
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Advances in Metabolic Reprogramming and Immune Regulatory Mechanisms in Lung Cancer
1 Department of Respiratory Intervention, The Affiliated Cancer Hospital of Zhengzhou University & Henan Cancer Hospital, No. 127, Dongming Road, Jinshui District, Zhengzhou, China
2 Department of Basic Medicine Science, North Henan Medical University, Xinxiang, China
* Corresponding Authors: Hongbo Wu. Email: ; Rui Li. Email:
(This article belongs to the Special Issue: Direct and Paracrine Interactions within the Tumor or Tumor and its Microenvironment)
Oncology Research 2026, 34(4), 11 https://doi.org/10.32604/or.2026.076176
Received 15 November 2025; Accepted 03 February 2026; Issue published 23 March 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Lung cancer remains the leading cause of cancer-related mortality worldwide, primarily driven by metabolic reprogramming and immune evasion mechanisms within tumor cells. To adapt to the nutrient-deprived tumor microenvironment (TME), lung cancer cells undergo profound metabolic reprogramming, characterized by enhanced glycolysis (the Warburg effect), increased glutamine dependency (mediated by GLS1), and accelerated lipid synthesis (involving enzymes such as FASN). These metabolic alterations not only remodel the TME but also dampen antitumor immune responses by promoting immunosuppressive cell populations (e.g., Tregs and M2 macrophages) and inhibiting effector functions of CD8+ T cells and natural killer (NK) cells. Critically, a bidirectional crosstalk operates between tumor cell metabolism and the immunosuppressive TME: metabolic reprogramming drives immune suppression through metabolite accumulation, whereas the immunosuppressive TME, in turn, promotes tumor cell adaptability—thus forming a positive feedback loop that reinforces immune evasion and therapy resistance. This review elucidates key molecular pathways governing metabolic reprogramming in lung cancer—spanning glucose, amino acid, and lipid metabolism—and their dynamic crosstalk with immune regulation, including epigenetic modifications and non-coding RNA-mediated mechanisms. Additionally, it evaluates emerging therapeutic strategies targeting the metabolic-immune axis, such as inhibitors of HK2 or GLS1 combined with anti-PD-1/PD-L1 agents, which aim to reverse immunosuppression and improve clinical outcomes. By synthesizing recent advances, this work provides a theoretical framework for precision oncology interventions, highlighting the potential of metabolic immunotherapies and future directions integrating AI and multi-omics data to overcome resistance in lung cancer.Keywords
Lung cancer (LC) remains one of the most frequently diagnosed malignancies worldwide, accounting for approximately 1.76 million deaths annually [1]. In developed countries, long-term tobacco control policies have stabilized or reduced male lung cancer incidence, while female incidence continues to rise due to factors like secondhand smoke and air pollution. In developing countries, persistently high smoking rates, industrial pollution, occupational exposure (asbestos, arsenic compounds), and indoor coal burning pollution have driven continued increases in incidence. Non-small cell lung cancer (NSCLC) accounts for approximately 85% of all lung cancer cases, while small cell lung cancer (SCLC) accounts for 15%. Overall, lung cancer patients have a poor prognosis, which is regulated by multiple factors—including stage, pathological type, molecular characteristics, and patient status—and shows significant heterogeneity.
Lung cancer treatment has evolved from the traditional single-modality approach of surgery plus chemoradiotherapy to individualized multimodal therapy, with core goals of extending survival and improving quality of life. For early-stage disease, treatment centers on radical surgery. Unresectable stage III NSCLC relies on concurrent chemoradiotherapy (CCRT); the PACIFIC regimen (CCRT followed by durvalumab, a PD-L1 inhibitor) boosts 5-year survival from 29% to 42.9%, establishing it as the new standard. Limited-stage SCLC uses CCRT as the only potentially curative option (>95% technical success, 20%–25% 5-year survival). Extensive-stage disease employs chemotherapy (EP regimen) plus immunotherapy (atezolizumab/durvalumab), achieving a median survival of 12–13 months.
In recent years, immunotherapy has reshaped advanced NSCLC treatment, emerging as a pivotal first-line standard using PD-1/PD-L1 inhibitors as monotherapy or in chemotherapy combinations. For PD-L1 expression ≥50% tumors, pembrolizumab monotherapy yields 45% objective response rate (ORR), while KEYNOTE-189 combination regimens extend overall survival (OS) from 11.3 to 22 months. In SCLC, atezolizumab-plus-chemo regimens prolong median OS from 10.3 to 12.3 months. However, despite their transformative impact via PD-1/PD-L1 axis blockade, a substantial subset of patients develops primary or acquired resistance. To address therapeutic resistance, combination strategies integrating immune checkpoint inhibitors (ICIs) with radiotherapy, targeted agents (e.g., anti-angiogenic drugs), microbiome modulation, or metabolic interventions (such as JAK inhibitors) exhibit synergistic efficacy. These approaches collectively remodel the tumor microenvironment (TME), enhance antigen presentation, and reverse T-cell exhaustion, thereby overcoming key resistance barriers.
The tumor microenvironment of lung cancer constitutes a dynamic ecosystem composed of diverse immune cells, stromal components, extracellular matrix, and bioactive molecules. As a critical niche supporting tumor growth and survival, the TME exerts profound influences on immune cell function and activity due to its intricate composition and unique physicochemical properties. These characteristics are closely associated with lung cancer progression and critically shape the efficacy of immunotherapeutic interventions. Central to this interplay is the TME’s metabolic landscape, which serves as a key regulator of immune cell functionality. Tumor and stromal cells undergo metabolic reprogramming, leading to the accumulation of metabolites that directly suppress anti-tumor immunity; concurrently, immune cells within the TME adapt metabolically, and their dynamic crosstalk with cancer cells modulates immune evasion and responses to immunotherapy. To sustain survival and proliferation in this hostile microenvironment—marked by hypoxia and nutrient scarcity—tumor cells rely on such metabolic rewiring [2]. This adaptation not only drives tumor growth but also remodels immune responses within the TME [3], a niche pivotal to tumor initiation, progression, invasion, and metastasis [4].
Emerging evidence links therapeutic resistance to three interconnected factors: the metabolic state of tumor cells, metabolic alterations in immune populations, and the TME’s complex regulatory network [5,6]. Against this backdrop, investigating the crosstalk between lung cancer metabolism and immune regulation is critically important for elucidating the pathogenesis, optimizing therapeutic strategies, and improving patient survival (Fig. 1). This review aims to elucidate the metabolic-immune crosstalk in lung cancer by summarizing advances in how metabolic reprogramming drives immune suppression, critically evaluating emerging metabolic-targeted therapies, and providing a theoretical framework for precision immunotherapy combinations.
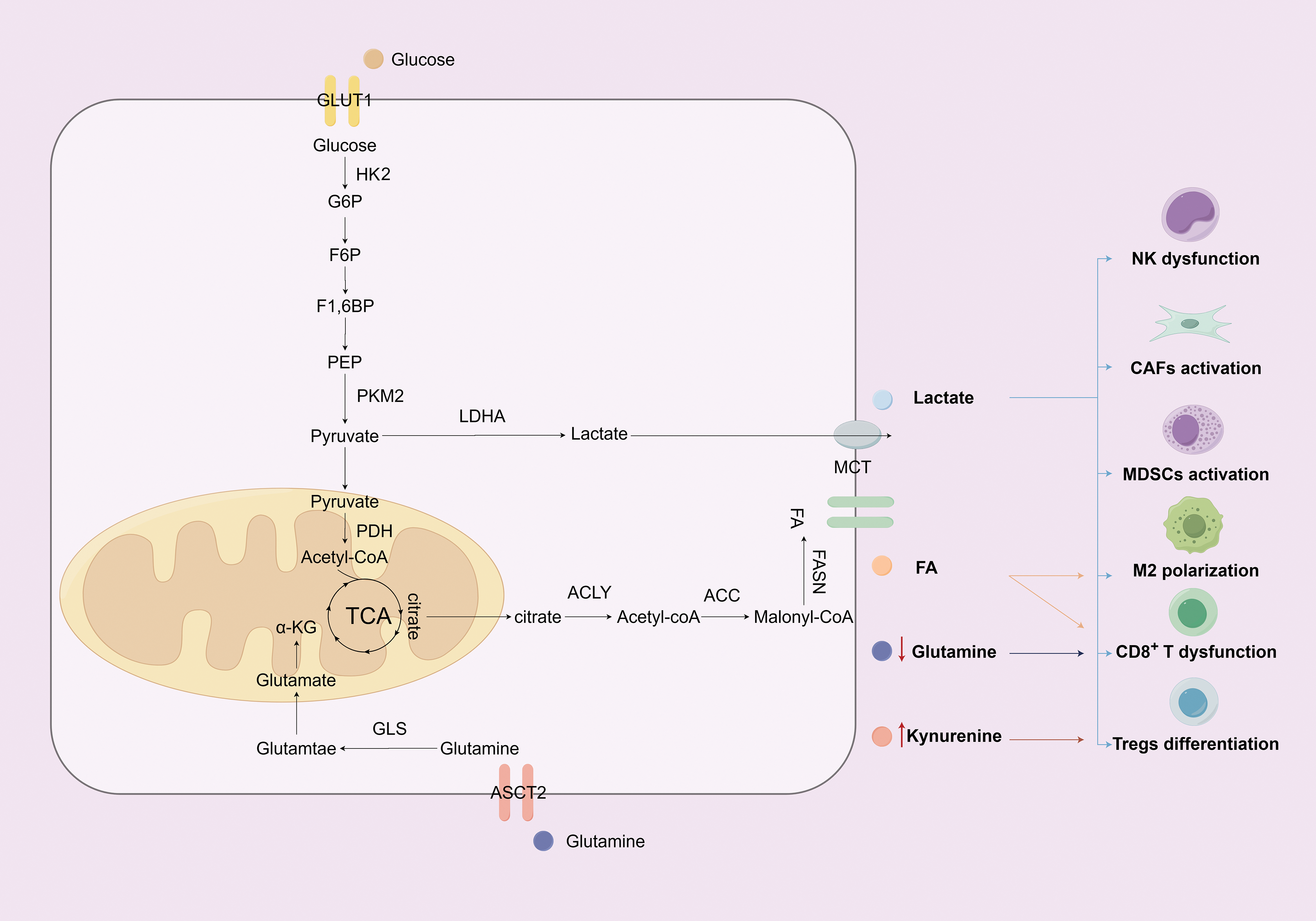
Figure 1: The impact of glucose, amino acid, and fatty acid metabolic reprogramming in lung cancer cells on the TME (Figure created with figdraw). This figure shows that lung cancer cells drive immune evasion through metabolic reprogramming and TME modulation. They enhance glycolysis via key molecules (GLUT1, HK2, PKM2, LDHA), export lactate to acidify the microenvironment—suppressing CD8+ T/NK cells while promoting M2 macrophages, CAFs, and MDSCs. In amino acid metabolism, ASCT2 uptakes glutamine for anabolism; its depletion and kynurenine (tryptophan metabolite) suppress CD8+ T cells and promote Tregs. For fatty acids, upregulated ACC/FASN boost synthesis, aiding proliferation and M2 activation while suppressing CD8+ T cells. These pathways synergize to drive immune evasion. Note: Abbs.: GLUT1, Glucose Transporter 1; HK2, Hexokinase 2; G6P, Glucose-6-Phosphate; F6P, Fructose-6-Phosphate; F1,6BP, Fructose-1,6-Bisphosphate; PEP, Phosphoenolpyruvate; PKM2, Pyruvate Kinase M2; LDHA, Lactate Dehydrogenase A; PDH, Pyruvate Dehydrogenase; TCA, Tricarboxylic Acid Cycle; α-KG, α-ketoglutarate; GLS, Glutaminase; ASCT2, Alanine-Serine-Cysteine Transporter 2; MCT, Monocarboxylate Transporter; FASN, Fatty Acid Synthase; ACLY, ATP-Citrate Lyase; ACC, Acetyl-CoA Carboxylase;NK, Natural killer cells; CAFs, Cancer-associated fibroblasts; MDSCs, Myeloid-derived suppressor cells; Tregs, Regulatory T cells.
2 Metabolic Reprogramming of Lung Cancer Cells
2.1 Glucose Metabolism in Lung Cancer
Glucose metabolism underpins cell proliferation and the establishment of terminal cellular states [7]. Tumor cells exhibit significant glucose metabolic reprogramming, marked by enhanced glucose uptake compared to normal cells and preferential redirection of glycolytic pyruvate to lactate production even under oxygen-replete conditions, a distinctive phenomenon referred to as the “Warburg effect” [8]. Although this glycolytic pathway results in inefficient ATP production due to lactate generation, it compensates for increased energy demands by significantly accelerating glycolytic flux, thereby facilitating rapid tumor cell proliferation and migration [9,10]. The glycolytic pathway comprises ten enzymatic reactions, among which the steps mediated by hexokinase (HK), phosphofructokinase (PFK), and pyruvate kinase (PK) function as irreversible rate-limiting nodes that are druggable targets for effectively blocking glycolysis, thereby reducing energy supply and suppressing tumor cell proliferation [11].
Glucose uptake into cells via glucose transporter proteins (GLUTs) is a prerequisite for glycolysis. In lung cancer, the upregulated expression of GLUT1 facilitates glucose uptake, thereby supplying sufficient energy for rapid tumor cell proliferation [12,13]. Jang et al. [14] observed that tobacco-induced hyperglycemia drives a metabolic-immune axis centered on GLUT1 in lung adenocarcinoma (LUAD), where GLUT1 overexpression enhances glucose uptake in both tumor cells and tumor-associated macrophages (TAMs), directly inducing glycolytic metabolic reprogramming. This metabolic dysregulation promotes TAMs to secrete insulin-like growth factor 2 (IGF2) in a paracrine manner, activating the insulin receptor (IR) signaling pathway in lung cancer cells. The activated IR undergoes nuclear translocation and forms a complex with nucleophosmin (NPM1), thereby upregulating PD-L1 expression. This GLUT1-dominated axis not only remodels the TME—notably by increasing M2-like TAM infiltration and suppressing T cell function—but also synergistically promotes immune evasion and tumor progression, highlighting GLUT’s pivotal role in linking glycolysis to TME regulation in LUAD. Zhou et al. [15] found that GLUT1 is overexpressed at both transcriptional and translational levels in LUAD, and its high expression is positively correlated with advanced clinical stage, poor differentiation, and lymph node metastasis. In LUAD patients, higher GLUT1 expression predicts poorer prognosis, which is further exacerbated when combined with Epidermal Growth Factor Receptor (EGFR) 19DEL or L858R mutations. Mechanistically, GLUT1 directly interacts with phospho-EGFR (p-EGFR) and inhibits EGFR protein degradation via the ubiquitin-proteasome pathway. Notably, GLUT1 inhibitors can enhance the sensitivity of LUAD cells to gefitinib. These findings suggest that targeting GLUT1 alone or in combination with EGFR-TKIs may represent a promising therapeutic strategy for LUAD.
Glucose is phosphorylated to glucose-6-phosphate (G6P) by hexokinase 2 (HK2), an enzyme frequently overexpressed in multiple malignancies [16]. HK2 binds to the voltage-dependent anion channel (VDAC) on the outer mitochondrial membrane, and this localization not only prevents feedback inhibition by G6P, but also enhances glycolytic flux [17]. HK2 promotes stemness properties in tumor cells, and its overexpression in NSCLC is related to poor patient prognosis [18,19]. Daurisoline, a bis-benzylisoquinoline alkaloid isolated from the traditional Chinese herb Menispermum dauricum, was demonstrated by Tan et al. [20] to suppress glycolysis and remodel the TME in lung cancer through modulation of the AKT-HK2 axis. Mechanistically, daurisoline directly binds to AKT, inhibiting its phosphorylation at Ser473; this reduction in AKT activity subsequently decreases GSK3β phosphorylation at Ser9, which enhances the ubiquitination and proteasomal degradation of c-Myc—a critical transcription factor that drives HK2 expression. Li et al. [21] proved that the circular RNA circRUNX1 acts as a molecular sponge to competitively bind miR-145, thereby relieving the post-transcriptional repression of HK2 mRNA by miR-145 and significantly upregulating HK2 protein expression. The elevated HK2 expression potently enhances the Warburg effect—characterized by increased glucose uptake, augmented lactate production, and elevated ATP generation—within tumor cells. The excessive lactate accumulated in the TME leads to microenvironmental acidification, which in turn promotes the infiltration and proliferation of regulatory T cells (Tregs) while suppressing the function of cytotoxic T cells, ultimately driving immune evasion in NSCLC. The findings highlight the critical role of the circRUNX1/miR-145/HK2 pathway in linking metabolic reprogramming to immune escape in NSCLC, providing a rationale for targeting this axis in cancer therapy.
Pyruvate kinase (PK) catalyzes the transformation of phosphoenolpyruvate and ADP into pyruvate and ATP [22]. During tumorigenesis, PKM1 or PKML/R gradually declines, while PKM2 is significantly upregulated, making PKM2 the tumor-specific isoform of pyruvate kinase [23]. Long et al. [24] demonstrated that PKM2 exhibits high expression in LUAD. Its elevated expression correlates with tumor invasiveness, positioning PKM2 as an independent prognostic marker that may guide therapeutic targeting to improve lung cancer outcomes. In the research conducted by Markowitz et al. [25], PKM2 regulates the differentiation of CD8+ T cells and their responsiveness to PD-1 checkpoint blockade. That is, by inhibiting glycolysis through PKM2, the activity of the pentose phosphate pathway (PPP) increases, leading to the enrichment of CD8+ T cells phenotype with TCF1high progenitor exhausted-like population, and enhancing the responsiveness to PD-1 blockade in vivo.
Hypoxia-inducible factor-1α (HIF-1α) modulates the expression of multiple critical glycolytic enzymes [26]. HIF-1α upregulates glycolytic enzyme activity by 90%, generates excessive lactate effluxing into the TME, induces metabolic competition with T cells, and ultimately suppresses lymphocyte antitumor function to promote immune evasion [27]. In patients with NSCLC, HIF-1α-positive tumor expression correlates with lower overall survival compared to HIF-1α-negative expression [28]. During NSCLC progression, aldolase A (ALDOA) expression positively correlates with nuclear HIF-1α levels. ALDOA overexpression enhances lactate secretion, which subsequently inhibits PHD enzymatic activities and contributes to HIF-1α protein stabilization [29], while reactive oxygen species (ROS) also stabilize HIF-1α by inhibiting PHD [30]. The tumor-suppressive effects of PHD2 on NSCLC proliferation, metabolic regulation, and ROS induction entirely rely on its enzymatic activity, as loss of activity fully abolishes these functions [31].
Notably, tumor cell metabolism demonstrates significant heterogeneity rather than a uniform pattern. Tumor cells adjacent to blood vessels predominantly utilize oxidative phosphorylation (OXPHOS) to generate energy, while hypoxic regions rely on glycolysis for glucose metabolism. This spatial metabolic heterogeneity fosters a symbiotic network, enabling tumor cells to adapt to the fluctuating hypoxic environment [32]. Neuroendocrine (NE) cells in small cell lung cancer (SCLC) exhibit electrophysiological excitability, with their action potential discharges contributing to tumor progression. This electrical activity not only promotes tumor expansion but also results in increased ATP demand and a strong reliance on OXPHOS [33]. NSCLC cells with CIP2A overexpression exhibit metabolic reprogramming characterized by increased OXPHOS and suppressed aerobic glycolysis [34]. Additionally, apoptosis-inducing factor (AIF) promotes tumor cell proliferation in KrasG12D-driven lung cancer by regulating OXPHOS, and high AIF expression correlates with unfavorable patient outcomes [35].
2.2 Amino Acid Metabolism in Lung Cancer
Glutamine (GLN), with its uptake and metabolic demand significantly increased in tumor cells under malignant contexts, acts as a conditionally essential amino acid under tumor metabolic reprogramming [36]. Lung cancer cells achieve efficient glutamine uptake through high expression of the alanine-serine-cysteine transporter-2 (ASCT2) [37]. Cancer cells exhibit elevated GLN consumption compared to other amino acids, promoting their rapid proliferation [36,38]. Once inside cells, glutaminase (GLS) catalyzes the conversion of GLN to glutamate, which is then transformed into α-ketoglutarate (α-KG) that enters the TCA cycle. Among GLS isoforms, GLS1 overexpression in various cancers promotes tumor stage progression [39]. GLS1 produces two splice variants—glutaminase C (GAC) and kidney-type glutaminase (KGA)—through alternative pre-mRNA splicing, both of which exhibit differential regulatory activities [40]. GAC is upregulated in lung cancer, a phenomenon closely linked to GLN metabolic reprogramming [41]. Increasing GLN levels in the TME may further enhance T cell activation and proliferation, thereby strengthening antitumor immunity [42].
Oncogene activation drives significant GLN dependence in lung cancer cells [43,44]. The transcription factor c-MYC, which regulates cell growth, proliferation, and metabolism, is frequently overexpressed in lung cancer and directly upregulates the key metabolic enzyme GLS1 [45,46]. c-MYC regulates GLN uptake and hydrolysis by modulating ASCT2 and GLS1 expression [47], while also regulating mitochondrial respiratory chain function and biogenesis, thereby influencing GLN metabolism [48]. In LUAD, the transcription factor TFDP1 transcriptionally activates the expression of SPC25 by binding to its promoter region, resulting in SPC25 overexpression [49]. This event markedly enhances the GLN metabolic pathway, which drives the synthesis and accumulation of Glutathione (GSH). Elevated GSH levels impair the antitumor immune function of natural killer (NK) cells through multiple mechanisms: suppressing the expression of activation receptors on NK cells, reducing cytotoxicity, and diminishing the secretion of key effector molecules such as perforin, granzyme B, and interferon-gamma (IFN-γ). Wu et al. [50] revealed that HHLA2 inhibits glutamine metabolism in CD8+ T cells via the KIR3DL3 receptor, driving immune escape in EGFR-mutant LUAD. Blocking the HHLA2-KIR3DL3 axis restores T cell function and exerts synergistic anti-tumor effects when combined with EGFR-TKIs. Thus, HHLA2 antibodies alone or in combination with EGFR-TKIs may serve as a novel therapeutic option for EGFR-mutant lung cancer patients, particularly those with immunotherapy resistance.
Despite the high uptake of glucose and glutamine by cancer cells, these nutrients often remain insufficient to fully meet the biosynthetic demands. Instead, serine and other similar amino acids provide a significant portion of the carbon and nitrogen needed for cancer cell survival [51]. Under serine-sufficient conditions, PKM2 is activated by serine to maintain efficient glycolysis; however, when serine is limited, reduced PKM2 activity redirects glycolytic intermediates toward the serine synthesis pathway (SSP) [52]. Within this pathway, 3-phosphoglycerate (3-PG), a glycolytic intermediate, is converted into 3-phosphohydroxypyruvate via phosphoglycerate dehydrogenase (PHGDH). It is subsequently transformed into 3-phosphoserine (3-PS) and α-KG via phosphoserine aminotransferase (PSAT1). Ultimately, 3-PS is transformed into serine via phosphoserine phosphatase (PSPH) [53,54]. In lung adenocarcinoma, high PHGDH protein expression and elevated mRNA levels of key SSP enzymes are linked to poor overall survival [53]. PSAT1 upregulation in NSCLC enhances cancer cell proliferation, metastasis, and chemotherapy resistance, leading to adverse outcomes [54].
2.3 Lipid Metabolism in Lung Cancer
Cancer cells also undergo lipid metabolic reprogramming to acquire energy reserves for rapid proliferation and membrane synthesis substrates [55]. Non-malignant cells preferentially utilize exogenous fatty acids to meet metabolic demands, whereas malignant cells activate de novo synthesis pathways through metabolic reprogramming to sustain accelerated proliferation and heightened biosynthetic requirements [56]. Lipids can enter cells via CD36 or passive diffusion, with fatty acid-binding proteins (FABPs) facilitating their uptake [57]. De novo fatty acid synthesis initiates with the transport of citrate from mitochondria to the cytosol via the citrate-pyruvate cycle; subsequently, citrate is cleaved by ATP-citrate lyase (ACLY) into cytosolic acetyl-CoA and oxaloacetate; acetyl-CoA carboxylase (ACC) then catalyzes the carboxylation of cytosolic acetyl-CoA to generate malonyl-CoA; finally, fatty acid synthase (FASN) condenses acetyl-CoA and malonyl-CoA into 16-carbon palmitate in the presence of NADPH. Through the elongase system, the resulting palmitate is extended to form long-chain fatty acids or is desaturated via stearoyl-CoA desaturase-1 (SCD1) [58–60]. In human lung cancer tissue, aberrantly high expression levels of ACLY and FASN drive tumor progression by promoting lipogenesis, which is significantly linked to unfavorable prognosis in patients [61]. Elevated SCD1 levels in lung cancer are closely associated with enhanced cancer cell invasiveness, poorer prognosis, and increased chemoresistance [62]. In NSCLC, EGFR-TKI-resistant cells exhibit significant lipid droplet accumulation and upregulated SCD1 expression compared to drug-sensitive cells, with this phenotype driving the maintenance of drug resistance [63]. Fatty acid oxidation (FAO) requires carnitine palmitoyltransferase1 (CPT1) to activate long-chain fatty acids [64,65]. CPT1C shows elevated expression in lung cancer, where it promotes FAO to increase ATP and ROS generation for metabolic stress adaptation [66], and enhances the chemoresistance of NSCLC cells to cisplatin [67]. Arachidonic acid (AA), a long-chain polyunsaturated fatty acid, plays multifaceted roles in tumor progression, including promoting cell cycle progression, enhancing proliferation, and facilitating metastasis. Additionally, AA contributes to malignant behavior by modulating the TME. Studies [68] have shown that PLA2G10-mediated AA secretion promotes LUAD progression by suppressing CD4+ T cell activation, with minimal impact on the proliferation and migration of tumor cells themselves. Mechanistically, PLA2G10 elevates AA levels in the TME, which inhibits the phosphorylation of LCK (Y394) and ZAP70 (Y391), thereby suppressing CD4+ T cell activation and fostering immunosuppression. Combination therapy with PLA2G10 inhibitors and anti-PD-1 antibodies has demonstrated efficacy in lung cancer treatment. These findings suggest that AA-centered metabolic profiling could serve as a prognostic indicator for LUAD and identify AA metabolism as a potential therapeutic target.
The epigenetic regulation of lung cancer is a sophisticated and dynamically evolving process [69,70], mainly involving DNA methylation, histone modification and non-coding RNA (ncRNA). Dysregulation of epigenetic mechanisms can drive tumor initiation and progression through metabolic reprogramming in cancer cells.
2.4.1 DNA Methylation and Demethylation
DNA methylation requires the involvement of DNA methyltransferases (DNMTs) [71], mainly including DNMT1, DNMT3A and DNMT3B. Aberrantly elevated expression of DNMTs is associated with lung cancer progression [72]. Among them, DNMT1 overexpression induces the silencing of tumor suppressor genes by promoting promoter hypermethylation, disrupts the synergistic regulatory network of the p53/Sp1 signaling pathway, and eventually facilitates lung cancer initiation, development, and poor patient prognosis [73]. Study [74] shows that DNMT1 inhibition reverses the hypermetabolic state of cancer cells by reducing glycolysis and antioxidant capacity. Mechanistically, DNMT1 drives metabolic reprogramming through epigenetic regulation of the SCARA5/AOX1 axis, offering a new therapeutic target for NSCLC. Meanwhile, the TET (Ten-eleven translocation) family—key mediators of DNA demethylation—dynamically reverses DNA methylation via stepwise oxidation of 5-methylcytosine (5mC) by its core members TET1, TET2, and TET3. Rahim et al. [75] identified TET1 as a critical tumor suppressor in LC, acting through dual mechanisms of epigenetic remodeling and immune regulation. Specifically, TET1 loss promotes tumor progression by upregulating translation- and metabolism-related genes, thereby enhancing metabolic and biosynthetic pathways. In contrast, TET1 overexpression drives epigenetic reprogramming via catalytic DNA demethylation and strengthens antitumor immunity by activating the TNFα/NF-κB signaling pathway.
Histone modifications encompass multiple forms, such as methylation, acetylation, and lactylation. Histone methylation occurs on the nitrogen atoms of side chains in lysine and arginine residues [76]. Mutations in the histone methyltransferase KMT2D are common in lung cancer. Loss of KMT2D impairs the super-enhancer of PER2, leading to upregulated glycolysis-related gene expression and promoting lung tumorigenesis [77]. SETD1A, a histone methyltransferase, is overexpressed in lung cancer. It activates the transcription of oncogenes such as MYC by catalyzing the H3K4me3 modification, thereby accelerating the proliferation of lung tumors [78]. Compared with adjacent normal lung tissue, the demethylase KDM2 is upregulated in tumor tissue from NSCLC patients [79]. Histone deacetylases (HDACs) are crucial in maintaining histone acetylation homeostasis [80]. Inhibition of HDACs reverses the silencing of tumor suppressor genes, induces cell cycle arrest, or triggers apoptosis [81]. HDAC1 overexpression in NSCLC drives TME remodeling via hypoxia-induced acidosis and enhanced lipogenesis, resulting in suppression of antitumor T cell immunity [82]. HDAC4 activates the glutaminase activity of GAC through deacetylation, thereby promoting glutamine metabolic reprogramming [83]. Lactylation modulates the expression of metabolic genes, in turn changing cellular energy metabolism pathways [84]. Lactate, a metabolite, regulates glycolysis and cell proliferation in NSCLC by mediating the expression of pertinent genes through histone lactylation [85].
NcRNAs drive metabolic reprogramming in cancer to facilitate tumor cell energy supply, biosynthesis, and microenvironmental adaptation, thereby promoting malignant tumor progression [86]. miR-144 is downregulated in lung cancer, and its loss of function activates glycolytic pathways by targeting GLUT1, thereby supplying energy for tumor cell proliferation [87]. miR-21 drives cell growth and migration in NSCLC by triggering the CD36-mediated fatty acid metabolic pathway [88]. lncRNAs participate in cell cycle regulation, gene regulation, immune response, and tumor metabolism [89]. lnc-IGFBP4-1 upregulates HK2, PDK1, and LDHA, thereby promoting lung cancer cell proliferation [90]. Accumulating evidence indicates that certain lncRNAs function as diagnostic staging biomarkers, prognostic predictors, and therapeutic targets during tumorigenesis and progression [91]. circRNAs modulate gene expression across the epigenetic and transcriptional levels [92,93]. The circ-PITX1/miR-1248/CCND2 axis modulates glycolysis and glutamine metabolism in NSCLC [94]. The inherent structural stability of circRNAs makes them ideal candidate biomarkers for cancer diagnosis [95]. circRNA_001846 is upregulated in NSCLC patients, and its serum levels may serve as a promising early diagnostic biomarker [96]. Furthermore, plasma circFARSA also demonstrates potential as a biomarker for NSCLC, providing research insights for the application of circRNAs in non-invasive cancer diagnosis [97].
3 Immune Metabolic Regulation and Immunosuppressive Network in the Lung Cancer Tumor Microenvironment
The tumor microenvironment, serving as a critical niche for tumor cell growth and survival, is a complex and dynamic ecosystem comprising tumor cells, immune cells, stromal cells, extracellular matrix (ECM) components, and various bioactive molecules. Within this ecosystem, metabolic and immune dysregulation plays a pivotal role in driving lung cancer progression and therapy resistance.
3.1 Metabolic Reprogramming of Immune Cells
Unlike cancer cells, the metabolic reprogramming of T cells requires co-stimulatory molecules and is initiated by TCR-mediated antigen recognition. This metabolic shift provides essential substrates and energy for T cells effector functions [98]. Naive T cells primarily rely on OXPHOS for energy production, whereas upon TCR-mediated activation, their metabolic phenotype shifts toward glycolysis to meet the heightened energy demands [99]. GLUT1 gene knockout or treatment with its inhibitors significantly impairs CD4+ T cell proliferation, effector differentiation, and inflammatory cytokine secretion [100]. Glucose deprivation in the TME restricts aerobic glycolysis in T cells, thereby impairing their antitumor capacity [101]. T cell activation is accompanied by significantly enhanced glycolysis and glutamine metabolism, whereas changes in FAO vary with T cell subsets and the microenvironment [102]. ASCT2 expression is low in naive or resting T cells but is markedly upregulated within hours of activation [103]. Intracellular L-arginine not only bolsters the survival capacity of both human and murine T cells but also promotes the differentiation of central memory-like T (Tcm) cells. In murine models, Tcm cells outperform effector memory T cells in tumor eradication [104]. Excessive lipid accumulation, commonly observed in CD8+ TILs, induces functional impairment and exhaustion, while in response to lipid-rich TME and nutrient stress, these cells upregulate lipid transporters to acquire lipids as alternative energy sources [105]. In the hypoxic and glucose-deprived TME, CD8+ TILs partially preserve their effector functions through enhanced PPAR-α signaling and fatty acid metabolism. This is reflected by the upregulated expression of lipid metabolism-related genes and accumulated metabolic intermediates [106].
Dendritic cells (DCs), the most potent antigen-presenting cells in the immune system, play pivotal roles in anti-tumor immunity and the TME. They are primarily categorized into two major subtypes: conventional dendritic cells (cDCs) and plasmacytoid dendritic cells (pDCs), with cDCs further subdivided into distinct functional subsets. Characterized by XCR1 and Clec9A expression, cDC1 specializes in cross-presenting antigens via the MHC-I pathway to recruit and activate CD8+ T cells, while cDC2—marked by CD11b and CD172a—primarily enhances the functions of CD4+ helper T cell 17 (Th17) and Th1 cells. Lobel et al. [107] found that GLN regulates the survival, proliferation, and differentiation of type 1 conventional dendritic cells (cDC1s) via the mTORC1 signaling pathway. cDC1s are highly dependent on GLN levels, and its deficiency suppresses mTOR signaling, triggering increased apoptosis and impaired differentiation—effects less pronounced in cDC2s. This leads to reduced cDC1 numbers and frequency within tumors, impairing antigen cross-presentation and T cell activation, thereby promoting immune evasion. Additionally, glycolysis serves as the metabolic foundation for DCs in anti-tumor immunity. Glycolysis-deficient DCs fail to suppress tumor growth effectively, accompanied by reduced infiltration and effector functions (e.g., IFN-γ and granzyme B production) of tumor-infiltrating CD4+ and CD8+ T cells. By elevating intracellular ATP levels in DCs—essential for STING phosphorylation—glycolysis drives STING signaling activation. STING activation further upregulates HIF-1α, which enhances accumulation of glycolytic key enzymes (e.g., HK2 and PKM2), forming a positive feedback loop. This metabolic reprogramming, particularly glycolysis-driven, acts as a DC activation driver: it promotes type I interferon responses and T cell activation via ATP-STING signaling, while STING reinforces glycolysis through HIF-1α [108]. Clinical studies confirm the universality of this axis in NSCLC, offering a novel strategy for DC metabolism-targeted immunotherapy.
Natural killer (NK) cells, as innate immune lymphocytes, have garnered significant research attention for their metabolic reprogramming during antitumor immune responses. The metabolic stress imposed by the TME critically governs NK cells survival, proliferation, cytokine production, cytotoxic capacity, and ultimately, their efficacy in cancer immunotherapy. In the TME of lung adenocarcinoma, significant downregulation of FABP4 and SPON2 disrupts lipid homeostasis in NK cells, reducing key effector molecule expression and markedly impairing cytotoxic function [109]. In vitro experiments confirmed that this metabolic reprogramming arrests NK cells in an immature state, compromising antitumor immunity. Huang et al. [49] revealed that TFDP1 transcriptionally activates SPC25 to upregulate glutamine metabolism, thereby suppressing NK cells antitumor immunity. Mechanistically, SPC25 overexpression enhances glutamine uptake and metabolite production (e.g., glutamate, α-ketoglutarate), resulting in reduced NK cells cytotoxicity, decreased expression of activating receptors, and diminished secretion of perforin, granzyme B, and IFN-γ. Knockdown of TFDP1 reverses this effect, whereas SPC25 overexpression reinstates NK cells suppression, confirming metabolic reprogramming as the core immunosuppressive mechanism. This offers a novel strategy to enhance immunotherapy by targeting the TFDP1/SPC25 axis.
3.2 Immune Suppressive Network Constructed by the Tumor Microenvironment
The immune suppression in the TME is not driven by a single pathway but rather constitutes a complex network resulting from the concerted action of multiple mechanisms, including metabolic competition, secretion of inhibitory factors, immune cell polarization, and immune checkpoint activation [110] (Fig. 2).
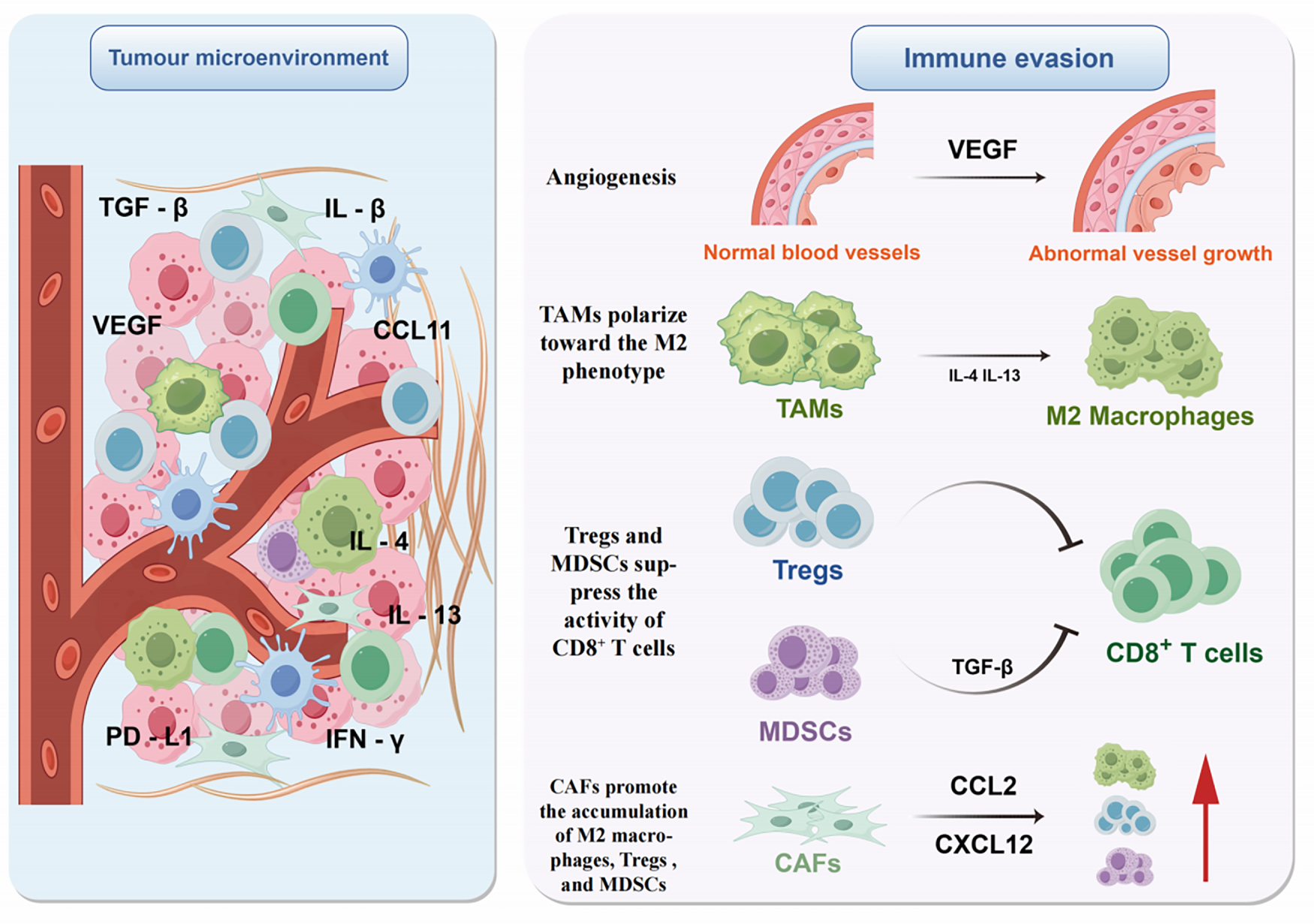
Figure 2: Immune evasion mediated by the tumor microenvironment (Figure created with figdraw). This figure illustrates key mechanisms by which the TME mediates immune evasion. Enriched with cytokines (e.g., TGF-β, VEGF, IL-1β) and diverse immune cells, the TME drives suppression via multiple pathways: VEGF promotes aberrant angiogenesis; IL-4/IL-13 polarizes TAMs to the M2 phenotype; Tregs and MDSCs inhibit CD8+ T cells via TGF-β; and CAFs recruit M2 macrophages, Tregs, and MDSCs by secreting CCL2/CXCL12. These mechanisms synergize to promote immune evasion. Note: Abbs.: TGF-β, Transforming Growth Factor-beta; IL-1β, Interleukin-1 beta; IL-4, Interleukin-4; IL-13, Interleukin-13; VEGF, Vascular Endothelial Growth Factor; CCL11, C-C motif chemokine ligand 11; PD-L1, Programmed Cell Death-Ligand 1; IFN-γ, Interferon-gamma; TAMs, Tumor-Associated Macrophages; Tregs, Regulatory T cells; MDSCs, Myeloid-derived suppressor cells; CAFs, Cancer-associated fibroblasts.
The hypoglycemic TME suppresses the PI3K-Akt-mTOR signaling pathway, downregulating glycolysis in CD4+ T cells and driving their metabolic reprogramming toward FAO and OXPHOS to meet biosynthetic demands for energy [111]. In LUAD patients, IL-38 overexpression reduces CD8+ TILs and suppresses antitumor immunity [112]. Tumor cells promote Tregs differentiation and suppress antitumor immune responses by generating kynurenine through the IDO-mediated tryptophan metabolic pathway [113]. Tumor-derived factors drive TAMs to secrete various cytokines, suppressing T cell-mediated immune responses and exacerbating the formation and maintenance of an immunosuppressive TME [114]. Lactate downregulates ATP6V0d2 expression in TAMs to promote HIF-2α-mediated tumor progression. Tumor tissue analysis from 35 lung cancer patients revealed a negative correlation between ATP6V0d2 and HIF-2α expression in TAMs. Moreover, high ATP6V0d2 expression correlated with longer survival, while high HIF-2α was associated with shorter survival [115]. Lung cancer cell-secreted macrophage colony-stimulating factor drives TAMs to M2 polarization by upregulating FASN expression and activating PPARβ/γ, thereby stimulating tumor angiogenesis and facilitating cancer cell invasion into the stroma [116]. Furthermore, by lowering amino acid levels and secreting lactate, M2 macrophages dampen the antitumor effector function of NK cells and T cells [117].
In the hypoxic TME, HIF-1α activation in NK cells upregulates HIF-1α-dependent genes that promote angiogenesis, such as VEGF and TGF-β [118]. Knockout of HIF-1α in NK cells enhances their antitumor response [119]. In the TME of KRAS-driven lung cancer, high TGF-β concentrations induce aberrant overexpression of FBP1 in NK cells, suppressing glycolysis and reducing cellular activity, ultimately compromising their antitumor function [120].
The TME promotes tumor immune evasion through PD-1/PD-L1 pathway activation [121]. In NSCLC, TGF-β1 reduces DNMT1 activity, leading to promoter demethylation of the PD-L1 gene and subsequent upregulation of PD-L1 expression [122]. Compared with adjacent non-tumor tissue, IL-1β expression is elevated in LUAD and contributes to cancer cell survival by upregulating PD-L1 expression [123]. Group 2 innate lymphoid cells (ILC2s) promote IL-4/IL-13 secretion through upregulated PD-1 signaling activity, driving M2 macrophage polarization and thereby enhancing the immunosuppressive TME [124]. VEGF-A enhances PD-1 expression on CD8+ T cells by binding to VEGFR2 on their cell surface [125]. Osteopontin (OPN) expressed by TAMs upregulates PD-L1 expression in NSCLC, thereby exacerbating tumor progression [126].
TAMs are functionally categorized into M1 and M2 subtypes, with M1 macrophages serving as critical effectors of antitumor immunity in early-stage cancer by employing multiple mechanisms to inhibit tumor growth and progression [127]. However, prolonged exposure to the TME, under the influence of external stimuli, drives TAMs to preferentially polarize to the M2 phenotype [128]. M2 macrophages, possessing anti-inflammatory and protumor properties, establish an immunosuppressive microenvironment by secreting anti-inflammatory cytokines. These cytokines simultaneously foster tumor angiogenesis while suppressing antitumor immunity [129–131]. M1 macrophages primarily rely on glycolysis and the pentose phosphate pathway to generate energy, whereas M2 macrophages meet their metabolic demands by enhancing FAO and OXPHOS [132]. Th1 cells drive M1 macrophage polarization by secreting IFN-γ, whereas Th2 cells promote M2 polarization by secreting IL-4 and IL-13 [133]. TAMs take up tumor cell-derived fatty acids via CD36, which promotes their polarization to the M2 macrophage phenotype and establishes an immunosuppressive TME [134]. This lipid accumulation drives a metabolic shift from glycolysis to FAO to sustain energy production [135]. Glutaminolysis-derived α-KG fuels the TCA cycle, enhancing FAO in M2 macrophages and thereby epigenetically driving M2 gene reprogramming in a histone methylation-dependent manner [136]. Suppressing glutamine synthetase (GS) activity in M2 macrophages promotes their transition to the M1 phenotype and enhances cytotoxic T cell recruitment [137].
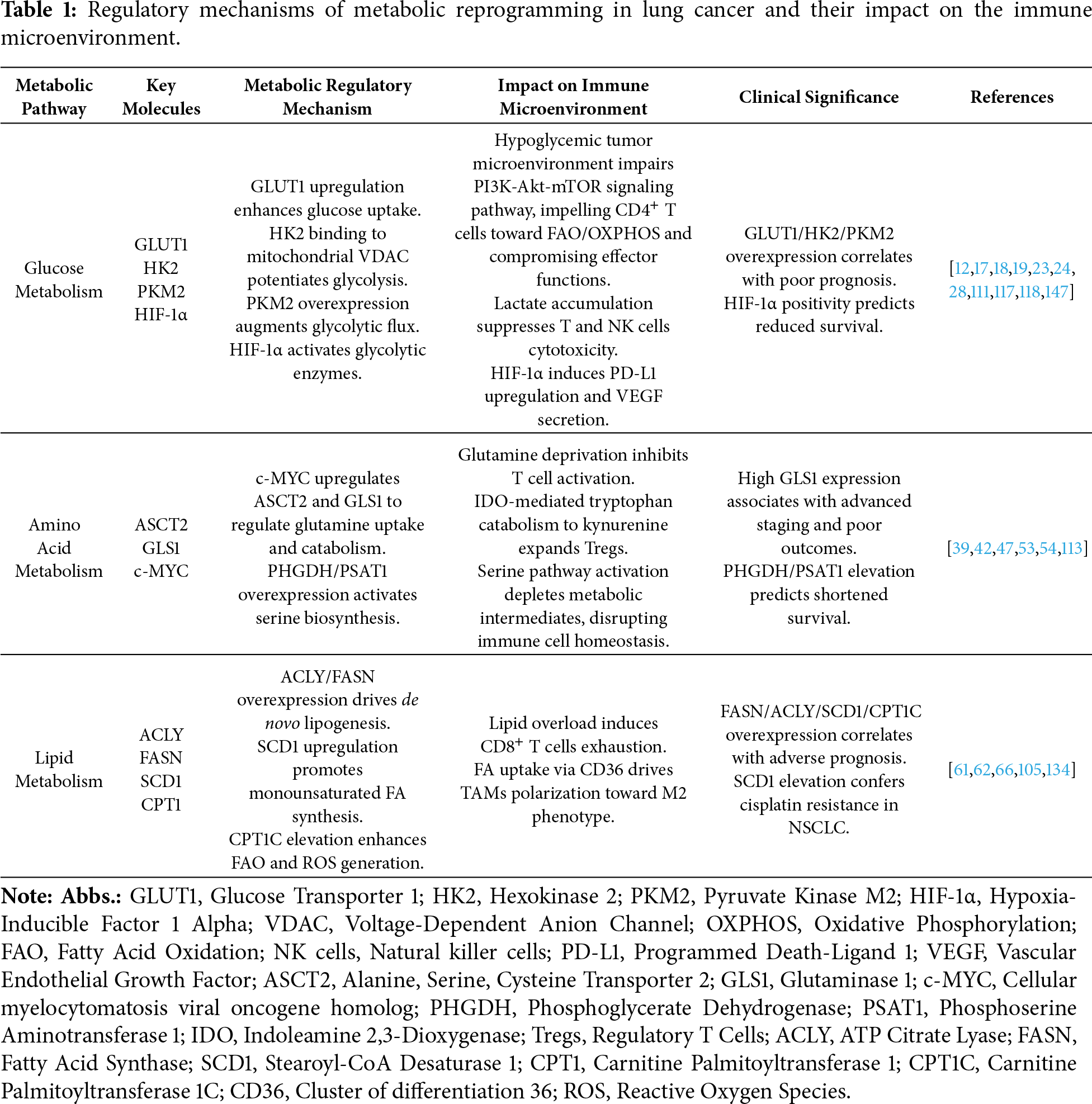
Myeloid-derived suppressor cells (MDSCs), originating in the bone marrow, accumulate gradually during tumor progression in tumor-bearing hosts and migrate to peripheral lymphoid organs and tumor sites [138,139]. NADPH oxidase 2 (NOX2) is expressed by MDSCs and produces ROS to support the immunosuppressive TME [140]. In lung cancer, MDSCs are significantly elevated, where they secrete abundant TGF-β to suppress the immune system, induce T cell dysfunction, and consequently facilitate tumor growth and metastatic dissemination [141]. MDSCs can also promote the metastasis of NSCLC by highly expressing chemokines such as CCL11 to activate AKT and ERK signaling pathways [142]. Table 1 summarizes the regulatory mechanisms of metabolic reprogramming in lung cancer and their impact on the immune microenvironment.
Regulatory T cells (Tregs), as a key immunosuppressive subgroup, play a crucial role in tumor immune evasion, angiogenesis, and metastasis by suppressing anti-tumor immunity [143]. Elevated levels of Tregs in lung cancer are closely associated with poor cellular differentiation, advanced disease stages, and enhanced metastatic potential, and are significantly linked to resistance against immune checkpoint inhibitor (ICI) therapy [144]. Liang et al.’s [145] study found that tobacco carcinogens activate the α7nAChR receptor and transcription factor c-Jun, induce upregulation of IDO1 expression, promote the metabolism of tryptophan (Trp) to kynurenine (Kyn), lower the Trp/Kyn ratio. This metabolic reprogramming directly affects the immune cell metabolic microenvironment: Trp depletion inhibits effector T cell proliferation, Kyn accumulation acts as a ligand for the aryl hydrocarbon receptor (AhR), and Kyn activates AhR signaling, inducing the differentiation of naive CD4+ T cells into Tregs, thereby enhancing immunosuppressive function. Kratzmeier et al. [146] revealed that IFN-γ derived from LUAD cells activates CD8+ T cells to upregulate CCR5 chemokines (e.g., CCL3/CCL4), promoting the migration of CD4+Foxp3+ Tregs into the TME. Tregs suppress anti-tumor immunity via mechanisms including metabolic disruption (e.g., interfering with nutrient competition of effector T cells) and secretion of immunosuppressive cytokines (such as IL-10 and TGF-β). Notably, CCR5 antagonists can block this cascade, restore immune activity, and enhance the efficacy of ICI therapy in lung cancer.
Cancer-associated fibroblasts (CAFs) drive tumor progression by remodeling the extracellular matrix, regulating metabolism, and mediating crosstalk between cancer cells and immune cells [148]. CAFs can significantly elevate the proportion of M2 macrophages, Tregs, and MDSCs, ultimately facilitating tumor immune evasion [149]. In NSCLC, two CAF subtypes are linked to T cell exclusion: MYH11+αSMA+CAF and FAP+αSMA+CAF, which both localize around tumor parenchyma and promote T cell exclusion by depositing collagen fibers [150]. CAFs derived from NSCLC not only suppress the immune response of cytokine-activated NK cells [151], but also promote lung cancer cell migration via Four and a half LIM domain protein 2 (FHL2) expression, while FHL2 knockout significantly attenuates this pro-tumorigenic effect [152].
Tumor-related metabolites, such as tryptophan, lactic acid, adenosine, chemokines, and others, can also contribute to an immunosuppressive TME, thereby limiting the function of immune cells and the efficacy of immunotherapy [153,154]. Wu et al. [155] revealed that NDRG1-driven lactate accumulation induces histone H3K18 lactylation (H3K18la) in macrophages, upregulating immunosuppressive genes (e.g., CD163, PD-L1, IL-10). This process reinforces the M2 phenotype, establishes an immunosuppressive microenvironment, and ultimately drives lung adenocarcinoma progression. Ding et al. [156] discovered that chemoresistant tumor cells upregulate the expression of CD39 and CD73, promoting the conversion of ATP to adenosine (Ado), which leads to elevated Ado levels in the TME. This accumulated Ado activates the A2AR/PKA/mTORC signaling axis, driving metabolic reprogramming in TAMs and upregulating IDO1 expression. IDO1 catalyzes the breakdown of tryptophan, depleting this essential amino acid in the microenvironment, thereby suppressing the activation and proliferation of CD8+ T cells and facilitating immune escape. TAMs induced by resistant tumors inhibit T-cell cytotoxic function via the Ado–A2AR axis. Targeting this signaling node can reverse drug resistance and offers a novel strategy for combination immunotherapy.
4 Therapeutic Approaches Directed at Metabolism and Immunity in Lung Cancer
Apoptin targets PKM2 to inhibit glycolysis in A549 lung cancer cells, while promoting autophagy and apoptosis through modulation of the PKM2/AMPK/mTOR pathway [157]. Pitavastatin inhibits NSCLC progression by blocking CD36 to disrupt the CD36/AKT/mTOR signaling pathway and reduce lipid accumulation [158]. ND-646, an allosteric inhibitor targeting ACC1, disrupts lung tumor growth by interfering with ACC1 protein subunit dimerization to block fatty acid synthesis [159]. Combined treatment with PKM2-IN-1 (a PKM2 inhibitor) and NCT-503 (a PHGDH inhibitor) synergistically decreases GLUT1 expression, effectively suppressing lung cancer cell proliferation [160]. The GLS inhibitor CB-839 not only enhances radiosensitivity in NSCLC patients by reducing intracellular GSH levels [161], but also synergizes with the targeted therapy selumetinib to potentiate antitumor activity against KRAS-mutant NSCLC [162].
Numerous emerging immune checkpoint molecules, including TIGIT, LAG-3, PD-1, TGF-β, CD73, TIM-3, and B7-H4, are presently under investigation as prospective therapeutic targets in cancer. T cell immunoglobulin and ITIM domain (TIGIT) is primarily expressed on T cells and NK cells [163]. Luo et al. [164] demonstrated that IL-15 stimulation enhances effector function in CD8+ TILs while concurrently upregulating TIGIT expression; combinatorial treatment with IL-15 and TIGIT blockade synergistically enhances CD8+ TILs cytotoxicity and augments antitumor immunity in LUAD. Although anti-TIGIT agents have demonstrated promising potential in lung cancer therapy, their clinical translation currently faces significant challenges [165]. CD73, also known as ecto-5′-nucleotidase or NT5E, is a GPI-anchored cell surface ectonucleotidase that plays a pivotal role in purinergic signaling by catalyzing the hydrolysis of extracellular AMP into immunosuppressive adenosine, and is highly expressed on tumor and immune cells. As the final mediator of extracellular adenosine production across multiple pathways, inhibiting CD73 directly counters immunosuppressive adenosine accumulation-mediated immune evasion, establishing it as a key therapeutic target in cancer immunotherapy [166,167]. In senescent tumor cells induced by radiotherapy or chemotherapy, IL-6 secretion activates the JAK/STAT3 pathway in macrophages, driving CD73 transcriptional upregulation that catalyzes adenosine accumulation in the TME and suppresses CD8+ T cell proliferation along with Granzyme B and IFN-γ secretion [168]. CD73 inhibition abrogates adenosine production, restores antitumor immunity, and synergizes with anti-PD-1 antibodies to significantly suppress tumor growth and prolong survival; this combination therapy elevates CD8+IFN-γ+ T cell proportions and PD-1 upregulation, indicating T cell activation, thereby positioning CD73 targeting as a viable approach to overcome immunosuppression in aged lung cancer TMEs, particularly when combined with PD-1/PD-L1 inhibitors.
4.3 Targeting Gut Microbiota and Metabolites
Gut microbiota-derived metabolites act as immune modulators, exerting their effects on the TME and anti-tumor therapeutic outcomes through diverse mechanisms [169]. Key metabolites include short-chain fatty acids (SCFAs, e.g., acetate, propionate, butyrate), tryptophan derivatives (e.g., indole-3-lactate, indole-3-propionate), secondary bile acids (e.g., deoxycholic acid), polyamines (e.g., spermidine), and trimethylamine N-oxide (TMAO) [170]. These molecules have been shown to critically influence lung cancer progression and drug response [171]. Zhu et al. [172] discovered that in a lung cancer mouse model, oral gavage of gut Akkermansia muciniphila (Akk) enriched tumor microbiota with Akkermansia and other intestinal microbes, indicating gut-derived Akk migrates to lung tumor sites to reshape intratumoral microbiome composition and reprogram metabolic pathways for anticancer effects. This process downregulated key metabolites like lactate and glutamate in the tumor microenvironment, inhibiting glycolysis and glutamine metabolism, which modulated immune cell activity via changes in enzymes such as LDHA and GLS, thereby indirectly enhancing antitumor immune responses. Camu-camu (CC) is an Amazonian berry rich in phytochemicals, and castalagin is a polyphenol extracted from it. Research by Messaoudene et al. [173] revealed that oral castalagin supplementation reshapes gut microbiota composition—enriching beneficial bacteria like Ruminococcaceaeand Alistipes, enhancing microbial diversity, and increasing the ratio of CD8+ T cells to Tregs in the TME. It also induces metabolic shifts, including elevated taurine-conjugated bile acids, thereby boosting anti-tumor immunity. In NSCLC models, castalagin overcomes anti-PD-1 resistance and enhances the efficacy of ICIs via a microbe-dependent mechanism. The gut microbiota, host metabolism, and immune function constitute an intricate regulatory network. Therefore, by leveraging patient-specific data including microbiome profiling, metabolomic signatures, and tumor immune subtypes, personalized therapeutic strategies can be developed. These tailored combinatorial regimens may involve direct microbial manipulation, metabolite-targeted approaches, dietary interventions, strategic metabolite supplementation, or inhibition of detrimental metabolic pathways. The ultimate objective is to reprogram the immunosuppressive TME into an anticancer immune-permissive state.
4.4 Metabolic-Immune Combination Therapy
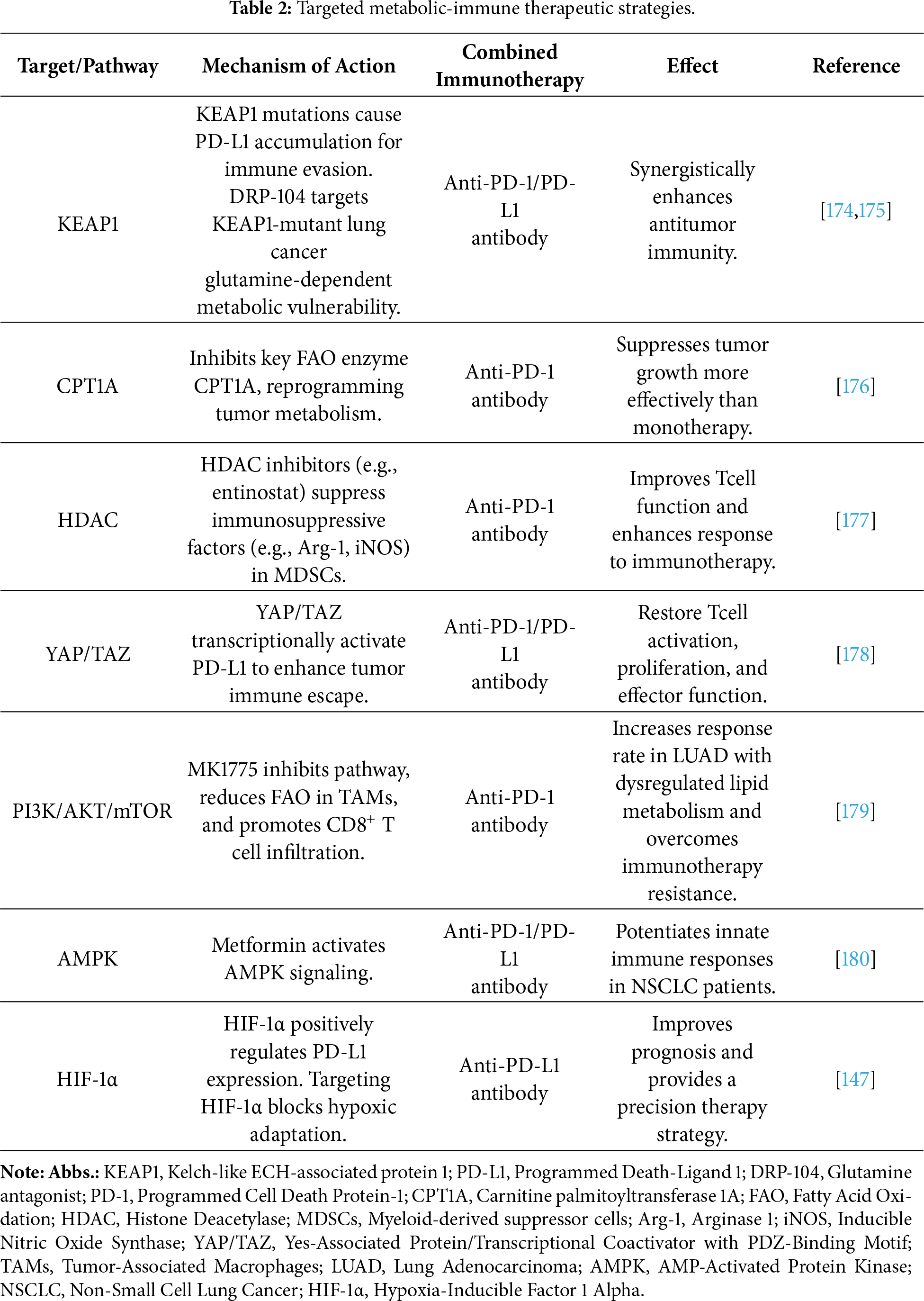
In NSCLC, KEAP1 mutations impair its ability to mediate ubiquitination-dependent PD-L1 degradation, resulting in PD-L1 accumulation and subsequent immune evasion. Thus, targeted restoration of KEAP1 activity to degrade PD-L1, combined with anti-PD-L1 therapy, synergistically enhances antitumor immunity [174]. DRP-104 targets the glutamine-dependent metabolic vulnerability of KEAP1-mutant lung cancer, inhibiting tumor growth and reversing T cell exhaustion. In combination with anti-PD-1 therapy, it exerts synergistic antitumor effects [175]. Targeted therapy against CPT1A combined with PD-1 antibody inhibits tumor growth, demonstrating superior efficacy compared with monotherapy [176]. HDAC inhibitors (e.g., entinostat) enhance the synergistic antitumor efficacy of anti-PD-1 therapy in murine lung cancer models by suppressing metabolism-linked immunosuppressive factors (e.g., arginase-1, iNOS) in MDSCs [177]. In NSCLC, YAP/TAZ transcriptionally upregulate PD-L1 expression to promote tumor immune evasion, while combined inhibition of YAP/TAZ and PD-1/PD-L1 synergistically boosts antitumor immune responses [178]. MK1775 inhibits the PI3K/AKT/mTOR pathway in tumor cells, reduces fatty acid oxidation in TAMs, and promotes CD8+ T cell infiltration, while its combination with anti-PD-1 therapy improves treatment response rates in lung adenocarcinoma patients with lipid metabolism abnormalities and overcomes immune resistance [179]. Anti-PD-1/PD-L1 therapy, when combined with metformin, enhances antitumor immune responses in peripheral immune cells from NSCLC patients [180]. In NSCLC tissue samples, PD-L1 expression correlates positively with HIF-1α levels, suggesting that combined targeting of PD-L1 and HIF-1α may represent a more effective precision therapeutic strategy for NSCLC [147].
Therefore, combining metabolic pathway targeting with immune checkpoint blockade represents a prospective therapeutic approach for lung cancer (Table 2).
Lung cancer progression is driven by a dynamic, bidirectional crosstalk between tumor cell metabolic reprogramming and immune suppression within the TME. This review has synthesized current evidence illustrating how metabolic adaptations in cancer cells (e.g., upregulated glycolysis, glutamine dependency, enhanced lipogenesis) not only fuel proliferation but also actively remodel the TME into an immunosuppressive niche. Conversely, immune cells within the TME undergo their own metabolic reprogramming, which often further supports tumor immune evasion. This reciprocal relationship establishes a positive feedback loop that promotes malignancy, therapeutic resistance, and poor patient outcomes. Despite substantial advancements in preclinical studies, numerous metabolic pathway-targeted therapies have yielded disappointing outcomes in LC clinical trials. This emphasizes the critical need for a more nuanced, systems-level comprehension of metabolism-immunity interactions.
5.1 Why Previous Metabolic-Targeted Therapies Mostly Failed in Clinical Trials
Early metabolic-targeted trials yielded disappointing results due to three critical oversights: (1) Tumor Heterogeneity: Metabolic heterogeneity presents a major therapeutic challenge in lung cancer [181]. Fundamental differences in metabolic profiles exist not only between subtypes [182] (LUAD/LUSC/SCLC) but also among tumors with similar clinical presentations, stemming from varied molecular drivers like genetic mutations and pathway activations [183]. This diversity directly undermines the effectiveness of metabolic-targeted approaches, resulting in disappointing clinical response rates. (2) Metabolic Adaptability: including metabolic flexibility and plasticity [184]. Metabolic flexibility denotes the ability of cancer cells to switch between different metabolic substrates in the nutrient-scarce TME. In contrast, metabolic plasticity refers to their capacity to bypass impairments in specific pathways, often by triggering compensatory mechanisms upon inhibition [185,186]. This adaptive response allows tumors to evade metabolic crises induced by targeted agents, ultimately limiting therapeutic benefit. (3) Clinical Strategy Gaps: Trials lacked patient selection based on metabolic vulnerability, faced on-target toxicities, and relied on inadequate biomarkers instead of direct TME validation. Future trials require spatial metabolomics and real-time metabolic imaging for patient stratification and target validation [187,188].
5.2 Controversies and Unresolved Questions in Metabolic-Immune Crosstalk
Persistent fundamental contradictions hinder understanding of lung cancer metabolism. The regulatory paradox of OXPHOS in cancer stems from the dichotomy of its dual “survival-function” roles. As an efficient energy supplier, it serves as both a key support for maintaining cancer stem cell (CSC) stemness and drug resistance, and a suppression target under microenvironmental stress (e.g., hypoxia). Meanwhile, it promotes tumor progression via ROS signaling yet may induce oxidative damage [189]. Similarly, glutamine supports tumor cell survival under hypoxic and glucose-deprived conditions by activating HIF-α and Akt/mTOR signaling pathways. In NSCLC, resistant cells rely on the glutamine metabolic enzyme GLUD1 to produce α-ketoglutarate (α-KG), which activates Snail expression to promote migration; targeting glutamine metabolism enhances sensitivity to EGFR-TKIs. Although glutamine predominantly promotes tumor progression, its deprivation or supplementation can suppress tumors in specific contexts. For example, dietary glutamine supplementation increases α-KG levels in the TME, reduces H3K4me3 methylation, and suppresses oncogenic pathways, while also reducing Treg differentiation and enhancing CD8+ T cell-mediated antitumor responses [190,191]. These paradoxes, shaped by tumor heterogeneity, metabolic plasticity, and dynamic microenvironmental changes, highlight that metabolic interventions are neither universally immunosuppressive nor stimulatory—their net effect depends on cell-specific metabolic wiring.
5.3 Clinical Translation: Overcoming Barriers to Precision Implementation
Successful translation from preclinical discovery to clinical application requires addressing three fundamental challenges. First, patient stratification must evolve beyond PD-L1 IHC to incorporate multi-omic signatures defining metabolic-immune TME subtypes, such as KEAP1-mutant NSCLC—a cohort with distinct glutamine dependency ideally suited for DRP-104 and anti-PD-1 combination therapy. Second, predictive biomarkers should extend beyond static genomics to include dynamic metabolic flux and spatial metabolomics [192–194]. Third, toxicity management for combinations demands innovative approaches like sequential scheduling (metabolic priming before ICI) and tumor-selective nanocarriers targeting specific receptors to minimize systemic toxicity while maximizing efficacy [195,196].
5.4 Refining the Framework for Metabolic-Immune Combination Therapies
We propose a stratified framework to stratify combination strategies by developmental maturity, addressing the current lack of stratification.
Clinically validated in lung cancer: Several combination therapies have reached clinical-stage testing in LC. Although grounded in a rational strategy of glutamine metabolic co-dependency, the combination of Telaglenastat (CB-839, 800 mg twice daily) and nivolumab exhibited a manageable safety profile in NSCLC patients; however, it showed no significant antitumor efficacy, potentially due to tumor heterogeneity or prior therapy resistance [197]. These findings inform future combination strategies targeting glutamine metabolism while necessitating further research to validate biomarker roles. The efficacy of such regimens, moreover, offers critical human data to validate the feasibility of metabolic immunotherapy.
Early-Phase Clinical: Strategies such as HDAC inhibitors (e.g., entinostat) combined with anti-PD-1 therapy are undergoing early-phase trials in multiple cancers [198,199]. Their potential lies in reversing epigenetic silencing of immune-related genes in T cells and myeloid cells. Yet, their efficacy in lung cancer hinges on biomarker identification—potentially derived from the metabolic subtypes outlined—to select responsive patients.
Preclinical: Numerous attractive metabolic nodes, such as PHGDH (serine pathway) [200] and CPT1A (fatty acid oxidation) [201], remain in the preclinical stage. While genetic knockdown studies confirm efficacy, the absence of specific, potent, and non-toxic small-molecule inhibitors for clinical application represents a significant barrier. Their role may be context-dependent, for instance, in tumors exhibiting defined lipid-metabolism signatures.
5.5 Precision Targeting through AI and Multi-Omics Integration
The integration of artificial intelligence (AI) with single-cell and spatial multi-omics is pivotal for decoding intratumoral metabolic-immune complexity, as AI/machine learning (ML) algorithms synthesize multi-omic data—genomic, transcriptomic, proteomic, and metabolomic—from single-cell RNA sequencing and spatial metabolomics to develop predictive models that optimize immunotherapy (e.g., ICIs + metabolically targeted agents) and guide combination therapies. Spatial transcriptomics reveals stark metabolic heterogeneity, where the hypoxic, glycolysis-dominated tumor core upregulates immunosuppressive markers (e.g., PD-L1), driving exhaustion of CTLs and NK cells and sparse immune infiltration, while the vascular-rich periphery exhibits greater infiltration of immune cells (e.g., CD8+ T cells, dendritic cells), forming an active interface often compromised by immunosuppressive cells like Tregs and MDSCs. This immune disparity, fueled by metabolic reprogramming, is addressed through metabolic-targeted therapies that modulate nutrient competition and reverse immunosuppression, with future efforts focused on integrating spatial multi-omics to devise personalized combinations—such as pairing immunotherapy with inhibitors of oxidative phosphorylation (OXPHOS) or lactate pathways—to improve prognostic outcomes [202,203].
Future research should now pivot from mechanism recapitulation to resolving key dilemmas: the optimal sequencing of metabolic modulators with immunotherapy, managing on-target toxicities against essential immune metabolic pathways, and expanding beyond Warburg effect to explore micronutrients and the microbiome. In conclusion, despite the heightened complexity revealed, the pursuit of targeting cancer metabolism remains sound. The failure of early monotherapies highlights that success will depend on a nuanced grasp of TME heterogeneity, the development of precise biomarkers, and the rational design of combination therapies, empowered by technological advances like AI and single-cell multi-omics to achieve clinical translation.
Lung cancer progression is governed by a dynamic metabolic-immune axis, defying simplistic intervention. Future advances in targeting the lung cancer metabolic landscape require a definitive shift from non-selective strategies to biomarker-guided precision. Success hinges on leveraging AI and multi-omics to decipher heterogeneity and stratify patients based on metabolic-immune phenotypes, ultimately unlocking the potential of metabolic-immunotherapy.
Acknowledgement: We acknowledge Figdraw for assistance in creating the figures.
Funding Statement: This work was supported by the Henan Provincial Science and Technology Research and Development Plan Joint Fund (Grant No. 242103810035).
Author Contributions: The authors confirm contribution to the paper as follows: Study conceptualization and design: Hongbo Wu and Rui Li; Literature investigation and data curation: Xiaomeng Li and Xuejiao Li; Visualization and figure preparation: Xiaomeng Li and Xuejiao Li; Writing—original draft preparation: Xiaomeng Li; Writing—review and editing: All authors (Xiaomeng Li, Xuejiao Li, Hongbo Wu, Rui Li); Supervision: Hongbo Wu and Rui Li; Funding acquisition: Hongbo Wu. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest.
References
1. Thai AA, Solomon BJ, Sequist LV, Gainor JF, Heist RS. Lung cancer. Lancet. 2021;398(10299):535–54. doi:10.1016/S0140-6736(21)00312-3. [Google Scholar] [PubMed] [CrossRef]
2. Zhang X, Song W, Gao Y, Zhang Y, Zhao Y, Hao S, et al. The role of tumor metabolic reprogramming in tumor immunity. Int J Mol Sci. 2023;24(24):17422. doi:10.3390/ijms242417422. [Google Scholar] [PubMed] [CrossRef]
3. Lin J, Rao D, Zhang M, Gao Q. Metabolic reprogramming in the tumor microenvironment of liver cancer. J Hematol Oncol. 2024;17(1):6. doi:10.1186/s13045-024-01527-8. [Google Scholar] [PubMed] [CrossRef]
4. Dias AS, Helguero L, Almeida CR, Duarte IF. Natural compounds as metabolic modulators of the tumor microenvironment. Molecules. 2021;26(12):3494. doi:10.3390/molecules26123494. [Google Scholar] [PubMed] [CrossRef]
5. Wang M, Zhu L, Yang X, Li J, Liu YE, Tang Y. Targeting immune cell types of tumor microenvironment to overcome resistance to PD-1/PD-L1 blockade in lung cancer. Front Pharmacol. 2023;14:1132158. doi:10.3389/fphar.2023.1132158. [Google Scholar] [PubMed] [CrossRef]
6. Luby A, Alves-Guerra MC. Targeting metabolism to control immune responses in cancer and improve checkpoint blockade immunotherapy. Cancers. 2021;13(23):5912. doi:10.3390/cancers13235912. [Google Scholar] [PubMed] [CrossRef]
7. Zhu J, Thompson CB. Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol. 2019;20(7):436–50. doi:10.1038/s41580-019-0123-5. [Google Scholar] [PubMed] [CrossRef]
8. Zhu K, Cai Y, Lan L, Luo N. Tumor metabolic reprogramming and ferroptosis: the impact of glucose, protein, and lipid metabolism. Int J Mol Sci. 2024;25(24):13413. doi:10.3390/ijms252413413. [Google Scholar] [PubMed] [CrossRef]
9. Yan L, Tan Y, Chen G, Fan J, Zhang J. Harnessing metabolic reprogramming to improve cancer immunotherapy. Int J Mol Sci. 2021;22(19):10268. doi:10.3390/ijms221910268. [Google Scholar] [PubMed] [CrossRef]
10. Yang Y, Gao Y, Xiong Y, Gong Y, Lu J, Zhang Y, et al. Research progress of Warburg effect in hepatocellular carcinoma. Front Biosci. 2024;29(5):178. doi:10.31083/j.fbl2905178. [Google Scholar] [PubMed] [CrossRef]
11. Nong S, Han X, Xiang Y, Qian Y, Wei Y, Zhang T, et al. Metabolic reprogramming in cancer: mechanisms and therapeutics. MedComm. 2023;4(2):e218. doi:10.1002/mco2.218. [Google Scholar] [PubMed] [CrossRef]
12. Yadav D, Yadav A, Bhattacharya S, Dagar A, Kumar V, Rani R. GLUT and HK: two primary and essential key players in tumor glycolysis. Semin Cancer Biol. 2024;100:17–27. doi:10.1016/j.semcancer.2024.03.001. [Google Scholar] [PubMed] [CrossRef]
13. Kokeza J, Strikic A, Ogorevc M, Kelam N, Vukoja M, Dilber I, et al. The effect of GLUT1 and HIF-1α expressions on glucose uptake and patient survival in non-small-cell lung carcinoma. Int J Mol Sci. 2023;24(13):10575. doi:10.3390/ijms241310575. [Google Scholar] [PubMed] [CrossRef]
14. Jang HJ, Min HY, Kang YP, Boo HJ, Kim J, Ahn JH, et al. Tobacco-induced hyperglycemia promotes lung cancer progression via cancer cell-macrophage interaction through paracrine IGF2/IR/NPM1-driven PD-L1 expression. Nat Commun. 2024;15(1):4909. doi:10.1038/s41467-024-49199-9. [Google Scholar] [PubMed] [CrossRef]
15. Zhou Z, Li Y, Chen S, Xie Z, Du Y, Liu Y, et al. GLUT1 promotes cell proliferation via binds and stabilizes phosphorylated EGFR in lung adenocarcinoma. Cell Commun Signal. 2024;22(1):303. doi:10.1186/s12964-024-01678-8. [Google Scholar] [PubMed] [CrossRef]
16. Huang Y, Ouyang F, Yang F, Zhang N, Zhao W, Xu H, et al. The expression of Hexokinase 2 and its hub genes are correlated with the prognosis in glioma. BMC Cancer. 2022;22(1):900. doi:10.1186/s12885-022-10001-y. [Google Scholar] [PubMed] [CrossRef]
17. Lis P, Dyląg M, Niedźwiecka K, Ko YH, Pedersen PL, Goffeau A, et al. The HK2 dependent “Warburg effect” and mitochondrial oxidative phosphorylation in cancer: targets for effective therapy with 3-bromopyruvate. Molecules. 2016;21(12):1730. doi:10.3390/molecules21121730. [Google Scholar] [PubMed] [CrossRef]
18. Wang J, Shao F, Yang Y, Wang W, Yang X, Li R, et al. A non-metabolic function of hexokinase 2 in small cell lung cancer: promotes cancer cell stemness by increasing USP11-mediated CD133 stability. Cancer Commun. 2022;42(10):1008–27. doi:10.1002/cac2.12351. [Google Scholar] [PubMed] [CrossRef]
19. Liu W, Yu X, Zhou L, Li J, Li M, Li W, et al. Sinomenine inhibits non-small cell lung cancer via downregulation of hexokinases II-mediated aerobic glycolysis. Onco Targets Ther. 2020;13:3209–21. doi:10.2147/OTT.S243212. [Google Scholar] [PubMed] [CrossRef]
20. Tan SM, Luo L, He YF, Li W, Wan XX. Daurisoline inhibits glycolysis of lung cancer by targeting the AKT-HK2 axis. Cancer Biol Ther. 2025;26(1):2442556. doi:10.1080/15384047.2024.2442556. [Google Scholar] [PubMed] [CrossRef]
21. Li J, Xu S, Zhan Y, Lv X, Sun Z, Man L, et al. CircRUNX1 enhances the Warburg effect and immune evasion in non-small cell lung cancer through the miR-145/HK2 pathway. Cancer Lett. 2025;620:217639. doi:10.1016/j.canlet.2025.217639. [Google Scholar] [PubMed] [CrossRef]
22. She F, Anderson BW, Khana DB, Zhang S, Steinchen W, Fung DK, et al. Allosteric regulation of pyruvate kinase enables efficient and robust gluconeogenesis by preventing metabolic conflicts and carbon overflow. mSystems. 2025;10(2):e01131–24. doi:10.1128/msystems.01131-24. [Google Scholar] [PubMed] [CrossRef]
23. Chen M, Liu H, Li Z, Ming AL, Chen H. Mechanism of PKM2 affecting cancer immunity and metabolism in Tumor Microenvironment. J Cancer. 2021;12(12):3566–74. doi:10.7150/jca.54430. [Google Scholar] [PubMed] [CrossRef]
24. Long L, Chen M, Yuan Y, Ming AL, Guo W, Wu K, et al. High expression of PKM2 synergizes with PD-L1 in tumor cells and immune cells to predict worse survival in human lung adenocarcinoma. J Cancer. 2020;11(15):4442–52. doi:10.7150/jca.42610. [Google Scholar] [PubMed] [CrossRef]
25. Markowitz GJ, Ban Y, Tavarez DA, Yoffe L, Podaza E, He Y, et al. Deficiency of metabolic regulator PKM2 activates the pentose phosphate pathway and generates TCF1+ progenitor CD8+ T cells to improve immunotherapy. Nat Immunol. 2024;25(10):1884–99. doi:10.1038/s41590-024-01963-1. [Google Scholar] [PubMed] [CrossRef]
26. Qannita RA, Alalami AI, Harb AA, Aleidi SM, Taneera J, Abu-Gharbieh E, et al. Targeting hypoxia-inducible factor-1 (HIF-1) in cancer: emerging therapeutic strategies and pathway regulation. Pharmaceuticals. 2024;17(2):195. doi:10.3390/ph17020195. [Google Scholar] [PubMed] [CrossRef]
27. Zhou L, Zhao Y, Pan LC, Wang J, Shi XJ, Du GS, et al. Sirolimus increases the anti-cancer effect of Huai Er by regulating hypoxia inducible factor-1α-mediated glycolysis in hepatocellular carcinoma. World J Gastroenterol. 2022;28(32):4600–19. doi:10.3748/wjg.v28.i32.4600. [Google Scholar] [PubMed] [CrossRef]
28. Zheng S, Mo J, Zhang J, Chen Y. HIF-1α inhibits ferroptosis and promotes malignant progression in non-small cell lung cancer by activating the Hippo-YAP signalling pathway. Oncol Lett. 2023;25(3):90. doi:10.3892/ol.2023.13676. [Google Scholar] [PubMed] [CrossRef]
29. Chang YC, Chan YC, Chang WM, Lin YF, Yang CJ, Su CY, et al. Feedback regulation of ALDOA activates the HIF-1α/MMP9 axis to promote lung cancer progression. Cancer Lett. 2017;403:28–36. doi:10.1016/j.canlet.2017.06.001. [Google Scholar] [PubMed] [CrossRef]
30. Chowdhury R, Godoy LC, Thiantanawat A, Trudel LJ, Deen WM, Wogan GN. Nitric oxide produced endogenously is responsible for hypoxia-induced HIF-1α stabilization in colon carcinoma cells. Chem Res Toxicol. 2012;25(10):2194–202. doi:10.1021/tx300274a. [Google Scholar] [PubMed] [CrossRef]
31. Deng H, Wang Z, Zhu C, Chen Z. Prolyl hydroxylase domain enzyme PHD2 inhibits proliferation and metabolism in non-small cell lung cancer cells in HIF-dependent and HIF-independent manners. Front Oncol. 2024;14:1370393. doi:10.3389/fonc.2024.1370393. [Google Scholar] [PubMed] [CrossRef]
32. Shi Z, Hu C, Zheng X, Sun C, Li Q. Feedback loop between hypoxia and energy metabolic reprogramming aggravates the radioresistance of cancer cells. Exp Hematol Oncol. 2024;13(1):55. doi:10.1186/s40164-024-00519-1. [Google Scholar] [PubMed] [CrossRef]
33. Peinado P, Stazi M, Ballabio C, Margineanu MB, Li Z, Colón CI, et al. Intrinsic electrical activity drives small-cell lung cancer progression. Nature. 2025;639(8055):765–75. doi:10.1038/s41586-024-08575-7. [Google Scholar] [PubMed] [CrossRef]
34. Liang LJ, Yang FY, Wang D, Zhang YF, Yu H, Wang Z, et al. CIP2A induces PKM2 tetramer formation and oxidative phosphorylation in non-small cell lung cancer. Cell Discov. 2024;10(1):13. doi:10.1038/s41421-023-00633-0. [Google Scholar] [PubMed] [CrossRef]
35. Rao S, Mondragón L, Pranjic B, Hanada T, Stoll G, Köcher T, et al. AIF-regulated oxidative phosphorylation supports lung cancer development. Cell Res. 2019;29(7):579–91. doi:10.1038/s41422-019-0181-4. [Google Scholar] [PubMed] [CrossRef]
36. Li X, Peng X, Li Y, Wei S, He G, Liu J, et al. Glutamine addiction in tumor cell: oncogene regulation and clinical treatment. Cell Commun Signal. 2024;22(1):12. doi:10.1186/s12964-023-01449-x. [Google Scholar] [PubMed] [CrossRef]
37. Vanhove K, Derveaux E, Graulus GJ, Mesotten L, Thomeer M, Noben JP, et al. Glutamine addiction and therapeutic strategies in lung cancer. Int J Mol Sci. 2019;20(2):252. doi:10.3390/ijms20020252. [Google Scholar] [PubMed] [CrossRef]
38. Ni R, Li Z, Li L, Peng D, Ming Y, Li L, et al. Rethinking glutamine metabolism and the regulation of glutamine addiction by oncogenes in cancer. Front Oncol. 2023;13:1143798. doi:10.3389/fonc.2023.1143798. [Google Scholar] [PubMed] [CrossRef]
39. Choi YK, Park KG. Targeting glutamine metabolism for cancer treatment. Biomol Ther. 2018;26(1):19–28. doi:10.4062/biomolther.2017.178. [Google Scholar] [PubMed] [CrossRef]
40. Cederkvist H, Kolan SS, Wik JA, Sener Z, Skålhegg BS. Identification and characterization of a novel glutaminase inhibitor. FEBS Open Bio. 2022;12(1):163–74. doi:10.1002/2211-5463.13319. [Google Scholar] [PubMed] [CrossRef]
41. Han T, Zhan W, Gan M, Liu F, Yu B, Chin YE, et al. Phosphorylation of glutaminase by PKCε is essential for its enzymatic activity and critically contributes to tumorigenesis. Cell Res. 2018;28(6):655–69. doi:10.1038/s41422-018-0021-y. [Google Scholar] [PubMed] [CrossRef]
42. Soth MJ, Le K, Di Francesco ME, Hamilton MM, Liu G, Burke JP, et al. Discovery of IPN60090, a clinical stage selective glutaminase-1 (GLS-1) inhibitor with excellent pharmacokinetic and physicochemical properties. J Med Chem. 2020;63(21):12957–77. doi:10.1021/acs.jmedchem.0c01398. [Google Scholar] [PubMed] [CrossRef]
43. Jin J, Byun JK, Choi YK, Park KG. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp Mol Med. 2023;55(4):706–15. doi:10.1038/s12276-023-00971-9. [Google Scholar] [PubMed] [CrossRef]
44. Nan D, Yao W, Huang L, Liu R, Chen X, Xia W, et al. Glutamine and cancer: metabolism, immune microenvironment, and therapeutic targets. Cell Commun Signal. 2025;23(1):45. doi:10.1186/s12964-024-02018-6. [Google Scholar] [PubMed] [CrossRef]
45. Elbadawy M, Usui T, Yamawaki H, Sasaki K. Emerging roles of C-myc in cancer stem cell-related signaling and resistance to cancer chemotherapy: a potential therapeutic target against colorectal cancer. Int J Mol Sci. 2019;20(9):2340. doi:10.3390/ijms20092340. [Google Scholar] [PubMed] [CrossRef]
46. Wallbillich NJ, Lu H. Role of c-Myc in lung cancer: progress, challenges, and prospects. Chin Med J Pulm Crit Care Med. 2023;1(3):129–38. doi:10.1016/j.pccm.2023.07.001. [Google Scholar] [PubMed] [CrossRef]
47. Huang J, Zhang X, Zhang H, Li Y, Huang H, Li Z, et al. Addressing clinical limitations of glutaminase inhibitors: novel strategies for osimertinib-resistant lung cancer by exploiting glutamine metabolic dependency. Adv Sci. 2025;12(6):2411479. doi:10.1002/advs.202411479. [Google Scholar] [PubMed] [CrossRef]
48. Tambay V, Raymond VA, Bilodeau M. MYC rules: leading glutamine metabolism toward a distinct cancer cell phenotype. Cancers. 2021;13(17):4484. doi:10.3390/cancers13174484. [Google Scholar] [PubMed] [CrossRef]
49. Huang B, Chen K, Huang M, Yin Z, He W, Peng X, et al. TFDP1 activates SPC25-mediated glutamine metabolism to repress anti-tumor immunity of NK cells in lung adenocarcinoma. Expert Rev Clin Immunol. 2025;21(8):1171–81. doi:10.1080/1744666X.2025.2524469. [Google Scholar] [PubMed] [CrossRef]
50. Wu F, Shen J, Zhao Z, Chen Y, Ren B, Li M, et al. HHLA2 promotes immune evasion in EGFR-mutant lung cancer by inhibiting CD8+ T cell glutamine metabolism via KIR3DL3 interaction. Cancer Lett. 2026;639:218219. doi:10.1016/j.canlet.2025.218219. [Google Scholar] [PubMed] [CrossRef]
51. Hosios AM, Hecht VC, Danai LV, Johnson MO, Rathmell JC, Steinhauser ML, et al. Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev Cell. 2016;36(5):540–9. doi:10.1016/j.devcel.2016.02.012. [Google Scholar] [PubMed] [CrossRef]
52. Tajan M, Hennequart M, Cheung EC, Zani F, Hock AK, Legrave N, et al. Serine synthesis pathway inhibition cooperates with dietary serine and glycine limitation for cancer therapy. Nat Commun. 2021;12(1):366. doi:10.1038/s41467-020-20223-y. [Google Scholar] [PubMed] [CrossRef]
53. Haitzmann T, Schindlmaier K, Frech T, Mondal A, Bubalo V, Konrad B, et al. Serine synthesis and catabolism in starved lung cancer and primary bronchial epithelial cells. Cancer Metab. 2024;12(1):9. doi:10.1186/s40170-024-00337-3. [Google Scholar] [PubMed] [CrossRef]
54. Pan S, Fan M, Liu Z, Li X, Wang H. Serine, glycine and one-carbon metabolism in cancer (Review). Int J Oncol. 2020;58(2):158–70. doi:10.3892/ijo.2020.5158. [Google Scholar] [PubMed] [CrossRef]
55. Fu Y, Zou T, Shen X, Nelson PJ, Li J, Wu C, et al. Lipid metabolism in cancer progression and therapeutic strategies. MedComm. 2020;2(1):27–59. doi:10.1002/mco2.27. [Google Scholar] [PubMed] [CrossRef]
56. Paunovic M, Stojanovic A, Pokimica B, Martacic JD, Cvetkovic Z, Ivanovic N, et al. Metabolic reprogramming of phospholipid fatty acids as a signature of lung cancer type. Cancers. 2024;16(19):3320. doi:10.3390/cancers16193320. [Google Scholar] [PubMed] [CrossRef]
57. Wu Y, Pu X, Wang X, Xu M. Reprogramming of lipid metabolism in the tumor microenvironment: a strategy for tumor immunotherapy. Lipids Health Dis. 2024;23(1):35. doi:10.1186/s12944-024-02024-0. [Google Scholar] [PubMed] [CrossRef]
58. Ding H, Liu J, Wang C, Su Y. NONO promotes hepatocellular carcinoma progression by enhancing fatty acids biosynthesis through interacting with ACLY mRNA. Cancer Cell Int. 2020;20:425. doi:10.1186/s12935-020-01520-4. [Google Scholar] [PubMed] [CrossRef]
59. Eltayeb K, La Monica S, Tiseo M, Alfieri R, Fumarola C. Reprogramming of lipid metabolism in lung cancer: an overview with focus on EGFR-mutated non-small cell lung cancer. Cells. 2022;11(3):413. doi:10.3390/cells11030413. [Google Scholar] [PubMed] [CrossRef]
60. Zhou X, Zhu X, Zeng H. Fatty acid metabolism in adaptive immunity. FEBS J. 2023;290(3):584–99. doi:10.1111/febs.16296. [Google Scholar] [PubMed] [CrossRef]
61. Wise JTF, Kondo K. Increased lipogenesis is important for hexavalent chromium-transformed lung cells and xenograft tumor growth. Int J Mol Sci. 2023;24(23):17060. doi:10.3390/ijms242317060. [Google Scholar] [PubMed] [CrossRef]
62. Yang J, Shay C, Saba NF, Teng Y. Cancer metabolism and carcinogenesis. Exp Hematol Oncol. 2024;13(1):10. doi:10.1186/s40164-024-00482-x. [Google Scholar] [PubMed] [CrossRef]
63. Huang Q, Wang Q, Li D, Wei X, Jia Y, Zhang Z, et al. Co-administration of 20(S)-protopanaxatriol (g-PPT) and EGFR-TKI overcomes EGFR-TKI resistance by decreasing SCD1 induced lipid accumulation in non-small cell lung cancer. J Exp Clin Cancer Res. 2019;38(1):129. doi:10.1186/s13046-019-1120-4. [Google Scholar] [PubMed] [CrossRef]
64. Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang XY, et al. Fatty acid oxidation: an emerging facet of metabolic transformation in cancer. Cancer Lett. 2018;435:92–100. doi:10.1016/j.canlet.2018.08.006. [Google Scholar] [PubMed] [CrossRef]
65. Shao H, Mohamed EM, Xu GG, Waters M, Jing K, Ma Y, et al. Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer. Oncotarget. 2016;7(4):3832–46. doi:10.18632/oncotarget.6757. [Google Scholar] [PubMed] [CrossRef]
66. Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011;25(10):1041–51. doi:10.1101/gad.1987211. [Google Scholar] [PubMed] [CrossRef]
67. Chen R, Wang J, Huang S, Hu X, He X, Zhang T, et al. Carnitine palmitoyltransferase 1C promotes EMT-associated cisplatin resistance in non-small cell lung cancer cells. Cancer Biol Med. 2025;22(1):48–66. doi:10.20892/j.issn.2095-3941.2024.0459. [Google Scholar] [PubMed] [CrossRef]
68. Tian Y, Ma QZ, Wang H, Shi MY, Zhao Y, Chen XH, et al. Tumor PLA2G10 restrains CD4+ T cell activation in lung adenocarcinoma via enhancing arachidonic acid secretion. Int J Biol Macromol. 2025;334:149147. doi:10.1016/j.ijbiomac.2025.149147. [Google Scholar] [PubMed] [CrossRef]
69. Zhang A, Luo X, Li Y, Yan L, Lai X, Yang Q, et al. Epigenetic changes driven by environmental pollutants in lung carcinogenesis: a comprehensive review. Front Public Health. 2024;12:1420933. doi:10.3389/fpubh.2024.1420933. [Google Scholar] [PubMed] [CrossRef]
70. Yang M, Lei X, Ren D, Qin D, Xia X. Epigenetic regulation of the tumor microenvironment in lung cancer: mechanism insights and therapeutic prospects. Front Immunol. 2025;16:1697428. doi:10.3389/fimmu.2025.1697428. [Google Scholar] [PubMed] [CrossRef]
71. Li Q, Zhang Z, Fan Y, Zhang Q. Epigenetic alterations in renal cell cancer with TKIs resistance: from mechanisms to clinical applications. Front Genet. 2021;11:562868. doi:10.3389/fgene.2020.562868. [Google Scholar] [PubMed] [CrossRef]
72. Lai Q, Xu YH, Chen Q, Tang L, Li AG, Zhang LF, et al. The loss-of-function of DNA methyltransferase 1 by siRNA impairs the growth of non-small cell lung cancer with alleviated side effects via reactivation of RASSF1A and APC in vitro and vivo. Oncotarget. 2017;8(35):59301–11. doi:10.18632/oncotarget.19573. [Google Scholar] [PubMed] [CrossRef]
73. Mohd Kamal K, Ghazali AR, Ab Mutalib NS, Abu N, Chua EW, Masre SF. The role of DNA methylation and DNA methyltransferases (DNMTs) as potential biomarker and therapeutic target in non-small cell lung cancer (NSCLC). Heliyon. 2024;10(19):e38663. doi:10.1016/j.heliyon.2024.e38663. [Google Scholar] [PubMed] [CrossRef]
74. Exposito F, Redrado M, Serrano D, Calabuig-Fariñas S, Bao-Caamano A, Gallach S, et al. G9a/DNMT1 co-targeting inhibits non-small cell lung cancer growth and reprograms tumor cells to respond to cancer-drugs through SCARA5 and AOX1. Cell Death Dis. 2024;15(11):787. doi:10.1038/s41419-024-07156-w. [Google Scholar] [PubMed] [CrossRef]
75. Rahim A, Ruis BL, Rajczewski AT, Kruk ME, Tretyakova NY. TET1 functions as a tumor suppressor in lung adenocarcinoma through epigenetic remodeling and immune modulation. Epigenetics Chromatin. 2025;18(1):53. doi:10.1186/s13072-025-00617-2. [Google Scholar] [PubMed] [CrossRef]
76. Zhao Z, Shilatifard A. Epigenetic modifications of histones in cancer. Genome Biol. 2019;20(1):245. doi:10.1186/s13059-019-1870-5. [Google Scholar] [PubMed] [CrossRef]
77. Alam H, Tang M, Maitituoheti M, Dhar SS, Kumar M, Han CY, et al. KMT2D deficiency impairs super-enhancers to confer a glycolytic vulnerability in lung cancer. Cancer Cell. 2020;37(4):599–617.e7. doi:10.1016/j.ccell.2020.03.005. [Google Scholar] [PubMed] [CrossRef]
78. Du M, Gong P, Zhang Y, Liu Y, Liu X, Zhang F, et al. Histone methyltransferase SETD1A participates in lung cancer progression. Thorac Cancer. 2021;12(16):2247–57. doi:10.1111/1759-7714.14065. [Google Scholar] [PubMed] [CrossRef]
79. Chao YL, Pecot CV. Targeting epigenetics in lung cancer. Cold Spring Harb Perspect Med. 2021;11(6):a038000. doi:10.1101/cshperspect.a038000. [Google Scholar] [PubMed] [CrossRef]
80. Shi MQ, Xu Y, Fu X, Pan DS, Lu XP, Xiao Y, et al. Advances in targeting histone deacetylase for treatment of solid tumors. J Hematol Oncol. 2024;17(1):37. doi:10.1186/s13045-024-01551-8. [Google Scholar] [PubMed] [CrossRef]
81. Wang KL, Yeh TY, Hsu PC, Wong TH, Liu JR, Chern JW, et al. Discovery of novel anaplastic lymphoma kinase (ALK) and histone deacetylase (HDAC) dual inhibitors exhibiting antiproliferative activity against non-small cell lung cancer. J Enzyme Inhib Med Chem. 2024;39(1):2318645. doi:10.1080/14756366.2024.2318645. [Google Scholar] [PubMed] [CrossRef]
82. Fan Y, Ji X, Yuan K, Wu Q, Lou M. HDAC1 mediates immunosuppression of the tumor microenvironment in non-small cell lung cancer. J Inflamm Res. 2025;18:3333–47. doi:10.2147/JIR.S509316. [Google Scholar] [PubMed] [CrossRef]
83. Wang T, Lu Z, Han T, Wang Y, Gan M, Wang JB. Deacetylation of glutaminase by HDAC4 contributes to lung cancer tumorigenesis. Int J Biol Sci. 2022;18(11):4452–65. doi:10.7150/ijbs.69882. [Google Scholar] [PubMed] [CrossRef]
84. Al-Malsi K, Xie S, Cai Y, Mohammed N, Xie K, Lan T, et al. The role of lactylation in tumor growth and cancer progression. Front Oncol. 2025;15:1516785. doi:10.3389/fonc.2025.1516785. [Google Scholar] [PubMed] [CrossRef]
85. Jiang J, Huang D, Jiang Y, Hou J, Tian M, Li J, et al. Lactate modulates cellular metabolism through histone lactylation-mediated gene expression in non-small cell lung cancer. Front Oncol. 2021;11:647559. doi:10.3389/fonc.2021.647559. [Google Scholar] [PubMed] [CrossRef]
86. Tomasetti M, Amati M, Santarelli L, Neuzil J. microRNA in metabolic re-programming and their role in tumorigenesis. Int J Mol Sci. 2016;17(5):754. doi:10.3390/ijms17050754. [Google Scholar] [PubMed] [CrossRef]
87. Liu M, Gao J, Huang Q, Jin Y, Wei Z. Downregulating microRNA-144 mediates a metabolic shift in lung cancer cells by regulating GLUT1 expression. Oncol Lett. 2016;11(6):3772–6. doi:10.3892/ol.2016.4468. [Google Scholar] [PubMed] [CrossRef]
88. Ni K, Wang D, Xu H, Mei F, Wu C, Liu Z, et al. miR-21 promotes non-small cell lung cancer cells growth by regulating fatty acid metabolism. Cancer Cell Int. 2019;19:219. doi:10.1186/s12935-019-0941-8. [Google Scholar] [PubMed] [CrossRef]
89. Hu Q, Li Y, Li D, Yuan Y, Wang K, Yao L, et al. Amino acid metabolism regulated by lncRNAs: the propellant behind cancer metabolic reprogramming. Cell Commun Signal. 2023;21(1):87. doi:10.1186/s12964-023-01116-1. [Google Scholar] [PubMed] [CrossRef]
90. Yang B, Zhang L, Cao Y, Chen S, Cao J, Wu D, et al. Overexpression of lncRNA IGFBP4-1 reprograms energy metabolism to promote lung cancer progression. Mol Cancer. 2017;16(1):154. doi:10.1186/s12943-017-0722-8. [Google Scholar] [PubMed] [CrossRef]
91. Gilyazova I, Gimalova G, Nizamova A, Galimova E, Ishbulatova E, Pavlov V, et al. Non-coding RNAs as key regulators in lung cancer. Int J Mol Sci. 2024;25(1):560. doi:10.3390/ijms25010560. [Google Scholar] [PubMed] [CrossRef]
92. Shao Y, Yu X, Hu M, Yan J, Miao M, Ye G, et al. Acting mechanism and clinical significance of hsa_circ_0005927 in the invasion and metastasis of gastric cancer. J Cancer. 2024;15(13):4081–94. doi:10.7150/jca.96749. [Google Scholar] [PubMed] [CrossRef]
93. Lou Y, Yan J, Liu Q, Miao M, Shao Y. Biological functions and molecular mechanisms of exosome-derived circular RNAs and their clinical implications in digestive malignancies: the vintage in the bottle. Ann Med. 2024;56(1):2420861. doi:10.1080/07853890.2024.2420861. [Google Scholar] [PubMed] [CrossRef]
94. Yue Q, Xu Y, Deng X, Wang S, Qiu J, Qian B, et al. Circ-PITX1 promotes the progression of non-small cell lung cancer through regulating the miR-1248/CCND2 axis. Onco Targets Ther. 2021;14:1807–19. doi:10.2147/OTT.S286820. [Google Scholar] [PubMed] [CrossRef]
95. Yan J, Ye G, Jin Y, Miao M, Li Q, Zhou H. Identification of novel prognostic circRNA biomarkers in circRNA-miRNA-mRNA regulatory network in gastric cancer and immune infiltration analysis. BMC Genomics. 2023;24(1):323. doi:10.1186/s12864-023-09421-2. [Google Scholar] [PubMed] [CrossRef]
96. Yang F, Ma C, Qiu J, Feng X, Yang K. Identification of circRNA_001846 as putative non-small cell lung cancer biomarker. Bioengineered. 2021;12(1):8690–7. doi:10.1080/21655979.2021.1991161. [Google Scholar] [PubMed] [CrossRef]
97. Hang D, Zhou J, Qin N, Zhou W, Ma H, Jin G, et al. A novel plasma circular RNA circFARSA is a potential biomarker for non-small cell lung cancer. Cancer Med. 2018;7(6):2783–91. doi:10.1002/cam4.1514. [Google Scholar] [PubMed] [CrossRef]
98. Yin Z, Bai L, Li W, Zeng T, Tian H, Cui J. Targeting T cell metabolism in the tumor microenvironment: an anti-cancer therapeutic strategy. J Exp Clin Cancer Res. 2019;38(1):403. doi:10.1186/s13046-019-1409-3. [Google Scholar] [PubMed] [CrossRef]
99. Herbel C, Patsoukis N, Bardhan K, Seth P, Weaver JD, Boussiotis VA. Clinical significance of T cell metabolic reprogramming in cancer. Clin Transl Med. 2016;5(1):29. doi:10.1186/s40169-016-0110-9. [Google Scholar] [PubMed] [CrossRef]
100. Guerrero JA, Klysz DD, Chen Y, Malipatlolla M, Lone J, Fowler C, et al. GLUT1 overexpression in CAR-T cells induces metabolic reprogramming and enhances potency. Nat Commun. 2024;15:8658. doi:10.1038/s41467-024-52666-y. [Google Scholar] [PubMed] [CrossRef]
101. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol. 2016;17(1):95–103. doi:10.1038/ni.3313. [Google Scholar] [PubMed] [CrossRef]
102. Dabi YT, Andualem H, Degechisa ST, Gizaw ST. Targeting metabolic reprogramming of T-cells for enhanced anti-tumor response. Biologics. 2022;16:35–45. doi:10.2147/BTT.S365490. [Google Scholar] [PubMed] [CrossRef]
103. Patel CH, Powell JD. Targeting T cell metabolism to regulate T cell activation, differentiation and function in disease. Curr Opin Immunol. 2017;46:82–8. doi:10.1016/j.coi.2017.04.006. [Google Scholar] [PubMed] [CrossRef]
104. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167(3):829–42.e13. doi:10.1016/j.cell.2016.09.031. [Google Scholar] [PubMed] [CrossRef]
105. Tang Y, Chen Z, Zuo Q, Kang Y. Regulation of CD8+ T cells by lipid metabolism in cancer progression. Cell Mol Immunol. 2024;21(11):1215–30. doi:10.1038/s41423-024-01224-z. [Google Scholar] [PubMed] [CrossRef]
106. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8+ T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell. 2017;32(3):377–91.e9. doi:10.1016/j.ccell.2017.08.004. [Google Scholar] [PubMed] [CrossRef]
107. Lobel GP, Han N, Molina Arocho WA, Silber M, Shoush J, Noji MC, et al. Glutamine is critical for the maintenance of type 1 conventional dendritic cells in normal tissue and the tumor microenvironment. Proc Natl Acad Sci U S A. 2024;121(50):e2412157121. doi:10.1073/pnas.2412157121. [Google Scholar] [PubMed] [CrossRef]
108. Hu Z, Yu X, Ding R, Liu B, Gu C, Pan XW, et al. Glycolysis drives STING signaling to facilitate dendritic cell antitumor function. J Clin Investig. 2023;133(7):e166031. doi:10.1172/jci166031. [Google Scholar] [PubMed] [CrossRef]
109. Tian H, Li K, Chen J, Guo W, Guo Z, Zhang S, et al. Impaired natural killer cell maturation in lung adenocarcinoma driven by FABP4 and SPON2 downregulation through disrupted lipid metabolism. Transl Lung Cancer Res. 2025;14(5):1660–76. doi:10.21037/tlcr-24-1027. [Google Scholar] [PubMed] [CrossRef]
110. Du Q, An Q, Zhang J, Liu C, Hu Q. Unravelling immune microenvironment features underlying tumor progression in the single-cell era. Cancer Cell Int. 2024;24(1):143. doi:10.1186/s12935-024-03335-z. [Google Scholar] [PubMed] [CrossRef]
111. Wang J, He Y, Hu F, Hu C, Sun Y, Yang K, et al. Metabolic reprogramming of immune cells in the tumor microenvironment. Int J Mol Sci. 2024;25(22):12223. doi:10.3390/ijms252212223. [Google Scholar] [PubMed] [CrossRef]
112. Kinoshita F, Tagawa T, Akamine T, Takada K, Yamada Y, Oku Y, et al. Interleukin-38 promotes tumor growth through regulation of CD8+ tumor-infiltrating lymphocytes in lung cancer tumor microenvironment. Cancer Immunol Immunother. 2021;70(1):123–35. doi:10.1007/s00262-020-02659-9. [Google Scholar] [PubMed] [CrossRef]
113. Lim AR, Rathmell WK, Rathmell JC. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife. 2020;9:e55185. doi:10.7554/eLife.55185. [Google Scholar] [PubMed] [CrossRef]
114. Li X, Liu R, Su X, Pan Y, Han X, Shao C, et al. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer. 2019;18(1):177. doi:10.1186/s12943-019-1102-3. [Google Scholar] [PubMed] [CrossRef]
115. Liu N, Luo J, Kuang D, Xu S, Duan Y, Xia Y, et al. Lactate inhibits ATP6V0d2 expression in tumor-associated macrophages to promote HIF-2α-mediated tumor progression. J Clin Invest. 2019;129(2):631–46. doi:10.1172/JCI123027. [Google Scholar] [PubMed] [CrossRef]
116. Shao N, Qiu H, Liu J, Xiao D, Zhao J, Chen C, et al. Targeting lipid metabolism of macrophages: a new strategy for tumor therapy. J Adv Res. 2025;68:99–114. doi:10.1016/j.jare.2024.02.009. [Google Scholar] [PubMed] [CrossRef]
117. Shen L, Zhou Y, He H, Chen W, Lenahan C, Li X, et al. Crosstalk between macrophages, T cells, and iron metabolism in tumor microenvironment. Oxid Med Cell Longev. 2021;2021:8865791. doi:10.1155/2021/8865791. [Google Scholar] [PubMed] [CrossRef]
118. Shin MH, Kim J, Lim SA, Kim J, Kim SJ, Lee KM. NK cell-based immunotherapies in cancer. Immune Netw. 2020;20(2):e14. doi:10.4110/in.2020.20.e14. [Google Scholar] [PubMed] [CrossRef]
119. Choi C, Finlay DK. Optimising NK cell metabolism to increase the efficacy of cancer immunotherapy. Stem Cell Res Ther. 2021;12(1):320. doi:10.1186/s13287-021-02377-8. [Google Scholar] [PubMed] [CrossRef]
120. Zeng Y, Lv X, Du J. Natural killer cell-based immunotherapy for lung cancer: challenges and perspectives (Review). Oncol Rep. 2021;46(5):232. doi:10.3892/or.2021.8183. [Google Scholar] [PubMed] [CrossRef]
121. Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms controlling PD-L1 expression in cancer. Mol Cell. 2019;76(3):359–70. doi:10.1016/j.molcel.2019.09.030. [Google Scholar] [PubMed] [CrossRef]
122. Asgarova A, Asgarov K, Godet Y, Peixoto P, Nadaradjane A, Boyer-Guittaut M, et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology. 2018;7(5):e1423170. doi:10.1080/2162402X.2017.1423170. [Google Scholar] [PubMed] [CrossRef]
123. Nawas AF, Solmonson A, Gao B, DeBerardinis RJ, Minna JD, Conacci-Sorrell M, et al. IL-1β mediates the induction of immune checkpoint regulators IDO1 and PD-L1 in lung adenocarcinoma cells. Cell Commun Signal. 2023;21(1):331. doi:10.1186/s12964-023-01348-1. [Google Scholar] [PubMed] [CrossRef]
124. Shen C, Liu C, Zhang Z, Ping Y, Shao J, Tian Y, et al. PD-1 affects the immunosuppressive function of group 2 innate lymphoid cells in human non-small cell lung cancer. Front Immunol. 2021;12:680055. doi:10.3389/fimmu.2021.680055. [Google Scholar] [PubMed] [CrossRef]
125. Zhao Y, Guo S, Deng J, Shen J, Du F, Wu X, et al. VEGF/VEGFR-targeted therapy and immunotherapy in non-small cell lung cancer: targeting the tumor microenvironment. Int J Biol Sci. 2022;18(9):3845–58. doi:10.7150/ijbs.70958. [Google Scholar] [PubMed] [CrossRef]
126. Li Y, Liu H, Zhao Y, Yue D, Chen C, Li C, et al. Tumor-associated macrophages (TAMs)-derived osteopontin (OPN) upregulates PD-L1 expression and predicts poor prognosis in non-small cell lung cancer (NSCLC). Thorac Cancer. 2021;12(20):2698–709. doi:10.1111/1759-7714.14108. [Google Scholar] [PubMed] [CrossRef]
127. Munir MT, Kay MK, Kang MH, Rahman MM, Al-Harrasi A, Choudhury M, et al. Tumor-associated macrophages as multifaceted regulators of breast tumor growth. Int J Mol Sci. 2021;22(12):6526. doi:10.3390/ijms22126526. [Google Scholar] [PubMed] [CrossRef]
128. Salmaninejad A, Layeghi SM, Falakian Z, Golestani S, Kobravi S, Talebi S, et al. An update to experimental and clinical aspects of tumor-associated macrophages in cancer development: hopes and pitfalls. Clin Exp Med. 2024;24(1):156. doi:10.1007/s10238-024-01417-w. [Google Scholar] [PubMed] [CrossRef]
129. Albini A, Bruno A, Noonan DM, Mortara L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: implications for immunotherapy. Front Immunol. 2018;9:527. doi:10.3389/fimmu.2018.00527. [Google Scholar] [PubMed] [CrossRef]
130. Liu J, Geng X, Hou J, Wu G. New insights into M1/M2 macrophages: key modulators in cancer progression. Cancer Cell Int. 2021;21(1):389. doi:10.1186/s12935-021-02089-2. [Google Scholar] [PubMed] [CrossRef]
131. Lai X, Zhong J, Zhang B, Zhu T, Liao R. Exosomal non-coding RNAs: novel regulators of macrophage-linked intercellular communication in lung cancer and inflammatory lung diseases. Biomolecules. 2023;13(3):536. doi:10.3390/biom13030536. [Google Scholar] [PubMed] [CrossRef]
132. Li Z, Chen S, He X, Gong S, Sun L, Weng L. SLC3A2 promotes tumor-associated macrophage polarization through metabolic reprogramming in lung cancer. Cancer Sci. 2023;114(6):2306–17. doi:10.1111/cas.15760. [Google Scholar] [PubMed] [CrossRef]
133. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci. 2021;22(13):6995. doi:10.3390/ijms22136995. [Google Scholar] [PubMed] [CrossRef]
134. Yang P, Qin H, Li Y, Xiao A, Zheng E, Zeng H, et al. CD36-mediated metabolic crosstalk between tumor cells and macrophages affects liver metastasis. Nat Commun. 2022;13:5782. doi:10.1038/s41467-022-33349-y. [Google Scholar] [PubMed] [CrossRef]
135. Su P, Wang Q, Bi E, Ma X, Liu L, Yang M, et al. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res. 2020;80(7):1438–50. doi:10.1158/0008-5472.CAN-19-2994. [Google Scholar] [PubMed] [CrossRef]
136. Zeng W, Li F, Jin S, Ho PC, Liu PS, Xie X. Functional polarization of tumor-associated macrophages dictated by metabolic reprogramming. J Exp Clin Cancer Res. 2023;42(1):245. doi:10.1186/s13046-023-02832-9. [Google Scholar] [PubMed] [CrossRef]
137. Palmieri EM, Menga A, Martín-Pérez R, Quinto A, Riera-Domingo C, De Tullio G, et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep. 2017;20(7):1654–66. doi:10.1016/j.celrep.2017.07.054. [Google Scholar] [PubMed] [CrossRef]
138. Tian X, Ma J, Wang T, Tian J, Zheng Y, Peng R, et al. Long non-coding RNA RUNXOR accelerates MDSC-mediated immunosuppression in lung cancer. BMC Cancer. 2018;18(1):660. doi:10.1186/s12885-018-4564-6. [Google Scholar] [PubMed] [CrossRef]
139. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208–20. doi:10.1016/j.it.2016.01.004. [Google Scholar] [PubMed] [CrossRef]
140. Wong KY, Cheung AH, Chen B, Chan WN, Yu J, Lo KW, et al. Cancer-associated fibroblasts in nonsmall cell lung cancer: from molecular mechanisms to clinical implications. Int J Cancer. 2022;151(8):1195–215. doi:10.1002/ijc.34127. [Google Scholar] [PubMed] [CrossRef]
141. Bronte G, Calabrò L, Olivieri F, Procopio AD, Crinò L. The prognostic effects of circulating myeloid-derived suppressor cells in non-small cell lung cancer: systematic review and meta-analysis. Clin Exp Med. 2023;23(5):1551–61. doi:10.1007/s10238-022-00946-6. [Google Scholar] [PubMed] [CrossRef]
142. He ZN, Zhang CY, Zhao YW, He SL, Li Y, Shi BL, et al. Regulation of T cells by myeloid-derived suppressor cells: emerging immunosuppressor in lung cancer. Discov Oncol. 2023;14(1):185. doi:10.1007/s12672-023-00793-1. [Google Scholar] [PubMed] [CrossRef]
143. Basurto-Olvera P, Serrano H, Maldonado-Bernal C. Regulatory T cells in cancer: from immunosuppression to therapeutic targeting. Front Immunol. 2025;16:1703211. doi:10.3389/fimmu.2025.1703211. [Google Scholar] [PubMed] [CrossRef]
144. Zhu S, Zhou N, Li Q, Liu X. Rewiring immune suppression in NSCLC: roles and plasticity of Tregs and Th17 cells. Front Immunol. 2025;16:1658848. doi:10.3389/fimmu.2025.1658848. [Google Scholar] [PubMed] [CrossRef]
145. Liang F, Wang GZ, Wang Y, Yang YN, Wen ZS, Chen DN, et al. Tobacco carcinogen induces tryptophan metabolism and immune suppression via induction of indoleamine 2,3-dioxygenase 1. Signal Transduct Target Ther. 2022;7(1):311. doi:10.1038/s41392-022-01127-3. [Google Scholar] [PubMed] [CrossRef]
146. Kratzmeier C, Taheri M, Mei Z, Lim I, Khalil MA, Carter-Cooper B, et al. Lung adenocarcinoma–derived IFN-γ promotes growth by modulating CD8+ T cell production of CCR5 chemokines. J Clin Investig. 2025;135(17):e191070. doi:10.1172/jci191070. [Google Scholar] [PubMed] [CrossRef]
147. Zheng H, Ning Y, Zhan Y, Liu S, Yang Y, Wen Q, et al. Co-expression of PD-L1 and HIF-1α predicts poor prognosis in patients with non-small cell lung cancer after surgery. J Cancer. 2021;12(7):2065–72. doi:10.7150/jca.53119. [Google Scholar] [PubMed] [CrossRef]
148. Zhang H, Yue X, Chen Z, Liu C, Wu W, Zhang N, et al. Define cancer-associated fibroblasts (CAFs) in the tumor microenvironment: new opportunities in cancer immunotherapy and advances in clinical trials. Mol Cancer. 2023;22(1):159. doi:10.1186/s12943-023-01860-5. [Google Scholar] [PubMed] [CrossRef]
149. Kundu M, Butti R, Panda VK, Malhotra D, Das S, Mitra T, et al. Modulation of the tumor microenvironment and mechanism of immunotherapy-based drug resistance in breast cancer. Mol Cancer. 2024;23(1):92. doi:10.1186/s12943-024-01990-4. [Google Scholar] [PubMed] [CrossRef]
150. Grout JA, Sirven P, Leader AM, Maskey S, Hector E, Puisieux I, et al. Spatial positioning and matrix programs of cancer-associated fibroblasts promote T-cell exclusion in human lung tumors. Cancer Discov. 2022;12(11):2606–25. doi:10.1158/2159-8290.CD-21-1714. [Google Scholar] [PubMed] [CrossRef]
151. Yang N, Lode K, Berzaghi R, Islam A, Martinez-Zubiaurre I, Hellevik T. Irradiated tumor fibroblasts avoid immune recognition and retain immunosuppressive functions over natural killer cells. Front Immunol. 2021;11:602530. doi:10.3389/fimmu.2020.602530. [Google Scholar] [PubMed] [CrossRef]
152. Kanzaki R, Reid S, Bolivar P, Sjölund J, Staaf J, Larsson S, et al. FHL2 expression by cancer-associated fibroblasts promotes metastasis and angiogenesis in lung adenocarcinoma. Int J Cancer. 2025;156(2):431–46. doi:10.1002/ijc.35174. [Google Scholar] [PubMed] [CrossRef]
153. Wang Q, Sun Y, Li J, Li Z, Yuan F, Xia Z, et al. Targeting LINC01711 in FAP+ cancer-associated fibroblasts overcomes lactate-mediated immunosuppression and enhances anti-PD-1 efficacy in lung adenocarcinoma. Cell Death Dis. 2025;16(1):642. doi:10.1038/s41419-025-07974-6. [Google Scholar] [PubMed] [CrossRef]
154. Zeng Y, Huang Y, Tan Q, Peng L, Wang J, Tong F, et al. Influence of lactate in resistance to anti-PD-1/PD-L1 therapy: mechanisms and clinical applications (Review). Mol Med Rep. 2025;31(2):48. doi:10.3892/mmr.2024.13413. [Google Scholar] [PubMed] [CrossRef]
155. Wu G, Cheng H, Yin J, Zheng Y, Shi H, Pan B, et al. NDRG1-driven lactate accumulation promotes lung adenocarcinoma progression through the induction of an immunosuppressive microenvironment. Adv Sci. 2025;12(33):e01238. doi:10.1002/advs.202501238. [Google Scholar] [PubMed] [CrossRef]
156. Ding W, Mo J, Su Y, Zhang Q, Sun D, Yao X, et al. Metabolic reprogramming of tumor-associated macrophages via adenosine-A2AR signaling drives cross-resistance in non-small cell lung cancer. Drug Resist Updat. 2025;82:101272. doi:10.1016/j.drup.2025.101272. [Google Scholar] [PubMed] [CrossRef]
157. Song G, Shang C, Zhu Y, Xiu Z, Li Y, Yang X, et al. Apoptin inhibits glycolysis and regulates autophagy by targeting pyruvate kinase M2 (PKM2) in lung cancer A549 cells. Curr Cancer Drug Targets. 2024;24(4):411–24. doi:10.2174/1568009623666221025150239. [Google Scholar] [PubMed] [CrossRef]
158. Liu H, Guo W, Wang T, Cao P, Zou T, Peng Y, et al. CD36 inhibition reduces non-small-cell lung cancer development through AKT-mTOR pathway. Cell Biol Toxicol. 2024;40(1):10. doi:10.1007/s10565-024-09848-7. [Google Scholar] [PubMed] [CrossRef]
159. Yu Y, Nie Q, Wang Z, Di Y, Chen X, Ren K. Targeting acetyl-CoA carboxylase 1 for cancer therapy. Front Pharmacol. 2023;14:1129010. doi:10.3389/fphar.2023.1129010. [Google Scholar] [PubMed] [CrossRef]
160. Wang K, Lu H, Wang X, Liu Q, Hu J, Liu Y, et al. Simultaneous suppression of PKM2 and PHGDH elicits synergistic anti-cancer effect in NSCLC. Front Pharmacol. 2023;14:1200538. doi:10.3389/fphar.2023.1200538. [Google Scholar] [PubMed] [CrossRef]
161. Boysen G, Jamshidi-Parsian A, Davis MA, Siegel ER, Simecka CM, Kore RA, et al. Glutaminase inhibitor CB-839 increases radiation sensitivity of lung tumor cells and human lung tumor xenografts in mice. Int J Radiat Biol. 2019;95(4):436–42. doi:10.1080/09553002.2018.1558299. [Google Scholar] [PubMed] [CrossRef]
162. Xia M, Li X, Diao Y, Du B, Li Y. Targeted inhibition of glutamine metabolism enhances the antitumor effect of selumetinib in KRAS-mutant NSCLC. Transl Oncol. 2021;14(1):100920. doi:10.1016/j.tranon.2020.100920. [Google Scholar] [PubMed] [CrossRef]
163. Yeo J, Ko M, Lee DH, Park Y, Jin HS. TIGIT/CD226 axis regulates anti-tumor immunity. Pharmaceuticals. 2021;14(3):200. doi:10.3390/ph14030200. [Google Scholar] [PubMed] [CrossRef]
164. Luo B, Sun Y, Zhan Q, Luo Y, Chen Y, Fu T, et al. Combining TIGIT blockade with IL-15 stimulation is a promising immunotherapy strategy for lung adenocarcinoma. Clin Transl Med. 2024;14(1):e1553. doi:10.1002/ctm2.1553. [Google Scholar] [PubMed] [CrossRef]
165. Zhang P, Liu X, Gu Z, Jiang Z, Zhao S, Song Y, et al. Targeting TIGIT for cancer immunotherapy: recent advances and future directions. Biomark Res. 2024;12(1):7. doi:10.1186/s40364-023-00543-z. [Google Scholar] [PubMed] [CrossRef]
166. Shen J, Liao B, Gong L, Li S, Zhao J, Yang H, et al. CD39 and CD73: biological functions, diseases and therapy. Mol Biomed. 2025;6(1):97. doi:10.1186/s43556-025-00345-9. [Google Scholar] [PubMed] [CrossRef]
167. Azimnasab-Sorkhabi P, Soltani-Asl M, Kfoury Junior JR, Ansa-Addo EA. Hybrid regulatory T cells: camouflaged architects of tumor immunity. Front Immunol. 2025;16:1658576. doi:10.3389/fimmu.2025.1658576. [Google Scholar] [PubMed] [CrossRef]
168. Deng Y, Chen Q, Yang X, Sun Y, Zhang B, Wei W, et al. Tumor cell senescence-induced macrophage CD73 expression is a critical metabolic immune checkpoint in the aging tumor microenvironment. Theranostics. 2024;14(3):1224–40. doi:10.7150/thno.91119. [Google Scholar] [PubMed] [CrossRef]
169. Liu H, Xiong X, Zhu W, Wang S, Huang W, Zhu G, et al. Gut microbial metabolites in cancer immunomodulation. Mol Cancer. 2025;25(1):8. doi:10.1186/s12943-025-02521-5. [Google Scholar] [PubMed] [CrossRef]
170. Bishoyi AK, Al-Hasnaawei S, Salem KH, Ganesan S, Shankhyan A, Nanda A, et al. Gut microbiome metabolites in lung cancer: the emerging importance of short-chain fatty acids. Int Immunopharmacol. 2026;168(Pt 1):115821. doi:10.1016/j.intimp.2025.115821. [Google Scholar] [PubMed] [CrossRef]
171. He J, Shu X, Pan H, Wang M, Song Y, Zhou F, et al. Ginseng polysaccharides inhibit Aspergillus sydowii-driven lung adenocarcinoma via modulating gut microbiota-bile acid metabolism axis. Cancers. 2025;17(19):3134. doi:10.3390/cancers17193134. [Google Scholar] [PubMed] [CrossRef]
172. Zhu Z, Cai J, Hou W, Xu K, Wu X, Song Y, et al. Microbiome and spatially resolved metabolomics analysis reveal the anticancer role of gut Akkermansia muciniphila by crosstalk with intratumoral microbiota and reprogramming tumoral metabolism in mice. Gut Microbes. 2023;15(1):2166700. doi:10.1080/19490976.2023.2166700. [Google Scholar] [PubMed] [CrossRef]
173. Messaoudene M, Pidgeon R, Richard C, Ponce M, Diop K, Benlaifaoui M, et al. A natural polyphenol exerts antitumor activity and circumvents anti-PD-1 resistance through effects on the gut microbiota. Cancer Discov. 2022;12(4):1070–87. doi:10.1158/2159-8290.CD-21-0808. [Google Scholar] [PubMed] [CrossRef]
174. Li J, Shi D, Li S, Shi X, Liu Y, Zhang Y, et al. KEAP1 promotes anti-tumor immunity by inhibiting PD-L1 expression in NSCLC. Cell Death Dis. 2024;15:175. doi:10.1038/s41419-024-06563-3. [Google Scholar] [PubMed] [CrossRef]
175. Pillai R, LeBoeuf SE, Hao Y, New C, Blum JLE, Rashidfarrokhi A, et al. Glutamine antagonist DRP-104 suppresses tumor growth and enhances response to checkpoint blockade in KEAP1 mutant lung cancer. Sci Adv. 2024;10(13):eadm9859. doi:10.1126/sciadv.adm9859. [Google Scholar] [PubMed] [CrossRef]
176. Ma L, Chen C, Zhao C, Li T, Ma L, Jiang J, et al. Targeting carnitine palmitoyl transferase 1A (CPT1A) induces ferroptosis and synergizes with immunotherapy in lung cancer. Signal Transduct Target Ther. 2024;9(1):64. doi:10.1038/s41392-024-01772-w. [Google Scholar] [PubMed] [CrossRef]
177. Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res. 2017;23(17):5187–201. doi:10.1158/1078-0432.CCR-17-0741. [Google Scholar] [PubMed] [CrossRef]
178. Papavassiliou KA, Marinos G, Papavassiliou AG. Targeting YAP/TAZ in combination with PD-L1 immune checkpoint inhibitors in non-small cell lung cancer (NSCLC). Cells. 2023;12(6):871. doi:10.3390/cells12060871. [Google Scholar] [PubMed] [CrossRef]
179. Chen Y, Zhou Y, Ren R, Chen Y, Lei J, Li Y. Harnessing lipid metabolism modulation for improved immunotherapy outcomes in lung adenocarcinoma. J Immunother Cancer. 2024;12(7):e008811. doi:10.1136/jitc-2024-008811. [Google Scholar] [PubMed] [CrossRef]
180. Amato L, De Rosa C, Di Guida G, Sepe F, Ariano A, Capaldo S, et al. Addition of metformin to anti-PD-1/PD-L1 drugs activates anti-tumor immune response in peripheral immune cells of NSCLC patients. Cell Death Dis. 2025;16(1):286. doi:10.1038/s41419-025-07636-7. [Google Scholar] [PubMed] [CrossRef]
181. Liu L, Yang L, Li H, Shang T, Liu L. The tumor microenvironment in lung cancer: heterogeneity, therapeutic resistance and emerging treatment strategies (Review). Int J Oncol. 2026;68(1):1–16. doi:10.3892/ijo.2025.5824. [Google Scholar] [PubMed] [CrossRef]
182. Kim J, DeBerardinis RJ. Mechanisms and implications of metabolic heterogeneity in cancer. Cell Metab. 2019;30(3):434–46. doi:10.1016/j.cmet.2019.08.013. [Google Scholar] [PubMed] [CrossRef]
183. Santo V, Brunetti L, Pecci F, Peroni M, Barnini G, Paoloni F, et al. Host-related determinants of response to immunotherapy in non-small cell lung cancer: the interplay of body composition, metabolism, sex and immune regulation. Curr Oncol Rep. 2025;27(12):1427–47. doi:10.1007/s11912-025-01718-7. [Google Scholar] [PubMed] [CrossRef]
184. Fendt SM, Frezza C, Erez A. Targeting metabolic plasticity and flexibility dynamics for cancer therapy. Cancer Discov. 2020;10(12):1797–807. doi:10.1158/2159-8290.CD-20-0844. [Google Scholar] [PubMed] [CrossRef]
185. Lau LYE, Skanderup AJ, Tan AC. Lineage plasticity and histologic transformation in EGFR-TKI resistant lung cancer. Int J Mol Sci. 2025;27(1):445. doi:10.3390/ijms27010445. [Google Scholar] [PubMed] [CrossRef]
186. Gai X, Liu Y, Lan X, Chen L, Yuan T, Xu J, et al. Oncogenic KRAS induces arginine auxotrophy and confers a therapeutic vulnerability to SLC7A1 inhibition in non-small cell lung cancer. Cancer Res. 2024;84(12):1963–77. doi:10.1158/0008-5472.CAN-23-2095. [Google Scholar] [PubMed] [CrossRef]
187. Tomassini L, Pacifico T, Monteleone G, Stolfi C, Laudisi F. Targeting cancer cell energy metabolism in colorectal cancer: opportunities and challenges from drug repositioning. Cells. 2025;14(24):1968. doi:10.3390/cells14241968. [Google Scholar] [PubMed] [CrossRef]
188. Ren Y, Zhang Z, Lei X, Shi L. Long non-coding RNAs in cancer glycolysis and metabolism: mechanisms and translational opportunities. Cell Death Dis. 2025;17(1):71. doi:10.1038/s41419-025-08289-2. [Google Scholar] [PubMed] [CrossRef]
189. Ahuja S, Zaheer S. Molecular mediators of metabolic reprogramming in cancer: mechanisms, regulatory networks, and therapeutic strategies. Immunology. 2026;177(1):1–43. doi:10.1111/imm.70045. [Google Scholar] [PubMed] [CrossRef]
190. Fang L, Gao D, Jiang Z, Li G, Li M. Glutamine’s double-edged sword: fueling tumor growth and offering therapeutic hope. Front Immunol. 2025;16:1578940. doi:10.3389/fimmu.2025.1578940. [Google Scholar] [PubMed] [CrossRef]
191. Zou W, Han Z, Wang Z, Liu Q. Targeting glutamine metabolism as a potential target for cancer treatment. J Exp Clin Cancer Res. 2025;44(1):180. doi:10.1186/s13046-025-03430-7. [Google Scholar] [PubMed] [CrossRef]
192. Tan Y, Tan W, Liang Y, Long Y, Chen S, Hu Q, et al. Machine learning-enabled spatial multi-omics uncovers lactate-driven targets and tumor microenvironmental reprogramming in cancer. npj Digit Med. 2026;9:109. doi:10.1038/s41746-025-02286-7. [Google Scholar] [PubMed] [CrossRef]
193. Chen B, Kiang KM, Liu F, Li C, Li X, Chen W, et al. Neutrophil extracellular trap reprograms cancer metabolism to form a metastatic niche promoting non-small cell lung cancer brain metastasis. Adv Sci. 2026;13(5):e08478. doi:10.1002/advs.202508478. [Google Scholar] [PubMed] [CrossRef]
194. Wu H, Zhang Q, Cao Z, Cao H, Wu M, Fu M, et al. Integrated spatial omics of metabolic reprogramming and the tumor microenvironment in pancreatic cancer. iScience. 2025;28(6):112681. doi:10.1016/j.isci.2025.112681. [Google Scholar] [PubMed] [CrossRef]
195. Gong L, Li S, Sun J, Liu R, Wang J, Wang S, et al. Targeted degradation of hexokinase 2 by a novel engineered bispecific chimeric liposome-based Nano-PROTACs for enhancing tumor chemotherapy. Biomaterials. 2026;326:123651. doi:10.1016/j.biomaterials.2025.123651. [Google Scholar] [PubMed] [CrossRef]
196. Liu H, Mao J, Wang M, Tian Q, Cai Q, An L. Copper-oxamate coordination polymers reverse cuproptosis resistance in nonsmall cell lung cancer via glycolysis reprogramming. ACS Nano. 2026;20(1):892–904. doi:10.1021/acsnano.5c15968. [Google Scholar] [PubMed] [CrossRef]
197. Gouda MA, Voss MH, Tawbi H, Gordon M, Tykodi SS, Lam ET, et al. A phase I/II study of the safety and efficacy of telaglenastat (CB-839) in combination with nivolumab in patients with metastatic melanoma, renal cell carcinoma, and non-small-cell lung cancer. ESMO Open. 2025;10(5):104536. doi:10.1016/j.esmoop.2025.104536. [Google Scholar] [PubMed] [CrossRef]
198. Ny L, Jespersen H, Karlsson J, Alsén S, Filges S, All-Eriksson C, et al. The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat Commun. 2021;12:5155. doi:10.1038/s41467-021-25332-w. [Google Scholar] [PubMed] [CrossRef]
199. Hellmann MD, Jänne PA, Opyrchal M, Hafez N, Raez LE, Gabrilovich DI, et al. Entinostat plus pembrolizumab in patients with metastatic NSCLC previously treated with anti-PD-(L)1 therapy. Clin Cancer Res. 2021;27(4):1019–28. doi:10.1158/1078-0432.ccr-20-3305. [Google Scholar] [PubMed] [CrossRef]
200. Zhang C, Yu JJ, Yang C, Yuan ZL, Zeng H, Wang JJ, et al. Wild-type IDH1 maintains NSCLC stemness and chemoresistance through activation of the serine biosynthetic pathway. Sci Transl Med. 2023;15(726):eade4113. doi:10.1126/scitranslmed.ade4113. [Google Scholar] [PubMed] [CrossRef]
201. Zhang Y, Zhang X, Yang X, Chen X, Wang Y, Hu J, et al. EGFR-TKIs induced DPP4 drives metabolic reprogramming of persister cells in lung cancer. Adv Sci. 2025;12(31):e06950. doi:10.1002/advs.202506950. [Google Scholar] [PubMed] [CrossRef]
202. Guan A, Quek C. Single-cell multi-omics: insights into therapeutic innovations to advance treatment in cancer. Int J Mol Sci. 2025;26(6):2447. doi:10.3390/ijms26062447. [Google Scholar] [PubMed] [CrossRef]
203. Sabit H, Arneth B, Pawlik TM, Abdel-Ghany S, Ghazy A, Abdelazeem RM, et al. Leveraging single-cell multi-omics to decode tumor microenvironment diversity and therapeutic resistance. Pharmaceuticals. 2025;18(1):75. doi:10.3390/ph18010075. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools