
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Dissecting the effects of androgen deprivation therapy on cadherin switching in advanced prostate cancer: A molecular perspective
1 Department of Molecular Biology and Genetics, Science Faculty, Dicle University, Diyarbakir, 21280, Turkey
2 Cancer Research Center, Dicle University, Diyarbakir, 21280, Turkey
3 Department of Chemical and Biomolecular Engineering, University of Illinois, Urbana, IL, 61801, USA
4 First Department of Pediatrics, National and Kapodistrian University of Athens, Athens, 11527, Greece
5 Department of Microbiology and Immunology, Feinberg School of Medicine, Northwestern University, Chicago, IL, 60611, USA
6 Department of Natural Sciences and Engineering, John Wood College, Quincy, IL, 62305, USA
* Corresponding Author: OSMAN CEN. Email:
Oncology Research 2022, 30(3), 137-155. https://doi.org/10.32604/or.2022.026074
Received 15 August 2022; Accepted 23 December 2022; Issue published 12 January 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Prostate cancer is one of the most often diagnosed malignancies in males and its prevalence is rising in both developed and developing countries. Androgen deprivation therapy has been used as a standard treatment approach for advanced prostate cancer for more than 80 years. The primary aim of androgen deprivation therapy is to decrease circulatory androgen and block androgen signaling. Although a partly remediation is accomplished at the beginning of treatment, some cell populations become refractory to androgen deprivation therapy and continue to metastasize. Recent evidences suggest that androgen deprivation therapy may cause cadherin switching, from E-cadherin to N-cadherin, which is the hallmark of epithelial-mesenchymal transition. Diverse direct and indirect mechanisms are involved in this switching and consequently, the cadherin pool changes from E-cadherin to N-cadherin in the epithelial cells. Since E-cadherin represses invasive and migrative behaviors of the tumor cells, the loss of E-cadherin disrupts epithelial tissue structure leading to the release of tumor cells into surrounding tissues and circulation. In this study, we review the androgen deprivation therapy-dependent cadherin switching in advanced prostate cancer with emphasis on its molecular basis especially the transcriptional factors regulated through TFG-β pathway.Keywords
Prostate cancer (PCa) is one of the most frequently diagnosed malignancies in males with an increasing prevalence in both developed and developing countries and is one of the major causes of cancer-related death in men [1,2]. Although the standard treatment for local PCa is surgery and radiotherapy, some other treatment protocols, such as brachytherapy and androgen deprivation therapy (ADT) especially in patients with advanced PCa are commonly used [3,4].
The primary purpose of ADT is to reduce circulating androgen levels and block androgen signaling, since androgen has an important role in cancer cell proliferation and consequently disease progression [5]. Indeed, ADT is a powerful and useful treatment approach that causes a decrease in prostate-specific antigen (PSA) levels and tumor volume. It provides a temporary relief to patients although some patients may experience severe to moderate side effects such as hot flashes and sexual dysfunction during treatment [6,7].
The response to the ADT may be observed in three phases: early, developing, and late phases (Fig. 1). The early phase is characteristic with decreased level of PSA, tumor regression, and increased quality of life while the late phase is characteristic with castration resistance, relapse, and highly metastatic and aggressive tumors. In the intervening period, which can be considered as a developing phase, homeostatic response of the cells leads to a progressive development of alteration in various cellular signaling mechanisms leading to the late phase response (Fig. 1). The cellular responses include reactivation of androgen receptor (AR) signaling through alternative mechanisms, decreased adhesion, increased motility and invasion, and altered tonic cell survival signaling. Mechanisms involving re-activation of androgen signaling include intra-tumor de novo androgen production, amplification of AR gene, mutations in the ligand-binding domain of AR (AR-LBD), expression of some constitutively active AR isoforms, overexpression of AR co-regulators, and androgen-independent AR activation [8,9]. These events lead to a more aggressive phenotype of the disease called castration-resistant PCa (CRPC), generally 24–36 months after the initiation of ADT [10]. In this stage, metastatic spread is observed in most of the patients and the average life expectancy is 18–24 months afterward [10].
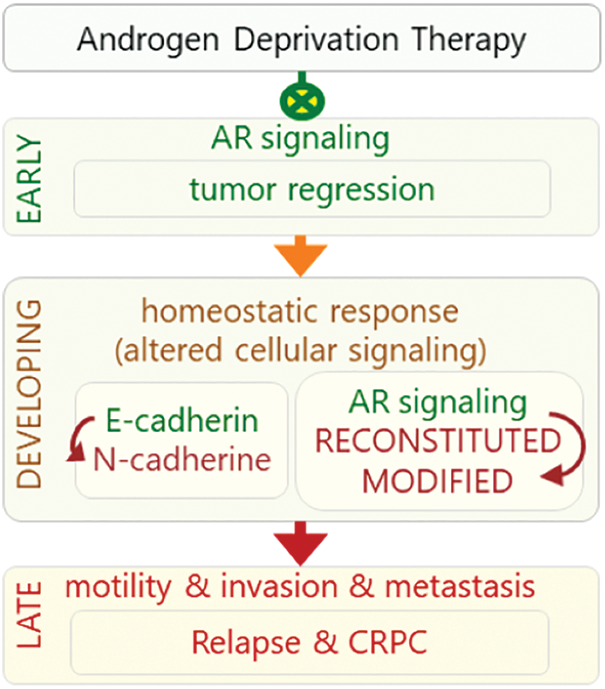
Figure 1: Response to ADT can be viewed in three phases: early, developing, and late phases. In early phase, ADT inhibits (green crossed-circles) AR signaling that leads to tumor regression. In the developing phase, cell’s homeostatic response results in re-activation of AR signaling in unconventional manners. Altered activities of signaling mechanisms lead to late phase (red arrow), in which castration resistance and more aggressive and highly metastatic AR-independent tumors develop.
Another altered mechanism is epithelial-mesenchymal transition (EMT), which decreases adhesion and increases motility and invasion. Growing evidence links EMT to ADT in advanced PCa despite the lack of consensus regarding the effect of androgen signaling in the regulation of EMT [11]. Indeed, initial studies have reported that ADT may induce EMT in advanced PCa at least through the “cadherin switching” mechanism [12–18]. These reports led to further investigation, which pointed to an aberrant activation of transforming growth factor β1 (TGF-β) signaling [19,20], a multifunctional pathway regulating proliferation and differentiation of cells and cellular signaling molecules involved in cadherin switching [21]. The term “cadherin switching” generally refers to diminished expression of E-cadherin, encoded by Cdh1, and increased expression of N-cadherin, encoded by Cdh2, in cells [22]. Cadherin switching is a normal process during embryogenesis and cannot initiate tumorigenesis alone in healthy cells but may confer further migration and increased invasive capabilities on tumor cells [22–24]. Decreased Cdh1 expression disrupts epithelial tissue structure causing cells to detach from one another and move freely [25]. Consistently, decreased expression of Cdh1 depends on cadherin switching, a process closely associated with tumor invasion and metastasis, two common characteristics of advanced cancers [26]. N-cadherin confers enhanced migratory and invasive abilities on tumor cells, which contrasts the inhibitory role of E-cadherin on migration and invasion [27].
In this study, we aim to review the current literature to dissect the role of ADT on cadherin switching in advanced PCa from a molecular perspective.
PCa usually develops from pre-malignant lesions such as proliferative inflammatory atrophy (PIA) and prostatic intraepithelial neoplasia (PIN) to androgen-dependent local tumors and eventually progresses to CRPC [28]. The androgens are steroid hormones that play crucial roles in all steps of PCa from benign local tumors to malignant CRPC [29,30]. They are mainly synthesized in Leydig cells in testes through enzymatic conversion reactions from cholesterol to testosterone (T). Following synthesis, T is secreted into the blood where it is mostly carried as bound to serum albumin or serum hormone-binding globulin [31]. The circulating level of T is strictly regulated by the hypothalamus-pituitary–gonadal (HPG) axis feedback system [32]. T is a low potency hormone and after entering prostate cells it is converted to dihydrotestosterone (DHT) by 5-α-reductase [33]. Compared to T, DHT is more potent in activating AR signaling [33,34]. Although T can activate AR in prostate cells, conversion to DHT is necessary for the development of the normal prostate gland [35]. The necessity of DHT for the development of prostate has also been confirmed in the treatment of benign prostatic hyperplasia (BPH) by 5-α-reductase inhibitors [36,37].
The action of androgen is mainly through AR [38]. In the absence of androgens, AR is mostly located in the cytoplasm, forming a heterocomplex with heat shock proteins (HSPs) comprised of HSP27, HSP40, HSP56, HSP70, and HSP90 [39,40]. Following ligand binding, AR can activate various metabolic cellular responses at genomic and non-genomic levels. At the genomic level, also known as the canonical pathway, activated AR leaves the HSP complex, dimerizes, and then is translocated to the nucleus (Fig. 2) [41]. In the nucleus, AR dimers bind to androgen response elements (ARE) on the promotors of the target genes, such as kallikrein related peptidase 3 (Klk3), NK3 homeobox 1 (Nkx3.1), hematological and neurological expressed sequence 1 (Hn1), and Cdh1, a process that controls the expression of these genes through recruiting various co-regulators such as transcriptional activators or suppressors [42–46].
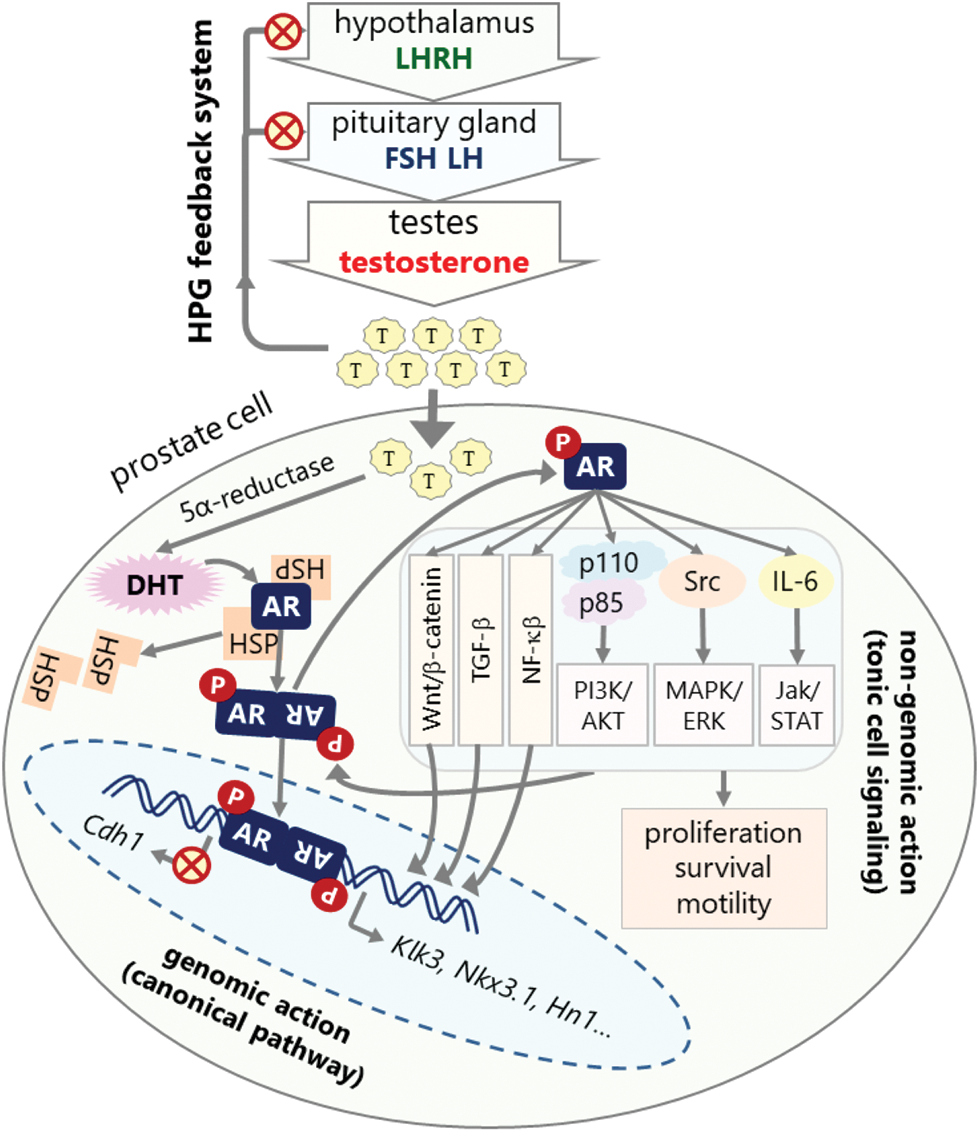
Figure 2: HPG axis feedback system for androgen synthesis and mechanism of androgen signaling. Androgens are mostly synthesized in testes under the control of the HPG feedback system. High androgen level inhibits hypothalamus and pituitary glands to decrease testosterone synthesis (red crossed-circles). Androgens exert their action in the prostate cells through canonical or non-genomic pathways. The canonical pathway involves transcriptional regulation of target genes. In this pathway, AR is sequestered in the cytoplasm by HSPs. Androgen binding activates and dimerizes AR, which is then translocated to the nucleus and binds to the AR elements in the promoters of target genes such as Klk3, Nkx3.1, Hn1, and Cdh1 leading to the upregulation or downregulation of their expressions depending on the presence of other regulatory factors. Non-genomic action involves the direct activation of cellular signaling pathways, in which activated AR interacts with and phosphorylates (P in red circles) the components of various signaling pathways such as PI3K/AKT, MAPK/Raf/ERK, Jak/STAT, TGF-β, NF-κB, and Wnt/β-catenin pathways leading to the activation of these pathways that promote survival, proliferation, and metastasis of cell (red crossed-circles: inhibition, gray arrows: activation and interaction).
At the non-genomic level, AR regulates diverse tonic cellular signaling mechanisms independent from AR nuclear transactivation (Fig. 2) [47,48]. One of the cellular kinases activated through AR signaling is Src kinase [49], which is highly active in PCa and is involved in increasing malignant behaviors of the tumor cells [49,50]. Phosphorylated Src is activated and can activate the downstream MAPK/ERK1-2 signaling pathway resulting in increased cell proliferation [49]. PI3K/AKT signaling pathway is also activated by AR in a non-genomic manner [51]. Activated AR interacts with the SH2 domain of the regulatory subunit (p85) of phosphatidylinositol 3-kinase (PI3K) in the cytoplasm, which promotes activation of the catalytic subunit (p110) of PI3K [52]. Consequently, PI3K activates serine/threonine kinase (AKT), a key molecule in the induction and maintenance of cell activation and survival signals. AR is also activated by Jak/STAT signaling pathway through IL-6 and possibly other cytokines [53–55]. MAP kinase and cytokine signaling pathways also activates the nuclear factor “kappa-light-chain-enhancer” of activated B-cells (NF-κB) pathway, which has been implicated in various cancers including prostate cancer. NF-κB is highly active in PCa that leads to increased AR expression and progression into androgen independent growth and CRPC [56,57]. Another deregulated pathway in PCa is Wnt/β-catenin signaling pathway, which is strictly regulated in normal cells. However, it has been dysregulated in various cancers including prostate cancer. While Wnt/β-catenin is inactive in early PCa, it is highly activated in late stages [58,59].
Overactivation of aforementioned signaling pathways and likely others, promote AR transactivation in a ligand independent manner via regulating interactions between AR and various co-regulators [51,53,54,60]. Consequently, this alternative activation of AR further activates cell proliferation and survival signaling, forming a feed-forward activation cycle [54]. Androgen-independent AR activation is a commonly observed mechanism in CRPC, which is further discussed below.
ADT in the Treatment of Advanced PCa
While the treatment of choice for local PCa is usually surgery, radiotherapy, or brachytherapy, these treatments are often replaced with ADT in patients with advanced recurring PCa after prostatectomy [3,4]. The primary aim of ADT is to reduce circulating androgen levels and therefore inhibits or attenuate AR signaling [61]. The newly developed some third generation agents, such as enzalutamide, act as competitive androgen inhibitors by binding to androgen binding site of AR and inhibiting its androgen-induced transactivation [61]. It is estimated that about 40% of PCa patients will receive ADT within six months of diagnosis [5,62]. Indeed, ADT was proposed about 80 years ago to be a powerful treatment option for advanced PCa [63]. Although the original form of ADT was bilateral orchiectomy this procedure has later been replaced with certain drugs that block androgen production and AR signaling in PCa cells [4]. Currently, a combination of luteinizing hormone-releasing hormone (LHRH) agonists and anti-androgen agents are used to block androgen production [64].
At the beginning of treatment, ADT has a strong effect on the inhibition of the progression of the disease and leads to partial remediation such as regression in tumor volume and decreased serum PSA level, but this effect is temporary and some cell populations become refractory to ADT after a while [6]. Consequently, the disease begins progressing with an increase in the tumor volume and serum PSA level, leading to metastasis into new sites 24–36 months after ADT started [10]. The new phenotype of PCa is characteristic with stem cell-like features [65] and referred to as CRPC, which is associated with a poor prognosis [6,66]. The elevated serum PSA level is an important indicator of restored AR activity because activated AR directly promotes the transcription of PSA encoding gene Klk3 [67].
Multiple AR re-activation mechanisms occur in CRPC including amplification of AR gene, the gain of function mutations in the AR-LBD, intra-tumoral de novo androgen production, increased transcriptional activation of AR, altered expressions of some AR co-regulators, growth factors- or cytokine-dependent AR activation, and expression of some constitutively active AR isoforms [68–74]. These mechanisms lead to AR stabilization and unconventional re-activation of AR signaling that ends up with increased transcriptional activity in CRPC even when there is a low androgen level.
AR gene amplification is the most common genetic change in CRPC patients. It has been shown that 20%–30% of the patients harbor amplified AR in their genomes in CRPC cells [75–77]. However, this amplification is not common in primary tumors and is reported in just 2% of PCa patients who had not undergone ADT [78]. Another cause of increased AR signaling is due to increased level of AR protein via post-transcriptional or post-translational deregulation, which may lead to increased stabilization of AR mRNA and/or protein [79,80]. Consequently, the increased level of AR protein in CRPC cells enables the cells to be hypersensitive to the low level of androgens [79,81,82].
Another mechanism for the re-activation of AR signaling in CRPC is AR mutations. While some mutations have been identified in the amino-terminal (NTD) and DNA-binding (DBD) domains of AR, most of the activating mutations are localized to the ligand-binding domain (LBD) of AR [79,83]. Although mutations in LBD are rare in hormone-responsive PCa cells, they have been detected in about 20% of CRPC tumor cells [84]. Mutations in LBD alters the ligand specificity of AR and facilitate its activation with other steroid hormones, in the absence of androgen [70,79]. Furthermore, it has been shown that anti-androgens may act as agonist on some AR with mutated LBD [83].
Although ADT largely suppresses the serum T level, intra-tumoral DHT level may still be high [85]. Intra-tumoral androgen production may be regulated by various mechanisms including de novo production of DHT and conversion of adrenal androgens or cholesterol to T [71,86,87]. Consequently, insufficient suppression of intra-tumoral androgens causes continued activation of AR signaling in the tumor cells even in castrate conditions [88].
Expression of constitutively active AR variants is an important mechanism that drive castration sensitive cells to CRPC [89,90]. These variants are expressed at later stages of PCa as a response to low androgen environment under ADT conditions [91,92]. Although more than 20 AR splice variants are expressed in PCa cells, the constitutively active variants AR-V3, AR-V4, AR-V7 and AR-V12 have been shown to be important in reconstituting the AR function [93,94]. Among these variants, the AR-V7 is the most extensively characterized AR variants [95,96]. AR7 mutant has truncated LBD due to the aberrant splicing events and its constitutive activity is ligand independent [95]. Lack of LBD in the AR-V7 renders AR-7 resistant to anti-androgens such as enzalutamide and abiraterone in CRPC cells. Therefore, AR-V7 is considered as a CRPC-specific AR variant [97–100]. AR-V7 is also associated with worse prognosis in PCa as the presence of AR-V7 is associated with poor survival in metastatic CRPC patients [89].
AR functions as a transcription factor in canonical signaling pathway and over 250 co-regulators (co-activators or co-repressors) have been shown to contribute to the regulation of AR transcriptional activity [101,102]. The term AR co-regulators represents a wide group of molecules that include proteins and RNAs that regulate AR activity [72] and the relationship between AR and co-regulators affects development, progression, and treatment outcome of PCa [103]. Indeed, AR co-regulators are differentially expressed in all stages of PCa including CRPC [104]. Furthermore, it has been suggested that differential expressions of AR co-regulators contribute to the development of CRPC [105] and altered levels of co-activators and co-repressors result in induction of AR transcriptional activity in CRPC, even at extremely low concentrations of androgen [104].
Growth factors and cytokines have been shown to activate AR even in the absence of or in the presence of low androgen concentration. For example, insulin-like growth factor-1 (IGF-1), epidermal growth factor (EGF) and keratinocyte growth factor can activate AR in androgen deprived conditions [73]. On the other hand, interleukin-6 (IL-6) can activate AR via Stat3, PI3K/AKT, and MAPK signaling in PCa cells [106,107]. Similarly, NF-κB induces increased AR transcription and protein [108] and promote androgen independent growth of cancer cells [56]. IL-8 can also induce androgen independent activation of AR in a FAK and Src dependent manner [109].
Activation of β-catenin in PCa after ADT
Wnt/β-catenin signaling is a conserved pathway that regulates various physiological functions including differentiation, embryonic development, adhesion, migration, apoptosis, and tissue homeostasis. Binding of Wnt to its G-protein coupled receptor activates a signaling cascade leading to the accumulation and translocation of β-catenin into the nucleus where it recruits transcription factors leading the transcription of target genes. The Wnt/β-catenin signaling is strictly regulated in normal cells; however, it has been dysregulated in various cancers including prostate cancer [58,59]. One of the EMT-promoting factor Cripto-1 (CR-1), a member of epidermal growth factor family, is upregulated in metastatic prostate cancers and is associated with decreased membrane associated β-catenin and increased nuclear β-catenin [110,111] and knocking down its expression inhibits cell proliferation, migration, and invasion [112]. CR-1 has a central role in regulating and cross-talking with various cellular signaling mechanisms including TGF-β, MAPK, and Wnt/β-catenin [113]. Overexpressed CR-1 is a poor prognostic indicator in prostate cancer [114].
In addition, a number of microRNAs (miRNAs) have been shown to modulate Wnt/β-catenin pathway in cancer cells [115,116]. For example, miRNA miR-744 promotes prostate cancer progression through aberrant activation of Wnt/β-catenin pathway [117] while miR-15A, miR-138, and miR-320 suppresses Wnt/β-catenin pathway [118–120].
TGF-β in PCa development and metastasis after ADT
TGF-β signaling is a multifunctional pathway regulating various cellular functions including embryonic development, tissue repair, differentiation, inflammation, migration, proliferation, and cell cycle [121]. TGF-β exerts its signaling through its heterodimeric receptors TβRI and TβRII leading to activation of its canonical (SMADs—transcriptional regulation) and non-canonical (TRAFs, PI3K/Akt/mTOR, and CDC42—regulation of tonic) cellular signaling mechanisms [122]. Aberrations in TGF-β signaling have been indicated in various diseases including cancers [21]. In PCa, TGF-β signaling has a dual role: while it represses survival signaling and promotes cell cycle arrest and pro-apoptotic signals in the normal prostate cells, it promotes oncogenic signals and treatment-resistant phenotypes in malignant and advanced tumor cells [123–125] [126]. TGF-β inhibits cell cycle progression in early-stage PCa cells, and TGF-β treatment together with androgen leads to a strong cell cycle arrest and apoptotic induction in the androgen-dependent PCa cells, it promotes invasion, angiogenesis, and metastasis in advanced PCa [127–129]. This contradicting effect may be related to the structure of its promoter, at least in the context of androgen signaling. The Tgf-β1 promoter has multiple AREs that group into positive and negative AREs that respectively recruit positive and negative regulatory factors, and binding of AR to these elements regulates Tgf-β1 expression positively or negatively depending on the combination of other regulatory factors [73]. In the normal prostate gland cells, activated AR binds to negatively regulating AREs on the TGF-β1 promoter and inhibits its expression [130]. However, in the metastatic PCa cells, androgen stimulation leads to the binding of activated AR to positively regulating AREs on the Tgf-β1 promoter and increases its expression [131]. Tgf-β1 expression is consistently high in the metastatic PCa cells and elevated TGF-β1 level increases Bcl-2 level rendering cells resistant to chemotherapeutic agent Docetaxel [132,133].
The effect of ADT on the TGF-β signaling has also been investigated. Interestingly, it has been shown that ADT causes an increase in the levels of TGF-β ligand, TGF-β receptor I (TβRI), TGF-β receptor II (TβRII), and further activation of transcription factor SMAD3 [134–136]. It is likely that ADT may also regulate mRNA and protein levels of TGF-β1 by post-transcriptional mechanisms [137]. Another mechanism has also been proposed for androgen and ADT-dependent TGF-β1 regulation during PCa progression. This suggests that TGF-β1 regulation may be through SAM Pointed Domain Containing ETS Transcription Factor (SPDEF) that inhibits EMT and thereby metastatic behaviors of PCa cells, as described below [138].
Cadherin Switching in the Progression of PCa and Its Relationship with Androgen Signaling
Cadherin switching usually refers to repression of Cdh1 expression, which encodes for E-cadherin, and induction of Cdh2 expression, which encodes for N-cadherin, and as a result, E-cadherin level decreases while N-cadherin level increases in the cells. E-cadherin is an important adhesion molecule for the formation and stability of epithelial tissue. Its loss impairs prostatic glandular formation and induces PIN in normal prostate luminal cells [139]. Cdh1 expression is inversely regulated by androgen signaling [140]. Increased androgen activity suppresses Cdh1 expression, which leads to decreased E-cadherin level, and consequently weaker cell-cell adhesion in androgen-dependent PCa cells [141]. Since ADT inhibits androgen signaling, the level of E-cadherin would be expected to increase upon ADT. In contrast, Cdh1 expression is further inhibited in ADT, in an AR-independent manner via the activities of a group of transcription factors and other proteins that induce cadherin switching [142,143]. The loss of Cdh1 expression impairs adhesion of primary tumor cells in many cancers, including PCa [15,27,144–146]. The association between decreased E-cadherin levels and metastasis has been shown in prostate tumors [147]. Furthermore, loss of E-cadherin in PCa has been shown to cause its metastasis into lymph nodes and bones, indication of poor prognosis [148–150].
N-cadherin, like E-cadherin, is a calcium-dependent adhesion molecule but it has opposite roles in tumorigenesis [151]. Cdh2 is highly expressed in mesenchymal and neural cells while its expression is absent or at a very low level in epithelial cells [145,152]. However, its level is increased in many advanced tumors, including PCa, and is associated with the progression of the disease [27,153]. Indeed, N-cadherin level is increased in CRPC cells compared to the castration sensitive PCa cells [14]. Likewise, increased Cdh2 expression causes castration resistance in PCa cells and targeting N-cadherin with monoclonal antibody inhibits the invasive and metastatic behaviors of the cells and renders them castration sensitive [14,153]. It is known that elevated Cdh2 expression increases migrative and invasive abilities of cancer cells [27]. Indeed, it has been shown that N-cadherin induces EMT and thereby increases the metastatic potential of PCa cells [154].
Androgen signaling has an important regulatory function on the level of N-cadherin in PCa cells. Indeed, it has been shown that androgen treatment causes a decrease in the N-cadherin level and inhibition of AR signaling upregulates N-cadherin expression and induces progression of PCa [12,16]. High Cdh2 expression after ADT is also associated with a higher Gleason score and metastasis [12,155,156]. The data suggest that ADT may cause cadherin switching in PCa, which switches the cell growth from androgen-dependent state to androgen-independent state and consequently increases migrative and metastatic abilities of the cells [15,16,155]. Although many signaling mechanisms may play roles in the cadherin switching, TGF-β signaling has a key role in this transition (Fig. 3) [21,157]. Depending on cellular micromilieu, it may act as a strong EMT promoter, which leads to loss of cellular polarity and tight junctions due to decreased expression of E-cadherin and increased expression of N-cadherin that renders a highly migratory phenotype on the cells [21]. TGF-β pathway also crosstalk with NF-κβ and Wnt/β-catenin pathways in EMT modulation. The interaction involves use of common signaling molecules and transcription factors. For example, CR-1 induces EMT through activating Wnt/β-catenin pathway as well as SMADs in the canonical TGF-β signaling [113]. Likewise, CR-1 can activate SNAIL1 through Src/Akt axis [158].
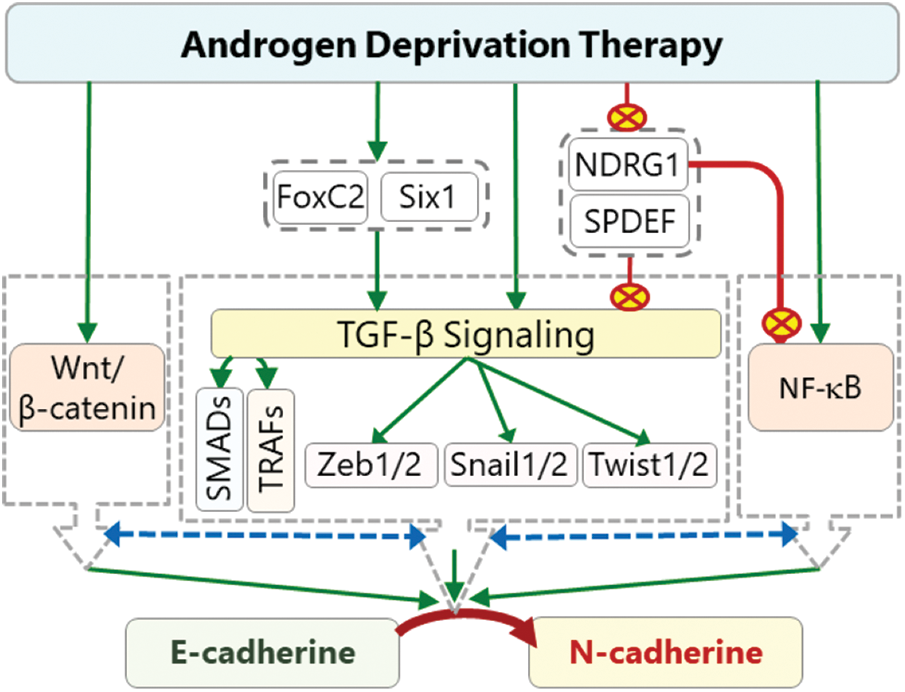
Figure 3: The effects of ADT on cadherin switching. ADT can activate TGF-β signaling directly or indirectly through activation (green arrows) of Six1 and inhibition (red crossed-circles) of SPDEF and NDRG1. Activated TGF-β induces the expression of Zeb1, Twist, and Snail transcription factors through SMADs (canonical) and TRAFs (non-canonical). These transcription factors promote cadherin switching by inhibiting Cdh1 expression and activating Cdh2 expression. Some of these factors can also induce the expression of FoxC2 which also promote cadherin switching. ADT-dependent activated Six1 can also directly or indirectly contribute to cadherin switching. ADT-induced repression of SPDEF and NDRG1 indirectly causes further increase in the ZEB1, TWIST, and SNAIL levels, and consequently results in a further decrease in Cdh1 and increase in Cdh2 expressions. The interaction (dashed two-way blue arrows) of TGF-β pathway with NF-κB and Wnt/β-catenin pathways further increase the intensity of cadherin switching (red crossed-circle: inhibition, green arrows: activation, dashed two-way blue arrows: interaction).
TGF-β-mediated EMT also involves microRNAs in regulating transcriptional factors involving cadherin switching. TGF-β downregulates miR-200 family of microRNAs, which normally repress the ZEB1/2, but their downregulation by TGF-β leads to indirect increase in the level of ZEB1/2 [159]. Likewise, miR-34 and miR-1 microRNAs repress expression of SNAIL1 and SNAIL2, respectively, and downregulation of these microRNAs by TGF-β leads to their increased expression [160,161]. Both ZEBs and SNAILs promote cadherin switching. ZEB and SNAIL proteins also downregulate the expression of these microRNAs further promoting their own expression and amplifying the cadherin switching mechanism [21]. A long list of microRNAs beyond the scope of this review has been indicated in the PCa–CRPC transition but we have focused on those important in connecting TGF-β with the EMT. In addition to those mediating TGF-β-induced EMT, various miRNAs have been implicated in modulating the EMT through NF-κβ and Wnt/β-catenin pathways. For example, NF-κβ-induced miR-230 promotes EMT and bone metastasis of advanced prostate cancer [162]. miR-96-5p inhibits NDRG1, which inhibits NF-κB [163].
Although many direct and indirect transcriptional targets of TGF-β signaling have been identified in this process, Zeb1, Twist1, Snail1, Snail2, FoxC2 and Six1 play important roles and worth further discussion in the cadherin switching process [21]. Indeed, increased expressions of these factors have been shown to lead to cadherin switching in various cancers, including PCa [164–171]. The major factors involved in the ADT-induced cadherin switching are listed in Table 1 and discussed below.

Zinc finger e-box binding homeobox 1 (Zeb1)
Zeb1 has seminal roles in embryogenesis as Zeb1 null mice die due to multiple lethal defects [172]. Altered expression of Zeb1 has been reported in many cancers including PCa, and elevated Zeb1 expression confers further malignant behaviors on the tumor cells [173–175]. In concordance, silencing Zeb1 inhibits proliferation, migration, and invasion of Pca cells [174,176].
ZEB1 protein is a strong EMT inducer. Increased Zeb1 gene expression induced by TGF-β, IGF-I or hypoxia promotes EMT in Pca cells [177,178], which is associated with poor prognosis [174,175]. In concordance, increased ZEB1 level has been shown to inhibit the epithelial phenotype of prostate cells and thereby increase their transendothelial migration [179]. ZEB1 level is also associated with vasculogenic mimicry and elevated ZEB1 is linked to a higher Gleason score and lymph node metastasis in Pca cells [180]. ZEB1 may influence the migrative and invasive behaviors of cells in multiple ways. The best known and probably the dominant mechanism is direct inhibition of Cdh1 expression. In this mechanism, ZEB1 directly binds to the Cdh1 promoter and recruits histone deacetylases HDAC1/HDAC2 and co-repressors to the Cdh1 promoter and consequently represses its expression [164,181–184]. ZEB1 may also contribute to epigenetic silencing of Cdh1 by recruiting DNA methyltransferase 1 (DNMT1) to Cdh1 promoter [185]. In addition, suppression of Zeb1 expression reverses the suppression of E-cadherin and upregulation of N-cadherin protein levels in Pca cells [177].
ZEB1 may also influence the expression of other cell adhesion molecules. For example, ZEB1 represses the expression of Syndecan 1 (SDC-1), a cell surface proteoglycan that has roles in cell adhesion [186]. It also represses the expression of glycosyl transferase LARGE2 and thereby causes hypo-glycosylation of cell surface glycoprotein alpha dystroglycan (αDG), which leads to increased invasion and metastasis [187].
High ZEB1 level is also associated with chemotherapy and radiotherapy resistance of cancer cells [188–190], ZEB1 level is higher in docetaxel-resistant cells compared to sensitive cells, and silencing of Zeb1 in docetaxel-resistant cells renders them docetaxel-sensitive [191,192]. Furthermore, it has been shown that ZEB1 may induce cancer stem cell features in various cancers and silencing of Zeb1 decreases treatment resistance and stem cell-like characteristics in Pca [193,194].
There is a negative correlation between AR activity and ZEB1 level in Pca cells. Indeed, inhibition of androgen signaling using enzalutamide has been shown to cause an increase in the ZEB1 level in Pca cells [195]. In concordance, ZEB1 level is also increased in androgen dependent LNCaP Pca cells when cultured in androgen deprived conditions [16]. Although it is controversial whether Zeb1 is a direct target of AR under physiological conditions, it is clear that AR and ZEB1 reciprocally regulate each other’s levels and activities in androgen-sensitive cells [196–199]. ZEB1 directly binds to AR promoter [199,200] and the importance of this binding in the presence or absence of androgen in prostate cells needs further investigation. However, it is clear that high ZEB1 promotes androgen independence via induction of stem cell-like properties while silencing Zeb1 renders Pca cells sensitive to androgen [201].
Enzalutamide treatment may induce neurodifferentiation of Pca cells by activating a positive feedback loop between ZEB1 and the calcium-sensitive potassium channel SK3 [202]. Furthermore, this feedback loop between ZEB1 and SK3 promotes calcium entry and cellular migration of Pca cells [178,203].
Twist family BHLH transcription factor 1 (Twist1)
Twist1 is an important transcription factor in health and cancer. It is important in EMT both during embryonic development and in cancer progression [168,204]. Although Twist1 exerts multiple effects in all stages of carcinogenesis including initiation, invasion, metastasis, angiogenesis, and therapy resistance, here, we focus only on the role of Twist1 in the cadherin switching mechanism [205,206]. The relationship between the cellular Twist1 level and PCa progression is relatively well documented [205]. It has been shown that Twist1 expression is higher in PCa compared to normal prostate and there is a positive correlation between Twist1 expression level and Gleason score in PCa [207]. Consistently, increased Twist1 expression is associated with metastasis, and silencing of Twist1 represses cell invasiveness in androgen independent PCa cells [208,209]. TWIST1 can directly bind to both Cdh1 promoter and the first intron of Cdh2 [210] and regulate their expression resulting in decreased E-cadherin and increased N cadherin protein levels in the cells [166,210]. Twist1 can also represses Cdh2 expression, indirectly, through inducing the expressions of other cadherin switching transcription factors, such as Snail 2 [211]. Furthermore, elevated Twist1 level is implicated in bone metastasis in PCa, an indication of poor prognosis [208].
Twist1 expression is negatively regulated by androgen and inhibition of androgen signaling by enzalutamide causes an increase in the Twist1 expression [195,212,213]. Accordingly, Twist1 expression increases following ADT [16,214,215]. This regulation of Twist1 by AR is indirect as AR does not seem to directly bind to its promotor [212,213]. Indeed, although an early study has proposed that this indirect regulation is through NKX3.1, which is direct target of AR, a recent study has proposed that this regulation is mediated by ETS variant 1 (ETV1), another direct target of AR [212,213]. Furthermore, the effects of androgen on Twist1 promoter activity may be activatory or inhibitory depending on the recruited co-regulators with ETV1 [213]. Shiota et al. [215] has reported that AR and TWIST1 reciprocally control the expressions of each other, and castration-induced oxidative stress may promote AR overexpression through the increase of Twist1 expression leading to the development of castration resistance [215]. Interestingly, it was shown that inhibition of AR signaling induced Twist1 expression via PKC activation, which led to castration resistance through upregulation of AR [216]. In this mechanism, NF-κB signaling has been shown to be responsible for PKC-dependent increased Twist1 expression [217]. This association may be a critical node in explaining Twist1-dependent PCa growth in androgen-dependent state.
Snail family transcriptional repressor 1 (Snail1) and Snail2
Increased expressions of Snail1 (Snail) and Snail2 (Slug) have been reported in various cancers and their elevated expressions are associated with further motility, invasiveness, and metastasis of cancer cells [195,218–221]. Concordantly, both Snail1 and Snail2 are overexpressed in PCa, which is associated with a higher Gleason score [222–224]. Furthermore, silencing of Snail1 or Snail2 represses the migrative and invasive abilities of PCa cells [218,225]. In fact, the SNAIL proteins inhibit the expression of not only Cdh1 but also many other genes associated with the epithelial structure and function such as claudins, occludin, and other components of tight junctions [226]. On the other hand, they activate the genes of mesenchymal markers including Cdh2, vimentin (Vim), and matrix metalloproteinases (MMPs) [227–229]. Indeed, it has been shown that both Snail1 and Snail2 directly bind to Cdh1 promoter and represses its expression [165,169].
The expressions of both Snail1 and Snail2 are regulated by androgens [195,230]. Enzalutamide-mediated inhibition of androgen signaling leads to an increase in the expression of Snail1, and this increased expression promotes resistance to ADT [195,231]. Similarly, elevated Snail2 expression confers a growth advantage on androgen-deprived PCa cells, and the loss of Snail2 expression correlates with a better clinical response to ADT [230,232]. On the other hand, Snail2 overexpression causes an increase in the AR protein level, with which it forms a complex to cooperatively regulate downstream gene expression leading to castration resistance [230].
Forkhead box protein C2 (FoxC2)
FOXC2 is a member of FOX family transcription factors, and its increased expression is associated with metastasis and poor prognosis in various cancers, including PCa [233–235]. Although it is not known whether androgen directly regulates FoxC2 expression, inhibition of androgen signaling by enzalutamide causes increased FoxC2 expression [195,236]. On the other hand, it has been shown that increased FOXC2 inhibits AR expression in a Zeb1-dependent manner, thereby contributing to castration-resistance [237]. Increased FOXC2 level has been shown to cause a decrease in the E-cadherin and an increase in the N-cadherin levels [237]. However, direct binding of FOXC2 to E-cadherin promoter could not be demonstrated and therefore the negative effect of FOXC2 on E-cadherin level is likely indirect. However, FOXC2 has been shown to directly bind to the promoter of p120-catenin, whose protein product β-catenin protects E-cadherin from endocytosis and proteasomal degradation [238,239]. Furthermore, increased FOXC2 protein level causes an increase in the ZEB1 and SNAIL protein levels [237]. Indeed, it was shown that FOXC2 can bind to Zeb1 promoter when phosphorylated on serine 367 by p38 kinase [240]. This event may explain another mechanism for FOXC2 dependent indirect inhibition of E-cadherin since Zeb1 is a strong E-cadherin repressor. However, it seems that the effects of FOXC2 on cellular N-cadherin level is direct. Indeed, FOXC2 has been shown to directly bind to the cdh2 promoter and induce its expression [241]. In concordance, inhibition of FOXC2 through a small-molecule inhibitor results in the reversal of cadherin switching and inhibition of metastasis [242]. Furthermore, inhibition of FOXC2 in vivo restores epithelial phenotype in metastatic Pca cells [237]. Therefore, it seems that FOXC2 plays a seminal role in establishing a positive loop among the transcription factors involved in cadherin switching. Indeed, SNAIL1 and TWIST1 can bind to the FoxC2 promoter and increase its expression [235] feeding into the positive loop of cadherin switching.
Sine oculis homeobox homolog 1 (Six1)
SIX1 is a homeobox transcription factor important in regulating genes involved in development. It is overexpressed in various cancers including PCa where it induces migration and invasion [243]. Although it is not yet clear whether the expression of Six1 gene is directly regulated by androgen, enzalutamide-mediated inhibition of androgen signaling has been shown to increase Six1 expression and promote castration resistance in PCa cells [244]. However, increasing evidence has been suggesting that this effect is not at the transcriptional level but is directly related to the increased protein stability of SIX1 [244]. In concordance, it was shown that triggering SIX1 degradation using a chemical inducer results in inhibition of cell growth and sensitization of PCa cells to enzalutamide-mediated castration [245]. In addition, increased SIX1 protein level causes a decrease in the E-cadherin and an increase in the N-cadherin levels [246,247]. However, the effects of SIX1 on E-cadherin and N-cadherin levels are probably regulated by indirect mechanisms [248]. Overexpression of Six1 has been shown to promote tumor growth and metastasis in the breast cancer via both increasing TGF-β signaling and converting it from suppressive to supportive mode for tumor growth [246,249–251]. Six1 overexpression also promotes EMT in colorectal cancer through ZEB1 activation [251]. Furthermore, its overexpression promotes EMT and stem/progenitor cell phenotype in mammary tumor cells [252]. This tumor and EMT inducive role of Six1 seems to be through increased levels of ZEB1, SNAIL1, and TWIST1 [250,251].
SAM pointed domain containing ETS transcription factor (SPDEF)
SPDEF, also known as PDEF—prostate-derived ETS factor, is an androgen-regulated transcription factor that is dysregulated in many tumors. Contradicting results have been reported about its role in cancers. It has been reported to inhibit metastatic behaviors of PCa cells and its knockdown in mice results in increased metastasis [138]. Its metastasis-inhibiting role seems to be through inhibition of EMT via induction of epithelial/luminal phenotype [253]. However, a survey of more than 9,000 prostate tumor samples shows that expression of SPDEF is detected in 80% of these samples and its positivity is associated with the Gleason grade tumor stage and poor prognosis [254]. These contradicting reports might be related to the combination of other factors, tumor microenvironment, and the stages of the tumors.
ADT has been reported to decrease the SPDEF level that is associated with decreased Cdh1 and increased Cdh2 expression [255,256]. SPDEF can directly bind to the Cdh1 promoter and promotes its expression [256]. However, it has been shown that this effect of SPDEF on Cdh1 expression may be also regulated indirectly by controlling the Twist1 expression, but independent of Snail1 and Snail2 activities [138,257]. In fact, SPDEF is considered a key molecule in the ADT-induced cadherin switching as its cellular level is directly controlled by AR and it directly regulates various cellular mechanisms [258]. For example, it is involved in the regulation of Ccl2 expression, a chemokine involved in cadherin switching cooperatively via activating STAT3-TWIST1 signaling [257]. Consequently, SPDEF inhibits Ccl2 expression and thereby cadherin switching in the presence of androgen, in PCa cells [255]. Conversely, ADT causes an increase in the Ccl2 expression due to the inhibition of androgen signaling and subsequent repression of AR-SPDEF axis [255]. On the other hand, it was shown that ADT causes an increase in the TGF-βI level due to inhibition of the inhibitory AR-SPDEF axis [19]. Increased TGF-β signaling leads to increased expression of transcription factors Zeb1, Snail1, Snail2, and Twist1, leading to the induction of cadherin switching [19,137]. More studies is needed to elucidate the detailed role of SPDEF in the tumor progression and EMT in prostate cancer.
N-myc downstream-regulated gene 1 (Ndrg1)
Ndrg1 is an androgen-regulated gene, which is linked to the androgen network through interacting with β-catenin and heat shock protein 90 (HSP90) [259,260]. A recent study has shown that NDRG1 may regulate androgen signaling directly in PCa cells with important roles in both androgen-dependent and androgen-independent AR signaling [261]. NDRG1 protein is a suppressor of metastasis through inhibiting oncogenic TGF-β signaling [262–265]. Although NDRG1 protein is not a transcription factor, it has a crucial role in repressing the levels of transcription factors that induce cadherin switching, such as ZEB1, SNAIL1, and SNAIL2, through inhibition of TGF-β signaling [266,267]. In concordance, it has been shown that ADT causes a decrease in the Ndrg1 expression, with a consequence of decreased Cdh1 and increased Cdh2 expressions [266]. Furthermore, ADT-induced increased N-cadherin inhibits the AR-NDRG1 axis in a c-Jun-dependent manner, creating a feedback loop that further induces Cdh2 expression [266].
Androgens have pivotal roles in the development and differentiation of the prostate gland. However, they also have critical roles in the pathogenesis of PCa. Although various treatment approaches are used for localized PCa tumors, ADT is the widely used standard treatment for advanced tumors. ADT is a powerful approach used to inhibit androgen signaling which leads to a successful response at the initial phase of treatment. However, androgen refractory tumors and metastases develop 24–36 months after ADT in many patients. Recent reports propose that ADT may also induce cadherin switching and ensuing metastasis in PCa. Indeed, growing pieces of evidence show that although ADT relieves the patients at the beginning of treatment, in advanced PCa, it may be the main cause for the development of metastasis via inducing cadherin switching.
ADT exerts a negative survival pressure on androgen-dependent PCa cells, inducing a homeostatic cellular response through activation of evolutionarily imprinted alternative survival mechanisms in cells, which may provide an advantage for growth of PCa cells with mutations leading to their androgen-independent survival. This is an evolutionary homeostatic response that in the long-range favors development of more aggressive CRPC. In this homeostatic response, decreased E-cadherin in the cells encourages anchorage independence and increased motility while increased N-cadherin encourages de-differentiation towards more stem cell-like characteristics in the cells. Meanwhile, other accumulated possible alterations, such as those leading to increased tonic signaling in the Wnt/β-catenin, TGF-β, PI3K/AKT, MAPK, and other pathways, fortify this transition leading to more aggressive and treatment-resistant CRPC. Developing approaches employing combination treatment strategies, such as targeting motility and survival, along with ADT to help prevent survival of initial mutants that will otherwise develop into CRPC may prove beneficial.
Acknowledgement: The authors disclose no acknowledgement.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: LV, OC, SV; data collection: LV, OC; analysis and interpretation of results: LV, OC, SV, VT, JHC; draft manuscript preparation: LV, OC. All authors reviewed the results and approved the final version of the manuscript.
Ethics Approval: This study did not involve use of animal or human subjects.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Siegel, R. L., Miller, K. D., Fuchs, H. E., Jemal, A. (2021). Cancer statistics, 2021. CA: A Cancer Journal for Clinicians, 71(1), 7–33. [Google Scholar]
2. Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A. et al. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 68(6), 394–424. DOI 10.3322/caac.21492. [Google Scholar] [CrossRef]
3. Zerbib, M., Zelefsky, M. J., Higano, C. S., Carroll, P. R. (2008). Conventional treatments of localized prostate cancer. Urology, 72(6), S25–S35. DOI 10.1016/j.urology.2008.10.005. [Google Scholar] [CrossRef]
4. Crawford, E. D., Heidenreich, A., Lawrentschuk, N., Tombal, B., Pompeo, A. C. L. et al. (2019). Androgen-targeted therapy in men with prostate cancer: Evolving practice and future considerations. Prostate Cancer and Prostatic Diseases, 22(1), 24–38. DOI 10.1038/s41391-018-0079-0. [Google Scholar] [CrossRef]
5. Oefelein, M. G., Feng, A., Scolieri, M. J., Ricchiutti, D., Resnick, M. I. (2000). Reassessment of the definition of castrate levels of testosterone: Implications for clinical decision making. Urology, 56(6), 1021–1024. DOI 10.1016/S0090-4295(00)00793-7. [Google Scholar] [CrossRef]
6. Harris, W. P., Mostaghel, E. A., Nelson, P. S., Montgomery, B. (2009). Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nature Clinical Practice: Urology, 6(2), 76–85. DOI 10.1038/ncpuro1296. [Google Scholar] [CrossRef]
7. Kiratli, B. J., Srinivas, S., Perkash, I., Terris, M. K. (2001). Progressive decrease in bone density over 10 years of androgen deprivation therapy in patients with prostate cancer. Urology, 57(1), 127–132. DOI 10.1016/S0090-4295(00)00895-5. [Google Scholar] [CrossRef]
8. Crona, D. J., Whang, Y. E. (2017). Androgen receptor-dependent and -independent mechanisms involved in prostate cancer therapy resistance. Cancers, 9(6), 67. DOI 10.3390/cancers9060067. [Google Scholar] [CrossRef]
9. Shi, X. B., Ma, A. H., Xia, L., Kung, H. J., de Vere White, R. W., (2002). Functional analysis of 44 mutant androgen receptors from human prostate cancer. Cancer Research, 62(5), 1496–1502. [Google Scholar]
10. Karantanos, T., Corn, P. G., Thompson, T. C. (2013). Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene, 32(49), 5501–5511. DOI 10.1038/onc.2013.206. [Google Scholar] [CrossRef]
11. Khan, T., Scott, K. F., Becker, T. M., Lock, J., Nimir, M. et al. (2020). The prospect of identifying resistance mechanisms for castrate-resistant prostate cancer using circulating tumor cells: Is epithelial-to-mesenchymal transition a key player? Prostate Cancer, 2020, 7938280–7938295. [Google Scholar]
12. Jennbacken, K., Tesan, T., Wang, W., Gustavsson, H., Damber, J. E. et al. (2010). N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocrine-Related Cancer, 17(2), 469–479. [Google Scholar]
13. Zhu, M. L., Kyprianou, N. (2010). Role of androgens and the androgen receptor in epithelial-mesenchymal transition and invasion of prostate cancer cells. FASEB Journal, 24(3), 769–777. [Google Scholar]
14. Tanaka, H., Kono, E., Tran, C. P., Miyazaki, H., Yamashiro, J. et al. (2010). Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nature Medicine, 16(12), 1414–1420. [Google Scholar]
15. Tomita, K., van Bokhoven, A., van Leenders, G. J., Ruijter, E. T., Jansen, C. F. et al. (2000). Cadherin switching in human prostate cancer progression. Cancer Research, 60(13), 3650–3654. [Google Scholar]
16. Sun, Y., Wang, B. E., Leong, K. G., Yue, P., Li, L. et al. (2012). Androgen deprivation causes epithelial-mesenchymal transition in the prostate: Implications for androgen-deprivation therapy. Cancer Research, 72(2), 527–536. DOI 10.1158/0008-5472.CAN-11-3004. [Google Scholar] [CrossRef]
17. Lee, Y. C., Cheng, C. J., Huang, M., Bilen, M. A., Ye, X. et al. (2010). Androgen depletion up-regulates cadherin-11 expression in prostate cancer. Journal of Pathology, 221(1), 68–76. DOI 10.1002/path.2687. [Google Scholar] [CrossRef]
18. Byrne, N. M., Nesbitt, H., Ming, L., McKeown, S. R., Worthington, J. et al. (2016). Androgen deprivation in LNCaP prostate tumour xenografts induces vascular changes and hypoxic stress, resulting in promotion of epithelial-to-mesenchymal transition. British Journal of Cancer, 114(6), 659–668. DOI 10.1038/bjc.2016.29. [Google Scholar] [CrossRef]
19. Chen, W. Y., Tsai, Y. C., Yeh, H. L., Suau, F., Jiang, K. C. et al. (2017). Loss of SPDEF and gain of TGFBI activity after androgen deprivation therapy promote EMT and bone metastasis of prostate cancer. Science Signaling, 10(492), 338. DOI 10.1126/scisignal.aam6826. [Google Scholar] [CrossRef]
20. Huang, P. A., Price, D. K., Figg, W. D. (2018). Molecular drivers of metastatic castrate-resistant prostate cancer: New roads to resistance. Cancer Biology & Therapy, 19(10), 869–870. DOI 10.1080/15384047.2018.1449618. [Google Scholar] [CrossRef]
21. Hao, Y., Baker, D., Ten Dijke, P. (2019). TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. International Journal of Molecular Sciences, 20(11), 2767. DOI 10.3390/ijms20112767. [Google Scholar] [CrossRef]
22. Wheelock, M. J., Shintani, Y., Maeda, M., Fukumoto, Y., Johnson, K. R. (2008). Cadherin switching. Journal of Cell Science, 121, 727–735. DOI 10.1242/jcs.000455. [Google Scholar] [CrossRef]
23. Knudsen, K. A., Sauer, C., Johnson, K. R., Wheelock, M. J. (2005). Effect of N-cadherin misexpression by the mammary epithelium in mice. Journal of Cellular Biochemistry, 95(6), 1093–1107. DOI 10.1002/(ISSN)1097-4644. [Google Scholar] [CrossRef]
24. Chen, A., Beetham, H., Black, M. A., Priya, R., Telford, B. J. et al. (2014). E-cadherin loss alters cytoskeletal organization and adhesion in non-malignant breast cells but is insufficient to induce an epithelial-mesenchymal transition. BMC Cancer, 14(1), 552. DOI 10.1186/1471-2407-14-552. [Google Scholar] [CrossRef]
25. Kaszak, I., Witkowska-Pilaszewicz, O., Niewiadomska, Z., Dworecka-Kaszak, B., Ngosa Toka, F. et al. (2020). Role of cadherins in cancer—A review. International Journal of Molecular Sciences, 21(20), 7624. DOI 10.3390/ijms21207624. [Google Scholar] [CrossRef]
26. Onder, T. T., Gupta, P. B., Mani, S. A., Yang, J., Lander, E. S. et al. (2008). Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Research, 68(10), 3645–3654. DOI 10.1158/0008-5472.CAN-07-2938. [Google Scholar] [CrossRef]
27. Hazan, R. B., Qiao, R., Keren, R., Badano, I., Suyama, K. (2004). Cadherin switch in tumor progression. Annals of the New York Academy of Sciences, 1014, 155–163. [Google Scholar]
28. de Marzo, A. M., Platz, E. A., Sutcliffe, S., Xu, J., Gronberg, H. et al. (2007). Inflammation in prostate carcinogenesis. Nature Reviews: Cancer, 7(4), 256–269. DOI 10.1038/nrc2090. [Google Scholar] [CrossRef]
29. Unni, E., Sun, S., Nan, B., McPhaul, M. J., Cheskis, B. et al. (2004). Changes in androgen receptor nongenotropic signaling correlate with transition of LNCaP cells to androgen independence. Cancer Research, 64(19), 7156–7168. DOI 10.1158/0008-5472.CAN-04-1121. [Google Scholar] [CrossRef]
30. Fujita, K., Nonomura, N. (2019). Role of androgen receptor in prostate cancer: A review. The World Journal of Men’s Health, 37(3), 288–295. DOI 10.5534/wjmh.180040. [Google Scholar] [CrossRef]
31. Hammond, G. L. (2016). Plasma steroid-binding proteins: Primary gatekeepers of steroid hormone action. Journal of Endocrinology, 230(1), R13–R25. DOI 10.1530/JOE-16-0070. [Google Scholar] [CrossRef]
32. Corradi, P. F., Corradi, R. B., Greene, L. W. (2016). Physiology of the hypothalamic pituitary gonadal axis in the male. Urologic Clinics of North America, 43(2), 151–162. DOI 10.1016/j.ucl.2016.01.001. [Google Scholar] [CrossRef]
33. Bruchovsky, N., Wilson, J. D. (1968). The conversion of testosterone to 5-α-androstan-17-β-ol-3-one by rat prostate in vivo and in vitro. Journal of Biological Chemistry, 243(8), 2012–2021. DOI 10.1016/S0021-9258(18)93542-8. [Google Scholar] [CrossRef]
34. Deslypere, J. P., Young, M., Wilson, J. D., McPhaul, M. J. (1992). Testosterone and 5 alpha-dihydrotestosterone interact differently with the androgen receptor to enhance transcription of the MMTV-CAT reporter gene. Molecular and Cellular Endocrinology, 88(1–3), 15–22. DOI 10.1016/0303-7207(92)90004-P. [Google Scholar] [CrossRef]
35. Wilson, J. D. (2001). The role of 5 alpha-reduction in steroid hormone physiology. Reproduction, Fertility, and Development, 13(7–8), 673–678. DOI 10.1071/RD01074. [Google Scholar] [CrossRef]
36. Rittmaster, R., Hahn, R. G., Ray, P., Shannon, J. B., Wurzel, R. (2008). Effect of dutasteride on intraprostatic androgen levels in men with benign prostatic hyperplasia or prostate cancer. Urology, 72(4), 808–812. DOI 10.1016/j.urology.2008.06.032. [Google Scholar] [CrossRef]
37. Steers, W. D. (2001). 5alpha-reductase activity in the prostate. Urology, 58(6), 17–24. DOI 10.1016/S0090-4295(01)01299-7. [Google Scholar] [CrossRef]
38. Truong, T. H., Lange, C. A. (2018). Deciphering steroid receptor crosstalk in hormone-driven cancers. Endocrinology, 159(12), 3897–3907. DOI 10.1210/en.2018-00831. [Google Scholar] [CrossRef]
39. Smith, D. F., Toft, D. O. (2008). Minireview: The intersection of steroid receptors with molecular chaperones: Observations and questions. Molecular Endocrinology, 22(10), 2229–2240. DOI 10.1210/me.2008-0089. [Google Scholar] [CrossRef]
40. Pratt, W. B., Sanchez, E. R., Bresnick, E. H., Meshinchi, S., Scherrer, L. C. et al. (1989). Interaction of the glucocorticoid receptor with the Mr 90,000 heat shock protein: An evolving model of ligand-mediated receptor transformation and translocation. Cancer Research, 49(8 Suppl), 2222s–2229s. [Google Scholar]
41. Tan, M. H., Li, J., Xu, H. E., Melcher, K., Yong, E. L. (2015). Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacologica Sinica, 36(1), 3–23. DOI 10.1038/aps.2014.18. [Google Scholar] [CrossRef]
42. Young, C. Y., Andrews, P. E., Montgomery, B. T., Tindall, D. J. (1992). Tissue-specific and hormonal regulation of human prostate-specific glandular kallikrein. Biochemistry, 31(3), 818–824. DOI 10.1021/bi00118a026. [Google Scholar] [CrossRef]
43. He, W. W., Sciavolino, P. J., Wing, J., Augustus, M., Hudson, P. et al. (1997). A novel human prostate-specific, androgen-regulated homeobox gene (NKX3.1) that maps to 8p21, a region frequently deleted in prostate cancer. Genomics, 43(1), 69–77. DOI 10.1006/geno.1997.4715. [Google Scholar] [CrossRef]
44. Prescott, J. L., Blok, L., Tindall, D. J. (1998). Isolation and androgen regulation of the human homeobox cDNA, NKX3.1. Prostate, 35(1), 71–80. DOI 10.1002/(ISSN)1097-0045. [Google Scholar] [CrossRef]
45. Varisli, L., Gonen-Korkmaz, C., Syed, H. M., Bogurcu, N., Debelec-Butuner, B. et al. (2012). Androgen regulated HN1 leads proteosomal degradation of androgen receptor (AR) and negatively influences AR mediated transactivation in prostate cells. Molecular and Cellular Endocrinology, 350(1), 107–117. DOI 10.1016/j.mce.2011.11.027. [Google Scholar] [CrossRef]
46. Shang, Y., Myers, M., Brown, M. (2002). Formation of the androgen receptor transcription complex. Molecular Cell, 9(3), 601–610. DOI 10.1016/S1097-2765(02)00471-9. [Google Scholar] [CrossRef]
47. Castoria, G., Auricchio, F., Migliaccio, A. (2017). Extranuclear partners of androgen receptor: At the crossroads of proliferation, migration, and neuritogenesis. FASEB Journal, 31(4), 1289–1300. DOI 10.1096/fj.201601047R. [Google Scholar] [CrossRef]
48. Deng, Q., Zhang, Z., Wu, Y., Yu, W. Y., Zhang, J. et al. (2017). Non-genomic action of androgens is mediated by rapid phosphorylation and regulation of androgen receptor trafficking. Cellular Physiology and Biochemistry, 43(1), 223–236. DOI 10.1159/000480343. [Google Scholar] [CrossRef]
49. Migliaccio, A., Castoria, G., Di Domenico, M., de Falco, A., Bilancio, A. et al. (2000). Steroid-induced androgen receptor-oestradiol receptor β-Src complex triggers prostate cancer cell proliferation. EMBO Journal, 19(20), 5406–5417. DOI 10.1093/emboj/19.20.5406. [Google Scholar] [CrossRef]
50. Zarif, J. C., Lamb, L. E., Schulz, V. V., Nollet, E. A., Miranti, C. K. (2015). Androgen receptor non-nuclear regulation of prostate cancer cell invasion mediated by Src and matriptase. Oncotarget, 6(9), 6862–6876. DOI 10.18632/oncotarget.3119. [Google Scholar] [CrossRef]
51. Leung, J. K., Sadar, M. D. (2017). Non-genomic actions of the androgen receptor in prostate cancer. Frontiers in Endocrinology, 8(2), 2. DOI 10.3389/fendo.2017.00002. [Google Scholar] [CrossRef]
52. Baron, S., Manin, M., Beaudoin, C., Leotoing, L., Communal, Y. et al. (2004). Androgen receptor mediates non-genomic activation of phosphatidylinositol 3-OH kinase in androgen-sensitive epithelial cells. Journal of Biological Chemistry, 279(15), 14579–14586. DOI 10.1074/jbc.M306143200. [Google Scholar] [CrossRef]
53. Ueda, T., Bruchovsky, N., Sadar, M. D. (2002). Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. Journal of Biological Chemistry, 277(9), 7076–7085. DOI 10.1074/jbc.M108255200. [Google Scholar] [CrossRef]
54. Miller, K. J., Asim, M. (2022). Unravelling the role of kinases that underpin androgen signalling in prostate cancer. Cells, 11(6), 952. DOI 10.3390/cells11060952. [Google Scholar] [CrossRef]
55. Culig, Z. (2021). Response to androgens and androgen receptor antagonists in the presence of cytokines in prostate cancer. Cancers, 13(12), 2944. DOI 10.3390/cancers13122944. [Google Scholar] [CrossRef]
56. Jin, R. J., Lho, Y., Connelly, L., Wang, Y., Yu, X. et al. (2008). The nuclear factor-κB pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Research, 68(16), 6762–6769. DOI 10.1158/0008-5472.CAN-08-0107. [Google Scholar] [CrossRef]
57. Witte, K. E., Pfitzenmaier, J., Storm, J., Lutkemeyer, M., Wimmer, C. et al. (2021). Analysis of several pathways for efficient killing of prostate cancer stem cells: A central role of NF-κB RELA. International Journal of Molecular Sciences, 22(16), 8901. DOI 10.3390/ijms22168901. [Google Scholar] [CrossRef]
58. Wang, C., Chen, Q., Xu, H. (2021). Wnt/β-catenin signal transduction pathway in prostate cancer and associated drug resistance. Discover Oncology, 12(1), 40. DOI 10.1007/s12672-021-00433-6. [Google Scholar] [CrossRef]
59. Yu, X., Wang, Y., DeGraff, D. J., Wills, M. L., Matusik, R. J. (2011). Wnt/β-catenin activation promotes prostate tumor progression in a mouse model. Oncogene, 30(16), 1868–1879. DOI 10.1038/onc.2010.560. [Google Scholar] [CrossRef]
60. Wen, Y., Hu, M. C., Makino, K., Spohn, B., Bartholomeusz, G. et al. (2000). HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Research, 60(24), 6841–6845. [Google Scholar]
61. Choi, E., Buie, J., Camacho, J., Sharma, P., de Riese, W. T. W., (2022). Evolution of androgen deprivation therapy (ADT) and its new emerging modalities in prostate cancer: An update for practicing urologists, clinicians and medical providers. Research and Reports in Urology, 14, 87–108. DOI 10.2147/RRU.S303215. [Google Scholar] [CrossRef]
62. Wagle, D. G. (2011). Reimbursement policy and androgen-deprivation therapy for prostate cancer. New England Journal of Medicine, 364(6), 579–580. DOI 10.1056/NEJMc1013967. [Google Scholar] [CrossRef]
63. Huggins, C., Hodges, C. V. (2002). Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. Journal of Urology, 168(1), 9–12. DOI 10.1016/S0022-5347(05)64820-3. [Google Scholar] [CrossRef]
64. Heidenreich, A., Bastian, P. J., Bellmunt, J., Bolla, M., Joniau, S. et al. (2014). EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. European Urology, 65(2), 467–479. DOI 10.1016/j.eururo.2013.11.002. [Google Scholar] [CrossRef]
65. Verma, S., Shankar, E., Kalayci, F. N. C., Mukunda, A., Alassfar, M. et al. (2020). Androgen deprivation induces transcriptional reprogramming in prostate cancer cells to develop stem cell-like characteristics. International Journal of Molecular Sciences, 21(24), 9568. DOI 10.3390/ijms21249568. [Google Scholar] [CrossRef]
66. Marques, R. B., Dits, N. F., Erkens-Schulze, S., van Weerden, W. M., Jenster, G. (2010). Bypass mechanisms of the androgen receptor pathway in therapy-resistant prostate cancer cell models. PLoS One, 5(10), e13500. DOI 10.1371/journal.pone.0013500. [Google Scholar] [CrossRef]
67. Shafi, A. A., Yen, A. E., Weigel, N. L. (2013). Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacology and Therapeutics, 140(3), 223–238. DOI 10.1016/j.pharmthera.2013.07.003. [Google Scholar] [CrossRef]
68. Feldman, B. J., Feldman, D. (2001). The development of androgen-independent prostate cancer. Nature Reviews: Cancer, 1(1), 34–45. DOI 10.1038/35094009. [Google Scholar] [CrossRef]
69. Hu, J., Wang, G., Sun, T. (2017). Dissecting the roles of the androgen receptor in prostate cancer from molecular perspectives. Tumour Biology, 39(5), 1010428317692259. DOI 10.1177/1010428317692259. [Google Scholar] [CrossRef]
70. Steinkamp, M. P., O'Mahony, O. A., Brogley, M., Rehman, H., Lapensee, E. W. et al. (2009). Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy. Cancer Research, 69(10), 4434–4442. DOI 10.1158/0008-5472.CAN-08-3605. [Google Scholar] [CrossRef]
71. Locke, J. A., Guns, E. S., Lubik, A. A., Adomat, H. H., Hendy, S. C. et al. (2008). Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Research, 68(15), 6407–6415. DOI 10.1158/0008-5472.CAN-07-5997. [Google Scholar] [CrossRef]
72. Heemers, H. V., Tindall, D. J. (2007). Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocrine Reviews, 28(7), 778–808. DOI 10.1210/er.2007-0019. [Google Scholar] [CrossRef]
73. Zhu, M. L., Kyprianou, N. (2008). Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocrine-Related Cancer, 15(4), 841–849. DOI 10.1677/ERC-08-0084. [Google Scholar] [CrossRef]
74. Bergerat, J. P., Ceraline, J. (2009). Pleiotropic functional properties of androgen receptor mutants in prostate cancer. Human Mutation, 30(2), 145–157. DOI 10.1002/humu.20848. [Google Scholar] [CrossRef]
75. Kumar, A., Coleman, I., Morrissey, C., Zhang, X., True, L. D. et al. (2016). Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nature Medicine, 22(4), 369–378. DOI 10.1038/nm.4053. [Google Scholar] [CrossRef]
76. Robinson, D., Van Allen, E. M., Wu, Y. M., Schultz, N., Lonigro, R. J. et al. (2015). Integrative clinical genomics of advanced prostate cancer. Cell, 161(5), 1215–1228. DOI 10.1016/j.cell.2015.05.001. [Google Scholar] [CrossRef]
77. Crowley, F., Sterpi, M., Buckley, C., Margetich, L., Handa, S. et al. (2021). A review of the pathophysiological mechanisms underlying castration-resistant prostate cancer. Research and Reports in Urology, 13, 457–472. DOI 10.2147/RRU.S264722. [Google Scholar] [CrossRef]
78. Bubendorf, L., Kononen, J., Koivisto, P., Schraml, P., Moch, H. et al. (1999). Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Research, 59(4), 803–806. [Google Scholar]
79. Mao, Y., Yang, G., Li, Y., Liang, G., Xu, W. et al. (2022). Advances in the current understanding of the mechanisms governing the acquisition of castration-resistant prostate cancer. Cancers, 14(15), 3744. DOI 10.3390/cancers14153744. [Google Scholar] [CrossRef]
80. Likos, E., Bhattarai, A., Weyman, C. M., Shukla, G. C. (2022). The androgen receptor messenger RNA: What do we know? RNA Biology, 19(1), 819–828. DOI 10.1080/15476286.2022.2084839. [Google Scholar] [CrossRef]
81. Gregory, C. W., Johnson, R. T.Jr, Mohler, J. L., French, F. S., Wilson, E. M. (2001). Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Research, 61(7), 2892–2898. [Google Scholar]
82. Wadosky, K. M., Koochekpour, S. (2016). Molecular mechanisms underlying resistance to androgen deprivation therapy in prostate cancer. Oncotarget, 7(39), 64447–64470. DOI 10.18632/oncotarget.10901. [Google Scholar] [CrossRef]
83. Shiota, M., Akamatsu, S., Tsukahara, S., Nagakawa, S., Matsumoto, T. et al. (2022). Androgen receptor mutations for precision medicine in prostate cancer. Endocrine-Related Cancer, 29(10), R143–R155. DOI 10.1530/ERC-22-0140. [Google Scholar] [CrossRef]
84. Beltran, H., Yelensky, R., Frampton, G. M., Park, K., Downing, S. R. et al. (2013). Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. European Urology, 63(5), 920–926. DOI 10.1016/j.eururo.2012.08.053. [Google Scholar] [CrossRef]
85. Montgomery, R. B., Mostaghel, E. A., Vessella, R., Hess, D. L., Kalhorn, T. F. et al. (2008). Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resistant tumor growth. Cancer Research, 68(11), 4447–4454. DOI 10.1158/0008-5472.CAN-08-0249. [Google Scholar] [CrossRef]
86. Stanbrough, M., Bubley, G. J., Ross, K., Golub, T. R., Rubin, M. A. et al. (2006). Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Research, 66(5), 2815–2825. DOI 10.1158/0008-5472.CAN-05-4000. [Google Scholar] [CrossRef]
87. Dillard, P. R., Lin, M. F., Khan, S. A. (2008). Androgen-independent prostate cancer cells acquire the complete steroidogenic potential of synthesizing testosterone from cholesterol. Molecular and Cellular Endocrinology, 295(1–2), 115–120. DOI 10.1016/j.mce.2008.08.013. [Google Scholar] [CrossRef]
88. Mostaghel, E. A., Page, S. T., Lin, D. W., Fazli, L., Coleman, I. M. et al. (2007). Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: Therapeutic implications for castration-resistant prostate cancer. Cancer Research, 67(10), 5033–5041. DOI 10.1158/0008-5472.CAN-06-3332. [Google Scholar] [CrossRef]
89. Qu, Y., Dai, B., Ye, D., Kong, Y., Chang, K. et al. (2015). Constitutively active AR-V7 plays an essential role in the development and progression of castration-resistant prostate cancer. Scientific Reports, 5(1), 7654. DOI 10.1038/srep07654. [Google Scholar] [CrossRef]
90. Zhu, Y., Luo, J. (2020). Regulation of androgen receptor variants in prostate cancer. Asian Journal of Urology, 7(3), 251–257. DOI 10.1016/j.ajur.2020.01.001. [Google Scholar] [CrossRef]
91. Yu, Z., Chen, S., Sowalsky, A. G., Voznesensky, O. S., Mostaghel, E. A. et al. (2014). Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clinical Cancer Research, 20(6), 1590–1600. DOI 10.1158/1078-0432.CCR-13-1863. [Google Scholar] [CrossRef]
92. Kanayama, M., Lu, C., Luo, J., Antonarakis, E. S. (2021). AR splicing variants and resistance to AR targeting agents. Cancers, 13(11), 2563. DOI 10.3390/cancers13112563. [Google Scholar] [CrossRef]
93. Lu, C., Luo, J. (2013). Decoding the androgen receptor splice variants. Translational Andrology and Urology, 2(3), 178–186. [Google Scholar]
94. Cancer Genome Atlas Research, N. (2015). The molecular taxonomy of primary prostate cancer. Cell, 163(4), 1011–1025. DOI 10.1016/j.cell.2015.10.025. [Google Scholar] [CrossRef]
95. Hu, R., Dunn, T. A., Wei, S., Isharwal, S., Veltri, R. W. et al. (2009). Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Research, 69(1), 16–22. DOI 10.1158/0008-5472.CAN-08-2764. [Google Scholar] [CrossRef]
96. Guo, Z., Yang, X., Sun, F., Jiang, R., Linn, D. E. et al. (2009). A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Research, 69(6), 2305–2313. DOI 10.1158/0008-5472.CAN-08-3795. [Google Scholar] [CrossRef]
97. Antonarakis, E. S., Lu, C., Wang, H., Luber, B., Nakazawa, M. et al. (2014). AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. New England Journal of Medicine, 371(11), 1028–1038. DOI 10.1056/NEJMoa1315815. [Google Scholar] [CrossRef]
98. Schmidt, K. T., Huitema, A. D. R., Chau, C. H., Figg, W. D. (2021). Resistance to second-generation androgen receptor antagonists in prostate cancer. Nature Reviews Urology, 18(4), 209–226. DOI 10.1038/s41585-021-00438-4. [Google Scholar] [CrossRef]
99. Zhu, Y., Sharp, A., Anderson, C. M., Silberstein, J. L., Taylor, M. et al. (2018). Novel junction-specific and quantifiable in situ detection of AR-V7 and its clinical correlates in metastatic castration-resistant prostate cancer. European Urology, 73(5), 727–735. DOI 10.1016/j.eururo.2017.08.009. [Google Scholar] [CrossRef]
100. Sharp, A., Coleman, I., Yuan, W., Sprenger, C., Dolling, D. et al. (2019). Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. Journal of Clinical Investigation, 129(1), 192–208. DOI 10.1172/JCI122819. [Google Scholar] [CrossRef]
101. Ozturan, D., Morova, T., Lack, N. A. (2022). Androgen receptor-mediated transcription in prostate cancer. Cells, 11(5), 898. DOI 10.3390/cells11050898. [Google Scholar] [CrossRef]
102. DePriest, A. D., Fiandalo, M. V., Schlanger, S., Heemers, F., Mohler, J. L. et al. (2016). Regulators of androgen action resource: A one-stop shop for the comprehensive study of androgen receptor action. Database: The Journal of Biological Databases and Curation, 2016. [Google Scholar]
103. Buchanan, G., Need, E. F., Barrett, J. M., Bianco-Miotto, T., Thompson, V. C. et al. (2011). Corepressor effect on androgen receptor activity varies with the length of the CAG encoded polyglutamine repeat and is dependent on receptor/corepressor ratio in prostate cancer cells. Molecular and Cellular Endocrinology, 342(1–2), 20–31. DOI 10.1016/j.mce.2011.05.023. [Google Scholar] [CrossRef]
104. Leach, D. A., Fernandes, R. C., Bevan, C. L. (2022). Cellular specificity of androgen receptor, coregulators, and pioneer factors in prostate cancer. Endocrine Oncology, 2(1), R112–R131. DOI 10.1530/EO-22-0065. [Google Scholar] [CrossRef]
105. Chen, C. D., Welsbie, D. S., Tran, C., Baek, S. H., Chen, R. et al. (2004). Molecular determinants of resistance to antiandrogen therapy. Nature Medicine, 10(1), 33–39. DOI 10.1038/nm972. [Google Scholar] [CrossRef]
106. Chen, T., Wang, L. H., Farrar, W. L. (2000). Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells. Cancer Research, 60(8), 2132–2135. [Google Scholar]
107. Yang, L., Wang, L., Lin, H. K., Kan, P. Y., Xie, S. et al. (2003). Interleukin-6 differentially regulates androgen receptor transactivation via PI3K-Akt, STAT3, and MAPK, three distinct signal pathways in prostate cancer cells. Biochemical and Biophysical Research Communications, 305(3), 462–469. DOI 10.1016/S0006-291X(03)00792-7. [Google Scholar] [CrossRef]
108. Zhang, L., Altuwaijri, S., Deng, F., Chen, L., Lal, P. et al. (2009). NF-κB regulates androgen receptor expression and prostate cancer growth. American Journal of Pathology, 175(2), 489–499. DOI 10.2353/ajpath.2009.080727. [Google Scholar] [CrossRef]
109. Lee, L. F., Louie, M. C., Desai, S. J., Yang, J., Chen, H. W. et al. (2004). Interleukin-8 confers androgen-independent growth and migration of LNCaP: Differential effects of tyrosine kinases Src and FAK. Oncogene, 23(12), 2197–2205. DOI 10.1038/sj.onc.1207344. [Google Scholar] [CrossRef]
110. Liu, Y., Qin, Z., Yang, K., Liu, R., Xu, Y. (2017). Cripto-1 promotes epithelial-mesenchymal transition in prostate cancer via Wnt/β-catenin signaling. Oncology Reports, 37(3), 1521–1528. DOI 10.3892/or.2017.5378. [Google Scholar] [CrossRef]
111. Arnouk, H., Yum, G., Shah, D. (2021). Cripto-1 as a key factor in tumor progression, epithelial to mesenchymal transition and cancer stem cells. International Journal of Molecular Sciences, 22(17), 9280. DOI 10.3390/ijms22179280. [Google Scholar] [CrossRef]
112. Wu, D., Shi, Z., Xu, H., Chen, R., Xue, S. et al. (2017). Knockdown of Cripto-1 inhibits the proliferation, migration, invasion, and angiogenesis in prostate carcinoma cells. Journal of Biosciences, 42(3), 405–416. DOI 10.1007/s12038-017-9700-y. [Google Scholar] [CrossRef]
113. Nagaoka, T., Karasawa, H., Castro, N. P., Rangel, M. C., Salomon, D. S. et al. (2012). An evolving web of signaling networks regulated by Cripto-1. Growth Factors, 30(1), 13–21. DOI 10.3109/08977194.2011.641962. [Google Scholar] [CrossRef]
114. Liu, Y., Wang, J., Yang, T., Liu, R., Xu, Y. (2019). Overexpression levels of cripto-1 predict poor prognosis in patients with prostate cancer following radical prostatectomy. Oncology Letters, 18(3), 2584–2591. DOI 10.3892/ol.2019.10555. [Google Scholar] [CrossRef]
115. Pashaei, E., Pashaei, E., Ahmady, M., Ozen, M., Aydin, N. (2017). Meta-analysis of miRNA expression profiles for prostate cancer recurrence following radical prostatectomy. PLoS One, 12(6), e0179543. DOI 10.1371/journal.pone.0179543. [Google Scholar] [CrossRef]
116. Akoto, T., Bhagirath, D., Saini, S. (2020). MicroRNAs in treatment-induced neuroendocrine differentiation in prostate cancer. Cancer Drug Resistance, 3(4), 804–818. DOI 10.20517/cdr.2020.30. [Google Scholar] [CrossRef]
117. Guan, H., Liu, C., Fang, F., Huang, Y., Tao, T. et al. (2017). MicroRNA-744 promotes prostate cancer progression through aberrantly activating Wnt/β-catenin signaling. Oncotarget, 8(9), 14693–14707. DOI 10.18632/oncotarget.14711. [Google Scholar] [CrossRef]
118. Cui, Y., Yang, Y., Ren, L., Yang, J., Wang, B. et al. (2019). miR-15a-3p suppresses prostate cancer cell proliferation and invasion by targeting SLC39A7 via downregulating Wnt/β-catenin signaling pathway. Cancer Biotherapy & Radiopharmaceuticals, 34(7), 472–479. DOI 10.1089/cbr.2018.2722. [Google Scholar] [CrossRef]
119. Hsieh, I. S., Chang, K. C., Tsai, Y. T., Ke, J. Y., Lu, P. J. et al. (2013). MicroRNA-320 suppresses the stem cell-like characteristics of prostate cancer cells by downregulating the Wnt/β-catenin signaling pathway. Carcinogenesis, 34(3), 530–538. DOI 10.1093/carcin/bgs371. [Google Scholar] [CrossRef]
120. Yu, Z., Wang, Z., Li, F., Yang, J., Tang, L. (2018). miR‑138 modulates prostate cancer cell invasion and migration via Wnt/β‑catenin pathway. Molecular Medicine Reports, 17(2), 3140–3145. [Google Scholar]
121. Liu, H., Chen, Y. G. (2022). The interplay between TGF-β signaling and cell metabolism. Frontiers in Cell and Developmental Biology, 10, 846723. DOI 10.3389/fcell.2022.846723. [Google Scholar] [CrossRef]
122. Aashaq, S., Batool, A., Mir, S. A., Beigh, M. A., Andrabi, K. I. et al. (2022). TGF-β signaling: A recap of SMAD-independent and SMAD-dependent pathways. Journal of Cellular Physiology, 237(1), 59–85. DOI 10.1002/jcp.30529. [Google Scholar] [CrossRef]
123. Massague, J. (2012). TGFβ signalling in context. Nature Reviews: Molecular Cell Biology, 13(10), 616–630. DOI 10.1038/nrm3434. [Google Scholar] [CrossRef]
124. Cao, Z., Kyprianou, N. (2015). Mechanisms navigating the TGF-β pathway in prostate cancer. Asian Journal of Urology, 2(1), 11–18. DOI 10.1016/j.ajur.2015.04.011. [Google Scholar] [CrossRef]
125. Zhang, M., Zhang, Y. Y., Chen, Y., Wang, J., Wang, Q. et al. (2021). TGF-β signaling and resistance to cancer therapy. Frontiers in Cell and Developmental Biology, 9, 786728. DOI 10.3389/fcell.2021.786728. [Google Scholar] [CrossRef]
126. Massague, J., Blain, S. W., Lo, R. S. (2000). TGFβ signaling in growth control, cancer, and heritable disorders. Cell, 103(2), 295–309. DOI 10.1016/S0092-8674(00)00121-5. [Google Scholar] [CrossRef]
127. Mirzaei, S., Paskeh, M. D. A., Saghari, Y., Zarrabi, A., Hamblin, M. R. et al. (2022). Transforming growth factor-β (TGF-β) in prostate cancer: A dual function mediator? International Journal of Biological Macromolecules, 206(11), 435–452. DOI 10.1016/j.ijbiomac.2022.02.094. [Google Scholar] [CrossRef]
128. Bruckheimer, E. M., Kyprianou, N. (2001). Dihydrotestosterone enhances transforming growth factor-β-induced apoptosis in hormone-sensitive prostate cancer cells. Endocrinology, 142(6), 2419–2426. DOI 10.1210/endo.142.6.8218. [Google Scholar] [CrossRef]
129. Bruckheimer, E. M., Kyprianou, N. (2002). Bcl-2 antagonizes the combined apoptotic effect of transforming growth factor-β and dihydrotestosterone in prostate cancer cells. Prostate, 53(2), 133–142. DOI 10.1002/(ISSN)1097-0045. [Google Scholar] [CrossRef]
130. Qi, W., Gao, S., Chu, J., Zhou, L., Wang, Z. (2012). Negative androgen-response elements mediate androgen-dependent transcriptional inhibition of TGF-β1 and CDK2 promoters in the prostate gland. Journal of Andrology, 33(1), 27–36. DOI 10.2164/jandrol.110.011999. [Google Scholar] [CrossRef]
131. Qi, W., Gao, S., Wang, Z. (2008). Transcriptional regulation of the TGF-β1 promoter by androgen receptor. Biochemical Journal, 416(3), 453–462. DOI 10.1042/BJ20080651. [Google Scholar] [CrossRef]
132. Zhu, B., Kyprianou, N. (2005). Transforming growth factor β and prostate cancer. Cancer Treatment and Research, 126, 157–173. DOI 10.1007/b105234. [Google Scholar] [CrossRef]
133. Li, Y., Zhang, B., Xiang, L., Xia, S., Kucuk, O. et al. (2020). TGF-β causes docetaxel resistance in prostate cancer via the induction of Bcl-2 by acetylated KLF5 and protein stabilization. Theranostics, 10(17), 7656–7670. DOI 10.7150/thno.44567. [Google Scholar] [CrossRef]
134. Kyprianou, N., Isaacs, J. T. (1989). Expression of transforming growth factor-β in the rat ventral prostate during castration-induced programmed cell death. Molecular Endocrinology, 3(10), 1515–1522. DOI 10.1210/mend-3-10-1515. [Google Scholar] [CrossRef]
135. Kim, I. Y., Ahn, H. J., Zelner, D. J., Park, L., Sensibar, J. A. et al. (1996). Expression and localization of transforming growth factor-β receptors type I and type II in the rat ventral prostate during regression. Molecular Endocrinology, 10(1), 107–115. [Google Scholar]
136. Chipuk, J. E., Cornelius, S. C., Pultz, N. J., Jorgensen, J. S., Bonham, M. J. et al. (2002). The androgen receptor represses transforming growth factor-β signaling through interaction with Smad3. Journal of Biological Chemistry, 277(2), 1240–1248. DOI 10.1074/jbc.M108855200. [Google Scholar] [CrossRef]
137. Fuzio, P., Ditonno, P., Rutigliano, M., Battaglia, M., Bettocchi, C. et al. (2012). Regulation of TGF-β1 expression by androgen deprivation therapy of prostate cancer. Cancer Letters, 318(2), 135–144. DOI 10.1016/j.canlet.2011.08.034. [Google Scholar] [CrossRef]
138. Steffan, J. J., Koul, S., Meacham, R. B., Koul, H. K. (2012). The transcription factor SPDEF suppresses prostate tumor metastasis. Journal of Biological Chemistry, 287(35), 29968–29978. DOI 10.1074/jbc.M112.379396. [Google Scholar] [CrossRef]
139. Olson, A., Le, V., Aldahl, J., Yu, E. J., Hooker, E. et al. (2019). The comprehensive role of E-cadherin in maintaining prostatic epithelial integrity during oncogenic transformation and tumor progression. PLos Genetics, 15(10), e1008451. DOI 10.1371/journal.pgen.1008451. [Google Scholar] [CrossRef]
140. Liu, Y. N., Liu, Y., Lee, H. J., Hsu, Y. H., Chen, J. H. (2008). Activated androgen receptor downregulates E-cadherin gene expression and promotes tumor metastasis. Molecular and Cellular Biology, 28(23), 7096–7108. DOI 10.1128/MCB.00449-08. [Google Scholar] [CrossRef]
141. Nightingale, J., Chaudhary, K. S., Abel, P. D., Stubbs, A. P., Romanska, H. M. et al. (2003). Ligand activation of the androgen receptor downregulates E-cadherin-mediated cell adhesion and promotes apoptosis of prostatic cancer cells. Neoplasia, 5(4), 347–361. DOI 10.1016/S1476-5586(03)80028-3. [Google Scholar] [CrossRef]
142. Wong, T. S., Gao, W., Chan, J. Y. (2014). Transcription regulation of E-cadherin by zinc finger E-box binding homeobox proteins in solid tumors. BioMed Research International, 2014(4), 921564. DOI 10.1155/2014/921564. [Google Scholar] [CrossRef]
143. Thiery, J. P., Acloque, H., Huang, R. Y., Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell, 139(5), 871–890. DOI 10.1016/j.cell.2009.11.007. [Google Scholar] [CrossRef]
144. Pecina-Slaus, N. (2003). Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell International, 3(1), 17. DOI 10.1186/1475-2867-3-17. [Google Scholar] [CrossRef]
145. Yu, W., Yang, L., Li, T., Zhang, Y. (2019). Cadherin signaling in cancer: Its functions and role as a therapeutic target. Frontiers in Oncology, 9, 989. DOI 10.3389/fonc.2019.00989. [Google Scholar] [CrossRef]
146. Loric, S., Paradis, V., Gala, J. L., Berteau, P., Bedossa, P. et al. (2001). Abnormal E-cadherin expression and prostate cell blood dissemination as markers of biological recurrence in cancer. European Journal of Cancer, 37(12), 1475–1481. DOI 10.1016/S0959-8049(01)00143-5. [Google Scholar] [CrossRef]
147. Umbas, R., Schalken, J. A., Aalders, T. W., Carter, B. S., Karthaus, H. F. et al. (1992). Expression of the cellular adhesion molecule E-cadherin is reduced or absent in high-grade prostate cancer. Cancer Research, 52(18), 5104–5109. [Google Scholar]
148. Gravdal, K., Halvorsen, O. J., Haukaas, S. A., Akslen, L. A. (2007). A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clinical Cancer Research, 13(23), 7003–7011. DOI 10.1158/1078-0432.CCR-07-1263. [Google Scholar] [CrossRef]
149. Putzke, A. P., Ventura, A. P., Bailey, A. M., Akture, C., Opoku-Ansah, J. et al. (2011). Metastatic progression of prostate cancer and E-cadherin regulation by ZEB1 and SRC family kinases. American Journal of Pathology, 179(1), 400–410. DOI 10.1016/j.ajpath.2011.03.028. [Google Scholar] [CrossRef]
150. Mason, M. D., Davies, G., Jiang, W. G. (2002). Cell adhesion molecules and adhesion abnormalities in prostate cancer. Critical Reviews in Oncology/Hematology, 41(1), 11–28. DOI 10.1016/S1040-8428(01)00171-8. [Google Scholar] [CrossRef]
151. Cao, Z. Q., Wang, Z., Leng, P. (2019). Aberrant N-cadherin expression in cancer. Biomedicine and Pharmacotherapy, 118(4), 109320. DOI 10.1016/j.biopha.2019.109320. [Google Scholar] [CrossRef]
152. Radice, G. L., Rayburn, H., Matsunami, H., Knudsen, K. A., Takeichi, M. et al. (1997). Developmental defects in mouse embryos lacking N-cadherin. Developmental Biology, 181(1), 64–78. DOI 10.1006/dbio.1996.8443. [Google Scholar] [CrossRef]
153. Quan, Y., Zhang, X., Butler, W., Du, Z., Wang, M. et al. (2021). The role of N-cadherin/c-Jun/NDRG1 axis in the progression of prostate cancer. International Journal of Biological Sciences, 17(13), 3288–3304. DOI 10.7150/ijbs.63300. [Google Scholar] [CrossRef]
154. Wang, M., Ren, D., Guo, W., Huang, S., Wang, Z. et al. (2016). N-cadherin promotes epithelial-mesenchymal transition and cancer stem cell-like traits via ErbB signaling in prostate cancer cells. International Journal of Oncology, 48(2), 595–606. DOI 10.3892/ijo.2015.3270. [Google Scholar] [CrossRef]
155. Jennbacken, K., Gustavsson, H., Welen, K., Vallbo, C., Damber, J. E. (2006). Prostate cancer progression into androgen independency is associated with alterations in cell adhesion and invasivity. Prostate, 66(15), 1631–1640. DOI 10.1002/(ISSN)1097-0045. [Google Scholar] [CrossRef]
156. Jaggi, M., Nazemi, T., Abrahams, N. A., Baker, J. J., Galich, A. et al. (2006). N-cadherin switching occurs in high Gleason grade prostate cancer. Prostate, 66(2), 193–199. DOI 10.1002/(ISSN)1097-0045. [Google Scholar] [CrossRef]
157. Miyazono, K. (2009). Transforming growth factor-β signaling in epithelial-mesenchymal transition and progression of cancer. Proceedings of the Japan Academy. Series B: Physical and Biological Sciences, 85(8), 314–323. DOI 10.2183/pjab.85.314. [Google Scholar] [CrossRef]
158. Rangel, M. C., Karasawa, H., Castro, N. P., Nagaoka, T., Salomon, D. S. et al. (2012). Role of Cripto-1 during epithelial-to-mesenchymal transition in development and cancer. American Journal of Pathology, 180(6), 2188–2200. DOI 10.1016/j.ajpath.2012.02.031. [Google Scholar] [CrossRef]
159. Gregory, P. A., Bert, A. G., Paterson, E. L., Barry, S. C., Tsykin, A. et al. (2008). The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nature Cell Biology, 10(5), 593–601. DOI 10.1038/ncb1722. [Google Scholar] [CrossRef]
160. Kim, N. H., Kim, H. S., Li, X. Y., Lee, I., Choi, H. S. et al. (2011). A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. Journal of Cell Biology, 195(3), 417–433. DOI 10.1083/jcb.201103097. [Google Scholar] [CrossRef]
161. Liu, Y. N., Yin, J. J., Abou-Kheir, W., Hynes, P. G., Casey, O. M. et al. (2013). MiR-1 and miR-200 inhibit EMT via Slug-dependent and tumorigenesis via Slug-independent mechanisms. Oncogene, 32(3), 296–306. DOI 10.1038/onc.2012.58. [Google Scholar] [CrossRef]
162. Ren, D., Yang, Q., Dai, Y., Guo, W., Du, H. et al. (2017). Oncogenic miR-210-3p promotes prostate cancer cell EMT and bone metastasis via NF-κB signaling pathway. Molecular Cancer, 16(1), 117. DOI 10.1186/s12943-017-0688-6. [Google Scholar] [CrossRef]
163. Lian, Z., Chang, T., Ma, S., Li, J., Zhang, H. et al. (2022). MiR-96-5p induced NDRG1 deficiency promotes prostate cancer migration and invasion through regulating the NF-κB signaling pathway. Cancer Biomarkers, 35(1), 83–98. DOI 10.3233/CBM-210072. [Google Scholar] [CrossRef]
164. Sanchez-Tillo, E., Lazaro, A., Torrent, R., Cuatrecasas, M., Vaquero, E. C. et al. (2010). ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene, 29(24), 3490–3500. DOI 10.1038/onc.2010.102. [Google Scholar] [CrossRef]
165. Bolos, V., Peinado, H., Perez-Moreno, M. A., Fraga, M. F., Esteller, M. et al. (2003). The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with Snail and E47 repressors. Journal of Cell Science, 116, 499–511. DOI 10.1242/jcs.00224. [Google Scholar] [CrossRef]
166. Vesuna, F., van Diest, P., Chen, J. H., Raman, V. (2008). Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochemical and Biophysical Research Communications, 367(2), 235–241. DOI 10.1016/j.bbrc.2007.11.151. [Google Scholar] [CrossRef]
167. Sasaki, K., Natsugoe, S., Ishigami, S., Matsumoto, M., Okumura, H. et al. (2009). Significance of twist expression and its association with E-cadherin in esophageal squamous cell carcinoma. Journal of Experimental and Clinical Cancer Research, 28(1), 158. DOI 10.1186/1756-9966-28-158. [Google Scholar] [CrossRef]
168. Yang, J., Mani, S. A., Donaher, J. L., Ramaswamy, S., Itzykson, R. A. et al. (2004). Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell, 117(7), 927–939. DOI 10.1016/j.cell.2004.06.006. [Google Scholar] [CrossRef]
169. Batlle, E., Sancho, E., Franci, C., Dominguez, D., Monfar, M. et al. (2000). The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nature Cell Biology, 2(2), 84–89. DOI 10.1038/35000034. [Google Scholar] [CrossRef]
170. Cano, A., Perez-Moreno, M. A., Rodrigo, I., Locascio, A., Blanco, M. J. et al. (2000). The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nature Cell Biology, 2(2), 76–83. DOI 10.1038/35000025. [Google Scholar] [CrossRef]
171. Lamouille, S., Xu, J., Derynck, R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nature Reviews: Molecular Cell Biology, 15(3), 178–196. DOI 10.1038/nrm3758. [Google Scholar] [CrossRef]
172. Takagi, T., Moribe, H., Kondoh, H., Higashi, Y. (1998). DeltaEF1, a zinc finger and homeodomain transcription factor, is required for skeleton patterning in multiple lineages. Development, 125(1), 21–31. DOI 10.1242/dev.125.1.21. [Google Scholar] [CrossRef]
173. Zhang, P., Sun, Y., Ma, L. (2015). ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle, 14(4), 481–487. DOI 10.1080/15384101.2015.1006048. [Google Scholar] [CrossRef]
174. Orellana-Serradell, O., Herrera, D., Castellon, E. A., Contreras, H. R. (2018). The transcription factor ZEB1 promotes an aggressive phenotype in prostate cancer cell lines. Asian Journal of Andrology, 20(3), 294–299. DOI 10.4103/aja.aja_61_17. [Google Scholar] [CrossRef]
175. Figiel, S., Vasseur, C., Bruyere, F., Rozet, F., Maheo, K. et al. (2017). Clinical significance of epithelial-mesenchymal transition markers in prostate cancer. Human Pathology, 61, 26–32. DOI 10.1016/j.humpath.2016.10.013. [Google Scholar] [CrossRef]
176. Herrera, D., Orellana-Serradell, O., Villar, P., Torres, M. J., Paciucci, R. et al. (2019). Silencing of the transcriptional factor ZEB1 alters the steroidogenic pathway, and increases the concentration of testosterone and DHT in DU145 cells. Oncology Reports, 41(2), 1275–1283. [Google Scholar]
177. Graham, T. R., Zhau, H. E., Odero-Marah, V. A., Osunkoya, A. O., Kimbro, K. S. et al. (2008). Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Research, 68(7), 2479–2488. DOI 10.1158/0008-5472.CAN-07-2559. [Google Scholar] [CrossRef]
178. Bery, F., Figiel, S., Kouba, S., Fontaine, D., Gueguinou, M. et al. (2020). Hypoxia promotes prostate cancer aggressiveness by upregulating EMT-activator Zeb1 and SK3 channel expression. International Journal of Molecular Sciences, 21(13), 4786. DOI 10.3390/ijms21134786. [Google Scholar] [CrossRef]
179. Drake, J. M., Strohbehn, G., Bair, T. B., Moreland, J. G., Henry, M. D. (2009). ZEB1 enhances transendothelial migration and represses the epithelial phenotype of prostate cancer cells. Molecular Biology of the Cell, 20(8), 2207–2217. DOI 10.1091/mbc.e08-10-1076. [Google Scholar] [CrossRef]
180. Wang, H., Huang, B., Li, B. M., Cao, K. Y., Mo, C. Q. et al. (2018). ZEB1-mediated vasculogenic mimicry formation associates with epithelial-mesenchymal transition and cancer stem cell phenotypes in prostate cancer. Journal of Cellular and Molecular Medicine, 22(8), 3768–3781. DOI 10.1111/jcmm.13637. [Google Scholar] [CrossRef]
181. Eger, A., Aigner, K., Sonderegger, S., Dampier, B., Oehler, S. et al. (2005). DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene, 24(14), 2375–2385. DOI 10.1038/sj.onc.1208429. [Google Scholar] [CrossRef]
182. Shi, Y., Sawada, J., Sui, G., Affar, E. B., Whetstine, J. R. et al. (2003). Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature, 422(6933), 735–738. DOI 10.1038/nature01550. [Google Scholar] [CrossRef]
183. Aghdassi, A., Sendler, M., Guenther, A., Mayerle, J., Behn, C. O. et al. (2012). Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut, 61(3), 439–448. DOI 10.1136/gutjnl-2011-300060. [Google Scholar] [CrossRef]
184. Aigner, K., Dampier, B., Descovich, L., Mikula, M., Sultan, A. et al. (2007). The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene, 26(49), 6979–6988. DOI 10.1038/sj.onc.1210508. [Google Scholar] [CrossRef]
185. Fukagawa, A., Ishii, H., Miyazawa, K., Saitoh, M. (2015). deltaEF1 associates with DNMT1 and maintains DNA methylation of the E-cadherin promoter in breast cancer cells. Cancer Medicine, 4(1), 125–135. DOI 10.1002/cam4.347. [Google Scholar] [CrossRef]
186. Farfan, N., Ocarez, N., Castellon, E. A., Mejia, N., de Herreros, A. G., et al. (2018). The transcriptional factor ZEB1 represses Syndecan 1 expression in prostate cancer. Scientific Reports, 8(1), 11467. DOI 10.1038/s41598-018-29829-1. [Google Scholar] [CrossRef]
187. Huang, Q., Miller, M. R., Schappet, J., Henry, M. D. (2015). The glycosyltransferase LARGE2 is repressed by Snail and ZEB1 in prostate cancer. Cancer Biology & Therapy, 16(1), 125–136. DOI 10.4161/15384047.2014.987078. [Google Scholar] [CrossRef]
188. Lazarova, D., Bordonaro, M. (2017). ZEB1 mediates drug resistance and EMT in p300-deficient CRC. Journal of Cancer, 8(8), 1453–1459. DOI 10.7150/jca.18762. [Google Scholar] [CrossRef]
189. Zhang, P., Wei, Y., Wang, L., Debeb, B. G., Yuan, Y. et al. (2014). ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nature Cell Biology, 16(9), 864–875. DOI 10.1038/ncb3013. [Google Scholar] [CrossRef]
190. Guo, C., Ma, J., Deng, G., Qu, Y., Yin, L. et al. (2017). ZEB1 promotes oxaliplatin resistance through the induction of epithelial-mesenchymal transition in colon cancer cells. Journal of Cancer, 8(17), 3555–3566. DOI 10.7150/jca.20952. [Google Scholar] [CrossRef]
191. Orellana-Serradell, O., Herrera, D., Castellon, E. A., Contreras, H. R. (2019). The transcription factor ZEB1 promotes chemoresistance in prostate cancer cell lines. Asian Journal of Andrology, 21(5), 460–467. DOI 10.4103/aja.aja_1_19. [Google Scholar] [CrossRef]
192. Hanrahan, K., O'Neill, A., Prencipe, M., Bugler, J., Murphy, L. et al. (2017). The role of epithelial-mesenchymal transition drivers ZEB1 and ZEB2 in mediating docetaxel-resistant prostate cancer. Molecular Oncology, 11(3), 251–265. DOI 10.1002/1878-0261.12030. [Google Scholar] [CrossRef]
193. Perez, G., Lopez-Moncada, F., Indo, S., Torres, M. J., Castellon, E. A. et al. (2021). Knockdown of ZEB1 reverses cancer stem cell properties in prostate cancer cells. Oncology Reports, 45(5), 58. DOI 10.3892/or.2021.8009. [Google Scholar] [CrossRef]
194. Zhou, C., Jiang, H., Zhang, Z., Zhang, G., Wang, H. et al. (2017). ZEB1 confers stem cell-like properties in breast cancer by targeting neurogenin-3. Oncotarget, 8(33), 54388–54401. DOI 10.18632/oncotarget.17077. [Google Scholar] [CrossRef]
195. Miao, L., Yang, L., Li, R., Rodrigues, D. N., Crespo, M. et al. (2017). Disrupting androgen receptor signaling induces snail-mediated epithelial-mesenchymal plasticity in prostate cancer. Cancer Research, 77(11), 3101–3112. DOI 10.1158/0008-5472.CAN-16-2169. [Google Scholar] [CrossRef]
196. Anose, B. M., Sanders, M. M. (2011). Androgen receptor regulates transcription of the ZEB1 transcription factor. International Journal of Endocrinology, 2011(5), 903918. DOI 10.1155/2011/903918. [Google Scholar] [CrossRef]
197. Mooney, S. M., Parsana, P., Hernandez, J. R., Liu, X., Verdone, J. E. et al. (2015). The presence of androgen receptor elements regulates ZEB1 expression in the absence of androgen receptor. Journal of Cellular Biochemistry, 116(1), 115–123. DOI 10.1002/jcb.24948. [Google Scholar] [CrossRef]
198. Cottard, F., Asmane, I., Erdmann, E., Bergerat, J. P., Kurtz, J. E. et al. (2013). Constitutively active androgen receptor variants upregulate expression of mesenchymal markers in prostate cancer cells. PLoS One, 8(5), e63466. DOI 10.1371/journal.pone.0063466. [Google Scholar] [CrossRef]
199. Graham, T. R., Yacoub, R., Taliaferro-Smith, L., Osunkoya, A. O., Odero-Marah, V. A. et al. (2010). Reciprocal regulation of ZEB1 and AR in triple negative breast cancer cells. Breast Cancer Research and Treatment, 123(1), 139–147. DOI 10.1007/s10549-009-0623-7. [Google Scholar] [CrossRef]
200. Qiao, L., Tasian, G. E., Zhang, H., Cao, M., Ferretti, M. et al. (2012). Androgen receptor is overexpressed in boys with severe hypospadias, and ZEB1 regulates androgen receptor expression in human foreskin cells. Pediatric Research, 71(4), 393–398. DOI 10.1038/pr.2011.49. [Google Scholar] [CrossRef]
201. Li, P., Wang, J., Chu, M., Zhang, K., Yang, R. et al. (2014). Zeb1 promotes androgen independence of prostate cancer via induction of stem cell-like properties. Experimental Biology and Medicine, 239(7), 813–822. DOI 10.1177/1535370214538727. [Google Scholar] [CrossRef]
202. Bery, F., Cancel, M., Gueguinou, M., Potier-Cartereau, M., Vandier, C. et al. (2021). Zeb1 and SK3 channel are up-regulated in castration-resistant prostate cancer and promote neuroendocrine differentiation. Cancers, 13(12), 2947. DOI 10.3390/cancers13122947. [Google Scholar] [CrossRef]
203. Figiel, S., Bery, F., Chantome, A., Fontaine, D., Pasqualin, C. et al. (2019). A novel calcium-mediated EMT pathway controlled by lipids: An opportunity for prostate cancer adjuvant therapy. Cancers, 11(11), 1814. DOI 10.3390/cancers11111814. [Google Scholar] [CrossRef]
204. Sozen, B., Pehlivanoglu, S., Demir, N. (2016). Differential expression pattern of Twist1 in mouse preimplantation embryos suggests its multiple roles during early development. Journal of Assisted Reproduction and Genetics, 33(11), 1533–1540. DOI 10.1007/s10815-016-0794-1. [Google Scholar] [CrossRef]
205. Zhao, Z., Rahman, M. A., Chen, Z. G., Shin, D. M. (2017). Multiple biological functions of Twist1 in various cancers. Oncotarget, 8(12), 20380–20393. DOI 10.18632/oncotarget.14608. [Google Scholar] [CrossRef]
206. Yuen, H. F., Chua, C. W., Chan, Y. P., Wong, Y. C., Wang, X. et al. (2007). Significance of TWIST and E-cadherin expression in the metastatic progression of prostatic cancer. Histopathology, 50(5), 648–658. DOI 10.1111/j.1365-2559.2007.02665.x. [Google Scholar] [CrossRef]
207. Gajula, R. P., Chettiar, S. T., Williams, R. D., Thiyagarajan, S., Kato, Y. et al. (2013). The twist box domain is required for Twist1-induced prostate cancer metastasis. Molecular Cancer Research, 11(11), 1387–1400. DOI 10.1158/1541-7786.MCR-13-0218-T. [Google Scholar] [CrossRef]
208. Yuen, H. F., Kwok, W. K., Chan, K. K., Chua, C. W., Chan, Y. P. et al. (2008). TWIST modulates prostate cancer cell-mediated bone cell activity and is upregulated by osteogenic induction. Carcinogenesis, 29(8), 1509–1518. DOI 10.1093/carcin/bgn105. [Google Scholar] [CrossRef]
209. Kwok, W. K., Ling, M. T., Lee, T. W., Lau, T. C., Zhou, C. et al. (2005). Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Research, 65(12), 5153–5162. DOI 10.1158/0008-5472.CAN-04-3785. [Google Scholar] [CrossRef]
210. Alexander, N. R., Tran, N. L., Rekapally, H., Summers, C. E., Glackin, C. et al. (2006). N-cadherin gene expression in prostate carcinoma is modulated by integrin-dependent nuclear translocation of Twist1. Cancer Research, 66(7), 3365–3369. DOI 10.1158/0008-5472.CAN-05-3401. [Google Scholar] [CrossRef]
211. Casas, E., Kim, J., Bendesky, A., Ohno-Machado, L., Wolfe, C. J. et al. (2011). Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Research, 71(1), 245–254. DOI 10.1158/0008-5472.CAN-10-2330. [Google Scholar] [CrossRef]
212. Eide, T., Ramberg, H., Glackin, C., Tindall, D., Tasken, K. A. (2013). TWIST1, A novel androgen-regulated gene, is a target for NKX3-1 in prostate cancer cells. Cancer Cell International, 13(1), 4. DOI 10.1186/1475-2867-13-4. [Google Scholar] [CrossRef]
213. Khatiwada, P., Kannan, A., Malla, M., Dreier, M., Shemshedini, L. (2020). Androgen up-regulation of Twist1 gene expression is mediated by ETV1. PeerJ, 8(3), e8921. DOI 10.7717/peerj.8921. [Google Scholar] [CrossRef]
214. Shiota, M., Song, Y., Takeuchi, A., Yokomizo, A., Kashiwagi, E. et al. (2012). Antioxidant therapy alleviates oxidative stress by androgen deprivation and prevents conversion from androgen dependent to castration resistant prostate cancer. Journal of Urology, 187(2), 707–714. DOI 10.1016/j.juro.2011.09.147. [Google Scholar] [CrossRef]
215. Shiota, M., Yokomizo, A., Tada, Y., Inokuchi, J., Kashiwagi, E. et al. (2010). Castration resistance of prostate cancer cells caused by castration-induced oxidative stress through Twist1 and androgen receptor overexpression. Oncogene, 29(2), 237–250. DOI 10.1038/onc.2009.322. [Google Scholar] [CrossRef]
216. Shiota, M., Yokomizo, A., Takeuchi, A., Imada, K., Kashiwagi, E. et al. (2014). Inhibition of protein kinase C/Twist1 signaling augments anticancer effects of androgen deprivation and enzalutamide in prostate cancer. Clinical Cancer Research, 20(4), 951–961. DOI 10.1158/1078-0432.CCR-13-1809. [Google Scholar] [CrossRef]
217. Shiota, M., Yokomizo, A., Takeuchi, A., Kashiwagi, E., Dejima, T. et al. (2017). Protein kinase C regulates Twist1 expression via NF-κB in prostate cancer. Endocrine-Related Cancer, 24(4), 171–180. DOI 10.1530/ERC-16-0384. [Google Scholar] [CrossRef]
218. Osorio, L. A., Farfan, N. M., Castellon, E. A., Contreras, H. R. (2016). SNAIL transcription factor increases the motility and invasive capacity of prostate cancer cells. Molecular Medicine Reports, 13(1), 778–786. DOI 10.3892/mmr.2015.4585. [Google Scholar] [CrossRef]
219. Liu, J., Uygur, B., Zhang, Z., Shao, L., Romero, D. et al. (2010). Slug inhibits proliferation of human prostate cancer cells via downregulation of cyclin D1 expression. Prostate, 70(16), 1768–1777. DOI 10.1002/pros.21213. [Google Scholar] [CrossRef]
220. Esposito, S., Russo, M. V., Airoldi, I., Tupone, M. G., Sorrentino, C. et al. (2015). SNAI2/Slug gene is silenced in prostate cancer and regulates neuroendocrine differentiation, metastasis-suppressor and pluripotency gene expression. Oncotarget, 6(19), 17121–17134. DOI 10.18632/oncotarget.2736. [Google Scholar] [CrossRef]
221. Wang, Y., Shi, J., Chai, K., Ying, X., Zhou, B. P. (2013). The role of snail in EMT and tumorigenesis. Current Cancer Drug Targets, 13(9), 963–972. DOI 10.2174/15680096113136660102. [Google Scholar] [CrossRef]
222. Heeboll, S., Borre, M., Ottosen, P. D., Dyrskjot, L., Orntoft, T. F. et al. (2009). Snail1 is over-expressed in prostate cancer. APMIS, 117(3), 196–204. DOI 10.1111/j.1600-0463.2008.00007.x. [Google Scholar] [CrossRef]
223. Poblete, C. E., Fulla, J., Gallardo, M., Munoz, V., Castellon, E. A. et al. (2014). Increased SNAIL expression and low syndecan levels are associated with high Gleason grade in prostate cancer. International Journal of Oncology, 44(3), 647–654. DOI 10.3892/ijo.2014.2254. [Google Scholar] [CrossRef]
224. Mickova, A., Kharaishvili, G., Kurfurstova, D., Gachechiladze, M., Kral, M. et al. (2021). Skp2 and slug are coexpressed in aggressive prostate cancer and inhibited by neddylation blockade. International Journal of Molecular Sciences, 22(6), 2844. DOI 10.3390/ijms22062844. [Google Scholar] [CrossRef]
225. Uygur, B., Wu, W. S. (2011). SLUG promotes prostate cancer cell migration and invasion via CXCR4/CXCL12 axis. Molecular Cancer, 10(1), 139. DOI 10.1186/1476-4598-10-139. [Google Scholar] [CrossRef]
226. Xu, J., Lamouille, S., Derynck, R. (2009). TGF-β-induced epithelial to mesenchymal transition. Cell Research, 19(2), 156–172. DOI 10.1038/cr.2009.5. [Google Scholar] [CrossRef]
227. Jorda, M., Olmeda, D., Vinyals, A., Valero, E., Cubillo, E. et al. (2005). Upregulation of MMP-9 in MDCK epithelial cell line in response to expression of the Snail transcription factor. Journal of Cell Science, 118, 3371–3385. DOI 10.1242/jcs.02465. [Google Scholar] [CrossRef]
228. Yokoyama, K., Kamata, N., Fujimoto, R., Tsutsumi, S., Tomonari, M. et al. (2003). Increased invasion and matrix metalloproteinase-2 expression by Snail-induced mesenchymal transition in squamous cell carcinomas. International Journal of Oncology, 22(4), 891–898. DOI 10.3892/ijo.22.4.891. [Google Scholar] [CrossRef]
229. Moreno-Bueno, G., Cubillo, E., Sarrio, D., Peinado, H., Rodriguez-Pinilla, S. M. et al. (2006). Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial-mesenchymal transition. Cancer Research, 66(19), 9543–9556. DOI 10.1158/0008-5472.CAN-06-0479. [Google Scholar] [CrossRef]
230. Wu, K., Gore, C., Yang, L., Fazli, L., Gleave, M. et al. (2012). Slug, a unique androgen-regulated transcription factor, coordinates androgen receptor to facilitate castration resistance in prostate cancer. Molecular Endocrinology, 26(9), 1496–1507. DOI 10.1210/me.2011-1360. [Google Scholar] [CrossRef]
231. Ware, K. E., Somarelli, J. A., Schaeffer, D., Li, J., Zhang, T. et al. (2016). Snail promotes resistance to enzalutamide through regulation of androgen receptor activity in prostate cancer. Oncotarget, 7(31), 50507–50521. DOI 10.18632/oncotarget.10476. [Google Scholar] [CrossRef]
232. Cmero, M., Kurganovs, N. J., Stuchbery, R., McCoy, P., Grima, C. et al. (2021). Loss of SNAI2 in prostate cancer correlates with clinical response to androgen deprivation therapy. JCO Precision Oncology, (5), 1048–1059. DOI 10.1200/PO.20.00337. [Google Scholar] [CrossRef]
233. Wang, T., Zheng, L., Wang, Q., Hu, Y. W. (2018). Emerging roles and mechanisms of FOXC2 in cancer. Clinica Chimica Acta, 479(5), 84–93. DOI 10.1016/j.cca.2018.01.019. [Google Scholar] [CrossRef]
234. Borretzen, A., Gravdal, K., Haukaas, S. A., Beisland, C., Akslen, L. A. et al. (2019). FOXC2 expression and epithelial-mesenchymal phenotypes are associated with castration resistance, metastasis and survival in prostate cancer. The Journal of Pathology: Clinical Research, 5(4), 272–286. DOI 10.1002/cjp2.142. [Google Scholar] [CrossRef]
235. Mani, S. A., Yang, J., Brooks, M., Schwaninger, G., Zhou, A. et al. (2007). Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. PNAS, 104(24), 10069–10074. DOI 10.1073/pnas.0703900104. [Google Scholar] [CrossRef]
236. Sarkar, P. L., Lee, W., Williams, E. D., Lubik, A. A., Stylianou, N. et al. (2019). Insulin enhances migration and invasion in prostate cancer cells by up-regulation of FOXC2. Frontiers in Endocrinology, 10, 481. DOI 10.3389/fendo.2019.00481. [Google Scholar] [CrossRef]
237. Paranjape, A. N., Soundararajan, R., Werden, S. J., Joseph, R., Taube, J. H. et al. (2016). Inhibition of FOXC2 restores epithelial phenotype and drug sensitivity in prostate cancer cells with stem-cell properties. Oncogene, 35(46), 5963–5976. DOI 10.1038/onc.2015.498. [Google Scholar] [CrossRef]
238. Mortazavi, F., An, J., Dubinett, S., Rettig, M. (2010). p120-catenin is transcriptionally downregulated by FOXC2 in non-small cell lung cancer cells. Molecular Cancer Research, 8(5), 762–774. DOI 10.1158/1541-7786.MCR-10-0004. [Google Scholar] [CrossRef]
239. Kourtidis, A., Ngok, S. P., Anastasiadis, P. Z. (2013). p120 catenin: An essential regulator of cadherin stability, adhesion-induced signaling, and cancer progression. Progress in Molecular Biology and Translational Science, 116, 409–432. DOI 10.1016/B978-0-12-394311-8.00018-2. [Google Scholar] [CrossRef]
240. Werden, S. J., Sphyris, N., Sarkar, T. R., Paranjape, A. N., LaBaff, A. M. et al. (2016). Phosphorylation of serine 367 of FOXC2 by p38 regulates ZEB1 and breast cancer metastasis, without impacting primary tumor growth. Oncogene, 35(46), 5977–5988. DOI 10.1038/onc.2016.203. [Google Scholar] [CrossRef]
241. Ren, Y. H., Liu, K. J., Wang, M., Yu, Y. N., Yang, K. et al. (2014). De-SUMOylation of FOXC2 by SENP3 promotes the epithelial-mesenchymal transition in gastric cancer cells. Oncotarget, 5(16), 7093–7104. DOI 10.18632/oncotarget.2197. [Google Scholar] [CrossRef]
242. Castaneda, M., Chen, L., Pradhan, L., Li, S., Zein, R. et al. (2018). A forkhead box protein C2 inhibitor: Targeting epithelial-mesenchymal transition and cancer metastasis. ChemBioChem, 19(13), 1359–1364. DOI 10.1002/cbic.201800022. [Google Scholar] [CrossRef]
243. Zeng, J., Shi, R., Cai, C. X., Liu, X. R., Song, Y. B. et al. (2015). Increased expression of Six1 correlates with progression and prognosis of prostate cancer. Cancer Cell International, 15(1), 63. DOI 10.1186/s12935-015-0215-z. [Google Scholar] [CrossRef]
244. Liao, Y., Liu, Y., Shao, Z., Xia, X., Deng, Y. et al. (2021). A new role of GRP75-USP1-SIX1 protein complex in driving prostate cancer progression and castration resistance. Oncogene, 40(25), 4291–4306. DOI 10.1038/s41388-021-01851-0. [Google Scholar] [CrossRef]
245. Liao, Y., Sun, W., Shao, Z., Liu, Y., Zhong, X. et al. (2022). A SIX1 degradation inducer blocks excessive proliferation of prostate cancer. International Journal of Biological Sciences, 18(6), 2439–2451. DOI 10.7150/ijbs.67873. [Google Scholar] [CrossRef]
246. Micalizzi, D. S., Christensen, K. L., Jedlicka, P., Coletta, R. D., Baron, A. E. et al. (2009). The Six1 homeoprotein induces human mammary carcinoma cells to undergo epithelial-mesenchymal transition and metastasis in mice through increasing TGF-β signaling. Journal of Clinical Investigation, 119(9), 2678–2690. DOI 10.1172/JCI37815. [Google Scholar] [CrossRef]
247. Min, W. P., Wei, X. F. (2021). Silencing SIX1 inhibits epithelial mesenchymal transition through regulating TGF-β/Smad2/3 signaling pathway in papillary thyroid carcinoma. Auris, Nasus, Larynx, 48(3), 487–495. DOI 10.1016/j.anl.2020.10.002. [Google Scholar] [CrossRef]
248. Wu, W., Ren, Z., Li, P., Yu, D., Chen, J. et al. (2015). Six1: A critical transcription factor in tumorigenesis. International Journal of Cancer, 136(6), 1245–1253. DOI 10.1002/ijc.28755. [Google Scholar] [CrossRef]
249. Micalizzi, D. S., Wang, C. A., Farabaugh, S. M., Schiemann, W. P., Ford, H. L. (2010). Homeoprotein Six1 increases TGF-β type I receptor and converts TGF-β signaling from suppressive to supportive for tumor growth. Cancer Research, 70(24), 10371–10380. DOI 10.1158/0008-5472.CAN-10-1354. [Google Scholar] [CrossRef]
250. Xu, H., Zhang, Y., Altomare, D., Pena, M. M., Wan, F. et al. (2014). Six1 promotes epithelial-mesenchymal transition and malignant conversion in human papillomavirus type 16-immortalized human keratinocytes. Carcinogenesis, 35(6), 1379–1388. DOI 10.1093/carcin/bgu050. [Google Scholar] [CrossRef]
251. Ono, H., Imoto, I., Kozaki, K., Tsuda, H., Matsui, T. et al. (2012). SIX1 promotes epithelial-mesenchymal transition in colorectal cancer through ZEB1 activation. Oncogene, 31(47), 4923–4934. DOI 10.1038/onc.2011.646. [Google Scholar] [CrossRef]
252. McCoy, E. L., Iwanaga, R., Jedlicka, P., Abbey, N. S., Chodosh, L. A. et al. (2009). Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial-mesenchymal transition. Journal of Clinical Investigation, 119(9), 2663–2677. DOI 10.1172/JCI37691. [Google Scholar] [CrossRef]
253. Wang, F., Koul, S., Shanmugam, P. S. T., Dong, Q., Koul, H. K. (2018). Prostate-Derived Ets Factor (PDEF) inhibits metastasis by inducing epithelial/luminal phenotype in prostate cancer cells. Molecular Cancer Research, 16(9), 1430–1440. DOI 10.1158/1541-7786.MCR-18-0010. [Google Scholar] [CrossRef]
254. Meiners, J., Schulz, K., Moller, K., Hoflmayer, D., Burdelski, C. et al. (2019). Upregulation of SPDEF is associated with poor prognosis in prostate cancer. Oncology Letters, 18(5), 5107–5118. DOI 10.3892/ol.2019.10885. [Google Scholar] [CrossRef]
255. Tsai, Y. C., Chen, W. Y., Abou-Kheir, W., Zeng, T., Yin, J. J. et al. (2018). Androgen deprivation therapy-induced epithelial-mesenchymal transition of prostate cancer through downregulating SPDEF and activating CCL2. Biochimica et Biophysica Acta, 1864(5), 1717–1727. DOI 10.1016/j.bbadis.2018.02.016. [Google Scholar] [CrossRef]
256. Osisami, M., Keller, E. T. (2013). SPDEF: A molecular switch for E-cadherin expression that promotes prostate cancer metastasis. Asian Journal of Andrology, 15(5), 584–585. DOI 10.1038/aja.2013.64. [Google Scholar] [CrossRef]
257. Chen, W., Gao, Q., Han, S., Pan, F., Fan, W. (2015). The CCL2/CCR2 axis enhances IL-6-induced epithelial-mesenchymal transition by cooperatively activating STAT3-Twist signaling. Tumour Biology, 36(2), 973–981. DOI 10.1007/s13277-014-2717-z. [Google Scholar] [CrossRef]
258. Fararjeh, A. S., Liu, Y. N. (2019). ZBTB46, SPDEF, and ETV6: Novel potential biomarkers and therapeutic targets in castration-resistant prostate cancer. International Journal of Molecular Sciences, 20(11), 2802. DOI 10.3390/ijms20112802. [Google Scholar] [CrossRef]
259. Tu, L. C., Yan, X., Hood, L., Lin, B. (2007). Proteomics analysis of the interactome of N-myc downstream regulated gene 1 and its interactions with the androgen response program in prostate cancer cells. Molecular & Cellular Proteomics, 6(4), 575–588. DOI 10.1074/mcp.M600249-MCP200. [Google Scholar] [CrossRef]
260. Segawa, T., Nau, M. E., Xu, L. L., Chilukuri, R. N., Makarem, M. et al. (2002). Androgen-induced expression of endoplasmic reticulum (ER) stress response genes in prostate cancer cells. Oncogene, 21(57), 8749–8758. DOI 10.1038/sj.onc.1205992. [Google Scholar] [CrossRef]
261. Lim, S. C., Geleta, B., Maleki, S., Richardson, D. R., Kovacevic, Z. (2021). The metastasis suppressor NDRG1 directly regulates androgen receptor signaling in prostate cancer. Journal of Biological Chemistry, 297(6), 101414. DOI 10.1016/j.jbc.2021.101414. [Google Scholar] [CrossRef]
262. Masuda, K., Werner, T., Maheshwari, S., Frisch, M., Oh, S. et al. (2005). Androgen receptor binding sites identified by a GREF_GATA model. Journal of Molecular Biology, 353(4), 763–771. DOI 10.1016/j.jmb.2005.09.009. [Google Scholar] [CrossRef]
263. Bandyopadhyay, S., Pai, S. K., Gross, S. C., Hirota, S., Hosobe, S. et al. (2003). The Drg-1 gene suppresses tumor metastasis in prostate cancer. Cancer Research, 63(8), 1731–1736. [Google Scholar]
264. Sharma, A., Mendonca, J., Ying, J., Kim, H. S., Verdone, J. E. et al. (2017). The prostate metastasis suppressor gene NDRG1 differentially regulates cell motility and invasion. Molecular Oncology, 11(6), 655–669. DOI 10.1002/1878-0261.12059. [Google Scholar] [CrossRef]
265. Chen, Z., Zhang, D., Yue, F., Zheng, M., Kovacevic, Z. et al. (2012). The iron chelators Dp44mT and DFO inhibit TGF-β-induced epithelial-mesenchymal transition via up-regulation of N-Myc downstream-regulated gene 1 (NDRG1). Journal of Biological Chemistry, 287(21), 17016–17028. DOI 10.1074/jbc.M112.350470. [Google Scholar] [CrossRef]
266. Quan, Y., Zhang, X., Butler, W., Du, Z., Wang, M. et al. (2021). The role of N-cadherin/c-Jun/NDRG1 axis in the progression of prostate cancer. International Journal of Biological Sciences, 17(13), 3288–3304. DOI 10.7150/ijbs.63300. [Google Scholar] [CrossRef]
267. Menezes, S. V., Fouani, L., Huang, M. L. H., Geleta, B., Maleki, S. et al. (2019). The metastasis suppressor, NDRG1, attenuates oncogenic TGF-β and NF-κB signaling to enhance membrane E-cadherin expression in pancreatic cancer cells. Carcinogenesis, 40(6), 805–818. DOI 10.1093/carcin/bgy178. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2022 The Author(s). Published by Tech Science Press.
Copyright © 2022 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools