
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
Detection and Characterization of an Isolate of Cucumber Mosaic Virus Infecting Catharanthus roseus Using Deep Sequencing
1 Department of Biology, Faculty of Science, University of Tabuk, Tabuk, Saudi Arabia
2 Plant Molecular Virology Lab, Department of Botany, School of Chemical and Life Sciences, Jamia Hamdard (A Deemed-to-Be University), New Delhi, India
3 Centre for Virology, School of Interdisciplinary Sciences & Technology, Jamia Hamdard (A Deemed-to-Be University), New Delhi, India
4 Department of Botany and Microbiology, College of Science, King Saud University, Riyadh, Saudi Arabia
5 Department of Studies and Basic Sciences, Applied College, University of Tabuk, Tabuk, Saudi Arabia
6 Department of Science and Basic Studies, Applied College, University of Tabuk, Tabuk, Saudi Arabia
7 Biodiversity Genomic Unit, Faculty of Science, University of Tabuk, Tabuk, Saudi Arabia
* Corresponding Authors: Zahid Hameed Siddiqui. Email: ; Md Salik Noorani. Email:
# These authors contributed equally to this work
(This article belongs to the Special Issue: Technological Advances for Sustainable Management and Biological Control of Plant Pests and Diseases)
Phyton-International Journal of Experimental Botany 2026, 95(4), 8 https://doi.org/10.32604/phyton.2026.076432
Received 20 November 2025; Accepted 11 March 2026; Issue published 28 April 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
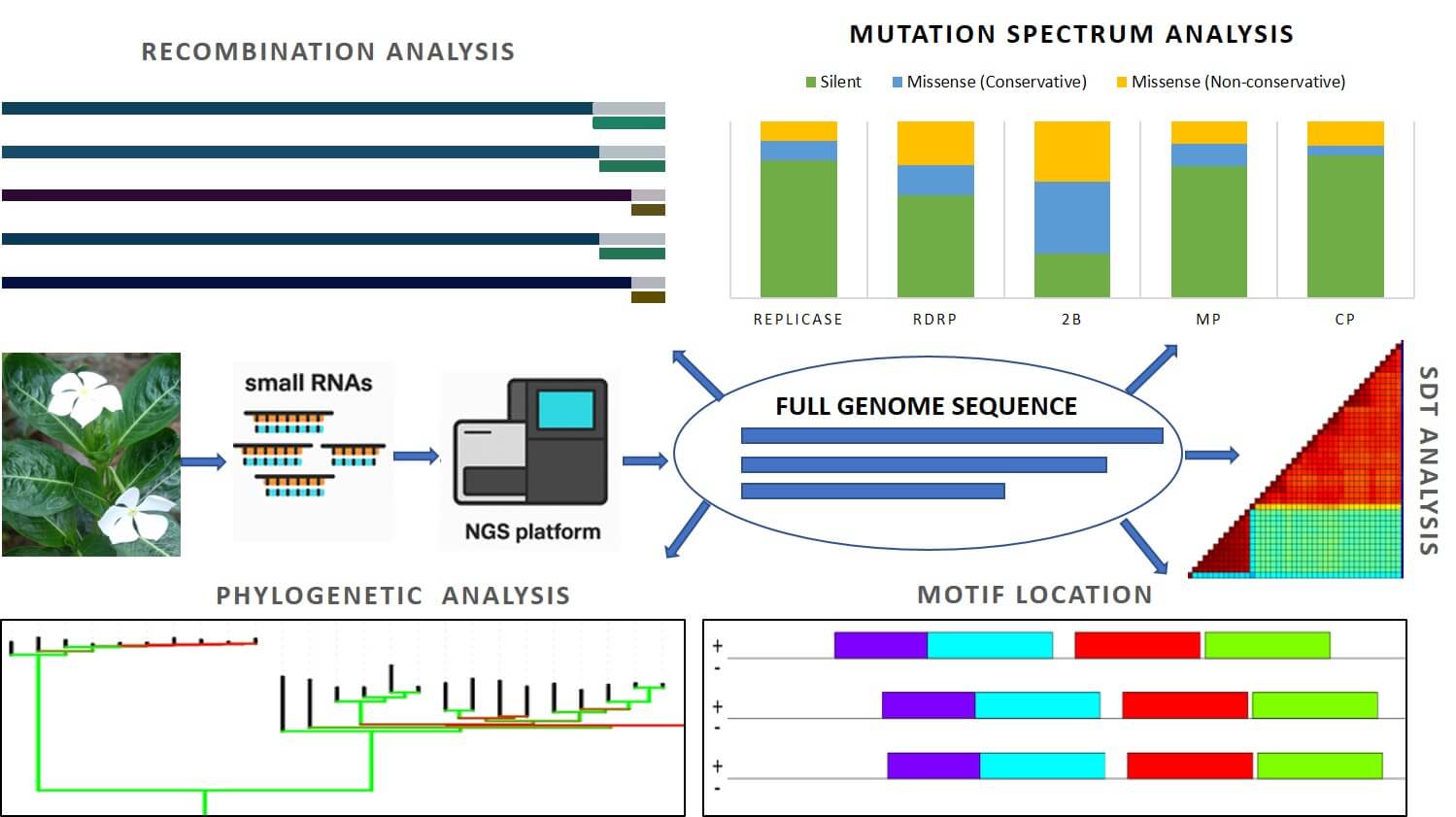
Cucumber mosaic virus (CMV) is among the most widespread plant viruses, infecting over a thousand plant species, including Catharanthus roseus, a medicinal plant valued for producing the anticancer alkaloids vincristine and vinblastine. Despite its economic significance, genomic information on CMV infecting C. roseus in India has been lacking. In this study, we employed small RNA deep sequencing integrated with advanced bioinformatics to generate the first complete genome of CMV infecting C. roseus in India, followed by validation through RT-PCR and Sanger sequencing. The reconstructed tripartite CMV genome encodes replication, silencing suppressor, movement, and coat proteins, consistent with known genomic organization. Sequence identity and phylogenetic analyses revealed a close relationship with Iranian tobacco isolates, placing the Indian isolate (CR8-JH) within subgroup IB. Recombination analysis indicated inter-fragmental recombination events, while mutational profiling highlighted natural variation, particularly in genes associated with host interaction. Conserved UTR motifs were identified, suggesting functional roles in viral replication and genome stability. By applying next-generation sequencing for accurate viral detection and characterization, this study enhances diagnostic capacity and contributes to technology-driven, sustainable plant disease management.Graphic Abstract
Keywords
Supplementary Material
Supplementary Material FileCucumber mosaic virus (CMV: Cucumovirus CMV) belongs to the genus Cucumovirus within the family Bromoviridae. CMV has icosahedral particles, but forms hexagonal inclusion bodies inside infected cells. It is a positive-sense, single-stranded RNA virus about 28–30 nm in diameter [1,2]. Its tripartite genome consists of RNA 1, RNA 2, and RNA 3, encoding five open reading frames (ORFs) [3,4]. RNA 1 encodes the 1a protein (replicase), while RNA 2 encodes the 2a (RdRP) and 2b (silencing suppressor) proteins. RNA3 encodes the 3a (movement protein) and 3b (coat protein) proteins, with the coat protein (CP) being expressed from subgenomic RNA 4. The MP plays a crucial role in virus movement between cells, while the CP is essential for genomic RNA encapsidation, aphid transmission, and symptom expression [5,6,7].
CMV is classified into subgroups I and II based on phylogenetic analysis of nucleotide sequences of RNA segments. Subgroup I is further divided into IA and IB based on the 5′ non-coding region of RNA3 and sequence similarity [5,6,7,8,9]. Subgroup IA and II have a global distribution, whereas subgroup IB is primarily found in Asian countries. In India, broad host range of CMV from subgroup I and II have been identified [6]. Comparatively, subgroup I strains tend to be more virulent than those in subgroup II. The high genetic diversity of CMV provides insights into the role of specific genes in viral virulence [1].
In addition to infecting several crop species, CMV also infects Catharanthus roseus (Madagascar periwinkle) [10,11,12,13,14], which is widely cultivated for both its ornamental value and medicinal importance [15]. The plant is a natural source of diverse bioactive compounds, particularly monoterpenoid indole alkaloids such as vinblastine and vincristine, which serve as essential chemotherapeutic agents [16,17,18]. Beyond its anticancer applications, C. roseus also demonstrates antioxidant, antimicrobial, and antidiabetic properties [19]. Despite its significance, the species is highly susceptible to a variety of diseases caused by fungi, bacteria, nematodes, phytoplasmas, and especially viral pathogens, which pose a major threat to its cultivation and global production [10].
Although C. roseus is economically important, genomic information on CMV infecting this host in India is limited. During field surveys, mild to severe virus-like symptoms were observed on several C. roseus plants within the study area, suggesting a possible localized infection of epidemiological concern [15]. As CMV spreads rapidly through aphids and mechanical contact, even a single infection source can act as a reservoir for transmission to nearby ornamental and medicinal crops [3,20].
Therefore, an accurate and early detection of viruses in plants is vital for effective disease management and epidemiological studies. Conventional methods, including serological assays, RT-PCR, and microscopy, have contributed significantly to virus identification but are limited in sensitivity, throughput, and their ability to detect mixed or novel infections [21,22,23]. In contrast, high-throughput sequencing (HTS), particularly small RNA (sRNA) sequencing, has transformed plant virology by enabling unbiased detection of both known and previously unreported viruses without prior sequence information [15,24,25,26,27]. This approach exploits the host RNA silencing defense pathway, in which virus-derived small interfering RNAs (siRNAs) are generated during infection, allowing reconstruction of viral genomes from short RNA fragments [28,29].
This study reports the detection and complete genome characterization of an isolate CMV through small RNA deep sequencing and validation by RT-PCR and Sanger sequencing. The results provides detailed information about genome structure, sequence identity, recombination events, mutation pattern and selective pressure analysis of Indian CMV isolate. Conserved motifs within the 5′ and 3′ regions of the viral genome were also identified.
2.1 Plant Material and Sample Preparation
Leaf samples were collected from six different C. roseus plants exhibiting viral symptoms such as mosaic, mottling, leaf curl, necrosis, and deformation from the Jamia Hamdard campus for virome analysis (Fig. 1). Young symptomatic leaves were cut into small pieces, mixed thoroughly, and divided into two equal portions to obtain technical replicates. Total RNA was extracted from one gram of tissue from each portion using a slightly modified CTAB method [30]. The extracted RNA was treated with RNase-free DNase A to remove any residual genomic DNA. RNA quantity and purity were assessed using a NanoDrop spectrophotometer (Thermo Fisher Scientific, USA), and RNA integrity was checked by 1% agarose gel electrophoresis. High-quality RNA was then sent to Centyle Biotech Private Limited (New Delhi, India) for small RNA isolation and deep sequencing.

Figure 1: Various symptoms appeared on Catharanthus roseus under field conditions. Infected plants exhibited a range of viral or virus like symptoms including leaf distortion with mild mosaic (A), severe leaf deformation with rugose (wrinkled) surface and vein banding (B), mosaic and chlorotic mottling on mature leaves (C), vein banding with interveinal yellow mosaic (D), stunting accompanied by necrosis and leaf drying (E), and mild mosaic with narrowing and malformation of young leaves (F).
2.2 Bioinformatics Analysis and Data Availability
Paired-end raw reads in FASTQ format, generated on the Illumina NovaSeq 6000 platform, were analyzed using Genome Detective Virus Tools v2.13.3 (https://www.genomedetective.com/app/typingtool/virus/) for virus identification and genome reconstruction. The pipeline automatically performs quality filtering, removal of non-viral reads, de novo contig assembly, and subsequent reference-guided refinement. During the reference-guided step, assembled contigs and reads were aligned against CMV reference genome segments available in GenBank: NC_002034.1 (RNA1), NC_002035.1 (RNA2), and NC_001440.1 (RNA3). To validate the results, the same datasets were independently analyzed using the Galaxy Europe platform (https://usegalaxy.org.au/), following a comparable workflow. The raw sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject accession number PRJNA1019943. Two datasets are available: SRR26156703 (sample CR1-8; BioSample SAMN37518972) and SRR26156704 (sample CR1-7; BioSample SAMN37505367). Among these, the complete genome obtained from sample CR1-8 was used for subsequent analyses, including genome annotation, phylogenetic, recombination, and mutation studies.
2.3 Experimental Validation of HTS Findings
RT-PCR was performed to validate the viral identifications obtained from HTS data. First-strand cDNA was synthesized from total RNA (~1 μg) extracted from symptomatic C. roseus leaf samples using M-MLV reverse transcriptase (Promega, USA). Reverse transcription was carried out in a 25 μL reaction containing total RNA, a mixture of random hexamers and oligo (dT) primers, dNTPs, reaction buffer, and M-MLV reverse transcriptase, with incubation at 25°C for 10 min and 42°C for 60 min, followed by enzyme inactivation at 80°C for 5 min.
The synthesized cDNA was used as a template to amplify a partial region of the RNA-dependent RNA polymerase (RdRp) gene of CMV using the primer pair described by Grieco et al. (2000): forward primer TAACCTCCCAGTTCTCACCGT and reverse primer CATCACCTTAGCTTCCATGT, yielding an expected product of 513 bp [31]. PCR amplification was carried out in a 25 μL reaction mixture containing Taq buffer, dNTPs, primers, and Taq DNA polymerase (GENEI, India). The PCR conditions included an initial denaturation at 95°C for 3 min, followed by 32 cycles of denaturation at 94°C for 30 s, annealing at 50°C for 30 s, and extension at 72°C for 30 s, with a final extension at 72°C for 5 min using a Thermal Cycler (Bio-Rad T100™).
The amplified products were resolved on a 2% agarose gel, purified, sequenced by the Sanger method, and the obtained sequences were confirmed by BLAST analysis at NCBI.
2.4 Sequence Identity Analysis
Pairwise nucleotide identity among CMV isolates was calculated using the Sequence Demarcation Tool v1.2 (SDTv1.2) [32]. A FASTA file having 126 (42 sequences for each RNA 1, RNA 2 and RNA 3) sequences were used as input to determine percentage identity values, providing a quantitative measure of genetic relatedness among isolates.
Genetic variations within the 5′ untranslated regions (UTRs), coding sequences (CDS), and 3′ UTRs of RNA 1, RNA 2, and RNA 3 were examined using CSV files generated by the Genome Detective Virus Tool. Reference sequences NC_002034.1 (RNA 1), NC_002035.1 (RNA 2), and NC_001440.1 (RNA 3) were employed for comparative analysis. Further, mutation classification were conducted using R Statistical Software (version 4.1.2).
2.6 Selective Pressure Analyses (dN/dS Ratios)
Selective pressure acting on the viral coding region was evaluated using MEGA11 [33]. The complete coding sequences of the Indian isolate (CR8-JH) and the reference strain (as mention above) were aligned in codon-aware mode using the MUSCLE algorithm to preserve reading frames. Pairwise estimates of synonymous (dS) and nonsynonymous (dN) substitution rates were calculated with the Nei–Gojobori method applying the Jukes–Cantor correction and pairwise deletion for gaps [34]. A codon-based Z-test of selection was conducted to assess the null hypothesis of neutral evolution (dN = dS). The ratio ω = dN/dS was used to infer selection pressure, where ω < 1 indicates purifying selection, ω = 1 neutral evolution, and ω > 1 positive selection.
Potential recombination events within CMV genomes were examined using the Recombination Detection Program v5 (RDP5) [35] from sequence alignments generated in MEGA11 [34]. The analysis includes seven independent algorithms (RDP, GENECONV, Bootscan, MaxChi, Chimaera, SisScan and 3Seq) applied with default parameters. To ensure reliability, only recombination events detected by at least four different methods and supported by a Bonferroni-corrected p-value < 0.05 were considered significant. This criterion helped to reduce false positives and confirm true recombination signals. The program identified putative recombination breakpoints and possible parental sequences by comparing the Indian CMV isolate with representative global CMV genomes retrieved from the NCBI database.
Phylogenetic analysis was carried out using MEGA11 [34] to examine evolutionary relationships among CMV isolates. The Neighbor-Joining method with the Maximum Composite Likelihood model was used to calculate evolutionary distances. A total of 162 nucleotide sequences from different geographical regions were analyzed, with RNA1, RNA2, and RNA3 sequences of Tobacco streak virus (TSV) used as the out-group. All codon positions (1st, 2nd, 3rd, and non-coding) were included, and ambiguous sites were removed using pairwise deletion. The reliability of the tree topology was tested with 1000 bootstrap replicates. The trees were drawn to scale and visualized using the iTOL online tool (https://itol.embl.de).
2.9 Prediction of Conserved Motifs and Their Secondary Structures
Conserved motifs within the 5′ and 3′ untranslated regions (UTRs) of RNA 1 (3357 nt), RNA 2 (3050 nt), and RNA 3 (2212 nt) of Indian isolate (CR8-JH) of CMV were identified using MEME Suite v5.5.7 (https://meme-suite.org/meme/tools/meme). Analyses were performed in DNA mode with reverse complement enabled, allowing up to five motifs (6–50 nt) under the ZOOPS model. Motif significance was assessed using site-specific p-values. Secondary structures of the predicted motifs were analyzed with the RNAfold server (ViennaRNA Package; http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). Minimum Free Energy (MFE) structures, ensemble free energy, and centroid conformations were obtained. Graphical outputs, color-coded for base-pairing probabilities and positional entropy, provided insights into motif stability and structural diversity.
3.1 Small RNA Sequencing Output, Read-Length Distribution and Quality
Small RNA sequencing generated a substantial volume of high-quality data from C. roseus samples. Paired-end sequencing with a read length of 161 bp yielded high sequencing depth across the analyzed samples (Table S1). For sample CR1-7, a total of 20,019,073 reads were obtained for each read orientation (R1 and R2), whereas CR1-8 yielded 17,865,087 reads per orientation. The average GC content of raw reads ranged from 49% to 53%, with a consistent average read length of 63 bp across samples, indicating uniform sequencing output.
After data processing, reads corresponding to the small RNA size range (17–30 nt) were retained. CR1-7 produced 2,046,991 reads per orientation within this size range, while CR1-8 yielded 185,725 reads per orientation. The GC content of the processed small RNA reads remained stable at approximately 53%. The read-length distribution showed enrichment of small RNA populations within the expected size range for virus-derived small RNAs, confirming the suitability of the sequencing data for reliable virome identification and downstream genomic analyses.
3.2 Identification of CMV through HTS-Based Bioinformatics Analysis
Sequencing analysis of samples CR1-7 and CR1-8 using the Genome Detective Virus Tool confirmed the presence of CMV. In sample CR1-7 yielded only partial genome assemblies, supported by 151,545 CMV-specific reads with an average coverage depth of 1261×. RNA 1 was represented by three contigs (2896 nt; 54,386 reads; 1124× depth), RNA 2 by two contigs (2279 nt; 52,650 reads; 1355× depth), and RNA 3 by two contigs (1941 nt; 44,509 reads; 1357× depth). In contrast, sample CR1-8, near complete genome assemblies for all three RNAs were obtained with high coverage: RNA 1 (3266 nt) was reconstructed from 167,676 reads at an average depth of 3216×, RNA 2 (3024 nt) from 89,484 reads at 1834× depth, and RNA 3 (2124 nt) from 136,403 reads at 4033× depth, resulting in a total of 403,468 CMV-specific reads. Further analysis using the Galaxy Europe Platform produced complete genome assemblies of all three RNAs from CR1-8 (Fig. S1). The complete CMV genome sequences (RNA 1-3357, RNA 2-3050 and RNA 3-2212) (Table 1) obtained from sample CR1-8 were deposited in GenBank under accession numbers OR826804–OR826806, while partial genome sequences from sample CR1-7 were submitted under accession numbers OR826808–OR826810. Further, a complete CMV genome obtained from CR1-8 was selected for subsequent downstream analyses.
Table 1: Genome organization of CMV isolate CR8-JH, showing 5′/3′ UTRs, ORFs with nucleotide positions, and encoded proteins in RNA 1, RNA 2, and RNA 3.
| 5′UTR | ORFs Name | ORF Location | ORF Product | 3′UTR | |
|---|---|---|---|---|---|
| RNA 1 | 1-95 | ORF1a | 96-3077 | Replicase | 3078-3357 |
| RNA 2 | 1-78 | ORF2a | 79-2658 | RdRp | 2753-3050 |
| ORF2b | 2417-2752 | 2b | |||
| RNA 3 | 1-121 | ORF3a | 122-961 | MP | 1911-2212 |
| ORF3b | 1254-1910 | CP |
3.3 Validation of CMV by RT-PCR and Sanger Sequencing
RT-PCR amplification of the RdRp region produced a ~513 bp fragment from both CR1-7 and CR1-8 (Fig. 2). Sanger sequencing of the amplified products, followed by BLAST analysis, confirmed the presence and identity of CMV in the samples (Fig. S2). The resulting 460 bp sequences were submitted to GenBank under accession numbers OR827714 and OR827715.

Figure 2: Detection of CMV by RT-PCR. Agarose gel electrophoresis showing amplification of a 513 bp fragment of the RdRp gene from CMV in samples CR1-7 and CR1-8 (Lanes 2 and 3, respectively). Lane M: 100 bp DNA ladder (GeneDireX, Taiwan); Lane 1: water as a negative control.
3.4 Sequence Identity Analysis
A pairwise sequence identity done by SDT tool showed that Indian C. roseus isolates (CR8-JH) of CMV (OR826804: RNA 1, OR826805: RNA 2, OR826806: RNA 3) revealed high similarity with both Indian and Iranian isolates. RNA 1 shared 93.0–94.7% identity with isolates from Nicotiana tabacum (Iran), Solanum melongena (India), and Piper nigrum (India), while RNA 2 showed 92.6–93.6% identity with isolates from Nicotiana tabacum (Iran), Cucumis sativus (India), and Capsicum annuum (India) at nucleotide level. RNA 3 exhibited the highest conservation, with 95.0–96.6% identity to isolates from Nicotiana tabacum (Iran), Cucumis sativus (India), and Capsicum annuum (India). These results indicate that the Catharanthus CR8-JH isolate is genetically closest to Iranian tobacco isolates across all three RNAs, while also clustering with diverse Indian host isolates (Fig. 3), suggesting regional circulation and possible reassortment of CMV segments among different host species.

Figure 3: Comparison of pairwise sequence identities between CMV isolates using the sequence demarcation tool SDT version 1.2. The color coding represents the percentage of similarity between the sequences of Indian C. roseus isolates and those found in the database. The Indian isolate is highlighted with a red rectangular box.
Mutation analysis was carried out by comparing the Indian CMV isolate CR8-JH (OR826804-RNA 1, OR826805-RNA 2, OR826806-RNA 3) with the corresponding reference sequences NC_002035 (RNA 1), NC_002034 (RNA 2), and NC_001440 (RNA 3). The analysis revealed region-specific and protein-specific patterns of nucleotide substitutions and indels across the three genomic RNAs.
Nucleotide substitutions and indels were recorded across all three RNAs. RNA 1 showed the highest number of substitutions in the coding region (Fig. 4A), while RNA 2 displayed mutation accumulation in both coding and non-coding regions (Fig. 4B). RNA 3 showed variable mutation distribution, with the intergenic spacer being the most indel-prone region (Fig. 4C). At the coding sequence level, variations included silent, conservative, and non-conservative mutations across all viral proteins (Fig. 4D).

Figure 4: Distribution of nucleotide substitutions (transitions and transversions) and indels in RNA 1 (A), RNA 2 (B), and RNA 3 (C), and mutation types in CDS regions (D), classified as silent or missense (conservative and non-conservative), in the Indian CMV isolate CR8-JH compared with the reference sequence.
3.6 Selective Pressure Analyses (dN/dS Ratios)
Codon-based Z-tests of selection performed in MEGA 11 revealed significantly negative Z-values for all viral proteins, indicating that nonsynonymous substitutions occur at a much lower rate than synonymous ones (Table 2). The replicase (Z = −18.64, p < 0.001) and RdRp (Z = −12.02, p < 0.001) showed the strongest purifying selection, reflecting functional constraints associated with replication. The movement protein (Z = −7.35, p < 0.001) and coat protein (Z = −6.76, p < 0.001) were also under strong purifying selection, consistent with the conserved roles in virus transport and encapsidation. In contrast, the 2b gene displayed a relatively weaker but still significant purifying signal (Z = −2.23, p = 0.027), suggesting partial relaxation or adaptive variation associated with host-virus interaction.
Table 2: Gene-wise selective pressure analysis showing Z-statistics, significance, and biological interpretation.
| Protein/Gene | Z-Statistic | p-Value | Interpretation | Biological Explanation |
|---|---|---|---|---|
| Replicase | −18.64 | 0.0000000001 | Strong purifying selection | Highly conserved replication enzyme; amino acid changes are deleterious and removed by selection. |
| RdRp | −12.02 | 0.0000000001 | Strong purifying selection | Catalytic domain essential for genome replication; intolerant to amino acid substitutions. |
| 2b | −2.23 | 0.0273918044 | Moderate purifying selection (significant) | Some amino acid variation tolerated; possible mild adaptation due to host-virus interaction. |
| MP | −7.35 | 0.0000000001 | Strong purifying selection | Conserved role in viral cell-to-cell movement; functional constraints limit amino acid change. |
| CP | −6.76 | 0.0000000005 | Strong purifying selection | Structural protein; amino acid conservation critical for virion assembly and host recognition. |
Recombination analysis of the Indian CMV isolate CR8-JH using RDP5 detected two significant recombination events supported by multiple algorithms (RDP, GENECONV, Bootscan, MaxChi, Chimaera, SisScan and 3Seq) with highly significant p-values (1.421 × 10−20 to 2.526 × 10−4) (Fig. 5).
The first recombination event was identified in RNA 2 (OR826805), where the South Korean isolate MH594045 was inferred as the major parent and the Chinese isolate OM719713 as the minor parent. The recombinant fragment was located between nucleotide positions ~2725 and ~2990, spanning the C-terminal region of the 2b gene and extending into the 3′ untranslated region (3′UTR) (Fig. S3).
The second event occurred in RNA 3 (OR826806), with the South Korean isolate MH594046 acting as the major parent and the Iranian isolate OR607755 as the minor parent. The recombination region was mapped between ~1907 and ~2181 nt, covering the terminal portion of the coat protein (CP) gene and continuing into the 3′UTR (Fig. S4). Overall, both recombination events were concentrated near the 3′ terminal region of the genome.

Figure 5: Recombination events detected in CMV isolate CR8-JH (India) by various detection methods of RDP5. RNA 2 (OR826805) originated from a South Korean (major) and Chinese (minor) isolate, while RNA 3 (OR826806) originated from a South Korean (major) and Iranian (minor) isolate. These events were strongly supported by highly significant p-values (10−4–10−20) across multiple detection methods. Abbreviations: CR = Catharanthus roseus; AM = Atractylodes macrocephala; NT = Nicotiana tabacum; MS = Musa acuminata.
Phylogenetic analysis based on full-length RNA1, RNA2, and RNA3 sequences separated CMV isolates into three major subgroups: IA, IB, and II. The Indian isolate CR8-JH from C. roseus consistently clustered within subgroup IB along with other CMV isolates reported in GenBank. This grouping was maintained across analyses of each genomic RNA segment as well as the combined dataset. The tree showed well-supported clustering within subgroup IB, as indicated by bootstrap support at the major nodes (Fig. 6).

Figure 6: Rooted circular phylogenetic tree of CMV isolates based on complete nucleotide sequences of RNA1, RNA2, and RNA3 (53 isolate each). The Indian isolate CR8-JH from C. roseus is highlighted in red and clusters within subgroup IB in all three genome segments. Different background colours represent CMV subgroups IA, IB, and II. Tobacco streak virus (TSV) sequences were used as the out-group. Branch lengths indicate nucleotide substitutions per site, and yellow circles at the nodes represent bootstrap support values.
3.9 Prediction of Conserved Motifs in UTRs
Using MEME Suite v5.5.7, five conserved motifs were identified within the 5′ and 3′ untranslated regions (UTRs) of RNA 1 (3357 nt), RNA 2 (3050 nt), and RNA 3 (2212 nt) of the Indian CMV isolate CR8-JH. Four motifs (Motifs 1, 2, 4, and 5) were consistently located in the 3′ UTRs of all three RNA segments, while Motif 3 was detected exclusively in the 5′ UTR of RNA 1 and RNA 2 but was absent in RNA 3, indicating segment-specific variability (Fig. S5). The identified motifs ranged in length from 29–50 nt and displayed highly significant p-values (10−18 to 10−31), supporting their conserved nature across the genomic RNAs (Table 3). Sequence logos revealed that the motifs were enriched with A/U-rich regions interspersed with conserved G/C residues, a typical feature of RNA regulatory elements.
Table 3: Genomic location and p-values of Identified Motifs across RNA 1, RNA 2, and RNA 3 Segments.
| Motif | Location | RNA 1 | RNA 2 | RNA 3 | |||
|---|---|---|---|---|---|---|---|
| Position | p-Value | Position | p-Value | Position | p-Value | ||
| 1 | 3′UTR | 3231-3280 | 6.13 × 10−31 | 2925-2974 | 1.15 × 10−30 | 2085-2134 | 5.07 × 10−31 |
| 2 | 3′UTR | 3151-3200 | 1.54 × 10−30 | 2845-2894 | 1.00 × 10−30 | 2005-2054 | 5.35 × 10−31 |
| 3 | 5′UTR | 3-45 | 1.92 × 10−26 | 2-44 | 8.85 × 10−27 | - | - |
| 4 | 3′UTR | 3281-3327 | 2.81 × 10−29 | 2975-3021 | 8.40 × 10−27 | 2135-2181 | 2.81 × 10−29 |
| 5 | 3′UTR | 3201-3229 | 8.54 × 10−18 | 2895-2930 | 3.98 × 10−18 | 2055-2083 | 3.98 × 10−18 |
3.10 Prediction of Secondary Structures of Conserved Motifs
Secondary structure predictions of these motifs were performed using RNA fold to assess their thermodynamic stability and base-pairing confidence (Table 4). Motif 1 exhibited the most stable conformation with a minimum free energy (MFE) of −16.7 kcal/mol and formed a long hairpin stem-loop with high base-pairing probability. Motif 2 also formed a stable hairpin but included multiple bulges, whereas Motif 3 folded into a shorter stem-loop with an internal bulge. Motif 4 showed a multi-branched junction with moderate stability (MFE −5.3 kcal/mol), while Motif 5, though shorter in length, formed a simple hairpin with high confidence (Fig. S6). Ensemble diversity values suggested that Motifs 1, 2, and 3 had well-defined stable structures, whereas Motif 4 was more structurally variable.
Table 4: Summary of Thermodynamic Parameters, Base-Pairing Confidence, and Structural Characteristics of predicted RNA secondary structures of motifs.
| Motif ID | Sequence Length (nt) | MFE (kcal/mol) | Ensemble Energy (kcal/mol) | MFE Frequency (%) | Ensemble Diversity | Base-Pairing Confidence | Structure Features |
|---|---|---|---|---|---|---|---|
| Motif 1 | 50 | −16.70 | −16.76 | 90.55 | 0.71 | High | Long stem-loop (hairpin) |
| Motif 2 | 50 | −11.00 | −11.58 | 38.74 | 3.88 | High | Hairpin with bulges |
| Motif 3 | 43 | −10.70 | −11.05 | 56.85 | 1.61 | High | Short stem-loop with internal bulge |
| Motif 4 | 47 | −5.30 | −6.26 | 21.19 | 8.28 | Moderate | Multi-branched junction |
| Motif 5 | 29 | −3.40 | −3.76 | 55.54 | 1.30 | High | Simple hairpin |
Comparative sequence and evolutionary analyses of an Indian CMV isolate CR8-JH revealed clear genetic relatedness with subgroup IB isolates and highlighted multiple evolutionary forces shaping its genome. SDT (Fig. 3) and phylogenetic analysis (Fig. 6) confirmed a close relationship with Indian and Iranian isolates, suggesting regional circulation and genetic exchange among CMV populations [5,6,36,37].
The mutation pattern across the three genomic RNAs showed a predominance of transition over transversion substitutions, a typical bias of RNA viruses [38,39]. Most substitutions were concentrated within coding regions, whereas indels were largely confined to the UTRs and intergenic regions, reflecting strong selective constraints on protein-coding sequences and greater structural flexibility in non-coding regions [40,41]. Gene-wise mutation profiling indicated that replication-related proteins (Replicase and RdRp) contained mostly silent substitutions, confirming their functional conservation, while the movement and interaction related proteins (2b, MP, and CP) showed a higher proportion of nonsynonymous changes, suggesting adaptive variation linked to host interaction and transmission (Fig. 4) [42,43].
The selective pressure (dN/dS) analysis further supported these findings. All proteins showed significantly negative Z-values (Replicase = −18.64, RdRp = −12.02, MP = −7.35, CP = −6.76, 2b = −2.23; p < 0.05), confirming that purifying selection predominates across the CMV genome (Table 2). The relatively weaker selection acting on the 2b gene suggests mild adaptive evolution, consistent with its role in host defense suppression and virulence modulation [44,45]. Collectively, these observations indicate that CMV maintains a dual evolutionary pattern, high conservation in replication and structural proteins to preserve vital functions, combined with flexibility in genes mediating host adaptation [46,47].
The recombination events detected in RNA2 and RNA3 of the CMV isolate CR8-JH were located near the 3′ terminal region of the genome, spanning coding sequences and the 3′UTR. These regions are functionally important for replication, translation efficiency, and host interaction [48,49]. Recombination involving the 2b gene may affect RNA silencing suppression and symptom development [50], whereas recombination at the coat protein-3′UTR junction could influence virion assembly and vector transmission [47]. Such events support previous observations that recombination, together with point mutations, plays a major role in CMV genetic variability and adaptive evolution [27,46]. Exchange of genomic fragments in regulatory regions can modify viral interactions and contribute to viral fitness and diversification in RNA viruses [45,51].
Motif analysis of untranslated regions further revealed conserved RNA structural elements characteristic of CMV. Stable stem-loop and tRNA-like motifs predicted in the 3′UTRs of all three RNAs likely act as cis-regulatory elements involved in replication, translation, and encapsidation [8,48,52,53,54]. The 5′UTRs of RNA 1 and RNA 2 contained predicted hairpin structures that may assist ribosome binding, while RNA 3 lacked a distinct 5′UTR motif, consistent with subgroup I isolates [49,55]. These conserved RNA elements probably contribute to maintaining replication efficiency and genome stability. However, as these structures were identified through in-silico prediction, experimental validation is required to confirm their biological significance.
In summary, an Indian CMV isolate CR8-JH shows a stable and well-conserved genome shaped by the combined effects of purifying selection, limited adaptive variation, and occasional recombination. The strong conservation of replication and structural genes ensures efficient viral multiplication and genome integrity, while moderate flexibility in host-interaction proteins such as 2b, MP, and CP allows the virus to adapt to different hosts and environmental conditions. Overall, these findings are based on in-silico analysis which highlight CMV evolution in India reflects a balance between genetic stability and adaptive plasticity, which enables the virus to maintain its persistence, broad host range, and ecological success.
Acknowledgement:
Funding Statement: Deanship of Research and Graduate Studies, University of Tabuk, Kingdom of Saudi Arabia (0176-1442-S); Research, Development, and Innovation Authority (RDIA)–Kingdom of Saudi Arabia (13445-Tabuk-2023-UT-R-3-1-SE).
Author Contributions: Conceptualization: Zahid Khorshid Abbas, Anjana Singh, Md Salik Noorani and Zahid Hameed Siddiqui; Data curation: Anjana Singh, Md Salik Noorani and Zahid Hameed Siddiqui; Formal analysis: Anjana Singh and Md Salik Noorani; Funding acquisition: Zahid Khorshid Abbas, Md Salik Noorani, Sulaiman Ali Alharbi, Yussri M. Mahrous, Naif Abdulrhman Zabin Alnefiei and Zahid Hameed Siddiqui; Investigation: Anjana Singh and Md Salik Noorani; Methodology: Zahid Khorshid Abbas, Anjana Singh, Md Salik Noorani and Zahid Hameed Siddiqui; Project administration: Zahid Khorshid Abbas, Md Salik Noorani and Zahid Hameed Siddiqui; Resources: Zahid Khorshid Abbas and Md Salik Noorani; Software: Md Salik Noorani and Mirza Sarwar Baig; Supervision: Md Salik Noorani and Zahid Hameed Siddiqui; Validation: Zahid Khorshid Abbas, Anjana Singh and Md Salik Noorani; Visualization: Zahid Khorshid Abbas, Md Salik Noorani, Mirza Sarwar Baig, Naif Abdulrhman Zabin Alnefiei and Zahid Hameed Siddiqui; Writing—original draft: Zahid Khorshid Abbas, Anjana Singh and Md Salik Noorani; Writing—review & editing: Md Salik Noorani, Sulaiman Ali Alharbi, Yussri M. Mahrous, M. Nasir Khan, Moawia Mukhtar Hassan and Zahid Hameed Siddiqui. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: Sequence data that supports the findings of this study have been deposited in the NCBI Sequence Read Archive (SRA) under the accession numbers SRR26156703 and SRR26156704. These samples were part of BioSamples SAMN37505367 and SAMN37518972 within the BioProject PRJNA1019943. After analysis, the discovered viral sequences were deposited in the GenBank database with accession numbers OR826804–OR826806 and OR826808–OR826810.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest.
Supplementary Materials: The supplementary material is available online at https://www.techscience.com/doi/10.32604/phyton.2026.076432/s1.
Abbreviations
| BLAST | Basic Local Alignment Search Tool |
| C. roseus | Catharanthus roseus |
| CDS | Coding sequence |
| CMV | Cucumber mosaic virus |
| CP | Coat protein |
| CR8-JH | Indian CMV isolate CR8-JH (isolate name) |
| CTAB | Cetyltrimethylammonium bromide |
| HTS | High-throughput sequencing |
| IGS | Intergenic spacer |
| iTOL | Interactive Tree Of Life |
| MFE | Minimum free energy |
| MEGA | Molecular Evolutionary Genetics Analysis |
| MP | Movement protein |
| NCBI | National Center for Biotechnology Information |
| NJ | Neighbor-Joining |
| ORF/ORFs | Open reading frame(s) |
| RdRp | RNA-dependent RNA polymerase |
| RDP | Recombination Detection Program |
| RT-PCR | Reverse transcription polymerase chain reaction |
| SRA | Sequence Read Archive |
| SDT | Sequence Demarcation Tool |
| siRNA | small interfering RNA(s) |
| sgRNA | subgenomic RNA |
| UTR | Untranslated region |
| VSR | Viral suppressor(s) of RNA silencing |
References
1. Mochizuki T , Ohki ST . Cucumber mosaic virus: Viral genes as virulence determinants. Mol Plant Pathol. 2012; 13( 3): 217– 25. doi:10.1111/j.1364-3703.2011.00749.x. [Google Scholar] [CrossRef]
2. Ganchozo-Mendoza E , Quadros AFF , Zerbini FM , Flores FJ , Garcés-Fiallos FR . Characterization and genome analysis of Cucumber mosaic virus on commercial tobacco plants in Ecuador. Trop Plant Pathol. 2024; 49( 4): 480– 8. doi:10.1007/s40858-024-00643-7. [Google Scholar] [CrossRef]
3. Palukaitis P , García-Arenal F . Cucumoviruses. In: Advances in virus research. Amsterdam, The Netherlands: Elsevier; 2003. p. 241– 323. doi:10.1016/s0065-3527(03)62005-1. [Google Scholar] [CrossRef]
4. Palukaitis P , García-Arenal F . Cucumber mosaic virus. Minneapolis, MN, USA: APS Press; 2019. doi:10.1094/9780890546109. [Google Scholar] [CrossRef]
5. Revathy KA , Bhat AI . Complete genome sequencing of cucumber mosaic virus from black pepper revealed rare deletion in the methyltransferase domain of 1a gene. Virus Dis. 2017; 28( 3): 309– 14. doi:10.1007/s13337-017-0386-4. [Google Scholar] [CrossRef]
6. Kumari R , Bhardwaj P , Singh L , Zaidi AA , Hallan V . Biological and molecular characterization of a novel isolate of cucumber mosaic virus associated with severe mosaic in gerbera (Gerbera jamesonii). Crop Prot. 2013; 45: 71– 6. doi:10.1007/s13337-012-0125-9. [Google Scholar] [CrossRef]
7. Vinaykumar HD , Hiremath S , Nandan M , Muttappagol M , Reddy M , Venkataravanappa V , et al. Genome sequencing of cucumber mosaic virus (CMV) isolates infecting chilli and its interaction with host ferredoxin protein of different host for causing mosaic symptoms. 3 Biotech. 2023; 13( 11): 361. doi:10.1007/s13205-023-03777-8. [Google Scholar] [CrossRef]
8. Roossinck MJ . Cucumber mosaic virus, a model for RNA virus evolution. Mol Plant Pathol. 2001; 2( 2): 59– 63. doi:10.1046/j.1364-3703.2001.00058.x. [Google Scholar] [CrossRef]
9. Nagendran K , Priyanka R , Aravintharaj R , Balaji CG , Prashant S , Basavaraj B , et al. Characterization of Cucumber mosaic virus infecting snake gourd and bottle gourd in India. Physiol Mol Plant Pathol. 2018; 103: 102– 6. doi:10.1016/j.pmpp.2018.05.010. [Google Scholar] [CrossRef]
10. Elbeshehy EKF , Al-Zahrani HSM , Aldhebiani AY , Elbeaino T . Viruses infecting periwinkle (Catharanthus roseus L.) in Western Saudi Arabia. Phytopathol Mediterr. 2017; 56( 3): 479– 85. [Google Scholar]
11. Choi SK , Cho IS , Choi GS , Yoon JY . First Report of Cucumber mosaic virus in Catharanthus roseus in Korea. Plant Dis. 2014; 98( 9): 1283. doi:10.1094/pdis-03-14-0230-pdn. [Google Scholar] [CrossRef]
12. Mazidah M , Yusoff K , Habibuddin H , Tan YH , Lau WH . Characterization of cucumber mosaic virus (CMV) causing mosaic symptom on Catharanthus roseus (L.) G. Don in Malaysia. Pertanika J Trop Agric Sci. 2012; 35( 1): 41– 53. [Google Scholar]
13. Samad A , Ajayakumar PV , Gupta MK , Shukla AK , Darokar MP , Somkuwar B , et al. Natural infection of periwinkle (Catharanthus roseus) withCucumber mosaic virus, subgroup IB. Australas Plant Dis Notes. 2008; 3( 1): 30– 4. doi:10.1007/BF03211229. [Google Scholar] [CrossRef]
14. Dabiri S , Moradi Z , Mehrvar M , Zakiaghl M . Analysis of the complete genome sequence of cucumber mosaic virus from Vinca minor and Wisteria sinensis in Iran. J Plant Pathol. 2020; 102( 4): 1263– 8. doi:10.1007/s42161-020-00650-y. [Google Scholar] [CrossRef]
15. Abbas ZK , Singh A , Noorani MS , Baig MS , Ali Alharbi S , Mahrous YMY , et al. Molecular characterization and genetic diversity of broad bean wilt virus 2 (BBWV-2) from Catharanthus roseus. ACS Omega. 2025; 10( 29): 32433– 43. doi:10.1021/acsomega.5c04600. [Google Scholar] [CrossRef]
16. Siddiqui ZH , Mujib A . Accumulation of vincristine in calcium chloride elicitated Catharanthus roseus cultures. Nat Prod J. 2012; 2( 4): 307– 15. doi:10.2174/2210315511202040307. [Google Scholar] [CrossRef]
17. Sahi N , Mostajeran A , Ghanadian M . Changing in the production of anticancer drugs (vinblastine and vincristine) in Catharanthus roseus (L.) G. Don by potassium and ascorbic acid treatments. Plant Soil Environ. 2022; 68( 1): 18– 28. doi:10.17221/121/2021-pse. [Google Scholar] [CrossRef]
18. Siddiqui ZH , Mujib A , Abbas ZK , Noorani MS , Khan S . Vinblastine synthesis under the influence of CaCl2 elicitation in embryogenic cell suspension culture of Catharanthus roseus. S Afr N J Bot. 2023; 154: 319– 29. doi:10.1016/j.sajb.2023.01.046. [Google Scholar] [CrossRef]
19. Aslam J , Khan SH , Siddiqui ZH , Fatima Z , Maqsood M , Bhat MA , et al. Catharanthus roseus (L.) G. Don. An important drug: Its applications and production. Pharm Glob (Int J Compr Pharm). 2010; 4( 12): 1– 16. [Google Scholar]
20. Shi X , Gao Y , Yan S , Tang X , Zhou X , Zhang D , et al. Aphid performance changes with plant defense mediated by Cucumber mosaic virus titer. Virol J. 2016; 13( 1): 70. doi:10.1186/s12985-016-0524-4. [Google Scholar] [CrossRef]
21. Webster CG , Wylie JS , Jones MGK . Diagnosis of plant viral pathogens. Curr Sci. 2004; 86: 1604– 7. [Google Scholar]
22. Makkouk KM , Kumari SG . Molecular diagnosis of plant viruses. Arab J Plant Prot. 2006; 24: 135– 8. [Google Scholar]
23. López-Martín M , Sifres A , Gómez-Guillamón ML , Picó B , Pérez-de-Castro A . Incidence and genetic diversity of cucurbit viruses in the Spanish Mediterranean area. Plant Pathol. 2024; 73( 2): 431– 43. doi:10.1111/ppa.13825. [Google Scholar] [CrossRef]
24. Roossinck MJ , Martin DP , Roumagnac P . Plant virus metagenomics: Advances in virus discovery. Phytopathology. 2015; 105( 6): 716– 27. doi:10.1094/phyto-12-14-0356-rvw. [Google Scholar] [CrossRef]
25. Massart S , Candresse T , Gil J , Lacomme C , Predajna L , Ravnikar M , et al. A framework for the evaluation of biosecurity, commercial, regulatory, and scientific impacts of plant viruses and viroids identified by NGS technologies. Front Microbiol. 2017; 8: 45. doi:10.3389/fmicb.2017.00045. [Google Scholar] [CrossRef]
26. Mrkvová M , Hančinský R , Predajňa L , Alaxin P , Achs A , Tomašechová J , et al. High-throughput sequencing discloses the cucumber mosaic virus (CMV) diversity in Slovakia and reveals new hosts of CMV from the Papaveraceae family. Plants. 2022; 11( 13): 1665. doi:10.3390/plants11131665. [Google Scholar] [CrossRef]
27. Singhal P , Prajapati MR , Diksha D , Baranwal VK , Singh J . Genomic characterization of recombinant cucumber mosaic virus isolates infecting mustard species via HTS in India. J Phytopathol. 2023; 171( 7–8): 333– 43. doi:10.1111/jph.13186. [Google Scholar] [CrossRef]
28. Zhan J , Meyers BC . Plant small RNAs: Their biogenesis, regulatory roles, and functions. Annu Rev Plant Biol. 2023; 74: 21– 51. doi:10.1146/annurev-arplant-070122-035226. [Google Scholar] [CrossRef]
29. Laura E-G , Blevins T , Heinlein M . Small RNA mobility and plant virus diseases. J Exp Bot. 2025; 77( 3): 732– 45. doi:10.1093/jxb/eraf226. [Google Scholar] [CrossRef]
30. Noorani MS , Awasthi P , Sharma MP , Ram R , Zaidi AA , Hallan V . Simultaneous detection and identification of four cherry viruses by two step multiplex RT-PCR with an internal control of plant nad5 mRNA. J Virol Meth. 2013; 193( 1): 103– 7. doi:10.1016/j.jviromet.2013.05.006. [Google Scholar] [CrossRef]
31. Grieco F , Alkowni R , Saponari M , Savino V , Martelli GP . Molecular detection of olive viruses. EPPO Bull. 2000; 30( 3–4): 469– 73. doi:10.1111/j.1365-2338.2000.tb00931.x. [Google Scholar] [CrossRef]
32. Muhire BM , Varsani A , Martin DP . SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS One. 2014; 9( 9): e108277. doi:10.1371/journal.pone.0108277. [Google Scholar] [CrossRef]
33. Tamura K , Stecher G , Kumar S . MEGA11: Molecular evolutionary genetics analysis version 11. Mol Biol Evol. 2021; 38( 7): 3022– 7. doi:10.1093/molbev/msab120. [Google Scholar] [CrossRef]
34. Nei M , Gojobori T . Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986; 3: 418– 26. [Google Scholar]
35. Martin DP , Varsani A , Roumagnac P , Botha G , Maslamoney S , Schwab T , et al. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021; 7: veaa087. doi:10.1093/ve/veaa087. [Google Scholar] [CrossRef]
36. Madhubala R , Bhadramurthy V , Bhat AI , Hareesh PS , Retheesh ST , Bhai RS . Occurrence of cucumber mosaic virus on vanilla (Vanilla planifolia Andrews) in India. J Biosci. 2005; 30( 3): 339– 50. doi:10.1007/BF02703671. [Google Scholar] [CrossRef]
37. Verma N , Singh AK , Singh L , Kulshreshtha S , Raikhy G , Hallan V , et al. Occurrence of cucumber mosaic virus in Gerbera jamesonii in India. Plant Dis. 2004; 88( 10): 1161. doi:10.1094/PDIS.2004.88.10.1161C. [Google Scholar] [CrossRef]
38. Duffy S , Shackelton LA , Holmes EC . Rates of evolutionary change in viruses: Patterns and determinants. Nat Rev Genet. 2008; 9( 4): 267– 76. doi:10.1038/nrg2323. [Google Scholar] [CrossRef]
39. Sanjuán R , Nebot MR , Chirico N , Mansky LM , Belshaw R . Viral mutation rates. J Virol. 2010; 84( 19): 9733– 48. doi:10.1128/jvi.00694-10. [Google Scholar] [CrossRef]
40. Kao CC , Sivakumaran K . Brome mosaic virus, good for an RNA virologist’s basic needs. Mol Plant Pathol. 2000; 1( 2): 91– 7. doi:10.1046/j.1364-3703.2000.00017.x. [Google Scholar] [CrossRef]
41. Domingo E , Perales C . Viral quasispecies. PLoS Genet. 2019; 15( 10): e1008271. doi:10.1371/journal.pgen.1008271. [Google Scholar] [CrossRef]
42. Shi BJ , Palukaitis P , Symons RH . Differential virulence by strains of Cucumber mosaic virus is mediated by the 2b gene. Mol Plant Microbe Interact. 2002; 15( 9): 947– 55. doi:10.1094/mpmi.2002.15.9.947. [Google Scholar] [CrossRef]
43. Melcher U . The ‘30K’ superfamily of viral movement proteins. Microbiology. 2000; 81( 1): 257– 66. doi:10.1099/0022-1317-81-1-257. [Google Scholar] [CrossRef]
44. Jacquemond M . Cucumber mosaic virus. In: Viruses and virus diseases of vegetables in the mediterranean basin. Amsterdam, The Netherlands: Elsevier; 2012. p. 439– 504. doi:10.1016/b978-0-12-394314-9.00013-0. [Google Scholar] [CrossRef]
45. Simon-Loriere E , Holmes EC . Why do RNA viruses recombine? Nat Rev Microbiol. 2011; 9( 8): 617– 26. doi:10.1038/nrmicro2614. [Google Scholar] [CrossRef]
46. Apalowo OA , Adediji AO , Balogun OS , Fakolujo TI , Archibong JM , Izuogu NB , et al. Genetic structure of cucumber mosaic virus from natural hosts in Nigeria reveals high diversity and occurrence of putative novel recombinant strains. Front Microbiol. 2022; 13: 753054. doi:10.3389/fmicb.2022.753054. [Google Scholar] [CrossRef]
47. Moury B . Differential selection of genes of cucumber mosaic virus subgroups. Mol Biol Evol. 2004; 21( 8): 1602– 11. doi:10.1093/molbev/msh164. [Google Scholar] [CrossRef]
48. Rasekhian M , Roohvand F , Habtemariam S , Marzbany M , Kazemimanesh M . The role of 3′UTR of RNA viruses on mRNA stability and translation enhancement. Mini Rev Med Chem. 2021; 21( 16): 2389– 98. doi:10.2174/1389557521666210217092305. [Google Scholar] [CrossRef]
49. Wang Y , Liu C , Liu S , Wang Z , Hao K , Wu Y , et al. Replicase components and the untranslated region of RNA2 synergistically regulate pathogenicity differentiation among different isolates of cucumber mosaic virus. Int J Biol Macromol. 2025; 294: 139076. doi:10.1016/j.ijbiomac.2024.139076. [Google Scholar] [CrossRef]
50. Takahashi H , Tabara M , Miyashita S , Ando S , Kawano S , Kanayama Y , et al. Cucumber mosaic virus infection in Arabidopsis: A conditional mutualistic symbiont? Front Microbiol. 2022; 12: 770925. doi:10.3389/fmicb.2021.770925. [Google Scholar] [CrossRef]
51. Chare ER , Holmes EC . A phylogenetic survey of recombination frequency in plant RNA viruses. Arch Virol. 2006; 151( 5): 933– 46. doi:10.1007/s00705-005-0675-x. [Google Scholar] [CrossRef]
52. Liu S , Mu J , Yu C , Geng G , Su C , Yuan X . Multiple levels of triggered factors and the obligated requirement of cell-to-cell movement in the mutation repair of cucumber mosaic virus with defects in the tRNA-like structure. Biology. 2022; 11( 7): 1051. doi:10.3390/biology11071051. [Google Scholar] [CrossRef]
53. Leppek K , Das R , Barna M . Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol. 2018; 19( 3): 158– 74. doi:10.1038/nrm.2017.103. [Google Scholar] [CrossRef]
54. Farzadfar S , Al-Waeli M , Pourrrahim R . Biological and molecular characterization of recombinant cucumber mosaic virus (Cucumovirus CMV) isolates from rapeseed in Southern-Eurasia Iraq. J Plant Pathol. 2025; 107( 2): 1245– 53. doi:10.1007/s42161-025-01870-w. [Google Scholar] [CrossRef]
55. Watters KE , Choudhary K , Aviran S , Lucks JB , Perry KL , Thompson JR . Probing of RNA structures in a positive sense RNA virus reveals selection pressures for structural elements. Nucleic Acids Res. 2018; 46( 5): 2573– 84. doi:10.1093/nar/gkx1273. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools