
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Semaphorin 7A promotes human vascular smooth muscle cell proliferation and migration through the β-catenin signaling pathway
Department of Cardiology, The Second Hospital of Shanxi Medical University, Taiyuan, 030001, China
* Corresponding Author: HUIYU YANG. Email:
(This article belongs to the Special Issue: Biochemical and Epigenetics Changes in Health and Disease)
BIOCELL 2023, 47(4), 849-858. https://doi.org/10.32604/biocell.2023.026545
Received 12 September 2022; Accepted 28 November 2022; Issue published 08 March 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Vascular smooth muscle cells (VSMCs) undergo a conversion from a contractile phenotype to a proliferative synthetic phenotype, contributing to the pathogenesis of cardiovascular diseases. Semaphorin 7A (SEMA7A) is a glycosylphosphatidylinositol-anchored membrane protein that plays an important role in vascular homeostasis by regulating endothelial cell behaviors. However, the expression and role of SEMA7A in VSMCs remain unclear.Methods: In this study, we screened for VSMC-regulating genes in publicly available datasets and analyzed the expression of SEMA7A in human coronary artery smooth muscle cells (hCASMCs) treated with platelet-derived growth factor-BB (PDGF-BB). The effects of SEMA7A overexpression and knockdown on hCASMC proliferation and migration were examined. The signaling pathways involved in the action of SEMA7A in hCASMCs were determined.Results: Bioinformatic analysis showed that SEMA7A was significantly dysregulated in VSMCs treated with oxidized low-density lipoprotein or overexpressing progerin, a pro-atherogenic gene. The PDGF-BB stimulation led to a concentration- and time-dependent induction of SEMA7A. Depletion of SEMA7A attenuated PDGF-BB-induced hCASMC proliferation and migration. Conversely, overexpression of SEMA7A enhanced hCASMC proliferation and migration. Mechanistically, SEMA7A stimulated the activation of the β-catenin pathway and upregulated c-Myc, CCND1, and MMP7. Knockdown of β-catenin impaired SEMA7A-induced hCASMC proliferation and migration.Conclusions: SEMA7A triggers phenotype switching in VSMCs through the β-catenin signaling pathway and may serve as a potential therapeutic target for cardiovascular diseases.Keywords
Vascular smooth muscle cells (VSMCs) are a major cell type of the arterial wall and play a key role in the development of various cardiovascular diseases including atherosclerosis (Pan et al., 2022). While VSMCs in healthy arteries remain quiescent and provide contractile function and structural support, they exhibit altered phenotypes under pathological conditions (Bennett et al., 2016). For example, in atherosclerotic plaques, contractile VSMCs switch to a pro-atherogenic phenotype with elevated proliferation, migration, and extracellular matrix production (Lagrange et al., 2022; Zheng et al., 2022). The transition of VSMCs from a contractile phenotype to a proliferative synthetic phenotype leads to pathological vascular remodeling and lesion formation (Sakic et al., 2022). Platelet-derived growth factor-BB (PDGF-BB) can promote VSMC proliferation and migration and is widely used as a stimulator of VSMC phenotype transition (Li et al., 2021; Ran et al., 2021). Understanding the mechanisms involved in the regulation of VSMC phenotype plasticity is of importance in developing effective therapies for the treatment of vascular lesions.
Semaphorin 7A (SEMA7A; also known as CD108) is a glycosylphosphatidylinositol (GPI)-anchored membrane protein that participates in a variety of biological processes including neural development, immune regulation, cardiovascular diseases, and cancer (You et al., 2019; Crump et al., 2021; Loy et al., 2021). Plexin C1 and β1 integrin have been identified as the receptors for Sema7A. Inoue et al. (2018) reported that Sema7A mediates olfactory synapse formation through interaction with plexin C1. Further, Sema7A has been found to modulate the metabolic reprogramming of macrophages by activating the mTOR- and AKT2-signaling pathways, leading to the resolution of acute inflammation and maintenance of tissue homeostasis (Körner et al., 2021). Suzuki et al. (2007) demonstrated that the interaction between Sema7A and α1β1 integrin triggers T-cell-mediated inflammatory responses. Recent studies have indicated the role of Sema7A in regulating vascular homeostasis and atherosclerotic plaque formation (Hu et al., 2018b). Overexpression of Sema7A in endothelial cells promotes the expression of cell adhesion molecules and monocyte adhesion through the β1 integrin receptor (Hu et al., 2018a). However, the expression and role of SEMA7A in VSMCs are poorly understood.
In this study, we screened for differentially expressed genes (DEGs) in publicly available datasets and identified SEMA7A as a DEG in human VSMCs exposed to pro-atherogenic stimuli. Moreover, we uncovered the role and molecular mechanisms of SEMA7A in mediating VSMC phenotype switching.
To search for key genes involved in the regulation of VSMCs, we analyzed 2 publicly available Gene Expression Omnibus (GEO) datasets, GSE68021 and GSE198983. Microarray data deposited in the GSE68021 dataset show gene expression changes in human VSMCs exposed to native and oxidized low-density lipoprotein (oxLDL). The oxLDL plays an important role in vascular remodeling by promoting phenotype switching in VSMCs (Wang et al., 2021a). Data on RNA sequencing in the GSE198983 dataset reveal differential expression of genes in VSMCs overexpressing progerin, a pro-atherogenic gene (Hamczyk et al., 2019). We used GEO2R software (https://www.ncbi.nlm.nih.gov/geo/geo2r/) to identify DEGs from microarray data, and NetworkAnalyst software (https://www.networkanalyst.ca/) to detect DEGs from RNA sequencing data. Statistically significant DEGs were defined at an adjusted p-value of <0.05 and log2(fold change) >1.
Human coronary artery smooth muscle cells (hCASMCs) were obtained from Lonza (Basel, Switzerland) and maintained in smooth muscle growth medium-2 (Lonza) containing 10% fetal bovine serum (FBS; Lonza). Cells in passages 2–6 were used in this study. For PDGF-BB treatment, hCASMCs were serum starved for 24 h and then exposed to recombinant human PDGF-BB (Peprotech, Rocky Hill, NJ, USA). Unless stated otherwise, hCASMCs were treated with 40 ng/mL of PDGF-BB for 24 h (Wang et al., 2021b). For inhibitor experiments, hCASMCs with overexpression of SEMA7A were exposed to 20 μM of LY29402 (a PI3K/Akt inhibitor; Sigma-Aldrich, St. Louis, MO, USA) or 100 nM of PD0325901 (an MEK inhibitor; Sigma-Aldrich) for 24 h.
RNA extraction and quantitative real-time PCR (qRT-PCR) analysis
Total RNA was extracted from hCASMCs using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. RNA samples were reverse transcribed to cDNA using oligo (dT) primers. The qRT-PCR was performed on an ABI Prism 7500 Sequence Detection System (Applied Biosystems, Foster City, CA) using the SYBR Premix Ex Taq (TaKaRa, Tokyo, Japan). The PCR primers used are summarized as follows: SEMA7A forward, 5′-CAGACTCCTCAAACTTGACTA-3′ and SEMA7A reverse, 5′-GTCTTCAGGAACACCAGCCAC-3′; c-Myc forward, 5′-GTCAAGAGGCGAACACACAAC-3′ and c-Myc reverse, 5′-TTGGACGGACAGGATGTATGC-3′; CCND1 forward, 5′-CGTGGCCTCTAAGATGAAGG-3′ and CCND1 reverse, 5′-CTGGCATTTTGGAGAGGAAG-3′; MMP7 forward, 5′-TGTATGGGGAACTGCTGACA-3′ and MMP7 reverse, 5′-GCGTTCATCCTCATCGAAGT-3′; CTNNB1 forward, 5′-AAAGCGGCTGTTAGTCACTGG-3′ and CTNNB1 reverse, 5′-CGAGTCATTGCATACTGTCCAT-3′; GAPDH forward, 5′-GGAGCGAGATCCCTCCAAAAT-3′ and GAPDH reverse, 5′-GGCTGTTGTCATACTTCTCATGG-3′. For an internal control, GAPDH was used. The relative gene expression was quantified using the comparative cycle threshold (CT) method (2−ΔΔCT method).
Small interfering RNAs (siRNAs), plasmids, and cell transfection
Two specific siRNAs targeting different sites of the SEMA7A transcripts were used in this study: siSEMA7A#1 sense, 5′-GUUCACUGUCAGUCUCCAAAG-3′; siSEMA7A#2 sense, 5′-CCACACUCACUCUUGGCUUAA-3′. A siRNA targeting β-catenin mRNA (siCTNNB1) was purchased from Dharmacon (Lafayette, CO, USA), with the sense sequence given to be 5′-AAGUCCUGUAUGAGUGGGAAC-3′ (Deng et al., 2002). For the SEMA7A overexpression study, full-length SEMA7A-expressing sequence (NM_003612) was amplified by PCR and cloned to pcDNA3.1(+) vector. The inserts were validated by sequencing.
hCASMCs at 85% confluence were transfected with the pcDNA3.1/SEMA7A plasmid or indicated siRNAs (a final concentration of 40–50 nM for each) using Lipofectamine 3000 (Invitrogen). Twenty-four hours later, transfected cells were examined for gene expression, proliferation, and migration.
Cell proliferation was evaluated by 5-ethynyl-2′-deoxyuridine (EdU) labeling assays. In brief, hCASMCs were seeded on coverslips at 5 × 104 cells per well in 12-well plates. After indicated treatments, the cells were labeled with EdU (10 μM; Beyotime Biotechnology, Shanghai, China) for 2 h. Following fixation, the cells were incubated with the Click Additive Solution for 30 min in the dark. Nuclei were counterstained with Hoechst 33342. Cell images were obtained using a fluorescence microscope. The proliferation index was expressed as a percentage of the EdU-positive cells.
hCASMCs were plated in 24-well plates (1 × 104 cells per well) and cultured for 72 h in the presence or absence of PDGF-BB (40 ng/mL). Viable cells were examined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. In brief, cells were incubated with MTT solution (0.5 mg/mL) at 37°C for 4 h. The purple precipitates were dissolved in dimethyl sulfoxide, and absorbance was recorded at 540 nm. The blank control was measured at 720 nm. Results are expressed as the corrected absorbance, which was obtained by subtracting the background absorbance for assay absorbance.
Cell migration capacity was determined using in vitro wound healing assays, as described previously (Jin et al., 2020). The hCASMCs transfected with indicated constructs were plated in 6-well plates (1 × 106 cells/well) and grew to confluence. A sterile 100-μL pipette tip was used to generate a scratch on the cell monolayer. After incubation with or without PDGF-BB (40 ng/mL) for 24 h, cell migration was evaluated under a microscope. Percentage of wound closure was calculated using the formula: ([(wound area at 0 h—wound area at 24 h)/(wound area at 0 h)] × 100).
The hCASMCs were lysed in radio-immunoprecipitation assay lysis buffer supplemented with protease and phosphatase inhibitor cocktails (Thermo Fisher Scientific, Waltham, MA, USA). Protein samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Sigma-Aldrich). Membranes were blocked for 1 h in 5% fat-free milk. After blocking, membranes were probed with primary antibodies at 4°C overnight. The primary antibodies used are listed as follows: anti-SEMA7A (#PA5-28971, Thermo Fisher Scientific), anti-active β-catenin (#8814, Cell Signaling Technology, Danvers, MA, USA), anti-β-catenin (#9562, Cell Signaling Technology), anti-phospho-ERK (#9101, Cell Signaling Technology), anti-ERK (#9102, Cell Signaling Technology), anti-phospho-Akt (#9275, Cell Signaling Technology), anti-Akt (#9272, Cell Signaling Technology), and anti-GAPDH (#2118, Cell Signaling Technology). The membranes were then incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse IgG (Cell Signaling Technology). Protein bands were visualized using chemiluminescence (Amersham Biosciences/GE Healthcare, Piscataway, NJ, USA) and quantified using Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA).
TOPFlash/FOPFlash reporter assay
TOPFlash/FOPFlash reporter assay was performed to evaluate β-catenin-dependent transcriptional activity (Chen et al., 2019). In brief, hCASMCs were co-transfected with pcDNA3.1/SEMA7A together with TOPFlash or FOPFlash luciferase reporter constructs (Beyotime, Shanghai, China). To control for transfection efficiency, the cells were co-transfected with the pRL-TK Renilla luciferase plasmid (Beyotime). Twenty-four hours after transfection, cells were lysed with lysis buffer and examined for luciferase activities using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA). The TOPFlash or FOPFlash luciferase reporter was normalized to Renilla luciferase activity. The normalized TOPFlash to FOPFlash ratio was then calculated.
Data are presented as mean ± standard deviation (SD). All experiments were repeated 3 times (3 biological replicates for each). Statistical significance was analyzed using Student’s t-test or one-way analysis of variance followed by the Tukey post-hoc test. Values with p < 0.05 were considered to be statistically significant.
Identification of VSMC-regulating genes
We analyzed the publicly available datasets GSE68021 and GSE198983 that were obtained from human VSMCs exposed to oxLDL or overexpressing progerin. There were 507 DEGs (upregulated or downregulated) in GSE68021 and 373 in GSE198983. A Venn diagram revealed 20 common genes between the 2 sets of DEGs (Fig. 1A). Among the 20 overlapping genes, we focused on SEMA7A because of its involvement in the regulation of vascular homeostasis and atherosclerotic plaque formation (Hu et al., 2018a).
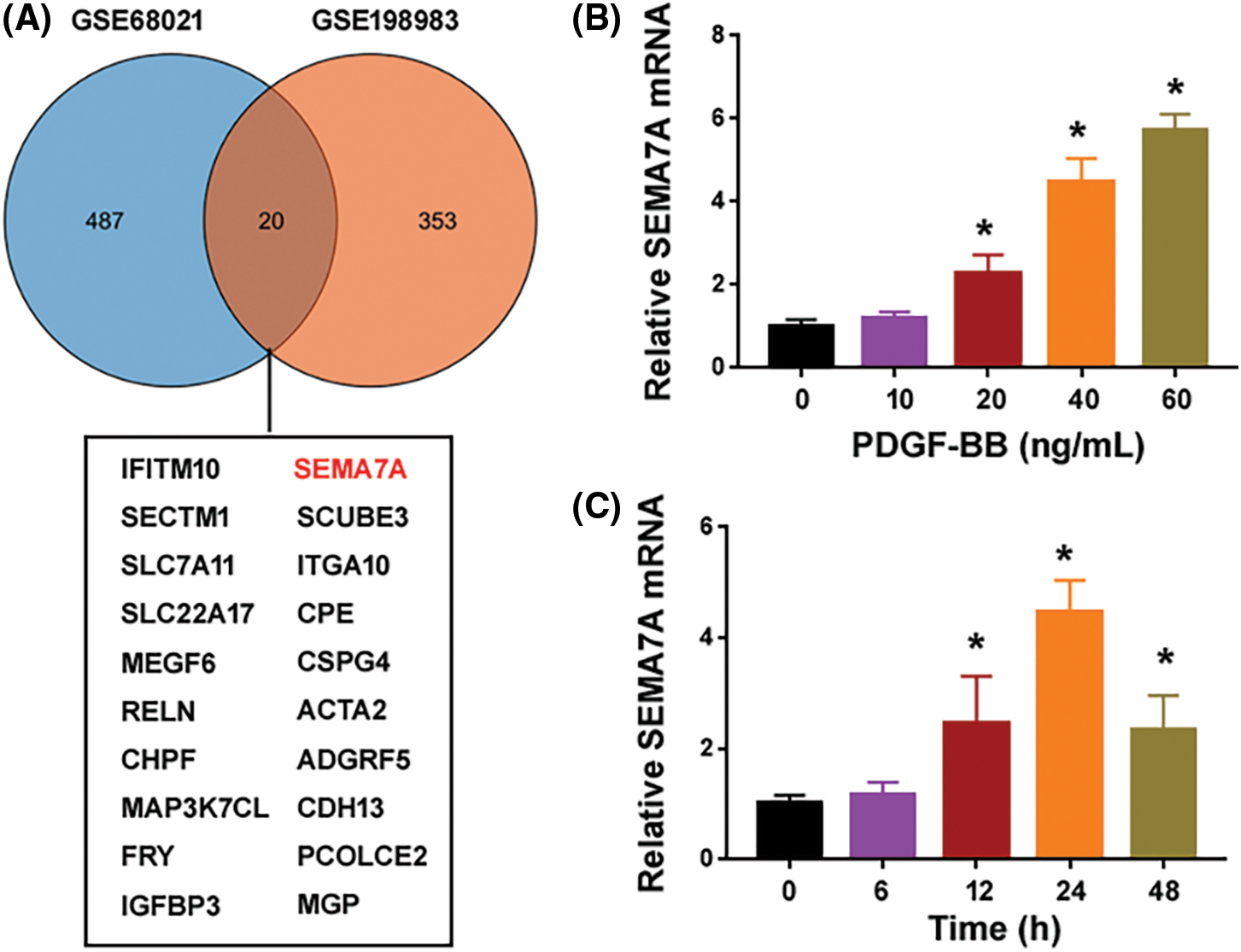
Figure 1: PDGF-BB induces the expression of SEMA7A in hCASMCs. (A) A Venn diagram showing differentially expressed genes identified from GSE68021 and GSE198983 datasets. A list of 20 common genes are summarized below. (B) Quantification of SEMA7A transcripts in hCASMCs exposed to different concentrations of PDGF-BB for 24 h. (C) Quantification of SEMA7A transcripts in hCASMCs exposed to 40 ng/mL of PDGF-BB for different times. *p < 0.05 vs. untreated cells. Each assay was repeated 3 times, each with 3 biological replicates.
PDGF-BB induces the expression of SEMA7A in hCASMCs
To investigate the regulation of SEMA7A in proliferating hCASMCs, we used PDGF-BB to stimulate serum-starved hCASMCs. Compared to control cells cultured in 0.2% FBS, PDGF-BB-treated hCASMCs showed a profound elevation in the expression of the SEMA7A transcript (Figs. 1B and 1C). The upregulation of SEMA7A was in a time- and concentration-dependent manner. Treatment with PDGF-BB at the concentrations of 20–60 ng/mL for 24 h led to a 2-6-fold induction of SEMA7A mRNA expression (Fig. 1B). When hCASMCs were exposed to 40 ng/mL of PDGF-BB, the expression of SEMA7A started to increase at 12 h and peaked at 24 h (Fig. 1C). These results suggest that SEMA7A may be involved in VSMC phenotype switching.
SEMA7A depletion impairs the effects of PDGF-BB on VSMC proliferation and migration
To ascertain the role of SEMA7A in regulating VSMC phenotypes, we knocked down SEMA7A with siRNA technology. Transfection with 2 different siRNAs caused a significant reduction of SEMA7A mRNA expression in hCASMCs (Fig. 2A). The EdU cell proliferation assays demonstrated that SEMA7A-silenced and control hCASMCs showed a comparable low proliferation index in a low serum (0.2% FBS) condition (Fig. 2B). When cultured in media containing 40 ng/mL PDGF-BB, SEMA7A-silenced cells displayed a significantly lower proliferation index than control cells. Consistently, MTT assays indicated a significant decline in viable cell number in the SEMA7A knockdown group when cultures were stimulated with PDGF-BB for 72 h (Fig. 2C). Additionally, PDGF-BB-induced hCASMC migration was blocked by SEMA7A silencing, as evidenced by in vitro wound-healing assays (Fig. 2D).
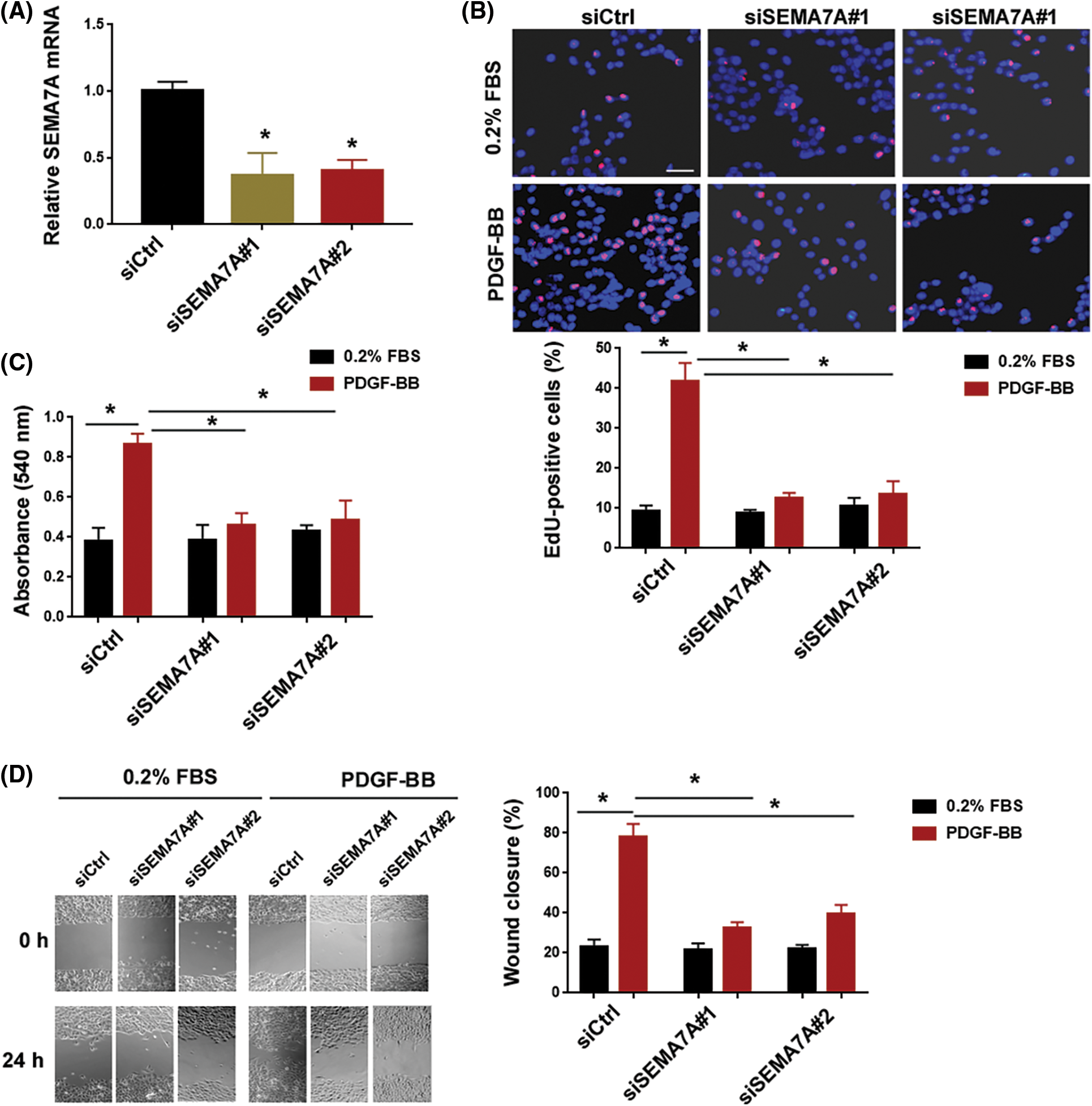
Figure 2: SEMA7A depletion impairs the effects of PDGF-BB on VSMC proliferation and migration. (A) Quantification of SEMA7A transcripts in hCASMCs transfected with indicated siRNAs. (B) EdU cell proliferation assay. hCASMCs transfected with indicated siRNAs were cultured in the presence of 0.2% FBS or 40 ng/mL of PDGF-BB. Nuclei were counterstained with Hoechst 33342 (blue). Scale bar = 50 μm. (C) hCASMCs transfected with indicated siRNAs were cultured for 72 h in the presence or absence of PDGF-BB (40 ng/mL). Cell viability was assessed by MTT assays. (D) In vitro wound-healing assays were performed to evaluate the migration capacity of hCASMCs after indicated treatments. *p < 0.05. Each assay was repeated 3 times, each with 3 biological replicates.
SEMA7A overexpression promotes the proliferation and migration of VSMCs
Next, we examined the impact of enforced expression of SEMA7A on VSMC phenotypes. Western blot analysis validated overexpression of SEMA7A in hCASMCs after transfection with the SEMA7A-expressing plasmid (Fig. 3A). Of note, ectopic expression of SEMA7A enhanced the proliferation and migration of hCASMCs (Figs. 3B–3D). These results indicate that SEMA7A has the capacity to drive VSMC proliferation and migration.
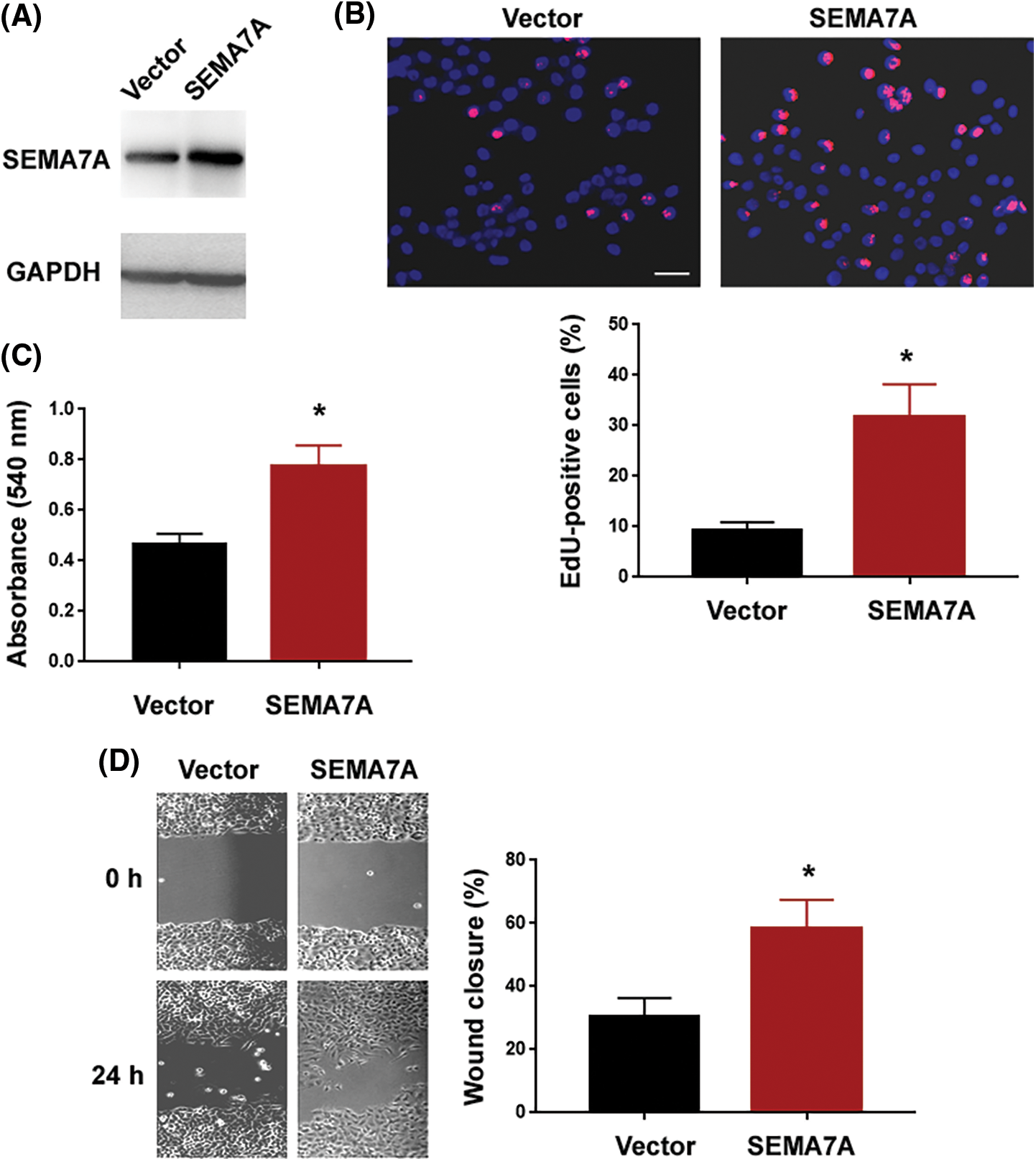
Figure 3: SEMA7A overexpression promotes the proliferation and migration of VSMCs. (A) Western blot analysis of SEMA7A protein levels in hCASMCs transfected with indicated constructs. Numbers indicate fold change relative to the vector group. (B) EdU cell proliferation assays performed in hCASMCs transfected with indicated constructs. Nuclei were counterstained with Hoechst 33342 (blue). Scale bar = 50 μm. (C) hCASMCs were transfected with indicated constructs, and viability was assessed by MTT assays. (D) In vitro wound-healing assays performed in hCASMCs transfected with indicated constructs. *p < 0.05. Each assay was repeated 3 times, each with 3 biological replicates.
SEMA7A induces the activation of the β-catenin pathway
Having determined that SEMA7A acts as an inducer of VSMC phenotype switching, we explored the mechanism underlying SEMA7A action. Several signaling pathways including ERK, PI3K/Akt, and β-catenin have been found to be involved in the regulation of VSMC phenotypes (Shen et al., 2014; An et al., 2022). Interestingly, SEMA7A overexpression increased the level of active β-catenin in hCASMCs (Figs. 4A and 4B). However, as determined by Western blot analysis, the phosphorylated and total protein levels for Akt and ERK remained unaffected in SEMA7A-overexpressing hCASMCs (Suppl. Fig. S1A). Immunofluorescent staining of β-catenin revealed that SEMA7A-overexpressing hCASMCs displayed an enhancement of β-catenin nuclear translocation (Fig. 4C). The TOPFlash/FOPFlash reporter assays indicated that SEMA7A-overexpressing hCASMCs had a 4.2-fold increase in the normalized TOPFlash reporter activity compared to control cells (p < 0.05; Fig. 4D). We also measured the effect of SEMA7A overexpression on the expression of several β-catenin signaling downstream targets, i.e., c-Myc, CCND1, and MMP7. Notably, ectopic expression of SEMA7A led to an induction of c-Myc, CCND1, and MMP7 in hCASMCs (Fig. 4E). All these data demonstrate that SEMA7A contributes to the activation of the β-catenin signaling pathway in VSMCs.
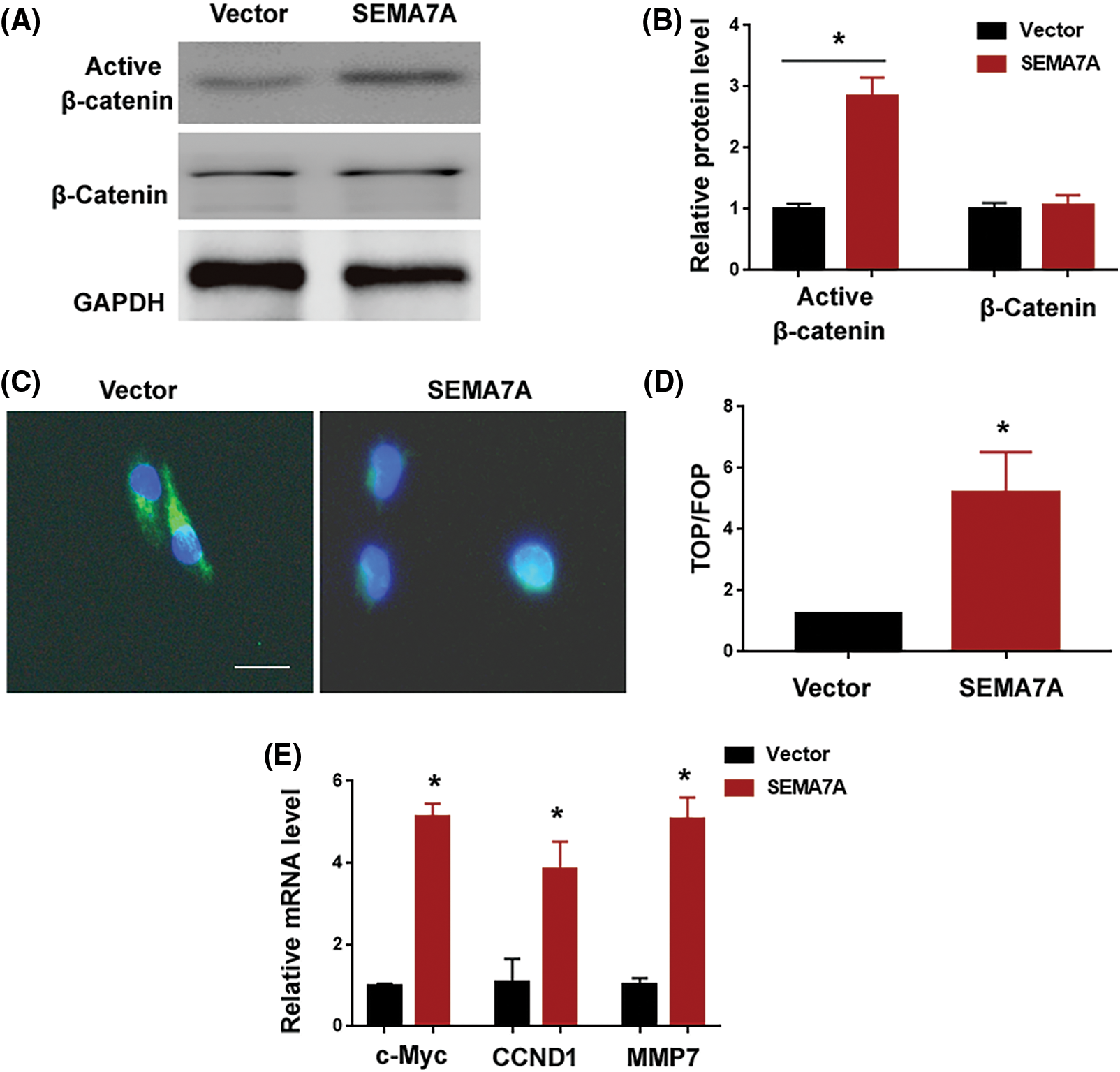
Figure 4: SEMA7A induces the activation of the β-catenin pathway. (A) Western blot analysis of protein levels in hCASMCs transfected with indicated constructs. (B) Bar graphs show quantification of active and total β-catenin levels. Results are expressed as fold change relative to the vector group. (C) Immunofluorescent staining of β-catenin (green) in SEMA7A-overexpressing hCASMCs. Nuclei were counterstained with Hoechst 33342 (blue). Scale bar = 20 μm. (D) TOPFlash/FOPFlash reporter assays. Data are expressed as fold change relative to the vector group. (E) Quantification of c-Myc, CCND1, and MMP7 transcripts. *p < 0.05. Each assay was repeated 3 times, each with 3 biological replicates.
The β-catenin signaling pathway is required for SEMA7A-induced phenotype conversion in VSMCs
Next, we asked whether SEMA7A triggered VSMC phenotype conversion through the β-catenin signaling pathway. To address this, we knocked down β-catenin in SEMA7A-overexpressing hCASMCs (Fig. 5A). Importantly, depletion of β-catenin attenuated the induction of hCASMC proliferation and migration by SEMA7A overexpression (Figs. 5B–5D). Moreover, the upregulation of c-Myc, CCND1, and MMP7 by SEMA7A was reversed when β-catenin was silenced (Fig. 5E). We also checked the effect of chemical inhibition of PI3K/Akt and ERK pathways on SEMA7A-mediated phenotype in hCASMCs. As shown in Suppl. Figs. S1B–S1D, treatment with 20 μM of LY29402 or 100 nM of PD0325901 for 24 h had no significant impact on SEMA7A-indced proliferation and migration in hCASMCs. Taken together, activation of the β-catenin signaling pathway is essential for SEMA7A action in VSMCs.
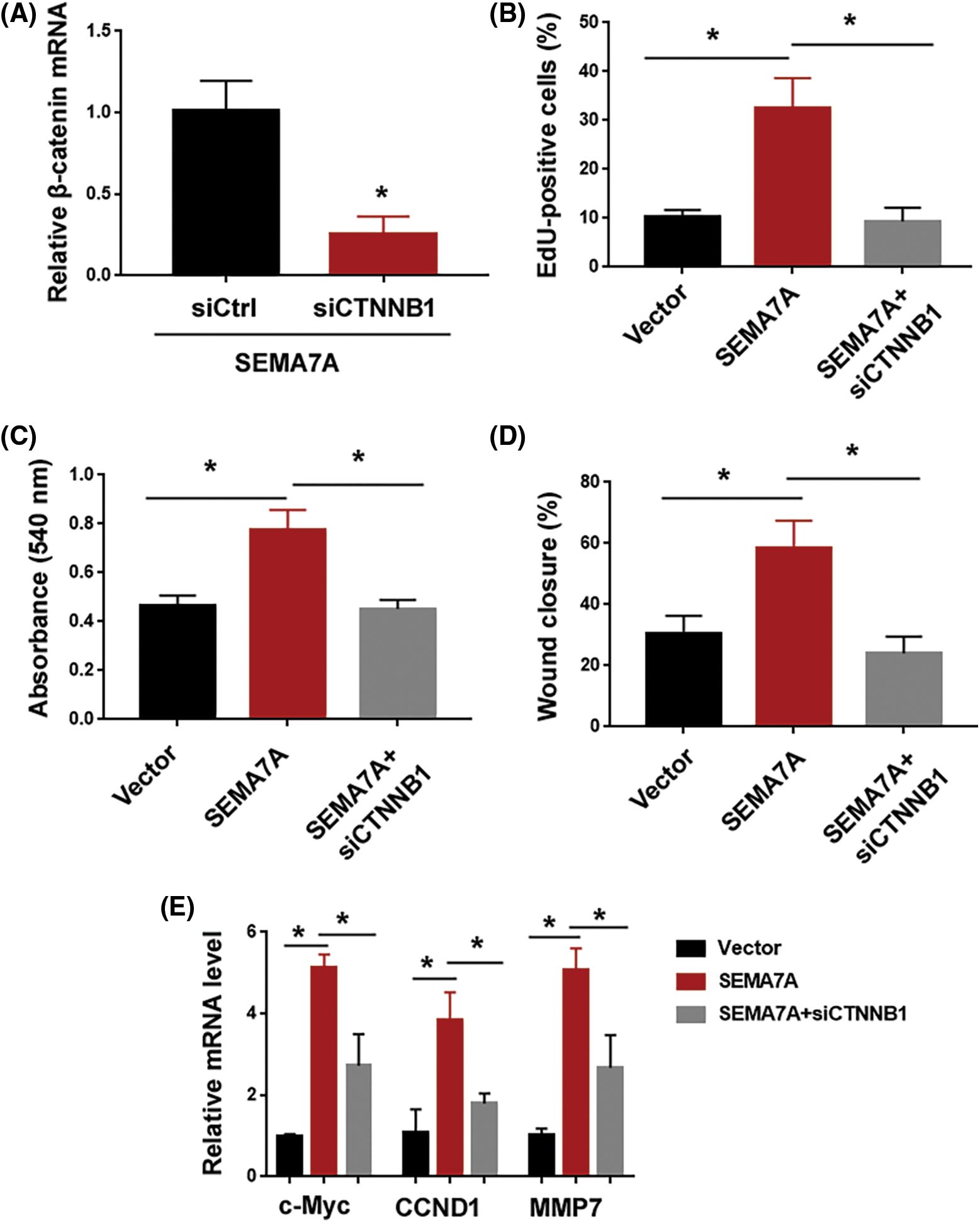
Figure 5: The β-catenin signaling pathway is required for SEMA7A-induced phenotype conversion in VSMCs. (A) Quantification of β-catenin transcripts in SEMA7A-overexpressing hCASMCs transfected with indicated siRNAs. (B) EdU cell proliferation assays performed in hCASMCs transfected with indicated constructs. (C) hCASMCs were transfected with indicated constructs, and viability was assessed by MTT assays. (D) In vitro wound-healing assays performed in hCASMCs transfected with indicated constructs. (E) Quantification of c-Myc, CCND1, and MMP7 transcripts in hCASMCs transfected with indicated constructs. *p < 0.05. Each assay was repeated 3 times, each with 3 biological replicates.
Aberrant vascular remodeling is a key event linked to the development of various cardiovascular diseases (Bennett et al., 2016). VSMCs exhibit a high plasticity and can transform from a contractile phenotype to a proliferative, migratory phenotype. Mounting evidence indicates that VSMCs play a critical role in pathological vascular remodeling (Chakraborty et al., 2021). In this study, we identify SEMA7A as an important regulator of VSMC phenotypes. Specially, overexpression of SEMA7A promotes CAMSC proliferation and migration. Moreover, the knockdown of SEMA7A blocks PDGF-BB-induced proliferation and migration of hCASMCs. Our data suggest that SEMA7A represents a potential therapeutic target for cardiovascular diseases.
We analyzed GEO datasets and found that SEMA7A is dysregulated in VSMCs in response to pro-atherogenic stimuli oxLDL and progerin (Chen et al., 2019; Jin et al., 2020). Consistently, our results show that SEMA7A is significantly upregulated in PDGF-BB-treated VSMCs. PDGF-BB has been reported to mediate arterial stiffening in response to aging and metabolic stress (Santhanam et al., 2021). These results suggest that SEMA7A may act as a stress-responsive gene and contribute to cardiovascular diseases. In support of our hypothesis, Köhler et al. (2020) reported that SEMA7A can increase platelet thrombo-inflammatory activity, thus aggravating post-ischemic myocardial tissue injury. Most interestingly, clinical evidence shows that a high serum SEMA7A level is an independent risk factor of acute atherothrombotic stroke (You et al., 2019). Therefore, SEMA7A may be a biomarker for cardiovascular diseases.
Semaphorin signaling is required for vascular patterning and heart development (Gitler et al., 2004). Several semaphorin proteins have been found to modulate VSMC phenotypes. For example, semaphorins 3A and 3E can suppress VSMC proliferation and migration (Wu et al., 2017; Wu et al., 2019). Moreover, the number of semaphorin-3E-positive VSMCs is reduced in atherosclerotic plaques (Wu et al., 2017), suggesting a protective role for semaphorin-3E during atherosclerosis. In contrast to semaphorins 3A and 3E, our data show that SEMA7A overexpression facilitates VSMC proliferation and migration. These findings suggest that semaphorin proteins play opposite roles in regulating the phenotypes of VSMCs. A previous study demonstrated that vascular SEMA7A induces atherosclerosis by modulating endothelial cell function (Hu et al., 2018a). VSMC phenotype conversion is causally linked to atherosclerotic lesion formation (Sakic et al., 2022). Thus, our results suggest that SEMA7A might contribute to atherosclerosis by promoting VSMC transformation to a proliferative, migratory phenotype.
Plexin C1 and β1 integrin are 2 established receptors for SEMA7A (Suzuki et al., 2007; Inoue et al., 2018). However, the blockade of plexin C1 and β1 integrin using neutralizing antibodies had no significant impact on SEMA7A-induced phenotype switching in hCASMCs (data not shown). Instead, we found that SEMA7A overexpression enhances the activation of β-catenin signaling in hCASMCs, as evidenced by increased levels of active β-catenin and β-catenin-dependent transcriptional activities. Multiple β-catenin downstream targets (c-Myc, CCND1, and MMP7) are upregulated by SEMA7A overexpression. These β-catenin target genes play important roles in mediating VSMC proliferation and migration. Luo et al. (2019) reported that the downregulation of c-Myc contributes to the ligustilide-mediated suppression of rat VSMC migration. In addition, c-Myc is reported to bind to the DNMT1 promoter to initiate DNMT1 expression, consequently facilitating VSMC proliferation (Xu et al., 2019). Upregulation of CCND1 is involved in oxidized low-density lipoprotein-stimulated VSMC proliferation and migration (Zhang et al., 2021). Therefore, we speculated that SEMA7A might promote phenotype conversion of VSMCs through the β-catenin signaling pathway. Interestingly, we demonstrate that knockdown of β-catenin impairs SEMA7A-induced hCASMC proliferation and migration. Moreover, the upregulation of c-Myc, CCND1, and MMP7 by SEMA7A is blocked when β-catenin is silenced. Taken together, our results suggest that SEMA7A upregulation induces the activation of β-catenin signaling to promote VSMC proliferation and migration, thereby contributing to vascular remodeling (Fig. 6).
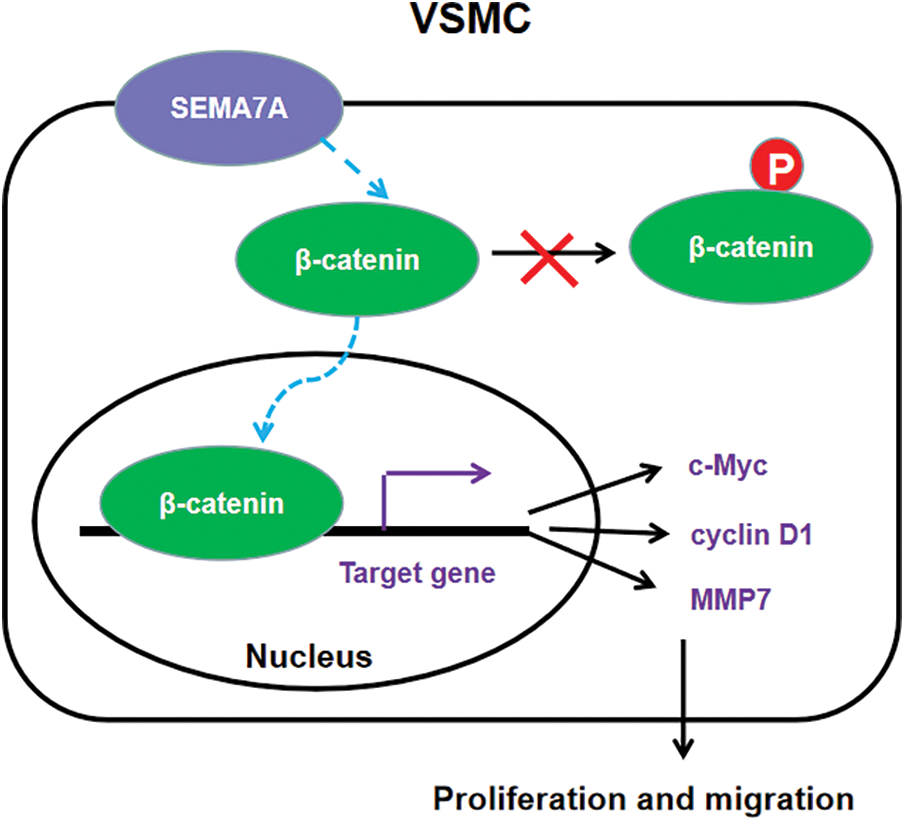
Figure 6: A schematic model of the mechanism for SEMA7A-induced VSMC proliferation and migration through activation of the β-catenin pathway.
Although our data confirm the capacity of SEMA7A in inducing VSMC phenotype switching, the exact role of SEMA7A in the pathogenesis of cardiovascular diseases warrants further investigation. Additionally, it remains to be determined how SEMA7A activates the β-catenin signaling pathway in VSMCs.
In conclusion, our work identifies SEMA7A as a novel inducer of VSMC proliferation and migration. The β-catenin signaling pathway mediates the effects of SEMA7A overexpression on phenotype conversion of VSMCs. These findings warrant further exploration of the value of SEMA7A as a therapeutic target in treating cardiovascular diseases.
Funding Statement: This work was supported by the Basic Research Program of Shanxi Province (Free Exploration) of China (20210302124416), Science and Technology Grant for Selected Returned Chinese Scholars of Shanxi Province of China (20220043), and Four “Batches” Innovation Project of Invigorating Medical through Science and Technology of Shanxi Province of China (2022XM08).
Author Contributions: X.S.S. and H.Y.Y. designed the study and analyzed the data. X.S.S., F.G., H.L., W.W.Q., C.J.C. and G.J.S. performed the experiments and collected the data. H.Y.Y. prepared the manuscript. All authors read and approved the final manuscript.
Availability of Data and Materials: The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
An W, Luong LA, Bowden NP, Yang M, Wu W et al. (2022). Cezanne is a critical regulator of pathological arterial remodelling by targeting beta-catenin signalling. Cardiovascular Research 118: 638–653. https://doi.org/10.1093/cvr/cvab056 [Google Scholar] [PubMed] [CrossRef]
Bennett MR, Sinha S, Owens GK (2016). Vascular smooth muscle cells in atherosclerosis. Circulation Research 118: 692–702. https://doi.org/10.1161/CIRCRESAHA.115.306361 [Google Scholar] [PubMed] [CrossRef]
Chakraborty R, Chatterjee P, Dave JM, Ostriker AC, Greif DM, Rzucidlo EM, Martin KA (2021). Targeting smooth muscle cell phenotypic switching in vascular disease. JVS-Vascular Science 2: 79–94. https://doi.org/10.1016/j.jvssci.2021.04.001 [Google Scholar] [PubMed] [CrossRef]
Chen L, Wu X, Xie H, Yao N, Xia Y et al. (2019). ZFP57 suppress proliferation of breast cancer cells through down-regulation of MEST-mediated Wnt/β-catenin signalling pathway. Cell Death & Disease 10: 169. https://doi.org/10.1038/s41419-019-1335-5 [Google Scholar] [PubMed] [CrossRef]
Crump LS, Wyatt GL, Rutherford TR, Richer JK, Porter WW, Lyons TR (2021). Hormonal regulation of semaphorin 7a in ER+ breast cancer drives therapeutic resistance. Cancer Research 81: 187–198. https://doi.org/10.1158/0008-5472.CAN-20-1601 [Google Scholar] [PubMed] [CrossRef]
Deng J, Miller SA, Wang HY, Xia W, Wen Y, Zhou BP, Li Y, Lin SY, Hung MC (2002). β-catenin interacts with and inhibits NF-κB in human colon and breast cancer. Cancer Cell 2: 323–334. https://doi.org/10.1016/S1535-6108(02)00154-X [Google Scholar] [PubMed] [CrossRef]
Gitler AD, Lu MM, Epstein JA (2004). PlexinD1 and semaphorin signaling are required in endothelial cells for cardiovascular development. Developmental Cell 7: 107–116. https://doi.org/10.1016/j.devcel.2004.06.002 [Google Scholar] [PubMed] [CrossRef]
Hamczyk MR, Villa-Bellosta R, Quesada V, Gonzalo P, Vidak S, Nevado RM, Andrés-Manzano MJ, Misteli T, López-Otín C, Andrés V (2019). Progerin accelerates atherosclerosis by inducing endoplasmic reticulum stress in vascular smooth muscle cells. EMBO Molecular Medicine 11: e9736. https://doi.org/10.15252/emmm.201809736 [Google Scholar] [PubMed] [CrossRef]
Hu S, Liu Y, You T, Heath J, Xu L et al. (2018a). Vascular semaphorin 7A upregulation by disturbed flow promotes atherosclerosis through endothelial β1 integrin. Arteriosclerosis, Thrombosis, and Vascular Biology 38: 335–343. https://doi.org/10.1161/ATVBAHA.117.310491 [Google Scholar] [PubMed] [CrossRef]
Hu S, Liu Y, You T, Zhu L (2018b). Semaphorin 7A promotes VEGFA/VEGFR2-mediated angiogenesis and intraplaque neovascularization in ApoE-/- mice. Frontiers in Physiology 9: 1718. https://doi.org/10.3389/fphys.2018.01718 [Google Scholar] [PubMed] [CrossRef]
Inoue N, Nishizumi H, Naritsuka H, Kiyonari H, Sakano H (2018). Sema7A/PlxnCl signaling triggers activity-dependent olfactory synapse formation. Nature Communications 9: 1842. https://doi.org/10.1038/s41467-018-04239-z [Google Scholar] [PubMed] [CrossRef]
Jin X, Xu Y, Guo M, Sun Y, Ding J, Li L, Zheng X, Li S, Yuan D, Li SS (2020). hsa_circNFXL1_009 modulates apoptosis, proliferation, migration, and potassium channel activation in pulmonary hypertension. Molecular Therapy. Nucleic Acids 23: 1007–1019. https://doi.org/10.1016/j.omtn.2020.09.029 [Google Scholar] [PubMed] [CrossRef]
Köhler D, Granja T, Volz J, Koeppen M, Langer HF et al. (2020). Red blood cell-derived semaphorin 7A promotes thrombo-inflammation in myocardial ischemia-reperfusion injury through platelet GPIb. Nature Communications 11: 1315. https://doi.org/10.1038/s41467-020-14958-x [Google Scholar] [PubMed] [CrossRef]
Körner A, Bernard A, Fitzgerald JC, Alarcon-Barrera JC, Kostidis S, Kaussen T, Giera M, Mirakaj V (2021). Sema7A is crucial for resolution of severe inflammation. Proceedings of the National Academy of Sciences 118: e2017527118. https://doi.org/10.1073/pnas.2017527118 [Google Scholar] [PubMed] [CrossRef]
Lagrange J, Worou ME, Michel JB, Raoul A, Didelot M et al. (2022). The VWF/LRP4/alphaVbeta3-axis represents a novel pathway regulating proliferation of human vascular smooth muscle cells. Cardiovascular Research 118: 622–637. https://doi.org/10.1093/cvr/cvab042 [Google Scholar] [PubMed] [CrossRef]
Li S, Zhao X, Cheng S, Li J, Bai X, Meng X (2021). Downregulating long non-coding RNA PVT1 expression inhibited the viability, migration and phenotypic switch of PDGF-BB-treated human aortic smooth muscle cells via targeting miR-27b-3p. Human Cell 34: 335–348. https://doi.org/10.1007/s13577-020-00452-5 [Google Scholar] [PubMed] [CrossRef]
Loy K, Fourneau J, Meng N, Denecke C, Locatelli G, Bareyre FM (2021). Semaphorin 7A restricts serotonergic innervation and ensures recovery after spinal cord injury. Cellular and Molecular Life Sciences 78: 2911–2927. https://doi.org/10.1007/s00018-020-03682-w [Google Scholar] [PubMed] [CrossRef]
Luo Z, Deng H, Fang Z, Zeng A, Chen Y, Zhang W, Lu Q (2019). Ligustilide inhibited rat vascular smooth muscle cells migration via c-Myc/MMP2 and ROCK/JNK signaling pathway. Journal of Food Science 84: 3573–3583. https://doi.org/10.1111/1750-3841.14936 [Google Scholar] [PubMed] [CrossRef]
Pan L, Bai P, Weng X, Liu J, Chen Y, Chen S, Ma X, Hu K, Sun A, Ge J (2022). Legumain is an endogenous modulator of integrin αvβ3 triggering vascular degeneration, dissection, and rupture. Circulation 145: 659–674. https://doi.org/10.1161/CIRCULATIONAHA.121.056640 [Google Scholar] [PubMed] [CrossRef]
Ran R, Cai D, King SD, Que X, Bath JM, Chen SY (2021). Surfactant protein A, a novel regulator for smooth muscle phenotypic modulation and vascular remodeling-brief report. Arteriosclerosis, Thrombosis, and Vascular Biology 41: 808–814. https://doi.org/10.1161/ATVBAHA.120.314622 [Google Scholar] [PubMed] [CrossRef]
Sakic A, Chaabane C, Ambartsumian N, Klingelhöfer J, Lemeille S, Kwak BR, Grigorian M, Bochaton-Piallat ML (2022). Neutralization of S100A4 induces stabilization of atherosclerotic plaques: Role of smooth muscle cells. Cardiovascular Research 118: 141–155. https://doi.org/10.1093/cvr/cvaa311 [Google Scholar] [PubMed] [CrossRef]
Santhanam L, Liu G, Jandu S, Su W, Wodu BP et al. (2021). Skeleton-secreted PDGF-BB mediates arterial stiffening. The Journal of Clinical Investigation 131: e147116. https://doi.org/10.1172/JCI147116 [Google Scholar] [PubMed] [CrossRef]
Shen YJ, Zhu XX, Yang X, Jin B, Lu JJ, Ding B, Ding ZS, Chen SH (2014). Cardamonin inhibits angiotensin II-induced vascular smooth muscle cell proliferation and migration by downregulating p38 MAPK, Akt, and ERK phosphorylation. Journal of Natural Medicines 68: 623–629. https://doi.org/10.1007/s11418-014-0825-0 [Google Scholar] [PubMed] [CrossRef]
Suzuki K, Okuno T, Yamamoto M, Pasterkamp RJ, Takegahara N et al. (2007). Semaphorin 7A initiates T-cell-mediated inflammatory responses through alpha1beta1 integrin. Nature 446: 680–684. https://doi.org/10.1038/nature05652 [Google Scholar] [PubMed] [CrossRef]
Wang X, Li H, Zhang Y, Liu Q, Sun X, He X, Yang Q, Yuan P, Zhou X (2021a). Suppression of miR-4463 promotes phenotypic switching in VSMCs treated with Ox-LDL. Cell and Tissue Research 383: 1155–1165. https://doi.org/10.1007/s00441-020-03338-y [Google Scholar] [PubMed] [CrossRef]
Wang H, Zhong B, Geng Y, Hao J, Jin Q, Zhang Y, Dong L, Gao D, Li J, Hou W (2021b). TIPE2 inhibits PDGF-BB-induced phenotype switching in airway smooth muscle cells through the PI3K/Akt signaling pathway. Respiratory Research 22: 238. https://doi.org/10.1186/s12931-021-01826-5 [Google Scholar] [PubMed] [CrossRef]
Wu JH, Li Y, Zhou YF, Haslam J, Elvis ON, Mao L, Xia YP, Hu B (2017). Semaphorin-3E attenuates neointimal formation via suppressing VSMCs migration and proliferation. Cardiovascular Research 113: 1763–1775. https://doi.org/10.1093/cvr/cvx190 [Google Scholar] [PubMed] [CrossRef]
Wu JH, Zhou YF, Hong CD, Chen AQ, Luo Y et al. (2019). Semaphorin-3A protects against neointimal hyperplasia after vascular injury. eBioMedicine 39: 95–108. https://doi.org/10.1016/j.ebiom.2018.12.023 [Google Scholar] [PubMed] [CrossRef]
Xu L, Hao H, Hao Y, Wei G, Li G et al. (2019). Aberrant MFN2 transcription facilitates homocysteine-induced VSMCs proliferation via the increased binding of c-Myc to DNMT1 in atherosclerosis. Journal of Cellular and Molecular Medicine 23: 4611–4626. https://doi.org/10.1111/jcmm.14341 [Google Scholar] [PubMed] [CrossRef]
You T, Zhu Z, Zheng X, Zeng N, Hu S et al. (2019). Serum semaphorin 7A is associated with the risk of acute atherothrombotic stroke. Journal of Cellular and Molecular Medicine 23: 2901–2906. https://doi.org/10.1111/jcmm.14186 [Google Scholar] [PubMed] [CrossRef]
Zhang P, Wang W, Li M (2021). Circ_0010283/miR-377-3p/Cyclin D1 axis is associated with proliferation, apoptosis, migration, and inflammation of oxidized low-density lipoprotein-stimulated vascular smooth muscle cells. Journal of Cardiovascular Pharmacology 78: 437–447. https://doi.org/10.1097/FJC.0000000000001076 [Google Scholar] [PubMed] [CrossRef]
Zheng X, Yu Q, Shang D, Yin C, Xie D et al. (2022). TAK1 accelerates transplant arteriosclerosis in rat aortic allografts by inducing autophagy in vascular smooth muscle cells. Atherosclerosis 343: 10–19. https://doi.org/10.1016/j.atherosclerosis.2022.01.009 [Google Scholar] [PubMed] [CrossRef]
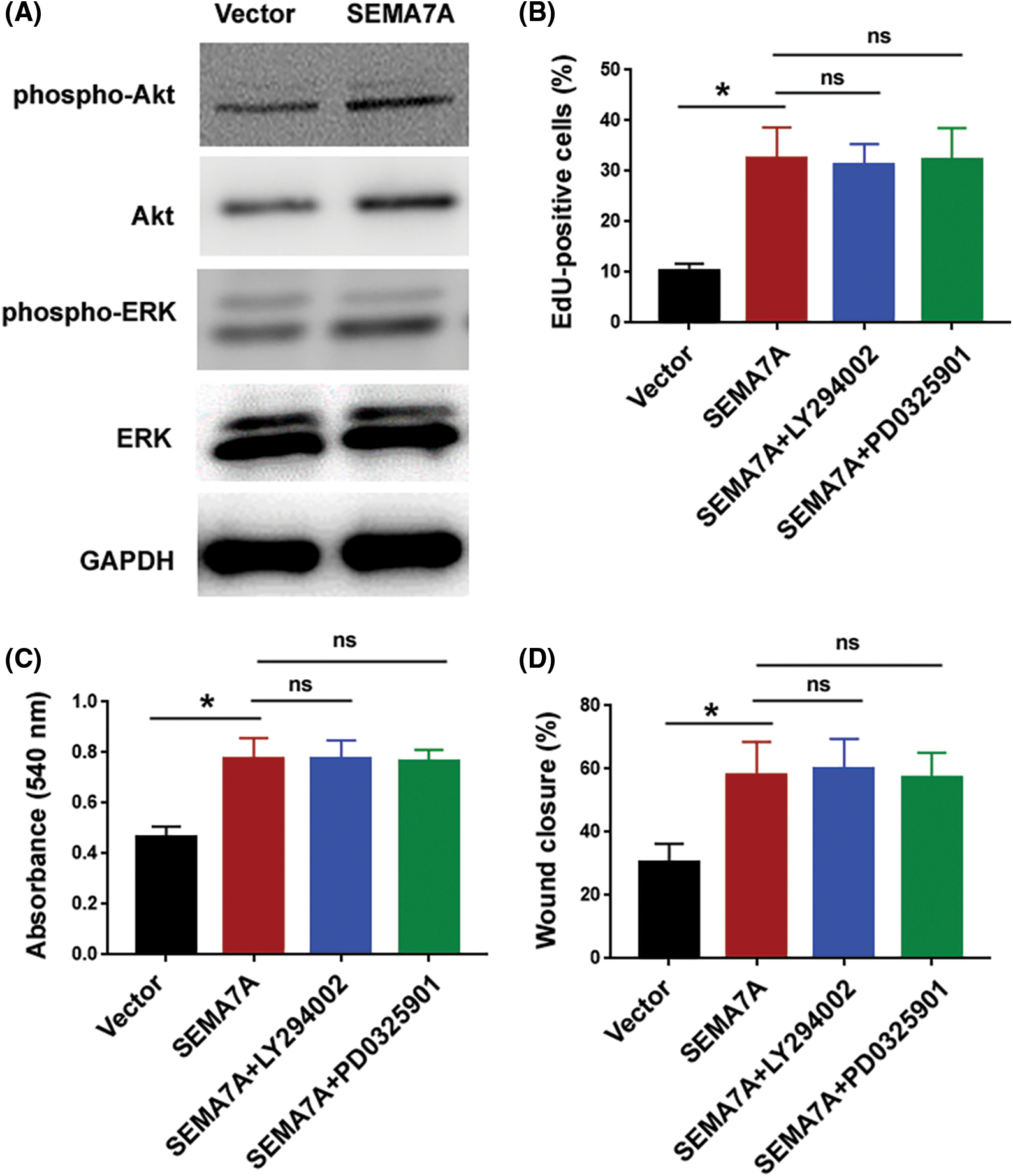
Figure S1: Inhibition of PI3K/Akt and ERK pathways has no significant effect on SEMA7A-mediated phenotype in hCASMCs. (A) Western blot analysis of indicated proteins in hCASMCs transfected with SEMA7A-expressing plasmids or empty vector. (B–D) hCASMCs with overexpression of SEMA7A were exposed to 20 μM of LY29402 or 100 nM PD0325901 for 24 h. The cells were subjected to (B) EdU cell proliferation assay, (C) MTT assay, and (D) in-vitro wound healing assay. *p < 0.05; ns, no significance.
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools