
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access

Anisodine hydrobromide alleviates oxidative stress caused by hypoxia/reoxygenation in human cerebral microvascular endothelial cells predominantly via inhibition of muscarinic acetylcholine receptor 4
1 Institute of Biomedical Engineering, West China School of Basic Medical Sciences & Forensic Medicine, Sichuan University, Chengdu, 610041, China
2 Department of Biomedical Engineering, The City College of the City University of New York, New York, 10031, USA
* Corresponding Author: YE ZENG. Email:
# Co-first authors
BIOCELL 2023, 47(10), 2255-2263. https://doi.org/10.32604/biocell.2023.030880
Received 29 April 2023; Accepted 31 July 2023; Issue published 08 November 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Anisodine hydrobromide (AT3), an anti-cholinergic agent, could be delivered to the brain across the blood-brain barrier and has been used clinically for the treatment of cerebral ischemia/reperfusion injury. Endothelial dysfunction can be caused by hypoxia/reoxygenation (H/R) via oxidative stress and metabolic alterations. The present study investigated whether AT3 regulates the production of nitric oxide (NO) and reactive oxygen species (ROS), and the HIF-1α pathway via regulation of muscarinic acetylcholine receptors (mAChRs) in brain microvascular endothelial cells after H/R exposure. Methods: Under H/R conditions, hCMEC/D3 cerebral microvascular endothelial cells were treated with AT3. Specific inhibitors of M2- and M4- mAChRs were used to explore the mechanism by which AT3 influences oxidative stress in endothelial cells. Then, mAChRs expression was detected by western blotting and NO production was detected by Greiss reaction. The intracellular ROS level was measured using DCFH-DA probes. The expression of hypoxia-inducible transcription factor 1α (HIF-1α) was also detected. Results: While H/R induced the expression of M2- and M4-mAChRs, AT3 suppressed the H/R-upregulated M2- and M4-mAChRs. H/R also induced the production of NO, ROS, and apoptosis. AT3 and M4-mAChR inhibitors inhibited the H/R-induced production of NO and ROS and apoptosis. HIF-1α was induced by H/R, but was suppressed by AT3. Conclusion: Thus, the in vitro evidence shows that AT3 protects against H/R injury in cerebral microvascular endothelial cells via inhibition of HIF-1α, NO and ROS, predominantly through the downregulation of M4-mAChR. The findings offer novel understandings regarding AT3-mediated attenuation of endothelial cell apoptosis and cerebral ischemia/reperfusion injury.Graphic Abstract
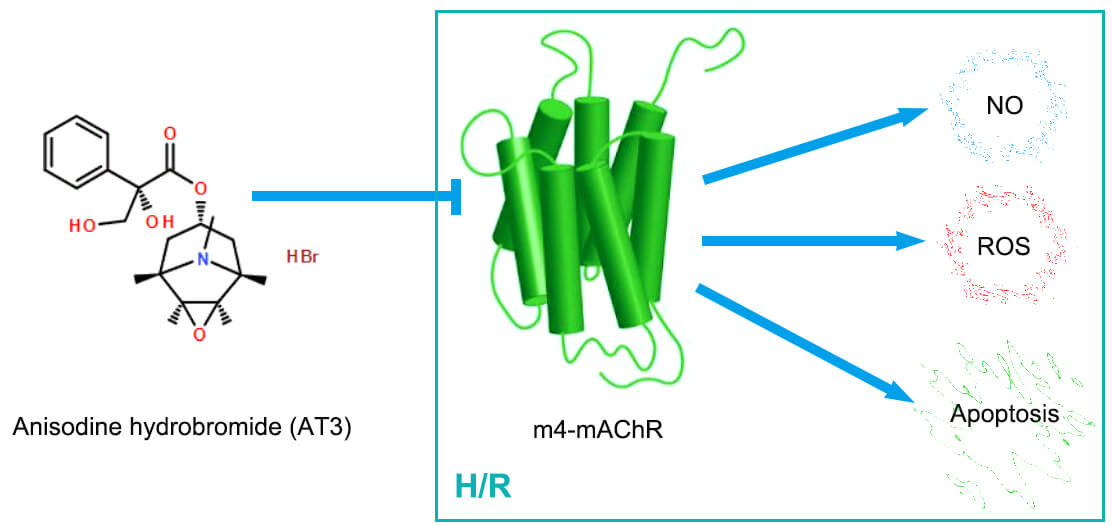
Keywords
Ischemic stroke is a major cause of disability and mortality and places a huge burden on society and families worldwide (Jackson-Weaver et al., 2019; Wu et al., 2019). The most effective treatment for acute ischemic stroke is to restore cerebral perfusion by thrombolysis (Campbell and Khatri, 2020; Kong et al., 2019). However, reperfusion after ischemia commonly leads to secondary damage to the brain due to the increased production of free radicals, excitatory amino acids and pro-inflammatory cytokines (Kong et al., 2019). Disruption of the blood-brain barrier (BBB) is regarded as an important pathological basis for ischemia-reperfusion (I/R) injury (Luo et al., 2017). The BBB comprises brain microvessel endothelial cells, pericytes, astrocytes, and extracellular matrix. Among these components, endothelial cells contribute the most in maintaining the integrity and function of BBB, and are susceptible to external harmful stimuli. During I/R, oxidative stress can damage the cell membrane, protein, and DNA, disrupting mitochondrial membrane potentials (Xie et al., 2023), thus leading to vascular endothelial cell dysfunction. Therefore, attenuating oxidative stress and endothelial cell injury is a promising treatment strategy to protect the BBB.
The activation of muscarinic acetylcholine receptors (mAChRs) by acetylcholine (Ach) has been widely known to elicit the anti-oxidative effects and prevents I/R injury in the heart (Bi et al., 2015). In one study, activation of M2-mAChR reduced cardiac I/R injury (Liao et al., 2015). M3-mAChR also activates the anti-apoptotic signaling cascades, enhancing the endogenous antioxidant capacity, and diminishing the intracellular Ca2+ overload, contributing to protecting the heart against ischemic injuries (Liu et al., 2011). M4-mAChR has anti-psychotic effects in the central nervous system, and improves cognitive impairment and motor dysfunction (Takai and Enomoto, 2018). M5-mAChR is located mainly in the substantia nigra, ventral tegmental area, cerebral cortex, and the striatum area of the central nervous system. Previous studies have shown the association between cerebral blood flow responses and the M3-mAChR levels in blood vessels (Feng et al., 2019). Activation of M2-mAChR has been reported to reduce cardiac I/R injury. However, the knockdown of the M2-mAChR or the ACh co-treatment with atropine abolished the antioxidant and cardioprotective effect of ACh (Miao et al., 2013). Therefore, it was believed that atropine and bioactive belladonna alkaloid are not suitable for the treatment of the cardiac I/R injury. However, the anti-cholinergic agent, anisodine hydrobromide (AT3) has shown favorable therapeutic outcomes in the clinical and preclinical trials for the ischemia stroke treatment (Dong et al., 2021; Xu and Deng, 1996; Zhang, 2022). The effect of AT3 on mAChRs is queried and attracted widespread interest.
Both anisodamine hydrobromide (Ani HBr) and AT3 are atropine (ATP)-like agents and active ingredients derived from the roots of a Chinese specialty plant Scopolia tangutica maxim (Qiu et al., 2022; Zhong et al., 2023). AT3 was thought to be readily transported across the BBB, but Ani HBr was not, due to its structure, and exerts its effects mainly by improving the microcirculation function and scavenging the ROS (Chen et al., 2019; Liu et al., 2015). AT3 has also been reported to exert neuroprotective effects in the cerebral I/R rat model (Chen et al., 2017; Xu and Deng, 1996). Therefore, whether AT3 acts on the brain endothelial cells as a specific antagonist of mAChRs is of particular interest.
Considering the harmful role of anti-cholinergic agents in cardiac I/R injury (Miao et al., 2013), understanding the molecular mechanisms by which AT3 protects the brain endothelial cell injury following hypoxia/reoxygenation (H/R) exposure will aid to the clinical use of AT3. Hypoxia enhances tissue injury via hypoxia-inducible factor-1α (HIF-1α)-dependent reactive oxygen species (ROS) production (Xie et al., 2023). HIF-1α is mediated by mAChRs (Xie et al., 2023). The nitric oxide (NO) synthase uncoupling induces massive production of ROS (Wierońska et al., 2021). During hypoxia, both NO and ROS were produced and caused oxidative stress in the brain endothelial cells (Meng et al., 2021; Robertson et al., 2011). Increased brain endothelial NO stabilized HIF-1α in the astrocytes, resulting in increased expression of a “hypoxic program” (Lopez-Ramirez et al., 2021). In the present study, we tested the hypothesis that AT3 protects brain endothelial cells against H/R-induced injury via downregulation of the specific mAChR subtypes and downstream HIF-1α, and suppression of NO and ROS; we used an in vitro endothelial cell H/R injury model to simulate the brain I/R injury in vivo and explored the potential mAChRs using the specific inhibitors of mAChRs.
Cell culture and hypoxia/reoxygenation treatment
The immortalized human cerebral microvascular endothelial cell (hCMEC/D3) was obtained from BNBIO, China. The hCMEC/D3 were grown at 37°C and 5% CO2 in endothelial cell medium (ECM; cat.1001, Sciencell, San Diego, USA) containing 10% fetal bovine serum (FBS; cat.0025, Sciencell, San Diego, USA), 1% penicillin/streptomycin (cat.0503, Sciencell, San Diego, USA) and 1% endothelial cell growth supplement (cat.1052, Sciencell, San Diego, USA). AT3 was obtained from Chengdu No.1 Pharmaceutical Co., Ltd., Chengdu, China, dissolved in phosphate-buffered saline (PBS) at 20 mg/mL and filtered through a 0.22 μm filter membrane (Millipore, Boston, USA). The final concentrations of AT3 at 10 and 20 µg/mL were prepared with ECM. For treatment, the hCMEC/D3 cells were maintained in 1% serum, and treated in hypoxia using the AnaeroPouch pack (C-11, Mitsubishi Gas Chemical Company, Inc., Tokyo, Japan) for 3 h with or without AT3 (10 and 20 µg/mL), M2 inhibitor gallamine triethiodide (68 μM, Selleck, Shanghai, China), or M4 inhibitor tropicamide (10 nM, MedChemExpress, Shanghai, China), respectively. Then, cells were transferred back to the normoxic conditions (95% air and 5% CO2) for 3 h for reoxygenation, without changing the culture medium. The cells cultured in normoxic conditions were used as controls. The concentrations of AT3 were selected by pre-experimental screening based on cell viability.
The hCMEC/D3 cell lysates were extracted with RIPA buffer (Beyotime, Shanghai, China) containing protease and phosphatase inhibitors on ice. Protein extract was centrifuged at 10000×g for 15 min at 4°C. The protein concentrations were determined by BCA Protein Assay Kit (Beyotime, Shanghai, China). The lysates containing proteins (30 μg) were denatured at 100°C for 5 min and then separated by gel electrophoresis, electro-transferred onto polyvinylidene difluoride membranes (Millipore, Boston, USA), blocked with 5% (w/v) non-fat milk (Solarbio, Beijing, China) for 1 h at room temperature, and then probed with the primary and horseradish peroxidase (HRP)-conjugated secondary antibodies. Finally, the gel bands were detected by the chemiluminescence of the applied ECL solution (Affinity, Changzhou, China). The bands were quantified by the ImageJ software (version 1.53 v, National Institutes of Health, Bethesda, USA). The antibodies used included the cholinergic receptor, muscarinic 1 (CHRM1) polyclonal antibody (1:1000, A16819; Abclonal, Wuhan, China), CHRM2 polyclonal antibody (1:1000, A1567, Abclonal, Wuhan, China), CHRM3 polyclonal antibody (1:1000, A1602; Abclonal, Wuhan, China), CHRM4 polyclonal antibody (1:1000, A2866; Abclonal, Wuhan, China) CHRM5 polyclonal antibody (1:1000, A5367; Abclonal, Wuhan, China), HIF-1α antibody (1:1000; Abclonal, Wuhan, China), GAPDH antibody (1:5000; Cell Signaling Technology, Boston, USA), and HRP-conjugated secondary antibody (1:5000; Cell Signaling Technology, Boston, USA).
Detection of nitric oxide production
To determine the effect of AT3 on NO release, the NO levels in hCMEC/D3 cells were measured using the Griess reaction system (Nanjing Jiancheng, Nanjing, China) according to the manufacturer’s instruction. After the H/R treatment, cells were immediately washed thrice with PBS. The supernatants of cell lysate (100 μL) were collected from each sample (n = 3), and then incubated with 100 μL of Griess reagent. Absorbance was measured at 550 nm by a microplate reader (Labserv K3 Touch, Thermo Fisher, Waltham, USA) and presented as a fold of control.
Measurement of intracellular reactive oxygen species and apoptosis
Cellular ROS production was measured using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; Beyotime, Shanghai, China) probes. Apoptotic cells were detected by Hoechst 33342 (Beyotime, Shanghai, China). After H/R, cells were incubated with 5 μM DCFH-DA in ECM with no FBS at 37°C for 30 min. After washing thrice with PBS, cells were stained with 1X Hoechst 33342 for 10 min in the dark. DCFH-DA and Hochest 33342-stained cells were detected using the IX73 inverted fluorescence microscopy (Olympus, Tokyo, Japan). The images were quantified using ImageJ software.
Experimental data are shown as mean ± standard deviation (SD). All statistical analyses were performed using SPSS software (v25.0; IBM, Armonk, USA). For data collected from more than two groups, statistical significance was determined by the one-way ANOVA; p < 0.05 was considered as statistically significant.
Anisodine hydrobromide inhibits hypoxia/reoxygenation-induced expression of muscarinic acetylcholine receptors in HCMEC/D3 cells
AT3 was earlier believed to exert its effect via mAChR (Han and Chen, 1984; Pan and Han, 2004), but the expression of mAChRs in endothelial cells remained unclear. To identify which subtype of mAChRs is responsible for the protection of AT3 against H/R injury in endothelial cells, the M1-M5 mAChRs were detected (Figs. 1a-1f). In control cells, the levels of mAChR subtypes were expressed in the order of M3 > M2 > M5 > M4 > M1. AT3 treatment (10 and 20 μg/mL) significantly inhibited the M2- and M4-mAChRs in a dose-dependent manner (Figs. 1c and 1e). Following H/R exposure, expressions of M2- and M4-mAChRs were significantly increased (p < 0.001), and were significantly inhibited by AT3 in a dose-dependent manner. Interestingly, in normoxic endothelial cells, atropine inhibited the expression of M2-mAChR, but induced their expression in endothelial cells after H/R exposure (Fig. 1c). Similar to AT3, Ani HBr significantly inhibited M2- and M4-mAChRs in normoxic cells and significantly inhibited the H/R-induced M2- and M4-mAChRs toward the baseline levels (Figs. 1c and 1e). These results suggest that AT3 suppressed the H/R-induced M2- and M4-mAChRs, and that the alteration of mAChRs and protective mechanism of AT3 in endothelial cells are rather different from atropine. The subsequent investigations were focused on the M2- and M4-mAChRs.
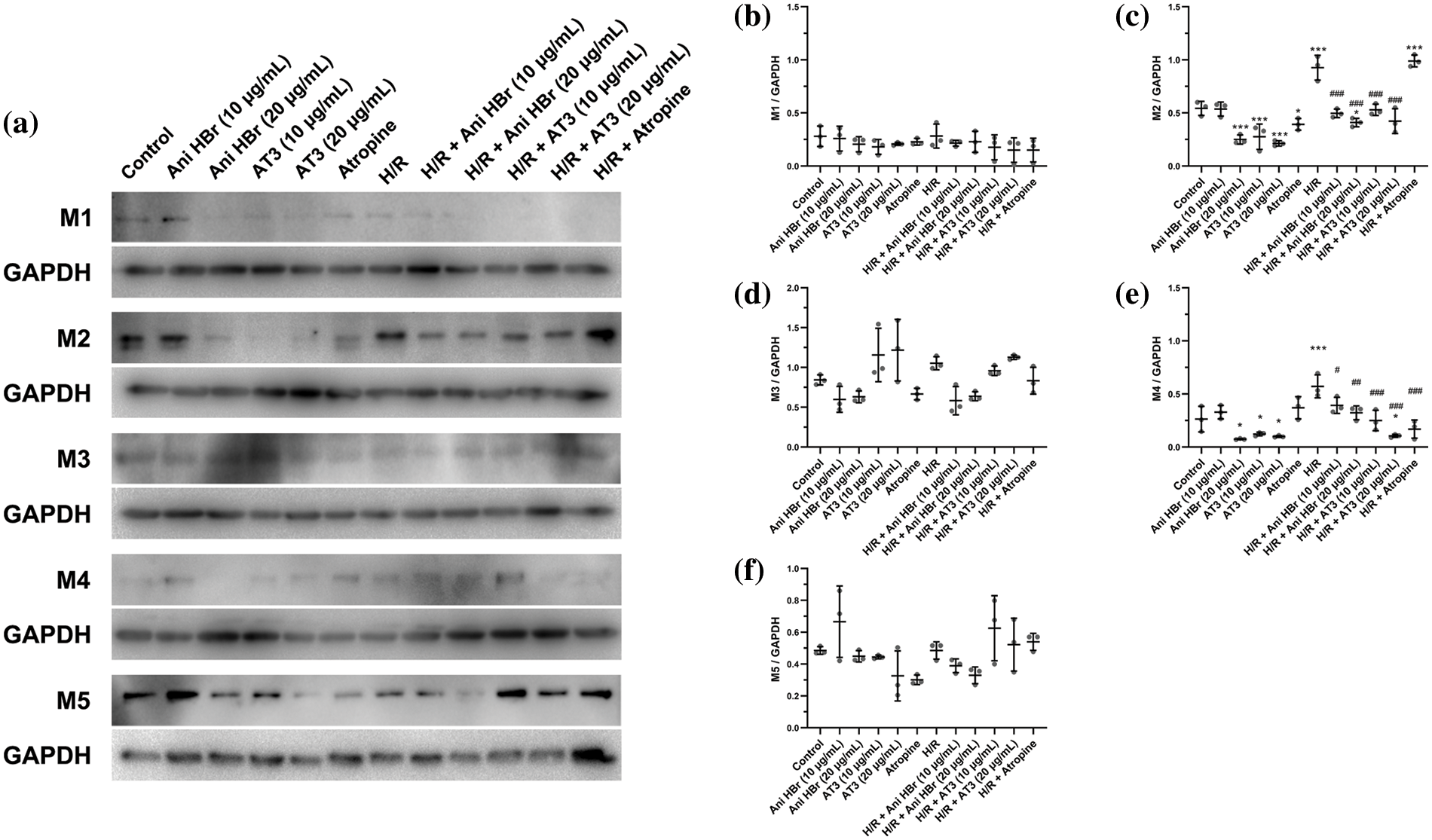
Figure 1: Effect of anisodine hydrobromide (AT3) and inhibitors of muscarinic acetylcholine receptors (mAChRs) on mAChRs expression in hCMEC/D3 cells. Cells were treated with or without AT3 (10 and 20 µg/mL) followed by normoxic (Control) or H/R (hypoxia 3 h and reoxygenation 3 h) exposures. The anti-cholinergic agents Ani HBr (10 and 20 µg/mL) and atropine were used as pharmaceutical controls. Then, the expression of M1-M5 mAChRs in the cells were detected by western blotting. (a) Detection of the protein levels of M1-M5 receptors in the hCMEC/D3 cells by western blotting. (b–f) Quantification of the ratios of M1-M5 mAChRs to GAPDH based on immunoblot images. Results are shown as mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 vs. control; #p < 0.05; ##p < 0.01; ###p < 0.001 vs. H/R. (d and f) ANOVA with Tamhane’s T2 test; (b, c and e) ANOVA with the least significant difference test.
Anisodine hydrobromide and muscarinic acetylcholine receptors inhibitors decrease hypoxia/reoxygenation-induced nitric oxide production in hCMEC/D3 cells
To investigate the role of AT3 in the regulation and the production of NO and its release after H/R exposure, Greiss reaction was performed using selective antagonists for mAChRs (Fig. 2). H/R exposure significantly increased the production of NO in hCMEC/D3 cells by 1.2-fold (Fig. 2; p < 0.01 vs. control). AT3 at 10 and 20 μg/mL significantly reduced the H/R-induced NO (p < 0.01 vs. H/R) to levels lower than baseline, showing that AT3 at 10 and 20 μg/mL abolished the H/R-induced NO production of endothelial cells.
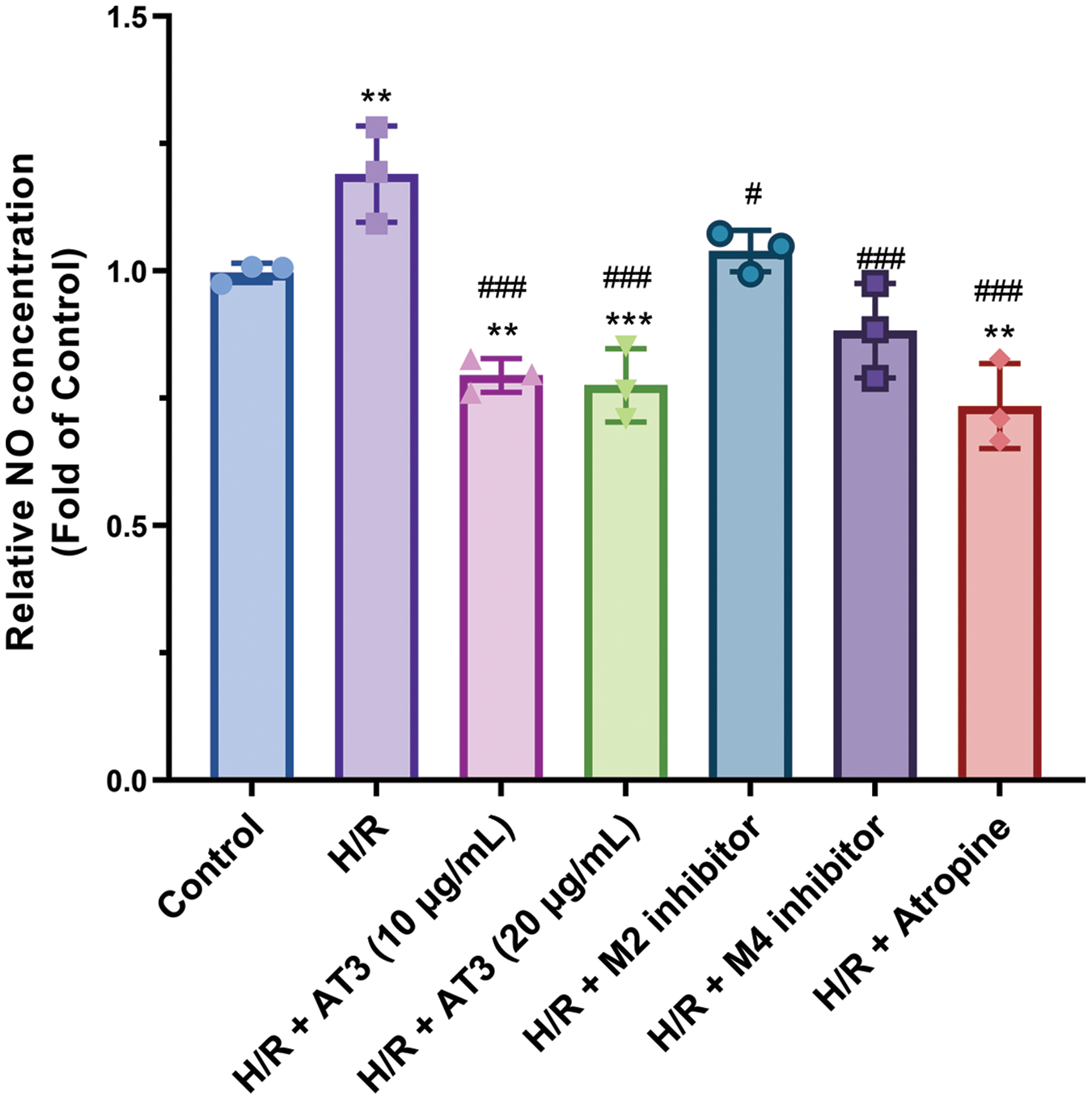
Figure 2: Effect of anisodine hydrobromide (AT3) and muscarinic acetylcholine receptors (mAChRs) inhibitors on nitric oxide (NO) production in hCMEC/D3 cells. Cells were treated with or without AT3 (10 and 20 µg/mL), M2 inhibitor gallamine triethiodide, and M4 inhibitor tropicamide followed by normoxic (Control) or H/R (hypoxia 3 h and reoxygenation 3 h) exposures. The anti-cholinergic agent atropine was used as pharmaceutical control. Then, the NO levels in supernatants of cell lysates were detected using the Greiss reaction. Data are presented as mean ± SD. **p < 0.01, ***p < 0.001 vs. control; #p < 0.05; ###p < 0.001 vs. H/R. ANOVA with the least significant difference test.
After treatment with the M4-mAChR inhibitor tropicamide, the NO expression induced by H/R was significantly reduced. However, M2 inhibitor gallamine triethiodide only slightly but not significantly decreased the production of NO after H/R exposure. In addition, other mAChRs inhibitors, including M1 inhibitor biperiden hydrochloride, M3 inhibitor darifenacin HBr, and M5 inhibitor aclidinium bromide were also used, but they could not suppress the H/R-induced NO production (data not shown).
Overall, these results show that AT3 treatment and M4-mAChR inhibitor attenuated the NO release of endothelial cells following H/R exposure. AT3-mediated downregulation of the M4-mAChR indicates that AT3 suppresses the NO production through downregulation of M4-mAChR in hCMEC/D3 cells after H/R exposure.
Anisodine hydrobromide and muscarinic acetylcholine receptors inhibitors attenuate hypoxia/reoxygenation-induced reactive oxygen species in hCMEC/D3 cells
H/R could cause excessive ROS production (Granger and Kvietys, 2015). Considering that intracellular ROS plays a critical role in brain I/R injury, we assessed ROS levels in hCMEC/D3 cells using DCFH-DA fluorescence probes (Fig. 3). Our results showed that while the normoxic endothelial cells produced a few ROS, and H/R exposure significantly elevated the ROS level (Fig. 3), consistent with the previous study (Jackson-Weaver et al., 2019). After treatment with 10 and 20 μg/mL AT3, the H/R-induced ROS production was significantly suppressed, and 20 μg/mL AT3 showed lower ROS level, indicating a better scavenging effect of 20 μg/mL AT3 on ROS and exerts a better protective effect on endothelial cells.
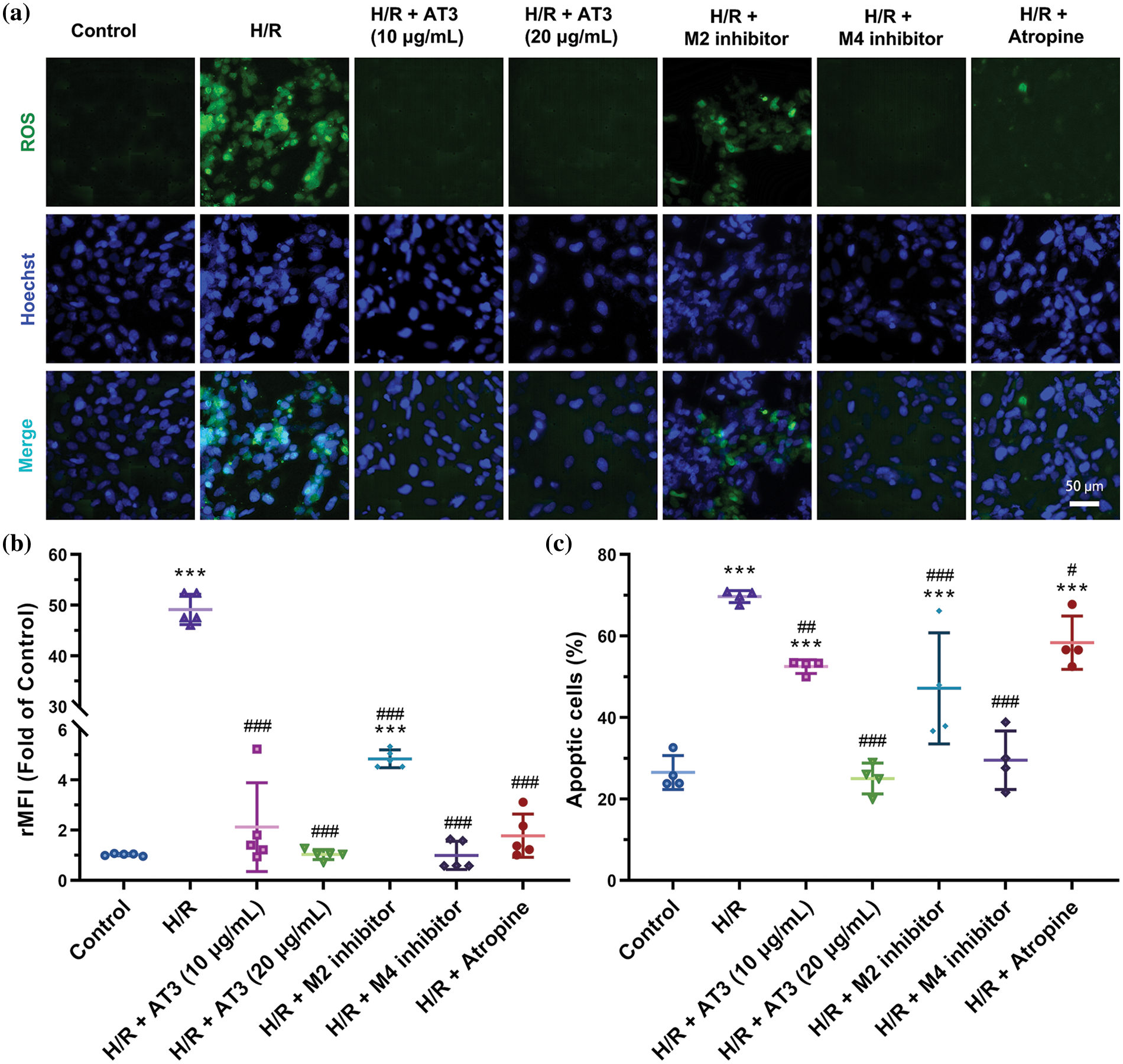
Figure 3: Effect of anisodine hydrobromide (AT3) and muscarinic acetylcholine receptors (mAChRs) inhibitors on reactive oxygen species (ROS) in hCMEC/D3 cells. Cells were treated with or without AT3 (10 and 20 µg/mL), M2 inhibitor gallamine triethiodide, and M4 inhibitor tropicamide followed by normoxic (Control) or H/R (hypoxia 3 h and reoxygenation 3 h) exposures. The anti-cholinergic agent atropine was used as pharmaceutical control. Then, the ROS levels of cells were estimated using DCFH-DA probes, and apoptotic cells were labeled using Hoechst 33342. (a) The fluorescence images. (b) Relative mean fluorescence intensity (rMFI) of ROS to that of controls. Data are presented as mean ± SD (n = 5). ***p < 0.001 vs. control; ###p < 0.001 vs. H/R. ANOVA with Tamhane’s T2 test. (c) Percentage of apoptotic cells in per field. Data were presented as mean ± SD (n = 4). ***p < 0.001 vs. control; #p < 0.05; ##p < 0.01; ###p < 0.001 vs. H/R. ANOVA with the least significant difference test.
The potential mechanism by which AT3 reduces ROS production induced by H/R was investigated and the roles of M2- and M4-mAChRs were also assessed by using gallamine triethiodide and tropicamide, respectively (Figs. 3a and 3b). M2-mAChR inhibitors significantly reduced the production of ROS (p < 0.001), while M4-mAChR inhibitors reduced more ROS compared to the M2-mAChR inhibitors (p < 0.001), to a level close to that in the normoxic cells. Other mAChRs inhibitors, including M1, M3, and M5 did not suppress the H/R-induced ROS production (data not shown). These results indicate that AT3 can inhibit H/R-induced ROS via mAChRs, especially M4-mAChR.
Anisodine hydrobromide and muscarinic acetylcholine receptors inhibitors attenuate hypoxia/reoxygenation-induced apoptosis in hCMEC/D3 cells
High levels of ROS cause apoptosis in endothelial cells (Dharmashekar et al., 2023; Jin et al., 2023). Consistently, Hoechst staining showed that H/R exposure induced cell apoptosis (Figs. 3a and 3c). The apoptotic cells were reduced by AT3 in a dose-dependent manner. Although atropine dramatically suppressed the production of ROS, the number of apoptotic cells were only slightly (16%) reduced (p < 0.05). Both M2- and M4-mAChR inhibitors significantly inhibited cell apoptosis after H/R exposure, and M4-mAChR inhibitors reduced the level more towards that of the control. Our results supported that AT3 had an anti-apoptotic effect against hypoxia-induced damage predominantly via downregulation of M4-mAChR.
Anisodine hydrobromide and muscarinic acetylcholine receptors inhibitors inhibit hypoxia/reoxygenation-induced hypoxia-inducible factor-1α expression in hCMEC/D3 cells
Given that HIF-1α plays a vital role in various hypoxic events, the effect of AT3 on the inflammation regulator HIF-1α was evaluated by western blotting (Figs. 4a and 4b). After H/R exposure, the HIF-1α level increased significantly to 3.6-fold of normoxic cells (p < 0.001). After treatment with 10 and 20 μg/mL AT3, H/R-induced HIF-1α decreased significantly (p < 0.05 at 10 and p < 0.01 at 20 μg/mL). After M2-mAChR inhibitors treatment, the H/R-induced HIF-1α was also reduced significantly (p < 0.05). After treatment with the M4-mAChR inhibitor, the H/R-induced HIF-1α level was reduced more and returned to basal level (p < 0.001). The results indicate the important role of AT3 in protecting the H/R-induced endothelial cell injury through the M2- and M4-mAChRs-HIF-1α signaling axis.
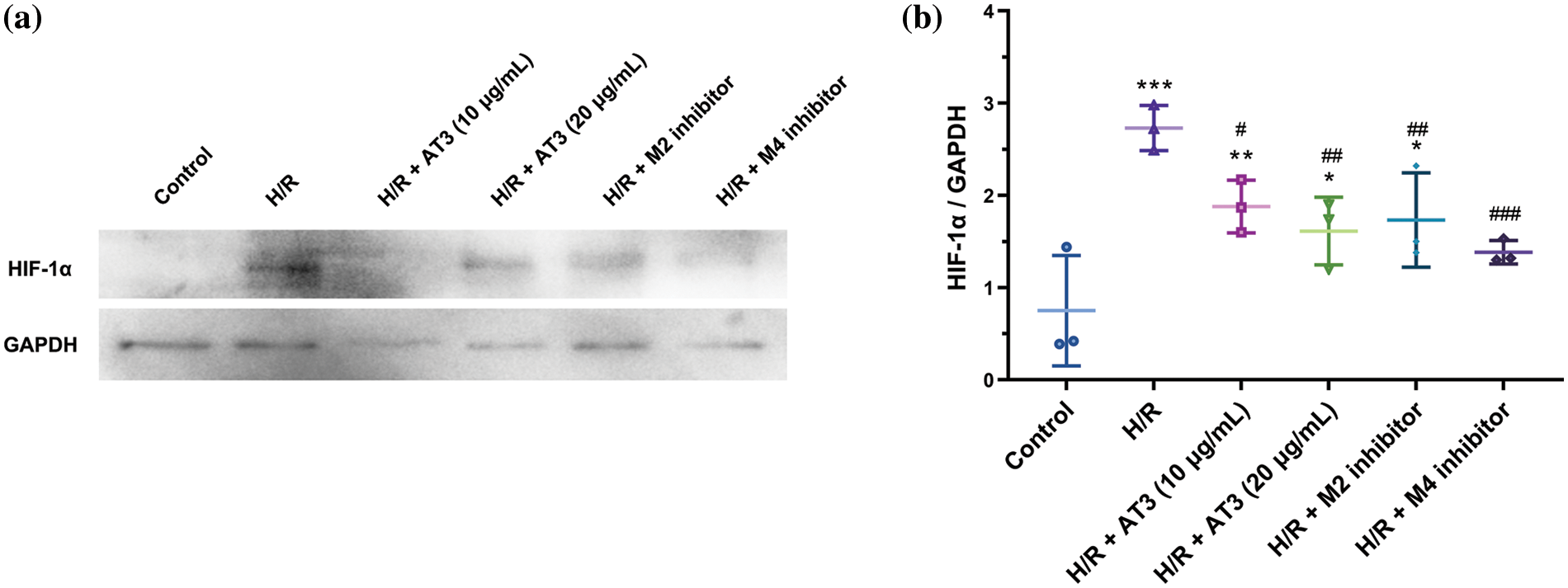
Figure 4: Effect of anisodine hydrobromide (AT3) and muscarinic acetylcholine receptors (mAChRs) inhibitors on hypoxia-inducible factor (HIF)-1α expression in hCMEC/D3 cells. Cells were treated with or without AT3 (10 and 20 µg/mL), M2 inhibitor gallamine triethiodide, and M4 inhibitor tropicamide followed by normoxic (Control) or H/R (hypoxia 3 h and reoxygenation 3 h) exposures. Then, the cell lysates were collected and HIF-1α levels were determined by western blotting (a). (b) The quantification was performed. Data were presented as mean ± SD (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001 vs. control; #p < 0.05; ##p < 0.01; ###p < 0.001 vs. H/R. ANOVA with the least significant difference test.
In the present study, we found that the AT3 downregulated the M2- and M4-mAChRs in the human cerebral microvascular endothelial cell hCMEC/D3 after H/R exposure. After treatment with AT3 or selective inhibitors of M4-mAChR, the H/R-induced ROS production, NO release, and cell apoptosis were abolished. Our findings suggest that AT3 protects the endothelial cells from H/R injury through M4-mAChR inhibition.
The mAChRs are a sub-class of the G-protein-coupled receptors, comprising five subtypes (M1–M5). It was demonstrated that all subtypes of mAChRs are expressed in mouse brain microvascular endothelial cells (BMVECs and bEnd.3), and that their mRNA levels were in the order of M3 > M4 > M1 > M5 > M2 (Radu et al., 2017). In the hCMEC/D3 cells, we found the order of M3 > M2 > M5 > M4 > M1, indicating the expression of mAChRs is cell specific. In the ischemic lesion of the mouse middle cerebral artery occlusion model, the mAChRs, such as M3 was upregulated in the microglia in the ischemic lesion; meanwhile, a protective role of M3-mAChR activation was shown in myocardial injury and motor coordination of microglia and monocytes (Costa et al., 2021). Suppression of M2-mAChR by moxibustion or the receptor antagonist methotramine hemihydrate attenuated the neurogenic detrusor overactivity in spinal cord injury (Wang et al., 2022). Rather different from the activation of mAChRs, however, in the present study, we found that downregulation of M2- and M4-mAChRs by AT3, and M2- and M4-mAChR inhibitors attenuated the H/R-induced cell apoptosis, showing a protective effect of AT3 and M2- and M4-mAChRs antagonists against the H/R injury of hCMEC/D3 cells. Furthermore, the protective role of AT3 in H/R-induced endothelial cell apoptosis is rather different from the effect of atropine, which only slightly attenuated cell apoptosis. Considering the difference in mAChRs expression between AT3 and atropine, M2-mAChR was downregulated by AT3 but not atropine, implying that M2-mAChR is a critical player in the apoptosis of endothelial cells after H/R exposure. In the present work, we tested whether the role of AT3 is similar to that of M2 or M4 inhibitors, thus we did not pretreat the cells with the M2 or M4 inhibitors prior to AT3 application. Also, we could not distinguish the effects of hypoxia or reoxygenation on M2 and M4-mAChR in the present work. The present only proved the AT3 could regulate muscarinic receptors, and the role of AT3 in muscarinic receptors should be further studied in detail.
The M1-, M3-, and M5-mAChRs are coupled with the Gq protein to generate cytosolic calcium transients via phospholipase C signaling pathway, and M2- and M4-mAChRs are coupled with the Gi protein to inhibit the adenylyl cyclase (Radu et al., 2017). The inhibition of cAMP by the activation of myocardial M2-mAChR could activate the NO synthase (iNOS)/NO-cGMP pathway (Ganzinelli et al., 2009). Enhanced NO production could inhibit I/R-induced cell death (Minato et al., 2020). However, a paradoxical result has also been reported, which showed that the inhibition of NO production could attenuate I/R-induced brain injury (Xia et al., 2023). Excessive NO production is harmful for the BBB in ischemia. Once overproduced NO reacts with superoxide anions to form the peroxynitrite, cell necrosis and apoptosis increase and thus aggravate the oxidative stress damage (Radi, 2018).
In H/R-treated rat cardiomyocytes, the activity and expression of iNOS are upregulated, while eNOS expression is decreased (Chen et al., 2001), suggesting iNOS is responsible for increased NO production under H/R. Anisodamine was found to downregulate the expression of iNOS and enhance the expression of eNOS under the I/R condition (Yao et al., 2018). Downregulation of iNOS expression is a possible mechanism of AT3-inhibited NO production in endothelial cells, which should be tested in the future. Although the precise mechanism of NO production reduced by AT3 in endothelial cells after H/R exposure remains to be explored, our results confirmed that AT3 and the M4-mAChR antagonist could inhibit the NO release, along with protection against endothelial cell apoptosis.
Hypoxia causes a substantial expression of NO and ROS, leading to oxidative stress, and eventually, endothelial cell apoptosis and dysfunction (Janaszak-Jasiecka et al., 2018; Dharmashekar et al., 2023; Jin et al., 2023). During H/R exposure, excessive ROS may be derived from the xanthine oxidase, NADPH oxidase, mitochondria, and uncoupled NO synthase (Granger and Kvietys, 2015; Wierońska et al., 2021). Inhibition of NO and ROS production had a protective effect on the H/R-induced endothelial cell injury (Janaszak-Jasiecka et al., 2018; Zhao et al., 2018). Our results showed that the excess of both NO and ROS in the hCMEC/D3 cells after H/R exposure were abolished by AT3. Also, both specific inhibitors of M2- and M4-mAChRs could suppress the H/R-induced ROS, and specific inhibitor of M4-mAChR yielded the largest inhibition. Taken together, our results suggest that AT3 inhibits the NO and ROS production predominantly via the inhibition of M4-mAChR to protect the cerebral microvascular endothelial cells. M2 and M4 may exert synergistic effects on NO production.
Hypoxia enhances ROS in a HIF-1α-dependent pathway (Xie et al., 2023). Hypoxia could induce accumulation of HIF-1α in endothelial cells, with maximum levels at 6 ± 0.5 and 4 ± 0.3 h in vascular and microvascular endothelial cells, respectively (Bartoszewski et al., 2019). Although we possibly missed the maximal levels of HIF-1α in HCMEC/D3 cells after 3 h hypoxia and 3 h reoxygenation, the HIF-1α level after reoxygenation remained 1.2-fold higher than that in normoxic cells. The muscarinic signaling pathway inhibits the hydroxylation and degradation of HIF-1α and induces the synthesis of HIF-1α protein, suggesting that HIF-1α is a downstream critical molecule of mAChR pathways (Hirota et al., 2004; Xie et al., 2023). Consistent with the downregulation of M2- and M4-mAChRs by AT3, AT3 inhibited the H/R-induced expression of HIF-1α. Also, both M4- and M2-mAChR specific inhibitors inhibited the H/R-induced HIF-1α, implying that AT3 attenuates oxidative stress via downregulation of M4- and M2-mAChRs and downstream HIF-1α.
In summary, following H/R exposure, M2- and M4-mAChRs and downstream HIF-1α were upregulated in hCMEC/D3 cells with concomitant excess levels of ROS and NO, which were recovered by AT3 and the corresponding specific mAChR inhibitors. Our results provide novel mechanistic evidence for the application of AT3 for the treatment of brain I/R injury. The expression of mAChRs in the endothelial cells in the ischemia-reperfusion lesions and their alterations by AT3 exposure should be further clarified by in vivo investigations in the future.
Acknowledgement: None.
Funding Statement: Y.Z. received funding from the National Natural Science Foundation of China (12272246) and the Key Research and Development Projects of Sichuan Province (2023YFS0075).
Author Contributions: Conceptualization and methodology Y.Z.; data curation, Y.Z., WL.J., JY.S., and XQ.D.; writing-original draft preparation, Y.Z., WL.J., JY.S., Y.Q., and XQ.D.; funding acquisition, Y.Z. All authors contributed to the review and editing. All authors have read and agreed to the published version of the manuscript.
Availability of Data and Materials: The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors have no conflicts of interest to report regarding the present study.
References
Bartoszewski R, Moszynska A, Serocki M, Cabaj A, Polten A et al. (2019). Primary endothelial cell-specific regulation of hypoxia-inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. Federation of American Societies for Experimental Biology Journal 33: 7929–7941. https://doi.org/10.1096/fj.201802650RR [Google Scholar] [PubMed] [CrossRef]
Bi X, He X, Xu M, Zhao M, Yu X, Lu X, Zang W (2015). Acetylcholine ameliorates endoplasmic reticulum stress in endothelial cells after hypoxia/reoxygenation via M3 AChR-AMPK signaling. Cell Cycle 14: 2461–2472. https://doi.org/10.1080/15384101.2015.1060383 [Google Scholar] [PubMed] [CrossRef]
Campbell BCV, Khatri P (2020). Stroke. Lancet 396: 129–142. https://doi.org/10.1016/S0140-6736(20)31179-X [Google Scholar] [PubMed] [CrossRef]
Chen JY, Brockmoller J, Tzvetkov MV, Wang LJ, Chen XJ (2019). An in vitro study on interaction of anisodine and monocrotaline with organic cation transporters of the SLC22 and SLC47 families. Chinese Journal of Natural Medicines 17: 490–497. https://doi.org/10.1016/S1875-5364(19)30070-6 [Google Scholar] [PubMed] [CrossRef]
Chen H, Li D, Saldeen T, Mehta JL (2001). TGF-β1 modulates NOS expression and phosphorylation of Akt/PKB in rat myocytes exposed to hypoxia-reoxygenation. American Journal of Physiology. Heart and Circulatory Physiology 281: H1035–1039. https://doi.org/10.1152/ajpheart.2001.281.3.H1035 [Google Scholar] [PubMed] [CrossRef]
Chen D, Peng C, Xie X, Chen Q, Liu H, Zhang S, Wan F, Ao H (2017). Low dose of anisodine hydrobromide induced neuroprotective effects in chronic cerebral hypoperfusion rats. CNS & Neurological Disorders Drug Targets 16: 1111–1119. https://doi.org/10.2174/1871527316666171026114043 [Google Scholar] [PubMed] [CrossRef]
Costa A, Haage V, Yang S, Wegner S, Ersoy B et al. (2021). Deletion of muscarinic acetylcholine receptor 3 in microglia impacts brain ischemic injury. Brain, Behavior, and Immunity 91: 89–104. https://doi.org/10.1016/j.bbi.2020.09.008 [Google Scholar] [PubMed] [CrossRef]
Dharmashekar C, Shreevatsa B, Jain AS, Harendra B, Pradeep S et al. (2023). Evaluating the antimicrobial and anti-hemolytic activity of synthesized pseudopeptide against leptospiral species: In silico and in vitro approach. Molecules 28: 1106. https://doi.org/10.3390/molecules28031106 [Google Scholar] [PubMed] [CrossRef]
Dong Z, Han L, Duan P, Liang J, Dong R, Liu N, Liu H, Hu C (2021). Anisodine hydrobromide injection for the treatment of acute ischemic stroke: A randomized controlled trail. Chinese Journal of Pharmacoepidemiology 30: 296–300. https://doi.org/10.19960/j.cnki.issn1005-0698.2021.05.002 [Google Scholar] [CrossRef]
Feng W, Liu S, Zhang C, Xia Q, Yu T, Zhu D (2019). Comparison of cerebral and cutaneous microvascular dysfunction with the development of type 1 diabetes. Theranostics 9: 5854–5868. https://doi.org/10.7150/thno.33738 [Google Scholar] [PubMed] [CrossRef]
Ganzinelli S, Borda E, Joensen L, Sterin-Borda L (2009). Chagasic antibodies induce cardiac COX-2/iNOS mRNA expression with PGE2/NO production. International Journal of Cardiology 134: 212–223. https://doi.org/10.1016/j.ijcard.2008.02.008 [Google Scholar] [PubMed] [CrossRef]
Granger DN, Kvietys PR (2015). Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biology 6: 524–551. https://doi.org/10.1016/j.redox.2015.08.020 [Google Scholar] [PubMed] [CrossRef]
Han YF, Chen XY (1984). Effect of anisodine and other cholinergic drugs on conditioned response of the hippocampal theta rhythm in rabbits. Zhongguo Yao Li Xue Bao 5: 166–170. [Google Scholar] [PubMed]
Hirota K, Fukuda R, Takabuchi S, Kizaka-Kondoh S, Adachi T, Fukuda K, Semenza GL (2004). Induction of hypoxia-inducible factor 1 activity by muscarinic acetylcholine receptor signaling. The Journal of Biological Chemistry 279: 41521–41528. https://doi.org/10.1074/jbc.M405164200 [Google Scholar] [PubMed] [CrossRef]
Jackson-Weaver O, Friedman JK, Rodriguez LA, Hoof MA, Drury RH, Packer JT, Smith A, Guidry C, Duchesne JC (2019). Hypoxia/reoxygenation decreases endothelial glycocalyx via reactive oxygen species and calcium signaling in a cellular model for shock. The Journal of Trauma and Acute Care Surgery 87: 1070–1076. https://doi.org/10.1097/TA.0000000000002427 [Google Scholar] [PubMed] [CrossRef]
Janaszak-Jasiecka A, Siekierzycka A, Bartoszewska S, Serocki M, Dobrucki LW, Collawn JF, Kalinowski L, Bartoszewski R (2018). eNOS expression and NO release during hypoxia is inhibited by miR-200b in human endothelial cells. Angiogenesis 21: 711–724. https://doi.org/10.1007/s10456-018-9620-y [Google Scholar] [PubMed] [CrossRef]
Jin P, Pan Q, Lin Y, Dong Y, Zhu J, Liu T, Zhu W, Cheng B (2023). Platelets facilitate wound healing by mitochondrial transfer and reducing oxidative stress in endothelial cells. Oxidative Medicine and Cellular Longevity 2023: 2345279. https://doi.org/10.1155/2023/2345279 [Google Scholar] [PubMed] [CrossRef]
Kong T, Liu M, Ji B, Bai B, Cheng B, Wang C (2019). Role of the extracellular signal-regulated kinase 1/2 signaling pathway in ischemia-reperfusion injury. Frontiers in Physiology 10: 1038. https://doi.org/10.3389/fphys.2019.01038 [Google Scholar] [PubMed] [CrossRef]
Liao F, Zheng Y, Cai J, Fan J, Wang J, Yang J, Cui Q, Xu G, Tang C, Geng B (2015). Catestatin attenuates endoplasmic reticulum induced cell apoptosis by activation type 2 muscarinic acetylcholine receptor in cardiac ischemia/reperfusion. Scientific Reports 5: 16590. https://doi.org/10.1038/srep16590 [Google Scholar] [PubMed] [CrossRef]
Liu WD, Chen LL, Shen CY, Jiang LB (2015). Neuroprotective effect of compound anisodine in a mouse model with chronic ocular hypertension. Chinese Medical Journal 128: 2652–2657. https://doi.org/10.4103/0366-6999.166043 [Google Scholar] [PubMed] [CrossRef]
Liu Y, Sun L, Pan Z, Bai Y, Wang N et al. (2011). Overexpression of M3 muscarinic receptor is a novel strategy for preventing sudden cardiac death in transgenic mice. Molecular Medicine 17: 1179–1187. https://doi.org/10.2119/molmed.2011.00093 [Google Scholar] [PubMed] [CrossRef]
Lopez-Ramirez MA, Lai CC, Soliman SI, Hale P, Pham A et al. (2021). Astrocytes propel neurovascular dysfunction during cerebral cavernous malformation lesion formation. The Journal of Clinical Investigation 131: e139570. https://doi.org/10.1172/JCI139570 [Google Scholar] [PubMed] [CrossRef]
Luo SY, Li R, Le ZY, Li QL, Chen ZW (2017). Anfibatide protects against rat cerebral ischemia/reperfusion injury via TLR4/JNK/caspase-3 pathway. European Journal of Pharmacology 807: 127–137. https://doi.org/10.1016/j.ejphar.2017.04.002 [Google Scholar] [PubMed] [CrossRef]
Meng P, Yang R, Jiang F, Guo J, Lu X, Yang T, He Q (2021). Molecular mechanism of astragaloside IV in improving endothelial dysfunction of cardiovascular diseases mediated by oxidative stress. Oxidative Medicine and Cellular Longevity 2021: 1481236. https://doi.org/10.1155/2021/1481236 [Google Scholar] [PubMed] [CrossRef]
Miao Y, Zhou J, Zhao M, Liu J, Sun L et al. (2013). Acetylcholine attenuates hypoxia/reoxygenation-induced mitochondrial and cytosolic ROS formation in H9c2 cells via M2 acetylcholine receptor. Cellular Physiology and Biochemistry 31: 189–198. https://doi.org/10.1159/000343360 [Google Scholar] [PubMed] [CrossRef]
Minato H, Hisatome I, Kurata Y, Notsu T, Nakasone N et al. (2020). Pretreatment with cilnidipine attenuates hypoxia/reoxygenation injury in HL-1 cardiomyocytes through enhanced NO production and action potential shortening. Hypertension Research 43: 380–388. https://doi.org/10.1038/s41440-019-0391-7 [Google Scholar] [PubMed] [CrossRef]
Pan SY, Han YF (2004). Comparison of the inhibitory efficacy of four belladonna drugs on gastrointestinal movement and cognitive function in food-deprived mice. Pharmacology 72: 177–183. https://doi.org/10.1159/000080102 [Google Scholar] [PubMed] [CrossRef]
Qiu Y, Ouyang Z, Zhong J, Shen J, Jiang W, Liu Y, Wan F, Zeng Y (2022). Anisodamine hydrobromide attenuates oxidative stress and proinflammatory cytokines in septic rats induced by cecal ligation and puncture. Cellular and Molecular Biology 68: 54–60. https://doi.org/10.14715/cmb/2022.68.12.11 [Google Scholar] [PubMed] [CrossRef]
Radi R (2018). Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proceedings of the National Academy of Sciences of the United States of America 115: 5839–5848. https://doi.org/10.1073/pnas.1804932115 [Google Scholar] [PubMed] [CrossRef]
Radu BM, Osculati AMM, Suku E, Banciu A, Tognoli C et al. (2017). All muscarinic acetylcholine receptors (M1-M5) are expressed in murine brain microvascular endothelium. Scientific Reports 7: 5083. https://doi.org/10.1038/s41598-017-05384-z [Google Scholar] [PubMed] [CrossRef]
Robertson SJ, Mokgokong R, Kania KD, Guedj AS, Hladky SB, Barrand MA (2011). Nitric oxide contributes to hypoxia-reoxygenation-induced P-glycoprotein expression in rat brain endothelial cells. Cellular and Molecular Neurobiology 31: 1103–1111. https://doi.org/10.1007/s10571-011-9711-4 [Google Scholar] [PubMed] [CrossRef]
Takai K, Enomoto T (2018). Discovery and development of muscarinic acetylcholine M4 activators as promising therapeutic agents for CNS diseases. Chemical & Pharmaceutical Bulletin 66: 37–44. https://doi.org/10.1248/cpb.c17-00413 [Google Scholar] [PubMed] [CrossRef]
Wang L, Fu YB, Liu Y, Yang NN, Ma SM, Wang XR, Huang J, Shi GX, Yang JW, Liu CZ (2022). Moxibustion attenuates neurogenic detrusor overactivity in spinal cord injury rats by inhibiting M2/ATP/P2X3 pathway. Brain Research 1788: 147926. https://doi.org/10.1016/j.brainres.2022.147926 [Google Scholar] [PubMed] [CrossRef]
Wierońska JM, Cieślik P, Kalinowski L (2021). Nitric oxide-dependent pathways as critical factors in the consequences and recovery after brain ischemic hypoxia. Biomolecules 11: 1097. https://doi.org/10.3390/biom11081097 [Google Scholar] [PubMed] [CrossRef]
Wu S, Wu B, Liu M, Chen Z, Wang S et al. (2019). Stroke in China: Advances and challenges in epidemiology, prevention, and management. The Lancet Neurology 18: 394–405. https://doi.org/10.1016/S1474-4422(18)30500-3 [Google Scholar] [PubMed] [CrossRef]
Xia B, Li Q, Zheng K, Wu J, Huang C, Liu K, You Q, Yuan X (2023). Down-regulation of Hrd1 protects against myocardial ischemia-reperfusion injury by regulating PPARα to prevent oxidative stress, endoplasmic reticulum stress, and cellular apoptosis. European Journal of Pharmacology 954: 175864. https://doi.org/10.1016/j.ejphar.2023.175864 [Google Scholar] [PubMed] [CrossRef]
Xie L, Wang Q, Ma J, Zeng Y (2023). Hypoxia-induced reactive oxygen species in organ and tissue fibrosis. BIOCELL 47: 261–267. https://doi.org/10.32604/biocell.2023.024738 [Google Scholar] [CrossRef]
Xu W, Deng YF (1996). Effect of anisodine on acute forebrain ischemia-reperfusion damage in rats. Zhongguo Yao Li Xue Bao 17: 161–163. [Google Scholar] [PubMed]
Yao BJ, He XQ, Lin YH, Dai WJ (2018). Cardioprotective effects of anisodamine against myocardial ischemia/reperfusion injury through the inhibition of oxidative stress, inflammation and apoptosis. Molecular Medicine Reports 17: 1253–1260. https://doi.org/10.3892/mmr.2017.8009 [Google Scholar] [PubMed] [CrossRef]
Zhang Y (2022). Effect and safety of anisodine hydrobromide injection for treating patients with acute ischemic stroke. Smart Healthcare 8: 23–25. https://doi.org/10.19335/j.cnki.2096-1219.2022.08.008 [Google Scholar] [CrossRef]
Zhao S, Wang Y, Zhang X, Zheng L, Zhu B, Yao S, Yang L, Du J (2018). Melatonin protects against hypoxia/reoxygenation-induced dysfunction of human umbilical vein endothelial cells through inhibiting reactive oxygen species generation. Acta Cardiologica Sinica 34: 424–431. https://doi.org/10.6515/ACS.201809_34(5).20180708A [Google Scholar] [PubMed] [CrossRef]
Zhong J, Ouyang Z, Shen J, Zeng Y (2023). Improvement of hemodynamics in mesenteric microcirculation in septic shock rats by anisodamine and anisodine. Mechanobiology in Medicine 1: 100006. https://doi.org/10.1016/j.mbm.2023.100006 [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools