
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Mechanistic Insights into N-Oleoylethanolamide-Mediated Hepatoprotection via PPAR-α
A.V. Zhirmunsky National Scientific Center of Marine Biology, Far Eastern Branch, Russian Academy of Sciences, Vladivostok, 119334, Russia
* Corresponding Author: Arina Ponomarenko. Email:
BIOCELL 2025, 49(4), 607-627. https://doi.org/10.32604/biocell.2025.061606
Received 28 November 2024; Accepted 08 February 2025; Issue published 30 April 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
The high prevalence of obesity and associated nonalcoholic fatty liver disease (NAFLD) in the population determines the increased interest in identifying molecular targets for regulating the processes underlying these pathologies. The search for new endogenous bioregulators of lipid metabolism and their inclusion in therapeutic regimens for the treatment of patients is becoming a potentially promising direction in science and medicine. Oleoylethanolamide (OEA) is an endogenous lipid mediator capable of exerting multiple hypolipidemic, anti-inflammatory, and hepatoprotective effects mediated by agonism with receptors of the peroxisome proliferator-activated receptor (PPAR) family (PPAR-α and PPAR-γ). This review focuses on a detailed description of the PPAR-dependent mechanisms of the hepatoprotective activity of OEA in the development of NAFLD. The main attention is paid to such topics as reduction of oxidative stress and inflammation, inhibition of liver fibrogenesis, suppression of hepatocyte death, and changes in various parameters of lipid metabolism.Keywords
Due to the worldwide obesity epidemic and the ongoing rise in the incidence of associated metabolic disorders, nonalcoholic fatty liver disease (NAFLD) is becoming a major worry for the healthcare system. According to recent meta-analysis data, the global prevalence of NAFLD is significantly higher than previously estimated and continues to increase at an alarming rate. The global prevalence of NAFLD is estimated to be 32.4% in 2022, significantly higher than in 2005 (25.5%). In addition, the incidence and prevalence of NAFLD are significantly higher in men than in women [1]. In addition, NAFLD is projected to become the leading cause of cirrhosis requiring liver transplantation in the next decade [2]. It is commonly established that NAFLD has a strong correlation with the onset of obesity [3] as well as with metabolic conditions that directly coexist with obesity, including hypertension [4], insulin resistance [5], and hypercholesterolemia [6]. NAFLD is often defined by a fairly broad range of liver lesions, including damage to hepatocytes, which are functioning liver cells, the growth of the inflammatory process, and the creation of cirrhosis and fibrosis on top of the inflammation [7].
Based on its histologic characteristics, NAFLD is commonly divided into nonalcoholic steatohepatitis (NASH) and nonalcoholic fatty liver (NAFL) [8]. With a more benign course, NAFL is characterized by fatty liver dystrophy and mild inflammation of the hepatic lobules [9]. In addition to more severe diffuse lobular inflammation progressing to cirrhosis and fibrosis, NASH is associated with significant hepatocyte destruction, including fat necrosis [10].
In the development of obesity, under conditions of increased caloric intake, de novo fat synthesis increases, with the main source of material for triacylglycerides (TAG) synthesis coming from white adipose tissue [11]. Also, one of the most significant pathogenetic aspects of NAFLD is insulin resistance, which, as a rule, also develops in the background of obesity [12].
In the context of excessive sugar intake, insulin has an antilipolytic effect, encourages the synthesis and storage of TAGs in adipose tissue, and increases the fatty acid synthesis and esterification processes [13]. In adipocytes, fatty acids (FA) are mostly stored as TAGs [14]. In instances of low energy experienced by the body due to insulin resistance, TAGs are known to respond to counterregulatory hormones, such as cortisol and epinephrine, by undergoing a process of breakdown into glycerol and free fatty acids (FAs). Sharp increases in FAs intake in the liver can produce mitochondrial dysfunction and poor beta-oxidation, which can then cause inflammation and concurrent activation of lipid peroxidation, which can ultimately lead to oxidative stress, damage, and hepatocyte death [15,16].
There are two key pathogenetic phases in the development of NAFLD [17]. Insulin resistance and the accumulation of excess fat in the liver are features of the first stage. As a result, insulin resistance increases even more, starting a vicious loop [18]. Molecular and cellular changes that eventually result in the development of chronic, long-lasting inflammation and fibrosis are the hallmarks of the second stage. These molecular alterations include immune cells producing proinflammatory cytokines, lipid peroxidation being activated [19], oxidative stress developing [20], lipotoxicity, and extracellular matrix remodeling [21].
Hepatocytes overexposed to free fatty acids (FFA) undergo oxidative stress that damages their mitochondria, produces more reactive oxygen species (ROS) [22,23], stresses the endoplasmic reticulum (ER) and ultimately leads to persistent inflammation [24] (Fig. 1).
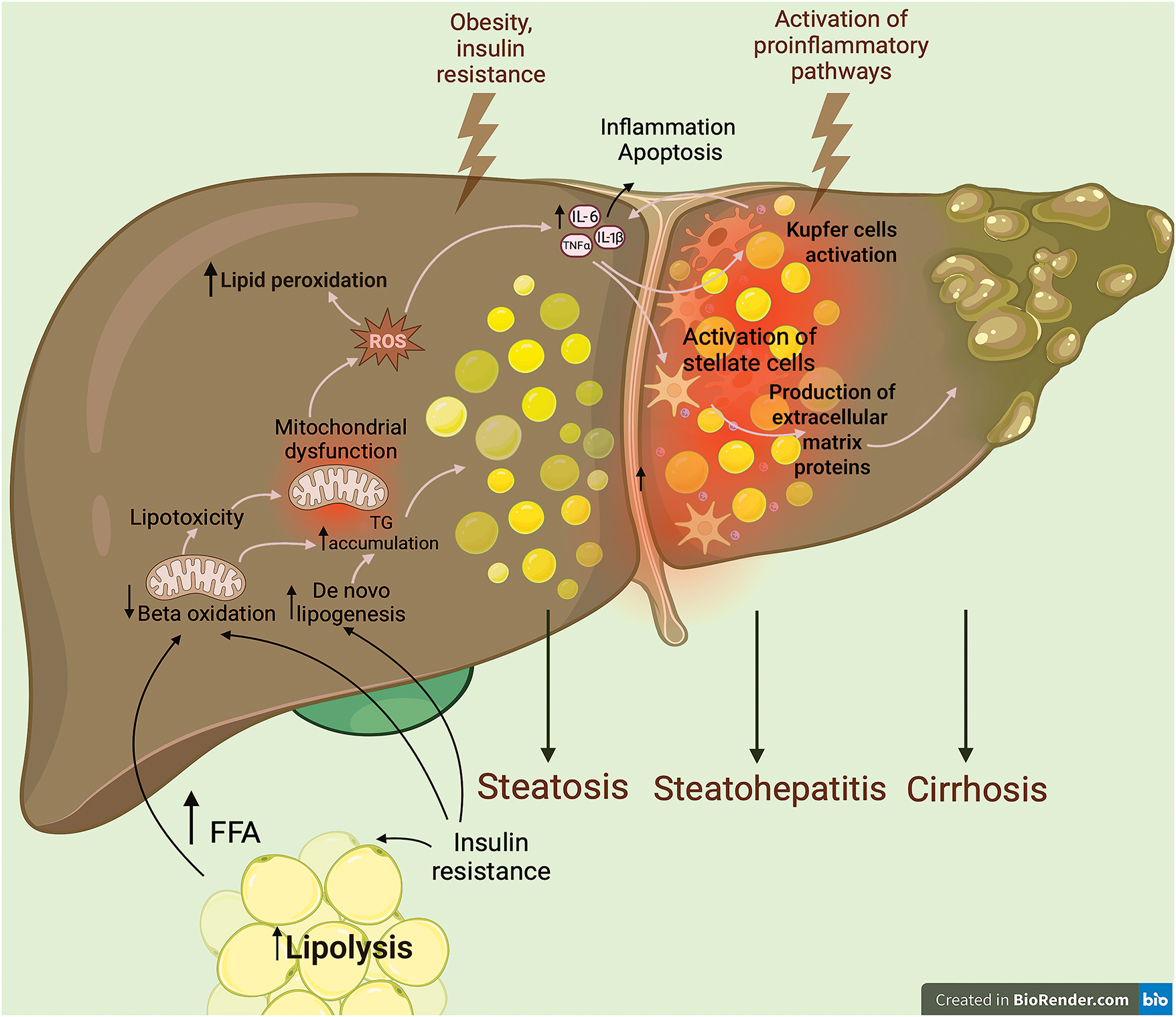
Figure 1: The main pathogenetic stages of NAFLD. In the initial phase, the presence of insulin resistance results in the influx of excessive free fatty acids into the liver. Concurrently, insulin triggers de novo lipogenesis and suppresses beta-oxidation of fatty acids. The interplay of these phenomena culminates in the induction of mitochondrial dysfunction, lipotoxicity, and lipid accumulation within the cytoplasm of hepatocytes. At the second stage, the development of a full-fledged inflammatory process accompanied by increased production of proinflammatory cytokines and activation of Kupffer cells—resident liver macrophages—is initiated. This contributes to the maintenance of the vicious circle of inflammation and apoptosis of hepatocytes. Consequently, the proinflammatory microenvironment contributes to the activation of Ito cells, manifesting as an increased production of extracellular matrix proteins. This, in turn, results in the replacement of functional liver tissue by connective tissue. FFA—free fatty acids; TG—triglycerides; ROS—reactive oxygen species. Created in BioRender.com
Peroxisome proliferator-activated receptor (PPAR) agonists are currently one of the most promising options for the comprehensive treatment of NAFLD and are attracting increasing research attention. The therapeutic areas related to lipid and glucose metabolism, inflammation, obesity [25,26], insulin resistance, and atherosclerosis [27,28], which often lead to secondary liver damage, are well aware of the potential benefits of PPAR agonists. The versatility of their therapeutic effects determines their advantage over other pharmacological targets.
As a lipid mediator, oleoylethanolamide (OEA) belongs to the class of N-acylethanolamines (NAE) and acts by activating the nuclear receptor PPAR-α [29]. OEA is implicated in the pathophysiology of appetite regulation, lipid metabolism, and carbohydrate metabolism, according to an increasing body of studies [30]. Numerous studies have demonstrated that OEA is a promising pharmacological target for the treatment of obesity and eating disorders [31]. It has been demonstrated to increase fullness and reduce appetite in both obese humans [32] and animals [33]. A growing body of experimental data indicates that the hepatoprotective benefits of OEA might be attributed to its capacity to activate PPAR-α.
The aim of this review is to describe point by point the mechanisms of NAFLD formation in the context of potential OEA-mediated therapeutic effects. The main focus is on PPAR-dependent pathways for the implementation of the hepatoprotective effect of OEA.
2 A Key Role of PPAR Receptors in the Hepatoprotective Effect of OEA
It is well-known that members of the PPARs family function as transcription factors that are induced by ligands [34]. To date, three PPAR isoforms have been identified: PPAR-α, PPAR-β/δ, and PPAR-γ [35]. The distribution of PPARs isoforms in tissues is diverse: PPAR-α is abundantly expressed in metabolically active tissues, PPAR-β/δ is expressed ubiquitously, and PPAR-γ is predominantly found in adipose tissue and immune cells [36,37]. The multiple functions of PPARs are determined by transcriptional activity; receptors in their activated form are able to modulate the expression of a large number of target genes. PPAR target genes include enzymes of cholesterol metabolism, fat synthesis and breakdown, homeostasis enzymes, as well as genes involved in the development of obesity and inflammation [38]. It should be noted that the regulation of gene expression by PPARs can be both positive and negative, manifested in the ability to inhibit gene expression [39].
Among all the above-mentioned subtypes of PPARs, the PPAR-α receptor contributes most importantly to the regulation of metabolic pathways in the liver. These metabolic pathways include: triglyceride synthesis and cleavage; lipoprotein and apoprotein metabolism; gluconeogenesis; bile acid metabolism; microsomal, peroxisomal, and mitochondrial fatty acids (FA) oxidation; FA binding and activation; FA elongation and desaturation and other related pathways [40]. Key regulatory enzymes that are targets of PPAR-α include FA beta-oxidation enzymes such as acyl-Coenzyme A (CoA) dehydrogenase and acyl-CoA oxidase 1 (ACOX1) [41]. PPAR-α activation enhances FA beta-oxidation, subsequent adenosine triphosphate (ATP) production, and ketogenesis, indicating a dominant role of PPAR-α in the control of FA oxidation and energy production under nutrient-deficient conditions [42].
To date, there are numerous studies showing that PPAR-α activation helps in liver damage. It has been demonstrated that prolonged PPAR-α activation can ameliorate the course of NAFLD by reducing lipotoxicity [43,44], oxidative stress, and ROS generation [40]. PPAR-α signaling has been shown to reduce the severity of hyperlipidemia and fatty liver degeneration by preventing mitochondrial and ER stress [45]. In animal studies, PPAR-α agonists stopped the development of liver fibrosis [46,47]. Gene expression of hepatic PPAR-α negatively correlates with the severity of NASH and fibrosis in humans [48], confirming the critical importance of hepatic PPAR-α for lipid homeostasis [49].
PPAR-β/δ also plays a critical role in liver metabolism, where it is mainly expressed in hepatocytes, Kupffer cells, sinusoidal endothelial cells, and human stellate cells (HSCs) [50]. Although the functions of PPAR-α and PPAR-β/δ in the liver appear to be similar, PPAR-β/δ cannot compensate for all the functions of PPAR-α, as shown in PPAR-α knockout experiments [51]. In addition, PPAR-β/δ also plays an important role in the modulation of inflammation. Ligands that bind to PPAR-β/δ induce the initiation of anti-inflammatory signals in liver-resident macrophages, and Kupffer cells [52]. However, the more detailed mechanism of the anti-inflammatory role of PPAR-β/δ is not yet fully understood.
The primary localization and functionality of PPAR-γ is exclusively concentrated in adipose tissue [53]. However, in individuals with NAFLD, PPAR-γ expression levels in the liver are significantly elevated [54], suggesting that PPAR-γ also has intrahepatic functions. PPAR-γ regulates several target genes in adipocytes that are responsible for lipid uptake and storage, production of inflammatory cytokines, and secretion of adipokines that increase insulin sensitivity [55]. In the liver, PPAR-γ has also been shown to stimulate free fatty acid uptake through the expression of fatty acid synthase (FASN) and is involved in activating the conversion of pyruvate to fatty acids in hepatocytes [56]. PPAR-α and PPAR-γ regulate lipid metabolism in opposite ways, but due to their localization in different tissues, PPAR-γ, although promoting fat accumulation in the liver, has a beneficial effect on NAFLD by reducing insulin resistance [38].
Currently, many potential endogenous ligands for PPAR-α have been described, including the aforementioned group of NAE, as well as FAs themselves and some of their derivatives, such as eicosanoids [57]. Also, these receptors can be activated by pharmacological agents used in the therapy of diabetes mellitus [58] and atherosclerosis [59], which indicates the important role of PPAR-α in the normalization of metabolic processes closely related to liver function. The anti-inflammatory properties of PPAR-α agonists are due to their ability to limit cytokine expression in the liver through different transcriptional mechanisms and targets [60]. Thus, the presence of multidirectional transcriptional activity for this receptor makes its agonists, including OEA, promising pharmacological targets for complex therapy of NAFLD.
Recent studies have shown that fenofibrate, a PPAR-α agonist, lowers lipid levels in an mTOR-independent manner by activating autophagy and transcription factors [61]. Unfortunately, fenofibrate has minimal activity in lowering blood glucose and regulating insulin sensitivity [62]. Pemafibrate, a newer and more specific PPAR-α modulator, has shown better efficacy than its predecessor both in preclinical models of NAFLD and in humans with diabetes and dyslipidemia [63]. However, all PPAR-α agonists show greater therapeutic benefit when combined with PPAR-γ agonists than when used alone [64]. At the same time, some researchers believe that pan-PPAR agonism appears to be necessary to achieve significant results on histologic endpoints in NAFLD treatment [65]. Therefore, OEA with activity against both PPAR-α and PPAR-γ, as shown in our previous work [66], is a promising candidate for the treatment of NAFLD.
The selective PPAR-β/δ agonist Seladelpar demonstrated improved insulin sensitivity in steatohepatitis in patients with NASH [67]. However, clinical trials of Seladelpar were prematurely terminated due to alarming findings of portal inflammation as well as hepatitis and localized biliary abnormalities at the end of treatment in patients with NASH [68].
The PPAR-α potential as a target for the treatment and prevention of NAFLD is clear due to its functional placement at the interface of lipid metabolism, energy balance, and inflammation. In the following, this review will discuss the various mechanisms, mainly PPAR-α-dependent, that enable OEA may reduce hepatocellular damage and ameliorate the progression of NAFLD.
3 OEA Reduces the Inflammatory Response in the Liver and Total Oxidative Stress
It is generally accepted that the inflammatory response and the emergence of general oxidative stress play a key role in the pathophysiology of acute liver injury [69]. In the process of liver injury, immune system cells produce a variety of proinflammatory cytokines, which contribute to further activation of immune cells, the development of a vicious circle of inflammation, hepatocyte damage, and eventually remodeling of the extracellular matrix and fibrosis development. At last, because overexpression of proinflammatory markers increases insulin resistance, it is critical to the pathophysiology of NAFLD.
It has been proven that exogenous administration of OEA through agonism with PPAR-α, is able to suppress the production of pro-inflammatory factors in the liver. In this instance, the drop in cytokine production was also accompanied by a rise in PPAR-α expression and a decrease in nuclear factor NF-κB and the other pro-inflammatory factor activator protein 1 (AP-1). Mechanisms by which PPAR-α can inhibit the transcriptional activity of these factors have also been described [70] thereby suppressing the production of pro-inflammatory cytokines [71]. That is, by reducing the expression level of NF-κB, a key mediator of the inflammatory response, the production of inflammatory factors such as TNFα, IL-1β, and IL-6 is ultimately suppressed. In a study by Payahoo et al., it was demonstrated that OEA treatment leads to a reduction in inflammation in obese patients by decreasing serum IL-6 and TNFα concentrations [72]. In a study by Yang et al., OEA inhibited lipopolysaccharide (LPS)-induced activation of NF-κB, AP-1, and signal transducer and activator of transcription 3 (STAT-3) [73]. In the same study, OEA significantly reduced the expression of TNFα, IL-1β, and IL-6 in the liver, brain, and spleen of mice treated with LPS. In a study by Tutunchi et al., a significant decrease in the expression levels of NF-κB and IL-6 was observed in obese and NASH patients after OEA supplementation with a simultaneous increase in the level of the anti-inflammatory cytokine IL-10. In a randomized clinical trial, treatment with OEA along with calorie restriction reduced systemic inflammatory factors in obese patients with NAFLD [74]. In the study by Hu et al., OEA markedly reduced the mRNA expression of pro-inflammatory factors including TNFα, IL-6, MCP1, and RANTES. In addition, OEA decreased IL-1β expression in the liver and plasma [75]. Also, in the same study, it was shown that the targeted inhibition of the inflammasome NLRP3 may underlie the anti-inflammatory action of OEA. In a mouse model of liver inflammation induced by LPS/D-Gal, OEA protected against acute liver injury through inhibition of NLRP3 inflammasome components. Given the fact that NLRP3 can be activated in response to increased expression of pro-inflammatory factors [76], and its activation is also a contributing factor to the pathogenesis of NAFLD [77], the effect of OEA on this signaling pathway may also depend on PPAR-α.
In the pathogenesis of NAFLD, inflammation is intimately connected with the development of oxidative stress the production of free radicals, and the activation of lipid peroxidation. OEA works as an antioxidant by interacting with PPAR-α, it can activate several links of the antioxidant defense system at once.
OEA, by enhancing PPAR-α signaling, inhibited LPS-induced oxidative stress; this effect was accompanied by a decrease in ROS production in obese humans [78]. Moreover, Giudetti et al., in an animal model of high-fat diet (HFD)-induced liver injury, demonstrated that OEA concurrently boosted the activity of antioxidant enzymes such as glutathione peroxidase (GSH-Px), catalase, and superoxide dismutase (SOD) in addition to lowering the formation of ROS [79]. A 12-week administration of OEA to subjects with obesity-associated NAFLD resulted in a decrease in markers of lipid peroxidation, such as malondialdehyde MDA and oxidized low-density lipoprotein (ox-LDL), in serum. Concurrently, the study observed an increase in total antioxidant capacity (TAC) and superoxide dismutase (SOD) serum levels [78]. OEA also reduced the hepatic MDA levels while boosting the activities of glutathione peroxidase (GSH-PX) and SOD in a mouse model of acute liver damage caused by LPS/D-Gal. This effect was accompanied by normalization of the levels of antioxidant factors Nrf-2 and HO-1, which were reduced during inflammation, indicating that OEA has several mechanisms for antioxidant activity (Fig. 2) [75].
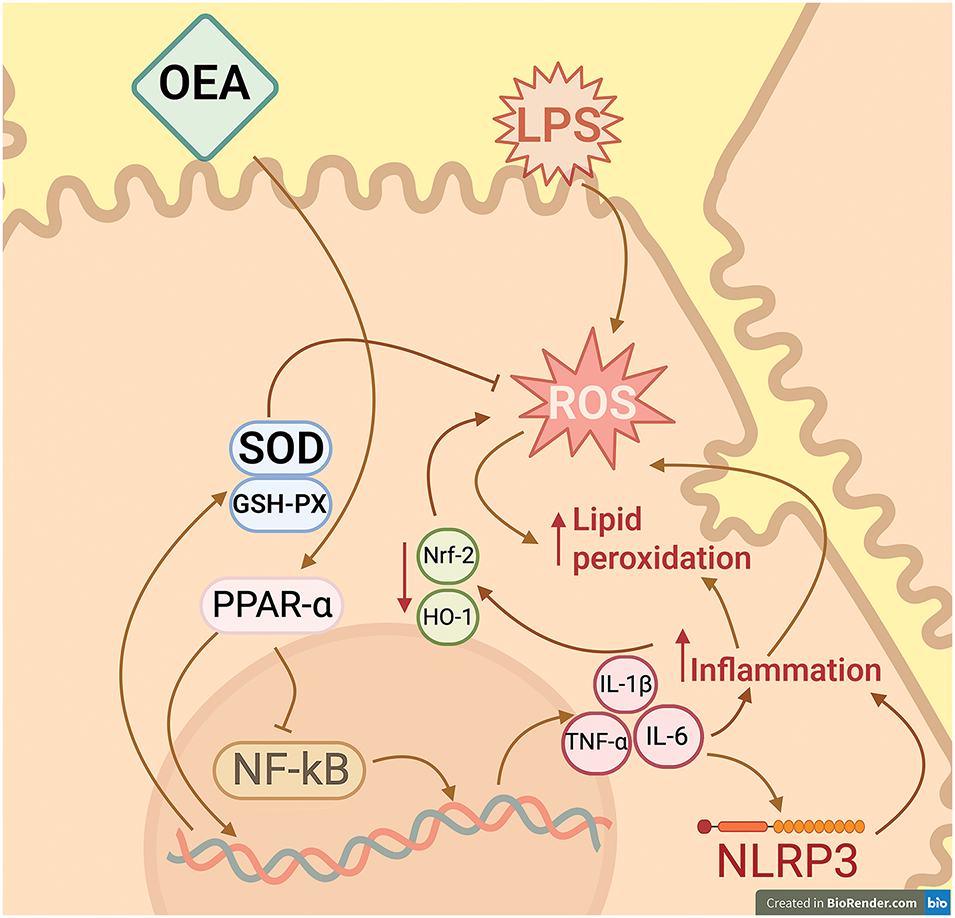
Figure 2: Effect of OEA on the development of inflammation and oxidative stress in hepatocytes. OEA has been shown to act as an agonist for the peroxisome proliferator-activated receptor alpha (PPAR-α). Activation of this receptor results in its function as a transcription regulator, capable of inducing or repressing the transcription of target genes. Notably, through a process known as transrepression, PPAR-α functions to impede the nuclear factor NF-κB, a pivotal component in inflammatory responses that plays a crucial role in the transcription of inflammatory cytokine genes. In the absence of OEA, when liver cells are exposed to LPS as an inflammation-inducing agent, NF-κB-dependent pathways are activated, which promotes inflammation and, consequently, oxidative stress. OEA has been shown to mitigate these effects by inhibiting the NF-κB pathway and by activating the transcription of antioxidant defense system enzymes, a process that is dependent on the activation of PPAR-α. SOD—superoxide dismutase; GSH-PX—glutathione peroxidase, NF-κB—nuclear factor κB; LPS—lipopolysaccharide; ROS—reactive oxygen species; NRF2—nuclear factor erythroid 2-related factor 2; HO-1—heme-oxygenase 1. Created in BioRender.com
4 OEA Prevents Hepatocyte Apoptosis and Also Regulates the Activity of Some Types of Non-Parenchymal Liver Cells
Reduced activity of antioxidant systems caused by the development of inflammation usually leads to irreversible damage to critical cellular structures and, as a consequence, to cell death [80]. The pathogenesis of NAFLD, in addition to the above factors, is based on excessive intake of FA into the liver and overload of hepatocytes with FA exceeding their enzymatic capacity. As a result, unmetabolized (unoxidized) FAs accumulate in hepatocytes, lipotoxicity, fatty degeneration, and subsequent apoptosis.
During the development of liver inflammation and oxidative stress, various signaling molecules activate resident liver macrophages (Kupffer cells) [81]. These cells acquire a pro-inflammatory phenotype (M1) and secondarily produce more and more pro-inflammatory cytokines such as TNFα and IL-6 [82], which promotes a vicious cycle of inflammation and further damage to hepatocytes [83]. In addition, being one of the main sources of ROS, activated macrophages contribute to the oxidative stress that develops in NAFLD [84,85]. Thus, the combination of increased ROS production, loss of antioxidant activity, activation of liver macrophages, and, as a consequence, increased lipid peroxidation (LPO) leads to the initiation of the apoptosis program in hepatocytes [86].
OEA administration can prevent cell damage by reducing the activity of apoptosis signaling pathways and inhibiting the expression of pro-apoptotic markers. A study by Hu et al. showed that OEA significantly reduced the levels of hepatocyte damage indicators such as alanine aminotransferase (ALT), aspartate aminotransferase (AST), and lactate dehydrogenase (LDH), as well as the synthesis of Bax, Bcl-2 and cleaved caspase-3. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining also confirmed OEA-mediated attenuation of hepatocyte apoptosis. Moreover, these processes occurred against the background of a significant increase in PPAR-α expression in the liver [75]. Hu et al. also revealed a pattern of increased activated Kupffer cells, which was accompanied by an increased hepatic expression of proinflammatory factors such as TNFα, IL-6, MCP1, and RANTES. OEA therapy against the background of increased hepatic levels of PPAR-α decreased both the number of activated Kupffer cells and the expression of proinflammatory cytokines.
The same study demonstrated the ability of OEA to suppress the expression of F4/80, which is a well-known marker of activated macrophages. These results suggest that OEA via PPAR-α can modulate immune cell activity in the liver, reducing inflammation and hepatocyte damage. In addition, OEA administration leads to the polarization of macrophages towards an anti-inflammatory M2 phenotype. Studies on BV2 microglia culture showed that OEA shifts the balance towards the M2 phenotype in LPS-induced inflammation, accompanied by downregulation of M1 phenotype markers (Iba-1, CD16, CD32) and upregulation of M2 phenotype markers (CD206, Arg, YM1) [87].
In an animal model of middle cerebral artery occlusion, similar changes in M1- and M2-specific markers were observed with OEA therapy; however, PPAR-α knockout mice did not show similar effects, suggesting a PPAR-α-dependent polarization mechanism [87]. Furthermore, in the THP-1 macrophage cell line, OEA supplementation promoted the expression of M2 phenotype macrophage markers (CD206 and TGFβ), while the expression of M1 markers (iNOS) was reduced. At the same time, blocking PPAR-α significantly reduced the expression of M2 markers [88].
Ito cells, or hepatic stellate cells, are also crucial for the pathophysiology of NASH and subsequent liver fibrosis. In the normal state, Ito cells function as antigen-presenting cells in the liver and are also responsible for storing vitamin A [89]. In chronic liver inflammation, these cells are activated, accompanied by enhanced production of extracellular matrix proteins, which underlies the pathogenesis of fibrotic liver injury. The profibrotic functions of activated Ito cells are based on the TGF-B1/Smad pathway, which is triggered by proinflammatory factors [90,91].
OEA, like other PPAR-α agonists in liver fibrosis models, can inhibit Smad protein phosphorylation and reduce TGFβ1 transcription, thereby inhibiting the proinflammatory JNK p38 and MAPK pathways. Numerous studies have shown that by preventing the activation of hepatic stellate cells, PPAR-α activation can reduce liver fibrosis associated with NAFLD [92,93]. In a study by Chen et al., OEA slowed the progression of liver fibrosis in mouse models by inhibiting collagen matrix, α-SMA (alpha-smooth muscle actin), and hepatic stellate cell activation, as well as genes involved in extracellular matrix remodeling and inflammation, including TIMP1, MMP2, and MMP9. In vitro studies showed that OEA inhibited the TGFβ1 pathway by suppressing phosphorylation of factor Smad2/3, α-SMA expression, and inhibiting the transformation of stellate cells into myofibroblasts. In addition, OEA administration did not induce antifibrotic effects in PPAR-α mutant animals, indicating that PPAR-α activation is the mechanism underlying all the above-mentioned actions of OEA in vivo and in vitro [94]. In the same study, the antifibrotic effect of OEA was accompanied by a decrease in serum levels of ALT, AST, and hepatic triglycerides (TGs), indicating an overall improvement in the metabolic profile and a reduction in hepatocyte damage.
In combination with anti-inflammatory activity, these effects of OEA allow us to speak about its effectiveness in complex therapy both at the initial stages of NAFLD and at the progression of the disease and the beginning of fibrotic processes.
5 Hepatoprotective Effects of OEA Caused by Regulation of Lipid Metabolism in the Liver
Numerous experimental studies confirm that OEA, via its association with PPAR-α, impacts multiple hepatic lipid metabolic pathways [95] that are crucial in the development of NAFLD [96]. A study by Drover et al. showed that the expression of PPAR receptors, including PPAR-α, was significantly increased in CD36-null mice [97]. Perhaps the regulation of the uptake of FAs by liver cells is also associated with the activation of PPAR, and exogenous activation of PPAR promotes the reduction of FA transport into hepatocytes and influences the pathogenesis of the development of NAFLD [40].
The ability of OEA to activate lipolysis in the liver by enhancing the expression of beta-oxidation enzymes has been repeatedly demonstrated in experimental studies in animal models. A study by Li et al. demonstrated that OEA in an HFD rat model stimulated β-lipid oxidation and simultaneously inhibited de novo lipogenesis [98]. OEA treatment markedly boosted PPAR-α and carnitine palmitoyltransferase I (CPT-1) mRNA levels and decreased sterol regulatory element binding protein 1c (SREBP-1c) and stearoyl-CoA desaturase-1 (SCD-1) expression, indicating that OEA may alter fatty acid metabolism in rats on a normal diet as well as in rats on an HFD [99]. A study of SCD-1 enzyme activity in the liver showed that OEA, by inhibiting SCD-1 activity, reduced the desaturation index in an HFD model. Cells derived from PPAR-α null mice did not exhibit the lipolytic effects of OEA, confirming the critical role of OEA in FAs metabolism by activating PPAR-α [100].
A study by Pan et al. showed that OEA inhibited triacylglycerol synthesis and secretion, apolipoprotein B (apoB) secretion, as well as microsomal triglyceride transfer protein (MTP) expression and activity in human hepatoma cell cultures Huh-7 and HepG2. Through PPAR-α-dependent processes, OEA also decreased lipoprotein secretion, glycerolipid production, and MTP expression in hepatocyte culture. Moreover, PPAR-α-deficient hepatocytes did not respond to exogenous OEA, suggesting a role for this receptor also in lipoprotein metabolism [100]. Taking into account the above-mentioned lipolytic activity of PPAR-α, we can conclude that OEA promotes the FAs redirection to a beta-oxidation pathway by inhibiting lipoprotein assembly in the liver.
OEA treatment has previously been shown to enhance ketone body production in rats [101]. Misto et al. then detailed the mechanism of OEA-mediated enhancement of fasting-induced hepatic ketogenesis by activating PPAR-α. Their findings indicate that histamine is secreted by extrahepatic mast cells in response to fasting. Histamine enters the liver via the portal vein, where it interacts with PPAR-α to activate G-protein coupled H1 receptors, cause local endogenous OEA production, and stimulate the transcription of ketogenesis enzymes genes including Acat-1 and Hmgcs-2. In the meantime, starvation-induced ketogenesis has been significantly reduced due to interfering genetic or pharmaceutical modifications such as mast cell eradication, the ablation of histamine or OEA synthesizing enzymes, and the H1 blockade. These results imply that endogenous histamine activates the H1 receptor and PPAR-α to promote OEA production in the liver [102]. Further Lin et al. showed that alimentary-induced obesity disrupts this mechanism and fasting does not cause histamine release and cannot trigger biosynthesis of hepatic OEA (Fig. 3) [103].
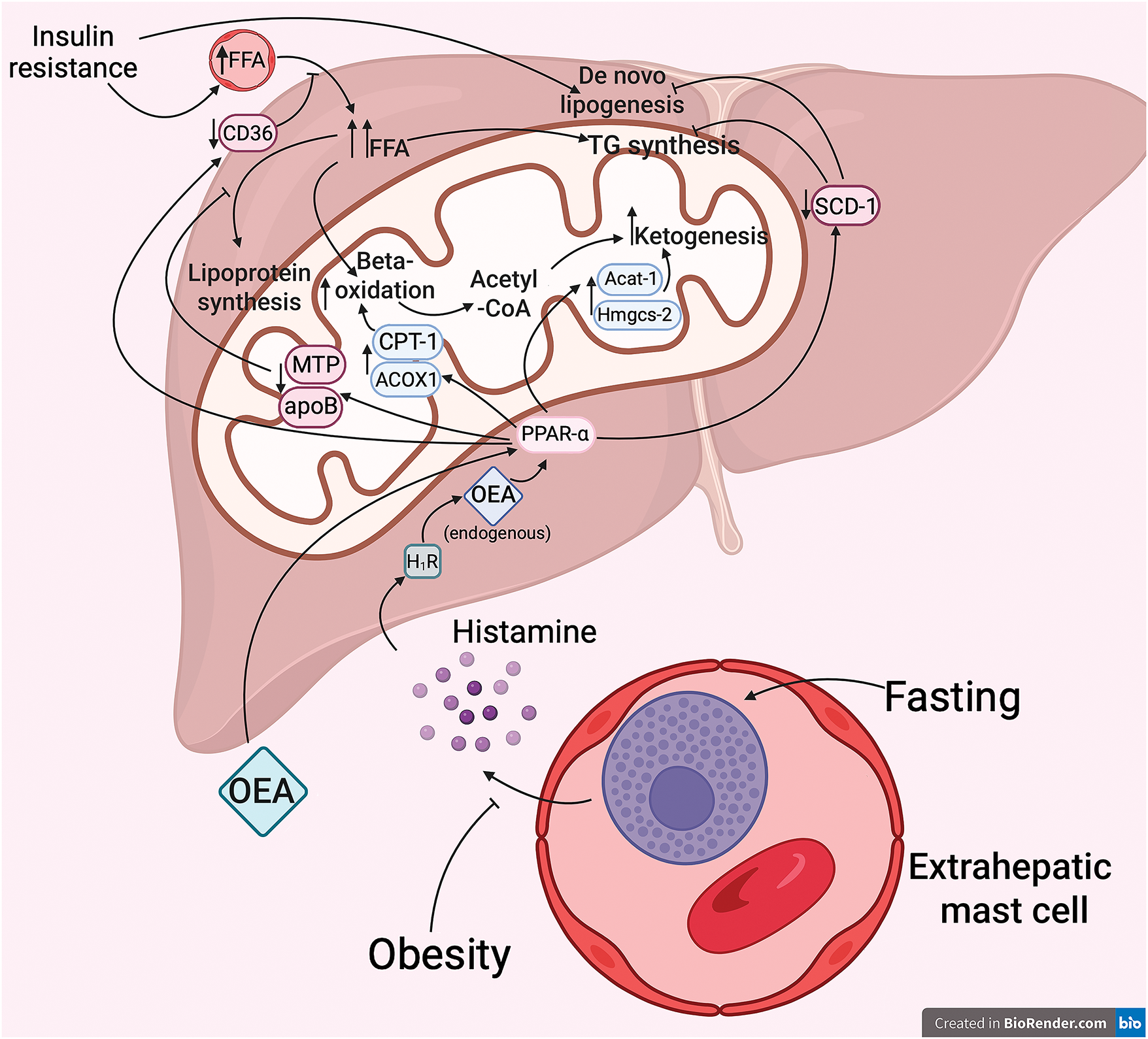
Figure 3: Effects of OEA on lipid metabolism in the liver. Fasting has been demonstrated to promote the release of histamine by extrahepatic mast cells. This, in turn, has been shown to interact with histamine 1 receptors (H1R) in the liver, thereby promoting the formation of endogenous OEA. The subsequent formation of endogenous OEA, in turn, activates the transcription of key enzymes involved in fatty acid beta-oxidation and ketogenesis, a process that is dependent on the presence of a specific mechanism that involves PPAR-α. Concurrently, the activation of PPAR-α results in a reduction in hepatic lipogenesis and a limitation of free fatty acid flux. Consequently, the exogenous administration of OEA may serve as a preventative measure against mitochondrial overload in conditions of insulin resistance and obesity, particularly when the endogenous production of OEA is impaired. ACOX1—acyl-Coenzyme A oxidase I; CPT-I—carnitine palmitoyltransferase I; Acat-1—acetyl-CoA acetyltransferase I; Hmgcs-2—hydroxymethylglutaryl-CoA synthase-2; SCD-1—stearoyl-CoA desaturase-1; MTP—microsomal triglyceride transfer protein; apoB—apolipoprotein B; FFA—free fatty acids; TG—triglycerides. Created in BioRender.com
Taken together, the results of these studies suggest that impaired mobilization of endogenous OEA in the liver caused by a high-fat diet may contribute to the development of NAFLD due to impaired lipid utilization. In this case, administration of exogenous OEA may contribute to the correction of these metabolic disorders.
6 Other Potential Mechanisms of Hepato-Protective Effect of OEA, Which May Be Mediated by Interaction with PPAR Receptors
6.1 OEA Regulates PCSK9 Protein Expression, Contributing to the Reduction of Cholesterol and LDL Levels
The low-density lipoprotein (LDL) receptor is naturally inhibited by the proprotein convertase subtilisin/kexin type 9 (PCSK9). Whether it is secreted or cellular, mature PCSK9 inhibits low-density lipoprotein receptor (LDLr) recycling by binding to its domain and promoting further degradation, much like a chaperone. Therefore, by decreasing LDL synthesis and resulting in hypercholesterolemia, overexpression of PCSK9 strongly contributes to the pathogenesis of NAFLD [104]. Moreover, insulin can stimulate PCSK9 transcription [105], supporting the link between liver pathology and problems with the metabolism of glucose and cholesterol. Conversely, a defect in PCSK9 decreases plasma cholesterol levels and offers protection against heart disease. Therefore, combination illnesses that do not react well to conventional therapy can be treated with drugs that inhibit the synthesis, processing, or binding of PCSK9 to LDL.
It has been demonstrated that PPAR-α agonists, which lower cholesterol, decrease PCSK9 mRNA and protein in the liver of mice. Fibrate, a traditional PPAR-α activator, affects the expression of many genes, which reduces macrophage activation [106], inhibits the growth of vascular smooth muscle cells [107], and speeds up the macrophages’ clearance of lipids [108]. By modifying the activity of its promoter, fibrates have been shown in numerous studies to be capable of controlling transcription factors including PPAR-α and sterol regulatory element-binding proteins (SREBP-1/2) [109,110]. Therefore, by binding to the appropriate locations on the PCSK9 gene promoter, PPAR-α activation suppresses PCSK9 gene expression.
Additionally, the study by Kourimate et al. showed that furin and PC5/6A, which are in charge of PCSK9’s proteolytic inactivation in a PPAR-α-dependent way, are positively regulated by fibrates. Therefore, posttranslational downregulation of PCSK9 also takes place [109]. Additionally, by triggering vascular endothelial lipoprotein lipase, which includes furin and PC5/6A, PPAR-α activation can lower the amount of lipids in plasma [111]. As per Jin et al., cleavage and inhibition of endothelial synthase triggered by furin and PC5/6A results in elevated amounts of antiatherogenic high-density lipoproteins (HDL-C) [112].
OEA may also help LDLr levels return to normal via a PPAR-dependent mechanism. By upregulating LDLr expression, activation of PPAR-α would improve cellular absorption of cholesterol and hence lower plasma cholesterol levels. Accordingly, exogenous OEA-mediated regulation of PPAR-α may have a complex impact on cholesterol metabolism and help lower hypercholesterolemia. It’s also critical to emphasize that fibrates do not have the same anorexigenic effects as OEA because they concurrently boost the expression of the lipolysis and lipogenesis enzymes [113]. For NAFLD linked to obesity and hypercholesterolemia, OEA is a more attractive therapeutic target.
6.2 OEA Stimulates GLP-1 Secretion, Contributing to the Reduction of Insulin Resistance
Several subtypes of PPARs are known to be involved in the regulation of glucose metabolism. The most studied modulators of carbohydrate metabolism are considered to be thiazolidinediones, which are pharmacological activators of PPAR-γ and are used for the treatment of type 2 diabetes mellitus (T2DM), helping to reduce insulin resistance [114].
In several studies, activation of PPAR-γ by drugs from the thiazolidinedione group led to a reduction in the intensity of inflammation in NAFLD [115,116], as well as improved insulin sensitivity in the liver [117].
However, studies conducted in mouse models of insulin resistance have shown that PPAR-α activation also improves glucose homeostasis by decreasing endogenous glucose production, reducing lipid content in adipose and non-adipose tissues, and increasing insulin sensitivity [118,119], its overexpression in mouse models of obesity improved insulin sensitivity [120].
In addition to its known agonism with PPAR-α, OEA also has the ability to bind to G-protein-coupled receptor 119 (GPR119), which is found in intestinal L-cells and pancreatic β-cells [121]. Activation of GPR119 is also a promising strategy in the therapy of NAFLD [122].
Other GPR119 agonists have been shown to increase the release of glucagon-like peptide-1 (GLP-1) [123,124]. GLP-1 regulates glucose metabolism by significantly increasing insulin sensitivity and glucose uptake in insulin-dependent tissues [125]. It has been suggested that GLP-1, in addition to enhancing glucose uptake, may also reduce insulin resistance [126]. Thus, GLP-1 agonists are a promising pharmacological target in the comprehensive therapy of NAFLD [127].
Thus, OEA can potentially modulate glucose homeostasis by interacting with several receptor targets at once.
6.3 OEA through PPAR-α Activation Can Enhance the Expression of Thermogenesis Proteins UCP1 and UCP2
Activation of uncoupling proteins (UCP) family thermogenesis proteins is another strategy to reduce inflammation and modulate immune cells in liver injury. UCP1-deficient mice exhibited succinate receptor 1 (SUCNR1)-dependent activation of stellate cells and macrophages in the liver, which promoted inflammation [128]. It has been shown that one aspect of the anorexigenic action of OEA may be the enhancement of thermogenesis in adipose tissue, including through UCP1 activation [129].
Treatment of OEA in obese and NAFLD patients resulted in a marked increase in the expression levels of PPAR-α, UCP1, and UCP2 genes in peripheral blood mononuclear cells (PBMCs) in a triple-blind placebo-controlled randomized clinical trial [130]. In addition, when comparing the OEA group with the placebo group, a decrease in anthropometric parameters, energy, and carbohydrate intake, and glycemic parameters other than hemoglobin A1c concentration was observed. When taking OEA, there was also a significant decrease in TG, ALT, AST, and serum ALT/AST index, as well as an increase in HDL levels. In animal models of alimentary-induced obesity, OEA administration led to an increase in the level of hepatic PPAR-α and UCP2 [131], and even in the adipose tissue of rats without obesity [101]. In addition, lipolysis enzymes were activated, but not in PPAR-α null animals.
Thus, OEA promotes the expression of PPAR-α, UCP1, and UCP2 genes in PBMCs during the treatment of NAFLD, which is reflected in increased energy expenditure, decreased de novo fat synthesis, inflammation, and overall weight loss.
6.4 OEA Reduces Hepatic Lipid Accumulation and Apoptosis through Enhanced Adiponectin Production
An important aspect of the pathogenesis of nonalcoholic fatty liver disease is adipokine regulation. The most common adipokine that has been shown to directly reduce inflammation and insulin resistance while also controlling hepatic lipid and glucose metabolism is adiponectin [132]. Adiponectin expression is directly proportional to HDL levels and inversely linked with cardiovascular risk factors [133,134].
According to certain theories, adiponectin may both promote FAs beta-oxidation and prevent de novo lipogenesis, which would result in the metabolic consequences listed above [135]. Furthermore, upregulation of adiponectin receptor expression in adipose tissue can result from agonist-induced activation of PPAR-α. On the other hand, serum adiponectin rises in response to PPAR-γ activation [136]. Reduced inflammation and enhanced insulin sensitivity accompanied both outcomes. Consequently, medication for metabolic diseases targeted at raising adiponectin levels may be essential to reducing NAFLD progression [137,138].
Apart from its metabolic functions, adiponectin possesses a range of anti-apoptotic properties. Adiponectin inhibited diabetic apoptosis in an animal model of diabetes in vivo and in vitro using a cardiomyocyte cell culture H9c2 by suppressing the TLR4/NF-κB signaling pathway [139]. Adiponectin reduced ER-stress-driven apoptosis in adipose tissue in a mouse model of tunicamycin-induced ER stress; this effect was mediated through interaction with the PPAR-α receptor. Adiponectin treatment was associated with an increase in the anti-apoptotic protein Bcl-2 and a decrease in the proapoptotic markers Bax, Chop, GRP78, ATF2, cleaved caspase 3/9, and Apaf-1 [140]. Lastly, another possible mechanism of OEA’s hypocholesterolemiс action may be adiponectin, which has antiatherosclerotic activity of its own. Experimental evidence has demonstrated its capacity to inhibit monocyte adhesion to endothelial cells by decreasing TNF-α-induced endothelial adhesion molecules. Additionally, it has been shown to inhibit macrophage conversion into foam cells and prevent endothelial cell activation [141]. Moreover, adiponectin caused endothelial cells to produce more nitric oxide (NO) and lower C-reactive protein (CRP) [142]. Additionally, it suppressed the generation of ROS and cell proliferation brought on by low-density lipoprotein (LDL) oxidase during the development of atherosclerotic plaque [143].
Thus, OEA, being an agonist of PPAR-α, upregulating adiponectin, may have a complex effect on several pathogenetic factors in the development of NAFLD.
7 Bioavailability and Side Effects of OEA
In vivo, studies of the beneficial effects of OEAs have focused primarily on intraperitoneal or subcutaneous administration of the drug. However, oral administration is considered to be the most convenient route of administration for the patient, which involves additional metabolic events that should be discussed in this review. The main disadvantage of fatty acid ethanolamides as a class of therapeutic agents is their poor metabolic stability in vivo due to their rapid hydrolysis by a number of hydrolytic enzymes such as fatty acid amide hydrolase (FAAH), N-acylethanolamic acid amidase (NAAA), and monoacylglycerol lipase (MAGL) [144]. Therefore, it is necessary to take these metabolic characteristics into account when calculating the dosage of the drug. It has also been found that significant hypophagic effects with oral administration of OEA occur only at the highest doses (100 and 200 mg/kg) [145–147]. In a study of the bioavailability of oral OEA, it was found that the drug is actively degraded along the gastrointestinal tract, resulting in only 0.48% of the administered OEA dose being converted unchanged into tissues. The ratio of intact OEA to hydrolyzed OEA decreases along the gastrointestinal tract, indicating that OEA is gradually catabolized [147].
In a randomized, double-blind, placebo-controlled study in humans, OEA at doses of 300 and 600 mg/day also showed significant anti-inflammatory efficacy, but side effects such as nausea, vomiting, dyspepsia, and headache were noted. It should be noted, however, that no statistically significant differences in reported adverse events were found between the different groups (treatment and placebo) [148].
In general, to expand the use of OEA in medical practice, it is necessary to search for new approaches, one of which may be to reduce the degradation of OEA through the possible development of FAAH inhibitors. On the other hand, an alternative strategy may be the development of OEA analogues that are more stable for enzymatic inactivation.
It is well known that the PPAR-α receptor regulates the expression of many genes that are crucial in the pathogenesis of nonalcoholic fatty liver disease. One of the main mechanisms responsible for the hepatoprotective effect of OEA is probably an agonistic action on the PPAR-α receptor. Thus, OEA may be used for the treatment of NAFLD due to the growing evidence supporting its efficacy in this disease, especially if NAFLD is accompanied by obesity, hypercholesterolemia, and insulin resistance. Alo, the possibility that the effect of OEA on lipid and carbohydrate metabolism is mediated by other target receptors cannot be excluded (Fig. 4), but a thorough investigation of these pathways is still needed.
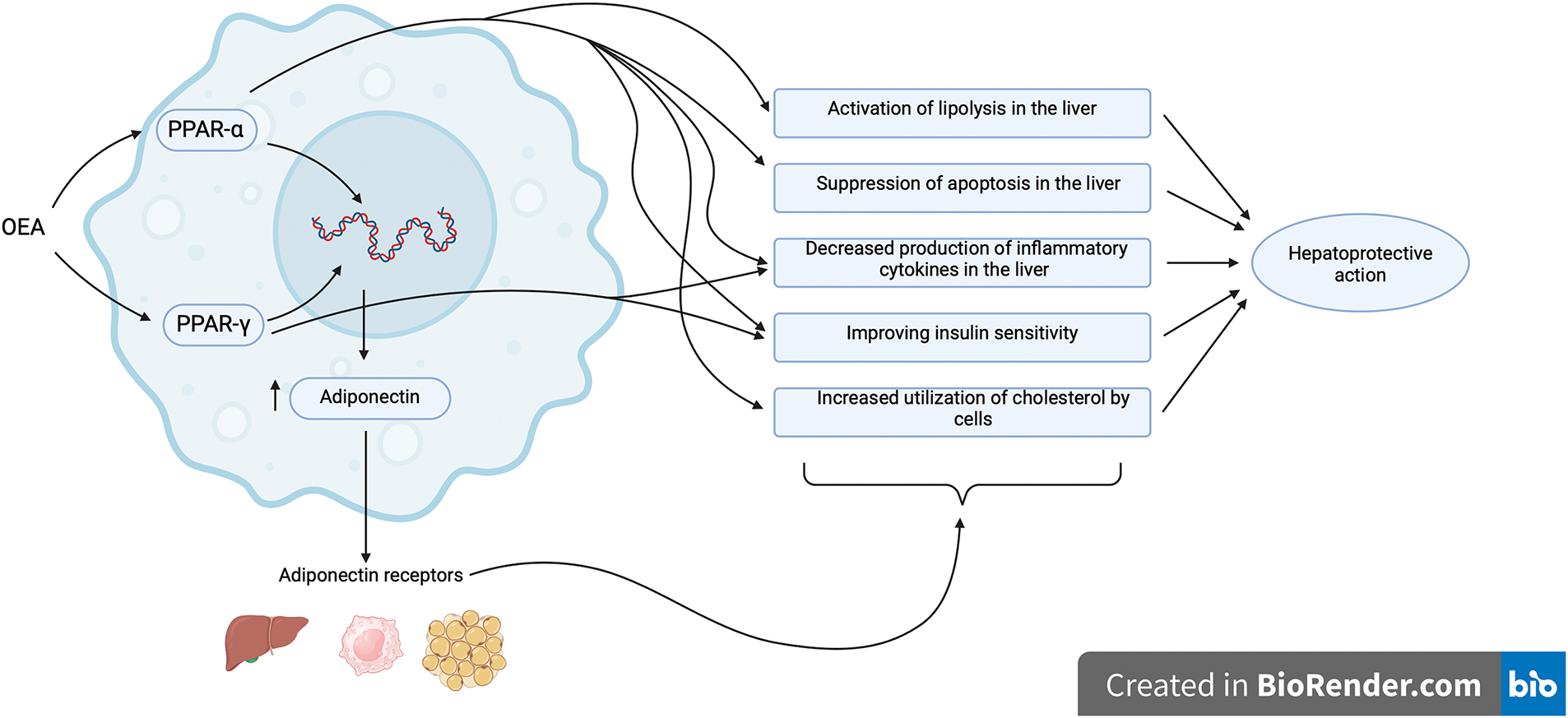
Figure 4: General scheme of receptor-mediated actions of OEA through which its hepatoprotective effect may be realized. Peroxisome proliferator-activated receptors alpha (PPAR-α) and gamma (PPAR-γ) are nuclear receptors that regulate the transcription of a wide range of target genes. The greatest contribution to the realization of the effects of OEA is made by the PPAR-α type, which is expressed to the greatest extent in the liver. However, it is noteworthy that both the PPAR-γ and G-protein-coupled receptor 119 (GPR119) may also contribute significantly to the complex hepatoprotective effect of OEA due to their ability to regulate carbohydrate metabolism. Created in BioRender.com
Acknowledgement: Investigations were carried out at the Core Shared Research Facility Resource Collection “Marine Biobank”, A.V. Zhirmunsky National Scientific Center of Marine Biology, Far Eastern Branch, Russian Academy of Sciences (http://marbank.dvo.ru/index.php/en/) (accessed on 07 February 2025).
Funding Statement: The study was supported by the Russian Science Foundation (project 24-75-00072).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Darya Ivashkevich, Inessa Dyuizen; data collection: Darya Ivashkevich; analysis and interpretation of results: Darya Ivashkevich, Igor Manzhulo, Arina Ponomarenko; draft manuscript preparation: Darya Ivashkevich, Arina Ponomarenko. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Riazi K, Azhari H, Charette JH, Underwood FE, King JA, Afshar EE, et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol. 2022;7(9):851–61. doi:10.1016/S2468-1253(22)00165-0. [Google Scholar] [PubMed] [CrossRef]
2. Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S. Nonalcoholic steatohepatitis: a review. JAMA. 2020;323(12):1175–83. doi:10.1001/jama.2020.2298. [Google Scholar] [PubMed] [CrossRef]
3. Nagarajan SR, Cross E, Sanna F, Hodson L. Dysregulation of hepatic metabolism with obesity: factors influencing glucose and lipid metabolism. Proc Nuxtr Soc. 2022;81(1):1–11. doi:10.1017/S0029665121003761. [Google Scholar] [PubMed] [CrossRef]
4. Ng CH, Wong ZY, Chew NWS, Chan KE, Xiao J, Sayed N, et al. Hypertension is prevalent in non-alcoholic fatty liver disease and increases all-cause and cardiovascular mortality. Front Cardiovasc Med. 2022;9:942753. doi:10.3389/fcvm.2022.942753. [Google Scholar] [PubMed] [CrossRef]
5. Jung I, Koo DJ, Lee WY. Insulin resistance, non-alcoholic fatty liver disease and type 2 diabetes mellitus: clinical and experimental perspective. Diabetes Metab J. 2024;48(3):327–39. doi:10.4093/dmj.2023.0350. [Google Scholar] [PubMed] [CrossRef]
6. Sposito AC. Accelerated pace of atherosclerosis in steatotic liver disease: implications for risk stratification. Circ Cardiovasc Imaging. 2024;17(9):e017376. doi:10.1161/CIRCIMAGING.124.017376. [Google Scholar] [PubMed] [CrossRef]
7. Pan Q, Fan JG, Yilmaz Y. Pathogenetic pathways in nonalcoholic fatty liver disease: an incomplete jigsaw puzzle. Clin Liver Dis. 2023;27(2):317–32. doi:10.1016/j.cld.2023.01.013. [Google Scholar] [PubMed] [CrossRef]
8. Mundi MS, Velapati S, Patel J, Kellogg TA, Abu Dayyeh BK, Hurt RT. Evolution of NAFLD and its management. Nutr Clin Pract. 2020;35(1):72–84. doi:10.1002/ncp.10449. [Google Scholar] [PubMed] [CrossRef]
9. Han SK, Baik SK, Kim MY. Non-alcoholic fatty liver disease: definition and subtypes. Clin Mol Hepatol. 2023;29(suppl):S5–16. doi:10.3350/cmh.2022.0424. [Google Scholar] [PubMed] [CrossRef]
10. Schattenberg JM, Anstee QM, Caussy C, Bugianesi E, Popovic B. Differences between current clinical guidelines for screening, diagnosis and management of nonalcoholic fatty liver disease and real-world practice: a targeted literature review. Expert Rev Gastroenterol Hepatol. 2021;15(11):1253–66. doi:10.1080/17474124.2021.1974295. [Google Scholar] [PubMed] [CrossRef]
11. Wallace M, Metallo CM. Tracing insights into de novo lipogenesis in liver and adipose tissues. Semin Cell Dev Biol. 2020;108:65–71. doi:10.1016/j.semcdb.2020.02.012. [Google Scholar] [PubMed] [CrossRef]
12. Ristic-Medic D, Bajerska J, Vucic V. Crosstalk between dietary patterns, obesity and nonalcoholic fatty liver disease. World J Gastroenterol. 2022;28(27):3314–33. doi:10.3748/wjg.v28.i27.3314. [Google Scholar] [PubMed] [CrossRef]
13. Smith GI, Shankaran M, Yoshino M, Schweitzer GG, Chondronikola M, Beals JW, et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest. 2020;130(3):1453–60. doi:10.1172/JCI134165. [Google Scholar] [PubMed] [CrossRef]
14. Engin A. Lipid storage, lipolysis, and lipotoxicity in obesity. Adv Exp Med Biol. 2024;1460:97–129. doi:10.1007/978-3-031-63657-8_4. [Google Scholar] [PubMed] [CrossRef]
15. Dornas W, Schuppan D. Mitochondrial oxidative injury: a key player in nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol. 2020;319(3):G400–11. doi:10.1152/ajpgi.00121.2020. [Google Scholar] [PubMed] [CrossRef]
16. Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med. 2020;152:116–41. doi:10.1016/j.freeradbiomed.2020.02.025. [Google Scholar] [PubMed] [CrossRef]
17. Rikhi R, Singh T, Modaresi Esfeh J. Work up of fatty liver by primary care physicians, review. Ann Med Surg. 2020;50:41–8. doi:10.1016/j.amsu.2020.01.001. [Google Scholar] [PubMed] [CrossRef]
18. Passos E, Pereira C, Gonçalves IO, Faria A, Ascensão A, Monteiro R et al. Physical exercise positively modulates nonalcoholic steatohepatitis-related hepatic endoplasmic reticulum stress. J Cell Biochem. 2022;123(10):1647–62. doi:10.1002/jcb.30250. [Google Scholar] [PubMed] [CrossRef]
19. Martín-Fernández M, Arroyo V, Carnicero C, Sigüenza R, Busta R, Mora N, et al. Role of oxidative stress and lipid peroxidation in the pathophysiology of NAFLD. Antioxid. 2022;11(11):2217. doi:10.3390/antiox11112217. [Google Scholar] [PubMed] [CrossRef]
20. Ramachandran A, Jaeschke H. Oxidative stress and acute hepatic injury. Curr Opin Toxicol. 2018;7:17–21. doi:10.1016/j.cotox.2017.10.011. [Google Scholar] [PubMed] [CrossRef]
21. Parthasarathy G, Malhi H. Assessment of lipotoxic Endoplasmic Reticulum (ER) stress in Nonalcoholic Steatohepatitis (NASH). Methods Mol Biol. 2022;2455:243–54. doi:10.1007/978-1-0716-2128-8. [Google Scholar] [CrossRef]
22. Zeng C, Chen M. Progress in nonalcoholic fatty liver disease: SIRT family regulates mitochondrial biogenesis. Biomolecules. 2022;12(8):1079. doi:10.3390/biom12081079. [Google Scholar] [PubMed] [CrossRef]
23. Zarei M, Aguilar-Recarte D, Palomer X, Vázquez-Carrera M. Revealing the role of peroxisome proliferator-activated receptor β/δ in nonalcoholic fatty liver disease. Metabolism. 2021;114:154342. doi:10.1016/j.metabol.2020.154342. [Google Scholar] [PubMed] [CrossRef]
24. Fromenty B, Roden M. Mitochondrial alterations in fatty liver diseases. J Hepatol. 2023;78(2):415–29. doi:10.1016/j.jhep.2022.09.020. [Google Scholar] [PubMed] [CrossRef]
25. Jeeyavudeen MS, Khan SKA, Fouda S, Pappachan JM. Management of metabolic-associated fatty liver disease: the diabetology perspective. World J Gastroenterol. 2023;29(1):126–43. doi:10.3748/wjg.v29.i1.126. [Google Scholar] [PubMed] [CrossRef]
26. Kim MJ, Park CH, Kim DH, Park MH, Park KC, Hyun MK et al. Hepatoprotective effects of MHY3200 on high-fat, diet-induced, non-alcoholic fatty liver disease in rats. Molecules. 2018;23(8):2057. doi:10.3390/molecules23082057. [Google Scholar] [PubMed] [CrossRef]
27. Nakamura A, Kagaya Y, Saito H, Kanazawa M, Sato K, Miura M, et al. Impact of pemafibrate on lipid profile and insulin resistance in hypertriglyceridemic patients with coronary artery disease and metabolic syndrome. Heart Vessels. 2024;39(6):486–95. doi:10.1007/s00380-024-02363-z. [Google Scholar] [PubMed] [CrossRef]
28. Honda A, Kamata S, Satta C, Machida Y, Uchii K, Terasawa K, et al. Structural basis for anti-non-alcoholic fatty liver disease and diabetic dyslipidemia drug saroglitazar as a PPAR α/γ dual agonist. Biol Pharm Bull. 2021;44(9):1210–9. doi:10.1248/bpb.b21-00232. [Google Scholar] [PubMed] [CrossRef]
29. Romano A, Coccurello R, Giacovazzo G, Bedse G, Moles A, Gaetani S. Oleoylethanolamide: a novel potential pharmacological alternative to cannabinoid antagonists for the control of appetite. Biomed Res Int. 2014;2014:203425. doi:10.1155/2014/203425. [Google Scholar] [PubMed] [CrossRef]
30. Tutunchi H, Saghafi-Asl M, Ostadrahimi A. A systematic review of the effects of oleoylethanolamide, a high-affinity endogenous ligand of PPAR-α, on the management and prevention of obesity. Clin Exp Pharmacol Physiol. 2020;47(4):543–52. doi:10.1111/1440-1681.13238. [Google Scholar] [PubMed] [CrossRef]
31. Laleh P, Yaser K, Alireza O. Oleoylethanolamide: a novel pharmaceutical agent in the management of obesity—an updated review. J Cell Physiol. 2019;234(6):7893–902. doi:10.1002/jcp.27913. [Google Scholar] [PubMed] [CrossRef]
32. Laleh P, Yaser K, Abolfazl B, Shahriar A, Mohammad AJ, Nazila F, et al. Oleoylethanolamide increases the expression of PPAR-A and reduces appetite and body weight in obese people: a clinical trial. Appetite. 2018;128:44–9. doi:10.1016/j.appet.2018.05.129. [Google Scholar] [PubMed] [CrossRef]
33. Caillon A, Duszka K, Wahli W, Rohner-Jeanrenaud F, Altirriba J. The OEA effect on food intake is independent from the presence of PPARα in the intestine and the nodose ganglion, while the impact of OEA on energy expenditure requires the presence of PPARα in mice. Metabolism. 2018;87:13–7. doi:10.1016/j.metabol.2018.06.005. [Google Scholar] [PubMed] [CrossRef]
34. Vázquez-Carrera M, Wahli W. PPARs as key transcription regulators at the crossroads of metabolism and inflammation. Int J Mol Sci. 2024;25(8):4467. doi:10.3390/ijms25084467. [Google Scholar] [PubMed] [CrossRef]
35. Wagner N, Wagner KD. Peroxisome proliferator-activated receptors and the hallmarks of cancer. Cells. 2022;11(15):2432. doi:10.3390/cells11152432. [Google Scholar] [PubMed] [CrossRef]
36. Torres JL, Usategui-Martín R, Hernández-Cosido L, Bernardo E, Manzanedo-Bueno L, Hernández-García I, et al. PPAR-γ gene expression in human adipose tissue is associated with weight loss after sleeve gastrectomy. J Gastrointest Surg. 2022;26(2):286–97. doi:10.1007/s11605-021-05216-6. [Google Scholar] [PubMed] [CrossRef]
37. da Silva CAT, Clemente-Napimoga JT, Abdalla HB, Basting RT, Napimoga MH. Peroxisome proliferator-activated receptor-gamma (PPARγ) and its immunomodulation function: current understanding and future therapeutic implications. Expert Rev Clin Pharmacol. 2022;15(3):295–303. doi:10.1080/17512433.2022.2071697. [Google Scholar] [PubMed] [CrossRef]
38. Changizi Z, Kajbaf F, Moslehi A. An overview of the role of peroxisome proliferator-activated receptors in liver diseases. J Clin Transl Hepatol. 2023;11(7):1542–52. doi:10.14218/JCTH.2023.00334. [Google Scholar] [PubMed] [CrossRef]
39. Berthier A, Johanns M, Zummo FP, Lefebvre P, Staels B. PPARs in liver physiology. Biochim Biophys Acta Mol Basis Dis. 2021;1867(5):166097. doi:10.1016/j.bbadis.2021.166097. [Google Scholar] [PubMed] [CrossRef]
40. Todisco S, Santarsiero A, Convertini P, De Stefano G, Gilio M, Iacobazzi V, et al. PPAR alpha as a metabolic modulator of the liver: role in the pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology. 2022;11(5):792. doi:10.3390/biology11050792. [Google Scholar] [PubMed] [CrossRef]
41. Tahri-Joutey M, Andreoletti P, Surapureddi S, Nasser B, Cherkaoui-Malki M, Latruffe N. Mechanisms mediating the regulation of peroxisomal fatty acid beta-oxidation by PPARα. Int J Mol Sci. 2021;22(16):8969. doi:10.3390/ijms22168969. [Google Scholar] [PubMed] [CrossRef]
42. Nakagawa Y, Satoh A, Tezuka H, Han SI, Takei K, Iwasaki H, et al. CREB3L3 controls fatty acid oxidation and ketogenesis in synergy with PPARα. Sci Rep. 2016;6:39182. doi:10.1038/srep39182. [Google Scholar] [PubMed] [CrossRef]
43. Feng X, Zhang R, Yang Z, Zhang K, Xing J. Mechanism of metabolic dysfunction-associated steatotic liver disease: important role of lipid metabolism. J Clin Transl Hepatol. 2024;12(9):815–26. doi:10.14218/JCTH.2024.00019. [Google Scholar] [PubMed] [CrossRef]
44. Bentanachs R, Velázquez AM, Sánchez RM, Alegret M, Laguna JC, Roglans N. Bempedoic acid as a PPARα activator: new perspectives for hepatic steatosis treatment in a female rat experimental model. El ácido bempedoico como activador PPARα: nuevas perspectivas para el tratamiento de la esteatosis hepática en un modelo experimental de rata hembra. Clin Investig Arterioscler. 2022;34(2):57–67. doi:10.1016/j.arteri.2021.09.004. [Google Scholar] [PubMed] [CrossRef]
45. van der Krieken SE, Popeijus HE, Mensink RP, Plat J. Link between ER-stress, PPAR-alpha activation, and BET inhibition in relation to apolipoprotein A-I transcription in HepG2 cells. J Cell Biochem. 2017;118(8):2161–7. doi:10.1002/jcb.25858. [Google Scholar] [PubMed] [CrossRef]
46. Ibrahim SA, Mohamed MZ, El-Tahawy NF, Abdelrahman AM. Antifibrotic effects of bezafibrate and pioglitazone against thioacetamide-induced liver fibrosis in albino rats. Can J Physiol Pharmacol. 2021;99(3):313–20. doi:10.1139/cjpp-2020-0159. [Google Scholar] [PubMed] [CrossRef]
47. Jain MR, Giri SR, Bhoi B, Trivedi C, Rath A, Rathod R, et al. Dual PPARα/γ agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int. 2018;38(6):1084–94. doi:10.1111/liv.13634. [Google Scholar] [PubMed] [CrossRef]
48. Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J Hepatol. 2015;63(1):164–73. doi:10.1016/j.jhep.2015.02.019. [Google Scholar] [PubMed] [CrossRef]
49. Régnier M, Polizzi A, Smati S, Lukowicz C, Fougerat A, Lippi Y, et al. Hepatocyte-specific deletion of Pparα promotes NAFLD in the context of obesity. Sci Rep. 2020;10(1):6489. doi:10.1038/s41598-020-63579-3. [Google Scholar] [PubMed] [CrossRef]
50. Tailleux A, Wouters K, Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys Acta. 2012;1821(5):809–18. doi:10.1016/j.bbalip.2011.10.016. [Google Scholar] [PubMed] [CrossRef]
51. Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor α mediates the adaptive response to fasting. J Clin Invest. 1999;103(11):1489–98. doi:10.1172/JCI6223. [Google Scholar] [PubMed] [CrossRef]
52. Sanderson LM, Boekschoten MV, Desvergne B, Müller M, Kersten S. Transcriptional profiling reveals divergent roles of PPARα and PPARβ/δ in regulation of gene expression in mouse liver. Physiol Genomics. 2010;41(1):42–52. doi:10.1152/physiolgenomics.00127.2009. [Google Scholar] [PubMed] [CrossRef]
53. Chen H, Tan H, Wan J, Zeng Y, Wang J, Wang H, et al. PPAR-γ signaling in nonalcoholic fatty liver disease: pathogenesis and therapeutic targets. Pharmacol Ther. 2023;245:108391. doi:10.1016/j.pharmthera.2023.108391. [Google Scholar] [PubMed] [CrossRef]
54. Skat-Rørdam J, Højland Ipsen D, Lykkesfeldt J, Tveden-Nyborg P. A role of peroxisome proliferator-activated receptor γ in non-alcoholic fatty liver disease. Basic Clin Pharmacol Toxicol. 2019;124(5):528–37. doi:10.1111/bcpt.13190. [Google Scholar] [PubMed] [CrossRef]
55. Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARγ. Annu Rev Biochem. 2008;77:289–312. doi:10.1146/annurev.biochem.77.061307.091829. [Google Scholar] [PubMed] [CrossRef]
56. Lee YK, Park JE, Lee M, Hardwick JP. Hepatic lipid homeostasis by peroxisome proliferator-activated receptor gamma 2. Liver Res. 2018;2(4):209–15. doi:10.1016/j.livres.2018.12.001. [Google Scholar] [PubMed] [CrossRef]
57. Miyachi H. Structural biology-based exploration of subtype-selective agonists for peroxisome proliferator-activated receptors. Int J Mol Sci. 2021;22(17):9223. doi:10.3390/ijms22179223. [Google Scholar] [PubMed] [CrossRef]
58. Singh G, Kumar R, Desna SD, Chaudhary M, Kaur C, Khurrana N. Thiazolidinedione as a promising medicinal scaffold for the treatment of type 2 diabetes. Curr Diabetes Rev. 2024;20(6):e201023222411. doi:10.2174/0115733998254798231005095627. [Google Scholar] [PubMed] [CrossRef]
59. Yamashita S, Masuda D, Matsuzawa Y. Clinical applications of a novel selective PPARα modulator, pemafibrate, in dyslipidemia and metabolic diseases. J Atheroscler Thromb. 2019;26(5):389–402. doi:10.5551/jat.48918. [Google Scholar] [PubMed] [CrossRef]
60. Kleemann R, Gervois PP, Verschuren L, Staels B, Princen HM, Kooistra T. Fibrates down-regulate IL-1-stimulated C-reactive protein gene expression in hepatocytes by reducing nuclear p50-NFκB–C/EBP-β complex formation. Blood. 2003;101(2):545–51. doi:10.1182/blood-2002-06-1762. [Google Scholar] [PubMed] [CrossRef]
61. Yoo J, Jeong IK, Ahn KJ, Chung HY, Hwang YC. Fenofibrate, a PPARα agonist, reduces hepatic fat accumulation through the upregulation of TFEB-mediated lipophagy. Metabolism. 2020;120:154798. doi:10.1016/j.metabol.2021.154798. [Google Scholar] [PubMed] [CrossRef]
62. Fabbrini E, Mohammed BS, Korenblat KM, Magkos F, McCrea J, Patterson BW, et al. Effect of fenofibrate and niacin on intrahepatic triglyceride content, very low-density lipoprotein kinetics, and insulin action in obese subjects with nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2010;95(6):2727–35. doi:10.1210/jc.2009-2622. [Google Scholar] [PubMed] [CrossRef]
63. Ginsberg HN, Hounslow NJ, Senko Y, Suganami H, Bogdanski P, Ceska R, et al. Efficacy and Safety of K-877 (pemafibratea selective PPARα modulator, in european patients on statin therapy. Diabetes Care. 2022;45(4):898–908. doi:10.2337/dc21-1288. [Google Scholar] [PubMed] [CrossRef]
64. Mahmoudi A, Jamialahmadi T, Johnston TP, Sahebkar A. Impact of fenofibrate on NAFLD/NASH: a genetic perspective. Drug Discov Today. 2022;27(8):2363–72. doi:10.1016/j.drudis.2022.05.007. [Google Scholar] [PubMed] [CrossRef]
65. Staels B, Butruille L, Francque S. Treating NASH by targeting peroxisome proliferator-activated receptors. J Hepatol. 2023;79(5):1302–16. doi:10.1016/j.jhep.2023.07.004. [Google Scholar] [PubMed] [CrossRef]
66. Ivashkevich D, Ponomarenko A, Manzhulo I, Sultanov R, Dyuizen I. Effect of oleoylethanolamide-based dietary supplement on systemic inflammation in the development of alimentary-induced obesity in mice. Nutrients. 2023;15(20):4345. doi:10.3390/nu15204345. [Google Scholar] [PubMed] [CrossRef]
67. Haczeyni F, Wang H, Barn V, Mridha AR, Yeh MM, Haigh WG, et al. The selective peroxisome proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol Commun. 2017;1(7):663–74. doi:10.1002/hep4.1072. [Google Scholar] [PubMed] [CrossRef]
68. Dhingra S, Mahadik JD, Tarabishy Y, May SB, Vierling JM. Prevalence and clinical significance of portal inflammation, portal plasma cells, interface hepatitis and biliary injury in liver biopsies from patients with non-alcoholic steatohepatitis. Pathology. 2022;54(6):686–93. doi:10.1016/j.pathol.2022.01.009. [Google Scholar] [PubMed] [CrossRef]
69. Allameh A, Niayesh-Mehr R, Aliarab A, Sebastiani G, Pantopoulos K. Oxidative stress in liver pathophysiology and disease. Antioxidants. 2023;12(9):1653. doi:10.3390/antiox12091653. [Google Scholar] [PubMed] [CrossRef]
70. Takada I, Makishima M. Peroxisome proliferator-activated receptor agonists and antagonists: a patent review (2014-present). Expert Opin Ther Pat. 2020;30(1):1–13. doi:10.1080/13543776.2020.1703952. [Google Scholar] [PubMed] [CrossRef]
71. Christofides A, Konstantinidou E, Jani C, Boussiotis VA. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism. 2021;114:154338. doi:10.1016/j.metabol.2020.154338. [Google Scholar] [PubMed] [CrossRef]
72. Payahoo L, Khajebishak Y, Asghari Jafarabadi M, Ostadrahimi A. Oleoylethanolamide supplementation reduces inflammation and oxidative stress in obese people: a clinical trial. Adv Pharm Bull. 2018;8(3):479–87. doi:10.15171/apb.2018.056. [Google Scholar] [PubMed] [CrossRef]
73. Yang L, Guo H, Li Y, Meng X, Yan L, Zhang D et al. Oleoylethanolamide exerts anti-inflammatory effects on LPS-induced THP-1 cells by enhancing PPARα signaling and inhibiting the NF-κB and ERK1/2/AP-1/STAT3 pathways. Sci Rep. 2016;6:34611. doi:10.1038/srep34611. [Google Scholar] [PubMed] [CrossRef]
74. Tutunchi H, Ostadrahimi A, Saghafi-Asl M, Roshanravan N, Shakeri-Bavil A, Asghari-Jafarabadi M, et al. Expression of NF-κB, IL-6, and IL-10 genes, body composition, and hepatic fibrosis in obese patients with NAFLD—combined effects of oleoylethanolamide supplementation and calorie restriction: a triple-blind randomized controlled clinical trial. J Cell Physiol. 2021;236(1):417–26. doi:10.1002/jcp.29870. [Google Scholar] [PubMed] [CrossRef]
75. Hu J, Zhu Z, Ying H, Yao J, Ma H, Li L, et al. Oleoylethanolamide protects against acute liver injury by regulating Nrf-2/HO-1 and NLRP3 pathways in mice. Front Pharmacol. 2021;11:605065. doi:10.3389/fphar.2020.605065. [Google Scholar] [PubMed] [CrossRef]
76. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–89. doi:10.1038/s41577-019-0165-0. [Google Scholar] [PubMed] [CrossRef]
77. Liu Q, Niu C, Zhang Q, Sun S, Chen Y, Shi Y. Amitriptyline inhibits NLRP3 inflammasome activation via the ASM/CE pathway in a cell model of NAFLD. BIOCELL. 2024;48(5):759–69. doi:10.32604/biocell.2024.048551. [Google Scholar] [CrossRef]
78. Tutunchi H, Zolrahim F, Nikbaf-Shandiz M, Naeini F, Ostadrahimi A, Naghshi S, et al. Effects of oleoylethanolamide supplementation on inflammatory biomarkers, oxidative stress and antioxidant parameters of obese patients with NAFLD on a calorie-restricted diet: a randomized controlled trial. Front Pharmacol. 2023;14:1144550. doi:10.3389/fphar.2023.1144550. [Google Scholar] [PubMed] [CrossRef]
79. Giudetti AM, Vergara D, Longo S, Friuli M, Eramo B, Tacconi S, et al. Oleoylethanolamide reduces hepatic oxidative stress and endoplasmic reticulum stress in high-fat diet-fed rats. Antioxidants. 2021;10(8):1289. doi:10.3390/antiox10081289. [Google Scholar] [PubMed] [CrossRef]
80. Mooli RGR, Mukhi D, Ramakrishnan SK. Oxidative stress and redox signaling in the pathophysiology of liver diseases. Compr Physiol. 2022;12(2):3167–92. doi:10.1002/cphy.c200021. [Google Scholar] [PubMed] [CrossRef]
81. Dou L, Shi X, He X, Gao Y. Macrophage phenotype and function in liver disorder. Front Immunol. 2020;10:3112. doi:10.3389/fimmu.2019.03112. [Google Scholar] [PubMed] [CrossRef]
82. Yu Y, Zhang Y, Zhang J, Guan C, Liu L, Ren L. Cantharidin-induced acute hepatotoxicity: the role of TNF-α, IKK-α, Bcl-2, Bax and caspase3. J Appl Toxicol. 2020;40(11):1526–33. doi:10.1002/jat.4003. [Google Scholar] [PubMed] [CrossRef]
83. Park SJ, Garcia Diaz J, Um E, Hahn YS. Major roles of kupffer cells and macrophages in NAFLD development. Front Endocrinol. 2023;14:1150118. doi:10.3389/fendo.2023.1150118. [Google Scholar] [PubMed] [CrossRef]
84. Ma Y, Lee G, Heo SY, Roh YS. Oxidative stress is a key modulator in the development of nonalcoholic fatty liver disease. Antioxidants. 2021;11(1):91. doi:10.3390/antiox11010091. [Google Scholar] [PubMed] [CrossRef]
85. Xu HL, Wan SR, An Y, Wu Q, Xing YH, Deng CH, et al. Targeting cell death in NAFLD: mechanisms and targeted therapies. Cell Death Discov. 2024;10(1):399. doi:10.1038/s41420-024-02168-z. [Google Scholar] [PubMed] [CrossRef]
86. LeFort KR, Rungratanawanich W, Song BJ. Contributing roles of mitochondrial dysfunction and hepatocyte apoptosis in liver diseases through oxidative stress, post-translational modifications, inflammation, and intestinal barrier dysfunction. Cell Mol Life Sci. 2024;81(1):34. doi:10.1007/s00018-023-05061-7. [Google Scholar] [PubMed] [CrossRef]
87. Li Y, Zhang Y, Wang Q, Wu C, Du G, Yang L. Oleoylethanolamide protects against acute ischemic stroke by promoting PPARα-mediated Microglia/Macrophage M2 polarization. Pharmaceuticals. 2023 Apr 20;16(4):621. doi:10.3390/ph16040621. [Google Scholar] [PubMed] [CrossRef]
88. Chen Z, Zhuo R, Zhao Y, Yang L, Zhou Y, Cheng X, et al. Oleoylethanolamide stabilizes atherosclerotic plaque through regulating macrophage polarization via AMPK-PPARα pathway. Biochem Biophys Res Commun. 2020;524(2):308–16. doi:10.1016/j.bbrc.2020.01.103. [Google Scholar] [PubMed] [CrossRef]
89. Kamm DR, McCommis KS. Hepatic stellate cells in physiology and pathology. J Physiol. 2022 Apr;600(8):1825–37. doi:10.1113/JP281061. [Google Scholar] [PubMed] [CrossRef]
90. Xu F, Liu C, Zhou D, Zhang L. TGF-β/SMAD pathway and its regulation in hepatic fibrosis. J Histochem Cytochem. 2016;64(3):157–67. doi:10.1369/0022155415627681. [Google Scholar] [PubMed] [CrossRef]
91. Yoshida K, Murata M, Yamaguchi T, Matsuzaki K. TGF-β/Smad signaling during hepatic fibro-carcinogenesis (review). Int J Oncol. 2014;45(4):1363–71. doi:10.3892/ijo.2014.2552. [Google Scholar] [PubMed] [CrossRef]
92. Rodríguez-Vilarrupla A, Laviña B, García-Calderó H, Russo L, Rosado E, Roglans N, et al. PPARα activation improves endothelial dysfunction and reduces fibrosis and portal pressure in cirrhotic rats. J Hepatol. 2012;56(5):1033–9. doi:10.1016/j.jhep.2011.12.008. [Google Scholar] [PubMed] [CrossRef]
93. Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARα agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39(5):1286–96. doi:10.1002/hep.20170. [Google Scholar] [PubMed] [CrossRef]
94. Chen L, Li L, Chen J, Li L, Zheng Z, Ren J, et al. Oleoylethanolamide, an endogenous PPAR-α ligand, attenuates liver fibrosis targeting hepatic stellate cells. Oncotarget. 2015;6(40):42530–40. doi:10.18632/oncotarget.6466. [Google Scholar] [PubMed] [CrossRef]
95. Romano A, Friuli M, Del Coco L, Longo S, Vergara D, Del Boccio P, et al. Chronic oleoylethanolamide treatment decreases hepatic triacylglycerol level in rat liver by a PPARγ/SREBP-mediated suppression of fatty acid and triacylglycerol synthesis. Nutrients. 2021;13(2):394. doi:10.3390/nu13020394. [Google Scholar] [PubMed] [CrossRef]
96. Shao M, Ye Z, Qin Y, Wu T. Abnormal metabolic processes involved in the pathogenesis of non-alcoholic fatty liver disease (Review). Exp Ther Med. 2020;20(5):26. doi:10.3892/etm.2020.9154. [Google Scholar] [PubMed] [CrossRef]
97. Drover VA, Abumrad NA. CD36-dependent fatty acid uptake regulates expression of peroxisome proliferator activated receptors. Biochem Soc Trans. 2005 Feb;33(Pt 1):311–5. doi:10.1042/BST0330311. [Google Scholar] [PubMed] [CrossRef]
98. Li L, Li L, Chen L, Lin X, Xu Y, Ren J, et al. Effect of oleoylethanolamide on diet-induced nonalcoholic fatty liver in rats. J Pharmacol Sci. 2015;127(3):244–50. doi:10.1016/j.jphs.2014.12.001. [Google Scholar] [PubMed] [CrossRef]
99. AlNafea HM, Korish AA. Activation of the peroxisome proliferator-activated receptors (PPAR-α/γ) and the fatty acid metabolizing enzyme protein CPT1A by camel milk treatment counteracts the high-fat diet-induced nonalcoholic fatty liver disease. PPAR Res. 2021;2021:5558731. doi:10.1155/2021/5558731. [Google Scholar] [PubMed] [CrossRef]
100. Pan X, Schwartz GJ, Hussain MM. Oleoylethanolamide differentially regulates glycerolipid synthesis and lipoprotein secretion in intestine and liver. J Lipid Res. 2018;59(12):2349–59. doi:10.1194/jlr.M089250. [Google Scholar] [PubMed] [CrossRef]
101. Guzmán M, Lo Verme J, Fu J, Oveisi F, Blázquez C, Piomelli D. Oleoylethanolamide stimulates lipolysis by activating the nuclear receptor peroxisome proliferator-activated receptor alpha (PPAR-α). J Biol Chem. 2004;279(27):27849–54. doi:10.1074/jbc.M404087200. [Google Scholar] [PubMed] [CrossRef]
102. Misto A, Provensi G, Vozella V, Passani MB, Piomelli D. Mast cell-derived histamine regulates liver ketogenesis via oleoylethanolamide signaling. Cell Metab. 2019;29(1):91–102. doi:10.1016/j.cmet.2018.09.014. [Google Scholar] [PubMed] [CrossRef]
103. Lin L, Mabou Tagne A, Squire EN, Lee HL, Fotio Y, Ramirez J, et al. Diet-induced obesity disrupts histamine-dependent oleoylethanolamide signaling in the mouse liver. Pharmacology. 2022;107(7–8):423–32. doi:10.1159/000524753. [Google Scholar] [PubMed] [CrossRef]
104. Li H, Yu XH, Ou X, Ouyang XP, Tang CK. Hepatic cholesterol transport and its role in non-alcoholic fatty liver disease and atherosclerosis. Prog Lipid Res. 2021;83:101109. doi:10.1016/j.plipres.2021.101109. [Google Scholar] [PubMed] [CrossRef]
105. Miao J, Manthena PV, Haas ME, Ling AV, Shin DJ, Graham MJ, et al. Role of insulin in the regulation of proprotein convertase subtilisin/kexin type 9. Arterioscler Thromb Vasc Biol. 2015;35(7):1589–96. doi:10.1161/ATVBAHA.115.305688. [Google Scholar] [PubMed] [CrossRef]
106. Wagner N, Wagner KD. Pharmacological utility of PPAR modulation for angiogenesis in cardiovascular disease. Int J Mol Sci. 2023;24(3):2345. doi:10.3390/ijms24032345. [Google Scholar] [PubMed] [CrossRef]
107. Gizard F, Amant C, Barbier O, Bellosta S, Robillard R, Percevault F, et al. PPARα inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16INK4a. J Clin Invest. 2005;115(11):3228–38. doi:10.1172/JCI22756. [Google Scholar] [PubMed] [CrossRef]
108. Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, et al. PPAR-α and PPAR-γ activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. 2001;7(1):53–8. doi:10.1038/83348. [Google Scholar] [PubMed] [CrossRef]
109. Kourimate S, Le May C, Langhi C, Jarnoux AL, Ouguerram K, Zaïr Y, et al. Dual mechanisms for the fibrate-mediated repression of proprotein convertase subtilisin/kexin type 9. J Biol Chem. 2008;283(15):9666–73. doi:10.1074/jbc.M705831200. [Google Scholar] [PubMed] [CrossRef]
110. Krysa JA, Ooi TC, Proctor SD, Vine DF. Nutritional and lipid modulation of PCSK9: effects on cardiometabolic risk factors. J Nutr. 2017;147(4):473–81. doi:10.3945/jn.116.235069. [Google Scholar] [PubMed] [CrossRef]
111. Seidah NG, Abifadel M, Prost S, Boileau C, Prat A. The proprotein convertases in hypercholesterolemia and cardiovascular diseases: emphasis on proprotein convertase subtilisin/kexin 9. Pharmacol Rev. 2017;69(1):33–52. doi:10.1124/pr.116.012989. [Google Scholar] [PubMed] [CrossRef]
112. Jin W, Wang X, Millar JS, Quertermous T, Rothblat GH et al. Hepatic proprotein convertases modulate HDL metabolism. Cell Metab. 2007;6(2):129–36. doi:10.1016/j.cmet.2007.07.009. [Google Scholar] [PubMed] [CrossRef]
113. Oosterveer MH, Grefhorst A, Van Dijk TH, Havinga R, Staels B, Kuipers F, et al. Fenofibrate simultaneously induces hepatic fatty acid oxidation, synthesis, and elongation in mice. J Biol Chem. 2009;284(49):34036–44. doi:10.1074/jbc.M109.051052. [Google Scholar] [PubMed] [CrossRef]
114. Ahsan W. The journey of thiazolidinediones as modulators of PPARs for the management of diabetes: a current perspective. Curr Pharm Des. 2019;25(23):2540–54. doi:10.2174/1381612825666190716094852. [Google Scholar] [PubMed] [CrossRef]
115. Rojas Ortiz V, Nieves J, Fernandez EC. Clinical management of nonalcoholic steatohepatitis (NASH) with the use of thiazolidinediones and the additive effect of thiazolidinediones and a GLP-1 agonist: case series. Cureus. 2021;13(3):e14082. doi:10.7759/cureus.14082. [Google Scholar] [PubMed] [CrossRef]
116. Thomas JA, Gupta R, Aithal GP. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis. Ann Intern Med. 2017;166(3):229–30. doi:10.7326/L16-0628. [Google Scholar] [PubMed] [CrossRef]
117. Li J, Xue YM, Zhu B, Pan YH, Zhang Y, Wang C et al. Rosiglitazone elicits an adiponectin-mediated insulin-sensitizing action at the adipose tissue-liver axis in otsuka long-evans tokushima fatty rats. J Diabetes Res. 2018;2018:4627842. doi:10.1155/2018/4627842. [Google Scholar] [PubMed] [CrossRef]
118. Araki M, Nakagawa Y, Oishi A, Han SI, Wang Y, Kumagai K, et al. The peroxisome proliferator-activated receptor α (PPARα) agonist pemafibrate protects against diet-induced obesity in mice. Int J Mol Sci. 2018;19(7):2148. doi:10.3390/ijms19072148. [Google Scholar] [PubMed] [CrossRef]
119. Koh EH, Kim MS, Park JY, Kim HS, Youn JY, Park HS, et al. Peroxisome proliferator-activated receptor (PPAR)-α activation prevents diabetes in OLETF rats: comparison with PPAR-γ activation. Diabetes. 2003;52(9):2331–7. doi:10.2337/diabetes.52.9.2331. [Google Scholar] [PubMed] [CrossRef]
120. Takahashi H, Sanada K, Nagai H, Li Y, Aoki Y, Ara T, et al. Over-expression of PPARα in obese mice adipose tissue improves insulin sensitivity. Biochem Biophys Res Commun. 2017;493(1):108–14. doi:10.1016/j.bbrc.2017.09.067. [Google Scholar] [PubMed] [CrossRef]
121. Yang JW, Kim HS, Choi YW, Kim YM, Kang KW. Therapeutic application of GPR119 ligands in metabolic disorders. Diabetes Obes Metab. 2018;20(2):257–69. doi:10.1111/dom.13062. [Google Scholar] [PubMed] [CrossRef]
122. Zhao J, Zhao Y, Hu Y, Peng J. Targeting the GPR119/incretin axis: a promising new therapy for metabolic-associated fatty liver disease. Cell Mol Biol Lett. 2021;26(1):32. doi:10.1186/s11658-021-00276-7. [Google Scholar] [PubMed] [CrossRef]
123. Brown JD, McAnally D, Ayala JE, Burmeister MA, Morfa C, Smith L, et al. Oleoylethanolamide modulates glucagon-like peptide-1 receptor agonist signaling and enhances exendin-4-mediated weight loss in obese mice. Am J Physiol Regul Integr Comp Physiol. 2018;315(4):R595–608. doi:10.1152/ajpregu.00459.2017. [Google Scholar] [PubMed] [CrossRef]
124. Lauffer LM, Iakoubov R, Brubaker PL. GPR119 is essential for oleoylethanolamide-induced glucagon-like peptide-1 secretion from the intestinal enteroendocrine L-cell. Diabetes. 2009;58(5):1058–66. doi:10.2337/db08-1237. [Google Scholar] [PubMed] [CrossRef]
125. Abdulla H, Phillips B, Wilkinson D, Gates A, Limb M, Jandova T, et al. Effects of GLP-1 infusion upon whole-body glucose uptake and skeletal muscle perfusion during fed-state in older men. J Clin Endocrinol Metab. 2023;108(4):971–8. doi:10.1210/clinem/dgac613. [Google Scholar] [PubMed] [CrossRef]
126. Jiang Y, Wang Z, Ma B, Fan L, Yi N, Lu B, et al. GLP-1 improves adipocyte insulin sensitivity following induction of endoplasmic reticulum stress. Front Pharmacol. 2018;9:1168. doi:10.3389/fphar.2018.01168. [Google Scholar] [PubMed] [CrossRef]
127. Nevola R, Epifani R, Imbriani S, Tortorella G, Aprea C, Galiero R, et al. GLP-1 receptor agonists in non-alcoholic fatty liver disease: current evidence and future perspectives. Int J Mol Sci. 2023;24(2):1703. doi:10.3390/ijms24021703. [Google Scholar] [PubMed] [CrossRef]
128. Mills EL, Harmon C, Jedrychowski MP, Xiao H, Garrity R, Tran NV, et al. UCP1 governs liver extracellular succinate and inflammatory pathogenesis. Nat Metab. 2021;3(5):604–17. doi:10.1038/s42255-021-00389-5. [Google Scholar] [PubMed] [CrossRef]
129. Suárez J, Rivera P, Arrabal S, Crespillo A, Serrano A, Baixeras E, et al. Oleoylethanolamide enhances β-adrenergic-mediated thermogenesis and white-to-brown adipocyte phenotype in epididymal white adipose tissue in rat. Dis Model Mech. 2014;7(1):129–41. doi:10.1242/dmm.013110. [Google Scholar] [PubMed] [CrossRef]
130. Tutunchi H, Ostadrahimi A, Saghafi-Asl M, Hosseinzadeh-Attar MJ, Shakeri A, Asghari-Jafarabadi M, et al. Oleoylethanolamide supplementation in obese patients newly diagnosed with non-alcoholic fatty liver disease: effects on metabolic parameters, anthropometric indices, and expression of PPAR-α, UCP1, and UCP2 genes. Pharmacol Res. 2020;156:104770. doi:10.1016/j.phrs.2020.104770. [Google Scholar] [PubMed] [CrossRef]
131. Fu J, Oveisi F, Gaetani S, Lin E, Piomelli D. Oleoylethanolamide, an endogenous PPAR-α agonist, lowers body weight and hyperlipidemia in obese rats. Neuropharmacol. 2005;48(8):1147–53. doi:10.1016/j.neuropharm.2005.02.013. [Google Scholar] [PubMed] [CrossRef]
132. Achari AE, Jain SK. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int J Mol Sci. 2017;18(6):1321. doi:10.3390/ijms18061321. [Google Scholar] [PubMed] [CrossRef]
133. Kou H, Deng J, Gao D, Song A, Han Z, Wei J, et al. Relationship among adiponectin, insulin resistance and atherosclerosis in non-diabetic hypertensive patients and healthy adults. Clin Exp Hypertens. 2018;40(7):656–63. doi:10.1080/10641963.2018.1425414. [Google Scholar] [PubMed] [CrossRef]
134. Adolph TE, Grander C, Grabherr F, Tilg H. Adipokines and non-alcoholic fatty liver disease: multiple interactions. Int J Mol Sci. 2017;18(8):1649. doi:10.3390/ijms18081649. [Google Scholar] [PubMed] [CrossRef]
135. Jung D, Bucher F, Ryu S, Jeong J, Lee BY, Jeong Y, et al. An adiponectin receptor agonist antibody stimulates glucose uptake and fatty-acid oxidation by activating AMP-activated protein kinase. Cytokine. 2020;126:154863. doi:10.1016/j.cyto.2019.154863. [Google Scholar] [PubMed] [CrossRef]
136. Tsuchida A, Yamauchi T, Takekawa S, Hada Y, Ito Y, Maki T, et al. Peroxisome proliferator-activated receptor (PPAR)α activation increases adiponectin receptors and reduces obesity-related inflammation in adipose tissue: comparison of activation of PPARα, PPARγ, and their combination. Diabetes. 2005;54(12):3358–70. doi:10.2337/diabetes.54.12.3358. [Google Scholar] [PubMed] [CrossRef]
137. Boutari C, Perakakis N, Mantzoros CS. Association of adipokines with development and progression of nonalcoholic fatty liver disease. Endocrinol Metab. 2018;33(1):33–43. doi:10.3803/EnM.2018.33.1.33. [Google Scholar] [PubMed] [CrossRef]
138. Ishtiaq SM, Rashid H, Hussain Z, Arshad MI, Khan JA. Adiponectin and PPAR: a setup for intricate crosstalk between obesity and non-alcoholic fatty liver disease. Rev Endocr Metab Disord. 2019;20(3):253–61. doi:10.1007/s11154-019-09510-2. [Google Scholar] [PubMed] [CrossRef]
139. Zuo Y, Xiao T, Qiu X, Liu Z, Zhang S, Zhou N. Adiponectin reduces apoptosis of diabetic cardiomyocytes by regulating miR-711/TLR4 axis. Diabetol Metab Syndr. 2022;14(1):131. doi:10.1186/s13098-022-00904-y. [Google Scholar] [PubMed] [CrossRef]
140. Liu Z, Gan L, Wu T, Feng F, Luo D, Gu H et al. Adiponectin reduces ER stress-induced apoptosis through PPARα transcriptional regulation of ATF2 in mouse adipose. Cell Death Dis. 2016;7(11):e2487. doi:10.1038/cddis.2016.388. [Google Scholar] [PubMed] [CrossRef]
141. Ouchi N, Kihara S, Funahashi T, Matsuzawa Y, Walsh K. Obesity, adiponectin and vascular inflammatory disease. Curr Opin Lipidol. 2003;14(6):561–6. doi:10.1097/00041433-200312000-00003. [Google Scholar] [PubMed] [CrossRef]
142. Ouedraogo R, Gong Y, Berzins B, Wu X, Mahadev K, Hough K, et al. Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. J Clin Invest. 2007;117(6):1718–26. doi:10.1172/JCI29623. [Google Scholar] [PubMed] [CrossRef]
143. Recasens M, Ricart W, Fernández-Real JM. Obesidad e inflamación [Obesity and inflammation]. Rev Med Univ Navarra. 2004;48(2):49–54. [Google Scholar] [PubMed]
144. D’Aloia A, Arrigoni F, Tisi R, Palmioli A, Ceriani M, Artusa V, et al. Synthesis, molecular modeling and biological evaluation of metabolically stable analogues of the endogenous fatty acid amide palmitoylethanolamide. Int J Mol Sci. 2020;21(23):9074. doi:10.3390/ijms21239074. [Google Scholar] [PubMed] [CrossRef]
145. Oveisi F, Gaetani S, Eng KT, Piomelli D. Oleoylethanolamide inhibits food intake in free-feeding rats after oral administration. Pharmacol Res. 2004;49(5):461–6. doi:10.1016/j.phrs.2003.12.006. [Google Scholar] [PubMed] [CrossRef]
146. Thabuis C, Destaillats F, Lambert DM, Muccioli GG, Maillot M, Harach T, et al. Lipid transport function is the main target of oral oleoylethanolamide to reduce adiposity in high-fat-fed mice. J Lipid Res. 2011;52(7):1373–82. doi:10.1194/jlr.M013391. [Google Scholar] [PubMed] [CrossRef]
147. Nielsen MJ, Petersen G, Astrup A, Hansen HS. Food intake is inhibited by oral oleoylethanolamide. J Lipid Res. 2004;45(6):1027–9. doi:10.1194/jlr.C300008-JLR200. [Google Scholar] [PubMed] [CrossRef]
148. Sabahi M, Ahmadi SA, Kazemi A, Mehrpooya M, Khazaei M, Ranjbar A et al. The effect of Oleoylethanolamide (OEA) add-on treatment on inflammatory, oxidative stress, lipid, and biochemical parameters in the acute ischemic stroke patients: randomized double-blind placebo-controlled study. Oxid Med Cell Longev. 2022;2022:5721167. doi:10.1155/2022/5721167. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools