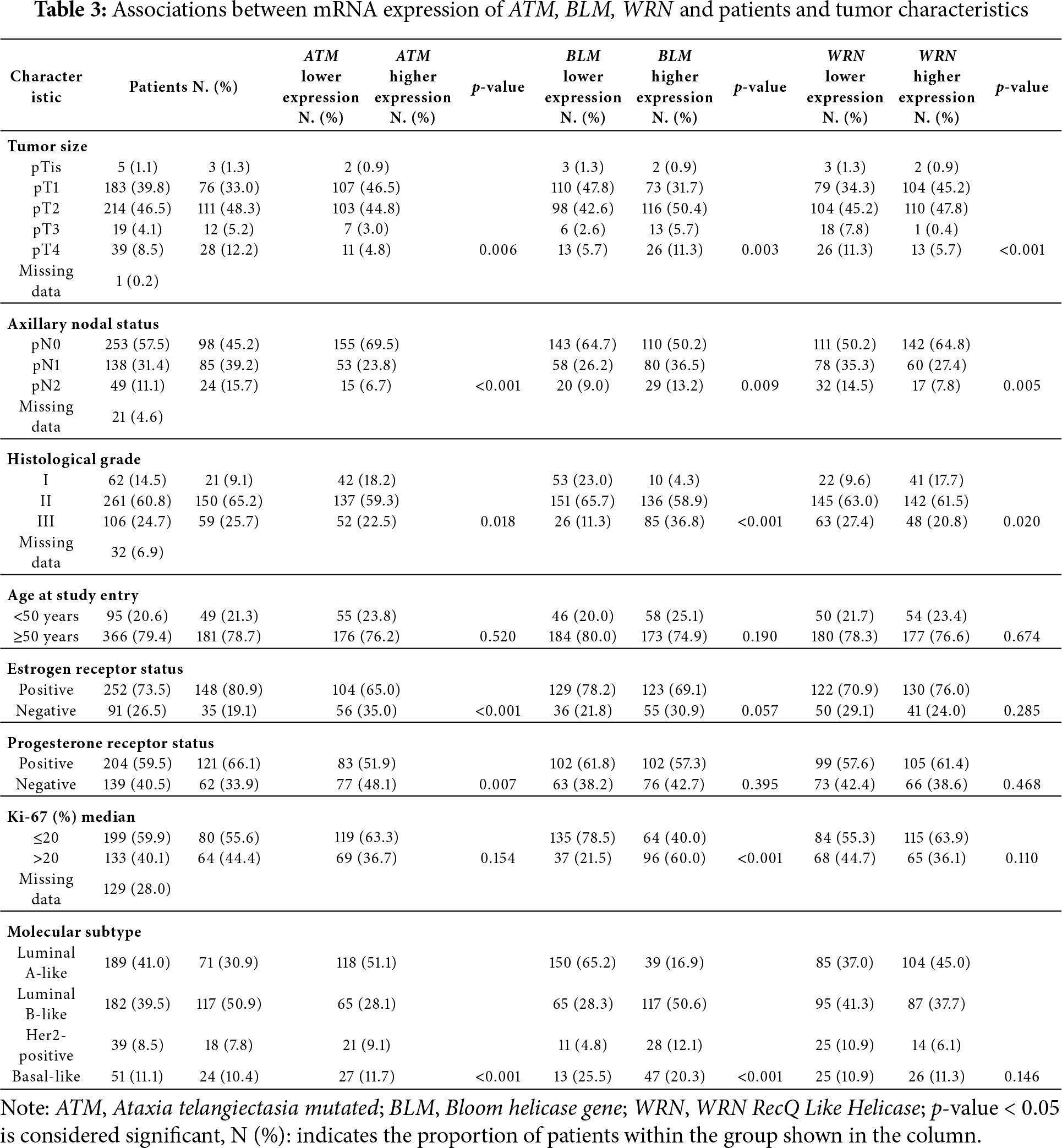
3.2 Patients and Tumor Characteristics
The median age at initial diagnosis of the patients included in our study was 62 years (range: 30–93). Patients and tumor characteristics, as well as their association with mRNA expression of ATM, BLM, and WRN, are outlined in Table 3.
3.3 Impact of ATM mRNA Expression on the Survival of eBC Patients
Kaplan-Meier survival analysis indicated that high mRNA expression of ATM correlated with longer MFS in the whole cohort (p = 0.022; Fig. 1). The median MFS was 148.0 and 103.0 months in each subgroup, respectively. In a multivariate Cox regression analysis, tumor size and histological grading were identified as independent prognostic factors. The multivariate Cox regression analysis, presented in Table 4 shows that ATM mRNA expression levels did not maintain independent significance after adjusting for other clinical variables (HR 0.913; 95% CI 0.579–1.441; p = 0.697).
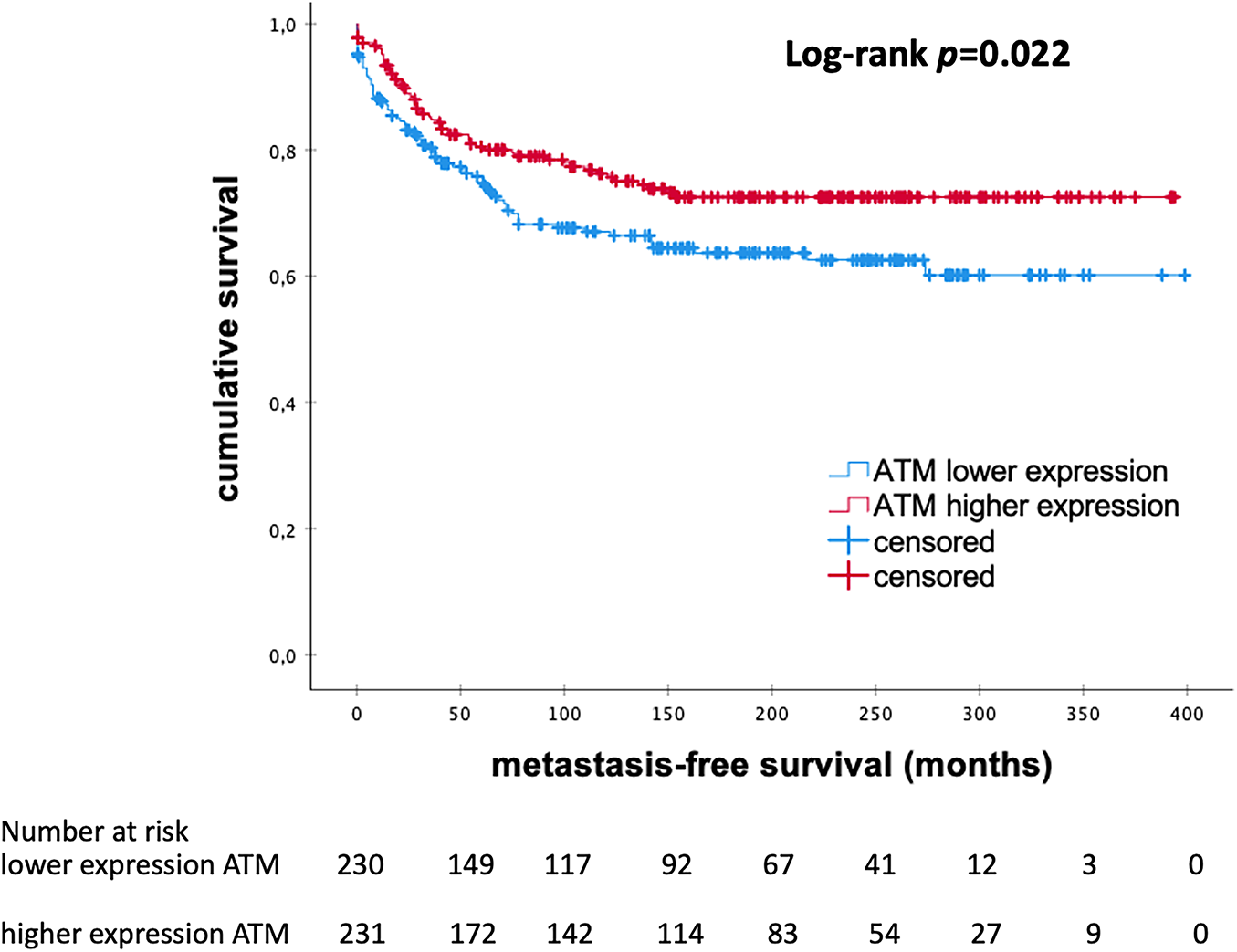
Figure 1: Kaplan–Meier analysis of MFS in patients with eBC according to levels of ATM mRNA expression. ATM, Ataxia Telangiectasia Mutated; p-value < 0.05 is considered significant
3.4 Impact of ATM mRNA Expression on Survival among Different Molecular Subtypes of eBC
Further, subgroup analyses demonstrated a significant impact on MFS in patients with Luminal-B-like eBC. Kaplan Meier analysis indicated that elevated mRNA expression levels of ATM correlate with longer MFS in Luminal-B-like subtype (p = 0.036; Fig. 2). No significant difference was noticed in other molecular subtypes (all p > 0.050). These findings suggest that high mRNA expression levels of ATM are associated with better outcomes, especially in patients with Luminal-B-like eBC.
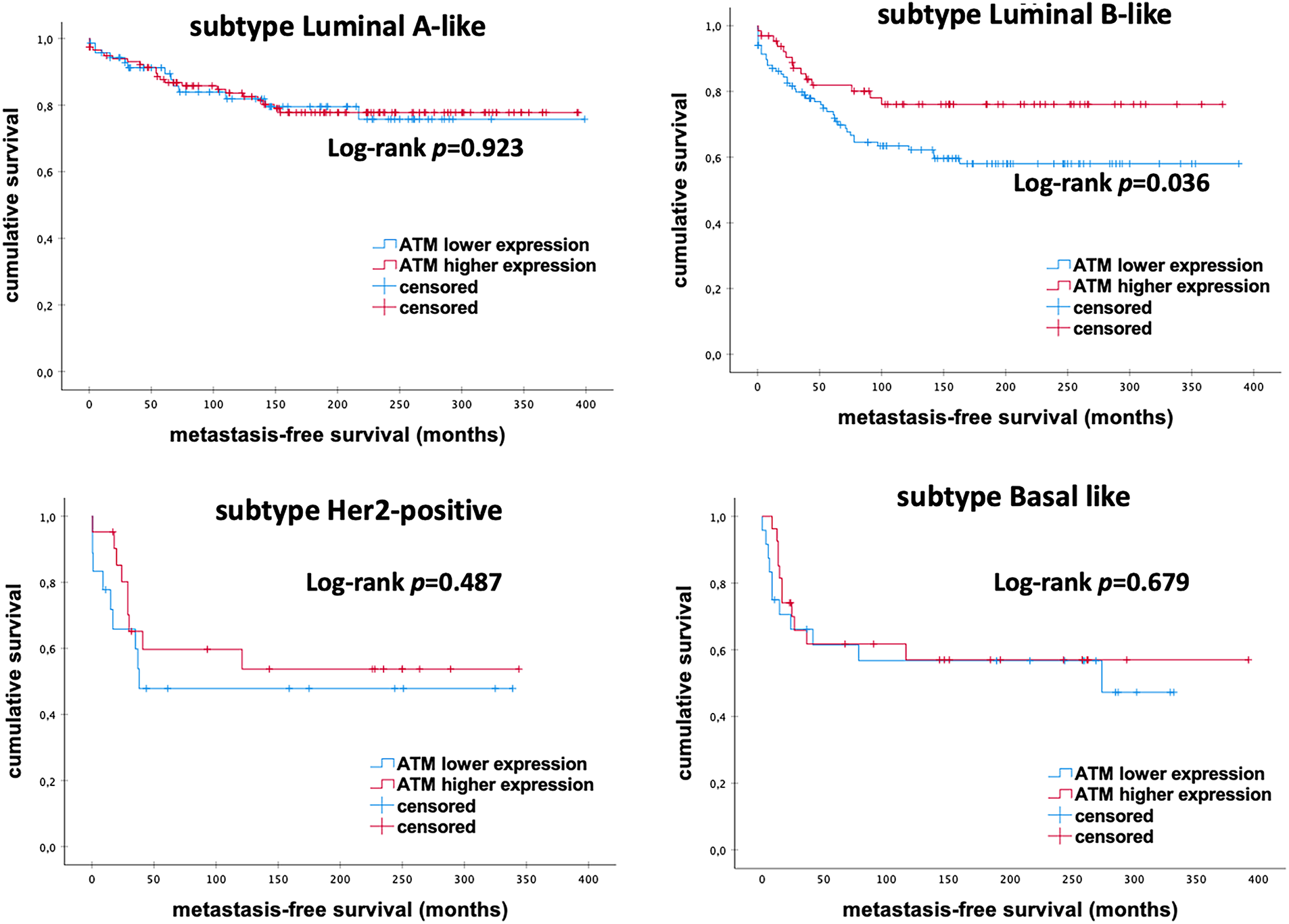
Figure 2: Kaplan–Meier analysis of MFS in eBC patients among different intrinsic molecular subtypes according to ATM mRNA expression levels. ATM, Ataxia Telangiectasia Mutated; p-value < 0.05 is considered significant
3.5 Impact of BLM mRNA Expression on the Survival of eBC Patients
The Kaplan–Meier analysis showed a significant prognostic effect on the levels of BLM mRNA expression among eBC patients. There was a trend in the entire cohort towards a favorable outcome associated with lower mRNA expression of BLM (p = 0.011; Fig. 3). The median MFS was 146.0 months in the subgroup with low levels of BLM expression and 111 months in the high BLM expression group.
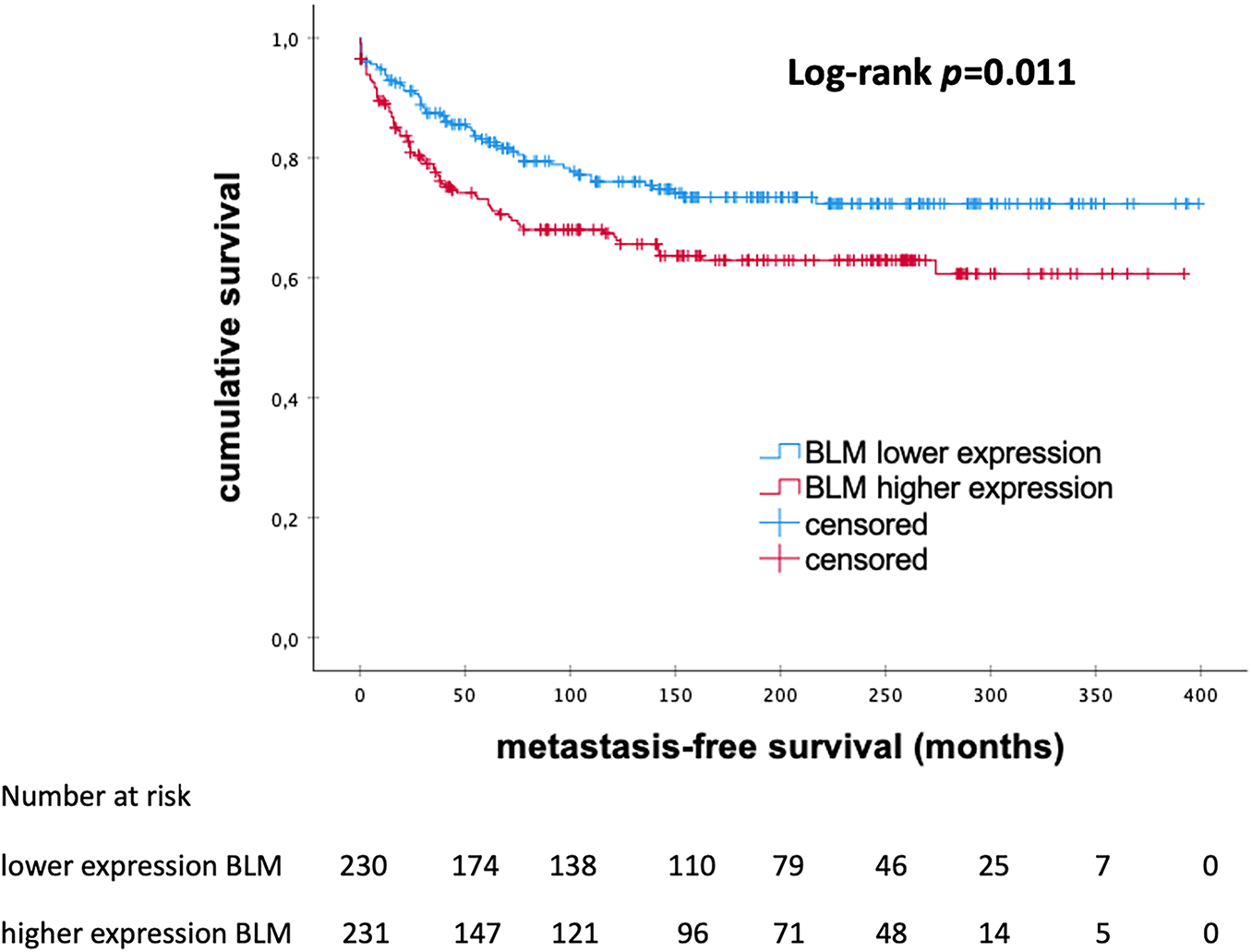
Figure 3: Kaplan–Meier analysis of metastasis-free survival in patients with early breast cancer according to the levels of BLM mRNA expression, BLM, Bloom helicase; p-value < 0.05 is considered significant
The multivariate Cox regression analysis for the BLM mRNA expression (Table 5) indicates that the levels of BLM mRNA expression did not retain independent significance when adjusted for other clinical variables.
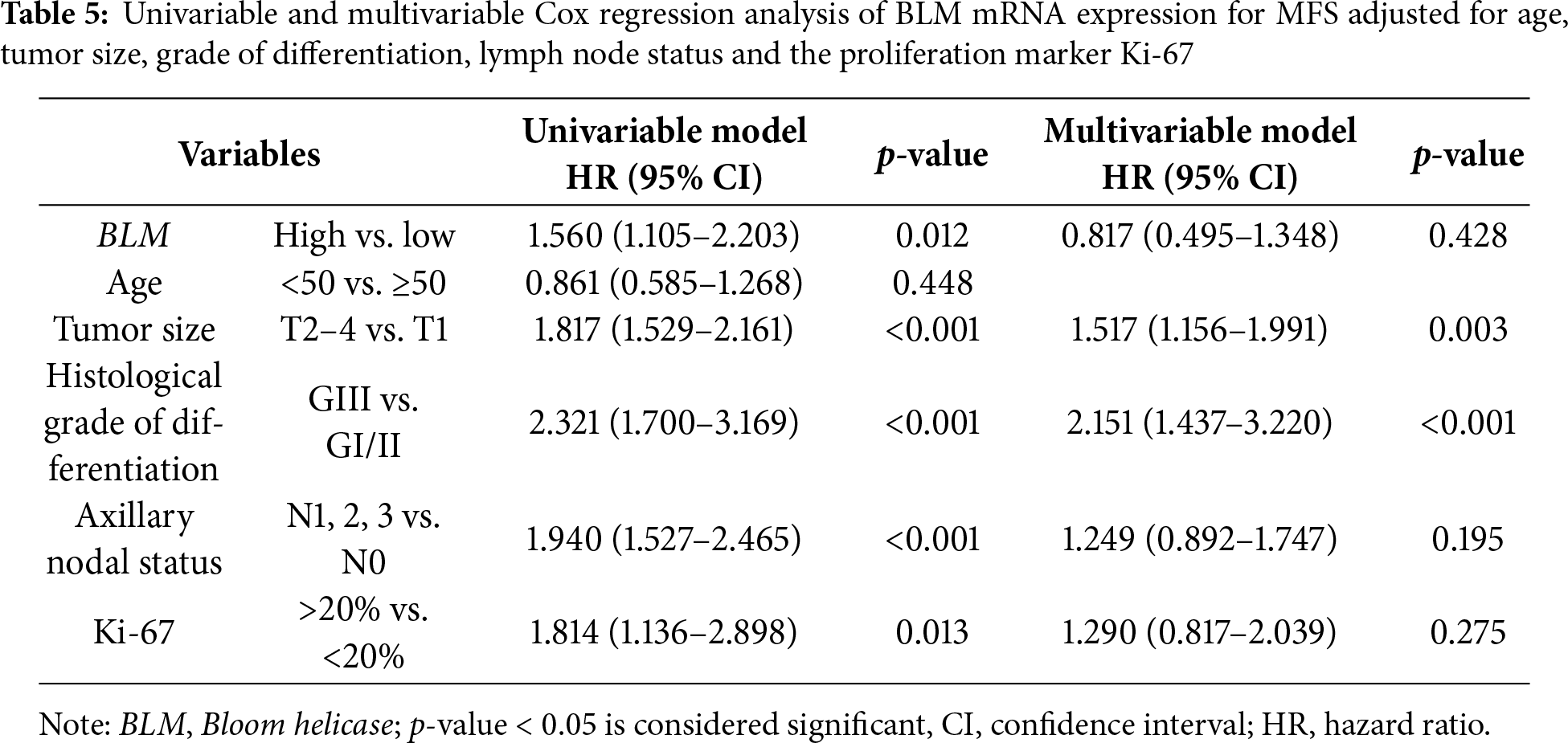
3.6 Impact of BLM mRNA Expression on Survival among Different Molecular Subtypes of eBC
Overall, we detected an association of low BLM mRNA expression with better prognosis in eBC patients (Fig. 3). The level of BLM mRNA expression differed between different intrinsic subtypes of eBC. Stratified subgroup analyses revealed significant survival differences, surprisingly in favor of high mRNA expression among Basal-like tumors. However, the power of the analysis is limited by the very small sample size (p = 0.030; Fig. 4).
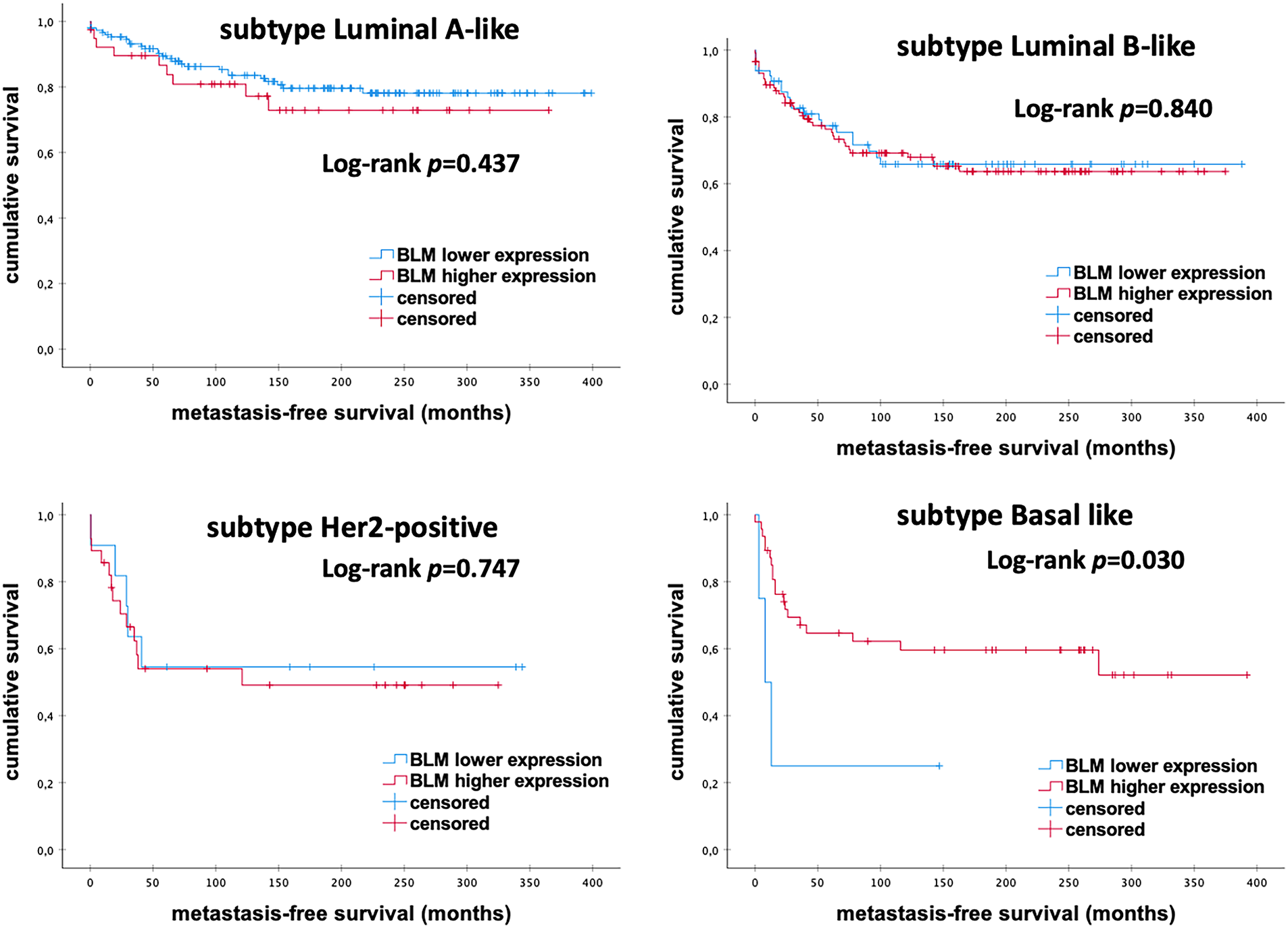
Figure 4: Kaplan–Meier analysis of MFS in eBC patients among different intrinsic molecular subtypes according to the levels of BLM mRNA expression. BLM, Bloom helicase; p-value < 0.05 is considered significant
3.7 Impact of WRN mRNA Expression on the Survival of eBC Patients
The Kaplan–Meier analysis showed that patients with high levels of WRN mRNA expression had significantly longer MFS than those with low mRNA expression (145.0 vs. 102.5 months; p = 0.009) (Fig. 5).
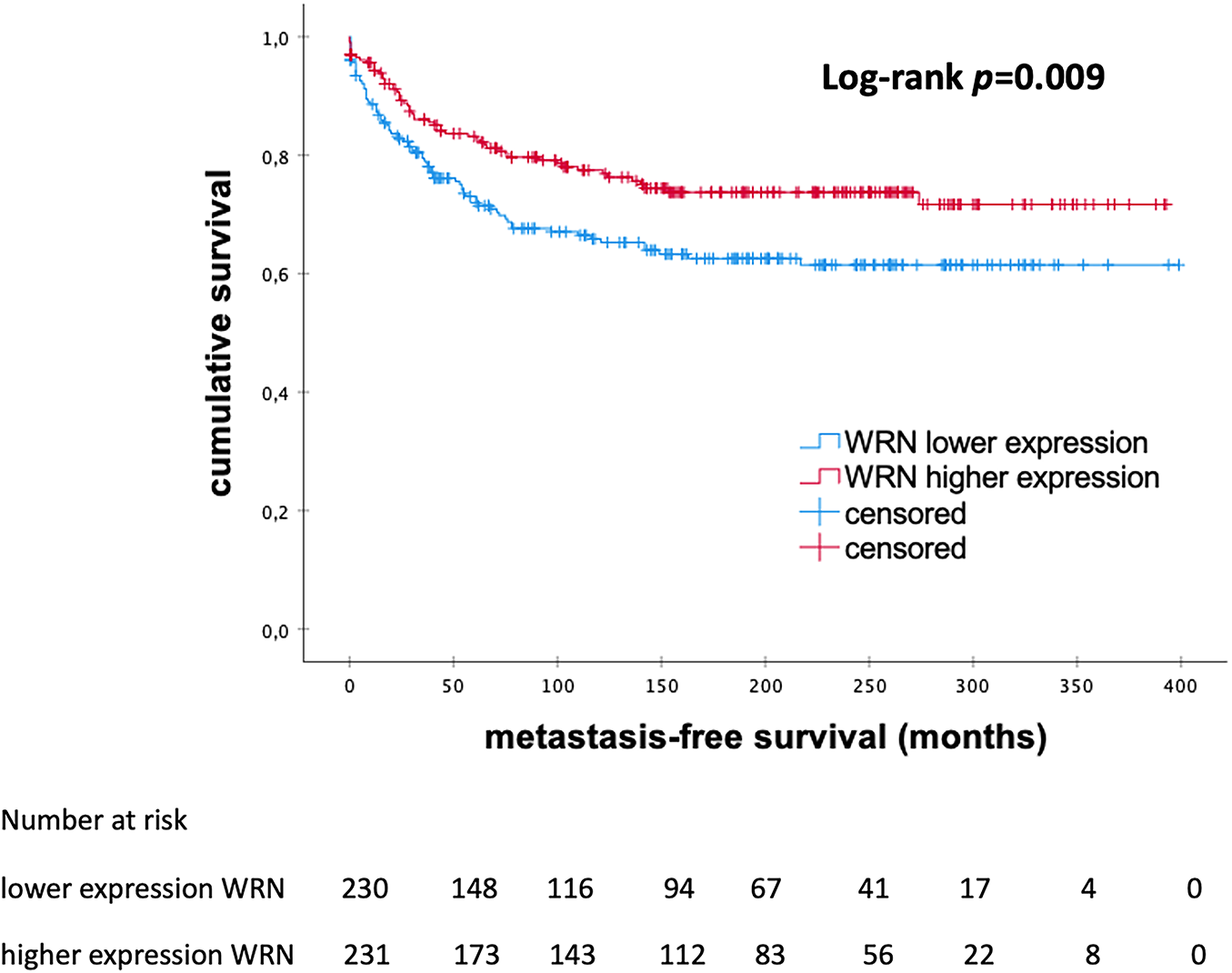
Figure 5: Kaplan–Meier analysis of MFS in patients with eBC according to the levels of WRN mRNA expression. WRN, WRN RecQ Like Helicase; p-value < 0.05 is considered significant
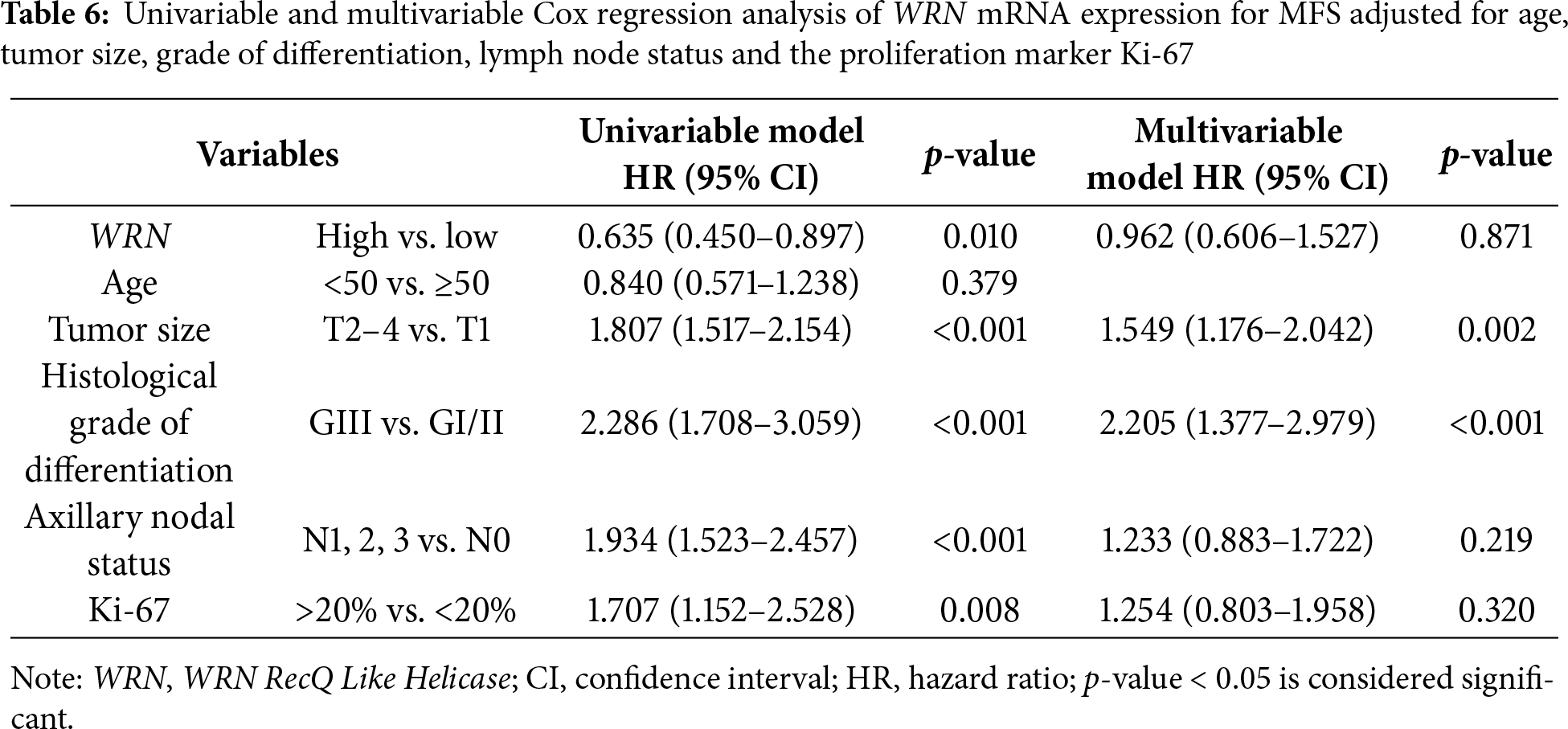
In the multivariable analysis, mRNA expression of WRN could not be identified as an independent prognostic factor (HR 0.962; 95% CI 0.606–1.527; p = 0.871). As shown in Table 6, tumor size and grading were identified as independent prognostic factors.
3.8 Impact of WRN mRNA Expression on Survival among Different Molecular Subtypes of eBC
As shown in Fig. 6, further subgroup analyses revealed a significant increase in MFS rates in patients with Luminal-A-like eBC and high levels of WRN mRNA expression (p = 0.048). However, there were no significant differences in MFS among other molecular subtypes (all p > 0.050).
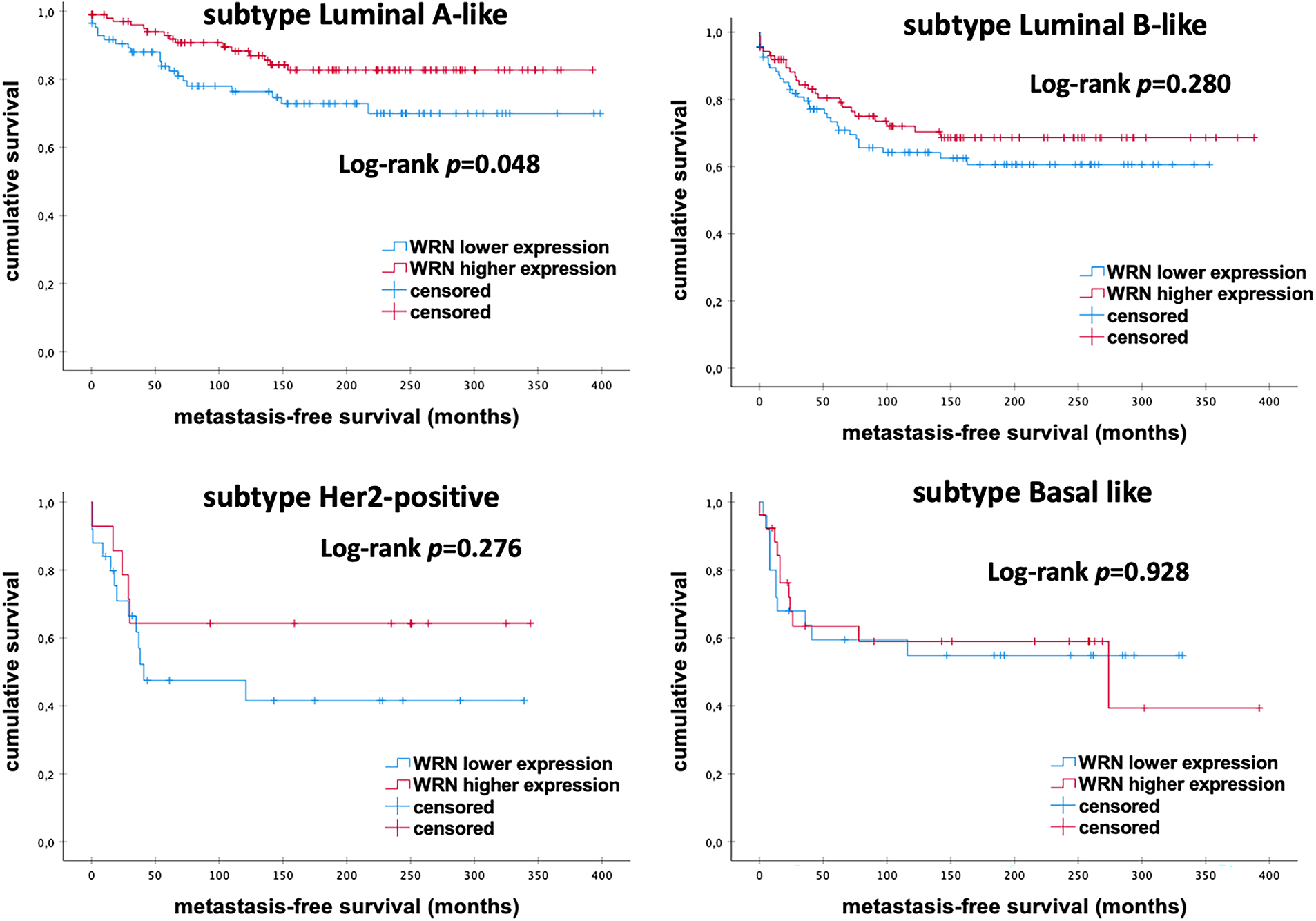
Figure 6: Kaplan–Meier analysis of MFS in eBC patients among different intrinsic molecular subtypes according to the levels of WRN mRNA expression. WRN, WRN RecQ Like Helicase; p-value < 0.05 is considered significant
3.9 The Prognostic Effect of mRNA Expression of ATM, BLM and WRN Genes in eBC Patients without Adjuvant Therapy
We performed further survival analysis in order to investigate the prognostic impact of the mRNA Expression of ATM, BLM and WRN in eBC patients (N = 200), who did not receive any adjuvant therapy. Similar to the entire cohort of patients, even within the untreated cohort, we demonstrated that the mRNA expression level was significantly associated with MFS in Kaplan-Meier and univariable Cox regression analyses (Table 7).
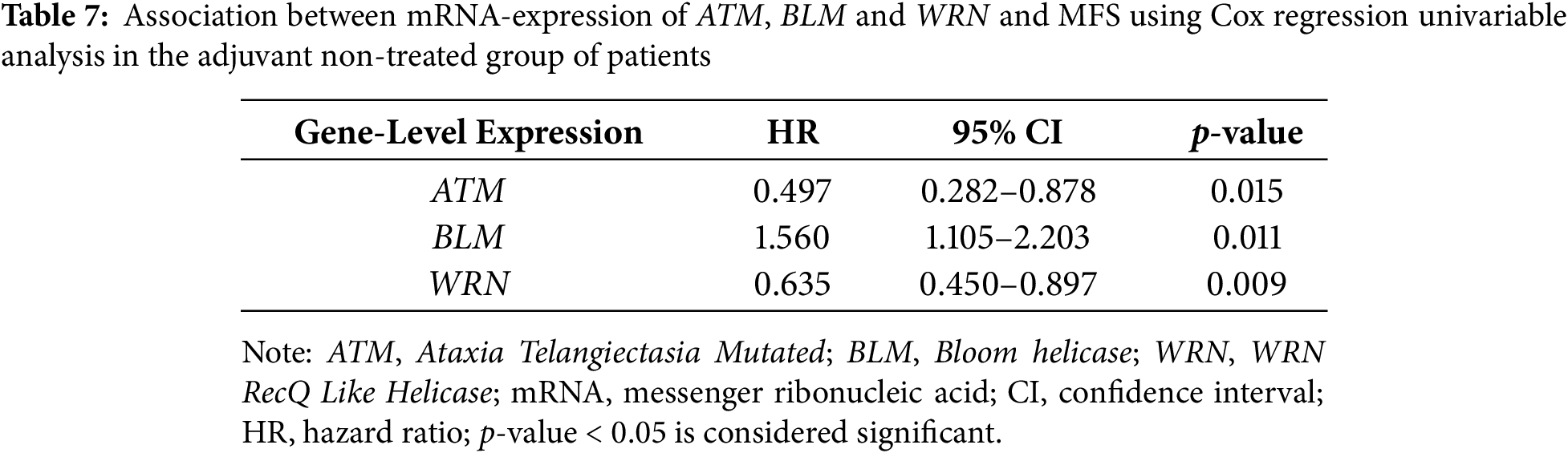
In the multivariable analysis, mRNA expression of ATM, BLM and WRN could not be identified as an independent prognostic factor. The Kaplan-Meier curves can be found as Supplementary Material Figs. S1–S3.
Gene expression analyses in eBC have shown that mRNA expression levels of particular genes are strong independent prognostic biomarkers. In this study, we examined the expression of mRNA levels of 20 non-BRCA genes in a cohort of 461 patients with eBC to assess their prognostic impact. Among the 20 analyzed genes, only the mRNA expression of ATM, BLM and WRN significantly correlated with prognosis in eBC patients. The other non-BRCA genes exhibited no predictive value for MFS.
4.1 The Prognostic Role of ATM mRNA Expression and Its Clinical Impact
Previous studies have established that ATM plays a crucial role in the development and progression of early breast cancer (eBC). Notably, down-regulated ATM mRNA expression has been observed in breast cancer tissues [9,19,20]. However, limited clinical studies have investigated the prognostic significance of ATM expression levels in eBC. Our study indicates that patients with higher ATM expression levels tend to have better survival rates compared to those with lower levels, particularly within the Luminal-B-like eBC subgroup. These findings align with the results of a study. By Ye et al., which involved 471 eBC patients and demonstrated that high ATM mRNA expression correlates with improved disease-free survival (DFS) (HR 0.66; 95% CI 0.36–1.24) and overall survival (OS) (HR 0.80; 95% CI 0.42–1.51) compared to low ATM mRNA expression [9]. Similarly, Bueno et al. found that high ATM expression is associated with better prognosis in eBC (HR 0.554; 95% CI 0.3991–0.784) [10]. Furthermore, Rondeau et al. confirmed ATM expression serves as an independent prognostic marker in breast cancer (N = 454 patients). Their study showed that low levels of ATM protein were linked to poorer MFS (p < 0.001). Patients with lower ATM expression exhibited a 5-year MFS of 70.4 ± 2.5% and a 10-year MFS of 59.0 ± 2.8%. In contrast, those with higher ATM expression had significantly better 5-year (85.7 ± 3.2%) and 10-year (78.7 ± 3.9%) [19].
4.2 The Prognostic Role of BLM mRNA Expression and Its Clinical Impact
Alterations in the mRNA expression of BLM and WRN, both members of the RecQ family, have also been linked to the prognosis of eBC [21,22]. Multiple studies have shown a correlation between expression of the five RECQL genes and breast cancer tumorigenesis [23–25].
Notably, there is increasing evidence regarding the impact of BLM mutations on eBC prognosis [13,25]. Arora et al. investigated BLM mRNA expression in the Molecular Taxonomy of Breast Cancer International Consortium cohort, which included 1650 breast tumors [25]. This study was the first to demonstrate, within a large cohort, that BLM mRNA overexpression is associated with poor breast cancer-specific survival (p < 0.001), supporting the role of BLM as a potential biomarker for eBC. At the protein level, BLM also independently influenced eBC survival; altered subcellular localization and high cytoplasmic BLM were linked to aggressive phenotypes [25].
Similarly, Zhu et al. reported that increased BLM expression is associated with reduced distant MFS across all breast cancer patients [13]. Their conclusions were based on the analysis of prognostic values of RecQ-family mRNA expression in various intrinsic subtypes of breast cancer, utilizing the Kaplan–Meier Plotter database (http://kmplot.com/analysis).
Supporting these findings, our study demonstrates a significant correlation between increased mRNA expression of BLM and poor MFS through both Kaplan-Meier and Cox-regression univariable analyses. Additionally, high BLM mRNA levels were associated with aggressive clinicopathologic characteristics such as larger tumor size, positive axillary node status, and higher histological grade of differentiation. Given BLM’s proposed role in replication and proliferation, it is not surprising that high BLM mRNA levels were more frequently observed in eBC cases with a worse prognosis [26]. Subgroup analysis among different molecular subtypes revealed that low BLM mRNA expression was linked to poor prognosis, specifically in the triple-negative subgroup. These results are consistent with Arora et al.’s findings, which indicated a significant survival advantage associated with high mRNA expression in the ER-negative subgroup (p = 0.049) [25].
Moreover, overexpression of BLM and WRN may contribute to increased resistance of cancer cells to conventional chemotherapy [23]. Another study confirmed the relationship between BLM expression levels and platinum sensitivity; specifically, it examined platinum-sensitive compared to platinum-resistant triple-negative (Basal-like) eBC in two cohorts of patients receiving neoadjuvant cisplatin treatment [27]. Using an integrated genomic approach that combined differential analysis of gene expression and DNA copy number, Birkbak et al. demonstrated that overexpression of BLM enhances sensitivity to cisplatin. This indicates that BLM expression levels may serve as a valuable biomarker for predicting platinum sensitivity in Basal-like breast cancer [27].
4.3 The Prognostic Role of WRN mRNA Expression and Its Clinical Impact
Given the essential role of WRN in DNA repair and replication, several studies have investigated the prognostic potential of WRN expression levels in patients with early-stage breast cancer (eBC) [24]. Shamanna et al. investigated mRNA levels of WRN and Topoisomerase 1 (TOP1) in the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) cohort and found that low WRN mRNA expression (16.5% of tumors) was significantly associated with aggressive clinicopathological characteristics such as: high histological grade, larger tumor size, high risk Nottingham prognostic index (NPI) > 3.4, Her2 over expression, ER- and PR-tumors as well as poor DFS (p < 0.05) [28].
Similarly, Zhu et al. demonstrated in their study using the Kaplan–Meier plotter database to assess the prognostic value of mRNA expression of the five RecQ DNA helicase genes in connection with clinical outcomes in women with eBC that increased WRN mRNA expression correlates with improved overall survival (OS) (HR 0.76; 95% CI 0.61–0.94; p = 0.011) and enhanced relapse-free survival (RFS) (HR 0.81; 95% CI 0.73–0.91; p < 0.001) [13]. In ER-negative eBC patients, higher WRN mRNA expression was associated with better OS (HR 0.64; 95% CI 0.4–1.02; p = 0.056) [13].
These findings are consistent with our own findings, which show that lower mRNA expression of WRN correlates with worse MFS. Further subgroup analyses revealed significantly longer MFS in patients with Luminal-A-like eBC who exhibited high levels of WRN mRNA expression. However, contrary to the findings of Zhu et al., we did not observe significant differences among other molecular subtypes.
Our study highlights the contrasting prognostic implications of BLM and WRN, despite both being members of the RecQ helicase family. This difference likely reflects their distinct roles in genome maintenance and context-dependent cellular functions.
4.4 Controversy in the Prognostic Impact of BLM and WRN
BLM overexpression has been linked to increased genomic instability, which may arise from inappropriate or excessive helicase activity that leads to replication stress. This overactivity of BLM could support tumor cell proliferation and survival in the face of genotoxic stress, potentially explaining its association with poor prognosis [27,29]. In contrast, WRN possesses both helicase and exonuclease activity, playing a crucial role in maintaining telomere integrity. The loss or reduced expression of WRN can impair DNA repair mechanisms, promote chromosomal abnormalities, and accelerate tumor progression [30,31]. Additionally, WRN deficiency has been associated with microsatellite instability and could impact immune surveillance, potentially resulting in more aggressive tumors [32,33]. Thus, the contrasting prognostic roles of BLM and WRN may reflect a complex balance: overactive repair mechanisms (as seen with BLM) may promote the survival of genetically unstable tumor cells, while the loss of essential repair pathways (as with WRN) can increase genomic damage and drive tumor evolution.
Tumors with high BLM expression may demonstrate resistance to DNA-damaging agents due to their enhanced repair capabilities, suggesting that patients might benefit from alternative treatment regimens or combination therapies [27]. Conversely, the loss of WRN expression may make cells more sensitive to genotoxic chemotherapy, indicating that WRN levels could serve as a predictive marker for treatment response [16,34].
4.5 Strengths, Limitations and the Association with New Biomarkers
Our study has several strengths, including a long follow-up period and the availability of mRNA microarray data for 20 non-BRCA genes. This adds further weight to the observed impact of the expression levels of ATM, BLM and WRN on the survival of eBC patients. Importantly, the patients included in the study did not receive any therapy prior to surgery, which eliminates potential treatment-related effects on gene expression levels. Notably, when excluding patients who received adjuvant therapy, the results remained consistent. This suggests that the prognostic value of mRNA expression of ATM, BLM and WRN was not influenced by the adjuvant therapy. However, the study has some limitations. These include its retrospective unicentric design, a relatively small sample size, and the heterogeneity of the cohort. Additionally, there was a lack of information on non-BRCA gene germline mutations among the patients due to the historical nature of the study. As it was an observational study, some selection bias, including unmeasured confounders, could not be entirely avoided. In our study, the mRNA expression of ATM, BLM and WRN did not maintain its independent prognostic significance when adjusted for other clinical variables. This suggests that their impact may be context-dependent and influenced by additional factors.
One of the limitations of our study is the lack of information regarding any potential correlation between breast cancer stem cells and important biomarkers such as Cluster of Differentiation 44 (CD44), b-series ganglioside (GD2+) and the mRNA expression levels of ATM, BLM and WRN. Many patients with breast cancer develop resistance to chemotherapy and experience tumor recurrence, which is primarily driven by breast cancer stem cells (BCSCs) [35]. It is now well established that BCSCs, characterized by markers such as CD44+ and Cluster of Differentiation 24 negative/low (CD24−/low), as well as GD2+, exhibit behavior similar to that of stem cells. These cells have the ability to self-renew and differentiate into mature tumor cells, which allows the cancer to regrow and metastasize [36,37]. Currently, there is no published data directly investigating the correlation between mRNA expression levels of ATM, BLM and WRN and specific markers. However, indirect associations can be observed. For instance, high BLM expression is linked to poorer survival outcomes and correlates with aggressive subtypes of breast cancer, such as triple-negative breast cancer, which are known to be enriched for BCSCs characterized by the CD44+CD24−/low phenotype [38]. The diversity of BCSC populations complicates their eradication and further analysis [35].
4.6 Genetic Background and Clinical Impact
Guidelines for genetic testing have been established to help identify which women should undergo multigene panel testing. Both germline and somatic alterations in the ATM, BLM and WRN genes have been associated with both hereditary and sporadic cases of breast cancer [39–41]. These alterations may interact with tumor subtypes, hormone receptor statuses, and the immune landscape, further influencing prognosis and treatment responses. ATM plays a central role in the DNA damage response following double-strand breaks and in cell cycle checkpoint control [42,43]. Germline heterozygous mutations in the ATM gene, which are found in 0.7% of the population, are associated with an increased risk of breast cancer. Among these mutations, the rare missense variant c.7271T>G (p.V2424G) is linked to a particularly high breast cancer risk [44]. Female carriers of the ATM gene have about a twofold increased lifetime risk of developing ER-positive breast cancer, with a penetrance of 20% to 30% [41,45,46]. The BLM gene is currently being studied as a potential breast cancer susceptibility gene and has shown a connection to survival rates following immunotherapy across various cancers. However, clear evidence indicates that mutations in the BLM gene, when present in a homozygous state, increase the risk of eBC, with an average age of diagnosis around 33 years [47]. In contrast, Kluzniak et al. and other recent studies have indicated that heterozygous BLM mutations do not significantly increase the risk of eBC and likely do not elevate the risk of other cancers either [48]. Research indicates that WRN may be a breast cancer susceptibility gene; however, most studies are based on small cohorts, highlighting the need for validation of these findings [49,50].
We only investigated the prognostic role of mRNA expression of these genes. This study did not examine the association between their mutations and prognosis or mRNA expression.
4.7 Future Perspectives on the Prognostic Impact of ATM, BLM and WRN mRNA Expression
The identification of altered mRNA expression levels in DNA repair genes may not only serve as a prognostic marker but could also guide the selection of patients for targeted therapeutic interventions in eBC. However, the potential therapeutic implications of mRNA Expression of non-BRCA DNA repair genes have been investigated in only a few studies related to eBC.
The association of mRNA expression of ATM, BLM and WRN with prognosis in eBC supports the development of personalized treatment strategies and therapeutic stratification. This could help identify patients who are at a higher risk of disease progression. High mRNA expression of BLM, along with low expression of ATM and WRN, may serve as prognostic biomarkers for pinpointing patients with worse survival outcomes. Incorporating these findings into molecular profiling panels for eBC could aid in personalizing standard treatment approaches and optimizing therapeutic outcomes. Further validation in larger cohorts and clinical trials is essential before these findings can be routinely implemented.
Understanding the role of ATM, BLM and WRN mRNA expression not only provides insights into potential prognostic biomarkers for eBC but also contributes to the development of genomic-based prognostic models. While these approaches necessitate further clinical validation, integrating DNA repair gene expression data into therapeutic decision-making holds promise for advancing precision oncology in eBC.
Future research should focus on validating these results in larger multicenter cohorts and exploring the underlying mechanisms to develop new genomic-based strategies for selecting eBC patients prior to treatment.
This study identifies high levels of ATM and WRN mRNA expression, along with low levels of BLM mRNA expression, as potential favorable prognostic markers for patients with eBC. Incorporating these biomarkers into clinical practice could enhance treatment and follow-up strategies for patients with eBC. Our analysis represents a significant step toward developing improved genomic-based prognostic algorithms.
Acknowledgement: The authors acknowledge Martina Seehase for excellent administrative and Deborah Bennett for their technical support throughout the preparation of this manuscript.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm contribution to the paper as follows: conceptualization, Anne-Sophie Heimes and Marcus Schmidt; methodology, Ina Shehaj; software, Ina Shehaj; validation, Anne-Sophie Heimes, Marcus Schmidt and Ina Shehaj; formal analysis, Ina Shehaj; investigation, Marcus Schmidt; resources, Anne-Sophie Heimes; data curation, Ina Shehaj; writing—original draft preparation, Ina Shehaj; writing—review and editing, Ina Shehaj, Slavomir Krajnak, Katrin Almstedt, Yaman Degirmenci, Roxana Schwab, Kathrin Stewen, Walburgis Brenner, Annette Hasenburg; visualization, Ina Shehaj; supervision, Anne-Sophie Heimes; project administration, Marcus Schmidt. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The main data supporting the findings of this study are available within the paper and its Supplementary Information. The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval: Written informed consent was obtained from all patients, and all clinical investigations were conducted ethically in accordance with ethical and legal standards and in consideration of the Declaration of Helsinki. The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board of the Ethics Committee of Rhineland-Palatinate, Germany [No. 837.139.05 (4797), date 20 October 2005].
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://www.techscience.com/doi/10.32604/or.2025.072222/s1.
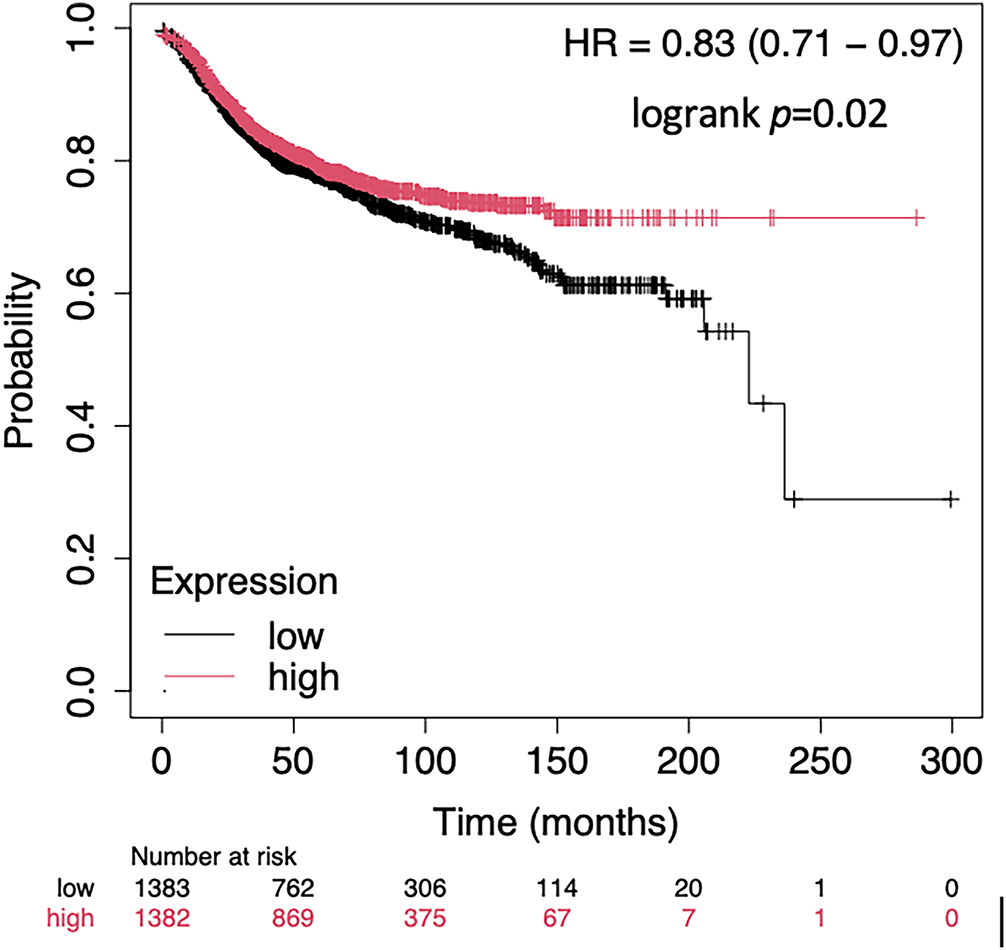
Figure A1: Kaplan Meier Plot of the ATM mRNA expression in terms of MFS of an unselected breast cancer cohort using publicly available gene expression data [18]
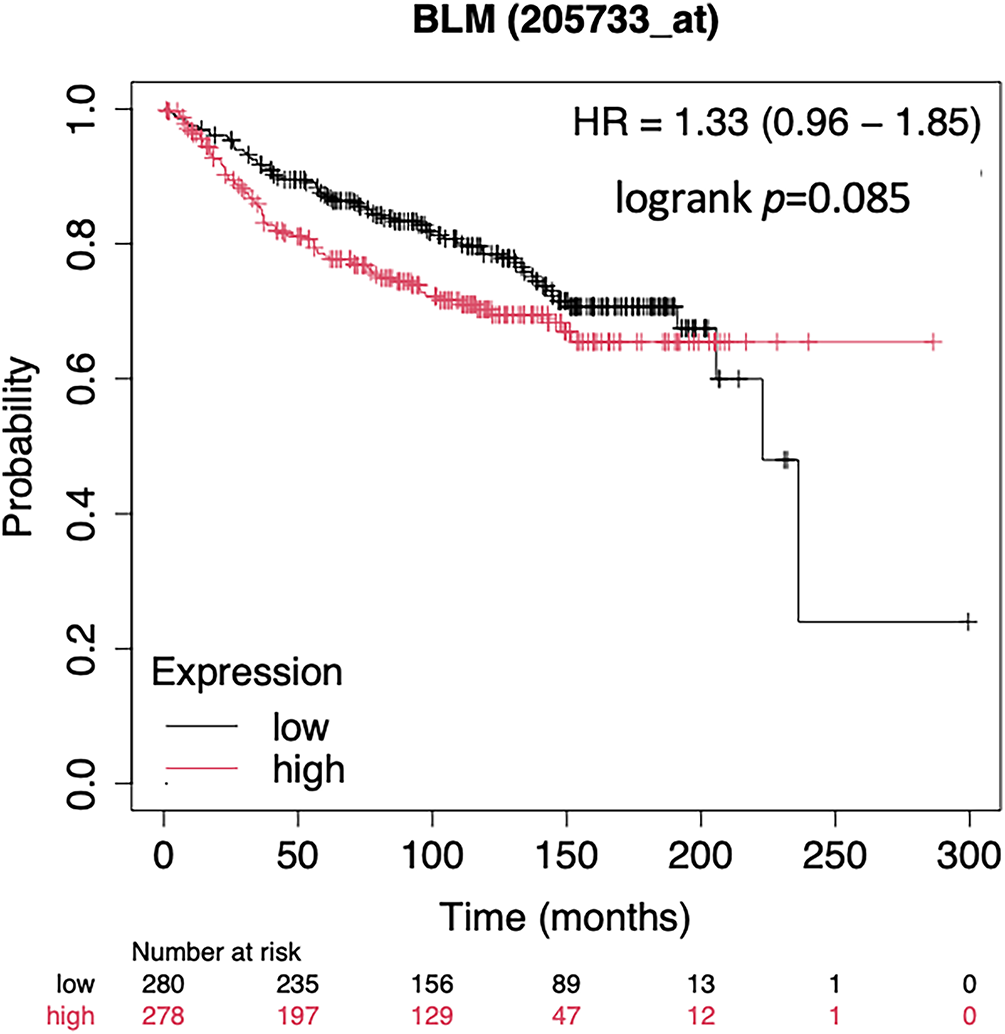
Figure A2: Kaplan Meier Plot of the BLM mRNA expression in terms of MFS of an unselected breast cancer cohort using publicly available gene expression data [18]
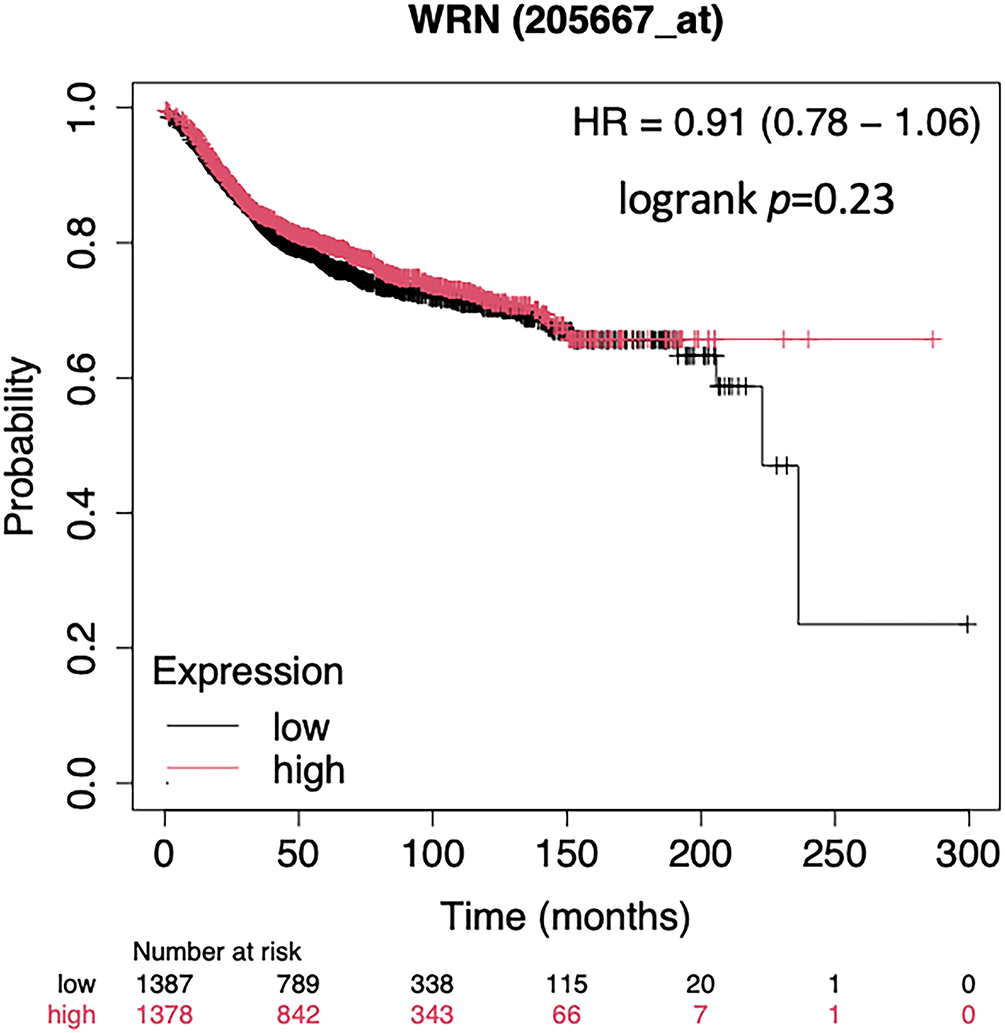
Figure A3: Kaplan Meier Plot of the WRN mRNA expression in terms of MFS of an unselected breast cancer cohort using publicly available gene expression data [18]
References
1. Haibe-Kains B, Desmedt C, Loi S, Culhane AC, Bontempi G, Quackenbush J, et al. A three-gene model to robustly identify breast cancer molecular subtypes. J Natl Cancer Inst. 2012;104(4):311–25. doi:10.1093/jnci/djr545. [Google Scholar] [PubMed] [CrossRef]
2. Heimes AS, Almstedt K, Krajnak S, Runkel A, Droste A, Schwab R, et al. Prognostic impact of LAG-3 mRNA expression in early breast cancer. Biomedicines. 2022;10(10):2656. doi:10.3390/biomedicines10102656. [Google Scholar] [PubMed] [CrossRef]
3. Schmidt M, Böhm D, von Törne C, Steiner E, Puhl A, Pilch H, et al. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008;68(13):5405–13. doi:10.1158/0008-5472.CAN-07-5206. [Google Scholar] [PubMed] [CrossRef]
4. Kurebayashi J, Yamamoto Y, Kurosumi M, Okubo S, Nomura T, Tanaka K, et al. Loss of BRCA1 expression may predict shorter time-to-progression in metastatic breast cancer patients treated with taxanes. Anticancer Res. 2006;26(1B):695–701. [Google Scholar] [PubMed]
5. Shehaj I, Krajnak S, Almstedt K, Degirmenci Y, Herzog S, Lebrecht A, et al. BRCA1, BRCA2 and PALB2 mRNA expression as prognostic markers in patients with early breast cancer. Biomedicines. 2024;12(6):1361. doi:10.3390/biomedicines12061361. [Google Scholar] [PubMed] [CrossRef]
6. Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308(5721):551–4. doi:10.1126/science.1108297. [Google Scholar] [PubMed] [CrossRef]
7. Stucci LS, Internò V, Tucci M, Perrone M, Mannavola F, Palmirotta R, et al. The ATM gene in breast cancer: its relevance in clinical practice. Genes. 2021;12(5):727. doi:10.3390/genes12050727. [Google Scholar] [PubMed] [CrossRef]
8. Miser-Salihoglu E, Demokan S, Karanlik H, Karahalil B, Önder S, Cömert S, et al. Investigation of mRNA expression levels of Tip60 and related DNA repair genes in molecular subtypes of breast cancer. Clin Breast Cancer. 2023;23(2):125–34. doi:10.1016/j.clbc.2022.10.012. [Google Scholar] [PubMed] [CrossRef]
9. Ye C, Cai Q, Dai Q, Shu XO, Shin A, Gao YT, et al. Expression patterns of the ATM gene in mammary tissues and their associations with breast cancer survival. Cancer. 2007;109(9):1729–35. doi:10.1002/cncr.22592. [Google Scholar] [PubMed] [CrossRef]
10. Bueno RC, Canevari RA, Villacis RAR, Domingues MAC, Caldeira JRF, Rocha RM, et al. ATM down-regulation is associated with poor prognosis in sporadic breast carcinomas. Ann Oncol. 2014;25(1):69–75. doi:10.1093/annonc/mdt421. [Google Scholar] [PubMed] [CrossRef]
11. Tommiska J, Bartkova J, Heinonen M, Hautala L, Kilpivaara O, Eerola H, et al. The DNA damage signalling kinase ATM is aberrantly reduced or lost in BRCA1/BRCA2-deficient and ER/PR/ERBB2-triple-negative breast cancer. Oncogene. 2008;27(17):2501–6. doi:10.1038/sj.onc.1210885. [Google Scholar] [PubMed] [CrossRef]
12. Chen CF, Brill SJ. Multimerization domains are associated with apparent strand exchange activity in BLM and WRN DNA helicases. DNA Repair. 2014;22:137–46. doi:10.1016/j.dnarep.2014.07.015. [Google Scholar] [PubMed] [CrossRef]
13. Zhu X, Chen H, Yang Y, Xu C, Zhou J, Zhou J, et al. Distinct prognosis of mRNA expression of the five RecQ DNA-helicase family members—RECQL, BLM, WRN, RECQL4, and RECQL5—in patients with breast cancer. Cancer Manag Res. 2018;10:6649–68. doi:10.2147/CMAR.S185769. [Google Scholar] [PubMed] [CrossRef]
14. Opresko PL, Calvo JP, von Kobbe C. Role for the Werner syndrome protein in the promotion of tumor cell growth. Mech Ageing Dev. 2007;128(7–8):423–36. doi:10.1016/j.mad.2007.05.009. [Google Scholar] [PubMed] [CrossRef]
15. Rossi ML, Ghosh AK, Bohr VA. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair. 2010;9(3):331–44. doi:10.1016/j.dnarep.2009.12.011. [Google Scholar] [PubMed] [CrossRef]
16. Agrelo R, Cheng WH, Setien F, Ropero S, Espada J, Fraga MF, et al. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc Natl Acad Sci U S A. 2006;103(23):8822–7. doi:10.1073/pnas.0600645103. [Google Scholar] [PubMed] [CrossRef]
17. Heimes AS, Härtner F, Almstedt K, Krajnak S, Lebrecht A, Battista MJ, et al. Prognostic significance of interferon-γ and its signaling pathway in early breast cancer depends on the molecular subtypes. Int J Mol Sci. 2020;21(19):7178. doi:10.3390/ijms21197178. [Google Scholar] [PubMed] [CrossRef]
18. Győrffy B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput Struct Biotechnol J. 2021;19:4101–9. doi:10.1016/j.csbj.2021.07.014. [Google Scholar] [PubMed] [CrossRef]
19. Rondeau S, Vacher S, De Koning L, Briaux A, Schnitzler A, Chemlali W, et al. ATM has a major role in the double-strand break repair pathway dysregulation in sporadic breast carcinomas and is an independent prognostic marker at both mRNA and protein levels. Br J Cancer. 2015;112(6):1059–66. doi:10.1038/bjc.2015.60. [Google Scholar] [PubMed] [CrossRef]
20. Savva C, De Souza K, Ali R, Rakha EA, Green AR, Madhusudan S. Clinicopathological significance of ataxia telangiectasia-mutated (ATM) kinase and ataxia telangiectasia-mutated and Rad3-related (ATR) kinase in MYC overexpressed breast cancers. Breast Cancer Res Treat. 2019;175(1):105–15. doi:10.1007/s10549-018-05113-8. [Google Scholar] [PubMed] [CrossRef]
21. Dahlstrom JE, Rakha EA, Lakhani SR, Schnitt SJ. Current topics in breast pathology: expert perspectives. Pathology. 2017;49(2):109–10. doi:10.1016/j.pathol.2016.12.001. [Google Scholar] [PubMed] [CrossRef]
22. Yiu TC, Tu J, Cheung HH. An overview of RecQ helicases and related diseases. Aging. 2025;17(7):1881–907. doi:10.18632/aging.206291. [Google Scholar] [PubMed] [CrossRef]
23. Li XL, Lu X, Parvathaneni S, Bilke S, Zhang H, Thangavel S, et al. Identification of RECQ1-regulated transcriptome uncovers a role of RECQ1 in regulation of cancer cell migration and invasion. Cell Cycle. 2014;13(15):2431–45. doi:10.4161/cc.29419. [Google Scholar] [PubMed] [CrossRef]
24. Thakkar MK, Lee J, Meyer S, Chang VY. RecQ helicase somatic alterations in cancer. Front Mol Biosci. 2022;9:887758. doi:10.3389/fmolb.2022.887758. [Google Scholar] [PubMed] [CrossRef]
25. Arora A, Abdel-Fatah TMA, Agarwal D, Doherty R, Moseley PM, Aleskandarany MA, et al. Transcriptomic and protein expression analysis reveals clinicopathological significance of bloom syndrome helicase (BLM) in breast cancer. Mol Cancer Ther. 2015;14(4):1057–65. doi:10.1158/1535-7163.MCT-14-0939. [Google Scholar] [PubMed] [CrossRef]
26. Kawabe T, Tsuyama N, Kitao S, Nishikawa K, Shimamoto A, Shiratori M, et al. Differential regulation of human RecQ family helicases in cell transformation and cell cycle. Oncogene. 2000;19(41):4764–72. doi:10.1038/sj.onc.1203841. [Google Scholar] [PubMed] [CrossRef]
27. Birkbak NJ, Li Y, Pathania S, Greene-Colozzi A, Dreze M, Bowman-Colin C, et al. Overexpression of BLM promotes DNA damage and increased sensitivity to platinum salts in triple-negative breast and serous ovarian cancers. Ann Oncol. 2018;29(4):903–9. doi:10.1093/annonc/mdy049. [Google Scholar] [PubMed] [CrossRef]
28. Shamanna RA, Lu H, Croteau DL, Arora A, Agarwal D, Ball G, et al. Camptothecin targets WRN protein: mechanism and relevance in clinical breast cancer. Oncotarget. 2016;7(12):13269–84. doi:10.18632/oncotarget.7906. [Google Scholar] [PubMed] [CrossRef]
29. Uechi Y, Fujikane R, Morita S, Tamaoki S, Hidaka M. Bloom syndrome DNA helicase mitigates mismatch repair-dependent apoptosis. Biochem Biophys Res Commun. 2024;723:150214. doi:10.1016/j.bbrc.2024.150214. [Google Scholar] [PubMed] [CrossRef]
30. Multani AS, Chang S. WRN at telomeres: implications for aging and cancer. J Cell Sci. 2007;120(5):713–21. doi:10.1242/jcs.03397. [Google Scholar] [PubMed] [CrossRef]
31. Orren DK, Machwe A. Response to replication stress and maintenance of genome stability by WRN, the Werner syndrome protein. Int J Mol Sci. 2024;25(15):8300. doi:10.3390/ijms25158300. [Google Scholar] [PubMed] [CrossRef]
32. Futami K, Ishikawa Y, Goto M, Furuichi Y, Sugimoto M. Role of Werner syndrome gene product helicase in carcinogenesis and in resistance to genotoxins by cancer cells. Cancer Sci. 2008;99(5):843–8. doi:10.1111/j.1349-7006.2008.00778.x. [Google Scholar] [PubMed] [CrossRef]
33. Kategaya L, Perumal SK, Hager JH, Belmont LD. Werner syndrome helicase is required for the survival of cancer cells with microsatellite instability. iScience. 2019;13:488–97. doi:10.1016/j.isci.2019.02.006. [Google Scholar] [PubMed] [CrossRef]
34. Arai A, Chano T, Futami K, Furuichi Y, Ikebuchi K, Inui T, et al. RECQL1 and WRN proteins are potential therapeutic targets in head and neck squamous cell carcinoma. Cancer Res. 2011;71(13):4598–607. doi:10.1158/0008-5472.CAN-11-0320. [Google Scholar] [PubMed] [CrossRef]
35. Huang AV, Kong Y, Wang K, Brown ML, Mu D. Protein marker-dependent drug discovery targeting breast cancer stem cells. Int J Mol Sci. 2025;26(16):7935. doi:10.3390/ijms26167935. [Google Scholar] [PubMed] [CrossRef]
36. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–8. doi:10.1073/pnas.0530291100. [Google Scholar] [PubMed] [CrossRef]
37. Battula VL, Shi Y, Evans KW, Wang RY, Spaeth EL, Jacamo RO, et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest. 2012;122(6):2066–78. doi:10.1172/jci59735. [Google Scholar] [PubMed] [CrossRef]
38. Ricardo S, Vieira AF, Gerhard R, Leitão D, Pinto R, Cameselle-Teijeiro JF, et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J Clin Pathol. 2011;64(11):937–46. doi:10.1136/jcp.2011.090456. [Google Scholar] [PubMed] [CrossRef]
39. Weigelt B, Bi R, Kumar R, Blecua P, Mandelker DL, Geyer FC, et al. The landscape of somatic genetic alterations in breast cancers from ATM germline mutation carriers. J Natl Cancer Inst. 2018;110(9):1030–4. doi:10.1093/jnci/djy028. [Google Scholar] [PubMed] [CrossRef]
40. Goldgar DE, Healey S, Dowty JG, Da Silva L, Chen X, Spurdle AB, et al. Rare variants in the ATM gene and risk of breast cancer. Breast Cancer Res. 2011;13(4):1–9. doi:10.1186/bcr2919. [Google Scholar] [PubMed] [CrossRef]
41. Hu C, Hart SN, Gnanaolivu R, Huang H, Lee KY, Na J, et al. A population-based study of genes previously implicated in breast cancer. N Engl J Med. 2021;384(5):440–51. doi:10.1056/nejmoa2005936. [Google Scholar] [PubMed] [CrossRef]
42. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14(4):197–210. doi:10.1038/nrm3546. [Google Scholar] [PubMed] [CrossRef]
43. Sinha S, Ng V, Novaj A, Zhu Y, Yazaki S, Pei X, et al. The cold immunological landscape of ATM-deficient cancers. J Immunother Cancer. 2025;13(5):e010548. doi:10.1136/jitc-2024-010548. [Google Scholar] [PubMed] [CrossRef]
44. Chenevix-Trench G. Dominant negative ATM mutations in breast cancer families. J Natl Cancer Inst. 2002;94(3):205–15. doi:10.1093/jnci/94.3.205. [Google Scholar] [PubMed] [CrossRef]
45. Llinares-Burguet I, Sanoguera-Miralles L, García-Álvarez A, Esteban-Sánchez A, Caloca MJ, de la Hoya M, et al. Unproductive alternative splicing of ATM exon 7: mapping of critical regulatory elements and identification of 34 spliceogenic variants. J Mol Med. 2025;103(11):1447–60. doi:10.1007/s00109-025-02595-0. [Google Scholar] [PubMed] [CrossRef]
46. Narod SA. Which genes for hereditary breast cancer? N Engl J Med. 2021;384(5):471–3. doi:10.1056/nejme2035083. [Google Scholar] [PubMed] [CrossRef]
47. Cunniff C, Djavid AR, Carrubba S, Cohen B, Ellis NA, Levy CF, et al. Health supervision for people with Bloom syndrome. Am J Med Genet Part A. 2018;176(9):1872–81. doi:10.1002/ajmg.a.40374. [Google Scholar] [PubMed] [CrossRef]
48. Kluźniak W, Wokołorczyk D, Rusak B, Huzarski T, Kashyap A, Stempa K, et al. Inherited variants in BLM and the risk and clinical characteristics of breast cancer. Cancers. 2019;11(10):1548. doi:10.3390/cancers11101548. [Google Scholar] [PubMed] [CrossRef]
49. Bonache S, Esteban I, Moles-Fernández A, Tenés A, Duran-Lozano L, Montalban G, et al. Multigene panel testing beyond BRCA1/2 in breast/ovarian cancer Spanish families and clinical actionability of findings. J Cancer Res Clin Oncol. 2018;144(12):2495–513. doi:10.1007/s00432-018-2763-9. [Google Scholar] [PubMed] [CrossRef]
50. Tedaldi G, Tebaldi M, Zampiga V, Danesi R, Arcangeli V, Ravegnani M, et al. Multiple-gene panel analysis in a case series of 255 women with hereditary breast and ovarian cancer. Oncotarget. 2017;8(29):47064–75. doi:10.18632/oncotarget.16791. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools