
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Igniting the Tumor: Targeting Mitochondrial Stress to Prime Breast Cancer for Immunotherapy
1
Department of Post-Baccalaureate Medicine, College of Medicine, National Chung Hsing University, Taichung 402, Taiwan
2
Research Assistant Center, Show Chwan Memorial Hospital, Changhua 500, Taiwan
3
Department of Nursing, Jenteh Junior College of Medicine, Nursing and Management, Miaoli 356, Taiwan
4
Department of Pathology, Show Chwan Memorial Hospital, Changhua 500, Taiwan
5
National Institute of Cancer Research, National Health Research Institutes, Tainan 704, Taiwan
†
These authors contributed equally to this work
* Corresponding Author: Pei-Yi Chu,
European Cytokine Network 2025, 36(3), 24-37. https://doi.org/10.1684/ecn.2025.0504
Accepted 04 January 2026; Issue published 28 February 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Immunotherapy has demonstrated limited efficacy in immunologically “cold” breast cancers characterized by absent T-cell infiltration and inadequate interferon signaling. The purpose of this work is to propose and articulate a mechanistic and therapeutic framework in which mitochondrial stress is deliberately harnessed to convert immunologically “cold” breast tumors into “hot,” T cell–inflamed, immunotherapy-responsive lesions. This review synthesizes emerging evidence positioning mitochondrial stress as a strategic lever to transform these immune-excluded tumors into inflamed, therapy-responsive lesions. We examine how mitochondrial dysfunction triggers cytosolic release of mitochondrial DNA (mtDNA), a potent damage-associated molecular pattern that activates the cGAS-STING pathway, initiating type I interferon responses and secretion of T-cell-recruiting chemokines such as CCL5 and CXCL10. This axis functions as a “double-edged sword”—while acute activation converts “cold” tumors into “hot” immune-responsive states, chronic engagement drives immunosuppressive cytokine networks and therapeutic resistance, with outcomes varying across breast cancer subtypes. We explore six combination therapeutic strategies: mitochondrial poisons, radiotherapy/chemotherapy, PARP/ATR inhibitors, metabolic reprogramming agents, mitochondrial quality control modulators, and localized mitochondrial stress induction, each paired with immune checkpoint blockade. The review emphasizes “controlled ignition” as a paradigm whereby precisely dosed mitochondrial stress amplifies tumor antigenicity and favorable cytokine landscapes while avoiding chronic immunosuppression. Cytokine networks emerge as both integrators and therapeutic targets of mitochondrial-immune crosstalk. Future advances require mapping subtype-specific thresholds, developing tumor-restricted delivery systems, and implementing biomarker-guided trials to safely harness mitochondrial stress, potentially redefining these organelles as programmable immunological adjuvants in breast cancer therapy.Keywords
Breast cancer remains a leading cause of mortality worldwide [1], with triple-negative breast cancer (TNBC) being particularly aggressive due to its lack of hormone receptors [2]. While immunotherapy has revolutionized treatment for some, baseline response rates in breast cancer remain low because many tumors are immunologically “cold”, characterized by T-cell exclusion and mitochondrial dysfunction [3, 4]. Priming strategies are essential to overcome these resistance mechanisms and improve survival outcomes across all molecular subtypes.
Immunologically “cold” breast tumors, particularly hormone receptor–positive and many HER2-enriched and triple-negative breast cancers (TNBC), show limited benefit from current immune checkpoint inhibitors because they lack robust T-cell infiltration, interferon signaling, and effective antigen presentation [5, 6]. This therapeutic ceiling has prompted a shift toward understanding and therapeutically exploiting tumor-intrinsic and microenvironmental mechanisms that govern immune exclusion, among which mitochondrial signaling has emerged as a central and druggable node [7].
Across current trials, only a minority of breast cancer patients—most notably a subset of PD L1–positive TNBC—achieve durable responses to checkpoint blockade [8–12], with luminal and many HER2 positive tumors remaining largely non responsive [13–15]. TNBC typically displays higher mutational burden, greater chromosomal instability [2, 16, 17], and more frequent baseline activation of the cGAS–STING axis, leading to partially inflamed, ‘hot leaning’ immune phenotypes [18, 19], whereas hormone receptor–positive luminal tumors are often immune excluded with low TILs and cytokine milieus dominated by TGF-β and IL-10 [6, 13, 20, 21]. These subtype specific immune and cytokine landscapes imply that strategies harnessing mitochondrial stress to prime immunotherapy must be tailored—using relatively modest perturbation in TNBC to avoid chronic immunosuppression, and more intensive or repeated priming regimens, often combined with cytokine/myeloid targeted agents, in luminal and HER2 enriched disease.
Traditionally viewed as bioenergetic engines, mitochondria in breast cancer cells and infiltrating immune cells are now recognized as hubs integrating metabolism, redox state, cell death pathways, and innate immune sensing to shape the tumor immune microenvironment [22–25]. Recent work shows that mitochondrial dysfunction, altered dynamics, and metabolic rewiring in breast tumors influence antigen presentation, oxidative stress, and susceptibility to immunogenic cell death [7], while mitochondrial fitness in T cells, NK cells, and myeloid cells critically determines their effector function within the hostile breast tumor niche [26].
A key conceptual advance is that mitochondrial stress can convert organelles into platforms for innate immune activation through release of mitochondrial DNA (mtDNA) and other damage-associated molecular patterns (DAMPs) into the cytosol [27–29]. These mitochondrial signals engage pathways such as cGAS–STING to induce type I interferons and chemokines that orchestrate dendritic cell activation and T-cell recruitment, suggesting that controlled mitochondrial perturbation in breast cancer may help drive “cold-to-hot” transition required for effective immunotherapy.
Within this framework, mitochondria move from passive metabolic supporters to master regulators of breast tumor immunity whose stress responses can be pharmacologically tuned. The following sections will dissect how mtDNA–cGAS–STING signaling, its downstream cytokine and chemokine networks, and the context-dependent consequences of mitochondrial stress can be leveraged—alone and in combination with checkpoint blockade—to “ignite” non-immunogenic breast cancers and improve the depth and durability of immunotherapy responses (figure 1).
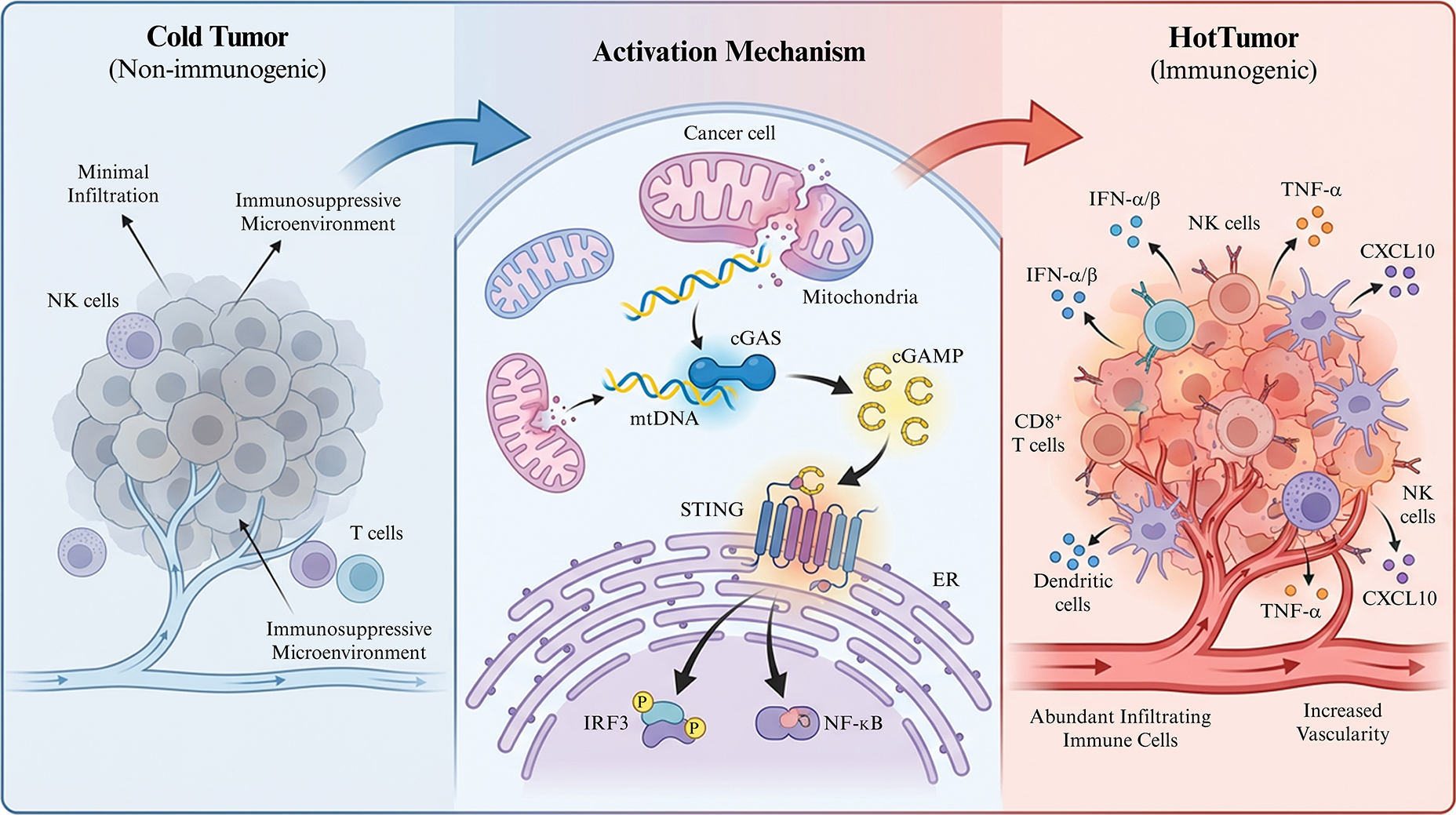
Figure 1.
The mtDNA-cGAS-STING pathway bridges cold and hot tumor phenotypes. Left: immune excluded cold tumor with poor T cell infiltration and immunosuppressive microenvironment. Center: mitochondrial damage, mtDNA release, cGAS activation and cGAMP mediated STING signaling. Right: type I interferon and chemokine induction (e.g., CXCL10, CCL5) promoting CD8 T cells, NK cells and dendritic cell recruitment and hot tumor phenotype. mtDNA: mitochondrial DNA. cGAS: cyclic GMP–AMP synthase. cGAMP: 2’3’-cyclic GMP–AMP. STING: stimulator of interferon genes. IFN: interferon (typically type I interferons, IFN-α/β, in this context). CCL5: C–C motif chemokine ligand 5. CXCL10: C–X–C motif chemokine ligand 10. CD8 T cells: CD8-positive T lymphocytes. NK cells: natural killer cells.
MITOCHONDRIA AS MASTER REGULATORS OF THE TUMOR IMMUNE MICROENVIRONMENT
Mitochondria shape the tumor immune microenvironment (TIME) through their control of cancer cell metabolism, redox balance, organelle quality, and innate immune signaling, thereby influencing how visible tumor cells are to the immune system and how effective antitumor effector cells can be [26]. In breast cancer, dysregulated mitochondrial dynamics, biogenesis, and oxidative phosphorylation (OXPHOS) not only fuel proliferation and metastasis but also remodel antigen presentation, cytokine production, and susceptibility to T-cell–mediated killing, while mitochondrial fitness in tumor-infiltrating lymphocytes and macrophages critically determines whether the TIME is “hot” or “cold” [26].
Mitochondrial dynamics and cancer cell immunogenicity
In breast cancer, metastatic and stem-like populations typically display enhanced mitochondrial fission and fragmented networks driven by factors such as DRP1 and related fission mediators, whereas more fused mitochondrial architectures are associated with lower metastatic potential and reduced aggressiveness [30]. Imbalanced dynamics have dual effects on immunogenicity: excessive fission promotes mitochondrial reactive oxygen species (mtROS), mtDNA damage, and neoantigen generation, yet also downregulates MHC-I antigen presentation and favors secretion of immunosuppressive mediators, enabling immune escape despite increased mutational load [31].
Biogenesis, mitophagy, and danger signaling
Coordinated mitochondrial biogenesis and mitophagy maintain organelle quality, whereas defective turnover permits accumulation of damaged mitochondria that leak mtROS and mtDNA, acting as DAMPs [32]. These signals can engage pathways such as cGAS–STING and NF-κB to induce type I interferons and inflammatory cytokines that, in principle, enhance dendritic cell activation and T-cell priming, but chronic, unrestrained stress skews toward immunosuppressive transcriptional programs and checkpoint upregulation, shaping a tolerant TIME [33].
OXPHOS, ROS, and antigen presentation
Breast cancer cells flexibly toggle between glycolysis and OXPHOS; high OXPHOS states provide ATP and anabolic intermediates but also generate mtROS that influence both cell death and immune visibility [34]. Moderate mitochondrial stress and ROS can promote immunogenic cell death with exposure of calreticulin, ATP release, and cGAS–STING-dependent interferon signaling, whereas excessive ROS and fragmented mitochondria impair MHC-I expression, reduce antigen presentation, and enhance secretion of factors such as IL-10 that dampen cytotoxic T-cell function [26].
Mitochondrial control of T cells in the TIME
Tumor-infiltrating CD8+ T cells in solid tumors frequently exhibit mitochondrial defects, including reduced mitochondrial mass, impaired OXPHOS, and dysfunctional dynamics, which collectively drive exhaustion and limit effector cytokine production [35, 36]. Interventions that restore mitochondrial fitness—such as improving mitochondrial content and respiratory capacity through metabolic conditioning or exercise—enhance T-cell persistence, granzyme production, and tumor control, highlighting mitochondria as central regulators of T-cell functionality in “hot” versus “cold” environments [37].
Macrophages, ROS, and polarization
Tumor-associated macrophages (TAMs) integrate environmental cues via mitochondrial metabolism, with OXPHOS- and fatty acid oxidation–driven programs favoring M2-like, immunosuppressive phenotypes and more glycolytic, ROS-producing states supporting pro-inflammatory, M1-like functions [36]. Mitochondrial reprogramming in TAMs, influenced by tumor-derived metabolites and acidity, can thus either sustain immune evasion or, when redirected, promote antigen presentation, cytokine production, and cross-priming of CD8+ T cells within the breast TIME [36, 38].
Bidirectional mitochondrial crosstalk in the tumor microenvironment
Beyond cell-autonomous effects, tumor and immune cell mitochondria engage in bidirectional metabolic crosstalk. Tumor-derived lactate, kynurenine, and adenosine suppress T-cell OXPHOS and promote exhaustion [39, 40], while robust mitochondrial fitness in infiltrating T cells and NK cells sustains granzyme and IFN-γ production that, in turn, induces tumor mitochondrial stress, mtDNA release, and cGAS–STING activation [41, 42]—creating a reinforcing loop.
Integrating cancer and immune cell mitochondria
Collectively, mitochondrial dynamics, biogenesis, and OXPHOS in breast cancer cells dictate the balance between immunogenic stress signals and immune evasion, while mitochondrial health in T cells and macrophages governs effector capacity and polarization. This multi-compartment mitochondrial network positions mitochondria as master regulators of the TIME and provides a mechanistic basis for strategies that deliberately impose “controlled” mitochondrial stress to enhance antigen presentation, type I interferon and chemokine production, and ultimately the efficacy of breast cancer immunotherapy.
THE MTDNA–CGAS–STING AXIS: A PIVOTAL INNATE IMMUNE SENSING PATHWAY IN CANCER
The mitochondrial DNA (mtDNA)–cGAS–STING axis has emerged as a central innate immune sensing pathway that links organelle stress to inflammatory and antitumor signaling in cancer [43, 44]. In breast tumors and other solid malignancies, diverse mitochondrial insults can drive leakage of mtDNA into the cytosol, where it is recognized as a foreign-like nucleic acid, thereby triggering cGAS–STING–dependent type I interferon and chemokine responses that shape the tumor immune microenvironment [44]. Figure 2 illustrates mechanism of cytosolic DNA sensing via cGAS-STING triggers a coordinated innate immune response.
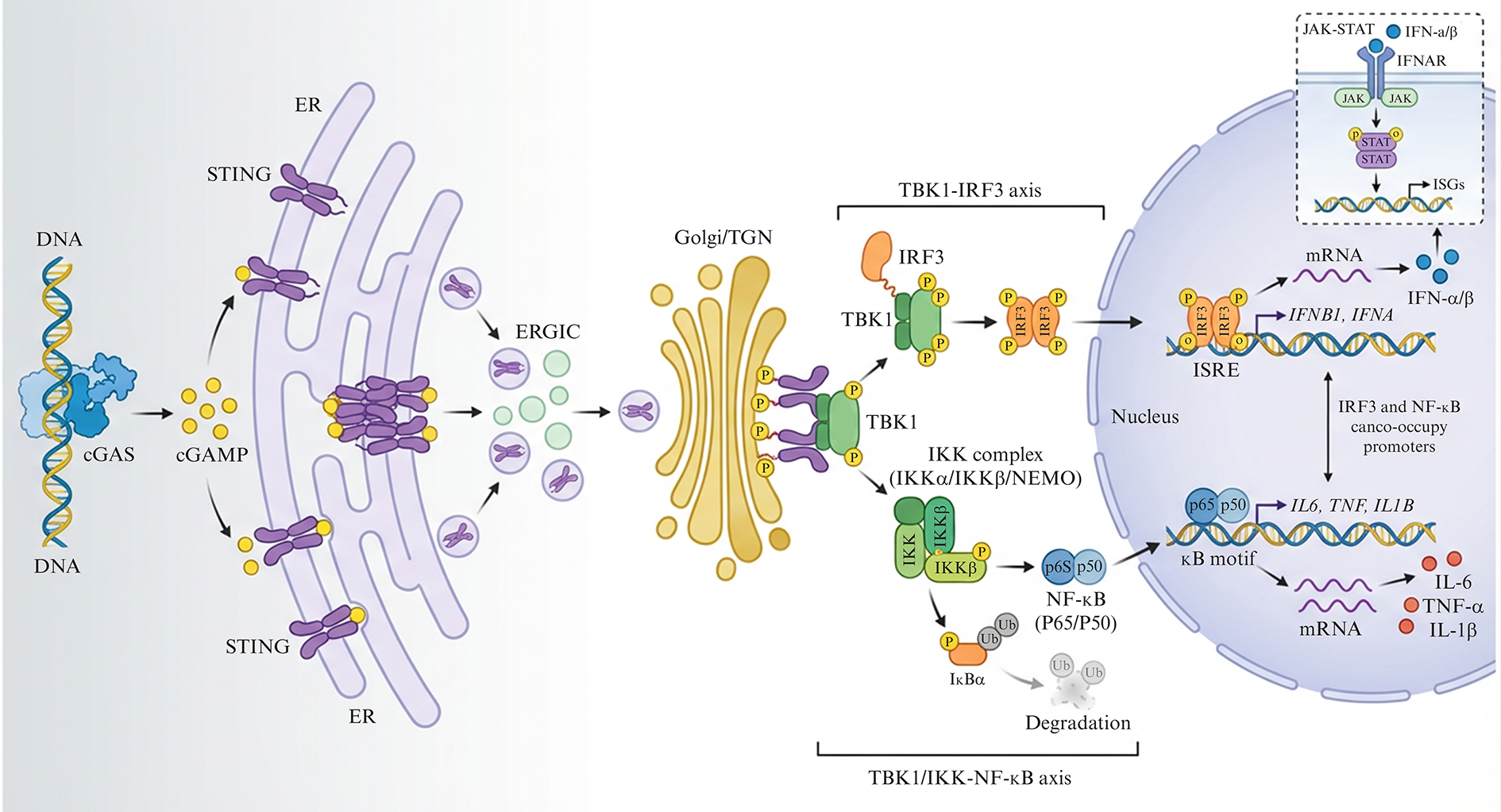
Figure 2.
Downstream signaling of cGAS–STING. Cytosolic DNA activates cGAS to produce cGAMP, which binds ER‑resident STING and drives its trafficking to Golgi compartments. STING then engages TBK1–IRF3 to induce type I interferons and IKK–NF‑κB to induce pro‑inflammatory cytokines such as IL‑6 and TNF‑α. cGAS: cyclic GMP–AMP synthase. cGAMP: 2’3’-cyclic GMP–AMP. STING: stimulator of interferon genes. ER: endoplasmic reticulum. TBK1: TANK-binding kinase 1. IRF3: interferon regulatory factor 3. IKK: IκB kinase. NF-κB: nuclear factor-κB. IL-6: interleukin-6. TNF-α: tumor necrosis factor alpha. IFN: interferon (type I interferons, IFN-α/β).
Mitochondrial stress and mtDNA destabilization
Oncogenic signaling, hypoxia, metabolic overload, and therapy-induced damage all impose chronic stress on tumor cell mitochondria, leading to elevated ROS, impaired replication, and accumulation of oxidatively damaged mtDNA [32]. When mitophagy and mitochondrial quality control are compromised, these damaged genomes persist, increasing the likelihood that nucleoids will be mispackaged, clustered, or exposed at sites of membrane instability that predispose them to escape the organelle [45].
Routes of mtDNA release into the cytosol
Multiple, partially overlapping mechanisms mediate mtDNA efflux from stressed mitochondria. BAX/BAK macropores formed during mitochondrial outer membrane permeabilization can allow inner membrane “herniations” that carry nucleoids through the outer membrane, releasing mtDNA into the cytosol even under sublethal, minority mitochondrial outer membrane permeabilization (MOMP) conditions [45]. In parallel, opening of the mitochondrial permeability transition pore and voltage‑dependent anion channel (VDAC)-dependent permeabilization increase inner membrane leakage, while impaired mitophagy and mitochondrial-derived vesicles can misdirect mtDNA-containing material to the cytosol rather than to lysosomal degradation, sustaining low-level DNA leakage without overt cell death [45, 46].
Cytosolic mtDNA as a DAMP sensed by cGAS
Once in the cytoplasm, mtDNA behaves as a potent damage-associated molecular pattern because of its bacterial ancestry, circular form, and relative CpG enrichment, features that distinguish it from well-packaged nuclear chromatin. The cytosolic DNA sensor cyclic GMP–AMP synthase (cGAS) binds double-stranded mtDNA in a largely sequence-independent manner, and DNA binding promotes cGAS dimerization and higher-order oligomerization, which in turn catalyzes synthesis of the cyclic dinucleotide 2′3′-cGAMP from ATP and GTP [45]. This enzymatic step converts the presence of mtDNA into a diffusible second messenger that can act cell-autonomously or spread to neighboring cells and immune populations via transport mechanisms such as gap junctions or extracellular vesicles [45, 46].
STING engagement and downstream signaling
STING, an adaptor protein residing on the endoplasmic reticulum, binds cGAMP and undergoes conformational changes that drive its oligomerization and trafficking from the ER to perinuclear compartments, including the ER–Golgi intermediate compartment and Golgi [47]. In these locations, STING recruits and activates TBK1 and IKK kinases, leading to phosphorylation and nuclear translocation of IRF3, together with NF-κB activation, thereby inducing a transcriptional program dominated by type I interferons, interferon-stimulated genes, and inflammatory chemokines such as CXCL10 and CCL5 that are critical for dendritic cell activation and effector T-cell recruitment [27, 43, 48].
In cancer biology, the mtDNA–cGAS–STING pathway functions as a molecular bridge between mitochondrial integrity, cell death pathways, and adaptive immune priming. Transient, therapy- or stress-induced mtDNA release can promote immunogenic cell death, enhance antigen presentation, and convert poorly infiltrated “cold” tumors into inflamed lesions more amenable to checkpoint blockade, whereas chronic or deregulated activation may drive tolerogenic or immunosuppressive feedback, contributing to immune evasion [43]. Understanding how distinct forms and magnitudes of mitochondrial stress control mtDNA leakage and cGAS–STING activation is therefore pivotal for rationally designing interventions that harness this axis to ignite productive antitumor immunity in breast cancer.
A DOUBLE-EDGED SWORD: CONTEXT-DEPENDENT OUTCOMES OF STING ACTIVATION
Downstream of the cGAS-STING axis (detailed in Figure 2), the immunological outcome is dictated by the temporal dynamics of signaling. Transient activation successfully ‘ignites’ the tumor by triggering an acute type I interferon wave that is crucial for dendritic cell cross-priming. In breast cancer, this context dependence is particularly evident across molecular subtypes, where STING can either ignite productive antitumor immunity or, when chronically engaged, foster immune evasion, stromal remodeling, and therapeutic resistance [49].
Igniting antitumor immunity: from “cold” to “hot”
Acute or well-timed STING activation in the tumor microenvironment triggers a robust type I interferon program that enhances dendritic cell maturation, cross-priming of CD8+ T cells, and natural killer (NK) cell activation [50–52]. Downstream of IRF3 and NF-κB, STING stimulation induces chemokines such as CXCL9, CXCL10, and CCL5, which drive recruitment and retention of effector T cells and NK cells, facilitating conversion of immune-desert or immune-excluded lesions into inflamed, T cell–infiltrated tumors [53–57]. Preclinical models show that intratumoral or systemic STING agonists can induce IFN-I–dependent tumor regression, promote trafficking of antigen-bearing myeloid cells to draining lymph nodes, and synergize with checkpoint blockade to deepen and prolong responses [58–60]. In breast cancer, transcriptomic and immunologic analyses indicate that tumors with intact cGAS–STING signaling and high STING-driven chemokine signatures are more likely to display “hot” immune phenotypes and enhanced sensitivity to immunotherapy, particularly in subsets of triple-negative disease [61–63].
Fueling resistance and immunosuppression
In contrast, chronic or dysregulated STING activation can skew cytokine output toward protumor inflammation, immunosuppression, and tissue remodeling [64]. Sustained NF-κB and inflammasome engagement downstream of STING promotes production of IL-6, TNF-α, and TGF-β, which support myeloid-derived suppressor cell and regulatory T-cell expansion, drive fibrosis and aberrant angiogenesis, and ultimately dampen effective cytotoxic T-cell function [49, 65, 66].
Breast cancer studies highlight that STING pathway status and output differ across luminal, HER2+, and TNBC subtypes, with prolonged or maladaptive activation linked to epithelial–mesenchymal transition, resistance to HER2-targeted therapy, and upregulation of checkpoints such as PD-L1 [67, 68]. Moreover, tumor-intrinsic mechanisms (for example, MYC-driven repression or selective loss of cGAS/STING components) can either silence beneficial signaling or bias it toward tolerogenic cytokine profiles, underscoring that therapeutic strategies must carefully calibrate the intensity and duration of STING engagement to avoid tipping from immune activation into immune escape [29, 43, 69].
Despite the therapeutic potential of the STING pathway, its systemic activation poses significant safety risks, including cytokine release syndrome and T-cell apoptosis driven by excessive type I interferons. Clinical data from first-generation STING agonists revealed that unconstrained signaling can lead to dose-limiting systemic inflammation and autoimmunity [70]. To mitigate these ‘dark side’ effects, current strategies employ tumor-restricted delivery systems, such as mitochondria-targeted nanocarriers or pH-responsive polymers, which localize the ‘ignition’ signal to the tumor microenvironment. This spatial control is critical to uncouple the beneficial anti-tumor immunity from detrimental systemic toxicity, ensuring that mitochondrial stress serves as a precise adjuvant rather than a systemic toxin [70, 71].
Although preclinical models clearly distinguish acute, beneficial mitochondrial stress from chronic, suppressive stress, this threshold is not yet quantitatively defined in clinical settings. In practice, it will likely need to be operationalized using dynamic pharmacodynamic readouts rather than fixed dose or time cut-offs. Short-lived surges in type I IFNs and T-cell–recruiting chemokines such as CXCL9, CXCL10, and CCL5 [49, 72–76], without sustained elevation of IL6, IL8, IL10, or TGFβ [77, 78], may represent a desirable ‘ignition’ pattern, whereas persistent pro-tumor inflammatory and immunosuppressive cytokine signatures would signal a shift into detrimental chronic stress. Early-phase trials of mitochondrial stress–based regimens should therefore incorporate serial cytokine profiling and interferon-stimulated gene signatures, together with careful toxicity monitoring, to empirically define safe and effective activation windows.
CYTOKINE NETWORKS: FUNCTIONAL READOUTS AND INTEGRATORS OF MITOCHONDRIAL-IMMUNE CROSSTALK
Cytokine networks sit at the interface of mitochondrial stress, cGAS–STING activation, and the emergent immune phenotype of breast tumors, acting both as readouts of underlying organelle-immune crosstalk and as active sculptors of the tumor microenvironment [79]. In this context, distinct cytokine signatures—ranging from interferon- and chemokine-dominated pro-inflammatory profiles to IL-6/IL-10/TGF-β–rich immunosuppressive states—provide mechanistic insight into mitochondrial and STING pathway activity (figure 3), while serving as prognostic biomarkers and therapeutic targets in breast cancer.
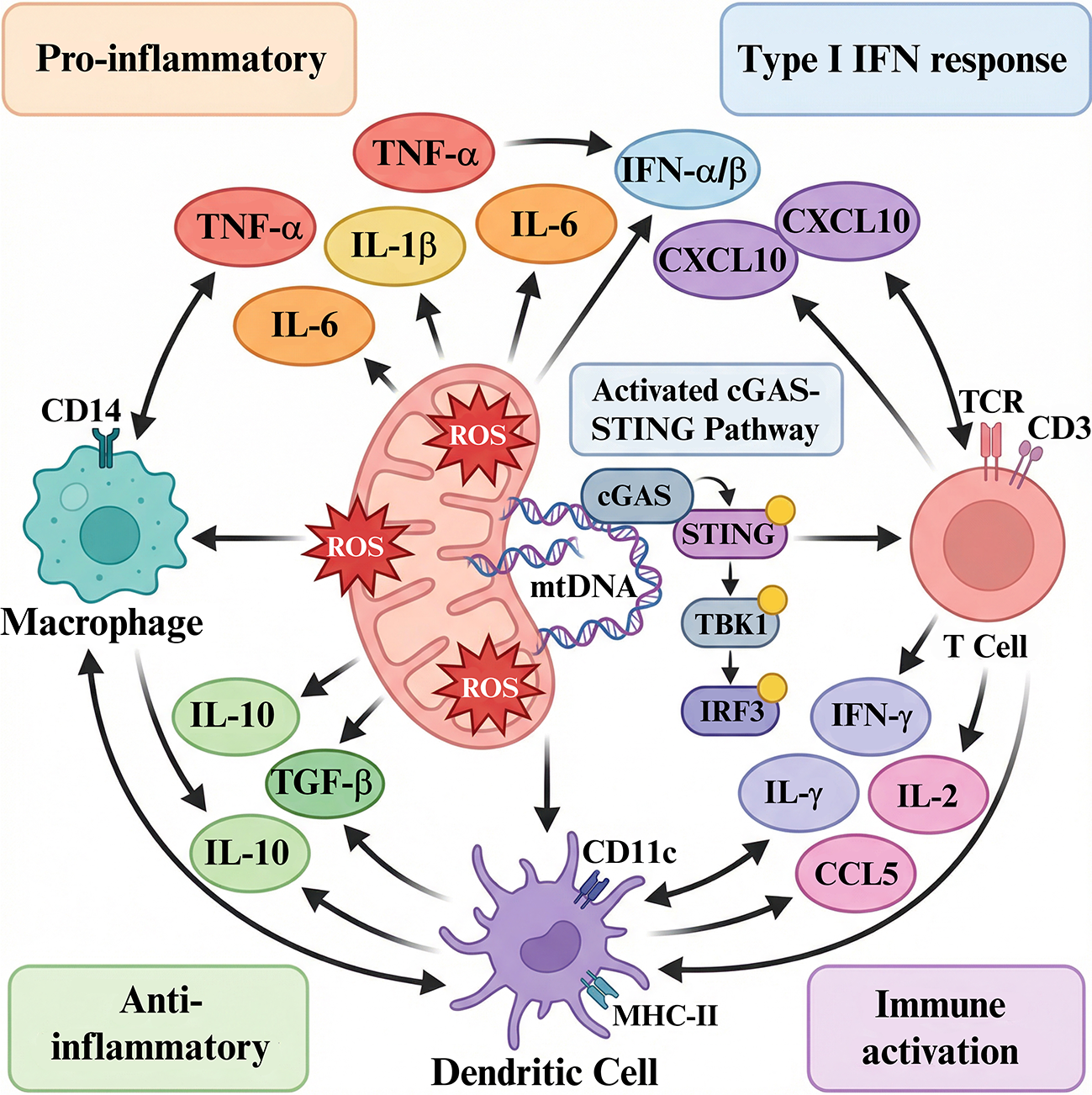
Figure 3.
Cytokine networks integrating mitochondrial stress and cGAS–STING activation. Mitochondrial ROS and mtDNA release trigger cGAS–STING signaling in tumor and immune cells, shaping production of interferons, chemokines (e.g., CXCL10) and immunosuppressive cytokines (e.g., IL 10, TGF β). The balance of these cytokines determines whether the breast tumor microenvironment is pro inflammatory or immunosuppressive. ROS: reactive oxygen species. mtDNA: mitochondrial DNA. cGAS: cyclic GMP–AMP synthase. STING: stimulator of interferon genes. IFN: interferon (mainly type I interferons, IFN α/β). CXCL10: C X C motif chemokine ligand 10. CCL5: C C motif chemokine ligand 5. IL 10: interleukin 10. TGF β: transforming growth factor beta. IL 6: interleukin 6. IL 1β: interleukin 1 beta. TNF α: tumor necrosis factor alpha. TCR: T cell receptor. TBK1: TANK binding kinase 1. TBK3: TANK binding kinase 3 (also known as IKK ε).
Cytokines as readouts of mitochondrial–STING signaling
Acute mitochondrial stress with controlled mtDNA release typically engages cGAS–STING and drives a type I interferon program characterized by IFN-α/β, interferon-stimulated genes, and T-cell–recruiting chemokines such as CXCL9, CXCL10, and CCL5 [43, 44, 80]. These cytokines correlate with enhanced dendritic cell activation, improved antigen presentation, and higher densities of cytotoxic and memory T cells in the tumor, reflecting a “hot,” inflamed microenvironment that is more permissive to checkpoint blockade. In contrast, chronic or dysregulated STING and mitochondrial stress can shift the cytokine output toward IL-6, IL-1β, TNF-α, IL-8, IL-10, and TGF-β, reflecting an exhausted or rewired STING axis and establishing a milieu that supports myeloid-derived suppressor cells, regulatory T cells, and tumor-promoting inflammation [81].
Pro-inflammatory versus immunosuppressive cytokine profiles in breast cancer
Recent systematic and multiplex analyses in breast cancer show that elevated pro-tumor inflammatory and immunosuppressive cytokines—especially IL-6, TNF-α, IL-1β, IL-8, IL-10, and TGF-β—associate with higher stage, increased metastasis, and poorer survival, underscoring their value as negative prognostic biomarkers [20, 79, 82]. Conversely, signatures enriched for IL-12, IFN-γ, and interferon-induced chemokines correlate with more effective antitumor immunity, higher tumor-infiltrating lymphocyte scores, and better outcomes or improved response to immunotherapy in selected breast cancer cohorts [83]. Spatial and transcriptomic studies further reveal that many breast tumors display “mixed” cytokine niches, where immunostimulatory and suppressive factors coexist, highlighting that the net functional state of the cytokine network—rather than any single mediator—captures the integrated output of mitochondrial, STING, and cellular stress signaling [84, 85].
Cytokine signatures as biomarkers and therapeutic targets
Because cytokine patterns reflect upstream mitochondrial integrity and cGAS–STING activity, composite cytokine signatures are increasingly explored as biomarkers to stratify patients, forecast immunotherapy benefit, and monitor pharmacodynamic responses to mitochondrial- or STING-targeted agents [20, 69]. Clinical and translational studies support the development of cytokine-based indices (for example, IL-6/IFN-γ ratios or multi-cytokine panels) and systemic inflammation scores as predictors of prognosis and treatment response in breast cancer, including inflammatory and immune-enriched subtypes [20, 86]. At the same time, cytokines themselves are being targeted or harnessed therapeutically: blockade of IL-6, IL-1β, or TGF-β aims to dismantle mitochondrial stress–driven immunosuppressive circuits, while agonistic strategies or engineered delivery of IL-12, IFN-α/β, or IFN-γ seek to amplify STING-induced pro-inflammatory signaling and consolidate cold-to-hot conversion [87–89].
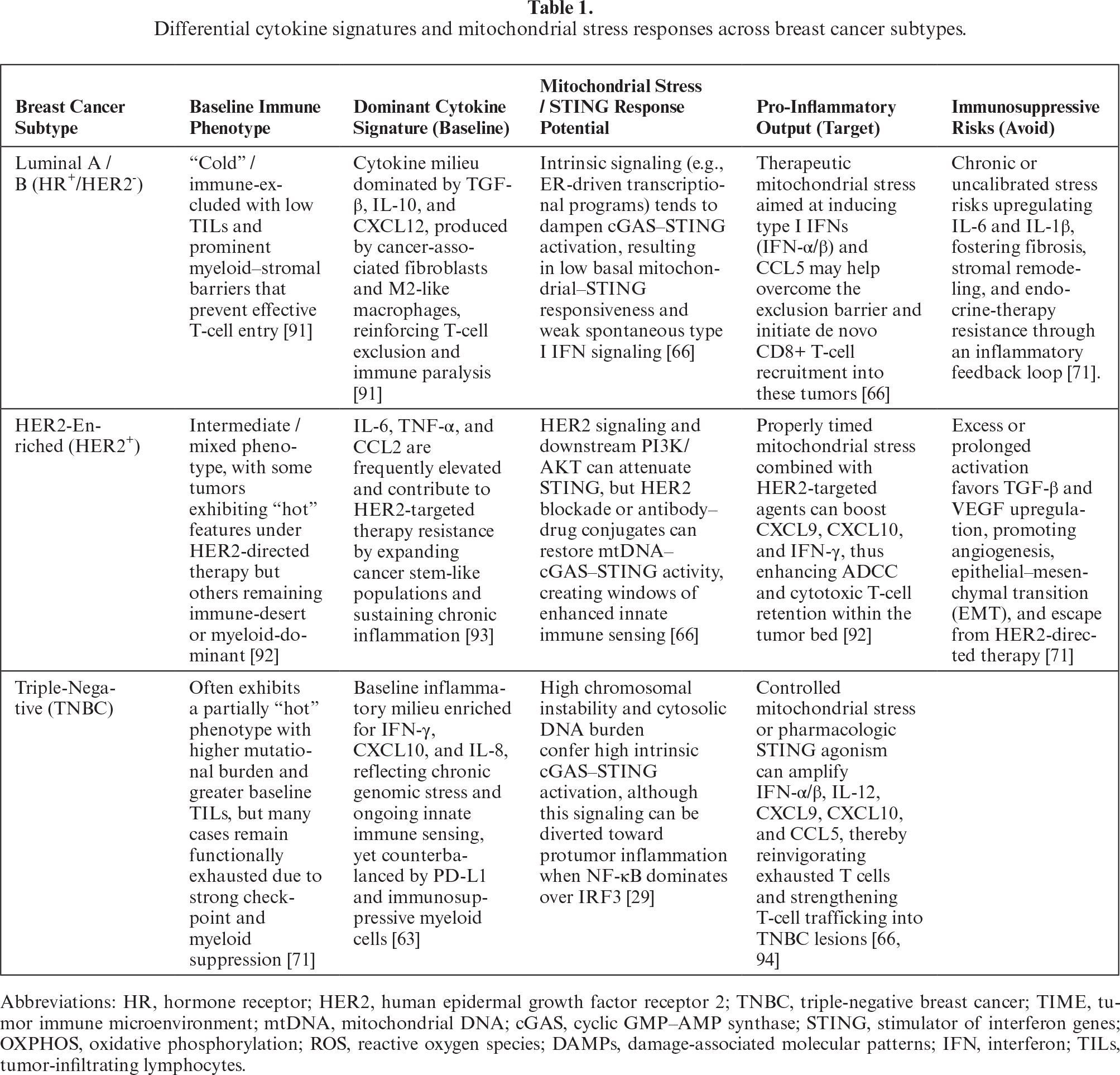
While STING activation is essential for priming anti-tumor immunity, its consequences are highly subtype-dependent. In Triple-Negative Breast Cancer (TNBC), DNA damage-induced STING signaling predominantly drives a type I interferon response (IFN-α/β) and the secretion of CXCL10 and CCL5, which correlates with prolonged progression-free survival by recruiting CD8+ cytotoxic T lymphocytes [29]. Conversely, in hormone receptor-positive (Luminal A/B) subtypes, chronic low-level STING activation is frequently linked to an immunosuppressive cytokine milieu rich in IL-6 and TGF-β, which promotes macrophage polarization toward an M2-like phenotype and facilitates therapeutic resistance [66]. Furthermore, recent profiling of “hot” versus “cold” tumors confirms that a sustained pro-inflammatory cytokine signature (IFN-γ, IL-12, CXCL9] is the primary determinant of cytolytic activity, whereas “cold” tumors exhibit elevated TGF-β and IL-10 levels that blunt mitochondrial stress signals [71]. Table 1 summarizes differential cytokine signatures and mitochondrial stress responses across breast cancer subtypes.
Taken together, cytokine networks can be viewed as dynamic integrators that encode the balance between beneficial and detrimental consequences of mitochondrial stress and cGAS–STING signaling in breast tumors. By reading out these cytokine states—and selectively modulating them with antibodies, receptor traps, or cytokine/chemokine agonists—future therapies may both report on and recalibrate mitochondrial-immune crosstalk, enabling rational combination regimens that align controlled mitochondrial stress, STING activation, and a favorable cytokine landscape to support durable antitumor immunity.
THERAPEUTIC STRATEGIES TO EXPLOIT MITOCHONDRIAL STRESS FOR IMMUNOTHERAPY
Mitochondrial stress represents a tractable lever to convert breast tumors from immune-deserted to inflamed states, but it must be engaged in a controlled, context-sensitive manner to avoid tipping into chronic immunosuppression. Figure 4 demonstrates mitochondria-centric strategies of combination therapy. Targeting mitochondrial function, mtDNA–cGAS–STING signaling, and associated cytokine networks has therefore become a focus of translational efforts to improve immunotherapy responses in solid tumors, including breast cancer [43, 69, 90].
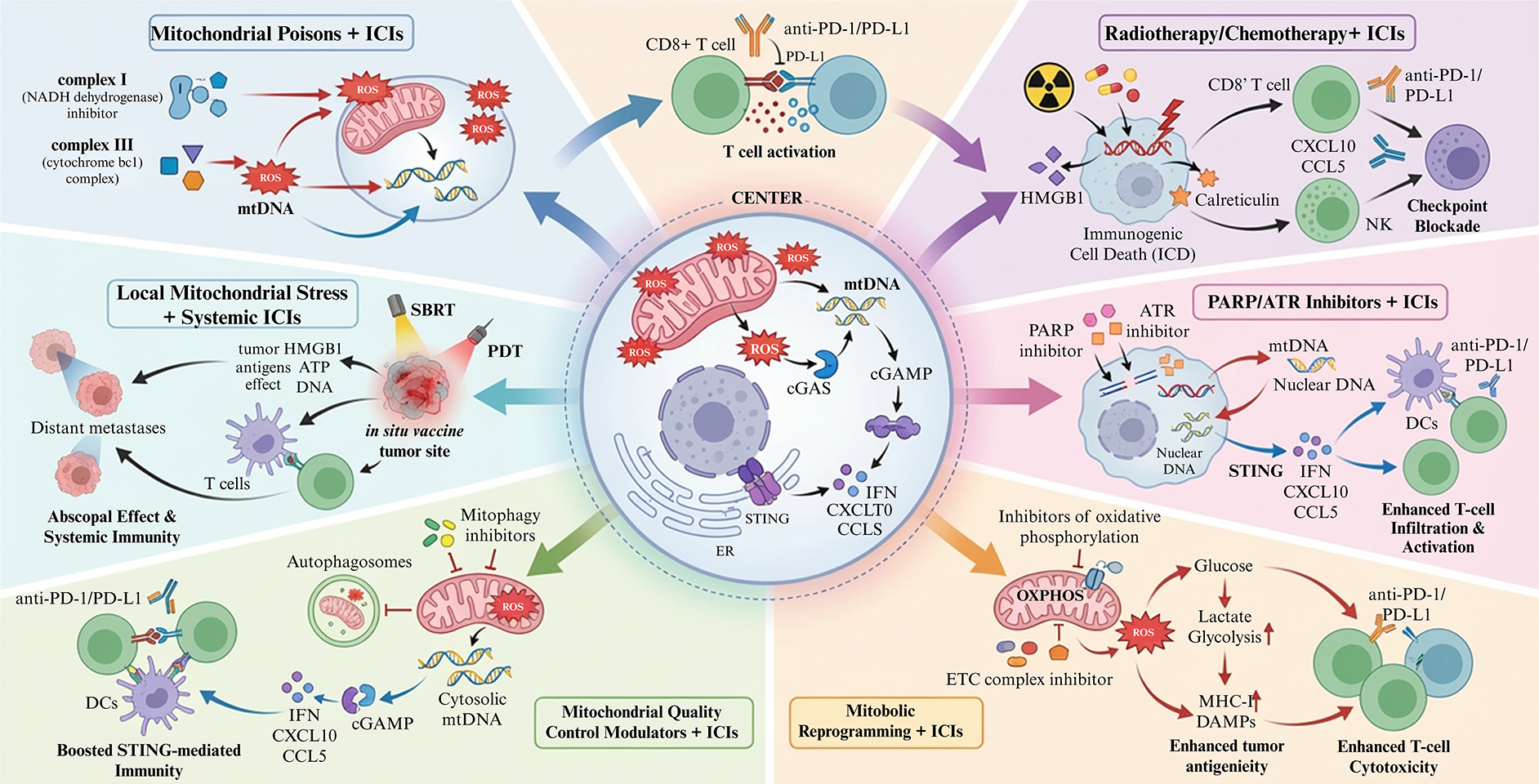
Figure 4.
Mitochondria targeting combination strategies to enhance immunotherapy. Center: mitochondrial stress in cancer cells increases ROS, mtDNA release and cGAS–STING–dependent interferon and chemokine production. Surrounding panels: examples of strategies including low dose mitochondrial poisons, radiotherapy/chemotherapy, PARP/ATR inhibitors, metabolic reprogramming agents, mitophagy modulators and local mitochondrial stress combined with systemic checkpoint blockade. mtDNA: mitochondrial DNA. ROS: reactive oxygen species. cGAS: cyclic GMP–AMP synthase. STING: stimulator of interferon genes. IFN: interferon (here primarily type I interferons, IFN‑α/β). CXCL9: C‑X‑C motif chemokine ligand 9. CXCL10: C‑X‑C motif chemokine ligand 10. CCL5: C‑C motif chemokine ligand 5. IL‑6: interleukin‑6. IL‑8: interleukin‑8. IL‑10: interleukin‑10. IL‑12: interleukin‑12. TNF‑α: tumor necrosis factor alpha. TGF‑β: transforming growth factor beta. OXPHOS: oxidative phosphorylation. FAO: fatty acid oxidation. DDR: DNA damage response. DAMPs: damage‑associated molecular patterns. ISG: interferon‑stimulated gene. MDSC: myeloid‑derived suppressor cell. TAM: tumor‑associated macrophage.
Inducing controlled mitochondrial stress to trigger immunity
Several classes of agents can induce mitochondrial stress in a way that favors acute, immunogenic signaling rather than catastrophic organelle failure or chronic inflammation [95]. Low-dose mitochondrial poisons (for example, complex I or III inhibitors, mild uncouplers) and selected radiotherapy or chemotherapy regimens can enhance mtROS, promote mtDNA oxidation and release, and stimulate cGAS–STING–dependent type I interferon and chemokine production without immediately triggering irreversible cell death in all tumor cells [27, 28, 89]. However, because mitochondria are essential for neurons, cardiomyocytes, and hematopoietic cells, systemic or prolonged inhibition of respiratory complexes or mitophagy carries substantial risks of neurotoxicity, cardiotoxicity, and myelosuppression [96–104]. In a ‘controlled ignition’ framework, these agents are therefore best deployed as short, sublethal priming pulses, ideally using tumorrestricted delivery systems or local administration [105–110], with pharmacodynamic monitoring of both cytokine output and organ toxicity to avoid sustained offtumor mitochondrial damage. Precision dosing and scheduling are crucial to maintain “sublethal” stress that allows sufficient antigen processing and cytokine elaboration, rather than overwhelming necrosis that floods the microenvironment with tolerogenic DAMPs and suppressive cytokines [34, 88].
A second strategy exploits targeted modulation of mitochondrial quality-control pathways. Pharmacologic or genetic interference with mitophagy and nucleoid maintenance (for example, via modulation of TFAM, PINK1/Parkin signaling, or mitochondrial proteases) can increase the pool of damaged mitochondria and facilitate mtDNA leakage into the cytosol, thereby amplifying cGAS–STING activation and type I interferon responses [32, 45, 46]. When carefully titrated, such interventions enhance immunogenic signaling, but excessive or prolonged blockade risks accumulation of dysfunctional mitochondria, chronic NFκB–biased output, and a shift toward IL6/IL10/TGFβ–dominated immunosuppressive states [43, 81, 111].
Third, mitochondria-targeted delivery systems and nanomedicines are being developed to impose spatially restricted stress. Conjugating chemotherapeutics, photosensitizers, or redox-active molecules to mitochondria-targeting moieties (such as triphenylphosphonium) or encapsulating STING-stimulating payloads in mitochondria-accumulating nanocarriers enables direct organelle engagement at lower systemic doses, enhancing immunogenic cell death, mtDNA release, and STING-driven chemokine production within the tumor while limiting off-tumor toxicity [87, 89, 90]. Photosensitizer- or radiotherapy-based mitochondria-targeted approaches are particularly attractive for localized disease or oligometastatic settings, where temporal control of light or dose can be synced with immunotherapy cycles to maximize acute immune activation [49, 112].
Finally, metabolic reprogramming agents—such as inhibitors of OXPHOS, fatty acid oxidation, or glutamine metabolism—can be used to reshape mitochondrial function in both tumor and immune compartments. In tumor cells, transient OXPHOS inhibition can augment ROS, enhance antigenicity, and increase susceptibility to T-cell killing [26, 34]. In T cells, interventions that improve mitochondrial biogenesis and spare respiratory capacity—through exercise-mimetic strategies, PGC1α activation, or cytokine support—can restore effector function and resilience in the nutrient-poor tumor microenvironment [35–37]. Here, a central challenge is separating beneficial stress in tumor cells from detrimental exhaustion in effector cells, necessitating careful attention to timing, dosing, and cell-type specificity.
Rational combination therapies: pairing mitochondrial modulators with checkpoint blockade
Because mitochondrial stress and cGAS–STING activation principally function as priming and inflaming signals, they naturally complement checkpoint blockade, which acts downstream to unleash pre-existing or nascent T-cell responses [64]. A central concept is to use mitochondrial modulators as “ignition” agents that increase tumor antigenicity, dendritic cell activation, and chemokine-driven T-cell trafficking, while PD1/PDL1 or CTLA4 blockade prevents exhaustion of the recruited effector pool [43, 69, 113]. In preclinical breast and other solid tumor models, sequencing mitochondrial stress–inducing chemotherapy, radiation, or targeted agents before or concurrent with checkpoint inhibitors enhances infiltration of CD8+ T-cells and NK cells, elevates CXCL9/CXCL10/CCL5 levels, and improves response rates compared with checkpoint blockade alone [53, 58–60].
Rational combinations can be organized along several axes. Mechanistically, agents that induce mtDNA release or immunogenic cell death—such as selected DNA-damaging drugs, PARP or ATR inhibitors, and mitochondrial complex inhibitors—are combined with checkpoint inhibitors to couple antigen/IFN/chemokine induction with relief of T-cell inhibition, an approach supported by recent ATR–cGAS–STING data and PARP inhibitor–ICI combinations [28, 47]. Spatial and temporal integration is achieved by using local mitochondrial stress (for example, via mitochondria-targeted photodynamic therapy or stereotactic radiotherapy) to create an “in situ vaccine” effect at the primary tumor or oligometastatic sites, followed by systemic checkpoint blockade to control microscopic or distant disease [49, 88, 112]. Optimizing the interval between mitochondrial perturbation and checkpoint dosing is key to align peak antigen presentation and chemokine production with maximal T-cell reinvigoration.
Given the risk that chronic STING activation and mitochondrial stress promote IL6/TGFβ–dominated immunosuppression, combinations that add cytokine or myeloid-targeted therapies are gaining interest. Pairing mitochondrial stress–inducing regimens and checkpoint blockade with IL6 or TGFβ inhibitors, CSF1R or CXCR2 antagonists, or modulators of myeloid metabolism may tilt the balance toward a durable pro-inflammatory cytokine landscape and reduce myeloid-derived suppressor cell and M2-like TAM accumulation [20, 49, 66, 79].
Subtype specific considerations
Subtype-specific considerations are likely to be critical for mitochondrial stress based combinations. In TNBC, where where genomic instability and baseline STING activity can be relatively higher and tumor-infiltrating lymphocytes (TILs) are more abundant, milder mitochondrial perturbation or intermittent dosing may suffice to amplify type I interferons and CXCL9/10/CCL5 without provoking sustained IL 6/TGF β–dominated immunosuppression [49, 61, 62]. In hormone receptor–positive and many HER2-enriched tumors, which commonly exhibit immune exclusion and TGF-β rich cytokine milieus, more intensive or cyclic priming strategies—potentially combined with blockade of dominant suppressive cytokines or myeloid pathways—may be required to first convert the tumor into a T cell–permissive state before or during checkpoint blockade (Table 1) [67, 68, 114]. Recent evidence further highlights that resistance to hormonal and targeted therapies in breast cancer can be driven by suppression of the NR6A1/DNMT3A axis [115], reinforcing the need to tailor metabolic and epigenetic interventions to subtype-specific vulnerabilities.
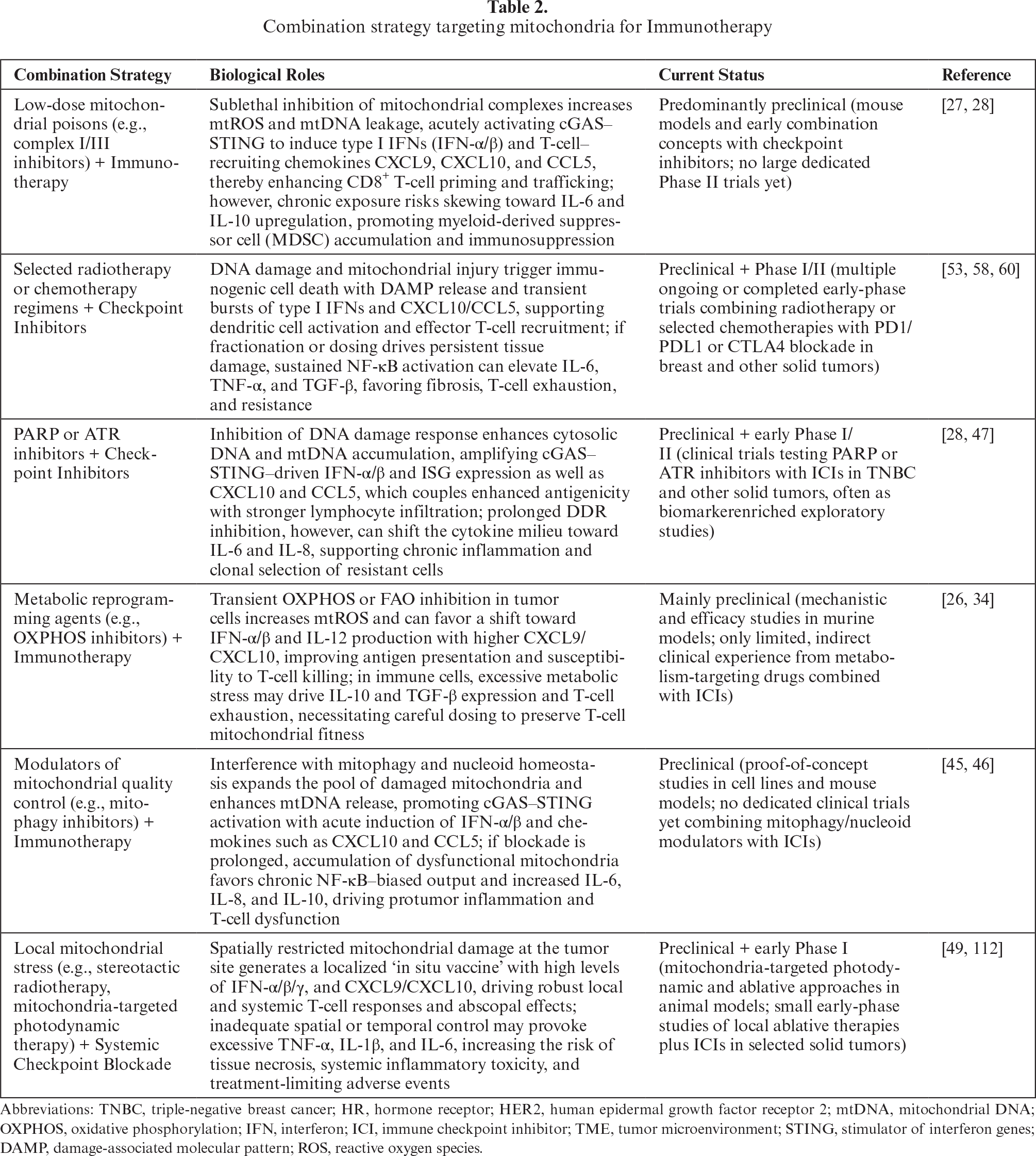
Overall, the most compelling therapeutic vision is a multi-layered regimen in which mitochondrial modulators are used not simply as cytotoxic agents but as programmable “danger signal” generators. By calibrating the intensity, duration, and cellular targets of mitochondrial stress, and embedding this within a framework of checkpoint inhibition and cytokine/myeloid control, future therapies may reliably ignite and sustain productive antitumor immunity in breast cancer while minimizing the risk that the same pathways are co-opted to drive resistance and immune escape. Table 2 summarizes the combination strategy targeting mitochondria for Immunotherapy.
CONCLUSION AND FUTURE PERSPECTIVES
Mitochondrial stress has emerged as a unifying framework to understand and therapeutically exploit how breast tumors interact with the immune system, repositioning mitochondria from passive metabolic organelles to programmable hubs of innate immune sensing and cytokine control. By linking mtDNA leakage, cGAS–STING activation, and cytokine network remodeling to cold-to-hot tumor conversion, this paradigm offers a mechanistic basis for designing combination strategies that move beyond empiric cytotoxicity toward deliberate immune priming in otherwise immunologically barren breast cancers [116].
This review highlights three central advances: first, that mitochondrial architecture, quality control, and metabolic wiring in cancer and immune cells collectively determine whether mitochondrial stress yields immunogenic interferon–chemokine programs or entrenched immunosuppression. Second, mtDNA–cGAS–STING signaling operates as a dose- and context-dependent rheostat whose acute activation can ignite T cell–inflamed phenotypes, whereas chronic engagement fuels resistance, stromal remodeling, and checkpoint upregulation. Third, cytokine networks function as both readouts and effectors of this mitochondrial–immune crosstalk, integrating upstream organelle stress into prognostic signatures and actionable therapeutic targets.
Therapeutically, these insights converge on a strategy of “controlled ignition,” in which mitochondrial stress is imposed in a spatially and temporally constrained manner to amplify antigenicity, type I interferons, and T cell–recruiting chemokines while avoiding sustained NF-κB-biased, IL-6/IL-10/TGF-β–dominated states. Emerging platforms—including mitochondria-targeted small molecules and nanomedicines, rationally dosed OXPHOS and mitophagy modulators, and STING agonists or epigenetic restorers of cGAS–STING—provide a growing toolbox for such programmable danger signaling, particularly when layered onto checkpoint blockade. As multiplex cytokine profiling and immune gene signatures mature clinically, they are poised to guide patient selection, monitor pharmacodynamic responses, and adapt combination regimens in real time.
Several priorities must be addressed to safely and effectively bring mitochondrial stress–based immuno-oncology into the clinic. Mechanistic studies should dissect cell type–specific and subtype-specific thresholds for beneficial versus deleterious mitochondrial stress, including how luminal, HER2-enriched, and triple-negative tumors differentially tune cGAS–STING output and cytokine landscapes. Systems-level approaches integrating single-cell and spatial multi-omics with metabolic and mitochondrial profiling will be critical to map how mitochondrial reprogramming in tumor, stromal, and immune compartments co-evolves under therapy and shapes response or resistance to immunotherapy. Parallel translational work should focus on rational trial designs that sequence and dose mitochondrial modulators as priming agents around immune checkpoint inhibitors, with built-in biomarker programs capturing mtDNA–STING activity, cytokine states, and immune cell fitness. Finally, safety frameworks must anticipate off-tumor inflammation and chronic STING-driven toxicities, motivating development of tumor-restricted delivery systems, reversible agonists, and combination strategies that simultaneously ignite anti-tumor immunity and restrain maladaptive cytokine circuits. If these challenges can be met, targeting mitochondrial stress has the potential to transform immunologically cold breast cancers into consistently treatable, inflamed diseases and to redefine mitochondria as programmable adjuvants at the core of breast cancer immunotherapy.
This review is primarily conceptual and therefore has several important limitations. First, most of the discussed mitochondrial stress–cGAS–STING mechanisms and combination strategies are derived from preclinical models, with limited validation in large, prospective breast cancer trials, so their translational robustness and safety remain uncertain. Second, subtype-specific differences in mitochondrial wiring, cytokine networks, and STING pathway status are still incompletely mapped, which constrains precise patient stratification and may oversimplify the heterogeneity of luminal, HER2-enriched, and TNBC tumors. Third, the proposed “controlled ignition” paradigm does not yet incorporate quantitative thresholds for beneficial versus deleterious mitochondrial stress or fully account for systemic toxicities observed with STING agonists, highlighting the need for biomarker-guided dosing, longitudinal cytokine monitoring, and tumor-restricted delivery platforms before these concepts can be reliably applied in the clinic.
DISCLOSURE
Acknowledgments: The authors extend their sincere appreciation to the Research Assistant Center, Show Chwan Memorial Hospital, Taiwan, for providing essential technical support throughout this study. We are grateful to all personnel who assisted in this research.
Funding Statement: This research was funded by the National Science and Technology Council, Taiwan; NSTC 112-2314-B-442-003, NSTC 113-2314-B-442-002, and 114-2314-B-442 -001-MY3 [recipient: Hung-Yu Lin]; NSTC 112-2314-B-442-001 and 113-2314-B-442-001-MY2 [recipient: Pei-Yi Chu]), and the National Health Research Institutes (NHRI-109BCCO-MF-202015-01 [recipient: Pei-Yi Chu]) and by the Show Chwan Memorial Hospital (SRD-112021 [recipient: Hsing-Ju Wu]).
Author Contributions: The authors confirm contribution to the paper as follows: Conceptualization, Hung-Yu Lin and Hsing-Ju Wu; methodology, Hung-Yu Lin; software, Hsing-Ju Wu; validation, Pei-Yi Chu; formal analysis, Hsing-Ju Wu; investigation, Hsing-Ju Wu; resources, Pei-Yi Chu; data curation, Hung-Yu Lin; writing—original draft preparation, Hung-Yu Lin and Hsing-Ju Wu; writing—review and editing, Pei-Yi Chu; visualization, Hung-Yu Lin; supervision, Pei-Yi Chu; project administration, Hsing-Ju Wu; funding acquisition, Hsing-Ju Wu. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
| cGAS | Cyclic GMP-AMP synthase |
| STING | Stimulator of interferon genes |
| mtDNA | Mitochondrial DNA |
| DAMPs | Damage-associated molecular patterns |
| TNBC | Triple-negative breast cancer |
| TIME | Tumor immune microenvironment |
| OXPHOS | Oxidative phosphorylation |
| mtROS | Mitochondrial reactive oxygen species |
| MHC-I | Major histocompatibility complex class I |
| DRP1 | Dynamin-related protein 1 |
| IFN | Interferon (IFN-α/β for type I interferons) |
| IRF3 | Interferon regulatory factor 3 |
| NF-κB | Nuclear factor-kappa B |
| cGAMP | 2’3’-cyclic GMP-AMP |
| TNF-α | Tumor necrosis factor alpha |
| CCL5 | C-C motif chemokine ligand 5 |
| CXCL10 | C-X-C motif chemokine ligand 10 |
| PD-L1 | Programmed death-ligand 1 |
| NK cells | Natural killer cells |
| ER | Endoplasmic reticulum |
| HER2 | Human epidermal growth factor receptor 2 |
REFERENCES
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2024;74:229–63. [Google Scholar]
2. Bianchini G, Balko JM, Mayer IA, et al. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol 2016;13:674–90. [Google Scholar]
3. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov 2019;18:197–218. [Google Scholar]
4. Gruosso T, Gigoux M, Manem VSK, et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J Clin Invest 2019;129:1785–800. [Google Scholar]
5. Campbell MJ, Wolf DM, Yau C, et al. Multi-platform biomarkers of response to an immune checkpoint inhibitor in the neoadjuvant I-SPY 2 trial for early-stage breast cancer. Cell Rep Med 2024;5:101799. [Google Scholar]
6. Imani S, Farghadani R, Roozitalab G, et al. Reprogramming the breast tumor immune microenvironment: cold-to-hot transition for enhanced immunotherapy. J Exp Clin Cancer Res 2025;44:131. [Google Scholar]
7. Zhang Y, Yan H, Wei Y, et al. Decoding mitochondria’s role in immunity and cancer therapy. Biochim Biophys Acta Rev Cancer 2024;1879:189107. [Google Scholar]
8. Dent R, Cortes J, Pusztai L, et al. Neoadjuvant pembrolizumab plus chemotherapy/adjuvant pembrolizumab for early-stage triple-negative breast cancer: quality-of-life results from the randomized KEYNOTE-522 study. J Natl Cancer Inst 2024;116:1654–63. [Google Scholar]
9. Elmakaty I, Abdo R, Elsabagh A, et al. Comparative efficacy and safety of PD-1/PD-L1 inhibitors in triple-negative breast cancer: a systematic review and network meta-analysis of randomized controlled trials. Cancer Cell Int 2023;23:90. [Google Scholar]
10. Latif F, Bint Abdul Jabbar H, Malik H, et al. Atezolizumab and pembrolizumab in triple-negative breast cancer: a meta-analysis. Expert Rev Anticancer Ther 2022;22:229–35. [Google Scholar]
11. Qi Y, Zhang W, Jiang R, et al. Efficacy and safety of PD-1 and PD-L1 inhibitors combined with chemotherapy in randomized clinical trials among triple-negative breast cancer. Front Pharmacol 2022;13:960323. [Google Scholar]
12. Zhu Y, Zhu X, Tang C, et al. Progress and challenges of immunotherapy in triple-negative breast cancer. Biochim Biophys Acta Rev Cancer 2021;1876:188593. [Google Scholar]
13. Goldberg J, Pastorello RG, Vallius T, et al. The immunology of hormone receptor positive breast cancer. Front Immunol 2021;12:674192. [Google Scholar]
14. Melissari MT, Papatheodoridi A, Argyriadis A, et al. Immunotherapy in primary hormone-receptor positive breast cancer: a systematic review. Crit Rev Oncol Hematol 2025;212:104768. [Google Scholar]
15. Solinas C, Fumagalli D, Dieci MV. Immune checkpoint blockade in HER2-positive breast cancer: what role in early disease setting? Cancers (Basel) 2021; 13. [Google Scholar]
16. Asleh K, Riaz N, Nielsen TO. Heterogeneity of triple negative breast cancer: current advances in subtyping and treatment implications. J Exp Clin Cancer Res 2022;41:265. [Google Scholar]
17. Sabit H, Adel A, Abdelfattah MM, et al. The role of tumor microenvironment and immune cell crosstalk in triple-negative breast cancer (TNBCemerging therapeutic opportunities. Cancer Lett 2025;628:217865. [Google Scholar]
18. Hu Y, Manasrah BK, McGregor SM, et al. Paclitaxel induces micronucleation and activates pro-inflammatory cGAS-STING signaling in triple-negative breast cancer. Mol Cancer Ther 2021;20:2553–67. [Google Scholar]
19. Parkes EE, Savage KI, Lioe T, et al. Activation of a cGAS-STING-mediated immune response predicts response to neoadjuvant chemotherapy in early breast cancer. Br J Cancer 2022;126:247–58. [Google Scholar]
20. Ciurescu S, Buciu V, Serban D, et al. Role of cytokines in breast cancer: a systematic review and meta-analysis. Biomedicines 2025; 13. [Google Scholar]
21. Nicolini A, Rossi G, Ferrari P. Experimental and clinical evidence in favour of an effective immune stimulation in ER-positive, endocrine-dependent metastatic breast cancer. Front Immunol 2023;14:1225175. [Google Scholar]
22. Ahn M, Ali A, Seo JH. Mitochondrial regulation in the tumor microenvironment: targeting mitochondria for immunotherapy. Front Immunol 2024;15:1453886. [Google Scholar]
23. Huang A, Xue H, Xie T, et al. A review of the pathogenesis of mitochondria in breast cancer and progress of targeting mitochondria for breast cancer treatment. J Transl Med 2025;23:70. [Google Scholar]
24. Lin HY, Chu PY. Mitochondrial calcium uniporter as biomarker and therapeutic target for breast cancer: prognostication, immune microenvironment, epigenetic regulation and precision medicine. J Adv Res 2025;70:445–61. [Google Scholar]
25. Lin HY, Wu HJ, Chu PY. Multi-omics and experimental analysis unveil theragnostic value and immunological roles of inner membrane mitochondrial protein (IMMT) in breast cancer. J Transl Med 2023;21:189. [Google Scholar]
26. Du H, Xu T, Yu S, et al. Mitochondrial metabolism and cancer therapeutic innovation. Signal Transduct Target Ther 2025;10:245. [Google Scholar]
27. Li Z, Zheng W, Liu Y, et al. Bridging innate and adaptive tumor immunity: cGAS-STING pathway activation to potentiate immune checkpoint blockade. J Exp Clin Cancer Res 2025;44:303. [Google Scholar]
28. Taniguchi H, Chakraborty S, Takahashi N, et al. ATR inhibition activates cancer cell cGAS/STING-interferon signaling and promotes antitumor immunity in small-cell lung cancer. Sci Adv 2024;10:eado4618. [Google Scholar]
29. Xiong H, Li R, Yang L, et al. Dual regulation of the cGAS-STING pathway: new targets and challenges for subtype-specific immunotherapy in breast cancer. Front Oncol 2025;15:1619097. [Google Scholar]
30. Minarrieta L, Annis MG, Audet-Delage Y, et al. Mitochondrial elongation impairs breast cancer metastasis. Sci Adv 2024;10:eadm8212. [Google Scholar]
31. Jiang J, Yan Y, Yang C, et al. Immunogenic cell death and metabolic reprogramming in cancer: mechanisms, synergies, and innovative therapeutic strategies. Biomedicines 2025; 13. [Google Scholar]
32. Newman LE, Shadel GS. Mitochondrial DNA release in innate immune signaling. Annu Rev Biochem 2023;92:299–332. [Google Scholar]
33. Cheng X, Wang Y, Johnson B, et al. Mitochondria-targeted strategies in tumor immunity. Front Immunol 2025;16:1646138. [Google Scholar]
34. Weinberg SE, Chandel NS. Mitochondria reactive oxygen species signaling-dependent immune responses in macrophages and T cells. Immunity 2025;58:1904–21. [Google Scholar]
35. Mukherjee S, Ghosh A, Chakraborty S, et al. Tumor-infiltrating lymphocytes and tumor-associated macrophages in cancer immunoediting: a targetable therapeutic axis. Front Immunol 2025;16:1655176. [Google Scholar]
36. Zhao J, Hu S, Qi Z, et al. Mitochondrial metabolic reprogramming of macrophages and T cells enhances CD47 antibody-engineered oncolytic virus antitumor immunity. J Immunother Cancer 2024; 12. [Google Scholar]
37. Voltarelli VA, Amano MT, Tobias GC, et al. Moderate-intensity aerobic exercise training improves CD8(+) tumor-infiltrating lymphocytes effector function by reducing mitochondrial loss. iScience 2024;27:110121. [Google Scholar]
38. Hsu WJ, Hsu MC, Chu CY, et al. Metastatic breast cancer cells are vulnerable to fatty acid oxidation inhibition through DDX3-DRP1-mediated mitochondrial plasticity. Redox Biol 2025;86:103845. [Google Scholar]
39. Chen C, Guo W, Wang H, et al. Metabolic-immune axis in the tumor microenvironment: a new strategy for prognostic assessment and precision therapy in DLBCL and FL. Front Immunol 2025;16:1659011. [Google Scholar]
40. Qiu Y, Xu Y, Ding X, et al. Bi-directional metabolic reprogramming between cancer cells and T cells reshapes the anti-tumor immune response. PLoS Biol 2025;23:e3003284. [Google Scholar]
41. Akter Z, Salamat N, Ali MY, et al. The promise of targeting heme and mitochondrial respiration in normalizing tumor microenvironment and potentiating immunotherapy. Front Oncol 2022;12:1072739. [Google Scholar]
42. Chen X, Lin P, Lu Y, et al. Mitochondrial regulation of CD8(+) T cells: mechanisms and therapeutic modulation. Adv Sci (Weinh) 2025;12:e03095. [Google Scholar]
43. Shen M, Jiang X, Peng Q, et al. The cGAS-STING pathway in cancer immunity: mechanisms, challenges, and therapeutic implications. J Hematol Oncol 2025;18:40. [Google Scholar]
44. Aloraini GS. Mitochondrial DNA release and cGAS-STING activation: emerging insights into anti-tumor immunity. Pathol Res Pract 2025;273:156158. [Google Scholar]
45. VanPortfliet JJ, Chute C, Lei Y, et al. Mitochondrial DNA release and sensing in innate immune responses. Hum Mol Genet 2024;33:R80–91. [Google Scholar]
46. Gogvadze V, Zhivotovsky B. Mitochondrial DNA: how does it leave mitochondria? Trends Cell Biol 2025;35:819–22. [Google Scholar]
47. Zhang X, Chen Y, Liu X, et al. STING in cancer immunoediting: modeling tumor-immune dynamics throughout cancer development. Cancer Lett 2025;612:217410. [Google Scholar]
48. Cryer AM, Dosta P, Dion MZ, et al. Restoration of cGAS in cancer cells promotes antitumor immunity via transfer of cancer cell-generated cGAMP. Proc Natl Acad Sci U S A 2025;122:e2409556122. [Google Scholar]
49. Li J, Yang H, Zhu M, et al. Unlocking the therapeutic potential of the STING signaling pathway in anti-tumor treatment. Clin Exp Med 2025;25:290. [Google Scholar]
50. Berger G, Knelson EH, Jimenez-Macias JL, et al. STING activation promotes robust immune response and NK cell-mediated tumor regression in glioblastoma models. Proc Natl Acad Sci U S A 2022;119:e2111003119. [Google Scholar]
51. Knelson EH, Ivanova EV, Tarannum M, et al. Activation of tumor-cell STING primes NK-cell therapy. Cancer Immunol Res 2022;10:947–61. [Google Scholar]
52. Yan X, Yao C, Fang C, et al. Rocaglamide promotes the infiltration and antitumor immunity of NK cells by activating cGAS-STING signaling in non-small cell lung cancer. Int J Biol Sci 2022;18:585–98. [Google Scholar]
53. Sen T, Rodriguez BL, Chen L, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov 2019;9:646–61. [Google Scholar]
54. Song Y, Liu Y, Teo HY, et al. Manganese enhances the antitumor function of CD8(+) T cells by inducing type I interferon production. Cell Mol Immunol 2021;18:1571–4. [Google Scholar]
55. Xu N, Palmer DC, Robeson AC, et al. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J Exp Med 2021;218. [Google Scholar]
56. Xu Y, Sun F, Tian Y, et al. Enhanced NK cell activation via eEF2K-mediated potentiation of the cGAS-STING pathway in hepatocellular carcinoma. Int Immunopharmacol 2024;129:111628. [Google Scholar]
57. Yi M, Niu M, Wu Y, et al. Combination of oral STING agonist MSA-2 and anti-TGF-beta/PD-L1 bispecific antibody YM101: a novel immune cocktail therapy for non-inflamed tumors. J Hematol Oncol 2022;15:142. [Google Scholar]
58. Ager CR, Reilley MJ, Nicholas C, et al. Intratumoral STING activation with T-cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol Res 2017;5:676–84. [Google Scholar]
59. Girard M, Yu T, Batista NV, et al. STING agonists drive recruitment and intrinsic type I interferon responses in monocytic lineage cells for optimal anti-tumor immunity. J Immunol 2025. [Google Scholar]
60. Wu YT, Fang Y, Wei Q, et al. Tumor-targeted delivery of a STING agonist improves cancer immunotherapy. Proc Natl Acad Sci U S A 2022;119:e2214278119. [Google Scholar]
61. Chen M, Yu S, van der Sluis T, et al. cGAS-STING pathway expression correlates with genomic instability and immune cell infiltration in breast cancer. NPJ Breast Cancer 2024;10:1. [Google Scholar]
62. Dai Z, Gu Z, Shen R, et al. An immunotherapy guide constructed by cGAS-STING signature for breast cancer and the biofunction validation of the pivotal gene HOXC13 via in vitro experiments. Front Immunol 2025;16:1586877. [Google Scholar]
63. Zimmerli D, Brambillasca CS, Talens F, et al. MYC promotes immune-suppression in triple-negative breast cancer via inhibition of interferon signaling. Nat Commun 2022;13:6579. [Google Scholar]
64. Li A, Yi M, Qin S, et al. Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J Hematol Oncol 2019;12:35. [Google Scholar]
65. Massague J, Sheppard D. TGF-beta signaling in health and disease. Cell 2023;186:4007–37. [Google Scholar]
66. Zhang Y, Wang Y, Mu P, et al. Bidirectional regulation of the cGAS-STING pathway in the immunosuppressive tumor microenvironment and its association with immunotherapy. Front Immunol 2024;15:1470468. [Google Scholar]
67. Saha S, Mahapatra S, Khanra S, et al. Decoding breast cancer treatment resistance through genetic, epigenetic, and immune-regulatory mechanisms: from molecular insights to translational perspectives. Cancer Drug Resist 2025;8:36. [Google Scholar]
68. Wang L, Wang Y, Li Y, et al. Resistance mechanisms and prospects of trastuzumab. Front Oncol 2024;14:1389390. [Google Scholar]
69. Yan Y, Tan X, Song B, et al. Breaking barriers: the cGAS-STING pathway as a novel frontier in cancer immunotherapy. Cancer Commun (Lond) 2025;45:1513–46. [Google Scholar]
70. Xu Y, An D, Zhang T, et al. Mitochondrion-targeted type I photodynamic therapy for agonist independent cGAS-STING activation. Adv Mater 2025;37:e2418894. [Google Scholar]
71. Wu B, Zhang B, Li B, et al. Cold and hot tumors: from molecular mechanisms to targeted therapy. Signal Transduct Target Ther 2024;9:274. [Google Scholar]
72. Araujo JM, Gomez AC, Aguilar A, et al. Effect of CCL5 expression in the recruitment of immune cells in triple negative breast cancer. Sci Rep 2018;8:4899. [Google Scholar]
73. House IG, Savas P, Lai J, et al. Macrophage-derived CXCL9 and CXCL10 are required for antitumor immune responses following immune checkpoint blockade. Clin Cancer Res 2020;26:487–504. [Google Scholar]
74. Lim J, Kang I, La J, et al. Harnessing type I interferon-mediated immunity to target malignant brain tumors. Front Immunol 2023;14:1203929. [Google Scholar]
75. Santana-Hernandez S, Suarez-Olmos J, Servitja S, et al. NK cell-triggered CCL5/IFNgamma-CXCL9/10 axis underlies the clinical efficacy of neoadjuvant anti-HER2 antibodies in breast cancer. J Exp Clin Cancer Res 2024;43:10. [Google Scholar]
76. Tokunaga R, Zhang W, Naseem M, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation – a target for novel cancer therapy. Cancer Treat Rev 2018;63:40–7. [Google Scholar]
77. Liu C, Yin Q, Wu Z, et al. Inflammation and immune escape in ovarian cancer: pathways and therapeutic opportunities. J Inflamm Res 2025;18:895–909. [Google Scholar]
78. Zhao H, Wu L, Yan G, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther 2021;6:263. [Google Scholar]
79. Xu S, Wang Q, Ma W. Cytokines and soluble mediators as architects of tumor microenvironment reprogramming in cancer therapy. Cytokine Growth Factor Rev 2024;76:12–21. [Google Scholar]
80. Maekawa H, Inoue T, Ouchi H, et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep 2019;29:1261–73. [Google Scholar]
81. Li K, Shi H, Zhang B, et al. Myeloidderived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther 2021;6:362. [Google Scholar]
82. Raju A, Thriveni K, Bharatnur SS. Comprehensive cytokine gene expression and multiplex analysis in breast cancer patients: insights into immune profiles and prognostic factors’ association. Cureus 2025;17:e85288. [Google Scholar]
83. Liang YK, Deng ZK, Chen MT, et al. CXCL9 is a potential biomarker of immune infiltration associated with favorable prognosis in ERnegative breast cancer. Front Oncol 2021;11:710286. [Google Scholar]
84. JimenezSantos MJ, GarciaMartin S, RubioFernandez M, et al. Spatial transcriptomics in breast cancer reveals tumour microenvironmentdriven drug responses and clonal therapeutic heterogeneity. NAR Cancer 2024;6:zcae046. [Google Scholar]
85. Qian J, Shao X, Bao H, et al. Identification and characterization of cell niches in tissue from spatial omics data at singlecell resolution. Nat Commun 2025;16:1693. [Google Scholar]
86. CastroEspin C, Cairat M, Navionis AS, et al. Prognostic role of prediagnostic circulating inflammatory biomarkers in breast cancer survival: evidence from the EPIC cohort study. Br J Cancer 2024;131:1496505. [Google Scholar]
87. Wang B, Tang M, Chen Q, et al. Delivery of mRNA encoding interleukin12 and a stimulator of interferon genes agonist potentiates antitumor efficacy through reversing T cell exhaustion. ACS Nano 2024;18:15499516 [Google Scholar]
88. Wang M, Zhai X, Li J, et al. The role of cytokines in predicting the response and adverse events related to immune checkpoint inhibitors. Front Immunol 2021;12:670391. [Google Scholar]
89. Zhao J, Tong A, Liu J, et al. Tumortargeting nanocarriers amplified immunotherapy of cold tumors by STING activation and inhibiting immune evasion. Sci Adv 2025;11:eadr1728 [Google Scholar]
90. Li S, Yuan H, Yang XZ, et al. Synergistic antitumor immunotherapy via mitochondria regulation in macrophages and tumor cells by an iridium photosensitizer. ACS Cent Sci 2025;11:44151. [Google Scholar]
91. Tang Y, Cui G, Liu H, et al. Converting “cold” to “hot”: epigenetics strategies to improve immune therapy effect by regulating tumorassociated immune suppressive cells. Cancer Commun (Lond) 2024;44:60136. [Google Scholar]
92. Ouyang P, Wang L, Wu J, et al. Overcoming cold tumors: a combination strategy of immune checkpoint inhibitors. Front Immunol 2024;15:1344272. [Google Scholar]
93. Korkaya H, Kim GI, Davis A, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell 2012;47:57084. [Google Scholar]
94. Liu X, Ding C, Lu J, et al. cGASSTING: from immunology and oncology view. Chin Med J (Engl) 2025;138:305068. [Google Scholar]
95. Oresta B, Pozzi C, Braga D, et al. Mitochondrial metabolic reprogramming controls the induction of immunogenic cell death and efficacy of chemotherapy in bladder cancer. Sci Transl Med 2021; 13. [Google Scholar]
96. Amato I, Meurant S, Renard P. The key role of mitochondria in somatic stem cell differentiation: from mitochondrial asymmetric apportioning to cell fate. Int J Mol Sci 2023;24. [Google Scholar]
97. Heinz S, Freyberger A, Lawrenz B, et al. Mechanistic investigations of the mitochondrial complex I inhibitor rotenone in the context of pharmacological and safety evaluation. Sci Rep 2017;7:45465. [Google Scholar]
98. Joshi A, Kundu M. Mitophagy in hematopoietic stem cells: the case for exploration. Autophagy 2013;9:173749. [Google Scholar]
99. Lin Q, Chen J, Gu L, et al. New insights into mitophagy and stem cells. Stem Cell Res Ther 2021;12:452. [Google Scholar]
100. Morelli MB, Bongiovanni C, Da Pra S, et al. Cardiotoxicity of anticancer drugs: molecular mechanisms and strategies for cardioprotection. Front Cardiovasc Med 2022;9:847012. [Google Scholar]
101. Varga ZV, Ferdinandy P, Liaudet L, et al. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am J Physiol Heart Circ Physiol 2015;309:H145367. [Google Scholar]
102. Vuda M, Kamath A. Drug induced mitochondrial dysfunction: mechanisms and adverse clinical consequences. Mitochondrion 2016;31:6374. [Google Scholar]
103. Xu S, Wang Z, Guo F, et al. Mitophagy in ischemic heart disease: molecular mechanisms and clinical management. Cell Death Dis 2024;15:934. [Google Scholar]
104. Yin Y, Shen H. Advances in cardiotoxicity induced by altered mitochondrial dynamics and mitophagy. Front Cardiovasc Med 2021;8:739095. [Google Scholar]
105. Boudreau CE, Najem H, Ott M, et al. Intratumoral delivery of STING agonist results in clinical responses in canine glioblastoma. Clin Cancer Res 2021;27:552835. [Google Scholar]
106. Dou Y, Chen R, Liu S, et al. Optogenetic engineering of STING signaling allows remote immunomodulation to enhance cancer immunotherapy. Nat Commun 2023;14:5461. [Google Scholar]
107. Li K, Wang J, Zhang R, et al. Overcome the challenge for intratumoral injection of STING agonist for pancreatic cancer by systemic administration. J Hematol Oncol 2024;17:62. [Google Scholar]
108. Li L. Stimulating STING for cancer therapy: taking the extracellular route. Cell Chem Biol 2024;31:85161. [Google Scholar]
109. Pulliam T, Jani S, Goff PH, et al. Intratumoral STING agonist reverses immune evasion in PD-(L)1-refractory Merkel cell carcinoma: mechanistic insights from detailed biomarker analyses. J Immunother Cancer 2024; 12. [Google Scholar]
110. Wang J, Meng F, Yeo Y. Delivery of STING agonists for cancer immunotherapy. Curr Opin Biotechnol 2024;87:103105. [Google Scholar]
111. Li J, Hubisz MJ, Earlie EM, et al. Non-cell-autonomous cancer progression from chromosomal instability. Nature 2023;620:10808. [Google Scholar]
112. Fenton M, Yoneyama M, Wennerberg E, et al. The untapped potential of radiation and immunotherapy for hormone receptor-positive breast cancer. NPJ Breast Cancer 2025;11:77. [Google Scholar]
113. Guo Z, Liu T, Gao Q, et al. Targeting the cGAS-STING pathway for cancer immunotherapy: from small-molecule agonists to advanced nanomaterials. Mol Pharm 2025;22:726284. [Google Scholar]
114. Tang L, Wang D, Hu T, et al. Current applications of tumor local ablation (TLA) combined with immune checkpoint inhibitors in breast cancer treatment. Cancer Drug Resist 2024;7:33. [Google Scholar]
115. Andreeva OE, Sorokin DV, Vinokurova SV, et al. Breast cancer cell resistance to hormonal and targeted therapeutics is correlated with the inactivation of the NR6A1 axis. Cancer Drug Resist 2024;7:48. [Google Scholar]
116. Kuo CL, Lin YC, Lo YK, et al. The mitochondrial stress signaling tunes immunity from a view of systemic tumor microenvironment and ecosystem. iScience 2024;27:110710. [Google Scholar]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools