
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Melatonin Ameliorates Hippocampal Excitotoxicity and Neuroinflammation in Permanent MCAO by Targeting NMDA/AMPA Receptors and the NLRP3 Inflammasome via Nrf2/PPARγ/JNK/NF-κB Crosstalk
1 Department of Pharmacology and Toxicology, Faculty of Pharmacy, University of Tabuk, Tabuk, 71491, Saudi Arabia
2 Department of Pharmacology and Toxicology, College of Pharmacy, Addiction and Neuroscience Taif University, Taif, 21944, Saudi Arabia
3 Department of Pharmaceutics, Faculty of Pharmacy, University of Tabuk, Tabuk, 71491, Saudi Arabia
4 Department of Pharmacology, Faculty of Medicine, University of Tabuk, Tabuk, 71491, Saudi Arabia
5 Swat College of Pharmaceutical Sciences (SCPS), Gogdara, Swat, 19200, Khyber Pakhtunkhwa, Pakistan
6 Riphah Institute of Pharmaceutical Sciences, Riphah International University, Islamabad, 44000, Pakistan
* Corresponding Author: Fawad Ali Shah. Email:
(This article belongs to the Special Issue: Neuroinflammation and Neuroprotection in CNS Diseases: From Mechanisms to Therapeutic Targets)
BIOCELL 2026, 50(3), 8 https://doi.org/10.32604/biocell.2026.074865
Received 20 October 2025; Accepted 04 January 2026; Issue published 23 March 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Objectives: Permanent middle cerebral artery occlusion (pMCAO) can lead to hippocampal damage through multiple linked pathways such as reactive oxidative stress (ROS), neuroinflammation mediated by NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3), tumour necrosis factor-alpha (TNF-α), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and glutamate excitotoxicity involving N-methyl-D-aspartate receptor subunits 2a and 2b (NR2a/NR2b) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR/GluR1). The hippocampus, which is essential for memory and cognition, is at a substantial risk of ischemic degeneration. The aim of this study was to investigate the neuroprotective potential of melatonin in regulating these pathways. Method: Male adult rats were subjected to pMCAO, and melatonin (5 mg/kg) was administered just prior to ischemia, while sham-operated animals underwent surgery without nylon insertion. Hippocampal samples were collected 24 h after ischemia, Results: Hippocampal tissues showed NLRP3 inflammasome activation, increased TNF-α and p-NF-κB, and decreased peroxisome proliferator-activated receptor (PPARγ) after pMCAO. Melatonin-modulated ischemia-induced glutamatergic receptor subunits (NR2a, NR2b, GluR1) dysregulation, which possibly stimulated the prosurvival pathways and reduced collagen response-mediated protein (CRMP2) and its phosphorylation. Melatonin also modulated the expression of the postsynaptic protein (PSD95) and inhibited inflammatory cascades while upregulating antioxidant proteins. Further, melatonin reduced inflammation triggered by NLRP3, restored synaptic integrity, possibly by enhancing nuclear factor erythroid 2-related factor 2 (Nrf2) expression. Conclusion: These results demonstrated the dual role of melatonin by protecting ischemic brain damage both as a modulator of excitotoxicity and neuroinflammation/oxidative stress.Graphic Abstract
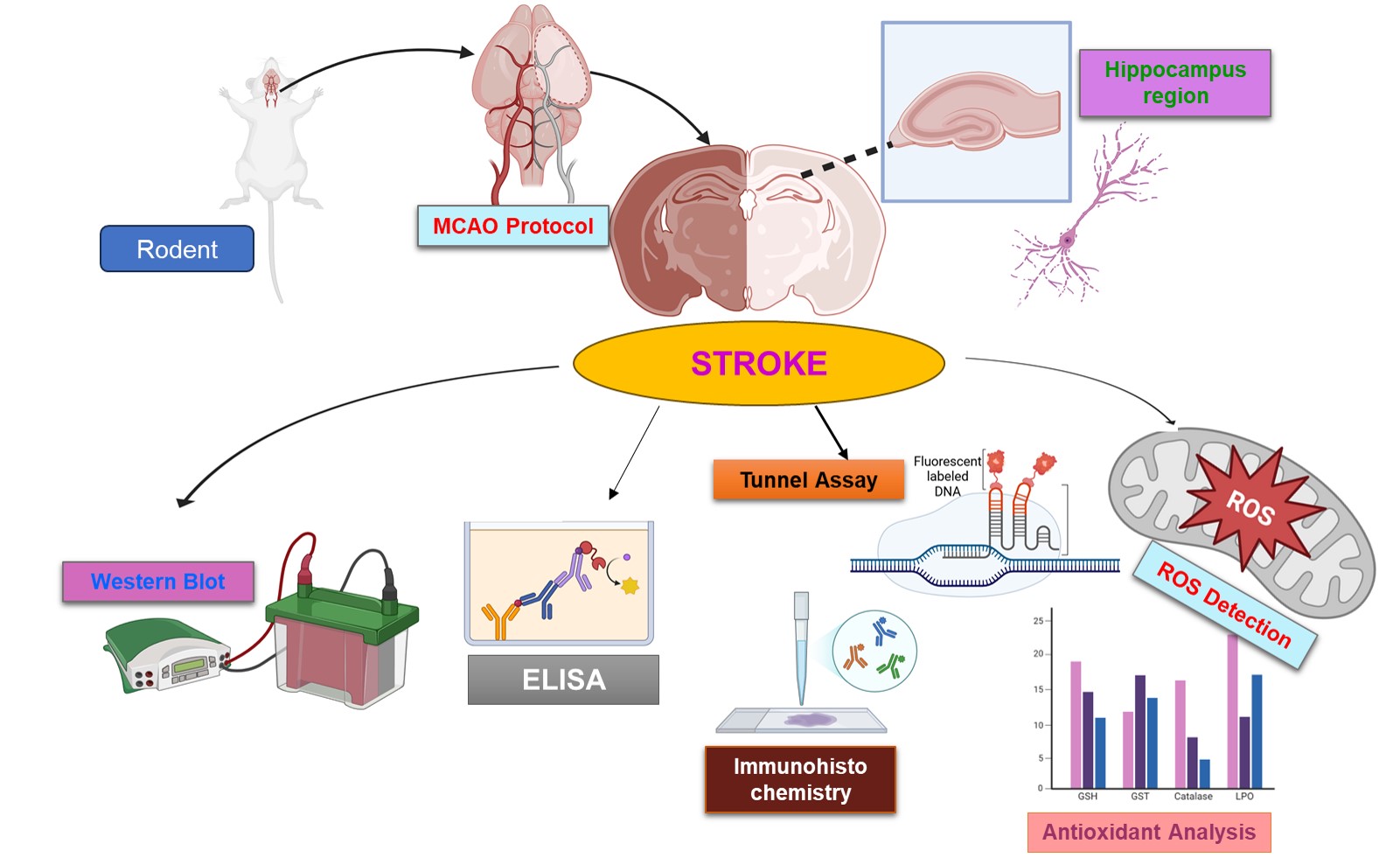
Keywords
Researchers have identified stroke as a significant global cause of disability and mortality, and even with appropriate treatment, ischemic stroke is still a significant contributor to stroke recurrence [1]. Despite a decade of extensive research on the etiology, effective treatment options remain scarce. Clinicians currently employ anticoagulants and endovascular intracranial therapy to treat stroke. However, their severe adverse effects and extensive contraindications restrict their clinical application [2].
Brain ischemia sets off a chain of events such as excitotoxicity, redox dysregulation, inflammation, and calcium overload, and all these biochemical events within minutes activate the apoptotic cascade, leading to the loss of neurons in the ischemic core. However, restoring the cerebral blood supply promptly can salvage the penumbra region, rendering it a prospective area of interest for the prevention of post-stroke disabilities [3]. Ischemic injury leads to neuronal death in multiple brain regions, such as the cortex, striatum, thalamus, and hypothalamus. In mice, the infarct additionally affects the hippocampus, a critical area for learning and memory [4,5]. Both the posterior hippocampal artery and the anterior choroid artery supply blood to the hippocampal region in rodents. Middle cerebral artery occlusion (MCAO) exclusively impedes blood flow to the hippocampus via the choroid artery [6]. Additional factors, including the duration of occlusion and the type of occluding material (e.g., nylon silk), affect blood flow in the hippocampus [7–10]. For several years, stroke research has been centered on excitotoxicity, which is associated with excessive glutamate release. Glutamate activates N-methyl-D-aspartate (NMDA) receptors, resulting in calcium accumulation and cell death [11]. Additionally, cerebral ischemia was not effectively treated by NMDA antagonists as a result of their significant adverse effects and lack of efficacy. Raïch et al., 2024 [12] described NMDA receptors into two subtypes according to their localization, NMDA receptor subunit 2a and 2b (NR2a/NR2b). NR2a is situated at the synapse and contributes to neuronal survival, whereas NR2b is located extrasynaptically and induces excitotoxicity, which expedites neuronal mortality [13,14].
Melatonin is an endogenous neuroprotectant and a naturally occurring hormone, and is widely known for its potent antioxidant and anti-inflammatory properties [15]. Repeated research has consistently shown melatonin’s protective effects in various disease models [15,16]. In ischemic brain injury, it reduces infarct size, restores blood-brain barrier (BBB) integrity, and improves behavioural outcomes. Previously, the differential expression of various signalling proteins was demonstrated in the hippocampus [17].
This research aimed to investigate the mechanisms by which melatonin protects against hippocampal damage in the pMCAO model by modulating neuroinflammation, oxidative stress, and glutamate excitotoxicity.
2.1 Animal Grouping and Drug Treatment
Sixty Sprague-Dawley male rats weighing 230–280 g and aged 8–12 weeks were obtained from the in-house breeding facility of Riphah Institute of Pharmaceutical Sciences (RIPS), Islamabad, and maintained in standard polypropylene cages with stainless-steel lids (n = 4/cage). The ambient temperature was maintained at 22°C–25°C, with standard conditions including a 12 h light/dark cycle and 35%–60% humidity levels. Rats were provided with a standard pellet diet and water ad libitum, in compliance with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines to mitigate animal suffering. The handling of animals was in accordance with the guidelines outlined in the “Guide for Care and Use of Laboratory Animals”, published by the National Institutes of Health (NIH Publications No. 8023, revised 1978). Furthermore, all experimental procedures were conducted as per protocol approved by the Institutional Animal Care and Committee of Riphah Institute of Pharmaceutical Sciences (Ref No.: REC/RIPS/2019/016/Cology was allocated), in line with ARRIVE guidelines. No human data was used to prepare this manuscript. The rats were randomized into four groups: Sham group, pMCAO group (MCAO), Melatonin+pMCAO (Mela+MCAO), and Melatonin+Sham group (Mela+Sham) (n = 15 per group). Melatonin (5 mg/kg, Cat# M5250-1G, CAS No.: 73-31-4, Sigma-Aldrich, Darmstadt, Germany) or a vehicle (dimethyl sulfoxide (DMSO) which was not higher than 5.0% in the final solution) was administered intraperitoneally to the animals 30 min prior to ischemia.
Rats were anesthetized with a combination of ketamine and xylazine (ratio 1:10), which corresponds to ketamine (90 mg/kg; CAS No.: 1867-66-9, Sigma-Aldrich, Darmstadt, Germany) and xylazine (9 mg/kg, CAS No.: 23076-35-9, Sigma-Aldrich, Darmstadt, Germany) in accordance with Gim et al., 2015 the previously published data [18], and a heating pad was used to maintain the rodents’ body temperatures at 37°C. On the ventral side, a cervical incision was made, and it was kept lateral to the right area. By separating the surrounding tissues, the vagus nerve—which passes laterally to the right common carotid artery (CCA)—was liberated. The external carotid artery (CA) was permanently knotted, and a thick nylon silk measuring 3/0 and 3 cm in length was inserted. The silk progressed into the internal CA until it encountered resistance, indicating that the middle cerebral artery (MCA) had been successfully occluded. The tissue samples were collected by euthanizing all animals 24 h after the occlusion. Similar procedures were performed on the sham group, but without the use of nylon. Throughout the trial, 10 rats died, four from the pMCAO group and three from the Mela+pMCAO group.
The brain hippocampal regions were separated and in order to assess the apoptosis in the CA1 and DG regions of the hippocampal, Tunel staining was carried out as suggested using TUNEL kit (Oncogene Research Products, Cambridge, MA, USA). Briefly, slides were deparaffinized and permeated with with proteinase K. After three washes with PBS for 5 min each, 100 μL of equilibration buffer was added and allowed to equilibrate at room temperature for 30 min. Subsequently, 50 μL of TdT working solution was introduced, and the slides were incubated in a humidified, light-shielded chamber at 37°C for 60 min. They were subsequently incubated in a with 3,3′-Diaminobenzidine (DAB) (Cat# SC-209686, CAS: 868272-85-9 Santa Cruz Biotechnology, Dallas, TX, USA) staining at room temperature and the development time was monitored until the desired brownish colour was noticed on tissue surface and it typically ranges from 1 min to 10 min depending upon whether DAB was freshly prepared or recycled. The reaction was stopped by putting the slides immediately in xylene solution (triplet rinsing). Images were captured with a light microscope (Olympus BX53, Olympus Corporation, Tokyo, Japan).
The hippocampus tissue was dissolved in lysis buffer with PMSF as a protease inhibitor. The homogenate underwent centrifugation, and the protein concentration was quantified using a bicinchoninic acid (BCA) kit (Cat# 23225, Pierce, Rockford, IL, USA) in accordance with the manufacturer’s instructions. An equivalent quantity of protein (30 μg per sample) was subjected to electrophoresis on 10% SDS-PAGE gels, followed by immunoblotting to transfer the protein to polyvinylidene fluoride (PVDF) membranes (Cat# 88520, Millipore, Billerica, MA, USA). To reduce nonspecific antibody binding, we incubated the PVDF membranes with skim milk for 1 h at ambient temperature. The PVDF membranes were rinsed in tris-buffered saline with 0.1% Tween 20 (TBST) and subsequently incubated with primary antibodies overnight at 4°C. The antibodies used were nuclear factor erythroid 2-related factor 2 (Nrf2) (SC-722), phospho-P38 (p-P38/Tyr 182) (SC-7973), phospho-c-Jun N-terminal kinase (p-JNK) (SC-6254), NR2a (SC-1468), NR2b (SC-1469), glutamate receptor (GluR1) (SC-55509), phospho-Ak strain transforming (p-AKT) (SC-7985), phospho-glycogen synthase kinase 3 beta (p-GSK3β) (SC-11757), p-GluR1 serine845 (SC-135699) p-NF-κB (SC-271908), collagen response-mediated protein (CRMP2, SC-376739), γ-enolase (SC-71046), and β-actin from Santa Cruz Biotechnology (Cat# SC-58673, Dallas, TX, USA) and were used at 1:1000 dilution ratio. Phospho-CRMP2 (p-CRMP2/Thr 514) (Cat# 9397) was obtained from Cell Signaling Technology (Danvers, MA, USA) and was used at a dilution of 1:500. The following morning, PVDF were incubated with an appropriate horseradish peroxidase (HRP) conjugated anti-rabbit IgG secondary antibody (Cat# 7074) from Cell Signaling Technology (Danvers, MA, USA) and anti-mouse IgG-HRP (SC-2005) from Santa Cruz Biotechnology (Dallas, TX, USA) (1:1000 dilution ratio) for 2 h at room temperature, and ECL detection kit (Cat# 32132 Amersham Pharmecia Biotech, Uppsala, Sweden) was used for signal amplification. We determined the band optical densities using the ImageJ software (Version 1.53, NIH, Bethesda, MD, USA).
2.5 Reactive Oxygen Species (ROS) Measurement
Tissue samples from the hippocampus were homogenized in ice-cold Tris-HCl buffer (40 mM, pH 7.4) at a 1:10 w/v ratio, and the homogenates were centrifuged at 1000× g for 10 min at 4°C. The resultant supernatant was subsequently divided into two equal portions, with 2′,7′-dichlorofluorescein diacetate (DCFH) (Cat# D6883, Sigma-Aldrich) (10 μM) added to one portion for reactive oxygen species (ROS) detection in the dark, while the other portion served as a control. Subsequently, samples were incubated for 15 to 30 min at 37°C, and the fluorescence intensity of the sample was determined using a FLUOstar® Omega multifunctional microplate reader (BMG LABTECH, Ortenberg, Germany) (λexcitation 485 nm and λemission 525 nm). The raw fluorescence of each sample’s blank was subtracted from the fluorescence of its corresponding probe-incubated aliquot to yield a corrected relative fluorescence unit (RFU) value. Further, we took three replicates per sample from three different sample groups. Percent of relative ROS levels for each sample were then calculated as the fold-change over the control mean using the following formula:
% Relative ROS = (Corrected RFU of Sample)/(Mean Corrected RFU of Control Group) ∗ 100
2.6 Levels of Reduced Glutathione (GSH)
GSH level in the hippocampus was determined by mixing 2 mL of 0.6 mM 5,5′-dithio-bis-2-nitrobenzoic acid (DTNB) with 0.2 mL of supernatant. Volume was made up to 3 mL with sodium phosphate buffer, followed by 10 min of incubation at room temperature, and to protect from light, samples were wrapped in aluminum foil. The supernatant was carefully separated after lysis of hippocampal tissues in the 5% metaphosphoric acid (at a 1:10 weight/volume ratio), followed by centrifugation (10,000× g for 15 min at 4°C). In a separate set, protein concentration was determined by the BCA kit (Cat# 23225, Pierce, Rockford, IL, USA) by lysis of the tissue in phosphate buffer. Furthermore, we measured absorbance in μmol/mg of proteins at 412 nm wavelength via a spectrophotometer (Shimadzu UV-1800, Shimadzu, Kyoto, Japan). We procured our desired readings after subtraction of control values from sample values, preceded by taking DTNB and phosphate buffer values as control. A standard curve for GSH was generated, and the sample absorbance was derived from this curve. Subsequently, normalization was performed, and the concentration was measured as μmol/mg of proteins. Further, we took three replicates per sample from three different sample groups.
2.7 Catalase Content in Tissue Lysate
Catalase level in the hippocampus was measured by lysis of hippocampal tissues in the phosphate buffer (at a 1:10 weight/volume ratio, followed by centrifugation (10,000× g for 15 min at 4°C), and the protein concentration was determined by the BCA kit (Cat# 23225, Pierce, Rockford, IL, USA). The assay involved mixing 0.05 mL of the tissue supernatant with 3 mL of a freshly prepared 60 mM H2O2 solution in phosphate buffer (pH 7.0). Catalase levels were represented in units of μmol H2O2/min/mg of protein in line with the absorbance measurement at 240 nm wavelength after the addition of supernatant and H2O2. The decrease in absorbance at 240 nm was immediately recorded for 60 s (1 min) at room temperature (25°C) using a spectrophotometer. A blank cuvette containing only the H2O2 substrate solution (without supernatant) was used to determine the initial absorbance. The catalase activity was calculated using the extinction coefficient of the product formed and expressed as μmol H2O2/min/mg of protein. Further, we took three replicates per sample from three different sample groups.
2.8 Glutathione-S-Transferase (GST) Levels
GST levels in the hippocampus were measured by lysis of hippocampal tissues in the phosphate buffer (at a 1:10 weight/volume ratio, followed by centrifugation (10,000× g for 15 min at 4°C), and the protein concentration was determined by the BCA kit (Cat# 23225, Pierce, Rockford, IL, USA). We prepared a homogeneous solution by blending 5 mM GSH (CAS Number: 70-18-8, Sigma-Aldrich, Darmstadt, Germany) and 1 mM 1-chloro-2,4-dinitrobenzene (CDNB) (CAS Number: 97-00-7, Sigma-Aldrich, Darmstadt, Germany) in 0.1 M phosphate buffer to a final volume of 1.2 mL. We added GSH from a 100 mM stock solution prepared with phosphate buffer and CDNB from a 50 mM stock solution prepared in ethanol.
Three replicates of 1.2 mL reaction mixture were placed in a glass vial. Tissue supernatant (0.2 mL) was then added to the reaction mixture and wrapped to protect from the light. Three blanks were also made, which did not contain tissue lysate. Aliquots of 210 μL from the reaction mixture were pipetted into a microtiter plate, and absorbance was recorded at 340 nm for 5 min at 23°C using an ELISA microplate reader (BioTek ELx808, BioTek Instruments, Winooski, VT, USA). The absorbance of the blank was subtracted from the sample readings to account for the non-enzymatic. The GST activity was calculated using the extinction coefficient of the product formed and expressed as μmol of CDNB/min/mg of protein.
2.9 Expression of Lipid Peroxidation (LPO) in Tissue Homogenate
LPO levels in the hippocampus were measured by lysis of hippocampal tissues in the phosphate buffer, followed by centrifugation (10,000× g for 15 min at 4°C), and the protein concentration was determined by the BCA kit (Cat# 23225, Pierce, Rockford, IL, USA). LPO was assessed by quantifying thiobarbituric acid reactive substances (TBARS). The assay mixture comprises 580 μL of 0.1 M phosphate buffer (pH 7.4), 200 μL of supernatant, 20 μL of 100 mM ferric chloride, and 200 μL of 100 mM ascorbic acid, which is incubated in a water bath at 37°C for 60 min. Following this incubation, the reaction was ended by adding 1000 μL of 10% trichloroacetic acid (TCA) and 1000 μL of 0.66% thiobarbituric acid (TBA) to the samples. The tubes were maintained in the water bath for 20 min, then chilled in an ice bath, and subsequently centrifuged at 3000× g for 10 min. The absorbance of the supernatant and the blank (containing all reagents except the supernatant) were measured at 535 nm via a spectrophotometer (Shimadzu UV-1800). A standard curve for MDA was generated, and the sample absorbance was derived from this curve. Subsequently, normalization was performed, and the concentration was measured as TBARS in nmol/mg protein.
2.10 Enzyme-Linked Immunosorbent Assay (ELISA)
ELISA kits of PPARγ (CAS# A19676) and NLRP3 (catalog number: A-5652) were obtained from MLBIO Biotechnology Co., Ltd. (Shanghai, China), and ELISA analysis was performed as per the manufacturer’s instructions using an ELISA microplate reader (BioTek ELx808, BioTek Instruments, Winooski, VT, USA).
2.11 Immunohistochemistry and Immunofluorescence
Tissue blocks were sliced into coronal sections measuring 4 μm. Slides containing embedded tissue were subjected to deparaffinization (xylene treatment) and rehydration (alcohol treatment starting from 100% to 70% and finally with water), followed by the antigen retrieval process with proteinase kinase (Proteinase-K with CAS number 39450-01-6, Sigma-Aldrich, Darmstadt, Germany) 1.3 mg/mL final concentration for10 min. Following antigen retrieval and the neutralization of peroxidase activity with 3% methanolic H2O2. Slides were incubated with 5% normal goat serum (Cat# G9023, Sigma-Aldrich, Darmstadt, Germany) containing 0.1% Triton X-100 for a minimum of 1 h in a humidified chamber. A sequence of steps was executed, beginning with the application of primary antibodies of the following: Tumour necrosis factor-alpha (TNF)-α (SC-12744, Santa Cruz Biotechnology, Dallas, TX, USA), p-JNK (SC-6254, Santa Cruz Biotechnology, Dallas, TX, USA), and nuclear factor kappa-light-chain-enhancer of activated B cells (p-NF-κB) (SC-271908) with a dilution of 1:100 for overnight incubation at 4°C, followed by the introduction of secondary antibody (Cat# 62-6520, Thermo Fisher Scientific, Waltham, MA, USA), dilution factor (1:50) in a humidified chamber, at room temperature for 90 mint followed by ABC treatment (SC-516216, Santa Cruz Biotechnology, Dallas, TX, USA) and concluding with 3,3′-Diaminobenzidine (DAB) (Cat# SC-209686, CAS: 868272-85-9 Santa Cruz Biotechnology, Dallas, TX, USA) staining at room temperature and the development time was monitored until the desired brownish colour was noticed on tissue surface and it typically ranges from 1 min to 10 min depending upon whether DAB was freshly prepared or recycled. The reaction was stopped by putting the slides immediately in xylene solution (triplet rinsing). Images were captured with a light microscope (Olympus BX53, Olympus Corporation, Tokyo, Japan).
Immunofluorescence was conducted using HSP70 (SC-66048) and PSD95 (SC-71933) antibodies were obtained from Santa Cruz Biotechnology, (Dallas, TX, USA) and were used in a dilution of 1:100 for a whole night incubations at 4°C in a humdified chamber, the slides were treated with fluorescent-labeled secondary antibody tetramethylrhodamine isothiocyanate (TRITC, SC-3796, 1:100), Santa Cruz Biotechnology, (Dallas, TX, USA) for signal amplification, incubated for 1 h in a dark chamber in humidified chamber. The TRITC fluorophore has peak excitation and emission wavelengths of approximately 552 nm and 570 nm. The slides were cover-slipped using Ultra Cruz mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Cat# SC-3598 and CAS: 28718-90-3), Santa Cruz Biotechnology, (Dallas, TX, USA). Images were captured by a confocal laser scanning microscope (CLSM, FV 1000MPE-B, Olympus, Tokyo, Japan).
The sample size was calculated as per previously reported similar literature. We assumed medium to large effect (f ≥ 0.25), α (significance) < 0.05, and power (1 − β) 0.8 for our groups. The normality of the numerical data from the four groups was assessed using the Shapiro-Wilk test. ImageJ software (Version 1.53, NIH) was used to measure Western blotting and histological data. GraphPad Prism 8 (GraphPad Software Inc., San Diego, CA, USA) was used for the preparation of graphs and statistical analysis. Statistical differences among groups were analyzed using one-way ANOVA, followed by Tukey’s post hoc test for multiple comparisons. A statistically significant value was defined as p < 0.05. The following definitions were given to statistical symbols: #p < 0.05, ##p < 0.01, ###p < 0.001 compared to the sham group; *p < 0.05, *p < 0.01, **p < 0.001 compared to treated group.
3.1 Evaluation of Neurodegeneration in the Hippocampus and the Protective Role of Melatonin
To investigate neuronal death in the hippocampus, tunel staining was performed. Our results showed that the hippocampus of MCAO animals (Fig. 1A) had significant tunnel-positive cells, compactly labeled and accompanied by fragmented apoptotic bodies (Fig. 1A), and melatonin partially reversed the effects or melatonin reduced the fragmented apoptotic bodies (**p < 0.01, Fig. 1A). We also performed an immunofluorescence analysis of heat shock proteins (HSPs), which could characterize the degree of injury in the core and other penumbral regions. It was demonstrated by our findings that the ischemic group exhibited significantly higher levels of HSP70 expression than the sham group, which suggests that the peri-infarct waves extended to the hippocampus sporadically after the ischemia (Fig. 1B). Melatonin reversed HSP70 upregulation in several hippocampal regions (Fig. 1B, *p < 0.05).
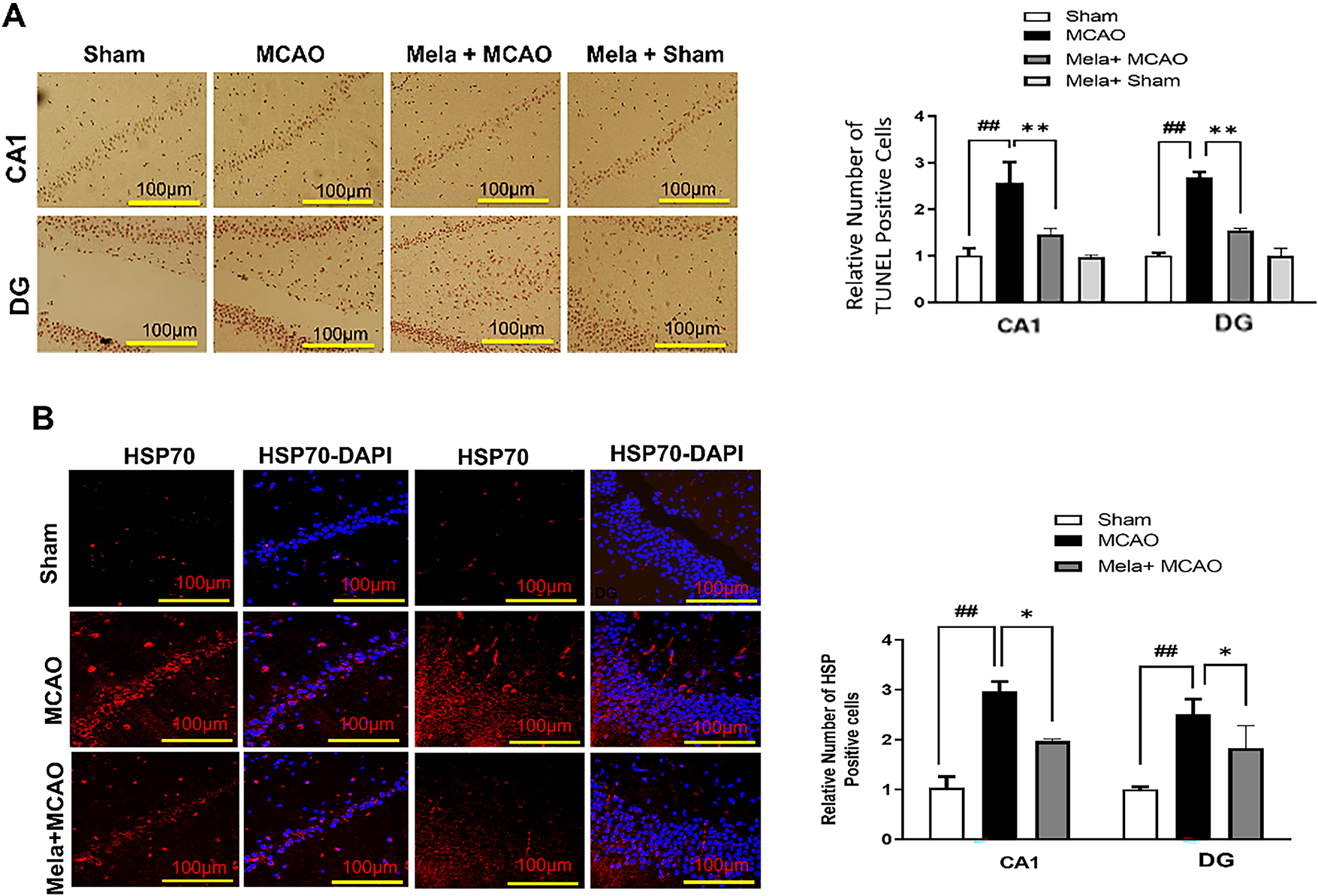
Figure 1: Evaluation of neurodegeneration and neuroprotective role of melatonin. (A) TUNEL histochemistry was performed in the CA1 and DG regions of the hippocampus (n = 5/group), with a scale bar of 100 μm and magnification 10 X. ##p < 0.01 represents relative to the sham group, and symbols **p < 0.01 represent relative to the middle cerebral artery occlusion (MCAO) group. (B) HSP70 immunofluorescence staining was conducted in the CA1 and DG regions of the hippocampus (n = 5/group), also with a scale bar of 100 μm and magnification 10 X. ##p < 0.01 represents relative to sham group and symbols *p < 0.05 represents relative to the MCAO group. The blue represents the DAPI, while the HP70 positive cells were visualized by TRITC
3.2 Effect of Melatonin on NMDA Receptor Signaling in the Hippocampus
Here, we showed the effect of melatonin on hippocampal NR2 subunit expression. Our results showed low expression of NR2a and NR2b in the disease/ischemic group (Fig. 2A), similar to previous observations [19,20]. This decrease may possibly be calpain-mediated, and interestingly, these changes could be reversed by administering melatonin (Fig. 2A,B). Furthermore, enhanced PSD95/NR2a interaction may avert calpain-mediated stimulation-induced NR2a cleavage, which is possibly responsible for NR2a cleavage in MCAO. Our immunofluorescence analysis showed the effect of melatonin on PSD95/NR2a interaction (Fig. 2C,D), and the results demonstrated that ischemic stroke increased PSD95 expression, and this hyperexpression may be linked to the NR2a survival mechanism. Consistent with previous studies, we observed cleavage of AMPAR (GluR1) and its phosphorylation at serine 845 (p-GluR1 Ser 845, Fig. 2A,B) relative to the sham, and these differences were restored by melatonin.
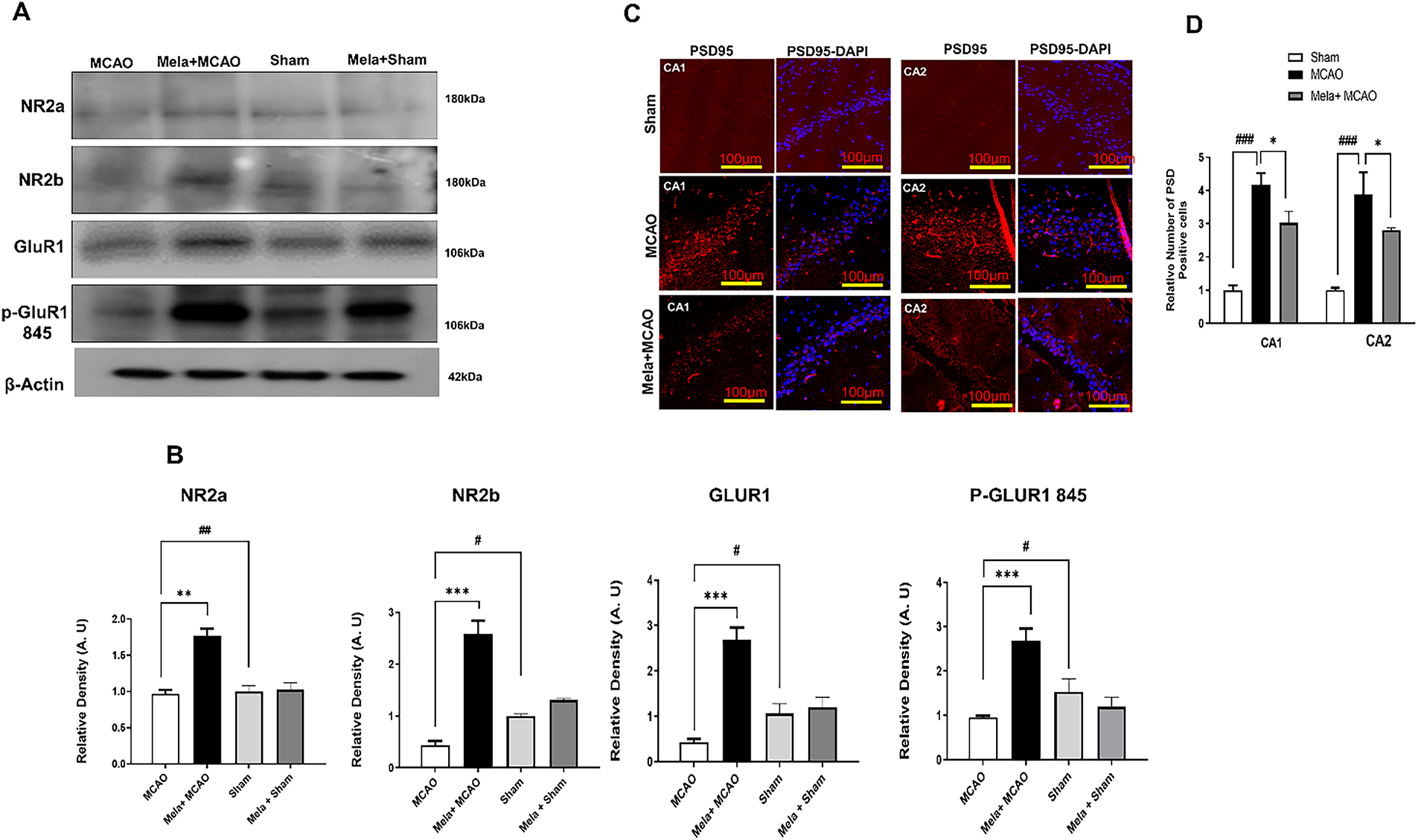
Figure 2: NMDA receptor signaling and the effect of melatonin. (A) Western blot analysis (n = 5 rats per group) from the hippocampal tissue. The bands were quantified using ImageJ and analyzed by GraphPad Prism software. β-actin was used as a control. (B) Represents the quantification of A. ##p < 0.01 or #p < 0.05 represents relative to sham group and symbols **p < 0.01 or ***p < 0.001 represents relative to the MCAO group. (C) Immunofluorescence reactivity of PSD95 (n = 5 rats/group) with a scale bar = 100 μm and magnification 10 X was shown in CA1 and CA2 region of the hippocampus. PSD95 shows cytoplasmic localization and was visualized by TRITC. (D) Represents the quantification of C. ###p < 0.001 represents relative to sham group and symbols *p < 0.05 represents relative to the MCAO group
3.3 Effect of Melatonin on MAPK Signaling in the Hippocampus
Numerous studies have shown a link between MAPK and neuronal death. The ischemia group (MCAO) (Fig. 3A) exhibited substantially higher levels of these activated kinases than the MCAO group. Here, we demonstrated the protein expression of p-JNK and p-P38 by western blot (Fig. 3A,B) and by immunohistochemistry staining (Fig. 3C,D), showing hyper-elevation in the ischemic (MCAO) group. Treatment with melatonin decreased these protein levels in the hippocampus.
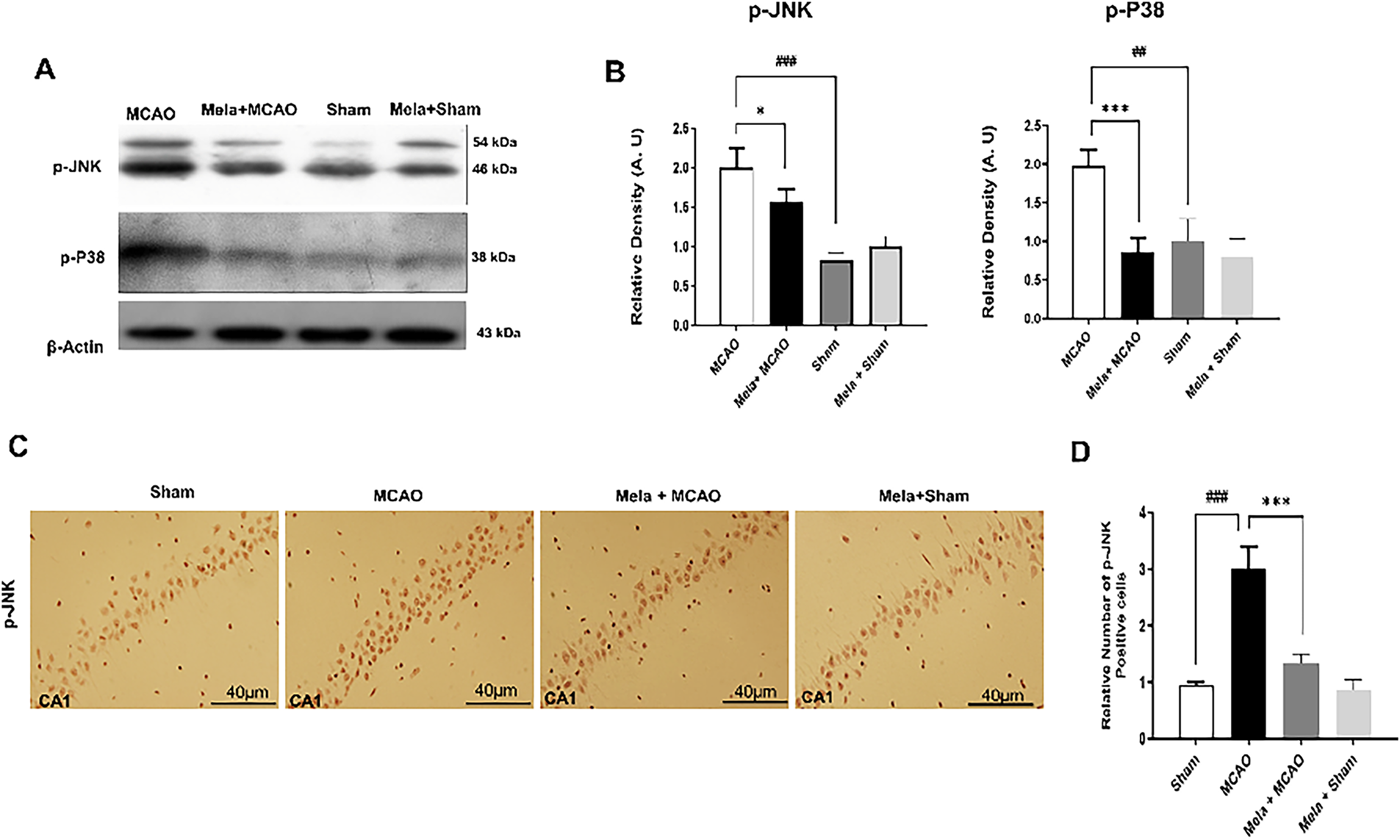
Figure 3: Expression of MAPK family protein. (A) Western blots of p-P38 and p-JNK (n = 5/group) from the hippocampal tissue. The bands were quantified using ImageJ and analyzed by GraphPad Prism software, and β-actin was used as a control. (B) Represents the quantification of A. ##p < 0.01 or ###p < 0.001 represents relative to sham group and symbols *p < 0.05 or ***p < 0.001 represents relative to the MCAO group. (C) p-JNK staining was conducted in the CA1 region of the hippocampus (n = 5/group), also with a scale bar of 40 μm. p-JNK shows cytoplasmic localization. (D) Represents the quantification of C. ###p < 0.001 represents relative to the sham group, and symbols ***p < 0.001 represent relative to the MCAO group
3.4 Melatonin-Promoted AKT/GSK-3β/CRMP2 Pathway
Evidence suggests the pro-survival role of γ-enolase in ischemic stroke, and it can target the downstream PI3K/AKT signaling pathways [21]. Our western blot results demonstrated no significant effect of ischemia on enolase expression (Fig. 4A). Moreover, our findings suggest the protective effects of melatonin on the AKT pathway in the hippocampus. GSK3β, which is downstream of AKT, can modulate CRMP2 expression, and CRMP2 exhibited a characteristic pattern of bands (Fig. 4), where the CRMP2 (66–70 kD) degraded into 62 and 55 kD proteins in the ischemic group. Melatonin notably reduced the intensity of this cleavage. Additionally, the phosphorylation of CRMP2 (CRMP2 Thr514) displayed a consistent expression pattern with that of CRMP2 (Fig. 4).
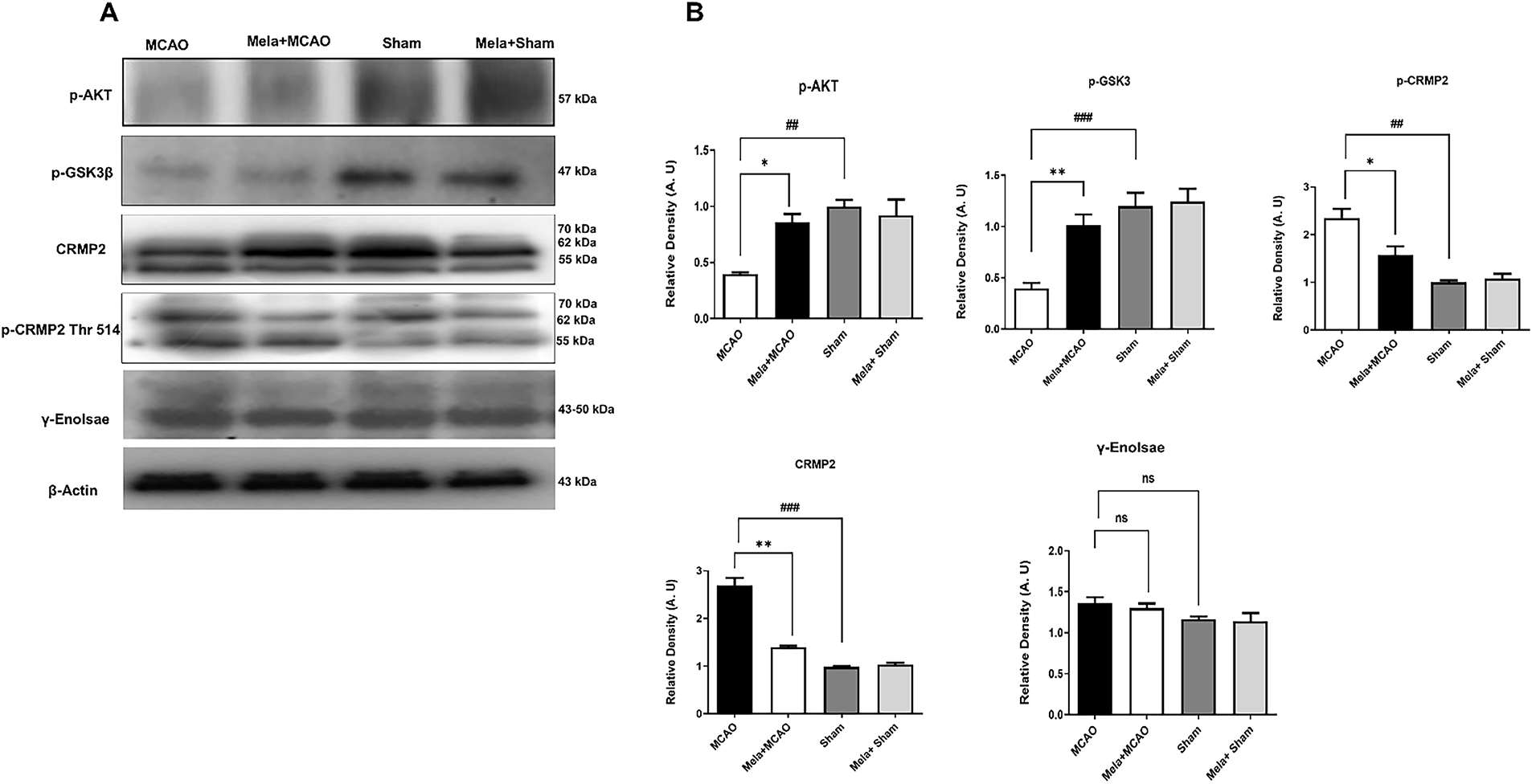
Figure 4: Melatonin promotes the AKT/GSK-3β/CRMP2 pathway. (A) Western blot analysis of p-AKT, p-GSK3β, CRMP2, p-CRMP2, γ-enolase (n = 5/rats per group). Protein samples were extracted from the homogenate of the whole hippocampus. (B) Represents the quantification of A. Data was analysed by ImageJ. ##p < 0.01 or ###p < 0.001 represents relative to sham group and symbols *p < 0.05 or **p < 0.01 represents relative to the MCAO group. Ns means non significant when compared to the MCAO group
3.5 Melatonin Attenuated NLRP3-Dependent Inflammatory Cascades and Augmented PPARγ and Nrf2 Expression
Protein levels of NLRP3 and other inflammatory mediators in the hippocampus were assessed using western blot, ELISA, and immunohistochemistry analysis (Fig. 5). Compared to the sham group, the MCAO group exhibited an increased level of NLRP3 (Fig. 5A), whereas melatonin treatment significantly reduced the NLRP3 level. Additionally, post-hoc analysis indicated statistical significance (*p < 0.05, ##p < 0.01, Fig. 5A). We conducted western blot (p-NFκB and Nrf2, Fig. 5B,C) and immunohistochemical analyses of TNF-α (Fig. 5D,E) and p-NFκB to further validate our findings (Fig. 5F,G). Nrf2 protein levels were significantly elevated in the Mela+MCAO group, as determined by western blot analysis (Fig. 5C, *p < 0.05), in comparison to the MCAO group. The MCAO group increased the levels of p-NFκB and TNF-α, whereas melatonin reduced this hyperexpression, as shown by western blot and immunohistochemistry analyses. Further, PPARγ and Nrf2 are transcription factors that mitigate oxidative stress and inflammation, and the concurrent activation of PPAR-γ and Nrf2 may represent a crucial therapeutic strategy to reduce ischemic damage. We conducted ELISA analyses to assess the impact of melatonin on PPARγ level (Fig. 5H). The level of PPAR-γ was assessed using ELISA analysis, yielding results consistent with those of Nrf2 (*p < 0.05, Fig. 5H).
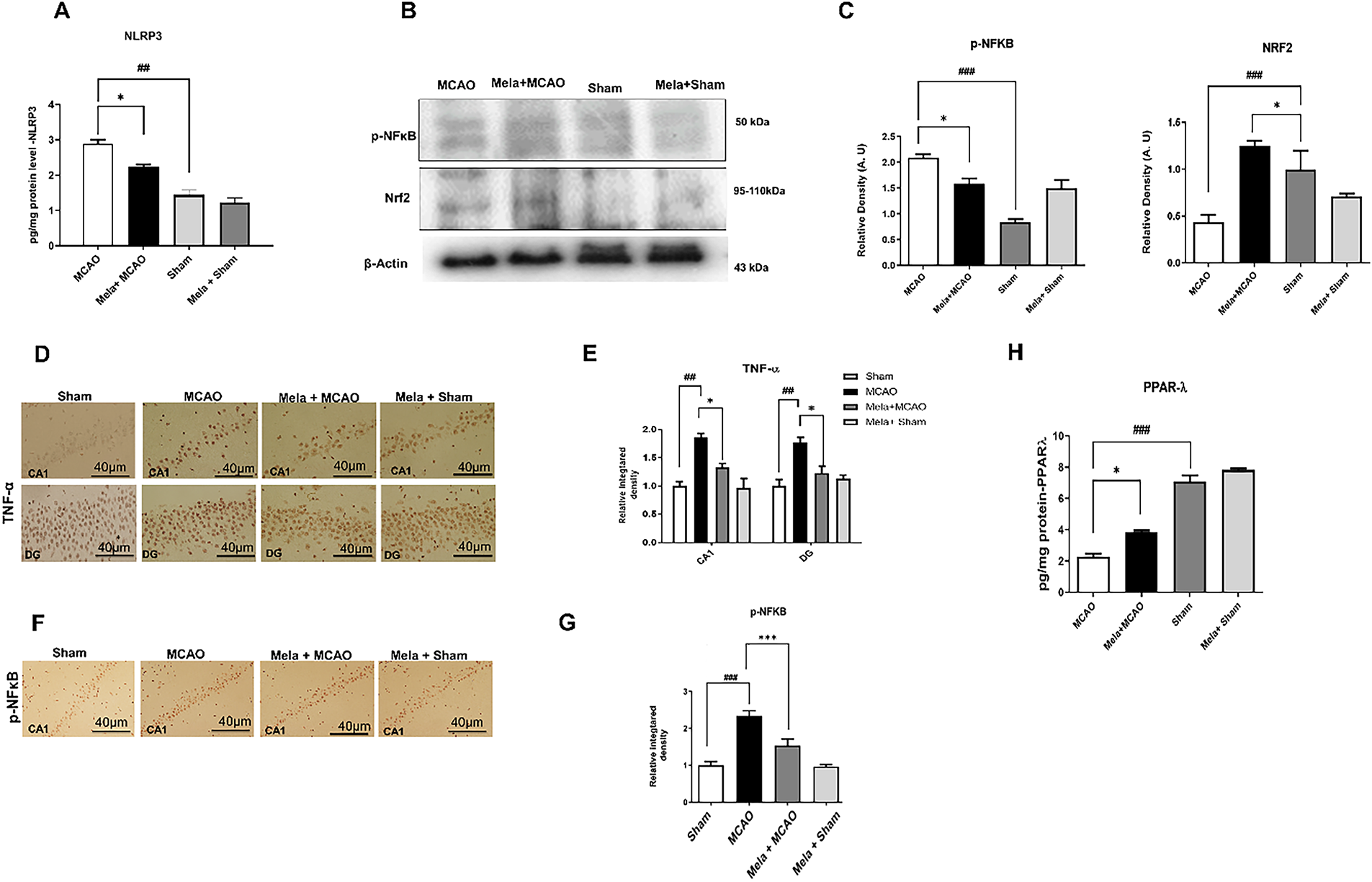
Figure 5: Effect on inflammatory and oxidative markers. (A) Enzyme-linked immunosorbent assay (ELISA) analysis of NLRP3 (n = 5/group) in the whole hippocampus. ##p < 0.01 represents relative to sham group and symbol *p < 0.05 represents relative to the MCAO group. (B) Western blot analysis of p-NF-κB, Nrf2 (n = 5/group). (C) Represents the quantification of B. ###p < 0.001 represents relative to the sham group, and symbol *p < 0.05 represents relative to the MCAO group. (D) TNF-α was conducted in the CA1 and DG regions of the hippocampus (n = 5/group), with a scale bar of 40 μm, and shows cytoplasmic localization. (E) Represents the quantification of D. ##p < 0.01 represents relative to the sham group, and symbols *p < 0.05 represent relative to the MCAO group. (F) p-NF-κB was conducted in the CA1 region of the hippocampus (n = 5/group), with a scale bar of 40 μm, and shows nuclear localization. (G) Represents the quantification of F. ###p < 0.001 represents relative to the sham group, and symbols ***p < 0.001 represent relative to the MCAO group. (H) ELISA analysis of PPARγ in the whole hippocampus. ###p < 0.001 represents relative to sham group and symbol *p < 0.05 represents relative to the MCAO group
3.6 Melatonin Downregulated ROS and Upregulated Antioxidant Enzymes
The levels of reactive oxygen species (ROS) and the effect of melatonin were assessed through the percentage of DCFH-DA fluorescence, and a significant difference in outcomes among the treatment groups was observed (Fig. 6A). Additionally, the post-hoc test demonstrated that MCAO increased ROS levels in the hippocampus (Fig. 6A, ##p < 0.05), while melatonin mitigated ROS generation (*p < 0.05). The protective effects of melatonin on levels of various endogenous antioxidant enzymes were assessed. The MCAO group exhibited a significant downregulation of hippocampal levels of GSH, GST, and CAT as indicated (Fig. 6B, ###p < 0.001; Fig. 6C, ##p < 0.01; Fig. 6D, ##p < 0.01). Conversely, the level of LPO was significantly increased in the MCAO group (Fig. 6E, ###p < 0.001). Melatonin reinstated the downregulated antioxidant enzymes GST, GSH, and CAT, while reducing the LPO level.
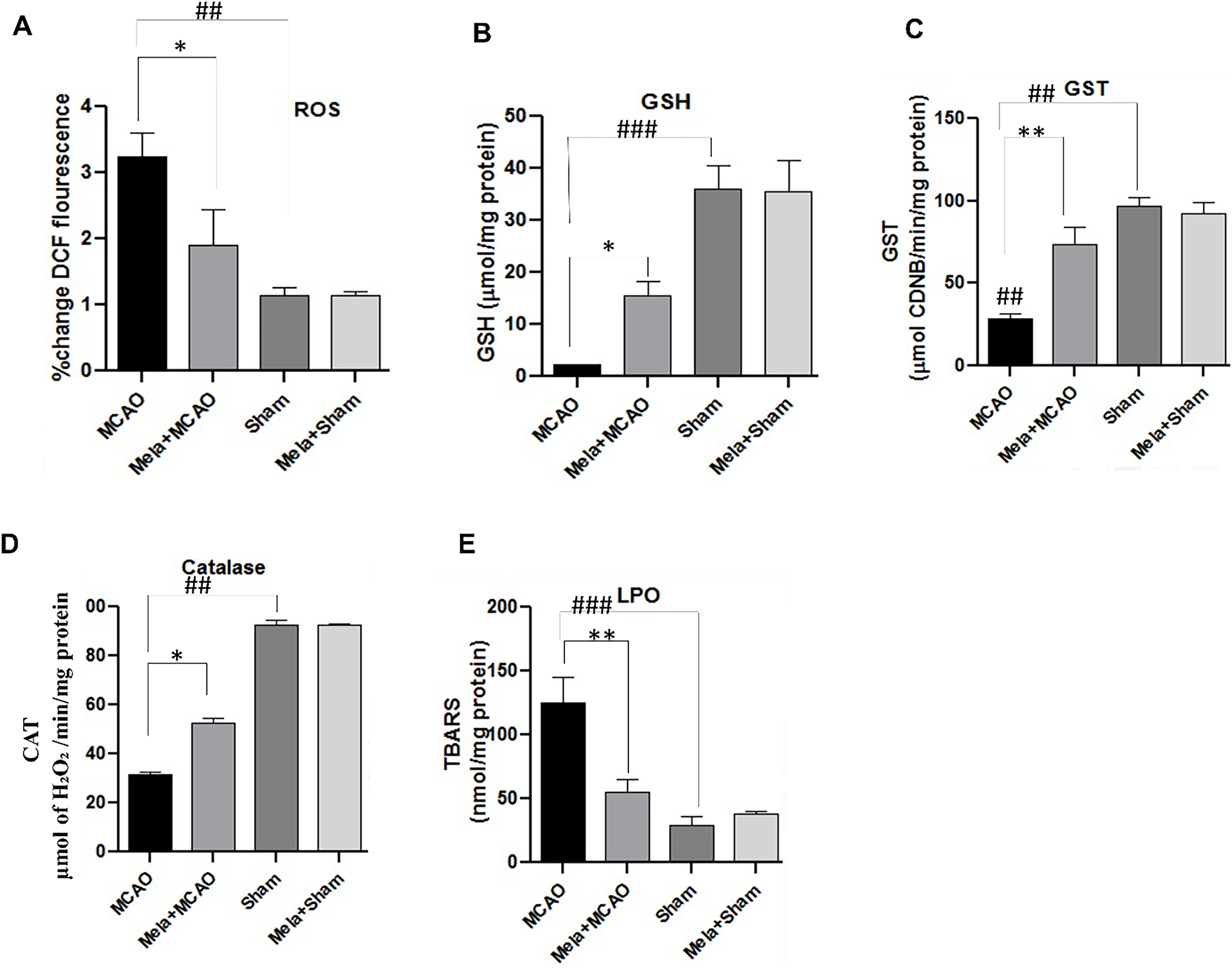
Figure 6: Effect of melatonin on oxidative stress and endogenous antioxidant enzymes. (A) % change of DCF fluorescence in ethanol-induced memory impairment model in the hippocampus, (B) GSH level in the hippocampus, (C) GST in the hippocampus, (D) Catalase in the hippocampus, (E) LPO level in the hippocampus. ###p < 0.001 or ##p < 0.01 represents relative to sham group and symbols *p < 0.05 or **p < 0.01 represents relative to the MCAO group
The permanent middle cerebral artery occlusion (pMCAO) model leads to complex hippocampal damage characterized by oxidative stress, neuroinflammation, synaptic dysfunction, and cognitive deficits. Our findings demonstrated that melatonin provides neuroprotection by modulating several interconnected pathways. The possible mechanisms might be the suppression of the NLRP3 inflammasome, activation of PPARγ and Nrf2-related proteins, stabilization of glutamate receptors, and preservation of synaptic proteins, along with effects on the JNK/p38 MAPK and AKT/GSK3β/CRMP2 pathways.
MCAO induces the most consistent infarction in the neocortex and striatum between 3 and 12 h of permanent blockage. Recently, numerous studies have indicated that ischemic damage can be extended to other remote areas of the brain, such as the hippocampus, where it exerts secondary damage [22,23]. Consistent with these studies, we also demonstrated significant structural alterations in the hippocampus, along with significant neuronal inflammation and apoptotic events. Numerous studies have demonstrated the neuroprotective role of HSP70 in various neurodegenerative models [24]. HSP70 is a cytoprotective chaperone and among the first response proteins towards stroke cytotoxicity, and it remains hyper-elevated, even for days [25]. Consistent studies indicated the overexpression of HSP70 in the penumbra region compared to the core region, where cells are under stress and salvageable. HSP70 executed its protective effects in numerous ways by inhibiting inflammation, apoptosis, and improving BBB integrity, all of which help in mitigating protein misfolding and aggregation [25–27]. Our study also demonstrated that ischemia triggered the overexpressed of HSP70 in the hippocampus, and this overexpression was reduced by melatonin. The alteration of HSP70 by melatonin implies that the hippocampus undergoes significant changes during permanent MCAO, which was reversed by melatonin. HSP70 was expressed differentially in the ischemic models depending upon the dose, model, and sample collection time. In some studies, melatonin hyper-elevated the HSP70, possibly by transcriptional regulation of the shock factors that upregulate HSP70 [28]. In this way, melatonin not only executes the antioxidant effect but also promotes the endogenous cellular response to this stress. In our study, we demonstrated the downregulation of HSP70 in the melatonin group, possibly by directly mitigating the cellular stress and augmenting the endogenous antioxidant enzymes such as glutathione (GSH), as glutathione directly regulates HSP70 activity, and by reducing the glutamate excitotoxicity. This demonstrates a paradoxical response of HSP70 to ischemia: an initial overexpression as a stress response, succeeded by a reduction influenced by melatonin. This implies that melatonin facilitates the restoration of cellular homeostasis rather than merely inhibiting HSP70.
It has been determined that excitotoxicity is the main pathogenic mechanism responsible for ischemic stroke. Excessive release of glutamate during stroke triggers numerous pathogenic cascades, ranging from calcium overloading to ROS production, which in turn dysregulate glutamate receptors. ROS oxidizes NR2b subunits and triggers amassing of calcium due to calcium channel opening. Various NMDAR receptor subtypes have been found to have distinct functional effects in response to ischemic insults. While the extra synaptic NR2b subunit stimulates excitotoxicity and death, the synaptic NR2a subunit is essential for neuronal survival and neuroprotection [13,14,20]. Considering the twofold activity of NMDARs, the optimal strategy would be to inhibit NR2b without disrupting NR2a [29]. Nevertheless, the widespread distribution of both subunits may not facilitate the replication of this approach in humans. Our findings coincide entirely with these prior studies and demonstrate that melatonin treatment influenced the protein activity of NR2a, indicating that melatonin may, to some extent, promote its neuroprotective effects by controlling the expression and phosphorylation patterns of the NMDAR subunits. NRb2 subunit of NMDA intensifies ROS generation during ischemic stroke by triggering mitochondrial dysfunction and calcium overload. Melatonin, a potent antioxidant, stabilizes this and improves mitochondrial stability. Moreover, NLRP3 can be activated by pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPS) such as ROS and calcium signalling. Both ROS and calcium bursts from NMDA/AMPA overactivation are the primary triggers for the NLRP3 inflammasome assembly [30,31]. Consistent previous studies demonstrated the antioxidative and anti-excitotoxic effect of melatonin in other models of inflammation, and it quenches the triggering signals of NLRP3 activation [32].
Furthermore, glutamate stimulates ligand-gated cation channels, notably α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), with subunits GluR1 to GluR4, which play a vital role in facilitating excitotoxicity [33]. GluR1 is mostly localized to the hippocampus, and several studies suggest that its phosphorylation is crucial not only for the recruitment of AMPAR channels, but also for the localization of synapses, the storage of memories, and the acquisition of spatial knowledge [34–36]. In particular, the GluR1 subunit’s serine 845 phosphorylation is crucial in regulating cognitive processes [37]. Consistent with prior studies, our results indicate that melatonin may impact cognitive functioning by stabilizing AMPARs and facilitating the phosphorylation of GluR1 at serine 845.PSD95 is a synaptic marker that controls the process of synapse remodelling and synaptogenesis in the hippocampus. Numerous studies suggest that synaptic remodelling in the hippocampus is linked to cognitive functions, and its disruption may lead to cognitive deficits [38,39]. The findings of our study revealed an increase in the levels of these proteins in the MCAO model. However, melatonin effectively reduces the level to a considerable degree, thereby restoring synaptic plasticity and safeguarding the hippocampus from ischemic injury. In another study by Juan et al. in 2014, PSD95 levels were upregulated by melatonin relative to the sham group. This discrepancy in the results may be attributable to the sample collection timeframe and the severity of the model. (pMCAO vs tMCAO) [40]. Reactive oxygen species (ROS) induce the nitrosylation of PSD95 through peroxynitrite, leading to the destabilization of its interaction with NMDA and AMPA receptors, resulting in synaptic collapse. The degradation of PSD95 disrupts NMDAR-mediated neuroprotective signaling pathways, such as AKT and CREB.
Simultaneously, we noted a significant downregulation of PPARγ after pMCAO, which typically functions as an essential regulator of anti-inflammatory responses. The reduction of PPARγ activity eliminates a critical inhibitory mechanism on NF-κB signaling, facilitating prolonged inflammation and enhanced NLRP3 activation [41]. This finding aligns with the research conducted by previous researchers, which illustrates PPARγ’s capacity to inhibit NF-κB-mediated inflammation. Our data indicate that melatonin treatment effectively restores PPARγ expression, thereby reinstating this endogenous anti-inflammatory mechanism.
The Nrf2 pathway modulates the oxidative stress-inflammation axis, which was found to be impaired after pMCAO. Under physiological conditions, the translocation of Nrf2 to the nucleus activates the transcription of antioxidant proteins such as HO-1, thereby offering cellular protection against oxidative damage [42]. The suppression of this pathway observed in our study likely contributes to sustained oxidative stress and subsequent neuronal damage. Melatonin activates Nrf2 signaling, which is a crucial neuroprotective mechanism, demonstrated by decreased oxidative markers and enhanced neuronal survival in treated animals.
Neurotrophic factor γ-enolase serves as a mediator for neuronal growth and differentiation [43]. Enolases are glycolytic enzymes having widespread roles ranging from cell proliferation to differentiation and are present in three isoforms: α-, β-, and γ-enolase [43]. γ-enolase, also known as neuronal-specific enolase (NSE) or enolase 2, is abundantly present in nervous tissue. γ-enolase serves as a mediator for neuronal growth and differentiation through PI3K pathways. Moreover, this isoform demonstrates neurotrophic and neuroprotective properties via PI3K/Akt and MAPK/ERK signalling pathways [43]. Previously, natural agents exhibited neuroprotective properties and mitigated ischemic stroke by modulating enolase expression. Researchers have found dysregulation of CRMP2 in neurodegenerative models involving NMDAR binding [44]. Moreover, excitotoxicity disassembles CRMP2 through a calpain-dependent mechanism into the smaller 58 kDa subunit and reduces the activity of NMDAR [44,45]. The regulation of NMDAR trafficking and function by CRMP2 has been evidenced in multiple studies through the downregulation of NR2b subunit expression of NMDAR, thereby supporting neuronal survival. Our findings also indicated an increased expression of CRMP2, consistent with previous studies, and established a mechanism by which melatonin may improve neuropathological impairments in the hippocampus after an ischemic stroke.
There are certain limitations in this study. We measure the effects in the pMCAO model, and these effects need to be validated in the transient tMCAO model with various time durations. Further, to rule out the role of melatonin on NMDA and AMPA receptors in the hippocampus, a mechanistic study should be carried out by blocking the NR2a and NR2b, and these should be linked to the behavioural deficits. We have calculated in this manuscript the phosphorylated level of JNK, P38, GSK3β, and AKT; their values should be relatively expressed to the total protein level as p-JNK/JNK, p-p38/p38, p-GSK3β/GSK3β.
In summary, this study offers insights into the neuroprotective properties of melatonin in the hippocampal region of the brain subsequent to an ischemic stroke (Fig. 7). This investigation implies that the neuroprotective properties of melatonin may be because of its capacity to modulate a variety of proteins, which can mitigate the loss of neurons induced by pMCAO. These findings suggest that conducting additional research into the functions of pertinent proteins and selectively targeting them can create new opportunities to treat a diverse range of neurodegenerative disorders, such as stroke.
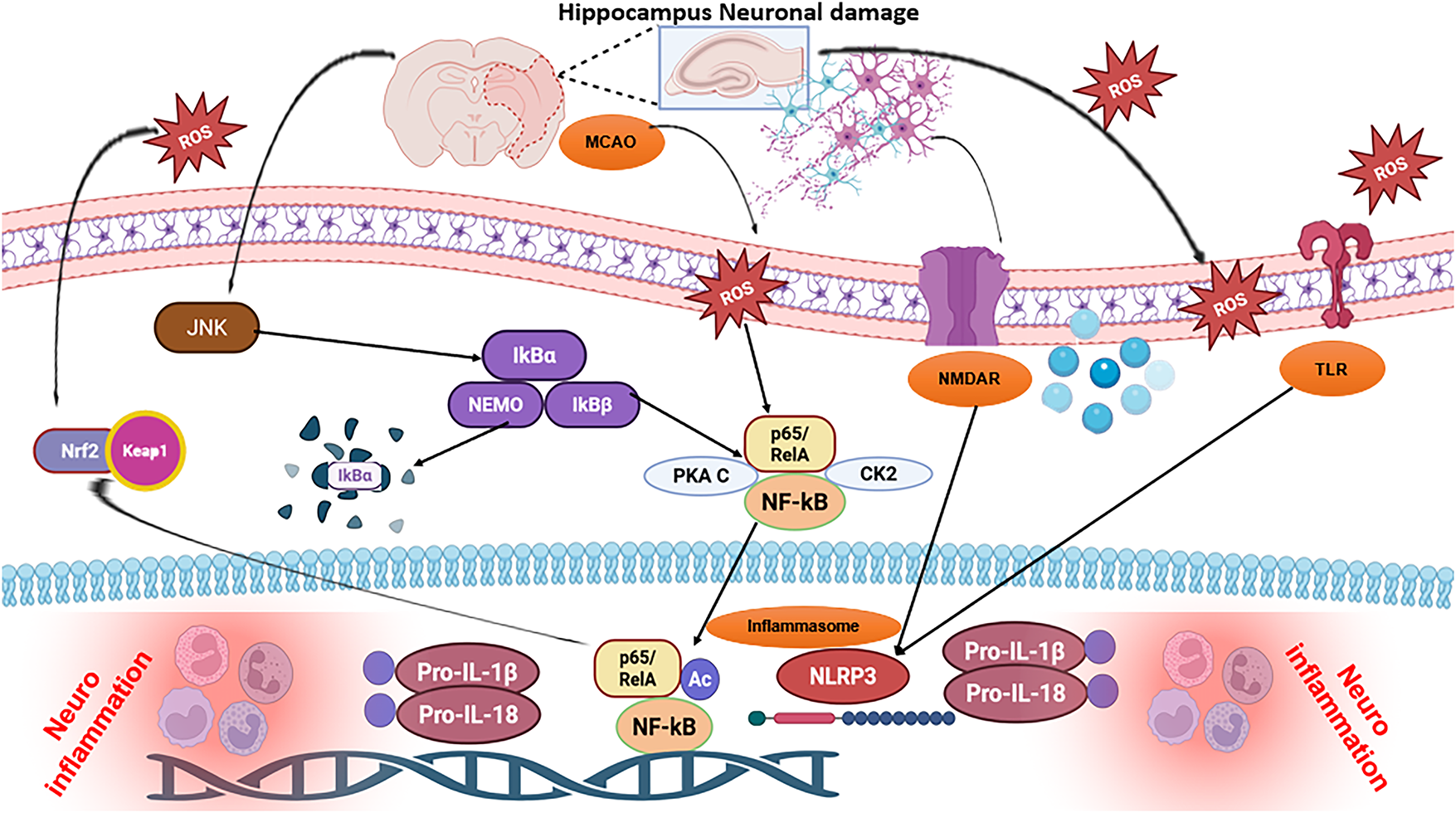
Figure 7: The possible mechanism of melatonin attenuating MCAO stroke. Abb: Pro-IL-1β: Pro-Interleukin-1 Beta; ROS: Reactive Oxygen Species; JNK: c-Jun N-Terminal Kinase; IKBα: Inhibitor of Kappa B Alpha; IKKβ: Inhibitor of Nuclear Factor Kappa-B Kinase Beta; NLRP3: NOD-Like Receptor Family, Pyrin Domain Containing 3 Inflammasome; TLR: Toll-Like Receptor; PKA C: Protein Kinase A Catalytic Subunit; CK2: Casein Kinase 2; NMDAR: N-Methyl-D-Aspartate Receptor; NF-κB: Nuclear Factor Kappa B; Nrf2: Nuclear Factor Erythroid 2-Related Factor 2
Acknowledgement: None.
Funding Statement: The authors wish to acknowledge the financial support for this work from the Deanship of Scientific Research (DSR) at the University of Tabuk, Tabuk, Saudi Arabia (grant No. S-1442-0164).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Fawad Ali Shah, Abdullah Alattar, Reem Alshaman; data collection: Fawad Ali Shah; analysis and interpretation of results: Abdullah Alattar, Reem Alshaman, Fawaz E. Alanazi, Yusuf S. Althobaiti; draft manuscript preparation: Ghareb M. Soliman, Howaida S. Ali, and subsequently edited by Waleed Salman Khubrni. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets used and/or analysed during the current study are available from the corresponding author [Fawad Ali Shah] on reasonable request.
Ethics Approval: All experimental procedures were conducted as per protocol approved by the Institutional Animal Care and Committee of Riphah Institute of Pharmaceutical Sciences (Ref No.: REC/RIPS/2019/016/Cology was allocated), in line with ARRIVE guidelines. No human data was used to prepare this manuscript.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
Abbreviations
| MCAO | Middle Cerebral Artery Occlusion |
| ROS | Reactive Oxygen Species |
| JNK | c-Jun N-Terminal Kinase |
| NF-κB | Nuclear Factor Kappa B |
| Pro-IL-1β | Pro-Interleukin-1 Beta |
| IL-1β | Interleukin-1 Beta |
| NLRP3 | Inflammasome |
| TLR | Toll-Like Receptor |
| NMDAR | N-Methyl-D-Aspartate Receptor |
| CK2 | Casein Kinase 2 |
| RF2 | Nuclear Factor Erythroid 2-Related Factor 2 |
| IKBα | Inhibitor of Kappa B Alpha |
| IKKβ | Inhibitor of Nuclear Factor Kappa-B Kinase Beta |
| PKA C | Protein Kinase A Catalytic Subunit |
References
1. Flach C, Muruet W, Wolfe CDA, Bhalla A, Douiri A. Risk and secondary prevention of stroke recurrence: a population-base cohort study. Stroke. 2020;51(8):2435–44. doi:10.1161/STROKEAHA.120.028992. [Google Scholar] [PubMed] [CrossRef]
2. Alobaida M, Lip GYH, Lane DA, Sagris D, Hill A, Harrison SL. Endovascular treatment for ischemic stroke patients with and without atrial fibrillation, and the effects of adjunctive pharmacotherapy: a narrative review. Expert Opin Pharmacother. 2023;24(3):377–88. doi:10.1080/14656566.2022.2161362. [Google Scholar] [PubMed] [CrossRef]
3. Gulyaeva NV. Functional neurochemistry of the ventral and dorsal hippocampus: stress, depression, dementia and remote hippocampal damage. Neurochem Res. 2019;44(6):1306–22. doi:10.1007/s11064-018-2662-0. [Google Scholar] [PubMed] [CrossRef]
4. Yu CL, Li JN, Gan P, Wang LP, Yang YX, Yu DF, et al. Developing of focal ischemia in the hippocampus or the amygdala reveals a regional compensation rule for fear memory acquisition. eNeuro. 2021;8(2):1–11. doi:10.1523/eneuro.0398-20.2021. [Google Scholar] [PubMed] [CrossRef]
5. Yavas E, Gonzalez S, Fanselow MS. Interactions between the hippocampus, prefrontal cortex, and amygdala support complex learning and memory. F1000Res. 2019;8:1–6. doi:10.12688/f1000research.19317.1. [Google Scholar] [PubMed] [CrossRef]
6. Ozdemir YG, Bolay H, Erdem E, Dalkara T. Occlusion of the MCA by an intraluminal filament may cause disturbances in the hippocampal blood flow due to anomalies of circle of Willis and filament thickness. Brain Res. 1999;822(1–2):260–4. doi:10.1016/s0006-8993(99)01175-0. [Google Scholar] [PubMed] [CrossRef]
7. Block F, Dihné M, Loos M. Inflammation in areas of remote changes following focal brain lesion. Prog Neurobiol. 2005;75(5):342–65. doi:10.1016/j.pneurobio.2005.03.004. [Google Scholar] [PubMed] [CrossRef]
8. Garcia JH, Liu KF, Ye ZR, Gutierrez JA. Incomplete infarct and delayed neuronal death after transient middle cerebral artery occlusion in rats. Stroke. 1997;28(11):2303–10. doi:10.1161/01.str.28.11.2303. [Google Scholar] [PubMed] [CrossRef]
9. Türeyen K, Vemuganti R, Sailor KA, Dempsey RJ. Ideal suture diameter is critical for consistent middle cerebral artery occlusion in mice. Neurosurgery. 2005;56(1):196–200. doi:10.1227/01.neu.0000144490.92966.59. [Google Scholar] [PubMed] [CrossRef]
10. Zarow GJ, Karibe H, States BA, Graham SH, Weinstein PR. Endovascular suture occlusion of the middle cerebral artery in rats: effect of suture insertion distance on cerebral blood flow, infarct distribution and infarct volume. Neurol Res. 1997;19(4):409–16. doi:10.1080/01616412.1997.11740834. [Google Scholar] [PubMed] [CrossRef]
11. Ludhiadch A, Sharma R, Muriki A, Munshi A. Role of calcium homeostasis in ischemic stroke: a review. CNS Neurol Disord Drug Targets. 2022;21(1):52–61. doi:10.2174/1871527320666210212141232. [Google Scholar] [PubMed] [CrossRef]
12. Raïch I, Lillo J, Rebassa JB, Capó T, Cordomí A, Reyes-Resina I, et al. Dual role of NMDAR containing NR2A and NR2B subunits in Alzheimer’s disease. Int J Mol Sci. 2024;25(9):4757. doi:10.3390/ijms25094757. [Google Scholar] [PubMed] [CrossRef]
13. Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27(11):2846–57. doi:10.1523/JNEUROSCI.0116-07.2007. [Google Scholar] [PubMed] [CrossRef]
14. Sun Y, Zhang L, Chen Y, Zhan L, Gao Z. Therapeutic targets for cerebral ischemia based on the signaling pathways of the GluN2B C terminus. Stroke. 2015;46(8):2347–53. doi:10.1161/STROKEAHA.115.009314. [Google Scholar] [PubMed] [CrossRef]
15. Sohail S, Ali Shah F, Zaman SU, Almari AH, Malik I, Ahmad Khan S, et al. Melatonin delivered in solid lipid nanoparticles ameliorated its neuroprotective effects in cerebral ischemia. Heliyon. 2023;9(9):e19779. doi:10.1016/j.heliyon.2023.e19779. [Google Scholar] [PubMed] [CrossRef]
16. Ullah U, Badshah H, Malik Z, Uddin Z, Alam M, Sarwar S, et al. Hepatoprotective effects of melatonin and celecoxib against ethanol-induced hepatotoxicity in rats. Immunopharmacol Immunotoxicol. 2020;42(3):255–63. doi:10.1080/08923973.2020.1746802. [Google Scholar] [PubMed] [CrossRef]
17. Jin X, Imai T, Morais A, Sasaki Y, Chung DY, Ayata C. Hippocampal infarction and generalized seizures predict early mortality after endovascular middle cerebral artery occlusion in mice. Exp Neurol. 2024;380:114903. doi:10.1016/j.expneurol.2024.114903. [Google Scholar] [PubMed] [CrossRef]
18. Gim SA, Lee SR, Shah FA, Koh PO. Curcumin attenuates the middle cerebral artery occlusion-induced reduction in γ-enolase expression in an animal model. Lab Anim Res. 2015;31(4):198–203. doi:10.5625/lar.2015.31.4.198. [Google Scholar] [PubMed] [CrossRef]
19. Chen M, Lu TJ, Chen XJ, Zhou Y, Chen Q, Feng XY, et al. Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke. 2008;39(11):3042–8. doi:10.1161/STROKEAHA.108.521898. [Google Scholar] [PubMed] [CrossRef]
20. Gascón S, Sobrado M, Roda JM, Rodríguez-Peña A, Díaz-Guerra M. Excitotoxicity and focal cerebral ischemia induce truncation of the NR2A and NR2B subunits of the NMDA receptor and cleavage of the scaffolding protein PSD-95. Mol Psychiatry. 2008;13(1):99–114. doi:10.1038/sj.mp.4002017. [Google Scholar] [PubMed] [CrossRef]
21. Hafner A, Obermajer N, Kos J. γ-Enolase C-terminal peptide promotes cell survival and neurite outgrowth by activation of the PI3K/Akt and MAPK/ERK signalling pathways. Biochem J. 2012;443(2):439–50. doi:10.1042/BJ20111351. [Google Scholar] [PubMed] [CrossRef]
22. López-Morales MA, Escobar I, Saul I, Jackson CW, Ferrier FJ, Fagerli EA, et al. Resveratrol preconditioning mitigates ischemia-induced septal cholinergic cell loss and memory impairments. Stroke. 2023;54(4):1099–109. doi:10.1161/STROKEAHA.122.040899. [Google Scholar] [PubMed] [CrossRef]
23. Gulyaeva NV, Onufriev MV, Moiseeva YV. Ischemic stroke, glucocorticoids, and remote hippocampal damage: a translational outlook and implications for modeling. Front Neurosci. 2021;15:781964. doi:10.3389/fnins.2021.781964. [Google Scholar] [PubMed] [CrossRef]
24. Su Y, Zheng X. HSP70-mediated autophagy-apoptosis-inflammation network and neuroprotection induced by heat acclimatization. Biology. 2025;14(7):774. doi:10.3390/biology14070774. [Google Scholar] [PubMed] [CrossRef]
25. Soldatov V, Venediktov A, Belykh A, Piavchenko G, Naimzada MD, Ogneva N, et al. Chaperones vs. oxidative stress in the pathobiology of ischemic stroke. Front Mol Neurosci. 2024;17:1513084. doi:10.3389/fnmol.2024.1513084. [Google Scholar] [PubMed] [CrossRef]
26. Alharbi BM, Albinhassan TH, Ali Alzahrani R, Bouchama A, Mohammad S, Alomari AA, et al. Profiling the Hsp70 chaperone network in heat-induced proteotoxic stress models of human neurons. Biol. 2023;12(3):416. doi:10.3390/biology12030416. [Google Scholar] [PubMed] [CrossRef]
27. Porto RR, Dutra FD, Crestani AP, Damian Holsinger RM, Quillfeldt JA, Homem de Bittencourt PI, et al. HSP70 facilitates memory consolidation of fear conditioning through MAPK pathway in the hippocampus. Neuroscience. 2018;375(2):108–18. doi:10.1016/j.neuroscience.2018.01.028. [Google Scholar] [PubMed] [CrossRef]
28. Belenichev IF, Pavlov SV, Bukhtiayarova NV, Samura IB, Egorov AN, Semenov DM. Expression of HSP70 in the brain of rats during experimental cerebral ischemia modeling and on the background of neuroprotection. Biol Markers Guid Ther. 2017;4(1):105–11. doi:10.12988/bmgt.2017.7911. [Google Scholar] [CrossRef]
29. Shu S, Pei L, Lu Y. Promising targets of cell death signaling of NR2B receptor subunit in stroke pathogenesis. Regen Med Res. 2014;2(1):8. doi:10.1186/2050-490X-2-8. [Google Scholar] [PubMed] [CrossRef]
30. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–89. doi:10.1038/s41577-019-0165-0. [Google Scholar] [PubMed] [CrossRef]
31. Jung H, Kim B, Jang G, Kim H, Lee AR, Yoon SH, et al. The NLRP3 inflammasome in microglia regulates repetitive behavior by modulating NMDA glutamate receptor functions. Cell Rep. 2025;44(5):115656. doi:10.1016/j.celrep.2025.115656. [Google Scholar] [PubMed] [CrossRef]
32. Ma S, Chen J, Feng J, Zhang R, Fan M, Han D, et al. Melatonin ameliorates the progression of atherosclerosis via mitophagy activation and NLRP3 inflammasome inhibition. Oxid Med Cell Longev. 2018;2018(1):9286458. doi:10.1155/2018/9286458. [Google Scholar] [PubMed] [CrossRef]
33. Zhang F, Huang G, Zhu X. Effect of different charges of modified electroconvulsive seizure on the cognitive behavior in stressed rats: effects of GluR1 phosphorylation and CaMKIIα activity. Exp Ther Med. 2019;17(1):748–58. doi:10.3892/etm.2018.7022. [Google Scholar] [PubMed] [CrossRef]
34. Wang T, Wang L, Li L, Ma L, Liu X. Effects of perampanel on cognitive behavior and GluR1 expression in immature mice of temporal lobe epilepsy. Biochem Biophys Res Commun. 2022;588(5):68–74. doi:10.1016/j.bbrc.2021.12.038. [Google Scholar] [PubMed] [CrossRef]
35. Díaz-Alonso J, Nicoll RA. AMPA receptor trafficking and LTP: carboxy-termini, amino-termini and TARPs. Neuropharmacology. 2021;197:108710. doi:10.1016/j.neuropharm.2021.108710. [Google Scholar] [PubMed] [CrossRef]
36. Park P, Kang H, Sanderson TM, Bortolotto ZA, Georgiou J, Zhuo M, et al. The role of calcium-permeable AMPARs in long-term potentiation at principal neurons in the rodent hippocampus. Front Synaptic Neurosci. 2018;10:42. doi:10.3389/fnsyn.2018.00042. [Google Scholar] [PubMed] [CrossRef]
37. Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, et al. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112(5):631–43. doi:10.1016/s0092-8674(03)00122-3. [Google Scholar] [PubMed] [CrossRef]
38. Mostafa M, Disouky A, Lazarov O. Therapeutic modulation of neurogenesis to improve hippocampal plasticity and cognition in aging and Alzheimer’s disease. Neurotherapeutics. 2025;22(3):e00580. doi:10.1016/j.neurot.2025.e00580. [Google Scholar] [PubMed] [CrossRef]
39. Han PP, Han Y, Shen XY, Gao ZK, Bi X. Enriched environment-induced neuroplasticity in ischemic stroke and its underlying mechanisms. Front Cell Neurosci. 2023;17:1210361. doi:10.3389/fncel.2023.1210361. [Google Scholar] [PubMed] [CrossRef]
40. Juan WS, Huang SY, Chang CC, Hung YC, Lin YW, Chen TY, et al. Melatonin improves neuroplasticity by upregulating the growth-associated protein-43 (GAP-43) and NMDAR postsynaptic density-95 (PSD-95) proteins in cultured neurons exposed to glutamate excitotoxicity and in rats subjected to transient focal cerebral ischemia even during a long-term recovery period. J Pineal Res. 2014;56(2):213–23. doi:10.1111/jpi.12114. [Google Scholar] [PubMed] [CrossRef]
41. Lu H, Guo T, Zhang Y, Liu D, Hou L, Ma C, et al. Endoplasmic reticulum stress-induced NLRP3 inflammasome activation as a novel mechanism of polystyrene microplastics (PS-MPs)-induced pulmonary inflammation in chickens. J Zhejiang Univ Sci B. 2024;25(3):233–43. doi:10.1631/jzus.B2300409. [Google Scholar] [PubMed] [CrossRef]
42. Wang D, Yin K, Zhang Y, Lu H, Hou L, Zhao H, et al. Novel pathways of fluoride-induced hepatotoxicity: P53-dependent ferroptosis induced by the SIRT1/FOXOs pathway and Nrf2/HO-1 pathway. Comp Biochem Physiol C Toxicol Pharmacol. 2023;264:109526. doi:10.1016/j.cbpc.2022.109526. [Google Scholar] [PubMed] [CrossRef]
43. Polcyn R, Capone M, Hossain A, Matzelle D, Banik NL, Haque A. Neuron specific enolase is a potential target for regulating neuronal cell survival and death: implications in neurodegeneration and regeneration. Neuroimmunol Neuroinflamm. 2017;4(12):254–7. doi:10.20517/2347-8659.2017.59. [Google Scholar] [PubMed] [CrossRef]
44. Castillo C, Martinez JC, Longart M, García L, Hernández M, Carballo J, et al. Extracellular application of CRMP2 increases cytoplasmic calcium through NMDA receptors. Neuroscience. 2018;376(M110):204–23. doi:10.1016/j.neuroscience.2018.02.002. [Google Scholar] [PubMed] [CrossRef]
45. Brittain JM, Pan R, You H, Brustovetsky T, Brustovetsky N, Zamponi GW, et al. Disruption of NMDAR-CRMP-2 signaling protects against focal cerebral ischemic damage in the rat middle cerebral artery occlusion model. Channels. 2012;6(1):52–9. doi:10.4161/chan.18919. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools