
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

A comprehensive analysis of the role of molecular docking in the development of anticancer agents against the cell cycle CDK enzyme
1 Department of Chemistry, Pandit Deendayal Energy University, Gandhinagar, 382426, India
2 School of Applied Material Sciences, Central University of Gujarat, Gandhinagar, 382030, India
* Corresponding Author: ANU MANHAS. Email:
(This article belongs to the Special Issue: Bioinformatics Study of Diseases)
BIOCELL 2023, 47(4), 707-729. https://doi.org/10.32604/biocell.2023.026615
Received 15 September 2022; Accepted 26 December 2022; Issue published 08 March 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Cancer is considered one of the most lethal diseases responsible for causing deaths worldwide. Although there have been many breakthroughs in anticancer development, cancer remains the major cause of death globally. In this regard, targeting cancer-causing enzymes is one of the efficient therapeutic strategies. Biological functions like cell cycle, transcription, metabolism, apoptosis, and other depend primarily on cyclin-dependent kinases (CDKs). These enzymes help in the replication of DNA in the normal cell cycle process, and deregulation in the functioning of any CDK can cause abnormal cell growth, which leads to cancer. This review is focused on anticancer drug discovery against cell cycle CDK enzyme using an in silico technique, i.e., molecular docking studies. Molecular docking helps in deciphering the key interactions formed within the inhibitor and the respective enzyme. This concise study provides an overview of the most current in silico research advancements made in the field of anticancer drug discovery. The findings presented in the current review article can help in understanding the nature of inhibitor-target interactions and provide information on the structural and molecular prerequisites for the inhibition of cell cycle CDKs.Keywords
Cancer is a lethal and complex disease that occurs due to an alteration in cellular growth. In the case of cancer disease, the cell grows abnormally. This transformation from normal to cancer cells has been the subject of a large number of research made in the field of biomedical sciences. The long-term cure for cancer is still as challenging as it was at the start of the disease (Seyfried and Shelton, 2010). According to the WHO report of 2022, in 2021, around 1.9 million new cases and 609,360 deaths were observed around the world, along with a ratio of 1 among the 6 deaths due to cancer (American Cancer Society, 2022). It is predicted that if cancer remains untreated, these numbers will rise and reach 29.5 million new cases and 16.4 million deaths in the next twenty-five years (Shah et al., 2019). Studies have revealed the fact that the most common form of cancer is breast, colon, rectum, and lung cancer. Major factors that were identified as the reason for cancer are low fiber and low nutrient intake, high tobacco use, uncontrollable alcohol consumption, and low physical activity. According to the Global Cancer Statistics-2019 presented by WHO, cancer was the leading cause of mortality till 70 years of age in 119 countries out of 183 (Siegel et al., 2019). The reflection on impetuous death is highly visible in the central magnitude of economic and social development. These figures and predictions represent the substantial burden caused by cancer globally. In absolute terms, the treatment of cancer becomes important. Considering the importance, many research groups are working in this field to eradicate cancer. Generally, the main cause of cancer is mutagens or carcinogens; however, the development of the carcinogens and replications are facilitated by cell cycles, various enzymes, and biomolecules. Among them, a class of kinase enzymes, i.e., cyclin-dependent kinase (CDKs), are responsible for causing cell duplication and enhancing the normal step of the cell cycle. In a normal cell cycle, kinase proteins combine with their specific cyclin unit to get phosphorylated by another activating kinase protein. In the cell cycle, cyclin acts as a regulatory molecule, whereas CDK acts as the catalytic collaborator. In different stages of the cell cycle, the activated CDK proteins function distinctively with their corresponding cyclin unit. In eukaryotes, the cell cycle is controlled by the CDK proteins, and dysregulation in the functioning of the CDKs causes the triggering of cancer (instigation and evolution) (Asghar et al., 2015). In this respect, various researchers are involved in designing new anticancer drugs that can target CDK enzymes specifically. Several published studies report the inhibition of CDK enzymes via experimental and computational studies (Tutone and Almerico, 2017; Marak et al., 2020; Shaikh et al., 2022). We have observed that with time, researchers are shifting towards the use of computer-aided drug-designing tools for the drug discovery process. Due to the development of high-performance computing facilities, improved algorithms, and easy accessibility of the protein 3D structure, academia and pharma sectors are using in silico studies to overcome the challenges posed by traditional drug discovery methods. In the current scenario, in silico studies have now become a crucial tool for reducing the chances of drug failure in the drug discovery process. Given the importance of computational drug design, the current work aimed to discuss the research work conducted on the CDK enzymes, which incorporate the in silico in general, and molecular docking studies in particular, along with the experimental or other computational techniques. Molecular docking is one of the most captivating tools among other in silico techniques used in the field of medicine and drug designing domain. This technique is based on the mathematical algorithms which help in deciphering the binding pose and interactions of the proposed/synthesized molecule pose within the binding cavity of the macromolecule (de Ruyck et al., 2016). Early, the rigid docking technique was implemented to study the binding pose based on the lock and key theory. This technique discards the flexibility of the protein and ligand as per induced-fit theory (Meng et al., 2011). The flexibility concept was introduced at the ligand level, and the technique was used to conduct docking for a long time. In recent years, efforts have been made to develop algorithms to deal with the flexibility of the protein (active site residues only) (Meng et al., 2011). However, protein backbone flexibility is still a challenge in the present docking algorithms (Meng et al., 2011). This was overcome by molecular dynamics simulations. Simulation calculations study the stability of the interaction formed in the solvent medium with respect to time. Hence, considering the drawback of the docking studies, these calculations are performed in combination with the experimental and/or molecular dynamics simulation. In the drug designing process, molecular docking can be used as a tool to identify the structural requirement for efficient protein-ligand binding, virtual screening, drug repositioning, identification of the compounds that can target more than one protein of the same disease and prediction of drug off-target activity, and many more (Pinzi and Rastelli, 2019). Ample studies are reported that have implemented the use of molecular docking and/or hybrid with experimental studies to search for various anticancer (Lone et al., 2017; Tutone and Almerico, 2017; Bhattacharya et al., 2022; El-Sayed et al., 2022a, 2022b, 2022c; Ghosh et al., 2022; Khanam et al., 2022; Obakachi et al., 2022), antimalarial (Manhas et al., 2016, 2018, 2019a, 2019b; Benjamin et al., 2022), antitubercular (Lone et al., 2018; Akki et al., 2022; Kaur and Singh, 2022; Modi et al., 2022; Sanka et al., 2022), etc., molecules. This depicts the importance of docking in the field of rational drug design. Thus, in this review, we have focused on the implementation of molecular docking (in combination with experimental and/or simulation studies) on the cell cycle CDK enzyme in the last two years.
Cyclin-dependent kinases (CDKs)
CDKs belong to the threonine/serine protein kinases family, which are responsible for causing normal cell division. Based on their function, they are broadly classified into two categories, i.e., cell cycle and transcriptional CDKs. CDKs are considered essential in many complex processes, like, angiogenesis, spermatogenesis, hematopoiesis, gene transcription, DNA repair, metabolism, etc. The dysregulation in the functioning of the CDK enzyme leads to the development of cancer. Many CDK inhibitors are under clinical trials; however, most of the drugs fail because of the lack of CDK specificity. Mostly, CDK inhibitors are known as ATP-competitive inhibitors. They have limited therapeutic effects as they inhibit the phosphorylation mechanism of other non-target kinases. Also, one of the renowned CDK2/7/9 inhibitors, roscovitine, failed in clinical trials due to toxicity and low potency. However, few CDK4/6 inhibitors, like, palbociclib, ribociclib, and abemaciclib, have successfully passed the clinical trials (Marak et al., 2020). The sequence and structure across all CDKs are identical as they contain a two-lobed structure. The active site of CDKs is sandwiched between the carboxy-terminal, which is rich in α-helices, and the amino-terminal lobe, which contains beta-sheets. The secondary structural elements of CDKs are highly preserved, but there are minor deviations in the activation segment, length of the chain, and nature of surface amino acids. These factors are crucial for the CDK enzymes to identify the respective inhibitors (Marak et al., 2020). There are two types of CDK inhibitors, i.e., exogenous and endogenous, that are lower molecular weight proteins and small molecules inhibitors, respectively. Endogenous inhibitors are small molecules, like, P21, P27, and P57 which are active against CDK4/6 (INK4), and CIP/KIP group, which contain P16, P15, P18, and P19 active against CDK enzymes. They inhibit the catalytic mechanism of CDKs by phosphorylation of the unbound CDKs or CDK-cyclin complex. Studies have shown that CDK inhibitors reduce the expression of CDKs and cyclins (Marak et al., 2020). For instance, melanoma, breast, colorectal, and lung cancers were all associated with P16 deficiency. P27 protein deficiency is typically linked to gastrointestinal, colon, breast, and prostate cancer. Because of this, the tumor can be identified based on the lack of endogenous CDK inhibitors (Marak et al., 2020).
Cell cycle cyclin-dependent kinases
CDK1 enzyme works in the presence of cyclin B1 to facilitate the transition from the G2 phase into mitosis (cell cycle progression). Overexpression of CDK1 causes various types of cancers, like, gastric, ovarian, colorectal, liver, oral squamous cell carcinoma, and breast cancer (Kourea et al., 2003). Therefore, it is important to design potent CDK1 inhibitors. In this regard, Sofi et al. (2022) target the CDK1 enzyme using dinaciclib analogs by employing molecular docking (AutoDock 4.2.6) and molecular dynamics simulations (Desmond 2020.1, Schrödinger). They retrieved 100 dinaciclib analogs from the online PubChem database, which were further analyzed for their drug-likeness and ADMET properties. The filtered compounds were subject to molecular docking, and it was reported that compounds 1 and 2 (Fig. 1) displayed higher docking scores of −9.3 and −9.2 kcal/mol in comparison to the reference dinaciclib with inhibitory constants (Ki) of 1.07 and 2.1 µM, respectively. These compounds were selected to conduct molecular dynamics simulations. From the docked interaction plots, compound 1 displayed hydrogen bond interaction with Ile10, Ser84, and Asp86 residues. Compound 2 formed hydrogen bonding with Ser84, Asp86, and Gln132 residues present in the active site. Based on the stable simulation outcome, their novelty was checked using ChemSpider. Thus, they utilized in silico methods to validate the CDK1 as a therapeutic target and evaluate the inhibitory activity of the dinaciclib analogs (Sofi et al., 2022).
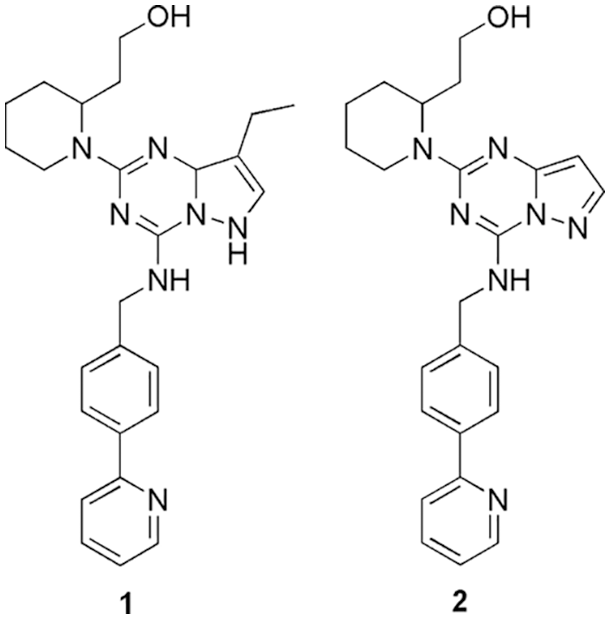
Figure 1: The chemical structures of the CDK1 inhibitor shortlisted via inhibitory activity evaluation.
Zhao and his group discovered small drug-like molecules using a transcriptome-based computational drug repositioning method for the treatment of ovarian cancer (Zhao et al., 2022). They screened out the bottleneck protein (CDK1) via protein-protein interaction (PPI) network analysis (String database v 11.5) and used the protein to conduct the molecular docking (AutoDock 4.2.6) utilizing the dataset retrieved from the PubChem database. From the outcome of interaction studies, they predicted that pidorubicine (3), PHA-793887 (4), and lestaurtinib (5) (Fig. 2) can target the cell cycle regulator with the binding energy of −7.67, −7.75, and −9.13 kcal/mol for topoisomerase IIα (TOP2A), CDK1, and aurora kinase A (AURKA) enzyme respectively. They observed that IC50 values of 3, 4, and 5 on the OC-314 human cancer cell line were lower than the reported ovarian cancer drugs palbociclib and vorinostat. However, among these three, only compound 4 binds with the CDK1 via hydrogen bonding with Glu81, Leu83, Asn133, and Asp146. Here in silico methods were performed to explore the interactions between candidate drugs and the proteins encoded by the bottleneck gene, which were screened out via PPI network analysis (Zhao et al., 2022).
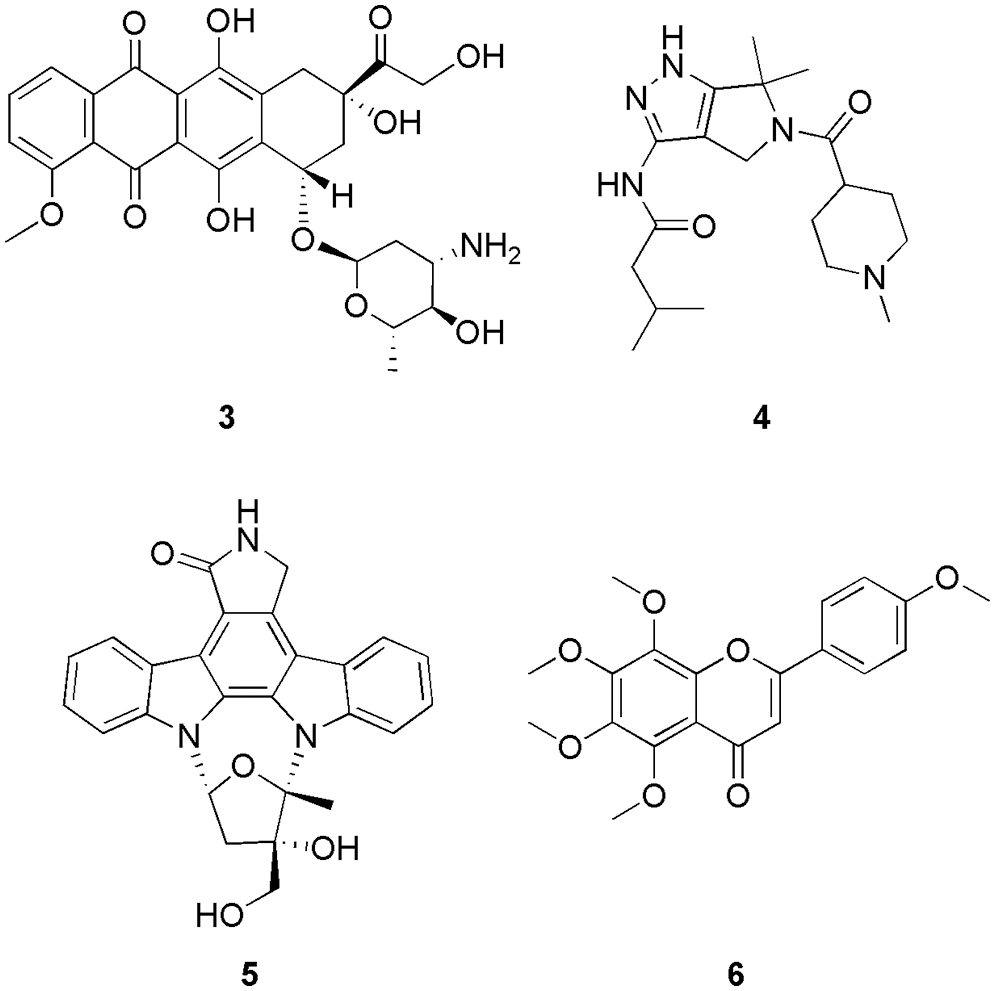
Figure 2: The chemical structures of CDK1 inhibitor derived from PPI network analysis.
Yu et al. (2022) discovered the natural plant herb tangeretin (6) (Fig. 2) that can be used to treat oral squamous cell carcinoma (oral cancer) by using network pharmacology and molecular docking studies. Similar to Zhao et al. (2022), Yu and the group also conducted a PPI network analysis (String data platform) to retrieve CDK1 as one of the bottleneck proteins. They conducted docking (AutoDock Vina 1.1.2) on CDK1 protein using the nine ingredients of natural plant herbs (five from the traditional Chinese medicinal database and four from the literature) (Yu et al., 2022). With CDK1, hydrophobic contacts were predominated; however, no hydrogen bond interactions were observed. The binding energy of 6 with CDK1 was −8.7 kcal/mol. Also, in vitro studies were conducted on 6 (obtained from the literature), which showed the inhibition of the cell proliferation process. In conclusion, they predicted that natural plant herbs, can be used in the treatment of oral cancer, and in silico methods were used to confirm the results of network pharmacology (Yu et al., 2022). Serag et al. (2021) proposed that hybrid ionic vanillyl-azole-Schiff bases (IVASBs) can be synthesized in three new multifunctional bioactive IVASBs (7, 8, and 9) (Fig. 3). The synthesized compounds were characterized by FTIR, NMR, and MS analysis. They also conducted molecular docking studies (AutoDock 4) on the CDK1 enzyme using synthesized IVASBs three compounds. With CDK1, molecule 7 displayed interactions via two different forms of bindings, i.e., hydrogen bonding between the hydroxyl groups of the amino acids (Thr25, Tyr 26, and Arg47) and the nitrogen atoms of the azomethine and hydrophobic binding between the carbon chains of compound 7 and phenyl rings of Glu32. Molecule 8 has lower binding energy than 7 and displayed binding via hydrophobic and hydrogen bonding in different binding models. Interestingly the binding between 9 and CDK1 enzyme involves three different kinds of interactions, like, hydrogen bonding between the N-atom of diamine-thiazole and the OH group of Gly24; π-stacking between the phenyl rings of diamine-thiazole and Tyr26; and hydrophobic binding, between the phenyl ring and carbon chain of 9 and Glu32, Thr25, and Lys138, respectively. Thereafter, they conducted in vitro studies on these compounds, and it was depicted that out of three, 7 and 9 displayed better inhibition (cell proliferation) of 13.5 ± 1.14 and 3.73 ± 0.2 μg/mL, respectively. Also, the binding energy values of 7 (−3.52 kcal/mol) and 9 (−3.43 kcal/mol) make them the most potent anti-breast cancer agents. Further, they conducted flow-cytometric analysis and in vivo studies and concluded that 9 can be used as a starting scaffold for the treatment of breast cancer (Serag et al., 2021).
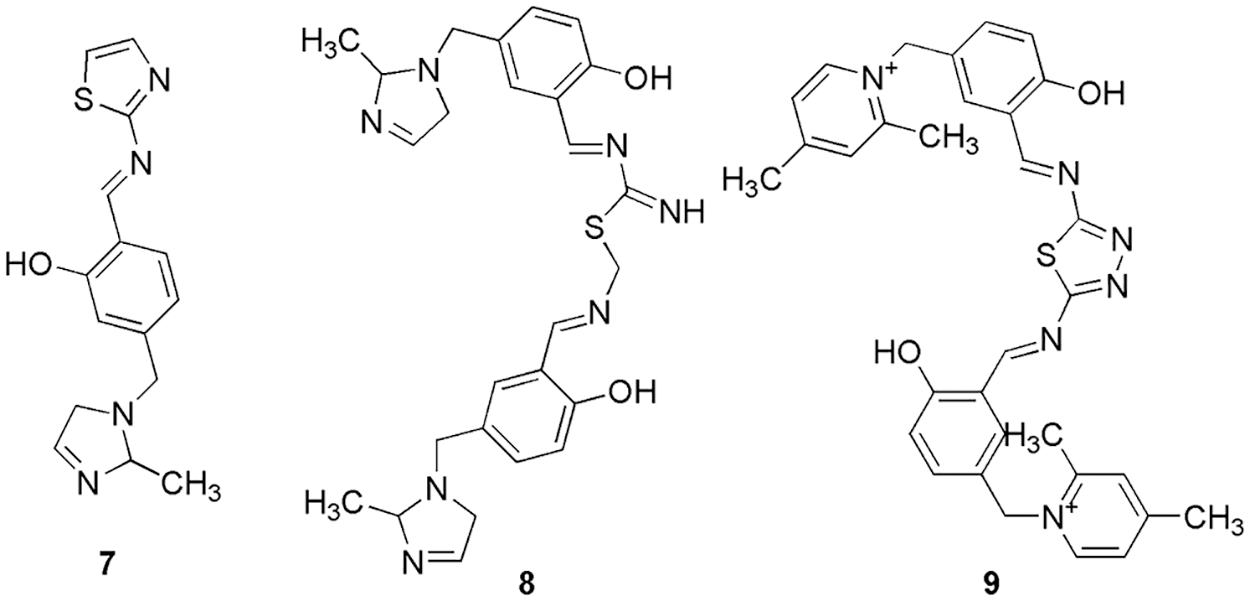
Figure 3: The chemical structures of CDK1 inhibitory of hybrid ionic vanillyl-azole-Schiff bases (IVASBs).
Elkamhawy and his research group synthesized twelve in-house N-substituted indole-based analogs that inhibited both CDK1 and HER2 (Elkamhawy et al., 2021). Among them, 10 (Fig. 4) displayed the highest inhibitory activity against both of the enzymes. They docked (Discovery Studio Client 21) molecule 10 in CDK1 and HER2 to check the possible binding modes. The interaction analysis revealed that the main scaffold (indole), right-handed aryl halide (3-fluorophenyl), and heteroaryl ring (pyridazine ring) of the molecule occupy the hinge binding region, back room, and solvent-exposed surface, respectively. They suggested that the chemical scaffold of the molecules has a significant role in modulating the position of the compound in their respective grooves, and it can be used in further drug development processes. In the CDK1 binding sites, 10 displayed interactions via hydrogen bonding with Asp86. Thus, in silico molecular docking was utilized to investigate the possible hypothetical binding modes of 10 in the CDK1 enzyme, which was screened out from the in vitro kinase inhibitory assay (Elkamhawy et al., 2021).
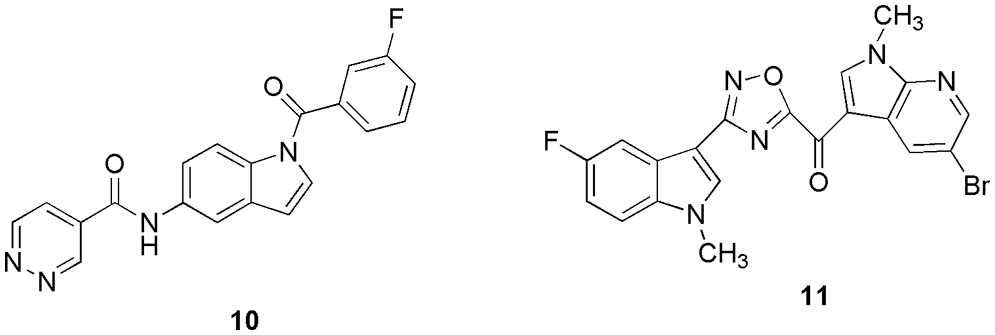
Figure 4: The chemical structures of the most potent CDK1 inhibitors from indole-based analogs (10) and oxadiazole molecules (11).
Pecoraro et al. (2021) reported the inhibition of the CDK1 enzyme via six newly synthesized oxadiazole molecules. Among them, compound 11 (Fig. 4) was found to be an efficient CDK1 inhibitor. From the in vitro anti-proliferative activity studies, they confirmed that 11 is the most active compound ranging from 5.7 to 10.7 µM, and it obeys the ADME prediction too. After in vitro studies, they conducted docking calculations on 11. Molecular docking studies revealed that 11 interacts in the nucleotide-binding pocket by establishing a hydrogen bond with the backbone of the Gly11 residue and with a docking score of −6.99 kcal/mol. In addition, the water-mediated interaction was also reported via Gln132 with the nitrogen group of the 7-azaindole moiety. Finally, they conclude that their compound displayed cytotoxicity activity, induced apoptosis, and targets the CDK1 enzyme, thus, can be used as the starting compound to treat cancer. With the help of molecular docking studies, they deciphered the ability of the compound to interact with the adenosine triphosphate binding pocket of CDK1 enzyme (Pecoraro et al., 2021).
CDK 2 being a part of the cell cycle, emulate a crucial role since it is involved in a series of different biological processes and cell cycle regulation. Intercellular pathways, DNA and RNA (metabolism and translation), signal transduction, DNA damage, etc., are some of the routes via which CDK 2 phosphorylates and hence interacts with proteins. Overexpression of CDK2 causes breast, colorectal, ovarian, lung, and pancreatic cancer (Peyressatre et al., 2015). El-Sayed et al. (2022b) designed and synthesized fifteen dichlorophenoxymethyl-based derivatives and estimated their antiproliferative activity against the cancer cell lines (HCT-116 and MCF). Out of fifteen, four (12, 13, 14, and 15) (Fig. 5) displayed the most promising cytotoxicity (IC50 = 3.78−11.46 μM) (13 > 14 > 15 > 12), and from kinase profiling, they retrieved IC50 of 0.21 to 0.88 μM. They selected these molecules to conduct molecular docking (MOE-Dock V 2014) in the binding pocket of CDK2. Similar to the reference molecule roscovitine, the docking data of our synthesized compounds 12, 13, 14, and 15 showed good fitting within the binding site. They displayed favorable interactions with the important amino acid Leu83. Molecules 12, 13, 14, and 15 display binding scores of −10.55, −13.50, −10.88, and −10.83 kcal/mol, respectively, with CDK2 enzyme. In 12, the backbone of Leu83 formed one hydrogen bond acceptor interaction with the NH of 2-thioxopyrido-[3,2-d]pyrimidin-4-one moiety. Along with that, it also displayed three hydrogen bond donor interactions with two sulfonic group oxygens of 12. Moreover, a hydrogen bond donor interaction between the Lys89 sidechain and the 2,4-dichlorophenoxy moiety in 12 and 13 were also observed. Interestingly, the ATP-bonding pocket of CDK-2/cyclin A kinase was shown to have comparable binding mechanisms for the derivatives 14 and 15. The backbone of Leu83 formed hydrogen bonds with the NH of the amide group and the nitrogen atom of benzo[d]thiazole or benzo[d]oxazole in 14 and 15, respectively. Additionally, the essential amino acid Lys89 forms contact between the centroid of the 2,4-dichlorophenoxy ring and arene-cation. From the docking studies, they found that the presence of amino-substituted nitrogenous bi-or tri-heterocyclic scaffolds with the core 2,4-dichloro-phenoxy-methyl fragment can be responsible for showing the inhibitory activity against cancer, and this scaffold can be used for further research. Thus, in silico method helped to clarify the hypothetical binding modes of the compounds that showed promising cytotoxic activity (El-Sayed et al., 2022b).
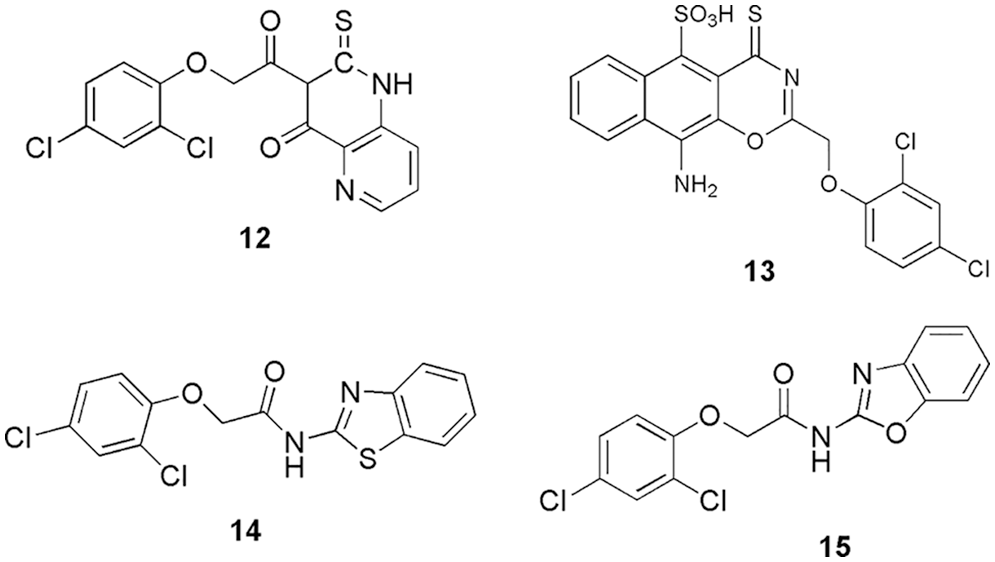
Figure 5: The chemical structures of dichlorophenoxymethyl-based derivatives as the most potent CDK2 inhibitor.
Nurhayati and his group conducted studies on known anticancer compounds of tris-indoline derivatives (16, 17, and 18) (Fig. 6) to evaluate their anticancer potency against CDK2, caspase-9, and p53 proteins (Nurhayati et al., 2022). They conducted molecular docking (Autodock Vina) on the three proteins with the respective three molecules separately. As per their studies, all the compounds were successfully docked in the binding site of CDK2. The ligands 16, 17, and 18 had the greatest binding affinity values of −7.3, −7.7, and −6.6 kcal/mol, respectively, with the CDK2 enzyme. MD simulations revealed that compound 17 binds with CDK2 via interaction with Tyr178, Ser180, Ser231, and Asn271. Moreover, on checking their cytotoxicity, they observed that 16 and 17 possess a good cytotoxicity effect and are more potent in inducing apoptosis in breast cancer cell lines in comparison to the chemotherapy agent doxorubicin. Compounds 16 and 18 displayed promising results against MCF-7 with IC50 2.059 and 3.9759 µM. Thus, in silico analysis and cytotoxicity assay confirmed that 16, 17, and 18 can act as potent CDK2 inhibitors (Nurhayati et al., 2022). El-Sayed et al. (2022a) synthesized eighteen novel scaffolds of tri-azolecoumarin-glycosyl hybrids and their tetrazole hybrid analogues as potent anticancer agents. Based on the outcome of their antiproliferative activity against the PC-3, Paca-2, A-375, Mel-501, and Caco-2 cancer cell lines, three candidates (19, 20, 21) (Fig. 7) showed promising results in comparison to the reference doxorubicin. Compound 19 showed IC50 of 10.7, 8.8, 25.4, and 10.9 µM against the Paca-2, Mel-501, PC-3, A-375, and Caco-2 cancer cell lines. Compound 20 displayed IC50 values of 14.6 and 16.7 µM for cell lines Paca-2 and Mel-501, respectively. Compound 21 showed IC50 values of 16.9, 4.1, 12.5, and 9.9 µM for Paca-2, Mel-501, and A-375 cell lines, respectively. Further, the mechanism of action of these compounds with the CDK2, epidermal growth factor receptor (EGFR), and vascular epidermal growth factor receptor (VEGFR2) enzymes were assessed. The outcomes revealed that 19 displayed potent activity against EGFR, VEGFR-2, and CDK-2, and compounds 20 and 21 displayed potent activity against CDK2. Overall, they suggest that 19 can be used as a multitarget drug against VEGFR-2, EGFR, and CDK-2 (binding energy of −11.30 kcal/mol), and 20 and 21 can be used as CDK2 inhibitors. Finally, to understand the binding pattern and affinity between 19, 20, 21, and the selected proteins, molecular docking studies (MOE-Dock) were conducted. The coumarin moiety in compounds 19, 20, and 21 played a crucial part in fitting within the CDK2 enzyme through hydrogen bond formation with the important amino acid Leu83. Additionally, the acetylated oxygens displayed hydrogen bonding with Gln131 in 20 and Lys129 in 21, in addition to other interactions. They retrieve binding energy of −10.76 and −10.85 kcal/mol for each derivative was promising. As a final assessment, it was noted that, in comparison to the other derivatives 20 and 21, bearing large glycoside moieties, 19 with its 2,3-dihydroxypropyl fragment (small size) enhanced good fitting and binding with the important amino acids within the active sites of EGFR, VEGFR-2, and CDK-2/cyclin A2. Furthermore, the coumarin nucleus plays a crucial part in fixing within the CDK2 active site in the screened derivatives 19, 20, 21. Thus, based on the kinase inhibitory results of the target, the in silico study was conducted in an attempt to provide a correlation between their activities and the possible binding modes (El-Sayed et al., 2022a).
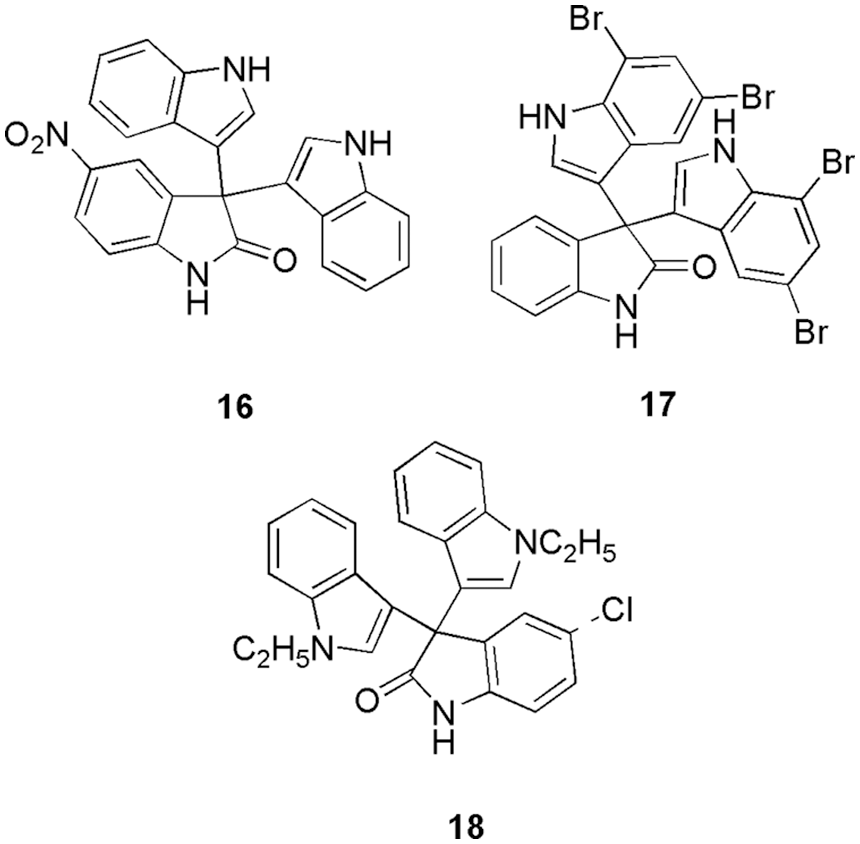
Figure 6: The chemical structures of tris-indoline derivatives having potent CDK2 inhibitory activity.
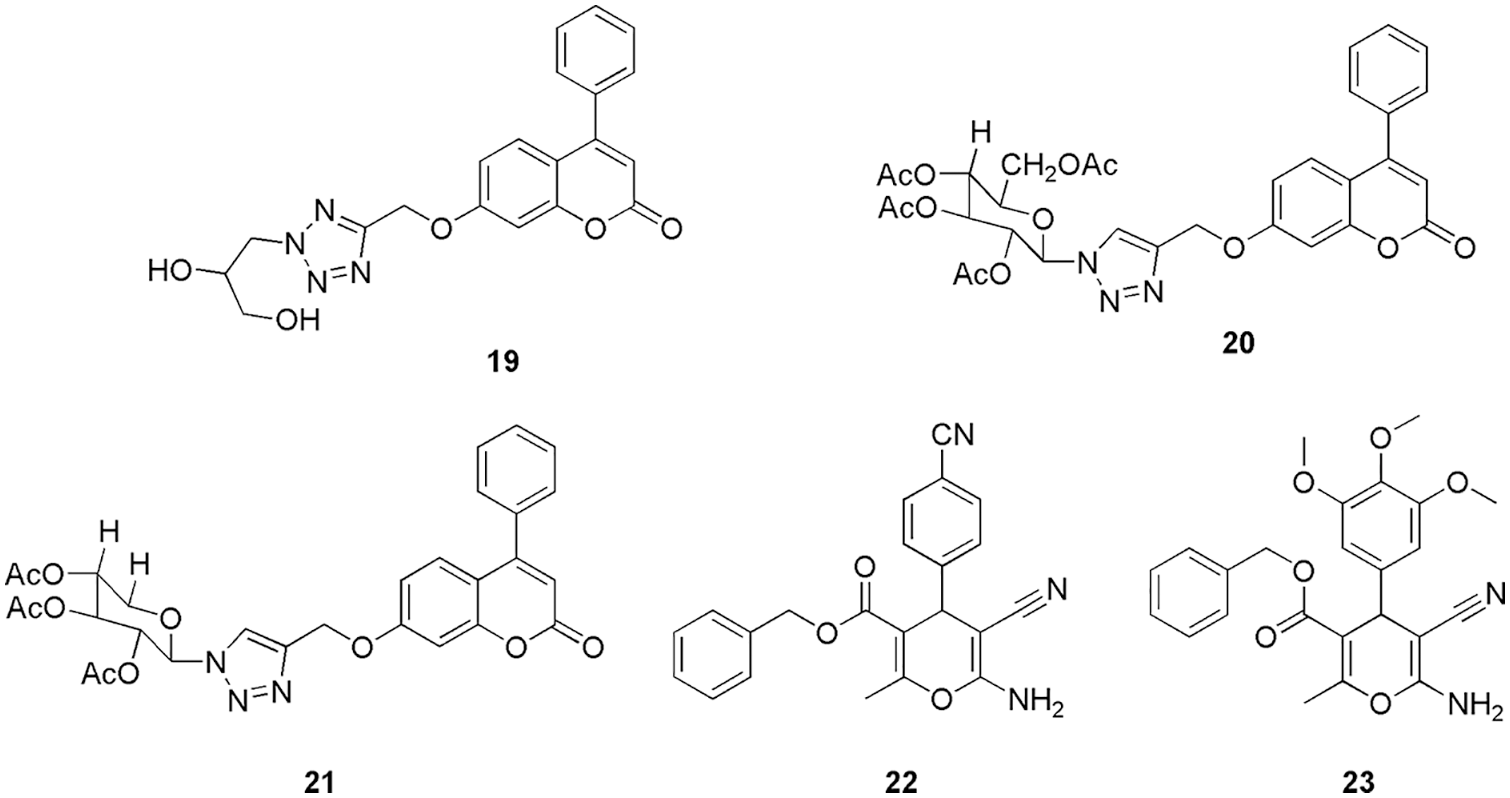
Figure 7: The chemical structures of Tri-azolecoumarin-glycosyl hybrids (19, 20, 21) and 2-amino-4H-pyran and one 2,4-dihydropyrano[2,3-c]pyrazole derivatives (22, 23) as the potent CDK2 inhibitor.
El-Sayed et al. (2022c) synthesized and characterized thirteen new derivatives of 2-amino-4H-pyran and one 2,4-dihydropyrano[2,3-c]pyrazole and further checked their antibacterial and antioxidant activities on the HCT-116 cell line (colorectal cancer). Based on better antioxidant and antiproliferative activity, they concluded that 22 and 23 (Fig. 7) can be used as anti-colorectal cancer agents. They reported the IC50 values of cytotoxicity on HCT-116 cells for compounds 22 and 23 as 75.10 and 85.88 μM, respectively. Further, to check their interaction pattern, they conducted molecular docking studies (Glide, Schrodinger) on the CDK2 enzyme. Pyran derivative 22 forms three hydrogen bonds with Asp86, Gln131, and Asn132. In the interaction plot, Asp86, through its amino group, acts as a donor, Gln131 act as an acceptor through the 3-methoxyl group, and Asn132 act as an acceptor through the 5-methoxyl group. These binding interactions demonstrated the significance of the amino group and the methoxy groups at the m-positions (3,5-positions) for future optimization. The ligand 22 also showed lipophilic interactions with the amino acids Ile10, Val18, Phe80, and Leu148. The binding energy of 22 with CDK2 protein was −48.70 kcal/mol. Additionally, the interactions between Phe80 and the phenyl ring in π-π stacking were reported. The investigation of the binding interactions between CDK2 and compound 23 further showed that two hydrogen bonds were formed between the amino acid residues Asp86 and Lys129 in the active pocket. The docking score of compound 23 was reported to be −43.31 kcal/mol. The benzyl moiety of the compound also showed lipophilic interactions with the amino acids Ile10, Val18, Val64, Phe80, Leu148, and Leu134. From the docking outcome, they concluded that a CDK2 inhibitor needs a larger ring to be connected to the primary scaffold (like a pyran ring) by a flexible chain of two to three atoms. Additionally, it must be located at the molecule’s periphery to effectively enter the ATP binding site and generate lipophilic and mildly polar interactions. Additionally, docking studies suggested that hydrogen bond donors or acceptors on the main scaffold could significantly improve binding to the receptor. Further, they conducted a kinase inhibitory assay, followed by quantitative measurement and real-time polymerase chain reaction (PCR) profiling of the CDK2 enzyme along with the gene. The outcome indicates that 10 mg/mL of 22 and 23 is optimum for causing apoptosis of HCT-116 cells of mitochondria of caspase-3 protein (El-Sayed et al., 2022c). Chaube and Bhatt (2022) reported non-toxic, potent, and CDK2 selective inhibitors. They generate the four common feature pharmacophores (GASP, Sybyl X) from the reported six CDK2 inhibitors, and then based on the Dmean score, they shortlisted one model to screen out the 20,542 molecules (Lipinski filtered) from the NCI database. Further, the top five screened/filtered molecules were selected to conduct molecular docking studies. Finally, they shortlisted one candidate, 24 (Fig. 8), that displayed interactions similar to the reference drug staurosporine. Thereafter, they used a knowledge-based approach (bio-isosteric replacement) to further modify the purine scaffold of 24. The modified five candidates were again subjected to molecular docking calculations. Based on the docking score and presence of important hydrogen bonding interactions with Glu81, Leu83, and Asp86, they shortlisted 24 for simulation studies (Sybyl X). Further, they checked their toxicity and ADMET properties which revealed their non-toxic nature (Chaube and Bhatt, 2022). Belal and his group synthesized novel fused pyrroles, pyrrolo-pyrimidines, and pyrrolo-diazepine derivatives as CDK2/EGFR inhibitors (Belal et al., 2022). Further, the synthesized derivatives were evaluated for their inhibitory activities against Hep3B, HCT116, and MCF-7 cell lines. Among all the derivatives, 25 (Fig. 8) exhibited the highest activity in comparison to the reference doxorubicin. They conducted molecular docking (MOE software) and ADMET studies to check their binding pattern and study their toxicity. On docking, compound 25 displayed binding energy of −24.52 kcal/mol, which was similar to that of the reference AZD543. The adenine pocket was occupied by the 1H-pyrrole-3-carbonitrile scaffold that form hydrogen bonding with Leu134 and Val18 along with hydrophobic contact with Lys89, Asp86, Gln85 Leu820, Leu694, and Val702. Moreover, the piperidine moiety of 25 interacts with Ile10. Also, they reported one more hydrophobic interaction within p-tolyl moiety and ribose binding pocket (Belal et al., 2022).
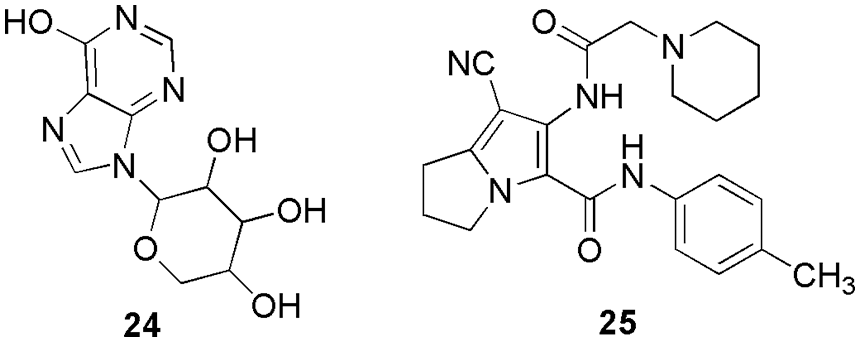
Figure 8: The chemical structures of the most potent CDK2 inhibitors shortlisted based on docking score.
Eltamany et al. (2022) performed a comparative analysis between Plicosepalus acacia and P. curviflorus to check their antioxidant and anticancer activities. On conducting the metabolomics profiling, they observed that P. curviflorus showed higher anticancer, and antioxidant properties owing to the nature of the compound (phenolic contents). The cytotoxicity value on PC-3 cell lines for P. curviflorus was 25.83 μg/mL. Further, they conducted molecular docking studies (MOE 2019) to decipher the reason behind the superiority of P. curviflorus. They observed that owing to the binding pattern and the presence of interactions similar to the experimental studies, the molecule showed better inhibitory activity. Molecular docking studies revealed that the majority of the investigated compounds, particularly those from P. curviflorus, had significant binding affinities towards both proteins. Their binding energies ranged from −10.69 to −16.39 kcal/mol with CDK2 and −14.68 to −18.69 kcal/mol with EGFR. Furthermore, they formed interactions with the key amino acids Leu83 and Lys89 within the CDK2 protein. Finally, they concluded with an emphasis on the validation of their outcome via experimental studies (Eltamany et al., 2022). Raut and his group synthesized azafavanone derivative, which can be used to treat prostate cancer (Raut et al., 2022b). Compound 26 (Fig. 9) was selected for further studies based on its lower inhibitory activity value (0.4 µM). Further, they conducted a cell cycle analysis for the treatment of the compound, which showed an arrest at the sub-G1 phase. They observed that the use of the compound enhances ROS generation and cell death (DU145 cells), thus, explaining the ROS-mediated mechanism responsible for the death. From molecular docking studies (SwissDock), they observed that 26 binds well in the active site of the CDK2 protein by forming hydrogen bonding interactions with Lys33, and Asp86 and displaying binding energy of −7.4 kcal/mol. In their work, they highlighted the ROS-mediated mechanism causing DU145 cell death by the aza derivative compound, and the compound can be a potent molecule in anticancer therapeutics development. Here, in silico studies were employed to find out the reason behind the cytotoxic property of the drug (Raut et al., 2022b). Mandour and his co-worker synthesized twelve pyrazolopyrimidine derivatives and checked their anti-proliferative activities against HCT-116, MCF-7, and HepG-2 cell lines, which was further used to check their CDK2 inhibition (Mandour et al., 2022). As per their outcome, all the compounds displayed greater toxicity against MCF-7, and HCT-116, while for HepG-2, only three compounds showed potent activity. On analyzing their CDK2 inhibitory activity, they observed that 27 (Fig. 8) displayed the most remarkable activity (IC50 0.061 ± 0.003 µM) and lower toxicity against the normal cell lines. Also, it causes apoptosis in HCT cells and observable alteration in the S and pre-G1 phases of the cell cycle. Further, they checked their binding pattern by molecular docking (Discovery Studio 4.0), from which they observed that all the compounds formed stable interactions with Leu83 in the binding pocket, and compound 27 interacted via Glu81, Leu83, and Asp86 by forming hydrogen bonding. The binding interaction energy ranged from −60.89 to −50.85 kcal/mol for the lead compound roscovitine (E = −55.75 kcal/mol), and 27 demonstrated a binding score of −59.85 kcal/mol and a similar binding mode. Also, the compound showed an acceptable range of ADMET parameters and drug-likeness studies. The molecular docking study revealed that all the potent anti-proliferative tested compounds were of comparable binding mode to that of the reference ligand (Mandour et al., 2022).
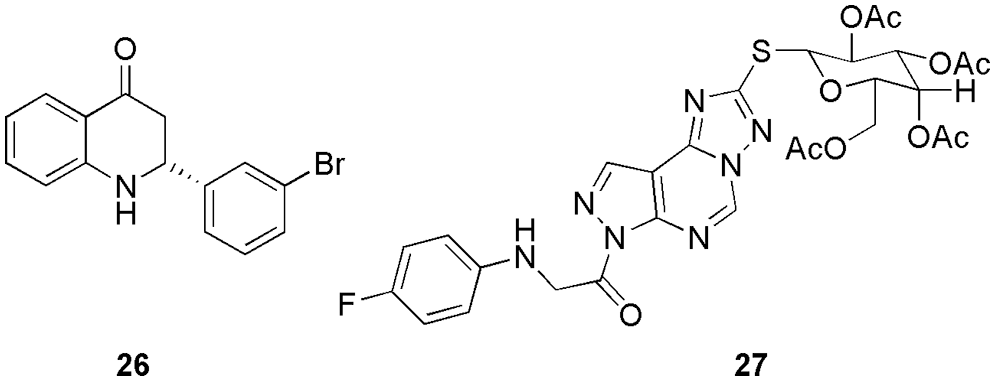
Figure 9: The chemical structures of potent CDK2 inhibitors of P. curviflorus (26) and pyrazolopyrimidine derivative (27).
Eldehna et al. (2022) synthesized 3-(piperazinyl-methyl) benzofuran derivatives as CDK2 inhibitors. On conducting the inhibitory activity, few of the compounds displayed better inhibitory activity in comparison to the reference staurosporine against CDK2 enzyme (Eldehna et al., 2022). Along with this, compounds 28 and 29 (Fig. 10) displayed better antiproliferative activities against different cancer cell lines, like, pancreatic (Panc-1), breast (MCF-7), and lung (A549) cancer cell lines. Compound 28 displayed efficient antiproliferative activity with IC50 values of 0.94, 2.92, and 1.71 μM for cell lines Panc-1, MCF-7, and A549, respectively. Compound 29 displayed antiproliferative activity with IC50 values 2.22, 5.57, and 2.99 μM for cell lines Panc-1, MCF-7, and A549, respectively. Also, they display infinitesimal cytotoxicity against the human lung fibroblast normal cell line (MRC-5). They conducted docking on DFG out conformation of CDK2 enzyme with the synthesized compounds 28 and 29 to check their binding pattern. From the outcome, they observed that common benzofuran moiety binds in the binding cavity of the CDK2 protein. Molecular docking simulations revealed that the designed compounds follow the common binding pattern of type II inhibitors. The thiosemicarbazide, semicarbazide, and acylhyrazone linkers in the designed 3-(piperazinylmethyl) benzofuran binds in the kinase binding site at the interface between the gate area and the allosteric back pocket. They form interaction via hydrogen bonding with the Glu51 side chain carboxylate and DFG Asp145 backbone NH. The peripheral (un)substituted phenyl moiety is directed towards the hydrophobic region between the gate area and the hinge region on one side, and the benzofuran ring is directed towards the hydrophobic allosteric back pocket. They form hydrophobic interactions with the hydrophobic side chains of the amino acids lining the back pocket on the other. Furthermore, at the 3-position of the benzofuran ring, the phenylpiperazine moiety is extended towards a hydrophobic region surrounded by the hydrophobic side chains of the amino acids Leu54, Leu58, Ile63, Cys118, Val123, and Leu143. The most prominent interactions of 28 and 29 were observed with Glu51 and Asp145. In short, in silico molecular docking studies were performed to explore the ability of target benzofurans to adopt the common binding pattern of CDK2 type II inhibitors (Eldehna et al., 2022). Mohammed and Elmasry (2022) synthesized quinazolinone-based derivatives as CDK2 inhibitors. They perform in vitro studies on these compounds using sixty tumor cell lines of CNS, melanoma, renal, prostate, breast, leukemia, lung, ovarian, and colon cancer. From the outcome, it was observed that among all, molecule 30 (Fig. 10) was the most active and displayed the highest activity against all the cell lines. Further, they tested 30 against the cell cycle and observed that it disrupts the functioning of the S phase and G2/M phase, thus can inhibit the CDK2 enzyme. Further, they selected this compound to conduct molecular docking (MOE) study on CDK2 and check its drug-likeness properties (SwissADME server). The results of the molecular docking study revealed that 30 binds most effectively within the binding pocket of CDK2 kinase with a binding score of −13.8 kcal/mo1. Compound 30 forms two hydrogen bonds between the quinazolinone moiety and the hinge region of the protein. The quinazolinone carbonyl oxygen, in particular, forms two water-mediated hydrogen bonds with the Leu83-NH and Glu81-CO. Furthermore, the planar aromatic core, like the co-crystallized ligand, makes extensive hydrophobic contacts with Ile10, Val18, Ala31, Phe80, Leu134, and Ala144. Other hydrogen π interactions were observed between the phenyl rings attached to the quinazolinone rings and Glu12 and Gln85, respectively. Moreover, as per its drug-likeness studies, the compound showed acceptable pharmacokinetic and physicochemical properties. The most important aspect of their research was that antiproliferative activity analysis, cell cycle analysis, docking, and ADME showed the same result confirming 30 as a potent CDK2 inhibitor (Mohammed and Elmasry, 2022). Shamsiya and Bahulayan (2022) synthesized a series of solid-state emitters based on oxazolone-coumarin-triazole (six) via multi-component reaction as anticancer compounds (breast cancer). They calculated the electronic structures (Gaussian 09) of their compounds and revealed that the nonlinear optical characteristic causes emission. Further from molecular electrostatic potential (MEP) maps, they observed that their molecules contain negative and positive potential on oxygen and carbon atoms, respectively. From the MEP study, they understand the reactive site present in their molecules for causing the reaction. Further, they conducted molecular docking studies (Auto Dock Vina 1.1.2) on all six synthesized compounds (31 to 36) (Fig. 11) with the CDK2 enzyme. It was observed that the molecules interact via hydrogen bonding, van der Waals, π-cation, and π-π interactions. Only compounds 31, 32, and 36 formed hydrogen bonds with the protein. The protein formed a hydrogen bond with Lys89 and the coumarin ring’s carbonyl oxygen, a π-π interaction between the coumarin ring and Phe82, two π-cation interactions between the triazole ring and Lys89, and benzene ring and Lys20. It displayed the docking score of −10 kcal/mol. Compound 32 exhibited two hydrogen bonds, one between the oxazolone’s carbonyl oxygen and Lys89 and the other between Gln131 and the triazole ring. In compound 36, hydrogen bonding interaction was observed between the carbonyl group of the coumarin ring and Asp86, as well as between the carbonyl group of the oxazolone and Thr14. The molecules also interacted via hydrophobic interactions. Compound 33 displayed the highest binding affinity with a binding score of −10.7 kcal/mol. All the compounds could bind well in the active site of the protein, and they relate the docking mode with the MEP of the compounds. They observed that the negative and positive potential obtained in the MEP map are responsible for showing interaction in the binding studies. Further, a cytotoxicity study depicted the potent activity of these molecules against the cervical cancer cell line (HeLa) (Shamsiya and Bahulayan, 2022). Zhang and his group explained the anticancer mechanism of 6-methoxy flavone (37) (Fig. 12) on cell lines of SiHa, C33A, HaCaT, and HeLa and cell cycle assay (Zhang et al., 2022). From the outcome, they revealed that molecule 37 inducted S-phase disruption in the HeLa cell line and in the cyclin A2 (CCNA2)/CDK2/cyclin-dependent kinase inhibitor 1 (p21CIP1) pathway too (PCR and western blotting). Based on the outcome, they conducted molecular docking (AutoDock Vina) on the CDK2, CCNA2, p21CIP1, and combined CDK2_CCNA2 enzyme. They observed that 37 has the highest affinity (−8.6 kcal/M) for the CDK2 enzyme. However, the hybrid CDK2_CCNA2 enhances the affinity of the 37. They also observed no covalent interaction with p21CIP1. Further, they analyzed the clinical characteristics, which revealed that the compound may be related to six clinic-pathological parameters of cervical cancer patients, and they propose that the compounds can be used to treat cervical cancer (Zhang et al., 2022).
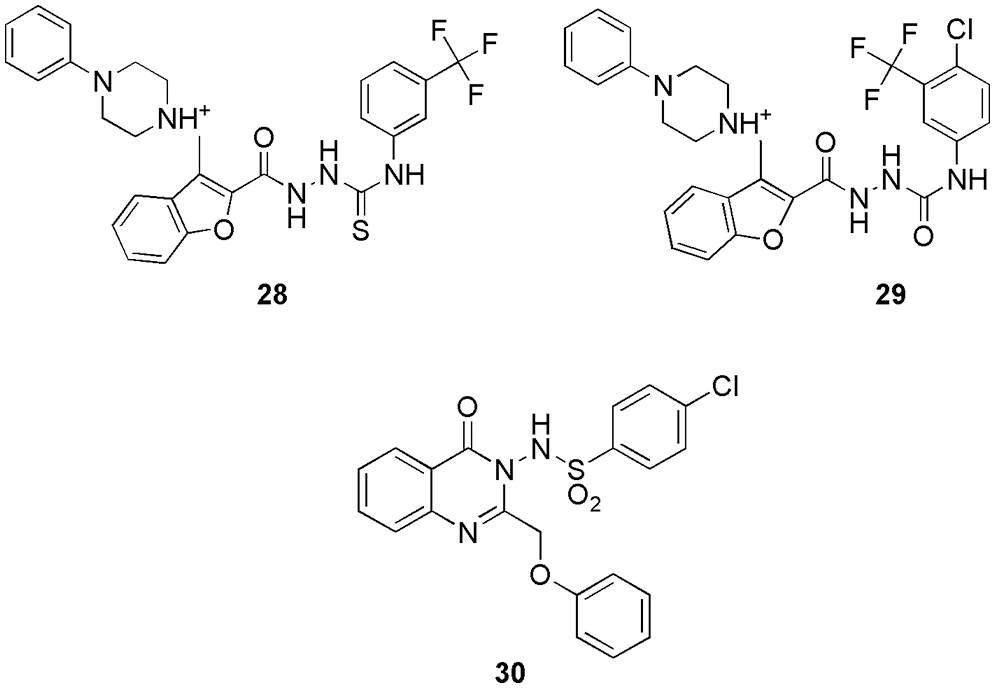
Figure 10: The chemical structures of 3-(piperazinyl-methyl) benzofuran derivatives (28, 29) and Quinazolinone-based derivatives (30) as CDK2 inhibitors.
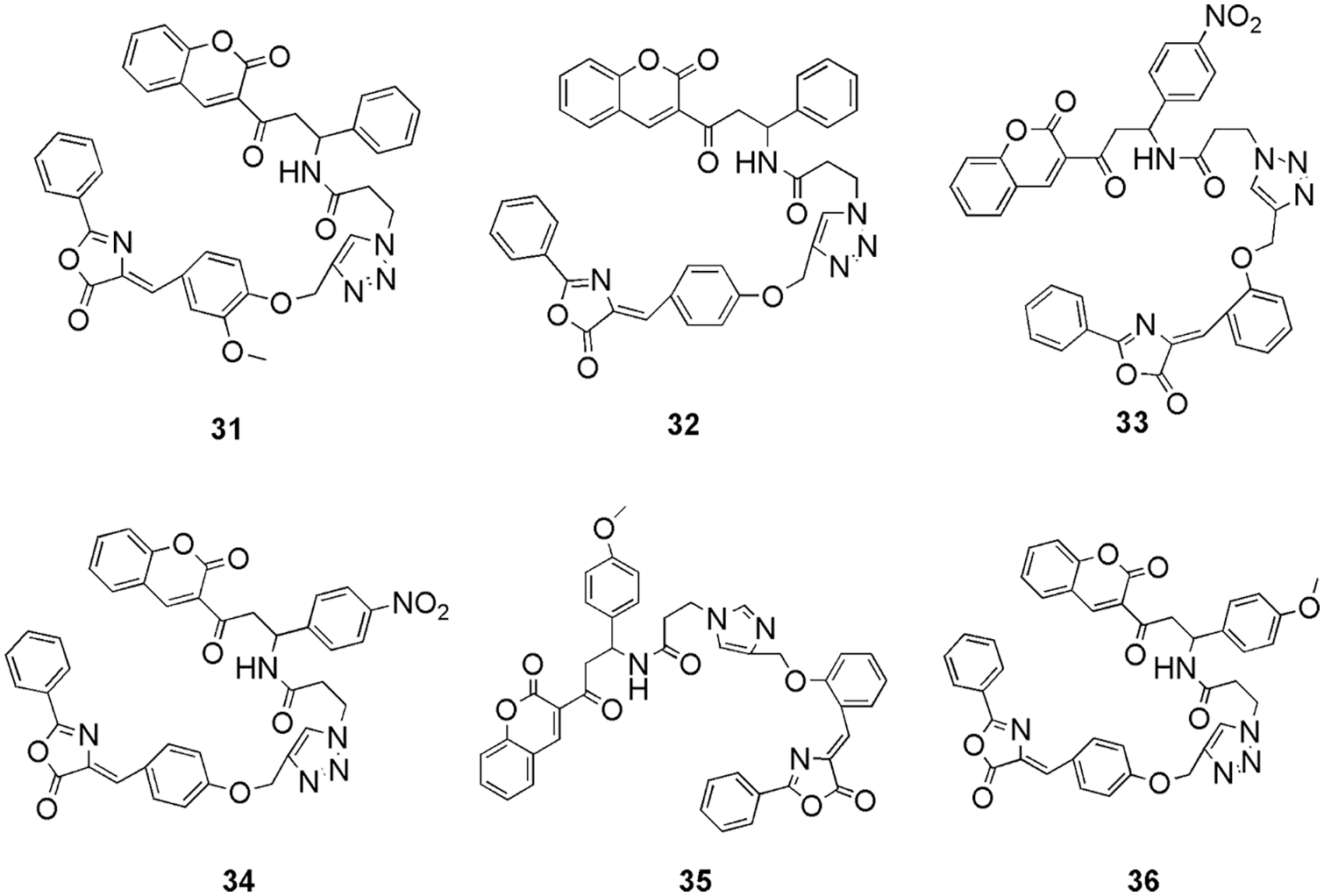
Figure 11: The chemical structures of oxazolone-coumarin-triazole derivatives as the potent CDK2 inhibitor.
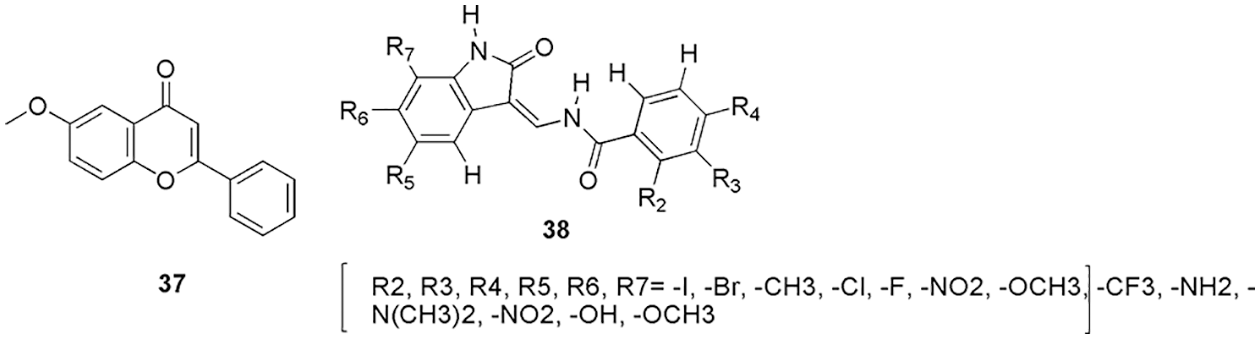
Figure 12: The chemical structures of 6-methoxy flavone (37) and isatin derivative (38) as the most active CDK2 inhibitor.
Many groups utilized docking to decipher the binding pattern in the CDK2 enzyme. For instance, Czeleń and his group’s work on isatin derivatives 38 (Czeleń et al., 2022) (Fig. 12), Nassar et al. (2022) synthesized pyrazolo-pyrimidine and pyrazolo-triazolo-pyrimidine compounds (39, 40, and 41) (Fig. 13) and Joshi et al. (2022) reported cnnamamide-chalcone derivatives (42, 43, 44, and 45) (Fig. 13). Metwally et al. (2021) synthesized and characterized pyrazolo[1,5-a]pyrimidine derivatives as anticancer compounds against CDK2 enzyme. They conducted in vitro studies using three human cancer cell lines (HepG-2, HeLa, and MCF-7). Among them, the most potent compounds were selected to assess their activity against the CDK2 enzyme, and their outcome was compared with the reference compounds, roscovitine, and dinaciclib. Out of five compounds 46, 47, and 48 (Fig. 13) showed the highest inhibitory activity against the CDK2 enzyme. Further, these compounds were docked within the binding site of the CDK2 enzyme and assessed via drug-likeness studies. From docking studies, they reported all the crucial interactions as observed in the reference compound. Compound 46 revealed three interactions with the enzyme, i.e., two hydrogen bonds and one arene-cation interaction with the binding energy of −14.6729 kcal/mol. In addition to that, basic arene-cation interaction occurs within the active site of the enzyme between the phenyl ring and Lys 89. Compound 47 formed two hydrogen bonds, between the CN of CH=CN and Lys 89, with a binding energy of −15.287 kcal/mol. Compound 48 had one hydrogen bond and two basic arene-cation interaction bonds with a binding energy of −12.486 kcal/mol. These theoretical molecular docking results agree well with the practical outcome. Finally, compiling all the results of in vitro and in silico studies, they concluded that 48 is a suitable candidate for anticancer discovery (Metwally et al., 2021). Fatahala and her group synthesized 2-thiouracil-5-sulfonamides derivatives as anticancer agents (Fatahala et al., 2021). Among the synthesized compounds, 49, 50–53, and 54 (Fig. 13) displayed significant anticancer activity against the CDK2 enzyme. Further analyses, like checking cell growth arrest at G1/S, S, and G2/M phase, 51 showed the highest inhibition of the CDK2. Finally, they conducted docking studies (MOE software) on 49, 50–53, and 54 and concluded that 51 displayed proper interaction within the binding pocket, which explains its highest potency against the CDK2 enzyme.The most important residues found in the CDK2-ATP binding pocket for 49 were Gly13, Lys33, Lys129, Gln131, those for 51 were Gly13, Lys33, Phe82, Gln131, for 52 were Gly13, Lys33, Asp86, Lys89, for 53 were Ile10, Lys33, Gln13, and those for 54 were Lys33, Gln131. Compound 51, the most promising anticancer agent, formed one hydrophobic bond with Gly 13 and three hydrogen bonds with Lys33, Lys89, and Asp86, whereas reference TPHP formed three hydrogen bonds with Lys33, Leu83, and Leu83. Compound 51 displayed the highest docking score of −5.48 kcal/mol, and a lower root-mean-square deviation (RMSD) of 1.05 Ǻ than the reference TPHP, which had a lower docking score (−4.52 kcal/mol) and a higher RMSD (1.86 Ǻ). The docking study explained medium to high activity of compound 51 against four cell lines over the remaining active compounds. Thus, molecular docking confirmed the final compound 51 as a potent CDK2 inhibitor (Fatahala et al., 2021). Goda et al. (2021) conducted in vivo analysis along with in vitro studies to check the anticancer (lung, A59 cell line) activity of Artemesia judaica L. It was observed that the 55 (Fig. 14) gives IC50 of 14.2 µg/mL compared to the reference doxorubicin. For in vivo studies, they selected the xenograft model and reference doxorubicin and found a reduction in the solid tumor mass with an inhibition ratio of 54%. When they conducted docking studies on the CDK2 enzyme, they observed that the binding affinity of 55 to the CDK2 enzyme explains the cytotoxicity activity obtained, which makes it a suitable cytotoxic compound against lung cancer. The most important interactions of 55 with CDK2 were Leu83 and Lys89, with binding energy in the range of −9.58 to −19.89 kcal/mol. Herein, the in silico docking analysis proved the cytotoxic activity of the detected metabolites that may contribute to the cytotoxic activity of the crude extract of Artemesia judaica L. (Goda et al., 2021).
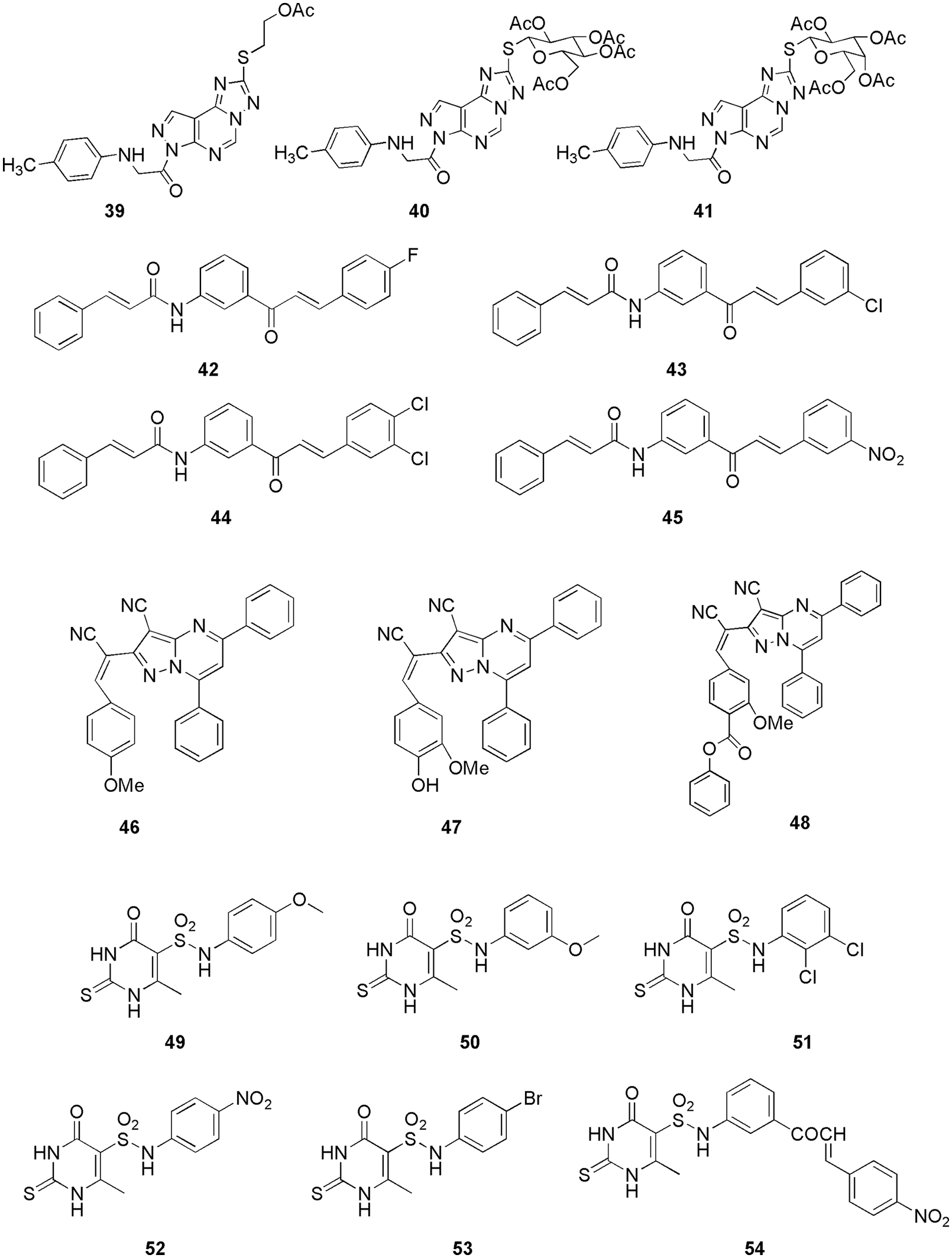
Figure 13: The chemical structures of pyrazolo-pyrimidine, pyrazolo-triazolo-pyrimidine compounds (39, 40, 41), cnnamamide-chalcone derivatives (42, 43, 44, 45), pyrazolo[1,5-a]pyrimidine derivatives (46, 47, 48) and 2-thiouracil-5-sulfonamides derivatives (49–54) as the potent CDK2 inhibitors.
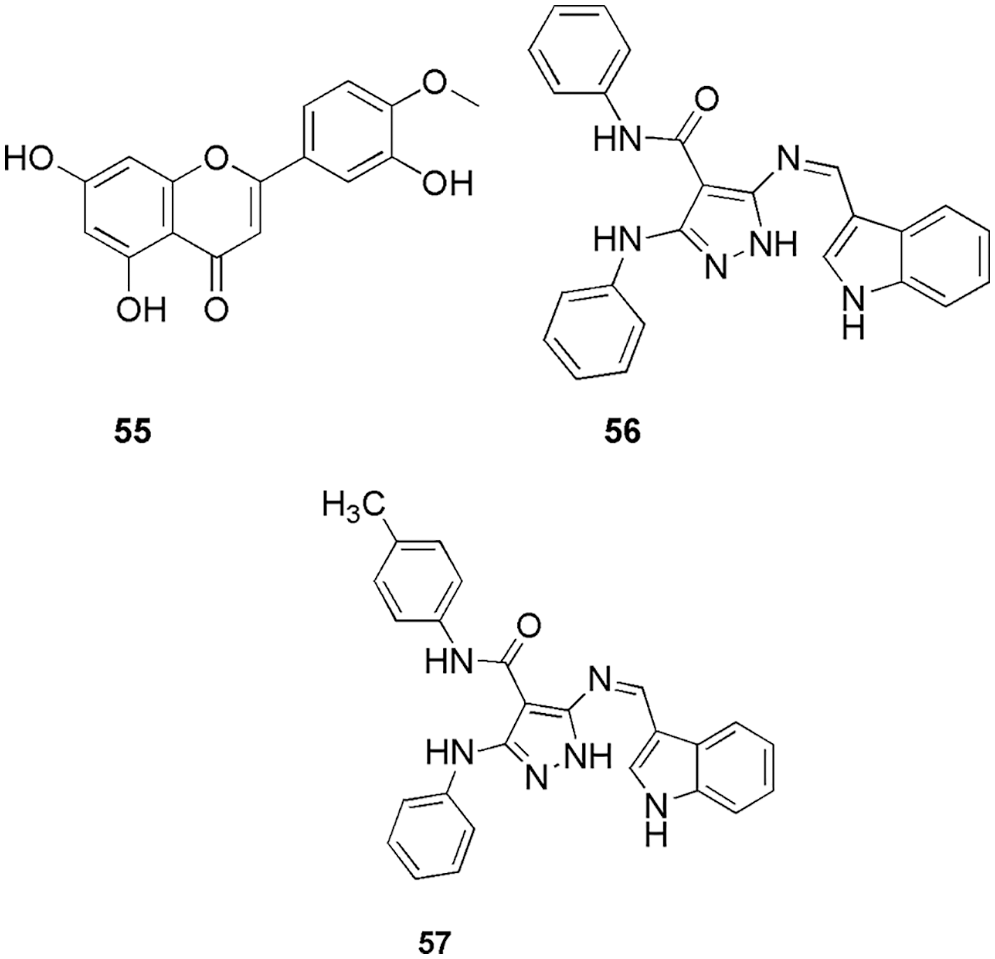
Figure 14: The chemical structures of Artemesia judaica L (55) and pyrazole-indole hybrids (56, 57) displaying potent CDK2 inhibition.
Hassan et al. (2021b) synthesized novel pyrazole-indole hybrids and checked their anticancer activities against MCF-7, HCT-116, A549, and HepG2 cell lines. Out of all the synthesized compounds, 56 and 57 (Fig. 14) displayed the highest activity, which was further selected to explore their mechanism in the cell cycle and apoptosis process. Further, the enzymatic assays were checked against caspase-3, CDK2, Bcl-2, and Bax protein. To study their (56 and 57) interaction pattern within the protein CDK2, they conducted molecular docking studies (MOE) and found that the presence of indole and pyrazole moiety in the molecule is responsible for fitting well in the active site and displaying the interactions. Higher negative energy scores of −13.68 and −12.55 kcal/mol for both inhibitors indicate higher predicted binding affinity than the reference ligand. The docked derivatives 56 and 57 were found to fit within the active site of the enzyme by forming interactions with Lys89 via two arene-cation interactions with indole centroids and hydrogen bonding with the pyrazole moiety. It was also discovered that the pyrazole scaffold of 56 supported the binding via hydrogen bond donor with the side chain of Lys89. Finally, they predicted that the two compounds (56 and 57), which contain indole and pyrazole moieties, would bind to CDK-2 via different interactions with the key amino acid Lys89. Furthermore, the obtained binding pattern investigated the superior CDK2 inhibitory activity of these compounds over the co-crystalized inhibitor (Roscovitine) (Hassan et al., 2021b).
Gosh and his team used in silico studies to retrieve alkyl and aryl-substituted isatin-triazole ligands as potential inhibitors of CDK2 and EGFR enzyme (Ghosh et al., 2021). First, they performed the density functional theory studies on the ligand, then selected them for docking studies on the two enzymes, and concluded with simulations studies. Finally, based on the outcome, they selected two ligands, 58 and 59 (Fig. 15), as the ones that showed the most preferred binding with the two proteins (Ghosh et al., 2021). The most important residues involved in the interaction of CDK2 for 58 and 59 were Leu83, Phe82, Ile10, Lys33, Leu134, Asp145, Asn132, Gln131, Lys129, Gly13, Thr14, Glu162, and Val163. They displayed the binding energies of −8.90 and −8.60 kcal/mol for 58 and 59, respectively (Ghosh et al., 2021).
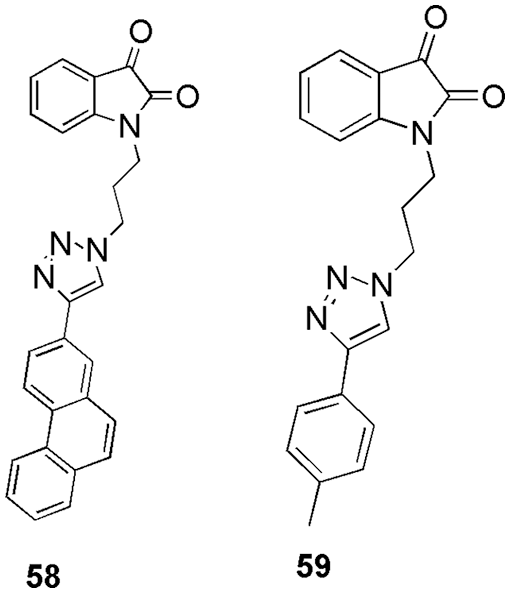
Figure 15: The chemical structures of aryl (58) and alkyl (59) substituted isatin-triazole ligands as efficient CDK2 inhibitors.
Ismail et al. (2021) synthesized new arylazo-pyrazoles derivatives and used the MTT technique to examine their antiproliferative activity. They observed that two compounds with IC50 of 3.0 and 4.0 µM displayed higher cytotoxicity compared with the reference standard Imatinib (IC50 of 7.0 µM). Further, cytometric analysis was carried out for biological assessment, and compound 60 (Fig. 16) exhibited 62.5% inhibition of CDK2 in comparison to imatinib. They confirm the affinity of 60 to the CDK2 via molecular docking and pharmacophore modeling (Discovery Studio 2.5). Docking studies revealed that compound 60 had a higher binding affinity in the active site of CDK2 with a binding energy of −5.6 kcal/mol than the reference ligand, which has a binding energy of −4.4 kcal/mol. The interaction plot revealed that 60, which contains a pyrazole scaffold, forms three hydrogen bonds with the Leu83 and Glu81 residues of the active site of CDK2., The aryl moiety attached to the pyrazole ring at position 5 forms hydrophobic interactions with the side chains of Ile10, Lys 89, and Gln131 residues. The acetophenone moiety formed more hydrophobic interactions with the amino acids Asp145 and Leu134. After docking studies, they performed ADME analysis by SwissADME, Molsoft, Pre-ADME websites, and DataWarrior software, which also revealed that compound 60 is orally bioavailable (Ismail et al., 2021).
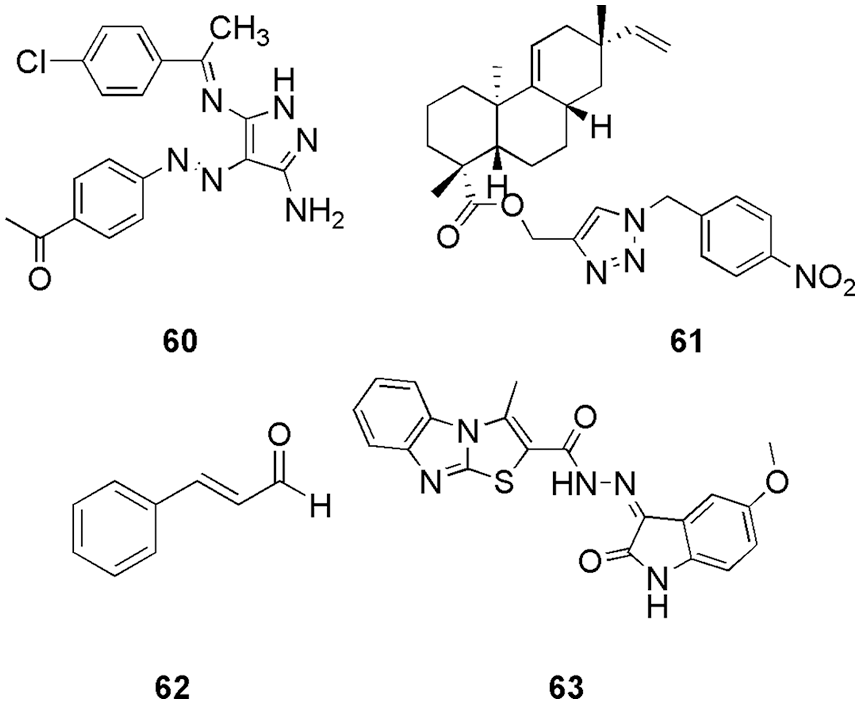
Figure 16: The chemical structures of arylazo-pyrazoles derivative (60), acanthoic acid analog (61), cinnamaldehyde (62), hybrid of isatin-thiazolo-benzimidazole (63) as the potent CDK2 inhibitor.
Kasemsuk et al. (2021) synthesized thirteen sets of acanthoic acid analogs containing triazole moiety and studied their cytotoxicity. Among the synthesized, compound 61 (Fig. 16) exhibited the strongest biological activity against four cell lines of cholangiocarcinoma cells with an IC50 of 18 µM. Molecular docking (AutoDock 4.2) studies suggest a favorable binding energy, which further suggests 61 as a potent CDK2 inhibitor. Interestingly, 61 had the lowest binding energy of −12.7 kcal/mol with CDK2 and demonstrated the formation of three hydrogen bonds of the nitro group with Gln85, Aps86, and Lys89 residues. Furthermore, the triazole and aromatic rings interacted with Ala31, Leu83, Leu134, Ile10, and Phe80 residues via-alkyl interactions, whereas the diterpene ring interacted with Val18, Ala31, Lys33, Ala144, and Leu134 residues via hydrophobic interactions. The triazole ring and the nitro group of compound 61 formed a strong hydrogen bond, while the diterpene ring formed a good hydrophobic interaction, leading to a better understanding of the cytotoxicity against CCA cell lines provided by these interactions. Compound 61 61 (Fig. 16) showed the most potent activity among 13 synthesized compounds and had maximum binding energy of −12.7 kcal/mol. Thus, in vitro and in silico both had similar results (Kasemsuk et al., 2021). Hermawan et al. (2021) detected the targets and mechanisms of 62 (cinnamaldehyde) in overcoming fulvestrant-resistant breast cancer using a computational approach. They used molecular docking analysis and observed that 62 could bind to six potential protein targets, including CDK2, which makes this compound an anticancer. Cinnamaldehyde (62) had a higher docking score than the selected reference of CDK2, i.e., AZD5438 and Erα. They form hydrogen bonding with Lys33 or Arg394 with a binding energy of −12.01 kcal/mol. Compound 62 had an additional hydrogen bond with Thr74 due to its higher docking score than the reference ligand (Hermawan et al., 2021). Eldehna et al. (2021b) proposed that a hybrid of isatin-thiazolo-benzimidazole can be used as a potent CDK2 inhibitor. They conducted the biological evaluation and in silico studies like ADME prediction (SwissADME) followed by docking (Autodock Vina), MD simulation (GROMACS 2020.3), and MM-PBSA calculations. Three compounds displayed potent dual activity against cell lines MDA-MB-231 and MCF-7. By docking analysis, one potent compound 63 (Fig. 16) was selected, which formed the most stable complex with CDK2 enzyme with a binding score of −11.2 kcal/mol. Particularly, the substitution of the oxindole moiety in hybrid 63 with a 5-methoxy group resulted in an extra hydrogen bond interaction with Thr14, which explained its superior in vitro CDK2 inhibitory activity (IC50 = 26.24 nM) over hybrid other two compounds (IC50 = 42.95 and 96.46 nM). Thus, with the help of molecular docking, they were able to select the most potential candidate for inhibition of CDK2 (Eldehna et al., 2021b). Mansour et al. (2021a) synthesized E/Z-diastereomers of a series of 3-benzylideneindolin-2-ones and 5-nitro-3-benzylidene-indolin-2-ones. With the help of in silico docking studies (MOE), they confirmed better binding affinity of synthesized Z-diastereomer (64–71) (Fig. 17) with CDK2 in comparison to the reference Sunitinib. Among them, important residues for 64 were Lys89, and Ile20 with a binding energy of −7.89 kcal/mol (Mansour et al., 2021a). Rouchal et al. (2021). revealed that the compounds with 2,6,9-trisubstituted purines scaffold (72) (Fig. 17) can act as good anticancer drugs. However, on the downside, they have low solubility in water, which can be improved by appropriate derivatization of the compounds with the adamantane moiety (Rouchal et al., 2021). They also performed molecular docking (AutoDock Vina) and in vitro analysis and found that purine derivatives with adamantane cages can easily bind inside the CDK2 to form complexes. They performed a docking study to clarify the inactivity of previously published adamantylated purines. Docking studies revealed that the inactivity was not related to the presence of the bulky adamantane moiety, which was believed to prevent the entrance of the inhibitor to the active site (Rouchal et al., 2021). The most promising residues for purine core were Ile11, Leu84, His85, Asp87, and Ls90.
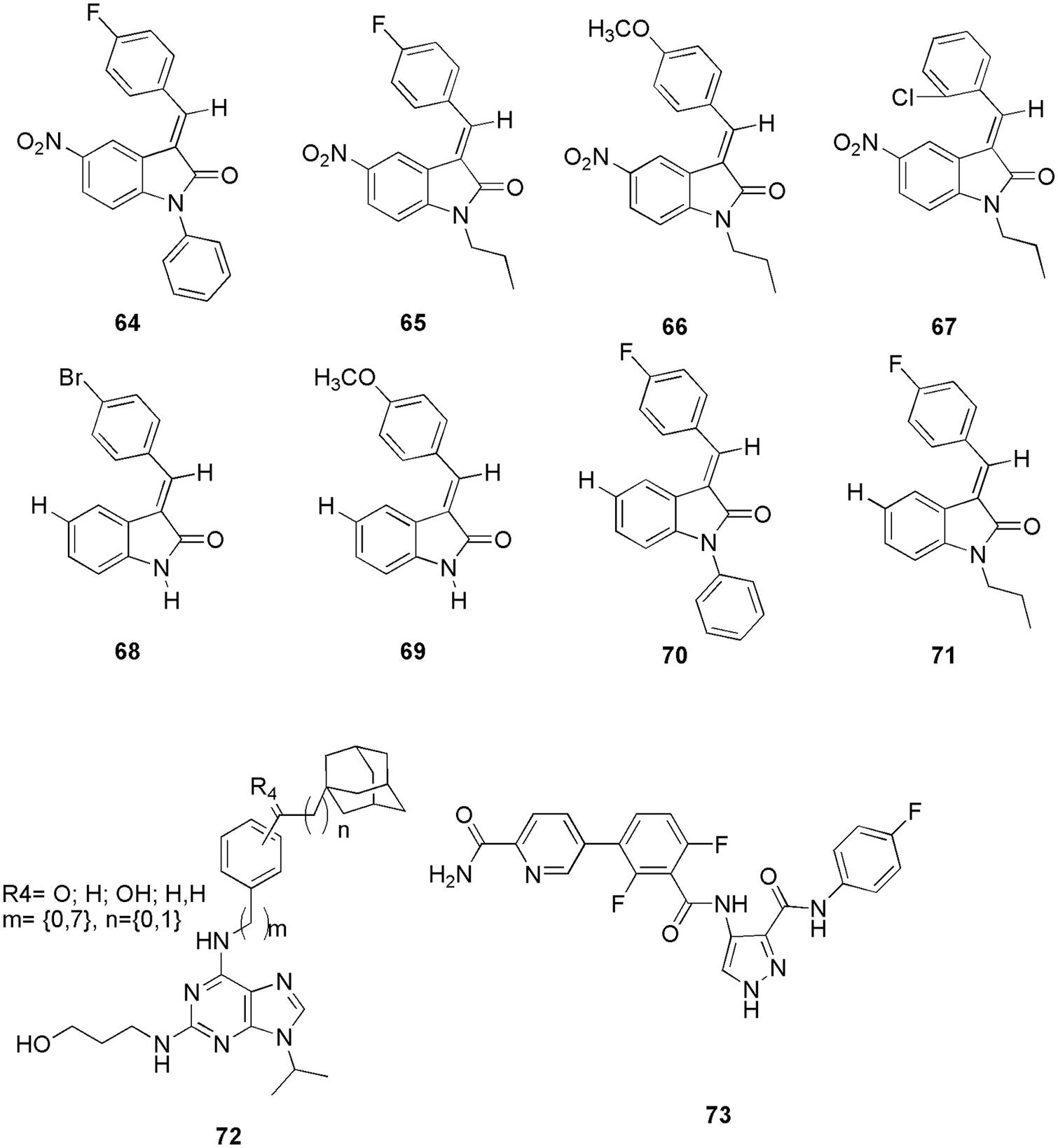
Figure 17: The chemical structures of 3-benzylideneindolin-2-ones, 5-nitro-3-benzylidene-indolin-2-ones-diastereomers (64–71), 2,6,9-trisubstituted purines scaffold (72) and 4-benzoyl-amino-1H-pyrazole-3-carboxamide derivative (73) showing most promising results for CDK2 inhibition.
Lin et al. (2021) synthesized a series of 4-benzoyl-amino-1H-pyrazole-3-carboxamide derivatives, and checked their anticancer activities. They conducted kinase assays, cell cultures, viability assay, western blot, flow cytometric analysis, siRNA transfection, and molecular docking (Glide, Schrodinger). Compound 73 (Fig. 17) exhibited effective activity against CDK2 with an IC50 value of 58 nM. Molecular docking illustrated that 73 had a similar binding mode with CDK2 as the reference C-73, the most selective inhibitor reported so far for CDK2. Compound 73 had a U-type conformation similar to C-73 and overlapped with it in the same region. In the hinge region of CDK2, the pyrazole ring formed three hydrogen bonds with Glu81 and Leu83. The 4-(6-carbamoyl-3-pyridyl)-2,6-difluorophenyl moiety of 73, mimics the position of the C-73 biphenyl. The selectivity of 73 may be explained by the similar binding mode of C-73 (Lin et al., 2021). Hassan et al. (2021a) synthesized a novel set of diphenyl-1H-pyrazoles and pyrazolo-pyridines and evaluated their antiproliferative activity. The IC50 was observed to be 1.29 and 0.93 µM for compounds 74 and 75, respectively, against MCF7, and the IC50 of 1.57 and 1.33 µM for compounds 76 and 77 against the HepG2 cell line. Further, molecular docking (MOE), 2D-QSAR studies, and ADMET study (SwissADME) were performed, and finally, three compounds (74, 76, and 77) (Fig. 18) were selected out of four that showed efficient anti-proliferative activity as potential CDK inhibitors. The interactions of 74 (−15.97 kcal/mol) showed hydrogen bonding interaction with Ile10, Gly13, and Leu83. Compound 75 (Fig. 18) (−15.07 kcal/mol) showed hydrogen bonding with essential residue Leu83. Other than hydrogen bonding, they also observed hydrophobic interactions. Compound 77 (−13.53 kcal/mol) exhibited two hydrogen bonding contacts with Leu83 via the CN group and aromatic C atom, as well as another with Lys89 via the pyrazole N atom. They reported that the involvement of the CN group indicates its implications for ligand-target interactions. Additional hydrophobic interactions were observed with Val18, Phe80, Lys89, and Gln131, residues (Hassan et al., 2021a). Sharma and her group screened out schinilenol (78) (Fig. 18) as a potential candidate from 5284 compounds of Taiwan indigenous plants database for CDK2 inbitiors (Sharma et al., 2021). For this, they performed in silico investigation along with pharmacophore modeling, virtual screening, mapping, MD simulations, and metabolic site reactivity analysis (molecular docking) (Sharma et al., 2021). They filtered the database of 5282 compounds and retrieved ten hits with the help of molecular docking. Among these ten hits, 78 showed the best results with a binding energy of −8.1 kcal/mol. Compound 78 is a coumarin that belongs to the benzopyrones and lactones families and has an alpha-pyrone ring combined with a benzene ring. These classes of compounds possess increased bioavailability, effective pharmacological features, reduced drug resilience and toxicity, and improved therapeutic effects on a variety of ailments. Docking results displayed the essential amino acid residues for 78 were Thr14, Gln31, Lys33, Asp86, Lys129, and Asn132. Thus, molecular docking was utilized to screen out the molecules and suggested a favorable CDK2 lead phyto inhibitor for further in vitro and in vivo studies. Many groups have worked on CDK2 inhibition with the help of molecular docking. For instance, (Mansour et al., 2021b, Elmorsy et al., 2022) reported novel pyridine-3-carbonitrile derivatives as anticancer compounds against the CDK2 enzyme (79–82) (Fig. 19), Eldehna and his group studied three series of oxindole-benzofuran hybrids (83–85) (Eldehna et al., 2021a) (Fig. 19), Al-Sanea et al. (2021) studied 3-hydrazonoindolin-2-one scaffold (86) (Fig. 19), Shawky et al. (2021) synthesized two new series of pyrrolizines bearing 3,4,5-tri-methoxyphenyl moiety (87–89) (Fig. 19). Roy et al. (2021) synthesized flavonol-based analogs of fisetin as anticancer agents (90, 91, 92) (Fig. 19). Utilizing inverse-docking, in vitro kinase activity, and various cell-based anticancer screening assays, new potential inhibitors of receptor tyrosine kinases (c-KITs), CDK2 and mTOR, were discovered. These proteins were known as attractive therapeutic targets for the treatment of melanoma and non-melanoma skin cancers. Eleven compounds, with IC50 values ranging from 0.12 to <15 µM, showed substantial inhibitory activity against four human skin cancer cell lines, including melanoma (A375 and SK-Mel-28) and NMSCs (A431 and UWBCC1). Inverse-docking (Glide) and screening against twelve known cancer targets, followed by kinase activity profiling, led to the identification of seven compounds as potentially effective single, dual or multi-kinase c-KITs, CDK2 and mTOR kinase inhibitors. Inverse docking results showed that the carbonyl on the flavone core moiety interacts as a hydrogen bond acceptor with the Leu83 backbone, and a majority of the compounds bind with the catalytic domain in the same pose as Fisetin (the reference compound). Additionally, the 2-phenyl group makes π-π interactions with Phe80 and Glu81 amino acids.
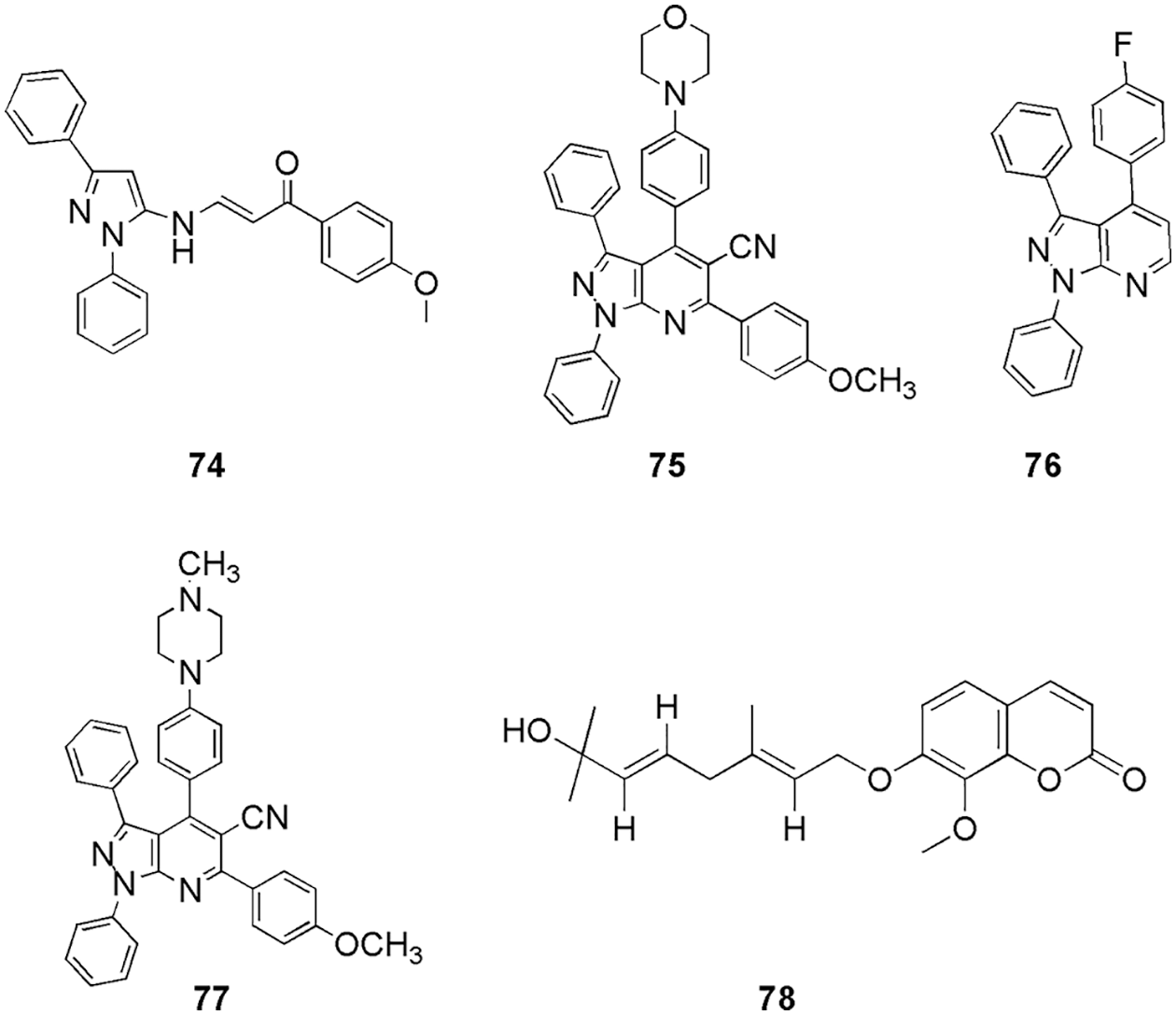
Figure 18: The chemical structures of diphenyl-1H-pyrazoles, pyrazolo-pyridines derivatives (74–77) showing excellent antiproliferative activity against CDK2 enzyme and schinileno (78) displaying best binding energy for CDK2 inhibition.
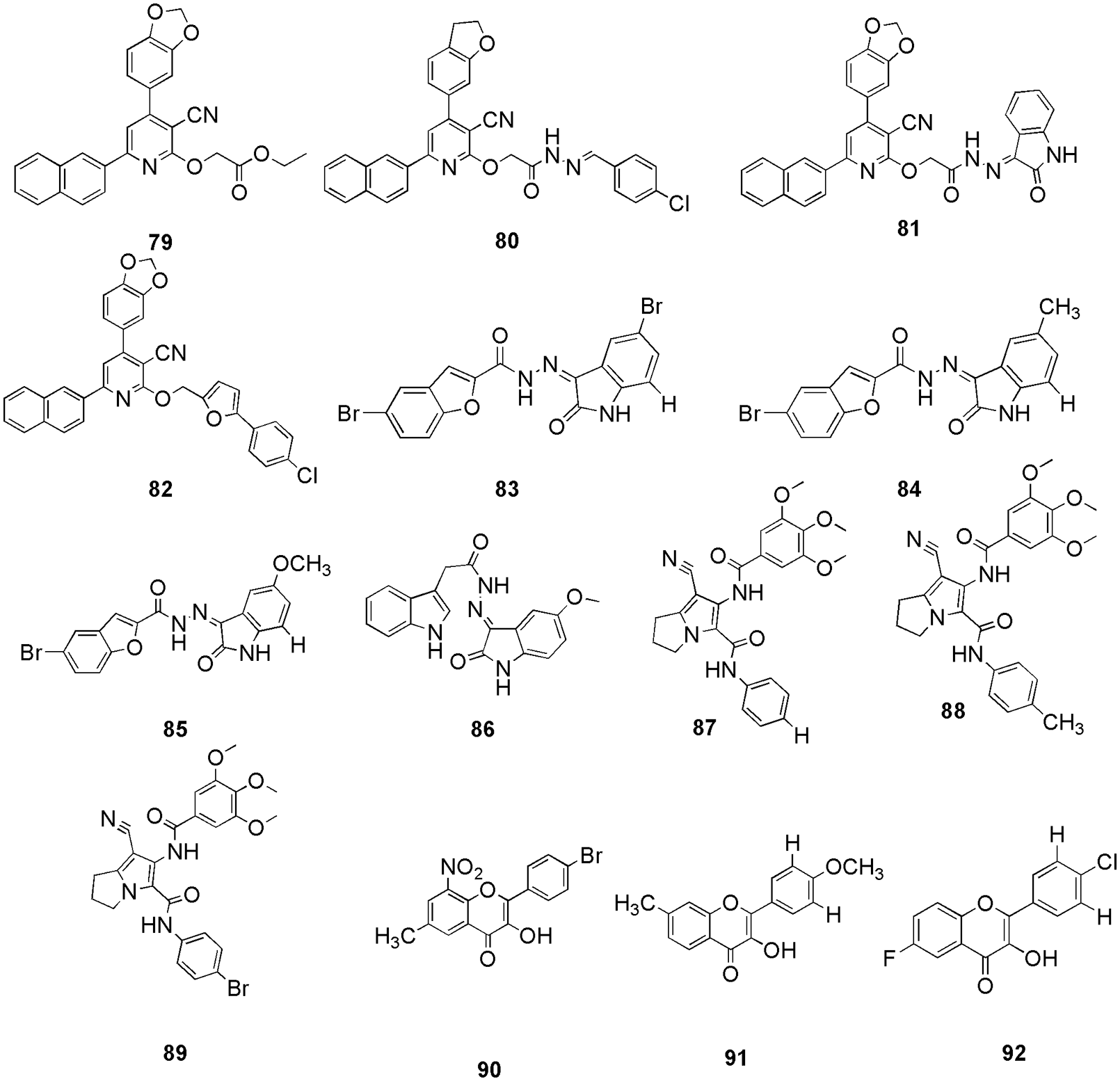
Figure 19: The chemical structures of pyridine-3-carbonitrile derivatives (79–82), oxindole-benzofuran hybrids (83–85), 3-hydrazonoindolin-2-one scaffold (86), pyrrolizines bearing 3,4,5-tri-methoxyphenyl moiety (87–89), and flavonol-based analogs (90–92) as the potent CDK2 inhibitor.
Overall, the computational evaluation showed that analogs with a single, small substituent were able to maintain poses similar to those of Fisetin. However, similar binding poses with analogs bearing multiple or bulky substituents on the ring were reported to display different poses. Important residues for compound 90 (−8.39 kcal/mol) were Leu83 and Asp145; for compound 91 (−9.14 kcal/mol) were Glu81, Leu83, and Asp145; for compound 92 (−8.81 kcal/mol) were Glu81 and Leu83.
Cyclin-dependent kinase 4 (CDK4)
CDK4 is known to control the G1-S phase of the cell cycle primarily by inactivating the tumor-suppressive protein retinoblastoma in cancer cells, thus validating it as an anti-tumor target. Qayoom et al. (2022) targeted the CDK4 enzyme using analogs of palbociclib by utilizing molecular docking (AutoDock 4.2.6) and molecular dynamic (MD) simulation (Schrodinger). They retrieved 100 compounds from the PubChem database and evaluated their drug-likeness via Lipinski’s rule of 5 and SwissADME. After screening, the selected 87 compounds were subjected to molecular docking; further, two compounds, 93 and 94 (Fig. 20), which exhibited the highest binding affinity, were considered for MD simulation studies. MD results revealed significant stable patterns during the entire simulation run. As per molecular docking results, compound 94 showed the lowest binding energy of −9.6 kcal/mol and was strongly bound with CDK4. According to the molecular surface view, the binding cavity of Thr117 residue of CDK4 participated in the formation of a hydrogen bond with the ligand 94. In addition to hydrogen bonding residues, alkyl interactions with Arg72, Lys112, and Ile56 and π-alyl interactions with Pro118 were also observed as hydrophobic contacts. With a binding energy of −9.3 kcal/mol, 93 demonstrated noticeably higher CDK4 protein binding affinity, indicating a strong binding affinity for human CDK4. At the binding cavity of the protein, Glu70 and Lys123 residues form hydrogen bonds with ligand 93 and apart from that it also showed some van der Waals interactions. Here, in silico methods were used to screen out two potent compounds from 100 compounds (Qayoom et al., 2022). Reddy et al. (2022) proposed that the methyl β-orsellinate conjugates of 1,2,3-triazole hybrids could be effective anticancer leads in breast cancer therapeutics. They synthesized and characterized methyl β-orsellinate-based 1,2,3-triazole hybrids. SwissADME was employed to calculate the ADME and pharmacokinetic properties of the synthesized compound, and further, the docking studies were done by Autodock Vina. Docking studies also suggested significant interaction at the ATP binding site of the CDK4, which indicates that it might act as a potent competitive inhibitor of ATP. Their studies suggest that methyl β-orsellinate-based 1,2,3-triazole hybrids 95 to 105 (Fig. 21) can be promising leads as CdK4/Cyclin D1 inhibitors. Docking results further showed that 101 attaches to the ATP-binding pocket of CDK4 and interacts with Glu144 residue with the binding energy of −7.7 kJ/mol. Other than Glu144, compounds 95 to 105 showed interactions with essential amino acid residues, i.e., Ile12, Val20, Ala33, Phe93, Glu144, Leu147, and Ala157. They reported that the methyl beta orsellinate ring of 101 connected with Lys142 and Glu144, and the interaction with Ala33 stabilized the oxygen of the substituted R group -OCH3. Further, they conducted the cell cytotoxicity and proliferation assay and found that among all the compounds, 101 showed the highest proliferation activity, and the same was confirmed by docking. Also, compound 101 displayed a binding energy of −7.7 KJ/mol, which was reported to be higher in comparison to the reference flavopiridol (−6.31 kJ/mol). Thus, based on the outcome, they predict that 101 can act as a potent ATP competitive inhibitor (Reddy et al., 2022). Ashraf et al. (2022) identified mangiferin (106) (Fig. 22), a xanthonoid extracted from the bark and leaves of the mango tree, as a potential anticancer drug that targets CDK4. They perform molecular docking (AutoDock Vina) and MD simulations (AMBER16) along with molecular mechanics generalized-Born and surface area (MM-GBSA) calculations to validate the affinity between 106 and CDK4. The CDK4-106 complex shows four hydrogen-bond interactions between the ligand and the receptor pocket residues. They formed interaction via Val14, Val93, and Glu141 and showed binding energy of −7.8 kcal/mol. The positively charged Glu141 maintained the positive charge contact with the Lys139 residue. The complex was further stabilized by the hydrophobic interactions between the Gly13 and Val14 pair and the residues Gln95, Ala33, Leu144, His92, Ile12, and Val20. Gurushankar et al. (2021) identified baimantuoluoamide A and baimantuoluoamide B (107) (Fig. 22) from six alkaloid compounds based on the geometric mean value using the ChemoSophia software for their high activity probability of CDK4 inhibition.
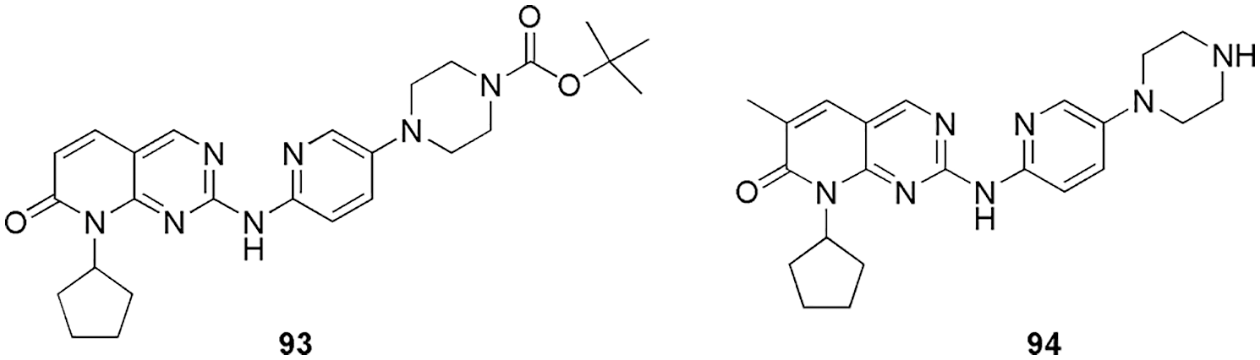
Figure 20: The chemical structures of analogs of Palbociclib which were selected after screening based on docking studies for CDK4.
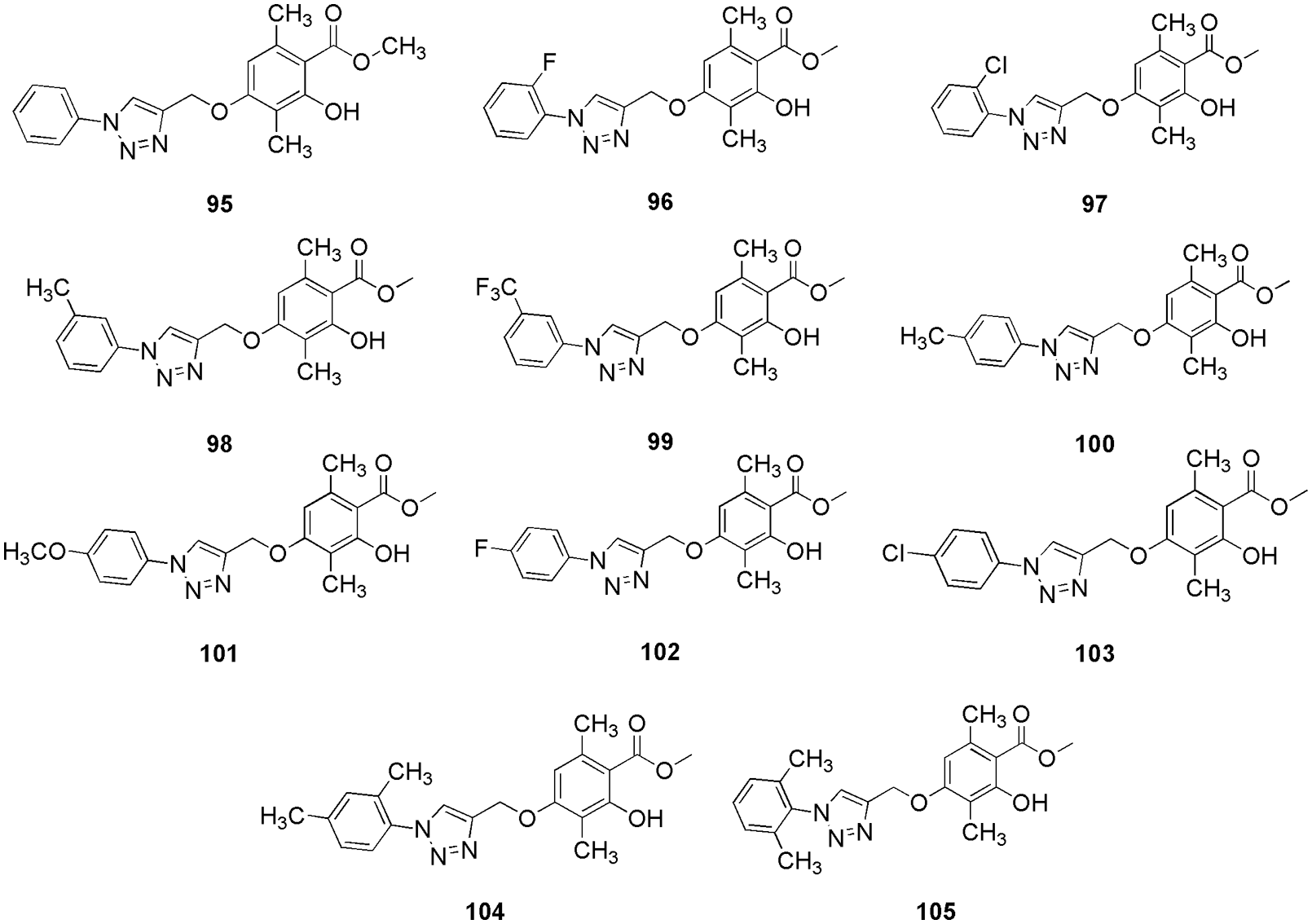
Figure 21: The chemical structures of methyl β-orsellinate-based 1,2,3-triazole hybrids which act as potent CDK4 inhibitor.
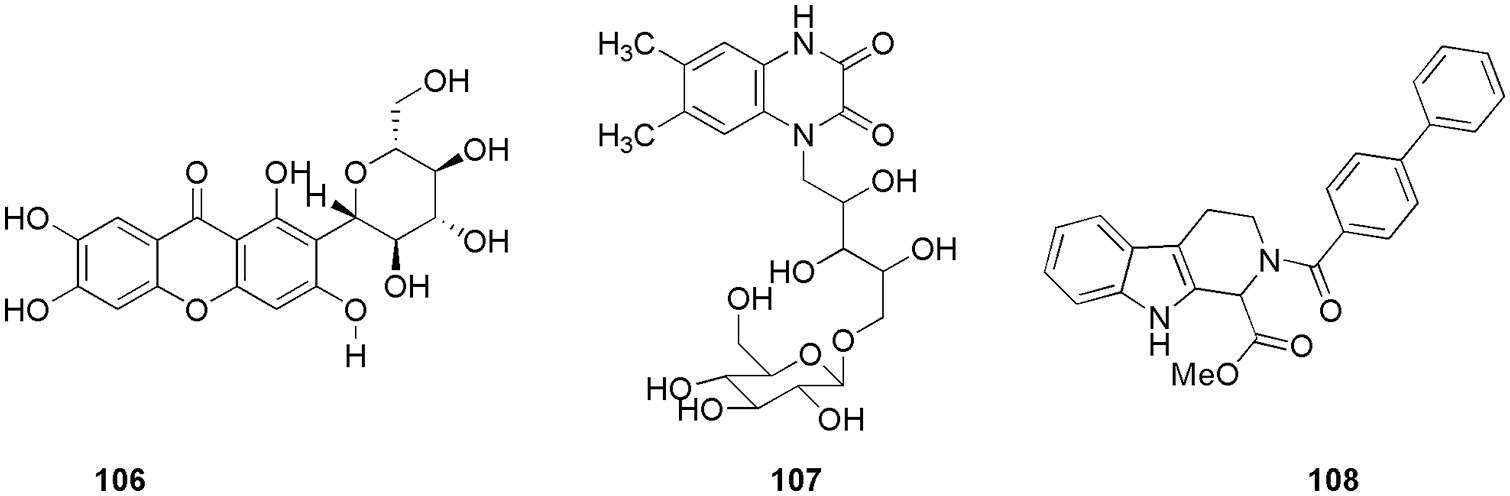
Figure 22: The chemical structures of most potent CDK4 inhibitor.
These two alkaloids were further subjected to molecular docking (AutoDock Vina), MD simulation (AMBER16), and MM-PBSA calculations (AMBER16). Based on the results of the in silico methods, 107 was found to be the hit molecule for CDK4 inhibition due to more negative binding energy, van der Waals and electrostatic interactions, and higher stability. The docking results for baimantuoluoamide A and B demonstrated that both ligands can bind in two distinct poses that are rotated by around 180 degrees to one another. The binding energies of the two positions for baimantuoluoamides A and B were −6.8 and −6.4 kcal/mol, respectively. They could retrieve similar binding energy and owing to that, they used both poses as the starting poses for conducting the MD studies. Compound 107 interacted with Val14, Val96, Leu147, His95, Val96, Gln98, Glu144 amino acid residues via hydrogen bonding. Liang et al. (2021) designed, synthesized, and characterized eighteen non-planar tetrahydro-β-carbolineanalogs based on the molecular skeleton of fascaplysin. Docking studies revealed that the spatial orientation of compound 108 (Fig. 22) at the binding pocket was quite similar to that of fascaplysin. Thus, they depict that the posed resemblance could be the primary factor for higher inhibitory effects than the other two produced compounds. The active pocket of fascaplysin contains amino acids His89, Val90, Asp91, Gln92, Ile9, Val17, Ala30, Val66, Phe87, Glu88, Asp93, and Leu141. Similarly, the binding site of compound 108 contained 14 residues, like, Val17, Ala30, Val66, Phe87, Ile9, Gly10, Val90, Asp91, Gln92, Asp93, Thr96, Glu138, Leu141, and Asp152. Ten residues that made up the binding site for compound 108 and fascaplysin were compared, and they observed the consistent binding site, which includes Ile9, Val17, Ala30, Val66, Phe87, Val90, Asp91, Gln92, Asp93, and Leu141. Further MTT assay indicated that compound 108 was a potential CDK4 inhibitor with IC50 of 1.03 ± 0.19 μM. The same outcome was observed in docking studies (Glide, Schrodinger) and MD simulation (AMBER16) (Liang et al., 2021). Li et al. (2021) synthesized and characterized a set of fluorine-substituted pyrrolo[2,3-d]-pyrimidine derivatives. These pyrimidine derivatives were tested for cytotoxic activity, and one of the compounds 109 (Fig. 23) showed superior activity on all the cancer cell lines and was further selected for in vitro and in vivo studies that showed improved preclinical in vitro and in vivo efficacy with the IC50 of 2.5 nM. Molecular docking, western blotting, and statistical analysis further confirmed compound 109 as a potent CDK4 inhibitor. They observed that in docking studies, Val96 and His95 were the backbone residues that showed hydrogen bonding interaction with the aminopyridine moiety of compound 109. They also revealed that the placement of carbamoyl substituents in the right places resulted in hydrogen bonding contact with the Arg101 residue. This deeper and closer binding of compound 109 to CDK4 resulted in a higher binding affinity for CDK4 than the reference ribociclib. From the docking outcome, they predicted that structural modification can alter the protein-ligand affinity (Li et al., 2021). Musa et al. (2021) isolated and characterized five flavonoids, i.e., luteolin, acacetin-7-O-β-D-glucoside, diosmin, kaempferol-3-O-rutinoside, and rutin from Bassia eriophora for their antiproliferative, and kinase inhibitory effects.
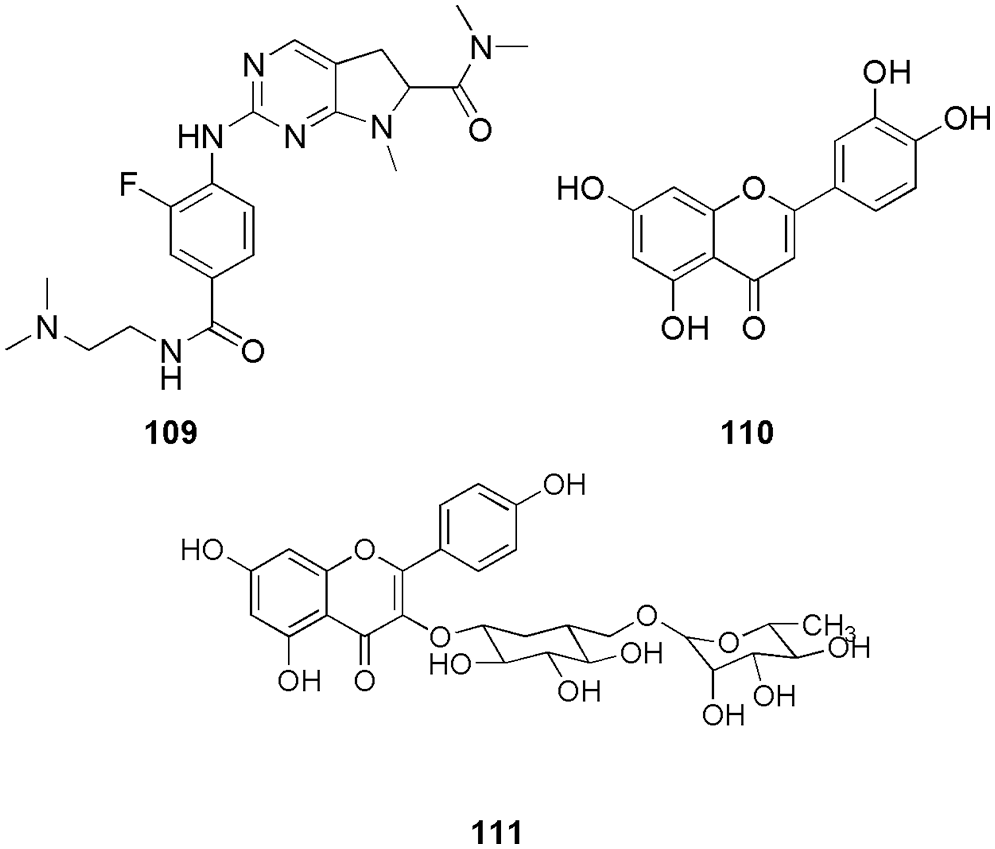
Figure 23: The chemical structures of fluorine-substituted pyrrolo[2,3-d]-pyrimidine derivative (109), Luteolin (110) and kaempferol-3-O-rutinoside (111) as the potent CDK4 inhibitor.
Among them, luteolin exhibited a strong antiproliferative effect against certain cell lines, including MCF-7, HCT-116, and HepG2, with IC50 of 33.24 ± 0.83, 31.62 ± 1.32, and 26.54 ± 1.02 μM, respectively, whereas, diosmin and kaempferol-3-O-rutinoside showed highest antiproliferative activity against MCF-7 with IC50 of 26.56 ± 1.12 and 28.72 ± 0.98 μM. These compounds were further assessed by molecular docking (MOE) with Aurora B and CDK4, and among them, 110 (Luteolin) and 111 (kaempferol-3-O-rutinoside) (Fig. 23) displayed a high affinity to CDK4. With scoring energies of −19.3755 and −23.9667 kcal/mol, 110 and 111 showed a strong affinity for CDK4/CycD1, and their binding modes involved interactions with the residues Val96 and Ala16 in compound 110 and Val96, Lys35, Asp97, and Thr102 in compound 111. The putative antiproliferative mechanism of 110 and 111 on Aurora B and CDK4/CycD1 kinase was predicted by in silico study (Musa et al., 2021).
Cyclin-dependent kinase 6 (CDK6)
CDK6, also known as the central node, is the enzyme that controls the G1 phase of the cell cycle. Overexpression of CDK6 is responsible for causing many types of cancer, like colon, pancreatic, bladder, and oral cancer. Huang et al. (2022) synthesized and characterized a series of new N4-alkyl-N2-phenylpyrrolo[3,2-d]pyrimidine-2,4-diamine derivatives. Further, they used the sulforhodamine B method for the determination of in vitro anti-proliferative activity. Among all compounds, 112 was the most potent CDK6 inhibitor, which showed super antitumor activities against MDA-MB-468, MCF-7, and MKN-28 with IC50 of 2.09, 2.1, and 2.61 µM, respectively. Further, molecular docking (Discovery Studio 2.5) was performed on 112 to know the probable binding modes. They reported that similar to the reference Palbociclib, 112 (Fig. 24) binds to the ATP-binding pocket of CDK and forms hydrogen bonds with Val101 in the hinge region. They observed that the ribose-binding pocket of the natural ligand ATP was occupied by the cyclohexyl substituent of 112. The 4-methylpiperazin substitution was pointing out of the binding pocket toward the solvent region of the binding pocket. They reported that compound 112 only formed two hydrogen bonds with CDK6, which was less than Palbociclib. They predicted that this could be the reason behind the poorer CDK6 inhibitory activity of 112 in comparison to palbociclib (Huang et al., 2022). Baig et al. (2022) proposed that selonserin (113) (Fig. 24) can be used as a potent inhibitor of CDK6. To investigate the inhibitory potential and binding affinity of 113, they performed molecular docking (InstaDock), MD simulation (GROMACS), MM-PBSA calculations, and enzyme inhibition assay. They reported that 113 binds with CDK6 via active site residues Ala17, Ile19, Val27, Ala41, Lys43, Val77, Val101, Gln103, Ala162, Lue152, and Asp163. The binding score of compound 113 was observed to be −10.9 kcal/mol. They also conducted molecular dynamics simulations, and from dynamic analysis and free energy calculation, they suggested stable behaviour of the 113-CDK6 complex. The result of fluorescence binding studies and enzyme inhibition assay indicated an acceptable binding with an IC50 value of 9.8 µM. Thus, by in silico analysis, fluorescence studies, and enzyme inhibition assay, they represented molecule 113 as a promising candidate against cancer (Baig et al., 2022).
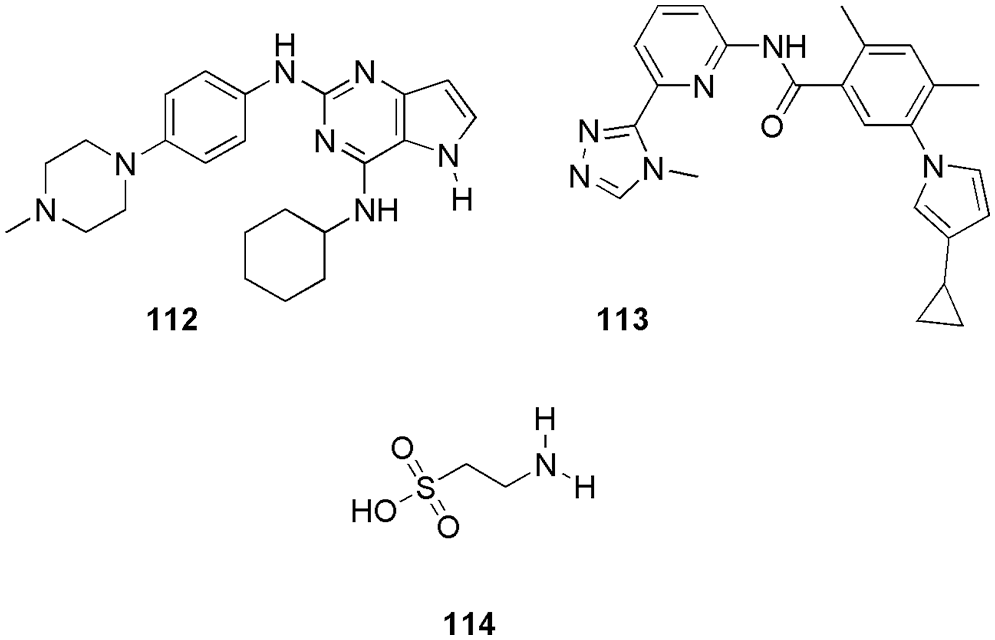
Figure 24: The chemical structures of new N4-alkyl-N2-phenylpyrrolo[3,2-d]pyrimidine-2,4-diamine derivative (112), selonserin (113), and taurine (114) as the potent CDK6 inhibitor.
Yousuf et al. (2022) identified taurine (114) (Fig. 24) as a potential CDK6 inhibitor using combined in silico and experimental studies. Molecular docking (AutoDock Vina) was performed to find out the bonding affinity between protein-ligand complexes. The significant binding of 114 with CDK6 showed a docking score of −3.3 kcal/mol. They observed that 114 occupied the CDK6 binding pocket and not the ATP-binding site. They noted that CDK6 residues Asp145, Asp163, and Leu166 form strong interactions with 114 via hydrogen bonding. In addition, 114 displayed Van der Waals interactions with Tyr24, Asn150, and Phe164, whereas His143 forms a carbon-hydrogen bond in the docked complex. The favorable interaction between 114 and CDK6 was considered the reason for showing excellent binding affinity. This result was further confirmed by fluorescence measurement studies for the CDK6-114 complex. Enzyme inhibition assay showed the IC50 value of 4.44 μM, and isothermal titration calorimetric analysis indicated the spontaneous binding of taurine with CDK6 (Yousuf et al., 2022). Lotlikar et al. (2021) synthesized and characterized 10 Schiff’s bases of 2,7-diamino-2′-oxospiro[chromene-4,3′-indoline]-3-carbonitrile. These ten derivatives were screened in vitro for antibacterial and anticancer activities. Out of ten, only eight molecules were selected for further in-silico docking analysis. Molecular docking (Biopredicta module of V life MDS 4.6) showed excellent binding between CDK6 and all eight derivatives. Among all eight docked molecules, they selected one compound, i.e., 115 (Fig. 25), based on the higher docking score (−14.21 kcal/mol) and presence of the crucial interactions. Compound 115 displayed hydrogen bond interactions via Asp104 and Gln149 residues, hydrophobic contacts with Gly20 and Lys29, and Van der Waals interactions with Ala17, Glu18, Ile19, Gly20, Glu21, Lys29, Thr107, Gln149, Asn150, Leu152, Ala162, and Asp163. Further ADME prediction (SwissADME) of these derivatives showed no Lipinski rule violation, which shows suitability for the oral route. Thus, in vitro studies screened out viable eight molecules out of ten synthesized molecules and molecule docking further selected the most efficient one out of that eight molecules as the potent anticancer molecule (Lotlikar et al., 2021). Roy and Bhatia (2021) proposed that plumbagin (116) (Fig. 25), a naphthoquinone derivative from plumbagozeylanica roots, can be used as an anticancer agent for the inhibition of CDK6. For that, they performed molecular docking (AutoDock 4.2), MD simulations, and ADME analysis (SwissADME). With the help of molecular docking, they retrieved the binding mechanism and molecular interactions of 116 against CDK6. The molecular docking score of 116 with CDK6 was found to be −6.18 kcal/mol. A reference inhibitor (4-(Pyrazol-4-yl)-pyrimidine) was utilized to compare docking scores (−6.48 kcal/mol). The docking score proved that 116 was quite similar to the reference CDK6 inhibitor. In the docked complex, 116 forms hydrogen bonds with Glu99 and Val101 and hydrophobic contacts with Ile19, Lys43, His100, and Asp163. Favorable interactions with each of these residues clearly show that 116 binds to ATP in the same location as the reference Palbociclib. Further, they conducted simulation studies, MD simulation showed stability between them, and ADMET analysis showed no Lipinski rule violation (Roy and Bhatia, 2021). Yousuf et al. (2021) identified vanillin (117) (Fig. 25) as a potent CDK6 inhibitor by implying combined computational and biochemical studies. The binding mechanism of CDK6 and 117 was studied by molecular docking (Glide, Schrodinger) and MD simulation (GROMACS), and MM-PBSA calculations. The docked complex of 117 formed two hydrogen bonds between the hydroxyl groups and side chains of Lys43 and Glu99, similar to the ATP binding position. The free energy of binding was around −4.53 kcal/mol, and the theoretical inhibition constant was 477.1 μM. The results showed the favorable binding of 117 to CDK6 and acceptable binding free energy. Further from enzyme inhibition and fluorescence binding, they obtained promising values of binding constant (K) of 4.1 × 107 M−1 and IC50 value of 4.99 μM (Yousuf et al., 2021). Luo et al. (2021) identified two novel CDK6 inhibitors 118 (Fig. 25) and 119 (Fig. 26) from the libraries ChemDiv and ChemBridge as CDK6 inhibitors via screening. The drug libraries contain 50,000 drug-like compounds, which were screened using pharmacophore-based virtual screening (LigPrep Schrodinger), molecular docking (Glide Schrödinger), ADME prediction (QikProp Schrodinger), in vitro evaluation, MM-GBSA-based free binding energy analysis (Prime VSGB 2.0), and MD simulation (Desmond Schrodinger) (Luo et al., 2021). Molecular docking revealed that compound 118 had essential interaction with residues Glu99 and Val101 and compound 119 showed interactions with Val101. Ates-Alagoz et al. (2021) synthesized sixteen indole-thiazolidinedione derivatives as anticancer compounds. Among the synthesized, three molecules were found to possess favorable cytotoxicity on the MCF-7 cell line with IC50 values of 8.52 and 14.60 µM for compounds 120 and 121 (Fig. 26), respectively. These three were further screened out by molecular docking studies. Compound 120 displayed a binding affinity of −8.8 kcal/mol, and it showed two hydrogen bonds with Tyr24 and two additional hydrophobic interactions, which stabilized the complex. In addition to that, it showed a hydrogen bond with Gly20. With a binding affinity of −8.6 kcal/mol, molecule 121 displayed hydrogen bonding with Gln149 and Lys147. In addition to that, it showed one hydrogen bond formation with Ile19 and Gly20 (Ates-Alagoz et al., 2021).
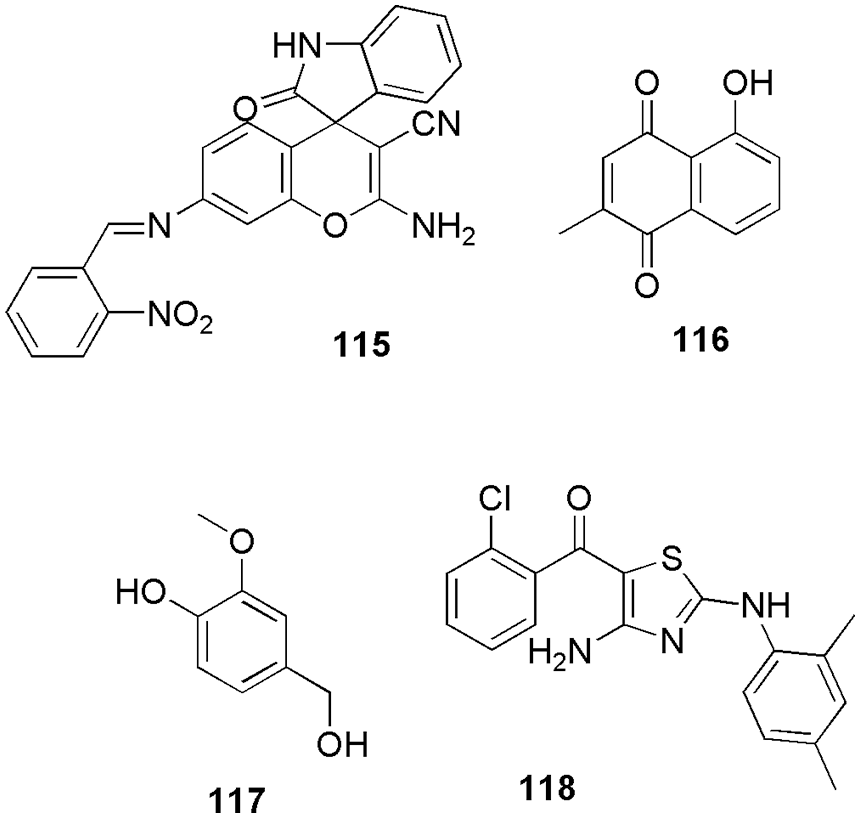
Figure 25: The chemical structures of the most potent CDK6 inhibitors.
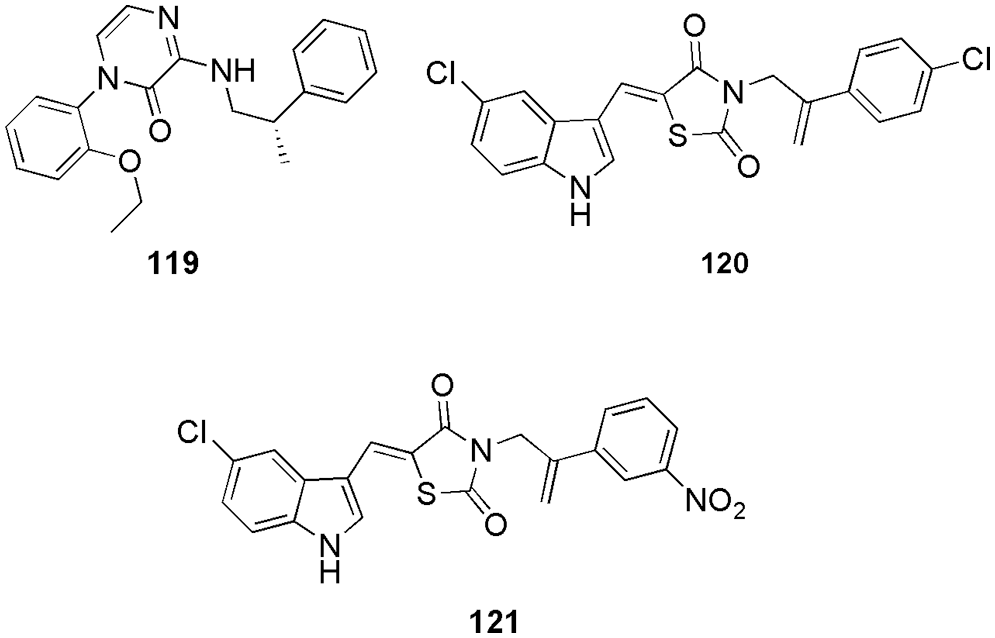
Figure 26: The chemical structures of the most potent CDK6 inhibitors.
Cancer has become a serious threat to human life. As per the cancer statistics, the number is increasing each year, thus, making it a grave problem in today’s world. Many groups are working in the direction of developing new drug molecules; however, the investigation of drugs at the molecular level and their interaction with enzymes is still unexplored, thus, making drug discovery a time taking and expensive process. Hence, the use of computational methods can give an edge to the research. Among various in silico tools, molecular docking is one of the popular tools which helps in the virtual screening, prediction of drug target, binding site, and binding pose of the molecule. Despite its drawback in the algorithm to treat protein backbone flexibility, the docking calculations are performed in combination with experimental and/or simulation studies. In recent years, both academic and professional contexts have used molecular docking extensively as a quick and affordable tool. There is still no easy and accurate way to quickly identify real ligands among a group of molecules or to precisely locate the ligand conformation inside the binding pocket of the target molecule. Despite this, new methods are always being created, and the number of publications is expanding quickly. Over the past years, significant progress has been made in the development of anti-proliferative drugs with CDKs as the prospective target. Some FDA-approved CDK4/6 and single isoform CDK9 inhibitors (third-generation) have shown selectivity in the inhibition of breast cancer-causing CDK enzyme. In the current review article, we observed that many researchers had implemented molecular docking calculations in finalizing the molecules as anticancer compounds. Also, the structural and functional properties of the recently developed CDK inhibitors have been summarized in this article. We observed that the inhibitors discussed in the current work can be categorized into groups based on the structure of their scaffolds. The group include pyrimidines, pyrazoles, purines, coumarins, flavonoids, indoles, quinazolines, and oxindoles as the common chemical moiety in the synthesized anticancer compounds. Few of the synthesized compounds like 1 (for CDK1), 26, 27, 28, 37, 40, 51, 73, and 75 (for CDK2), 108 (for CDK4), and 112, and 119 (for CDK6) displayed promising cancer inhibition and anti-proliferation activity. It is obvious from the structures of anticancer compounds that they contain polar groups like alcohols, amides, imine, amine, ester, and ether, along with the fused five to a six-membered aromatic ring containing nitrogen heterocycles. Moreover, along with these mentioned groups, some compounds contain pharmacophores of chloride, fluoride, bromide, and sulfur within the ring or linker or oxides. The substitution of the polar groups (amines, imines, esters, etc.) plays an important role in forming interaction with the receptor. Molecular docking studies established that these compounds form interactions in the ATP binding pocket of CDK enzymes. Inhibitors active against CDK1 enzyme display interactions with Asp86 amino acid, which is crucial for ATP binding. Apart from Asp86, the CDK1 inhibitors also showed hydrogen bonding interactions with the hydrophilic region (Glu12 to Tyr15), polar region (Asp128, Lys130, Gln132, and Asn133), and fragment region of CDK1 enzyme (Phe80 to Leu83). CDK2 displayed interaction via Leu83, Asp86 and Lys89. CDK4 form bonds with Val196, and CDK6 form a hydrogen bond with common Val101 and Glu99 residues. We believe that the current report will help the readers to understand the scaffolds implemented in anticancer drug discovery and the role of molecular docking in revealing their mechanism of action within the cell cycle CDK enzymes. We hope that the findings presented here will help readers to better understand the nature of inhibitor-target interactions and provide information on the structural and molecular requirements needed to achieve the necessary selectivity against CDKs over other types of kinases. We have also observed that CDK2 is a highly explored drug target when compared to other cell cycle enzymes. Moreover, several crystallized protein-ligands complexes reported in the RCSB database belong to the CDK2 enzyme only; thus, several in silico studies are reported on it. In comparison to CDK2, all other cell cycles CDKs are moderately explored, and very few crystallized complexes are reported, thus, limiting the in silico studies. To implement the computer-aided drug design, the issue of these scarcities must be resolved.
Acknowledgement: PS, NR and AM acknowledge the PDEU for infrastructure and facilities.
Funding Statement: This work was supported by the Teachers Associateship for Research Excellence (TARE) under Science and Engineering Research Board for Project Grant through Grant No. TAR/2021/000031.
Authors Contributions: Study conception and design: M. Anu; data collection: S. Priyanks, R. Rana; analysis and interpretation of results: M. Anu; S. Priyanks, R. Rana; draft manuscript preparation: M. Anu; S. Priyanks, R. Rana; supervison: AM and PCJ. All authors reviewed the results and approved the final version of the manuscript.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
Akki M, Reddy DS, Katagi KS, Kumar A, Devarajegowda HC, Babagond V, Mane S, Joshi SD (2022). Synthesis of coumarin-thioether conjugates as potential anti-tubercular agents: Their molecular docking and X-ray crystal studies. Journal of Molecular Structure 1266: 133452. https://doi.org/10.1016/j.molstruc.2022.133452 [Google Scholar] [CrossRef]
Al-Sanea MM, Obaidullah AJ, Shaker ME, Chilingaryan G, Alanazi MM, Alsaif NA, Alkahtani HM, Alsubaie SA, Abdelgawad MA (2021). A new CDK2 inhibitor with 3-hydrazonoindolin-2-one scaffold endowed with anti-breast cancer activity: Design, synthesis, biological evaluation, and in silico insights. Molecules 26: 412. https://doi.org/10.3390/molecules26020412 [Google Scholar] [PubMed] [CrossRef]
American Cancer Society (2022). Cancer facts & figures 2022. Atlanta: American Cancer Society. [Google Scholar]
Asghar U, Witkiewicz AK, Turner NC, Knudsen ES (2015). The history and future of targeting cyclin-dependent kinases in cancer therapy. Nature Reviews Drug Discovery 14: 130–146. https://doi.org/10.1038/nrd4504 [Google Scholar] [PubMed] [CrossRef]
Ashraf MA, Sayed S, Bello M, Hussain N, Chando RK, Alam S, Hasan MK (2022). CDK4 as a phytochemical based anticancer drug target. Informatics in Medicine Unlocked 28: 100826. https://doi.org/10.1016/j.imu.2021.100826 [Google Scholar] [CrossRef]
Ates-Alagoz Z, Kisla MM, Karadayi FZ, Baran S, Doğan TS, Mutlu P (2021). Design, synthesis, molecular docking and ADME studies of novel indole-thiazolidinedione derivatives and their antineoplastic activity as CDK6 inhibitors. New Journal of Chemistry 45: 18025–18038. https://doi.org/10.1039/D1NJ02808A [Google Scholar] [CrossRef]
Baig M, Yousuf M, Khan M, Khan I, Ahmad I, Alshahrani M, Hassan M, Dong J (2022). Investigating the mechanism of inhibition of cyclin-dependent kinase 6 inhibitory potential by selonsertib: Newer insights into drug repurposing. Frontiers in Oncology 12: 865454. https://doi.org/10.3389/fonc.2022.865454 [Google Scholar] [PubMed] [CrossRef]
Belal A, Abdel Gawad NM, Mehany AB, Abourehab MA, Elkady H, Al-Karmalawy AA, Ismael AS (2022). Design, synthesis and molecular docking of new fused 1 H-pyrroles, pyrrolo [3,2-d] pyrimidines and pyrrolo [3,2-e][1,4] diazepine derivatives as potent EGFR/CDK2 inhibitors. Journal of Enzyme Inhibition and Medicinal Chemistry 37: 1884–1902. https://doi.org/10.1080/14756366.2022.2096019 [Google Scholar] [PubMed] [CrossRef]
Benjamin I, Udoikono AD, Louis H, Agwamba EC, Unimuke TO, Owen AE, Adeyinka AS (2022). Antimalarial potential of naphthalene-sulfonic acid derivatives: Molecular electronic properties, vibrational assignments, and in-silico molecular docking studies. Journal of Molecular Structure 1264: 133298. https://doi.org/10.1016/j.molstruc.2022.133298 [Google Scholar] [CrossRef]
Bhattacharya A, Guha PS, Chowdhury N, Bagchi A, Guha D (2022). Virtual screening and molecular docking of flavone derivatives as a potential anticancer drug in the presence of dexamethasone. Biointerface Research in Applied Chemistry 13: 215. https://doi.org/10.33263/BRIAC133.215 [Google Scholar] [CrossRef]
Chaube U, Bhatt H (2022). Identification of potent, non-toxic, selective CDK2 inhibitor through the pharmacophore-based scaffold hopping, molecular dynamics simulation-assisted molecular docking study, Lee Richard contour map analysis, and ADMET properties. Structural Chemistry 33: 1–11. https://doi.org/10.1007/s11224-022-01958-4 [Google Scholar] [CrossRef]
Czeleń P, Skotnicka A, Szefler B (2022). Designing and synthesis of new isatin derivatives as potential CDK2 inhibitors. International Journal of Molecular Sciences 23: 8046. https://doi.org/10.3390/ijms23148046 [Google Scholar] [PubMed] [CrossRef]
de Ruyck J, Brysbaert G, Blossey R, Lensink MF (2016). Molecular docking as a popular tool in drug design, an in silico travel. Advances and Applications in Bioinformatics and Chemistry 9: 1–11. https://doi.org/10.2147/AABC [Google Scholar] [CrossRef]
El-Sayed WA, Alminderej FM, Mounier MM, Nossier ES, Saleh SM, Kassem AF (2022a). Novel 1, 2, 3-triazole-coumarin hybrid glycosides and their tetrazolyl analogues: Design, anticancer evaluation and molecular docking targeting EGFR, VEGFR-2 and CDK-2. Molecules 27: 2047. https://doi.org/10.3390/molecules27072047. [Google Scholar] [PubMed] [CrossRef]
El-Sayed AA, Nossier ES, Almehizia AA, Amr AEGE (2022b). Design, synthesis, anticancer evaluation and molecular docking study of novel 2,4-dichlorophenoxymethyl-based derivatives linked to nitrogenous heterocyclic ring systems as potential CDK-2 inhibitors. Journal of Molecular Structure 1247: 131285. https://doi.org/10.1016/j.molstruc.2021.131285 [Google Scholar] [CrossRef]
El-Sayed NN, Zaki ME, Al-Hussain SA, Ben Bacha A, Berredjem M, Masand VH, Almarhoon ZM, Omar HS (2022c). Synthesis and evaluation of some new 4 H-pyran derivatives as antioxidant, antibacterial and anti-HCT-116 cells of CRC, with molecular docking, antiproliferative, apoptotic and ADME investigations. Pharmaceuticals 15: 891. https://doi.org/10.3390/ph15070891 [Google Scholar] [PubMed] [CrossRef]
Eldehna WM, Al-Rashood ST, Al-Warhi T, Eskandrani RO, Alharbi A, El Kerdawy AM (2021a). Novel oxindole/benzofuran hybrids as potential dual CDK2/GSK-3β inhibitors targeting breast cancer: Design, synthesis, biological evaluation, and in silico studies. Journal of Enzyme Inhibition and Medicinal Chemistry 36: 271–286. https://doi.org/10.1080/14756366.2020.1862101 [Google Scholar] [PubMed] [CrossRef]
Eldehna WM, El Hassab MA, Abo-Ashour MF, Al-Warhi T, Elaasser MM, Safwat NA, Suliman H, Ahmed MF, Al-Rashood ST, Abdel-Aziz HA (2021b). Development of isatin-thiazolo [3, 2-a] benzimidazole hybrids as novel CDK2 inhibitors with potent in vitro apoptotic anti-proliferative activity: Synthesis, biological and molecular dynamics investigations. Bioorganic Chemistry 110: 104748. https://doi.org/10.1016/j.bioorg.2021.104748 [Google Scholar] [PubMed] [CrossRef]
Eldehna WM, Maklad RM, Almahli H, Al-Warhi T, Elkaeed EB, Abourehab MA, Abdel-Aziz HA, El Kerdawy AM (2022). Identification of 3-(piperazinylmethyl) benzofuran derivatives as novel type II CDK2 inhibitors: Design, synthesis, biological evaluation, and in silico insights. Journal of Enzyme Inhibition and Medicinal Chemistry 37: 1227–1240. https://doi.org/10.1080/14756366.2022.2062337 [Google Scholar] [PubMed] [CrossRef]
Elkamhawy A, Ammar UM, Paik S, Abdellattif MH, Elsherbeny MH, Lee K, Roh EJ (2021). Scaffold repurposing of in-house small molecule candidates leads to discovery of first-in-class CDK-1/HER-2 dual inhibitors: In vitro and in silico screening. Molecules 26: 5324. https://doi.org/10.3390/molecules26175324 [Google Scholar] [PubMed] [CrossRef]
Elmorsy MR, Mahmoud SE, Fadda AA, Abdel-Latif E, Abdelmoaz MA (2022). Synthesis, biological evaluation and molecular docking of new triphenylamine-linked pyridine, thiazole and pyrazole analogues as anticancer agents. BMC Chemistry 16: 1–20. https://doi.org/10.1186/s13065-022-00879-x [Google Scholar] [PubMed] [CrossRef]
Eltamany EE, Goda MS, Nafie MS, Abu-Elsaoud AM, Hareeri RH, Aldurdunji MM, Elhady SS, Badr JM, Eltahawy NA (2022). Comparative assessment of the antioxidant and anticancer activities of Plicosepalus acacia and Plicosepalus curviflorus: Metabolomic profiling and in silico studies. Antioxidants 11: 1249. https://doi.org/10.3390/antiox11071249 [Google Scholar] [PubMed] [CrossRef]
Fatahala SS, Sayed AI, Mahgoub S, Taha H, El-Sayed M-IK, El-Shehry MF, Awad SM, Abd El-Hameed RH (2021). Synthesis of novel 2-Thiouracil-5-sulfonamide derivatives as potent inducers of cell cycle arrest and CDK2A inhibition supported by molecular docking. International Journal of Molecular Sciences 22: 11957. https://doi.org/10.3390/ijms222111957 [Google Scholar] [PubMed] [CrossRef]
Ghosh A, Manhas A, Jha PC (2022). Computational studies to identify the common type-I and type-II inhibitors against the CDK8 enzyme. Journal of Cellular Biochemistry 123: 628–643. https://doi.org/10.1002/jcb.30209 [Google Scholar] [PubMed] [CrossRef]
Ghosh S, Ramarao TA, Samanta PK, Jha A, Satpati P, Sen A (2021). Triazole based isatin derivatives as potential inhibitor of key cancer promoting kinases-insight from electronic structure, docking and molecular dynamics simulations. Journal of Molecular Graphics and Modelling 107: 107944. https://doi.org/10.1016/j.jmgm.2021.107944 [Google Scholar] [PubMed] [CrossRef]
Goda MS, Nafie MS, Awad BM, Abdel-Kader MS, Ibrahim AK, Badr JM, Eltamany EE (2021). In vitro and in vivo studies of anti-lung cancer activity of Artemesia judaica L. crude extract combined with LC-MS/MS metabolic profiling, docking simulation and HPLC-DAD quantification. Antioxidants 11: 17. https://doi.org/10.3390/antiox11010017 [Google Scholar] [PubMed] [CrossRef]
Gurushankar K, Rimac H, Potemkin V, Grishina M (2021). Investigation of the newly characterized baimantuoluoamide a and baimantuoluoamide b alkaloids as potential cyclin-dependent kinase 4 (CDK4) inhibitors using molecular docking and molecular dynamics simulations. Journal of Molecular Structure 1230: 129925. https://doi.org/10.1016/j.molstruc.2021.129925 [Google Scholar] [CrossRef]
Hassan GS, Georgey HH, Mohammed EZ, George RF, Mahmoud WR, Omar FA (2021a). Mechanistic selectivity investigation and 2D-QSAR study of some new antiproliferative pyrazoles and pyrazolopyridines as potential CDK2 inhibitors. European Journal of Medicinal Chemistry 218: 113389. https://doi.org/10.1016/j.ejmech.2021.113389 [Google Scholar] [PubMed] [CrossRef]
Hassan AS, Moustafa GO, Awad HM, Nossier ES, Mady MF (2021b). Design, synthesis, anticancer evaluation, enzymatic assays, and a molecular modeling study of novel pyrazole-indole hybrids. ACS Omega 6: 12361–12374. https://doi.org/10.1021/acsomega.1c01604 [Google Scholar] [PubMed] [CrossRef]
Hermawan A, Putri H, Utomo RY (2021). Exploration of targets and molecular mechanisms of cinnamaldehyde in overcoming fulvestrant-resistant breast cancer: A bioinformatics study. Network Modeling Analysis in Health Informatics and Bioinformatics 10: 1–14. https://doi.org/10.1007/s13721-021-00303-9 [Google Scholar] [CrossRef]
Huang C, Wang S, Ma W (2022). Design, synthesis, and antitumor activity of new N4-Alkyl-N2-Phenyl-Pyrrolo[3,2-d] Pyrimidine-2,4-Diamine derivatives as CDK6 Inhibitors. Russian Journal of Bioorganic Chemistry 48: 557–564. https://doi.org/10.1134/S1068162022030062 [Google Scholar] [CrossRef]
Ismail MM, Soliman DH, Sabour R, Farrag AM (2021). Synthesis of new arylazopyrazoles as apoptosis inducers: Candidates to inhibit proliferation of MCF-7 cells. Archiv der Pharmazie 354: 2000214. https://doi.org/10.1002/ardp.202000214 [Google Scholar] [PubMed] [CrossRef]
Joshi AJ, Bhojwani HR, Joshi U, Begwani KV, Wagal OS, Sathaye SS, Kanchan DM (2021). Cinnamamide-chalcone derivatives as CDK2 inhibitors: Synthesis, pharmacological evaluation, and molecular modelling study. Journal of the Iranian Chemical Society 19: 4445–4455. https://doi.org/10.1007/s13738-022-02610-y [Google Scholar] [CrossRef]
Joshi AJ, Bhojwani HR, Joshi UJ, Begwani KV, Wagal OS, Sathaye SS, Kanchan DM (2022). Cinnamamide-chalcone derivatives as CDK2 inhibitors: Synthesis, pharmacological evaluation, and molecular modelling study. Journal of the Iranian Chemical Society 19: 4445–4455. https://doi.org/10.1007/s13738-022-02610-y [Google Scholar] [CrossRef]
Kasemsuk T, Saehlim N, Arsakhant P, Sittithumcharee G, Okada S, Saeeng R (2021). A novel synthetic acanthoic acid analogues and their cytotoxic activity in cholangiocarcinoma cells. Bioorganic & Medicinal Chemistry 29: 115886. https://doi.org/10.1016/j.bmc.2020.115886 [Google Scholar] [PubMed] [CrossRef]
Kaur H, Singh R (2022). Synthesis, molecular docking, and antitubercular evaluation of triazole-chalcone conjugates. Russian Journal of Organic Chemistry 58: 518–525. https://doi.org/10.1134/S107042802204008X [Google Scholar] [CrossRef]
Khanam M, Moin AT, Ahmed KA, Patil RB, Khalipha ABR, Ahmed N, Bagchi R, Ullah MA, Ferdoush J, Islam S (2022). Computational modeling of potential milciclib derivatives inhibitor-CDK2 binding through global docking and accelerated molecular dynamics simulations. Informatics in Medicine Unlocked 33: 101069. https://doi.org/10.1016/j.imu.2022.101069 [Google Scholar] [CrossRef]
Kourea H, Koutras A, Scopa C, Marangos M, Tzoracoeleftherakis E, Koukouras D, Kalofonos H (2003). Expression of the cell cycle regulatory proteins p34cdc2, p21waf1, and p53 in node negative invasive ductal breast carcinoma. Molecular Pathology 56: 328–335. https://doi.org/10.1136/mp.56.6.328 [Google Scholar] [PubMed] [CrossRef]
Li Y, Du R, Nie Y, Wang T, Ma Y, Fan Y (2021). Design, synthesis and biological assessment of novel CDK4 inhibitor with potent anticancer activity. Bioorganic Chemistry 109: 104717. https://doi.org/10.1016/j.bioorg.2021.104717 [Google Scholar] [PubMed] [CrossRef]
Liang Y, Quan H, Bu T, Li X, Liu X, Wang S, He D, Jia Q, Zhang Y (2021). Comparison of the inhibitory binding modes between the planar fascaplysin and its nonplanar tetrahydro-β-carboline analogs in CDK4. Frontiers in Chemistry 9: 614154. https://doi.org/10.3389/fchem.2021.614154 [Google Scholar] [PubMed] [CrossRef]
Lin T, Li J, Liu L, Li Y, Jiang H, Chen K, Xu P, Luo C, Zhou B (2021). Design, synthesis, and biological evaluation of 4-benzoylamino-1H-pyrazole-3-carboxamide derivatives as potent CDK2 inhibitors. European Journal of Medicinal Chemistry 215: 113281. https://doi.org/10.1016/j.ejmech.2021.113281 [Google Scholar] [PubMed] [CrossRef]
Lone MY, Athar M, Manhas A, Jha PC, Bhatt S, Shah A (2017). In silico exploration of vinca domain tubulin inhibitors: A combination of 3D-QSAR-based pharmacophore modeling, docking and molecular dynamics simulations. ChemistrySelect 2: 10848–10853. https://doi.org/10.1002/slct.201701971 [Google Scholar] [CrossRef]
Lone MY, Manhas A, Athar M, Jha PC (2018). Identification of InhA inhibitors: A combination of virtual screening, molecular dynamics simulations and quantum chemical studies. Journal of Biomolecular Structure and Dynamics 36: 2951–2965. https://doi.org/10.1080/07391102.2017.1372313 [Google Scholar] [PubMed] [CrossRef]
Lotlikar O, Dandekar S, Ramana M, Rathod S (2021). Synthesis, molecular docking, in vitro anti-bacterial, and anti-cancer activities of some novel oxo-spiro chromene Schiff’s bases. Russian Journal of Bioorganic Chemistry 47: 199–207. https://doi.org/10.1134/S1068162021010131 [Google Scholar] [CrossRef]
Luo X, Zhao Y, Tang P, Du X, Li F, Wang Q, Li R, He J (2021). Discovery of new small-molecule cyclin-dependent kinase 6 inhibitors through computational approaches. Molecular Diversity 25: 367–382. https://doi.org/10.1007/s11030-020-10120-3 [Google Scholar] [PubMed] [CrossRef]
Mandour AA, Nassar IF, Abdel Aal MT, Shahin MA, El-Sayed WA, Hegazy M, Yehia AM, Ismail A, Hagras M, Elkaeed EB (2022). Synthesis, biological evaluation, and in silico studies of new CDK2 inhibitors based on pyrazolo[3,4-d] pyrimidine and pyrazolo[4,3-e][1,2,4] triazolo[1,5-c] pyrimidine scaffold with apoptotic activity. Journal of Enzyme Inhibition and Medicinal Chemistry 37: 1957–1973. https://doi.org/10.1080/14756366.2022.2086866 [Google Scholar] [PubMed] [CrossRef]
Manhas A, Kumar SP, Jha PC (2016). Molecular modeling of Plasmodium falciparum peptide deformylase and structure-based pharmacophore screening for inhibitors. RSC Advances 6: 29466–29485. https://doi.org/10.1039/C6RA01071G [Google Scholar] [CrossRef]
Manhas A, Lone MY, Jha PC (2019a). Multicomplex-based pharmacophore modeling in conjunction with multi-target docking and molecular dynamics simulations for the identification of Pf DHFR inhibitors. Journal of Biomolecular Structure and Dynamics 37: 4181–4199. https://doi.org/10.1080/07391102.2018.1540362 [Google Scholar] [PubMed] [CrossRef]
Manhas A, Patel D, Lone MY, Jha PC (2019b). Identification of natural compound inhibitors against PfDXR: A hybrid structure-based molecular modeling approach and molecular dynamics simulation studies. Journal of Cellular Biochemistry 120: 14531–14543. https://doi.org/10.1002/jcb.28714 [Google Scholar] [PubMed] [CrossRef]
Manhas A, Patel A, Lone MY, Jha PK, Jha PC (2018). Identification of PfENR inhibitors: A hybrid structure-based approach in conjunction with molecular dynamics simulations. Journal of Cellular Biochemistry 119: 8490–8500. https://doi.org/10.1002/jcb.27075 [Google Scholar] [PubMed] [CrossRef]
Mansour HS, Abd El-wahab HA, Ali AM, Aboul-Fadl T (2021a). Inversion kinetics of some E/Z 3-(benzylidene)-2-oxo-indoline derivatives and their in silico CDK2 docking studies. RSC Advances 11: 7839–7850. https://doi.org/10.1039/D0RA10672K [Google Scholar] [PubMed] [CrossRef]
Mansour E, Abd-Rabou AA, Nassar IF, Elewa SI (2021b). Synthesis, docking and anticancer evaluation of new pyridine-3-carbonitrile derivatives. Polycyclic Aromatic Compounds 42: 3523–3544. https://doi.org/10.1080/10406638.2020.1870507 [Google Scholar] [CrossRef]
Marak BN, Dowarah J, Khiangte L, Singh VP (2020). A comprehensive insight on the recent development of cyclic dependent kinase inhibitors as anticancer agents. European Journal of Medicinal Chemistry 203: 112571. https://doi.org/10.1016/j.ejmech.2020.112571 [Google Scholar] [PubMed] [CrossRef]
Meng XY, Zhang HX, Mezei M, Cui M (2011). Molecular docking: A powerful approach for structure-based drug discovery. Current Computer-aided Drug Design 7: 146–157. https://doi.org/10.2174/157340911795677602 [Google Scholar] [PubMed] [CrossRef]
Metwally NH, Mohamed MS, Deeb EA (2021). Synthesis, anticancer evaluation, CDK2 inhibition, and apoptotic activity assessment with molecular docking modeling of new class of pyrazolo [1,5-a] pyrimidines. Research on Chemical Intermediates 47: 5027–5060. https://doi.org/10.1007/s11164-021-04564-x [Google Scholar] [CrossRef]
Modi P, Patel S, Chhabria M (2022). Discovery of newer pyrazole derivatives with potential anti-tubercular activity via 3D-QSAR based pharmacophore modelling, virtual screening, molecular docking and molecular dynamics simulation studies. Molecular Diversity: 1–20. https://doi.org/10.1007/s11030-022-10511-8 [Google Scholar] [PubMed] [CrossRef]
Mohammed ER, Elmasry GF (2022). Development of newly synthesised quinazolinone-based CDK2 inhibitors with potent efficacy against melanoma. Journal of Enzyme Inhibition and Medicinal Chemistry 37: 686–700. https://doi.org/10.1080/14756366.2022.2036985 [Google Scholar] [PubMed] [CrossRef]
Musa A, Al-Sanea MM, Alotaibi NH, Alnusaire TS, Ahmed SR, Mostafa EM (2021). In silico study, protein kinase inhibition and antiproliferative potential of flavonoids isolated from Bassia eriophora (Schrad.) growing in KSA. Indian Journal of Pharmaceutical Education and Research 55: 483–490. https://doi.org/10.5530/ijper.55.2.86 [Google Scholar] [CrossRef]
Nassar IF, Aal MTA, El-Sayed WA, Shahin MA, Elsakka EG, Mokhtar MM, Hegazy M, Hagras M, Mandour AA, Ismail NS (2022). Discovery of pyrazolo[3,4-d] pyrimidine and pyrazolo[4,3-e][1,2,4] triazolo [1,5-c] pyrimidine derivatives as novel CDK2 inhibitors: Synthesis, biological and molecular modeling investigations. RSC Advances 12: 14865–14882. https://doi.org/10.1039/D2RA01968J [Google Scholar] [PubMed] [CrossRef]
Nurhayati APD, Rihandoko A, Fadlan A, Ghaissani SS, Jadid N, Setiawan E (2022). Anti-cancer potency by induced apoptosis by molecular docking P53, caspase, cyclin D1, cytotoxicity analysis and phagocytosis activity of trisindoline 1,3 and 4. Saudi Pharmaceutical Journal 30: 1345–1359. https://doi.org/10.1016/j.jsps.2022.06.012 [Google Scholar] [PubMed] [CrossRef]
Obakachi VA, Kehinde I, Kushwaha ND, Akinpelu OI, Kushwaha B, Merugu SR, Kayamba F, Kumalo HM, Karpoormath R (2022). Structural based investigation of novel pyrazole-thiazole Hybrids as dual CDK-1 and CDK-2 inhibitors for cancer chemotherapy. Molecular Simulation 48: 687–701. https://doi.org/10.1080/08927022.2022.2045016 [Google Scholar] [CrossRef]
Pecoraro C, Parrino B, Cascioferro S, Puerta A, Avan A, Peters GJ, Diana P, Giovannetti E, Carbone D (2021). A new oxadiazole-based topsentin derivative modulates cyclin-dependent kinase 1 expression and exerts cytotoxic effects on pancreatic cancer cells. Molecules 27: 19. https://doi.org/10.3390/molecules27010019 [Google Scholar] [PubMed] [CrossRef]
Peyressatre M, Prével C, Pellerano M, Morris MC (2015). Targeting cyclin-dependent kinases in human cancers: from small molecules to peptide inhibitors. Cancers 7: 179–237. https://doi.org/10.3390/cancers7010179 [Google Scholar] [PubMed] [CrossRef]
Pinzi L, Rastelli G (2019). Molecular docking: Shifting paradigms in drug discovery. International Journal of Molecular Sciences 20: 4331. https://doi.org/10.3390/ijms20184331 [Google Scholar] [PubMed] [CrossRef]
Qayoom H, Mehraj U, Sofi S, Aisha S, Almilaibary A, Alkhanani M, Mir MA (2022). Expression patterns and therapeutic implications of CDK4 across multiple carcinomas: A molecular docking and MD simulation study. Medical Oncology 39: 1–13. https://doi.org/10.1007/s12032-022-01779-9 [Google Scholar] [PubMed] [CrossRef]
Raut GK, Sukumar G, Chakrabarti M, Mendonza JJ, Pabbaraja S, Jagan Mohan Reddy B, Sistla R, Balaji Andugulapati S, Bhadra MP (2022a). Anticancer effect and apoptosis induction by azaflavanone derivative in human prostate cancer cells. Apoptosis 27: 1–15. https://doi.org/10.1007/s10495-022-01745-w [Google Scholar] [PubMed] [CrossRef]
Raut GK, Sukumar G, Chakrabarti M, Mendonza JJ, Pabbaraja S, Jagan Mohan Reddy B, Sistla R, Balaji Andugulapati S, Bhadra MP (2022b). Anticancer effect and apoptosis induction by azaflavanone derivative in human prostate cancer cells. Apoptosis 27: 825–839. https://doi.org/10.1007/s10495-022-01745-w [Google Scholar] [PubMed] [CrossRef]
Reddy ST, Ramakrishna M, Makani VKK, Mendonza JJ, Edathara PM, Bhadra MP, Uppuluri VM (2022). Synthesis of novel 1,2,3-triazole hybrids of methyl β-orsellinate with capabilities to arrest cell cycle and induce apoptosis in breast cancer cells (MCF-7). Monatshefte für Chemie-Chemical Monthly 153: 1–13. https://doi.org/10.1007/s00706-022-02922-y [Google Scholar] [CrossRef]
Rouchal M, Rudolfová J, Kryštof V, Vojáčková V, Čmelík R, Vícha R (2021). Adamantane-substituted purines and their β-Cyclodextrin complexes: Synthesis and biological activity. International Journal of Molecular Sciences 22: 12675. https://doi.org/10.3390/ijms222312675 [Google Scholar] [PubMed] [CrossRef]
Roy A, Bhatia KS (2021). In silico analysis of plumbagin against cyclin-dependent kinases receptor. Vegetos 34: 50–56. https://doi.org/10.1007/s42535-020-00169-8 [Google Scholar] [CrossRef]
Roy T, Boateng ST, Banang-Mbeumi S, Singh PK, Basnet P, Chamcheu R-CN, Ladu F, Chauvin I, Spiegelman VS, Hill RA (2021). Synthesis, inverse docking-assisted identification and in vitro biological characterization of Flavonol-based analogs of fisetin as c-Kit, CDK2 and mTOR inhibitors against melanoma and non-melanoma skin cancers. Bioorganic Chemistry 107: 104595. https://doi.org/10.1016/j.bioorg.2020.104595 [Google Scholar] [PubMed] [CrossRef]
Sanka BM, Tadesse DM, Bedada ET, Mengesha ET, Babu N (2022). Design, synthesis, biological screening and molecular docking studies of novel multifunctional 1,4-di (aryl/heteroaryl) substituted piperazine derivatives as potential antitubercular and antimicrobial agents. Bioorganic Chemistry 119: 105568. https://doi.org/10.1016/j.bioorg.2021.105568 [Google Scholar] [PubMed] [CrossRef]
Serag WM, Zahran F, Abdelghany YM, Elshaarawy RF, Abdelhamid MS (2021). Synthesis and molecular docking of hybrids ionic azole Schiff bases as novel CDK1 inhibitors and anti-breast cancer agents: In vitro and in vivo study. Journal of Molecular Structure 1245: 131041. https://doi.org/10.1016/j.molstruc.2021.131041 [Google Scholar] [CrossRef]
Seyfried TN, Shelton LM (2010). Cancer as a metabolic disease. Nutrition & Metabolism 7: 1–22. https://doi.org/10.1186/1743-7075-7-7 [Google Scholar] [PubMed] [CrossRef]
Shah SC, Kayamba V, Peek RM Jr, Heimburger D (2019). Cancer control in low-and middle-income countries: Is it time to consider screening? Journal of Global Oncology 5: 1–8. https://doi.org/10.1200/JGO.18.00200 [Google Scholar] [PubMed] [CrossRef]
Shaikh J, Patel K, Khan T (2022). Advances in pyrazole based scaffold as cyclin-dependent kinase 2 inhibitors for the treatment of cancer. Mini Reviews in Medicinal Chemistry 22: 1197–1215. https://doi.org/10.2174/1389557521666211027104957 [Google Scholar] [PubMed] [CrossRef]
Shamsiya A, Bahulayan D (2022). D-A systems based on oxazolone-coumarin triazoles as solid-state emitters and inhibitors of human cervical cancer cells (HeLa). New Journal of Chemistry 46: 480–489. https://doi.org/10.1039/D1NJ04151G [Google Scholar] [CrossRef]
Sharma M, Sharma N, Muddassir M, Rahman QI, Dwivedi U, Akhtar S (2021). Structure-based pharmacophore modeling, virtual screening and simulation studies for the identification of potent anticancerous phytochemical lead targeting cyclin-dependent kinase 2. Journal of Biomolecular Structure and Dynamics 40: 1–18. https://doi.org/10.1080/07391102.2021.1936178 [Google Scholar] [PubMed] [CrossRef]
Shawky AM, Ibrahim NA, Abdalla AN, Abourehab MA, Gouda AM (2021). Novel pyrrolizines bearing 3,4,5-trimethoxyphenyl moiety: design, synthesis, molecular docking, and biological evaluation as potential multi-target cytotoxic agents. Journal of Enzyme Inhibition and Medicinal Chemistry 36: 1312–1332. https://doi.org/10.1080/14756366.2021.1937618 [Google Scholar] [PubMed] [CrossRef]
Siegel RL, Miller KD, Jemal A (2019). Cancer statistics, 2019. CA: A Cancer Journal for Clinicians 69: 7–34. https://doi.org/10.3322/caac.21551 [Google Scholar] [PubMed]
Sofi S, Mehraj U, Qayoom H, Aisha S, Almilaibary A, Alkhanani M, Mir MA (2022). Targeting cyclin-dependent kinase 1 (CDK1) in cancer: Molecular docking and dynamic simulations of potential CDK1 inhibitors. Medical Oncology 39: 1–15. https://doi.org/10.1007/s12032-022-01748-2 [Google Scholar] [PubMed] [CrossRef]
Tutone M, Almerico AM (2017). Recent advances on CDK inhibitors: An insight by means of in silico methods. European Journal of Medicinal Chemistry 142: 300–315. https://doi.org/10.1016/j.ejmech.2017.07.067 [Google Scholar] [PubMed] [CrossRef]
Yousuf M, Shamsi A, Mohammad T, Azum N, Alfaifi SY, Asiri AM, Mohamed Elasbali A, Islam A, Hassan MI, Haque QMR (2022). Inhibiting cyclin-dependent kinase 6 by taurine: Implications in anticancer therapeutics. ACS Omega 7: 25844–25852. https://doi.org/10.1021/acsomega.2c03479 [Google Scholar] [PubMed] [CrossRef]
Yousuf M, Shamsi A, Queen A, Shahbaaz M, Khan P, Hussain A, Alajmi MF, Rizwanul Haque QM, Imtaiyaz Hassan M (2021). Targeting cyclin-dependent kinase 6 by vanillin inhibits proliferation of breast and lung cancer cells: Combined computational and biochemical studies. Journal of Cellular Biochemistry 122: 897–910. https://doi.org/10.1002/jcb.29921 [Google Scholar] [PubMed] [CrossRef]
Yu Z, Wu Y, Ma Y, Cheng Y, Song G, Zhang F (2022). Systematic analysis of the mechanism of aged citrus peel (Chenpi) in oral squamous cell carcinoma treatment via network pharmacology, molecular docking and experimental validation. Journal of Functional Foods 91: 105012. https://doi.org/10.1016/j.jff.2022.105012 [Google Scholar] [CrossRef]
Zhang C, Quan Y, Yang L, Bai Y, Yang Y (2022). 6-Methoxyflavone induces S-phase arrest through the CCNA2/CDK2/p21CIP1 signaling pathway in HeLa cells. Bioengineered 13: 7277–7292. https://doi.org/10.1080/21655979.2022.2047496 [Google Scholar] [PubMed] [CrossRef]
Zhao Y, Zuo J, Shen Y, Yan D, Chen J, Qi X (2022). Identification of novel drugs targeting cell cycle regulators for the treatment of high-grade serous ovarian cancer via integrated bioinformatics analysis. Symmetry 14: 1403. https://doi.org/10.3390/sym14071403 [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools