
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
Bone marrow mesenchymal stem cell-induced autophagy ameliorates TNBS-induced experimental colitis by downregulating the NLRP3 inflammasome
1 Department of Gastroenterology, The Affiliated Changzhou No. 2 People’s Hospital, Nanjing Medical University, Changzhou, China
2 Department of Gastroenterology, Jinling Hospital, Nanjing Medical University, Nanjing, China
* Corresponding Author: FANGYU WANG. Email:
# These authors contributed equally to this work
BIOCELL 2023, 47(12), 2627-2639. https://doi.org/10.32604/biocell.2023.042586
Received 05 June 2023; Accepted 11 October 2023; Issue published 27 December 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: This study aimed to elucidate the potential mechanisms through which bone marrow-derived mesenchymal stem cells (BM-MSCs) may be effective in alleviating experimental colitis induced by treatment with 2,4,6-trinitrobenzene-sulfonate acid (TNBS), specifically through autophagy modulation. Methods: BM-MSCs were collected from BALB/c mice for subsequent experiments. The study employed cell counting kits (CCK-8) to investigate the impact of the MSC-conditioned medium (M medium) on the proliferation of RAW264.7 macrophages. The GFP-mRFP-LC3 adenovirus was transfected into RAW264.7 to detect autophagic flux. The gene expression of cytokines was assessed through quantitative reverse transcription polymerase chain reaction (qRT-PCR). Western blot analysis was employed to determine the presence of a binding interaction between NOD-like receptor protein 3 (NLRP3) and autophagy. Furthermore, a colitis mouse model was established by TNBS induction. Clinical disease activity score was assessed regularly, and histological and morphometric analyses were performed on colonic tissues. Inflammatory serum cytokines were identified using an enzyme-linked immunosorbent assay. Results: BM-MSCs significantly promoted the proliferation of RAW264.7. In vitro lipopolysaccharide (LPS)-stimulated RAW264.7 cells, treated with BM-MSCs, triggered autophagy and inhibited cytokine mRNA expression. Additionally, in LPS-induced RAW264.7, BM-MSCs enhanced the Beclin1 protein expression and the microtubule-associated protein 1 light chain 3 (LC3)-II to LC3-I ratio while suppressing the protein levels of NLRP3 and apoptosis-associated speck-like protein (ASC). Nevertheless, 3-methyladenine (3-MA), an inhibitor of autophagy, prevented the impact of BM-MSCs by reducing the levels of NLRP3 and ASC proteins, suggesting that autophagy triggered the inhibition of the NLRP3 inflammasome. In comparison to the mice in the TNBS group, the mice in the TNBS+MSC group displayed a more acute form of colitis, and the IL1β and IL18 cytokines in their serum were lowered as well. In the meantime, 3-MA raised IL1β and IL18 cytokine levels and worsened TNBS-induced experimental colitis. Conclusions: BM-MSCs can suppress inflammation in TNBS-induced experimental mice by inhibiting the NLRP3 inflammasome, thereby enhancing autophagy.Keywords
Abbreviations
| ANOVA | a one-way analysis of variance |
| ASC | apoptosis-associated speck-like protein |
| ATP | adenosine triphosphate |
| BM-MSCs | bone marrow-derived mesenchymal stem cells |
| CCK-8 | cell counting kits |
| CM | conditioned medium |
| DAMPs | danger-associated molecular patterns |
| DAI | disease activity index |
| D media | RAW-conditioned medium |
| EDTA | ethylenediaminetetraacetic acid |
| ELISA | Enzyme-linked immunosorbent assay |
| FBS | fetal bovine serum |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GFP | green fluorescent protein |
| IBDs | Inflammatory bowel diseases |
| IL | interleukin |
| LC3 | light chain 3 |
| LPS | lipopolysaccharide |
| M medium | MSC-conditioned medium |
| MOI | multiplicity of infection |
| MSCs | Mesenchymal stem cells |
| NLRP3 | NOD-like receptor protein 3 |
| OD | Optical Density |
| PAMPs | pathogen-associated molecular patterns |
| PBS | phosphate-buffered saline |
| qRT-PCR | quantitative reverse transcription polymerase chain reaction |
| SD | standard deviation |
| TNBS | 2,4,6-trinitrobenzene-sulfonate acid |
| TNFα | tumor necrosis factor α |
| 3-MA | 3-methyladenine |
Inflammatory bowel diseases (IBDs), which include Crohn’s disease and ulcerative colitis, are progressive, multifactorial, and chronic immune-mediated inflammation of the gastrointestinal tract (Adolph et al., 2022). IBD is characterized by inflammation of the mucosal lining of the intestines. This condition presents with recurring abdominal pain associated with weight loss, diarrhea, bloody stools, and an increased neutrophil and macrophage count. These immune cells stimulate the release of proteolytic enzymes and cytokines with the generation of free radicals, resulting in the development of ulcers and inflammation (Flynn and Eisenstein, 2019). Current strategies that target the symptoms of IBD do not substantially alter its pathogenesis (Saez et al., 2023). Patients with IBD have an enhanced risk of thrombosis, bone complications, intestinal malignancies (colorectal dysplasia, small bowel, and/or other cancers), and a probable small risk of lymphoma (Nuñez et al., 2021; Quaglio et al., 2022). Globally, IBD imposes significant health and economic burdens while markedly diminishing patients’ quality of life (GBD 2017 Inflammatory Bowel Disease Collaborators, 2020). However, as the exact underlying pathophysiology of IBD is still undetermined, a cure remains elusive. Despite the unidentified cause, substantial progress has been made in recent years to elucidate its associated pathogenesis. A study suggests that the pathogenesis of IBD is related to the host’s genetic susceptibility, intestinal microbiota, other environmental factors, and immunological anomalies (Ramos and Papadakis, 2019). The assessment of IBD-related genes and their loci indicates the involvement of multiple pathways in regulating intestinal homeostasis, including epithelial barrier activity, innate mucosal defense, cell migration, immune regulation, autophagy, adaptive immunity, and cellular homeostatic metabolic pathways (Younis et al., 2020).
The NOD-like receptor protein 3 (NLRP3) is regarded as the canonical inflammasome protein complex, consisting of three main components: the NLRP3 sensor, the apoptotic speck-like protein CARD (ASC), and the effector caspase-1. Research has indicated that the accumulation of reactive components of the NLRP3 inflammasome occurs upon the recognition of certain pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). Following this, the assembly of the NLRP3 inflammasome complex triggers the activation of procaspase-1 to caspase-1, which in turn cleaves the precursor forms of IL1β and IL18. This process generates mature cytokines that play a vital role in inducing pyroptosis and cellular pyronecrosis, hence regulating the inflammatory response (Huang et al., 2021; Swanson et al., 2019). Growing evidence indicates that the NLRP3 inflammasome plays a critical role in developing IBD, as it triggers inflammation in the intestinal mucosa via caspase-1 activation (Zhen and Zhang, 2019; Chen et al., 2019). Murine colitis models indicate that colitis triggered by dextran sulfate sodium (DSS) is linked to NLPP3 inflammasome stimulation and enhanced IL1β generation (Gong et al., 2018). Inhibiting the NLRP3 inflammasome is one of the most promising therapeutic strategies to improve IBD (Chen et al., 2021). Zhang et al. (2014) revealed that the antagonist of the IL1β receptor and caspase-1 suppressor ameliorated IBD in mice. The inhibition of the NLRP3 inflammasome by the MCC950 is a highly potent and selective small-molecule inhibitor that targets both canonical and noncanonical activation of the NLRP3 inflammasome. Additionally, IBD mice treated with glyburide have also shown suppression of inflammasome, further emphasizing the significance of inflammasome in the course of IBD (Perera et al., 2018).
Mesenchymal stem cells (MSCs) are multipotent stem cells that originate from the mesoderm. They have an excellent potential for self-renewal and can develop into various cell types. Transplantation of MSCs has been shown to promote the repair of IBD by efficiently improving mucosal epithelium regeneration (Bergmann et al., 2022). In patients with IBD, evidence has revealed a link between autophagy impairment and an increased risk of IBD (Solà-Tapias et al., 2020). The transplantation of MSCs exerts a protective effect induced by modulating autophagy and decreasing inflammatory responses (Fig. 1) (Lin et al., 2022). Autophagy primarily serves a pro-survival function, facilitating the degradation or recycling of unnecessary cellular components to generate adenosine triphosphate (ATP) and produce proteins during periods of stress or nutrient deficiency. Autophagy is involved in various mechanisms, such as development, defense against pathogens, differentiation, adaptive and innate immunity, senescence, and cell death.
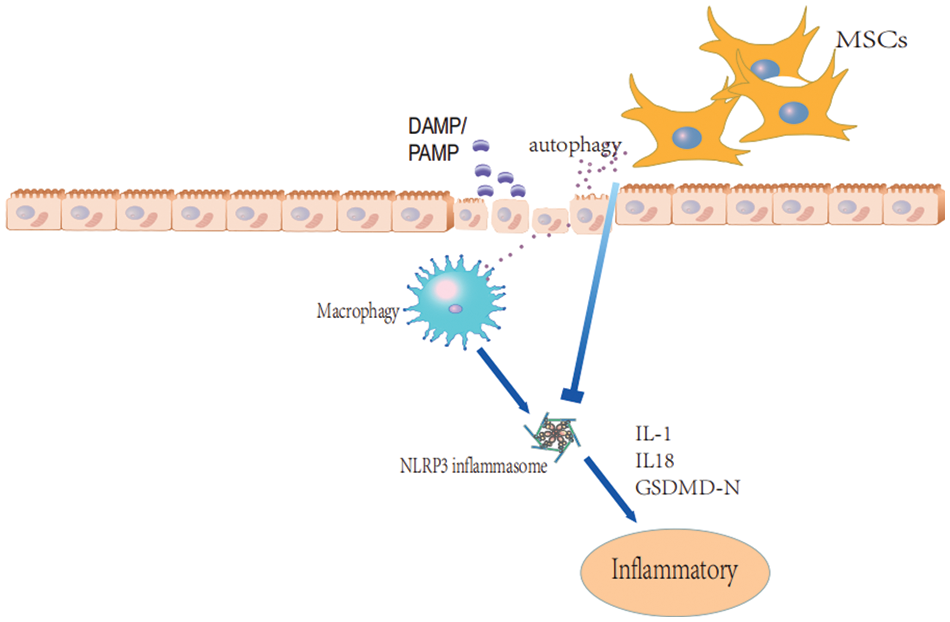
Figure 1: The potential mechanism through which bone marrow-derived mesenchymal stem cells (BM-MSCs) may decrease inflammation.
Autophagy activation has been demonstrated to impede NLRP3 inflammasome activation (Biasizzo and Kopitar-Jerala, 2020). Autophagy plays a vital role in regulating inflammasome activation, from eliminating inflammasome-induced endogenous signals to sequestering and degrading inflammasome components. Moreover, autophagy also influences the fate of IL1β, a cytokine abundant in autophagosomes. As a crucial modulator of inflammasome activation, autophagy serves as a subsequent stimulator of the IL-1 family of cytokines. However, the relationship between the anti-inflammatory and autophagic effects of MSCs remains unclear.
Given the pivotal role that the NLRP3 inflammasome plays in the development and progression of IBD, the efficacy of NLRP3 inflammasome inhibition, and the potential of MSCs to enhance autophagy in IBD treatment, we propose a hypothesis. We suggest that MSCs may provide protection against TNBS-induced colitis in mice by inhibiting the NLRP3 inflammasome, possibly through the mediation of enhanced autophagic responses. To validate this hypothesis, specific details are reported as follows.
Extraction, propagation, and identification of bone marrow-derived mesenchymal stem cells (BM-MSCs)
From the femurs of 8–10 week-old BALB/c mice, BM-MSCs were obtained (CAVENS.LA Technology, Changzhou, China) (Huang et al., 2022a). BM-MSCs were acquired by flushing the femurs using complete propagation media consisting of Dulbecco’s modified Eagle’s medium/Nutrient Mixture F-12 (DMEM/F12; Gibco, Carlsbad, California, USA), fetal bovine serum (10%; FBS; Gibco, Carlsbad, California, USA), penicillin, and streptomycin (Beyotime Bio, Nanjing, China). A single-cell suspension was propagated in an equal volume of complete DMEM/F12 medium in dishes and incubated in 5% CO2 at 37°C. The next day, non-adherent cells were rinsed out, and the remaining cells were allowed to grow. The medium was replaced every 3–4 days. Upon achieving 80%–90% population, the cells were extracted using 0.25% trypsin–ethylenediaminetetraacetic acid (EDTA) (Beyotime Bio, Nanjing, China), and the medium was changed on day 3.
Lipid-osteogenic differentiation of mouse BM-MSCs
The cultured cells were passaged when the cells reached 80%–90% confluency and osteogenic and adipogenic differentiation was induced in the third passage of cells. The evaluation of cell stemness involved the assessment of the differentiation potential of BM-MSCs into osteoblasts and adipocytes. The differentiation of BM-MSCs into adipocytes and osteocytes was induced using differentiation-induction kits (CTCC Bioscience, Beijing, China) according to the manufacturer’s protocols. Adipocytes and osteoblasts were assessed using Oil Red O and Alizarin Red dyes (Solarbio, Beijing, China), respectively (Huang et al., 2022a).
Flow cytometry evaluation of BM-MSC phenotypes
The propagated BM-MSCs were distinguished using the minimal functional and phenotypic parameters provided by the International Society for Cellular Therapy (ISCT) (Dominici et al., 2006). The characteristics of the three passaged BM-MSC cells were evaluated using a FACSCalibur flow cytometer (BD Biosciences, San Jose, California, USA), following a previously documented method (Stavely et al., 2015). The following primary antibodies utilized: CD105 monoclonal antibody (FITC, Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA), rat IgG2a kappa isotype control (FITC, Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA), CD90 monoclonal antibody (FITC, Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA), rat IgG1 κ isotype control (FITC, Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA), anti-mouse CD73 antibody (FITC, BioLegend, San Diego, California, USA), anti-mouse CD45 antibody (FITC, BioLegend, San Diego, California, USA), PE anti-mouse CD34 antibody (BioLegend, San Diego, California, USA), and PE rat IgG2a κ isotype control antibody (BioLegend, San Diego, California, USA). Cell assessment was carried out using FlowJo software (Becton, Dickinson and Company, Ashland, Oregon, USA).
RAW264.7 cell culture and conditioned medium (CM) collection
Macrophages are the most common inflammatory cells, so we selected RAW264.7 murine macrophages for subsequent investigations. RAW264.7 cells were provided by the Laboratory of General Surgery, the Affiliated Changzhou No. 2 People’s Hospital, Nanjing Medical University, and cultured in 10% FBS (Gibco, Carlsbad, California, USA) containing DMEM (Gibco, Carlsbad, California, USA). Lipopolysaccharides (LPS, Sigma-Aldrich, St. Louis, Missouri, USA) were prepared in distilled water at a 1 mg/mL stock concentration and diluted to 100 ng/mL for the working medium concentration.
MSC-conditioned medium (M medium) and RAW-conditioned medium (D media) were collected for in vitro experiments following 24 h of incubation with BM-MSCs and RAW264.7 (Chen et al., 2013).
RAW264.7 was categorized into four groups: control, LPS, LPS+MSCs, and LPS+MSCs+3-3-methyladenine (3-MA, a specific autophagy inhibitor of proliferation and colony formation). All the chemicals used in different treatments were pre-dissolved in DMEM. The cells were treated with a 100 ng/mL concentration of LPS for 24 h in three experimental groups: LPS, LPS+MSCs, and LPS+MSCs+3-MA. The LPS+MSCs+3-MA group received 5 mM 3-MA for 15 min, followed by 2 mL of M medium. The LPS+MSCs group received M medium. The control and LPS groups received an equal volume of D medium.
Detection of cell activity by Cell Counting Kit (CCK)-8
The viability of RAW264.7 cells was assessed using the CCK-8 in accordance with the manufacturer’s instructions (Dojindo Laboratories, Kanagawa Prefecture, Japan). RAW264.7 macrophages (1,000/well) were seeded into 96-well plates. RAW264.7 macrophages were kept in M and D media for 6, 12, 18, 24, 36, 48, and 60 h to detect the favorable impact of BM-MSCs on cell viability. At the prespecified time points, CCK-8 solution (10 μL; Beyotime Bio, Nanjing, China) was introduced into the cells. After 1 h of incubation, optical density (OD) was calculated at 450 nm via a microplate reader (TECAN, Zurich, Switzerland). Each group was tested in triplicate in three replicate wells.
Observation of autophagy flow of each group by using a double-label adenovirus
RAW264.7 macrophages were evenly incubated into six-well plates at a concentration of 1 × 105 cells/well at 24 h prior to transfection. When the confluency reached 50%–70%, the mRFP-GFP-LC3 adenovirus was introduced at a multiplicity of infection (MOI) of 100, as described by the manufacturer (HanBio, Shanghai, China). After a duration of 6 hours, the cells were replenished with fresh medium and subjected to different treatments as performed previously. The cells were provided with 100 ng/mL LPS for 24 h in the LPS, LPS+MSCs, and LPS+MSCs+3-MA groups. The LPS+MSCs+3-MA group received 5 mM 3-MA for 15 min, followed by 2 mL of M medium. The LPS+MSCs group received M medium. The control and LPS groups received an equal volume of D medium. After 24 h, the cells were photographed and analyzed using an Olympus IX71 fluorescence microscope (Olympus, Tokyo, Japan). Autophagic flux was assessed as mentioned before (Luo et al., 2023). Autophagy flux strength was observed clearly under the microscope by measuring different colored spots. Green/red puncta were viewed, and images were captured using Olympus IX71. When the lysosomes and autophagosomes were fused, green fluorescent protein (GFP) fluorescence was quenched, and only red fluorescence was visible. In the merged images, yellow fluorescent spots (merge) represent transfected cells (merge)/autophagosomes, red spots (mRFP) denote autophagic lysosomes and enhanced fluorescent dots suggest elevated autophagic flux.
RNA extraction and quantitative reverse transcription polymerase chain reaction (qRT-PCR)
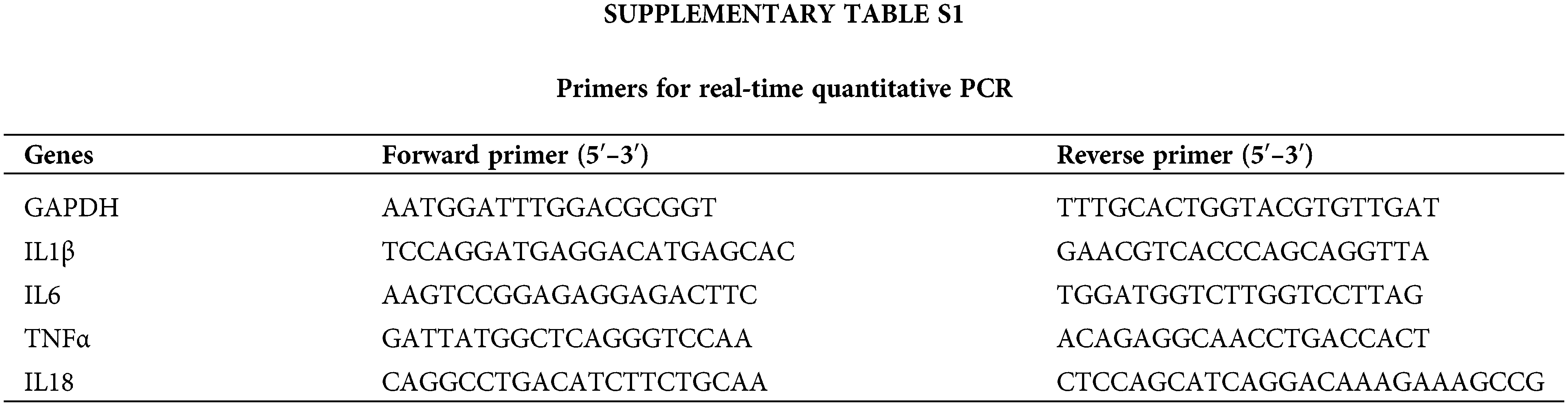
The Trizol reagent (Vazyme Biotech Co., Nanjing, China) was utilized to extract total RNA from Raw 264.7 macrophages. HiScript® IIQ RT SuperMix for qPCR (Vazyme Biotech Co., Nanjing, China) was applied for reverse transcription. Green Master Mix (Low ROX Premixed) (Vazyme Biotech Co., Nanjing, China) was utilized for RT-PCR as per the manufacturer’s recommendations. The gene expression analysis kits for mouse IL1β, IL18, tumor necrosis factor (TNFα), IL6, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (all from Sangon Biotech, Shanghai, China) were used for qRT-PCR. The primer sets specific to each gene are given in Suppl. Table S1. The normalization of gene expression was done using the expression of GAPDH.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analyses
Cultured cells were propagated and reaped using a lysis buffer with protease inhibitor (Beyotime Bio, Nanjing, China). Protein samples (20 mg/lane) were resolved by 5%–20% SDS-PAGE and subsequently transferred to a 0.22 mm polyvinylidene fluoride (PVDF) membrane (Millipore, Sigma, Louis, Missouri, USA) according to the kit’s protocol. The protein expression levels of NLRP3 ASC, Beclin1, LC3II/I ratio (all from Cell Signaling Technology, Danvers, Massachusetts, USA), and GAPDH (Beyotime Bio, Nanjing, China) were determined in RAW264.7. The quantification of each protein was performed using the ImageJ program (National Institutes of Health).
Animals and induction of TNBS-induced colitis
The study utilized male BABL/c mice, aged 8–10 weeks, with a body weight ranging from 20 to 22 g. The mice were procured from CAVENS. LA technology (Changzhou, China). The animals were kept at 22°C–24°C under a 12-h day/night cycle with ad libitum feeding. After fasting overnight, all animals were anesthetized through ether inhalation. The Animal Care and Use Committee at Nanjing Medical University granted approval for all experimental work involving the use of animals (Approval No. 2021DZGKJDWLS-00143).
TNBS modeling in mice was established according to internationally recognized Morris methods (Morris et al., 1989). In brief, 2.5 mg (100 μL) of 2,4,6-trinitrobenzene-sulfonate acid (TNBS) (Sigma-Aldrich Corp) in 50% ethanol was introduced into the rectum, 4 cm proximal to the anus by using a lubricated silicone catheter. The mice received BM-MSC therapy for 3 h post-TNBS delivery at the peak of tissue injury (Pontell et al., 2009). BM-MSCs were delivered by enema at 1 × 106 cells in 100 μL of sterile phosphate-buffered saline (PBS). The experimental group designated as 3-MA was administered an intraperitoneal injection of the autophagy inhibitor 3-MA at a dosage of 24 mg/kg 30 min prior to the administration of LPS. After the treatment, the mice were weighed daily and monitored. At 7 days post-treatment, the mice (n = 20) were euthanized by stunning and exsanguination and then randomly assigned into four groups (n = 5 mice/group): control, TNBS, TNBS+MSCs, and TNBS+MSCs+3MA.
Clinical score and evaluation of disease activity
The daily recording of body weight and disease activity index (DAI) was conducted following the induction of colitis since the DAI is a commonly employed measure for assessing the severity of colitis. The calculation of the DAI was performed by considering the weight loss, stool consistency, and presence of hematochezia, in line with the scoring criteria established in the existing literature (Shon et al., 2015; Huang et al., 2022b). Body weight alterations were indicated as a percentage loss of the baseline body weight.
Histology and morphometric analyses
After euthanasia, the colon was removed, and its length was measured. The general morphology and histology of colonic mucosa were graded. For performing paraffin embedding, a segment of the distal colon tissue was subjected to fixation using a 10% buffered formalin solution. The H&E staining procedure was performed in accordance with established guidelines.
Enzyme-linked immunosorbent assay (ELISA)
Blood samples were acquired from all mice, followed by serum separation and storage at −80°C until use. The serum concentrations of IL1β and IL18 (Multi Sciences Biotech Co., Ltd., Guangzhou, China) were measured using an ELISA kit (Camilo Bioengineering Co., Ltd., Guangzhou, China) according to the kit’s protocol.
The data were subjected to statistical analysis using GraphPad Prism (GraphPad Software, version 8.0.2, San Diego, CA, USA) and were represented as the mean ± standard deviation (SD). The statistically significant variabilities between two or several groups were elucidated by Student’s t-test or one-way analysis of variance (ANOVA). A p-value ≤ 0.05 was considered statistically significant.
Balb/c mouse culture and BM-MSC identification
BM-MSCs were successfully extracted and culture-expanded from Balb/c mice as described previously (Huang et al., 2022a). The cultured BM-MSCs were identified as round or oval after inoculation. As the adherence increased, the cells acquired the multi-fusiform shape and formed a monolayer. After propagation, BM-MSCs exhibited a uniform shape and a long, spindle-like fibroblast morphology (Fig. 2A), which is a common characteristic of MSCs.
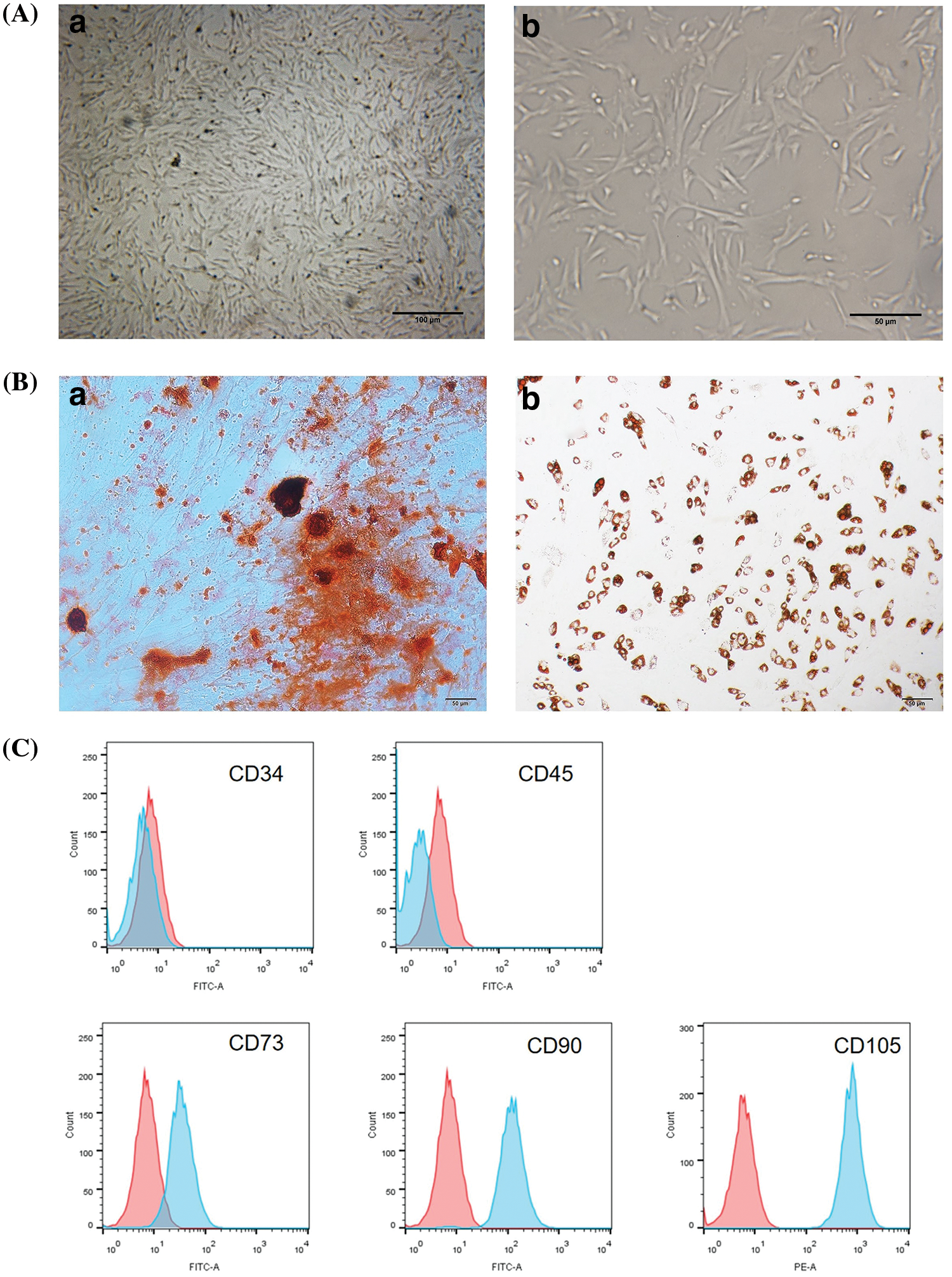
Figure 2: Characterization of BM-MSCs. (A) Under 10× (bar: 100 μm) (a) and 40× (bar: 50 μm) (b) microscope magnification, a characteristic spindle-shaped fibroblast-like morphology was observed. (B) Multilineage-transforming ability of BM-MSCs. The assessment of osteogenic differentiation involved the examination of calcium deposition using alizarin Red S staining (a); the accumulation of lipid vacuoles in the cells differentiated into adipocytes were evaluated by the oil red O staining (b). Scale bar = 50 µm. (C) Phenotypic analysis of BM-MSCs by flow cytometry. Histograms labeled with surface antigen expression (blue) and their corresponding isotype control (red). BM-MSCs expressed CD90, CD105, and CD73 but were negative for CD45 and CD34.
In vitro differentiation of BM-MSCs
MSCs in osteogenic media showed an altered morphology from spindle to cuboidal and formed large nodules by the 18th day of induction. After osteogenic induction, alizarin red staining revealed densely stained calcium salt deposition in the culture of BM-MSCs (red), indicating their osteogenic capacity (Fig. 2B).
Furthermore, oil red O staining showed the formation of an oil drop after adipogenic induction, which is indicative of BM-MSC differentiation into adipocytes (Fig. 2B). Adipogenic differentiation was demonstrated by lipid vacuole accumulation. These vacuoles were visible after induction and had increased size and number until they coalesced with increasing induction time. Oil Red O and Alizarin Red S staining indicated that the BM-MSCs differentiated into mature adipocytes and osteocytes (Fig. 2B).
Identification of surface markers for BM-MSCs
BM-MSCs are positive for endoglin receptors, namely, CD73, stem cell antigen-1 (Sca-1), CD105, and CD90, but are negative for hematopoietic cell line indices such as CD34, CD11b, major histocompatibility complex class II (MHCII), CD45, and endothelial marker CD31. The propagated mesenchymal cells of the third passage had a single phenotypic population, as assessed by flow cytometry identification of the surface-expressed antigens. The flow cytometry data indicated that BM-MSCs were negative for CD34 and CD45 and positive for CD90, CD73, and CD105 (Fig. 2C).
Therefore, we determined and selected BM-MSC populations based on their plastic adherence ability, phenotypic characteristics, and ability to transform into adipocytes and osteoblasts. Overall, BM-MSCs from Balb/c mice met the well-established parameters.
BM-MSCs promote RAW264.7 cell proliferation
RAW264.7 macrophages were cultured with M and D media for 6, 12, 24, 36, 48, or 60 h to determine whether BM-MSCs exert a proliferative effect. In order to investigate the impact of BM-MSCs on cellular viability, a CCK-8 assay was conducted. Cell proliferation in the control group did not significantly differ from that in the BM-MSCs-treated RAW264.7 macrophages after 6 or 60 h of culture. However, cell proliferation in the BM-MSCs group significantly increased following 12, 24, 36, and 48 h of culture with BM-MSCs in M medium (p < 0.05). As shown in Fig. 3, the proliferation of RAW 264.7 macrophages treated with M media was markedly enhanced in comparison to the control group. These results demonstrate that BM-MSCs promoted the proliferation of RAW264.7 macrophages.
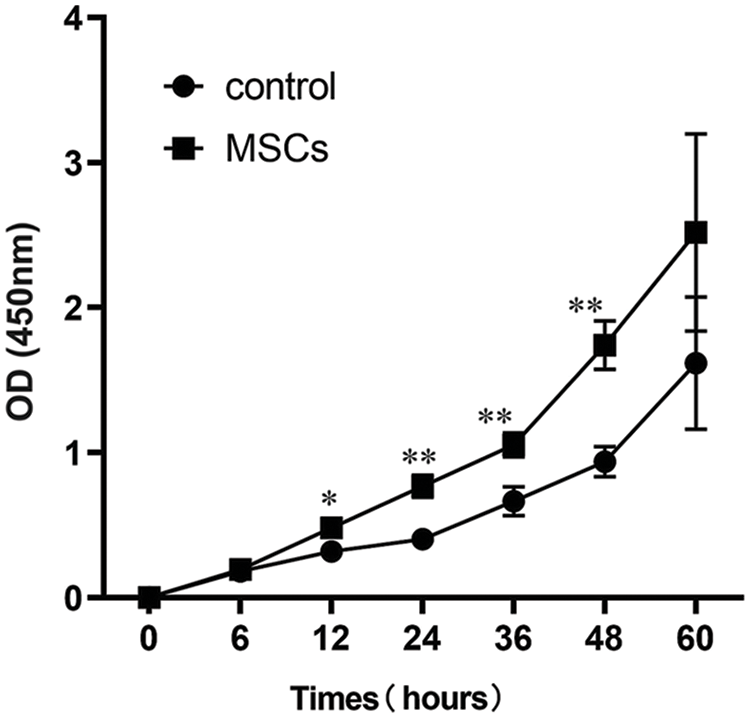
Figure 3: BM-MSC culture media induced the proliferation of RAW264.7 macrophages. OD450 values at 6, 12, 24, 36, 48, and 60 h after administration of BM-MSCs. Two-tailed Student’s t-test, Data shown are mean ± SD; n = 3. *p < 0.05; **p < 0.01.
BM-MSCs enhance autophagy in LPS-treated RAW264.7 cells
Autophagy exerts essential protective activity against stress or injury under disease conditions. RAW264.7 cells were transfected with GFP-RFP-LC3 adenovirus and then treated with LPS, M media, and 3-MA (5 mM) for 24 h to evaluate the effect of BM-MSC therapy on autophagic flux. As shown in Fig. 4, BM-MSC-treated cells had the reddest and yellow puncta compared with those in the three other groups. 3-MA, an autophagic inhibitor, largely decreased the quantity of yellow puncta in RAW264.7 macrophages relative to the BM-MSC group. This indicated that BM-MSCs enhance autophagy in LPS-treated RAW264.7 cells; 3-MA suppressed the autophagy.
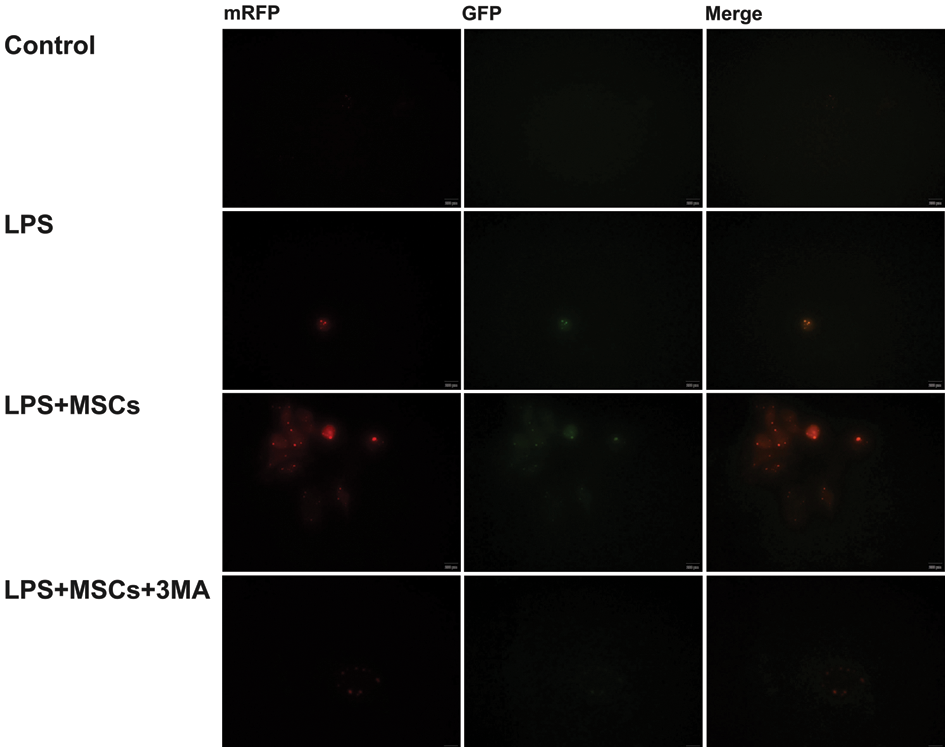
Figure 4: Autophagic flow of macrophages (RAW264.7) in each group. RAW264.7 macrophages were administered with GFP-RFP-LC3 adenovirus and then treated with the aforementioned chemicals. Red and green puncta were identified by fluorescence microscopy. Scale bar = 100 μm.
Impact of BM-MSCs on pro-inflammatory cytokine mRNA expression levels in RAW264.7 macrophages stimulated by LPS
qRT-PCR was performed on all four groups to examine the expression of various pro-inflammatory cytokines. Figs. 5A–5D display the mRNA levels of tumor necrosis factor-α (TNF-α) and inflammatory mediators interleukin IL1β, IL18, and IL6. The LPS-only group’s mRNA expression levels were significantly higher than that of the control group (p < 0.01). BM-MSCs exhibited a significant reduction in the production of pro-inflammatory cytokines in comparison to the LPS-only group (p < 0.01); however, the autophagy inhibitor 3-MA reversed the levels of the four mRNAs in the LPS+MSCs+3MA group in comparison to the LPS+MSCs group (p < 0.05). Therefore, we preliminarily inferred that BM-MSCs exerted their anti-inflammatory activities by enhancing autophagy induction.
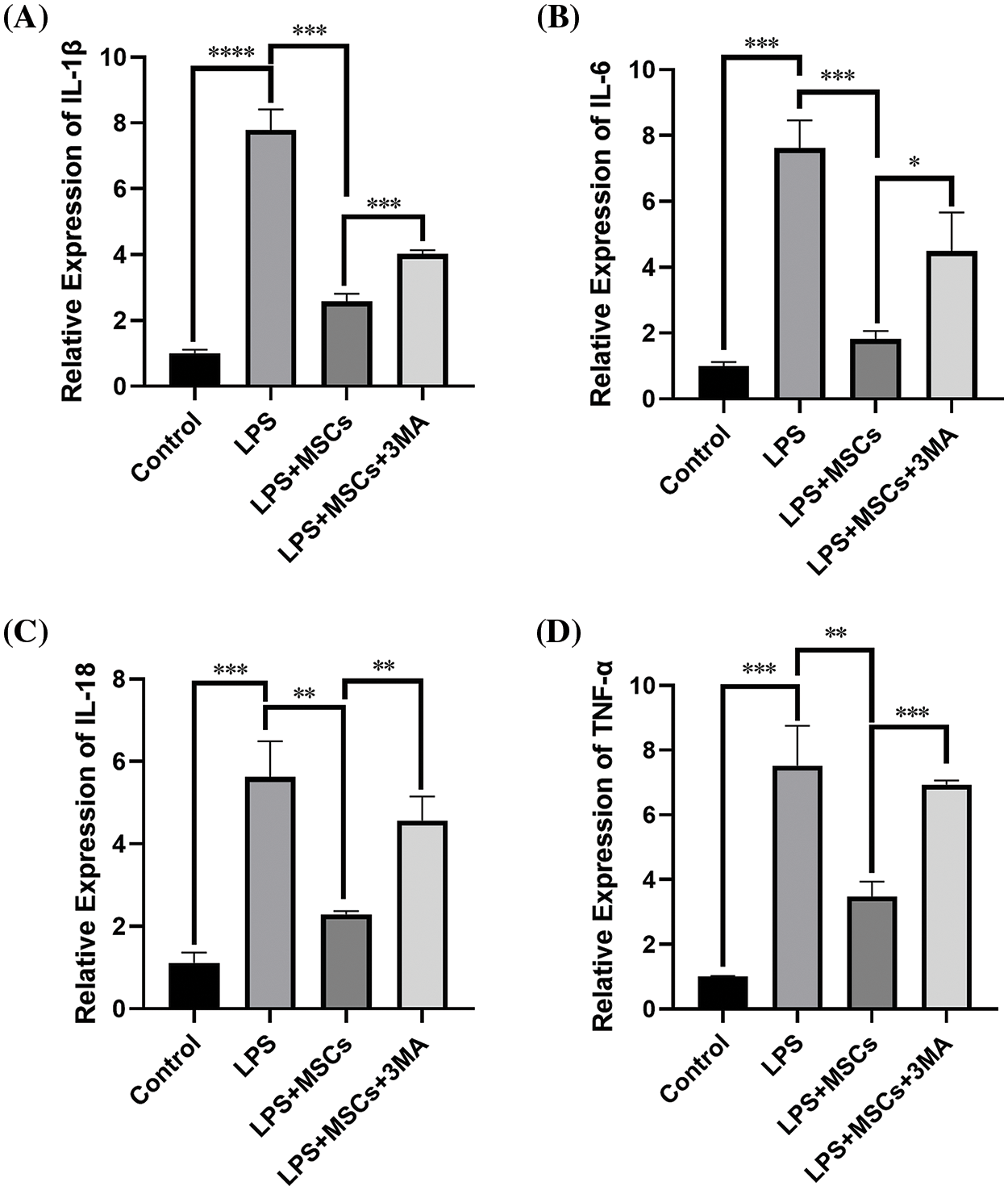
Figure 5: BM-MSCs decreased the relative mRNA expression levels of IL6, IL1β, TNF-α, and IL18 in LPS-stimulated RAW264.7 macrophages. The cells in each group were collected towards the completion of the experiment, and quantitative PCR was carried out to assess the mRNA levels of various cytokines. The mRNA levels of the target genes were normalized to the GAPDH gene as a reference. The relative mRNA levels of IL1β (A), IL6 (B), IL18 (C), and TNF-α (D) are shown. qRT-PCR results of BM-MSCs-suppressed cytokine genes: TNF-α, IL6, IL1β, and IL18. MSCs substantially alleviated the mRNA levels of inflammatory mediators. Data shown are mean ± SD. n = 3. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
BM-MSCs enhance autophagy induction by reducing NLRP3 in Raw 264.7 cells treated with LPS
The expression levels of two autophagic markers, Beclin1 and microtubule-linked protein 1 light chain 3 (LC3), were evaluated by Western blot analysis in the treatment group of RAW264.7 cells. Upon the addition of M medium, the protein levels of LC3-II and Beclin1 showed a significant increase in the group treated with LPS+MSCs, compared to the control and LPS-only groups (p < 0.01, Figs. 6A and B). These results showed that BM-MSCs are autophagy enhancers. However, the administration of 3-MA in the LPS+MSCs+3MA-treated group significantly decreased the levels of LC3II/I and Beclin1 compared with those in LPS+MSC-treated cells (Figs. 6A and 6B). These results indicate that autophagy induction that occurred after LPS treatment was mitigated by 3-MA.
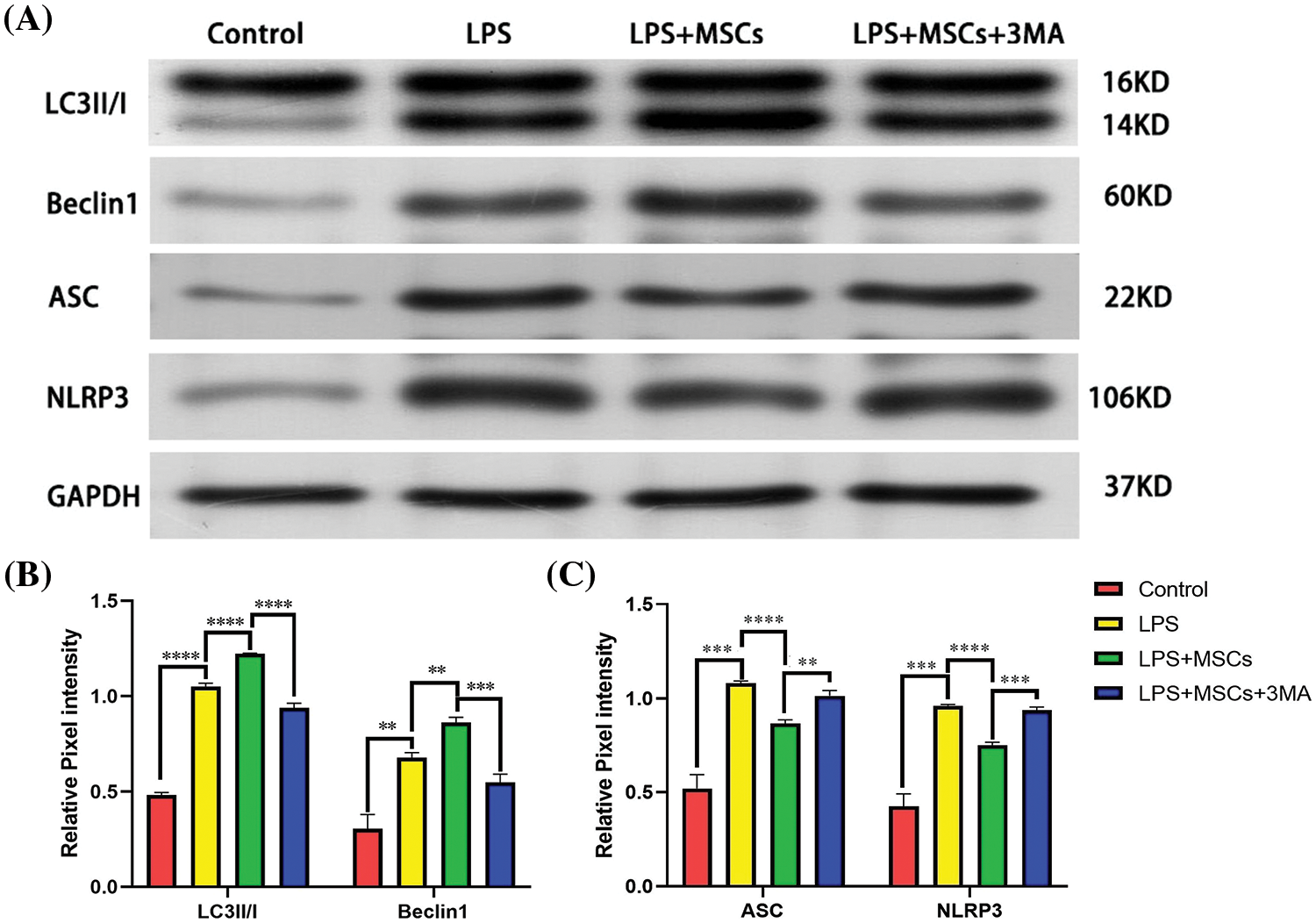
Figure 6: Western blot analysis of autophagy and NLRP3 inflammasome protein expression. (A) Western blot plots of LC3II/I, Beclin 1, NLRP3 and ASC. GAPDH was used for normalization of the cytoplasmic protein. (B) Relative protein expression levels of LC3II/I and Beclin 1; (C) Relative protein expression levels of ASC and NLRP3. Data shown are mean ± SD. n = 3. **p < 0.01; ***p < 0.001; ****p < 0.0001.
We applied Western blot analysis to assess whether BM-MSCs control autophagic function by modulating the NLRP3 inflammasome. We determined the protein expression levels of NLRP3 and ASC following BM-MSC application in vitro. The levels of cellular ASC and NLRP3 proteins increased in LPS-treated cells relative to the control, and BM-MSC administration in LPS-treated cells markedly decreased the ASC and NLRP3 protein levels (p < 0.01, Figs. 6A and 6C). In contrast to the LPS+MSCs group, the LPS+MSCs+3-MA group exhibited elevated expression levels of ASC and NLRP3, which were accompanied by a reduction in autophagic flux (Figs. 6A and 6C). The increased autophagy of BM-MSCs reduced the production of the NLRP3 inflammasome, confirming that BM-MSCs reduced NLRP3 inflammasome protein expression by enhancing autophagy.
Overall, BM-MSCs decreased the NLRP3 inflammasome protein level in an autophagy-dependent manner.
BM-MSCs ameliorate TNBS-induced colitis in mice
A TNBS-induced colitis mouse model was established to evaluate the potential anti-inflammatory effect of BM-MSCs (Fig. 7). During the modeling period, we measured the weights of the mice, observed their stool, and evaluated the DAI score daily at 19:00. The mice were killed on the 7th day, and the lengths of their colons were measured.
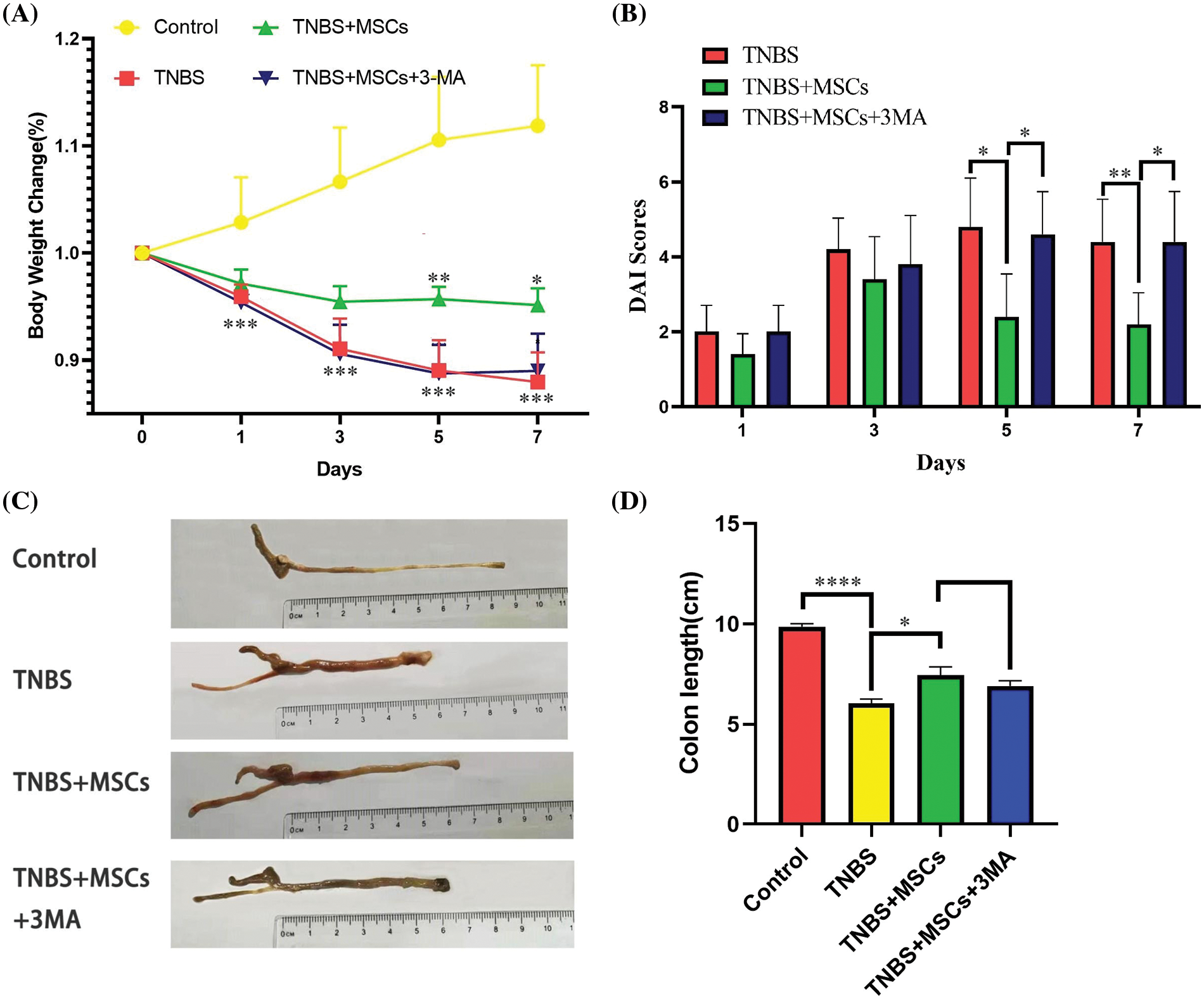
Figure 7: Reduction in colitis severity after BM-MSC treatment. (A) Weights of the mice in each group. (B) Disease activity index (DAI) in each group. (C) Representative images of colons in the normal, TNBS-treated, TNBS-MSCs-treated, and TNBS-MSCs-3MA-treated mice. (D) Colon lengths in each group. n = 5. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Fig. 7 shows the differences in the weights and the DAIs of mice. Mice in the TNBS group had significantly decreased body weight and enhanced DAI scores compared with normal mice. BM-MSC administration had a significant effect on body weight. The DAI was reduced considerably in the colitis group between days 5 and 7 in the TNBS+MSCs group relative to the TNBS group (p < 0.05). The body weight was significantly higher in the TNBS+MSCs group between days 5 and 7 of colitis induction relative to that in the TNBS group (p < 0.05). However, the body weight or DAI did not significantly change between TNBS+MSCs+3MA and TNBS groups (Figs. 7A and 7B).
Mice in the TNBS group showed a significantly shorter colon length than normal mice. BM-MSC administration had a significant effect on the colon length of mice in the TNBS+MSCs group. The colon exhibited a greater length in the group treated with MSCs compared to the TNBS-colitis group. However, no significant variability was identified between TNBS+MSCs and TNBS+MSCs+3MA groups (p < 0.05, Figs. 7C and 7D).
Colitis severity was determined histologically in the colonic tissue. TNBS mice demonstrated intense inflammatory infiltration in colon tissues, as evidenced by the H&E staining. As predicted, BM-MSC administration in TNBS mice resulted in decreased infiltration compared with that in TNBS mice without BM-MSC treatment. Importantly, BM-MSCs reduced the BM-MSC-stimulated inflammatory infiltration in TNBS mice (Fig. 8). However, the inflammation of mice in the TNBS+MSCs+3-MA group did not show decreased infiltration relative to TNBS-group mice. The conducted investigations validated that the administration of BM-MSCs resulted in an improvement in the histological characteristics of colitis. Additionally, it was shown that the protective effect of BM-MSCs was diminished upon administration of the autophagy inhibitor 3-MA.
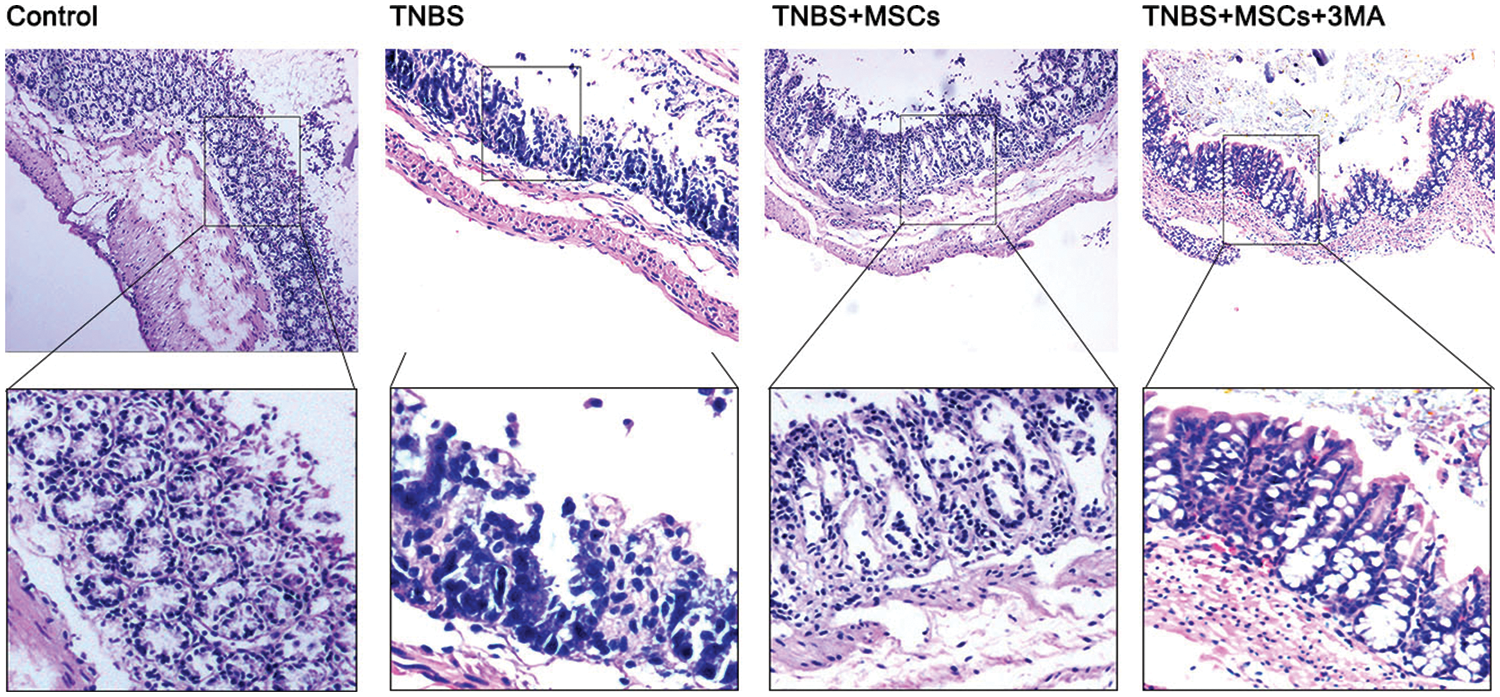
Figure 8: Decrease in the severity of colitis following BM-MSC treatment. Micrographs of colon histology on day 7 in the control, TNBS, TNBS+MSCs, and TNBS+MSCs+3MA groups. Colon histological specimens were stained with H&E (magnification, ×10).
BM-MSCs reduce caspase-1-dependent IL1β level in TNBS-colitis mice
Caspase-1-mediated cleavage can control the secretion of active IL1β and IL18. As such, we analyzed the cytokine concentrations in serum from the hearts of mice in each group after anesthesia. Multiplex analysis revealed that mice treated with TNBS in vivo secreted higher levels of IL1β and IL18 compared with normal mice (p < 0.01). The levels of serum pro-inflammatory cytokines were found to be significantly reduced in mice following intrarectal delivery of BM-MSCs compared to TNBS mice (p < 0.01). Nevertheless, the administration of the autophagy inhibitor 3-MA effectively counteracted the impact of BM-MSCs on cytokine levels. Specifically, the concentrations of IL1β and IL18 were shown to be higher in the TNBS+MSCs+3MA group compared to the TNBS+MSCs group (p < 0.01, Figs. 9A and 9B). Overall, the local administration of BM-MSCs in experimental colitis enhanced the secretion of IL1β and IL18 in an autophagy-dependent manner.
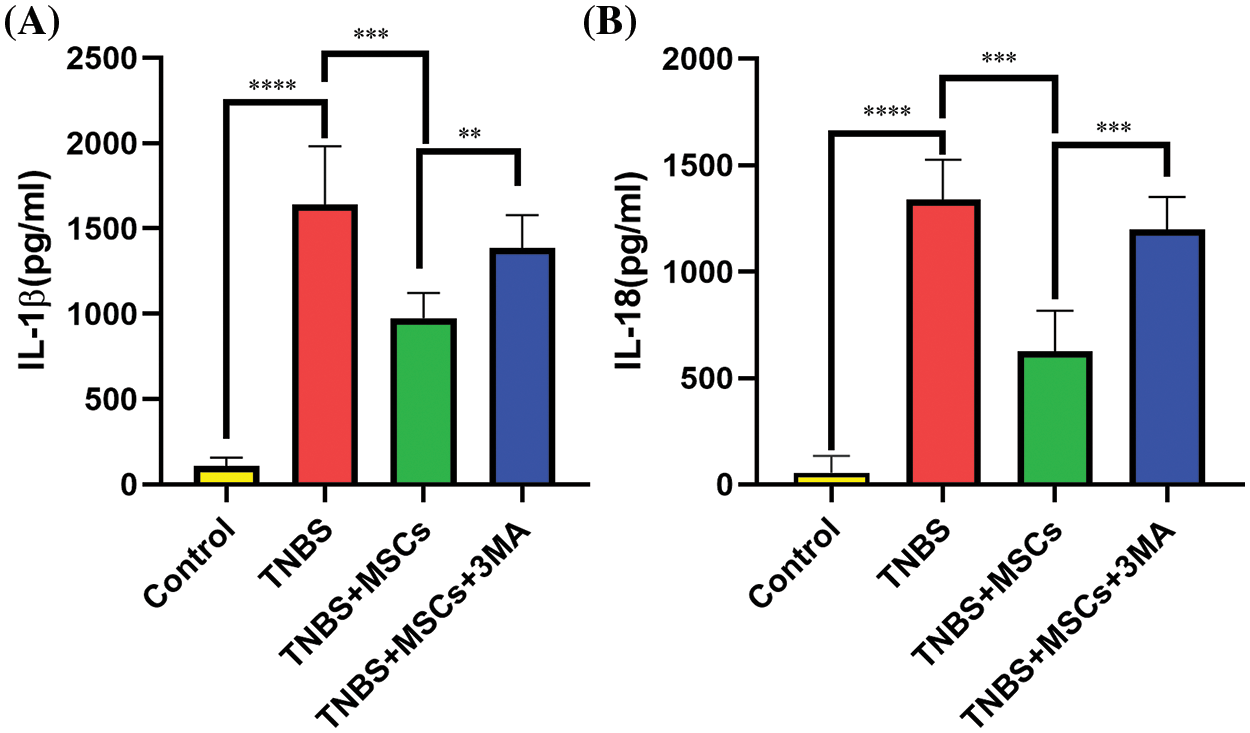
Figure 9: Effect of BM-MSC treatment on the cytokine profile in TNBS-treated mice. Blood serum specimens were assessed for IL1β (A) and IL18 (B) by using one-way ANOVA. Data are expressed as mean ± SD. n = 5. **p < 0.01; ***p < 0.001; ****p < 0.0001.
The current investigation elucidated the probable pathogenic mechanisms of IBD and understood the efficiency of BM-MSCs in IBD management to better control the disease with fewer adverse effects. Herein, we demonstrated the enhanced autophagic effect of BM-MSCs against inflammation in a murine model of TNBS and macrophages stimulated with LPS. It was observed that TNBS upregulated the production of IL1β and IL18 in TNBS-induced mice. In part, BM-MSCs attenuated inflammatory activity triggered by TNBS and LPS by increasing autophagy.
IBD is a severe, clinically chronic, inflammatory, relapsing gastrointestinal tract disease with unidentified etiology and pathology (Lee and Chang, 2021). Existing treatment can only provide temporary relief from the disease, and a cure remains elusive. Studies suggested that the development of IBD is related to infection and inflammation, which often cause damage to the intestinal mucosa and barrier function (Larabi et al., 2020). Current IBD therapies, including anti-inflammatory drugs and biologics, primarily depend on symptomatic therapies, which can involve a lifelong disorder, unsatisfactory long-term efficacy, and diverse side effects (Kucharzik et al., 2020). TNBS models of colitis in mice effectively mimic some of the characteristics of Crohn’s disease in humans and are considered ideal experimental models because they help to assess the mechanisms and therapeutic approaches of intestinal inflammation (Chang et al., 2022; Katsandegwaza et al., 2022). IBD development is related to perturbations in apoptosis (Li and Law, 2022) and NLRP3 inflammasome activation (Armstrong et al., 2023), consistent with the present data.
During infection or damage, inflammasomes are responsible for detecting signs of damage and subsequently initiating the activation of caspase-1. This activation, in turn, triggers the cleavage of pro-IL-18 and pro-IL-1β, leading to their activation. The activation of caspase-1 triggers programmed cell death and pyroptosis and modulates the advancement of many inflammatory diseases, including IBD. Increased levels of pro-inflammatory cytokines, such as IL6, IL1β, TNF α, and IL18, have been linked to severe inflammation associated with IBD. The IL1β levels increased in the colonic mucosa and peritoneal macrophages of the mouse colitis model and may thus be an initial intestinal inflammation trigger (Gong et al., 2018).
As a prominent inflammatory response mediator, NLRP3 inflammasomes perform essential roles in the pathogenic progression of IBD (Mao et al., 2018). The process of activation of the canonical inflammasome pathway involves the cleavage of pro-IL-1β and pro-IL-18 by caspase-1, resulting in the production of active IL1β and IL18. The involvement of NLRP3 inflammasomes in IBD pathophysiology has been shown in many studies. Moreover, the activation of NLRP3 has been observed to elevate the levels of IL1β in both animal models of colitis and individuals diagnosed with IBD (Chen et al., 2019; Gong et al., 2018). IL1β demonstrates potent inflammatory attributes, and its production is tightly controlled by transcriptional regulation and proteolytic cleavage mechanisms that can be both inflammasome-dependent and inflammasome-independent. Hence, the elevation in the IL1β gene levels in active ulcerative colitis and Crohn’s disease is consistent with early studies on human-derived materials (McAlindon et al., 1998).
In this study, we induced colitis in mice through an enema injection of TNBS. Compared with normal mice, TNBS-treated mice showed a significant reduction in body weight with increased DAI scores. Moreover, their colon lengths significantly decreased. Furthermore, TNBS mice demonstrated chronic inflammation in colon tissues, as evident from the H&E staining. Meanwhile, the serum concentrations of IL1β and IL18 cytokines increased.
Findings indicate that pro-inflammatory cytokine production from colonic macrophages and the aberrant activation of the inflammasome can lead to an imbalance in inflammatory activity and damage to the colon tissue, which in turn can induce the onset of IBD. Hence, inflammasomes are promising targets for IBD management. However, therapies with high efficacy and safety are yet to be developed.
Treatment with MSCs is a potential therapeutic method for IBD due to their immunoregulatory properties (Chinnadurai et al., 2015). The administration of BM-MSCs may attenuate intestinal inflammation and even reverse colitis in different experimental models (Robinson et al., 2017). The anti-inflammatory effect of MSC treatment was elucidated by inhibiting NLRP3 and ASC protein stimulation and reducing the levels of pro-inflammatory cytokines such as IL1β and IL18 (Zhou et al., 2022). We identified the effect of BM-MSC treatment using an in vivo mouse model and evaluated physiological effects after treatment. Treatment with BM-MSCs derived from Balb/c mice can stimulate the proliferation of RAW264.7 macrophages in vitro and accelerate the recovery of TNBS-induced colitis in Balb/c mice in vivo. After the injection of TNBS, the administration of BM-MSCs via enema demonstrated significant efficacy in expediting both functional and morphological recovery, as well as mitigating the severity of TNBS-induced colitis. This was evidenced by various disease parameters, such as the acceleration of functional and morphological recovery, reduction in body weight loss, and decreased infiltration of inflammatory cells. The clinical DAI score was reduced, and the colon length increased in MSCs+LPS-treated colitis mice compared with those in TNBS mice. Moreover, the titers of the inflammatory mediators IL1β and IL18 significantly decreased.
Autophagy is crucial for inflammation activity. Autophagy regulation in the colon has become a focus of research to determine novel strategies against colitis. Bergmann et al. (2022) revealed that autophagy modulates the therapeutic efficiency of BMCs, and the augmentation of this ability could be an implication for IBD treatment. The activation of MSCs is accompanied by elevated autophagic flux and inhibition, which partially inhibit inflammation. In the present study investigation, GFP-RFP-LC3 adenoviruses and autophagy modulators were used, and BM-MSCs enhanced the in vitro autophagic flow in LPS-induced RAW264.7 macrophages. LPS treatment resulted in a cellular inflammation model, as reported. LC3, the mammalian ortholog of yeast autophagy-related gene (Atg) 8, is commonly utilized to monitor autophagy. During autophagy stimulation, the soluble LC3 (LC3-I) form is transformed into LC3-II, which is linked to autophagosomal membranes and is critical for autophagosome formation. LC3-II indicates the number of autophagosomes and is a good index of autophagosome generation. Beclin1 expression is the principal marker for the modulation of autophagic activity, which promotes cell survival. In the present research, the protein levels of LC3-II and Beclin1 were markedly elevated in LPS+MSCs-treated RAW264.7 relative to LPS-treated cells. Hence, BM-MSCs enhanced autophagic flux in LPS-treated RAW264.7 macrophages. The study conducted by Li et al. (2018) showed that restricting the activation of inflammasome in macrophages might potentially serve as a vital mechanism by which MSCs can effectively counteract sepsis. According to the findings of Gholaminejhad et al. (2022), it has been proposed that the suppression of inflammatory pathways through the induction of autophagy by MSCs may have potential therapeutic benefits in terms of promoting functional restoration and mitigating the degradation of spinal cord tissue subsequent to spinal cord injury. The present investigation observed that the protein expression levels of NLRP3 and ASC exhibited a significant drop. Furthermore, the expression levels of four specific mRNAs, notably IL6, TNF-α, IL1β, and IL18, showed a substantial reduction in the LPS+MSCs treated group compared to the group treated with LPS alone. However, the decrease in NLRP3 and ASC protein expression levels in BM-MSCs-treated RAW264.7 was partially suppressed by 3-MA treatment in the LPS+MSCs+3MA group. Therefore, we hypothesized that BM-MSCs prevented NLRP3 inflammasome by enhancing autophagy. The induction of autophagy by BM-MSCs has been seen to exert a restraining effect on the mRNA expression of inflammatory cytokines, namely IL6, TNF-α, IL1β, and IL18, via modulation of the NLRP3 inflammasome pathway. To validate our results, mice were given with 3-MA. The findings indicated that the levels of pro-inflammatory factors IL1β and IL18 were increased in comparison to the LPS+MSCs group. Hence, autophagy induction that occurred after LPS treatment was mitigated by BM-MSC therapy.
Collectively, our findings provide more evidence supporting existing research indicating that autophagy has a role in influencing the therapeutic efficacy of MSCs. Additionally, our study demonstrates that enhancing the autophagic ability of BM-MSCs through the reduction of NLRP3 might potentially have therapeutic implications in treating IBD.
It has been shown that BM-MSCs have anti-inflammatory properties against various diseases; however, the mechanisms of BM-MSCs remain unclear. In summary, our study demonstrated that the increase in NLRP3 and IL1β levels may be crucial to the pathogenesis of IBD, and BM-MSCs can ameliorate TNBS-induced colitis by eliciting an autophagy effect and inhibiting NLRP3 and IL1β. These findings provide an essential perspective on the mechanisms of BM-MSC treatment in IBD.
Acknowledgement: None.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: JJF and XYF; data collection: XG and FDL; analysis and interpretation of results: JW, YG and JH; draft manuscript preparation: SHZ and FYW. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All experimental data used to support the findings of this study are available from the corresponding author upon request.
Ethics Approval: The animal experiment in this study was approved by the Ethics Committee of Jinling Hospital, Nanjing Medical University (No. 2021DZGKJDWLS-00143).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
Adolph TE, Meyer M, Schwärzler J, Mayr L, Grabherr F, Tilg H (2022). The metabolic nature of inflammatory bowel diseases. Nature Reviews. Gastroenterology & Hepatology 19: 753–767. [Google Scholar]
Armstrong HK, Bording-Jorgensen M, Santer DM, Zhang Z, Valcheva R et al. (2023). Unfermented β-fructan fibers fuel inflammation in select inflammatory bowel disease patients. Gastroenterology 164: 228–240. [Google Scholar] [PubMed]
Bergmann CA, Beltran S, Vega-Letter AM, Murgas P, Hernandez MF et al. (2022). The autophagy protein pacer positively regulates the therapeutic potential of mesenchymal stem cells in a mouse model of DSS-induced colitis. Cells 11: 1503. [Google Scholar] [PubMed]
Biasizzo M, Kopitar-Jerala N (2020). Interplay between NLRP3 inflammasome and autophagy. Frontiers in Immunology 11: 591803. [Google Scholar] [PubMed]
Chang CC, Liu CY, Su IC, Lee YJ, Yeh HJ, Chen WC, Yu CJ, Kao WY, Liu YC, Huang CJ (2022). Functional plasmon-activated water increases Akkermansia muciniphila abundance in gut microbiota to ameliorate inflammatory bowel disease. International Journal of Molecular Sciences 23: 11422. [Google Scholar] [PubMed]
Chen HW, Chen HY, Wang LT, Wang FH, Fang LW et al. (2013). Mesenchymal stem cells tune the development of monocyte-derived dendritic cells toward a myeloid-derived suppressive phenotype through growth-regulated oncogene chemokines. Journal of Immunology 190: 5065–5077. [Google Scholar]
Chen X, Liu G, Yuan Y, Wu G, Wang S, Yuan L (2019). NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling. Cell Death & Disease 10: 906. [Google Scholar]
Chen QL, Yin HR, He QY, Wang Y (2021). Targeting the NLRP3 inflammasome as new therapeutic avenue for inflammatory bowel disease. Biomedicine & Pharmacotherapy 138: 111442. [Google Scholar]
Chinnadurai R, Copland IB, Ng S, Garcia M, Prasad M, Arafat D, Gibson G, Kugathasan S, Galipeau J (2015). Mesenchymal stromal cells derived from Crohn’s patients deploy indoleamine 2,3-dioxygenase-mediated immune suppression, independent of autophagy. Molecular Therapy 23: 1248–1261. [Google Scholar] [PubMed]
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop DJ, Horwitz E (2006). Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 8: 315–317. [Google Scholar] [PubMed]
Flynn S, Eisenstein S (2019). Inflammatory bowel disease presentation and diagnosis. The Surgical Clinics of North America 99: 1051–1062. [Google Scholar] [PubMed]
GBD 2017 Inflammatory Bowel Disease Collaborators (2020). The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. The Lancet Gastroenterology & Hepatology 5: 17–30. [Google Scholar]
Gholaminejhad M, Jameie SB, Abdi M, Abolhassani F, Mohammed I, Hassanzadeh G (2022). All-trans retinoic acid-preconditioned mesenchymal stem cells improve motor function and alleviate tissue damage after spinal cord injury by inhibition of HMGB1/NF-κB/NLRP3 pathway through autophagy activation. Journal of Molecular Neuroscience 72: 947–962. [Google Scholar] [PubMed]
Gong Z, Zhao S, Zhou J, Yan J, Wang L, Du X, Li H, Chen Y, Cai W, Wu J (2018). Curcumin alleviates DSS-induced colitis via inhibiting NLRP3 inflammsome activation and IL-1β production. Molecular Immunology 104: 11–19. [Google Scholar] [PubMed]
Huang JQ, Huang HN, Wang XJ, Guo Y, Zhong YX, Hu YQ (2022a). Bie Jia Jian pill enhances the amelioration of bone mesenchymal stem cells on hepatocellular carcinoma progression. Journal of Natural Medicines 76: 49–58. [Google Scholar]
Huang C, Wang J, Liu H, Huang R, Yan X, Song M, Tan G, Zhi F (2022b). Ketone body β-hydroxybutyrate ameliorates colitis by promoting M2 macrophage polarization through the STAT6-dependent signaling pathway. BMC Medicine 20: 148. [Google Scholar] [PubMed]
Huang Y, Xu W, Zhou R (2021). NLRP3 inflammasome activation and cell death. Cellular & Molecular Immunology 18: 2114–2127. [Google Scholar]
Katsandegwaza B, Horsnell W, Smith K (2022). Inflammatory bowel disease: A review of pre-clinical murine models of human disease. International Journal of Molecular Sciences 23: 9344. [Google Scholar] [PubMed]
Kucharzik T, Koletzko S, Kannengiesser K, Dignass A (2020). Ulcerative colitis-diagnostic and therapeutic algorithms. Deutsches Arzteblatt International 117: 564–574. [Google Scholar] [PubMed]
Larabi A, Barnich N, Nguyen HTT (2020). New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 16: 38–51. [Google Scholar] [PubMed]
Lee M, Chang EB (2021). Inflammatory bowel diseases (IBD) and the microbiome-searching the crime scene for clues. Gastroenterology 160: 524–537. [Google Scholar] [PubMed]
Li Y, Law HKW (2022). Deciphering the role of autophagy in the immunopathogenesis of inflammatory bowel disease. Frontiers in Pharmacology 13: 1070184. [Google Scholar] [PubMed]
Li S, Wu H, Han D, Ma S, Fan W et al. (2018). A novel mechanism of mesenchymal stromal cell-mediated protection against sepsis: Restricting inflammasome activation in macrophages by increasing mitophagy and decreasing mitochondrial ROS. Oxidative Medicine and Cellular Longevity 2018: 3537609. [Google Scholar] [PubMed]
Lin D, Chen H, Xiong J, Zhang J, Hu Z et al. (2022). Mesenchymal stem cells exosomal let-7a-5p improve autophagic flux and alleviate liver injury in acute-on-chronic liver failure by promoting nuclear expression of TFEB. Cell Death & Disease 13: 865. [Google Scholar]
Luo D, Ye W, Chen L, Yuan X, Zhang Y, Chen C, Jin X, Zhou Y (2023). PPARα inhibits astrocyte inflammation activation by restoring autophagic flux after transient brain ischemia. Biomedicines 11: 973. [Google Scholar] [PubMed]
Mao L, Kitani A, Strober W, Fuss IJ (2018). The role of NLRP3 and IL-1β in the pathogenesis of inflammatory bowel disease. Frontiers in Immunology 9: 2566. [Google Scholar] [PubMed]
McAlindon ME, Hawkey CJ, Mahida YR (1998). Expression of interleukin 1β and interleukin 1β converting enzyme by intestinal macrophages in health and inflammatory bowel disease. Gut 42: 214–219. [Google Scholar] [PubMed]
Morris GP, Beck PL, Herridge MS, Depew WT, Szewczuk MR, Wallace JL (1989). Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology 96: 795–803. [Google Scholar] [PubMed]
Nuñez P, García Mateo S, Quera R, Gomollón F (2021). Inflammatory bowel disease and the risk of cardiovascular diseases. Gastroenterology & Hepatology 44: 236–242. [Google Scholar]
Perera AP, Fernando R, Shinde T, Gundamaraju R, Southam B, Sohal SS, Robertson AAB, Schroder K, Kunde D, Eri R (2018). MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Scientific Reports 8: 8618. [Google Scholar] [PubMed]
Pontell L, Castelucci P, Bagyánszki M, Jovic T, Thacker M, Nurgali K, Bron R, Furness JB (2009). Structural changes in the epithelium of the small intestine and immune cell infiltration of enteric ganglia following acute mucosal damage and local inflammation. Virchows Archiv 455: 55–65. [Google Scholar] [PubMed]
Quaglio AEV, Grillo TG, de Oliveira ECS, di Stasi LC, Sassaki LY (2022). Gut microbiota, inflammatory bowel disease and colorectal cancer. World Journal of Gastroenterology 28: 4053–4060. [Google Scholar] [PubMed]
Ramos GP, Papadakis KA (2019). Mechanisms of disease: Inflammatory bowel diseases. Mayo Clinic Proceedings 94: 155–165. https://doi.org/10.1016/j.mayocp.2018.09.013 [Google Scholar] [PubMed] [CrossRef]
Robinson AM, Rahman AA, Miller S, Stavely R, Sakkal S, Nurgali K (2017). The neuroprotective effects of human bone marrow mesenchymal stem cells are dose-dependent in TNBS colitis. Stem Cell Research & Therapy 8: 87. [Google Scholar]
Saez A, Herrero-Fernandez B, Gomez-Bris R, Sánchez-Martinez H, Gonzalez-Granado JM (2023). Pathophysiology of inflammatory bowel disease: Innate immune system. International Journal of Molecular Sciences 24: 1526. [Google Scholar] [PubMed]
Shon WJ, Lee YK, Shin JH, Choi EY, Shin DM (2015). Severity of DSS-induced colitis is reduced in Ido1-deficient mice with down-regulation of TLR-MyD88-NF-kB transcriptional networks. Scientific Reports 5: 17305. [Google Scholar] [PubMed]
Solà-Tapias N, Vergnolle N, Denadai-Souza A, Barreau F (2020). The interplay between genetic risk factors and proteolytic dysregulation in the pathophysiology of inflammatory bowel disease. Journal of Crohn’s Colitis 14: 1149–1161. [Google Scholar] [PubMed]
Stavely R, Robinson AM, Miller S, Boyd R, Sakkal S, Nurgali K (2015). Human adult stem cells derived from adipose tissue and bone marrow attenuate enteric neuropathy in the guinea-pig model of acute colitis. Stem Cell Research & Therapy 6: 244. [Google Scholar]
Swanson KV, Deng M, Ting JP (2019). The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nature Reviews Immunology 19: 477–489. [Google Scholar] [PubMed]
Younis N, Zarif R, Mahfouz R (2020). Inflammatory bowel disease: Between genetics and microbiota. Molecular Biology Reports 47: 3053–3063. [Google Scholar] [PubMed]
Zhang J, Fu S, Sun S, Li Z, Guo B (2014). Inflammasome activation has an important role in the development of spontaneous colitis. Mucosal Immunology 7: 1139–1150. [Google Scholar] [PubMed]
Zhen Y, Zhang H (2019). NLRP3 inflammasome and inflammatory bowel disease. Frontiers in Immunology 10: 276. [Google Scholar] [PubMed]
Zhou L, Wang X, Wang X, An J, Zheng X, Han D, Chen Z (2022). Neuroprotective effects of human umbilical cord mesenchymal stromal cells in PD mice via centrally and peripherally suppressing NLRP3 inflammasome-mediated inflammatory responses. Biomedicine & Pharmacotherapy 153: 113535. [Google Scholar]
Supplementary Materials
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools