
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Xenosialylation as immunological chimerism: a host-centered unifying model for viral and post-vaccination immune complications
1 Independent Researcher, Rome, Italy
2 School of Bioscience and Veterinary Medicine, University of Camerino, Macerata, Italy
* Corresponding Author: Fiorella Carnevali. Email:
(This article belongs to the Special Issue: Inflammation and Cytokine Biology in Chronic Diseases: Mechanisms and Therapeutic Targets)
European Cytokine Network 2026, 37(2), 55-77. https://doi.org/10.32604/ecn.2026.082188
Received 12 March 2026; Accepted 29 May 2026; Issue published 30 June 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Severe immune-mediated complications following viral infections and vaccinations, including COVID-19 and anti–SARS-CoV-2 immunization, display remarkable clinical overlap despite occurring in distinct biological contexts. In a previous hypothesis-driven work, we proposed that metabolic incorporation of the non-human sialic acid N-glycolylneuraminic acid (Neu5Gc) into human glycoconjugates—defined as xenosialylation—may contribute to post-infectious and post-vaccination immune dysregulation. We further suggest that this phenomenon may represent a form of “immunological chimerism”, in which host glycoconjugates incorporate non-self molecular structures that predispose the immune system to varying degrees of immune imbalance. In its most severe manifestation, this process may culminate in a profound state of immune dysregulation characterized by loss of immune tolerance, aberrant antibody responses, cytokine storm, and thrombo-inflammatory pathology, which we define as “immunological marasmus”. In the present paper, we extend this conceptual framework by integrating glycobiology, Fc immunoglobulin glycosylation, endothelial biology, and sex-dependent immune regulation into a unified, testable immunopathogenic model. We hypothesize that interindividual differences in the extent, tissue distribution, and persistence of xenosialylation may influence susceptibility to exaggerated innate and adaptive immune responses following antigenic challenge. In this context, immune activation may unmask pre-existing xeno-sialylated self-structures embedded within host glycans, promoting varying degrees of glycan dysregulation, autoantibody production, immunothrombosis and chronic inflammatory sequelae. We further propose circulating anti-Neu5Gc antibodies as functional biomarkers for risk stratification and outline preventive strategies based on dietary modulation of xenosialic acid exposure. Taken together, this expanded model provides a potential mechanistic framework for understanding the shared immunological features of post-viral syndromes and vaccine-related adverse immune reactions, while offering a basis for experimental validation and future approaches to personalized risk mitigation.Graphic Abstract
Keywords
The occurrence of severe immune-mediated complications following viral infections and vaccinations raises a fundamental question in immunopathology: why do similar inflammatory and thrombo-immune syndromes arise from biologically distinct antigenic stimuli? We propose that the answer may reside primarily in host-centered mechanisms rather than in the nature of the triggering antigen alone. In this perspective, the immunological state of the host—particularly when shaped by metabolic incorporation of non-human glycans—may represent a permissive condition that determines susceptibility to different degrees of immune dysregulation following antigenic challenge.
Recently, Carnevali et al. [1] proposed that xenosialylation—the metabolic incorporation of the non-human sialic acid N-glycolylneuraminic acid (Neu5Gc) into human glycoconjugates—may represent a critical but underappreciated contributor to post-infectious and post-vaccination immune complications. In the present work, we extend this hypothesis by proposing that xenosialylation may generate a state that can be conceptualized as immunological chimerism, in which host glycoconjugates incorporate non-self-molecular signatures capable of dysregulating immune recognition mechanisms. Under conditions of intense immune activation, recognition of these xeno-modified host glycoconjugates may lead to progressive immune dysregulation characterized by loss of immune tolerance, aberrant antibody responses, cytokine storm, and thrombo-inflammatory pathology. In its most severe manifestation, this cascade may culminate in a profound state of immune dysregulation that we define as “immunological marasmus”.
In this manuscript, we integrate recent advances in glycobiology, Fc immunoglobulin glycosylation, endothelial immunopathology, and sex-specific immune regulation to assess whether this unified mechanistic framework can explain why only a subset of individuals develop severe immune-mediated complications after viral infection or vaccination.
The SARS-CoV-2 pandemic has generated a growing body of glycobiological and immunological evidence on post-infectious and post-vaccination anti-glycan immune responses [2,3], consistent with key elements of the xenosialylation framework. We review autoimmune and immune-mediated manifestations associated with viral infections and vaccine reactions, interpreting them as aberrant anti-glycan immune responses directed against variably distributed xeno-sialylated glycans.
In this context, identifying individuals at increased risk of post-infectious or post-vaccination immune complications becomes a priority. This risk may be assessed by the detection of specific biomarkers using both established and emerging laboratory techniques providing experimentally testable strategies to evaluate the proposed hypothesis and do not exclude the contribution of additional immunological, genetic, metabolic, or environmental factors.
1.1 Autoimmune Disorders Associated with Viral Infections
Immunological diseases are classified as autoinflammatory, autoimmune, or mixed-pattern disorders [4]. Autoinflammatory diseases are primarily driven by dysregulated innate immune activation, leading to tissue damage independently of adaptive immunity, whereas autoimmune diseases result from loss of immune tolerance and abnormal activation of dendritic cells, B cells, and T cells [5].
However, most immune-mediated disorders reflect overlapping contributions of autoinflammatory and autoimmune mechanisms, forming a continuum rather than distinct categories [4]. This conceptual framework is particularly relevant for immune complications triggered by viral infections. Viral pathogens have been implicated in the development of autoimmune diseases [6], including SARS-CoV-2 [7]. For example, human cytomegalovirus has been associated with immune thrombocytopenic purpura and systemic lupus erythematosus [8,9], Epstein–Barr virus has been linked to systemic lupus erythematosus and rheumatoid arthritis [10,11] and enteroviruses and paramyxoviruses with islet autoimmunity and type 1 diabetes [12].
Antibodies against common human coronaviruses have been associated with several autoimmune diseases [13,14]. Given these associations and the sequence similarity between SARS-CoV-2 and endemic coronaviruses, a link with post-COVID autoimmune sequelae is biologically plausible [15].
1.2 Clinical Aspects of COVID-19
The clinical course of acute infection shows marked interindividual variability, ranging from asymptomatic or mild disease to life-threatening conditions, partly explained by host-specific immune factors such as autoantibodies against type I interferons [16]. Age is the strongest risk factor for mortality, and severe disease is more frequent in men than in women [17,18]. Severe COVID-19 typically progresses from an initial viral phase to a systemic inflammatory phase leading to respiratory failure and multi-organ dysfunction [19].
Although the precise mechanisms underlying Acute Respiratory Distress Syndrome (ARDS) and severe COVID-19 remain incompletely understood, advanced age is the strongest predictor of mortality, reaching 15–20% in individuals older than 80 years compared with <1% in those younger than 50 years [20,21]. In contrast, SARS-CoV-2 infection is generally mild or asymptomatic in children [22]; however, rare but severe post-infectious inflammatory syndromes have been described, including Pediatric Inflammatory Multisystem Syndrome temporally associated with COVID-19 (PIMS-TS) and Multisystem Inflammatory Syndrome in Children (MIS-C), typically occurring 4–6 weeks after infection [23–25]. The prevailing pathogenic model implicates virus-induced production of self-reactive antibodies, particularly IgA-producing plasma cells localized at mucosal sites and within arterial walls [26], a mechanism that has been subsequently integrated into contemporary clinical models of MIS-C pathogenesis [25]. Similar, though much rarer, multisystem inflammatory syndromes have also been reported in adults (MIS-A) [27,28].
Several pre-existing conditions, including diabetes, chronic lung and cardiovascular disease, obesity, and other chronic inflammatory disorders, significantly increase the risk of severe COVID-19 [29]. Although no specific food or dietary intervention has been proven to prevent or mitigate SARS-CoV-2 infection and outcomes, it is plausible that certain dietary components may exacerbate immune dysregulation during infection or following vaccination [30].
Consistent with previous coronavirus outbreaks, diabetes has emerged as a major independent predictor of COVID-19 mortality [31]. In contrast, evidence on asthma remains inconsistent: despite being a risk factor for respiratory infections, several studies reported no increased hospitalization risk in COVID-19 [32].
Cardiovascular complications are frequent in COVID-19 and include arrhythmias, myocarditis, acute myocardial injury, thrombosis, and cardiomyopathy, likely resulting from systemic inflammation and direct viral injury [33]. SARS-CoV-2 infection has also been associated with reactivation of latent viral infections, particularly varicella-zoster virus, likely facilitated by COVID-19–associated lymphopenia and T-cell dysfunction [34,35]. Co-infection or reactivation of influenza A virus has been reported [36], and severe influenza infections were already known, prior to the COVID-19 pandemic, to induce ARDS similar to that observed in COVID-19 [37].
1.3 Sars-CoV-2, Infection Mechanism
SARS.CoV-2 shares the angiotensin-converting enz yme 2 (ACE2) receptor with SARS-CoV [38]. Consequently, ACE2 tissue distribution may correlate with organ-specific disease severity [39,40]. Viral entry is mediated by ACE2 and the host serine protease TMPRSS2, which cleaves and activates the spike protein [41,42]. The presence of a polybasic furin cleavage site enables additional spike activation by ubiquitous furin proteases, contributing to broader cellular tropism, including vascular endothelial cells [43,44]. This expanded tropism likely underlies the increased infectivity and systemic manifestations of SARS-CoV-2 compared with SARS-CoV [45] other host factors contribute to viral adhesion and entry and particular, Neuropilin-1 has been identified as a key co-receptor that enhances SARS-CoV-2 infectivity [46,47]. Host proteases, including TMPRSS2 and furin, are essential for efficient viral membrane fusion in airway epithelial cells [48].
Beyond epithelial infection, SARS-CoV-2 directly targets the vascular endothelium, inducing endothelial dysfunction, endothelitis and widespread microvascular injury, which are central features of severe COVID-19 [49,50]. At the systemic level, severe disease is characterized by a dysregulated immune response, with an early imbalance between antiviral interferon signaling and excessive inflammatory cytokine production [51], followed by sustained immune misfiring and pathological immune activation over time [52], similarly to immune and influenza virus infection [37].
Structurally, the SARS-CoV-2 spike protein is extensively glycosylated, forming a complex glycan shield that modulates receptor binding, immune recognition, and viral pathogenicity [53,54]. Together, these findings support a model in which SARS-CoV-2 pathogenesis results from the integration of multi-receptor viral entry, protease-dependent spike activation, endothelial involvement, immune dysregulation, and glycan-mediated modulation of host–virus interactions.
1.4 Clinical Complications in COVID-19
Clinical complications, predominantly immune-mediated, are increasingly recognized in COVID-19 patients and involve multiple organ systems in both adults and children [15]. These manifestations have been linked to immune dysregulation pathways such as molecular mimicry, epitope spreading, and bystander activation, consistent with mechanisms classically implicated in post-infectious autoimmunity [15]. Recent clinical–immunological studies have identified distinct immuno-inflammatory profiles associated with disease severity, supporting the concept that exaggerated and dysregulated host immune responses—rather than direct viral cytopathology—are key determinants of adverse clinical outcomes and long-term sequelae in COVID-19 [55].
Many COVID-19 manifestations, particularly those associated with “long COVID”, resemble autoimmune disorders, suggesting that dysregulated autoantibody responses may contribute to disease pathogenesis [56,57]. Notably, antibodies directed against glycan epitopes have emerged as key mediators of immune-mediated pathology, as alterations in glycan structures or their immunological presentation can convert normally tolerated self-glycans into targets of pathogenic antibody responses [2,57].
Severe COVID-19 outcomes are frequently observed in patients with pre-existing autoimmune or chronic inflammatory conditions following SARS-CoV-2 exposure, supporting a link between viral infection and immune dysregulation [57,58]. Reported manifestations include antiphospholipid syndrome, systemic vasculitis, systemic autoimmune diseases, and multi-organ immune-mediated complications [58].
Neurological disorders such as Guillain–Barré syndrome (GBS) have been widely described in adults [59], while Kawasaki disease and multisystem inflammatory syndromes (MIS-C and MIS-A) have been reported in both children and adults [27,60]. Similar autoimmune sequelae have been documented following other viral infections, including influenza A virus [61].
COVID-19 has been associated with autoimmune-like hyperinflammatory vascular and neurological complications, including immune cytopenia, antiphospholipid syndrome–related thrombosis, arthritis, and connective tissue–like manifestations, supporting a role for immune dysregulation in severe disease [62]. Anti-phospholipid and anti-cardiolipin antibodies are frequently detected in severe COVID-19, mirroring findings in non–COVID-19 ARDS and autoimmune coagulopathies [63,64]. Immune-mediated endothelial damage promotes hypercoagulability, thrombosis, and endotheliitis, leading to vascular and neurological complications such as stroke and cerebral venous thrombosis [65,66]. Beyond classical protein autoantigens, immune recognition of glycan determinants has been implicated in vascular inflammation and thromboinflammatory processes, highlighting a potential pathogenic role for anti-glycan antibodies in endothelial dysfunction [2].
Neurological symptoms are common in COVID-19 and include anosmia, encephalopathy, and stroke [67]. These manifestations are unlikely to result from direct central nervous system infection, as SARS-CoV-2 is rarely detected in cerebrospinal fluid [66]. Autoimmune peripheral neuropathies, particularly Guillain–Barré syndrome (GBS), have been repeatedly associated with COVID-19 [68]. GBS is a post-infectious autoimmune polyneuropathy classically attributed to molecular mimicry between microbial antigens and gangliosides [69]. Notably, most COVID-19–associated GBS cases lack classical anti-ganglioside antibodies, but display elevated antibodies against multiple glycolipids, supporting a broader dysregulated autoimmune response rather than direct viral neurotropism [70].
Autoimmune endocrine conditions, particularly autoimmune thyroid diseases, have been reported following COVID-19 [71]. COVID-19 patients frequently exhibit a broad spectrum of autoantibodies, including ANA, antiphospholipid antibodies, and antibodies against type I interferons, supporting the concept of COVID-19–induced autoimmunity and antiphospholipid syndrome (APS)-related thrombotic events [63,72,73].
Cardiovascular complications such as myocarditis have been described after SARS-CoV-2 infection, particularly in young males [74,75]. Viral myocarditis is mediated by both direct myocardial injury and immune-driven inflammation, with interleukin-6 playing a central pathogenic role [76,77]. Persistent cardiac involvement has been documented months after infection, suggesting that COVID-19–related myocarditis may be underrecognized [78,79]. Finally, SARS-CoV-2 infection may promote reactivation of latent viruses, particularly Epstein–Barr virus, contributing to immune dysregulation and autoimmune manifestations in post-acute COVID-19 and long COVID [80].
1.5 Correlation between Post-Covid Complications and Autoimmune Diseases
Patients with severe COVID-19 frequently exhibit elevated levels of autoantibodies, including antibodies classically associated with autoimmune diseases and directed against multiple self-antigens [56], including cytokines (e.g., IFN-γ, GM-CSF, IL-6, IL-10) and neural proteins [16,81]. Notably, these autoantibodies were detected almost exclusively in severe disease and were largely absent in mild or asymptomatic cases, supporting a link between COVID-19 severity and immune dysregulation [15,56].
Additional contributors to sex-based immune differences include X-chromosome mosaicism in females and sex-specific immune regulatory pathways, which shape immune response profiles and influence susceptibility to autoimmune manifestations [82–84].
A similar female predominance has been reported in COVID-19–related autoimmune complications and long COVID, whereas no significant sex differences are observed in pediatric populations [85,86]. Overall, SARS-CoV-2 induces autoimmune pathological mechanisms comparable to those described for other viral infections [6,87]. However, COVID-19 appears to trigger these processes more frequently and with greater intensity, particularly in female patients [62,72,88,89].
1.6 Specific Post Viral Autoimmune Disorders: Coagulopathies
A hallmark of SARS-CoV-2 pathogenesis is COVID-19–associated coagulopathy, characterized by a marked prothrombotic state with high rates of thrombosis and microvascular complications [90]. In severe disease, this condition may progress to disseminated intravascular coagulation, largely driven by endothelial dysfunction and immunothrombosis [91,92].
The concept of immunothrombosis describes an innate immune defense mechanism that limits pathogen dissemination but, when dysregulated, promotes excessive coagulation and thrombo-inflammation [93,94].
COVID-19–associated coagulopathy shares key features with disseminated intravascular coagulation, notably endotheliopathy and intense inflammation, although these processes appear more pronounced in COVID-19 than in other infectious conditions [94]. Endothelial damage is central to COVID-19 pathology, but the precise mechanisms underlying thrombocytopathy and endotheliopathy remain incompletely defined [95,96]. Similar coagulation abnormalities have been reported in other viral infections, including influenza [97].
Venous thromboembolism and arterial thrombosis significantly contribute to COVID-19–related morbidity and mortality, particularly in critically ill patients [91]. Immune-mediated manifestations, including immune thrombocytopenia, further exacerbate thromboinflammatory processes and contribute to adverse outcomes in both adult and pediatric patients [58,92].
It is possible that elevated inflammatory and vascular biomarkers—including D-dimer and pro-inflammatory cytokines—are consistently associated with severe disease, supporting a mechanistic link between immune activation and endothelial dysfunction [98,99]. Patients in intensive care settings show markedly increased endothelial and platelet activation, indicating a central role for immunothrombosis in severe COVID-19 [90,94]. SARS-CoV-2 infection is strongly associated with thromboembolic events despite anticoagulation [100].
Endothelial activation following viral entry through ACE2 induces a procoagulant state with elevated circulating coagulation biomarkers and von Willebrand factor, reflecting endothelial dysfunction and correlating with disease severity [90].
Platelet hyperactivation and the formation of platelet–antibody complexes contribute to sustained thromboinflammation, consistent with mechanisms previously described in sepsis and now implicated in COVID-19 [101,102]. In addition, autoantibodies against annexin A2 have been detected in severe COVID-19 and may interfere with fibrinolysis, further promoting a prothrombotic state [103]. Finally, susceptibility to severe COVID-19 may be influenced by host autoantibodies, particularly those directed against type I interferons, predominantly observed in elderly male patients [16].
1.7 Main Post-Viral Complication: Long COVID
Long COVID affects a substantial proportion of individuals recovering from SARS-CoV-2 infection and is characterized by persistent symptoms such as fatigue, dyspnea, cognitive impairment, and cardiovascular and neurological disturbances [104,105]. Current integrative models indicate that long COVID results from the convergence of immune dysregulation, chronic inflammation, vascular dysfunction, and tissue-specific injury, operating upstream of heterogeneous clinical manifestations [106].
Women appear to be at higher risk of developing long COVID, likely reflecting sex-dependent immune differences [85], whereas no clear sex bias is observed in pediatric populations [86,107]. Similar post-viral syndromes have been described after other infections, supporting a shared biological basis for prolonged post-infectious illness [108]. Among proposed mechanisms, autoimmunity is currently considered central to long COVID pathogenesis, with evidence of persistent immune hyperactivation, sustained B-cell activation, and autoantibody production targeting multiple tissues [55,56,109,110]. These findings support an autoimmune contribution to long-term post-COVID immune sequelae.
2 Adverse Reactions after Vaccination
COVID-19 vaccines can be broadly classified into four main platforms: whole-virus vaccines, nucleic acid vaccines (mRNA and DNA), viral vector vaccines, and protein-based vaccines, which differ in their mechanisms of antigen delivery and immune activation [111].
In the mid-2021, multiple COVID-19 vaccines had been approved for global use [112] and, while early trials demonstrated favorable efficacy and safety profiles, increasing attention has been directed toward potential immediate, intermediate, and long-term adverse effects. Large-scale post-marketing surveillance and pharmacovigilance studies have confirmed that COVID-19 vaccines retain a favorable benefit–risk profile, while allowing the identification of rare adverse events of special interest, highlighting the importance of continuous safety monitoring [113]. The most frequently reported adverse reactions are mild and self-limiting, including local injection-site reactions and systemic symptoms such as headache, fever, fatigue, and myalgia [114,115]. These reactions typically occur within a few days after vaccination and are not associated with severe illness, resembling mild, non-respiratory symptoms of SARS-CoV-2 infection [116].
For the Comirnaty (BNT162b2) mRNA vaccine, the most common adverse reactions are mild to moderate and include injection-site pain and swelling, fatigue, headache, myalgia, chills, and fever, with systemic reactions more frequent after the second dose [117].
Rare but more severe post-vaccination reactions have been reported and may overlap with autoimmune-like, neurological, and vascular syndromes similar to those observed after SARS-CoV-2 infection [118–120]. Reactivation of latent viruses, particularly herpesviruses, has also been described, especially in individuals with pre-existing autoimmune conditions [121].
Sex- and age-related differences are well documented, with males showing higher rates of myocarditis and pericarditis, and females generally developing stronger immune responses and reporting more adverse reactions, including autoimmune manifestations [122]. These demographic patterns have been consistently confirmed in large global pharmacovigilance studies [113].
2.1 Cardiovascular Disorders Induced by Vaccination
Myocarditis is a rare adverse event following COVID-19 vaccination, with an estimated incidence of approximately 2 cases per 100,000 vaccinated individuals and the highest risk observed in males aged 16–29 years [123]. Population-based studies consistently show that myocarditis and other cardiovascular complications occur far more frequently after SARS-CoV-2 infection than after mRNA vaccination across age and sex strata [124,125]. However, epidemiological analyses indicate that myocarditis is observed more frequently after mRNA vaccines than after adenoviral vector vaccines [125].
Proposed pathogenic mechanisms include sex hormone influences, immune hyperreactivity to mRNA vaccines, and molecular mimicry between spike protein–induced antibodies and myocardial antigens [125].
2.2 Multisystem Inflammatory Syndrome Induced by Vaccination
Multisystem inflammatory syndromes resembling Kawasaki disease and MIS-C/MIS-A had been documented prior to the COVID-19 pandemic in temporal association with several viral vaccines, suggesting that these conditions reflect host-dependent hyperinflammatory immune responses rather than vaccine-specific entities [126]. MIS, originally described following SARS-CoV-2 infection, has been only rarely reported after COVID-19 vaccination. Standardized diagnostic criteria for MIS-C and MIS-A have been established by the Brighton Collaboration to support vaccine safety assessment [60]. To date, only isolated post-vaccination cases have been described, beginning with a well-documented MIS-A case following mRNA vaccination [127], and subsequent reviews have confirmed the extreme rarity of this phenomenon [128]. Large-scale pharmacovigilance studies have not identified an increased risk of MIS after vaccination, and most reported cases show evidence of prior SARS-CoV-2 infection rather than vaccination as the primary trigger [129].
Similarly, analyses of MIS cases reported after COVID-19 vaccination indicate that most affected individuals had evidence of prior SARS-CoV-2 infection, supporting infection-related immune priming rather than a direct vaccine effect [130]. Regulatory and pharmacovigilance assessments have consistently classified MIS as an extremely rare event following vaccination, with no evidence of vaccine platform–specific risk [113,131]. Collectively, these findings suggest that MIS temporally associated with vaccination is more plausibly explained by host-dependent immune dysregulation in predisposed individuals than by a direct pathogenic effect of the vaccine itself [129].
2.3 Vascular Disorders Induced by Vaccination
Thrombotic thrombocytopenia following vaccination is not a novel phenomenon in vaccinology, with early cases reported after influenza vaccination [132] and subsequently described following other vaccines, including H1N1 influenza, pneumococcal vaccines [133,134]. In 2021, atypical thrombotic events associated with thrombocytopenia were reported after administration of adenoviral vector–based COVID-19 vaccines, particularly ChAdOx1 nCoV-19 and Ad26.COV2.S, a condition later defined as vaccine-induced immune thrombotic thrombocytopenia (VITT) [135,136] also known as Thrombosis with Thrombocytopenia Syndrome (TTS) [137]. These safety signals led several European countries to restrict or discontinue the use of the ChAdOx1 vaccine [138].
Standardized diagnostic criteria for VITT have been established within the Brighton Collaboration framework, providing harmonized case definitions for surveillance and vaccine safety assessment [139]. The clinical features and pathogenic mechanisms of VITT have been comprehensively reviewed in recent literature [140]. Early reports documented high mortality rates, particularly in cases presenting with cerebral venous sinus thrombosis (CVST) [135,141]. Thrombotic events predominantly involve cerebral venous thrombosis but may also include arterial thrombosis and venous thromboembolism at unusual sites [135,136]. Clinically, VITT is characterized by severe thrombocytopenia and aggressive thrombosis, in some cases progressing to disseminated intravascular coagulation, closely resembling autoimmune heparin-induced thrombocytopenia (HIT) [141]. However, most affected patients had no prior exposure to heparin [135].
HIT is mediated by IgG autoantibodies against platelet factor 4 (PF4) complexes, leading to platelet activation and thrombin generation. Neutrophil extracellular traps (NETs) play a key amplifying role in this process, promoting immunothrombosis [142]. These observations support the hypothesis that VITT reflects an aberrant immune response involving anti-PF4 antibodies rather than direct vaccine toxicity [135]. A unifying two-step model proposes that initial PF4–vaccine interactions generate neoantigens, followed by anti-PF4–mediated platelet and granulocyte activation, NET release, and immune thrombosis 5–20 days after vaccination [135]. Current guidelines recommend that individuals who develop major thrombosis with thrombocytopenia after adenoviral vector COVID-19 vaccination should not receive a second dose of the same platform [143].
Several mechanisms have been proposed to explain VITT pathogenesis, all converging on aberrant immune activation against platelet factor 4 [PF4]. Vaccine-induced expression of spike protein may promote platelet activation and PF4 release, triggering anti-PF4 antibody production [144]. Alternatively, PF4 may form immunogenic complexes with negatively charged vaccine components, such as adenoviral proteins or nucleic acids, leading to activation of antigen-presenting cells and memory B cells [145].
Recent structural and immunological evidence suggests that adenoviral vector proteins may play a direct role in triggering anti-PF4 antibody formation in VITT through the formation of immunogenic complexes between platelet factor-4 and adenoviral components, followed by affinity-matured B-cell responses, particularly in genetically predisposed individuals carrying specific somatic mutations affecting the anti-PF4 B-cell repertoire [146]. This mechanism likely explains the markedly higher incidence of VITT observed after adenoviral vector vaccination compared with other vaccine platforms. Interestingly, the anatomical distribution of thrombosis in VITT is not random. Cerebral venous sinuses and splanchnic veins represent the most frequently affected vascular territories, suggesting that local vascular or endothelial factors, particularly the endothelial glycocalyx, may contribute to disease expression. The glycocalyx contains heparan-sulfate proteoglycans and sialic-acid–containing glycoconjugates that participate in endothelial mechano-transduction and vascular homeostasis [147,148].
Endothelial glycocalyx alterations have been documented in cerebrovascular disease and inflammatory vascular conditions, indicating that this structure plays a critical role in maintaining vascular integrity in the central nervous system [149,150]. Given the high density of glycosylated and sialylated structures on the endothelial surface, particularly in low-shear venous districts, modifications of the glycocalyx composition could influence local thrombo-inflammatory responses.
This anatomical predilection is reflected in early surveillance reports describing cases of cerebral venous sinus thrombosis with thrombocytopenia following Ad26.COV2.S vaccination, predominantly in young women [151].
VITT is now recognized as a distinct immune-mediated prothrombotic syndrome characterized by thrombocytopenia, anti-PF4 antibodies, and atypical thrombosis, including CVST [135]. Regulatory agencies have concluded that, although rare, this association is biologically plausible [152,153]. Population-level analyses indicate that the apparent female predominance is likely influenced by early reporting bias and vaccination strategies rather than true sex-specific susceptibility [154].
Venous thromboembolism occurs in the general population at an annual incidence of approximately 1 per 1000 adults [155]. Following administration of ChAdOx1 nCoV-19, thrombotic events have been reported at a low incidence, including rare cases of disseminated intravascular coagulation [153]. The predilection of VITT for cerebral and splanchnic vessels remains unexplained, but the syndrome closely resembles immune thrombocytopenia and autoimmune heparin-induced thrombocytopenia, where paradoxical coexistence of thrombosis and thrombocytopenia reflects systemic platelet activation and consumption [156,157]. Immune thrombocytopenia is a recognized autoimmune disorder occasionally triggered by vaccination, including influenza and herpes zoster vaccines [158].
2.4 Neurological Disorders Associated with Vaccination
Neurological complications reported after vaccination include headache, syncope, seizures, encephalomyelitis, transverse myelitis, meningitis, and polyneuritis, with post-vaccination encephalomyelitis representing a rare but potentially severe condition [159,160]. The most consistently documented neurological syndrome associated with vaccination is Guillain–Barré syndrome (GBS), classically observed after influenza vaccination campaigns, particularly in 1976 and during the 2009 H1N1 pandemic [161,162].
Influenza virus infection and vaccination can induce anti-ganglioside antibodies, including anti-GM1, likely through molecular mimicry involving viral surface glycoproteins such as hemagglutinin [HA], which binds sialic acid receptors, and neuraminidase (NA), which removes sialic acid residues to facilitate viral release [163,164]. It has been hypothesized that incomplete desialylation of HA may generate ganglioside-like glycan structures capable of eliciting cross-reactive antibodies, a mechanism proposed to underlie post-influenza Guillain–Barré syndrome, particularly in vaccine formulations with low NA content [163].
Despite the recurrent observation of Guillain–Barré syndrome following vaccination—most notably after influenza vaccines—the establishment of a definitive causal relationship has been historically cautious, owing to epidemiological limitations and confounding factors in surveillance studies [161,165,166]. Notwithstanding large cohort studies consistently showing that the risk of Guillain–Barré syndrome and other neurological complications is substantially higher following SARS-CoV-2 infection than after vaccination, population-level safety monitoring has documented a re-emergence of GBS after COVID-19 vaccination, both with adenoviral vector platforms [115,125] and following mRNA-based vaccines [167,168].
2.5 Vaccine-Induced Viral Reactivation
Herpes zoster (HZ) reactivation has been reported in temporal association with COVID-19 vaccination, particularly in immunologically vulnerable individuals and patients with autoimmune or inflammatory diseases. Recent systematic reviews and meta-analyses indicate that reactivation of latent herpesviruses—especially varicella-zoster virus—can occur after vaccination, although the absolute risk is low and appears to be primarily modulated by host immune status rather than vaccine-specific effects [169,170]. Observational cohort data further support this interpretation, showing rare HZ cases predominantly among patients with pre-existing autoimmune conditions and none in healthy controls [121].
Cell-mediated immunity is critical for preventing varicella-zoster virus (VZV) reactivation, and its decline with aging or immune-mediated disease is associated with reduced VZV-specific T-cell surveillance. Case series have documented herpes zoster reactivation shortly after COVID-19 vaccination, particularly in immunocompromised patients with autoimmune inflammatory diseases [121]. However, pooled epidemiological analyses indicate that the risk of herpes zoster reactivation is substantially higher following SARS-CoV-2 infection than after vaccination, supporting the notion that viral infection represents a stronger trigger for latent virus reactivation than vaccination itself [171].
Reactivation of other latent herpesviruses, including Epstein–Barr virus (EBV) and cytomegalovirus (CMV), has also been documented in COVID-19 and has been implicated in persistent immune activation and inflammatory complications, potentially contributing to long COVID [106,172,173]. Collectively, these findings suggest that viral reactivation reflects a host-dependent failure of immune surveillance unmasked by immunological stress, rather than a direct vaccine-specific effect.
3 Infection versus Vaccination: Similar Events, Different Incidence—What Is the Link?
As outlined above, the clinical entities observed after viral infection and vaccination largely overlap, as both are driven by host immune responses to antigenic stimulation. Viral infections such as influenza are well known to increase the risk of thrombotic events, including myocardial infarction and stroke [174], supporting the concept that immune-triggered thromboinflammation represents a general pathological mechanism rather than a pathogen-specific phenomenon.
SARS-CoV-2 infection and COVID-19 vaccination are associated with the same spectrum of thrombotic complications indicating a shared underlying pathophysiology [175,176]. The absolute incidence is substantially higher after infection than vaccination, but both exposures converge on similar immune-mediated thrombotic pathways, differing primarily in magnitude rather than in nature [175] (see also references cited in Section 2.3-Vascular disorders induced by vaccination). The substantial overlap between the most frequent COVID-19 complications and post-vaccination adverse reactions—supports the concept that these events arise primarily from host immune responses to antigenic stimulation rather than from direct viral cytopathology or ongoing viral replication. This is particularly evident in the context of vaccination, where viral replication is absent, and in post-acute COVID-19, which is characterized by persistent immune dysregulation [56,66]. Thrombotic manifestations such as cerebral venous thrombosis occur in both contexts, with higher incidence following infection but qualitatively similar clinical and immunopathological features [175].
COVID-19 is frequently associated with venous thromboembolic complications, including pulmonary embolism, deep vein thrombosis, and, more rarely, cerebral venous thrombosis (CVT), reflecting a systemic thromboinflammatory state. Although CVT has been reported in rare cases following COVID-19 vaccination—particularly with adenoviral vector platforms—these events are characterized by immune-mediated mechanisms involving platelet-reactive antibodies, with higher incidence following SARS-CoV-2 infection than vaccination [175,177].
Multisystem inflammatory syndrome (MIS) is a rare but severe hyperinflammatory condition that can lead to multi-organ dysfunction and has been reported both after viral infections and following vaccination, in children and adults. In the context of SARS-CoV-2, MIS typically occurs weeks after infection and within a few weeks after vaccination, frequently in individuals with prior COVID-19, suggesting immune priming as a key pathogenic factor [128,178]. Nevertheless, MIS has also been described in vaccine recipients without documented previous infection [179,180].
Neurological complications such as GBS have been reported following both COVID-19 infection and vaccination, as previously observed with influenza infection and vaccination [125,162,167].
A similar pattern has been observed for myocarditis and pericarditis following mRNA vaccination, particularly with BNT162b2, confirming that these immune-mediated reactions may occur even in the absence of viral circulation or direct tissue damage with myocarditis occurring at substantially higher rates than myocarditis following mRNA vaccination [125,181].
Growing evidence further indicates that both SARS-CoV-2 infection and vaccination may be associated with long-term symptoms, including cognitive impairment and thrombotic complications, described within the spectrum of post-acute sequelae of SARS-CoV-2 infection (Long COVID) as well as in persistent post-vaccination syndromes [159,182].
Altogether, these parallels, despite differences in incidence, support the existence of a common yet unidentified upstream mechanism underlying neurological, vascular, and immune-mediated complications following both the disease and vaccination (see Section 4).
A Host-Dependent Framework Underlying Immune Complications Following Infection and Vaccination
Infectious agents can represent a trigger of autoimmune diseases, and post-infectious autoimmunity is a well-established clinical entity. Infections can break immune tolerance through mechanisms such as molecular mimicry and immune cross-reactivity [183,184].
Severe immune-mediated complications of COVID-19 are strongly associated with pre-existing comorbidities, including diabetes, cardiovascular disease, and autoimmune or chronic inflammatory conditions. Reported manifestations include antiphospholipid syndrome, systemic vasculitis, Guillain–Barré syndrome, and a wide range of neurological, hematological, and endocrine disorders [58,63,70,185]. Similar host-dependent immune mechanisms have been proposed to underlie multisystem inflammatory syndromes and long-term effects of SARS-CoV-2 infection, where affected individuals frequently exhibit multiple autoantibodies and overlapping autoimmune phenotypes [185,186].
Despite this substantial body of evidence, a central question remains unresolved: why do only a minority of infected or vaccinated individuals develop autoimmune or severe immune-mediated complications? If molecular mimicry alone were sufficient to induce autoimmunity, the prevalence of autoimmune diseases would be far higher than approximately 5% observed in the general population [184]. This discrepancy indicates that additional host-dependent permissive factors critically modulate individual susceptibility to immune dysregulation. This question becomes particularly relevant in the context of COVID-19 and long COVID, where clinical manifestations are highly heterogeneous.
Several hypotheses have been proposed to explain interindividual variability in disease severity. Sardu et al. 2020 [95] suggested that age- and sex-related differences in the human sialome may influence viral interactions and immune responses. Similarly, the interplay between SARS-CoV-2–induced immune dysregulation and age-associated biological changes has been emphasized [45].
Striking similarities have been observed between ARDS induced by SARS-CoV-2 and influenza A virus, as well as with macrophage activation syndrome in autoimmune diseases, where shared antiviral and inflammatory pathways drive excessive immune activation and tissue damage [187]. However, while these models explain downstream inflammatory cascades, they do not clarify why only one subset of individuals develops severe immune-mediated complications. Importantly, they also fail to account for the occurrence of comparable or even more severe syndromes in children and young adults—such as MIS-C, Kawasaki disease, and long COVID—who lack age-related degenerative changes [27,188].
Overall, long COVID appears to result from the convergence of multiple, non-mutually exclusive mechanisms, including immune dysregulation, autoimmune responses, vascular and metabolic disturbances, and possibly viral persistence [189].
Similarly, although rare, severe adverse events associated with vaccination have been documented, with clinical manifestations including vascular, neurological, endocrine, and other organ-specific sequelae. The occurrence of immune-mediated complications even in the absence of active infection highlights an important limitation of explanations based exclusively on the pathogen—such as the molecular mimicry hypothesis—which fail to adequately account for the emergence of comparable clinical conditions following vaccination, where active viral replication and direct cytopathic damage are absent. Indeed, similar autoimmune and inflammatory syndromes have been reported after vaccination against SARS-CoV-2, influenza A, and other viruses, suggesting that such immune complications cannot be attributed solely to pathogen-induced tissue injury.
Importantly, excessive immune activation—characterized by early innate immune responses, cytokine release, and high antibody titers—has been observed in both severe COVID-19 and post-vaccination settings, suggesting the existence of a shared pathogenic pathway.
Further complicating interpretation, identical clinical entities are often classified differently depending on whether they arise after infection or vaccination. For example, Guillain–Barré syndrome, multisystem inflammatory syndromes, and thrombotic disorders are assigned distinct nomenclature and presumed mechanisms based on exposure context, despite substantial overlap in clinical and immunological features [175]. This fragmentation has contributed to conceptual confusion and hindered recognition of shared underlying mechanisms.
Collectively, evidence accumulated over the pandemic and the global vaccination campaign indicates that immune-mediated complications can be triggered by both viral infections and vaccination, even in individuals without prior exposure to the pathogen. These observations support the interpretation that post-infectious and post-vaccination immune-mediated manifestations are qualitatively similar and differ primarily in incidence, magnitude, timing, and host susceptibility, rather than in their fundamental immunopathological mechanisms.
Taken together, these observations indicate that pathogen-centered or molecular mimicry–based models alone are insufficient to explain the full spectrum and recurrence of immune-mediated complications. A unifying host-centered mechanism is therefore required to account for the marked heterogeneity observed across individuals and clinical contexts. We propose that such a mechanism operates at the level of individual susceptibility rather than pathogen specificity, thereby linking post-infectious and post-vaccination immune-mediated manifestations within a common biological framework. In the following section, we present our causal hypothesis based on xenosialylation as a form of immunological chimerism, which may culminate in a state of profound and widespread immune dysregulation that we define as immunological marasmus.
4 Sialic Acid: Sialylation vs. Xenosialylation
Sialylation is the enzymatic process by which sialic acid residues are added to the terminal positions of glycan chains on glycoproteins and glycolipids, modulating the structural and immunological properties of cell surfaces and secreted molecules. This modification is a defining feature of vertebrate glycoconjugates and plays a central role in shaping the glycocalyx [190–192].
Sialic acids constitute a family of nine-carbon acidic sugars located at the outermost positions of cell-surface glycans, where they generate a negatively charged interface that contributes to cell hydration, molecular stability, intercellular interactions, and immune recognition [193–195]. They are highly abundant in mucus, endothelial glycocalyx, and specialized neural structures such as gangliosides and myelin, resulting in a marked enrichment of total body sialic acid within the nervous system [196–198]. This heterogeneous tissue distribution suggests that perturbations of sialylation may produce organ-specific immunological consequences.
Beyond their structural roles, glycans are key mediators of molecular recognition in innate and adaptive immunity, acting as regulators of immune tolerance and activation [199,200]. Sialic acid–containing glycans function as self-associated molecular patterns (SAMPs), delivering inhibitory signals through receptors such as Siglecs and complement regulators, thereby actively suppressing excessive immune activation [199,201]. The human sialome undergoes age-dependent modifications, and progressive desialylation of cell surfaces and circulating glycoproteins encodes aspects of biological aging and chronic inflammation [202,203]. A paradigmatic example is the desialylation of senescent erythrocytes, which triggers their clearance via hepatic asialoglycoprotein receptors [204].
Sialylation of the IgG Fc glycan exerts a potent anti-inflammatory effect, whereas loss of terminal sialic acids markedly increases antibody-mediated immune activation and inflammatory signaling [200,205]. Sialic acid–mediated recognition is also exploited by numerous viruses, including influenza viruses and coronaviruses, which use sialylated host glycans for cell entry and release [53,54]. Because viral envelopes originate from host cell membranes, they incorporate host-derived glycoconjugates, including sialylated glycans that may influence immune recognition and antibody responses [206]. As a countermeasure, sialylated mucins form dense barriers on mucosal surfaces, acting as decoys that limit pathogen access to epithelial cells [195,207].
Together, these observations place sialylation at the core of immune homeostasis and highlight that qualitative alterations in sialic acid composition have profound implications for immune regulation.
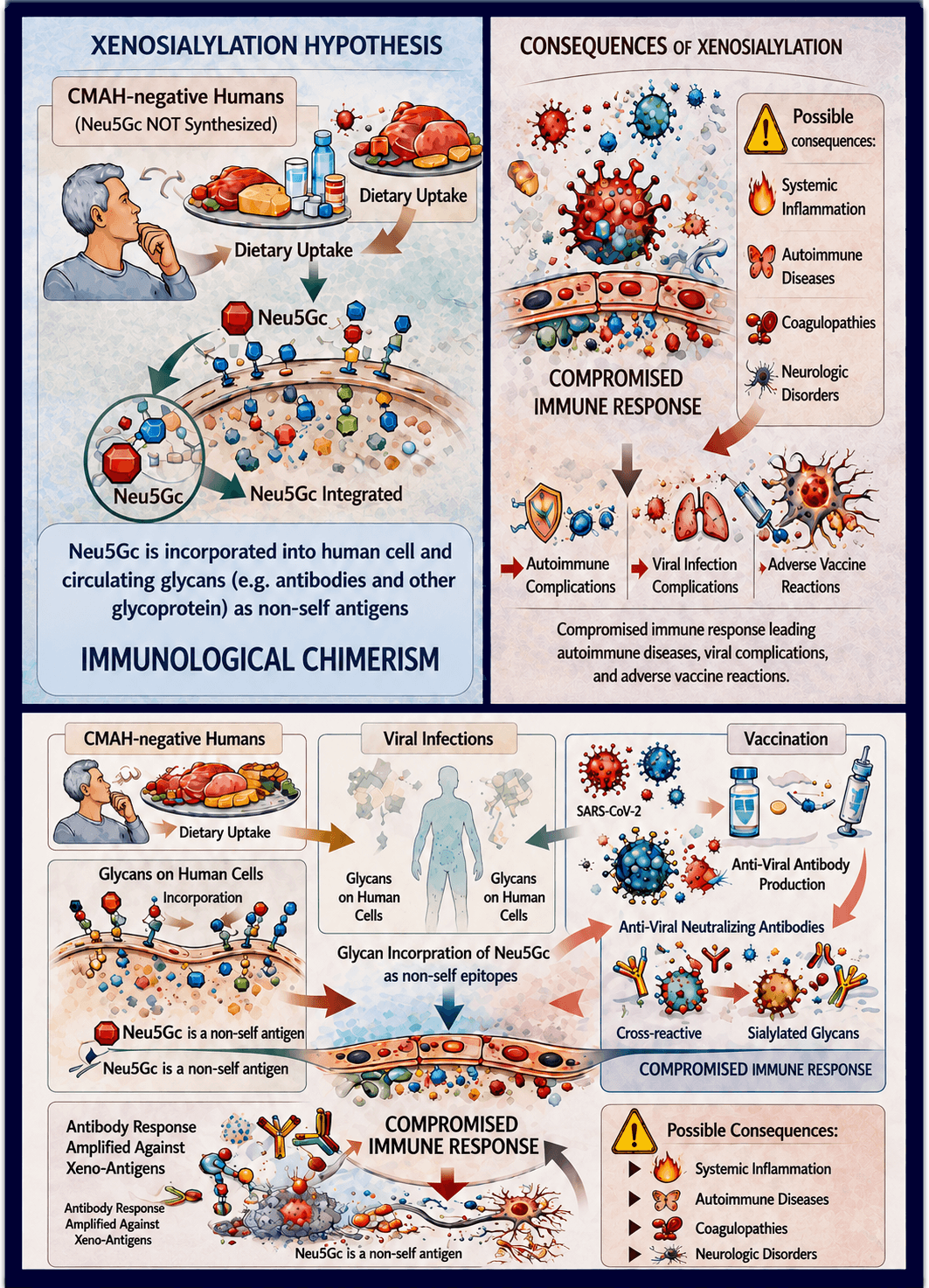
Xenosialylation refers to the metabolic incorporation and persistence of non-human sialic acids into human glycans, generating a form of immunological chimerism that results in qualitative alterations of the sialome and may profoundly affect immune recognition and inflammatory control [57,203]. We use the term here as a unifying framework encompassing the metabolic incorporation of non-human sialic acids and the resulting anti-glycan immune responses, as previously proposed [1].
Humans are genetically unable to synthesize N-glycolylneuraminic acid (Neu5Gc) due to a human-specific inactivating mutation of the cytidine monophospho-N-acetylneuraminic acid hydroxylase (CMAH) gene, rendering Neu5Gc immunologically foreign [208,209]. Nevertheless, humans are chronically exposed to Neu5Gc through dietary intake of edible CMAH+/+ mammalian products, primarily red meat, as well as fish eggs (e.g., caviar) and edible echinoderms (e.g., sea urchins) and through biotechnological and pharmaceutical products derived from non-human cell lines [210,211].
Because human metabolic pathways do not discriminate between Neu5Ac and Neu5Gc, exogenous Neu5Gc is incorporated into glycans on cell surfaces and secreted glycoconjugates [212,213]. Together, these observations indicate that qualitative alterations of the human sialome induced by xenosialylation may have profound immunopathological consequences.
Following dietary intake, Neu5Gc-containing glycoconjugates are taken up by enterocytes, transiently enter the circulation, and are subsequently incorporated into glycoproteins and glycolipids of peripheral tissues, particularly on endothelial and epithelial surfaces, as well as into circulating and secreted glycoproteins [212,214,215]. This implies that a broad range of tissues may become xenosialylated depending on cumulative dietary and/or iatrogenic exposure.
Given the central role of sialic acids in immune regulation, self-recognition, and inflammatory control, qualitative alterations of the human sialome are therefore expected to have profound immunopathological consequences.
From an immunological perspective, diet-derived Neu5Gc-contaminated epitopes are immunologically visible and specifically recognized by human antibodies, constituting non-self-determinants embedded within otherwise self-associated glycans [212,216]. Xenosialylation therefore converts normally tolerogenic self-glycans into mammalian-associated carbohydrate antigens (MCAs) [217], shifting their immunological interpretation from inhibitory self-patterns toward targets of immune recognition and activation. MCAs comprise three major antigenic specificities—α-Gal, Forssman (a heterophilic glycolipid present on the cell membranes of many animal species and bacteria, but generally absent in healthy humans, known as a heterophile antigen because it can induce the production of antibodies that react with tissues of different species), and Neu5Gc—but only Neu5Gc can be metabolically assimilated and stably incorporated into human glycans through dietary and exogenous exposure [217]. As a consequence, Neu5Gc uniquely generates xenosialylated structures that constitute an immunopathological state, which we define as immunological chimerism, in which non-self-antigenic determinants become structurally embedded within the human sialome, thereby blurring the classical boundary between self and non-self-recognition.
Alterations in the Fc glycan—particularly the replacement of endogenous sialic acids with non-human (xeno) sialic acids—may disrupt immune homeostasis and contribute to heightened immunoreactivity, potentially triggering or exacerbating heterogeneous inflammatory and autoimmune pathologies.
Strong viral antigenic stimulation has been shown to induce broad, polyreactive anti-glycan antibody repertoires with autoreactive properties. Antibodies generated against the SARS-CoV-2 nucleocapsid protein display reactivity toward multiple human self-antigens, indicating that virus-induced humoral responses may encompass normally cryptic self-epitopes [218]. In parallel, SARS-CoV-2 infection can induce broad anti-glycan antibody repertoires, providing a mechanistic link between viral immune activation and qualitative alterations of the host glycan landscape [56,57].
Together, these findings support the concept that pre-existing anti-Neu5Gc antibodies may represent a subset of a wider inducible anti-glycan immune response, whose magnitude and pathogenic relevance are amplified by strong antigenic stimuli such as viral infection or vaccination. The presence of these antibodies alone is not sufficient to induce pathology, but establishes a permissive immunological context in which additional inflammatory or antigenic triggers may precipitate disease.
Recent reviews on sialic acids in health and disease have introduced the concept of xenosialitis, referring to chronic inflammation driven by the metabolic incorporation of non-human sialic acids—particularly Neu5Gc—from dietary sources [215]. Dysregulated antibody responses against altered glycan epitopes are increasingly recognized as contributors to chronic inflammation and autoimmunity, especially when glycan structures deviate from physiological self-patterns [57].
Inter-individual variability in Neu5Gc incorporation and antibody repertoires likely underlies the marked heterogeneity of inflammatory and autoimmune phenotypes observed in humans. Polyclonal anti-Neu5Gc antibodies have been implicated in cancer, atherosclerosis, diabetes, and autoimmune disorders [219–221]. The frequent coexistence of multiple autoimmune diseases further supports the existence of a shared upstream mechanism involving broad anti-glycan immune reactivity [184,222].
In this context, glycan-array profiling in SARS-CoV-2–infected patients have shown that viral antigenic stimulation can induce broad polyclonal antibody responses against multiple self-carbohydrate epitopes [57,203]. When superimposed on metabolically incorporated Neu5Gc, such infection-induced antiglycan antibodies may provide a mechanistic link between viral immune activation and xenosialylation-driven immune dysregulation, a condition previously associated with chronic inflammatory states [215].
Marked inter-individual variability in anti-Neu5Gc antibody titers appears to be influenced by gut microbiota composition and its capacity to metabolize dietary xenoglycans [223]. Dietary Neu5Gc interacts with the gut microbiota, where commensal and pathogenic bacteria can scavenge and metabolize sialic acids, potentially amplifying the persistence and immunological visibility of Neu5Gc-containing epitopes and favoring molecular mimicry and chronic immune activation [224–227]. Neu5Gc-rich diets have been shown to alter gut microbial composition—particularly affecting Bacteroidales and Clostridiales—and to enhance xenosialylation of microbial glycans, increasing both the diversity and persistence of xeno-sialylated antigens and extending immune activation beyond the gut [223,227].
Induction of anti-Neu5Gc antibodies in humans is thought to occur during infancy, typically between 6 and 12 months of age, coinciding with the introduction of solid foods or milk-based formula and with the establishment of the gut microbiota, which critically shapes carbohydrate-specific antibody repertoires [225,228]. During neonatal life, human milk provides a contrasting physiological context: human milk oligosaccharides contain exclusively Neu5Ac and support immune tolerance and the development of a protective gut microbiome [229–231]. In contrast, infant formula and bovine milk contain Neu5Gc, and early dietary exposure has been associated with the appearance of circulating anti-Neu5Gc antibodies in infancy [225,232].
Together, these observations identify early life as a critical window in which exposure to non-human sialylated glycans, in conjunction with microbiota maturation, may shape long-term antiglycan immune repertoires. At this stage, xenosialylation can no longer be viewed as a purely host cell–restricted phenomenon, but rather as an emergent property of host–microbiome interactions.
When exposure to non-human sialylated glycans is sustained over time, the combined effects of metabolic incorporation and microbiota-mediated processing may progressively reshape the intestinal glycan landscape. Chronic exposure to a “xeno-enhanced microbiome”—defined as a microbial ecosystem enriched in or adapted to non-human sialic acids—has been associated with persistent intestinal inflammation, increased epithelial permeability, and an elevated risk of diet-related colorectal cancer, a condition already conceptualized as xenosialitis [215,224].
Similar observations have been made in dogs, which, like humans, lack Neu5Gc in their glycan repertoire; recent studies show that in canine IBD, Neu5Gc expression in colonic mucus and enterocytes is proportional to the degree of colitis and the severity of mucosal damage [233]. In this animal model, 90-day administration of a desialyzing probiotic mixture (characterized by bifidobacteria, lactobacilli, and yeasts such as Saccharomyces boulardii) is able to reduce fecal Neu5Gc shedding, reducing the clinical signs of colitis and inflammatory parameters [234].
Within this same pathogenic framework, interactions between metabolically incorporated Neu5Gc and circulating anti-Neu5Gc antibodies have been implicated in tumor progression, vascular inflammation, atherosclerosis, diabetes, and autoimmune disease, indicating that xenosialylation-driven immune dysregulation extends beyond the gut and manifests as systemic pathology [198,215,220]. Together, these observations support the existence of a self-reinforcing loop linking xenosialylation, microbiome-mediated amplification, and immune activation, consistent with emerging systems-level models of glycan-driven immune regulation [57,200,203].
Anti-glycan antibodies can actively contribute to immune dysregulation and tissue pathology, supporting the concept that antibodies directed against xeno-sialylated (Neu5Gc-contaminated) glycans may function as upstream amplifiers of inflammatory and autoimmune responses [2,200,203]. Recent analyses of the SARS-CoV-2 spike protein have shown that virus-associated glycan patterns, including site-specific O-glycosylation, can modulate host immune recognition and promote anti-glycan antibody responses, indicating that qualitative alterations of glycan structures—whether of viral or host origin—represent potent drivers of antiglycan reactivity independent of the underlying peptide antigen [53,54,235].
Metabolic incorporation of Neu5Gc establishes a state of immunological chimerism in which non-self-molecular determinants become structurally embedded within the self glycome, thereby promoting immune recognition of xeno-sialylated glycans. Immune-mediated targeting and removal of these modified structures can, in turn, expose cryptic antigenic epitopes—normally hidden or tolerogenic—thus reinforcing pathological immune responses through loss of immune tolerance, a mechanism well described in autoimmunity following post-translational molecular modifications [236,237]. Subsequent glycan turnover favors renewed incorporation of dietary Neu5Gc, perpetuating stochastic cycles of autoantigen exposure.
Over time, this process drives the emergence of sub-liminal, polyclonal antibody responses that contribute to the initiation, progression, and amplification of both organ-specific and systemic autoimmune diseases. Xenosialylation functions as a permissive immunological state that lowers the threshold for aberrant immune activation in response to strong antigenic challenges, including viral infection or vaccination. This host-dependent condition establishes a background of immune tension that shapes disease susceptibility and clinical heterogeneity across individuals.
This model offers a unifying mechanistic framework to explain the heterogeneous, multisystem, and delayed immune-mediated complications observed after SARS-CoV-2 infection, anti–SARS-CoV-2 vaccination, and other viral infections or immunizations. In this context, the preferential incorporation of Neu5Gc into endothelial glycans [212,215,219] represents a form of immunological chimerism at the vascular interface, predisposing the microvasculature to antibody-mediated inflammation and thrombosis. The coexistence of Neu5Gc-containing glycans and circulating anti-Neu5Gc antibodies may promote immune-complex formation, complement activation, and endothelial activation, thereby creating a pro-thromboinflammatory microenvironment. Because sialylated glycans play a key role in complement regulation and self-recognition at the endothelial surface [201], qualitative alterations of the vascular sialome may further amplify complement-mediated endothelial injury and immunothrombosis. These mechanisms may be particularly relevant in low-shear venous districts with high endothelial glycan density, such as the cerebral venous sinuses and the splanchnic circulation, where thrombo-inflammatory events have been widely documented in both acute and post-acute COVID-19 [94,96] and have occasionally been reported after vaccination [143–145], particularly in genetically predisposed individuals [146].
4.1 Tissue-Level Consequences of Xenosialylation: Breakdown of Immune Self-Tolerance
The heterogeneous autoimmune manifestations observed following viral infections and adverse vaccine reactions are unlikely to be explained solely by direct viral cytopathic effects, molecular mimicry, bystander activation, or occult viral persistence [66,184]. In post-vaccination complications, viral circulation is absent, and in SARS-CoV-2–associated neurological disorders, viral RNA is typically undetectable in cerebrospinal fluid, indicating that tissue damage is not directly mediated by viral presence in the affected organs [66].
Systematic analyses of new-onset autoimmune diseases following COVID-19 vaccination have highlighted the heterogeneity, variable latency, and organ specificity of post-vaccination manifestations, while explicitly acknowledging that currently proposed mechanisms do not explain why only a subset of individuals develops clinically overt disease [74].
Large population-based cohort studies have not identified a disproportionate increase in autoimmune diseases following COVID-19 vaccination at the population level; in contrast, SARS-CoV-2 infection itself has been associated with a higher risk of several autoimmune conditions [238].
We propose that a key determinant of severe immune-mediated complications following both infection and vaccination is the host immune response against xeno-sialic acids (Neu5Gc) metabolically incorporated into cellular glycans across multiple tissues. The individual pattern and extent of Neu5Gc distribution across organs determines the anatomical localization, severity, and clinical heterogeneity of immune-mediated manifestations.
Xenosialylation promotes persistent low-grade inflammation and is associated with elevated levels of circulating polyclonal anti-Neu5Gc antibodies. Immune targeting of xenosialylated tissues leads to removal of altered glycans and exposure of cryptic self-antigens, triggering secondary waves of autoantibody production. Such polyreactive antibody profiles have been documented following SARS-CoV-2 infection and are consistent with those reported in long COVID and post-vaccination immune complications [56,57].
At this stage, immune reactivity becomes systemically oriented toward xenosialylated structures as a molecular class, irrespective of their anatomical localization. Continued dietary or iatrogenic exposure to Neu5Gc may sustain a self-reinforcing inflammatory cycle characterized by lymphocytic infiltration, excessive cytokine release, microvascular inflammation, endothelial injury, and progressive tissue damage. Antibodies directed against glycan epitopes are increasingly recognized as active contributors to immune dysregulation and tissue pathology, including microvascular immune-mediated damage. Under specific immunological contexts, anti-glycan antibodies recognizing self-glycans can play a direct pathogenic role in autoimmune and neurological disorders, including Guillain–Barré syndrome [2,3], endothelial dysfunction in COVID-19 [94–96], or after anti–SARS-CoV-2 vaccination [142,143,146]. Aberrant anti-glycan immune responses can therefore directly drive immune-mediated pathology under permissive conditions [57].
Within this framework, the emergence of autoimmune disorders after infection or vaccination may reflect aberrant anti-glycan immune responses directed against xenosialylated tissues, followed by stochastic exposure of underlying self-antigens, ultimately contributing to chronic inflammation, endothelial dysfunction, and multisystem autoimmune, neurological, and vascular complications.
Widespread qualitative alterations of the glycocalyx may be interpreted by the immune system as evidence of pathogenic invasion [99,147,148], particularly since many enveloped viruses exploit sialylated receptors for host cell entry [194,207]. This is especially relevant for enveloped viruses, whose structural components are directly derived from host cell membranes [217], and may therefore incorporate xenosialylated features present on the infected cells.
Consequently, a host state of xenosialylation biases immune recognition toward mammalian-associated carbohydrate antigens (MCAs), triggering immune responses directed against xeno-contaminated tissues. This framework provides a mechanistic basis for understanding how identical antigenic challenges can elicit minimal, transient responses in some individuals but severe, multisystem immune-mediated pathology in others.
4.2 IgG Fc Homeostasis and Selective Clearance in Xenosialylated Hosts
Antibody responses to viral infections show marked interindividual variability and can result in either viral neutralization or excessive inflammation, depending largely on Fc domain structure and its interaction with Fcγ receptors (FcγRs) expressed by immune effector cells, including monocytes, macrophages, and natural killer cells [239,240]. The balance between activating and inhibitory FcγR engagement critically determines the magnitude and quality of downstream inflammatory responses.
IgG Fc glycosylation is a key regulator of antibody effector function. Variations in fucosylation, galactosylation, and sialylation profoundly modulate pro- and anti-inflammatory activity in infection, autoimmunity, and aging [241–244]. In particular, afucosylation of IgG1 Fc domains confers markedly higher affinity for activating FcγRIIIa receptors, thereby enhancing cytokine production and cytotoxic effector functions [244].
In COVID-19 and following anti–SARS-CoV-2 vaccination, distinct IgG Fc glycosylation repertoires have been described. Severe disease and severe vaccine-related adverse reactions are associated with IgG profiles enriched in IgG1 and IgG2 subclasses with reduced core fucosylation and reduced Fc sialylation, whereas milder disease correlates with higher levels of sialylated IgG3 Fc domains with lower inflammatory potential [245–247]. Hyperinflammatory responses typically emerge around the time of seroconversion, approximately two weeks after infection, and are frequently described as cytokine storms characterized by excessive pro-inflammatory cytokine release [240]. In this context, low Fc fucosylation correlates with enhanced anti-spike IgG–mediated inflammation and increased engagement of activating Fcγ receptors [245]. Afucosylated IgG responses are not unique to COVID-19, and have been reported in autoimmune diseases and in viral infections such as dengue and HIV, where they promote macrophage polarization toward a pro-inflammatory phenotype [244,248,249]. However, it remains unclear why Fc glycosylation abnormalities are particularly pronounced in severe COVID-19 and severe post-vaccination reactions, especially around seroconversion. Although Fc afucosylation appears to be a general feature of immune responses to enveloped viruses, its exaggerated magnitude in severe disease remains mechanistically unexplained [245,246]. Taken together, these observations indicate that current models of Fc glycosylation dysregulation describe the phenomenon but do not explain its upstream cause. Although population-level evidence consistently shows a shift from sialylated, immunoregulatory IgG profiles toward desialylated, pro-inflammatory IgGs in aging and disease, the biological driver of this transition remains undefined [244].
Notably, IgG3 represents the most heavily glycosylated IgG subclass, displaying canonical Fc N-glycosylation together with unique O-glycosylation sites in the hinge region [244]. While Fc sialylation can occur across all IgG subclasses, IgG3 is distinguished by additional hinge-associated glycosylation that may influence antibody effector functions [200,237].
We hypothesize that xenosialylation disrupts IgG Fc homeostasis by favoring the preferential loss or functional depletion of tolerogenic, highly sialylated antibody subsets, thereby amplifying FcγR-mediated inflammatory signaling and systemic cytokine production. The resulting depletion of regulatory IgGs promotes a self-reinforcing inflammatory loop characterized by chronic immune activation and progressive dysregulation of Fc glycosylation patterns. Through this antibody-centered mechanism, xenosialylation lowers the threshold for cytokine amplification, endothelial inflammation, and multisystem immune-mediated pathology following viral infection or vaccination. In this framework, inflammation arises not from the intrinsic pathogenicity of IgG3, but from its preferential loss and the consequent collapse of Fc sialylation-mediated immune regulation.
In heavily xenosialylated individuals, during the intense antibody response triggered by viral infection or vaccination, IgG synthesis occurs in the presence of abundant non-human sialic acids, potentially favoring the incorporation of these residues into the Fc glycans of newly produced antibodies. These modified IgGs may become preferential targets of immune effector mechanisms, resulting in depletion of anti-inflammatory, sialylated IgGs and relative enrichment of pro-inflammatory IgG fractions.
The resulting imbalance shifts immune regulation toward uncontrolled inflammation. Concurrently, innate immune targeting of xenosialylated cell surfaces exposes cryptic self-antigens and promotes autoantibody production, as described above. The convergence of these processes generates a self-amplifying immune escalation, a process we term xenosialylation-induced immunological marasmus, a functional immunological state characterized by progressive erosion of immune regulatory capacity. Under these conditions, even minimal antigenic stimulation—such as viral infection or vaccination—may trigger disproportionate inflammatory responses, manifesting either acutely (e.g., severe COVID-19 around seroconversion) or chronically (e.g., long COVID or post-vaccination immune syndromes).
Clinical heterogeneity—including differences in susceptibility, disease severity, and organ-specific outcomes such as coagulopathies, thrombosis, neurological syndromes (e.g., Guillain–Barré syndrome), autoimmune disease onset or reactivation, and latent viral reactivation—may reflect interindividual variation in the tissue distribution of xenosialylation, while overall disease severity correlates with the magnitude of the pre-existing xenosialylation-associated inflammatory state.
Within this framework, post-infectious and post-vaccination immune-mediated complications differ in magnitude and clinical manifestations, but share the same underlying pathophysiological mechanism. This host-centered model helps explain why identical immune challenges can produce divergent outcomes across individuals.
Importantly, it also accounts for the limited efficacy of antiviral therapies once immune dysregulation predominates, and for the clinical benefit of early anti-inflammatory interventions such as corticosteroids, including dexamethasone [250–252].
4.3 Sex as a Modulator of Xenosialylation-Associated Immune Outcomes
Pronounced sex-related differences characterize COVID-19 severity, post-infectious complications, and vaccine-related immune adverse events. Males more frequently develop severe acute disease, hyper-inflammatory syndromes, and cardiovascular complications, whereas females exhibit higher rates of autoimmune and post-acute immune-mediated sequelae [58,62,253,254], reflecting intrinsic biological dimorphisms in immune regulation rather than epidemiological factors alone.
Females typically mount stronger innate and adaptive immune responses, improving pathogen clearance and vaccine efficacy but increasing susceptibility to autoimmune disease [83,84,254]. These differences partly derive from the X chromosome, which is enriched in immune-related genes and subject to mosaic expression, providing greater immunological redundancy [255].
Hormonal regulation further shapes these effects. Estrogens enhance immune responsiveness and support endothelial homeostasis through modulation of ACE2, whose sex-dependent regulation contributes to differential endothelial resilience [18,256,257]. Consequently, males are more prone to endothelial dysfunction, thrombosis, and acute inflammatory injury [49,90], whereas females show greater protection from acute damage but increased susceptibility to immune escalation.
Within the xenosialylation framework, these sex-specific immune architectures produce divergent clinical trajectories. In males, endothelial xenosialylation combined with a pro-inflammatory immune bias favors exaggerated innate activation and acute hyper-inflammatory syndromes. In females, stronger humoral responses may limit acute tissue injury but increase immune targeting of xenosialylated self-structures, predisposing to delayed autoimmune manifestations [254].
Overall, male-predominant acute severity and female-predominant autoimmunity represent complementary outcomes of a shared upstream condition—xenosialylation—whose clinical expression is modulated by sex-specific genetic, hormonal, endothelial, and immunological factors [18,253–255].
5 Prediction and Prevention of Viral, Post-Vaccination, and Autoimmune Complications
If the xenosialylation hypothesis is correct, it becomes feasible to identify individuals at increased risk of severe viral, post-vaccination, and autoimmune complications, and to implement preventive strategies aimed at reducing both incidence and severity. A central predictive tool is the assessment of circulating anti-Neu5Gc antibodies, which have been implicated in chronic inflammatory and autoimmune diseases and proposed as mechanistic biomarkers of immune-mediated pathology [184,215].
Within this framework, anti-Neu5Gc antibodies could serve as biomarkers to stratify individual susceptibility to autoimmune diseases, viral comp lications—including COVID-19—and post- vaccination adverse immune reactions.
High-resolution glycan microarray technologies enable quantitative and qualitative profiling of anti-Neu5Gc and broader anti-glycan antibody repertoires, allowing assessment of antibody specificity, diversity, and binding strength across hundreds of defined glycan structures [219,258]. Using these platforms, human sera have been shown to contain heterogeneous IgG and IgA responses against Neu5Gc epitopes, supporting the feasibility of antiglycan serology as a biomarker of immune susceptibility [259,260]. Importantly, these approaches capture the breadth and polyreactivity of antiglycan immune responses that are not detectable with single-antigen assays, as demonstrated in SARS-CoV-2–infected individuals [57]. Highly sensitive methods for detecting Neu5Gc and anti-Neu5Gc antibodies in human sera are already available and can be applied to large-scale clinical and epidemiological studies. Affinity-based and glycan-array–based platforms allow high-resolution profiling of anti-Neu5Gc, and broader anti-glycan antibody repertoires, enabling detection of heterogeneous and poly-reactive immune responses [216,219].
Recent methodological advances have further improved the analytical sensitivity and specificity of anti-Neu5Gc detection, highlighting the need for standardized, harmonized serological platforms before clinical implementation [261]. This requirement is particularly critical given the complexity and interindividual variability of polyclonal antiglycan immune repertoires.
Several immunological methods are already available for detecting Neu5Gc and anti-Neu5Gc antibodies in biological samples. ELISA and immunohistochemistry have been applied to identify Neu5Gc-containing glycans in tissues [210], competitive or serological detection [262] or hemagglutination assays to evaluate anti-Neu5Gc antibody levels [219].
In addition, affinity-based assays capable of capturing overall anti-Neu5Gc reactivity have been proposed as screening tools [216] but still new analytical platforms for anti-Neu5Gc detection have been described [261].
It would also be of interest to quantify circulating levels of desialylated and defucosylated IgGs, since shifts in Fc glycosylation toward pro-inflammatory profiles are consistently associated with aging and immune-mediated disease [244]. Such integrated serological profiling may provide a quantitative readout of immune imbalance and help operationalize the concept of xenosialylation-associated immunological marasmus.
From a preventive perspective, individuals with autoimmune diseases, chronic inflammatory conditions, or relevant comorbidities could benefit from adopting a Neu5Gc–free diet to promote gradual clearance of xenosialylated glycans from cell surfaces. Similarly, in subjects at increased risk of post-vaccination adverse immune reactions, a strict Neu5Gc–free diet for approximately 2–3 months prior to vaccination may reduce the burden of incorporated Neu5Gc and circulating anti-Neu5Gc antibodies. This proposed clearance window is biologically plausible, as it is consistent with the turnover rates of erythrocytes, circulating immunoglobulins, endothelial glycocalyx components, and cell-surface glycoproteins, which together define the minimal systemic timescale required for substantial remodeling of the human sialome in the absence of exogenous Neu5Gc. On the other hand, the result of reducing the mucosal incorporation/absorption of Neu5Gc proposed in the spontaneous canine model of IBD-related colitis, through the use of specific probiotic supplements [233], also appears very interesting. This approach involves the use of appropriately assembled probiotic mixtures, containing high concentrations of sialidases + probiotic bacteria, capable of detaching Neu5Gc from the glicoconjugates and, through the metabolic mechanism of the cross-feeding [263,264] using Neu5Gc as a carbon source, and therefore eliminated from the intestinal content with a clearance of potentially xenosialyzing food molecules [265].
If strict long-term adherence to a xenosialic acid–free diet is not feasible, a more pragmatic strategy may be to limit Neu5Gc intake at least during critical immunological windows, particularly in the period immediately following vaccination and around seroconversion. This temporal restriction may reduce the incorporation of high levels of Neu5Gc into newly synthesized IgG molecules that are rapidly and abundantly produced in response to vaccine-induced antigenic stimulation.
Similarly, during periods of active circulation of highly prevalent viral pathogens (e.g., influenza viruses, coronaviruses), maintaining a low dietary intake of Neu5Gc-containing foods could potentially mitigate post-infectious immune complications and facilitate immune resolution and convalescence by limiting xenosialylation of both host tissues and virus-induced antibody repertoires.
Although causal relationships remain to be formally demonstrated, this framework enables the formulation of testable interventional hypotheses. Individuals with elevated anti-Neu5Gc antibody levels—particularly those with autoimmune or chronic inflammatory diseases—could be enrolled in interventional studies and stratified according to dietary exposure. Longitudinal monitoring of anti-Neu5Gc titers and clinical outcomes would allow evaluation of whether sustained elimination of Neu5Gc leads to progressive antibody reduction and clinical improvement.
Diets rich in Neu5Gc-containing foods, such as red meat and dairy products, may contribute to a pro-inflammatory immune milieu and should therefore be reconsidered in high-risk populations. In this context, nutritional protocols currently adopted in hospital and metabolic disease settings [266,267] may warrant re-evaluation in light of xenosialylation.
Although no dietary intervention has been proven to prevent SARS-CoV-2 infection or fully mitigate severe COVID-19 outcomes [30], it is plausible that specific dietary components may exacerbate immune dysregulation during infection or following vaccination. Timely reduction of Neu5Gc intake may therefore contribute to lowering the incidence and severity of post-viral and post-vaccination immune complications and improving the clinical course of chronic inflammatory and autoimmune diseases. Importantly, this model is intended to generate falsifiable and experimentally testable predictions, rather than to establish definitive causality at this stage.
6 Limitations and Controversies: Diet-Derived Neu5Gc and Human Disease
Despite the growing biological plausibility of Neu5Gc-mediated immune modulation within the broader biological roles of sialic acids and glycans in immune regulation [190,191,195], the contribution of dietary Neu5Gc to human inflammatory and autoimmune disease remains debated. Much of the mechanistic evidence derives from preclinical studies, particularly CMAH-deficient mouse models exposed to high Neu5Gc loads or passive transfer of anti-Neu5Gc antibodies [212,214,215,219]. Although these models clarified pathways linking xenosialylation to chronic inflammation and vascular pathology, they may not fully reflect the complexity, dose, and tissue distribution of Neu5Gc exposure in humans.
In humans, circulating anti-Neu5Gc antibodies are widely detectable but highly variable, and their presence does not consistently translate into overt disease [219,268]. This suggests that Neu5Gc-related immune effects are conditional and shaped by host factors such as glycan turnover, endothelial exposure, microbiome composition, and concurrent inflammatory or infectious stimuli [57,203,215,237].
Epidemiological associations between red meat consumption and chronic disease are further confounded by metabolic, lifestyle, and dietary variables, complicating isolation of the specific contribution of Neu5Gc [269]. These limitations were emphasized by Soulillou et al. 2020 [270], who questioned the strength of evidence linking dietary Neu5Gc and anti-Neu5Gc antibodies to specific diseases and highlighted methodological constraints and the absence of large prospective interventional studies.
Conversely, multiple experimental and translational studies support the biological plausibility of Neu5Gc-driven immune modulation, demonstrating metabolic incorporation of Neu5Gc into human tissues and its capacity to induce chronic inflammatory responses through anti-Neu5Gc antibodies [212,214,215,219,220]. Current objections therefore largely reflect limitations in detection and causal inference rather than a refutation of the mechanistic relevance of xenosialylation.
Methodological variability across antiglycan detection platforms has also contributed to inconsistent findings, underscoring the need for standardized high-resolution serological assays and glycan profiling technologies [258,260,261,271]. Accordingly, the present work does not claim definitive causality but proposes a hypothesis-generating framework in which dietary Neu5Gc acts as a contextual modifier whose pathogenic relevance emerges under permissive host conditions characterized by pre-existing immune dysregulation and inflammatory challenges.
This work extends our previous hypothesis on xenosialylation as a host-dependent modulator of post-infectious and post-vaccination immune complications [1], integrating convergent evidence from glycobiology and immunology into a unified mechanistic framework. The model links tissue-specific Neu5Gc incorporation, antibody glycosylation, host variability, and clinical heterogeneity within a coherent host-centered interpretation of immune dysregulation, in which xenosialylation establishes a state of immunological chimerism characterized by the incorporation of non-human sialic acids into host glycoconjugates.
Severe immune-mediated complications following SARS-CoV-2 infection and vaccination resemble those observed after other viral infections and immunizations and frequently occur in individuals with autoimmune or chronic inflammatory comorbidities. Rather than acting as a direct pathogenic trigger, xenosialylation is proposed to represent a permissive upstream condition that lowers the threshold for dysregulated immune activation in response to antigenic challenges. Within this framework, immunological chimerism reshapes immune recognition of host tissues and provides a biologically grounded explanation for delayed onset, multisystem involvement, and marked inter-individual variability of immune-mediated outcomes.
Circulating anti-Neu5Gc antibodies may serve as practical biomarkers for identifying individuals at increased risk, while dietary Neu5Gc reduction represents a testable strategy to attenuate inflammation and improve clinical outcomes. Longitudinal monitoring of anti-Neu5Gc repertoires and Fc glycosylation profiles may further enable individualized risk stratification and preventive intervention.
Importantly, the model is explicitly falsifiable and generates measurable predictions that can be tested in retrospective and prospective studies. By reframing immune adverse events as conditional outcomes arising from host-specific molecular susceptibility rather than stochastic anomalies, this framework links glycobiology to clinical heterogeneity and supports predictive and preventive strategies.
Finally, improved mechanistic understanding of adverse immune reactions can strengthen public health strategies by providing a rational basis for enhancing vaccine safety, trust, and long-term effectiveness. Identifying biological determinants of immune dysregulation is therefore essential to the scientific, ethical, and societal sustainability of vaccination.
Acknowledgement: The authors acknowledge the contributions of the many researchers whose work in glycobiology, immunology, and infectious diseases has informed the conceptual framework presented in this manuscript. AI Policy Statement: Artificial intelligence tools were used in this manuscript exclusively for linguistic revision of the text and for the generation of the images included in the Graphical Abstract. The scientific content, interpretation of the literature, and conceptual development of the manuscript were performed entirely by the authors, who take full responsibility for the accuracy and integrity of the work.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: Conceptualization: Fiorella Carnevali, Giacomo Rossi. Investigation and Literature Analysis: Fiorella Carnevali, Giacomo Rossi. Writing—Original Draft: Fiorella Carnevali, Giacomo Rossi. Writing—Review & Editing: Maria Chiara Muollo, Lucia Biagini. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: No new datasets were generated or analyzed in this study. The manuscript is based exclusively on previously published literature cited in the reference list.
Ethics Approval: Ethical approval was not required for this study as it is based solely on previously published literature and does not involve human participants, animals, or identifiable personal data.
Conflicts of Interest: The authors declare no conflicts of interest.
References
1. Carnevali F, Mangiaterra S, Rossi G. Role of xenosialylation in post-infectious and post-vaccination complications, including COVID-19 and anti-SARS-CoV-2 vaccination. J Inflamm Res 2024;17:8385–94. doi:10.2147/JIR.S471093. [Google Scholar] [PubMed] [CrossRef]
2. Temme JS, Butler DL, Gildersleeve JC. Anti-glycan antibodies: roles in human disease. Biochem J 2021;478(8):1485–509. doi:10.1042/bcj20200610. [Google Scholar] [PubMed] [CrossRef]
3. Gillmann KM, Temme JS, Marglous S, Brown CE, Gildersleeve JC. Anti-glycan monoclonal antibodies: basic research and clinical applications. Curr Opin Chem Biol 2023;74:102281. doi:10.1016/j.cbpa.2023.102281. [Google Scholar] [PubMed] [CrossRef]
4. McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med 2006;3(8):e297. doi:10.1371/journal.pmed.0030297. [Google Scholar] [PubMed] [CrossRef]
5. Davidson A, Diamond B. Autoimmune diseases. N Engl J Med 2001;345(5):340–50. doi:10.1056/NEJM200108023450506. [Google Scholar] [PubMed] [CrossRef]
6. Smatti MK, Cyprian FS, Nasrallah GK, Al Thani AA, Almishal RO, Yassine HM. Viruses and autoimmunity: a review on the potential interaction and molecular mechanisms. Viruses 2019;11(8):762. doi:10.3390/v11080762. [Google Scholar] [PubMed] [CrossRef]
7. Icenogle T. COVID-19: infection or autoimmunity. Front Immunol 2020;11:2055. doi:10.3389/fimmu.2020.02055. [Google Scholar] [PubMed] [CrossRef]
8. DiMaggio D, Anderson A, Bussel JB. Cytomegalovirus can make immune thrombocytopenic purpura refractory. Br J Haematol 2009;146(1):104–12. doi:10.1111/j.1365-2141.2009.07714.x. [Google Scholar] [PubMed] [CrossRef]
9. Chen J, Zhang H, Chen P, et al. Correlation between systemic lupus erythematosus and cytomegalovirus infection detected by different methods. Clin Rheumatol 2015;34(4):691–8. doi:10.1007/s10067-015-2868-3. [Google Scholar] [PubMed] [CrossRef]
10. Moon UY, Park SJ, Oh ST, et al. Patients with systemic lupus erythematosus have abnormally elevated Epstein-Barr virus load in blood. Arthritis Res Ther 2004;6(4):R295–302. doi:10.1186/ar1181. [Google Scholar] [PubMed] [CrossRef]
11. Yokochi T, Yanagawa A, Kimura Y, Mizushima Y. High titer of antibody to the Epstein-Barr virus membrane antigen in sera from patients with rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol 1989;16(8):1029–32. doi:10.1016/0165-2478(88)90037-5. [Google Scholar] [PubMed] [CrossRef]
12. Honkanen H, Oikarinen S, Nurminen N, et al. Detection of enteroviruses in stools precedes islet autoimmunity by several months: possible evidence for slowly operating mechanisms in virus-induced autoimmunity. Diabetologia 2017;60(3):424–31. doi:10.1007/s00125-016-4177-z. [Google Scholar] [PubMed] [CrossRef]
13. Arbour N, Day R, Newcombe J, Talbot PJ. Neuroinvasion by human respiratory coronaviruses. J Virol 2000;74(19):8913–21. doi:10.1128/jvi.74.19.8913-8921.2000. [Google Scholar] [PubMed] [CrossRef]
14. Magdi M, Rahil A. Severe immune thrombocytopenia complicated by intracerebral haemorrhage associated with coronavirus infection: a case report and literature review. Eur J Case Rep Intern Med 2019;6(7):001155. doi:10.12890/2019_001155. [Google Scholar] [PubMed] [CrossRef]
15. Moody R, Wilson K, Flanagan KL, Jaworowski A, Plebanski M. Adaptive immunity and the risk of autoreactivity in COVID-19. Int J Mol Sci 2021;22(16):8965. doi:10.3390/ijms22168965. [Google Scholar] [PubMed] [CrossRef]
16. Bastard P, Rosen LB, Zhang Q, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020;370(6515):eabd4585. doi:10.1126/science.abd4585. [Google Scholar] [PubMed] [CrossRef]
17. Levin AT, Hanage WP, Owusu-Boaitey N, Cochran KB, Walsh SP, Meyerowitz-Katz G. Assessing the age specificity of infection fatality rates for COVID-19: systematic review, meta-analysis, and public policy implications. Eur J Epidemiol 2020;35(12):1123–38. doi:10.1007/s10654-020-00698-1. [Google Scholar] [PubMed] [CrossRef]
18. Peckham H, de Gruijter NM, Raine C, et al. Male sex identified by global COVID-19 meta-analysis as a risk factor for death and ITU admission. Nat Commun 2020;11(1):6317. doi:10.1038/s41467-020-19741-6. [Google Scholar] [PubMed] [CrossRef]
19. Osuchowski MF, Winkler MS, Skirecki T, et al. The COVID-19 puzzle: deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir Med 2021;9(6):622–42. doi:10.1016/S2213-2600(21)00218-6. [Google Scholar] [PubMed] [CrossRef]
20. Chen G, Wu D, Guo W, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest 2020;130(5):2620–9. doi:10.1172/JCI137244. [Google Scholar] [PubMed] [CrossRef]
21. Onder G, Rezza G, Brusaferro S. Case-fatality rate and characteristics of patients dying in relation to COVID-19 in Italy. JAMA 2020;323(18):1775–6. doi:10.1001/jama.2020.4683. [Google Scholar] [PubMed] [CrossRef]
22. Snape MD, Viner RM. COVID-19 in children and young people. Science 2020;370(6514):286–8. doi:10.1126/science.abd6165. [Google Scholar] [PubMed] [CrossRef]
23. Whittaker E, Bamford A, Kenny J, et al. Clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2. JAMA 2020;324(3):259–69. doi:10.1001/jama.2020.10369. [Google Scholar] [PubMed] [CrossRef]
24. Feldstein LR, Rose EB, Horwitz SM, et al. Multisystem inflammatory syndrome in U.S. children and adolescents. N Engl J Med 2020;383(4):334–46. doi:10.1056/nejmoa2021680. [Google Scholar] [PubMed] [CrossRef]
25. Shyong O, Alfakhri N, Bates SV, et al. Multisystem inflammatory syndrome in children: a comprehensive review over the past five years. J Intensive Care Med 2026;41(4):284–308. doi:10.1177/08850666251320558. [Google Scholar] [PubMed] [CrossRef]
26. Rowley AH, Baker SC, Orenstein JM, Shulman ST. Searching for the cause of Kawasaki disease—cytoplasmic inclusion bodies provide new insight. Nat Rev Microbiol 2008;6(5):394–401. doi:10.1038/nrmicro1853. [Google Scholar] [PubMed] [CrossRef]
27. Viner RM, Whittaker E. Kawasaki-like disease: emerging complication during the COVID-19 pandemic. Lancet 2020;395(10239):1741–3. doi:10.1016/S0140-6736(20)31129-6. [Google Scholar] [PubMed] [CrossRef]
28. Auger N, Bégin P, Kang H, et al. Multisystem inflammatory syndrome in adults: comparison with other inflammatory conditions during the COVID-19 pandemic. Respir Med 2023;206:107084. doi:10.1016/j.rmed.2022.107084. [Google Scholar] [PubMed] [CrossRef]
29. Yang J, Zheng Y, Gou X, et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int J Infect Dis 2020;94:91–5. doi:10.1016/j.ijid.2020.03.017. [Google Scholar] [PubMed] [CrossRef]
30. Calder PC, Carr AC, Gombart AF, Eggersdorfer M. Optimal nutritional status for a well-functioning immune system is an important factor to protect against viral infections. Nutrients 2020;12(4):1181. doi:10.3390/nu12041181. [Google Scholar] [PubMed] [CrossRef]
31. Holman N, Knighton P, Kar P, et al. Risk factors for COVID-19-related mortality in people with type 1 and type 2 diabetes in England: a population-based cohort study. Lancet Diabetes Endocrinol 2020;8(10):823–33. doi:10.1016/S2213-8587(20)30271-0. [Google Scholar] [PubMed] [CrossRef]
32. The Lancet Respiratory Medicine. Reflecting on world asthma day in the era of COVID-19. Lancet Respir Med 2020;8(5):423. doi:10.1016/S2213-2600(20)30184-3. [Google Scholar] [PubMed] [CrossRef]
33. Maitz T, Parfianowicz D, Vojtek A, Rajeswaran Y, Vyas AV, Gupta R. COVID-19 cardiovascular connection: a review of cardiac manifestations in COVID-19 infection and treatment modalities. Curr Probl Cardiol 2023;48(8):101186. doi:10.1016/j.cpcardiol.2022.101186. [Google Scholar] [PubMed] [CrossRef]
34. Tartari F, Spadotto A, Zengarini C, et al. Herpes zoster in COVID-19-positive patients. Int J Dermatology 2020;59(8):1028–9. doi:10.1111/ijd.15001. [Google Scholar] [PubMed] [CrossRef]
35. Ferreira ACADF, Romão TT, Macedo YS, Pupe C, Nascimento OJM. COVID-19 and herpes zoster co-infection presenting with trigeminal neuropathy. Euro J Neurology 2020;27(9):1748–50. doi:10.1111/ene.14361. [Google Scholar] [PubMed] [CrossRef]
36. Cheng Y, Ma J, Wang H, et al. Co-infection of influenza A virus and SARS-CoV-2: a retrospective cohort study. J Med Virol 2021;93(5):2947–54. doi:10.1002/jmv.26817. [Google Scholar] [PubMed] [CrossRef]
37. Flerlage T, Boyd DF, Meliopoulos V, Thomas PG, Schultz-Cherry S. Influenza virus and SARS-CoV-2: pathogenesis and host responses in the respiratory tract. Nat Rev Microbiol 2021;19(7):425–41. doi:10.1038/s41579-021-00542-7. [Google Scholar] [PubMed] [CrossRef]
38. Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020;581(7807):215–20. doi:10.1038/s41586-020-2180-5. [Google Scholar] [PubMed] [CrossRef]
39. Gheblawi M, Wang K, Viveiros A, et al. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res 2020;126(10):1456–74. doi:10.1161/circresaha.120.317015. [Google Scholar] [PubMed] [CrossRef]
40. Perico L, Benigni A, Remuzzi G. Should COVID-19 concern nephrologists? Why and to what extent? The emerging impasse of angiotensin blockade. Nephron 2020;144(5):213–21. doi:10.1159/000507305. [Google Scholar] [PubMed] [CrossRef]
41. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020;181(2):271–80.e8. doi:10.1016/j.cell.2020.02.052. [Google Scholar] [PubMed] [CrossRef]
42. Matsuyama S, Nao N, Shirato K, et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc Natl Acad Sci U S A 2020;117(13):7001–3. doi:10.1073/pnas.2002589117. [Google Scholar] [PubMed] [CrossRef]
43. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020;181(2):281–92.e6. doi:10.1016/j.cell.2020.02.058. [Google Scholar] [PubMed] [CrossRef]
44. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004;203(2):631–7. doi:10.1002/path.1570. [Google Scholar] [PubMed] [CrossRef]
45. Torres Acosta MA, Singer BD. Pathogenesis of COVID-19-induced ARDS: implications for an ageing population. Eur Respir J 2020;56(3):2002049. doi:10.1183/13993003.02049-2020. [Google Scholar] [PubMed] [CrossRef]
46. Cantuti-Castelvetri L, Ojha R, Pedro LD, et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020;370(6518):856–60. doi:10.1126/science.abd2985. [Google Scholar] [PubMed] [CrossRef]
47. Daly JL, Simonetti B, Antón-Plágaro C, et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. bioRxiv 2020;20:533. doi:10.1101/2020.06.05.134114. [Google Scholar] [PubMed] [CrossRef]
48. Bestle D, Heindl MR, Limburg H, et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci Alliance 2020;3(9):e202000786. doi:10.26508/lsa.202000786. [Google Scholar] [PubMed] [CrossRef]
49. Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020;395(10234):1417–8. doi:10.1016/S0140-6736(20)30937-5. [Google Scholar] [PubMed] [CrossRef]
50. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N Engl J Med 2020;383(2):120–8. doi:10.1056/nejmoa2015432. [Google Scholar] [PubMed] [CrossRef]
51. Blanco-Melo D, Nilsson-Payant BE, Liu WC, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020;181(5):1036–45.e9. doi:10.1016/j.cell.2020.04.026. [Google Scholar] [PubMed] [CrossRef]
52. Lucas C, Wong P, Klein J, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020;584(7821):463–9. doi:10.1038/s41586-020-2588-y. [Google Scholar] [PubMed] [CrossRef]
53. Watanabe Y, Allen JD, Wrapp D, McLellan JS, Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020;369(6501):330–3. doi:10.1126/science.abb9983. [Google Scholar] [PubMed] [CrossRef]
54. Casalino L, Gaieb Z, Goldsmith JA, et al. Beyond shielding: the roles of glycans in the SARS-CoV-2 spike protein. ACS Cent Sci 2020;6(10):1722–34. doi:10.1021/acscentsci.0c01056. [Google Scholar] [PubMed] [CrossRef]
55. Talwar S, Harker JA, Openshaw PJM, Thwaites RS. Autoimmunity in long COVID. J Allergy Clin Immunol 2025;155(4):1082–94. doi:10.1016/j.jaci.2025.02.005. [Google Scholar] [PubMed] [CrossRef]
56. Jernbom AF, Skoglund L, Pin E, et al. Prevalent and persistent new-onset autoantibodies in mild to severe COVID-19. Nat Commun 2024;15(1):8941. doi:10.1038/s41467-024-53356-5. [Google Scholar] [PubMed] [CrossRef]
57. Butler DL, Imberti L, Quaresima V, et al. Abnormal antibodies to self-carbohydrates in SARS-CoV-2-infected patients. PNAS Nexus 2022;1(3):pgac062. doi:10.1093/pnasnexus/pgac062. [Google Scholar] [PubMed] [CrossRef]
58. Ramos-Casals M, Brito-Zerón P, Mariette X. Systemic and organ-specific immune-related manifestations of COVID-19. Nat Rev Rheumatol 2021;17(6):315–32. doi:10.1038/s41584-021-00608-z. [Google Scholar] [PubMed] [CrossRef]
59. Toscano G, Palmerini F, Ravaglia S, et al. Guillain-Barré syndrome associated with SARS-CoV-2. N Engl J Med 2020;382(26):2574–6. doi:10.1056/nejmc2009191. [Google Scholar] [PubMed] [CrossRef]
60. Vogel TP, Top KA, Karatzios C, et al. Multisystem inflammatory syndrome in children and adults (MIS-C/Acase definition & guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine 2021;39(22):3037–49. doi:10.1016/j.vaccine.2021.01.054. [Google Scholar] [PubMed] [CrossRef]
61. Lehmann HC, Hartung HP, Kieseier BC, Hughes RA. Guillain-Barré syndrome after exposure to influenza virus. Lancet Infect Dis 2010;10(9):643–51. doi:10.1016/S1473-3099(10)70140-7. [Google Scholar] [PubMed] [CrossRef]
62. Yazdanpanah N, Rezaei N. Autoimmune complications of COVID-19. J Med Virol 2022;94(1):54–62. doi:10.1002/jmv.27292. [Google Scholar] [PubMed] [CrossRef]
63. Casciola-Rosen L, Thiemann DR, Andrade F, et al. IgM anti-ACE2 autoantibodies in severe COVID-19 activate complement and perturb vascular endothelial function. JCI Insight 2022;7(9):e158362. doi:10.1172/jci.insight.158362. [Google Scholar] [PubMed] [CrossRef]
64. Helms J, Tacquard C, Severac F, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med 2020;46(6):1089–98. doi:10.1007/s00134-020-06062-x. [Google Scholar] [PubMed] [CrossRef]
65. Yang Y, Zhao J, Wu J, Teng Y, Xia X. A rare case of immune thrombocytopenic purpura, secondary to COVID-19. J Med Virol 2020;92(11):2358–60. doi:10.1002/jmv.26051. [Google Scholar] [PubMed] [CrossRef]
66. Dunai C, Collie C, Michael BD. Immune-mediated mechanisms of COVID-19 neuropathology. Front Neurol 2022;13:882905. doi:10.3389/fneur.2022.882905. [Google Scholar] [PubMed] [CrossRef]
67. Yavarpour-Bali H, Ghasemi-Kasman M. Update on neurological manifestations of COVID-19. Life Sci 2020;257(1):118063. doi:10.1016/j.lfs.2020.118063. [Google Scholar] [PubMed] [CrossRef]
68. Gutiérrez-Ortiz C, Méndez-Guerrero A, Rodrigo-Rey S, et al. Miller Fisher syndrome and polyneuritis cranialis in COVID-19. Neurology 2020;95(5):e601–5. doi:10.1212/wnl.0000000000009619. [Google Scholar] [PubMed] [CrossRef]
69. Hughes RA, Cornblath DR. Guillain-Barré syndrome. Lancet 2005;366(9497):1653–66. doi:10.1016/S0140-6736(05)67665-9. [Google Scholar] [PubMed] [CrossRef]
70. Abu-Rumeileh S, Abdelhak A, Foschi M, Tumani H, Otto M. Guillain-Barré syndrome spectrum associated with COVID-19: an up-to-date systematic review of 73 cases. J Neurol 2021;268(4):1133–70. doi:10.1007/s00415-020-10124-x. [Google Scholar] [PubMed] [CrossRef]
71. Anaya JM, Monsalve DM, Rojas M, et al. Latent rheumatic, thyroid and phospholipid autoimmunity in hospitalized patients with COVID-19. J Transl Autoimmun 2021;4:100091. doi:10.1016/j.jtauto.2021.100091. [Google Scholar] [PubMed] [CrossRef]
72. Vlachoyiannopoulos PG, Magira E, Alexopoulos H, et al. Autoantibodies related to systemic autoimmune rheumatic diseases in severely ill patients with COVID-19. Ann Rheum Dis 2020;79(12):1661–3. doi:10.1136/annrheumdis-2020-218009. [Google Scholar] [PubMed] [CrossRef]
73. Zuo Y, Estes SK, Ali RA, et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci Transl Med 2020;12(570):eabd3876. doi:10.1126/scitranslmed.abd3876. [Google Scholar] [PubMed] [CrossRef]
74. Guo T, Fan Y, Chen M, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol 2020;5(7):811. doi:10.1001/jamacardio.2020.1017. [Google Scholar] [PubMed] [CrossRef]
75. Kornowski R, Witberg G. Acute myocarditis caused by COVID-19 disease and following COVID-19 vaccination. Open Heart 2022;9(1):e001957. doi:10.1136/openhrt-2021-001957. [Google Scholar] [PubMed] [CrossRef]
76. Yajima T, Knowlton KU. Viral myocarditis: from the perspective of the virus. Circulation 2009;119(19):2615–24. doi:10.1161/CIRCULATIONAHA.108.766022. [Google Scholar] [PubMed] [CrossRef]
77. Siripanthong B, Nazarian S, Muser D, et al. Recognizing COVID-19-related myocarditis: the possible pathophysiology and proposed guideline for diagnosis and management. Heart Rhythm 2020;17(9):1463–71. doi:10.1016/j.hrthm.2020.05.001. [Google Scholar] [PubMed] [CrossRef]
78. Puntmann VO, Carerj ML, Wieters I, et al. Outcomes of cardiovascular magnetic resonance imaging in patients recently recovered from coronavirus disease 2019 (COVID-19). JAMA Cardiol 2020;5(11):1265–73. doi:10.1001/jamacardio.2020.3557. [Google Scholar] [PubMed] [CrossRef]
79. Daniels CJ, Rajpal S, Greenshields JT, et al. Prevalence of clinical and subclinical myocarditis in competitive athletes with recent SARS-CoV-2 infection: results from the big ten COVID-19 cardiac registry. JAMA Cardiol 2021;6(9):1078–87. doi:10.1001/jamacardio.2021.2065. [Google Scholar] [PubMed] [CrossRef]
80. Tarasco MC, Iacomino N, Mantegazza R, Cavalcante P. COVID-19, Epstein-Barr virus reactivation and autoimmunity: casual or causal liaisons? J Microbiol Immunol Infect 2025;58(5):508–16. doi:10.1016/j.jmii.2025.03.014. [Google Scholar] [PubMed] [CrossRef]
81. Zhou Y, Han T, Chen J, et al. Clinical and autoimmune characteristics of severe and critical cases of COVID-19. Clin Transl Sci 2020;13(6):1077–86. doi:10.1111/cts.12805. [Google Scholar] [PubMed] [CrossRef]
82. Souyris M, Cenac C, Azar P, et al. TLR7 escapes X chromosome inactivation in immune cells. Sci Immunol 2018;3(19):eaap8855. doi:10.1126/sciimmunol.aap8855. [Google Scholar] [PubMed] [CrossRef]
83. Takahashi T, Ellingson MK, Wong P, et al. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature 2020;588(7837):315–20. doi:10.1038/s41586-020-2700-3. [Google Scholar] [PubMed] [CrossRef]
84. Raza HA, Sen P, Bhatti OA, Gupta L. Sex hormones, autoimmunity and gender disparity in COVID-19. Rheumatol Int 2021;41(8):1375–86. doi:10.1007/s00296-021-04873-9. [Google Scholar] [PubMed] [CrossRef]
85. Ortona E, Buonsenso D, Carfi A, Malorni W. Long Covid Kids study group. Long COVID: an estrogen-associated autoimmune disease? Cell Death Discov 2021;7(1):77. doi:10.1038/s41420-021-00464-6. [Google Scholar] [PubMed] [CrossRef]
86. Osmanov IM, Spiridonova E, Bobkova P, et al. Risk factors for post-COVID-19 condition in previously hospitalised children using the ISARIC Global follow-up protocol: a prospective cohort study. Eur Respir J 2022;59(2):2101341. doi:10.1183/13993003.01341-2021. [Google Scholar] [PubMed] [CrossRef]
87. Ercolini AM, Miller SD. The role of infections in autoimmune disease. Clin Exp Immunol 2009;155(1):1–15. doi:10.1111/j.1365-2249.2008.03834.x. [Google Scholar] [PubMed] [CrossRef]
88. Ehrenfeld M, Tincani A, Andreoli L, et al. COVID-19 and autoimmunity. Autoimmun Rev 2020;19(8):102597. doi:10.1016/j.autrev.2020.102597. [Google Scholar] [PubMed] [CrossRef]
89. Galeotti C, Bayry J. Autoimmune and inflammatory diseases following COVID-19. Nat Rev Rheumatol 2020;16(8):413–4. doi:10.1038/s41584-020-0448-7. [Google Scholar] [PubMed] [CrossRef]
90. Goshua G, Pine AB, Meizlish ML, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol 2020;7(8):e575–82. doi:10.1016/S2352-3026(20)30216-7. [Google Scholar] [PubMed] [CrossRef]
91. Klok FA, Kruip MJHA, van der Meer NJM,et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: an updated analysis. Thromb Res 2020;191:148–50. doi:10.1016/j.thromres.2020.04.041. [Google Scholar] [PubMed] [CrossRef]
92. Middeldorp S, Coppens M, van Haaps TF,et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemost 2020;18(8):1995–2002. doi:10.1111/jth.14888. [Google Scholar] [PubMed] [CrossRef]
93. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol 2013;13(1):34–45. doi:10.1038/nri3345. [Google Scholar] [PubMed] [CrossRef]
94. Bonaventura A, Vecchié A, Dagna L, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol 2021;21(5):319–29. doi:10.1038/s41577-021-00536-9. [Google Scholar] [PubMed] [CrossRef]
95. Sardu C, Gambardella J, Morelli MB, Wang X, Marfella R, Santulli G. Hypertension, thrombosis, kidney failure, and diabetes: is COVID-19 an endothelial disease? A comprehensive evaluation of clinical and basic evidence. J Clin Med 2020;9(5):1417. doi:10.3390/jcm9051417. [Google Scholar] [PubMed] [CrossRef]
96. Otifi HM, Adiga BK. Endothelial dysfunction in COVID-19 infection. Am J Med Sci 2022;363(4):281–7. doi:10.1016/j.amjms.2021.12.010. [Google Scholar] [PubMed] [CrossRef]
97. Yang Y, Tang H. Aberrant coagulation causes a hyper-inflammatory response in severe influenza pneumonia. Cell Mol Immunol 2016;13(4):432–42. doi:10.1038/cmi.2016.1. [Google Scholar] [PubMed] [CrossRef]
98. Velavan TP, Meyer CG. Mild versus severe COVID-19: laboratory markers. Int J Infect Dis 2020;95:304–7. doi:10.1016/j.ijid.2020.04.061. [Google Scholar] [PubMed] [CrossRef]
99. Goonewardena SN, Grushko OG, Wells J, et al. Immune-mediated glycocalyx remodeling in hospitalized COVID-19 patients. Cardiovasc Drugs Ther 2023;37(2):307–13. doi:10.1007/s10557-021-07288-7. [Google Scholar] [PubMed] [CrossRef]
100. Tufano A, Rendina D, Abate V, et al. Venous thromboembolism in COVID-19 compared to non-COVID-19 cohorts: a systematic review with meta-analysis. J Clin Med 2021;10(21):4925. doi:10.3390/jcm10214925. [Google Scholar] [PubMed] [CrossRef]
101. Assinger A, Schrottmaier WC, Salzmann M, Rayes J. Platelets in sepsis: an update on experimental models and clinical data. Front Immunol 2019;10:1687. doi:10.3389/fimmu.2019.01687. [Google Scholar] [PubMed] [CrossRef]
102. Colling ME, Tourdot BE, Kanthi Y. Inflammation, infection and venous thromboembolism. Circ Res 2021;128(12):2017–36. doi:10.1161/CIRCRESAHA.121.318225. [Google Scholar] [PubMed] [CrossRef]
103. Zuniga M, Gomes C, Carsons SE, et al. Autoimmunity to annexin A2 predicts mortality among hospitalised COVID-19 patients. Eur Respir J 2021;58(4):2100918. doi:10.1183/13993003.00918-2021. [Google Scholar] [PubMed] [CrossRef]
104. Michelen M, Manoharan L, Elkheir N, et al. Characterising long COVID: a living systematic review. BMJ Glob Health 2021;6(9):e005427. doi:10.1136/bmjgh-2021-005427. [Google Scholar] [PubMed] [CrossRef]
105. Lopez-Leon S, Wegman-Ostrosky T, Ayuzo del Valle NC,et al. Long-COVID in children and adolescents: a systematic review and meta-analyses. Sci Rep 2022;12(1):9950. doi:10.1038/s41598-022-13495-5. [Google Scholar] [PubMed] [CrossRef]
106. Peluso MJ, Deeks SG. Mechanisms of long COVID and the path toward therapeutics. Cell 2024;187(20):5500–29. doi:10.1016/j.cell.2024.07.054. [Google Scholar] [PubMed] [CrossRef]
107. Sudre CH, Murray B, Varsavsky T, et al. Attributes and predictors of long COVID. Nat Med 2021;27(4):626–31. doi:10.1038/s41591-021-01292-y. [Google Scholar] [PubMed] [CrossRef]
108. Clark DV, Kibuuka H, Millard M, et al. Long-term sequelae after Ebola virus disease in Bundibugyo, Uganda: a retrospective cohort study. Lancet Infect Dis 2015;15(8):905–12. doi:10.1016/S1473-3099(15)70152-0. [Google Scholar] [PubMed] [CrossRef]
109. Ortona E, Malorni W. Long COVID: to investigate immunological mechanisms and sex/gender related aspects as fundamental steps for tailored therapy. Eur Respir J 2022;59(2):2102245. doi:10.1183/13993003.02245-2021. [Google Scholar] [PubMed] [CrossRef]
110. Wang EY, Mao T, Klein J, et al. Diverse functional autoantibodies in patients with COVID-19. Nature 2021;595(7866):283–8. doi:10.1038/s41586-021-03631-y. [Google Scholar] [PubMed] [CrossRef]
111. Nagy A, Alhatlani B. An overview of current COVID-19 vaccine platforms. Comput Struct Biotechnol J 2021;19:2508–17. doi:10.1016/j.csbj.2021.04.061. [Google Scholar] [PubMed] [CrossRef]
112. Ndwandwe D, Wiysonge CS. COVID-19 vaccines. Curr Opin Immunol 2021;71:111–6. doi:10.1016/j.coi.2021.07.003. [Google Scholar] [PubMed] [CrossRef]
113. Faksova K, Walsh D, Jiang Y, et al. COVID-19 vaccines and adverse events of special interest: a multinational Global Vaccine Data Network (GVDN) cohort study of 99 million vaccinated individuals. Vaccine 2024;42(9):2200–11. doi:10.1016/j.vaccine.2024.01.100. [Google Scholar] [PubMed] [CrossRef]
114. Centers for Disease Control and Prevention. COVID-19 vaccine safety: reported adverse events. Atlanta, GA, USA: CDC; 2025 [cited 2026 May 28]. Available from: https://www.cdc.gov/coronavirus/2019-ncov/vaccines/safety/adverse-events.html. [Google Scholar]
115. Gee J, Marquez P, Su J, et al. First month of COVID-19 vaccine safety monitoring-United States, December 14, 2020–January 13, 2021. MMWR Morb Mortal Wkly Rep 2021;70(8):283–8. doi:10.15585/mmwr.mm7008e3. [Google Scholar] [PubMed] [CrossRef]
116. Meo SA, Bukhari IA, Akram J, Meo AS, Klonoff DC. COVID-19 vaccines: comparison of biological, pharmacological characteristics and adverse effects of Pfizer/BioNTech and moderna vaccines. Eur Rev Med Pharmacol Sci 2021;25(3):1663–9. doi:10.26355/eurrev_202102_24877. [Google Scholar] [PubMed] [CrossRef]
117. Agenzia Italiana del Farmaco (AIFA). Comirnaty (BNT162b2Riassunto delle caratteristiche del prodotto. Roma, Italy: AIFA; 2021[cited 2026 May 28]. Available from: https://www.aifa.gov.it. [Google Scholar]
118. Dotan A, Shoenfeld Y. Perspectives on vaccine induced thrombotic thrombocytopenia. J Autoimmun 2021;121(8):102663. doi:10.1016/j.jaut.2021.102663. [Google Scholar] [PubMed] [CrossRef]
119. Segal Y, Shoenfeld Y. Vaccine-induced autoimmunity: the role of molecular mimicry and immune crossreaction. Cell Mol Immunol 2018;15(6):586–594. doi:10.1038/cmi.2017.151. [Google Scholar] [PubMed] [CrossRef]
120. Bidari A, Asgarian S, Pour Mohammad A, et al. Immune thrombocytopenic purpura secondary to COVID-19 vaccination: a systematic review. Eur J Haematol 2023;110(4):335–53. doi:10.1111/ejh.13917. [Google Scholar] [PubMed] [CrossRef]
121. Furer V, Zisman D, Kibari A, Rimar D, Paran Y, Elkayam O. Herpes zoster following BNT162b2 mRNA COVID-19 vaccination in patients with autoimmune inflammatory rheumatic diseases: a case series. Rheumatology 2021;60(SI):SI90–5. doi:10.1093/rheumatology/keab345. [Google Scholar] [PubMed] [CrossRef]
122. Flanagan KL, Fink AL, Plebanski M, Klein SL. Sex and gender differences in the outcomes of vaccination over the life course. Annu Rev Cell Dev Biol 2017;33(1):577–99. doi:10.1146/annurev-cellbio-100616-060718. [Google Scholar] [PubMed] [CrossRef]
123. Witberg G, Barda N, Hoss S, et al. Myocarditis after COVID-19 vaccination in a large health care organization. N Engl J Med 2021;385(23):2132–9. doi:10.1056/nejmoa2110737. [Google Scholar] [PubMed] [CrossRef]
124. Buoninfante A, Andeweg A, Genov G, Cavaleri M. Myocarditis associated with COVID-19 vaccination. npj Vaccin 2024;9(1):122. doi:10.1038/s41541-024-00893-1. [Google Scholar] [PubMed] [CrossRef]
125. Choi MJ, Na Y, Hyun HJ, et al. Comparative safety analysis of mRNA and adenoviral vector COVID-19 vaccines: a nationwide cohort study using an emulated target trial approach. Clin Microbiol Infect 2024;30(5):646–52. doi:10.1016/j.cmi.2023.12.010. [Google Scholar] [PubMed] [CrossRef]
126. Phuong LK, Bonetto C, Buttery J, et al. Kawasaki disease and immunisation: a systematic review. Vaccine 2017;35(14):1770–9. doi:10.1016/j.vaccine.2016.09.033. [Google Scholar] [PubMed] [CrossRef]
127. Uwaydah AK, Hassan NMM, Abu Ghoush MS, Shahin KMM. Adult multisystem inflammatory syndrome in a patient who recovered from COVID-19 postvaccination. BMJ Case Rep 2021;14(4):e242060. doi:10.1136/bcr-2021-242060. [Google Scholar] [PubMed] [CrossRef]
128. Bova C, Vigna E, Gentile M. Multisystem inflammatory syndrome after Ad26.COV2.S vaccination. IDCases 2022;27(4):e01411. doi:10.1016/j.idcr.2022.e01411. [Google Scholar] [PubMed] [CrossRef]
129. Klein NP, Lewis N, Goddard K, et al. Surveillance for adverse events after COVID-19 mRNA vaccination. JAMA 2021;326(14):1390–9. doi:10.1001/jama.2021.15072. [Google Scholar] [PubMed] [CrossRef]
130. Yousaf AR, Mak J, Gwynn L, et al. COVID-19 mRNA vaccination reduces the occurrence of post-COVID conditions in U.S. children aged 5-17 years following Omicron SARS-CoV-2 infection, July 2021–September 2022. Open Forum Infect Dis 2023;10(Supplement_2):ofad500.2466. doi:10.1093/ofid/ofad500.2466; [Google Scholar] [CrossRef]
131. European Medicines Agency. COVID-19 vaccines: PRAC assessment of multisystem inflammatory syndrome (MIS). Amsterdam, The Netherlands: EMA; 2022[cited 2026 May 28]. Available from: https://www.ema.europa.eu. [Google Scholar]
132. Brown RC, Blecher TE, French EA, Toghill PJ. Thrombotic thrombocytopenic purpura after influenza vaccination. Br Med J 1973;2(5861):303. doi:10.1136/bmj.2.5861.303-a. [Google Scholar] [PubMed] [CrossRef]
133. Ramakrishnan N, Parker LP. Thrombotic thrombocytopenic purpura following influenza vaccination—a brief case report. Conn Med 1998;62(10):587–8; [Google Scholar]
134. Kojima Y, Ohashi H, Nakamura T, et al. Acute thrombotic thrombocytopenic Purpura after pneumococcal vaccination. Blood Coagul Fibrinolysis 2014;25(5):512–4. doi:10.1097/MBC.0000000000000058. [Google Scholar] [PubMed] [CrossRef]
135. Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, Eichinger S. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N Engl J Med 2021;384(22):2092–101. doi:10.1056/nejmoa2104840. [Google Scholar] [PubMed] [CrossRef]
136. Cines DB, Bussel JB. SARS-CoV-2 vaccine-induced immune thrombotic thrombocytopenia. N Engl J Med 2021;384(23):2254–6. doi:10.1056/nejme2106315. [Google Scholar] [PubMed] [CrossRef]
137. Barnes G, Cuker A, Piazza G, Siegal D. Vaccine-induced thrombotic thrombocytopenia (VITT) and COVID-19 vaccines: what cardiovascular clinicians need to know. Cardiol Mag 2021. [Google Scholar]
138. Casucci G, Acanfora D. DIC-like syndrome following administration of ChAdOx1 nCov-19 vaccination. Viruses 2021;13(6):1046. doi:10.3390/v13061046. [Google Scholar] [PubMed] [CrossRef]
139. Brighton Collaboration. Case definitions and standardized tools to support vaccine safety surveillance. Basel, Switzerland: Brighton Collaboration; 2025[cited 2026 May 28]. Available from: https://brightoncollaboration.org/case-definitions/. [Google Scholar]
140. Sharifian-Dorche M, Bahmanyar M, Sharifian-Dorche A, Mohammadi P, Nomovi M, Mowla A. Vaccine-induced immune thrombotic thrombocytopenia and cerebral venous sinus thrombosis post COVID-19 vaccination; a systematic review. J Neurol Sci 2021;428(5):117607. doi:10.1016/j.jns.2021.117607. [Google Scholar] [PubMed] [CrossRef]
141. Scully M, Singh D, Lown R, et al. Pathologic antibodies to platelet factor 4 after ChAdOx1 nCoV-19 vaccination. N Engl J Med 2021;384(23):2202–11. doi:10.1056/NEJMoa2105385. [Google Scholar] [PubMed] [CrossRef]
142. Perdomo J, Leung HHL, Ahmadi Z, et al. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat Commun 2019;10(1):1322. doi:10.1038/s41467-019-09160-7. [Google Scholar] [PubMed] [CrossRef]
143. Sholzberg M, Arnold DM, Laupacis A. Recognizing, managing and reporting vaccine-induced immune thrombotic thrombocytopenia. CMAJ 2021;193(24):E913–5. doi:10.1503/cmaj.210882. [Google Scholar] [PubMed] [CrossRef]
144. Rzymski P, Perek B, Flisiak R. Thrombotic thrombocytopenia after COVID-19 vaccination: in search of the underlying mechanism. Vaccines 2021;9(6):559. doi:10.3390/vaccines9060559. [Google Scholar] [PubMed] [CrossRef]
145. McGonagle D, de Marco G, Bridgewood C. Mechanisms of immunothrombosis in vaccine-induced thrombotic thrombocytopenia (VITT) compared to natural SARS-CoV-2 infection. J Autoimmun 2021;121(3):102662. doi:10.1016/j.jaut.2021.102662. [Google Scholar] [PubMed] [CrossRef]
146. Wang JJ, Schönborn L, Warkentin TE, et al. Adenoviral inciting antigen and somatic hypermutation in VITT. N Engl J Med 2026;394(7):669–83. doi:10.1056/NEJMoa2514824. [Google Scholar] [PubMed] [CrossRef]
147. Moore KH, Murphy HA, George EM. The glycocalyx: a central regulator of vascular function. Am J Physiol Regul Integr Comp Physiol 2021;320(4):R508–18. doi:10.1152/ajpregu.00340.2020. [Google Scholar] [PubMed] [CrossRef]
148. Psefteli PM, Kitscha P, Vizcay-Barrena G, et al. Glycocalyx sialic acids regulate Nrf2-mediated signaling by fluid shear stress in human endothelial cells. Redox Biol 2021;38(10):101816. doi:10.1016/j.redox.2020.101816. [Google Scholar] [PubMed] [CrossRef]
149. Zou Z, Li L, Schäfer N, Huang Q, Maegele M, Gu Z. Endothelial glycocalyx in traumatic brain injury associated coagulopathy: potential mechanisms and impact. J Neuroinflamm 2021;18(1):134. doi:10.1186/s12974-021-02192-1. [Google Scholar] [PubMed] [CrossRef]
150. Wang J, Ma L, Fang Y, Ye T, Li H, Lan P. Factors influencing glycocalyx degradation: a narrative review. Front Immunol 2025;15:1490395. doi:10.3389/fimmu.2024.1490395. [Google Scholar] [PubMed] [CrossRef]
151. Muir KL, Kallam A, Koepsell SA, Gundabolu K. Thrombotic thrombocytopenia after Ad26.COV2.S vaccination. N Engl J Med 2021;384(20):1964–5. doi:10.1056/NEJMc2105869. [Google Scholar] [PubMed] [CrossRef]
152. World Health Organization. Statement of the COVID-19 subcommittee of the WHO global advisory committee on vaccine safety (GACVS) on rare adverse blood coagulation events with COVID-19 vaccines. Geneva, Switzerland: WHO; 2021 Apr 16[cited 2026 May 28]. Available from: https://www.who.int. [Google Scholar]
153. European Medicines Agency. COVID-19 vaccine AstraZeneca: benefits still outweigh the risks despite possible link to rare blood clots with low blood platelets. Amsterdam, The Netherlands: EMA; 2021[cited 2026 May 28]. Available from: https://www.ema.europa.eu. [Google Scholar]
154. de Gregorio C, Colarusso L, Calcaterra G, et al. Cerebral venous sinus thrombosis following COVID-19 vaccination: analysis of 552 worldwide cases. Vaccines 2022;10(2):232. doi:10.3390/vaccines10020232. [Google Scholar] [PubMed] [CrossRef]
155. Cushman M. Epidemiology and risk factors for venous thrombosis. Semin Hematol 2007;44(2):62–9. doi:10.1053/j.seminhematol.2007.02.004. [Google Scholar] [PubMed] [CrossRef]
156. Oldenburg J, Klamroth R, Langer F, et al. Diagnosis and management of vaccine-related thrombosis following AstraZeneca COVID-19 vaccination: guidance statement from the GTH. Hämostaseologie 2021;41(3):184–9. doi:10.1055/a-1469-7481. [Google Scholar] [PubMed] [CrossRef]
157. Greinacher A, Selleng K, Warkentin TE. Autoimmune heparin-induced thrombocytopenia. J Thromb Haemost 2017;15(11):2099–114. doi:10.1111/jth.13813. [Google Scholar] [PubMed] [CrossRef]
158. Perricone C, Ceccarelli F, Nesher G, et al. Immune thrombocytopenic Purpura (ITP) associated with vaccinations: a review of reported cases. Immunol Res 2014;60(2):226–35. doi:10.1007/s12026-014-8597-x. [Google Scholar] [PubMed] [CrossRef]
159. Finsterer J. Neurological side effects of SARS-CoV-2 vaccinations. Acta Neuro Scand 2022;145(1):5–9. doi:10.1111/ane.13550. [Google Scholar] [PubMed] [CrossRef]
160. Garg RK, Paliwal VK. Spectrum of neurological complications following COVID-19 vaccination. Neurol Sci 2022;43(1):3–40. doi:10.1007/s10072-021-05662-9. [Google Scholar] [PubMed] [CrossRef]
161. Schonberger LB, Bregman DJ, Sullivan-Bolyai JZ, et al. Guillain-Barre syndrome following vaccination in the National Influenza Immunization Program, United States, 1976–1977. Am J Epidemiol 1979;110(2):105–23. doi:10.1093/oxfordjournals.aje.a112795. [Google Scholar] [PubMed] [CrossRef]
162. Salmon DA, Proschan M, Forshee R, et al. Association between Guillain-Barré syndrome and influenza A (H1N1) 2009 monovalent inactivated vaccines in the USA: a meta-analysis. Lancet 2013;381(9876):1461–8. doi:10.1016/S0140-6736(12)62189-8. [Google Scholar] [PubMed] [CrossRef]
163. Nachamkin I, Shadomy SV, Moran AP, et al. Anti-ganglioside antibody induction by swine (A/NJ/1976/H1N1) and other influenza vaccines: insights into vaccine-associated guiflain-Barré syndrome. J Infect Dis 2008;198(2):226–33. doi:10.1086/589624. [Google Scholar] [PubMed] [CrossRef]
164. Saariaho AH, Vuorela A, Freitag TL, et al. Autoantibodies against ganglioside GM3 are associated with narcolepsy-cataplexy developing after Pandemrix vaccination against 2009 pandemic H1N1 type influenza virus. J Autoimmun 2015;63(3):68–75. doi:10.1016/j.jaut.2015.07.006. [Google Scholar] [PubMed] [CrossRef]
165. Andrews N, Stowe J, Al-Shahi Salman R, Miller E. Guillain-Barré syndrome and H1N1 (2009) pandemic influenza vaccination using an AS03 adjuvanted vaccine in the United Kingdom: self-controlled case series. Vaccine 2011;29(45):7878–82. doi:10.1016/j.vaccine.2011.08.069. [Google Scholar] [PubMed] [CrossRef]
166. Chang KH, Lyu RK, Lin WT, Huang YT, Lin HS, Chang SH. Gulllain-barre syndrome after trivalent influenza vaccination in adults. Front Neurol 2019;10:768. doi:10.3389/fneur.2019.00768. [Google Scholar] [PubMed] [CrossRef]
167. Anjum Z, Iyer C, Naz S, et al. Guillain-Barré syndrome after mRNA-1273 (Moderna) COVID-19 vaccination: a case report. Clin Case Rep 2022;10(4):e05733. doi:10.1002/ccr3.5733. [Google Scholar] [PubMed] [CrossRef]
168. Nasreen S, Jiang Y, Lu H, et al. Risk of Guillain-Barré syndrome after COVID-19 vaccination or SARS-CoV-2 infection: a multinational self-controlled case series study. Vaccine 2025;60(2):127291. doi:10.1016/j.vaccine.2025.127291. [Google Scholar] [PubMed] [CrossRef]
169. Shafiee A, Teymouri Athar MM, Amini MJ, et al. Reactivation of herpesviruses during COVID-19: a systematic review and meta-analysis. Rev Med Virol 2023;33(3):e2437. doi:10.1002/rmv.2437. [Google Scholar] [PubMed] [CrossRef]
170. Psichogiou M, Samarkos M, Mikos N, Hatzakis A. Reactivation of varicella zoster virus after vaccination for SARS-CoV-2. Vaccines 2021;9(6):572. doi:10.3390/vaccines9060572. [Google Scholar] [PubMed] [CrossRef]
171. Wang F, Gao Y, Wagner AL, Lu Y. A systematic review and meta-analysis of herpes zoster occurrence/recurrence after COVID-19 infection and vaccination. J Med Virol 2024;96(5):e29629. doi:10.1002/jmv.29629. [Google Scholar] [PubMed] [CrossRef]
172. Plüß M, Mese K, Kowallick JT, Schuster A, Tampe D, Tampe B. Case report: cytomegalovirus reactivation and pericarditis following ChAdOx1 nCoV-19 vaccination against SARS-CoV-2. Front Immunol 2021;12:784145. doi:10.3389/fimmu.2021.784145. [Google Scholar] [PubMed] [CrossRef]
173. Proal AD, VanElzakker MB. Long COVID or post-acute sequelae of COVID-19 (PASCan overview of biological factors that may contribute to persistent symptoms. Front Microbiol 2021;12:698169. doi:10.3389/fmicb.2021.698169. [Google Scholar] [PubMed] [CrossRef]
174. Kwong JC, Schwartz KL, Campitelli MA. Acute myocardial infarction after laboratory-confirmed influenza infection. N Engl J Med 2018;378(26):2540–1. doi:10.1056/NEJMc1805679. [Google Scholar] [PubMed] [CrossRef]
175. Taquet M, Husain M, Geddes JR, Luciano S, Harrison PJ. Cerebral venous thrombosis and portal vein thrombosis: a retrospective cohort study of 537, 913 COVID-19 cases. EClinicalMedicine 2021;39:101061. doi:10.1016/j.eclinm.2021.101061. [Google Scholar] [PubMed] [CrossRef]
176. Tu TM, Goh C, Tan YK, et al. Cerebral venous thrombosis in patients with COVID-19 infection: a case series and systematic review. J Stroke Cerebrovasc Dis 2020;29(12):105379. doi:10.1016/j.jstrokecerebrovasdis.2020.105379. [Google Scholar] [PubMed] [CrossRef]
177. Nicolson PL, Abrams ST, Amirthalingam G, et al. Understanding mechanisms of thrombosis and thrombocytopenia with adenoviral SARS-CoV-2 vaccines: a comprehensive synopsis. Efficacy Mech Eval 2025:1–36. doi:10.3310/ffss9010. [Google Scholar] [PubMed] [CrossRef]
178. Belay ED, Godfred-Cato S, Ak R, Al E. Multisystem inflammatory syndrome in adults after SARS-CoV-2 infection and COVID-19 vaccination. Clin Infect Dis 2022;75(1):e741–8. doi:10.1093/cid/ciab936. [Google Scholar] [PubMed] [CrossRef]
179. Nune A, Iyengar KP, Goddard C, Ahmed AE. Multisystem inflammatory syndrome in an adult following the SARS-CoV-2 vaccine (MIS-V). BMJ Case Rep 2021;14(7):e243888. doi:10.1136/bcr-2021-243888. [Google Scholar] [PubMed] [CrossRef]
180. Choi YK, Moon JY, Kim J, et al. Postvaccination multisystem inflammatory syndrome in adult with no evidence of prior SARS-CoV-2 infection. Emerg Infect Dis 2022;28(2):411–4. doi:10.3201/eid2802.211938. [Google Scholar] [PubMed] [CrossRef]
181. Heymans S, Cooper LT. Myocarditis after COVID-19 mRNA vaccination: clinical observations and potential mechanisms. Nat Rev Cardiol 2022;19(2):75–7. doi:10.1038/s41569-021-00662-w. [Google Scholar] [PubMed] [CrossRef]
182. Couzin-Frankel J, Vogel G. Vaccines may cause rare, long COVID-like symptoms. Science 2022;375(6579):364–6. doi:10.1126/science.ada0536. [Google Scholar] [PubMed] [CrossRef]
183. Fujinami RS, von Herrath MG, Christen U, Whitton JL. Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev 2006;19(1):80–94. doi:10.1128/cmr.19.1.80-94.2006. [Google Scholar] [PubMed] [CrossRef]
184. Rojas M, Restrepo-Jiménez P, Monsalve DM, et al. Molecular mimicry and autoimmunity. J Autoimmun 2018;95:100–23. doi:10.1016/j.jaut.2018.10.012. [Google Scholar] [PubMed] [CrossRef]
185. Chang SE, Feng A, Meng W, et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat Commun 2021;12(1):5417. doi:10.1038/s41467-021-25509-3. [Google Scholar] [PubMed] [CrossRef]
186. Dotan A, Muller S, Kanduc D, David P, Halpert G, Shoenfeld Y. The SARS-CoV-2 as an instrumental trigger of autoimmunity. Autoimmun Rev 2021;20(4):102792. doi:10.1016/j.autrev.2021.102792. [Google Scholar] [PubMed] [CrossRef]
187. Crayne CB, Albeituni S, Nichols KE, Cron RQ. The immunology of macrophage activation syndrome. Front Immunol 2019;10:119. doi:10.3389/fimmu.2019.00119. [Google Scholar] [PubMed] [CrossRef]
188. Verdoni L, Mazza A, Gervasoni A, et al. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet 2020;395(10239):1771–8. doi:10.1016/S0140-6736(20)31103-X. [Google Scholar] [PubMed] [CrossRef]
189. Castanares-Zapatero D, Chalon P, Kohn L, et al. Pathophysiology and mechanism of long COVID: a comprehensive review. Ann Med 2022;54(1):1473–87. doi:10.1080/07853890.2022.2076901. [Google Scholar] [PubMed] [CrossRef]
190. Schauer R. Chemistry, metabolism, and biological functions of sialic acids. Adv Carbohydr Chem Biochem 1982;40:131–234. doi:10.1016/s0065-2318(08)60109-2. [Google Scholar] [PubMed] [CrossRef]
191. Varki A. Diversity in the sialic acids. Glycobiology 1992;2(1):25–40. doi:10.1093/glycob/2.1.25. [Google Scholar] [PubMed] [CrossRef]
192. Angata T, Varki A. Chemical diversity in the sialic acids and related alpha-keto acids: an evolutionary perspective. Chem Rev 2002;102(2):439–69. doi:10.1021/cr000407m. [Google Scholar] [PubMed] [CrossRef]
193. Schauer R. Sialic acids: fascinating sugars in higher animals and man. Zoology 2004;107(1):49–64. doi:10.1016/j.zool.2003.10.002. [Google Scholar] [PubMed] [CrossRef]
194. Varki A. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature 2007;446(7139):1023–9. doi:10.1038/nature05816. [Google Scholar] [PubMed] [CrossRef]
195. Varki A. Biological roles of glycans. Glycobiology 2017;27(1):3–49. doi:10.1093/glycob/cww086. [Google Scholar] [PubMed] [CrossRef]
196. Brunngraber EG. Biochemistry, function, and neuropathology of the glycoproteins in brain tissue. In: Functional and structural proteins of the nervous system. Boston, MA, USA: Springer; 1972. p. 109–33. doi:10.1007/978-1-4684-6979-0_11. [Google Scholar] [CrossRef]
197. Wang B. Sialic acid is an essential nutrient for brain development and cognition. Annu Rev Nutr 2009;29(1):177–222. doi:10.1146/annurev.nutr.28.061807.155515. [Google Scholar] [PubMed] [CrossRef]
198. D’Addio M, Frey J, Otto VI. The manifold roles of sialic acid for the biological functions of endothelial glycoproteins. Glycobiology 2020;30(8):490–9. doi:10.1093/glycob/cwaa008. [Google Scholar] [PubMed] [CrossRef]
199. Chen HY, Fermin A, Vardhana S, et al. Galectin-3 negatively regulates TCR-mediated CD4+ T-cell activation at the immunological synapse. Proc Natl Acad Sci U S A 2009;106(34):14496–501. doi:10.1073/pnas.0903497106. [Google Scholar] [PubMed] [CrossRef]
200. Alter G, Ottenhoff THM, Joosten SA. Antibody glycosylation in inflammation, disease and vaccination. Semin Immunol 2018;39:102–10. doi:10.1016/j.smim.2018.05.003. [Google Scholar] [PubMed] [CrossRef]
201. Meri S, Pangburn MK. Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc Natl Acad Sci U S A 1990;87(10):3982–6. doi:10.1073/pnas.87.10.3982. [Google Scholar] [PubMed] [CrossRef]
202. Cohen M, Varki A. The sialome—far more than the sum of its parts. OMICS A J Integr Biol 2010;14(4):455–64. doi:10.1089/omi.2009.0148. [Google Scholar] [PubMed] [CrossRef]
203. Maverakis E, Kim K, Shimoda M, et al. Glycans in the immune system and the altered glycan theory of autoimmunity: a critical review. J Autoimmun 2015;57:1–13. doi:10.1016/j.jaut.2014.12.002. [Google Scholar] [PubMed] [CrossRef]
204. Ashwell G, Harford J. Carbohydrate-specific receptors of the liver. Annu Rev Biochem 1982;51(1):531–54. doi:10.1146/annurev.bi.51.070182.002531. [Google Scholar] [PubMed] [CrossRef]
205. Wong AH, Fukami Y, Sudo M, Kokubun N, Hamada S, Yuki N. Sialylated IgG-Fc: a novel biomarker of chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry 2016;87(3):275–9. doi:10.1136/jnnp-2014-309964. [Google Scholar] [PubMed] [CrossRef]
206. Schauer R, Kamerling JP. Exploration of the sialic acid world. Adv Carbohydr Chem Biochem 2018;75:1–213. doi:10.1016/bs.accb.2018.09.001. [Google Scholar] [PubMed] [CrossRef]
207. Morniroli D, Giannì ML, Consales A, Pietrasanta C, Mosca F. Human sialome and coronavirus disease-2019 (COVID-19) pandemic: an understated correlation? Front Immunol 2020;11:1480. doi:10.3389/fimmu.2020.01480. [Google Scholar] [PubMed] [CrossRef]
208. Chou HH, Takematsu H, Diaz S, et al. A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proc Natl Acad Sci U S A 1998;95(20):11751–6. doi:10.1073/pnas.95.20.11751. [Google Scholar] [PubMed] [CrossRef]
209. Irie A, Koyama S, Kozutsumi Y, Kawasaki T, Suzuki A. The molecular basis for the absence of N-glycolylneuraminic acid in humans. J Biol Chem 1998;273(25):15866–71. doi:10.1074/jbc.273.25.15866. [Google Scholar] [PubMed] [CrossRef]
210. Ghaderi D, Taylor RE, Padler-Karavani V, Diaz S, Varki A. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat Biotechnol 2010;28(8):863–7. doi:10.1038/nbt.1651. [Google Scholar] [PubMed] [CrossRef]
211. Decker EL, Parsons J, Reski R. Glyco-engineering for biopharmaceutical production in moss bioreactors. Front Plant Sci 2014;5:346. doi:10.3389/fpls.2014.00346. [Google Scholar] [PubMed] [CrossRef]
212. Tangvoranuntakul P, Gagneux P, Diaz S, et al. Human uptake and incorporation of an immunogenic nonhuman dietary sialic acid. Proc Natl Acad Sci U S A 2003;100(21):12045–50. doi:10.1073/pnas.2131556100. [Google Scholar] [PubMed] [CrossRef]
213. Hedlund M, Tangvoranuntakul P, Takematsu H, et al. N-glycolylneuraminic acid deficiency in mice: implications for human biology and evolution. Mol Cell Biol 2007;27(12):4340–6. doi:10.1128/MCB.00379-07. [Google Scholar] [PubMed] [CrossRef]
214. Bardor M, Nguyen DH, Diaz S, Varki A. Mechanism of uptake and incorporation of the non-human sialic acid N-glycolylneuraminic acid into human cells. J Biol Chem 2005;280(6):4228–37. doi:10.1074/jbc.M412040200. [Google Scholar] [PubMed] [CrossRef]
215. Dhar C, Sasmal A, Varki A. From serum sickness to xenosialitis: past, present, and future significance of the non-human sialic acid Neu5Gc. Front Immunol 2019;10:807. doi:10.3389/fimmu.2019.00807. [Google Scholar] [PubMed] [CrossRef]
216. Padler-Karavani V, Tremoulet AH, Yu H, Chen X, Burns JC, Varki A. A simple method for assessment of human anti-Neu5Gc antibodies applied to Kawasaki disease. PLoS One 2013;8(3):e58443. doi:10.1371/journal.pone.0058443. [Google Scholar] [PubMed] [CrossRef]
217. Galili U. Human natural antibodies to mammalian carbohydrate antigens as unsung heroes protecting against past, present, and future viral infections. Antibodies 2020;9(2):25. doi:10.3390/antib9020025. [Google Scholar] [PubMed] [CrossRef]
218. Rak A, Zabrodskaya Y, Wong PF, Isakova-Sivak I. Antibodies against SARS-CoV-2 nucleocapsid protein possess autoimmune properties. Antibodies 2025;15(1):2. doi:10.3390/antib15010002. [Google Scholar] [PubMed] [CrossRef]
219. Padler-Karavani V, Yu H, Cao H, et al. Diversity in specificity, abundance, and composition of anti-Neu5Gc antibodies in normal humans: potential implications for disease. Glycobiology 2008;18(10):818–30. doi:10.1093/glycob/cwn072. [Google Scholar] [PubMed] [CrossRef]
220. Samraj AN, Bertrand KA, Luben R, et al. Polyclonal human antibodies against glycans bearing red meat-derived non-human sialic acid N-glycolylneuraminic acid are stable, reproducible, complex and vary between individuals: total antibody levels are associated with colorectal cancer risk. PLoS One 2018;13(6):e0197464. doi:10.1371/journal.pone.0197464. [Google Scholar] [PubMed] [CrossRef]
221. Löfvenborg JE, Ahlqvist E, Alfredsson L, et al. Consumption of red meat, genetic susceptibility, and risk of LADA and type 2 diabetes. Eur J Nutr 2021;60(2):769–79. doi:10.1007/s00394-020-02285-2. [Google Scholar] [PubMed] [CrossRef]
222. Anaya JM. The diagnosis and clinical significance of polyautoimmunity. Autoimmun Rev 2014;13(4–5):423–6. doi:10.1016/j.autrev.2014.01.049. [Google Scholar] [PubMed] [CrossRef]
223. Zaramela LS, Martino C, Alisson-Silva F, et al. Gut bacteria responding to dietary change encode sialidases that exhibit preference for red meat-associated carbohydrates. Nat Microbiol 2019;4(12):2082–9. doi:10.1038/s41564-019-0564-9. [Google Scholar] [PubMed] [CrossRef]
224. Byres E, Paton AW, Paton JC, et al. Incorporation of a non-human glycan mediates human susceptibility to a bacterial toxin. Nature 2008;456(7222):648–52. doi:10.1038/nature07428. [Google Scholar] [PubMed] [CrossRef]
225. Taylor RE, Gregg CJ, Padler-Karavani V, et al. Novel mechanism for the generation of human xeno-autoantibodies against the nonhuman sialic acid N-glycolylneuraminic acid. J Exp Med 2010;207(8):1637–46. doi:10.1084/jem.20100575. [Google Scholar] [PubMed] [CrossRef]
226. Haines-Menges BL, Whitaker WB, Lubin JB, Boyd EF. Host sialic acids: a delicacy for the pathogen with discerning taste. Microbiol Spectr 2015;3(4):0005. doi:10.1128/microbiolspec.MBP-0005-2014. [Google Scholar] [PubMed] [CrossRef]
227. Guerrero-Flores GN, Butler FM, Martinez Marignac VL, Zhang G, Pacheco FJ, Boskovic DS. Sialic acids in health and disease. Biologics 2025;5(2):10. doi:10.3390/biologics5020010. [Google Scholar] [PubMed] [CrossRef]
228. Kappler K, Hennet T. Emergence and significance of carbohydrate-specific antibodies. Genes Immun 2020;21(4):224–39. doi:10.1038/s41435-020-0105-9. [Google Scholar] [PubMed] [CrossRef]
229. Newburg DS. Human milk glycoconjugates that inhibit pathogens. Curr Med Chem 1999;6(2):117–27. doi:10.2174/0929867306666220207212739; [Google Scholar] [CrossRef]
230. Wang B, Brand-Miller J, McVeagh P, Petocz P. Concentration and distribution of sialic acid in human milk and infant formulas 1 2 3. Am J Clin Nutr 2001;74(4):510–5. doi:10.1093/ajcn/74.4.510. [Google Scholar] [PubMed] [CrossRef]
231. Cacho NT, Lawrence RM. Innate immunity and breast milk. Front Immunol 2017;8:584. doi:10.3389/fimmu.2017.00584. [Google Scholar] [PubMed] [CrossRef]
232. Berger PK, Plows JF, Jones RB, et al. Human milk oligosaccharide 2′-fucosyllactose links feedings at 1 month to cognitive development at 24 months in infants of normal and overweight mothers. PLoS One 2020;15(2):e0228323. doi:10.1371/journal.pone.0228323. [Google Scholar] [PubMed] [CrossRef]
233. Rossi G, De Bellis D, Ricci S, et al. Neu5Gc expression in canine intestinal biopsies and its correlation with colitis severity, microbioma and cell apoptosis via lysosomal pathway activation. J Comp Pathol 2025;222:61. doi:10.1016/j.jcpa.2025.10.112. [Google Scholar] [PubMed] [CrossRef]
234. Rossi G, Biagini L, Ricci S, et al. Possible role of dietary N-glycolylneuraminic acid and dysbiosis in canine enteropathy pathogenesis. J Vet Intern Med 2024;38(Suppl 1):678–9; [Google Scholar]
235. Hanisch FG. Site-specific O-glycosylation of SARS-CoV-2 spike protein and its impact on immune and autoimmune responses. Cells 2024;13(2):107. doi:10.3390/cells13020107. [Google Scholar] [PubMed] [CrossRef]
236. Anderton SM. Post-translational modifications of self antigens: implications for autoimmunity. Curr Opin Immunol 2004;16(6):753–8. doi:10.1016/j.coi.2004.09.001. [Google Scholar] [PubMed] [CrossRef]
237. Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Glycosylation and the immune system. Science 2001;291(5512):2370–6. doi:10.1126/science.291.5512.2370. [Google Scholar] [PubMed] [CrossRef]
238. Peng K, Li X, Yang D, et al. Risk of autoimmune diseases following COVID-19 and the potential protective effect from vaccination: a population-based cohort study. EClinicalMedicine 2023;63(6):102154. doi:10.1016/j.eclinm.2023.102154. [Google Scholar] [PubMed] [CrossRef]
239. Bournazos S, Ravetch JV. Fcγ receptor pathways during active and passive immunization. Immunol Rev 2015;268(1):88–103. doi:10.1111/imr.12343. [Google Scholar] [PubMed] [CrossRef]
240. Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol 2020;20(6):363–74. doi:10.1038/s41577-020-0311-8. [Google Scholar] [PubMed] [CrossRef]
241. Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science 2005;310(5753):1510–2. doi:10.1126/science.1118948. [Google Scholar] [PubMed] [CrossRef]
242. Anthony RM, Nimmerjahn F. The role of differential IgG glycosylation in the interaction of antibodies with FcγRs in vivo. Curr Opin Organ Transplant 2011;16(1):7–14. doi:10.1097/MOT.0b013e328342538f. [Google Scholar] [PubMed] [CrossRef]
243. Dekkers G, Rispens T, Vidarsson G. Novel concepts of altered immunoglobulin G galactosylation in autoimmune diseases. Front Immunol 2018;9:553. doi:10.3389/fimmu.2018.00553. [Google Scholar] [PubMed] [CrossRef]
244. Krištić J, Lauc G. The importance of IgG glycosylation—What did we learn after analyzing over 100,000 individuals. Immunol Rev 2024;328(1):143–70. doi:10.1111/imr.13407. [Google Scholar] [PubMed] [CrossRef]
245. Hoepel W, Chen HJ, Geyer CE, et al. High titers and low fucosylation of early human anti-SARS-CoV-2 IgG promote inflammation by alveolar macrophages. Sci Transl Med 2021;13(596):eabf8654. doi:10.1126/scitranslmed.abf8654. [Google Scholar] [PubMed] [CrossRef]
246. Larsen MD, de Graaf EL, Sonneveld ME, et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Science 2021;371(6532):eabc8378. doi:10.1126/science.abc8378. [Google Scholar] [PubMed] [CrossRef]
247. Farkash I, Feferman T, Cohen-Saban N, et al. Anti-SARS-CoV-2 antibodies elicited by COVID-19 mRNA vaccine exhibit a unique glycosylation pattern. Cell Rep 2021;37(11):110114. doi:10.1016/j.celrep.2021.110114. [Google Scholar] [PubMed] [CrossRef]
248. Ackerman ME, Crispin M, Yu X, et al. Natural variation in Fc glycosylation of HIV-specific antibodies impacts antiviral activity. J Clin Invest 2013;123(5):2183–92. doi:10.1172/JCI65708. [Google Scholar] [PubMed] [CrossRef]
249. Vogelpoel LT, Hansen IS, Rispens T, et al. Fc gamma receptor-TLR cross-talk elicits pro-inflammatory cytokine production by human M2 macrophages. Nat Commun 2014;5(1):5444. doi:10.1038/ncomms6444. [Google Scholar] [PubMed] [CrossRef]
250. Group The RECOVERY Collaborative. Dexamethasone in hospitalized patients with covid-19. N Engl J Med 2021;384(8):693–704. doi:10.1056/nejmoa2021436. [Google Scholar] [PubMed] [CrossRef]
251. World Health Organization (WHO). WHO living guideline: therapeutics and COVID-19 [Internet]. Geneva, Switzerland: WHO; 2020[cited 2026 May 5]. Available from: https://www.who.int/publications/i/item/WHO-2019-nCoV-therapeutics-2020.1. [Google Scholar]
252. National Institutes of Health. COVID-19 Treatment Guidelines. Bethesda, MD, USA: NIH; 2024. [Google Scholar]
253. Bouman A, Heineman MJ, Faas MM. Sex hormones and the immune response in humans. Hum Reprod Update 2005;11(4):411–23. doi:10.1093/humupd/dmi008. [Google Scholar] [PubMed] [CrossRef]
254. Ghosh S, Klein RS. Sex drives dimorphic immune responses to viral infections. J Immunol 2017;198(5):1782–90. doi:10.4049/jimmunol.1601166. [Google Scholar] [PubMed] [CrossRef]
255. Schurz H, Salie M, Tromp G, Hoal EG, Kinnear CJ, Möller M. The X chromosome and sex-specific effects in infectious disease susceptibility. Hum Genomics 2019;13(1):2. doi:10.1186/s40246-018-0185-z. [Google Scholar] [PubMed] [CrossRef]
256. Bukowska A, Spiller L, Wolke C, et al. Protective regulation of the ACE2/ACE gene expression by estrogen in human atrial tissue from elderly men. Exp Biol Med 2017;242(14):1412–23. doi:10.1177/1535370217718808. [Google Scholar] [PubMed] [CrossRef]
257. Luo Y, Liu C, Guan T, et al. Association of ACE2 genetic polymorphisms with hypertension-related target organ damages in south Xinjiang. Hypertens Res 2019;42(5):681–9. doi:10.1038/s41440-018-0166-6. [Google Scholar] [PubMed] [CrossRef]
258. Muthana SM, Gildersleeve JC. Glycan microarrays: powerful tools for biomarker discovery. Chem Rev 2014;114(3):2170–202. doi:10.1021/cr400319x; [Google Scholar] [CrossRef]
259. Leviatan Ben-Arye S, Schneider C, Yu H, et al. Differential recognition of diet-derived Neu5Gc-neoantigens on glycan microarrays by carbohydrate-specific pooled human IgG and IgA antibodies. Bioconjug Chem 2019;30(5):1565–74. doi:10.1021/acs.bioconjchem.9b00273. [Google Scholar] [PubMed] [CrossRef]
260. Tran PMH, Dong F, Kim E, et al. Use of a glycomics array to establish the anti-carbohydrate antibody repertoire in type 1 diabetes. Nat Commun 2022;13(1):6527. doi:10.1038/s41467-022-34341-2. [Google Scholar] [PubMed] [CrossRef]
261. Hutton E, Uno Y, Scott E, Robson C, Fascione MA, Signoret N. A general and accessible approach to enrichment and characterisation of natural anti-Neu5Gc antibodies from human samples. RSC Chem Biol 2025;6(7):1135–47. doi:10.1039/d5cb00073d. [Google Scholar] [PubMed] [CrossRef]
262. Wang H, Wu ZC, Hu P, et al. Identification of chicken-derived scFv against N-glycolylneuraminic acid retrieved from an immune library by phage display. Protein Expr Purif 2021;182:105841. doi:10.1016/j.pep.2021.105841. [Google Scholar] [PubMed] [CrossRef]
263. Bell A, Severi E, Owen CD, Latousakis D, Juge N. Biochemical and structural basis of sialic acid utilization by gut microbes. J Biol Chem 2023;299(3):102989. doi:10.1016/j.jbc.2023.102989. [Google Scholar] [PubMed] [CrossRef]
264. Robinson LS, Lewis WG, Lewis AL. The sialate O-acetylesterase EstA from gut Bacteroidetes species enables sialidase-mediated cross-species foraging of 9-O-acetylated sialoglycans. J Biol Chem 2017;292(28):11861–72. doi:10.1074/jbc.M116.769232. [Google Scholar] [PubMed] [CrossRef]
265. Zhang Z, Shao X, Al-Dalali S, Xu B, Li P. Distinct mechanisms of N-glycolylneuraminic acid reduction by Lactiplantibacillus plantarum R2 and Staphylococcus carnosus C1: adsorption versus biodegradation. Food Biosci 2025;74(9):107937. doi:10.1016/j.fbio.2025.107937. [Google Scholar] [PubMed] [CrossRef]
266. Caccialanza R, Laviano A, Lobascio F, et al. Early nutritional supplementation in non-critically ill patients hospitalized for the 2019 novel coronavirus disease (COVID-19rationale and feasibility of a shared pragmatic protocol. Nutrition 2020;74:110835. doi:10.1016/j.nut.2020.110835. [Google Scholar] [PubMed] [CrossRef]
267. Hartmann-Boyce J, Morris E, Goyder C, et al. Diabetes and COVID-19: risks, management, and learnings from other national disasters. Diabetes Care 2020;43(8):1695–703. doi:10.2337/dc20-1192. [Google Scholar] [PubMed] [CrossRef]
268. Altman MO, Gagneux P. Absence of Neu5Gc and presence of anti-Neu5Gc antibodies in humans—an evolutionary perspective. Front Immunol 2019;10:789. doi:10.3389/fimmu.2019.00789. [Google Scholar] [PubMed] [CrossRef]
269. Kim Y, Je Y. Meat consumption and risk of metabolic syndrome: results from the Korean population and a meta-analysis of observational studies. Nutrients 2018;10(4):390. doi:10.3390/nu10040390. [Google Scholar] [PubMed] [CrossRef]
270. Soulillou JP, Cozzi E, Bach JM. Challenging the role of diet-induced anti-Neu5Gc antibodies in human pathologies. Front Immunol 2020;11:834. doi:10.3389/fimmu.2020.00834. [Google Scholar] [PubMed] [CrossRef]
271. Reddy R, Carpenter E, Halpin A, et al. Evaluation of multiplexed Liquid Glycan Array (LiGA) for serological detection of glycan-binding antibodies. Glycobiology 2025;35(11):cwaf042. doi:10.1093/glycob/cwaf042. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools