
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

ERK- and p53-Mediated ATF3 Expression Contributes to Cisplatin-Induced DNA Damage in Renal Epithelial Cells
1 Division of Applied Life Science, Gyeongsang National University, Jinju, Republic of Korea
2 Division of Bio & Medical Bigdata Department (BK4 Program), Gyeongsang National University, Jinju, Republic of Korea
3 Department of Neurosurgery, Duke University School of Medicine, Durham, NC, USA
4 Research Institute of Life Sciences, Gyeongsang National University, Jinju, Republic of Korea
5 Division of Life Science, Gyeongsang National University, Jinju, Republic of Korea
6 School of Biological Sciences, Seoul National University, Seoul, Republic of Korea
7 Institute of Molecular Biology and Genetics, Seoul National University, Seoul, Republic of Korea
8 College of Pharmacy and Research Institute of Pharmaceutical Sciences, Gyeongsang National University, Jinju, Republic of Korea
9 Department of Pharmacology, Institute of Medical Sciences, College of Medicine, Gyeongsang National University, Jinju, Republic of Korea
10 Department of Convergence Medical Science, College of Medicine, Gyeongsang National University, Jinju, Republic of Korea
11 KNU G-LAMP Project Group, KNU Institute of Basic Sciences, Kyungpook National University, Daegu, Republic of Korea
12 BK21 FOUR KNU Creative BioResearch Group, School of Life Science and Biotechnology, Kyungpook National University, Daegu, Republic of Korea
13 S&K Therapeutics, Ajou University, Suwon, Republic of Korea
14 Department of Molecular Science and Technology, Ajou University, Suwon, Republic of Korea
* Corresponding Authors: Dong Kyu Choi. Email: ; Sangdun Choi. Email:
; Hyuk-Kwon Kwon. Email:
# These authors contributed equally to this work
BIOCELL 2026, 50(3), 12 https://doi.org/10.32604/biocell.2026.074555
Received 13 October 2025; Accepted 19 January 2026; Issue published 23 March 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Objective: Cisplatin is a widely used chemotherapeutic agent due to its ability to damage DNA in the treatment of cancer. However, its clinical application is often limited by adverse effects on normal tissues, especially the kidneys. Understanding the molecular mechanisms of cisplatin-induced nephrotoxicity is crucial for developing strategies to mitigate its side effects. In this study, we aimed to elucidate the molecular mechanisms underlying cisplatin-induced DNA damage and apoptosis in human renal epithelial cells, with a focus on key signaling pathways and mediators that drive nephrotoxicity. Methods: To explore these mechanisms, human proximal tubule epithelial cells (HK-2) were treated with cisplatin. The study assessed DNA damage response (DDR) and stress-related protein expression, cell cycle distribution, and apoptosis. Activation of mitogen-activated protein kinases (MAPKs), particularly Extracellular signal-regulated Kinase (ERK), was analyzed, along with the expression and functional role of activating transcription factor 3 (ATF3) and tumor protein p53 (p53). Results: Cisplatin treatment upregulated DDR and stress response proteins, induced S phase arrest, and increased the SubG1 population, indicating apoptotic cell death. ERK was identified as a critical mediator of cisplatin-induced DNA damage and stress responses. ATF3 expression was significantly elevated in an ERK-dependent manner and required p53 activation. Knockdown of ATF3 reduced cisplatin-induced DNA damage, highlighting its role in the cytotoxic response. Conclusions: Cisplatin induces nephrotoxicity through ERK- and p53-dependent upregulation of ATF3, which is associated with DNA damage and cell death, suggesting a modulatory role in the cellular stress response. These findings provide novel insights into the molecular basis of cisplatin-induced renal injury and suggest potential therapeutic targets to alleviate its adverse effects.Keywords
Supplementary Material
Supplementary Material FileCisplatin, a platinum-based chemotherapeutic agent, is widely used for treating various malignancies, including testicular, ovarian, and cervical cancers, and other solid tumors [1,2]. Its anticancer effect primarily arises from the formation of intra- and inter-strand deoxyribonucleic acid (DNA) cross-links, especially between adjacent guanine bases. These DNA adducts interfere with essential cellular processes such as DNA replication and RNA transcription, ultimately leading to cell cycle arrest and apoptosis in rapidly dividing tumor cells [2,3]. The resulting DNA damage also activates canonical DNA damage response (DDR) pathways, such as H2AX at Ser139 (γ-H2AX), cleavage of poly (ADP-ribose) polymerase 1 (C-PARP1), phosphorylation of ataxia telangiectasia mutated (ATM) at Ser1981 (p-ATM), and phosphorylation of ataxia telangiectasia and Rad3-related protein (ATR) at Ser428 (p-ATR), which coordinate DNA repair, cell cycle control, MAPK activation, and programmed cell death [3–5].
Despite its clinical efficacy, cisplatin therapy is frequently limited by toxicity to normal tissues, with nephrotoxicity being the most prominent adverse effect. Cisplatin-induced kidney injury often manifests as acute kidney injury or long-term renal impairment [2,3,6–8]. Accumulation of cisplatin in proximal tubule cells, mediated by transporters like organic cation transporter 2 (OCT2), exacerbates its renal toxicity [9]. Mechanistically, nephrotoxicity involves oxidative stress, mitochondrial dysfunction, inflammatory signaling, and programmed cell death [10]. However, the therapeutic application of cisplatin is often hampered by dose-limiting toxicities to normal tissues [8]. Among its adverse effects, nephrotoxicity remains the most prominent and clinically relevant, frequently resulting in acute kidney injury and long-term renal impairment.
Key regulators of cisplatin nephrotoxicity include tumor protein p53 (p53) and mitogen-activated protein kinases (MAPKs). p53 senses DNA damage and orchestrates cell cycle arrest, DNA repair, and apoptosis in tumor and kidney cells [11–14]. Its activation can be supported by upstream signaling, such as the eEF2 kinase pathway, enhancing p53 stability in response to cisplatin-induced DNA lesions [15]. MAPKs, including extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinases (JNK), and p38 MAPKs (p38), are critically involved in mediating cellular responses to cisplatin-induced DNA damage [11,16,17].
Emerging evidence identifies activating transcription factor 3 (ATF3) as a nodal integrator of p53- and ERK-mediated DDR. ATF3 is transcriptionally induced by stress pathways and modulates apoptotic signaling and inflammatory response following DNA damage [18–20]. Its roles in ferroptosis, inflammation, and cancer biology further suggest a broader function in cellular stress regulation [21]. However, the contribution of ATF3 to cisplatin-induced renal injury remains largely unexplored.
Previous studies indicate that sustained p53 activation drives tubular apoptosis, whereas ERK signaling supports transcriptional reprogramming under stress and coordinates autophagy and apoptosis [10]. Therapeutic interventions targeting these pathways, including antioxidants or MAPK inhibitors, have shown potential in mitigating renal damage [22]. Nevertheless, the precise molecular regulators within these pathways, such as ATF3, have yet to be fully elucidated.
ATF3 belongs to the ATF/cAMP family and is rapidly induced by various cellular stresses, including DNA damage, oxidative stress, and inflammation [23–25]. It plays context-dependent roles in cell survival, apoptosis, and immune regulation, maintaining intracellular homeostasis under physiological and pathological conditions [19,23,26,27]. ATF3 also interacts with transcriptional regulators, such as nuclear factor-κB (NF-κB), thereby modulating inflammatory and apoptotic pathways. Prior studies have shown that ATF3 influences cancer cell growth, stemness, and ferroptosis, highlighting its potential as a therapeutic target beyond cancer biology [28–32].
In this study, we aimed to elucidate the molecular mechanisms underlying cisplatin-induced nephrotoxicity. Specifically, we investigated the roles of MAPKs and p53 in mediating DNA damage, cell cycle regulation, and cell death, and further examined the upstream regulatory mechanisms of ATF3 expression and its contribution to cisplatin-induced DNA damage in human proximal tubule epithelial cells.
HK-2 human normal kidney cells (CRL-2190, American Type Culture Collection [ATCC], Manassas, VA, USA), which were STR-authenticated and confirmed to be mycoplasma-free. Cells were cultured in 10 cm dishes using serum-free keratinocyte medium (ATCC) supplemented with 1% penicillin/streptomycin (P/S; Thermo Fisher Scientific, Waltham, MA, USA), in a humidified atmosphere containing 5% CO2 at 37°C. The culture medium was changed every other day, and cells were trypsinized using 0.05% trypsin-EDTA solution for 3 min to obtain a single-cell suspension. Dissociated cells were harvested by centrifugation at 200 × g at 4°C for 3 min using a centrifuge.
Cisplatin (15663-27-1, Sigma-Aldrich, St. Louis, MO, USA) was prepared as a 10 mM stock solution by dissolving the powder in dimethyl sulfoxide (DMSO; D8418, Sigma-Aldrich, St. Louis, MO, USA) and stored at −20°C in dark brown tubes. The pharmacological p53 inhibitor PFT-α (P4359, Sigma-Aldrich, St. Louis, MO, USA) was also prepared as a 10 mM stock solution in DMSO and stored under the same conditions. Pharmacological MAPK inhibitor—p38 inhibitor (SB203580; S8307, Sigma-Aldrich, St. Louis, MO, USA), JNK inhibitor (SP600125; S5567, Sigma-Aldrich, St. Louis, MO, USA), and ERK inhibitor (U0126; 662005, Sigma-Aldrich, St. Louis, MO, USA)—were each prepared as 10 mM stock solutions in DMSO.
HK-2 cells were seeded into 6 cm dishes at a density of 1 × 106 cells per dish. After overnight incubation, the medium was replaced, and PFT-α (20 μM), SP600125 (20 μM), U0126 (20 μM), or SB203580 (10 μM) were added to the fresh medium. One hour later, cells were treated with cisplatin at 10 or 50 μM for 24 or 72 h. For the dose-dependent experiment, cells were treated with cisplatin at 6.25, 12.5, 25, or 50 μM for 24 h. For the time-course experiment, cells were treated with 50 μM cisplatin for 6, 12, or 24 h.
Cell viability was assessed using the CellTiter 96® AQueousOne Solution Cell Proliferation Assay (Promega, Madison, WI, USA). Cells (1 × 104/well) were seeded into 96-well plates, allowed to stabilize overnight, and then treated with cisplatin for 24 h. At the end of the treatment period, 10 μL of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) solution was added to each well, and the cells were incubated for 3 to 4 h at 37°C. Optical density (OD) was measured at 490 nm using a microplate spectrophotometer (VersaMax, Molecular Devices, San Jose, CA, USA). The O.D. values were used to calculate relative cell viability (%) using the following formula:
Cells were plated at a density of 1 × 105 per 6 cm dish and allowed to grow to approximately 70% confluence. After replacing the culture with fresh medium (serum-free keratinocyte medium; 10744019, Thermo Fisher Scientific, Waltham, MA, USA), cells were treated with cisplatin under the indicated conditions (dose-dependent: 6.25–50 μM for 24 h; time-dependent: 50 μM for 6, 12, or 24 h). For the inhibitor experiment, cells were pretreated with the inhibitors described in Section 2.2 before cisplatin exposure (10 or 50 μM for 24 or 72 h). Following treatment, cells were collected, washed with 1 mL of phosphate-buffered saline, and centrifuged at 200× g at 4°C for 3 min using a benchtop centrifuge. The cell pellet was resuspended in a solution of 70% cold ethanol diluted in PBS for fixation, and the cells were stored at 4°C overnight. The following day, fixed cells were washed with 1 mL of PBS and centrifuged again at 200 × g at 4°C for 3 min. Cells were then resuspended in 1 mL of staining solution with propidium iodide (PI, 50 μg/mL; 537060, Sigma-Aldrich, St. Louis, MO, USA) and RNase A (500 μg/mL; 10109169001, Sigma-Aldrich, St. Louis, MO, USA) for 15 min at room temperature in the dark. For validation of cell cycle analysis, unstained controls and PI single-stained controls were included to set flow cytometry gates and exclude autofluorescence. RNase A treatment was applied to all samples to eliminate RNA-derived PI signals. In addition, cisplatin-treated cells were positive controls for cell cycle arrest. Flow cytometric analysis was performed using a FACSAria™ III system (BD Biosciences, San Jose, CA, USA) with Diva™ software (v8.0, BD Biosciences, San Jose, CA, USA). PE channel (excitation 488 nm, emission 585/42 nm). For each sample, at least 20,000 events were collected. Performed based on forward scatter (FSC) and side scatter (SSC) to exclude debris and select single-cell populations. Doublets were further excluded using FSC-A vs. FSC-H gating. After debris and doublet exclusion, 15,000 single–cell events per sample were used for analysis.
Cell cycle distribution (G0/G1, S, and G2/M) was analyzed using FlowJo software (Ver.10.0.7; BD Bioscience, San Jose, CA, USA). Peak fitting was performed using the Watson pragmatic model with automatic background subtraction and constrained fitting parameters. All experiments were performed using three independent biological replicates (n = 3).
2.5 Confocal Microscopy Analysis
Round coverslips (SPL Lifescience) were sterilized and placed in the wells of a 24-well plate. HK-2 cells (4 × 104 cells/well) were seeded onto the coverslips and incubated overnight in a humidified incubator at 37°C with 5% CO2. When cells reached approximately 70% confluence, the culture medium was replaced with serum-free keratinocyte medium (10744019, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 1% P/S (15070063, Thermo Fisher Scientific, Waltham, MA, USA), and cells were treated with cisplatin (50 μM) for 12 or 24 h. After the cisplatin treatment for 12 or 24 h, cells were fixed with 3.7% formaldehyde for 15 min at room temperature (25°C), followed by three washes with PBS. Fixed cells were permeabilized with 0.2% Triton X-100 for 15 min at room temperature and washed three times with PBS. They were then blocked with 10% fetal bovine serum (FBS; 26140079, Thermo Fisher Scientific, Waltham, MA, USA, diluted in PBS) for 1 h at room temperature, followed by three additional PBS washes. Cells were incubated with primary antibodies against γ-H2AX (1:500) and PARP1 (1:1000) in blocking buffer for 1 h at room temperature. After three PBS washes, cells were incubated with Alexa Fluor® 488- or 546-conjugated secondary antibodies (1:1000) in blocking buffer for 1 h at room temperature in the dark, protected from light. For nuclear staining, cells were incubated with Hoechst 33258 (94403, 5 μM; Sigma-Aldrich, St. Louis, MO, USA) for 10 min at room temperature. Images were acquired using a laser-scanning confocal microscope (LSM-700, Carl Zeiss Microscopy GmbH, Jena, Germany) using appropriate excitation wavelengths (405, 488, and 594 nm) at 600× magnification and analyzed with Zen 2009 software.
2.6 Cellomics ArrayScan HCS Reader Analysis
Cells (5 × 103/well) were seeded in black clear-bottom 96-well plates (Greiner Bio-One, Frickenhausen, Austria) and treated with 50 μM cisplatin for 12 h. Cells were fixed with 3.7% formaldehyde for 15 min and permeabilized with 0.2% Triton X-100 for 15 min. Subsequently, cells were blocked with 5% fetal bovine serum in PBS for 1 h and incubated with primary antibodies against γ-H2AX (1:1000) and phospho-ATM (1:1000) for 1 h at room temperature. After washing, cells were incubated with Alexa Fluor® 488- or 546-conjugated secondary antibodies (1:1000; Invitrogen) for 1 h at room temperature. Nuclei were stained with Hoechst 33258 reagent (94403, 5 μM; Sigma-Aldrich, St. Louis, MO, USA) for 15 min. Stained cells were imaged using a Cellomics ArrayScan VTI HCS Reader (Thermo Fisher Scientific), and fluorescence intensities were quantified using ArrayScan® VTI (Version 6.6.1.3, 600 series; Thermo Fisher Scientific) software in at least 200 cells per condition.
Cell pellets were lysed using M-PER extraction solution (78501, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with protease and phosphatase inhibitor cocktails (Thermo Fisher Scientific, Waltham, MA, USA) for 10 min at 4°C. The lysates were then centrifuged at 16,000 × g for 10 min at 4°C. Protein concentrations were determined using the Pierce™ BCA Protein Assay Kits (23225, Thermo Fisher Scientific, Waltham, MA, USA), and values were calculated based on a BSA standard curve. Equal amounts of protein (20–60 μg; 60 μg for phospho form targets and 20 μg for other targets) were separated by 10%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; 12% SDS-PAGE Gel for low-molecular-weight proteins and 10% SDS-PAGE Gels for high molecular weight proteins), followed by electrophoretic transfer to 0.45 μm nitrocellulose membranes. Membranes were blocked with 5% nonfat dry milk in deionized water for 1 h at room temperature (25°C), then incubated overnight at 4°C with primary antibodies specific to ERK (1:1000), p-ERK (1:1000), p-p38 (1:1000), and p-JNK (1:1000), or ATF3 (1:1000), γ-H2AX (1:1000), p-ATR (1:500), p-p53 (1:1000), p-ATM (1:500), PARP1 (1:1000), Bcl2 (1:1000), and β-actin (1:1000). After washing with Tris-buffered saline containing Tween® 20 (TBST), the membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit IgG secondary antibodies (1:1000) for 2 h at room temperature. Detailed information on the antibodies used for Western blot analysis, including sources, catalog numbers, and working dilutions, is provided in Supplementary Table S1.
Immunoreactive bands were visualized using ECL Western Blotting Substrate (34580, Thermo Fisher Scientific, Waltham, MA, USA) and imaged with the Fuji LAS-3000 system (Fuji Film Life Science, Tokyo, Japan). The band intensity ratio was normalized to β-actin for target proteins, and C-PARP1 was normalized to full-length PARP1, followed by quantification using ImageJ software (ImageJ v1.54p; Wayne Rasband, National Institutes of Health, Bethesda, MD, USA) [33].
2.8 ATF3 siRNA Design and Transfection
siRNA was designed and purchased from Genolution Pharmaceuticals (Seoul, Republic of Korea). The siRNA sequences used in this study have been previously reported and validated in the literature [34]. The siRNA sequences were as follows: control-siRNA, 5′-CTGAAGGCTCAGATTGAGGAGC-3′; ATF3-siRNA, 5′-CTGTGAGATAAGCGGGACTCAG-3′. G-Fectin reagent (Genolution Pharmaceuticals) was used for siRNA transfection. HK-2 cells were seeded at a density of 1 × 106 cells per 6 cm dish in 2 mL of serum-free keratinocyte medium and incubated for 12 h until the cells were fully adhered and reached approximately 70% confluence. A mixture of 10 nM siRNA and 4 μL of G-Fectin in 200 μL of PBS was incubated at room temperature for 10 min and then added to the cells. Transfection efficiency was evaluated by assessing the reduction of ATF3 protein expression using Western blot analysis. After 48 h of transfection, the medium was replaced with fresh serum-free keratinocyte medium (10744019, Thermo Fisher Scientific, Waltham, MA, USA), and the cells were used for subsequent experiments.
All statistical analyses were performed using GraphPad Prism, version 8.01 (GraphPad Software, San Diego, CA, USA). Depending on the experimental design, statistical significance was assessed by Student’s t-test, one-way ANOVA with Dunnett’s multiple comparisons test (variance homogeneity verified by Bartlett’s test), or two-way ANOVA with Bonferroni’s post hoc test. All experiments were independently repeated at least three times, and p < 0.05 was considered statistically significant.
3.1 Cisplatin Induces DNA Damage and Cell Cycle Arrest in a Dose-Dependent Manner
Based on previous findings, we investigated molecular alterations associated with DNA damage responses in human proximal tubule epithelial cells following cisplatin treatment. Cisplatin increased cytotoxicity in a dose-dependent manner, and a concentration of 50 μM approximately induced half-maximal inhibitory concentration (IC50) compared to the control (Fig. 1A). At low concentrations, cisplatin induced cell cycle arrest at the S phase, whereas increasing concentrations led to a rise in the SubG1 population, which is indicative of DNA damage and apoptotic cell death, accompanied by a decrease in the proportions of cells in the S and G2/M phases (Fig. 1B). Cisplatin increased the expression of DDR-associated proteins, including γ-H2AX, cleaved PARP1, and p-ATM, in a concentration-dependent manner, whereas p-ATR expression was reduced (Fig. 1C). Notably, lower concentrations of cisplatin enhanced ATM phosphorylation, but p-ATM levels decreased at 50 μM, in parallel with the induction of γ-H2AX and cleaved PARP1. These findings indicate that excessive DNA damage at this high dose triggers apoptosis, as evidenced by increased PARP1 cleavage, which may contribute to reduced ATM abundance and consequently attenuated ATM signaling. Consistent with these changes, γ-H2AX, a sensitive and early marker of DNA double-strand breaks, was shown to be upregulated within the nucleus (Fig. 1D). Phosphorylation of p38 (p-p38) and ERK (p-ERK), members of the MAPKs proteins, was induced by cisplatin treatment in a pattern similar to that of DDR-related proteins, whereas phosphorylation of JNK (p-JNK) was reduced. In addition, the expression of B-cell lymphoma 2 (Bcl2), a key protein located on the outer mitochondrial membrane that regulates apoptotic cell death, was decreased, while the expression of ATF3, a known cellular stress marker, was increased. Taken together, we identified that cisplatin, a DNA-damaging chemotherapeutic agent, induces DNA damage accompanied by cell cycle arrest.
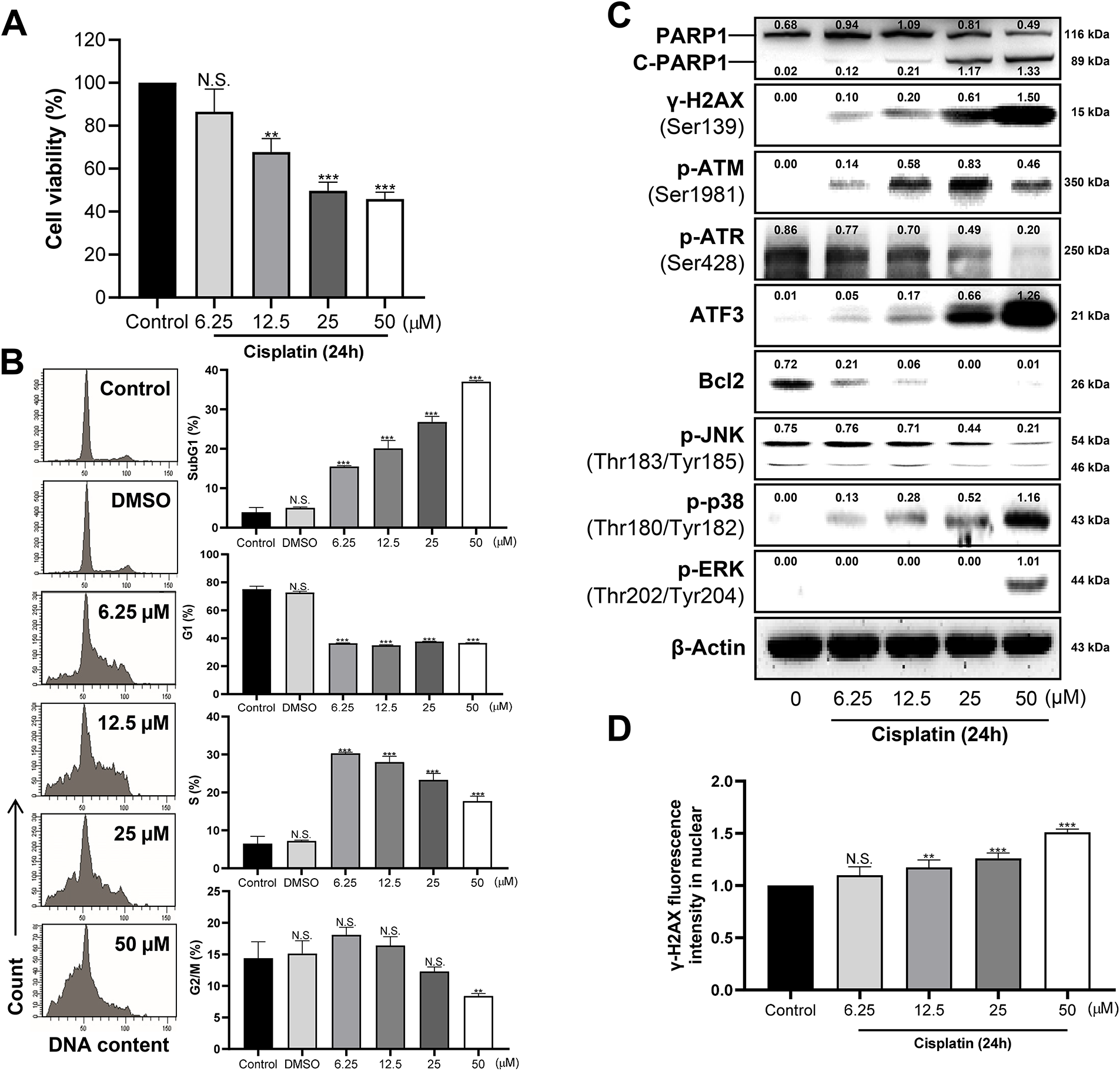
Figure 1: Dose-dependent effects of cisplatin on cell cycle arrest and DNA damage response (DDR). (A) Cell viability was decreased in a dose-dependent manner. (B) Cell cycle distribution changes with increasing cisplatin concentration. Histograms represent the mean percentage of cells in each cell cycle phase. (C) Whole-cell lysates were analyzed by western blotting for the expression of DDR and apoptosis-related proteins: Poly(ADP-ribose) polymerase1 (PARP1), cleaved PARP1 (C-PARP1), γ-H2A histone family member X (H2AX; Ser139), phospho-ataxia-telangiectasia mutated (p-ATM; Ser1981), phospho-ataxia telangiectasia and Rad3-related protein (p-ATR; Ser428), Activating transcription factor 3 (ATF3), B-cell lymphoma 2 (Bcl2), c-Jun N-terminal kinase (p-JNK; Thr183/Tyr185), phospho- mitogen-activated protein kinase 14 (p-p38; Thr180/Tyr182), and phospho-extracellular signal-regulated kinase (p-ERK; Thr202/Tyr204). β-Actin was used as a loading control. (D) Nuclear expression of γ-H2AX was quantified using the Cellomics ArrayScan HCS Reader following immunofluorescence staining (≥200 cells). Nuclear intensity was assessed using Hoechst 33258 staining. All experiments were independently performed in triplicate. Statistical significance was determined by one-way ANOVA with Dunnett’s test; variance homogeneity was assessed by Bartlett’s test. Statistical significance is indicated as follows: N.S., not significant; **p < 0.01; ***p < 0.001.
3.2 Cisplatin Induces the Expression of DDR Proteins and Causes S-Phase Arrest during Apoptotic Cell Death
Based on the dose-dependent experimental results, we selected 50 μM, corresponding to the IC50 concentration, and investigated DDR in a time-dependent manner. Consistent with the dose-dependent findings, cisplatin treatment resulted in a sustained increase in the proportions of cells in the SubG1 and S phases, while those in the G1 and G2/M phases gradually decreased (Fig. 2A). Similarly, the expression levels of DDR-associated proteins such as γ-H2AX, C-PARP1, and p-ATM, as well as MAPK proteins, including p-ERK and p-p38, were markedly elevated (Fig. 2B). These changes became significant at 12 h post-treatment and reached the highest levels observed within 24 h within the examined time course. (Fig. 2C,D). Notably, the increase in the SubG1 population, a hallmark of apoptotic cell death, was accompanied by elevated levels of C-PARP1, a substrate cleaved by caspase-3, further supporting apoptosis induction. In addition, expression of the cellular stress marker ATF3 was significantly increased, showing a temporal pattern similar to DDR-associated proteins. Taken together, we established an in vitro model using human proximal tubule epithelial cells to evaluate chemotherapeutic drug-induced nephrotoxicity, demonstrating that cisplatin induces DNA damage, cell cycle arrest, and apoptotic cell death.
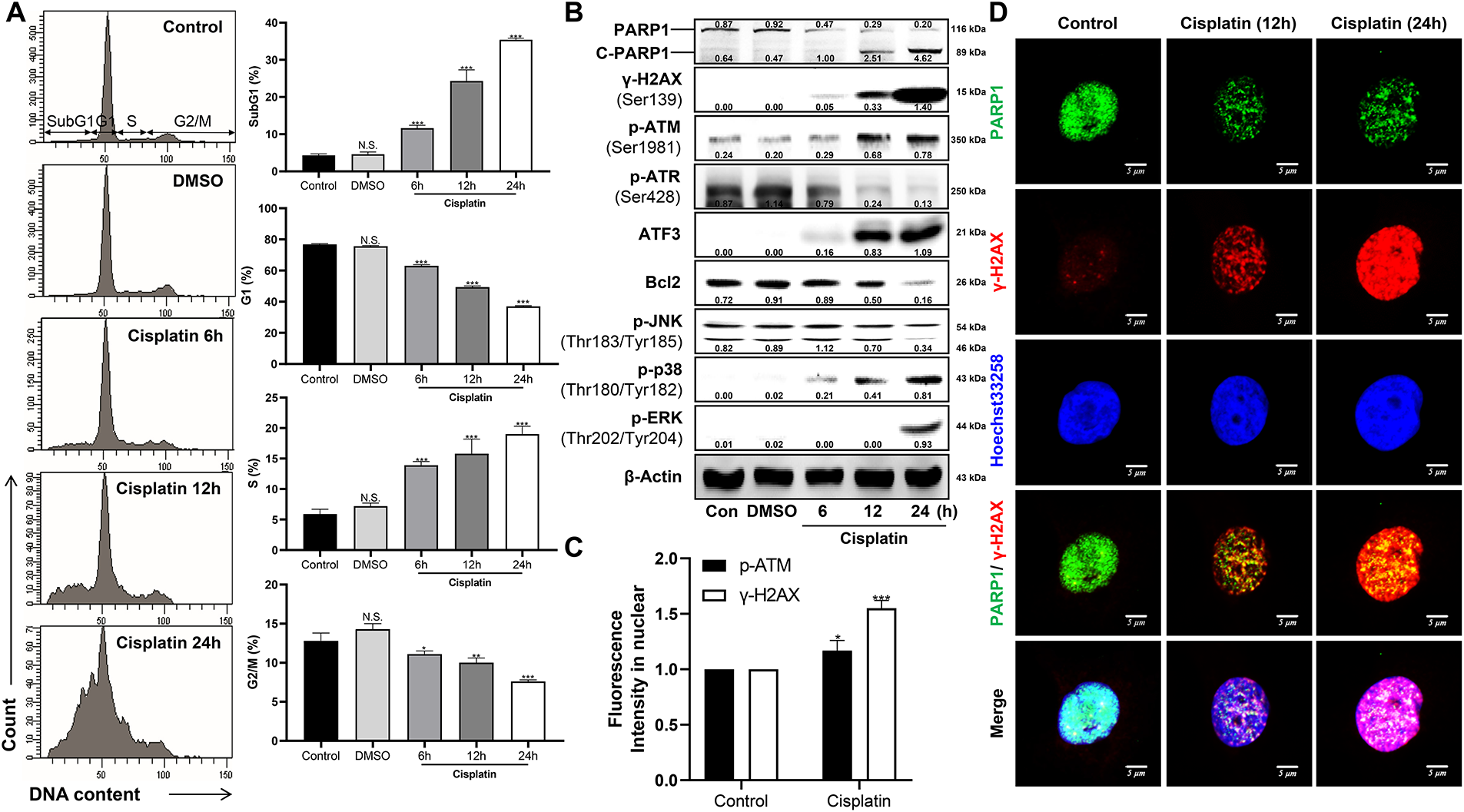
Figure 2: Time-dependent effects of cisplatin on cell cycle arrest, DDR, mitogen-activated protein kinases (MAPKs), and ATF3 expression. (A) Cell cycle distribution was analyzed by flow cytometry following propidium iodide staining. Histograms represent the mean percentage of cells in each phase of the cell cycle. (B) Whole-cell lysates were subjected to western blot analysis to detect the expression levels of PARP1, cleaved PARP1 (C-PARP1), γ-H2AX (Ser139), p-ATM (Ser1981), p-ATR (Ser428), ATF3, Bcl2, p-JNK (Thr183/Tyr185), p-p38 (Thr180/Tyr182), and p-ERK (Thr202/Tyr204). β-Actin was used as a loading control. (C) Nuclear expression levels of γ-H2AX and p-ATM were quantified using the Cellomics ArrayScan HCS Reader following immunofluorescence staining (≥200 cells). Nuclear intensity was measured using Hoechst 33258 staining. (D) Representative confocal images of immunofluorescent staining for γ-H2AX (red), PARP1 (green), and nuclei (Hoechst 33258, blue) following cisplatin treatment. All experiments were independently repeated at least three times. Statistical significance was determined by one-way ANOVA with Dunnett’s test; variance homogeneity was assessed by Bartlett’s test. Statistical significance is indicated as follows: N.S., not significant; *p < 0.05; **p < 0.01; ***p < 0.001.
3.3 Cisplatin Induces the Expression of DDR and ATF3, as well as Apoptotic Cell Death, in an ERK-Dependent Manner
Based on our previous findings that cisplatin induced the activation of MAPKs, including p-ERK and p-p38, we hypothesized that these pathways may contribute to cisplatin-induced responses, including DNA damage. Therefore, we investigated the effects of pharmacological MAPK inhibitors, including SP600125 (JNK inhibitor), SB203580 (p38 inhibitor), and U0126 (ERK inhibitor). The cisplatin-induced SubG1 population, indicative of DNA damage and apoptotic cell death, was generally reduced by MAPK inhibitors. In contrast, the increased S phase population was unaffected by JNK inhibition but was further elevated by the inhibition of p38 and ERK (Fig. 3A). Similarly, inhibitors of p38 and ERK suppressed the expression of cisplatin-induced markers, including γ-H2AX (a DNA damage marker), ATF3 (a cellular stress marker), and C-PARP1 (an apoptosis marker), whereas the JNK inhibitor had minimal effect (Fig. 3B,C). ERK inhibition most effectively reduced cisplatin-induced DNA damage, cellular stress, and apoptosis regardless of cisplatin concentration or treatment duration (Fig. 3D,E). Taken together, our findings suggest that cisplatin-induced DNA damage, cellular stress responses, and apoptotic cell death are mostly ERK-dependent, whereas S phase cell cycle arrest occurs independently of ERK signaling.
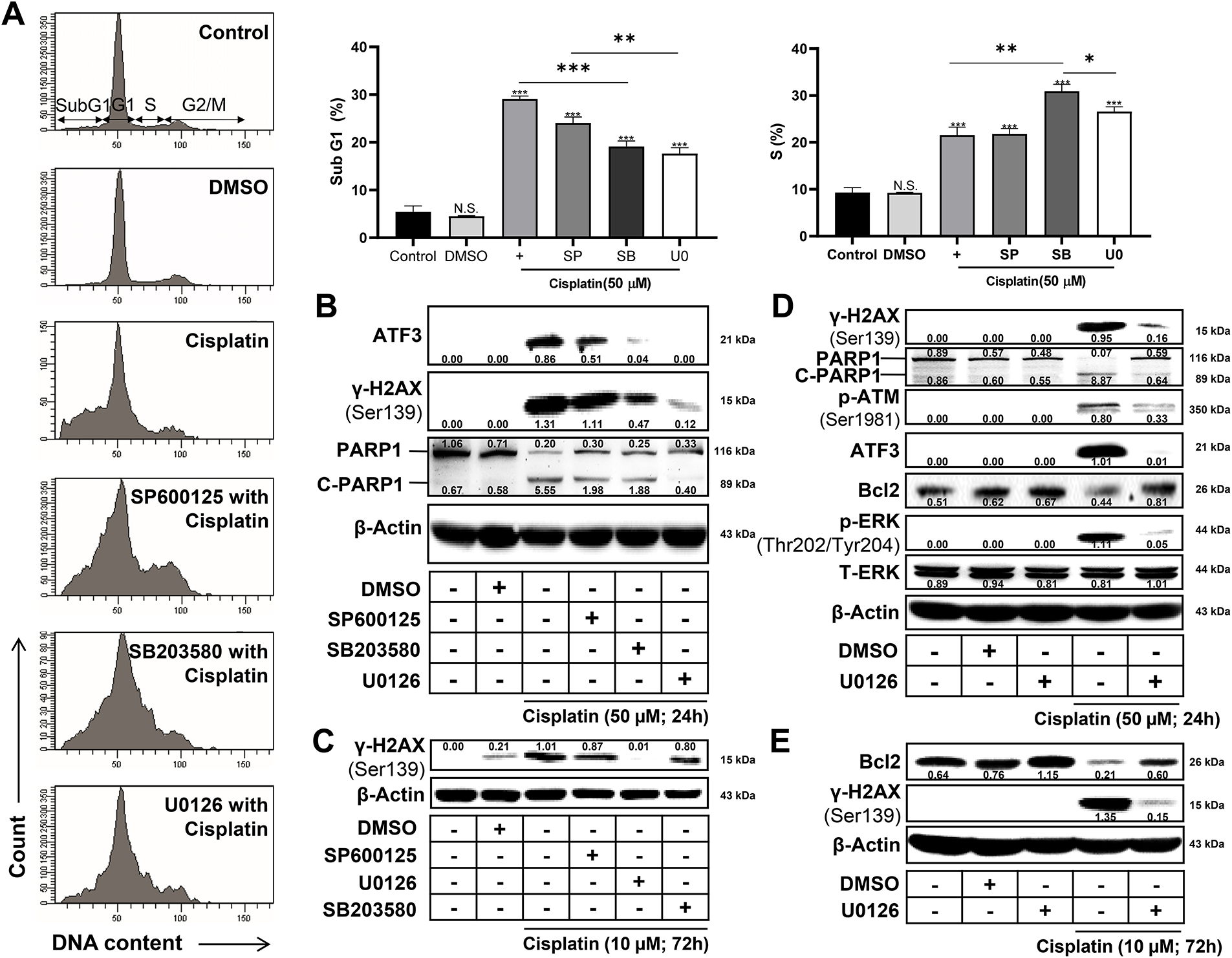
Figure 3: Inhibition of ERK attenuates cisplatin-induced DNA damage, ATF3 expression, and cell death. (A) Cell cycle distribution was analyzed by flow cytometry using propidium iodide staining, showing the mean percentage of cells in the SubG1 and S phases. (B) Western blot analysis of ATF3, γ-H2AX, and cleaved PARP1 after 50 μM cisplatin treatment for 24 h. ERK inhibition reduced the cisplatin-induced expression of DNA damage and apoptosis markers. (C) Western blot analysis of ATF3, γ-H2AX, and cleaved PARP1 after 10 μM cisplatin treatment for 72 h. U0126 suppressed late-stage DNA damage and apoptotic signaling. (D) Western blot analysis of DNA damage markers (γ-H2AX (Ser139), p-ATM (Ser1981)), apoptosis marker (cleaved PARP1), stress response marker (ATF3), anti-apoptotic marker (Bcl2), and ERK signaling proteins (p-ERK (Thr202/Tyr204), total ERK) in cells pretreated with an ERK inhibitor and subsequently treated with 50 μM cisplatin for 24 h. (E) Western blot analysis of DNA damage marker (γ-H2AX (Ser139)) and anti-apoptotic marker (Bcl2) in cells pretreated with an ERK inhibitor followed by 10 μM cisplatin for 72 h. β-Actin served as a loading control. All experiments were independently performed at least three times. Statistical significance was determined by one-way ANOVA with Dunnett’s test (variance homogeneity assessed by Bartlett’s test) or two-way ANOVA with Bonferroni’s post hoc test. Statistical significance is indicated as follows: N.S., not significant; *p < 0.05; **p < 0.01; ***p < 0.001.
3.4 Cisplatin Induces p53-Dependent DDR and Apoptotic Cell Death, along with the Upregulation of ATF3, Which Contributes to DDR
We investigated the role of p53 in cisplatin-induced cell cycle arrest, DDR, and apoptotic cell death using a pharmacological p53 inhibitor, pifithrin-α (PFT-α). Inhibition of p53 reduced the cisplatin-induced SubG1 population and the expression of C-PARP1, thereby suppressing apoptotic cell death, while not affecting cisplatin-induced S phase arrest (Fig. 4A,B). In addition, p53 inhibition increased both the G1 and G2/M population under cisplatin treatment, indicating altered cell cycle checkpoint control. Furthermore, p53 inhibition attenuated the expression of γ-H2AX and ATF3, markers of DNA damage and cellular stress, respectively.
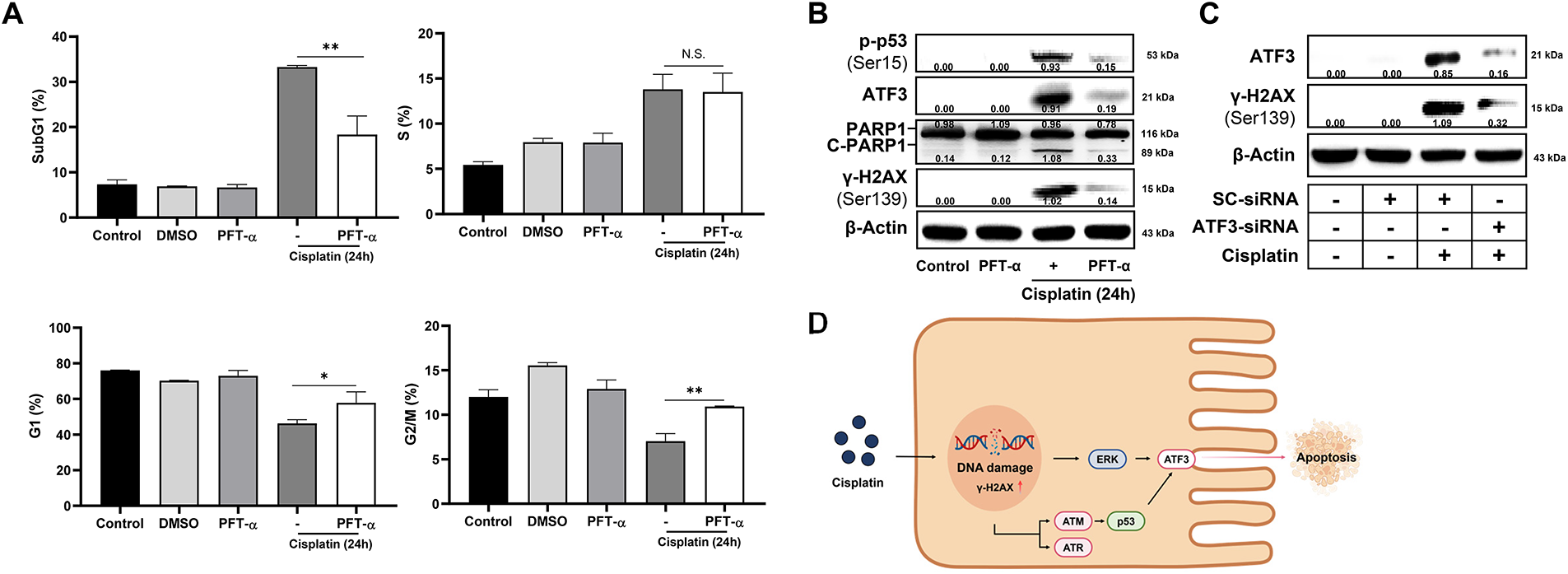
Figure 4: Cisplatin induces p53-dependent DDR and apoptotic cell death, accompanied by ATF3 upregulation. HK-2 cells were co-treated with cisplatin (50 μM) and PFT-α (p53 inhibitor, 20 μM) for 24 h. (A) Cell cycle distribution was analyzed by flow cytometry using propidium iodide staining. Histograms represent the mean percentage (%) of cells in each phase of the cell cycle. (B) Whole-cell lysates were subjected to western blot analysis for phosphorylated p53 (Ser15), ATF3, PARP1, cleaved PARP1 (C-PARP1), and γ-H2AX (Ser139). (C) HK-2 cells were transfected with either scrambled (SC)-siRNA or ATF3-siRNA for 36 h, followed by treatment with DMSO (control) or cisplatin (50 μM) for 24 h. Whole-cell lysates were analyzed by western blotting for ATF3 and γ-H2AX (Ser139). β-Actin was used as a loading control. (D) Diagram of cisplatin-induced DNA damage in renal epithelial cells via ERK- and p53-mediated ATF3. All experiments were independently performed three times. Statistical significance was determined by one-way ANOVA with Dunnett’s test (variance homogeneity assessed by Bartlett’s test) or two-way ANOVA with Bonferroni’s post hoc test. Statistical significance is indicated as follows: N.S., not significant; *p < 0.05; **p < 0.01. The schematic diagram in the panel was illustrated using Biorender.
Based on the finding that cisplatin-induced DNA damage, apoptosis, and ATF3 expression are modulated by both ERK and p53, we hypothesized that ATF3 may play a functional role in mediating cisplatin-induced DNA damage. Supporting this hypothesis, knockdown of ATF3 resulted in a marked reduction in γ-H2AX expression, indicating that ATF3 may modulate the cellular response to the DNA damage response (Fig. 4C). Furthermore, the schematic illustrated model that cisplatin-induced DNA damage activates ERK and p53, both of which enhance ATF3 expression, while ATF3 subsequently promotes apoptotic cell death in response to DNA damage (Fig. 4D). Taken together, these findings suggest that p53 mediates cisplatin-induced DNA damage and apoptotic cell death, at least in part, by promoting ATF3 expression, which appears to correlate with DNA damage rather than directly causing it.
In this study, we investigated the molecular mechanisms underlying cisplatin-induced nephrotoxicity in HK-2 cells, focusing on the coordinated activation of the p53 and ERK pathways, which converge on ATF3 to regulate DNA damage responses and apoptotic signaling. In our experiments, the IC50 of cisplatin in HK-2 cells was determined to be approximately 50 μM, a concentration that induced dose-dependent cytotoxicity, DNA damage, S phase arrest, and apoptotic cell death. We demonstrated that ATF3 is induced in a p53- and ERK-dependent manner and functions as a stress-responsive modulator of genotoxic stress, highlighting its role as a critical mediator of renal epithelial injury.
Cisplatin is known to cause DNA lesions by forming intra- and inter-strand crosslinks between adjacent guanine bases, leading to replication fork arrest, transcriptional inhibition, and ultimately, cell death [1–3,8]. Consistent with this, we observed dose- and time-dependent increases in DDR markers such as γ-H2AX, p-ATM, and C-PARP1, as well as cell cycle arrest and SubG1 accumulation in HK-2 cells. Recent studies have further elucidated the complexity of cisplatin-induced DDR, with eEF2 kinase supporting p53 activation and coordinating ERK-mediated response to DNA damage [15]. Our findings are consistent with these observations, as inhibition of p53 resulted in reduced γ-H2AX, C-PARP1, and ATF3 expression. This suggests that p53 plays a central role in orchestrating the DNA damage response and apoptotic signaling underlying cisplatin-induced nephrotoxicity.
Among the MAPKs, ERK played a predominant role in mediating cisplatin-induced cytotoxicity, as ERK inhibition significantly reduced DDR and apoptotic markers, including ATF3. In contrast, p38 inhibition had modest effects, while JNK activity was suppressed after cisplatin treatment, indicating a context-dependent MAPK response in renal epithelial cells. This is consistent with recent findings that ERK signaling modulates apoptosis in response to cisplatin, influencing cellular outcomes in renal cells [10]. Moreover, ERK’s role in cisplatin nephrotoxicity has been linked to its regulation of inflammatory and oxidative stress pathways, which are critical in renal tubular injury [22]. p53 is a key transcription factor that regulates various intracellular signaling pathways, including DDR, cell cycle, oxidative stress, mitochondrial respiration, and apoptosis [35–37]. Our previous study demonstrated that cisplatin treatment enhances p53 activation in renal epithelial cells, and inhibition of p53 suppresses cell death, including both necrosis and apoptosis, along with a reduction in ROS production [38]. Consistent with our study, cisplatin robustly activated p53, and pharmacological inhibition of p53 attenuated the expression of γ-H2AX, C-PARP1, and ATF3, along with a reduction in the SubG1 population. These findings support the role of both p53 and ERK as key signaling mediators in cisplatin-induced nephrotoxicity.
Importantly, our results suggest that ATF3 functions downstream of both p53 and ERK pathways under genotoxic stress, although the precise causal relationships remain to be fully elucidated. Knockdown of ATF3 reduced DDR marker expression, supporting its role as a stress-responsive modulator of DNA damage signaling in renal epithelial cells. As a rapid-response transcription factor induced by various cellular stresses, ATF3 may serve as a central integrator of DNA damage and apoptotic responses in renal epithelial cells [21,32]. Recent studies have highlighted ATF3’s multifaceted role in cellular stress response, including its regulation of ferroptosis and inflammation in cisplatin-treated cells, suggesting that ATF3 may amplify renal injury through multiple mechanisms [20,21]. Consistent with previous reports, our findings show that ATF3 induction accompanies cisplatin-induced DNA damage responses in renal epithelial cells. Therefore, ATF3 may act as a critical mediator of p53- and ERK-driven nephrotoxicity and represents a potential therapeutic target. Therapeutic approaches using antioxidants or MAPK inhibitors, such as piracetam or ketotifen, have shown promise in mitigating cisplatin-induced renal damage by targeting ERK and related pathways [22,39].
Despite the robust mechanistic insights provided, several limitations remain. First, the physiological relevance of our findings should be validated in animal models of cisplatin-induced acute kidney injury. Second, reliance on a single cell line, the lack of clear identification of ATF3 downstream targets, and the consequent reduction in transparency limit the generalizability of the conclusions. Third, the interplay between ATF3 and other stress pathways, including autophagy, lysosomal function, and ferroptosis, requires further exploration to clarify the broader regulatory networks involved in nephrotoxicity [40–42]. Recent evidence suggests that ATF3 modulates ferroptosis in cisplatin-treated cells, potentially contributing to renal tubular injury [20]. However, the downstream gene targets of ATF3 in renal epithelial cells remain unidentified and warrant comprehensive transcriptomic and promoter analyses. Moreover, ATF3 may serve as a central node integrating multiple forms of cell death and stress responses. By coordinating DNA damage-induced apoptosis with autophagy and ferroptosis, ATF3 could determine the overall cellular outcome under genotoxic stress. Future studies should investigate how ATF3 interfaces with autophagic machinery, lysosomal function, and lipid peroxidation processes to fully elucidate its role in renal injury and its potential as a therapeutic target for nephroprotection.
Moreover, ATF3 may serve as a central node integrating multiple forms of cell death and stress responses. By coordinating DNA damage-induced apoptosis with autophagy and ferroptosis, ATF3 could determine the overall cellular outcome under genotoxic stress. This positions ATF3 not merely as a downstream effector of p53 and ERK, but as a multifunctional regulator that links diverse stress pathways. Future studies should investigate how ATF3 interfaces with autophagic machinery, lysosomal function, and lipid peroxidation processes to fully elucidate its role in renal injury. Understanding these interactions would enhance the mechanistic insight and translational relevance of targeting ATF3 in nephroprotection.
In summary, our study demonstrates that ERK- and p53-mediated induction of ATF3 plays a key role in the DDR and apoptotic cell death triggered by cisplatin in renal epithelial cells. These findings provide mechanistic insights into cisplatin-induced nephrotoxicity and highlight potential molecular targets for the development of nephroprotective strategies in cancer therapy.
Acknowledgement: None.
Funding Statement: This work was supported by the research grant of Gyeongsang National University in 2023. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2025-00516213) and the Brain Pool Program of the National Research Foundation (NRF) of Korea funded by the Korea government (MSIT) (RS-2025-25439144). A Korea Basic Science Institute (National Research Facilities and Equipment Center) grant funded by the Ministry of Education (2022R1A6C10B724). This research was supported by the Regional Innovation System & Education (RISE) program through the RISE Center, Gyeongsangnam-do Provincial Government, Republic of Korea (2025-RISE-16-001). Learning & Academic research institution for Master’s·PhD students, and Postdocs (LAMP) Program of the National Research Foundation of Korea (NRF) grant funded by the Ministry of Education (RS-2023-00301974 and RS-2023-00301914).
Author Contributions: Writing—original draft: Semin Lee, MinJun Kim and Hyuk-kwon Kwon; Review & editing: Semin Lee, MinJun Kim, Seungmin Lee, Jiyun Yoo, Soo Seok Hwang, Seongchan Kim, Seung Pil Yun, Dong Kyu Choi, Sangdun Choi and Hyuk-Kwon Kwon; Conceptualization: Semin Lee, Jiyun Yoo, Soo Seok Hwang, Seung Pil Yun, Dong Kyu Choi, Sangdun Choi and Hyuk-Kwon Kwon; Methodology: Jiyun Yoo, Soo Seok Hwang, Seongchan Kim, Seung Pil Yun, Dong Kyu Choi and Sangdun Choi; Investigation: Semin Lee; Visualization: Semin Lee; Funding acquisition: Dong Kyu Choi, Sangdun Choi and Hyuk-kwon Kwon; Supervision: Hyuk-kwon Kwon; Project administration: Hyuk-kwon Kwon. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: The data that support the findings of this study are available from the Corresponding Author, Hyuk-kwon Kwon, upon reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest.
Supplementary Materials: Supplementary Table S1. Reagents, resources, and antibodies used in this study. The table lists cell lines, chemicals, reagents, instruments, and antibodies, along with their source, location, and catalog or identifier numbers. The supplementary material is available online at https://www.techscience.com/doi/10.32604/biocell.2026.074555/s1.
References
1. Cornelison TL, Reed E. Nephrotoxicity and hydration management for cisplatin, carboplatin, and ormaplatin. Gynecol Oncol. 1993;50(2):147–58. doi:10.1006/gyno.1993.1184. [Google Scholar] [PubMed] [CrossRef]
2. Ghosh S. Cisplatin: the first metal based anticancer drug. Bioorg Chem. 2019;88(1):102925. doi:10.1016/j.bioorg.2019.102925. [Google Scholar] [PubMed] [CrossRef]
3. Elmorsy EA, Saber S, Hamad RS, Abdel-Reheim MA, El-Kott AF, AlShehri MA, et al. Advances in understanding cisplatin-induced toxicity: molecular mechanisms and protective strategies. Eur J Pharm Sci. 2024;203:106939. doi:10.1016/j.ejps.2024.106939. [Google Scholar] [PubMed] [CrossRef]
4. Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25(5):409–33. doi:10.1101/gad.2021311. [Google Scholar] [PubMed] [CrossRef]
5. Huang R, Zhou PK. DNA damage repair: historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct Target Ther. 2021;6(1):254. doi:10.1038/s41392-021-00648-7. [Google Scholar] [PubMed] [CrossRef]
6. Ries F, Klastersky J. Nephrotoxicity induced by cancer chemotherapy with special emphasis on cisplatin toxicity. Am J Kidney Dis. 1986;8(5):368–79. doi:10.1016/s0272-6386(86)80112-3. [Google Scholar] [PubMed] [CrossRef]
7. Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4(4):307–20. doi:10.1038/nrd1691. [Google Scholar] [PubMed] [CrossRef]
8. Oun R, Moussa YE, Wheate NJ. The side effects of platinum-based chemotherapy drugs: a review for chemists. Dalton Trans. 2018;47(19):6645–53. doi:10.1039/c8dt00838h. [Google Scholar] [PubMed] [CrossRef]
9. Filipski KK, Mathijssen RH, Mikkelsen TS, Schinkel AH, Sparreboom A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin Pharmacol Ther. 2009;86(4):396–402. doi:10.1038/clpt.2009.139. [Google Scholar] [PubMed] [CrossRef]
10. Xu J, Gewirtz DA. Is autophagy always a barrier to cisplatin therapy? Biomolecules. 2022;12(3):463. doi:10.3390/biom12030463. [Google Scholar] [PubMed] [CrossRef]
11. Basu A, Krishnamurthy S. Cellular responses to cisplatin-induced DNA damage. J Nucleic Acids. 2010;2010(1):201367. doi:10.4061/2010/201367. [Google Scholar] [PubMed] [CrossRef]
12. Abuetabh Y, Wu HH, Chai C, Al Yousef H, Persad S, Sergi CM, et al. DNA damage response revisited: the p53 family and its regulators provide endless cancer therapy opportunities. Exp Mol Med. 2022;54(10):1658–69. doi:10.1038/s12276-022-00863-4. [Google Scholar] [PubMed] [CrossRef]
13. Zhu S, Pabla N, Tang C, He L, Dong Z. DNA damage response in cisplatin-induced nephrotoxicity. Arch Toxicol. 2015;89(12):2197–205. doi:10.1007/s00204-015-1633-3. [Google Scholar] [PubMed] [CrossRef]
14. Sengupta S, Harris CC. p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol. 2005;6(1):44–55. doi:10.1038/nrm1546. [Google Scholar] [PubMed] [CrossRef]
15. Lim JKM, Samiei A, Delaidelli A, De Santis JO, Brinkmann V, Carnie CJ, et al. The eEF2 kinase coordinates the DNA damage response to cisplatin by supporting p53 activation. Cell Death Dis. 2024;15(7):501. doi:10.1038/s41419-024-06891-4. [Google Scholar] [PubMed] [CrossRef]
16. Achkar IW, Abdulrahman N, Al-Sulaiti H, Joseph JM, Uddin S, Mraiche F. Cisplatin based therapy: the role of the mitogen activated protein kinase signaling pathway. J Transl Med. 2018;16(1):96. doi:10.1186/s12967-018-1471-1. [Google Scholar] [PubMed] [CrossRef]
17. Ou S, Kim TY, Jung E, Shin SY. p38 mitogen-activated protein kinase contributes to TNFα-induced endothelial tube formation of bone-marrow-derived mesenchymal stem cells by activating the JAK/STAT/TIE2 signaling axis. BMB Rep. 2024;57(5):238–43. doi:10.5483/BMBRep.2023-0152. [Google Scholar] [PubMed] [CrossRef]
18. Yan C, Boyd DD. ATF3 regulates the stability of p53: a link to cancer. Cell Cycle. 2006;5(9):926–9. doi:10.4161/cc.5.9.2714. [Google Scholar] [PubMed] [CrossRef]
19. Yan C, Lu D, Hai T, Boyd DD. Activating transcription factor 3, a stress sensor, activates p53 by blocking its ubiquitination. EMBO J. 2005;24(13):2425–35. doi:10.1038/sj.emboj.7600712. [Google Scholar] [PubMed] [CrossRef]
20. Jia M, Shi M, Zhao Y, Li Y, Liu X, Zhao L. The role of ATF3 in the crosstalk between cellular stress response and ferroptosis in tumors. Crit Rev Oncol Hematol. 2025;213(23):104791. doi:10.1016/j.critrevonc.2025.104791. [Google Scholar] [PubMed] [CrossRef]
21. Liu S, Li Z, Lan S, Hao H, Baz AA, Yan X, et al. The dual roles of activating transcription factor 3 (ATF3) in inflammation, apoptosis, ferroptosis, and pathogen infection responses. Int J Mol Sci. 2024;25(2):824. doi:10.3390/ijms25020824. [Google Scholar] [PubMed] [CrossRef]
22. El-Dessouki AM, Alzokaky AA, Raslan NA, Ibrahim S, Salama LA, Yousef EH. Piracetam mitigates nephrotoxicity induced by cisplatin via the AMPK-mediated PI3K/Akt and MAPK/JNK/ERK signaling pathways. Int Immunopharmacol. 2024;137(5):112511. doi:10.1016/j.intimp.2024.112511. [Google Scholar] [PubMed] [CrossRef]
23. Li D, Jing J, Dong X, Zhang C, Wang J, Wan X. Activating transcription factor 3: a potential therapeutic target for inflammatory pulmonary diseases. Immun Inflamm Dis. 2023;11(9):e1028. doi:10.1002/iid3.1028. [Google Scholar] [PubMed] [CrossRef]
24. Chen BP, Liang G, Whelan J, Hai T. ATF3 and ATF3 delta Zip. Transcriptional repression versus activation by alternatively spliced isoforms. J Biol Chem. 1994;269(22):15819–26. doi:10.1016/s0021-9258(17)40754-x. [Google Scholar] [CrossRef]
25. Liang G, Wolfgang CD, Chen BP, Chen TH, Hai T. ATF3 gene. Genomic organization, promoter, and regulation. J Biol Chem. 1996;271(3):1695–701. doi:10.1074/jbc.271.3.1695. [Google Scholar] [PubMed] [CrossRef]
26. Zhao J, Li X, Guo M, Yu J, Yan C. The common stress responsive transcription factor ATF3 binds genomic sites enriched with p300 and H3K27ac for transcriptional regulation. BMC Genomics. 2016;17(1):335. doi:10.1186/s12864-016-2664-8. [Google Scholar] [PubMed] [CrossRef]
27. Lu D, Wolfgang CD, Hai T. Activating transcription factor 3, a stress-inducible gene, suppresses Ras-stimulated tumorigenesis. J Biol Chem. 2006;281(15):10473–81. doi:10.1074/jbc.M509278200. [Google Scholar] [PubMed] [CrossRef]
28. Kim EY, Shin HY, Kim JY, Kim DG, Choi YM, Kwon HK, et al. ATF3 plays a key role in Kdo2-lipid A-induced TLR4-dependent gene expression via NF-κB activation. PLoS One. 2010;5(12):e14181. doi:10.1371/journal.pone.0014181. [Google Scholar] [PubMed] [CrossRef]
29. Zhao X, Chen C, Qiu H, Liu J, Shao N, Guo M, et al. The landscape of ATF3 in tumors: metabolism, expression regulation, therapy approach, and open concerns. Pharmacol Res. 2025;214(23):107666. doi:10.1016/j.phrs.2025.107666. [Google Scholar] [PubMed] [CrossRef]
30. Xu L, Zu T, Li T, Li M, Mi J, Bai F, et al. ATF3 downmodulates its new targets IFI6 and IFI27 to suppress the growth and migration of tongue squamous cell carcinoma cells. PLoS Genet. 2021;17(2):e1009283. doi:10.1371/journal.pgen.1009283. [Google Scholar] [PubMed] [CrossRef]
31. Qin XY, Su T, Yu WK, Kojima S. Lipid desaturation-associated endoplasmic reticulum stress regulates MYCN gene expression in hepatocellular carcinoma cells. Cell Death Dis. 2020;11(1):66. doi:10.1038/s41419-020-2257-y. [Google Scholar] [PubMed] [CrossRef]
32. Liu J, Lu X, Zeng S, Fu R, Wang X, Luo L, et al. ATF3-CBS signaling axis coordinates ferroptosis and tumorigenesis in colorectal cancer. Redox Biol. 2024;71(1):103118. doi:10.1016/j.redox.2024.103118. [Google Scholar] [PubMed] [CrossRef]
33. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–82. doi:10.1038/nmeth.2019. [Google Scholar] [PubMed] [CrossRef]
34. Yoshida T, Sugiura H, Mitobe M, Tsuchiya K, Shirota S, Nishimura S, et al. ATF3 protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. 2008;19(2):217–24. doi:10.1681/asn.2005111155. [Google Scholar] [PubMed] [CrossRef]
35. Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis—the p53 network. J Cell Sci. 2003;116(20):4077–85. doi:10.1242/jcs.00739. [Google Scholar] [PubMed] [CrossRef]
36. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science. 2006;312(5780):1650–3. doi:10.1126/science.1126863. [Google Scholar] [PubMed] [CrossRef]
37. Jänicke RU, Sohn D, Schulze-Osthoff K. The dark side of a tumor suppressor: anti-apoptotic p53. Cell Death Differ. 2008;15(6):959–76. doi:10.1038/cdd.2008.33. [Google Scholar] [PubMed] [CrossRef]
38. Choi YM, Kim HK, Shim W, Anwar MA, Kwon JW, Kwon HK, et al. Mechanism of cisplatin-induced cytotoxicity is correlated to impaired metabolism due to mitochondrial ROS generation. PLoS One. 2015;10(8):e0135083. doi:10.1371/journal.pone.0135083. [Google Scholar] [PubMed] [CrossRef]
39. Muñoz-Reyes D, Casanova AG, González-Paramás AM, Martín Á, Santos-Buelga C, Morales AI, et al. Protective effect of quercetin 3-O-glucuronide against cisplatin cytotoxicity in renal tubular cells. Molecules. 2022;27(4):1319. doi:10.3390/molecules27041319. [Google Scholar] [PubMed] [CrossRef]
40. Radyk MD, Spatz LB, Peña BL, Brown JW, Burclaff J, Cho CJ, et al. ATF3 induces RAB7 to govern autodegradation in paligenosis, a conserved cell plasticity program. EMBO Rep. 2021;22(9):e51806. doi:10.15252/embr.202051806. [Google Scholar] [PubMed] [CrossRef]
41. Sood V, Sharma KB, Gupta V, Saha D, Dhapola P, Sharma M, et al. ATF3 negatively regulates cellular antiviral signaling and autophagy in the absence of type I interferons. Sci Rep. 2017;7(1):8789. doi:10.1038/s41598-017-08584-9. [Google Scholar] [PubMed] [CrossRef]
42. Wang Y, Quan F, Cao Q, Lin Y, Yue C, Bi R, et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J Adv Res. 2021;28:231–43. doi:10.1016/j.jare.2020.07.007. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools