
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

The role of FZR1 in tumorigenesis: Focus on cell-cycle control
1 Guizhou Provincial College-Based Key Lab for Tumor Prevention and Treatment with Distinctive Medicines, Zunyi Medical University, Zunyi, 563000, China
2 Department of Hematology, Southwest Hospital, Third Military Medical University (Army Medical University), Chongqing, 400038, China
* Corresponding Authors: YUN LIU. Email: ; JIEPING CHEN. Email:
BIOCELL 2023, 47(10), 2177-2186. https://doi.org/10.32604/biocell.2023.029373
Received 15 February 2023; Accepted 21 June 2023; Issue published 08 November 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Fizzy-related protein homolog 1 (FZR1) mainly functions as a specific activator of the anaphase-promoting complex/cyclosome (APC/C) in the cell cycle and controls the G0 and G1 phases of the cell cycle. We highlight recent work that has studied the role of FZR1 in tumorigenesis, growth, differentiation, and genome stability through cell-cycle control. We summarize the current state of knowledge regarding FZR1 structure, function, and the distinct ways of APC/C dysregulation in solid tumors and hematologic malignancies. We also discuss novel approaches for targeting the FZR1 as a cancer therapy and research area for future work.Keywords
Abbreviations
| APC/C | The anaphase-promoting complex/cyclosome |
| AMPK | AMP-activated protein kinase |
| Cdh1 | Cell division cycle 20 related 1 |
| CDC20 | Cell division cycle 20 homolog |
| CDK | Cyclin-dependent kinase |
| CENP-T | Centromere protein T |
| CSC | Cancer stem-like cells |
| CPC | Chromosomal passenger complex |
| ECs | Endothelial cells |
| FZR1 | Fizzy-related protein homolog 1 |
| HSCs | Hematopoietic stem cell |
| MM | Multiple myeloma |
| MEKK2 | Mitogen-activated protein kinase 2 |
| PLK1 | Polo-like kinase 1 |
| pRb | Retinoblastoma protein |
| SAC | Spindle assembly checkpoint |
| SKP2 | S-phase kinase-associated protein 2 |
| topiα | Telomerase IIα |
| SCs | Satellite cells |
| SPOP | Speckle-type POZ protein |
| TBK1 | Tank binding kinase 1 |
Fizzy-related protein homolog1 (FZR1) is named after the Drosophila homolog fizzy, the anaphase-promoting complex/cyclosome (APC/C) coactivator, and is therefore also called CDC20 homolog1 (Cdh1). FZR1 mainly comprises a β-propeller with a seven-leaf WD40 repeat and a C-box containing important functional motifs (Pines, 2011; Primorac and Musacchio, 2013) (Figs. 1A and 1B). It can use its WD40 repeat domain to recruit to different APC/C (the anaphase-promoting complex/cyclosome) substrates for ubiquitination degradation of the substrates. The D box directly interacts with the evolutionarily conserved surface on the WD40 propeller structure of FZR1. It is activated by the binding of CDC27, allowing APC/C to recognize the substrates D box and KEN box (Harper et al., 2002; Kraft et al., 2005; Brown et al., 2015).
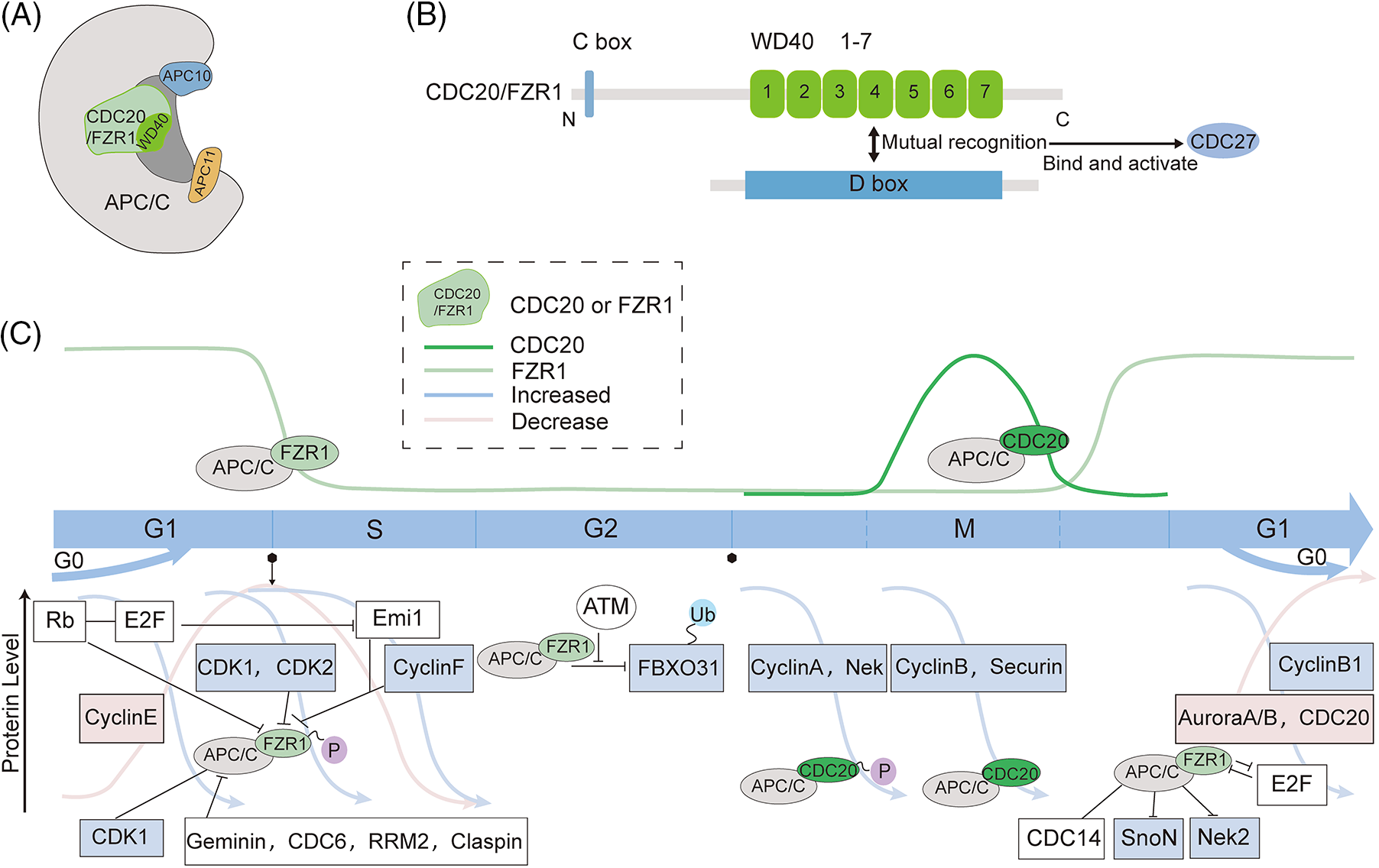
Figure 1: Schematic structure and intra-cycle regulation of fizzy-related protein homolog 1 (FZR1). (A to B) FZR1 mainly comprises a β-propeller with a seven-leaf WD40 repeat and a C-box containing significant functional motifs. (C) APC/CFZR1 activity regulates the cell cycle process and fluctuates throughout the cell cycle. APC/CFZR1 maintains its stability as a tumor suppressor after cells enter the G1 phase. In the G1 phase, Rb represses E2F by binding to transcription factors and regulates APC/CFZR1 through a double negative feedback mechanism of Emi1. Cyclin E accumulates and activates Cdk2, followed by CDK1 and Cdk2 to phosphorylate FZR1, and geminin, CDC6, RRM2, and claspin to inactivate APC/CFZR1, allowing the cell to enter the S phase. Upon inactivation of APC/CFZR1 by Cdk2, Emi1 is converted from a substrate of APC/C to its inhibitor. Although FZR1 levels are low during G2, it can still act through the ubiquitinated tumor suppressor FBXO31. However, this interaction is disrupted if FBXO31 is phosphorylated by the DNA damage response kinase ATM. Cyclin B degradation leads to CDK inhibition, dephosphorylation, and FZR1 activation. APC/CFZR1 initiates the degradation of various proteins, such as CDC20 and Aurora A/B, during prophase and telophase. At the end of mitosis, APC/CFZR1 inactivates APC/CCDC20 and regulates late spindle dynamics and cytoplasmic division. Thus, APC/CFZR1 is activated in late mitosis and controls proliferation and differentiation decisions during the G1 phase. The Wnt and TGF-β pathways are conserved extracellular signaling pathways that regulate different developmental and physiological processes in multicellular organisms. FZR1 regulates the pathway by regulating cellular SnoN transcription factors and Nek2 degradation. APC/CCDC20 content increases during mitosis and regulates the cell cycle through cytokinesis. This partially inhibits the substrate recruitment of APC/C activated by CDC20, but APC/CCDC20 can further degrade various pro-term and mid-term proteins such as cell cycle A, cell cycle B, and securin, thereby mediating chromosome segregation and initiating mitosis out.
The ligand for FZR1 Is APC/C, a conserved multi-subunit E3 ubiquitination ligase consisting of 14 proteins with 19 subunits and 3 associated adaptors. It contains a catalytic core, platform, and tetrapeptide repetition, including APC1~APC16, and is primarily known for its ability to trigger a mid-to-late transition in mitosis. Its crucial role in cell division makes it essential for survival and development, and is involved in abnormal regulation of tumorigenesis (Peters, 2006; Thornton and Toczyski, 2006; Wang et al., 2009; Schreiber et al., 2011; Uzunova et al., 2012; Chang et al., 2015; Yamaguchi et al., 2015). Ubiquitination of substrates by APC/C requires the formation of an APC/C-activator-substrate complex. FZR1 and CDC20 act as activators of APC/C to induce conformational changes in the active forms APC/CCDC20 and APC/CFZR1, which regulate the targeting of APC/C to specific substrates. The disruption box is a pattern frequently found in the APC substrate, but others may also be recognized by APC/CCDC20 or APC/CFZR1 (Harper et al., 2002) (Fig. 1A).
In this review, we present the role of FZR1 in tumors and discuss the feasibility of targeting FZR1 in cancer therapy. First, we introduce the function of FZR1 as an APC/C coactivator in regulating the tumor cell cycle, differentiation, genome stability, and DNA damage of tumor cells in recent years. The underlying mechanism and potential of FZR1 as target genes in tumor therapy are also discussed.
Functions of Fizzy-Related Protein Homolog 1
There is a high homology between Drosophila fizzy (fzy) and eukaryotic fizzy-related (fzr) (Sigrist and Lehner, 1997). The most classical function of FZR1 is to regulate the cell cycle in association with APC/C, and its general function is similar to that of CDC20, but it is mainly active in late mitosis, and G1 phase (Fig. 1C). CDC20 and FZR1 are substrate-specific adapters of APC/C during the cell cycle and can function as targets of the spindle assembly checkpoint (SAC) (Pines, 2011; Primorac and Musacchio, 2013). The substrate of CDC20 is mainly a mitogenic protein, which functions at the initiation of mitosis by APC/CCDC20, and phosphorylation of APC/C is critical for its mitogenic regulation (Kraft et al., 2003).
In cell progression, APC/CCDC20 is present at the onset of mitosis until the onset of anaphase (Fig. 1C). Early in mitosis, CDC20 becomes more active and binds to phosphorylated APC/C, leading to the degradation of prometaphase substrates cyclin A and Nek2 (Kramer et al., 2000; Kraft et al., 2003; Qiao et al., 2016; Zhang et al., 2016). Once all chromosomes have reached proper microtubule double orientation at metaphase, APC/CCDC20 degrades securin and cyclin B, facilitating the cell transition to anaphase (Fig. 1C). Degradation of cyclin B eliminates cyclin-CDK activity at anaphase and enables APC/CFZR1 activation, which disrupts other mitotic regulators, including CDC20 (Listovsky et al., 2004). APC/CFZR1 appears in late G1 and promotes efficient exit from mitosis and maintenance of the G1 state (Fig. 1C). In addition, FZR1 plays a crucial role in controlling proliferation, differentiation, and maintenance of genome integrity.
In early mitosis, CDC20 begins to become more active and binds to phosphorylated APC/C, dislocated from the C-box binding site of the inhibitory region Apc1-loop300, allowing CDC20 to bind, leading to degradation of the prometaphase substrates cyclin A and Nek2 (Kraft et al., 2003; Qiao et al., 2016; Zhang et al., 2016; Yamano, 2019). The autoinhibitory loop in APC1 prevents CDC20 from binding to APC/C, and phosphorylation alleviates this inhibition (Qiao et al., 2016). Interestingly, CDC20 binds to phosphorylated APC/C, whereas its phosphorylation status is dispensable for its binding to APC/C. However, due to competition from FZR1, APC/CCDC20 is maintained at a low level for most of the cell cycle (Fig. 1C); as a result, APC/C lacks sufficient phosphorylation to form an activated APC/CCDC20 complex, which makes FZR1 more competitive to bind APC/C (Fujimitsu et al., 2016). At the onset of anaphase, APC/C requires the activating protein CDC20 to target safety proteins and cyclin B1 for proteasome-dependent degradation but then relies on the CDC20-associated protein FZR1 to remain active until the onset of the next S phase (Sigl et al., 2009). Degradation of cyclin B eliminates cyclin-CDK activity at anaphase and allows APC/CFZR1 activation, disrupting the remaining mitotic regulators, including CDC20 (Brandeis and Hunt, 1996; García-Higuera et al., 2008). APC/CFZR1 appears in the G1 phase of late cells and promotes efficient exit from mitosis and maintenance of the G1 state.
Late in mitosis, dephosphorylation of FZR1 activates APC/C, allowing it to continue ubiquitinating mitotic cycle proteins and new targets in place of CDC20 to drive cell mitosis into G0/G1 (Peters, 2006; Skaar and Pagano, 2008; Pines, 2011). APC/CFZR1 activity is present throughout G1 until FZR1 is inactivated at the G1 to S transition through degradation, phosphorylation, and binding of Emi1. At the G1/S transition, FZR1 is phosphorylated, leading to its dissociation from APC/C. After completion of mitosis, APC/C remains active until the next S phase (Brandeis and Hunt, 1996; Skaar and Pagano, 2008); the inactivation of APC/CFZR1 is required for the G1 to S transition of cells (Li and Zhang, 2009). DNA replication in the S phase, requires cyclin-dependent kinase 2 (CDK2) activation. The changes in regulatory trigger mechanism, APC/CFZR1 inactivation, and Emi1 content are the key points to control permanent cell cycle entry. Emi1 performs rapid and irreversible inactivation of APC/CFZR1. After its inactivation, it cannot revert to quiescence (Cappell et al., 2016, 2018).
In mammals, the placenta is an essential organ of the embryo, and deletion of FZR1 can affect placental development. In the placenta, polyploid giant cells are acquired by endoreplication, and placental giant cells fail to form in the absence of FZR1, suggesting that APC/CFZR1 is required for endoreplication of DNA and that to undergo endoreplication, cells must not enter mitosis after one round of DNA synthesis (García-Higuera et al., 2008; Li et al., 2008).
Fizzy-Related Protein Homolog 1 Is a Master Regulator of the G1 Phase and the Quiescent G0 Phase in Tumor
Cell cycle control occupies a central place in the complex rules that govern the various processes within our cells, and the exit of mitosis requires the degradation of mitotic cyclins (Amon et al., 1994). In the past, many studies have shown the role of FZR1 in the cell cycle. Despite substrate overlap, APC/CCDC20 mainly regulates metaphase and postmitotic transitions and regulates mitotic exit, while APC/CFZR1 is active at the end of mitosis and G1 phase (Fig. 1C). FZR1 can regulate the rapid degradation of mitotic cyclin in the cell cycle (Sigrist and Lehner, 1997). Phosphorylation and dephosphorylation of FZR1 regulate the binding, activation, and separation of APC/C with CDC20 and FZR1 (Kramer et al., 2000) Phosphorylated FZR1 is found to be limited in binding to APC/C during the cell cycle, thereby blocking the activation of APC/C (Kramer et al., 2000). In FZR1 deficient cells, the G1 phase is shortened, and the S phase is prolonged. The retinoblastoma protein (pRb) is another G1/G0 regulatory factor. FZR1 has been confirmed to bind to pRb with synergistic effects in the G1/G0 phase (Kernan et al., 2018). The inactivation of FZR1 is critical for cells to enter the S phase (Cappell et al., 2018). During the G1 phase, retinoblastoma (Rb) inhibits E2F by binding to transcription factors (Yao et al., 2008). Accumulation of cyclin E activates Cdk2, which phosphorylates FZR1, inactivates APC/CFZR1, and causes cells to enter the S phase (Yao et al., 2008). After Cdk2 inactivates APC/CFZR1, Emi1 changes from a substrate of APC/C to its inhibitor, which inactivates Emi1 irrepressibly, making it unable to return to the G0 phase (Cappell et al., 2016, 2018), allowing cells to enter the S phase, CDK inhibitors cooperate with FZR1 to maintain the G0/G1 phase state (Kim and Bonni, 2007). The inactivation of CDK1 phosphorylates FZR1 and binds APC/C to degrade CDC20. APC/CFZR1 is a key factor in G1/G0 regulation by keeping cells in the G1 phase or entering the G0 phase (Kernan et al., 2018). Moreover, several replication regulators of APC/CFZR1 are disrupted during the G1/S transition, including Geminin (McGarry and Kirschner, 1998), CDC6 (Petersen et al., 2000), RRM2 (Chabes et al., 2003), and claspin (Bassermann et al., 2008), which can inactivate APC/CFZR1. Although the activity of APC/CFZR1 decreases after the G1/S transition, there is a role for FZR1 in the G2 phase, and FBXO31, an adapter for the E3 ligase SCF, has been suggested to function as a substrate for APC/CFZR1, with the two being dependent on both AKT and ATM kinases, among them, the oncogene AKT ubiquitinates FBXO31 via APC/CFZR1 (Choppara et al., 2018). AKT can regulate APC/CFZR1 recognition of SKP2 and cyclin F (Wei et al., 2004; Choudhury et al., 2016). In the G2 phase, FZR1 and CDC20 accumulate, but APC/CFZR1 remains inactive in late mitosis due to the inhibition of the protein Emi1 and phosphorylation of cell cycle protein-dependent kinases (CDKs), which inhibit FZR1 binding to APC/C in late prophase. Unlike Emi1 regulation, the accumulation of APC/C substrate proteins can be promoted by rapid dissociation of FZR1 after human cytomegalovirus (HCMV) infection of quiescent cells (Wiebusch et al., 2005). In FZR1-deficient cells, cyclin B1 is efficiently degraded during mitosis, with a shortened G1 phase and prolonged S phase, causing a proliferation defect, and is largely independent of p53 (Sigl et al., 2009) (Fig. 1C). APC/CFZR1 can conduct intracellular and extracellular cascade signals. For example, the SnoN transcription factor is thought to be a substrate of FZR1; FZR1 may directly interact with SnoN and Smad3 to induce SnoN degradation, which makes APC/C-dependent, and thus promotes TGF-β-induced transcription (Stroschein et al., 2001; Wan et al., 2001) (Fig. 1C).
The property of FZR1 is also seen in diseases. In breast cancer, FZR1 mediates cisplatin-induced apoptosis by weakening phosphorylation at the ser15 position involved in p53 stability, which can lead to breast cancer cell cycle arrest (Liu et al., 2020). While shortening the G1 phase, FZR1 knockout also leads to the accumulation of DNA damage in progenitor cells, and premature entry of cells into the S phase can also lead to p53-mediated apoptosis and a decrease in the number of progenitor cells (Delgado-Esteban et al., 2013; Eguren et al., 2013). FZR1 can be used as a substrate for CDK4/6, phosphorylating the Ser714 and Thr719 sites located between the A and B regions of the pocket to reverse the cytokinesis blocking effect induced by CDK4/6-specific inhibitors (The et al., 2015). In addition, FZR1 directly interacts with Src in breast cancer cells, enhances binding between the N-terminal and c-terminal structural domains of Src, locks Src in a closed conformation to inhibit Src, and can induce phosphorylation at the Y419 position with enhanced binding capacity in the G2 phase (Han et al., 2019). CDC27 is a core component of the APC/C that anchors FZR1 to the ubiquitinated substrate on APC/C (Peters, 2006). Concurrently, its dysregulation can lead to premature activation of the APC/CCDC20 complex and subsequent abnormal degradation of FZR1, resulting in abnormal mitosis and aneuploidy (Link et al., 2016). FZR1 binds directly to the LxCxE-binding vesicle of pRB via a cysteine residue in its LxCxD motif, but this motif does not affect the association of FZR1 with core APC/C. When this interaction is significantly disrupted, polo-like kinase 1 (PLK1) and S phase kinase-associated protein 2 (SKP2) accumulate, and the expression of the proteins p27Kip1 and p21Cip1 are down-regulated. Defects exist in the G1 cell cycle (Ramanujan et al., 2021). In neurological disorders, Cdk5 (cyclin-dependent kinase 5) can be transported to the cytoplasm in response to neuronal stress to eliminate its cell cycle inhibitory activity. Neuronal re-entry into the S phase is ubiquitinated by the E3 ligase APC/CFZR1. Ubiquitinated Cdk5 is then rapidly degraded by the proteasome, and subsequently, it is ubiquitinated by FZR1 and rapidly degraded, which plays a key role in the protective function of many neurodegenerative diseases (Zhang et al., 2012) (Fig. 1C). Moreover, APC/CFZR1 controls casein kinase 1δ (CK1δ) levels to regulate the proliferation and cell cycle exit of developing mouse cerebellar granule cell progenitors (GCPs) (Penas et al., 2015). In the hematopoietic system, Recent studies have revealed new roles of FZR1 in the blood system. FZR1 haploinsufficiency in Fzr1 mice impairs hematopoietic stem cell (HSCs) quiescence and self-renewal, and FZR1 deficiency impairs RUNX1 ubiquitination in patients with severe aplastic anemia. This leads to RUNX1 protein accumulation and disturbs the quiescence of hematopoietic stem cells in patients with severe aplastic anemia (Zhou et al., 2022). The progression of replication forks in FZR1-deficient fetal hepatocytes is slowed, resulting in a delayed G2/M cell cycle (Cuadrado et al., 2022). In the G1 phase of the cancer stem-like cells (CSC), hyperphosphorylation of FZR1 itself can decrease APC/CFZR1 activity, mainly because APC/CFZR1 destroys borealin protein during the G1 process. Throughout the cell cycle, the levels of boreal in Aurora B remained constant (De et al., 2019). Functional inhibition of tank binding kinase 1 (TBK1) increases the association of FZR1 with APC1, leading to increased mitotic aberrations (Maan et al., 2021). Progression of the replication fork is slowed in FZR1-deficient fetal hepatocytes, resulting in a G2/M cell cycle delay (Cuadrado et al., 2022). FoxM1 haploinsufficiency can improve muscle regeneration in Fzr1 knockout mice by addressing the depletion of quiescent muscle satellite cells (SCs) pool through the promotion of SCS to enter the cell cycle (Chen et al., 2020). These results confirm the role of FZR1 in the cell cycle, including its interaction with CDC20 protein interaction and other cycles, and change the content to influence the cell cycle process, and potentially lead to either delayed or accelerated cell cycle process depending on FZR1 (Fig. 1C).
Fizzy-Related Protein Homolog 1 Regulates Differentiation via the Cell Cycle
The process of cell differentiation occurs when the stem cells of multicellular animals divide and give rise to various cell types. During this process, the gene expression of the daughter cells is controlled. A critical component that regulates G1 residence on a bistable switch is also used to hold the differentiation component, causing G1 to be coordinately arrested and differentiated. As was previously mentioned, FZR1 inactivation during the G1 phase is crucial for starting a new cell cycle. Several mechanisms regulate cell cycle departure and differentiation. Some studies suggest that FZR1 not only participates in the cell cycle but may also encourages cell cycle exit and differentiation of specific cell types.
The role of FZR1 in differentiation has been described earlier. During yeast cell division, Cdk inhibitors are downregulated. FZR1 is activated by the pheromone signaling pathway and binds to and inhibits Cln-Cdc28 kinase, resulting in G1 cell cycle arrest (Peter and Herskowitz, 1994). When primary neural stem cells are differentiated into neurons, the presence of all-trans retinoic acid-induce an up-regulation of FZR1 expression and down-regulation of downstream substrate Id2 expression, indicating the control role of FZR1 in the differentiation of neural stem cells into neurons (Yao et al., 2010). Retinoic acid can stimulate neuroblastoma cells by controlling FZR1 nuclear accumulation, Skp2 degradation and p27 accumulation to cause cell cycle arrest and differentiation, and encourage the differentiation of human SH-SY5Y neuroblastoma cells (Cuende et al., 2008) (Fig. 2).
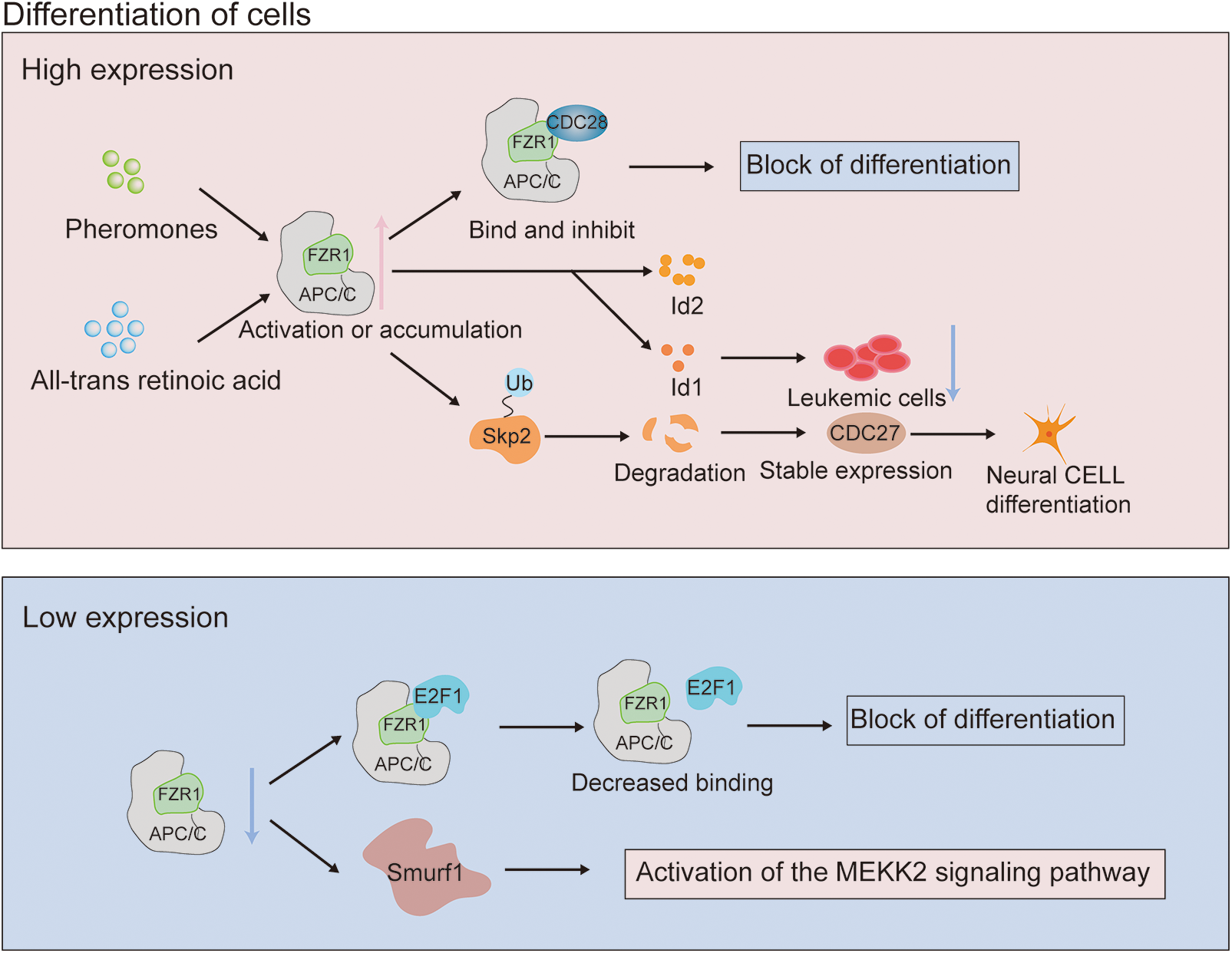
Figure 2: Mechanism of action of fizzy-related protein homolog 1 (FZR1) in tumor differentiation. Enhanced FZR1 expression is induced by CDK inhibitor and all-trans retinoic acid stimulation. Cdk inhibitors negatively modulate the role of FZR1 in differentiation, activated by the pheromone signal transduction pathway to bind and inhibit Cln-Cdc28 kinase. The role of FZR1 in differentiation upregulates the cyclin-dependent kinase p27. The interaction of FZR1 controls p27 stability by targeting Skp2 ubiquitination degradation, which promotes cell cycle exit and regulates differentiation. Inhibition of FZR1 prevents its binding to E2F1, which together regulate post-translational modification and down-regulate E2F1, producing a response to differentiation signals.
FZR1 may also be as crucial in differentiation as cyclin-dependent kinase p27 or the tumor suppressor retinoblastoma protein (pRb), primarily because pRb interacts with APC/CFZR1 to control the stability of p27 by targeting Skp2 ubiquitination degradation and promoting pRb-mediated cell cycle exit (Binné et al., 2007). The Id gene is expressed in human acute myeloid leukemia cells and inhibition of Id1 expression can prevent the growth of leukemia cell lines; therefore, FZR1 may be involved in hematopoietic stem cell differentiation by controlling Id protein and other likely targets. This is because Id is necessary for the proliferation of leukemia cells. Myeloid cancers may occur if the expression is dysregulated (Ji et al., 2008). Like FZR1, ras also interacts with pRb to influence differentiation, contribute to tumorigenicity, and may even lead to metastatic disease and increase the likelihood of overlapping transformation pathways (Fig. 2). FZR1 is a G0/G1 regulator, which can control the differentiation of hematopoietic stem cells into mature progenitor cells of the lineage and regulate the self-renewal of hematopoietic stem cells (Ishizawa et al., 2011). Abnormal expression of E2F1 can promote non-melanoma skin cancer, and FZR1 binds to E2F1 in keratinocytes. Inhibition of FZR1 prevents its binding to E2F1, co-regulating post-translational modification, and down-regulating E2F1, resulting in a response to differentiation signals (Singh and Dagnino, 2017).
In the Fzr1 knockout mouse model, genetic ablation of Fzr1 impaired the ability of APC/C to promote neurogenesis by delaying progenitor cell exit from the cell cycle (Delgado-Esteban et al., 2013). Loss of FZR1 can induce SmURF1-induced activation of the MEKK2 signaling pathway and induce osteoblast differentiation (Wan et al., 2011). While the significance of many of the described mechanisms has yet to be confirmed through in vivo models, FZR1 plays a crucial role in coordinating proliferation and differentiation.
Fizzy-Related Protein Homolog 1 Regulates the G2/M Checkpoint upon DNA Damage
High-fidelity DNA replication and chromosome separation are necessary to preserve genome integrity and prevent carcinogenesis. The regulatory role of FZR1 on the G2/M phase transition has been confirmed earlier. In G2 phase, CDK1 phosphorylates CDC20 and activates CDC20 (Kramer et al., 2000; Kraft et al., 2003), and APC/CCDC20 degrades Nek and cyclinA in prometaphase (Boekhout and Wolthuis, 2015), securin and cyclinB1 in metaphase (Fig. 1C). FZR1 remains silent during G2 phase and early mitosis due to phosphorylation (Kramer et al., 2000; Peters, 2002), whereas in anaphase, CDC14 dephosphorylates FZR1 (Visintin et al., 1998) (Fig. 1C) and ubiquitinates CDC20 (Huang et al., 2001), Aurora kinase (Littlepage and Ruderman, 2002), polo-like kinase 1 (PLK1) ensure mitotic exit.
In oocytes, CENR-T overexpression inactivates FZR1 and accelerates the G2/M1 phase transformation of oocytes (Wang et al., 2020). CenpH competed with APC/CFZR1 in the centromeric inner plate, affecting cyclin B1 stability, resulting in the downregulation of MPF activation and regulating G2/M transition (Zhang et al., 2017). In addition, Hec1 can stabilize cyclin B2, while APC/CFZR1 can destroy cyclin B2 and prevent cyclin from transforming into the G2/M phase (Gui and Homer, 2013). FZR1 was found to be an essential component of the G2 checkpoint induced by DNA damage in chicken B-cell lymphoma cell lines. After DNA damage, FZR1-APC complex was activated to induce G2/M transition regulator degradation and G2 arrest (Sudo et al., 2001) (Fig. 3A). At the same time, after DNA damage in the G2 phase, Cdc14B activates APC/CFZR1 and promotes the degradation of mitotic kinase Plk1, thereby regulating the G2 DNA damage response checkpoint (Bassermann et al., 2008). The deficiency of FZR1 can cause the accumulation of DNA damage in mature red blood cells (Cuadrado et al., 2022). The loss of Fzr1 in vivo increases the DNA damage in B-ALL cells, and long-term loss can lead to drug resistance (Ishizawa et al., 2017). After DNA damage, FZR1 is required at the G2 DNA damage checkpoint; its dephosphorylation and rebinding to APC/C lead to ubiquitination of PLK1, preventing entry into mitosis. As an adapter for APC/C (Fig. 3B), FZR1 targets the DNA terminal excision factor RBBP 8/CtIP for ubiquitination and subsequent proteasome degradation. The regulation of the turnover of RBBP 8/CtIP proteins can possibly play a role in DNA damage response by facilitating DNA double-stranded repair via error-prone non-homologous terminal junction compared to error-free RBBP 8-mediated homologous recombination (Fig. 3C). The discovery that FZR1 deficiency decreased dNTP levels and increased DNA fragmentation strongly suggested that abnormal replication was the cause of genomic instability in primary mouse embryo fibroblasts, as was the finding that acute depletion or permanent ablation of FZR1 limited the progression of replication fork mobility (Fig. 3C). These findings underscore the importance of FZR1 activity in reducing replication stress and the subsequent chromosomal breakage leading to tumor development (Garzón et al., 2017). Some studies have shown that decreased levels of silence information regulator 2 can lead to increased levels of the mitotic regulators Aurora-A and Aurora-B, which deacetylates APC/CFZR1 to regulate APC/C activity. Results in centrosome amplification, aneuploidy, and mitotic cell death, suggest that silence information regulator 2 acts to suppress tumors by regulating mitosis and genome integrity (Kim et al., 2011). In future work, it will be interesting to understand how oncogene-induced genomic instability is promoted when DNA damage caused by FZR1 deletion occurs (Fig. 3D).
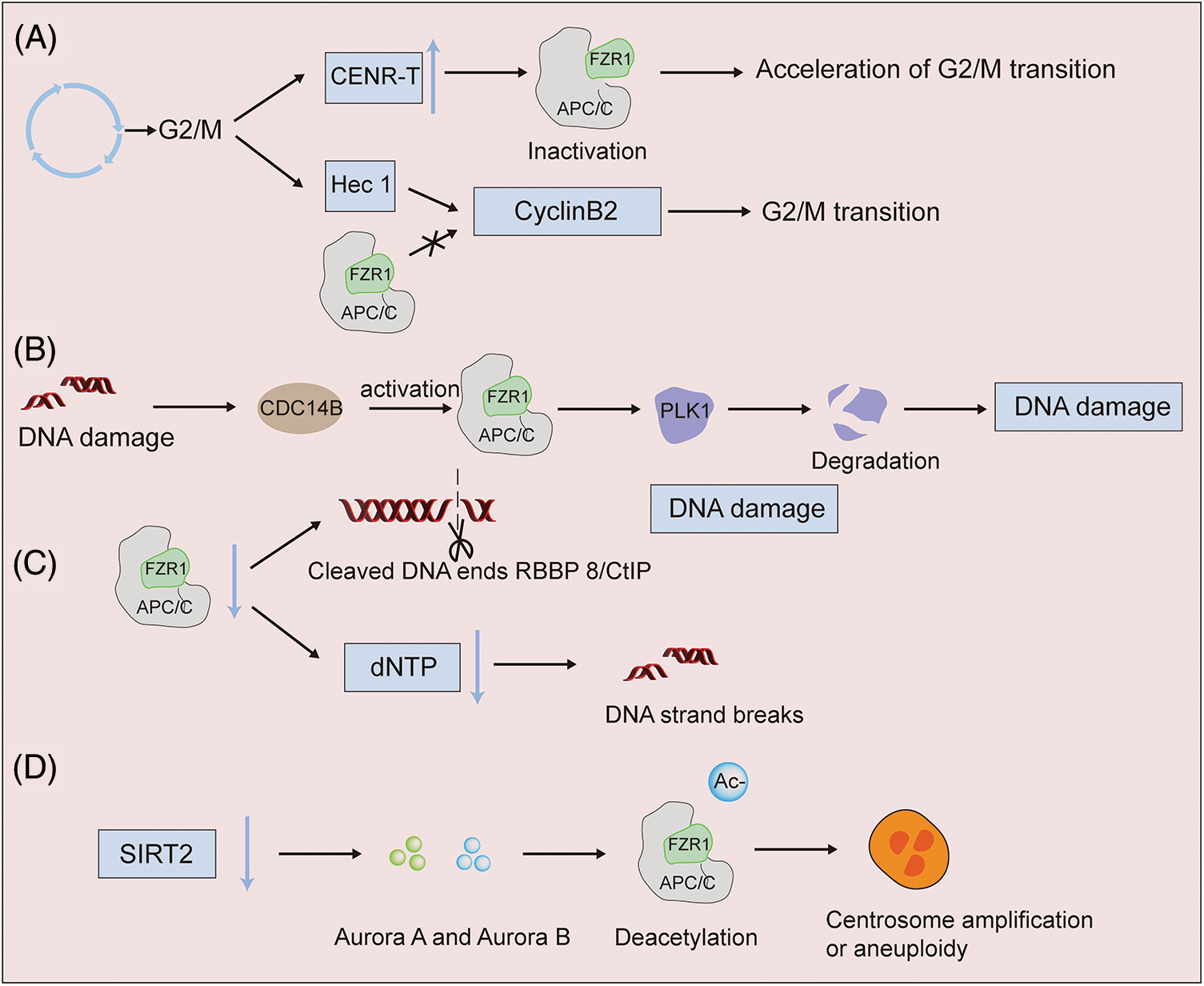
Figure 3: APC/CFZR1 activity has different expressions in DNA damage. (A) FZR1 regulates the G2/M phase transition. Overexpression of CENR-T inactivates FZR1 and accelerates the G2/M phase transition of oocytes. Hec1 antagonizes the stability of cyclin B2 with APC/CFZR1, preventing it from converting to the G2/M phase (B to D). High-fidelity DNA replication and chromosome segregation are required to maintain genome integrity and prevent carcinogenesis. Knockdown of FZR1 results in binucleated or multinucleated cell aberrations, and acute or permanent deletion of FZR1 increases DNA fragmentation and protein cleavage. After DNA damage, PLK1 ubiquitination is induced, which in turn affects cell differentiation.
FZR1 participates in a variety of processes, and the ability of FZR1 to regulate the cell cycle is well-known and has a clear connection with cancer. Since FZR1 regulates many events in the cell cycle, it is advantageous for cancer cells to maintain a certain level of activity. While some oncogenic targets of FZR1 can enhance activity through amplification or other FZR1-independent mechanisms, which may contribute to tumorigenesis, overall, combined deregulation of multiple pathways by partially reducing FZR1 function appear sufficient to create a permissive environment for tumor cell evolution. However, its function in regulating cancer is not limited, and here we discuss more recent publications and highlight current studies that have improved our understanding of FZR1 and cancer. Specifically, because FZR1 plays a role in tumorigenesis and progression, such as regulation of tumor quiescence, regulation of differentiation through the cycle and the G2/M checkpoint through ubiquitination of PLK1 upon DNA damage. Recently, the robust mechanism of APC/CFZR1 relevant to carcinogenesis was shown. As described earlier in this paper, low expression or low activity of FZR1 is associated with cell cycle dysregulation, cell differentiation, genomic instability, DNA damage, and the regulation of many cancer-related pathways. However, although FZR1 is generally considered to be a tumor suppressor, in several cancers, FZR1 may be overexpressed at the RNA and protein levels. Like reduced activity, elevated FZR1 activity is associated with DNA damage and regeneration, which may contribute to tumorigenesis. However, given that multiple mechanisms destabilize the FZR1 protein, elevated RNA may not imply high protein levels. Overall, the singular deficiency in FZR1 activity appears to be achieved in cancer through a potential combination of multiple mechanisms of activity, mainly post-translational, which complicates the development of strategies to use FZR1 activity as a cancer treatment. FZR1 expression is not necessarily an indication of activity in the context of the cell cycle. The expression of FZR1 peaks in G2 and M but remains inactive during anaphase of mitosis. Since the expression of many APC/C substrates also peaks during the G2 and M, the positive correlation between FZR1 protein and APC/C substrates observed in the TMA analysis may reflect the cell cycle status of tumor cell proliferation. Interestingly, in addition to the function of FZR1 in tumors, recent studies have also found that there are also some mutations or deletions that can lead to the gain or loss of FZR1 function during cancer growth. In the GS-Wnt model of gastric carcinoid, mutations in FZR1 lead to a structural shift to a grape-like form (Seidlitz et al., 2019). Mutated genes that initiate FZR1 in the Wnt subtype of medulloblastoma tumorigenesis (Robinson et al., 2012). However, increasing evidence suggests that enhancing APC/CFZR1 activity may contribute to the activity of current therapeutic strategies, including inhibition of Akt, Src, and potentially CDKs. CDC20 remains an important clinical target of APC/C because it is a coactivator required for mitotic exit.
The study of FZR1 for nearly 25 years since its discovery has revealed a conserved mechanism of APC/CFZR1-dependent cell cycle regulation from yeast to humans. It is now clear that FZR1 plays a key role in cell division, stem cell regulation, neuronal processes, DNA damage, and tumorigenesis. Further validation of the newly developed APC/CFZR1 small molecule inhibitors using various animal disease models based on basic research is expected to initiate a new era of APC/CFZR1, and a better understanding of these tumorigenic mechanisms could both lead to a better understanding of cancer and the development of more sophisticated targeted cancer therapies.
Acknowledgement: The authors thank the staff of the Hematology Center of Chongqing Southwest Hospital for their support in the author’s work.
Funding Statement: This study was partly supported by the National Key Scientific Research Project (2017YFC1001903), Provincial and Ministerial Level Projects (cstc2016shms-tzx10006), and the Guizhou Provincial Science & Technology Program (QKHZC [2020]4Y154).
Author Contributions: Conceptualization, JPC, YL, and HL; writing and original draft preparation, HL; writing—review and editing, and visualization, HL, CFZ, MK and JPC; supervision, JPC and YL. All authors have read and agreed to the published version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
Amon A, Irniger S, Nasmyth K (1994). Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle. Cell 77: 1037–1050. https://doi.org/10.1016/0092-8674(94)90443-X [Google Scholar] [PubMed] [CrossRef]
Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M (2008). The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 134: 256–267. https://doi.org/10.1016/j.cell.2008.05.043 [Google Scholar] [PubMed] [CrossRef]
Binné UK, Classon MK, Dick FA, Wei W, Rape M, KaelinJr WG, Näär AM, Dyson NJ (2007). Retinoblastoma protein and anaphase-promoting complex physically interact and functionally cooperate during cell-cycle exit. Nature Cell Biology 9: 225–232. https://doi.org/10.1038/ncb1532 [Google Scholar] [PubMed] [CrossRef]
Boekhout M, Wolthuis R (2015). Nek2A destruction marks APC/C activation at the prophase-to-prometaphase transition by spindle-checkpoint-restricted Cdc20. Journal of Cell Science 128: 1639–1653. https://doi.org/10.1242/jcs.163279 [Google Scholar] [PubMed] [CrossRef]
Brandeis M, Hunt T (1996). The proteolysis of mitotic cyclins in mammalian cells persists from the end of mitosis until the onset of S phase. The EMBO Journal 15: 5280–5289. https://doi.org/10.1002/j.1460-2075.1996.tb00913.x [Google Scholar] [CrossRef]
Brown NG, VanderLinden R, Watson ER, Qiao R, Grace CR, Yamaguchi M, Weissmann F, Frye JJ, Dube P, Ei Cho S et al. (2015). RING E3 mechanism for ubiquitin ligation to a disordered substrate visualized for human anaphase-promoting complex. Proceedings of the National Academy of Sciences 112: 5272–5279. https://doi.org/10.1073/pnas.1504161112 [Google Scholar] [PubMed] [CrossRef]
Cappell SD, Chung M, Jaimovich A, Spencer SL, Meyer T (2016). Irreversible APC(Cdh1) inactivation underlies the point of no return for cell-cycle entry. Cell 166: 167–180. https://doi.org/10.1016/j.cell.2016.05.077 [Google Scholar] [PubMed] [CrossRef]
Cappell SD, Mark KG, Garbett D, Pack LR, Rape M, Meyer T (2018). EMI1 switches from being a substrate to an inhibitor of APC/C (CDH1) to start the cell cycle. Nature 558: 313–317. https://doi.org/10.1038/s41586-018-0199-7 [Google Scholar] [PubMed] [CrossRef]
Chabes AL, Pfleger CM, Kirschner MW, Thelander L (2003). Mouse ribonucleotide reductase R2 protein: A new target for anaphase-promoting complex-Cdh1-mediated proteolysis. Proceedings of the National Academy of Sciences 100: 3925–3929. https://doi.org/10.1073/pnas.0330774100 [Google Scholar] [PubMed] [CrossRef]
Chang L, Zhang Z, Yang J, McLaughlin SH, Barford D (2015). Atomic structure of the APC/C and its mechanism of protein ubiquitination. Nature 522: 450–454. https://doi.org/10.1038/nature14471 [Google Scholar] [PubMed] [CrossRef]
Chen Z, Li L, Xu S, Liu Z, Zhou C et al. (2020). A Cdh1-FoxM1-Apc axis controls muscle development and regeneration. Cell Death & Disease 11: 180. https://doi.org/10.1038/s41419-020-2375-6 [Google Scholar] [PubMed] [CrossRef]
Choppara S, Malonia SK, Sankaran G, Green MR, Santra MK (2018). Degradation of FBXO31 by APC/C is regulated by AKT- and ATM-mediated phosphorylation. Proceedings of the National Academy of Sciences 115: 998–1003. https://doi.org/10.1073/pnas.1705954115 [Google Scholar] [PubMed] [CrossRef]
Choudhury R, Bonacci T, Arceci A, Lahiri D, Mills CA, Kernan JL, Branigan TB, DeCaprio JA, Burke DJ, Emanuele MJ (2016). APC/C and SCF (cyclin F) constitute a reciprocal feedback circuit controlling S-phase entry. Cell Reports 16: 3359–3372. https://doi.org/10.1016/j.celrep.2016.08.058 [Google Scholar] [PubMed] [CrossRef]
Cuadrado M, Garzon J, Moreno S, Garcia-Higuera I (2022). Efficient terminal erythroid differentiation requires the APC/C cofactor Cdh1 to limit replicative stress in erythroblasts. Scientific Reports 12: 10489. https://doi.org/10.1038/s41598-022-14331-6 [Google Scholar] [PubMed] [CrossRef]
Cuende J, Moreno S, Bolaños JP, Almeida A (2008). Retinoic acid downregulates Rae1 leading to APCCdh1 activation and neuroblastoma SH-SY5Y differentiation. Oncogene 27: 3339–3344. https://doi.org/10.1038/sj.onc.1210987 [Google Scholar] [PubMed] [CrossRef]
De K, Grubb TM, Zalenski AA, Pfaff KE, Pal D, Majumder S, Summers MK, Venere M (2019). Hyperphosphorylation of CDH1 in glioblastoma cancer stem cells attenuates APC/C (CDH1) activity and pharmacologic inhibition of APC/C (CDH1/CDC20) compromises viability. Molecular Cancer Research 17: 1519–1530. https://doi.org/10.1158/1541-7786.MCR-18-1361 [Google Scholar] [PubMed] [CrossRef]
Delgado-Esteban M, García-Higuera I, Maestre C, Moreno S, Almeida A (2013). APC/C-Cdh1 coordinates neurogenesis and cortical size during development. Nature Communications 4: 2879. https://doi.org/10.1038/ncomms3879 [Google Scholar] [PubMed] [CrossRef]
Eguren M, Porlan E, Manchado E, García-Higuera I, Cañamero M, Fariñas I, Malumbres M (2013). The APC/C cofactor Cdh1 prevents replicative stress and p53-dependent cell death in neural progenitors. Nature Communications 4: 2880. https://doi.org/10.1038/ncomms3880 [Google Scholar] [PubMed] [CrossRef]
Fujimitsu K, Grimaldi M, Yamano H (2016). Cyclin-dependent kinase 1-dependent activation of APC/C ubiquitin ligase. Science 352: 1121–1124. https://doi.org/10.1126/science.aad3925 [Google Scholar] [PubMed] [CrossRef]
García-Higuera I, Manchado E, Dubus P, Cañamero M, Méndez J, Moreno S, Malumbres M (2008). Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nature Cell Biology 10: 802–811. https://doi.org/10.1038/ncb1742 [Google Scholar] [PubMed] [CrossRef]
Garzón J, Rodríguez R, Kong Z, Chabes A, Rodríguez-Acebes S, Méndez J, Moreno S, García-Higuera I (2017). Shortage of dNTPs underlies altered replication dynamics and DNA breakage in the absence of the APC/C cofactor Cdh1. Oncogene 36: 5808–5818. https://doi.org/10.1038/onc.2017.186 [Google Scholar] [PubMed] [CrossRef]
Gui L, Homer H (2013). Hec1-dependent cyclin B2 stabilization regulates the G2-M transition and early prometaphase in mouse oocytes. Developmental Cell 25: 43–54. https://doi.org/10.1016/j.devcel.2013.02.008 [Google Scholar] [PubMed] [CrossRef]
Han T, Jiang S, Zheng H, Yin Q, Xie M et al. (2019). Interplay between c-Src and the APC/C co-activator Cdh1 regulates mammary tumorigenesis. Nature Communications 10: 3716. https://doi.org/10.1038/s41467-019-11618-7 [Google Scholar] [PubMed] [CrossRef]
Harper JW, Burton JL, Solomon MJ (2002). The anaphase-promoting complex: It’s not just for mitosis any more. Genes & Development 16: 2179–2206. https://doi.org/10.1101/gad.1013102 [Google Scholar] [PubMed] [CrossRef]
Huang JN, Park I, Ellingson E, Littlepage LE, Pellman D (2001). Activity of the APCCdh1 form of the anaphase-promoting complex persists until S phase and prevents the premature expression of Cdc20p. The Journal of Cell Biology 154: 85–94. https://doi.org/10.1083/jcb.200102007 [Google Scholar] [PubMed] [CrossRef]
Ishizawa J, Kuninaka S, Sugihara E, Naoe H, Kobayashi Y et al. (2011). The cell cycle regulator Cdh1 controls the pool sizes of hematopoietic stem cells and mature lineage progenitors by protecting from genotoxic stress. Cancer Science 102: 967–974. https://doi.org/10.1111/j.1349-7006.2011.01884.x [Google Scholar] [PubMed] [CrossRef]
Ishizawa J, Sugihara E, Kuninaka S, Mogushi K, Kojima K et al. (2017). FZR1 loss increases sensitivity to DNA damage and consequently promotes murine and human B-cell acute leukemia. Blood 129: 1958–1968. https://doi.org/10.1182/blood-2016-07-726216 [Google Scholar] [PubMed] [CrossRef]
Ji M, Li H, Suh HC, Klarmann KD, Yokota Y, Keller JR (2008). Id2 intrinsically regulates lymphoid and erythroid development via interaction with different target proteins. Blood 112: 1068–1077. https://doi.org/10.1182/blood-2008-01-133504 [Google Scholar] [PubMed] [CrossRef]
Kernan J, Bonacci T, Emanuele MJ (2018). Who guards the guardian? Mechanisms that restrain APC/C during the cell cycle. Biochimica et Biophysica Acta (BBA)–Molecular Cell Research 1865: 1924–1933. https://doi.org/10.1016/j.bbamcr.2018.09.011 [Google Scholar] [PubMed] [CrossRef]
Kim AH, Bonni A (2007). Thinking within the D box: Initial identification of Cdh1-APC substrates in the nervous system. Molecular and Cellular Neuroscience 34: 281–287. https://doi.org/10.1016/j.mcn.2006.11.019 [Google Scholar] [PubMed] [CrossRef]
Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z et al. (2011). SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 20: 487–499. https://doi.org/10.1016/j.ccr.2011.09.004 [Google Scholar] [PubMed] [CrossRef]
Kraft C, Herzog F, Gieffers C, Mechtler K, Hagting A, Pines J, Peters JM (2003). Mitotic regulation of the human anaphase-promoting complex by phosphorylation. The EMBO Journal 22: 6598–6609. https://doi.org/10.1093/emboj/cdg627 [Google Scholar] [PubMed] [CrossRef]
Kraft C, Vodermaier HC, Maurer-Stroh S, Eisenhaber F, Peters JM (2005). The WD40 propeller domain of Cdh1 functions as a destruction box receptor for APC/C substrates. Molecular Cell 18: 543–553. https://doi.org/10.1016/j.molcel.2005.04.023 [Google Scholar] [PubMed] [CrossRef]
Kramer ER, Scheuringer N, Podtelejnikov AV, Mann M, Peters JM (2000). Mitotic regulation of the APC activator proteins CDC20 and CDH1. Molecular Biology of the Cell 11: 1555–1569. https://doi.org/10.1091/mbc.11.5.1555 [Google Scholar] [PubMed] [CrossRef]
Li M, Shin YH, Hou L, Huang X, Wei Z, Klann E, Zhang P (2008). The adaptor protein of the anaphase promoting complex Cdh1 is essential in maintaining replicative lifespan and in learning and memory. Nature Cell Biology 10: 1083–1089. https://doi.org/10.1038/ncb1768 [Google Scholar] [PubMed] [CrossRef]
Li M, Zhang P (2009). The function of APC/CCdh1 in cell cycle and beyond. Cell Division 4: 2. https://doi.org/10.1186/1747-1028-4-2 [Google Scholar] [PubMed] [CrossRef]
Link LA, Howley BV, Hussey GS, Howe PH (2016). PCBP1/HNRNP E1 protects chromosomal integrity by translational regulation of CDC27. Molecular Cancer Research 14: 634–646. https://doi.org/10.1158/1541-7786.MCR-16-0018 [Google Scholar] [PubMed] [CrossRef]
Listovsky T, Oren YS, Yudkovsky Y, Mahbubani HM, Weiss AM, Lebendiker M, Brandeis M (2004). Mammalian Cdh1/Fzr mediates its own degradation. The EMBO Journal 23: 1619–1626. https://doi.org/10.1038/sj.emboj.7600149 [Google Scholar] [PubMed] [CrossRef]
Littlepage LE, Ruderman JV (2002). Identification of a new APC/C recognition domain, the A box, which is required for the Cdh1-dependent destruction of the kinase Aurora-A during mitotic exit. Genes & Development 16: 2274–2285. https://doi.org/10.1101/gad.1007302 [Google Scholar] [PubMed] [CrossRef]
Liu S, Wang H, Li J, Zhang J, Wu J, Li Y, Piao Y, Pan L, Xiang R, Yue S (2020). FZR1 as a novel biomarker for breast cancer neoadjuvant chemotherapy prediction. Cell Death & Disease 11: 804. https://doi.org/10.1038/s41419-020-03004-9 [Google Scholar] [PubMed] [CrossRef]
Maan M, Agrawal NJ, Padmanabhan J, Leitzinger CC, Rivera-Rivera Y, Saavedra HI, Chellappan SP (2021). Tank binding kinase 1 modulates spindle assembly checkpoint components to regulate mitosis in breast and lung cancer cells. Biochimica et Biophysica Acta (BBA)–Molecular Cell Research 1868: 118929. https://doi.org/10.1016/j.bbamcr.2020.118929 [Google Scholar] [PubMed] [CrossRef]
McGarry TJ, Kirschner MW (1998). Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 93: 1043–1053. https://doi.org/10.1016/S0092-8674(00)81209-X [Google Scholar] [PubMed] [CrossRef]
Penas C, Govek EE, Fang Y, Ramachandran V, Daniel M et al. (2015). Casein kinase 1δ is an APC/C(Cdh1) substrate that regulates cerebellar granule cell neurogenesis. Cell Reports 11: 249–260. https://doi.org/10.1016/j.celrep.2015.03.016 [Google Scholar] [PubMed] [CrossRef]
Peter M, Herskowitz I (1994). Joining the complex: Cyclin-dependent kinase inhibitory proteins and the cell cycle. Cell 79: 181–184. https://doi.org/10.1016/0092-8674(94)90186-4 [Google Scholar] [PubMed] [CrossRef]
Peters JM (2002). The anaphase-promoting complex: Proteolysis in mitosis and beyond. Molecular Cell 9: 931–943. https://doi.org/10.1016/S1097-2765(02)00540-3 [Google Scholar] [PubMed] [CrossRef]
Peters JM (2006). The anaphase promoting complex/cyclosome: A machine designed to destroy. Nature Reviews Molecular Cell Biology 7: 644–656. https://doi.org/10.1038/nrm1988 [Google Scholar] [PubMed] [CrossRef]
Petersen BO, Wagener C, Marinoni F, Kramer ER, Melixetian M, Denchi EL, Gieffers C, Matteucci C, Peters JM, Helin K (2000). Cell cycle-and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes & Development 14: 2330–2343. https://doi.org/10.1101/gad.832500 [Google Scholar] [PubMed] [CrossRef]
Pines J (2011). Cubism and the cell cycle: The many faces of the APC/C. Nature Reviews Molecular Cell Biology 12: 427–438. https://doi.org/10.1038/nrm3132 [Google Scholar] [PubMed] [CrossRef]
Primorac I, Musacchio A (2013). Panta rhei: The APC/C at steady state. Journal of Cell Biology 201: 177–189. https://doi.org/10.1083/jcb.201301130 [Google Scholar] [PubMed] [CrossRef]
Qiao R, Weissmann F, Yamaguchi M, Brown NG, VanderLinden R et al. (2016). Mechanism of APC/CCDC20 activation by mitotic phosphorylation. Proceedings of the National Academy of Sciences 113: E2570–2578. https://doi.org/10.1073/pnas.1604929113 [Google Scholar] [PubMed] [CrossRef]
Ramanujan A, Bansal S, Guha M, Pande NT, Tiwari S (2021). LxCxD motif of the APC/C coactivator subunit FZR1 is critical for interaction with the retinoblastoma protein. Experimental Cell Research 404: 112632. https://doi.org/10.1016/j.yexcr.2021.112632 [Google Scholar] [PubMed] [CrossRef]
Robinson G, Parker M, Kranenburg TA, Lu C, Chen X et al. (2012). Novel mutations target distinct subgroups of medulloblastoma. Nature 488: 43–48. https://doi.org/10.1038/nature11213 [Google Scholar] [PubMed] [CrossRef]
Schreiber A, Stengel F, Zhang Z, Enchev RI, Kong EH, Morris EP, Robinson CV, da Fonseca PC, Barford D (2011). Structural basis for the subunit assembly of the anaphase-promoting complex. Nature 470: 227–232. https://doi.org/10.1038/nature09756 [Google Scholar] [PubMed] [CrossRef]
Seidlitz T, Merker SR, Rothe A, Zakrzewski F, von Neubeck C et al. (2019). Human gastric cancer modelling using organoids. Gut 68: 207–217. https://doi.org/10.1136/gutjnl-2017-314549 [Google Scholar] [PubMed] [CrossRef]
Sigl R, Wandke C, Rauch V, Kirk J, Hunt T, Geley S (2009). Loss of the mammalian APC/C activator FZR1 shortens G1 and lengthens S phase but has little effect on exit from mitosis. Journal of Cell Science 122: 4208–4217. https://doi.org/10.1242/jcs.054197 [Google Scholar] [PubMed] [CrossRef]
Sigrist SJ, Lehner CF (1997). Drosophila fizzy-related down-regulates mitotic cyclins and is required for cell proliferation arrest and entry into endocycles. Cell 90: 671–681. https://doi.org/10.1016/S0092-8674(00)80528-0 [Google Scholar] [PubMed] [CrossRef]
Singh RK, Dagnino L (2017). CDH1 regulates E2F1 degradation in response to differentiation signals in keratinocytes. Oncotarget 8: 4977–4993. https://doi.org/10.18632/oncotarget.13636 [Google Scholar] [PubMed] [CrossRef]
Skaar JR, Pagano M (2008). Cdh1: A master G0/G1 regulator. Nature Cell Biology 10: 755–757. https://doi.org/10.1038/ncb0708-755 [Google Scholar] [PubMed] [CrossRef]
Stroschein SL, Bonni S, Wrana JL, Luo K (2001). Smad3 recruits the anaphase-promoting complex for ubiquitination and degradation of SnoN. Genes & Development 15: 2822–2836. https://doi.org/10.1101/gad.912901 [Google Scholar] [PubMed] [CrossRef]
Sudo T, Ota Y, Kotani S, Nakao M, Takami Y, Takeda S, Saya H (2001). Activation of Cdh1-dependent APC is required for G1 cell cycle arrest and DNA damage-induced G2 checkpoint in vertebrate cells. The EMBO Journal 20: 6499–6508. https://doi.org/10.1093/emboj/20.22.6499 [Google Scholar] [PubMed] [CrossRef]
The I, Ruijtenberg S, Bouchet BP, Cristobal A, Prinsen MB et al. (2015). Rb and FZR1/Cdh1 determine CDK4/6-cyclin D requirement in C. elegans and human cancer cells. Nature Communications 6: 5906. https://doi.org/10.1038/ncomms6906 [Google Scholar] [PubMed] [CrossRef]
Thornton BR, Toczyski DP (2006). Precise destruction: An emerging picture of the APC. Genes & Development 20: 3069–3078. https://doi.org/10.1101/gad.1478306 [Google Scholar] [PubMed] [CrossRef]
Uzunova K, Dye BT, Schutz H, Ladurner R, Petzold G et al. (2012). APC15 mediates CDC20 autoubiquitylation by APC/C (MCC) and disassembly of the mitotic checkpoint complex. Nature Structural & Molecular Biology 19: 1116–1123. https://doi.org/10.1038/nsmb.2412 [Google Scholar] [PubMed] [CrossRef]
Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, Amon A (1998). The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Molecular Cell 2: 709–718. https://doi.org/10.1016/S1097-2765(00)80286-5 [Google Scholar] [PubMed] [CrossRef]
Wan Y, Liu X, Kirschner MW (2001). The anaphase-promoting complex mediates TGF-β signaling by targeting SnoN for destruction. Molecular Cell 8: 1027–1039. https://doi.org/10.1016/S1097-2765(01)00382-3 [Google Scholar] [PubMed] [CrossRef]
Wan L, Zou W, Gao D, Inuzuka H, Fukushima H et al. (2011). Cdh1 regulates osteoblast function through an APC/C-independent modulation of Smurf1. Molecular Cell 44: 721–733. https://doi.org/10.1016/j.molcel.2011.09.024 [Google Scholar] [PubMed] [CrossRef]
Wang J, Dye BT, Rajashankar KR, Kurinov I, Schulman BA (2009). Insights into anaphase promoting complex TPR subdomain assembly from a CDC26-APC6 structure. Nature Structural & Molecular Biology 16: 987–989. https://doi.org/10.1038/nsmb.1645 [Google Scholar] [PubMed] [CrossRef]
Wang Y, Li J, Dong F, Yue W, Ouyang YC, Wang ZB, Hou Y, Schatten H, Sun QY (2020). CENP-T regulates both the G2/M transition and anaphase entry by acting through CDH1 in meiotic oocytes. Journal of Cell Science 133: jcs238105. https://doi.org/10.1242/jcs.238105 [Google Scholar] [PubMed] [CrossRef]
Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, KaelinJr WG (2004). Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 428: 194–198. https://doi.org/10.1038/nature02381 [Google Scholar] [PubMed] [CrossRef]
Wiebusch L, Bach M, Uecker R, Hagemeier C (2005). Human cytomegalovirus inactivates the G0/G1-APC/C ubiquitin ligase by Cdh1 dissociation. Cell Cycle 4: 1435–1439. https://doi.org/10.4161/cc.4.10.2077 [Google Scholar] [PubMed] [CrossRef]
Yamaguchi M, Yu S, Qiao R, Weissmann F, Miller DJ, VanderLinden R, Brown NG, Frye JJ, Peters JM, Schulman BA (2015). Structure of an APC3-APC16 complex: Insights into assembly of the anaphase-promoting complex/cyclosome. Journal of Molecular Biology 427: 1748–1764. https://doi.org/10.1016/j.jmb.2014.11.020 [Google Scholar] [PubMed] [CrossRef]
Yamano H (2019). APC/C: Current understanding and future perspectives. F1000Research 8: 725. https://doi.org/10.12688/f1000research.18582.1 [Google Scholar] [PubMed] [CrossRef]
Yao G, Lee TJ, Mori S, Nevins JR, You L (2008). A bistable Rb-E2F switch underlies the restriction point. Nature Cell Biology 10: 476–482. https://doi.org/10.1038/ncb1711 [Google Scholar] [PubMed] [CrossRef]
Yao W, Qian W, Zhu C, Gui L, Qiu J, Zhang C (2010). Cdh1-APC is involved in the differentiation of neural stem cells into neurons. NeuroReport 21: 39–44. https://doi.org/10.1097/WNR.0b013e32833312fe [Google Scholar] [PubMed] [CrossRef]
Zhang S, Chang L, Alfieri C, Zhang Z, Yang J, Maslen S, Skehel M, Barford D (2016). Molecular mechanism of APC/C activation by mitotic phosphorylation. Nature 533: 260–264. https://doi.org/10.1038/nature17973 [Google Scholar] [PubMed] [CrossRef]
Zhang J, Li H, Zhou T, Zhou J, Herrup K (2012). Cdk5 levels oscillate during the neuronal cell cycle: Cdh1 ubiquitination triggers proteosome-dependent degradation during S-phase. Journal of Biological Chemistry 287: 25985–25994. https://doi.org/10.1074/jbc.M112.343152 [Google Scholar] [PubMed] [CrossRef]
Zhang T, Zhou Y, Li L, Wang ZB, Shen W, Schatten H, Sun QY (2017). CenpH regulates meiotic G2/M transition by modulating the APC/CCdh1-cyclin B1 pathway in oocytes. Development 144: 305–312. https://doi.org/10.1242/dev.141135 [Google Scholar] [PubMed] [CrossRef]
Zhou C, Kuang M, Liu Z, Jia X, Chen Z et al. (2022). Insufficiency of FZR1 disturbs HSC quiescence by inhibiting ubiquitin-dependent degradation of RUNX1 in aplastic anemia. Leukemia 36: 834–846. https://doi.org/10.1038/s41375-021-01445-5 [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools