
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
E3 Ligases and COVID-19: Insights into Viral Control and Therapeutic Potential
1 Department of Cancer Biology, Abramson Family Cancer Research Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
2 Department of Pathology, Jawaharlal Nehru Medical College, Aligarh Muslim University, Aligarh, 202002, Uttar Pradesh, India
* Corresponding Author: Sehbanul Islam. Email:
(This article belongs to the Special Issue: Functional Interactions Between Viral Proteins and Cellular Mechanisms)
BIOCELL 2025, 49(1), 127-147. https://doi.org/10.32604/biocell.2025.058038
Received 03 September 2024; Accepted 09 December 2024; Issue published 24 January 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
The COVID-19 pandemic, caused by the newly emerged coronavirus SARS-CoV-2, has resulted in unprecedented global health challenges, including millions of infections and deaths. While the direct effects of the virus are critical, the interplay between SARS-CoV-2 and cellular host factors significantly impacts the replication cycle of the virus and the clinical severity of COVID-19. This review provides a comprehensive analysis of host-pathogen interactions, focusing on the functional roles and regulatory mechanisms of SARS-CoV-2 viral proteins. We systematically review the literature to detail how SARS-CoV-2 engages with host cellular machinery, with a specific emphasis on their modulation by E3 ubiquitin ligases. By dissecting these intricate interactions and the impact of E3 ligases on SARS-CoV-2 infection, we aim to uncover novel therapeutic opportunities and strategies to effectively combat COVID-19.Keywords
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the virus that causes COVID-19. In early 2020, the World Health Organization (WHO) classified the COVID-19 pandemic as a Public Health Emergency of International Concern [1]. In addition to SARS-CoV-2, seven human coronaviruses (HCoVs) are identified. The two human coronaviruses HCoV-229E and HCoV-OC43, discovered in the 1960s, are known to cause common colds [2]. SARS-CoV emerged in 2002, leading to severe acute respiratory syndrome (SARS). HCoV-NL63, identified in 2004, causes bronchiolitis and conjunctivitis, while HCoV-HKU1, discovered in 2005, is associated with acute respiratory distress syndrome. MERS-CoV, which appeared in the Middle East in 2012, also causes severe illness [3]. In December 2019, an outbreak of pneumonia in Wuhan, China, led to the discovery of a novel coronavirus, named SARS-CoV-2. The first COVID-19 death occurred on 11 January 2020, and the virus has since affected over 2.6 million people worldwide [4].
SARS-CoV-2 has a single-stranded, positive-sense RNA genome encoding several critical proteins: the spike (S) protein, envelope (E) protein, membrane (M) protein, and nucleocapsid (N) protein [5] (Fig. 1).
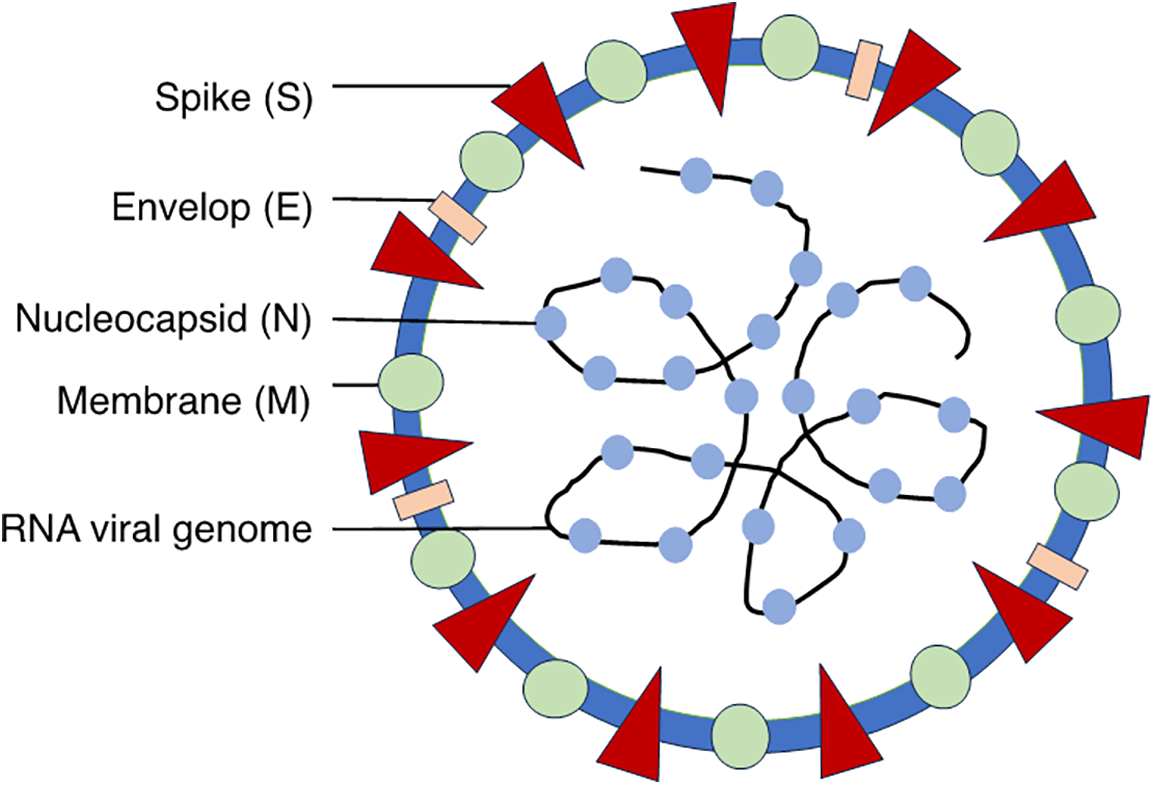
Figure 1: Schematic representation of SARS-CoV-2 illustrating the structural proteins: Spike (S) protein, Nucleocapsid (N) protein, Membrane (M) protein, Envelope (E) protein and genomic RNA
The spike protein facilitates viral entry by binding to the Angiotensin-converting enzyme 2 (ACE2) receptor on host cells, leading to virus uptake through endocytosis or direct membrane fusion. Inside the host cell, the viral RNA is translated into proteins and replicated using the host’s ribosomal machinery and RNA-dependent RNA polymerase. New viral particles are assembled in the endoplasmic reticulum (ER) and Golgi apparatus and released via exocytosis [6].
During COVID-19 infection, viral and host proteins are subject to ubiquitination by host E3 ligases, which affects viral replication and pathogenesis [7]. E3 ubiquitin ligases are pivotal elements within the ubiquitin-proteasome system (UPS), which regulates protein degradation, localization, and function within the host cell. E3 ubiquitin ligases play an essential role in various processes including cell cycle [8,9], signaling [10,11], apoptosis [12], viral pathogenesis [13], immune response [14], and neurodegeneration [15]. E3 ligases facilitate the attachment of ubiquitin to lysine (K) residues on target proteins that can occur singly (monoubiquitination) or in chains (polyubiquitination) [16]. Polyubiquitination of proteins is carried out in three steps through a sequential activity of three different classes of enzymes. The initial step in this process involves the activation of ubiquitin by ubiquitin-activating enzyme E1 (first step), followed by the transfer of the activated ubiquitin to the ubiquitin-conjugating enzyme E2 (second step), and then the activated ubiquitin is transferred from E2 conjugating enzymes to the substrate proteins (third step) through E3 ubiquitin ligase enzymes [17,18] (Fig. 2).
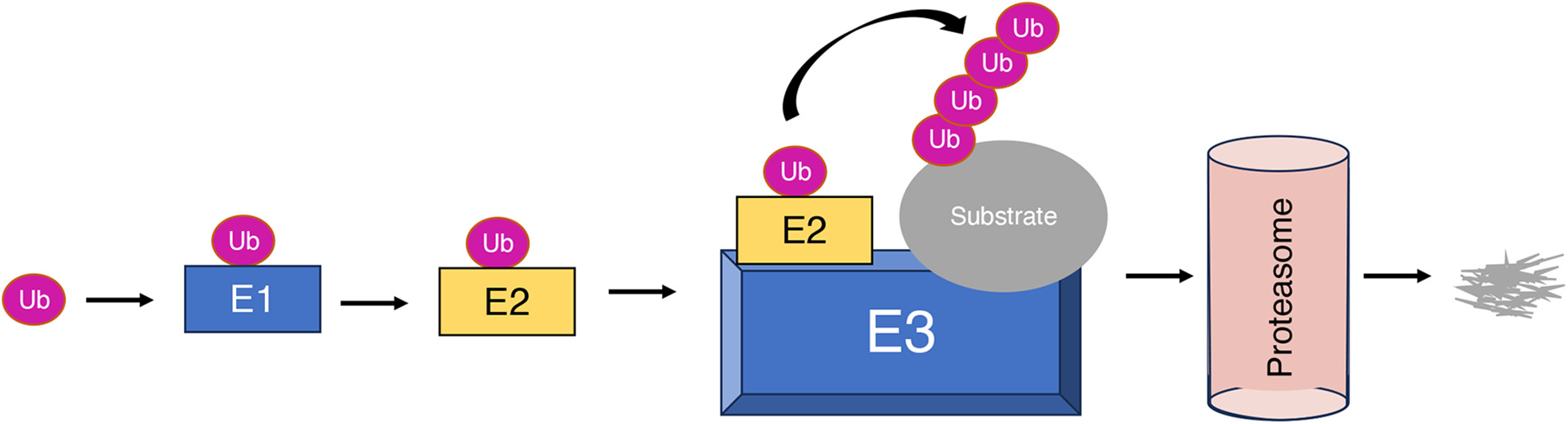
Figure 2: Overview of the cascade process of ubiquitination mediated by ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3)
The human genome encodes approximately 600 putative E3 ubiquitin ligase enzymes [19]. E3 ubiquitin ligases are classified into four major classes: RING finger-type, HECT-type, U-box-type, and RBR-finger-type [8,20]. HECT-type E3 ligases are categorized by their HECT (Homologous to the E6-AP Carboxyl Terminus) domain, which facilitates a two-step ubiquitin transfer: first, ubiquitin is transferred to a catalytic cysteine residue in the HECT domain, and then to the substrate protein. RING finger-type E3 ligases are identified by their RING (Really Interesting New Gene) finger domain, a zinc-binding motif that stabilizes the interaction between the E2 ubiquitin-conjugating enzyme and the substrate, thereby facilitating direct ubiquitin transfer. RING-finger-type E3 ubiquitin ligases are the largest family, comprised of monomeric, dimeric, or multi-subunit RINGs assembled around Cullin subunits [20] (Fig. 3A–D).
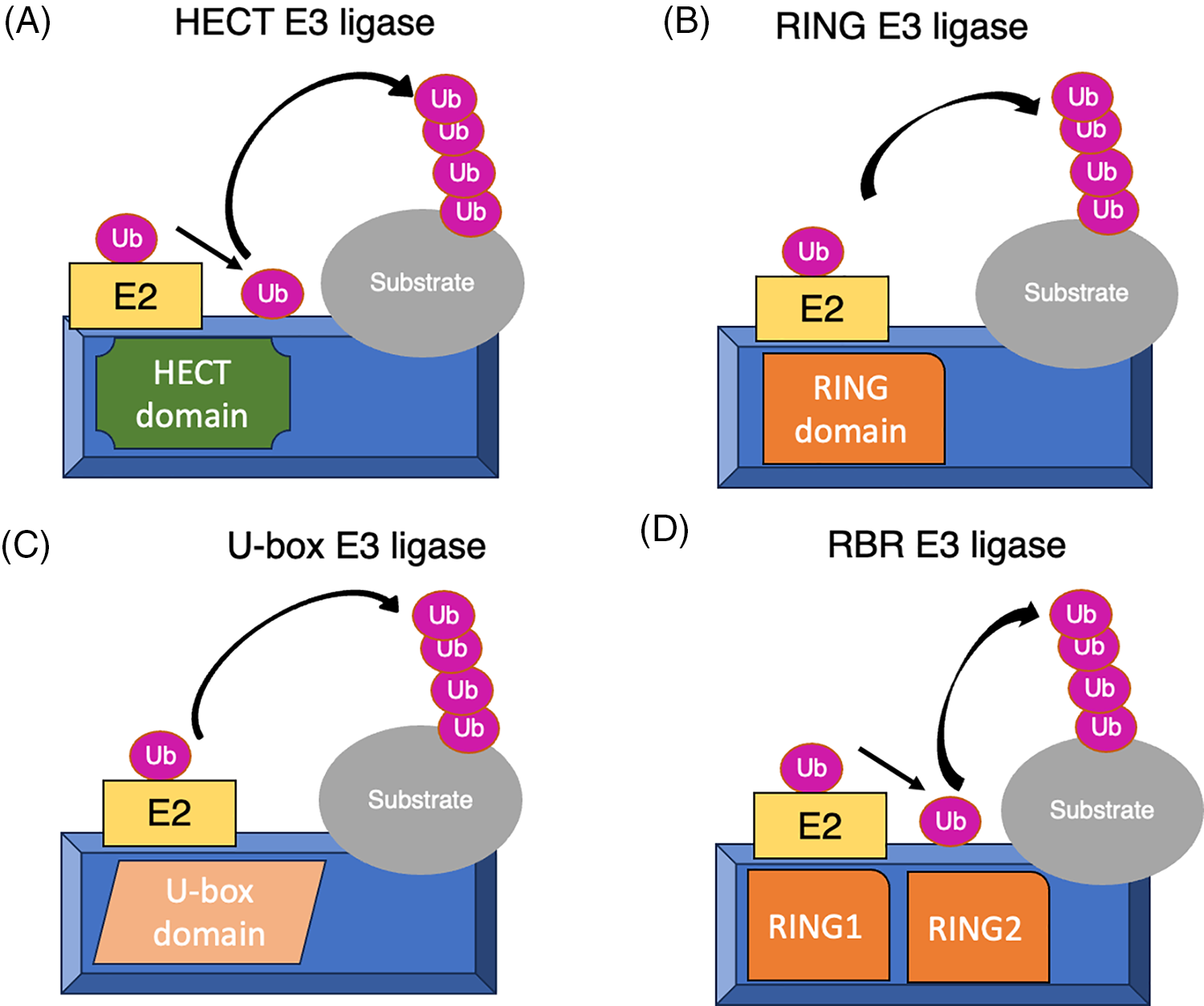
Figure 3: Illustration of the diverse mechanisms employed by (A)HECT-type: Transfer ubiquitin in two steps via a catalytic cysteine in the HECT domain. (B)RING-type: Mediate direct ubiquitin transfer from E2 to the substrate. (C)U-box-type: Use a U-box domain to bind E2 and facilitate ubiquitin transfer and (D)RBR-type: Perform in two-step where ubiquitin is first transferred to a catalytic RING2 domain on the E3 and then to the substrate
Cullins (1, 2, 3, 4, 5, 7 and 9) are integral components of E3 ubiquitin ligase complexes, acting as scaffolds to facilitate substrate ubiquitination and degradation [21]. Cullin-1 forms complexes with S-phase kinase-associated protein 1 (SKP1) and various substrate receptor F-box proteins, such as Fbxw1, Fbxw7, and Fbx8 [22,23]. Cullin-2 partners with Elongin B and C and the von Hippel-Lindau (VHL) protein [24]. Cullin-3 associates with BTB-domain proteins, including the potassium channel tetramerization domain (KCTD) family and the Kelch-like (KLHL) family proteins [25]. Cullin-4A and Cullin-4B both interact with Damaged DNA Binding proteins 1 and 2 (DDB1 and DDB2) and DDB1 Cullin4 associated factor (DCAF) proteins [26]. Lastly, Cullin-5 partners with Elongin B and C and a substrate receptor protein-Suppressors of Cytokine Signalling (SOCS) family [27] (Fig. 4). Each cullin’s interaction with specific substrate-binding proteins determines the selectivity of the ubiquitination process, thereby influencing various cellular pathways [21,28]. In this review, we summarized the E3 ligases involved in the infection, replication, and pathogenesis of SARS-CoV-2.
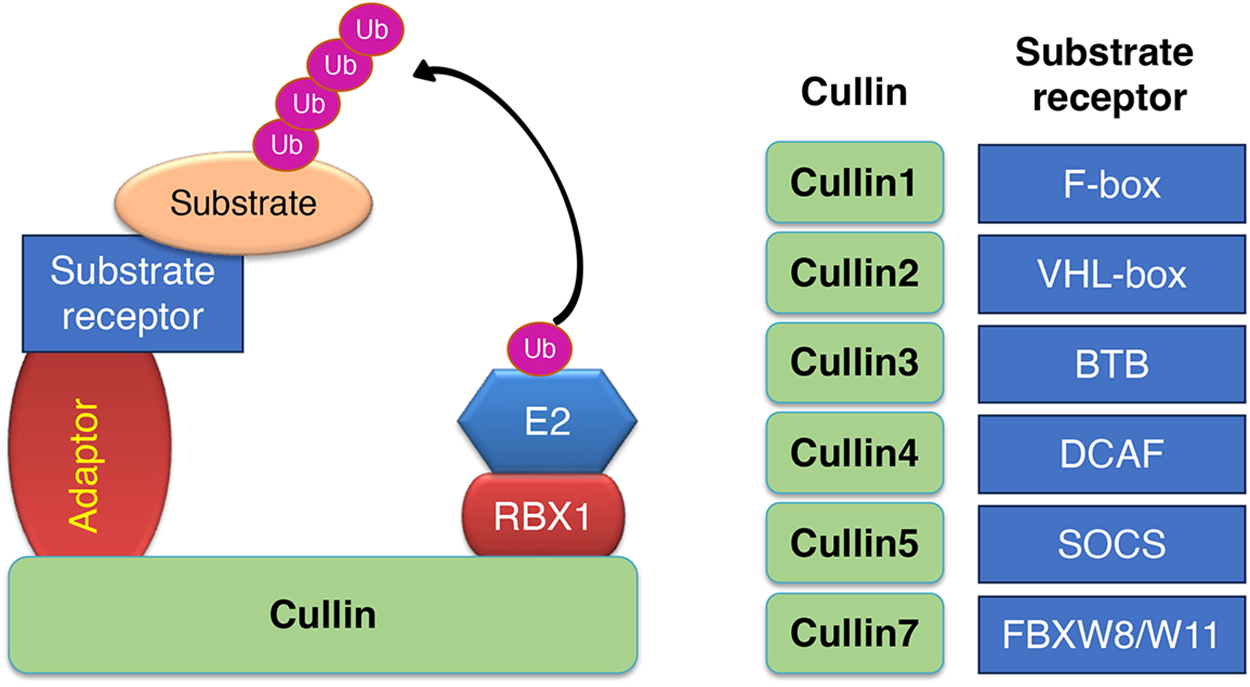
Figure 4: Schematic representation of different types of Cullin based E3 ubiquitin ligase. Cullins 1, 2, 3, 4A, 4B, 5, and 7 are scaffold proteins that assemble with RING finger proteins (RBX1/2), adaptor proteins (SKP1, EloC/EloB, BTB, DDB1) and substrate receptor proteins (F-box family, VHL family, BTB, DCAF family, SOCS family among others)
Coronaviruses have a genome size ranging from 26.4 to 31.7 kilobases, consisting of a positive-sense, single-stranded RNA genome, making it one of the largest among RNA viruses. For SARS-CoV-2, the genome includes untranslated regions (UTRs) at both ends with cis-acting secondary RNA structures essential for RNA synthesis. It has 14 open reading frames (ORFs) encoding 29 proteins, including two overlapping polyproteins (pp1a and pp1ab) [29]. Two polyproteins, pp1a and pp1ab, are translated from ORF1a and ORF1b. They undergo proteolytic cleavage by cysteine proteases Nsp3 (papain-like protease; PLpro) and Nsp5 (chymotrypsin-like protease), releasing 16 nonstructural proteins (Nsps) via co-translation and post-translation [30–32]. These Nsps form the viral replication and transcription complex (RTC) and are involved in RNA processing and proofreading [6]. The remaining genome encodes four structural proteins—spike (S), envelope (E), membrane (M), and nucleocapsid (N)—and several accessory proteins (e.g., ORF3a, ORF3b, ORF6, ORF7a) that modulate the host immune response and contribute to viral pathogenesis [33].
The coronavirus genome encodes structural and non-structural proteins crucial for its function. The 5′-region of the viral RNA genome encodes non-structural proteins (Nsps) Nsp1 to Nsp16. Nsp1 plays a crucial role in host translation inhibition by endonucleolytic cleavage of host mRNAs and helps in evading the host immune response [34]. Nsp3 features multiple domains, including the papain-like protease (PLpro) domain. Nsp5 is a 3C-like protease that cleaves the polyprotein to release Nsp4-Nsp16. Meanwhile, Nsp6 inhibits autophagosome formation, and Nsp7 partners with Nsp8 to form a hexadecamer assembly with primase activity. Nsp9 serves as a ss-RNA binding protein, while Nsp10 is crucial for viral mRNA cap methylation through its interaction with Nsp14 and Nsp16. Nsp12, with cofactors Nsp7 and Nsp8, is the RNA-dependent RNA polymerase (RdRp) essential for RNA synthesis. Nsp13 exhibits helicase activity with zinc-binding capabilities and unwinding abilities, whereas Nsp14 possesses both exoribonuclease and N7-guanine methyltransferase activities [35]. Lastly, Nsp15 acts as a uridylate-specific endoribonuclease, and Nsp16 functions as a 2’-O-ribose methyltransferase [36]. Additionally, nine accessory proteins (ORF3a, 3d, 6, 7a, 7b, 8, 9b, 14, and 10) arise from at least five Open Reading Frames (ORFs) [29]. These accessory proteins are crucial virulence factors that play a significant role in SARS-CoV-2 infection, aiding in viral replication and immune evasion. For example, ORF9c inhibits cytokine secretion, while ORF3b, ORF6, ORF7a, ORF8, and ORF9b counteract type I interferon responses. Additionally, ORF3a modulates autophagy and apoptosis, ORF3d influences mitochondrial function, and ORF9b is involved in inflammasome activation [37].
SARS-CoV-2 has four structural proteins known as Spike (S), Envelope (E), Membrane (M), and Nucleocapsid (N) proteins. S protein recognizes the host cell, binds to the ACE2 receptor, leads to the fusion of viral envelope and host membranes, and mediates entry of the SARS-CoV-2 virus. SARS-CoV-2 with mutations in the S protein such as Alpha, Beta, Gamma, Delta, and Omicron significantly enhance infectivity and contribute to immune evasion [38]. S protein consists of two subunits, S1 and S2. The S1 subunit is composed of a N-terminal domain (NTD) and a long C-terminal receptor-binding domain (RBD), which binds to ACE2 on the membrane of host cells [39]. The S2 subunit facilitates viral entry and includes a fusion peptide, two heptad repeats, a transmembrane domain, and a C-terminal tail. The S2 subunit mediates the viral and cellular membrane fusion [40].
The E protein is the smallest structural SARS-CoV-2 protein and plays a critical role in viral assembly, release, and pathogenesis. The E protein is highly conserved across evolution and shares 96% similarity between SARS-CoV-1 and SARS-CoV-2. It compromises three domains: N-terminus, transmembrane, and a C-terminus [41]. Only a small percentage of E protein is incorporated into the virions, while most of the protein is localized at the Golgi and endoplasmic reticulum–Golgi intermediate compartment (ERGIC), facilitating the viral assembly and release [42]. The SARS-CoV-2 E protein has two notable features. First, it forms cation-selective channels, or viroporins, which increase the pH in the ERGIC and lysosomes [43]. And it’s C-terminal DLLV motif binds to postsynaptic density-95 (PSD-95), discs-large, zona occludens 1 (ZO-1) (PDZ) domain-containing proteins, disrupting epithelial barriers and promoting viral spread [44].
The M protein is crucial for the viral assembly and release of SARS-CoV-2 [45]. It consists of a short N-terminal domain (NTD), three transmembrane domains, and a C-terminal domain (CTD) located within the virion [46]. The assembly of SARS-CoV-2 virions occurs in the ERGIC, where the M protein, carrying a trans-Golgi network localization signal, arrives slightly earlier than the S and E proteins, indicating its role in initiating virion assembly [47].
The SARS-CoV-2 nucleocapsid (N) protein packages viral RNA into a helical ribonucleoprotein complex within the viral capsid [48]. It aids RNA transcription and replication through liquid-liquid phase separation [49]. The N protein consists of an N-terminal domain (NTD), a linker region rich in serine and arginine, and a C-terminal domain (CTD). The NTD has a β-sheet core and a β-hairpin region, while the CTD forms a rectangular homodimer [50]. Both NTD and CTD are involved in RNA binding, with linker region phosphorylation affecting this interaction [51].
3 The Life Cycle of SARS-CoV-2
The life cycle of SARS-CoV-2 begins with the virus binding to the ACE2 receptor on human cells via its spike (S) protein, which consists of two subunits: S1, responsible for receptor binding, and S2, which facilitates membrane fusion [52]. Upon S1 binding to ACE2, a conformational change exposes the fusion peptide, allowing cellular proteases like Transmembrane Serine Protease 2 (TMPRSS2) and cathepsins to cleave S2. This cleavage triggers the fusion of the viral envelope with the host cell membrane, enabling the virus to enter the cytoplasm [53] (Fig. 5).
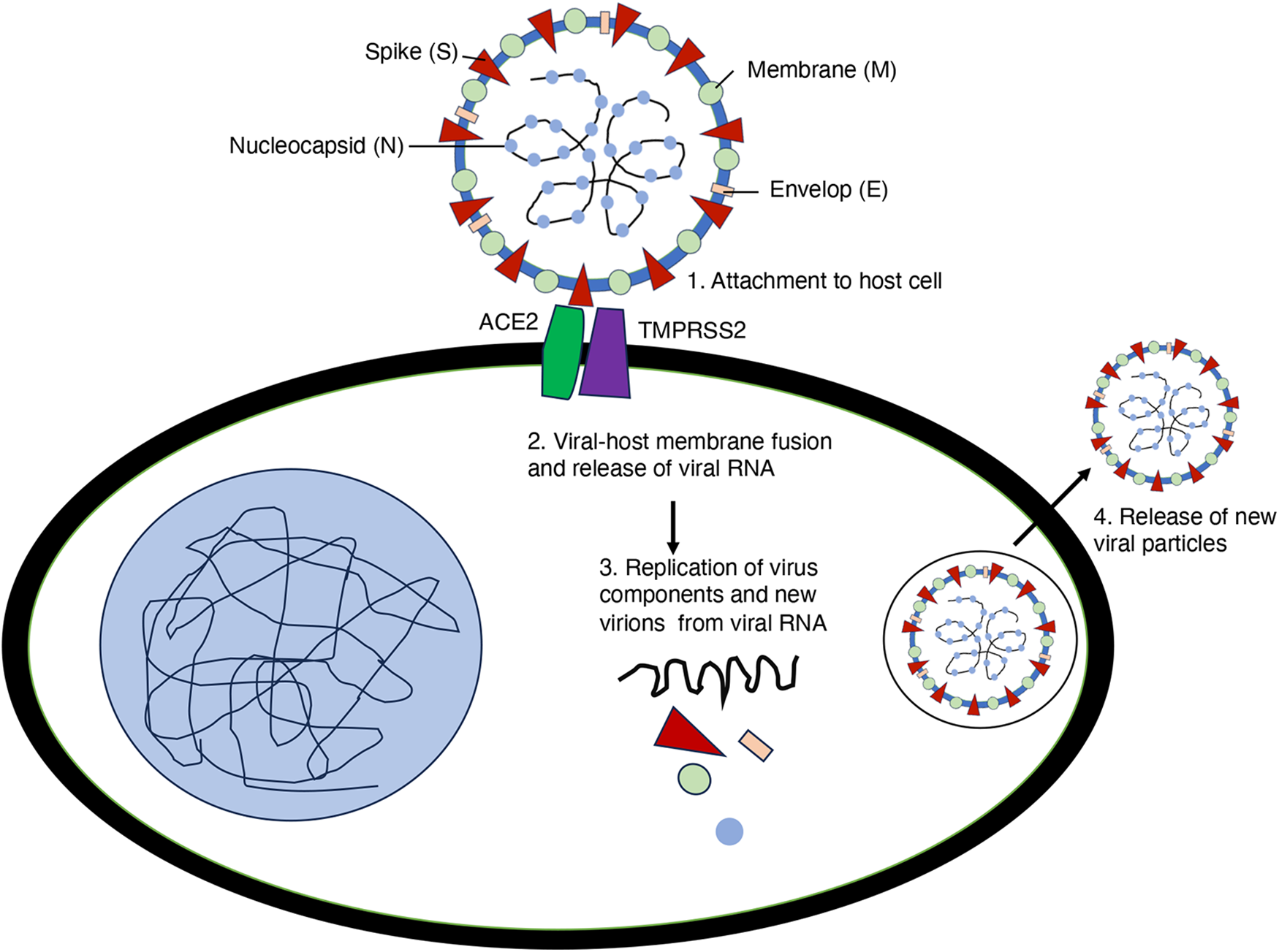
Figure 5: Life cycle of SARS-CoV-2. The spike protein binds to host cell surface receptors to mediate viral entry. The uncoated genomic RNA is subsequently translated, leading to the replication of viral components. Viral proteins are then assembled for maturation, and newly packaged viral particles are released from the host cell
In addition to ACE2, several alternative receptors for SARS-CoV and SARS-CoV-2 have been identified. Lectins and members of phosphatidylserine receptor families (TIM1 and AXL) facilitate viral entry through virus recognition but are non-specific and ineffective without ACE2. CD147 has been suggested as a receptor, but its role is disputed due to the lack of S protein binding despite some increases in viral entry. In addition, Neuropilin 1 (NRP1) can enhance TMPRSS2-mediated entry of wild-type SARS-CoV-2 and bind S1, promoting S1 shedding. Additionally, mutations in the spike protein, such as the E484D substitution, increase binding to TMEM106B, enhancing TMEM106B-mediated viral entry [54]. Upon entry into the cell, the virus releases its single-stranded RNA (ssRNA) genome, which is translated by host ribosomes into two large polyproteins, pp1a and pp1ab. These polyproteins are cleaved by viral proteases, papain-like protease (PL-PRO), and 3C-like protease (3CL-PRO), into functional non-structural proteins (Nsps) [32]. The Nsps form the replicase-transcriptase complex (RTC), which replicates the genomic RNA and transcribes subgenomic mRNAs. These mRNAs are then used to produce structural proteins S, E, M, and N, essential for new virion formation (Fig. 5) [6]. The structural proteins are synthesized and inserted into the endoplasmic reticulum (ER), where the nucleocapsid is formed by the binding of the N protein to the genomic RNA. The nucleocapsid buds into the ERGIC, acquiring the viral envelope [55]. The mature virions are transported to the cell surface via vesicles, which fuse with the plasma membrane to release new virions through exocytosis [47]. This cycle of infection, replication, assembly, and release enables the virus to spread to new cells and continue the infection process.
4 Cellular Host Factors for SARS-CoV-2 Infection
Multiple genome-wide and targeted CRISPR screens have been performed to uncover cellular host factors in SARS-CoV-2 infection, with ACE2 being the sole gene consistently identified in all screens [56–59]. SARS-CoV-2 recognizes the ACE-2 receptor present on the host cell membrane to gain entry inside the cell [52]. Importantly, CRISPR screens in lung adenocarcinoma Calu-3 and colorectal carcinoma Caco-2 cell lines identified TMPRSS2, indicating the involvement of factors that are involved in the early entry route, whereas screens in liver Huh7, non-small cell lung cancer A549, and kidney Vero E6 cell lines identified CTSL (encoding cathepsin L), indicating viral entry through the late endosomal route [60,61]. Other receptor candidates such as TMEM106B and EXT1 were identified in specific screens [62]. In addition to receptors and proteases, various complexes of vesicle biology, such as Rab GTPases (RAB7A, RAB10, and RAB14) were identified as essential for SARS-CoV-2 infection. A genome-wide CRISPR screen also highlighted several genes involved in cholesterol synthesis, phosphatidylinositol phosphate biosynthesis pathways, and transforming growth factor beta (TGF-β) signaling pathway as critical host factors [63]. Through proteomic analysis, it was revealed that SARS-CoV-2 inhibits the interferon (IFN) signaling pathway to suppress the host immune response. The virus specifically targets components of the Janus kinase/signal transducers and activators of the transcription (JAK-STAT) pathway, including JAK1, Tyk2, and IFNAR1. Additionally, inhibition of JAK kinases enhanced viral infection, suggesting that SARS-CoV-2 exploits this pathway to enhance its replication and survival in host cells [64].
SARS-CoV-2 infection profoundly disrupts cellular processes across multiple levels, including transcriptional, post-transcriptional, translation, and post-translational pathways [65]. RNA sequencing (RNA-seq) data reveal a significant reduction in cellular mRNA levels during infection, while viral mRNAs become predominant [66]. Additionally, the CRISPR screen also identified components and the proteins involved in the regulation of transcription, such as transcription factors, histone-modifying enzymes, and members of the SWI/SNF complex [56,57,59,67]. It is shown that SWI/SNF complex activity is crucial for ACE2 expression and promotes SARS-CoV-2 infection [68]. In addition, the ACE2 receptor is transcriptionally regulated by regulatory elements in the ACE2 locus, Interferon Stimulated Gene Factor 3 (ISGF3), Androgen receptor (AR), Nuclear factor erythroid 2-related factor 2 (Nrf2), and Bromodomain and extraterminal (BET) proteins [69–73]. Additionally, post-transcriptional modifications such as splicing, capping, and polyadenylation are also disrupted by SARS-CoV-2 [65]. For instance, Nsp16 binds to the spliceosome (U1/U2 snRNA) and inhibits global mRNA splicing to suppress interferon response upon SARS-CoV-2 infection [74].
SARS-CoV-2 disrupts the host immune response by altering microRNAs (miRNAs) expression. miRNAs are 18-25 nucleotide small non-coding RNA molecules important in regulating gene expression at the post-transcriptional level [75]. For example, miR-2392 was identified as being elevated in COVID-19 patients and targets mitochondrial and inflammatory pathways associated with SARS-CoV-2 [76]. Like other viruses, SARS-CoV-2 can hijack the miRNA pathway by producing its miRNAs, such as CoV2-miR-O7a and CoV2–miR–O7a.2 (generated by dicer–dependent cleavage of a stem–loop structure of ORF7a) associates with human Argonaute proteins and represses human targets. CoV2–miR–O7a.2 can target 3′UTRs of interferon–stimulated genes and repress their expression in a miRNA–like fashion [77,78]. Such dysregulation of miRNAs upon infection can lead to a compromised immune response, and targeting these miRNAs could offer potential therapeutic strategies.
The SARS-CoV-2 infection results in a global decrease in protein translation and an increase in the degradation of cytosolic cellular mRNA [66]. A recent study showed that the N protein directly binds to host mRNAs in the cell, and it is preferable to select 3′ UTR and regulate the stability of the target mRNA [79]. Notably, Nsp1 binds to 18S ribosomal RNA in the mRNA entry channel of the ribosome, leading to a widespread inhibition of mRNA translation [74]. The coronavirus infection triggers endoplasmic reticulum (ER) stress, which subsequently activates the unfolded protein response (UPR), a key regulator of the innate immune response [80]. SARS-CoV-2 hijacks the ER-Golgi intermediate compartment for virion assembly and release, leveraging ER stress to enhance its replication cycle. The ER stress response is primarily mediated by three main sensors—Protein kinase R-like endoplasmic reticulum kinase (PERK), Inositol requiring enzyme 1 (IRE1), and Activating transcription factor 6 (ATF6)—which become activated in SARS-CoV-2 infected cells, leading to protein synthesis inhibition and an increase in ER folding capacity [81]. Interestingly, the overexpression of SARS-CoV-2 ORF8 or spike (S) proteins alone is sufficient to trigger the UPR. Notably, pharmacological inhibition of the UPR significantly reduces SARS-CoV-2 replication, underscoring the crucial role of this pathway in viral replication [82]. Targeting the ER stress pathway has the potential to inhibit both viral release and the release of proinflammatory cytokines, thereby mitigating multiple pathological aspects of COVID-19 [82,83].
5 SARS-CoV-2 Proteins and E3 Ubiquitin Ligases
Research has highlighted the crucial function of E3 ubiquitin ligases in regulating SARS-CoV-2 within the host [84,85]. The battle for control over these E3 ligases plays a pivotal role in deciding if a virus can successfully invade its host or if the host can mount an effective defense. The virus invades by binding to host receptors, with the Spike (S) protein targeting ACE2 as a key binding site. However, recent findings suggest that the ubiquitin E3 ligase may disrupt this interaction. E3 ubiquitin ligase S-phase kinase-associated protein 2 (SKP2) has been found to regulate ACE2 levels [86]. Studies show that exposure to cigarette smoke extract (CSE) and the benzo(a)pyrene (BaP) a carcinogen promotes the degradation of ACE2 through SKP2. Mechanistically, BaP can increase the cellular levels of SKP2, which leads to the ubiquitination and subsequent breakdown of ACE2. This mechanism potentially has implications for understanding how environmental pollutants like BaP might influence susceptibility to COVID-19 [86]. Additionally, research has demonstrated that the Cyclin-dependent kinase 4/6 (CDK4/6) inhibitor palbociclib induces cell cycle arrest at the G0/G1 phase while increasing ACE2 mRNA and protein expression without altering its subcellular localization. Palbociclib achieves this by downregulating SKP2, thereby inhibiting the ubiquitin-proteasome and lysosomal degradation pathways of ACE2. Moreover, the elevated ACE2 levels resulting from palbociclib treatment, as well as from other compounds that cause cell cycle arrest, have been shown to enhance pseudotyped SARS-CoV-2 infection [87]. Together with previous findings, this underscores the complex role of E3 ubiquitin ligases like SKP2 in regulating ACE2 levels and their potential impact on SARS-CoV-2 infection dynamics (Table 1). The transmembrane serine protease type 2 (TMPRSS2) is vital for the virus to bind to ACE2 and enter host cells [52]. In 2021, Chen et al., using a library of 2560 FDA-approved or current clinical trial compounds, identify “halofuginone” a small molecule that reduces the surface expression of TMPRSS2. Interestingly, DDB1- and CUL4-associated factor 1 (DCAF1), an E3 ligase, ubiquitinates TMPRSS2 at four lysine residues (K70, K80, K82, K83) on its intracellular domain leading to its degradation, thereby impeding the binding of the S protein to ACE2 (Table 1) [88]. Furthermore, knockdown studies confirm that Tripartite motif-containing 28 (TRIM28) regulates the entry of SARS-CoV-2 by targeting ACE2 in non-small cell lung cancer A549 cells and primary pulmonary alveolar epithelial cells (PAEpiCs) [89]. Unlike ubiquitination, which often targets proteins for degradation, the E3 small ubiquitin-like modifier (SUMO) ligase PIAS4 stabilizes ACE2 through SUMOylation. Specifically, the attachment of SUMO3 to ACE2 at lysine (K) 187 prevents K48-linked ubiquitination of ACE2. This leads to inhibition of its autophagic degradation mediated by the cargo receptor TOLLIP, ultimately enhancing SARS-CoV-2 infection [90]. In conclusion, E3 ubiquitin ligases, such as TRIM28, and SKP2, mediated the degradation of ACE2, while DCAF1 promotes the degradation of TMPRSS2, and plays a crucial role in SARS-CoV-2 entry. Targeting these cell surface proteins and their associated pathways could significantly reduce infection risk and viral spread, underscoring the pivotal role of cellular mechanisms in antiviral defense strategies (Fig. 6).
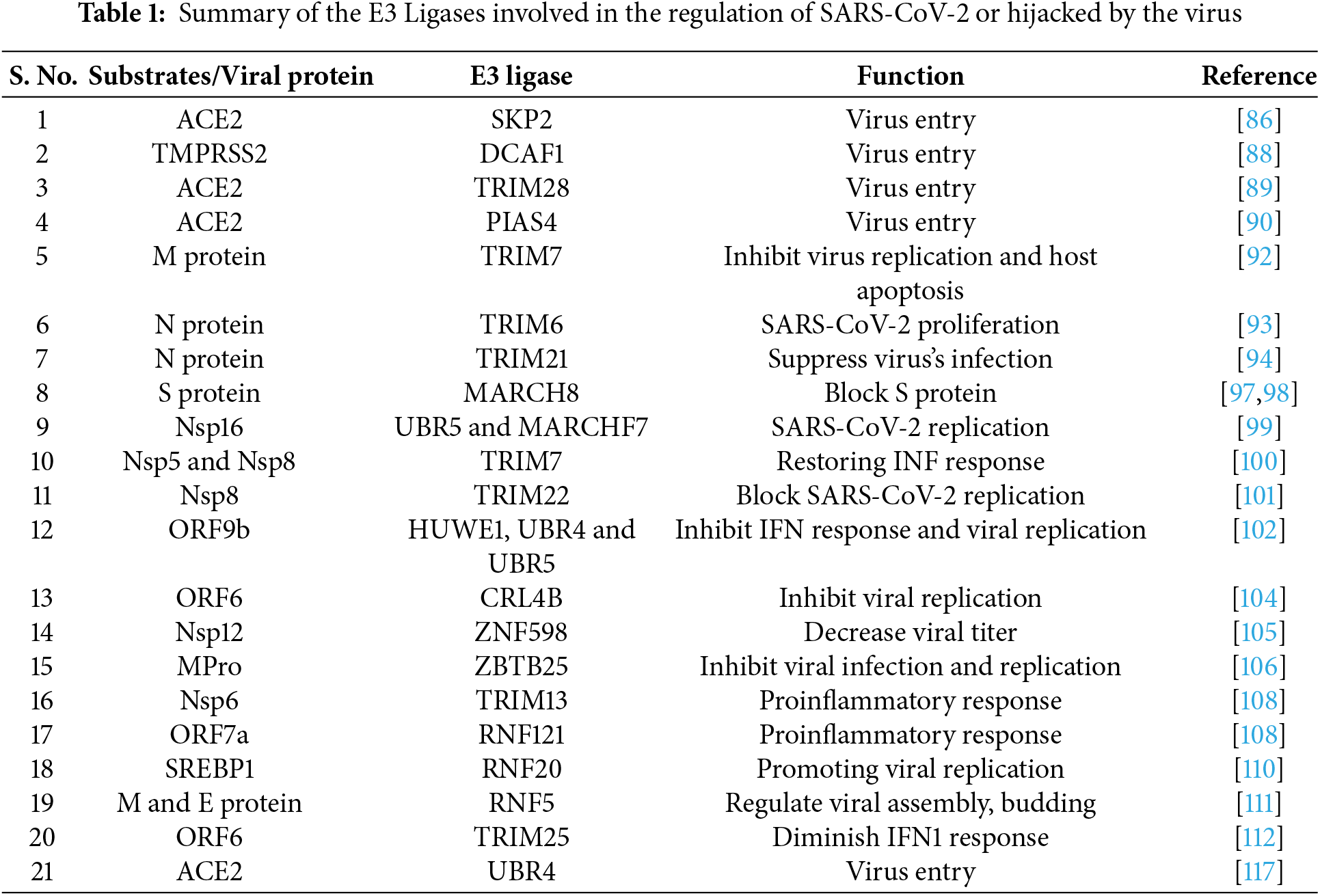
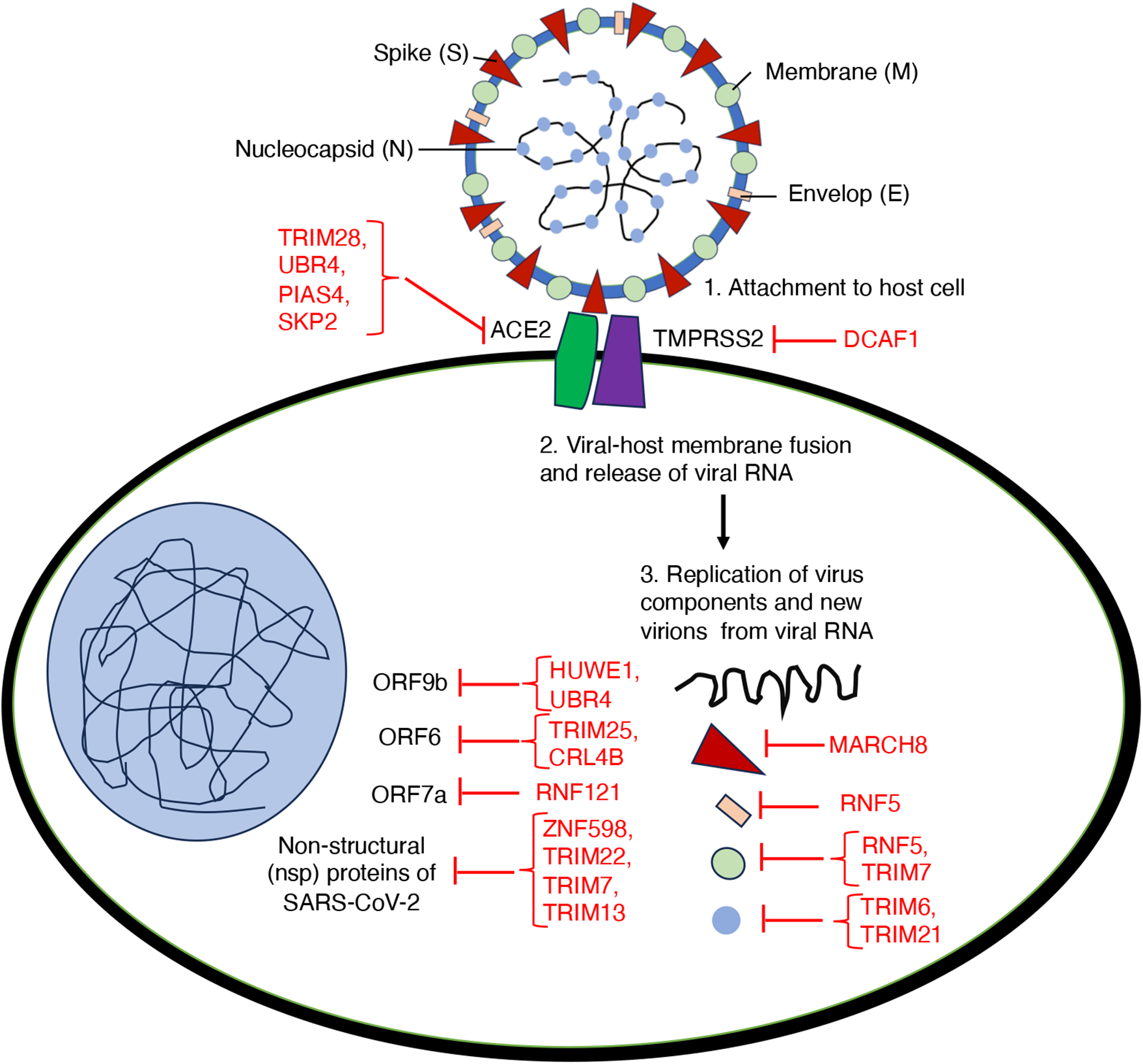
Figure 6: An overview on the role of E3 ubiquitin ligases involving in different aspects of host-virus interactions during SARS-CoV-2 infection
Further expanding on the contribution of E3 ligases in combating SARS-CoV-2, several ligases have been identified as crucial regulators of viral replication, assembly, protein degradation, and host defense mechanisms. For instance, RNF5 has been found to regulate viral replication by exerting an inhibitory effect through the K48-linked ubiquitination of the E protein on the 63rd lysine. Importantly, the pharmacological activator Analog-1 of RNF5 diminishes disease development in a mouse model. Moreover, RNF5 expression varies across age groups and among patients with differing levels of disease severity, suggesting its potential use as a prognostic marker for COVID-19 [91]. The E3-ligase TRIM7 acts as an antiviral factor by non-degradative ubiquitination of M-protein on the SARS-CoV-2 membrane to limit apoptosis and virus replication [92]. To explore the molecular mechanisms governing the degradation of viral proteins, a genetic screening was conducted to identify human E3 ligases responsible for destabilizing SARS-CoV-2 proteins. This screen revealed that the E3 ligase RNF185 is responsible for the regulation of SARS-CoV-2 envelope protein. RNF185 and the envelope protein co-localize within the endoplasmic reticulum (ER). RNF185 facilitates the degradation of the envelope protein, and depletion of RNF185 increases the viral titer of SARS-CoV-2 in cellular models [84]. TRIM6 promotes K29-linked ubiquitination of N protein at K102, K347, and K361 residues, increasing its binding to viral genomic RNA, and leading to the proliferation of SARS-CoV-2 [93]. TRIM21 interacts and promotes polyubiquitination-mediated degradation of N protein, regulating innate immunity [94]. E3 ubiquitin ligase MARCH8 inhibits SARS-CoV-2 virus replication through degrading N protein [95]. TRIM28-mediated SUMOylation of the N protein is essential for SARS-CoV-2 virulence, as it plays a pivotal role in suppressing the innate antiviral immune response [96]. The effective degradation of the viral S protein is also crucial for a robust antiviral response. In this regard, the E3 ubiquitin ligase MARCH8 has been shown to inhibit the SARS-CoV-2 S protein, potentially by retaining the S protein in the lysosome (Table 1) [97,98].
Beyond the Structural proteins, some nonstructural proteins have also been identified as targets for E3 enzymes. For example, UBR5 and MARCHF7 inhibit SARS-CoV-2 replication by inducing nsp16 degradation via K48-and K27-linked ubiquitination, respectively [99]. In addition, TRIM7 regulates the degradation of nsp5 and nsp8 by binding to their glutamine-end motifs, moderately restoring the interferon response [100]. The E3 ligase TRIM22 reduces viral RNA and protein levels and inhibits SARS-CoV-2 replication by promoting Nsp8 degradation. Upon viral infection, the IFN signaling pathway was activated and IFN-α stimulates TRIM22 expression to promote Nsp8 degradation. TRIM22 interacts with and promotes K48-linked polyubiquitination of Nsp8 at K97 position [101]. Yu et al. identified three E3 ubiquitin ligases (HUWE1, UBR4, and UBR5) inducing K48-linked ubiquitination and degradation of ORF9b, thereby diminishing ORF9b-dependent inhibition of the IFN response including SARS-CoV-2 replication [102]. CUL5-TOM70-HSP90α complex serves as a critical regulator of ORF9b stability in SARS-CoV-2 replication, elucidating the intricate host-virus immune response dynamics [103]. CRL4B E3 ligase recruited by Pre-mRNA-processing factor 19 (PRPF19) inhibits SARS-CoV-2 infection by promoting ORF6 ubiquitination and subsequent degradation [104]. Moreover, the E3 ubiquitin ligase Zinc Finger Protein 598 (ZNF598) plays a significant role in targeting Nsp12 for ubiquitination-mediated degradation under elevated temperature conditions. This degradation results in a reduction of viral RNA copies and a subsequent decrease in viral titer [105]. The E3 ubiquitin ligase ZBTB25 inhibits beta coronavirus infection by ubiquitinating the main viral protease, MPro [106]. The ubiquitin ligase Parkin modulates the stability of the SARS-CoV-2 main protease and inhibits viral replication [107]. TRIM13 and RNF121 mediated K63-linked polyubiquitination of Nsp6 and ORF7a, respectively, appears essential for Nuclear factor-kappa B (NF-κB) activation through associations with TAK1 [108]. Collectively, these findings emphasize the diverse and critical roles of various E3 ubiquitin ligases in regulating SARS-CoV-2 infection.
Viruses harness host machinery including ubiquitination modifications to amplify their virulence. Site-specific ubiquitination of the GTPase Rab7 disrupts its colocalization with the SARS-CoV-2 ORF3a protein, thereby reducing the virus’s ability to invade host cells [109]. In addition, E3 ligase ring finger protein 20 (RNF20) was found as a target of the SARS-CoV-2 3CLpro protease. The disruption of RNF20 function was shown to interfere with its role in regulating the ubiquitination of sterol regulatory element binding protein 1 (SREBP1), thereby promoting viral replication [110]. Conversely, a different study highlighted a beneficial role for RNF5, which facilitates the interaction between the M protein and E protein of the virus [111]. Furthermore, SARS-CoV-2 ORF6 protein targets TRIM25 for proteasomal degradation, thereby reducing K63-linked ubiquitination of Retinoic acid-inducible gene I (RIG-I) and inhibiting type-I interferon production [112]. Also, SARS-CoV-2 ORF10 exploits CUL2ZYG11B to degrade Intraflagellar Transport 46 (IFT46), resulting in cilia dysfunction [113]. Viruses can also leverage ubiquitination modifications to enhance their virulence. Israeli et al. identified CUL5 as a potential contributor to viral infection. CUL5 serves as a key component of multiple E3 ubiquitin-protein ligase complexes. This study suggests that SARS-CoV-2 may have adapted E3 enzyme-mediated ubiquitination to promote its pathogenicity under certain conditions [114]. Additionally, SARS-CoV-2 ubiquitinates ORF7a by utilizing the host’s system to disrupt IFN response and aid infected cells in evading apoptosis, thereby enabling viral replication (Table 2) [115,116].
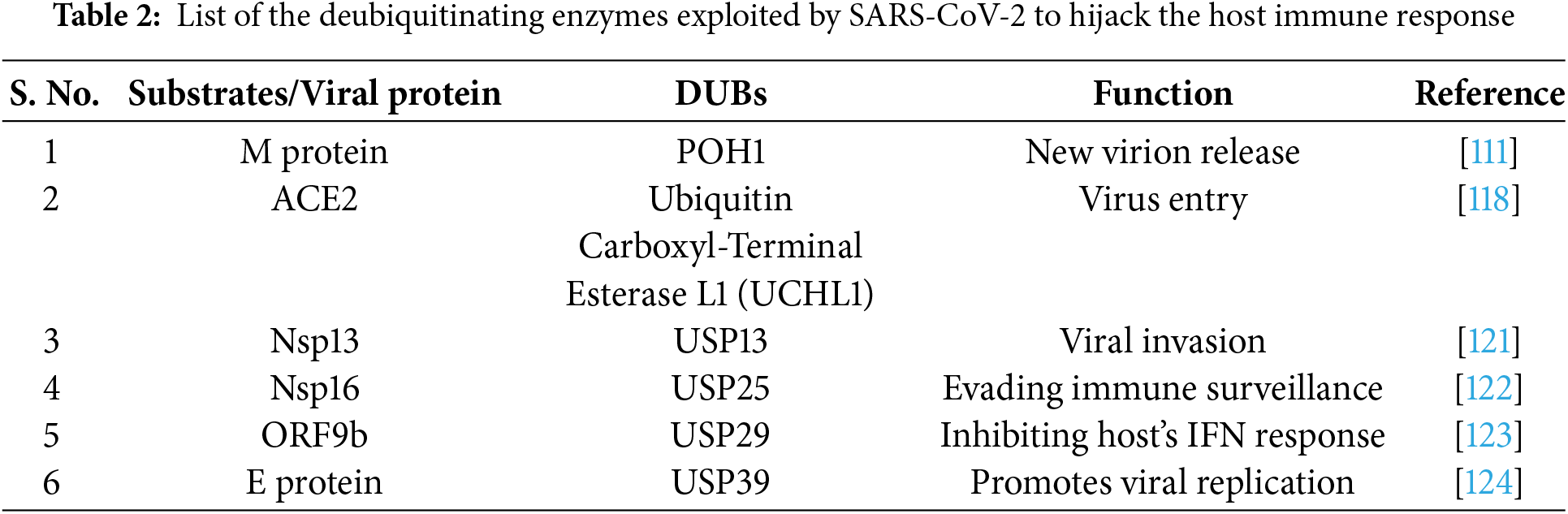
Viral invasion is influenced not only by the actions of E3 ligases but also by the activity of deubiquitinating enzymes (DUBs). The host’s immune system utilizes ubiquitination to degrade viral proteins and mount an antiviral response. However, SARS-CoV-2 can subvert this defense mechanism by hijacking the host’s deubiquitinating enzyme system. For example, the SARS-CoV-2 S protein can induce the alienation of ACE2 from the E3 ligase UBR box N-recognin-4 (UBR4), thereby stabilizing ACE2 levels [117]. Additionally, deubiquitination by ubiquitin carboxyl-terminal hydrolase 1 (UCHL1) further contributes to ACE2 stability [118]. The deubiquitinating enzyme ubiquitin-specific peptidase 13 (USP13), which interacts with SARS-CoV-2, shows a notably high frequency of genomic alterations within the immune cells of COVID-19 patients [119]. While USP13 is typically recognized for its antiviral role in the pathogenesis of SARS-CoV-2 [120], emerging evidence suggests that in certain circumstances, USP13 may contribute to viral invasion. This paradoxical function stems from findings that SARS-CoV-2 can exploit the host’s USP13 to prevent the ubiquitination and subsequent degradation of Nsp13. By stabilizing Nsp13, the virus disrupts the activity of antiviral components and suppresses type I interferon (IFN) production [121]. These studies suggest that SARS-CoV-2 employs complex strategies to manipulate host cellular mechanisms, enabling it to evade immune defenses and enhance viral replication.
Furthermore, other members of the USP family, such as USP25 and USP29, are crucial in the pathogenicity of SARS-CoV-2. USP25 contributes by deubiquitinating Nsp16 and its associated complex, thereby assisting viral RNA to evade immune surveillance with methylation modification [122]. Similarly, USP29 stabilizes ORF9b, which is essential for inhibiting the host’s interferon response to SARS-CoV-2 [123]. USP39, a deubiquitinase, enhances SARS-CoV-2 replication by removing ubiquitin from and stabilizing the envelope protein [124]. The studies also identified Pad-One-Homologue1 (POH1), a deubiquitinase, as a negative regulator of this ubiquitination process that helps in virus assembly and release [111]. These findings underscore the complex interplay between viral and host proteins in regulating the outcome of SARS-CoV-2 infection.
Host-directed therapeutics have emerged as promising strategies in combating COVID-19, with a focus on modulating pathogenic immune activation and directly targeting viral entry mechanisms. Among the key targets is ACE2, a crucial receptor for SARS-CoV-2 entry. Soluble ACE2, ACE2-Fc, and enzyme-inactive ACE2-Fc fusion proteins have shown efficacy in blocking SARS-CoV-2 and SARS-CoV infection [125–127]. Additionally, drugs like losartan and lisinopril, initially developed for hypertension, indirectly modulate ACE2 expression, potentially mitigating COVID-19 severity [128]. Moreover, targeting host proteases like TMPRSS2, furin, TMPRSS4, TMPR11D/HAT, and cathepsin L is crucial for preventing virus entry [129].
It has been suggested that SARS-CoV-2 mediated induction of proinflammatory cytokines is achieved through the activation of NF-κB and Interleukin 6(IL-6)–JAK–STAT3 signaling pathways. JAK2 inhibitors have emerged as a potential therapeutic option for managing cytokine release syndrome (CRS) associated with severe COVID-19. Unlike pan-JAK inhibitors, JAK2-selective inhibitors are advantageous because they do not interfere with type I and type II interferon responses, which are crucial for antiviral and antibacterial defenses. JAK2 inhibitors selectively inhibit signaling by inflammatory cytokines such as IL-6 and Granulocyte-macrophage colony-stimulating factor (GM-CSF) that drive CRS. This makes JAK2 inhibitors particularly promising in reducing severe inflammation in COVID-19 without compromising overall antiviral immunity [130].
Furthermore, chloroquine and hydroxychloroquine, which interfere with endosomal pH, have demonstrated inhibitory effects on SARS-CoV-2 entry [131]. Neutralization of the SARS-CoV-2 spike protein is achieved through various means, including neutralizing antibodies, recombinant or soluble ACE2, small peptides, and sera [132]. Additionally, small-molecule mSWI/SNF ATPase inhibitors or degraders have been shown to reduce ACE2 expression, rendering cells resistant to SARS-CoV-2 variants and a remdesivir-resistant virus [68]. Furthermore, studies have highlighted the potential of lenalidomide, an FDA-approved drug, in preventing SARS-CoV-2 infection by inactivating Speckle-type BTB–POZ protein (SPOP)/Casein kinase 1 (CK1)/ACE2 signaling [133]. These findings underscore the development of a class of broad-acting antivirals for combating emerging coronaviruses and drug-resistant variants, emphasizing the significance of ongoing research in optimizing efficacy and safety for potential clinical use.
7 Conclusion and Future Perspectives
The COVID-19 pandemic has revealed a spectrum of long-term complications, often referred to as long COVID or post-acute sequelae of SARS-CoV-2 infection (PASC), which can persist even after individuals recover from the acute phase of the illness. These complications significantly impact the quality of life of survivors and affect various organs including the lungs, heart, and brain [134,135]. Although vaccines have an important role in combating COVID-19, the challenge of viral infections underscores the need for further research and development of antiviral drugs. A comprehensive study is required to understand the role of SARS-CoV-2 regulators including E3 ubiquitin ligases that affect virus replication directly or indirectly by regulating expression levels of other proviral or antiviral factors. One promising approach involves proteasomal targeting both viral proteins and host factors essential for viral replication and infection. For viral proteins, innovative strategies such as Proteolysis Targeting Chimeras (PROTACs) might offer a potent means to selectively degrade specific viral proteins critical for the virus’s lifecycle. PROTACs are bifunctional degrader molecules, that function by recruiting a target protein to an E3 ligase. Several E3 ubiquitin ligases including Cereblon (CRBN), Von Hippel-Lindau (VHL), Kelch-like ECH-associated protein 1 (KEAP1) and Mouse double minute 2 homolog (MDM2) have been utilized to develop PROTAC molecules [136]. By harnessing the cellular ubiquitin-proteasome system, these degrader molecules can target viral proteins.
Indomethacin (INM), a nonsteroidal anti-inflammatory drug (NSAID) with antiviral properties, has been explored as a potential therapeutic for SARS-CoV-2 [137]. INM has been shown to interact with the host protein prostaglandin E synthase-2 (PGES-2), which further associates with the SARS-CoV-2 Nsp7 protein. This discovery led to the creation of INM-based degraders that recruit either VHL or CRBN E3 ligases [137]. By modifying the linker moiety, researchers expanded the INM-based PROTAC library, significantly enhancing its antiviral activity and demonstrating strong anti-SARS-CoV-2 effects in infected human lung cells [138]. While INM-PROTACs do not degrade human PGES-2, they promote the concentration-dependent degradation of the SARS-CoV-2 main protease, 3CLpro/Mpro, in infected cells. These INM-based PROTACs represent the first identified class of Mpro degraders, marking a significant step forward in the development of broad-spectrum, effective anti-coronavirus treatments [139]. 3CLpro/Mpro is a conserved enzyme critical for viral replication and pathogenicity. Building on this, Cheng et al. adapted the Mpro inhibitor GC376 to create novel PROTACs with broad-spectrum activity against human-infecting coronaviruses such as HCoV-229E, HCoV-OC43, and SARS-CoV-2. Mechanistic binding studies showed that these PROTACs effectively targeted Mpro in vitro, leading to its degradation within cells [140]. Another PROTAC, MPD2, developed by Alugubelli et al., efficiently degraded Mpro in 293T cells and significantly reduced Mpro protein levels in SARS-CoV-2-infected A549-ACE2 cells. MPD2 exhibited strong antiviral activity across multiple SARS-CoV-2 strains, with enhanced potency against nirmatrelvir-resistant variants. The targeted degradation of Mpro offers a promising strategy to combat drug-resistant viral variants [141].
Another innovative approach involves cell-based cysteine chemoproteomics, which identified a covalent degrader of SARS-CoV-2’s Nsp14. This degrader works by covalently modifying both Nsp14 and host chaperones, leading to compound-induced proteasomal degradation. These compounds also induce widespread changes in cellular proteostasis, including increased ubiquitylation and proteasome activation [142]. This targeted approach minimizes off-target effects and enhances specificity compared to traditional small molecule inhibitors. Another promising approach is targeting host factors involved in key processes like viral entry, replication, or assembly for antiviral drug development. By disrupting essential host-virus interactions, such as receptor binding or intracellular signaling pathways, novel therapeutics can prevent viral entry, replication, or assembly. In this context, Khramtsov et al. developed a modular nanotransporter (MNT), which contains an amino acid sequence capable of binding to the KEAP1 E3 ubiquitin ligase. It also functions as an antibody mimetic to SARS-CoV-2’s N-protein, leading to the degradation of the N-protein. The MNT can internalize into target cells, escape from the endosome into the cytosol, and bind to the N-protein. Significant degradation of the N-protein in non-small cell lung cancer A549 and epidermoid carcinoma A431 cell lines was observed through flow cytometry and western blotting after ectopic expression of the N-protein and incubation with the MNTs. This represents a novel approach to the treatment of viral diseases [143]. Additionally, targeted degradation of S protein via computationally optimized peptide fusions by the E3 enzyme TRIM21 inhibits viral infection [144]. These interventions not only inhibit viral spread but also minimize the likelihood of drug resistance, as targeting host factors limits the virus’s ability to adapt through mutations in viral proteins. By leveraging cutting-edge technologies and interdisciplinary collaborations, the development of antiviral drugs targeting both viral and host factors holds promise for improving patient outcomes bolstering our defenses against emerging viral threats and combatting COVID-19 and future viral outbreaks.
Acknowledgment: None.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm their contribution to the paper as follows: study conception and design: Mukul Mishra and Sehbanul Islam; draft manuscript preparation: Mukul Mishra and Sehbanul Islam; review and editing: Mukul Mishra, Deeba Khan and Sehbanul Islam; visualization: Mukul Mishra, Deeba Khan and Sehbanul Islam; supervision: Sehbanul Islam. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Sharma A, Tiwari S, Deb MK, Marty JL. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2a global pandemic and treatment strategies. Int J Antimicrob Agents. 2020;56:106054. doi:10.1016/j.ijantimicag.2020.106054. [Google Scholar] [PubMed] [CrossRef]
2. Kesheh MM, Hosseini P, Soltani S, Zandi M. An overview on the seven pathogenic human coronaviruses. Rev Med Virol. 2022;32:e2282. doi:10.1002/rmv.2282. [Google Scholar] [PubMed] [CrossRef]
3. Kin N, Miszczak F, Lin W, Gouilh MA, Vabret A, Consortium E. Genomic analysis of 15 Human Coronaviruses OC43 (HCoV-OC43s) circulating in France from 2001 to 2013 reveals a high intra-specific diversity with new recombinant genotypes. Viruses. 2015;7:2358–77. doi:10.3390/v7052358. [Google Scholar] [PubMed] [CrossRef]
4. Chauhan S. Comprehensive review of coronavirus disease 2019 (COVID-19). Biomed J. 2020;43:334–40. doi:10.1016/j.bj.2020.05.023. [Google Scholar] [PubMed] [CrossRef]
5. Naqvi AAT, Fatima K, Mohammad T, Fatima U, Singh IK, Singh A, et al. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165878. doi:10.1016/j.bbadis.2020.165878. [Google Scholar] [PubMed] [CrossRef]
6. V’kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 2021;19:155–70. doi:10.1038/s41579-020-00468-6. [Google Scholar] [PubMed] [CrossRef]
7. Xu G, Wu Y, Xiao T, Qi F, Fan L, Zhang S, et al. Multiomics approach reveals the ubiquitination-specific processes hijacked by SARS-CoV-2. Signal Transduct Target Ther. 2022;7:312. doi:10.1038/s41392-022-01156-y. [Google Scholar] [PubMed] [CrossRef]
8. Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–81. doi:10.1038/nrc1881. [Google Scholar] [PubMed] [CrossRef]
9. Islam S, Dutta P, Chopra K, Rapole S, Chauhan R, Santra MK. FBXW8 regulates G1 and S phases of cell cycle progression by restricting β-TrCP1 function. FEBS J. 2021;288:5474–97. doi:10.1111/febs.15828. [Google Scholar] [PubMed] [CrossRef]
10. Paul D, Islam S, Manne RK, Dinesh US, Malonia SK, Maity B, et al. F-box protein FBXO16 functions as a tumor suppressor by attenuating nuclear β-catenin function. J Pathol. 2019;248:266–79. doi:10.1002/path.5252. [Google Scholar] [PubMed] [CrossRef]
11. Islam S, Dutta P, Sahay O, Santra MK. β-TrCP1 facilitates cell cycle checkpoint activation, DNA repair, and cell survival through ablation of β-TrCP2 in response to genotoxic stress. J Biol Chem. 2021;296:100511. doi:10.1016/j.jbc.2021.100511. [Google Scholar] [PubMed] [CrossRef]
12. Sharma VK, Islam S, Borkar J, Mishra S, Panda D, Santra MK, et al. ATR facilitates the degradation of Api5 through the ubiquitin-proteasome pathway via FBXW2 to regulate apoptosis upon DNA damage. bioRxiv. 2021. doi:10.1101/2021.08.08.455545. [Google Scholar] [CrossRef]
13. Augustine T, Chaudhary P, Gupta K, Islam S, Ghosh P, Santra MK, et al. Cyclin F/FBXO1 interacts with HIV-1 viral infectivity factor (Vif) and restricts progeny virion infectivity by ubiquitination and proteasomal degradation of Vif protein through SCFcyclin F E3 ligase machinery. J Biol Chem. 2017;292:5349–63. doi:10.1074/jbc.M116.765842. [Google Scholar] [PubMed] [CrossRef]
14. Hu H, Sun SC. Ubiquitin signaling in immune responses. Cell Res. 2016;26:457–83. doi:10.1038/cr.2016.40. [Google Scholar] [PubMed] [CrossRef]
15. Upadhya SC, Hegde AN. Role of the ubiquitin proteasome system in Alzheimer’s disease. BMC Biochem. 2007;8 (Suppl 1):S12. [Google Scholar] [PubMed]
16. Kelley DR. E3 ubiquitin ligases: key regulators of hormone signaling in plants. Mol Cell Proteomics. 2018;17:1047–54. doi:10.1074/mcp.MR117.000476. [Google Scholar] [PubMed] [CrossRef]
17. Barik GK, Sahay O, Mukhopadhyay A, Manne RK, Islam S, Roy A, et al. FBXW2 suppresses breast tumorigenesis by targeting AKT-Moesin-SKP2 axis. Cell Death Dis. 2023;14:623. doi:10.1038/s41419-023-06127-x. [Google Scholar] [PubMed] [CrossRef]
18. Ebner P, Versteeg GA, Ikeda F. Ubiquitin enzymes in the regulation of immune responses. Crit Rev Biochem Mol Biol. 2017;52:425–60. doi:10.1080/10409238.2017.1325829. [Google Scholar] [PubMed] [CrossRef]
19. Barik GK, Sahay O, Islam S, Ghate NB, Kalita B, Alam A. Ubiquitination in cancer metastasis: emerging functions, underlying mechanisms, and clinical implications. Technol Cancer Res Treat. 2023;22:15330338231210720. doi:10.1177/15330338231210720. [Google Scholar] [PubMed] [CrossRef]
20. Yang Q, Zhao J, Chen D, Wang Y. E3 ubiquitin ligases: styles, structures and functions. Mol Biomed. 2021;2:23. doi:10.1186/s43556-021-00043-2. [Google Scholar] [PubMed] [CrossRef]
21. Jang SM, Redon CE, Aladjem MI. Chromatin-bound cullin-ring ligases: regulatory roles in DNA replication and potential targeting for cancer therapy. Front Mol Biosci. 2018;5:19. doi:10.3389/fmolb.2018.00019. [Google Scholar] [PubMed] [CrossRef]
22. Mishra M, Thacker G, Sharma A, Singh AK, Upadhyay V, Sanyal S, et al. FBW7 Inhibits Myeloid differentiation in Acute Myeloid Leukemia via GSK3-dependent ubiquitination of PU.1. Mol Cancer Res. 2021;19:261–73. doi:10.1158/1541-7786.MCR-20-0268. [Google Scholar] [PubMed] [CrossRef]
23. Islam S, Dutta P, Chopra K, Sahay O, Rapole S, Chauhan R, et al. Co-operative binding of SKP1, Cullin1 and Cullin7 to FBXW8 results in Cullin1-SKP1-FBXW8-Cullin7 functional complex formation that monitors cellular function of β-TrCP1. Int J Biol Macromol. 2021;190:233–43. doi:10.1016/j.ijbiomac.2021.08.195. [Google Scholar] [PubMed] [CrossRef]
24. Liu X, Zurlo G, Zhang Q. The roles of Cullin-2 E3 ubiquitin ligase complex in cancer. Adv Exp Med Biol. 2020;1217:173–86. doi:10.1007/978-981-15-1025-0. [Google Scholar] [CrossRef]
25. Jeong Y, Oh AR, Jung YH, Gi H, Kim YU, Kim K. Targeting E3 ubiquitin ligases and their adaptors as a therapeutic strategy for metabolic diseases. Exp Mol Med. 2023;55:2097–104. doi:10.1038/s12276-023-01087-w. [Google Scholar] [PubMed] [CrossRef]
26. Jackson S, Xiong Y. CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem Sci. 2009;34:562–70. doi:10.1016/j.tibs.2009.07.002. [Google Scholar] [PubMed] [CrossRef]
27. Okumura F, Joo-Okumura A, Nakatsukasa K, Kamura T. The role of cullin 5-containing ubiquitin ligases. Cell Div. 2016;11:1. doi:10.1186/s13008-016-0016-3. [Google Scholar] [PubMed] [CrossRef]
28. Chen Z, Sui J, Zhang F, Zhang C. Cullin family proteins and tumorigenesis: genetic association and molecular mechanisms. J Cancer. 2015;6:233–42. doi:10.7150/jca.11076. [Google Scholar] [PubMed] [CrossRef]
29. Kim D, Lee JY, Yang JS, Kim JW, Kim VN, Chang H. The architecture of SARS-CoV-2 transcriptome. Cell. 2020;181:914–21.e10. doi:10.1016/j.cell.2020.04.011. [Google Scholar] [PubMed] [CrossRef]
30. Finkel Y, Mizrahi O, Nachshon A, Weingarten-Gabbay S, Morgenstern D, Yahalom-Ronen Y, et al. The coding capacity of SARS-CoV-2. Nature. 2021;589:125–30. doi:10.1038/s41586-020-2739-1. [Google Scholar] [PubMed] [CrossRef]
31. Irigoyen N, Firth AE, Jones JD, Chung BY, Siddell SG, Brierley I. High-resolution analysis of coronavirus gene expression by RNA sequencing and ribosome profiling. PLoS Pathog. 2016;12:e1005473. doi:10.1371/journal.ppat.1005473. [Google Scholar] [PubMed] [CrossRef]
32. Mousavizadeh L, Ghasemi S. Genotype and phenotype of COVID-19: their roles in pathogenesis. J Microbiol Immunol Infect. 2021;54:159–63. doi:10.1016/j.jmii.2020.03.022. [Google Scholar] [PubMed] [CrossRef]
33. Masters PS. The molecular biology of coronaviruses. Adv Virus Res. 2006;66:193–292. doi:10.1016/S0065-3527(06)66005-3. [Google Scholar] [PubMed] [CrossRef]
34. Fisher T, Gluck A, Narayanan K, Kuroda M, Nachshon A, Hsu JC, et al. Parsing the role of NSP1 in SARS-CoV-2 infection. Cell Rep. 2022;39:110954. [Google Scholar] [PubMed]
35. Jahirul Islam M, Nawal Islam N, Siddik Alom M, Kabir M, Halim MA. A review on structural, non-structural, and accessory proteins of SARS-CoV-2: highlighting drug target sites. Immunobiology. 2023;228:152302. doi:10.1016/j.imbio.2022.152302. [Google Scholar] [PubMed] [CrossRef]
36. Saramago M, Costa VG, Souza CS, Bárria C, Domingues S, Viegas SC, et al. The nsp15 nuclease as a good target to combat SARS-CoV-2: mechanism of action and its inactivation with FDA-approved drugs. Microorganisms. 2022;10:342. doi:10.3390/microorganisms10020342. [Google Scholar] [PubMed] [CrossRef]
37. Redondo N, Zaldívar-López S, Garrido JJ, Montoya M. SARS-CoV-2 accessory proteins in viral pathogenesis: knowns and unknowns. Front Immunol. 2021;12:708264. doi:10.3389/fimmu.2021.708264. [Google Scholar] [PubMed] [CrossRef]
38. Willett BJ, Grove J, MacLean OA, Wilkie C, De Lorenzo G, Furnon W, et al. SARS-CoV-2 Omicron is an immune escape variant with an altered cell entry pathway. Nat Microbiol. 2022;7:1161–79. doi:10.1038/s41564-022-01143-7. [Google Scholar] [PubMed] [CrossRef]
39. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from wuhan: an analysis based on decade-long structural studies of SARS Coronavirus. J Virol. 2020;94:e00127-20. doi:10.1128/JVI.00127-20. [Google Scholar] [PubMed] [CrossRef]
40. Seyran M, Takayama K, Uversky VN, Lundstrom K, Palù G, Sherchan SP, et al. The structural basis of accelerated host cell entry by SARS-CoV-2. FEBS J. 2021;288:5010–20. doi:10.1111/febs.15651. [Google Scholar] [PubMed] [CrossRef]
41. Santos-Mendoza T. The envelope (E) protein of SARS-CoV-2 as a pharmacological target. Viruses. 2023;15:1000. doi:10.3390/v15041000. [Google Scholar] [PubMed] [CrossRef]
42. Schoeman D, Fielding BC. Coronavirus envelope protein: current knowledge. Virol J. 2019;16:69. doi:10.1186/s12985-019-1182-0. [Google Scholar] [PubMed] [CrossRef]
43. Zhou S, Lv P, Li M, Chen Z, Xin H, Reilly S, et al. SARS-CoV-2 E protein: pathogenesis and potential therapeutic development. Biomed Pharmacother. 2023;159:114242. doi:10.1016/j.biopha.2023.114242. [Google Scholar] [PubMed] [CrossRef]
44. Chai J, Cai Y, Pang C, Wang L, McSweeney S, Shanklin J, et al. Structural basis for SARS-CoV-2 envelope protein recognition of human cell junction protein PALS1. Nat Commun. 2021;12:3433. doi:10.1038/s41467-021-23533-x. [Google Scholar] [PubMed] [CrossRef]
45. Boson B, Legros V, Zhou B, Siret E, Mathieu C, Cosset FL, et al. The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles. J Biol Chem. 2021;296:100111. doi:10.1074/jbc.RA120.016175. [Google Scholar] [PubMed] [CrossRef]
46. Marques-Pereira C, Pires MN, Gouveia RP, Pereira NN, Caniceiro AB, Rosário-Ferreira N, et al. SARS-CoV-2 membrane protein: from genomic data to structural new insights. Int J Mol Sci. 2022;23:2986. doi:10.21203/rs.3.rs-702792/v2. [Google Scholar] [CrossRef]
47. Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015;1282:1–23. doi:10.1007/978-1-4939-2438-7_1. [Google Scholar] [PubMed] [CrossRef]
48. Wu W, Cheng Y, Zhou H, Sun C, Zhang S. The SARS-CoV-2 nucleocapsid protein: its role in the viral life cycle, structure and functions, and use as a potential target in the development of vaccines and diagnostics. Virol J. 2023;20:6. doi:10.1186/s12985-023-01968-6. [Google Scholar] [PubMed] [CrossRef]
49. Savastano A, Ibáñez de Opakua A, Rankovic M, Zweckstetter M. Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nat Commun. 2020;11:6041. doi:10.1038/s41467-020-19843-1. [Google Scholar] [PubMed] [CrossRef]
50. Peng Y, Du N, Lei Y, Dorje S, Qi J, Luo T, et al. Structures of the SARS-CoV-2 nucleocapsid and their perspectives for drug design. EMBO J. 2020;39:e105938. doi:10.15252/embj.2020105938. [Google Scholar] [PubMed] [CrossRef]
51. Wu C, Qavi AJ, Hachim A, Kavian N, Cole AR, Moyle AB, et al. Characterization of SARS-CoV-2 nucleocapsid protein reveals multiple functional consequences of the C-terminal domain. iScience. 2021;24:102681. doi:10.1016/j.isci.2021.102681. [Google Scholar] [PubMed] [CrossRef]
52. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–80.e8. doi:10.1016/j.cell.2020.02.052. [Google Scholar] [PubMed] [CrossRef]
53. Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol. 2010;84:12658–64. doi:10.1128/JVI.01542-10. [Google Scholar] [PubMed] [CrossRef]
54. Baggen J, Jacquemyn M, Persoons L, Vanstreels E, Pye VE, Wrobel AG, et al. TMEM106B is a receptor mediating ACE2-independent SARS-CoV-2 cell entry. Cell. 2023;186:3427–42.e22. doi:10.1016/j.cell.2023.06.005. [Google Scholar] [PubMed] [CrossRef]
55. Stertz S, Reichelt M, Spiegel M, Kuri T, Martínez-Sobrido L, García-Sastre A, et al. The intracellular sites of early replication and budding of SARS-coronavirus. Virology. 2007;361:304–15. doi:10.1016/j.virol.2006.11.027. [Google Scholar] [PubMed] [CrossRef]
56. Wei J, Alfajaro MM, DeWeirdt PC, Hanna RE, Lu-Culligan WJ, Cai WL, et al. Genome-wide CRISPR screens reveal host factors critical for SARS-CoV-2 infection. Cell. 2021;184:76–91.e13. doi:10.1016/j.cell.2020.10.028. [Google Scholar] [PubMed] [CrossRef]
57. Schneider WM, Luna JM, Hoffmann HH, Sánchez-Rivera FJ, Leal AA, Ashbrook AW, et al. Genome-scale identification of SARS-CoV-2 and pan-coronavirus host factor networks. Cell. 2021;184:120–32.e14. doi:10.1016/j.cell.2020.12.006. [Google Scholar] [PubMed] [CrossRef]
58. Hoffmann HH, Sánchez-Rivera FJ, Schneider WM, Luna JM, Soto-Feliciano YM, Ashbrook AW, et al. Functional interrogation of a SARS-CoV-2 host protein interactome identifies unique and shared coronavirus host factors. Cell Host Microbe. 2021;29:267–80.e5. doi:10.1016/j.chom.2020.12.009. [Google Scholar] [PubMed] [CrossRef]
59. Zhu Y, Feng F, Hu G, Wang Y, Yu Y, Xu W, et al. A genome-wide CRISPR screen identifies host factors that regulate SARS-CoV-2 entry. Nat Commun. 2021;12:961. doi:10.1038/s41467-021-21213-4. [Google Scholar] [PubMed] [CrossRef]
60. Biering SB, Sarnik SA, Wang E, Zengel JR, Leist SR, Schäfer A, et al. Genome-wide bidirectional CRISPR screens identify mucins as host factors modulating SARS-CoV-2 infection. Nat Genet. 2022;54:1078–89. doi:10.1038/s41588-022-01131-x. [Google Scholar] [PubMed] [CrossRef]
61. Rebendenne A, Roy P, Bonaventure B, Chaves Valadão AL, Desmarets L, Arnaud-Arnould M, et al. Bidirectional genome-wide CRISPR screens reveal host factors regulating SARS-CoV-2, MERS-CoV and seasonal HCoVs. Nat Genet. 2022;54:1090–102. doi:10.1038/s41588-022-01110-2. [Google Scholar] [PubMed] [CrossRef]
62. Baggen J, Persoons L, Vanstreels E, Jansen S, Van Looveren D, Boeckx B, et al. Genome-wide CRISPR screening identifies TMEM106B as a proviral host factor for SARS-CoV-2. Nat Genet. 2021;53:435–44. doi:10.1038/s41588-021-00805-2. [Google Scholar] [PubMed] [CrossRef]
63. Wang R, Simoneau CR, Kulsuptrakul J, Bouhaddou M, Travisano KA, Hayashi JM, et al. Genetic screens identify host factors for SARS-CoV-2 and common cold coronaviruses. Cell. 2021;184:106–19.e14. doi:10.1016/j.cell.2020.12.004. [Google Scholar] [PubMed] [CrossRef]
64. Chen DY, Khan N, Close BJ, Goel RK, Blum B, Tavares AH, et al. SARS-CoV-2 disrupts proximal elements in the JAK-STAT pathway. J Virol. 2021;95:e0086221. doi:10.1128/JVI.00862-21. [Google Scholar] [PubMed] [CrossRef]
65. Malvankar S, Singh A, Ravi Kumar YS, Sahu S, Shah M, Murghai Y, et al. Modulation of various host cellular machinery during COVID-19 infection. Rev Med Virol. 2023;33:e2481. doi:10.1002/rmv.2481. [Google Scholar] [PubMed] [CrossRef]
66. Finkel Y, Gluck A, Nachshon A, Winkler R, Fisher T, Rozman B, et al. SARS-CoV-2 uses a multipronged strategy to impede host protein synthesis. Nature. 2021;594:240–5. doi:10.1038/s41586-021-03610-3. [Google Scholar] [PubMed] [CrossRef]
67. Daniloski Z, Jordan TX, Wessels HH, Hoagland DA, Kasela S, Legut M, et al. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell. 2021;184:92–105.e16. doi:10.1016/j.cell.2020.10.030. [Google Scholar] [PubMed] [CrossRef]
68. Wei J, Patil A, Collings CK, Alfajaro MM, Liang Y, Cai WL, et al. Pharmacological disruption of mSWI/SNF complex activity restricts SARS-CoV-2 infection. Nat Genet. 2023;55:471–83. doi:10.1038/s41588-023-01307-z. [Google Scholar] [PubMed] [CrossRef]
69. Fadason T, Gokuladhas S, Golovina E, Ho D, Farrow S, Nyaga D, et al. A transcription regulatory network within the ACE2 locus may promote a pro-viral environment for SARS-CoV-2 by modulating expression of host factors. bioRxiv. 2020. doi:10.1101/2020.04.14.042002. [Google Scholar] [CrossRef]
70. Deng Q, Rasool RU, Russell RM, Natesan R, Asangani IA. Targeting androgen regulation of TMPRSS2 and ACE2 as a therapeutic strategy to combat COVID-19. iScience. 2021;24:102254. doi:10.1016/j.isci.2021.102254. [Google Scholar] [PubMed] [CrossRef]
71. Samelson AJ, Tran QD, Robinot R, Carrau L, Rezelj VV, Kain AM, et al. BRD2 inhibition blocks SARS-CoV-2 infection by reducing transcription of the host cell receptor ACE2. Nat Cell Biol. 2022;24:24–34. doi:10.1038/s41556-021-00821-8. [Google Scholar] [PubMed] [CrossRef]
72. Mendonca P, Soliman KFA. Flavonoids activation of the transcription factor Nrf2 as a hypothesis approach for the prevention and modulation of SARS-CoV-2 infection severity. Antioxidants. 2020;9. doi:10.3390/antiox9080659. [Google Scholar] [PubMed] [CrossRef]
73. Lodhi N, Singh R, Rajput SP, Saquib Q. SARS-CoV-2: understanding the transcriptional regulation of ACE2 and TMPRSS2 and the role of Single Nucleotide Polymorphism (SNP) at codon 72 of p53 in the innate immune response against virus infection. Int J Mol Sci. 2021;22:8660. doi:10.3390/ijms22168660. [Google Scholar] [PubMed] [CrossRef]
74. Banerjee AK, Blanco MR, Bruce EA, Honson DD, Chen LM, Chow A, et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell. 2020;183:1325–39.e21. doi:10.1016/j.cell.2020.10.004. [Google Scholar] [PubMed] [CrossRef]
75. Ahmad W, Gull B, Baby J, Panicker NG, Khader TA, Akhlaq S, et al. Differentially-regulated miRNAs in COVID-19: a systematic review. Rev Med Virol. 2023;33:e2449. doi:10.1002/rmv.2449. [Google Scholar] [PubMed] [CrossRef]
76. McDonald JT, Enguita FJ, Taylor D, Griffin RJ, Priebe W, Emmett MR, et al. Role of miR-2392 in driving SARS-CoV-2 infection. Cell Rep. 2021;37:109839. doi:10.1016/j.celrep.2021.109839. [Google Scholar] [PubMed] [CrossRef]
77. Pawlica P, Yario TA, White S, Wang J, Moss WN, Hui P, et al. SARS-CoV-2 expresses a microRNA-like small RNA able to selectively repress host genes. Proc Natl Acad Sci U S A. 2021;118:e2116668118. doi:10.1073/pnas.2116668118. [Google Scholar] [PubMed] [CrossRef]
78. Singh M, Chazal M, Quarato P, Bourdon L, Malabat C, Vallet T, et al. A virus-derived microRNA targets immune response genes during SARS-CoV-2 infection. EMBO Rep. 2022;23:e54341. doi:10.1073/pnas.2116668118. [Google Scholar] [CrossRef]
79. Nabeel-Shah S, Lee H, Ahmed N, Burke GL, Farhangmehr S, Ashraf K, et al. SARS-CoV-2 nucleocapsid protein binds host mRNAs and attenuates stress granules to impair host stress response. iScience. 2022;25:103562. doi:10.1016/j.isci.2021.103562. [Google Scholar] [PubMed] [CrossRef]
80. Fung TS, Huang M, Liu DX. Coronavirus-induced ER stress response and its involvement in regulation of coronavirus-host interactions. Virus Res. 2014;194:110–23. doi:10.1016/j.virusres.2014.09.016. [Google Scholar] [PubMed] [CrossRef]
81. Xue M, Feng L. The role of unfolded protein response in coronavirus infection and its implications for drug design. Front Microbiol. 2021;12:808593. doi:10.3389/fmicb.2021.808593. [Google Scholar] [PubMed] [CrossRef]
82. Echavarría-Consuegra L, Cook GM, Busnadiego I, Lefèvre C, Keep S, Brown K, et al. Manipulation of the unfolded protein response: a pharmacological strategy against coronavirus infection. PLoS Pathog. 2021;17:e1009644. doi:10.1371/journal.ppat.1009644. [Google Scholar] [PubMed] [CrossRef]
83. Upadhyay M, Gupta S. Endoplasmic reticulum secretory pathway: potential target against SARS-CoV-2. Virus Res. 2022;320:198897. doi:10.1016/j.virusres.2022.198897. [Google Scholar] [PubMed] [CrossRef]
84. Zou C, Yoon H, Park PMC, Patten JJ, Pellman J, Carreiro J, et al. The human E3 ligase RNF185 is a regulator of the SARS-CoV-2 envelope protein. iScience. 2023;26:106601. doi:10.1016/j.isci.2023.106601. [Google Scholar] [PubMed] [CrossRef]
85. Zhao M, Zhang M, Yang Z, Zhou Z, Huang J, Zhao B. Role of E3 ubiquitin ligases and deubiquitinating enzymes in SARS-CoV-2 infection. Front Cell Infect Microbiol. 2023;13:1217383. doi:10.3389/fcimb.2023.1217383. [Google Scholar] [PubMed] [CrossRef]
86. Wang G, Zhao Q, Zhang H, Liang F, Zhang C, Wang J, et al. Degradation of SARS-CoV-2 receptor ACE2 by the E3 ubiquitin ligase Skp2 in lung epithelial cells. Front Med. 2021;15:252–63. doi:10.1007/s11684-021-0837-6. [Google Scholar] [PubMed] [CrossRef]
87. Xiao Y, Yan Y, Chang L, Ji H, Sun H, Song S, et al. CDK4/6 inhibitor palbociclib promotes SARS-CoV-2 cell entry by down-regulating SKP2 dependent ACE2 degradation. Antiviral Res. 2023;212:105558. doi:10.1016/j.antiviral.2023.105558. [Google Scholar] [PubMed] [CrossRef]
88. Chen Y, Lear TB, Evankovich JW, Larsen MB, Lin B, Alfaras I, et al. A high-throughput screen for TMPRSS2 expression identifies FDA-approved compounds that can limit SARS-CoV-2 entry. Nat Commun. 2021;12:3907. doi:10.1038/s41467-021-24156-y. [Google Scholar] [PubMed] [CrossRef]
89. Wang Y, Fan Y, Huang Y, Du T, Liu Z, Huang D, et al. TRIM28 regulates SARS-CoV-2 cell entry by targeting ACE2. Cell Signal. 2021;85:110064. doi:10.1016/j.cellsig.2021.110064. [Google Scholar] [PubMed] [CrossRef]
90. Jin S, He X, Ma L, Zhuang Z, Wang Y, Lin M, et al. Suppression of ACE2 SUMOylation protects against SARS-CoV-2 infection through TOLLIP-mediated selective autophagy. Nat Commun. 2022;13:5204. doi:10.1038/s41467-022-32957-y. [Google Scholar] [PubMed] [CrossRef]
91. Li Z, Hao P, Zhao Z, Gao W, Huan C, Li L, et al. Correction To: the E3 ligase RNF5 restricts SARS-CoV-2 replication by targeting its envelope protein for degradation. Signal Transduct Target Ther. 2023;8:85. doi:10.1038/s41392-023-01374-y. [Google Scholar] [PubMed] [CrossRef]
92. Orozco MJG, Hage A, Xia H, Huante M, Sv Tol, Giraldo MI, et al. The E3-ligase TRIM7 acts as an antiviral factor by ubiquitinating the SARS-CoV-2 membrane protein to limit apoptosis and virus replication. J Immunol. 2022;208(1_Supplement):51.07. doi:10.1038/s41392-023-01335-5. [Google Scholar] [PubMed] [CrossRef]
93. Zhou J, Zhou Y, Wei XF, Fan L, Gao X, Li Y, et al. TRIM6 facilitates SARS-CoV-2 proliferation by catalyzing the K29-typed ubiquitination of NP to enhance the ability to bind viral genomes. J Med Virol. 2024;96:e29531. doi:10.1002/jmv.29531. [Google Scholar] [PubMed] [CrossRef]
94. Mao S, Cai X, Niu S, Wei J, Jiang N, Deng H, et al. TRIM21 promotes ubiquitination of SARS-CoV-2 nucleocapsid protein to regulate innate immunity. J Med Virol. 2023;95:e28719. doi:10.1002/jmv.28719. [Google Scholar] [PubMed] [CrossRef]
95. Zhao Y, Sui L, Wu P, Li L, Liu L, Ma B, et al. EGR1 functions as a new host restriction factor for SARS-CoV-2 to inhibit virus replication through the E3 ubiquitin ligase MARCH8. J Virol. 2023;97:e0102823. doi:10.1128/jvi.01028-23. [Google Scholar] [PubMed] [CrossRef]
96. Ren J, Wang S, Zong Z, Pan T, Liu S, Mao W, et al. TRIM28-mediated nucleocapsid protein SUMOylation enhances SARS-CoV-2 virulence. Nat Commun. 2024;15:244. doi:10.1038/s41467-023-44502-6. [Google Scholar] [PubMed] [CrossRef]
97. Zhang Y, Ozono S, Tada T, Tobiume M, Kameoka M, Kishigami S, et al. MARCH8 targets cytoplasmic lysine residues of various viral envelope glycoproteins. Microbiol Spectr. 2022;10:e0061821. doi:10.1128/spectrum.00618-21. [Google Scholar] [PubMed] [CrossRef]
98. Lun CM, Waheed AA, Majadly A, Powell N, Freed EO. Mechanism of Viral Glycoprotein Targeting by Membrane-associated-RING-CH Proteins. mBio. 2021;12:e00219-21. doi:10.1128/mbio.00219-21. [Google Scholar] [CrossRef]
99. Tian L, Liu Z, Gao W, Zhao Z, Li X, Zhang W, et al. SARS-CoV-2 nsp16 is regulated by host E3 ubiquitin ligases, UBR5 and MARCHF7. bioRxiv. 2024. doi:10.1101/2024.08.30.610469. [Google Scholar] [CrossRef]
100. Liang X, Xiao J, Li X, Liu Y, Lu Y, Wen Y, et al. A C-terminal glutamine recognition mechanism revealed by E3 ligase TRIM7 structures. Nat Chem Biol. 2022;18:1214–23. doi:10.1038/s41589-022-01128-x. [Google Scholar] [PubMed] [CrossRef]
101. Fan L, Zhou Y, Wei X, Feng W, Guo H, Li Y, et al. The E3 ligase TRIM22 restricts SARS-CoV-2 replication by promoting proteasomal degradation of NSP8. mBio. 2024;15:e0232023. doi:10.1128/mbio.02320-23. [Google Scholar] [PubMed] [CrossRef]
102. Yu M, Li J, Gao W, Li Z, Zhang W. Multiple E3 ligases act as antiviral factors against SARS-CoV-2 via inducing the ubiquitination and degradation of ORF9b. J Virol. 2024;98:e0162423. doi:10.1128/jvi.01624-23. [Google Scholar] [PubMed] [CrossRef]
103. Zhou Y, Chen Z, Liu S, Liao Y, Du A, Dong Z, et al. A Cullin 5-based complex serves as an essential modulator of ORF9b stability in SARS-CoV-2 replication. Signal Transduct Target Ther. 2024;9:159. doi:10.1038/s41392-024-01874-5. [Google Scholar] [PubMed] [CrossRef]
104. Zhang L, Hao P, Chen X, Lv S, Gao W, Li C, et al. CRL4B E3 ligase recruited by PRPF19 inhibits SARS-CoV-2 infection by targeting ORF6 for ubiquitin-dependent degradation. mBio. 2024;15:e0307123. doi:10.1128/mbio.03071-23. [Google Scholar] [PubMed] [CrossRef]
105. Maimaitiyiming Y, Yang T, Wang QQ, Feng Y, Chen Z, Björklund M, et al. Heat treatment promotes ubiquitin-mediated proteolysis of SARS-CoV-2 RNA polymerase and decreases viral load. Research. 2022;2022:9802969. doi:10.34133/2022/9802969. [Google Scholar] [PubMed] [CrossRef]
106. Lear TB, Boudreau Á, Lockwood KC, Chu E, Camarco DP, Cao Q, et al. E3 ubiquitin ligase ZBTB25 suppresses beta coronavirus infection through ubiquitination of the main viral protease MPro. J Biol Chem. 2023;299:105388. doi:10.1016/j.jbc.2023.105388. [Google Scholar] [PubMed] [CrossRef]
107. Zhou L, Liu R, Pathak H, Wang X, Jeong GH, Kumari P, et al. Ubiquitin ligase parkin regulates the stability of SARS-CoV-2 main protease and suppresses viral replication. ACS Infect Dis. 2024;10:879–89. doi:10.1021/acsinfecdis.3c00418. [Google Scholar] [PubMed] [CrossRef]
108. Nishitsuji H, Iwahori S, Ohmori M, Shimotohno K, Murata T. Ubiquitination of SARS-CoV-2 NSP6 and ORF7a facilitates NF-κB activation. mBio. 2022;13:e0097122. doi:10.1128/mbio.00971-22. [Google Scholar] [PubMed] [CrossRef]
109. Jung J, Baek J, Tae K, Shin D, Han S, Yang W, et al. Structural mechanism for regulation of Rab7 by site-specific monoubiquitination. Int J Biol Macromol. 2022;194:347–57. doi:10.1016/j.ijbiomac.2021.11.074. [Google Scholar] [PubMed] [CrossRef]
110. Zhang S, Wang J, Cheng G. Protease cleavage of RNF20 facilitates coronavirus replication via stabilization of SREBP1. Proc Natl Acad Sci U S A. 2021;118. doi:10.1073/pnas.2107108118. [Google Scholar] [PubMed] [CrossRef]
111. Yuan Z, Hu B, Xiao H, Tan X, Li Y, Tang K, et al. The E3 ubiquitin ligase RNF5 facilitates SARS-CoV-2 membrane protein-mediated virion release. mBio. 2021;13:e0316821. doi:10.1128/mbio.03168-21. [Google Scholar] [PubMed] [CrossRef]
112. Khatun O, Sharma M, Narayan R, Tripathi S. SARS-CoV-2 ORF6 protein targets TRIM25 for proteasomal degradation to diminish K63-linked RIG-I ubiquitination and type-I interferon induction. Cell Mol Life Sci. 2023;80:364. doi:10.1007/s00018-023-05011-3. [Google Scholar] [PubMed] [CrossRef]
113. Wang L, Liu C, Yang B, Zhang H, Jiao J, Zhang R, et al. SARS-CoV-2 ORF10 impairs cilia by enhancing CUL2ZYG11B activity. J Cell Biol. 2022;221. doi:10.1083/jcb.202108015. [Google Scholar] [PubMed] [CrossRef]
114. Israeli M, Finkel Y, Yahalom-Ronen Y, Paran N, Chitlaru T, Israeli O, et al. Genome-wide CRISPR screens identify GATA6 as a proviral host factor for SARS-CoV-2 via modulation of ACE2. Nat Commun. 2022;13:2237. doi:10.1038/s41467-022-29896-z. [Google Scholar] [PubMed] [CrossRef]
115. Cao Z, Xia H, Rajsbaum R, Xia X, Wang H, Shi PY. Ubiquitination of SARS-CoV-2 ORF7a promotes antagonism of interferon response. Cell Mol Immunol. 2021;18:746–8. doi:10.1038/s41423-020-00603-6. [Google Scholar] [PubMed] [CrossRef]
116. Liu Z, Fu Y, Huang Y, Zeng F, Rao J, Xiao X, et al. Ubiquitination of SARS-CoV-2 ORF7a prevents cell death induced by recruiting BclXL to activate ER stress. Microbiol Spectr. 2022;10:e0150922. doi:10.1128/spectrum.01509-22. [Google Scholar] [PubMed] [CrossRef]
117. Chuang HC, Hsueh CH, Hsu PM, Huang RH, Tsai CY, Chung NH, et al. SARS-CoV-2 spike protein enhances MAP4K3/GLK-induced ACE2 stability in COVID-19. EMBO Mol Med. 2022;14:e15904. doi:10.15252/emmm.202215904. [Google Scholar] [PubMed] [CrossRef]
118. Bednash JS, Johns F, Farkas D, Elhance A, Adair J, Cress K, et al. Inhibiting the deubiquitinase UCHL1 reduces SARS-CoV-2 viral uptake by ACE2. Am J Respir Cell Mol Biol. 2023;68:566–76. doi:10.1165/rcmb.2022-0331OC. [Google Scholar] [PubMed] [CrossRef]
119. Biterge Süt B. Molecular profiling of immune cell-enriched severe acute respiratory syndrome Coronavirus 2 (SARS-CoV-2) interacting protein USP13. Life Sci. 2020;258:118170. doi:10.1016/j.lfs.2020.118170. [Google Scholar] [PubMed] [CrossRef]
120. Ravindran V, Wagoner J, Athanasiadis P, Den Hartigh AB, Sidorova JM, Ianevski A, et al. Discovery of host-directed modulators of virus infection by probing the SARS-CoV-2-host protein-protein interaction network. Brief Bioinform. 2022;23:bbac456. doi:10.1093/bib/bbac456. [Google Scholar] [PubMed] [CrossRef]
121. Guo G, Gao M, Gao X, Zhu B, Huang J, Luo K, et al. SARS-CoV-2 non-structural protein 13 (nsp13) hijacks host deubiquitinase USP13 and counteracts host antiviral immune response. Signal Transduct Target Ther. 2021;6:119. doi:10.1038/s41392-021-00509-3. [Google Scholar] [PubMed] [CrossRef]
122. Alshiraihi IM, Klein GL, Brown MA. Targeting NSP16 Methyltransferase for the broad-spectrum clinical management of Coronaviruses: managing the next pandemic. Diseases. 2021;9:12. doi:10.3390/diseases9010012. [Google Scholar] [PubMed] [CrossRef]
123. Gao W, Wang L, Ju X, Zhao S, Li Z, Su M, et al. The deubiquitinase USP29 promotes SARS-CoV-2 virulence by preventing proteasome degradation of ORF9b. mBio. 2022;13:e0130022. doi:10.1128/mbio.01300-22. [Google Scholar] [PubMed] [CrossRef]
124. Chen X, Tian L, Zhang L, Gao W, Yu M, Li Z, et al. Deubiquitinase USP39 promotes SARS-CoV-2 replication by deubiquitinating and stabilizing the envelope protein. Antiviral Res. 2024;221:105790. doi:10.1016/j.antiviral.2023.105790. [Google Scholar] [PubMed] [CrossRef]
125. Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181:905–13.e7. doi:10.1016/j.cell.2020.04.004. [Google Scholar] [PubMed] [CrossRef]
126. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–9. doi:10.1038/nm1267. [Google Scholar] [PubMed] [CrossRef]
127. Lei C, Qian K, Li T, Zhang S, Fu W, Ding M, et al. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat Commun. 2020;11:2070. doi:10.1038/s41467-020-16048-4. [Google Scholar] [PubMed] [CrossRef]
128. Wösten-van Asperen RM, Lutter R, Specht PA, Moll GN, van Woensel JB, van der Loos CM, et al. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J Pathol. 2011;225:618–27. doi:10.1002/path.2987. [Google Scholar] [PubMed] [CrossRef]
129. Wu A, Shi K, Wang J, Zhang R, Wang Y. Targeting SARS-CoV-2 entry processes: the promising potential and future of host-targeted small-molecule inhibitors. Eur J Med Chem. 2024;263:115923. doi:10.1016/j.ejmech.2023.115923. [Google Scholar] [PubMed] [CrossRef]
130. Gajjela BK, Zhou MM. Calming the cytokine storm of COVID-19 through inhibition of JAK2/STAT3 signaling. Drug Discov Today. 2022;27:390–400. doi:10.1016/j.drudis.2021.10.016. [Google Scholar] [PubMed] [CrossRef]
131. Tayfun Acar MBA, Arvas B, Ucar B, Sahin Y. Adsorption and desorption of hydroxychloroquine onto sulphur doped graphene powders as a potential drug for COVID-19: physicochemical investigation, surface chemistry and in vitro cytotoxicity evaluation. Adsorption. 2024;30:1377–93. doi:10.1007/s10450-024-00506-2. [Google Scholar] [CrossRef]
132. Suvarnapathaki S, Chauhan D, Nguyen A, Ramalingam M, Camci-Unal G. Advances in targeting ACE2 for developing COVID-19 therapeutics. Ann Biomed Eng. 2022;50:1734–49. doi:10.1007/s10439-022-03094-w. [Google Scholar] [PubMed] [CrossRef]
133. Su S, Chen J, Wang Y, Wong LM, Zhu Z, Jiang G, et al. Lenalidomide downregulates ACE2 protein abundance to alleviate infection by SARS-CoV-2 spike protein conditioned pseudoviruses. Signal Transduct Target Ther. 2021;6:182. doi:10.1038/s41392-021-00608-1. [Google Scholar] [PubMed] [CrossRef]
134. Greenhalgh T, Knight M, A’Court C, Buxton M, Husain L. Management of post-acute COVID-19 in primary care. BMJ. 2020;370:m3026. doi:10.1136/bmj.m3026. [Google Scholar] [PubMed] [CrossRef]
135. Suvvari TK, Kutikuppala LVS, Tsagkaris C, Corriero AC, Kandi V. Post-COVID-19 complications: multisystemic approach. J Med Virol. 2021;93:6451–5. doi:10.1002/jmv.27222. [Google Scholar] [PubMed] [CrossRef]
136. Lee J, Lee Y, Jung YM, Park JH, Yoo HS, Park J. Discovery of E3 ligase ligands for target protein degradation. Molecules. 2022;27:6515. doi:10.3390/molecules27196515. [Google Scholar] [PubMed] [CrossRef]
137. Desantis J, Mercorelli B, Celegato M, Croci F, Bazzacco A, Baroni M, et al. Indomethacin-based PROTACs as pan-coronavirus antiviral agents. Eur J Med Chem. 2021;226:113814. doi:10.1016/j.ejmech.2021.113814. [Google Scholar] [PubMed] [CrossRef]
138. Khurshid R, Schulz JM, Hu J, Snowden TS, Reynolds RC, Schürer SC. Targeted degrader technologies as prospective SARS-CoV-2 therapies. Drug Discov Today. 2024;29:103847. doi:10.1016/j.drudis.2023.103847. [Google Scholar] [PubMed] [CrossRef]
139. Desantis J, Bazzacco A, Eleuteri M, Tuci S, Bianconi E, Macchiarulo A, et al. Design, synthesis, and biological evaluation of first-in-class indomethacin-based PROTACs degrading SARS-CoV-2 main protease and with broad-spectrum antiviral activity. Eur J Med Chem. 2024;268:116202. doi:10.1016/j.ejmech.2024.116202. [Google Scholar] [PubMed] [CrossRef]
140. Cheng S, Feng Y, Li W, Liu T, Lv X, Tong X, et al. Development of novel antivrial agents that induce the degradation of the main protease of human-infecting coronaviruses. Eur J Med Chem. 2024;275:116629. doi:10.1016/j.ejmech.2024.116629. [Google Scholar] [PubMed] [CrossRef]
141. Alugubelli YR, Xiao J, Khatua K, Kumar S, Sun L, Ma Y, et al. Discovery of first-in-class PROTAC degraders of SARS-CoV-2 main protease. J Med Chem. 2024;67:6495–507. doi:10.1021/acs.jmedchem.3c02416. [Google Scholar] [PubMed] [CrossRef]
142. Julio AR, Shikwana F, Truong C, Burton NR, Dominguez ER, Turmon AC, et al. Delineating cysteine-reactive compound modulation of cellular proteostasis processes. Nat Chem Biol. 2024. doi:10.1038/s41589-024-01760-9. Epub ahead of print. [Google Scholar] [PubMed] [CrossRef]
143. Khramtsov YV, Ulasov AV, Lupanova TN, Slastnikova TA, Rosenkranz AA, Bunin ES, et al. Intracellular degradation of SARS-CoV-2 N-protein caused by modular nanotransporters containing anti-N-protein monobody and a sequence that recruits the Keap1 E3 ligase. Pharmaceutics. 2023;16. doi:10.3390/pharmaceutics16010004. [Google Scholar] [PubMed] [CrossRef]
144. Chatterjee P, Ponnapati M, Kramme C, Plesa AM, Church GM, Jacobson JM. Targeted intracellular degradation of SARS-CoV-2 via computationally optimized peptide fusions. Commun Biol. 2020;3:715. doi:10.1038/s42003-020-01470-7. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools