
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
Machine Learning (ML) and Molecular Dynamics–Driven Optimization of VEGFR2 Ligands against Hepatocellular Carcinoma
1 Department of Molecular Science and Technology, Ajou University, Suwon, Republic of Korea
2 S&K Therapeutics, Suwon, Republic of Korea
* Corresponding Authors: Abdul Manan. Email: ; Sangdun Choi. Email:
# These authors contributed equally to this work
Oncology Research 2026, 34(5), 24 https://doi.org/10.32604/or.2026.076072
Received 13 November 2025; Accepted 20 January 2026; Issue published 22 April 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Objectives: Vascular endothelial growth factor receptor 2 (VEGFR2) is a critical therapeutic target in hepatocellular carcinoma (HCC) due to its role in angiogenesis and tumor progression. While several inhibitors are currently used, clinical utility is often limited by resistance and adverse effects, necessitating the discovery of novel therapeutic agents. The aim of this study was to identify and characterize novel, highly effective VEGFR2 inhibitors using an integrated computational pipeline to advance the development of new HCC treatments. Methods: A comprehensive dataset from the ChEMBL database was curated and standardized for Quantitative Structure-Activity Relationship (QSAR) modeling. A binary classification framework was employed, where a Light Gradient Boosting Machine (LGBM) model demonstrated superior predictive performance. Two lead compounds and a reference were selected for in-depth molecular modeling. Their binding poses were predicted via molecular docking and subsequently subjected to 200 ns Molecular Dynamics (MD) simulations to assess stability and conformational dynamics. Thermodynamic binding affinities were calculated using the Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA) method. Results: The LGBM model achieved high accuracy and a robust Matthews Correlation Coefficient (MCC) on an independent test set. MD analysis, including Root Mean Square Deviation (RMSD) and Radius of Gyration (Rg), confirmed stable binding throughout the 200 ns trajectory. MMPBSA calculations validated the binding affinities, identifying van der Waals and electrostatic interactions as the primary driving forces for complex stability. Conclusion: This study successfully bridges machine learning with advanced molecular simulations, offering a validated workflow for the rational design and optimization of novel small-molecule VEGFR2 inhibitors.Graphic Abstract
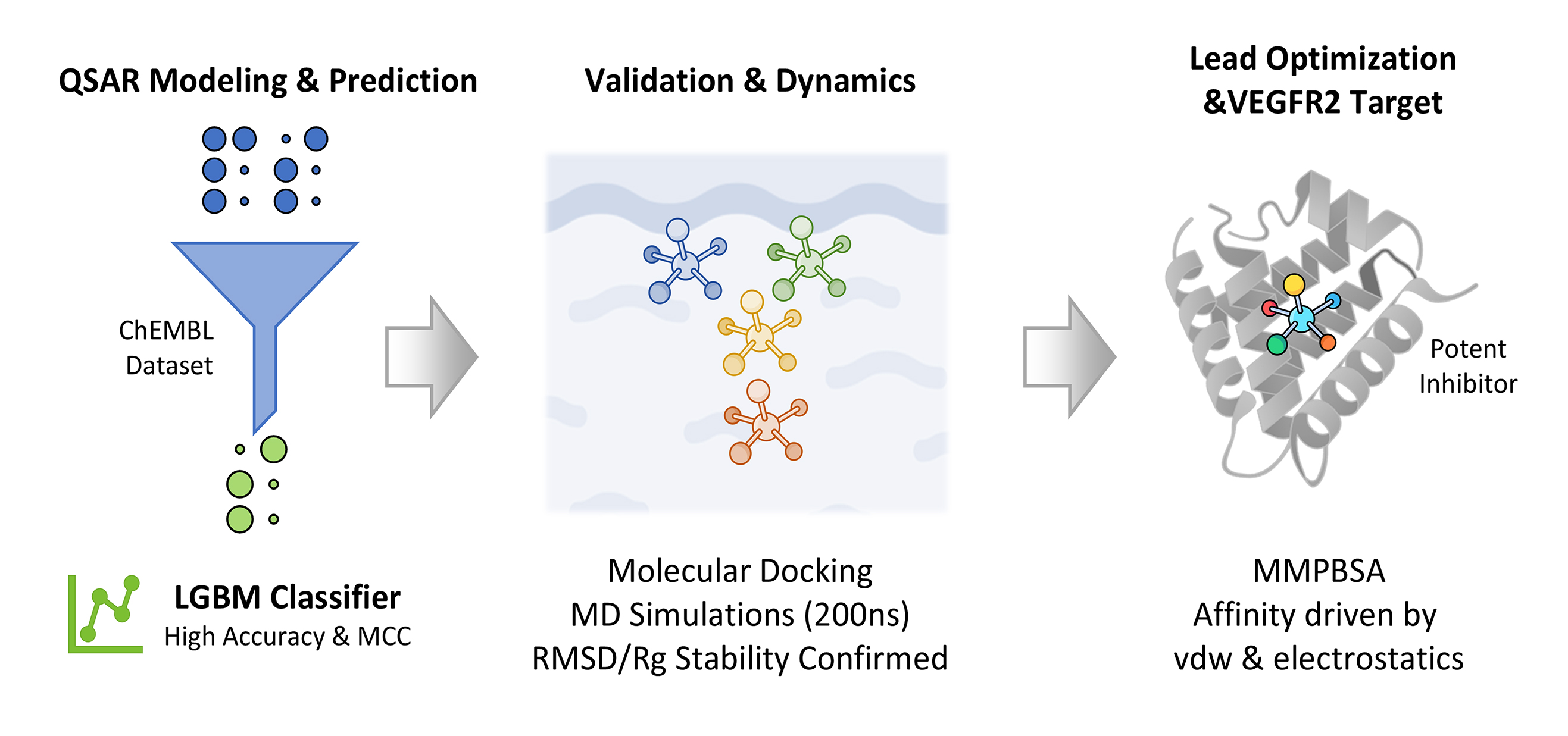
Keywords
Hepatocellular carcinoma (HCC); machine learning; quantitative structure-activity relationship (QSAR); vascular endothelial growth factor receptor 2 (VEGFR2); RDKit; molecular docking and dynamics
Supplementary Material
Supplementary Material FileCite This Article
APA Style
Yasmeen, F., Manan, A., Kim, W., Choi, S. (2026). Machine Learning (ML) and Molecular Dynamics–Driven Optimization of VEGFR2 Ligands against Hepatocellular Carcinoma. Oncology Research, 34(5), 24. https://doi.org/10.32604/or.2026.076072
Vancouver Style
Yasmeen F, Manan A, Kim W, Choi S. Machine Learning (ML) and Molecular Dynamics–Driven Optimization of VEGFR2 Ligands against Hepatocellular Carcinoma. Oncol Res. 2026;34(5):24. https://doi.org/10.32604/or.2026.076072
IEEE Style
F. Yasmeen, A. Manan, W. Kim, and S. Choi, “Machine Learning (ML) and Molecular Dynamics–Driven Optimization of VEGFR2 Ligands against Hepatocellular Carcinoma,” Oncol. Res., vol. 34, no. 5, pp. 24, 2026. https://doi.org/10.32604/or.2026.076072
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools