
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
The Role of miRNAs in Mechanotransduction Regulation and Cancer Development
1 Instituto de Ciencias de la Salud, Universidad Veracruzana, Xalapa, 91190, Veracruz, México
2 Maestría en Ciencias de la Salud, Instituto de Ciencias de la Salud, Universidad Veracruzana, Xalapa, 91190, Veracruz, México
* Corresponding Author: Elisa Tamariz. Email:
(This article belongs to the Special Issue: Non-coding RNAs (ncRNAs) in Human Diseases)
BIOCELL 2025, 49(9), 1663-1695. https://doi.org/10.32604/biocell.2025.066201
Received 01 April 2025; Accepted 21 July 2025; Issue published 25 September 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Cells are exposed to various mechanical forces, including extracellular and intracellular forces such as stiffness, tension, compression, viscosity, and shear stress, which regulate cell biology. The process of transducing mechanical stimuli into biochemical signals is termed mechanotransduction. These mechanical forces can regulate protein and gene expression, thereby impacting cell morphology, adhesion, proliferation, apoptosis, and migration. During cancer development, significant changes in extracellular and intracellular mechanical properties occur, resulting in altered mechanical inputs to which cells are exposed. MicroRNAs (miRNAs), key post-transcriptional regulators of gene and protein expression, are increasingly recognized as mechanosensitive molecules involved in cancer development. In this review, we summarize the primary cellular pathways involved in force sensing and mechanotransduction, emphasizing the role of forces in miRNA biogenesis and expression, as well as their influence on the regulation of key mechanotransducers. Furthermore, we focus on recent evidence regarding the induction or repression of miRNAs involved in cancer development by mechanical forces and their impact on the regulation of proteins that contribute to cancer progression.Keywords
Cancer ranks among the top ten causes of mortality in middle- and upper-income countries [1]. In 2022, there were more than 19 million new cases and more than 9 million deaths worldwide [2]. Cancer research has led to an understanding of some of the cellular and molecular mechanisms underlying the malignant transformation of cells. The complex process of cancer development involves several characteristics or hallmarks, including sustained proliferative signaling, evading growth suppressors, avoiding immune destruction, inducing vasculature formation, genome instability and mutation, and epigenetic reprogramming, among others [3]. However, the microenvironment is also essential for cancer development and progression. The microenvironment provides chemical and physical signaling to the cells; among the physical signals, mechanical forces are fundamental in modulating cell biology and tissue homeostasis. In solid tumors, the composition and structure of the extracellular matrix (ECM) are altered, transforming fibroblasts into cancer-associated fibroblasts that secrete growth factors and ECM components, leading to fibrosis and alterations in the ECM composition. The changes in ECM also modify the mechanical properties of the tissues, generating solid stress that compresses cells (compression force), blood, and lymphatic vessels, and elevates fluid pressure [4]. Cells in the tumor and surrounding tissue respond to environmental changes, establishing a loop that influences the malignancy of the cells and the progression of cancer. It is well established that cells can transduce forces into biochemical signals to regulate protein and gene expression. Elucidating the signaling pathways activated by mechanical forces is an active area of research aimed at understanding how external stimuli are sensed and transmitted from the ECM to the cell nucleus. However, conducting experimental studies in this domain presents significant challenges and requires a multidisciplinary approach. These studies demand precise control of physical forces at molecular, cellular, or tissue levels, and many processes remain to be understood and integrated due to the variety of molecular targets involved in the response to mechanical stimuli. MiRNAs, as key regulators of protein expression, have a fundamental role in cancer development. The role of mechanical forces in their regulation has also been reported, although it is not fully understood and is under active research. In this review, we intend to give a general panorama of the forces-driven cell signal transduction and its main pathways, and address how the forces regulate gene and protein expression, particularly by the miRNA biogenesis regulation; finally, we focus on the recently studied role of forces in miRNA regulation and cancer development, and some of the challenges and prospects for a translational and clinical application of this knowledge.
Cells respond to the environment’s mechanical stimulus by a complex process termed mechanotransduction, which converts mechanical forces into biochemical signals [5]. Understanding the process of transducing mechanical inputs into relevant signaling that modifies cellular and molecular responses is a complex task that has been the subject of intense research. Mechanical cues, such as stress, tension, compression, and shear forces, are dynamic signals that cells can sense and transduce (Fig. 1) to promote health or disease [5,6]. For example, shear forces generated by the bloodstream are essential for blood vessel formation and function. Modifying this force can promote atherosclerosis, alter vessel wall function, induce inflammation, disrupt wound healing, and facilitate cancer metastasis [7,8]. ECM stiffness and topography are also relevant mechanical cues. Cells interact with the ECM and apply tension forces; the resistance of the ECM to deformation is an important cue, and cells can modify it by synthesizing ECM components or remodeling its structure. An excess of ECM components, such as collagen type I and III, fibronectin, elastin deposition, the crosslink of collagens and the improper remodeling induces fibrosis and increase stiffness, altering the mechanical environment and triggering cell modifications that can promote inflammation, cell transformation, migration, and proliferation, among other effects [6,9].
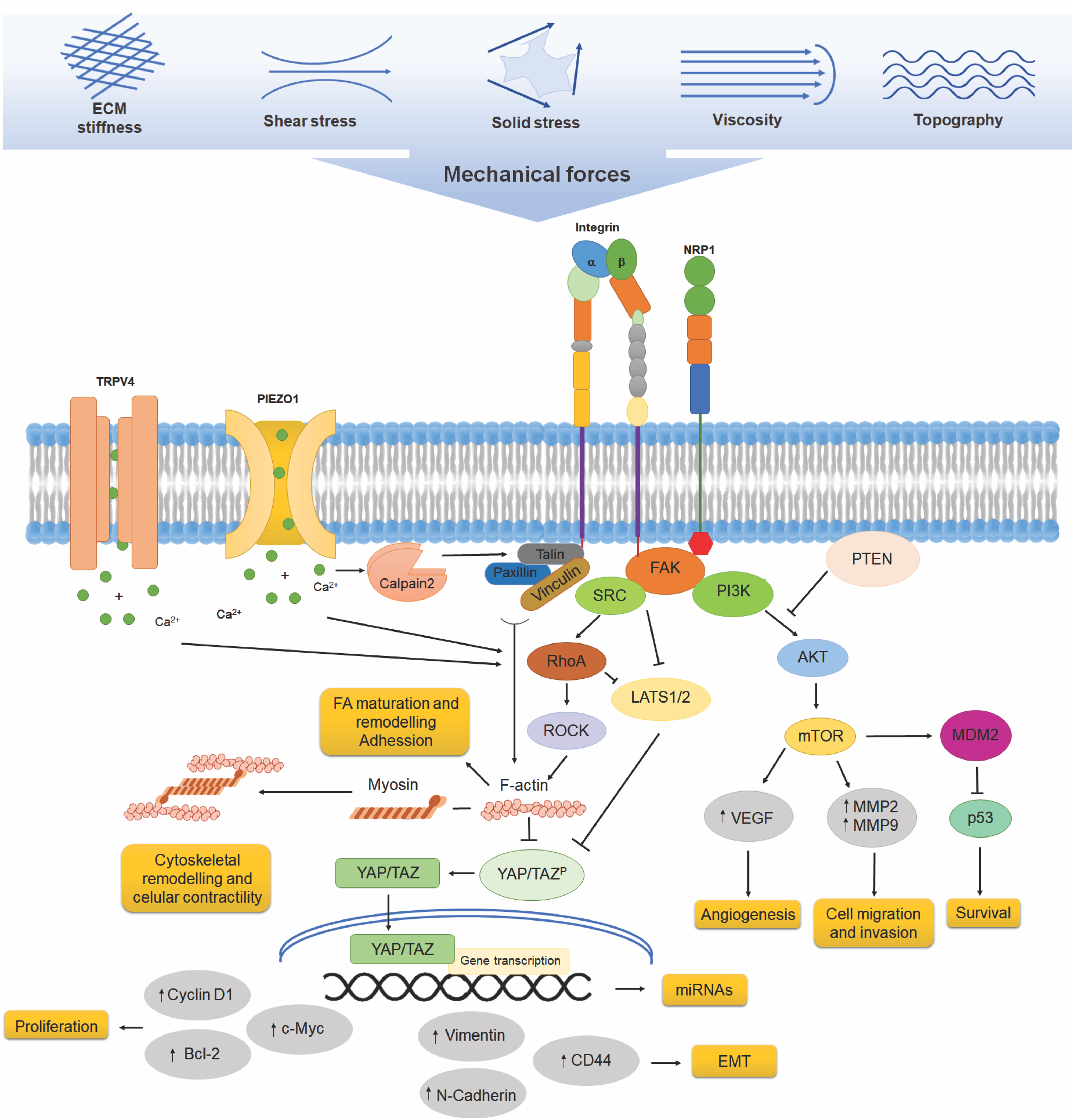
Figure 1: Mechanotransduction pathways in cancer cells. Cells experience a variety of mechanical forces, including extracellular matrix (ECM) stiffness, shear stress, viscosity, and topographical cues, which influence their behavior. Mechanosensors, including integrins, neuropilin-1 (NRP1), and mechanosensitive ion channels such as TRPV4 and PIEZO1, detect these forces. Integrins, in particular, interact with focal adhesion proteins such as vinculin, talin, and paxillin, leading to the activation of focal adhesion kinase (FAK) and downstream signaling pathways, including SRC, RhoA, and PI3K. RhoA signaling influences actin cytoskeleton remodeling through ROCK, impacting YAP/TAZ activation and nuclear translocation, which triggers gene transcription and increases the expression of CD44, N-cadherin, vimentin, cyclin D1, Bcl-2, and c-Myc, genes associated with EMT and survival, as well as miRNAs. Additionally, the PI3K/Akt/mTOR pathway promotes angiogenesis through VEGF upregulation, as well as cell migration and invasion, by increasing MMP2 and MMP9 expression. The inhibition of PTEN further amplifies PI3K/Akt signaling, contributing to oncogenic progression
Mechanical cues also arise from cell-cell interactions; cells can generate force through actomyosin contractility, transmit it to their neighborhoods via the actin cytoskeleton coupled to cell-cell junctions, and propagate it within the tissue to control morphogenesis, tissue repair, and collective cell migration [10]. As the detailed mechanotransduction process is out of the scope of this review, we focus on some of the most relevant steps involved in mechanotransduction, from the extracellular mechanical input and the primary mechanosensors in the cell membrane, towards the intracytoplasmic transmission until the cell nuclei that converge in gene expression regulation and particularly in the regulation of miRNAs.
2.1 Cell Membrane and Its Role as a Mechanical Force Sensor
A primary site for force sensing is the cell membrane, which adapts to tension, compression, and shear stress exerted by the surrounding environment, leading to membrane unfolding, bending, lipid reorganization, endocytosis, and exocytosis. The coupling of the cell membrane and cell cytoskeleton also contributes to membrane tension and force transmission. Additionally, proteins inserted in the cell membrane undergo conformational changes, relocate, and activate downstream signaling as part of the transduction of mechanical forces into biochemical signals within the cells [11].
Mechanosensing molecules, as proteins inserted in the membrane, can modify their structure or establish protein interactions in response to mechanical cues. Among the mechanosensors, integrins and stretch-activated ion channels are some of the most characterized [12]. Integrins are transmembrane receptors for ECM components. They mediate cell attachment by forming adhesion sites and connecting with the actin cytoskeleton [13]. Integrins transduce signals from the ECM into biochemical signaling, thereby regulating cellular functions such as proliferation, migration, apoptosis, ECM deposition, and remodeling [14].
Mechanical loads by ECM, such as increased stiffness, can induce conformational changes, like bending to an extended conformation that activate integrins, modifying their ECM binding affinity, facilitating nucleation, and clustering [14,15]. Upon activation, integrins recruit several proteins at their intracellular portion, such as talin, which, under stretch forces, is activated by a conformational change to interact with integrins, allowing vinculin binding. The association of talin-vinculin promotes actin interaction, stress fibers formation and adhesion growth by the recruitment of other adhesion and actin interacting proteins such as paxillin, α-actinin, proto-oncogene tyrosine-protein kinase Src (SRC) and Focal Adhesion Kinase (FAK), driving the maturation from nascent adhesions into more mature, multiprotein complexes and actin cytoskeleton-linked focal adhesions (FA) [16–19]. FA formation and cytoskeleton interaction induce the activation of small Rho GTPases, such as Ras homolog family member A (RhoA), to propagate force-induced signaling by regulating actin fiber assembly and actomyosin contractility [20] (Fig. 1). Actin fiber assembly and stress fiber formation promote the nuclear localization of proteins such as transcription factors by the direct interaction of actin fibers with the nuclear cytoskeleton, as will be described later.
Piezo are single-channel pore-forming transmembrane proteins that allow the flow of positive ions, sodium, potassium, and calcium, into cells [21]. Structurally, forms a propeller-like machinery with curved blades and long intracellular beams that act as lever-like apparatus that opens in response to membrane deformation induced by mechanical forces [22,23]. There are two isoforms of Piezo: Piezo1 and Piezo2. Piezo1 is expressed in various cell types, including blood cells, vascular endothelial cells, smooth muscle cells, and renal epithelial cells. At the same time, Piezo2 is present mainly in specialized sensory cells, such as dorsal root ganglion neurons, and regulates mechanosensory nociception and proprioception [24–26]. Several mechanical inputs, such as membrane tension induced by shear stress, the tension exerted by ECM stiffness, or membrane deformation due to extracellular topography, open the Piezo channel, allowing the permeation of cations [22,26,27]. Cell membrane deformation, resulting from modifications in local curvature and lateral tension, induces force-induced conformational transitions. During its activation, Piezo can change from a curved state into a flattened state in a force-dependent conformational change [28]; therefore, in response to mechanical forces, channels change from a resting-closed state to an activated-open state without second messengers, being the fastest transduction of mechanical forces into biochemical signals [29]. On the other hand, the cytoskeleton underlying the cell membrane can also modulate the channel; activation of Piezo1 is easier in bleb membrane protrusion that lacks attachment to the cortical cytoskeleton, and the inhibition of actomyosin contractility inhibits Piezo1 activity [27,29]. Opening the Piezo channels converts mechanical stimuli into electrical and chemical signaling to regulate a multitude of physiological processes such as blood flow [30–32], axonal growth regulation [33], and cardiovascular homeostasis [34]. The resulting increase in intracellular calcium by Piezo opening initiates downstream signaling cascades such as phosphoinositide 3-kinase (PI3K)/phosphatidylinositol (3,4,5)-trisphosphate (PIP3) and FAK/RhoA/Rho-associated coiled-coil containing protein kinase (ROCK), which drive focal adhesion assembly, activation of integrin focal adhesion signaling, and cytoskeletal remodeling [35]. Piezo1 interacts with integrins and is enriched in mature FA in a force-dependent recruitment [36]; the increase in intracellular calcium by Piezo1 activation leads to activation of calpain 2, a protease that activates and cleaves several of the FA proteins, such as talin, vinculin, and paxillin [37,38]. Once talin is cleaved by calpain, it interacts with integrins, inducing the linkage with the actin cytoskeleton [39] (Fig. 1). Additional evidence supports that Piezo1 acts as a mechanosensor during human T cell activation. Membrane stretch during immune synapse formation induces Piezo1 opening, resulting in increased calcium influx and subsequent activation of calpain, which facilitates the reorganization of the cortical actin cytoskeleton [40].
Piezo mutations are highly deleterious and are dysregulated in cancer cells [21]; tumor tissue stiffening promotes increased expression and activation of mechanical signaling in Piezo1. Piezo1 regulates the assembly of FA, activation of integrin signaling, tumor cell proliferation, and the expression of several genes involved in extracellular matrix remodeling, thereby enhancing the mechanosensory capabilities of tumor cells. These processes establish a reciprocal feedback loop between tumor cell mechanosignaling and aberrant tissue mechanics in gliomas, ultimately promoting malignancy [41].
Piezo2 upregulation has been related to good prognosis in breast cancer [42], although its overexpression has also been associated with gastric cancer [43] or breast cancer brain metastasis [44]. In BrM2 metastatic breast cancer cells, Piezo2 activation has been related to brain migration and invasion through the increase of RhoA activity by the Src tyrosine kinase Fyn, which activates LARG, a guanine nucleotide exchange factor (GEF), and calpain. RhoA activates mammalian diaphanous-related formin 1 (mDia1) and drives actin polymerization and FA formation, increasing cell migration and invasion [44].
2.2 The Actin Cytoskeleton as the Link between the Cell Membrane and the Cell Nucleus to Transduce Mechanical Forces
The actin cytoskeleton is connected to the nuclear envelope by the linker of nucleoskeleton cytoskeleton complex (LINC), a group of proteins, including Nesprins, inserted in the outer nuclear membrane, that interact with actin fibers and microtubules; the Sad1 and UNC-84 domain-containing proteins (SUN) on the inner side of the nuclear membrane; and the intermediate filament proteins, lamins, that form the nuclear lamina and provide anchoring sites for chromatin [45]. The interaction of the cytoskeleton with the LINC complex transmits forces from outside the cell to the nucleus, regulating conformational changes in chromatin, the translocation of transcription factors, and gene expression [46–48]. Force sensing at the integrin-ECM adhesion sites can modify the shape of the nucleus and chromatin, as well as influence chromatin remodeling and regulate acetylation and methylation [49–51]. Therefore, the cytoskeleton mediates a continuum from external forces to gene regulation; this force-driven signal transduction is a faster way to activate genes, allowing for millisecond activation.
2.3 Gene Expression Regulation by Mechanical Forces
Mechanical forces can modulate gene expression at multiple levels, including chromatin organization, epigenetic state modification, transcriptional activity regulation, and modulation of miRNA expression. Within the nucleus, chromatin is organized into two distinct regions: one comprising transcriptionally active euchromatin and another comprising transcriptionally silent heterochromatin, which is primarily localized at the nuclear periphery and interacts with the nuclear lamina via chromatin domains or lamina-associated domains (LADs). LADs are linked to the nuclear lamina via several proteins, including laminin-associated protein 2β (LAP2β), heterochromatin protein 1 (HP1), and CCCTC-binding factor (CTCF), a zinc finger protein involved in demarcating DNA loop domains and organizing gene regulatory elements in the loops. Mechanical forces transmitted to the nucleus can modify the interaction of chromatin with the nuclear lamina and promote LAD modifications, thereby changing the domains to a highly transcriptional activity region [52]. Upon alteration of chromatin compaction and supercoiling, there is an increased dissociation of LADs from the nuclear lamina, as shown in the CTCF loss-of-function melanoma cells [53].
External forces can deform the nucleus due to the mechanical coupling of the nuclear lamina with the cytoskeleton and cell adhesion proteins. Nuclear deformation leads to chromatin remodeling and changes in gene expression patterns [54]. Chromatin functions as a mechanosensory structure, becoming more relaxed in response to increased ECM stiffness or intracellular deformations caused by heightened tension, and this relaxation can modulate access to transcription factors and influence gene expression [55,56]. On the other hand, highly condensed heterochromatin and less condensed euchromatin regions can be regulated by several epigenetic modifications on the amino acid residues on histones, such as acetylation and methylation [57]. External mechanical forces, such as tension or shear, can modify the epigenetic state. For example, changes from unidirectional to oscillatory fluid flow alter epigenetic marks in endothelial cells, leading to DNA hypermethylation and gene silencing [58]. On the other hand, nuclear deformation and chromatin adaptation to mechanical forces are relevant to maintaining nuclear and DNA integrity. Mechanical stretch decreases the lamina-associated heterochromatin with H3K9me3 histone modifications and induces nuclei softening in stem/progenitor epithelial cells; this effect is mediated by calcium release from the endoplasmic reticulum through a Piezo1-mediated mechanism and maintains nuclear and DNA integrity [59]. Interestingly, loss of chromatin adaptation to mechanical forces by the inhibition of calcium influx or chromatin modifications induces DNA damage [59]. In the case of cancer cells, mechanical deformations of the cell nucleus induce DNA damage due to their rupture [60]; however, during cancer cell migration through confined spaces that induce nuclear deformation, cells use endosomal sorting complexes required for transport-III (ESCRT-III) machinery to repair their nuclear integrity [61].
Several pieces of evidence show that ECM stiffness modifies the DNA epigenetic state. An increase in the expression of histone deacetylases (HDACs) has been observed in mouse embryonic fibroblasts cultured on 0.5 kPa soft hydrogels compared to those cultured on 100 kPa stiff hydrogels [62]. On the other hand, 60 kPa soft matrices significantly enhanced the phosphorylation of H3 histone, promoting the mesoderm differentiation of human embryonic stem cells compared to 400 kPa stiff matrices [63]. Moreover, in rounded, unspread cells, a lysine methyltransferase that targets several histones and other non-histone proteins, as well as the vascular endothelial growth factor (VEGF) receptor, is translocated from the cytoplasm into the nucleus [64]. Additionally, when actomyosin contractility and the formation of actin fibers are disrupted, SET and MYND domain containing 3 (SMYD3) and histone deacetylase 3 (HDAC3) alter the cytoplasmic-to-nuclear redistribution [65].
One of the most characterized mechanisms of gene expression regulation by mechanical forces is the activation and translocation of transcriptional factors to the nucleus. Co-transcriptional factors Yes-associated protein (YAP) and the transcriptional coactivator with PDZ-binding motif (TAZ) are downstream effectors of the Hippo pathway, regulating the transcription of several transcription factors, including runt-related transcription factor 2 (RUNX2), p73, and T-box 5 (TBX5) [52]. YAP/TAZ is a key effector of mechanical forces, playing multiple roles in driving mechanical signals during development, regulating organ size, and promoting cancer progression [66].
Integrin β1 and FAK activation in high-stiffness cell cultures significantly increases YAP activation through the FAK/Src/PI3K pathway [67]. Moreover, YAP activates integrin and other FA protein genes that enhance FA strength [68]. In addition to the FAK-mediated pathway, YAP/TAZ activation may depend on the RhoA/Rho-ROCK signaling pathway, thus transmitting ECM stiffness signals to the nucleus [14] (Fig. 1). Mechanical forces transmitted to the nuclei induce their deformation and increase the permeability of proteins, such as YAP/TAZ. Forces exerted on ECM adhesions reach the nucleus through the actin cytoskeleton and the LINC complex. Talin, an adaptor protein that mediates the link between integrins and actin, unfolds under stretch and leads to vinculin interaction and focal adhesion and stress fiber formation, coupling ECM, focal adhesions, cytoskeleton, and nucleoskeleton, allowing forces to reach the nucleus and directly drive YAP translocation [69]. Furthermore, YAP/TAZ is activated by stress fibers (F-actin) resulting from stiffness changes, and this pathway is independent of large tumor suppressor kinase 1 and 2 (LATS1/2) regulation [70].
Interestingly, the crosstalk between integrin/FAK/myosin/YAP/TAZ has been mathematically modeled and experimentally validated using mouse embryonic fibroblasts 3T3 cells seeded on hydrogels with a gradient stiffness range of 1 to 125 kPa, which mimic physiological and pathological tissue conditions. The evidence shows the complex relationships between cell adhesions and the YAP/TAZ nuclear vs. cytoplasm location, the dependence on the amount of ligand-integrin interaction that reaches a saturation level, and the feedback with the ECM viscoelasticity activation [71].
2.4 Mechanical Forces and MiRNA Regulation
MiRNAs are a class of short, non-coding RNAs, approximately 22 to 25 nucleotides in length. MiRNAs can regulate the expression of numerous genes, and a target gene can be targeted by several miRNAs, creating a complex regulatory pathway. It has been estimated that miRNAs regulate approximately 60% of human genes. They are involved in various biological processes, including cell cycle control, apoptosis, metabolism, development, and differentiation. They are also involved in many diseases, such as neurodegenerative and metabolic disorders, and cancer [72]. MiRNAs can have promoter and terminator sequences [73]; however, some miRNAs are located within the introns of other genes and share the same regulatory elements [74,75].
The biogenesis of a miRNA involves a series of stages that occur in the nucleus and cytoplasm. In the nucleus, miRNAs are transcribed by RNA polymerase II enzymes, and then capped, spliced, and polyadenylated. The primary miRNA (pri-miRNA) is formed with one or more hairpin structures. Inside the nucleus, the pri-miRNA is processed by Drosha ribonuclease III (Drosha) and its cofactor DiGeorge syndrome critical region 80 (DGCR8), resulting in a 70- to 100-nt pre-miRNA that is transported to the cytoplasm by Exportin-5 through nuclear pores. Once outside the nucleus, Dicer, another RNase, cleaves the pre-miRNA into double-stranded RNA in the cytoplasm that encloses the miRNA strand and its complementary sequence. After this “clipping,” the helix continuously unwinds this duplex miRNA into a single, short strand of RNA called a mature miRNA, which is incorporated into the RNA-induced silencing complex (RISC) complex containing an Argonaute protein (AGO2). At this point, the miRNA is fully functional, and this miRNA-RISC complex targets the 3′ untranslated region (3′ UTR) of the target mRNA, resulting in mRNA cleavage (if homology is high) or translation inhibition (when homology is not as high, which is normal in mammals). There is another pathway by which miRNAs are also processed: from the intron of the protein-coding gene by the pre-mRNA splicing machinery, a Drosha/DGCR8-independent pathway. The expression of these miRNAs, called miRtrons, correlates with host gene expression based on their location in introns or at splice site junctions. MiRtrons are continuously transported into the cytoplasm and processed by Dicer [72,73,76,77].
The regulation of miRNAs by mechanical stimulus is a relatively novel subject. Some of the first reports showed that cycling stretching of human airway smooth muscle cells (HASMCs) for one h at 1 Hz every 12 h leads to the upregulation of miR-26a, which suppresses the translation of glycogen synthase kinase-3 (GSK-3) mRNA and promotes airway smooth muscle hypertrophy [78]. The miRNAs regulated by mechanical forces have been termed mechanomiRs and have been reported to regulate chondrocyte differentiation in a 3D collagen scaffold cell culture subjected to mechanical forces through uniaxial stretching, which induces 5% elongation at 60 cycles/min and 15 min/h, promoting chondrocyte hypertrophy and differentiation. Stretching induces the upregulation of miR-365 expression, which targets the 3′ UTR of histone deacetylase 4 (HDAC4) mRNA and suppresses its protein levels [79]. Moreover, it has been shown that mechanical stretching with a passive tension of ~0.4 N/cm for 15 min of ex-vivo diaphragm muscle of normal or muscular dystrophies mice models, induce an opposite regulation of miRNAs expression, particularly of let-7e-5p that target ECM genes Col1a1, Col1a2, Col3a1, Col24a1, Col127a1, Itga1, Itga4, Scd1, and Thbs1, involved in muscle fibrosis [80]. Additionally, it has been reported that unidirectional shear stress of 15 dynes/cm2 for 24 h upregulates miR-19a and miR-21; the former suppresses cyclin D1 protein expression in endothelial cells, and the latter downregulates phosphatase and tensin homolog (PTEN), thereby decreasing apoptosis and activating the nitric oxide pathway [81]. MiRNAs can also regulate genes related to the expression of the cell mechanosensing machinery in response to mechanical forces. In human endothelial cells cultured on high substrate stiffness (30 kPa), the expression of miRNAs related to mRNAs encoding actin- and microtubule-associated proteins, focal adhesion proteins, ECM proteins, and functionally related regulatory proteins increases. The lack of Drosha or AGO2 in endothelial cells and epidermal fibroblasts cultured in soft substrates (3 kPa) induces enhanced F-actin, cell contractility, and adhesion; moreover, zebrafish mutants in AGO2 have increased tissue stiffness, contractility, extracellular matrix deposition, and an impaired wound healing response [82]; suggesting that miRNAs have a buffer effect to prevent excessive contractility and maintain homeostasis of mechanical properties of the tissues [82,83].
3 Effect of Mechanical Forces in Cancer Development
The most recent definition of cancer states, “Cancer is a disease of uncontrolled proliferation by transformed cells subject to evolution by natural selection” [84]. Cancer progression is driven by genetic and epigenetic alterations that disrupt the regulation of cell proliferation, primarily through the activation of oncogenes [3] and the downregulation of tumor-suppressive microRNAs, which act as post-transcriptional regulators [85]. The imbalance between cell division and apoptosis favors tumor formation, which may eventually progress to metastasis (dissemination from the primary tumor to surrounding or distant tissues), which remains the primary cause of cancer-related mortality [86].
During cancer development, cells are exposed to mechanical stimuli that influence tumor initiation, progression, and metastasis. The impact of mechanical forces in this process can be analyzed from two perspectives: external and internal mechanical changes, as well as cellular responses. External changes include alterations in microarchitecture, referring to modifications in ECM topography; increased stiffness, resulting from abnormal ECM proteins deposition, such as collagen and fibronectin; and changes in fluid pressure, where the formation of hyperpermeable blood vessels and the compression of existing blood and lymphatic vessels lead to increased interstitial fluid pressure in peripheral tumor cells, while pressure remains relatively stable within the tumor [4,87].
On the other hand, cells perceive internal mechanical forces, like shear flow force, which arises as cells enter the bloodstream or interstitial fluid; contractile forces, generated by actomyosin cytoskeletal tension in response to the dense, cross-linked fibers of the ECM and its increased stiffness, leading to cellular stress as they pull and push within this network [88]. Compressive forces, induced by tumor expansion due to uncontrolled proliferation in a confined environment, result in intracellular compression that can modify the expression of autocrine and paracrine signaling molecules [55].
Recent studies have highlighted another mechanical property relevant to mechanotransduction and cancer development or progression: viscosity. An increase in extracellular viscosity, experimentally induced using methylcellulose or Ficoll to reach physiological ranges up to ~20 cP, can alter mechanotransduction signaling by activating the calcium ion channel TRPV4, driven by increased actin-related protein 2/3 complex (ARP2/3) activity. This, in turn, activates the RhoA/Hippo pathway, evidenced by increased phosphorylated myosin light chain 2 (pMLC2) levels and RhoA-GTP activity. These events lead to cytoskeletal remodeling, cellular contractility, and migration in breast cancer cells [89] (Fig. 1). Furthermore, high extracellular viscosity and viscoelasticity, modeled in vitro using hydrogels with tunable stiffness mimicking fibrotic liver ECM, has been shown to activate the integrin β1/tensin signaling pathway, inducing nuclear YAP translocation and thereby enhancing hepatocellular carcinoma (HCC) cell proliferation in colony formation assays and HCC progression in mouse models of pre-cirrhotic liver fibrosis [90]. At the same time, intracellular viscosity has emerged as a potential mechanical biomarker for metastatic potential. MDA-MB-231 breast cancer cells, which have high metastatic potential, exhibited lower cytoplasmic viscosity, measured using magnetic rotational spectroscopy (MRS), compared to MCF-7 cells, which were fivefold more viscous. This suggests that lower cytoplasmic viscosity may be linked to increased metastatic potential, as a less viscous cytoplasm could facilitate cell deformability, migration, and invasion [91].
During a tumorigenic process, cellular changes trigger the transformation, growth, and dissemination of cancer cells. ECM surrounding the tumor increases in stiffness and elasticity due to matrix deposition and reorganization, particularly by collagen deposition. This process, known as desmoplasia (excessive production of connective tissue), induces a fibrotic state that promotes tumor progression and is associated with a poor prognosis [92]. The fibrotic microenvironment contributes to the establishment and modification of cell-ECM interactions, which in turn promote signaling for the recruitment of other cells such as macrophages, lymphocytes, neutrophils, mesenchymal stem cells (MSCs), and cancer-associated fibroblasts (CAFs). These cells, in turn, produce additional ECM components, such as collagen I and IV, fibronectin, laminin, and proteoglycans, and growth factors like transforming growth factor beta (TGFβ), VEGF, epidermal growth factor (EGF), fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), and hepatocyte growth factor (HGF), that drive tumor hypervascularization and the loss of ECM elasticity, thereby facilitating subsequent tumor cell migration, invasion, and ultimately metastasis [93].
Specifically, the increase in tumor microenvironment (TME) stiffness results in changes in the patterns of protein expression and transcription factor activity. Enhanced extracellular signal-regulated kinase (ERK) signaling, which translocates to the nucleus upon activation to regulate transcription factor activity and the expression of mitogenic genes related to vascularization, such as VEGF, promotes proliferation and inhibits apoptosis by blocking p53 expression and ECM degradation through the secretion of matrix metalloproteinase 2 (MMP2) and metalloproteinase 9 (MMP9) (Fig. 1). These processes contribute to tumor invasion and metastasis [94]. In addition, increased substrate stiffness has been shown, in endothelial cells, to promote VEGF function through the PI3K/protein kinase B (Akt)/mechanistic target of rapamycin (mTOR) signaling pathway, enhancing angiogenic and increased survival responses [95] (Fig. 1), a mechanism relevant in cancer, where high collagen density substrate enhances mTOR dependent cancer stem cell metastasis in Erα+ breast tumors [96].
Additionally, ECM stiffness increases the expression of vinculin, a key regulator of cell adhesion, and of signal transducer and activator of transcription 3 (STAT3), a transcription factor that, upon phosphorylation, translocates to the nucleus to promote the overexpression of cyclin D1, c-Myc, and Bcl-2, which inhibit apoptosis. Stiffness also enhances the expression of tropomyosin 3 (TPM3), which stabilizes actin filaments, promoting cellular contractility and migration. Moreover, it induces the activation of the YAP/TAZ complex, which mediates the acquisition of stem-like characteristics during the epithelial-mesenchymal transition (EMT) [97–99].
These changes provide tumor cells with significant advantages for proliferation, migration, and metastasis; however, cancer cells may respond differently based on their genetic heterogeneity and the stage of tumor progression. For example, in a patient-derived organoid (PDO) model of colorectal cancer, cancer stem cells (CSCs) that express the leucine-rich repeat-containing G-protein coupled receptor 5 (LGR5) showed lower expression of ezrin, radixin, and moesin adaptative proteins (ERM), exhibiting more adhesiveness and enabling them to adhere to blood vessel walls and invade other tissues, thus establish metastatic sites. In contrast, LGR5-negative cells were softer, less adhesive, and moved faster, facilitating their detachment from the primary tumor [100]. Additionally, in spheroids of malignant (MDA-MB-231) and non-tumoral (MCF10A) breast cells under mechanical compression, malignant cells increased motility and invaded the collagen matrix, while non-tumor cells reduced motility with no invasion; this response may be mediated by the integrin/FAK/myosin/YAP/TAZ axis, a signaling pathway previously implicated in mechanotransduction and TME stiffness responses [101]. A mathematical model suggests that cellular heterogeneity may result from slow transcriptional regulation, where cells remain in intermediate gene expression states, similar to what occurs with epigenetic processes such as DNA methylation and histone modification. These intermediate states may be linked to mechanical forces, including intracellular viscosity and nuclear stiffness. This phenotypic plasticity may help cancer cells adapt to the TME in vivo [102].
3.1 MiRNAs in Cancer Mechanotransduction
Recent evidence suggests that miRNAs play a crucial role in regulating mechanosensing pathways. While their involvement in cancer has been widely studied [103], their specific role in mechanotransduction and their response to the mechanical properties and forces in the TME is an emerging field of research. Experimental evidence suggests that particular miRNAs are aberrantly expressed in various cancers, where they suppress the expression of genes involved in cell growth, angiogenesis, and metastasis. The same miRNA can either suppress or promote cell proliferation and invasion, leading to malignancy in different cells and cancer types [104]. Canonically, miRNAs control mRNA expression and can target non-coding RNAs, including long non-coding RNAs and miRNAs. The latter, called miRNA:miRNA interaction, constitutes a form of autoregulation. Some miRNAs act as oncogenes when overexpressed, such as miR-21, which inhibits tumor suppressor genes. Others, such as miR-34, function as suppressors, and their loss promotes tumor progression. In addition to regulating genes in tumor cells, miRNAs also affect the microenvironment, promoting angiogenesis, invasion, and metastasis [105,106].
To identify miRNA-mRNA regulatory interactions in the context of cancer and mechanical forces, the studies typically require both computational and experimental approaches. Several of the studies reviewed here relied on tools like miRNet, miRDB, TargetScan, TargetMiner, DIANA, miRanda, and PicTar [107–109] to forecast miRNA binding sites based on how well the miRNA seed region aligns with target sequences and whether those sites are conserved across species. However, since this in silico prediction alone can yield a high false-positive rate, most studies followed up with experimental validation. To confirm direct interactions between miRNAs and the 3′ UTRs of their target miRNAs, dual-luciferase reporter assays are commonly used. Likewise, these assays are often combined with qRT-PCR and western blotting to evaluate the regulatory effect at transcript and protein levels, and, in certain instances, this interaction has been experimentally validated using animal models [110–112]. This integration of bioinformatic predictions with experimental data strengthens the evidence for specific miRNAs acting as post-transcriptional regulators within signaling pathways related to mechanical forces in cancer.
MiRNAs regulate the ECM by directly targeting mRNAs encoding ECM proteins or indirectly modulating genes involved in the synthesis and degradation of ECM proteins. ECM stiffness, in turn, regulates miRNAs, creating a regulatory feedback loop [82,113,114]. For example, miR-21 is overexpressed in several tumors, promoting cancer progression [115,116]. Within the TME, miR-21 functions as a mechanical responder; increased stiffness induces its expression, which is associated with cytoskeletal reorganization through elevated TGFβ1 activation, thereby enhancing cellular contractility [117–120]. This process influences tumor cell adaptation, migration, and invasion of surrounding tissues through the PTEN pathway. Similarly, ECM stiffening upregulates miR-17-5p expression in HCC cells, which downregulates PTEN. This reduction leads to the activation of PI3K/Akt/MMP2 and MMP9 through increased Akt phosphorylation, promoting cell survival, proliferation, and motility [113]. MiR-30a is downregulated in anaplastic thyroid cancer and regulates cellular invasion, migration, and metastasis. Bioinformatic analysis and luciferase reporter assays have confirmed lysyl oxidase (LOX) as a direct target of miR-30a. Overexpression of LOX in human thyroid carcinoma cell lines, as well as in xenograft mouse model, has been associated with a reduction in metastatic lesions [111]. LOX is an ECM-remodeling enzyme, responsible for fiber cross-linking, and its upregulation has been linked to increased metastasis and decreased survival outcomes in patients with breast cancer and head and neck squamous cell carcinoma [121].
Recent studies on MSCs have shown that substrate stiffness influences the expression of miR-99b and miR-140. As stiffness decreases, so do their levels, and this downregulation persists over time. Stiff ECM environments reduce mTOR expression, a pathway that regulates MSC differentiation. Increased miR-99b expression enhances RhoA activity, a key regulator of cytoskeletal dynamics and contractility [20,122]. Regarding expression of miR-99b, an increase of this miRNA expression has been found in exosomes of LNCaP prostate cancer cells cultured over stiff substrates [123]. Similarly, increased substrate stiffness sensed through integrin-mediated FA and cytoskeletal tension via the RhoA/ROCK axis triggers osteogenic transducers. In this context, miR-100-5p and miR-143-3p act as post-transcriptional repressors of key components of the mTORC1 complex. miR-100-5p directly targets mTOR mRNA, while miR-143-3p suppresses regulatory-associated protein of mTOR (RPTOR), thereby reducing downstream phosphorylation of S6K1, a canonical marker of mTORC1 activity. This repression of mTOR signaling in response to increased stiffness promotes osteogenic differentiation while inhibiting adipogenic lineage commitment. miR-100 has been shown to promote osteogenic differentiation of MSCs in soft 3D polyethylene glycol hydrogels by repressing mTOR expression and downstream signaling, indicating a mechanosensitive role of this miRNA [124]. This mechanism is relevant not only in normal cells but also in cancer cells, where it can facilitate tumor migration, invasion, and metastasis.
Other mechanoresponsive miRNAs, such as miR-146a and miR-32-5p, have been indirectly implicated in mechanotransduction pathways. MiR-146a acts as a negative regulator of inflammation by suppressing nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling in response to pressure-induced mechanical stress in small airway epithelial cells. At the same time, miR-32-5p promotes vascular smooth muscle cell calcification by enhancing tumor necrosis factor alpha (TNF-α) expression, thus indirectly activating NF-κB and promoting osteogenic gene expression that contributes to pathological microenvironment remodeling [107,125]. Given the crucial role of inflammation and ECM stiffening in tumor progression, these miRNAs may serve as modulators in the TME, suggesting potential therapeutic value by either restoring homeostatic regulation or targeting miRNA-mediated cancer mechanotransduction. In the same way, the transcription factor hypoxia-inducible factor 1 alpha (HIF-1α), which is activated by hypoxia in the TME, regulates the expression of genes that enable the tumor to adapt to low oxygen conditions. The activation of HIF-1α induced the expression of tenascin-C (TNC), increasing ECM stiffness and creating a more aggressive tumor phenotype. In the HIF–1α–TNC feedback loop, it has been described that miR-203 is downregulated, thereby favoring glioblastoma progression, likely due to increased hypoxic adaptation and enhanced ECM remodeling [126].
In ovarian cancer, shear stress influences chemoresistance by downregulating miR-199a-3p. This regulation does not occur post-transcriptionally via the Dicer/Drosha/Exportin 5 machinery, but instead through the repression of the MIR199A1 gene transcription, which is located on chromosome 19. Shear stress activates the c-Met tyrosine kinase receptor, which stimulates the PI3K/Akt signaling pathway, leading to reduced miR-199a-3p expression through a positive feedback loop, ultimately promoting cell survival and resistance to paclitaxel and cisplatin [127].
Below, we will address some evidence regarding the role of mechanical forces in miRNA regulation and their impact on the expression or suppression of specific proteins relevant for cancer development and progression.
3.1.1 MiRNAs Regulation of Integrins by Mechanical Forces
As previously discussed, integrins mediate ECM-derived mechanical signals to regulate cellular function [13]. Extracellular and intracellular signals activate integrin, facilitate nucleation and clustering, and drive their maturation into focal adhesions [18]. Increased ECM stiffness, a hallmark of tumors, promotes integrin clustering [17], and altered integrin expression in stiffer tumors contributes to malignant phenotypes through strength-dependent regulation of integrin activity or adhesion [128,129].
In HCC, stiff ECM environments increase integrin β1 expression, activating the PI3K/Akt pathway, thus elevating VEGF levels, which correlate with poor prognosis [130]. Stiff substrates also upregulate miR-17-5p, negatively affecting the PTEN/PI3K/Akt signaling pathway via integrin β1. This mechanism significantly attenuates the inhibitory effect of metformin on HCC invasion and metastasis [113].
Other miRNAs modulate integrin expression. For instance, miR-29b inhibits collagen I and II expression while reducing integrin β1 levels in hepatic stellate cells, preventing their transformation into myofibroblast-like cells [131]. MiR-124 is downregulated in glioblastoma patients, while integrin β1 is overexpressed. Restoring miR-124 in glioblastoma cells reduces integrin β1 expression, suppressing tumor migration and invasion [132]. Similarly, in oral squamous cell carcinoma (OSCC), miR-124 overexpression decreases integrin β1 expression, reducing cell adhesion and motility by directly targeting the 3′ UTR of the integrin β1 mRNA, leading to diminished integrin β1 protein levels and subsequent impairment of cell-ECM interactions [133]. MiR-124 is considered a tumor suppressor; it is frequently decreased in several cancer types, including breast, prostate, ovarian, liver, and bladder cancer. This reduction is associated with increased cell proliferation, tumor migration, and invasion, as well as decreased overall survival in patients [134]. Recently, by using a bioengineered pre-miR-124, it has been shown in human lung carcinoma and osteosarcoma cells, the disruption of adherent junctions, focal adhesion plaques, and a decrease of plectin; leading to the downregulation of several proteins such as integrin β1, vimentin, talin1, IQ motif containing GTPase-activating protein 1 (IQGAP1), cadherin 2 or N-cadherin (CDH2), and junctional adhesion molecule A. The bioengineered pre-miR-124 also reduced cell proliferation and lung metastasis in a mouse model without adverse effects [135]. Although there is no direct evidence of mechanical forces regulating miR-124 in cancer cells, it has been reported that miR-124 was upregulated in glomerular podocytes under mechanical stretch for 24 h, and by luciferase assays, it has been shown that miR-124 targets and downregulates integrin α3 and therefore podocyte cell adhesion [110,136].
MiR-31 regulates genes involved in cancer development, acting as either a tumor suppressor or promoter [137]. MiR-31 acts as a metastatic suppressor by inhibiting the expression of multiple integrins, especially those that are part of the complex with integrin β1, essential in cell adhesion, these effects were observed mainly in triple-negative breast cancer and prostate cancer, luciferase assays demonstrate that miR-31 specifically targets and represses several integrin subunits simultaneously (α2, α5, αV, and β3), and indirectly β1 subunits, reducing cell adhesion, invasion, and metastasis [138]. Its expression is downregulated in breast, brain, and ovarian cancers [139]. In resected meningioma tissues from patients, miR-31-5p is significantly upregulated in stiffer tumors compared to their softer counterparts. Notably, miR-31-5p demonstrated a strong association with tumor stiffness, exhibiting a 71% and 83% sensitivity and specificity, respectively, in distinguishing high-stiffness tumors. Therefore, miR-31-5p may serve as a valuable biomarker for assessing tumor consistency and be useful for preoperative planning [140].
3.1.2 MiRNAs Regulation of Vascular Endothelial Growth Factor (VEGF) by Mechanical Forces
Angiogenesis is a key process in the malignant transformation, spread, and metastasis of tumors, influenced by mechanical factors such as ECM stiffness. Increased angiogenesis facilitates metastasis by expanding endothelial surfaces, allowing for greater tumor cell dissemination through the circulation and promoting ECM degradation through the action of proteases and growth factors. This process is tightly regulated by a balance between angiogenic stimulators and inhibitors [141–143].
VEGF, a principal angiogenic factor, binds to vascular endothelial growth factor receptor 2 (VEGFR-2), triggering tyrosine kinase signaling cascades that enhance vascular permeability, proliferation, survival, and migration [144]. Several factors regulate VEGF expression, including oncogenic mutations, cytokines, nitric oxide, and mitogen-activated protein kinase (MAPK) signaling [130], along with microenvironmental conditions such as hypoxia and acidosis [145,146]. ECM stiffness increases VEGFR-2 receptor internalization in endothelial cells, enhancing downstream signaling, including ERK1/2 phosphorylation and cell proliferation. This effect is mediated by actin cytoskeletal contractility, regulated by the RhoA/ROCK pathway [147].
Overexpression of miR-21 has been shown to induce tumor angiogenesis by targeting PTEN, which activates Akt/ERK signaling, ultimately enhancing HIF-1α and VEGF expression in DU145 and PC-3 prostate cancer cells transfected with miR-21 mimics, where increased angiogenic potential was confirmed [148]. This miRNA is also overexpressed in other cancers, such as breast, colon, lung, pancreatic, and stomach cancers [149]. Interestingly, a more recent study demonstrated that MSCs cultured on stiff polyacrylamide hydrogels (~20 kPa), coated with collagen I, exhibited increased miRNA-21 expression, contrary to cells on soft substrates (~1 kPa). With the use of stiffness-tunable matrices and qRT-PCR, it was shown that miR-21 mediates a mechanical memory effect that supports fibrotic phenotype even after removal from stiff environments. In addition, VEFG was identified among the target genes modulated by miR-21, suggesting a potential role in linking mechanical forces to angiogenic signaling [150]. Furthermore, miR-9 downregulates E-cadherin, activating β-catenin signaling, which enhances VEGF-A expression and promotes tumor angiogenesis. This was demonstrated in human breast cancer cell lines, MCF-7 and MDA-MB-231, where luciferase reporter assays confirmed that miR-9 directly targets CDH1. In vivo mouse model assays, overexpression or miR-9, promoted metastasis. At the same time, its inhibition reduced metastatic dissemination, linking miR-9 to MYC/MYCN signaling and more aggressive tumor behavior [151]. In breast cancer, mechanical compression in confined tissues downregulates miR-9 via DNA (cytosine-5)-methyltransferase 3 alpha (DNMT3A)-dependent promoter methylation; this was demonstrated by applying three dimensional static compression using a compression device with a piston system to MDA-MB-231 and BT-474 breast cancer cell lines, as well as in CAFs embedded in collagen matrices. This approach tried to mimicking the physical compression environment that surrounds cells in solid tumors; thus, miR-9 downregulation resulted in the overexpression of its target genes AMC2, ITGA6, and VEGFA, thereby enhancing tumor invasiveness and enhancing angiogenic potential [152].
3.1.3 MiRNAs Regulation of Neuropilin-1 Receptor by Mechanical Forces
Neuropilin 1 (NRP1) is a transmembrane glycoprotein that acts as a co-receptor for several extracellular ligands such as semaphorins, specific isoforms of VEGF, TGFβ, among others [153,154]. NRP1 also mediates cell adhesion to the extracellular matrix by acting as an integrin co-receptor [155,156]. In human dermal endothelial cells, mechanical compression (30 mmHg) increases NRP1 expression, indicating that mechanical forces can induce NRP1 expression [157].
Previous studies have reported that NRP1 promotes mechanical changes in the ECM that favor cancer progression by increased fibronectin fibril assembly through the glycosylation of serine 612 of the extracellular domain of NRP1 and intracellular associations between NRP1, the scaffolding protein GAIP-interacting protein, C-terminus (GIPC), and the non-receptor cellular Abelson tyrosine kinase (c-Abl) which increase fibronectin fibril assembly activity of integrin α5β1 [158]. In cancer, NRP1 is associated with tumor progression by inhibiting apoptosis, promoting angiogenesis, and increasing cell survival through interactions with VEGF [154] (Fig. 1).
Several miRNAs target NRP1 across different cancers. MiR-9-5p acts as both a promoter and suppressor in various cancers, and its overexpression in breast cancer, osteosarcoma, lung cancer, prostate cancer, and colorectal cancer promotes metastasis [159]. MiR-141-3p is associated with poor prognosis in cervical, colon, and prostate cancers [160–162]. In pancreatic cancer tissues and cell lines PANC-1 and BxPC-3, miR-141-3p acts as a tumor suppressor by targeting NRP1, while ectopic overexpression decreases NRP1 levels, thereby inhibiting cell proliferation and migration. Functional assays in vitro and xenograft models confirmed that the miR-141-3p/NRP1 axis plays a vital role in suppressing tumor growth [163].
Furthermore, miR-152-3p is downregulated in non-small cell lung cancer (NSCLC), leading to the upregulation of NRP1 mRNA expression and promoting EMT through the activation of the PI3K/Akt and TGFβ/Sma and Mad related protein (SMAD) signaling pathways [164]. Recent research has demonstrated its involvement in prostate cancer progression, where it negatively regulates Kruppel-like factor 4 (KLF4) in synergy with miR-148-3p, although it does not directly target NRP1 [165]. Similarly, in glioblastoma, miR-148-3p exerts an antitumor function through the modulation of DNA methyltransferase 1 (DNMT1), highlighting the context-specific role of these miRNAs in tumor progression [166].
Recently, a study using a polyacrylamide hydrogel model reported that increased substrate stiffness, comparable to that of prostate tumors (~80 kPa), upregulates NRP1 expression in non-cancerous prostate epithelial cells. This upregulation is associated with enhanced cell protrusion, suggesting a relevant link between substrate stiffness, NRP1 expression, and cancer progression [167]. Interestingly, NRP1 has been identified as one of the several cytoskeletal-actin-ECM-related proteins transcriptionally regulated by stiffness via miRNA expression. On soft substrate (3 kPa), the increase of NRP1 regulatory miRNAs suppressed NRP1 expression, whereas on stiffer substrate (30 kPa), this suppression is diminished [82]. However, despite these findings, the relationship between mechanical forces and miRNAs that regulate NRP1 remains unclear.
3.1.4 MiRNAs Regulation of YAP/TAZ by Mechanical Forces
YAP/TAZ overexpression is common in several cancers, regulating genes involved in the cell cycle, including cyclins, mitotic kinases, and those involved in DNA replication and repair [168–171]. The Hippo pathway regulates YAP activity based on cell density and ECM interactions. At low cell density, Hippo signaling is suppressed, allowing nuclear YAP translocation, thus promoting cell proliferation. Increased cell-cell contacts induce YAP phosphorylation, leading to its cytoplasmic retention via adherent junction proteins such as E-cadherin [172]. Additionally, YAP is activated in ECM-attached cells with actin polymerization but is inactivated in detached cells [70]. YAP also modulates miRNA biogenesis. At low cell density, nuclear YAP binds and sequesters p72, a microprocessor complex cofactor, reducing miRNA processing. At high density, Hippo signaling releases p72 and allows its association with the Drosha/DGCR8 heterodimer, enhancing miRNAs biogenesis. In tumors, inactivation of the Hippo pathway suppresses miRNA production, linking contact inhibition to miRNA regulation [173,174].
A recent study revealed that protein Ran-binding protein 1 (RANBP1) plays a key role in colorectal cancer (CRC) progression by regulating the nuclear export of pre-miRNA, creating a positive feedback loop with YAP. YAP enhances RANBP1 expression, facilitating the export of pre-miRNAs like miR-18a, miR-183, and miR-106 to the cytoplasm. This dysregulation of miRNAs promotes tumor growth and invasion [175]. These findings underscore the interplay between mechanotransduction, miRNA biogenesis, and the Hippo/YAP signaling pathway in the context of tumor progression. Additionally, recent findings suggest that exosomes derived from human bladder mesenchymal stromal cells (hBSC) carry miR-217, which modulates bladder cancer-cell survival through the Hippo/YAP pathway. MiR-217 has been shown to promote bladder cancer cell proliferation and migration by activating the YAP-mediated signaling pathway. Specifically, exosomal miR-217 derived from bladder mesenchymal stromal cells downregulates LATS1, and this suppression leads to increased nuclear localization of YAP, which in turn, enhances transcriptional activation of connective tissue growth factor (CTGF) and cysteine-rich angiogenic inducer 61 (CYR61) [176].
Supporting the regulatory role of miRNAs over YAP, it has been shown that miR-582-3p expression mitigates osteoarthritis progression by directly targeting YAP1; in response, chondrocyte apoptosis, inflammation, and ECM degradation diminished [177]. Although this study does not explicitly explore cancer pathologies, it highlights a broader mechanism by which miRNAs regulate YAP signaling in response to mechanical forces and how these mechanisms may be relevant in tumor contexts where ECM stiffening and altered biomechanics drive YAP activation, ultimately leading to cancer progression. Despite this, the relationship between mechanical forces and their specific role in modulating miRNA biogenesis and YAP signaling in the context of cancer progression requires further investigation.
3.1.5 MiRNAs Regulation of Piezo by Mechanical Forces
Piezo are evolutionarily conserved mechanosensitive ion channels [178], serving as primary mechanosensors, converting mechanical forces into electrochemical signals [179]. Piezo1 has also been implicated in osteoarthritis under conditions of increased mechanical stress, where its activation upregulates miR-155-5p, which represses GDF6 (Growth Differentiation Factor 6 or BMP13) expression and attenuates SMAD2/3 signaling, leading to chondrocyte senescence and ECM degradation, thus favoring the degenerative pathology [180].
In cancer, increased tumor tissue stiffness further enhances Piezo1 expression, which in turn increases the mechanosensory and mechanotransduction capacity of tumor cells. These processes form a reciprocal feedback loop between tumor cell mechanotransduction and aberrant tissue mechanics in gliomas and bladder fibrosis, thus promoting malignancy [35]. Moreover, it can activate YAP1 and its downstream effector CTGF in response to NF-κB-induced expression by H. pylori in gastric tissues, thereby promoting CAF activation and cancer progression in a positive feedback loop [181]. In HCC, an increase in ECM stiffness activates Piezo1, facilitating calcium influx, which stabilizes HIF-1α by inhibiting its ubiquitination and induces the transcription of VEGF and CXC chemokine ligand 16 (CXCL16), while downregulating miR-625-5p, a negative regulator of Piezo1 [182]. In breast cancer, bioinformatic research has shown that the downregulation of miR-10 b-5p is linked to increased Piezo1 expression. The upregulation of Piezo1 is associated with altered purine metabolism, involving key genes such as GUK1, POLD1, and APRT, which may contribute to cancer cell survival [108]. Further studies are required to fully understand how these miRNA-Piezo pathways are integrated across different cancer types and how they could be modulated by mechanotransduction to favor cancer progression.
Recent studies have shown that the mechanosensitive ion channel Piezo2 is the primary mechanosensor in several specialized mechanosensory cells, including proprioceptive neurons [25] and dorsal root ganglion neurons that react to touch [183], as well as Merkel cells in the skin [184]. As well as Piezo1, Piezo2 is a transmembrane protein that forms a cation-selective, mechanosensitive ion channel [179]. Several studies have reported that Piezo2 deregulation is associated with cancer proliferation, angiogenesis, and resistance to anticancer treatments [185–187]. In breast cancer, Piezo2 expression is downregulated due to the post-transcriptional regulation mediated by miRNAs such as miR-130b-3p, miR-196a-3p, miR-301a-3p, miR-421, and miR-454-3p. This downregulation is associated with the activation of the Hedgehog (Hh) signaling pathway, specifically affecting cell adhesion molecule-related/downregulated by oncogenes (CDON), a molecule related to cell adhesion whose reduction is correlated with a poor prognosis [42].
Piezo2 knockdown in tumor models resulted in reduced angiogenesis and decreased tumor vascular permeability, suggesting that Piezo2 has an anti-angiogenic role. Additionally, in non-tumoral models, Piezo2 is crucial for the activation of wingless/integrated (Wnt)/β-catenin and VEGF signaling pathways that regulate angiogenesis. The reduction of wingless-type MMTV integration site family member 11 (Wnt11) and VEGF-interleukin 1 beta (IL-1β, caused by Piezo2 knockdown, decreased endothelial cell migration, tube formation, and pathological angiogenesis, implying a potential role in suppressing tumor vascularization [187]. Moreover, evidence suggests that components of the Hh pathway, such as glioma-associated oncogene homolog 1 (GLI1), can cross-regulate the Wnt/β-catenin pathway by sharing regulatory nodes like glycogen synthase kinase 3 beta (GSK3β, casein kinase 1 alpha (CK1α, suppressor of fused homolog (Sufu), and downstream targets such as Snail and c-Myc, leading to the regulation of genes involved in EMT and tumor progression [188]. In this context, miRNA-mediated modulation of Piezo2 by mechanical forces may indirectly influence this cross-regulation between Hh-Wnt signaling. However, it is essential to note that, despite the extensive research on Piezo2’s role in tumor biology and its signaling pathways, no miRNAs have been identified to regulate Piezo2 specifically in the context of cancer-related angiogenesis due to mechanical forces.
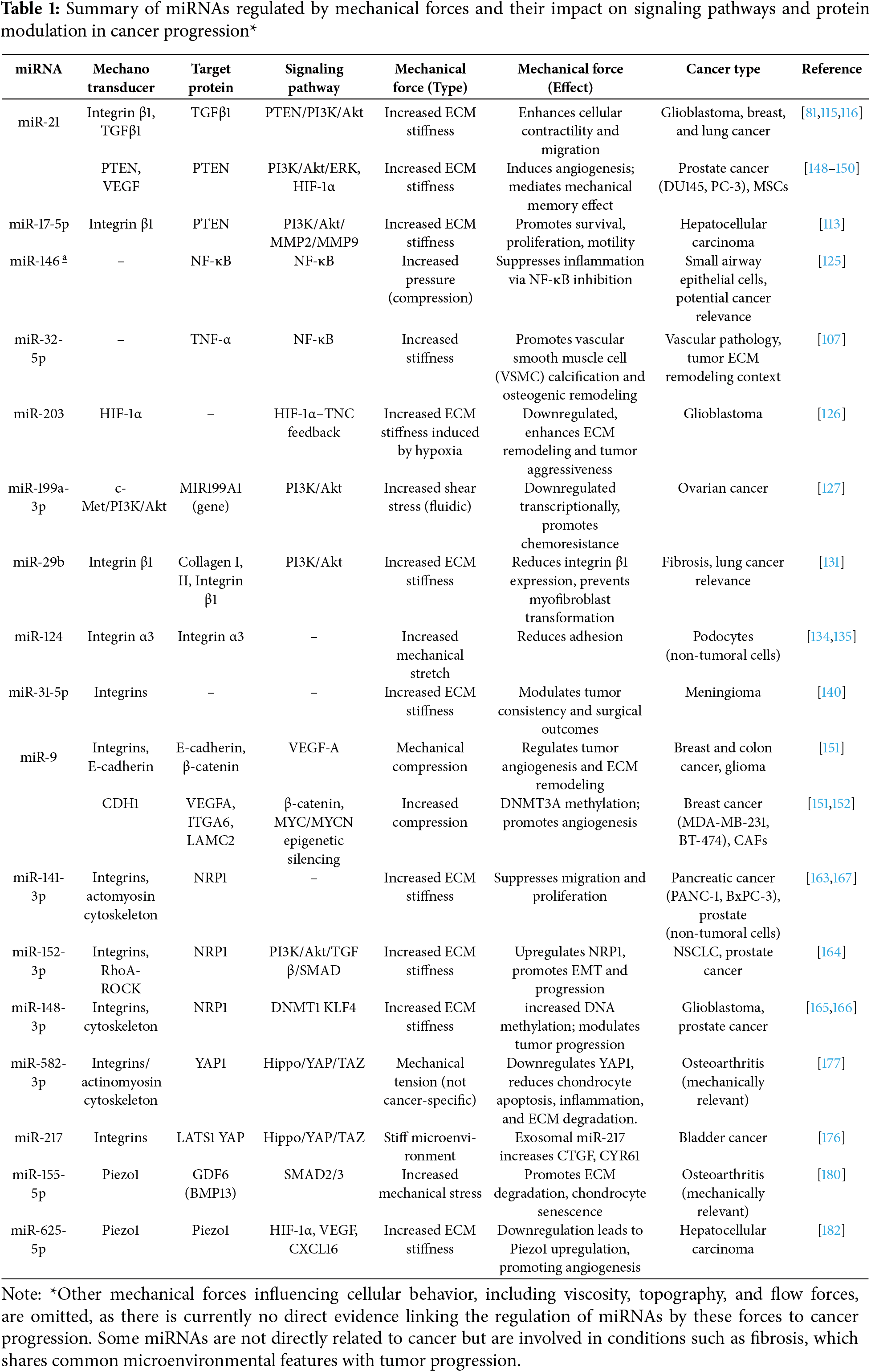
Although various miRNAs have been implicated in cancer, relatively few have been directly linked to regulation by mechanical forces, and even fewer have well-defined signaling pathways associated with these mechanical cues. Some of their downstream effector pathways, which contribute to key tumor-promoting processes including migration, survival, proliferation, angiogenesis, and chemoresistance, are illustrated in Fig. 2. Likewise, Table 1 summarizes miRNAs that are specifically modulated by mechanical forces, such as ECM stiffness, compression, and shear stress. Some of these miRNAs have been directly linked to cancer, while others are supported by indirect or non-cancer evidence but are involved in mechanotransduction pathways relevant to tumor biology.
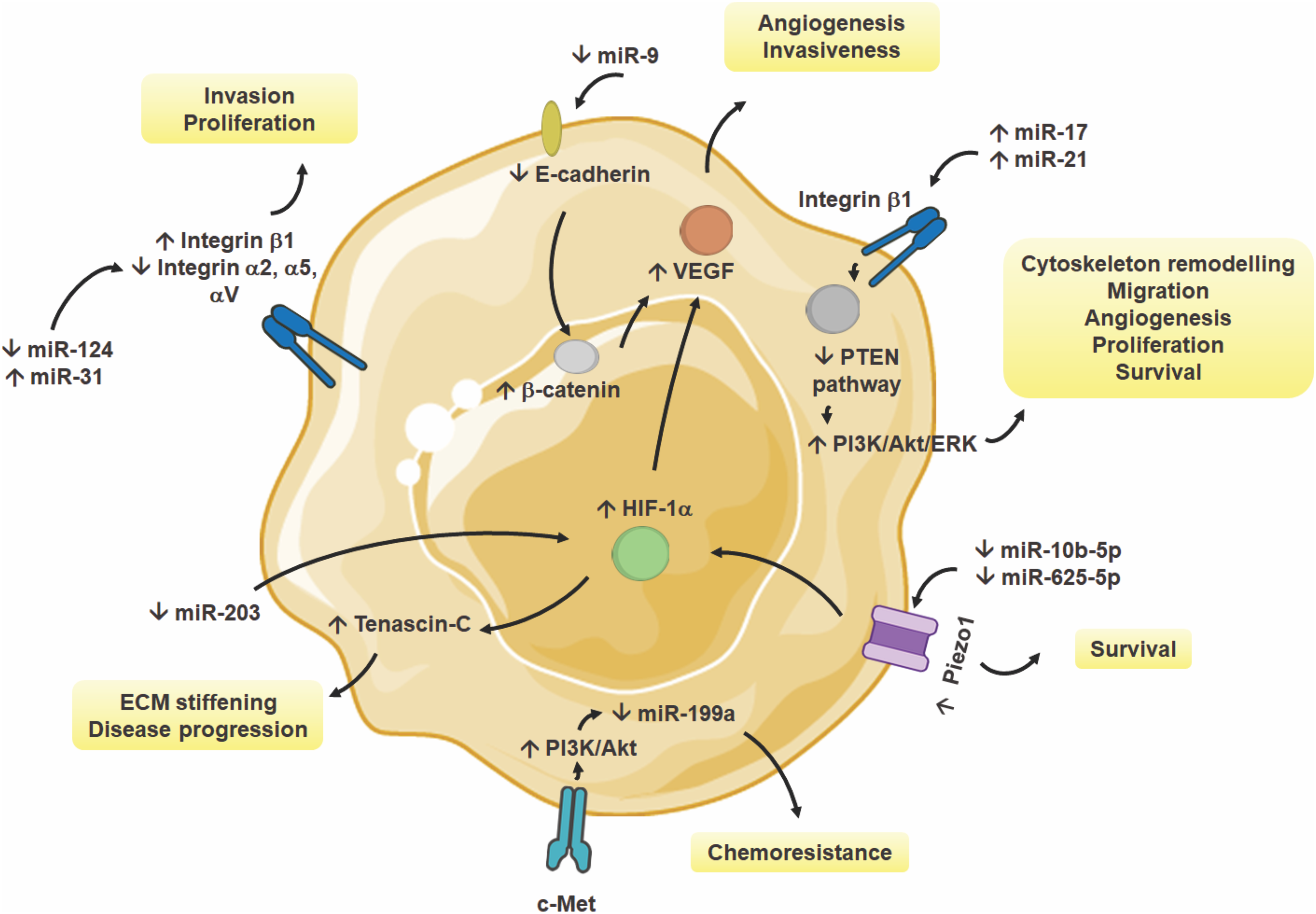
Figure 2: Regulation of miRNAs by mechanical forces in cancer cells. Various miRNAs modulate key signaling pathways, including PTEN, PI3K/Akt, and HIF-1α, in response to mechanical cues such as ECM stiffness, viscosity, solid stress or mechanical compression, and shear stress. Upregulation or downregulation of specific miRNAs influences critical cellular processes, including cytoskeleton remodeling, migration, invasion, angiogenesis, proliferation, and chemoresistance. These mechanosensitive miRNAs contribute to tumor progression by altering integrin signaling, VEGF-mediated angiogenesis, and mechanotransduction through Piezo1 and c-Met. The figure has been produced using an image from Servier Medical Art (http://smart.servier.com/, accessed on 20 March 2025 and vectors from https://bioart.niaid.nih.gov/, accessed on 26 March 2025) [81,108,113,115,116,126,127,134,135,138,139,151,182]
4 Clinical Significance, Translational Prospects, and Associated Challenges
Changes in the mechanical properties of tissues during the formation of solid tumors are linked to increased tumorigenesis and invasiveness, influencing factors such as cell proliferation, motility, and invasion. Research into how cells respond to external mechanical forces has highlighted the significance of mechanoresponses in cancer development and progression. Although the underlying mechanisms by which cells respond to and integrate mechanical stimuli remain not fully understood and still inconclusive, this research is gradually offering new insights that could enhance our understanding of, as well as strategies for preventing and treating cancer.
Tissue stiffness holds significant prognostic value in cancer. Non-invasive techniques, such as ultrasound and magnetic resonance elastography, can assess the mechanical properties and structure of tissues, revealing changes in tissue mechanics across various cancer types, including pancreatic, breast, and prostate cancers [189,190]. Several therapy development strategies and clinical trials are focusing on changes in tissue stiffness, such as those resulting from fibrotic tissue formation, which is a critical early step in tumor development. Approaches include the use of inhibitors that target collagen synthesis and crosslinking, as well as agents that affect fibronectin and hyaluronic acid. Additionally, inhibitors of TGFβ1 and its receptor are being explored. Other strategies involve targeting integrins or signaling molecules involved in mechanotransduction, such as FAK and Rho kinase inhibitors [93]. Moreover, modulation of stress and desmoplasia in tumors is a key target of several drugs, such as pirfenidone, metformin, losartan, or tranilast, which also improve sensitivity to chemotherapeutics [191–194].
In this context, miRNAs offer a valuable tool for diagnosis and treatment. Targeting miRNAs involved in mechanotransduction, or those induced by mechanical forces and engaged in TME changes, can be a focused therapeutic strategy to modify the early transformations of the ECM and its mechanical impact on cells, or to use as diagnostic and prognostic tools; however, mechanomiR research and its effects on cancer development and progression are still an emerging field.
For instance, several efforts are being reported, mainly in animal models, such as using an adeno-associated virus serotype two vector (AAV2) to deliver miR-19b and reduce liver fibrosis. In a recent study, miR-19b delivered via AAV2 significantly attenuated liver fibrosis in rats by inhibiting hepatic stellate cell activation and reducing collagen deposition [112]. Additionally, there is interest in targeting mechanically activated miRNAs, or mechanomiRs, such as members of the miR-17-92 cluster and miR-143, which regulate genes associated with ECM components and cellular proliferation in HCC, contributing to invasion and metastasis [195]. Notably, the miR-17-92 cluster is overexpressed in HCC tissues and liver cancer cell lines, and its induced overexpression in a liver-specific transgenic mouse model promotes hepatocarcinogenesis after exposure to diethylnitrosamine (DEN), a hepatocarcinogenic agent [196]. MiR-18a, a member of this cluster, is induced by increased stiffness in 3D culture models of mammary epithelial cells subjected to different matrix stiffness and downregulates the tumor suppressor PTEN by base-pairing with the 3′ UTR of PTEN, leading to PTEN suppression and increased cell proliferation [197]. In addition, screening of miRNAs regulated by changes in stiffness and modulating the cytoskeletal–actin–extracellular matrix (CAM) regulatory network identifies potential therapeutic targets that, although requiring extensive research and validation regarding their role in cancer development, may prove valuable in the future [82]. Although the results obtained in preclinical models indicate promising target, the clinical phase is a challenging step that can lead discrepancies due to the heterogeneity of tumor cells, the variability in responses to mechanical forces of the distinct type of tumors, and the dynamic modifications of ECM and mechanical forces during the different stages of cancer progression. The stage of treatment can impair the use of miRNAs to inhibit LOX and decrease fibrosis. For instance, miRNAs may be applied in the early stages to prevent malignant collagen crosslinking; however, once the tissue becomes fibrotic, with altered collagen fibers and desmoplasia, their effect is no longer relevant [93].
Moreover, the use of miRNAs in cancer treatment has numerous challenges that must be addressed before such approaches can be translated into clinical practice. MiRNAs can regulate multiple genes simultaneously, which raises the risk of undesired effects. An example of this is the miR-34a mimic, MRX34, which was withdrawn from clinical trials in liver cancer due to severe immune-related side effects, including cytokine release syndrome, that tragically led to patient fatalities [198,199]. Moreover, synthetic miRNA molecules can trigger innate immune responses by mimicking pathogen-associated molecular patterns (PAMPs), thus activating pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs), and inducing further immunological side effects [200]. Another significant challenge in developing mRNA therapies is determining the optimal dosage. Individual variability in responses complicates dose standardization, and extrapolating dosages from preclinical models to humans poses ethical concerns, as administering potentially inactive or toxic doses is considered unethical [201].
Efficient delivery of miRNAs to target cells is another major challenge. For miRNAs to exert their therapeutic effect, they must evade degradation in lysosomes and escape from endosomes into the cytoplasm. Current delivery strategies, such as pH-sensitive liposomes and nanoparticle-based carriers, have shown promise but still face significant limitations [199]. For instance, miRNAs chemical modifications, like the introduction of phosphorothioate bonds, are employed to improve their stability and facilitate cellular uptake; however, these modifications may elicit unintended side effects, such as the activation of the complement cascade, cytotoxic effects in immune cells, and a reduction in peripheral white blood cell counts [202]. Despite the difficulties, clinical trials are being carried out, some still ongoing and others completed about the use of miRNAs for cancer treatment and detection, such as miR-16 malignant pleural mesothelioma, non–small cell lung cancer, miR-193a-3p for advanced solid tumors, miR-155 for lymphoma and leukemia, miR-25 for pancreatic cancer detection, miR-10b for prognostic and detection of gliomas [203].
The challenges associated with miRNAs intervention and gaps in the knowledge about the miRNAs regulation by mechanical forces are numerous. Even though the field remains under intense research, further integration and understanding of the intricate mechanism by which cells sense and respond to mechanical forces, as well as how miRNAs act within this biomechanical landscape are still lacking. The advances in the field, however, will undoubtedly provide essential knowledge that should be scaled to the clinic to have effective therapeutic targets for the treatment of cancer patients.
The role of miRNAs in cellular regulation and cancer development is undeniable. However, regulating miRNAs during homeostasis and cancer is a complex task due to their context-dependent activity and impact on various types of tissues. On the other hand, cell responses to mechanical stimuli also involve complex regulatory networks that are still under intense study. In the context of regulating cancer development and progression, understanding the pathways by which mechanical forces can regulate miRNAs and, therefore, promote or inhibit cancer development is undoubtedly a challenge that is beginning to be addressed. As a relatively new aspect to study in the cancer research field, it will be essential to understand the role of mechanical forces in regulating miRNA biogenesis, and also how miRNAs can regulate the complex mechanical forces-response cell machinery, to have more robust evidence of both of these aspects that can lead to a valuable comprehension of the cell biology of cancer. Future efforts should focus on identifying specific mechanosensitive miRNAs that show consistent roles in tumor progression or suppression. Since many complex mechanisms of action remain to be understood and integrated in both fields, the translation of this knowledge to the clinic is still a considerable distance away. Furthermore, tumor heterogeneity and the dynamic alterations within the TME pose significant challenges in designing an optimal therapeutic intervention aimed at modulating mechanical forces that drive cellular malignancy and cancer progression. Additionally, concerns regarding the clinical application of miRNAs remain a critical obstacle that must be addressed. Despite the growing body of evidence highlighting the role of mechanical forces in modulating miRNA and their involvement in malignancy, research directed toward translational and clinical studies remains lacking. A comprehensive understanding of regulatory mechanisms will enable the development of more integrative novel strategies for effective diagnosis and cancer treatment in the future.
Acknowledgement: Not applicable.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design, Elisa Tamariz; data collection, Elisa Tamariz, Ana M. Vela-Alcántara and Diego J. Hernández-Sánchez; analysis and interpretation of results, Elisa Tamariz, Ana M. Vela-Alcántara and Diego J. Hernández-Sánchez; writing—original draft, Elisa Tamariz, Ana M. Vela-Alcántara and Diego J. Hernández-Sánchez; writing—review and editing, Elisa Tamariz and Ana M. Vela-Alcántara; funding acquisition, Elisa Tamariz. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
Nomenclature
| 3′ UTR | 3′ untranslated region |
| ARP2/3 | Actin-related protein 2/3 complex |
| AGO2 | Argonaute protein |
| CDH2 | Cadherin 2 or N-cadherin |
| CSCs | Cancer stem cells |
| CAFs | Cancer-associated fibroblasts |
| CK1α | Casein kinase 1 alpha |
| CTCF | CCCTC-binding factor |
| CDON | Cell adhesion molecule-related/downregulated by oncogenes |
| c-Abl | Cellular Abelson tyrosine kinase |
| CRC | Colorectal cancer |
| CTGF | Connective tissue growth factor |
| CXCL16 | CXC chemokine ligand 16 |
| CYR61 | Cysteine-rich angiogenic inducer 61 |
| CAM | Cytoskeletal-actin-extracellular matrix |
| DEN | Diethylnitrosamine |
| DGCR8 | DiGeorge syndrome critical region 8 |
| DNMT3A | DNA (cytosine-5)-methyltransferase 3 alpha |
| DNMT1 | DNA methyltransferase 1 |
| Drosha | Drosha ribonuclease III |
| ESCRT-III | Endosomal sorting complex required for transport III |
| EGF | Epidermal growth factor |
| EMT | Epithelial-mesenchymal transition |
| ECM | Extracellular matrix |
| ERK | Extracellular signal-regulated kinase |
| FGF | Fibroblast growth factor |
| FAK | Focal adhesion kinase |
| FA | Focal adhesions |
| GIPC | GAIP-interacting protein, C-terminus |
| GLI1 | Glioma-associated oncogene homolog 1 |
| GSK-3 | Glycogen synthase kinase 3 |
| GSK3β | Glycogen synthase kinase 3 beta |
| IQGAP1 | IQ motif containing GTPase-activating protein 1 |
| Hh | Hedgehog |
| HCC | Hepatocellular carcinoma |
| HGF | Hepatocyte growth factor |
| HP1 | Heterochromatin protein 1 |
| HDAC3 | Histone deacetylase 3 |
| HDAC4 | Histone deacetylase 4 |
| HDACs | Histone deacetylases |
| HASMCs | Human airway smooth muscle cells |
| hBSC | Human bladder mesenchymal stromal cells |
| HIF-1α | Hypoxia-inducible factor 1 alpha |
| IL-1β | Interleukin 1 beta |
| KLF4 | Krüppel-like factor 4 |
| LADs | Lamina-associated domains |
| LAP2β | Laminin-associated protein 2β |
| LATS1/2 | Large tumor suppressor kinase 1 and 2 |
| LGR5 | Leucine-rich repeat-containing G-protein coupled receptor 5 |
| LINC | Linker of nucleoskeleton and cytoskeleton complex |
| LOX | Lysyl oxidase |
| MRS | Magnetic rotational spectroscopy |
| MMP2 | Matrix metalloproteinase 2 |
| MMP9 | Matrix metalloproteinase 9 |
| mTOR | Mechanistic target of rapamycin |
| MSCs | Mesenchymal stem cells |
| MAPK | Mitogen-activated protein kinase |
| NRP1 | Neuropilin 1 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| OSCC | Oral squamous cell carcinoma |
| PAMPs | Pathogen-associated molecular patterns |
| PDO | Patient-derived organoid |
| PRRs | Pattern recognition receptors |
| PTEN | Phosphatase and tensin homolog |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate |
| PI3K | Phosphoinositide 3-kinase |
| pMLC2 | Phosphorylated myosin light chain 2 |
| PDGF | Platelet-derived growth factor |
| Akt | Protein kinase B |
| SRC | Proto-oncogene tyrosine-protein kinase Src |
| RANBP1 | Ran-binding protein 1 |
| RhoA | Ras homolog family member A |
| RPTOR | Regulatory-associated protein of mTOR |
| ROCK | Rho-associated coiled-coil containing protein kinase |
| RISC | RNA-induced silencing complex |
| RUNX2 | Runt-related transcription factor 2 |
| SUN | Sad1 and UNC-84 domain-containing proteins |
| SMYD3 | SET and MYND domain containing 3 |
| SMAD | Sma and Mad related protein |
| Sufu | Suppressor of fused homolog |
| TBX5 | T-box transcription factor 5 |
| TNC | Tenascin-C |
| TLRs | Toll-like receptors |
| TAZ | Transcriptional coactivator with PDZ-binding motif |
| TGFβ | Transforming growth factor beta |
| TPM3 | Tropomyosin 3 |
| TME | Tumor microenvironment |
| TNF-α | Tumor necrosis factor alpha |
| VEGF | Vascular endothelial growth factor |
| VEGFR-2 | Vascular endothelial growth factor receptor 2 |
| VSMC | Vascular smooth muscle cell |
| AAV2 | Adeno-associated virus serotype two vector |
| Wnt | Wingless/Integrated |
| Wnt11 | Wingless-type MMTV integration site family member 11 |
| YAP | Yes-associated protein |
References
1. World Health Organization. Top 10 Causes Death 2025 [Internet]. [cited 2025 Mar 11]. Available from: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death. [Google Scholar]
2. Ferlay J, Ervik M, Lam F, Laversanne M, Colombet M, Mery L, et al. Global cancer observatory cancer today 2024 [Internet]. [cited 2025 Mar 11]. Available from: https://gco.iarc.who.int/media/globocan/factsheets/populations/900-world-fact-sheet.pdf. [Google Scholar]
3. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12(1):31–46. doi:10.1158/2159-8290.CD-21-1059. [Google Scholar] [PubMed] [CrossRef]
4. Nia HT, Munn LL, Jain RK. Physical traits of cancer. Science. 2020;370(6516):eaaz0868. doi:10.1126/science.aaz0868. [Google Scholar] [PubMed] [CrossRef]
5. Ayad NME, Kaushik S, Weaver VM. Tissue mechanics, an important regulator of development and disease. Philos Trans R Soc Lond B Biol Sci. 2019;374(1779):20180215. doi:10.1098/rstb.2018.0215. [Google Scholar] [PubMed] [CrossRef]
6. Di X, Gao X, Peng L, Ai J, Jin X, Qi S, et al. Cellular mechanotransduction in health and diseases: from molecular mechanism to therapeutic targets. Signal Transduct Target Ther. 2023;8(1):282. doi:10.1038/s41392-023-01501-9. [Google Scholar] [PubMed] [CrossRef]
7. Perea Paizal J, Au SH, Bakal C. Squeezing through the microcirculation: survival adaptations of circulating tumour cells to seed metastasis. Br J Cancer. 2021;124(1):58–65. doi:10.1038/s41416-020-01176-x. [Google Scholar] [PubMed] [CrossRef]
8. Souilhol C, Serbanovic-Canic J, Fragiadaki M, Chico TJ, Ridger V, Roddie H, et al. Endothelial responses to shear stress in atherosclerosis: a novel role for developmental genes. Nat Rev Cardiol. 2020;17(1):52–63. doi:10.1038/s41569-019-0239-5. [Google Scholar] [PubMed] [CrossRef]
9. Karsdal MA, Nielsen SH, Leeming DJ, Langholm LL, Nielsen MJ, Manon-Jensen T, et al. The good and the bad collagens of fibrosis—their role in signaling and organ function. Adv Drug Deliv Rev. 2017;121:43–56. doi:10.1016/j.addr.2017.07.014. [Google Scholar] [PubMed] [CrossRef]
10. Yap AS, Duszyc K, Viasnoff V. Mechanosensing and mechanotransduction at cell-cell junctions. Cold Spring Harb Perspect Biol. 2018;10(8):a028761. doi:10.1101/cshperspect.a028761. [Google Scholar] [PubMed] [CrossRef]
11. Le Roux AL, Quiroga X, Walani N, Arroyo M, Roca-Cusachs P. The plasma membrane as a mechanochemical transducer. Philos Trans R Soc Lond B Biol Sci. 2019;374(1779):20180221. doi:10.1098/rstb.2018.0221. [Google Scholar] [PubMed] [CrossRef]
12. Martino F, Perestrelo AR, Vinarský V, Pagliari S, Forte G. Cellular mechanotransduction: from tension to function. Front Physiol. 2018;9:824. doi:10.3389/fphys.2018.00824. [Google Scholar] [PubMed] [CrossRef]
13. Silver FH, Deshmukh T. Do tensile and shear forces exerted on cells influence mechanotransduction through stored energy considerations? Biocell. 2024;48(4):525–40. doi:10.32604/biocell.2024.047965. [Google Scholar] [CrossRef]
14. Sun Z, Guo SS, Fässler R. Integrin-mediated mechanotransduction. J Cell Biol. 2016;215(4):445–56. doi:10.1083/jcb.201609037. [Google Scholar] [PubMed] [CrossRef]
15. Friedland JC, Lee MH, Boettiger D. Mechanically activated integrin switch controls α5β1 function. Science. 2009;323(5914):642–4. doi:10.1126/science.1168441. [Google Scholar] [PubMed] [CrossRef]
16. Riveline D, Zamir E, Balaban NQ, Schwarz US, Ishizaki T, Narumiya S, et al. Focal contacts as mechanosensors. J Cell Biol. 2001;153(6):1175–86. doi:10.1083/jcb.153.6.1175. [Google Scholar] [PubMed] [CrossRef]
17. Huang S, Ingber DE. Cell tension, matrix mechanics, and cancer development. Cancer Cell. 2005;8(3):175–6. doi:10.1016/j.ccr.2005.08.009. [Google Scholar] [PubMed] [CrossRef]
18. Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nat Rev Cancer. 2009;9(2):108–22. doi:10.1038/nrc2544. [Google Scholar] [PubMed] [CrossRef]
19. Lu F, Zhu L, Bromberger T, Yang J, Yang Q, Liu J, et al. Mechanism of integrin activation by talin and its cooperation with kindlin. Nat Commun. 2022;13(1):2362. doi:10.1038/s41467-022-30117-w. [Google Scholar] [PubMed] [CrossRef]
20. Burridge K, Monaghan-Benson E, Graham DM. Mechanotransduction: from the cell surface to the nucleus via RhoA. Philos Trans R Soc Lond B Biol Sci. 2019;374(1779):20180229. doi:10.1098/rstb.2018.0229. [Google Scholar] [PubMed] [CrossRef]
21. Wu J, Lewis AH, Grandl J. Touch, tension, and transduction—the function and regulation of piezo ion channels. Trends Biochem Sci. 2017;42(1):57–71. doi:10.1016/j.tibs.2016.09.004. [Google Scholar] [PubMed] [CrossRef]
22. Lewis AH, Grandl J. Mechanical sensitivity of Piezo1 ion channels can be tuned by cellular membrane tension. eLife. 2015;4:e12088. doi:10.7554/eLife.12088. [Google Scholar] [PubMed] [CrossRef]
23. Zhao Q, Zhou H, Chi S, Wang Y, Wang J, Geng J, et al. Structure and mechanogating mechanism of the Piezo1 channel. Nature. 2018;554(7693):487–92. doi:10.1038/nature25743. [Google Scholar] [PubMed] [CrossRef]
24. Faucherre A, Nargeot J, Mangoni ME, Jopling C. piezo2b regulates vertebrate light touch response. J Neurosci. 2013;33(43):17089–94. doi:10.1523/JNEUROSCI.0522-13.2013. [Google Scholar] [CrossRef]
25. Woo SH, Lukacs V, de Nooij JC, Zaytseva D, Criddle CR, Francisco A, et al. Piezo2 is the principal mechanotransduction channel for proprioception. Nat Neurosci. 2015;18(12):1756–62. doi:10.1038/nn.4162. [Google Scholar] [PubMed] [CrossRef]
26. Cox CD, Bae C, Ziegler L, Hartley S, Nikolova-Krstevski V, Rohde PR, et al. Removal of the mechanoprotective influence of the cytoskeleton reveals PIEZO1 is gated by bilayer tension. Nat Commun. 2016;7:10366. doi:10.1038/ncomms10366. [Google Scholar] [PubMed] [CrossRef]
27. Pathak MM, Nourse JL, Tran T, Hwe J, Arulmoli J, Le DTT, et al. Stretch-activated ion channel Piezo1 directs lineage choice in human neural stem cells. Proc Natl Acad Sci U S A. 2014;111(45):16148–53. doi:10.1073/pnas.1409802111. [Google Scholar] [PubMed] [CrossRef]
28. Yang X, Lin C, Chen X, Li S, Li X, Xiao B. Structure deformation and curvature sensing of PIEZO1 in lipid membranes. Nature. 2022;604(7905):377–83. doi:10.1038/s41586-022-04574-8. [Google Scholar] [PubMed] [CrossRef]
29. Nourse JL, Pathak MM. How cells channel their stress: interplay between Piezo1 and the cytoskeleton. Semin Cell Dev Biol. 2017;71:3–12. doi:10.1016/j.semcdb.2017.06.018. [Google Scholar] [PubMed] [CrossRef]
30. Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow MJ, et al. Piezo1 integration of vascular architecture with physiological force. Nature. 2014;515(7526):279–82. doi:10.1038/nature13701. [Google Scholar] [PubMed] [CrossRef]
31. Ranade SS, Qiu Z, Woo SH, Hur SS, Murthy SE, Cahalan SM, et al. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc Natl Acad Sci U S A. 2014;111(28):10347–52. doi:10.1073/pnas.1409233111. [Google Scholar] [PubMed] [CrossRef]
32. Wang S, Chennupati R, Kaur H, Iring A, Wettschureck N, Offermanns S. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J Clin Invest. 2016;126(12):4527–36. doi:10.1172/JCI87343. [Google Scholar] [PubMed] [CrossRef]
33. Koser DE, Thompson AJ, Foster SK, Dwivedy A, Pillai EK, Sheridan GK, et al. Mechanosensing is critical for axon growth in the developing brain. Nat Neurosci. 2016;19(12):1592–8. doi:10.1038/nn.4394. [Google Scholar] [PubMed] [CrossRef]
34. Rode B, Shi J, Endesh N, Drinkhill MJ, Webster PJ, Lotteau SJ, et al. Piezo1 channels sense whole body physical activity to reset cardiovascular homeostasis and enhance performance. Nat Commun. 2017;8(1):350. doi:10.1038/s41467-017-00429-3. [Google Scholar] [PubMed] [CrossRef]
35. Chen G, Gao X, Chen J, Peng L, Chen S, Tang C, et al. Actomyosin activity and Piezo1 activity synergistically drive urinary system fibroblast activation. Adv Sci. 2023;10(33):e2303369. doi:10.1002/advs.202303369. [Google Scholar] [PubMed] [CrossRef]
36. Yao M, Tijore A, Cheng D, Li JV, Hariharan A, Martinac B, et al. Force- and cell state-dependent recruitment of Piezo1 drives focal adhesion dynamics and calcium entry. Sci Adv. 2022;8(45):eabo1461. doi:10.1126/sciadv.abo1461. [Google Scholar] [PubMed] [CrossRef]
37. McHugh BJ, Buttery R, Lad Y, Banks S, Haslett C, Sethi T. Integrin activation by Fam38A uses a novel mechanism of R-Ras targeting to the endoplasmic reticulum. J Cell Sci. 2010;123(Pt 1):51–61. doi:10.1242/jcs.056424. [Google Scholar] [PubMed] [CrossRef]
38. Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR, et al. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat Cell Biol. 2004;6(10):977–83. doi:10.1038/ncb1175. [Google Scholar] [PubMed] [CrossRef]
39. Hemmings L, Rees DJ, Ohanian V, Bolton SJ, Gilmore AP, Patel B, et al. Talin contains three actin-binding sites each of which is adjacent to a vinculin-binding site. J Cell Sci. 1996;109(Pt 11):2715–26. doi:10.1242/jcs.109.11.2715. [Google Scholar] [PubMed] [CrossRef]
40. Liu CSC, Raychaudhuri D, Paul B, Chakrabarty Y, Ghosh AR, Rahaman O, et al. Cutting edge: piezo1 mechanosensors optimize human T cell activation. J Immunol. 2018;200(4):1255–60. doi:10.4049/jimmunol.1701118. [Google Scholar] [PubMed] [CrossRef]
41. Chen X, Wanggou S, Bodalia A, Zhu M, Dong W, Fan JJ, et al. A feedforward mechanism mediated by mechanosensitive ion channel PIEZO1 and tissue mechanics promotes glioma aggression. Neuron. 2018;100(4):799–815.e7. doi:10.1016/j.neuron.2018.09.046. [Google Scholar] [PubMed] [CrossRef]
42. Lou W, Liu J, Ding B, Jin L, Xu L, Li X, et al. Five miRNAs-mediated PIEZO2 downregulation, accompanied with activation of Hedgehog signaling pathway, predicts poor prognosis of breast cancer. Aging. 2019;11(9):2628–52. doi:10.18632/aging.101934. [Google Scholar] [PubMed] [CrossRef]
43. Zhang YC, Yang M, Lu CD, Li QY, Shi JN, Shi J. PIEZO2 expression is an independent biomarker prognostic for gastric cancer and represents a potential therapeutic target. Sci Rep. 2024;14(1):1206. doi:10.1038/s41598-023-48577-5. [Google Scholar] [PubMed] [CrossRef]
44. Pardo-Pastor C, Rubio-Moscardo F, Vogel-González M, Serra SA, Afthinos A, Mrkonjic S, et al. Piezo2 channel regulates RhoA and actin cytoskeleton to promote cell mechanobiological responses. Proc Natl Acad Sci U S A. 2018;115(8):1925–30. doi:10.1073/pnas.1718177115. [Google Scholar] [PubMed] [CrossRef]
45. Crisp M, Burke B. The nuclear envelope as an integrator of nuclear and cytoplasmic architecture. FEBS Lett. 2008;582(14):2023–32. doi:10.1016/j.febslet.2008.05.001. [Google Scholar] [PubMed] [CrossRef]
46. Banerjee I, Zhang J, Moore-Morris T, Pfeiffer E, Buchholz KS, Liu A, et al. Targeted ablation of nesprin 1 and nesprin 2 from murine myocardium results in cardiomyopathy, altered nuclear morphology and inhibition of the biomechanical gene response. PLoS Genet. 2014;10(2):e1004114. doi:10.1371/journal.pgen.1004114. [Google Scholar] [PubMed] [CrossRef]
47. Pennacchio FA, Nastały P, Poli A, Maiuri P. Tailoring cellular function: the contribution of the nucleus in mechanotransduction. Front Bioeng Biotechnol. 2021;8:596746. doi:10.3389/fbioe.2020.596746. [Google Scholar] [PubMed] [CrossRef]
48. Tajik A, Zhang Y, Wei F, Sun J, Jia Q, Zhou W, et al. Transcription upregulation via force-induced direct stretching of chromatin. Nat Mater. 2016;15(12):1287–96. doi:10.1038/nmat4729. [Google Scholar] [PubMed] [CrossRef]
49. Kim YB, Yu J, Lee SY, Lee MS, Ko SG, Ye SK, et al. Cell adhesion status-dependent histone acetylation is regulated through intracellular contractility-related signaling activities. J Biol Chem. 2005;280(31):28357–64. doi:10.1074/jbc.M412608200. [Google Scholar] [PubMed] [CrossRef]
50. Dalby MJ, Gadegaard N, Herzyk P, Sutherland D, Agheli H, Wilkinson CDW, et al. Nanomechanotransduction and interphase nuclear organization influence on genomic control. J Cell Biochem. 2007;102(5):1234–44. doi:10.1002/jcb.21354. [Google Scholar] [PubMed] [CrossRef]
51. Le Beyec J, Xu R, Lee SY, Nelson CM, Rizki A, Alcaraz J, et al. Cell shape regulates global histone acetylation in human mammary epithelial cells. Exp Cell Res. 2007;313(14):3066–75. doi:10.1016/j.yexcr.2007.04.022. [Google Scholar] [PubMed] [CrossRef]
52. Mishra J, Chakraborty S, Niharika, Roy A, Manna S, Baral T et al. Mechanotransduction and epigenetic modulations of chromatin: role of mechanical signals in gene regulation. J Cell Biochem. 2024;125(3):e30531. doi:10.1002/jcb.30531. [Google Scholar] [PubMed] [CrossRef]
53. Kaczmarczyk LS, Levi N, Segal T, Salmon-Divon M, Gerlitz G. CTCF supports preferentially short lamina-associated domains. Chromosome Res. 2022;30(1):123–36. doi:10.1007/s10577-022-09686-5. [Google Scholar] [PubMed] [CrossRef]
54. Miroshnikova YA, Nava MM, Wickström SA. Emerging roles of mechanical forces in chromatin regulation. J Cell Sci. 2017;130(14):2243–50. doi:10.1242/jcs.202192. [Google Scholar] [PubMed] [CrossRef]
55. Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. 2009;10(1):63–73. doi:10.1038/nrm2597. [Google Scholar] [PubMed] [CrossRef]
56. Walker CJ, Crocini C, Ramirez D, Killaars AR, Grim JC, Aguado BA, et al. Nuclear mechanosensing drives chromatin remodelling in persistently activated fibroblasts. Nat Biomed Eng. 2021;5(12):1485–99. doi:10.1038/s41551-021-00709-w. [Google Scholar] [PubMed] [CrossRef]
57. Grewal SIS. The molecular basis of heterochromatin assembly and epigenetic inheritance. Mol Cell. 2023;83(11):1767–85. doi:10.1016/j.molcel.2023.04.020. [Google Scholar] [PubMed] [CrossRef]
58. Zhou J, Li YS, Wang KC, Shu C. Epigenetic mechanism in regulation of endothelial function by disturbed flow: induction of DNA hypermethylation by DNMT1. Cell Mol Bioeng. 2014;7(2):218–24. doi:10.1007/s12195-014-0325-z. [Google Scholar] [PubMed] [CrossRef]
59. Nava MM, Miroshnikova YA, Biggs LC, Whitefield DB, Metge F, Boucas J, et al. Heterochromatin-driven nuclear softening protects the genome against mechanical stress-induced damage. Cell. 2020;181(4):800–17.e22. doi:10.1016/j.cell.2020.03.052. [Google Scholar] [PubMed] [CrossRef]
60. Xia Y, Ivanovska IL, Zhu K, Smith L, Irianto J, Pfeifer CR, et al. Nuclear rupture at sites of high curvature compromises retention of DNA repair factors. J Cell Biol. 2018;217(11):3796–808. doi:10.1083/jcb.201711161. [Google Scholar] [PubMed] [CrossRef]
61. Denais CM, Gilbert RM, Isermann P, McGregor AL, Te Lindert M, Weigelin B, et al. Nuclear envelope rupture and repair during cancer cell migration. Science. 2016;352(6283):353–8. doi:10.1126/science.aad7297. [Google Scholar] [PubMed] [CrossRef]
62. Li Y, Tang CB, Kilian KA. Matrix mechanics influence fibroblast-myofibroblast transition by directing the localization of histone deacetylase 4. Cell Mol Bioeng. 2017;10(5):405–15. doi:10.1007/s12195-017-0493-8. [Google Scholar] [PubMed] [CrossRef]
63. Przybyla L, Lakins JN, Weaver VM. Tissue mechanics orchestrate wnt-dependent human embryonic stem cell differentiation. Cell Stem Cell. 2016;19(4):462–75. doi:10.1016/j.stem.2016.06.018. [Google Scholar] [PubMed] [CrossRef]
64. Pereira D, Richert A, Medjkane S, Hénon S, Weitzman JB. Cell geometry and the cytoskeleton impact the nucleo-cytoplasmic localisation of the SMYD3 methyltransferase. Sci Rep. 2020;10(1):20598. doi:10.1038/s41598-020-75833-9. [Google Scholar] [PubMed] [CrossRef]
65. Jain N, Iyer KV, Kumar A, Shivashankar GV. Cell geometric constraints induce modular gene-expression patterns via redistribution of HDAC3 regulated by actomyosin contractility. Proc Natl Acad Sci U S A. 2013;110(28):11349–54. doi:10.1073/pnas.1300801110. [Google Scholar] [PubMed] [CrossRef]
66. Fu M, Hu Y, Lan T, Guan KL, Luo T, Luo M. Correction: the Hippo signalling pathway and its implications in human health and diseases. Signal Transduct Target Ther. 2024;9(1):5. doi:10.1038/s41392-023-01682-3. [Google Scholar] [PubMed] [CrossRef]
67. Kim NG, Gumbiner BM. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J Cell Biol. 2015;210(3):503–15. doi:10.1083/jcb.201501025. [Google Scholar] [PubMed] [CrossRef]
68. Nardone G, Oliver-De La Cruz J, Vrbsky J, Martini C, Pribyl J, Skládal P, et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat Commun. 2017;8:15321. doi:10.1038/ncomms15321. [Google Scholar] [PubMed] [CrossRef]
69. Elosegui-Artola A, Andreu I, Beedle AEM, Lezamiz A, Uroz M, Kosmalska AJ, et al. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell. 2017;171(6):1397–410.e14. doi:10.1016/j.cell.2017.10.008. [Google Scholar] [PubMed] [CrossRef]
70. Wada KI, Itoga K, Okano T, Yonemura S, Sasaki H. Hippo pathway regulation by cell morphology and stress fibers. Development. 2011;138(18):3907–14. doi:10.1242/dev.070987. [Google Scholar] [PubMed] [CrossRef]
71. Cheng B, Li M, Wan W, Guo H, Genin GM, Lin M, et al. Predicting YAP/TAZ nuclear translocation in response to ECM mechanosensing. Biophys J. 2023;122(1):43–53. doi:10.1016/j.bpj.2022.11.2943. [Google Scholar] [PubMed] [CrossRef]
72. Smolarz B, Durczyński A, Romanowicz H, Szyłło K, Hogendorf P. miRNAs in cancer (review of literature). Int J Mol Sci. 2022;23(5):2805. doi:10.3390/ijms23052805. [Google Scholar] [PubMed] [CrossRef]
73. Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. microRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23(20):4051–60. doi:10.1038/sj.emboj.7600385. [Google Scholar] [PubMed] [CrossRef]
74. Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14(10A):1902–10. doi:10.1101/gr.2722704. [Google Scholar] [PubMed] [CrossRef]
75. Lin SL, Miller JD, Ying SY. Intronic microRNA (miRNA). J Biomed Biotechnol. 2006;2006(4):26818. doi:10.1155/jbb/2006/26818. [Google Scholar] [PubMed] [CrossRef]
76. Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol. 2012;19(6):586–93. doi:10.1038/nsmb.2296. [Google Scholar] [PubMed] [CrossRef]
77. Ho PTB, Clark IM, Le LTT. microRNA-based diagnosis and therapy. Int J Mol Sci. 2022;23(13):7167. doi:10.3390/ijms23137167. [Google Scholar] [PubMed] [CrossRef]
78. Mohamed JS, Lopez MA, Boriek AM. Mechanical stretch up-regulates microRNA-26a and induces human airway smooth muscle hypertrophy by suppressing glycogen synthase kinase-3β. J Biol Chem. 2010;285(38):29336–47. doi:10.1074/jbc.M110.101147. [Google Scholar] [PubMed] [CrossRef]
79. Guan YJ, Yang X, Wei L, Chen Q. MiR-365: a mechanosensitive microRNA stimulates chondrocyte differentiation through targeting histone deacetylase 4. FASEB J. 2011;25(12):4457–66. doi:10.1096/fj.11-185132. [Google Scholar] [PubMed] [CrossRef]
80. Mohamed JS, Hajira A, Lopez MA, Boriek AM. Genome-wide mechanosensitive microRNA (MechanomiR) screen uncovers dysregulation of their regulatory networks in the mdm mouse model of muscular dystrophy. J Biol Chem. 2015;290(41):24986–5011. doi:10.1074/jbc.M115.659375. [Google Scholar] [PubMed] [CrossRef]
81. Weber M, Baker MB, Moore JP, Searles CD. MiR-21 is induced in endothelial cells by shear stress and modulates apoptosis and eNOS activity. Biochem Biophys Res Commun. 2010;393(4):643–8. doi:10.1016/j.bbrc.2010.02.045. [Google Scholar] [PubMed] [CrossRef]
82. Moro A, Driscoll TP, Boraas LC, Armero W, Kasper DM, Baeyens N, et al. microRNA-dependent regulation of biomechanical genes establishes tissue stiffness homeostasis. Nat Cell Biol. 2019;21(3):348–58. doi:10.1038/s41556-019-0272-y. [Google Scholar] [PubMed] [CrossRef]
83. D’Angelo A, Godeau AL, Solon J. A new player in tissue mechanics: microrna control of mechanical homeostasis. Dev Cell. 2019;48(5):596–8. doi:10.1016/j.devcel.2019.02.019. [Google Scholar] [PubMed] [CrossRef]
84. Brown JS, Amend SR, Austin RH, Gatenby RA, Hammarlund EU, Pienta KJ. Updating the definition of cancer. Mol Cancer Res. 2023;21(11):1142–7. doi:10.1158/1541-7786.MCR-23-0411. [Google Scholar] [CrossRef]
85. Kim T, Croce CM. microRNA: trends in clinical trials of cancer diagnosis and therapy strategies. Exp Mol Med. 2023;55(7):1314–21. doi:10.1038/s12276-023-01050-9. [Google Scholar] [PubMed] [CrossRef]
86. Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev Oncog. 2013;18(1–2):43–73. doi:10.1615/critrevoncog.v18.i1-2.40. [Google Scholar] [PubMed] [CrossRef]
87. Northcott JM, Dean IS, Mouw JK, Weaver VM. Feeling stress: the mechanics of cancer progression and aggression. Front Cell Dev Biol. 2018;6:17. doi:10.3389/fcell.2018.00017. [Google Scholar] [PubMed] [CrossRef]
88. Kumari A, Veena SM, Luha R, Tijore A. Mechanobiological strategies to augment cancer treatment. ACS Omega. 2023;8(45):42072–85. doi:10.1021/acsomega.3c06451. [Google Scholar] [PubMed] [CrossRef]
89. Bera K, Kiepas A, Godet I, Li Y, Mehta P, Ifemembi B, et al. Extracellular fluid viscosity enhances cell migration and cancer dissemination. Nature. 2022;611(7935):365–73. doi:10.1038/s41586-022-05394-6. [Google Scholar] [CrossRef]
90. Fan W, Adebowale K, Váncza L, Li Y, Rabbi MF, Kunimoto K, et al. Matrix viscoelasticity promotes liver cancer progression in the pre-cirrhotic liver. Nature. 2024;626(7999):635–42. doi:10.1038/s41586-023-06991-9. [Google Scholar] [PubMed] [CrossRef]
91. Dessard M, Manneville JB, Berret JF. Cytoplasmic viscosity is a potential biomarker for metastatic breast cancer cells. Nanoscale Adv. 2024;6(6):1727–38. doi:10.1039/d4na00003j. [Google Scholar] [CrossRef]
92. Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014;15(12):1243–53. doi:10.15252/embr.201439246. [Google Scholar] [PubMed] [CrossRef]
93. Huang J, Zhang L, Wan D, Zhou L, Zheng S, Lin S, et al. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct Target Ther. 2021;6(1):153. doi:10.1038/s41392-021-00544-0. [Google Scholar] [PubMed] [CrossRef]
94. Guo Y, Pan W, Liu S, Shen Z, Xu Y, Hu L. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp Ther Med. 2020;19:1997–2007. doi:10.3892/etm.2020.8454. [Google Scholar] [PubMed] [CrossRef]
95. Husain A, Khadka A, Ehrlicher A, Saint-Geniez M, Krishnan R. Substrate stiffening promotes VEGF-A functions via the PI3K/Akt/mTOR pathway. Biochem Biophys Res Commun. 2022;586:27–33. doi:10.1016/j.bbrc.2021.11.030. [Google Scholar] [PubMed] [CrossRef]
96. Shea MP, O’Leary KA, Wegner KA, Vezina CM, Schuler LA. High collagen density augments mTOR-dependent cancer stem cells in ERα+ mammary carcinomas, and increases mTOR-independent lung metastases. Cancer Lett. 2018;433:1–9. doi:10.1016/j.canlet.2018.06.025. [Google Scholar] [PubMed] [CrossRef]
97. Akrida I, Makrygianni M, Nikou S, Mulita F, Bravou V, Papadaki H. Hippo pathway effectors YAP, TAZ and TEAD are associated with EMT master regulators ZEB, Snail and with aggressive phenotype in phyllodes breast tumors. Pathol Res Pract. 2024;262:155551. doi:10.1016/j.prp.2024.155551. [Google Scholar] [PubMed] [CrossRef]
98. Zhou T, Li X, Liu J, Hao J. The hippo/YAP signaling pathway: the driver of cancer metastasis. Cancer Biol Med. 2023;20(7):483–9. doi:10.20892/j.issn.2095-3941.2023.0164. [Google Scholar] [PubMed] [CrossRef]
99. Cheng D, Jin L, Chen Y, Xi X, Guo Y. YAP promotes epithelial mesenchymal transition by upregulating Slug expression in human colorectal cancer cells. Int J Clin Exp Pathol. 2020;13(4):701–10. [Google Scholar] [PubMed]
100. Conti S, Venturini V, Cañellas-Socias A, Cortina C, Abenza JF, Stephan-Otto Attolini C, et al. Membrane to cortex attachment determines different mechanical phenotypes in LGR5+ and LGR5−colorectal cancer cells. Nat Commun. 2024;15(1):3363. doi:10.1038/s41467-024-47227-2. [Google Scholar] [PubMed] [CrossRef]
101. Pandey M, Suh YJ, Kim M, Davis HJ, Segall JE, Wu M. Mechanical compression regulates tumor spheroid invasion into a 3D collagen matrix. Phys Biol. 2024;21(3):036003. doi:10.1088/1478-3975/ad3ac5. [Google Scholar] [PubMed] [CrossRef]
102. Yu C, Liu Q, Wang J. A physical mechanism of heterogeneity and micro-metastasis in stem cells, cancer cells, and cancer stem cells. J Chem Phys. 2022;156(7):075103. doi:10.1063/5.0078196. [Google Scholar] [PubMed] [CrossRef]
103. Grzywa TM, Klicka K, Włodarski PK. Regulators at every step-how microRNAs drive tumor cell invasiveness and metastasis. Cancers. 2020;12(12):3709. doi:10.3390/cancers12123709. [Google Scholar] [PubMed] [CrossRef]
104. Khafaei M, Rezaie E, Mohammadi A, Shahnazi Gerdehsang P, Ghavidel S, Kadkhoda S, et al. miR-9: from function to therapeutic potential in cancer. J Cell Physiol. 2019;234(9):14651–65. doi:10.1002/jcp.28210. [Google Scholar] [PubMed] [CrossRef]
105. Sun Z, Shi K, Yang S, Liu J, Zhou Q, Wang G, et al. Effect of exosomal miRNA on cancer biology and clinical applications. Mol Cancer. 2018;17(1):147. doi:10.1186/s12943-018-0897-7. [Google Scholar] [PubMed] [CrossRef]
106. Zheng Q, Hou W. Regulation of angiogenesis by microRNAs in cancer. Mol Med Rep. 2021;24(2):583. doi:10.3892/mmr.2021.12222. [Google Scholar] [PubMed] [CrossRef]
107. Cao J, Chen L, Zhong X, Shen Y, Gao Y, Chen Q, et al. miR32-5p promoted vascular smooth muscle cell calcification by upregulating TNFα in the microenvironment. BMC Immunol. 2020;21(1):3. doi:10.1186/s12865-019-0324-x. [Google Scholar] [PubMed] [CrossRef]
108. Chen X, Chen J. miR-10b-5p-mediated upregulation of PIEZO1 predicts poor prognosis and links to purine metabolism in breast cancer. Genomics. 2022;114(3):110351. doi:10.1016/j.ygeno.2022.110351. [Google Scholar] [PubMed] [CrossRef]
Mansouri S, Khansarinejad B, Mosayebi G, Eghbali A, Mondanizadeh M. Alteration in expression of miR-32 and FBXW7 tumor suppressor in plasma samples of patients with T-cell acute lymphoblastic leukemia. Cancer Manag Res. 2020;12:1253–9. doi:10.2147/CMAR.S238470. [Google Scholar] [PubMed] [CrossRef]
110. Li D, Lu Z, Jia J, Zheng Z, Lin S. Curcumin ameliorates podocytic adhesive capacity damage under mechanical stress by inhibiting miR-124 expression. Kidney Blood Press Res. 2014;38(1):61–71. doi:10.1159/000355755. [Google Scholar] [PubMed] [CrossRef]
111. Boufraqech M, Nilubol N, Zhang L, Gara SK, Sadowski SM, Mehta A, et al. miR30a inhibits LOX expression and anaplastic thyroid cancer progression. Cancer Res. 2015;75(2):367–77. doi:10.1158/0008-5472.CAN-14-2304. [Google Scholar] [PubMed] [CrossRef]
112. Brandon-Warner E, Benbow JH, Swet JH, Feilen NA, Culberson CR, McKillop IH, et al. Adeno-associated virus serotype 2 vector-mediated reintroduction of microRNA-19b attenuates hepatic fibrosis. Hum Gene Ther. 2018;29(6):674–86. doi:10.1089/hum.2017.035. [Google Scholar] [PubMed] [CrossRef]
113. Gao X, Qiao X, Xing X, Huang J, Qian J, Wang Y, et al. Matrix stiffness-upregulated microRNA-17-5p attenuates the intervention effects of metformin on HCC invasion and metastasis by targeting the PTEN/PI3K/Akt pathway. Front Oncol. 2020;10:1563. doi:10.3389/fonc.2020.01563. [Google Scholar] [PubMed] [CrossRef]
114. Rutnam ZJ, Wight TN, Yang BB. miRNAs regulate expression and function of extracellular matrix molecules. Matrix Biol. 2013;32(2):74–85. doi:10.1016/j.matbio.2012.11.003. [Google Scholar] [PubMed] [CrossRef]
115. Dey N, Ghosh-Choudhury N, Kasinath BS, Choudhury GG. TGFβ-stimulated microRNA-21 utilizes PTEN to orchestrate AKT/mTORC1 signaling for mesangial cell hypertrophy and matrix expansion. PLoS One. 2012;7(8):e42316. doi:10.1371/journal.pone.0042316. [Google Scholar] [PubMed] [CrossRef]
116. Melnik BC. miR-21: an environmental driver of malignant melanoma? J Transl Med. 2015;13:202. doi:10.1186/s12967-015-0570-5. [Google Scholar] [PubMed] [CrossRef]
117. Lin CH, Pelissier FA, Zhang H, Lakins J, Weaver VM, Park C, et al. Microenvironment rigidity modulates responses to the HER2 receptor tyrosine kinase inhibitor lapatinib via YAP and TAZ transcription factors. Mol Biol Cell. 2015;26(22):3946–53. doi:10.1091/mbc.E15-07-0456. [Google Scholar] [PubMed] [CrossRef]
118. Schrader J, Gordon-Walker TT, Aucott RL, Van Deemter M, Quaas A, Walsh S, et al. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53(4):1192–205. doi:10.1002/hep.24108. [Google Scholar] [PubMed] [CrossRef]
119. Tilghman RW, Cowan CR, Mih JD, Koryakina Y, Gioeli D, Slack-Davis JK, et al. Matrix rigidity regulates cancer cell growth and cellular phenotype. PLoS One. 2010;5(9):e12905. doi:10.1371/journal.pone.0012905. [Google Scholar] [PubMed] [CrossRef]
120. Wolfenson H, Yang B, Sheetz MP. Steps in mechanotransduction pathways that control cell morphology. Annu Rev Physiol. 2019;81:585–605. doi:10.1146/annurev-physiol-021317-121245. [Google Scholar] [PubMed] [CrossRef]
121. Kirschmann DA, Seftor EA, Fong SFT, Nieva DRC, Sullivan CM, Edwards EM, et al. A molecular role for lysyl oxidase in breast cancer invasion. Cancer Res. 2002;62(15):4478–83. [Google Scholar] [PubMed]
122. Bhadriraju K, Yang M, Alom Ruiz S, Pirone D, Tan J, Chen CS. Activation of ROCK by RhoA is regulated by cell adhesion, shape, and cytoskeletal tension. Exp Cell Res. 2007;313(16):3616–23. doi:10.1016/j.yexcr.2007.07.002. [Google Scholar] [PubMed] [CrossRef]
123. Liu Z, Jin Q, Yan T, Wo Y, Liu H, Wang Y. Exosome-mediated transduction of mechanical force regulates prostate cancer migration via microRNA. Biochem Biophys Rep. 2022;31:101299. doi:10.1016/j.bbrep.2022.101299. [Google Scholar] [PubMed] [CrossRef]
124. Frith JE, Kusuma GD, Carthew J, Li F, Cloonan N, Gomez GA, et al. Mechanically-sensitive miRNAs bias human mesenchymal stem cell fate via mTOR signalling. Nat Commun. 2018;9(1):257. doi:10.1038/s41467-017-02486-0. [Google Scholar] [PubMed] [CrossRef]
125. Huang Y, Crawford M, Higuita-Castro N, Nana-Sinkam P, Ghadiali SN. miR-146a regulates mechanotransduction and pressure-induced inflammation in small airway epithelium. FASEB J. 2012;26(8):3351–64. doi:10.1096/fj.11-199240. [Google Scholar] [PubMed] [CrossRef]
126. Miroshnikova YA, Mouw JK, Barnes JM, Pickup MW, Lakins JN, Kim Y, et al. Tissue mechanics promote IDH1-dependent HIF1α–tenascin C feedback to regulate glioblastoma aggression. Nat Cell Biol. 2016;18(12):1336–45. doi:10.1038/ncb3429. [Google Scholar] [PubMed] [CrossRef]
127. Hassan AA, Artemenko M, Tang MKS, Shi Z, Chen LY, Lai HC, et al. Ascitic fluid shear stress in concert with hepatocyte growth factor drive stemness and chemoresistance of ovarian cancer cells via the c-Met-PI3K/Akt-miR-199a-3p signaling pathway. Cell Death Dis. 2022;13(6):537. doi:10.1038/s41419-022-04976-6. [Google Scholar] [PubMed] [CrossRef]
128. Li S, Sampson C, Liu C, Piao HL, Liu HX. Integrin signaling in cancer: bidirectional mechanisms and therapeutic opportunities. Cell Commun Signal. 2023;21(1):266. doi:10.1186/s12964-023-01264-4. [Google Scholar] [PubMed] [CrossRef]
129. Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8(3):241–54. doi:10.1016/j.ccr.2005.08.010. [Google Scholar] [PubMed] [CrossRef]
130. Dong Y, Xie X, Wang Z, Hu C, Zheng Q, Wang Y, et al. Increasing matrix stiffness upregulates vascular endothelial growth factor expression in hepatocellular carcinoma cells mediated by integrin β1. Biochem Biophys Res Commun. 2014;444(3):427–32. doi:10.1016/j.bbrc.2014.01.079. [Google Scholar] [PubMed] [CrossRef]
131. Sekiya Y, Ogawa T, Yoshizato K, Ikeda K, Kawada N. Suppression of hepatic stellate cell activation by microRNA-29b. Biochem Biophys Res Commun. 2011;412(1):74–9. doi:10.1016/j.bbrc.2011.07.041. [Google Scholar] [PubMed] [CrossRef]
132. Fowler A, Thomson D, Giles K, Maleki S, Mreich E, Wheeler H, et al. miR-124a is frequently down-regulated in glioblastoma and is involved in migration and invasion. Eur J Cancer. 2011;47(6):953–63. doi:10.1016/j.ejca.2010.11.026. [Google Scholar] [PubMed] [CrossRef]
133. Hunt S, Jones AV, Hinsley EE, Whawell SA, Lambert DW. microRNA-124 suppresses oral squamous cell carcinoma motility by targeting ITGB1. FEBS Lett. 2011;585(1):187–92. doi:10.1016/j.febslet.2010.11.038. [Google Scholar] [PubMed] [CrossRef]
134. Liu Y, Yang Y, Wang X, Yin S, Liang B, Zhang Y, et al. Function of microRNA-124 in the pathogenesis of cancer (review). Int J Oncol. 2024;64(1):6. doi:10.3892/ijo.2023.5594. [Google Scholar] [PubMed] [CrossRef]
135. Deng L, Petrek H, Tu MJ, Batra N, Yu AX, Yu AM. Bioengineered miR-124-3p prodrug selectively alters the proteome of human carcinoma cells to control multiple cellular components and lung metastasis in vivo. Acta Pharm Sin B. 2021;11(12):3950–65. doi:10.1016/j.apsb.2021.07.027. [Google Scholar] [CrossRef]
136. Li D, Lu Z, Jia J, Zheng Z, Lin S. Changes in microRNAs associated with podocytic adhesion damage under mechanical stress. J Renin Angiotensin Aldosterone Syst. 2013;14(2):97–102. doi:10.1177/1470320312460071. [Google Scholar] [PubMed] [CrossRef]
137. Stepicheva NA, Song JL. Function and regulation of microRNA-31 in development and disease. Mol Reprod Dev. 2016;83(8):654–74. doi:10.1002/mrd.22678. [Google Scholar] [PubMed] [CrossRef]
138. Augoff K, Das M, Bialkowska K, McCue B, Plow EF, Sossey-Alaoui K. miR-31 is a broad regulator of β1-integrin expression and function in cancer cells. Mol Cancer Res. 2011;9(11):1500–8. doi:10.1158/1541-7786.MCR-11-0311. [Google Scholar] [PubMed] [CrossRef]
139. Yu T, Ma P, Wu D, Shu Y, Gao W. Functions and mechanisms of microRNA-31 in human cancers. Biomed Pharmacother. 2018;108:1162–9. doi:10.1016/j.biopha.2018.09.132. [Google Scholar] [PubMed] [CrossRef]
140. Duba M, Al Tukmachi D, Samoilenko T, Vecera M, Ruckova M, Vankova T, et al. In reply: microrna analysis in meningiomas with different degrees of tissue stiffness: a potential tool for effective preoperative planning. Neurosurgery. 2025;97(2):e54–5. doi:10.1227/neu.0000000000003575. [Google Scholar] [PubMed] [CrossRef]
141. Liu ZL, Chen HH, Zheng LL, Sun LP, Shi L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct Target Ther. 2023;8(1):198. doi:10.1038/s41392-023-01460-1. [Google Scholar] [PubMed] [CrossRef]
142. Ribatti D, Vacca A, Nico B, Sansonno D, Dammacco F. Angiogenesis and anti-angiogenesis in hepatocellular carcinoma. Cancer Treat Rev. 2006;32(6):437–44. doi:10.1016/j.ctrv.2006.06.002. [Google Scholar] [PubMed] [CrossRef]
143. Wang YH, Dong YY, Wang WM, Xie XY, Wang ZM, Chen RX, et al. Vascular endothelial cells facilitated HCC invasion and metastasis through the Akt and NF-κB pathways induced by paracrine cytokines. J Exp Clin Cancer Res. 2013;32(1):51. doi:10.1186/1756-9966-32-51. [Google Scholar] [PubMed] [CrossRef]
144. Shaw P, Dwivedi SKD, Bhattacharya R, Mukherjee P, Rao G. VEGF signaling: role in angiogenesis and beyond. Biochim Biophys Acta Rev Cancer. 2024;1879(2):189079. doi:10.1016/j.bbcan.2024.189079. [Google Scholar] [CrossRef]
145. Chen J, Huang Z, Chen Y, Tian H, Chai P, Shen Y, et al. Lactate and lactylation in cancer. Signal Transduct Target Ther. 2025;10(1):38. doi:10.1038/s41392-024-02082-x. [Google Scholar] [PubMed] [CrossRef]
146. Rodriguez D, Watts D, Gaete D, Sormendi S, Wielockx B. Hypoxia pathway proteins and their impact on the blood vasculature. Int J Mol Sci. 2021;22(17):9191. doi:10.3390/ijms22179191. [Google Scholar] [PubMed] [CrossRef]
147. LaValley DJ, Zanotelli MR, Bordeleau F, Wang W, Schwager SC, Reinhart-King CA. Matrix stiffness enhances VEGFR-2 internalization, signaling, and proliferation in endothelial cells. Converg Sci Phys Oncol. 2017;3:044001. doi:10.1088/2057-1739/aa9263. [Google Scholar] [PubMed] [CrossRef]
148. Liu LZ, Li C, Chen Q, Jing Y, Carpenter R, Jiang Y, et al. miR-21 induced angiogenesis through AKT and ERK activation and HIF-1α expression. PLoS One. 2011;6(4):e19139. doi:10.1371/journal.pone.0019139. [Google Scholar] [PubMed] [CrossRef]
149. Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103(7):2257–61. doi:10.1073/pnas.0510565103. [Google Scholar] [PubMed] [CrossRef]
150. Li CX, Talele NP, Boo S, Koehler A, Knee-Walden E, Balestrini JL, et al. microRNA-21 preserves the fibrotic mechanical memory of mesenchymal stem cells. Nat Mater. 2017;16(3):379–89. doi:10.1038/nmat4780. [Google Scholar] [PubMed] [CrossRef]
151. Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12(3):247–56. doi:10.1038/ncb2024. [Google Scholar] [PubMed] [CrossRef]
152. Kim BG, Gao MQ, Kang S, Choi YP, Lee JH, Kim JE, et al. Mechanical compression induces VEGFA overexpression in breast cancer via DNMT3A-dependent miR-9 downregulation. Cell Death Dis. 2017;8(3):e2646. doi:10.1038/cddis.2017.73. [Google Scholar] [PubMed] [CrossRef]
153. Colotti G, Failla CM, Lacal PM, Ungarelli M, Ruffini F, Di Micco P, et al. Neuropilin-1 is required for endothelial cell adhesion to soluble vascular endothelial growth factor receptor 1. FEBS J. 2022;289(1):183–98. doi:10.1111/febs.16119. [Google Scholar] [PubMed] [CrossRef]
154. Chaudhary B, Khaled YS, Ammori BJ, Elkord E. Neuropilin 1: function and therapeutic potential in cancer. Cancer Immunol Immunother. 2014;63(2):81–99. doi:10.1007/s00262-013-1500-0. [Google Scholar] [PubMed] [CrossRef]
155. Fukasawa M, Matsushita A, Korc M. Neuropilin-1 interacts with integrin beta1 and modulates pancreatic cancer cell growth, survival and invasion. Cancer Biol Ther. 2007;6(8):1173–80. doi:10.4161/cbt.6.8.4363. [Google Scholar] [PubMed] [CrossRef]
156. Valdembri D, Caswell PT, Anderson KI, Schwarz JP, König I, Astanina E, et al. Neuropilin-1/GIPC1 signaling regulates alpha5beta1 integrin traffic and function in endothelial cells. PLoS Biol. 2009;7(1):e25. doi:10.1371/journal.pbio.1000025. [Google Scholar] [PubMed] [CrossRef]
157. Li M, Wang P, Li J, Zhou F, Huang S, Qi S, et al. NRP1 transduces mechanical stress inhibition via LATS1/YAP in hypertrophic scars. Cell Death Discov. 2023;9(1):341. doi:10.1038/s41420-023-01635-3. [Google Scholar] [PubMed] [CrossRef]
158. Yaqoob U, Cao S, Shergill U, Jagavelu K, Geng Z, Yin M, et al. Neuropilin-1 stimulates tumor growth by increasing fibronectin fibril assembly in the tumor microenvironment. Cancer Res. 2012;72(16):4047–59. doi:10.1158/0008-5472.CAN-11-3907. [Google Scholar] [PubMed] [CrossRef]
159. Liu Y, Zhao Q, Xi T, Zheng L, Li X. microRNA-9 as a paradoxical but critical regulator of cancer metastasis: implications in personalized medicine. Genes Dis. 2020;8(6):759–68. doi:10.1016/j.gendis.2020.10.005. [Google Scholar] [PubMed] [CrossRef]
160. Fang F, Cheng L, Wu X, Ye M, Zhang H. miR-141 promotes colon cancer cell proliferation by targeted PHLPP2 expression inhibitionn. Cancer Manage Res. 2020;12:11341–50. doi:10.2147/CMAR.S256670. [Google Scholar] [PubMed] [CrossRef]
161. Li JZ, Li J, Wang HQ, Li X, Wen B, Wang YJ. miR-141-3p promotes prostate cancer cell proliferation through inhibiting kruppel-like factor-9 expression. Biochem Biophys Res Commun. 2017;482(4):1381–6. doi:10.1016/j.bbrc.2016.12.045. [Google Scholar] [PubMed] [CrossRef]
162. Ni Z, Shen Y, Wang W, Cheng X, Fu Y. miR-141-5p affects the cell proliferation and apoptosis by targeting BTG1 in cervical cancer. Cancer Biother Radiopharm. 2024;39(6):395–405. doi:10.1089/cbr.2021.0227. [Google Scholar] [PubMed] [CrossRef]
163. Ma L, Zhai B, Zhu H, Li W, Jiang W, Lei L, et al. The miR-141/neuropilin-1 axis is associated with the clinicopathology and contributes to the growth and metastasis of pancreatic cancer. Cancer Cell Int. 2019;19:248. doi:10.1186/s12935-019-0963-2. [Google Scholar] [PubMed] [CrossRef]
164. Zhang YJ, Liu XC, Du J, Zhang YJ. miR-152 regulates metastases of non-small cell lung cancer cells by targeting neuropilin-1. Int J Clin Exp Pathol. 2015;8(11):14235–40. [Google Scholar] [PubMed]
165. Feng F, Liu H, Chen A, Xia Q, Zhao Y, Jin X, et al. miR-148-3p and miR-152-3p synergistically regulate prostate cancer progression via repressing KLF4. J Cell Biochem. 2019;120(10):17228–39. doi:10.1002/jcb.28984. [Google Scholar] [PubMed] [CrossRef]
166. Li Y, Chen F, Chu J, Wu C, Li Y, Li H, et al. miR-148-3p inhibits growth of glioblastoma targeting DNA methyltransferase-1 (DNMT1). Oncol Res. 2019;27(8):911–21. doi:10.3727/096504019X15516966905337. [Google Scholar] [PubMed] [CrossRef]
167. Vela-Alcántara AM, Santiago-García J, Barragán-Palacios M, León-Chacón A, Domínguez-Pantoja M, Barceinas-Dávila I, et al. Differential modulation of cell morphology, migration, and Neuropilin-1 expression in cancer and non-cancer cell lines by substrate stiffness. Front Cell Dev Biol. 2024;12:1352233. doi:10.3389/fcell.2024.1352233. [Google Scholar] [PubMed] [CrossRef]
168. Croci O, De Fazio S, Biagioni F, Donato E, Caganova M, Curti L, et al. Transcriptional integration of mitogenic and mechanical signals by Myc and YAP. Genes Dev. 2017;31(20):2017–22. doi:10.1101/gad.301184.117. [Google Scholar] [PubMed] [CrossRef]
169. Mizuno T, Murakami H, Fujii M, Ishiguro F, Tanaka I, Kondo Y, et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene. 2012;31(49):5117–22. doi:10.1038/onc.2012.5. [Google Scholar] [PubMed] [CrossRef]
170. Samji P, Rajendran MK, Warrier VP, Ganesh A, Devarajan K. Regulation of Hippo signaling pathway in cancer: a microRNA perspective. Cell Signal. 2021;78:109858. doi:10.1016/j.cellsig.2020.109858. [Google Scholar] [PubMed] [CrossRef]
171. Sha F, Xin D, Xu J, Zheng Z, Lin W, Cai X, et al. Curcumin inhibits colorectal cancer development by blocking the YAP/TAZ signaling axis. Biocell. 2024;48(3):443–51. doi:10.32604/biocell.2023.029188. [Google Scholar] [CrossRef]
172. Kim NG, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc Natl Acad Sci U S A. 2011;108(29):11930–5. doi:10.1073/pnas.1103345108. [Google Scholar] [PubMed] [CrossRef]
173. Mori M, Triboulet R, Mohseni M, Schlegelmilch K, Shrestha K, Camargo FD, et al. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell. 2014;156(5):893–906. doi:10.1016/j.cell.2013.12.043. [Google Scholar] [PubMed] [CrossRef]
174. Chaulk SG, Lattanzi VJ, Hiemer SE, Fahlman RP, Varelas X. The Hippo pathway effectors TAZ/YAP regulate dicer expression and microRNA biogenesis through Let-7. J Biol Chem. 2014;289(4):1886–91. doi:10.1074/jbc.C113.529362. [Google Scholar] [PubMed] [CrossRef]
175. Zheng D, Cao M, Zuo S, Xia X, Zhi C, Lin Y, et al. Correction: ranbp1 promotes colorectal cancer progression by regulating pre-miRNA nuclear export via a positive feedback loop with YAP. Oncogene. 2022;41(7):1070. doi:10.1038/s41388-021-02152-2. [Google Scholar] [PubMed] [CrossRef]
176. Huang ZM, Wang H, Ji ZG. Bladder mesenchymal stromal cell-derived exosomal miRNA-217 modulates bladder cancer cell survival through Hippo-YAP pathway. Inflamm Res. 2021;70(9):959–69. doi:10.1007/s00011-021-01494-7. [Google Scholar] [PubMed] [CrossRef]
177. He J, Su X, Xie W. miR-582-3p alleviates osteoarthritis progression by targeting YAP1. Mol Immunol. 2020;128:258–67. doi:10.1016/j.molimm.2020.10.022. [Google Scholar] [PubMed] [CrossRef]
178. Volkers L, Mechioukhi Y, Coste B. Piezo channels: from structure to function. Pflugers Arch. 2015;467(1):95–9. doi:10.1007/s00424-014-1578-z. [Google Scholar] [PubMed] [CrossRef]
179. Gao W, Hasan H, Anderson DE, Lee W. The role of mechanically-activated ion channels Piezo1, Piezo2, and TRPV4 in chondrocyte mechanotransduction and mechano-therapeutics for osteoarthritis. Front Cell Dev Biol. 2022;10:885224. doi:10.3389/fcell.2022.885224. [Google Scholar] [PubMed] [CrossRef]
180. Qin C, Feng Y, Yin Z, Wang C, Yin R, Li Y, et al. The PIEZO1/miR-155-5p/GDF6/SMAD2/3 signaling axis is involved in inducing the occurrence and progression of osteoarthritis under excessive mechanical stress. Cell Signal. 2024;118:111142. doi:10.1016/j.cellsig.2024.111142. [Google Scholar] [PubMed] [CrossRef]
181. Chen B, Liu X, Yu P, Xie F, Kwan JSH, Chan WN, et al. H. pylori-induced NF-κB-PIEZO1-YAP1-CTGF axis drives gastric cancer progression and cancer-associated fibroblast-mediated tumour microenvironment remodelling. Clin Transl Med. 2023;13(11):e1481. doi:10.1002/ctm2.1481. [Google Scholar] [PubMed] [CrossRef]
182. Li M, Zhang X, Wang M, Wang Y, Qian J, Xing X, et al. Activation of Piezo1 contributes to matrix stiffness-induced angiogenesis in hepatocellular carcinoma. Cancer Commun. 2022;42(11):1162–84. doi:10.1002/cac2.12364. [Google Scholar] [PubMed] [CrossRef]
183. Ranade SS, Woo SH, Dubin AE, Moshourab RA, Wetzel C, Petrus M, et al. Piezo2 is the major transducer of mechanical forces for touch sensation in mice. Nature. 2014;516(7529):121–5. doi:10.1038/nature13980. [Google Scholar] [PubMed] [CrossRef]
184. Ikeda R, Gu JG. Piezo2 channel conductance and localization domains in Merkel cells of rat whisker hair follicles. Neurosci Lett. 2014;583:210–5. doi:10.1016/j.neulet.2014.05.055. [Google Scholar] [PubMed] [CrossRef]
185. Etem EÖ, Ceylan GG, Özaydın S, Ceylan C, Özercan I, Kuloğlu T. The increased expression of Piezo1 and Piezo2 ion channels in human and mouse bladder carcinoma. Adv Clin Exp Med. 2018;27(8):1025–31. doi:10.17219/acem/71080. [Google Scholar] [PubMed] [CrossRef]
186. Moccia F. Endothelial Ca2+ signaling and the resistance to anticancer treatments: partners in crime. Int J Mol Sci. 2018;19(1):217. doi:10.3390/ijms19010217. [Google Scholar] [PubMed] [CrossRef]
187. Yang H, Liu C, Zhou RM, Yao J, Li XM, Shen Y, et al. Piezo2 protein: a novel regulator of tumor angiogenesis and hyperpermeability. Oncotarget. 2016;7(28):44630–43. doi:10.18632/oncotarget.10134. [Google Scholar] [PubMed] [CrossRef]
188. Song L, Li ZY, Liu WP, Zhao MR. Crosstalk between Wnt/β-catenin and Hedgehog/Gli signaling pathways in colon cancer and implications for therapy. Cancer Biol Ther. 2015;16(1):1–7. doi:10.4161/15384047.2014.972215. [Google Scholar] [PubMed] [CrossRef]
189. Itoh Y, Takehara Y, Kawase T, Terashima K, Ohkawa Y, Hirose Y, et al. Feasibility of magnetic resonance elastography for the pancreas at 3T. J Magn Reson Imaging. 2016;43(2):384–90. doi:10.1002/jmri.24995. [Google Scholar] [PubMed] [CrossRef]
190. Jamshidi MH, Karami A, Keshavarz A, Fatemi A, Ghanavati S. Magnetic resonance elastography for breast cancer diagnosis through the assessment of tissue biomechanical properties. Health Sci Rep. 2024;7(12):e70253. doi:10.1002/hsr2.70253. [Google Scholar] [PubMed] [CrossRef]
191. Zhao Y, Cao J, Melamed A, Worley M, Gockley A, Jones D, et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc Natl Acad Sci U S A. 2019;116(6):2210–9. doi:10.1073/pnas.1818357116. [Google Scholar] [PubMed] [CrossRef]
192. Papageorgis P, Polydorou C, Mpekris F, Voutouri C, Agathokleous E, Kapnissi-Christodoulou CP, et al. Tranilast-induced stress alleviation in solid tumors improves the efficacy of chemo- and nanotherapeutics in a size-independent manner. Sci Rep. 2017;7:46140. doi:10.1038/srep46140. [Google Scholar] [PubMed] [CrossRef]
193. Polydorou C, Mpekris F, Papageorgis P, Voutouri C, Stylianopoulos T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget. 2017;8(15):24506–17. doi:10.18632/oncotarget.15534. [Google Scholar] [PubMed] [CrossRef]
194. Incio J, Suboj P, Chin SM, Vardam-Kaur T, Liu H, Hato T, et al. Metformin reduces desmoplasia in pancreatic cancer by reprogramming stellate cells and tumor-associated macrophages. PLoS One. 2015;10(12):e0141392. doi:10.1371/journal.pone.0141392. [Google Scholar] [PubMed] [CrossRef]
195. Zhang X, Liu S, Hu T, Liu S, He Y, Sun S. Up-regulated microRNA-143 transcribed by nuclear factor kappa B enhances hepatocarcinoma metastasis by repressing fibronectin expression. Hepatology. 2009;50(2):490–9. doi:10.1002/hep.23008. [Google Scholar] [PubMed] [CrossRef]
196. Zhu H, Han C, Wu T. miR-17-92 cluster promotes hepatocarcinogenesis. Carcinogenesis. 2015;36(10):1213–22. doi:10.1093/carcin/bgv112. [Google Scholar] [PubMed] [CrossRef]
197. Mouw JK, Yui Y, Damiano L, Bainer RO, Lakins JN, Acerbi I, et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat Med. 2014;20(4):360–7. doi:10.1038/nm.3497. [Google Scholar] [PubMed] [CrossRef]
198. Beg MS, Brenner AJ, Sachdev J, Borad M, Kang YK, Stoudemire J, et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs. 2017;35(2):180–8. doi:10.1007/s10637-016-0407-y. [Google Scholar] [PubMed] [CrossRef]
199. Segal M, Slack FJ. Challenges identifying efficacious miRNA therapeutics for cancer. Expert Opin Drug Discov. 2020;15(9):987–92. doi:10.1080/17460441.2020.1765770. [Google Scholar] [PubMed] [CrossRef]
200. Bereczki Z, Benczik B, Balogh OM, Marton S, Puhl E, Pétervári M, et al. Mitigating off-target effects of small RNAs: conventional approaches, network theory and artificial intelligence. Br J Pharmacol. 2025;182(2):340–79. doi:10.1111/bph.17302. [Google Scholar] [PubMed] [CrossRef]
201. Reda El Sayed S, Cristante J, Guyon L, Denis J, Chabre O, Cherradi N. microRNA therapeutics in cancer: current advances and challenges. Cancers. 2021;13(11):2680. doi:10.3390/cancers13112680. [Google Scholar] [PubMed] [CrossRef]
202. Abou Madawi NA, Darwish ZE, Omar EM. Targeted gene therapy for cancer: the impact of microRNA multipotentiality. Med Oncol. 2024;41(9):214. doi:10.1007/s12032-024-02450-1. [Google Scholar] [PubMed] [CrossRef]
203. Seyhan AA. Trials and tribulations of microRNA therapeutics. Int J Mol Sci. 2024;25(3):1469. doi:10.3390/ijms25031469. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools