
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Placenta-derived mesenchymal stem cells attenuate secondary brain injury after controlled cortical impact in rats by inhibiting matrix metalloproteinases
1 Department of Neurosurgery, School of Clinical Medicine, Ningxia Medical University, Yinchuan, China
2 Key Laboratory of Ningxia Stem Cell and Regenerative Medicine, General Hospital of Ningxia Medical University, Yinchuan, China
3 Department of Neurosurgery, Xi’an International Medical Center Hospital, Xi’an, China
* Corresponding Author: HECHUN XIA. Email:
BIOCELL 2024, 48(1), 149-162. https://doi.org/10.32604/biocell.2023.042367
Received 27 May 2023; Accepted 03 November 2023; Issue published 30 January 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: As a form of biological therapy, placenta-derived mesenchymal stem cells (PDMSCs) exhibit considerable promise in addressing the complex pathological processes of traumaticbrain injury (TBI) due to their multi-target and multi-pathway mode of action. Material & Methods: This study investigates the protective mechanisms and benefits of PDMSCs in mitigating the effects of controlled cortical impact (CCI) in rats and glutamate-induced oxidative stress injury in HT22 cells in vitro. Our primary objective is to provide evidence supporting the clinical application of PDMSCs. Results: In the in vivo arm of our investigation, we observed a swift elevation of matrix metalloproteinase-9 (MMP-9) in the proximal cortex of injured brain tissues after CCI. PDMSCs, distinguished by their heightened expression of metalloproteinase tissue inhibitors-1 and -2 (TIMP-1 and TIMP-2): were intravenously administered via the caudal vein. This intervention yielded significant reductions in the permeability of the blood-brain barrier (BBB): the extent of brain edema, the levels of inflammatory cytokines IL-1β and TNF-α in damaged brain tissue, and the activation status of microglia in CCI-afflicted rats. In the realm of in vitro experiments, PDMSC-conditioned media demonstrated substantial reductions in mortality rates and cleaved caspase-3 levels in glutamate-induced HT22 cells compared with conventional media. Notably, this advantage was negated upon the introduction of neutralizing antibodies targeting TIMP-1 and TIMP-2. Conclusion: Collectively, our findings underscore the potential of PDMSCs in alleviating oxidative stress injury and secondary brain injury in the pathological process of TBI.Graphic Abstract
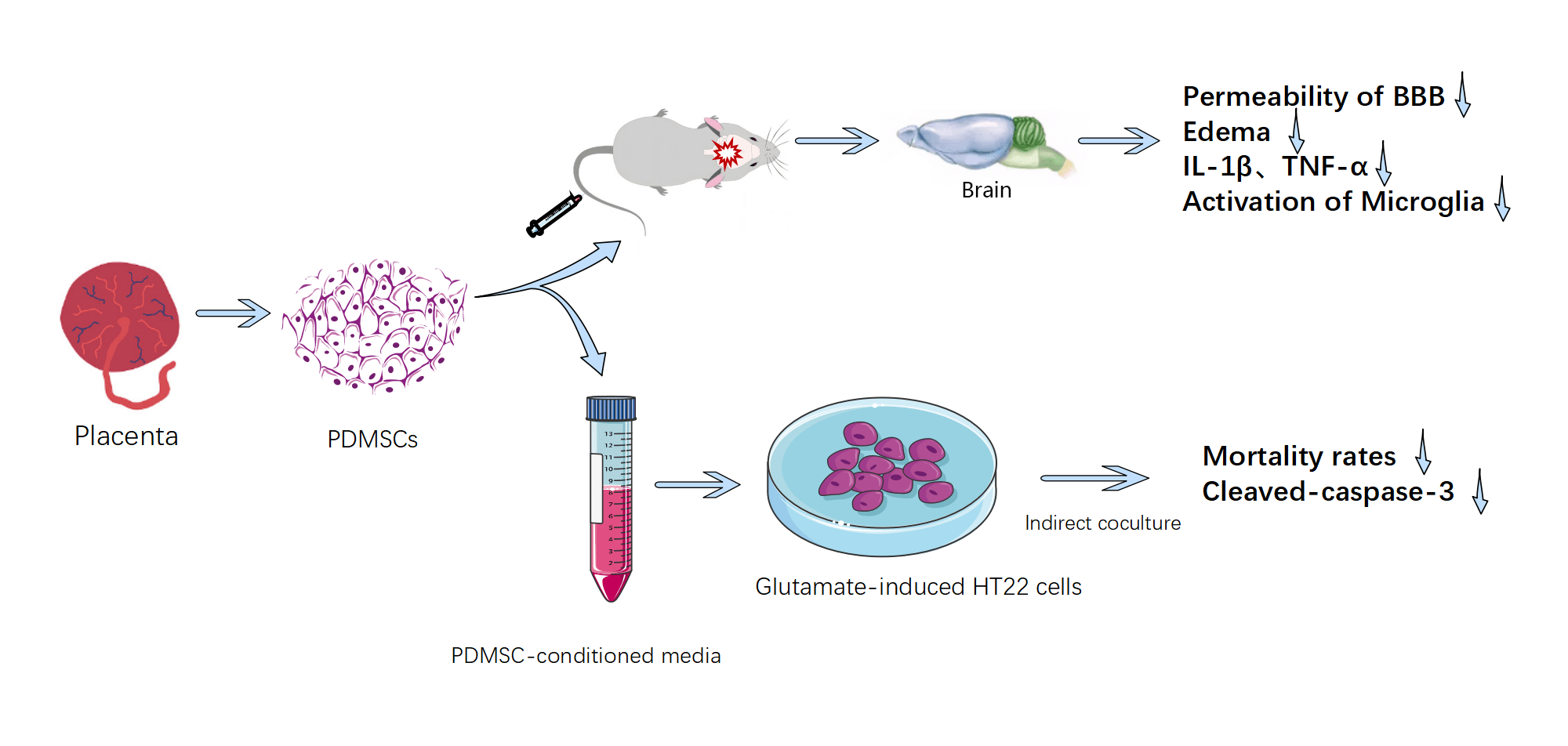
Keywords
Traumatic brain injury (TBI) has, to date, afflicted approximately 69 million individuals worldwide, with about 13 million enduring moderate to severe cases [1]. TBI stands as a formidable public health challenge, engendering severe disability, high mortality rates, substantial economic burdens, and diminished labor force productivity [2]. The pathological underpinnings of TBI encompass both primary and secondary injury mechanisms. Primary injury ensues from direct mechanical forces impacting the cranial and cerebral structures, resulting in skull fractures, cerebral contusions, and axonal injuries. Subsequent alterations in biochemical cascades and pathophysiological processes culminate in secondary brain injury, characterized by the disruption of the blood-brain barrier (BBB): increased permeability, cerebral edema, intracranial hypertension, cerebral ischemia, neuroinflammation, and neurodegeneration [3,4]. Secondary brain injury is the primary determinant of in-hospital mortality among TBI patients and a critical contributor to sustained neurological deficits after TBI [5]. The intricate nature of the pathological mechanisms underlying TBI, coupled with the diversity of treatment methods and their varying efficacies, underscores the considerable challenges associated with the formation of TBI treatment guidelines and the development of therapeutic options.
Matrix metalloproteinases (MMPs) constitute a family of zinc-dependent endopeptidases. These enzymes are initially secreted in an inactive proenzyme form (pro-MMPs) and subsequently activated by proteolytic enzymes (active-MMPs). The activity of MMPs is rigorously regulated by posttranslational metalloproteinase tissue inhibitors (TIMPs) [6]. MMPs and TIMPs form a tight, non-covalent 1:1 complex without cleaving TIMPs. Research has elucidated that MMP-9 is instrumental in precipitating BBB disruption, cerebral hemorrhage, neuroinflammation, and cell demise [6]. Following a TBI incident, physical trauma can trigger the upregulation of MMP-9 expression via the mitogen-activated protein kinase (MAPK) pathway [7]. Resulting in the degradation of the microvascular basement membranes, compromised endothelial cell stability, and increased BBB permeability [8]. This cascade subsequently induces or exacerbates cellular damage and angioedema [9,10]. In addition, activated MMPs induce apoptosis, exacerbate neural and tissue damage [11,12], participate in neurodegenerative processes, and promote the emergence of an inflammatory microenvironment [13,14].
Mesenchymal stem cells (MSCs) have emerged as focal points of bioengineering research due to their multifaceted therapeutic attributes involving various pathways and targets. MSCs can traverse the BBB, mitigate neuroinflammation, counteract apoptosis, promote angiogenesis and neurogenesis, and facilitate functional recuperation [15–18]. Influenced by diverse secretory factors, MSCs can migrate across endothelial cells to injury sites, differentiate into mature cell lineages, and thus promote tissue regeneration [19]. MSCs also regulate glial activation after TBI, attenuating glial scar formation around the injury site, thus contributing to the restoration of neural tissue recuperation and functional restoration [20]. The observed improvement in motor and cognitive deficits in certain animal models following MSC treatment may be attributed to endogenous neurogenesis and synaptogenesis [21,22].
In a previous investigation conducted at the Key Laboratory of Stem Cells and Regenerative Medicine of Ningxia Hui Autonomous Region, we detected elevated expression levels of TIMP-1 and TIMP-2 in human placental-derived mesenchymal stem cells (PDMSCs) using a protein microarray. Given the pivotal role of MMPs in TBI, it was hypothesized that PDMSCs inhibited MMP activity through their heightened TIMP expressions, thereby alleviating secondary brain injury in controlled cortical impact (CCI) rats. Given the prevalent occurrence of oxidative stress injury in TBI, an in vitro model of glutamate-induced oxidative stress injury in HT22 cells was established. An in-depth understanding of the protective effects and underlying mechanisms of PDMSCs against brain injury may provide further evidence for the clinical application of PDMSCs in therapeutic settings.
Approval for this study was granted by the Animal Ethics Committee of the General Hospital of Ningxia Medical University (Approval No. 2017-073) on February 13, 2017. All experimental procedures outlined herein strictly adhered to the National Institutes of Health Guide: Revised 1996 Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication No. 85–23). The rats were anesthetized by intramuscular injection of Zoletil TM50 (Tiletamine and Zolazepam; Virbac, Nice, France) at a dosage of 50 mg/kg. Euthanasia was carried out by CO2 asphyxiation, with a gradual displacement of 50% of the chamber volume per minute, at the conclusion of the study. Every conceivable measure was taken to minimize the pain and distress experienced by the animals. Female Sprague-Dawley rats, aged 9 ± 1 weeks, weighing 190–220 g, were procured from the Animal Experiment Center of Ningxia Medical University. These rats were individually housed in ventilated cages (n = 3 per cage) at 25°C ± 1°C with 50%–60% relative humidity. Rats were granted ad libitum access to both food and water and subjected to a 12-h light/dark cycle.
The rats were divided into three groups: the CCI group, the CCI + PDMSC group (at a dosage of 1 × 106 i.v. at 0.5 h and 24 h after CCI): and the sham group, which underwent craniotomy without any additional intervention. Unilateral brain injury was induced utilizing a CCI device (Impact One; Wuhan Yihong Technology Co., Ltd., Wuhan, Hubei, China): followed by a craniectomy with a diameter of 5–6 mm on the right cranial vault. The center of the craniectomy was placed equidistant between bregma and lambda, with a lateral deviation of 3 mm from the midline. A 4-mm-diameter flat impactor tip was used at an angle of 10° from the vertical plane. The rats received a single impact with a 4-mm deformation depth at an impact velocity of 5.5 m/s and with a residence time of 150 ms, resulting in a moderate-severe injury to the brain tissue surface.
Subsequent to a 72-h post-CCI period, the rats were humanely euthanized. Subsequently, the brains were promptly excised, with the olfactory bulb, cerebellum, and brain stem being excluded from the analysis. The cerebral hemisphere underwent prompt weight measurement in its hydrated state (wet weight) and subsequent desiccation within an oven at 110°C for 24 h (dry weight). To determine the percentage of water content, the following formula was applied: Percentage of wet content = [(wet weight − dry weight)/wet weight] × 100%.
BBB permeability was assessed using Evans blue staining. Rats received an intravenous injection of 0.5% Evans blue (3 mL/kg, Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) three days post-injury. After a 2-h interval, the rats were anesthetized, followed by transcardial perfusion with 200–250 mL of saline. Subsequently, the cerebral hemisphere on the injured side was swiftly homogenized, and the supernatant was collected after centrifugation at 3,000× g at 4°C for 10 min. The supernatant was then diluted with acetone (3:7) and incubated at room temperature for 24 h. Following this incubation period, the samples were centrifuged at 12,000× g at 4°C for 15 min, and the Evans blue content in the resulting supernatant was quantified through spectrophotometric analysis at 620 nm.
To quantify cytokine concentrations, we humanely euthanized rats four days after TBI or sham operation. Cerebral tissues were immediately harvested, and punch biopsies, measuring 6 mm in diameter, from the injured cortex were isolated. The tissue specimens were homogenized in a chilled extraction buffer supplemented with a protease inhibitor cocktail, with a tissue-to-buffer ratio of 1:10. This homogenate was agitated on ice for 60 min and subsequently subjected to centrifugation at 10,000× g at 4°C for 10 min. Total protein concentrations were measured using a BCA Protein Assay Kit (Thermo Fisher Scientific, Inc. Waltham, MA, USA). The samples were then diluted with double distilled water to ensure uniform final protein concentrations across all samples. The levels of pro-inflammatory cytokines, namely interleukin-1β (IL-1β; Proteintech Group, Inc., Chicago, IL, USA) and tumor necrosis factor-α (TNF-α; Proteintech Group, Inc., Chicago, IL, USA): were determined utilizing enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions.
To gauge systemic glutamate levels at various time points post-CCI, we anesthetized rats and extracted blood samples from the cardiac tissue. These blood samples were then centrifuged at 5,000× g for 30 min to isolate the serum component. The serum glutamate concentrations were quantified using a glutamic ELISA kit (Jiangsu Enzyme Marker Biology Co., Ltd., Jiangsu, China) following the manufacturer’s protocol.
Rats were anesthetized and underwent transcardial perfusion with PBS and 4% paraformaldehyde for brain tissue fixation. Following extraction, the brains were immersed in the same fixative solution, ensuring overnight fixation at 4°C, before they were embedded in paraffin. These brains were then coronally sliced at a thickness of 2 μm, and these slices were mounted on glass slides in preparation for immunohistochemical examinations. Prior to the analysis, the slices were deparaffinized through three xylene washes, followed by rehydration via graded alcohol solutions. Subsequently, they underwent three 5-min washes in PBS. Both primary and secondary antibodies were prepared in PBST with 2% bovine serum albumin and 1% goat serum. The incubation with primary antibodies was carried out overnight at 4°C, followed by a subsequent 2-h incubation with a secondary antibody at room temperature. Following each antibody incubation phase, the slices were thoroughly washed three times with PBST for 10 min each. To conclude the process, the slices were mounted onto slides, and a coverslip with DAPI Fluoromount-G was affixed to facilitate nucleus staining. The primary antibodies used were MMP-9 and CD11b (both diluted at 1:100, Boaosen Biotechnology Co., Ltd., Beijing, China): while the secondary antibody was a FITC-labeled goat anti-rabbit antibody (diluted at 1:100, Wuhan Servicebio Technology Co., Ltd., Wuhan, China).
All brain slices were restricted to the bregma coordinates ranging from −3.55 to −4.55 mm. Within each rat, a minimum of three brain slices, positioned at least 20 μm apart, were used. Mean gray values (Mean) within the target regions were quantified and analyzed using ImageJ (National Institutes of Health, Bethesda, MD, USA). Mean = Integrated Destity/Area. The Mean for each sample was computed based on data obtained from three brain slices.
Cell culture and conditioned media collection
PDMSCs, graciously provided by the Key Laboratory of Stem Cells and Regenerative Medicine of the Ningxia Hui Autonomous Region, merit special mention as their preparation methodology is patented (Patent No. ZL200 880115431.5). These PDMSCs were extracted from placental tissues with informed consent from healthy donors. Upon revival, PDMSCs were uniformly seeded in 10 cm Petri dishes and cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 4.5 g/L glucose, 110 mg/L sodium pyruvate, 1% penicillin/streptomycin, and 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). Culture conditions entailed maintenance at 37°C in a humidified incubator under a 5% CO2 atmosphere. Medium replenishment transpired every 48 h until passage. When cells achieved an 80% confluence in the fourth generation, enzymatic digestion using TrypLE (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) was performed. Subsequently, the cells were resuspended at a concentration of 1 × 106 cells/mL for tail-vein injection in rats.
HT22 cells, purchased from Bena Chuang Lian Biotechnology Co., Ltd. (Beijing, China): shared an identical culture milieu with PDMSCs. During the logarithmic growth phase, PDMSCs or HT22 cells were seeded at a density of 8 × 105/well in 10 cm dishes, permitting adhesion for 24 h. Following this adhesion period, the fresh medium replaced the existing culture medium, and cells were afforded an additional incubation of either 24 or 48 h. Post-incubation, the conditioned medium produced by PDMSCs was collected. A centrifugation step at 1,000× g for 10 min separated the supernatant, which was then carefully retrieved and stored at −20°C, awaiting subsequent use.
Cell viability evaluations were performed with distinct objectives in mind. The first objective centered on scrutinizing the influence of PDMSC-conditioned media on the growth of normal HT22 cells. Specifically, HT22 cells were seeded into 96-well plates at a density of 5 × 103 cells/well. On the ensuing day, these cells were exposed to either DMEM supplemented with 10% FBS or PDMSC-conditioned media from a 48-h culture. The second facet of the analysis aimed to assess the protective effects of the conditioned media. In this context, HT22 cells were individually subjected to conventional media or PDMSC-conditioned media. These treatments occurred in the presence or absence of neutralizing antibodies and were followed by co-incubation with glutamate (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China). Except for the control group, all other experimental groups were subjected to a uniform glutamate concentration of 5 mM.
The quantification of cell viability hinged on the introduction of 10 µL CCK-8 solution (Dojindo Molecular Technologies, Inc., Rockville, MD, USA) to each well. Depending on the specific experimental design, the incubation period was either 60 or 90 min, conducted at 37°C. Subsequently, absorbance measurements were recorded at 450 nm with a microplate reader (Amersham Image Quant 800uv, USA). The results from the cell viability assays were presented as optical density (OD) values or relative values, expressed as a percentage relative to the control group, which comprised cells devoid of glutamate treatment.
In vivo experiments, CCI rats were sacrificed at various time points, and homogenates of cerebral hemisphere tissue from the injured side were extracted. The homogenates were rocked at 4°C for 30 min and centrifuged at 12,000× g for 20 min. In vitro experiments, HT22 cells were seeded into 6-well plates at a density of 1.5 × 105 cells/well and incubated overnight. Following this incubation period, the culture medium was replaced, and the cells were further incubated for an additional 24 h. Post-incubation, the cells were harvested, subjected to a thorough washing with DPBS, and subsequently lysed in RIPA buffer enriched with a protease inhibitor cocktail to yield whole-cell extracts. Accurate quantification of protein concentrations within these samples was accomplished using a bicinchoninic acid protein assay kit (KaiJi Biological Technology Co., Ltd., Jiangsu, China).
Uniform quantities of protein from each sample underwent electrophoresis on 15% sodium dodecyl sulfate (SDS)–polyacrylamide gels before being methodically transferred onto polyvinylidene difluoride membranes. These membranes were incubated for 24 h at 4°C with the following primary antibodies: anti-MMP-9 (1:1000; Boaosen Biotechnology Co., Ltd., Beijing, China), anti-Caspase-3 (1:2000; Proteintech Group, Inc., USA), anti-β-actin (1:3000; Proteintech Group, Inc., USA) and anti-β-tubulin (1:5000; Proteintech Group, Inc., Chicago, IL, USA). The membranes were then incubated for 1 h at room temperature with the following secondary antibodies: goat anti-mouse (1:10000; Abbkine, Wuhan, China) and goat anti-mouse (1:3000, Cell Signaling Technology, lnc., Boston, MA, USA). Visualization was performed by chemiluminescence. Immunoblots were scanned using an Amersham Image Quant 800 uv reader, and the resultant images were subjected to analysis for densitometry using ImageJ software.
The concentrations of TIMP-1 and TIMP-2 within the PDMSC-conditioned media were quantified using ELISA kits (Proteintech Group, Inc., Chicago, IL, USA). This analysis was carried out following the manufacturer’s protocols. For comparative purposes, HT22-conditioned media and fresh DMEM containing 10% serum were employed as controls. The OD of each well was recorded at 450 nm within 15 min from the initiation of the analysis.
To determine the activity of the MMPs produced by HT22 cells in response to glutamate treatment, HT22 cells, seeded into 6-well plates at a density of 1.5 × 105 cells/well, were incubated overnight. Subsequently, these cells were exposed to various concentrations of glutamate (0, 1, 5, or 10 mM) for an additional 15 h. Following treatment, the cells were centrifuged at 3,000× g for 15 min at 4°C and stored at −20°C for gelatin zymography and western blotting analysis.
Gelatin zymography was performed using 8% Tris-glycine SDS–polyacrylamide gels containing 2 mg/mL gelatin. Equal volumes of the conditioned medium samples were combined with zymogram sample buffer (Shanghai Xinfan Biological Technology Co., Ltd., Shanghai, China) and separated using SDS–PAGE. The gels were then immersed in zymogram renaturation buffer, followed by incubation with zymogram development buffer, both provided by Shanghai Xinfan Biological Technology Co., Ltd., Shanghai, China, for 40 h at 37°C. Subsequently, the gels were stained with Coomassie Blue (0.25 g in 100 mL of a solution comprising 45% methanol and 10% glacial acetic acid in double-distilled water) for about 40 min. Gel images were captured using a Gelatin Imaging System (Bio-Rad Laboratories, Inc., CA, USA) and analyzed using Image Lab 3.0 (Bio-Rad Laboratories, Inc., CA, USA). It is pertinent to note that, for quantification purposes, we opted to determine the total MMP levels by aggregating the bands corresponding to pro-MMPs and active-MMPs. Each sample underwent a minimum of three replicates, and representative scans were presented.
The presentation of data adheres to the convention of the mean ± standard deviation. To establish statistical significance, we performed analyses using Student’s t-test and one-way analysis of variance for relevant multiple comparisons. Data analysis was conducted using GraphPad Prism version 7 (GraphPad Software, Inc., La Jolla, CA, USA). A predetermined significance threshold of p < 0.05 was steadfastly applied.
CCI induces MMP-9 accumulation in cortical tissue adjacent to the injured cortex
The expression of MMP-9 in CCI rats was investigated by Immunohistochemistry, Western blotting assay and ELISA (Fig. 1A). An evident elevation in MMP-9 expression was observed in the cortical bordering the CCI-affected cortex at 1, 2, and 3 days after injury in comparison to the sham controls (Figs. 1B and 1C). Statistical analysis revealed a significant time-dependent increase in MMP-9 immunoreactivity following CCI induction. MMP-9 upregulation gradually subsided by day 3 post-CCI. By day 5 following CCI, there was no significant (ns) difference in MMP-9 expression in the cortex proximate to the injured brain tissue compared with the control group (p > 0.05). Similar results were obtained in further western blot experiments (Figs. 1D and 1E). This temporal pattern suggests that interventions targeting MMP-9 following a TBI should be initiated in the acute phase.
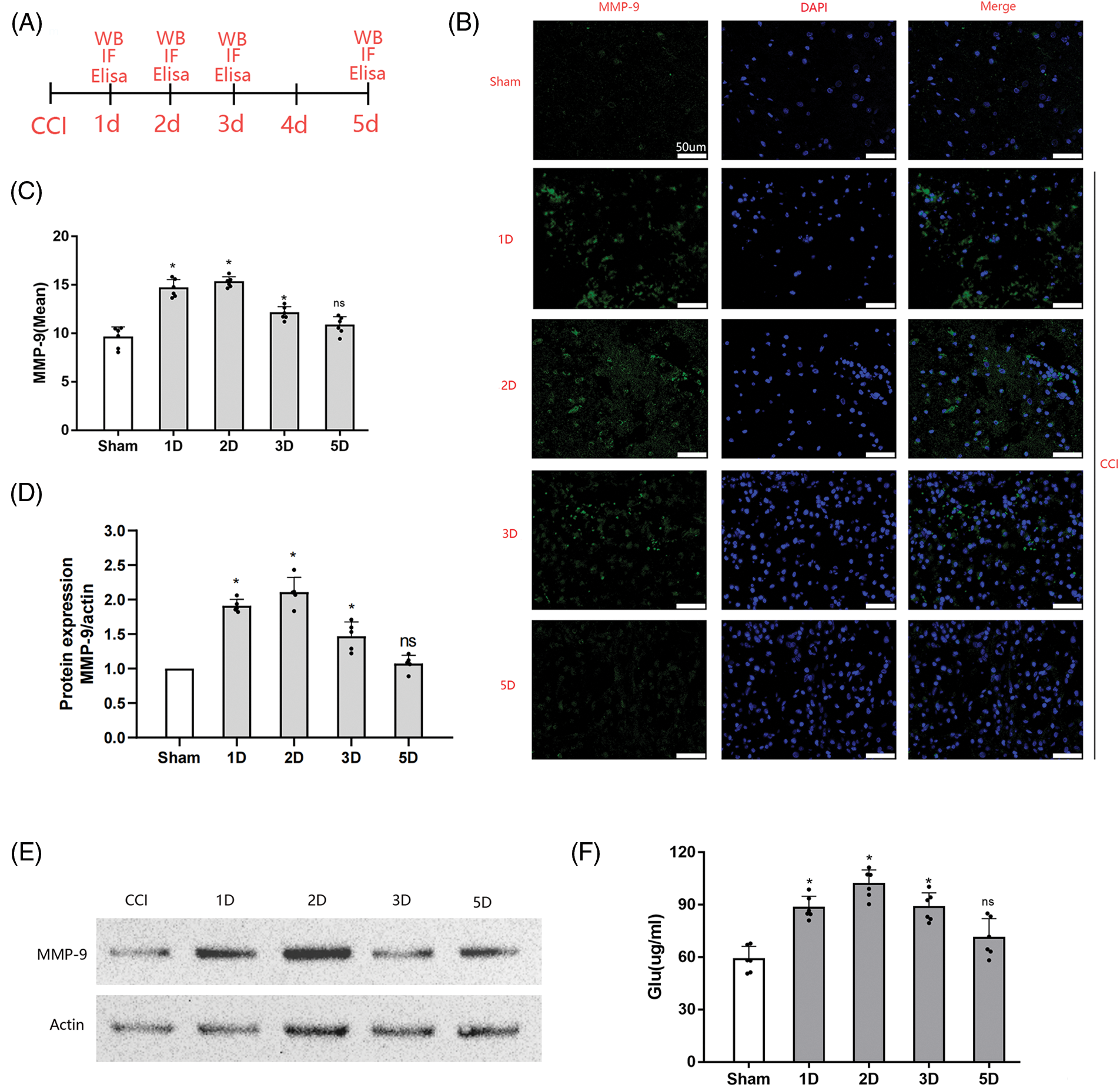
Figure 1: Cortical MMP-9 accumulation and systemic glutamate levels post-CCI. (A) Experimental design and timeline. A total of 30 female Sprague-Dawley rats, divided into the sham group and four treatment groups (1, 2, 3, and 5 days after CCI) for immunofluorescence assay. In addition, 25 rats were used for western blot experiments. Rats in the treatment groups received surgical procedures, while those in the sham group only received craniotomy. Blood samples were obtained from each rat for glutamate level measurements before pathological assessment; (B) Representative images of the cortex depict MMP-9 staining in different groups (scale bars = 50 μm); (C) Quantitative analyses reveal temporal changes in MMP-9 immunoreactivity (n = 3 different sections analyzed from 6 animals per group); (D) Relative expression analysis of MMP-9; (E) Representative images of Western blot analysis of MMP-9; (F) Systemic glutamate levels in rats at different time points after CCI; Data are presented as the mean ± standard deviation. *p < 0.05, ns = not significant (one-way ANOVA).
CCI increases systemic glutamate levels
Concurrently with MMP-9 assessment after CCI, we examined systemic glutamate levels using ELISA. The results demonstrated that CCI induced an elevation in systemic glutamate levels in rats, with significant differences observed on day 1 post-CCI (p < 0.05): peaking on day 2 post-CCI (p < 0.05). By day 3 post-CCI, glutamate levels gradually decreased (p < 0.05). On day 5 post-CCI, no significant difference in plasma glutamate levels was noted compared with sham-operated rats (p > 0.05): as shown in Fig. 1F. These fluctuations in systemic glutamate levels mirrored the trend observed for MMP-9 after CCI.
PDMSC treatment enhances BBB permeability
We designed the following experiments to investigate the protective effects of DPMSC on CCI rats (Fig. 2A). The direct physical impact of CCI leads to the disruption of BBB integrity, further exacerbated by the upregulation or activation of MMP-9. BBB permeability is often indirectly assessed by detecting Evans blue content in brain tissue via absorbance. Our results revealed that BBB breakdown and consequent brain permeability were significantly higher in rats subjected to CCI than those in the sham-operated rats at 72 h (Fig. 2B). However, rats treated with PDMSCs exhibited significantly lower brain permeability than CCI-only rats.
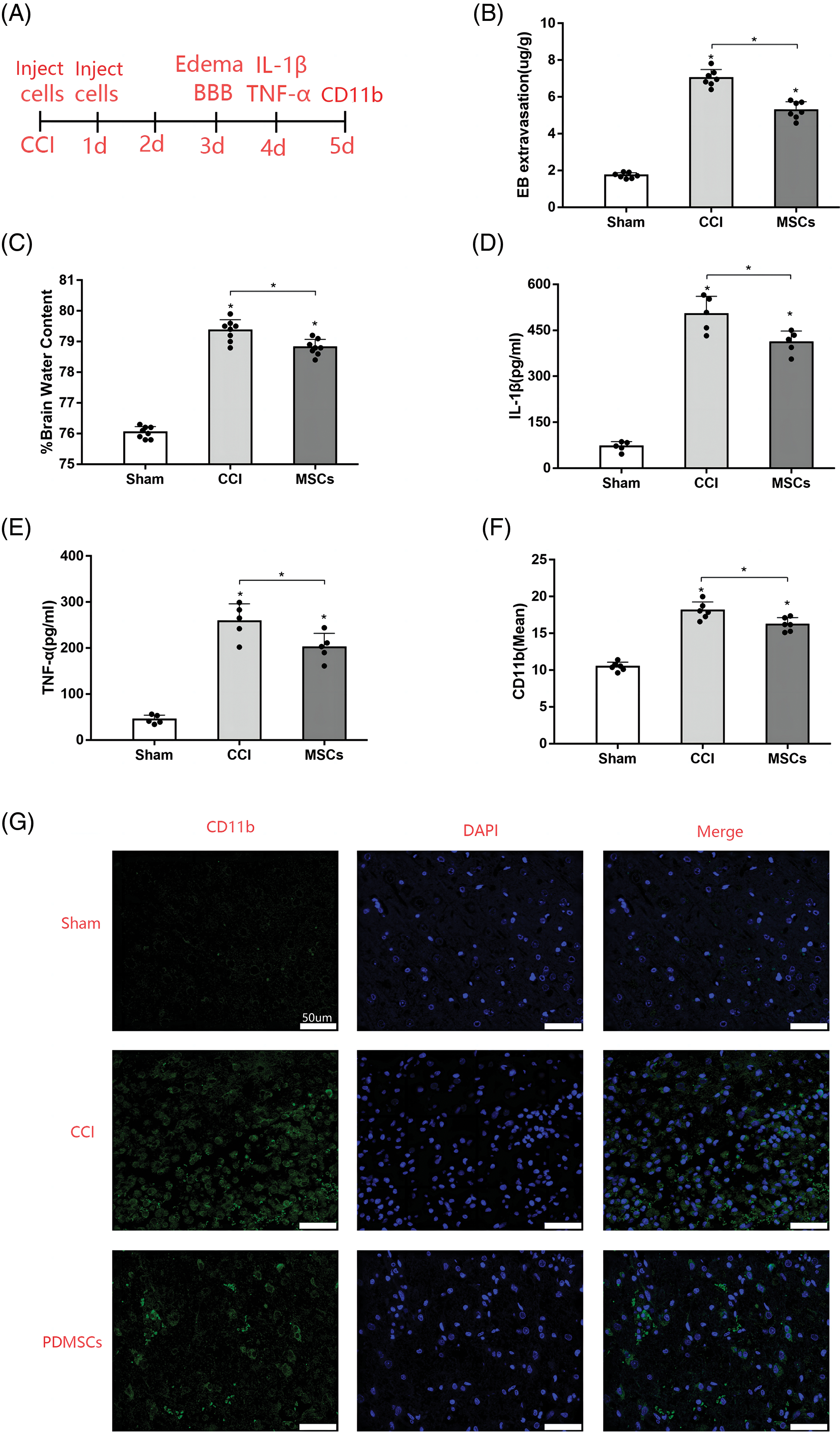
Figure 2: PDMSC treatment effects on secondary brain injury post-CCI. (A) Experimental design and timeline. Rats in the sham group underwent craniotomy only, while those in the treatment group were injected with 1 × 106 PDMSCs through the caudal vein within 30 min and 24 h post-CCI. An equivalent volume of saline was administered to the injured group. On days 3 and 4 post-CCI, selected rats were evaluated for cerebral edema, BBB permeability, and inflammatory cytokine levels, six rats in each group were used to measure the levels of inflammatory cytokines in brain tissues. On day 5 after CCI, six rats in each group were used to detect microglial activation; (B) Quantification of BBB permeability based on dye accumulation in the brain; (C) Measurement of water content in the cerebral hemisphere on the injured side 72 h post-CCI; Influence of PDMSC treatment on IL-1β (D) and TNG-α (E) in cortex homogenates; (F) Quantification of microglia in the cortex adjacent to the lesioned brain tissue of the rats in the sham group, CCI group, and CCI + PDMSC group (n = 3 different sections analyzed from 6 animals per group); (G) Representative images of the injured brain tissue samples stained for CD11b to visualize microglial activation (scale bars = 50 μm).; Data are presented as the mean ± standard deviation. *p < 0.05.
PDMSCs mitigate cerebral edema
Cerebral edema stands as a prominent hallmark of brain injury. We measured the water content in cerebral hemispheres using wet and dry gravimetry. The analysis unveiled a noteworthy increase in water content in the injured cerebral hemisphere on day 3 post-CCI. However, PDMSC treatment markedly mitigated brain tissue edema compared with the CCI group (Fig. 2C).
PDMSCs reduce inflammatory cytokine levels in the injured cortex
To investigate the anti-inflammatory properties of PDMSCs, we assessed the levels of inflammatory cytokines in cortex homogenates 96 h post-CCI. CCI substantially increased IL-1β and TNF-α levels in the damaged cortex compared with those in the sham group. However, the levels of IL-1β (Fig. 2D) and TNF-α (Fig. 2E) were significantly lower in the PDMSC-treatment group compared with those in the CCI group.
PDMSC treatment attenuates microglial activation
TBI is associated with neuroinflammatory responses, including microglial activation. We examined the microglia in rat brains after CCI using immunofluorescent staining with the microglial marker CD11b. Resting microglia exhibited minimal or no CD11b expression, while inflammatory conditions enhanced CD11b expression, reflecting the degree of microglial activation. As depicted in Figs. 2F and 2G, the increased expression of CD11b (p < 0.05) was observed in the cortex adjacent to the lesioned brain tissue on day 5 after CCI. PDMSC therapy attenuated CD11b expression intensity (p < 0.05) compared with the CCI group (scale bars = 20 μm). These results align with previous observations of microglial activation after TBI.
We employed FBS as the standard in our gelatinase assays, as mammalian serum contains substantial pro-MMP-2/9. Consequently, we detected pro-MMP-2 and pro-MMP-9 expressions in all groups, with results indicating no alterations with glutamate treatment (Fig. 3A). Active-MMP-2 was undetectable, and total MMP-2 expression remained unchanged compared with that in the uninjured control (Fig. 3B). Active-MMP-9 expression significantly increased after glutamate injury (10 and 5 mM): resulting in a notable difference in total MMP-9 expression, except in the group treated with 1 mM glutamate (Fig. 3C). Active-MMP-9 was positively associated with glutamate concentration. Active-MMPs are known to trigger apoptotic mechanisms in several cell models. Trypan blue staining enabled the enumeration of viable HT22 cells following glutamate treatment. Higher glutamate concentrations corresponded to increased active-MMP-9 expression, resulting in diminished cell viability (Fig. 3D). The levels of MMP-9 in the supernatant of HT22 cells induced by glutamate at different concentrations were further investigated by Western blot (Figs. 3E and 3F). The final data statistics yielded results similar to those obtained in gelatin zymography experiments. However, from a visual point of view, gelatin zymography experiments can visually visualize the zymogen as well as the activated form of gelatinase.
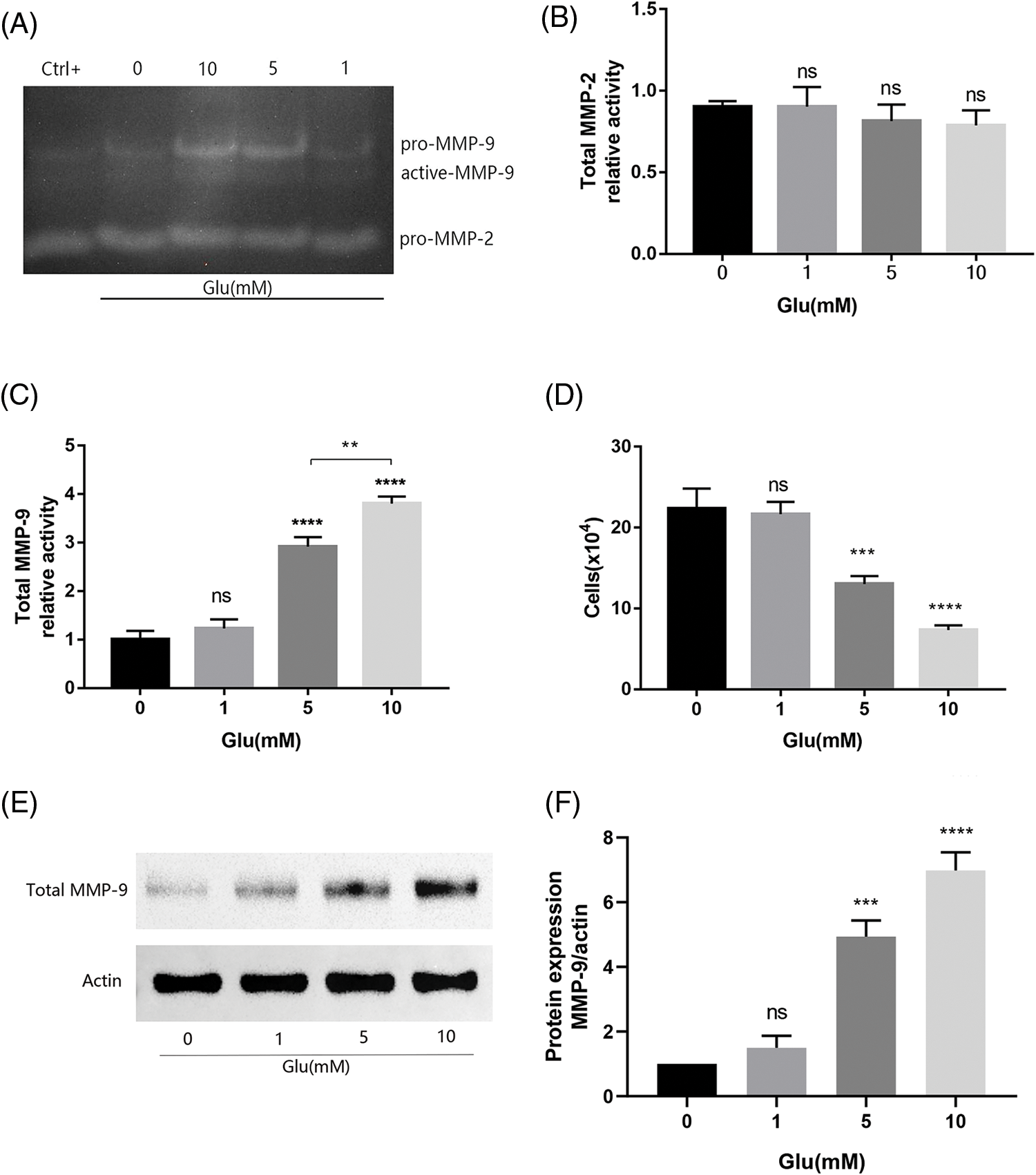
Figure 3: MMP levels in HT22 cell supernatant 15 h post-glutamate treatment. (A) MMP activity is visualized as pale bands where the gelatin has been degraded; Total MMP-2 (B) and total MMP-9 (C) Levels are quantified as the relative activity using Image Lab analysis software (n = 3 biological replicates); (D) Number of viable HT22 cells treated with glutamate based on Trypan blue staining; (E) Representative immunoblot plots of MMP-9 expression in HT22 cell supernatant induced by different concentrations of glutamate; (F) Relative expression analysis of MMP-9. **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. Glu-untreated cells, ns = not significant (one-way ANOVA).
PDMSCs secrete TIMP-1 and TIMP-2
TIMP-1 and TIMP-2 in PDMSC-conditioned media and HT22 cells at different incubation times were analyzed by ELISA, with fresh DMEM containing 10% FBS as the control. Both TIMP-1 and TIMP-2 were below the limit of detection in DMEM and the HT22 group, whether incubated for 1 or 2 days. The PDMSC group exhibited high concentrations of TIMP-1 and TIMP-2, with concentrations increasing over time (Figs. 4A and 4B).
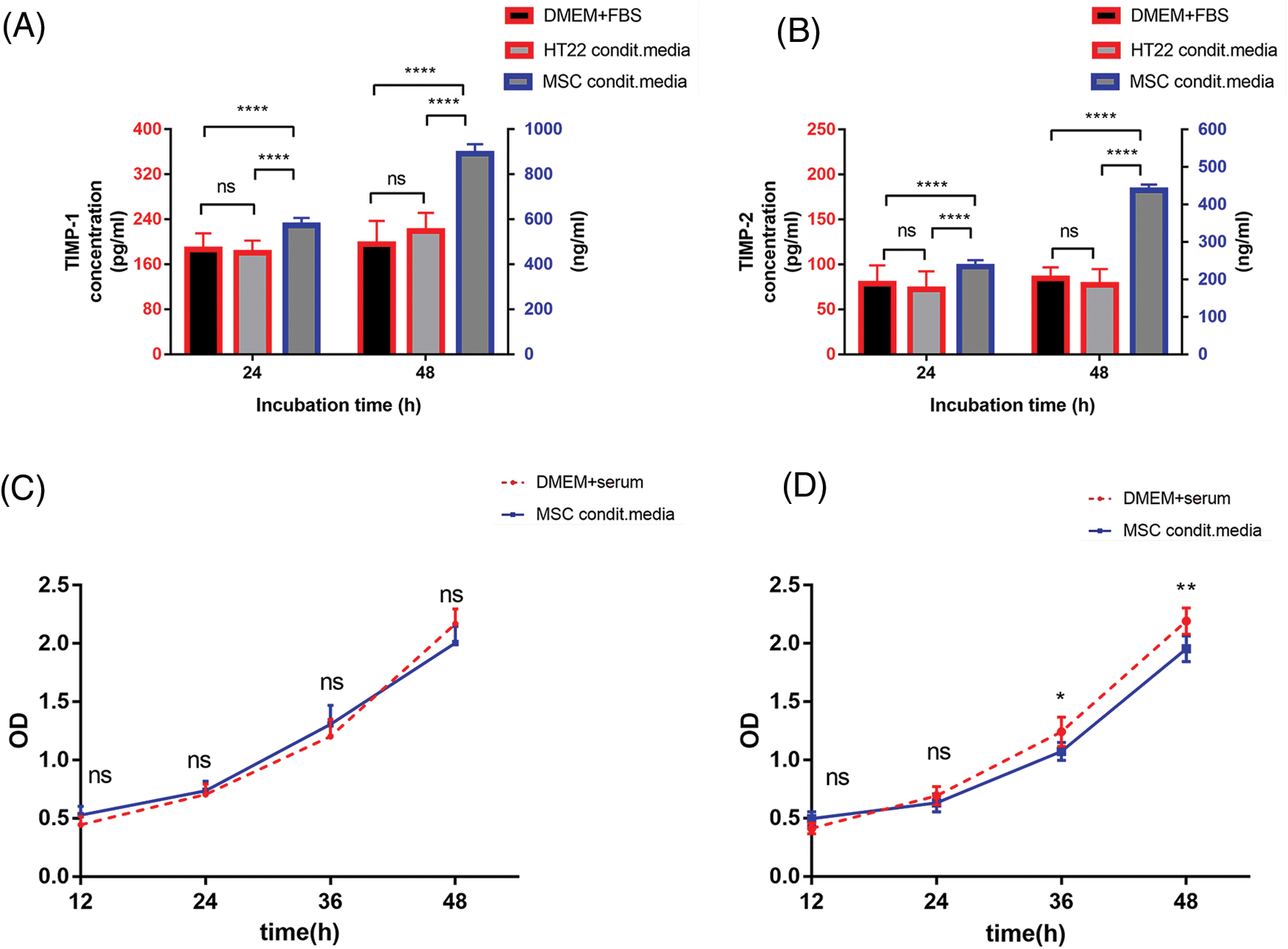
Figure 4: PDMSCs secrete TIMP-1 and TIMP-2. TIMP-1 (A) and TIMP-2 (B) concentrations in PDMSC-conditioned media and HT22 cells for 1 or 2 days compared with those in the fresh DMEM supplemented with 10% FBS; The effect of PDMSC-conditioned media from PDMSCs on HT22 cell viability for 24 (C) or 48 h (D) compared with fresh DMEM supplemented with 10% FBS; *p < 0.05, **p < 0.01, ****p < 0.0001, ns = not significant (one-way ANOVA).
PDMSC-conditioned media influence HT22 cell viability
One challenge with conditioned media is nutrient depletion. Therefore, we evaluated the impact of PDMSC-conditioned media at different incubation times on normal HT22 cell viability. 24-h conditioned media did not affect HT22 cell viability over the observed period compared with traditional media (Fig. 4C). However, 48-h conditioned media inhibited cell viability after 36 h of continuous incubation (Fig. 4D). Despite the higher TIMP-1 and -2 concentrations in the 48-h conditioned media, the 24-h conditioned media were chosen for subsequent experiments based on cell viability analysis results.
PDMSC-conditioned media decrease HT22 cell apoptosis
Glutamate-induced HT22 cells were continuously incubated in conventional or conditioned media for 36 h. Cell survival rates in the conditioned media group were significantly higher, showing a 20% increase (p < 0.0001) after 12 h and an 11% increase (p < 0.0001) after 24 h of exposure to glutamate, compared to the normal media group. However, this survival advantage was no longer evident after 36 h of incubation (p > 0.05, Fig. 5A). The likely explanation for this decline in viability was the depletion of TIMPs in the conditioned media.
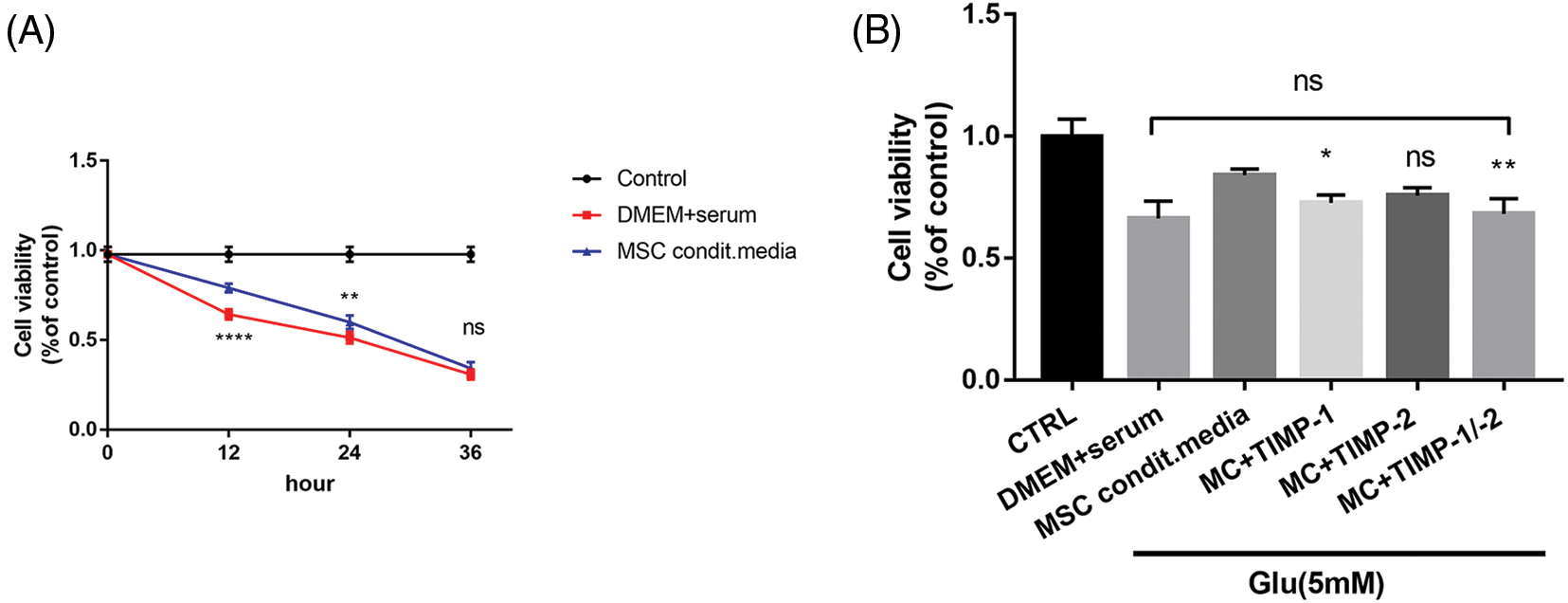
Figure 5: Protective effects of PDMSC-conditioned media on glutamate-treated HT22 cells. (A) PDMSC-conditioned media reduces glutamate-treated HT22 cell apoptosis; (B) Inhibition of TIMP-1 and TIMP-2 activity reverses the protective effects of PDMSC-conditioned media on glutamate-induced HT22 cells; *p < 0.05, **p < 0.01, ****p < 0.0001 vs. DMEM + serum group, ns = not significant (one-way ANOVA).
Inhibition of TIMP-1 and TIMP-2 reverses the protective effects of conditioned media on glutamate-treated HT22 cells
Following the collection of conditioned media, neutralizing antibody dosages were determined based on TIMP-1 and TIMP-2 concentrations in the conditioned media measured by ELISA. The addition of TIMP-1 neutralizing antibody (1 μg/mL) partially reversed the protective effects of conditioned media on HT22 cells (p < 0.05). The introduction of TIMP-2 neutralizing antibody (0.5 μg/mL) yielded a significant, albeit statistically insignificant, effect (p > 0.05). However, concurrent addition of both neutralizing antibodies (1 and 0.5 μg/mL) significantly increased cell apoptosis (p < 0.05). There was no significant difference in the cell survival rate between the neutralizing antibody group and the normal culture group (p > 0.05, Fig. 5B). In all these experiments involving neutralizing antibodies, cells were co-incubated with glutamate for 12 h.
PDMSC-conditioned media reduce the expression of cleaved-caspase-3 in glutamate-induced HT22 cells
Cleaved caspase-3 was significantly lower in the conditioned media group than that in the traditional media group (p < 0.01, Figs. 6A and 6B). There was no significant difference in cleaved-caspase-3 expression between the neutralizing antibody group and the DMEM + serum group (p > 0.05). Fig. 6C is a representative optical microscope image of cells grown under different conditions. These results underscore the pivotal role of active-MMP inhibition in safeguarding neurons against apoptosis.
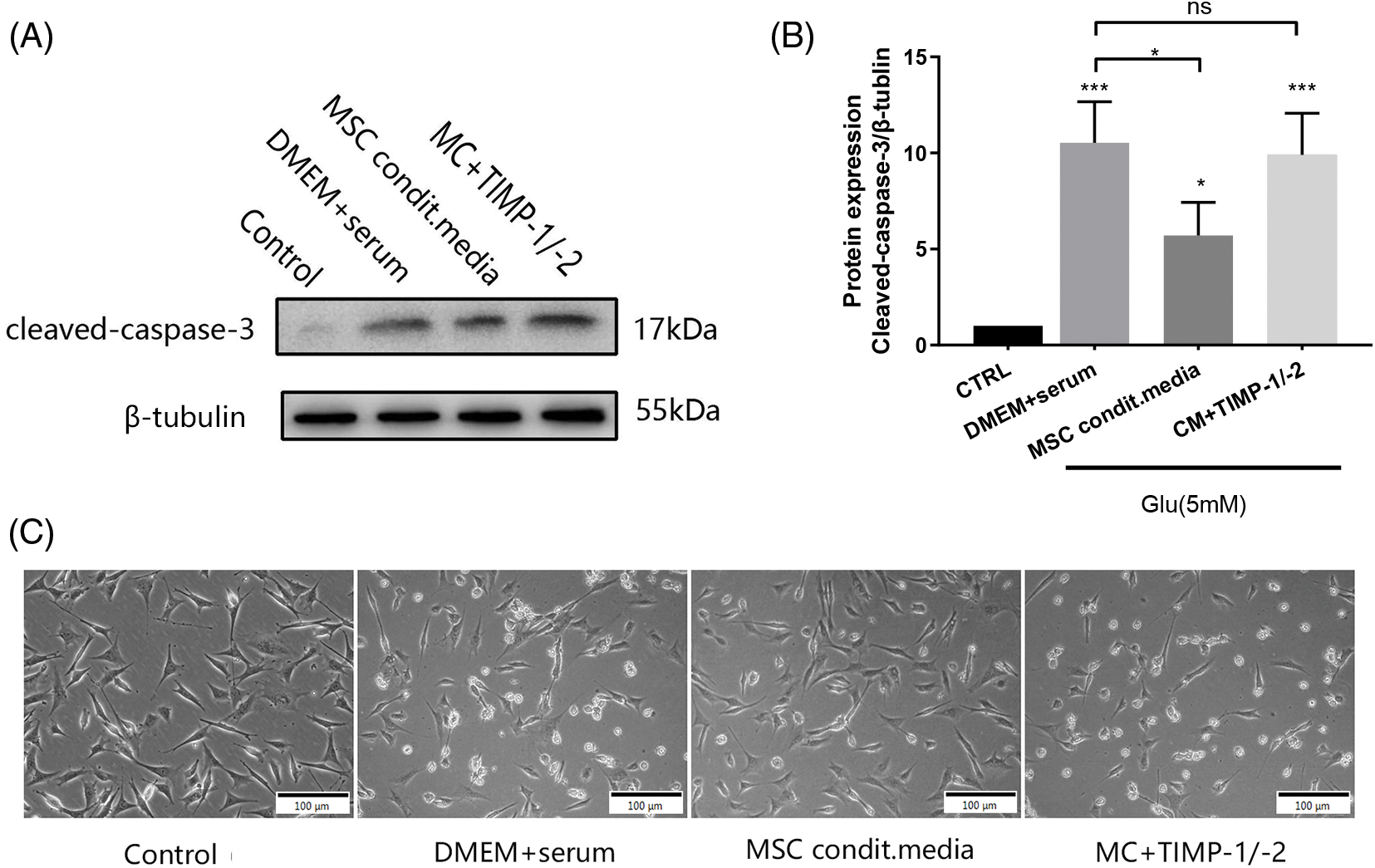
Figure 6: Expression of cleaved-caspase-3 in glutamate-induced HT22 cells after 24 h of incubation under different conditions. (A) Representative immunoblot of cleaved-caspase 3 expressions under different treatment conditions; (B) Densitometry analysis of cleaved-caspase-3 expressions (n = 3 biological replicates); (C) A representative optical microscope image of cells grown under different conditions; *p < 0.05, ***p < 0.001 vs. DMEM + serum group, ns = not significant (one-way ANOVA).
Our study revealed a rapid upregulation of MMP-9 in response to CCI, with a statistically significant difference observed 72 h post-CCI compared with the sham group. This aligns with findings from previous experiments on TBI in rats, where Hayashi et al. [23] detected a significant rise in the levels of MMP-9 24 h after TBI, persisting for at least 48 h as assessed by RT-qPCR. Furthermore, Hadass reported a significant increase in both pro-MMP-9 and active-MMP-9 levels in the injured cortex 24 h after TBI, as determined by gelatin zymography [24]. Although active-MMP-9 levels decreased rapidly after the first day, pro-MMP-9 levels remained elevated in the injured cortex 10 days after TBI. These findings emphasize the importance of initiating therapeutic strategies targeting MMPs during the acute phase of injury.
In parallel with our investigation into MMP-9 expression in rats with brain injury, we explored the concentration of glutamate in their blood. Our results demonstrated a significant and sustained increase in blood glutamate levels following CCI, peaking on day 2 post-injury and remaining significantly higher than those in the sham group for 2 to 5 days before gradually declining. Research has established that abnormally elevated glutamate concentrations in the cerebrospinal fluid or blood can occur in several acute nervous system diseases, including TBI, stroke, and bacterial meningitis [25,26]. TBI results in the stretching and tearing of brain fibers and uncontrolled release of glutamate into the brain parenchyma and extracellular fluid, increasing glutamate concentrations [27]. Elevated glutamate levels can induce oxidative stress injury and neuronal apoptosis, thus leading to cognitive decline [28]. High concentrations of glutamate in the peripheral circulation are associated with a poor prognosis, highlighting the need to mitigate glutamate-induced neurotoxicity and actively improve neurological outcomes [29,30].
In this study, we evaluated BBB integrity in CCI rats using Evans blue extravasation staining. The results showed that PDMSC therapy preserved BBB integrity in CCI rats. Disruption of the BBB results in a massive outflow of intravascular fluid into the brain parenchyma, causing angiogenic edema. Notably, early cerebral edema caused by TBI is mainly attributed to vasogenic cerebral edema. Our study revealed a significant reduction in cerebral edema in CCI rats following cell therapy. These findings align with previous research demonstrating the ability of MSCs to mitigate the degree of brain edema in rodent models of traumatic, ischemic, or hemorrhagic brain injuries [31–33]. Our study provides novel evidence supporting the therapeutic potential of PDMSCs for brain injury. The above studies collectively underscore that in the acute pathological phase of TBI characterized by upregulated expression of MMP-9, both endogenous and exogenous upregulation of TIMPs represent an opportune window for MMP inhibition. This balance between TIMPs and MMPs is essential for preserving an opportune window for and reducing the degree of brain edema. In a cortical injury model, TIMP-1 overexpression in mice led to significantly lower BBB permeability and reduced brain injury volume compared with the wild group [34]. Accordingly, the induction of an MMP-9 knockout model in the context of brain injury engendered a discernible reduction in BBB compromise [35]. Under physiological conditions, the repercussions, while simultaneous administration of TIMP-2 can inhibit this reaction [36].
TBI invariably accompanies neuroinflammatory processes within the central nervous system. A pivotal facet of this inflammatory cascade is the activation of microglia, which stands as a central player in this dynamic response. It is an established notion that the exacerbation and prolonged activation of microglia are detrimental to neurons and astrocytes. However, it is imperative to acknowledge that microglial responses remain indispensable for essential functions such as the clearance of necrotic tissue and the initiation of myelin repair [37]. One consequential outcome of immune activation is the release of a variety of pro-inflammatory cytokines, including IL-1β, IL-6, IL-12, TNF-α, and chemokines [38]. The unwarranted overproduction of these inflammatory cytokines heralds the onset of sustained brain edema, neurological deficits, and impaired cognitive function. It warrants mention that microglial activation can be initiated even in cases of mild to moderate TBI, thereby perpetuating and intensifying the presence of inflammatory signals in response to secondary stimuli [39]. Intriguingly, a clinical study by Xu et al. unveiled that TBI patients continued to exhibit heightened levels of inflammatory cytokines three months post-injury. Notably, certain cytokines, such as IFN-α, IL 1-β, IL-6, and TNF-α, bore predictive potential with regard to poor cognitive function [40]. Therefore, the inhibition of inflammatory cytokines after TBI has a positive effect on reducing secondary brain injury, reducing neurological impairment, and improving cognitive function. Within the scope of our investigation, we ascertained that TBI led to microglial activation in the cortex near the injury site, accompanied by elevations in the inflammatory cytokines IL-1β and TNF-α. However, the administration of PDMSCs inhibited microglial activation while concurrently diminishing the levels of inflammatory cytokines in the injured brain tissue. These findings align with the outcomes reported by Yan et al. whereby BMSC treatment manifested analogous inhibitory effects on microglial activation [41]. Furthermore, transient microglial depletion after brain injury reduces the expression of pro-inflammatory cytokines, reduces neuronal death, and prevents long-term cognitive impairment [42].
Oxidative stress injury generally assumes a prominent role in the pathological process of TBI. Glutamate, the principal excitatory neurotransmitter in the mammalian brain, occupies a pivotal position in learning and memory by maintaining synaptic plasticity. However, abnormal surges in glutamate levels can cause oxidative stress damage in the central nervous system. The neurotoxic effects of glutamate involve not only acute injury, including TBI and ischemic brain injury, but also chronic neuronal degeneration, such as Huntington’s disease, Parkinson’s disease, multiple sclerosis, and Amyotrophic lateral sclerosis [43]. In the animal experiments performed in the present study, employing the CCI rat model, we observed a substantial elevation in plasma glutamate levels in rats in the acute stage of brain injury. The HT22 cell line, immortalized neurons derived from murine hippocampal tissue, is a widely recognized in vitro model for investigating oxidative stress injury induced by glutamate [44,45]. Previous studies have established that the MAPK signaling pathway is involved in the upregulation of MMP-9 in rodent models of TBI. In the in vitro model of oxidative stress injury, the MAPK signaling pathway was involved in glutamate-induced damage to HT22 [46,47]. Furthermore, our investigations revealed a positive correlation between glutamate concentration and the expression of active-MMP-9 in HT22 cells, consistent with findings reported by Dziembowska et al. wherein glutamate stimulation of cultured hippocampal neurons rapidly upregulated the expression of MMP-9 [48]. It is noteworthy that MMPs are not confined to neurons but are secreted by a diverse array of cell types, including neurons, astrocytes, endothelial cells, fibroblasts, osteoblasts, and leukocytes. These proteases initially assume the form of proenzymes (pro-MMPs): which are then activated into their functional state (active-MMPs) by proteolytic enzymes such as serine proteases, furans, and plasminases [49]. The overexpression/activation of MMPs disrupts the equilibrium between TIMPs, a pathophysiological mechanism observed in various neurodegenerative diseases [50,51]. Compelling evidence substantiates the contribution of MMPs to neuronal demise in both in vivo and in vitro settings, as elucidated by the application of chemical inhibitors and genetic knockout models [52,53]. Therefore, MMPs are generally considered promising targets for pharmacological intervention.
MMP inhibitors (MMPIs) include endogenous tissue inhibitors and exogenous pharmacologically synthesized inhibitors. Among these, TIMPs have emerged as extensively studied representatives. TIMPs exhibit the remarkable capacity to restrain the activity of all MMPs, with the notable exception of MMP-14 [54]. TIMPs establish intricate interactions with active-MMPs, forming a tight, non-covalent 1:1 complex without cleaving TIMPs. The key mechanism through which TIMPs exert their inhibitory influence on active-MMPs resides in the binding of the wedge-shaped ridge structure within the N-domain of TIMP to the precise active cleavage site of the target MMP [55]. Several externally synthesized MMPIs have been developed, most of which inactivate MMPs by replacing zinc-bound water molecules with zinc-bound globulin [56]. However, they are nowhere near as effective as the TIMPs. Pro-MMPs are activated by the proteolytic removal of the N-terminal inhibitory domain [57]. Once activated, active-MMPs can be inhibited by TIMPs [58]. Within the purview of MMPIs, N-terminal TIMP (N-TIMP) derivatives, forged through the annals of protein engineering, have garnered significant attention. These stable peptides adhere to the same mechanism for inhibiting MMPs as endogenous TIMPs. In addition, due to their relatively small size, N-TIMPs are proficient at traversing cell membranes directly [59]. In the present study, PDMSCs secreted large quantities of TIMP-1 and TIMP-2. In the extracellular environment, these TIMPs exhibit a pronounced proclivity for selective binding to active-MMPs, thereby impeding their enzymatic activity and reducing cell apoptosis. Studies by Lozito et al. underscored that MSCs curtailed heightened exogenous MMP-2 and MMP-9 activity, leveraging the abundant reservoirs of TIMP-1 and TIMP-2 [60,61]. Beyond the confines of neurological contexts, MMPs are involved in the invasion of tumor cells in oncological diseases. To this end, the work of Mitchell has illuminated the capacity of MSCs to mitigate the migration and invasion of breast cancer cells by secreting TIMP-1 and TIMP-2 [62].
Numerous compounds have demonstrated the capacity to shield HT22 cells from glutamic acid-induced damage, with these substances primarily enhancing HT22 activity by inhibiting ROS generation and Ca2+ release, restoring the physiological mitochondrial membrane potential, increasing glutathione production activity, or inhibiting MAPK phosphorylation [19,63,64]. The present study focused on MMPs, a key downstream protein in the complex process of events accompanying oxidative stress injury. Our findings illuminated that glutamate exposure increased active-MMP-9 expression in HT22 cells. Furthermore, PDMSC-conditioned media featuring high levels of TIMP-1 and TIMP-2 significantly reduced the apoptotic rate and the expression of cleaved caspase-3 in glutamate-induced HT22 cells. The addition of neutralizing antibodies targeting TIMP-1 and TIMP-2 reversed the protective effect of conditioned media on HT22 cells. Our study provides compelling evidence that PDMSCs secrete TIMP-1 and TIMP-2, which in turn confer protection against oxidative stress-induced neuronal cell death. These findings introduce novel insights and therapeutic options for the management of central nervous system diseases that involve oxidative stress injury. Furthermore, our research lays the essential theoretical groundwork for the future development of targeted pharmacological interventions.
In summary, this study underscores the potential of PDMSCs as a promising treatment for alleviating secondary TBI. TBI is a complex condition involving excitotoxicity, oxidative stress, and inflammatory responses. Conventional neuroprotective drugs often target singular mechanisms, resulting in limited treatment options. Ideally, treatments aimed at protecting the brain in cases of TBI should be capable of addressing multiple aspects of TBI pathology simultaneously, including cell protection, neural nourishment, and regeneration promotion. Alternatively, if a single treatment cannot accomplish all these objectives, a combination of different treatments can be used to achieve the desired outcomes. Numerous studies have identified promising therapeutic targets in TBI. MSC therapy, either in isolation or as part of combination therapies, exhibits almost unlimited possibilities. The adaptable delivery modes of MSCs allow transplantation strategies to align with the brain’s inflammatory response. Local intracerebral delivery of MSCs is suitable for focal injuries as it directly targets the inflammatory area. In contrast, intravenous systemic administration can address diffuse brain injuries and impact peripheral components, such as the spleen, reducing inflammation from central and peripheral sources. MSCs deliver robust short-term neuroprotective effects while promoting long-term benefits by enhancing nerve regeneration and modulating immune responses. Genetic modification of MSCs has shown promise in TBI treatment, enhancing their viability, proliferation, migration, and therapeutic potential in a safe and targeted manner. Future research is expected to yield innovative translational applications of MSC-based therapies. MSCs remain a fertile field for exploration, offering numerous cell-based innovations and translational possibilities.
In conclusion, this study underscores the remarkable potential of PDMSCs in alleviating secondary brain injury following CCI in rats. Moreover, the pivotal role of PDMSC-secreted and TIMP-1 and TIMP-2 in protecting neurons against oxidative stress-induced demise has been elucidated. The remarkable multi-target and multi-pathway attributes of PDMSCs position them as promising candidates for addressing the intricate mechanisms in TBI. These findings offer fresh insights into the potential clinical interventions for TBI.
Acknowledgement: The authors would like to thank TopEdit (www.topeditsci.com) for its linguistic assistance during the preparation of this manuscript.
Funding Statement: This study was financially supported by the Key Research Projects of Ningxia Hui Autonomous Region of China under Grant No. 2018BCG01002 (to HCX). The funding sources assumed no role in the study’s conception and design, data analysis, interpretation, manuscript preparation, or the decision to submit it for publication.
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Hechun Xia; data collection: Ping Yang; analysis and interpretation of results: Yuanxiang Lan, Zhong Zeng, Yan Wang; draft manuscript preparation: Ping Yang. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics Approval: All procedures carried out in this study received ethical approval and adhered rigorously to the ethical standards stipulated by the Ethics Committee of the General Hospital of Ningxia Medical University (approval No. 2017-073).
Conflicts of Interest: The authors declare that there are no conflicts of interest.
References
1. Voormolen D, Polinder S, von Steinbuechel N, Feng Y, Wilson L, Oppe M, et al. Health-related quality of life after traumatic brain injury: deriving value sets for the QOLIBRI-OS for Italy, The Netherlands and The United Kingdom. Qual Life Res. 2020;29(11):3095–107. [Google Scholar] [PubMed]
2. Huijben J, Wiegers E, Lingsma H, Citerio G, Maas A, Menon D, et al. Changing care pathways and between-center practice variations in intensive care for traumatic brain injury across Europe: a CENTER-TBI analysis. Intensive Care Med. 2020;46(5):995–1004. [Google Scholar] [PubMed]
3. Hanscom M, Loane DJ, Shea-Donohue T. Brain-gut axis dysfunction in the pathogenesis of traumatic brain injury. J Clin Invest. 2021;131(12):e143777. [Google Scholar] [PubMed]
4. Zou Z, Li L, Li Q, Zhao P, Zhang K, Liu C, et al. The role of S100B/RAGE-enhanced ADAM17 activation in endothelial glycocalyx shedding after traumatic brain injury. J Neuroinflamm. 2022;19(1):46. [Google Scholar]
5. Holden SS, Grandi FC, Aboubakr O, Higashikubo B, Cho FS, Chang AH, et al. Complement factor C1q mediates sleep spindle loss and epileptic spikes after mild brain injury. Sci. 2021;373(6560):eabj2685. [Google Scholar]
6. Casault C, Al Sultan A, Banoei M, Couillard P, Kramer A, Winston BJ. Cytokine responses in severe traumatic brain injury: where there is smoke, is there fire? Neurocrit Care. 2019;30(1):22–32. [Google Scholar] [PubMed]
7. Haorah J, Ramirez S, Schall K, Smith D, Pandya R, Persidsky Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. Journal of Neurochemistry. 2007;101(2):566–76. [Google Scholar] [PubMed]
8. Robinson BD, Isbell CL, Melge AR, Lomas AM, Shaji CA, Mohan CG, et al. Doxycycline prevents blood-brain barrier dysfunction and microvascular hyperpermeability after traumatic brain injury. Sci Rep. 2022;12(1):11903. [Google Scholar]
9. Zhang S, Kojic L, Tsang M, Grewal P, Liu J, Namjoshi D, et al. Distinct roles for metalloproteinases during traumatic brain injury. Neurochem Int. 2016;96:46–55. [Google Scholar] [PubMed]
10. Wei P, Wang K, Luo C, Huang Y, Misilimu D, Wen H, et al. Cordycepin confers long-term neuroprotection via inhibiting neutrophil infiltration and neuroinflammation after traumatic brain injury. J Neuroinflamm. 2021;18(1):137. [Google Scholar]
11. Qiu Y, Buffonge S, Ramnath R, Jenner S, Fawaz S, Arkill KP, et al. Endothelial glycocalyx is damaged in diabetic cardiomyopathy: angiopoietin 1 restores glycocalyx and improves diastolic function in mice. Diabetologia. 2022;65(5):879–94. [Google Scholar] [PubMed]
12. Tiwari D, Martineau AR. Inflammation-mediated tissue damage in pulmonary tuberculosis and host-directed therapeutic strategies. Semin Immunol. 2023;65:101672. [Google Scholar] [PubMed]
13. Iemmolo M, Ghersi G, Bivona G. The cytokine CX3CL1 and ADAMs/MMPs in concerted cross-talk influencing neurodegenerative diseases. Int J Mol Sci. 2023;24(9):8026. [Google Scholar] [PubMed]
14. Simões G, Pereira T, Caseiro A. Matrix metaloproteinases in vascular pathology. Microvasc Res. 2022;143:104398. [Google Scholar]
15. Maeda Y, Otsuka T, Takeda M, Okazaki T, Shimizu K, Kuwabara M, et al. Transplantation of rat cranial bone-derived mesenchymal stem cells promotes functional recovery in rats with spinal cord injury. Sci Rep. 2021;11(1):21907. [Google Scholar] [PubMed]
16. Qi L, Xue X, Sun J, Wu Q, Wang H, Guo Y, et al. The promising effects of transplanted umbilical cord mesenchymal stem cells on the treatment in traumatic brain injury. J Craniofac Surg. 2018;29(7):1689–92. [Google Scholar] [PubMed]
17. Wang F, Tang H, Zhu J, Zhang JH. Transplanting mesenchymal stem cells for treatment of ischemic stroke. Cell Transplant. 2018;27(12):1825–34. [Google Scholar] [PubMed]
18. Pires A, Teixeira F, Mendes-Pinheiro B, Serra S, Sousa N, Salgado Aónio J. Old and new challenges in Parkinson’s disease therapeutics. Prog Neurobiol. 2017;156:69–89. [Google Scholar] [PubMed]
19. Walker P, Shah S, Harting M, Cox CS. Progenitor cell therapies for traumatic brain injury: barriers and opportunities in translation. Dis Model Mech. 2009;2(1–2):23–38. [Google Scholar] [PubMed]
20. Kodali M, Madhu LN, Reger RL, Milutinovic B, Upadhya R, Gonzalez JJ, et al. Intranasally administered human MSC-derived extracellular vesicles inhibit NLRP3-p38/MAPK signaling after TBI and prevent chronic brain dysfunction. Brain Behav Immun. 2023;108(1):118–34. [Google Scholar] [PubMed]
21. Wang D, Zhang S, Ge X, Yin Z, Li M, Guo M, et al. Mesenchymal stromal cell treatment attenuates repetitive mild traumatic brain injury-induced persistent cognitive deficits via suppressing ferroptosis. J Neuroinflamm. 2022;19(1):185. [Google Scholar]
22. Köhli P, Otto E, Jahn D, Reisener MJ, Appelt J, Rahmani A, et al. Future perspectives in spinal cord repair: brain as saviour? TSCI with concurrent TBI: pathophysiological interaction and impact on MSC treatment. Cells. 2021;10(11):2955. [Google Scholar]
23. Hayashi T, Kaneko Y, Yu S, Bae E, Stahl C, Kawase T, et al. Quantitative analyses of matrix metalloproteinase activity after traumatic brain injury in adult rats. Brain Res. 2009;1280:172–7. [Google Scholar] [PubMed]
24. Hadass O, Tomlinson B, Gooyit M, Chen S, Purdy J, Walker J, et al. Selective inhibition of matrix metalloproteinase-9 attenuates secondary damage resulting from severe traumatic brain injury. PLoS One. 2013;8(10):e76904. [Google Scholar] [PubMed]
25. Shen Z, Xiang M, Chen C, Ding F, Wang Y, Shang C, et al. Glutamate excitotoxicity: potential therapeutic target for ischemic stroke. Biomed Pharmacother. 2022;151(1):113125. [Google Scholar] [PubMed]
26. Verma M, Lizama BN, Chu CT. Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration. Transl Neurodegen. 2022;11(1):3. [Google Scholar]
27. Frank D, Gruenbaum BF, Shelef I, Zvenigorodsky V, Severynovska O, Fleidervish I, et al. Blood glutamate scavenging as a novel glutamate-based therapeutic approach for post-traumatic brain injury anxiety and social impairment. Transl Psychiatry. 2023;13(1):41. [Google Scholar] [PubMed]
28. Wilcox JM, Consoli DC, Tienda AA, Dixit S, Buchanan RA, May JM, et al. Altered synaptic glutamate homeostasis contributes to cognitive decline in young APP/PSEN1 mice. Neurobiol Dis. 2021;158(8):105486. [Google Scholar] [PubMed]
29. Neul JL, Percy AK, Benke TA, Berry-Kravis EM, Glaze DG, Marsh ED, et al. Trofinetide for the treatment of Rett syndrome: a randomized phase 3 study. Nat Med. 2023;29(6):1468–75. [Google Scholar] [PubMed]
30. Sun JY, Zhao SJ, Wang HB, Hou YJ, Mi QJ, Yang MF, et al. Ifenprodil improves long-term neurologic deficits through antagonizing glutamate-induced excitotoxicity after experimental subarachnoid hemorrhage. Transl Stroke Res. 2021;12(6):1067–80. [Google Scholar] [PubMed]
31. Wang C, Börger V, Mohamud Yusuf A, Tertel T, Stambouli O, Murke F, et al. Postischemic neuroprotection associated with anti-inflammatory effects by mesenchymal stromal cell-derived small extracellular vesicles in aged mice. Stroke. 2022;53(1):e14–8. [Google Scholar] [PubMed]
32. Do PT, Chuang DM, Wu CC, Huang CZ, Chen YH, Kang SJ, et al. Mesenchymal stem cells overexpressing FGF21 preserve blood-brain barrier integrity in experimental ischemic stroke. Transl Stroke Res. 2023. doi:https://doi.org/10.1007/s12975-023-01196-8 [Google Scholar] [PubMed] [CrossRef]
33. Ahn SY, Sung DK, Kim YE, Sung S, Chang YS, Park WS. Brain-derived neurotropic factor mediates neuroprotection of mesenchymal stem cell-derived extracellular vesicles against severe intraventricular hemorrhage in newborn rats. Stem Cells Transl Med. 2021;10(3):374–84. [Google Scholar] [PubMed]
34. Tejima E, Guo S, Murata Y, Arai K, Lok J, van Leyen K, et al. Neuroprotective effects of overexpressing tissue inhibitor of metalloproteinase TIMP-1. J Neurotrauma. 2009;26(11):1935–41. [Google Scholar] [PubMed]
35. Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20(12):1681–9. [Google Scholar] [PubMed]
36. Zhang Y, Liu H, Chen Z, Yu M, Li J, Dong H, et al. TLR4-mediated hippocampal MMP/TIMP imbalance contributes to the aggravation of perioperative neurocognitive disorder in db/db mice. Neurochem Int. 2020;140:104818. [Google Scholar] [PubMed]
37. Cunha MI, Su M, Cantuti-Castelvetri L, Müller SA, Schifferer M, Djannatian M, et al. Pro-inflammatory activation following demyelination is required for myelin clearance and oligodendrogenesis. J Exp Med. 2020;217(5):e20191390. [Google Scholar] [PubMed]
38. Bolte AC, Lukens JR. Neuroimmune cleanup crews in brain injury. Trends Immunol. 2021;42(6):480–94. [Google Scholar] [PubMed]
39. McKim D, Weber M, Niraula A, Sawicki C, Liu X, Jarrett B, et al. Microglial recruitment of IL-1β-producing monocytes to brain endothelium causes stress-induced anxiety. Mol Psychiatry. 2018;23(6):1421–31. [Google Scholar] [PubMed]
40. Xu W, Yue S, Wang P, Wen B, Zhang X. Systemic inflammation in traumatic brain injury predicts poor cognitive function. Immun Inflamm Dis. 2022;10(3):e577. [Google Scholar] [PubMed]
41. Yan K, Zhang R, Sun C, Chen L, Li P, Liu Y, et al. Bone marrow-derived mesenchymal stem cells maintain the resting phenotype of microglia and inhibit microglial activation. PLoS One. 2013;8(12):e84116. [Google Scholar] [PubMed]
42. Witcher K, Bray C, Chunchai T, Zhao F, O’Neil S, Gordillo A, et al. Traumatic brain injury causes chronic cortical inflammation and neuronal dysfunction mediated by microglia. The Journal of Neuroscience. 2021;41(7):1597–616. [Google Scholar] [PubMed]
43. Andersen JV, Markussen KH, Jakobsen E, Schousboe A, Waagepetersen HS, Rosenberg PA, et al. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology. 2021;196(1802):108719. [Google Scholar] [PubMed]
44. Chen S, Zou Q, Guo Q, Chen Y, Kuang X, Zhang Y, et al. SPARC knockdown reduces glutamate-induced HT22 hippocampal nerve cell damage by regulating autophagy. Front Neurosci. 2021;14:581441. [Google Scholar] [PubMed]
45. Jacques MT, Saso L, Farina M. LPS-activated microglial cell-derived conditioned medium protects HT22 neuronal cells against glutamate-induced ferroptosis. Int J Mol Sci. 2023;24(3):2910. [Google Scholar] [PubMed]
46. Ko J, Jang S, Kwon W, Kim SY, Jang S, Kim E, et al. Protective effect of gip against monosodium glutamate-induced ferroptosis in mouse hippocampal HT-22 cells through the MAPK signaling pathway. Antioxid. 2022;11(2):189. [Google Scholar]
47. Kwon OY, Lee SH. Ishige okamurae suppresses trimethyltin-induced neurodegeneration and glutamate-mediated excitotoxicity by regulating MAPKs/Nrf2/HO-1 antioxidant pathways. Antioxid. 2021;10(3):440. [Google Scholar]
48. Dziembowska M, Milek J, Janusz A, Rejmak E, Romanowska E, Gorkiewicz T, et al. Activity-dependent local translation of matrix metalloproteinase-9. The Journal of Neuroscience. 2012;32(42):14538–47. [Google Scholar] [PubMed]
49. Liu J, Khalil RJ. Matrix metalloproteinase inhibitors as investigational and therapeutic tools in unrestrained tissue remodeling and pathological disorders. Prog Mol Biol Transl Sci. 2017;148:355–420. [Google Scholar] [PubMed]
50. Engel C, Pirard B, Schimanski S, Kirsch R, Habermann J, Klingler O, et al. Structural basis for the highly selective inhibition of MMP-13. Chem Biol. 2005;12(2):181–9. [Google Scholar] [PubMed]
51. Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92(8):827–39. [Google Scholar] [PubMed]
52. Kiaei M, Kipiani K, Calingasan N, Wille E, Chen J, Heissig B, et al. Matrix metalloproteinase-9 regulates TNF-α and FasL expression in neuronal, glial cells and its absence extends life in a transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2007;205(1):74–81. [Google Scholar] [PubMed]
53. Guedez L, Stetler-Stevenson W, Wolff L, Wang J, Fukushima P, Mansoor A, et al. In vitro suppression of programmed cell death of B cells by tissue inhibitor of metalloproteinases-1. J Clin Investig. 1998;102(11):2002–10. [Google Scholar] [PubMed]
54. Hu J, Van den Steen P, Sang Q, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007;6(6):480–98. [Google Scholar] [PubMed]
55. Li K, Tay F, Yiu C. The past, present and future perspectives of matrix metalloproteinase inhibitors. Pharmacol Therapeut. 2020;207:107465. [Google Scholar]
56. Murphy G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011;12(11):233. [Google Scholar] [PubMed]
57. Ries C, Egea V, Karow M, Kolb H, Jochum M, Neth P. MMP-2, MT1-MMP, and TIMP-2 are essential for the invasive capacity of human mesenchymal stem cells: differential regulation by inflammatory cytokines. Blood. 2007;109(9):4055–63. [Google Scholar] [PubMed]
58. Rodríguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta (BBA)—Mol Cell Res. 2010;1803(1):39–54. [Google Scholar]
59. Hosseini A, Kumar S, Hedin K, Raeeszadeh‐Sarmazdeh M. Engineering minimal tissue inhibitors of metalloproteinase targeting MMPs via gene shuffling and yeast surface display. Protein Sci. 2023;32(12):e4795. [Google Scholar] [PubMed]
60. Lozito TP, Jackson WM, Nesti LJ, Tuan RS. Human mesenchymal stem cells generate a distinct pericellular zone of MMP activities via binding of MMPs and secretion of high levels of TIMPs. Matrix Biol. 2014;34:132–43. [Google Scholar] [PubMed]
61. Scuteri A, Ravasi M, Pasini S, Bossi M, Tredici GJN. Mesenchymal stem cells support dorsal root ganglion neurons survival by inhibiting the metalloproteinase pathway. Neurosci. 2011;172:12–9. [Google Scholar]
62. Clarke M, Imhoff F, Baird SK. Mesenchymal stem cells inhibit breast cancer cell migration and invasion through secretion of tissue inhibitor of metalloproteinase-1 and -2. Mol Carcinog. 2015;54(10):1214–9. [Google Scholar] [PubMed]
63. Wang L, Zhang R, Li J. Transplantation of neurotrophin-3-expressing bone mesenchymal stem cells improves recovery in a rat model of spinal cord injury. Acta Neurochir. 2014;156(7):1409–18. [Google Scholar] [PubMed]
64. Liu L, Cao J, Sun B, Li H, Xia Y, Wu Z, et al. Mesenchymal stem cells inhibition of chronic ethanol-induced oxidative damage via upregulation of phosphatidylinositol-3-kinase/Akt and modulation of extracellular signal-regulated kinase 1/2 activation in PC12 cells and neurons. Neurosci. 2010;167(4):1115–24. [Google Scholar]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools