
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Disulfidptosis: A Metabolic Cell Death Mechanism with Therapeutic Potential in Cancer
Department of Spine Surgery, Pingdingshan First People’s Hospital, Pingdingshan, China
* Corresponding Author: Jun Zhang. Email:
# These authors contributed equally to this work
(This article belongs to the Special Issue: Therapeutic Challenges in Targeting Cell Death)
Oncology Research 2026, 34(4), 12 https://doi.org/10.32604/or.2026.076406
Received 20 November 2025; Accepted 09 January 2026; Issue published 23 March 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Disulfidptosis is a newly identified form of regulated cell death (RCD) first described in 2023, representing a significant advance in understanding programmed cell death pathways. This unique cell death modality is characterized by abnormal intracellular accumulation of disulfide bonds and disruption of redox homeostasis, leading to cytoskeletal collapse without caspase activation. Disulfidptosis is primarily triggered by glucose deprivation in cells with high expression of solute carrier family 7 member 11 (SLC7A11). Under these conditions, insufficient NADPH supply prevents the effective reduction of accumulated cystine to cysteine, thereby inducing disulfide stress. Distinct from apoptosis, ferroptosis, cuproptosis, or pyroptosis, disulfidptosis exhibits unique metabolic dependencies and a hallmark feature of cytoskeletal disintegration. Current evidence indicates that this mechanism is operative in various tumor types, including hepatocellular carcinoma, colorectal cancer, and lung adenocarcinoma, suggesting its potential therapeutic relevance. Therapeutic strategies targeting disulfidptosis include modulation of metabolic pathways—such as the use of GLUT1 or G6PD inhibitors—to selectively induce this form of cell death in cancer cells. This review systematically summarizes current understanding, aiming to elucidate the unique mechanisms and therapeutic potential of disulfidptosis, and provides a foundational framework for future studies and the development of innovative strategies targeting tumor metabolic vulnerabilities.Keywords
1 Introduction and Overview of Disulfidptosis
Regulated cell death (RCD) refers to forms of cell death that are controlled by specific molecular pathways and can be modulated through genetic or pharmacological interventions, with its dysregulation being causally linked to various diseases, including cancer [1]. Given that cancer cells often develop drug resistance by evading cell death mechanisms [2], exploring alternative RCD mechanisms beyond traditional apoptosis is of significant importance for cancer therapy. In 2023, a novel form of cell death—disulfidptosis—was first identified and named [3]. Its initial discovery was based on the specific death of solute carrier family 7 member 11 (SLC7A11)-overexpressing cells under glucose deprivation, a form of cell death distinct from previously known types. The core mechanism of disulfidptosis lies in the abnormal accumulation of intracellular disulfide bonds and the disruption of redox homeostasis, which subsequently affects protein structure and function, ultimately leading to cell death. This form of cell death is independent of caspase activation; instead, it occurs through disulfide bond-mediated actin cross-linking that disrupts the cytoskeleton—mechanistically distinct from apoptosis (triggered by caspase activation) and ferroptosis (driven by the accumulation of lipid peroxides) [4,5]. Current studies indicate that disulfidptosis primarily occurs in cells with high expression of SLC7A11, particularly under conditions of glucose deprivation [6]. This suggests that disulfidptosis is not universally present in all cell types, but rather exhibits certain cell-type specificity. Moreover, studies have shown that lactate dehydrogenase B (LDHB) promotes disulfidptosis and thereby mediates the exhaustion of tumor-infiltrating CD8+ T cells, revealing the role of disulfidptosis in immune cells and its impact on the tumor microenvironment [7]. These findings indicate that disulfidptosis plays a crucial role in tumors. Beyond disulfidptosis, tumors also exhibit pronounced metabolic reprogramming. Within the tumor microenvironment, cancer cells display dependencies on specific nutrients or metabolic pathways. Targeting these aberrant metabolic features offers high specificity, rendering disulfidptosis a promising strategy for tumor-targeted therapy [8,9]. This is manifested as a dependency on specific nutrients or metabolic pathways. Targeting the aberrant metabolism of tumor cells offers high specificity, making disulfidptosis a promising strategy for tumor-targeted therapy [10,11].
In summary, studying disulfidptosis in the context of cancer holds significant importance. Given that many cancer cells can evade therapy-induced apoptosis, leading to drug resistance and disease recurrence [12,13], targeting disulfidptosis opens new avenues for developing novel cancer therapeutic strategies. Its unique metabolic sensitivity and impact on the actin cytoskeleton make it a promising target for cancer therapy.
The core objective of this review is to provide a timely and comprehensive synthesis of research on “disulfidptosis”—an emerging form of cell death—in the context of cancer. To this end, the article first aims to thoroughly elucidate its underlying molecular mechanisms, followed by a systematic summary of how disulfidptosis is regulated and functions across different tumor types. By comparing it with other cell death pathways, the review further clarifies its unique characteristics. Building upon this foundation, it consolidates current potential therapeutic strategies aimed at inducing disulfidptosis. Finally, by highlighting key unresolved questions in the field, the review offers a forward-looking perspective to guide future basic research and clinical translation efforts.
2 Molecular Mechanisms of Disulfidptosis
Disulfidptosis, as a novel form of programmed cell death, involves complex and intricate molecular mechanisms. It encompasses the synergistic actions of multiple critical metabolic pathways and organelles, collectively constructing a comprehensive pathway from initial triggers to cell death [14]. The core of this process lies in the vulnerability of cancer cells in maintaining redox homeostasis, and the induction of catastrophic disulfide stress under specific metabolic pressures [3]. Fig. 1 provides a brief summary of the molecular mechanisms of disulfidptosis.
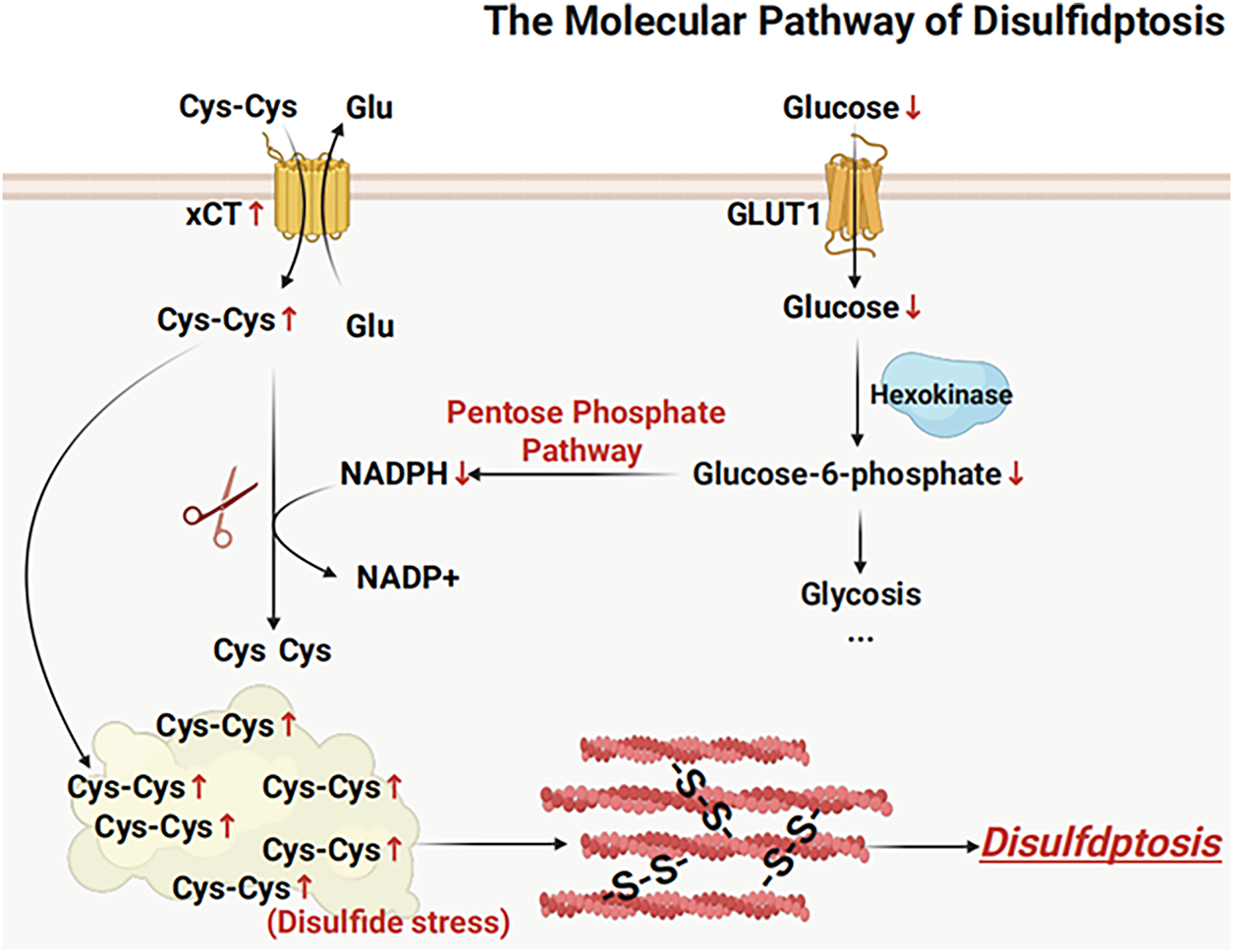
Figure 1: Basic mechanisms of disulfidptosis. Disulfidptosis is a novel form of cell death mediated by solute carrier family 7 member 11, which encodes the xCT transporter protein. Under glucose deprivation, cells with high SLC7A11 expression excessively import cystine. Due to insufficient NADPH supply, cystine cannot be efficiently reduced to cysteine, leading to abnormal accumulation of intracellular disulfides. This disulfide stress induces aberrant disulfide bonding and contraction within the actin cytoskeleton, ultimately resulting in cytoskeletal collapse and cell death. Abb: Glutamate (Glu), Cysteine (Cys), Cystine/Glutamate Antiporter (Xct) (Created with BioRender.com).
2.1 Triggers of Disulfidptosis: Glucose Deprivation and High Expression of SLC7A11
Disulfidptosis is initiated under specific conditions, including glucose deprivation and high expression of SLC7A11 [14,15]. Activating transcription factor 4 (ATF4) and nuclear factor erythroid 2-related factor 2 (NRF2) are two key factors responsible for activating SLC7A11 transcription [16]. Glucose deprivation promotes ATF4 translation by inducing endoplasmic reticulum stress. Glucose is a crucial source for cellular energy metabolism. In oral squamous cell carcinoma cells, glucose deprivation significantly enhances ATF4 enrichment in the SLC7A11 promoter region and upregulates SLC7A11 mRNA levels [17]. Under oxidative stress conditions, NRF2 binds to the antioxidant response element (ARE) in the SLC7A11 promoter region, thereby markedly enhancing its transcriptional activity [18]. When cells are under glucose deprivation, the glycolytic pathway is impaired, leading to reduced generation of NADPH [19]. NADPH is a crucial intracellular reductant that provides electrons for glutathione (GSH) reductase, thereby maintaining cellular redox homeostasis. Under conditions of insufficient NADPH supply, cells are unable to effectively eliminate excess reactive oxygen species (ROS) and disulfide bonds, leading to oxidative stress and disulfide stress [20–22]. SLC7A11, also known as xCT, is a cystine/glutamate antiporter primarily responsible for importing extracellular cystine into the cell [19]. After entering the cell, cystine is reduced to cysteine, which serves as a key precursor for the synthesis of GSH. GSH is the primary intracellular antioxidant that neutralizes ROS and alleviates oxidative stress. However, under glucose deprivation, high expression of SLC7A11 leads to continuous cystine uptake, further depleting the already limited NADPH pool. This exacerbates the accumulation of disulfide bonds, ultimately triggering disulfidptosis [19,23].
The redox balance of cells relies on the precise regulation of multiple factors, among which nicotinamide adenine dinucleotide phosphate (NADPH) plays a crucial role [24–28]. NADPH is primarily generated through the pentose phosphate pathway (PPP) [29]. It is the primary intracellular reductant, playing a key role in maintaining reduced GSH levels, scavenging ROS, and participating in various anabolic reactions [30]. The STAT3–LDHB–G6PD signaling axis has been identified as a key pathway regulating disulfidptosis and exhaustion in CD8+ T cells, highlighting the important role of G6PD in mediating disulfidptosis [7].
In tumor cells with high expression of SLC7A11, the cells take up large amounts of cystine and convert it into cysteine [23]. This process requires the consumption of large amounts of NADPH [19]. Under glucose deprivation conditions, the supply of NADPH in tumor cells is severely limited, leading to a disruption of the intracellular redox balance [15]. The depletion of NADPH directly impairs the reduction of disulfide bonds, causing intracellular disulfide bonds to fail to be properly reduced and leading to their abnormal accumulation [3]. This abnormal accumulation of disulfide bonds particularly affects proteins sensitive to redox state, such as actin cytoskeletal proteins, severely impairing their structure and function [23].
Therefore, NADPH depletion and the abnormal accumulation of disulfide bonds constitute the core mechanism underlying disulfidptosis [31]. On one hand, glucose deprivation limits NADPH production; on the other hand, excessive cystine uptake in tumor cells with high SLC7A11 expression further exacerbates NADPH consumption. This dual stress leads to intracellular redox imbalance and a burst of disulfide stress, ultimately triggering disulfidptosis. The following sections will further explore how this stress affects the cytoskeleton and the roles of other regulatory factors in this process.
2.3 Effector: Collapse of the Actin Cytoskeleton
Disulfidptosis, as a novel form of cell death triggered by disulfide stress, is characterized prominently by the collapse of the actin cytoskeleton [32]. The integrity of the cytoskeleton is crucial for maintaining cell morphology, motility, and intracellular transport [33]. Disulfidptosis exerts its cytotoxic effects precisely by disrupting the actin cytoskeleton. Under conditions such as glucose deprivation, cells with high SLC7A11 expression abnormally accumulate disulfides, leading to the formation of aberrant disulfide bonds within actin cytoskeletal proteins [3].
Specifically, the reduction state of intracellular cysteine residues is tightly regulated to maintain normal protein function [34–36]. However, during disulfidptosis, this balance is disrupted, leading to excessive formation of disulfide bonds within actin monomers and other cytoskeleton-associated proteins. These aberrant disulfide bonds interfere with the normal assembly and polymerization of actin, causing F-actin depolymerization and consequently disrupting the structural integrity of the entire cytoskeleton [3].
The collapse of the cytoskeleton directly impairs cell morphology and function. For instance, cells lose their normal shape, exhibit reduced adhesion, and impaired migration. More importantly, disruption of the cytoskeleton also interferes with intracellular signaling and transport, ultimately leading to cell death. This phenomenon is particularly evident in cancer cells with high SLC7A11 expression, especially when glucose uptake is limited [31,37,38].
Beyond direct structural damage, the collapse of the actin cytoskeleton may promote cell death through additional pathways. For example, the cytoskeleton interacts with receptors and ion channels on the cell membrane, modulating their activity. Disruption of the cytoskeleton could lead to dysfunction of these receptors and ion channels, further exacerbating cellular damage. Moreover, the cytoskeleton is involved in the regulation of processes such as autophagy and apoptosis. Therefore, cytoskeletal collapse may indirectly promote cell death by interfering with these pathways.
In summary, the collapse of the actin cytoskeleton is one of the key effectors of disulfidptosis. By forming aberrant disulfide bonds in actin and other cytoskeletal proteins, disulfidptosis rapidly disrupts the structure and function of the cytoskeleton, ultimately leading to cell death [32,39,40]. Understanding the molecular mechanisms of this process is crucial for developing anti-tumor therapeutic strategies that target disulfidptosis.
2.4 Other Regulatory Factors: WAVE Complex and Rac1
In addition to the two key triggering conditions—glucose deprivation and high SLC7A11 expression—the WAVE regulatory complex (WRC) and the Rac1 pathway also play important roles in regulating disulfidptosis (Fig. 2). These molecules are involved in the process of actin polymerization, thereby influencing cytoskeletal stability and ultimately determining the cell’s sensitivity to disulfidptosis [39].
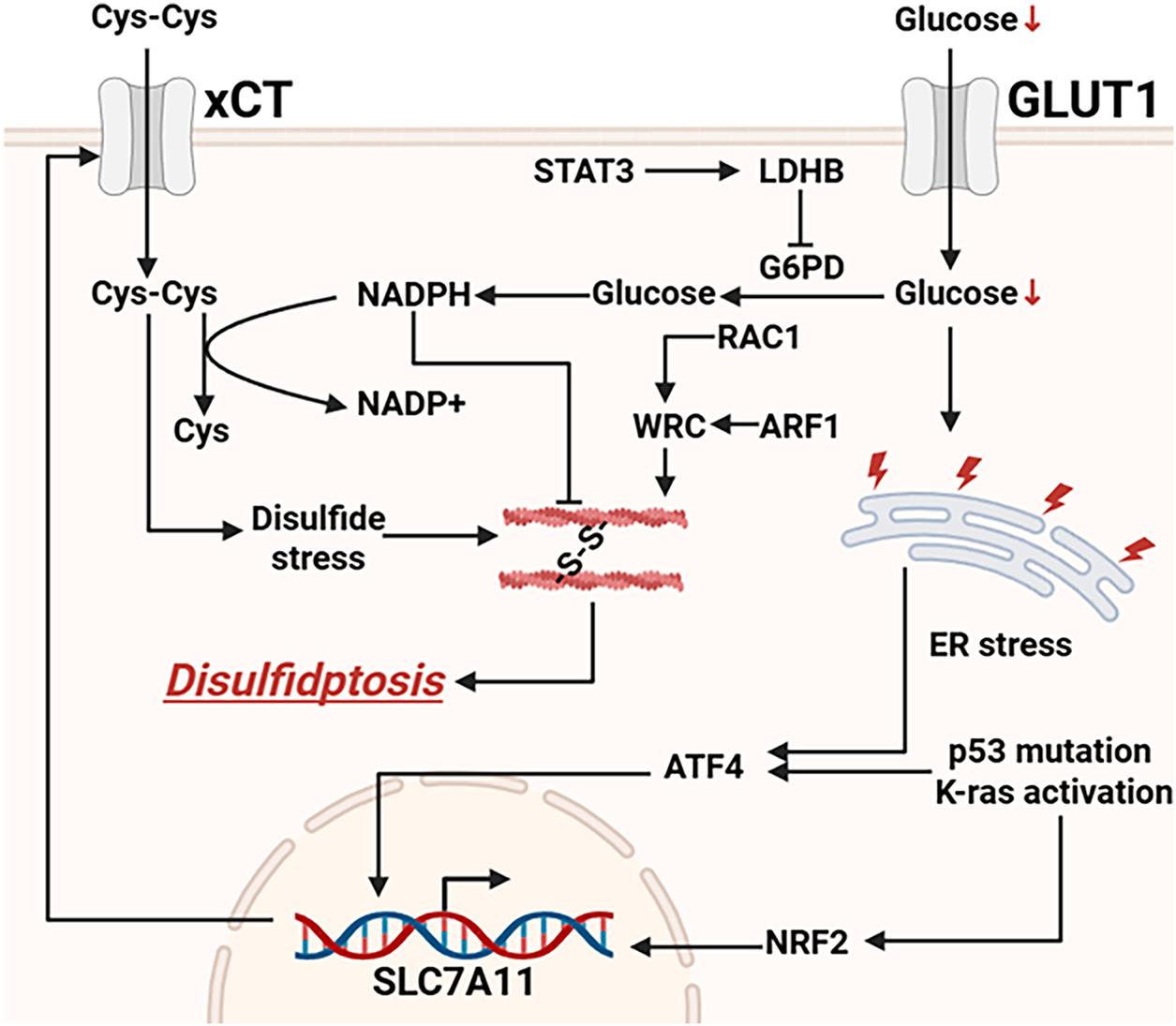
Figure 2: Currently known signaling pathways regulating disulfidptosis. LDHB inhibits NADPH production by suppressing G6PD. The transcription factors ATF4 and NRF2 can translocate into the nucleus to promote the transcription and expression of SLC7A11. Abb: Glucose Transporter 1 (GLUT1), Signal Transducer and Activator of Transcription 3 (STAT3), Lactate Dehydrogenase B (LDHB), Glucose-6-Phosphate Dehydrogenase (G6PD), Ras-related C3 botulinum toxin substrate 1 (RAC1), WAVE Regulatory Complex (WRC), ADP-Ribosylation Factor 1 (ARF1), Activating Transcription Factor 4 (ATF4), Nuclear factor erythroid 2–related factor 2 (NRF2), Solute Carrier Family 7 Member 11 (SLC7A11) (Created with BioRender.com).
The WRC is a multi-protein complex essential for actin polymerization and involved in the formation of lamellipodia. Studies have shown that inactivation of the WRC can suppress disulfidptosis [41,42]. This may be because WRC function is critical for maintaining normal actin dynamics, and its inactivation weakens the cell’s ability to cope with disulfide stress in the actin cytoskeleton. On the other hand, constitutive activation of the small GTPase Rac1 promotes disulfidptosis [43]. Rac1 is an upstream activator of the WRC [44]. It influences actin polymerization by regulating WRC activity. Therefore, activation of Rac1 may enhance disulfide bond formation within the actin network, making it more prone to collapse and thereby promoting disulfidptosis. Additionally, Arf1 acts as a WRC activator and can synergize with Rac1 to promote WRC-mediated disulfidptosis; however, the precise mechanism of this activation remains unclear, and thus this pathway is not discussed in detail in this review [45].
In summary, WRC and Rac1 modulate cellular sensitivity to disulfidptosis by regulating actin polymerization. Inactivation of WRC suppresses actin polymerization and thereby inhibits disulfidptosis, whereas activation of Rac1 promotes both actin polymerization and disulfidptosis. Targeting the Rac1-WRC signaling pathway may represent a novel strategy for modulating disulfidptosis in tumor cells.
3 Regulation and Roles of Disulfidptosis in Different Tumor Types
3.1 Pan-Cancer Analysis: The Universality and Specificity of Disulfidptosis-Related Genes
Pan-cancer analysis studies have generally revealed that disulfidptosis-related genes (DRGs) exhibit significant expression abnormalities across multiple tumor types [46,47]. These differences are not only evident between tumor tissues and normal tissues, but also exist among different tumor subtypes [48]. For example, analysis of 15 DRGs, including SLC7A11, INF2, and CD2AP, revealed significant differences in the expression levels of these genes across more than 9000 samples from over 30 cancer types [48]. Specifically, high expression of ACTB, ACTN4, and MYL6 in GBMLGG, LGG, MESO, and LAML is associated with poor prognosis, while high expression of INF2 in LIHC, LUAD, UVM, HNSC, GBM, LAML, and KIPAN is also linked to poor prognosis [49]. Moreover, as a key gene mediating disulfidptosis, SLC7A11 is significantly upregulated in 25 out of 27 tumor types in a pan-cancer analysis, highlighting the widespread activation of disulfidptosis [50]. Another pan-cancer study similarly reported broad upregulation of this gene across tumors [51]. Disulfidptosis has been more intensively studied across various tumors. Table 1 summarizes the mechanisms and pathways of disulfidptosis in different tumor types.
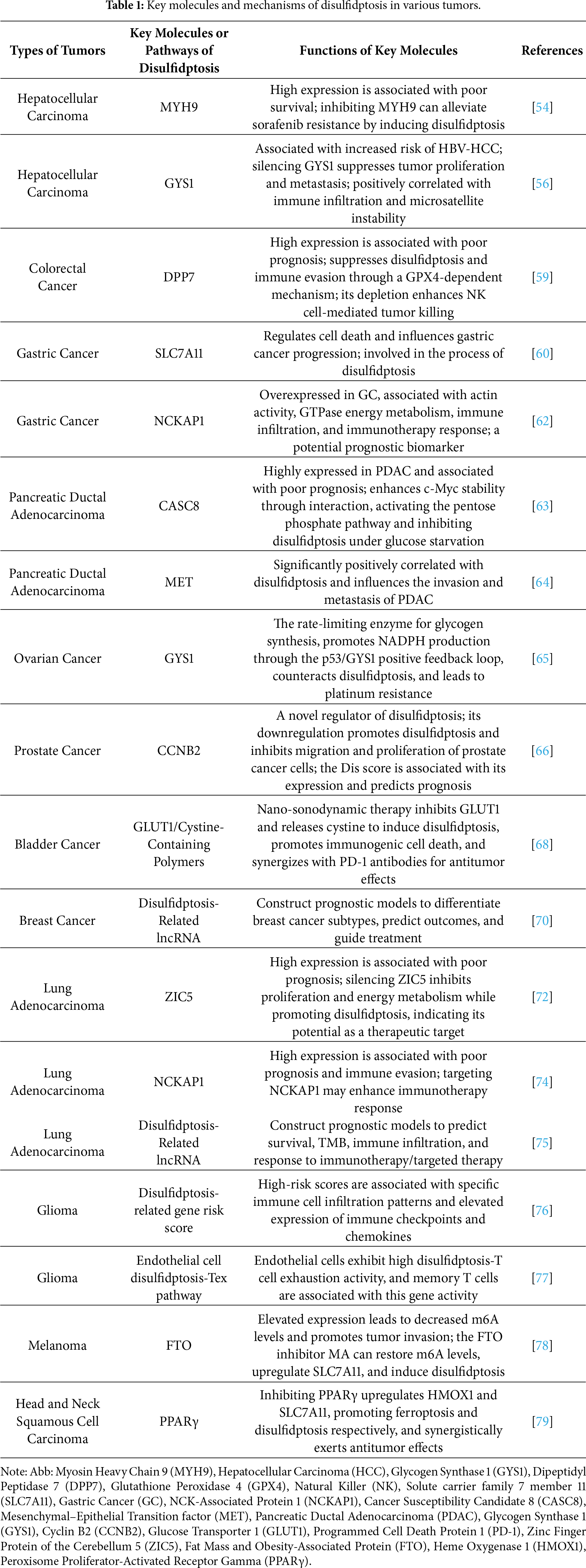
3.2 Disulfidptosis in Gastrointestinal Tumors
3.2.1 Hepatocellular Carcinoma (HCC)
In HCC, multi-omics analysis has been employed to explore the disulfidptosis landscape and establish a new model for predicting the prognosis of HCC patients [52]. Machine learning has also been used to identify disulfidptosis subtypes in HCC [53]. Based on the transcriptomic profiles of 31 disulfidptosis-related genes, HCC patients were stratified into two groups: C1, characterized by high disulfidptosis activity, and C2, characterized by low disulfidptosis activity. The C2 group, with lower disulfidptosis levels, generally exhibited better overall survival (OS) and progression-free survival (PFS), reduced infiltration of immunosuppressive cells, and activation of the glycine/serine/threonine metabolism pathway [53]. In mechanistic studies, research has found that high expression of disulfidptosis-related genes is associated with poor survival in liver cancer patients [54]. The MYH9 gene plays a key role, and inhibition of MYH9 can alleviate sorafenib resistance in HCC through disulfidptosis-like changes [54]. This provides a new strategy for enhancing tumor cell sensitivity to drugs. Additional evidence has revealed that the disulfidptosis-related gene RPN1 is markedly upregulated in both HCC tissues and peripheral blood mononuclear cells (PBMCs) from HCC patients, with its expression levels correlating positively with disease progression. Functional assays showed that RPN1 overexpression suppresses HCC cell proliferation, migration, and tumorigenicity in vivo, whereas RPN1 knockdown exerts the opposite effects. Mechanistically, RPN1 induces G0/G1 cell cycle arrest by modulating key cell cycle regulators—including CDK1, CDK2, Cyclin D1, and Cyclin E1—thereby impeding tumor progression [55]. These findings collectively suggest that RPN1 may serve as a potential therapeutic target and prognostic biomarker in HCC.
Regarding HBV-related hepatocellular carcinoma (HBV-HCC), studies have shown that increased genetic susceptibility to HBV increases the risk of developing HCC. Moreover, genetic variations in disulfidptosis are significantly associated with an increased risk of HBV-HCC [56]. GYS1 is considered a potential therapeutic target for HBV-HCC, showing a significant positive correlation with immune infiltration and microsatellite instability (MSI). Silencing GYS1 can effectively inhibit tumor proliferation and metastasis in HBV-HCC [56].
In the field of CRC, research on disulfidptosis primarily focuses on constructing prognostic models, evaluating the immune microenvironment, and exploring potential therapeutic targets. Studies generally suggest that disulfidptosis is closely associated with the prognosis of CRC patients [57,58], and it may act as a protective factor for CRC patients. Dipeptidyl peptidase 7 (DPP7) promotes colorectal cancer progression by inhibiting disulfidptosis and NK cell-mediated cytotoxicity in a GPX4-dependent manner. DPP7 overexpression reduces glucose deprivation-induced disulfide bond formation in cytoskeletal proteins (drebrin, FLNA, FLNB), protecting cells from disulfidptosis. Mechanistically, DPP7 physically interacts with and stabilizes GPX4 protein without affecting its mRNA. Restoring GPX4 in DPP7-depleted cells rescues resistance to disulfidptosis and immune killing, while GPX4 knockdown abolishes DPP7’s protective effect [59]. High expression of DPP7 is associated with poor prognosis in CRC patients, while the depletion of DPP7 can enhance the cytotoxicity of natural killer (NK) cells against tumor cells [59].
GC, as a common malignant tumor of the digestive system, is characterized by poor prognosis and high mortality. Studies have shown that SLC7A11 can regulate cell death and influence the progression of GC, suggesting that disulfidptosis may be involved in the pathological process of GC [60]. Studies have found that several disulfidptosis-related genes may be associated with the prognosis of GC, and a predictive model incorporating multiple genes has been constructed [61]. Clinical studies have shown that NCKAP1 and SLC7A11 are overexpressed in GC, and they are associated with actin activity, GTPase energy metabolism, immune infiltration, and immunotherapy [62]. These genes can serve as potential prognostic and diagnostic biomarkers for GC.
3.2.4 Pancreatic Ductal Adenocarcinoma (PDAC)
PDAC is a highly lethal malignancy. Studies have confirmed the presence of disulfidptosis in PDAC, and it has been found that the oncogene CASC8 plays a role in this process [63]. CASC8 is expressed at higher levels in PDAC tissues compared to normal pancreatic tissues, and high CASC8 expression is associated with poor prognosis in PDAC patients [63]. CASC8 interacts with c-Myc, enhancing the stability of the c-Myc protein and activating the pentose phosphate pathway, which lowers the NADP/NADPH ratio, thereby inhibiting disulfidptosis under glucose-starved conditions [63]. The study also constructed a prognosis-related signature associated with disulfidptosis, and found that the MET gene is significantly positively correlated with disulfidptosis and influences the invasion and metastasis of PAAD [64].
3.3 Disulfidptosis in Genitourinary Tumors
3.3.1 Disulfidptosis in Ovarian Cancer
Ovarian clear cell carcinoma exhibits unique metabolic characteristics, demonstrating higher glycogen levels and resistance to platinum-based drugs [65]. Research has found that the glycogen synthesis rate-limiting enzyme GYS1 is associated with poor prognosis and chemotherapy resistance in OCCC. It promotes the production of energetic NADPH through a p53/GYS1 positive feedback loop, thereby resisting disulfidptosis and enhancing OCCC’s resistance to platinum-based drugs [65].
3.3.2 Disulfidptosis in Prostate Cancer and Renal Cell Carcinoma
In the field of prostate cancer, CCNB2 has been identified as a novel regulator of disulfidptosis. A molecular subtyping framework based on disulfidptosis was constructed to classify prostate cancer into distinct subtypes, and the Dis score was used to assess the severity of each patient subtype. The results indicate that the Dis score is significantly associated with PCa prognosis, with higher Dis scores correlating with lower tumor mutational burden and improved outcomes [66]. Experimental results confirmed that downregulation of CCNB2 expression promotes disulfidptosis in prostate cancer cells, thereby inhibiting their migration and proliferation capabilities [66]. In clear cell renal cell carcinoma (ccRCC), a recent study similarly established a novel classification system—termed the Disulfidptosis-based Classifier System (DCS)—based on disulfidptosis-related genes, stratifying ccRCC into four distinct molecular subtypes. Among these, the DCS3 subtype exhibits hallmark features of suppressed disulfidptosis, insensitivity to immunotherapy, pronounced genomic instability, and poor clinical outcomes. Notably, the study further demonstrated that NU1025 significantly inhibits the malignant progression of DCS3-subtype tumors, thereby offering a promising strategy for precision therapy in ccRCC [67].
3.3.3 Disulfidptosis in Bladder Cancer
In bladder cancer, the combination of nanotechnology and sonodynamic therapy, utilizing GLUT1 inhibitors and cystine-containing polymers to induce disulfidptosis, has demonstrated improved efficacy in immunotherapy [68]. This approach effectively releases Bay-876, disrupts intracellular redox homeostasis, releases cystine to induce disulfidptosis, promotes immunogenic cell death, and synergizes with PD-1 monoclonal antibodies to suppress tumor growth [68].
3.4 Disulfidptosis in Breast Cancer and Lung Cancer.
3.4.1 Disulfidptosis in Breast Cancer
Breast cancer is a highly heterogeneous disease, classified into molecular subtypes such as Luminal A, Luminal B, HER2-positive, and Basal-like. Emerging research indicates that disulfidptosis plays a complex and critical role in the development and progression of breast cancer [69].
Long non-coding RNAs play a crucial role in gene expression regulation. Some studies have attempted to construct predictive models based on disulfidptosis-related lncRNAs to differentiate between molecular subtypes of breast cancer [70]. These models can help predict breast cancer prognosis and provide guidance for clinical treatment.
3.4.2 Disulfidptosis in Lung Cancer
Lung cancer is one of the leading causes of cancer-related deaths worldwide, with lung adenocarcinoma being the most common subtype. Growing evidence suggests that disulfidptosis plays a significant role in the development and progression of lung adenocarcinoma [71]. ZIC5, a member of the Zic family of transcription factors, is highly expressed in lung adenocarcinoma cells and is associated with poor patient prognosis [72]. Silencing ZIC5 suppresses proliferation and energy metabolism in lung adenocarcinoma cells while promoting disulfidptosis [72]. This suggests that targeting ZIC5 may represent a novel therapeutic strategy for lung adenocarcinoma. NCKAP1, a disulfidptosis-related gene, shows significant differential expression across various cancers [73]. In lung adenocarcinoma, high expression of NCKAP1 is associated with poor prognosis and immune evasion mechanisms [74]. Targeting NCKAP1 may enhance the response of lung adenocarcinoma patients to immunotherapy. Similar to breast cancer, researchers have also developed prognostic models for lung adenocarcinoma based on disulfidptosis-related lncRNAs [75]. These models can not only predict patient survival but also forecast tumor mutational burden, immune cell infiltration, and responses to immunotherapy and targeted therapy.
3.5 Disulfidptosis in Other Tumors
Disulfidptosis may influence the immune microenvironment in glioma. Studies have found that glioma patients with high-risk scores exhibit distinct immune cell infiltration patterns, and most immune checkpoints and chemokines show a positive correlation with risk scores [76]. Furthermore, studies have revealed elevated disulfidptosis-T cell exhaustion activity in glioma endothelial cells, and memory T cell populations are associated with these genes [77].
Melanoma is a malignant tumor originating from melanocytes, with uveal melanoma being the most common primary intraocular malignancy in adults and associated with an extremely poor prognosis. Research indicates that N6-methyladenosine modification plays a critical role in the development and progression of melanoma, while FTO acts as an m6A demethylase. Elevated FTO expression in melanoma tissues is correlated with reduced m6A levels, increased tumor aggressiveness, and poor prognosis [78]. The FTO inhibitor meclofenamic acid (MA) has been shown to restore m6A RNA methylation levels in uveal melanoma cells, leading to upregulation of SLC7A11 and triggering disulfidptosis, a form of cell death driven by glutathione depletion and NADPH consumption. In vitro and in vivo experiments demonstrated that MA treatment effectively induced disulfidptosis and suppressed tumor growth, supporting FTO inhibition as a potential therapeutic strategy [78].
In head and neck squamous cell carcinoma, studies indicate that PPARγ antagonists exert antitumor effects by modulating ferroptosis and disulfidptosis. In oral squamous cell carcinoma, PPARγ inhibition leads to upregulation of heme oxygenase 1, promoting ferroptosis, while also upregulating SLC7A11 to induce disulfidptosis [79].
3.6 Therapeutic Implications across Different Tumor Types
Based on current research, bladder cancer and ovarian clear cell carcinoma are the tumor types most likely to benefit from interventions targeting disulfidptosis. In bladder cancer, studies have successfully combined nanotechnology with sonodynamic therapy to deliver GLUT1 inhibitors, thereby directly creating the metabolic conditions that trigger disulfidptosis—such as glucose deprivation. This approach has been effectively integrated with immunotherapy (e.g., PD-1 antibodies), establishing a well-defined therapeutic paradigm [68]. Ovarian clear cell carcinoma (OCCC) represents an ideal therapeutic target for disulfidptosis-based strategies due to its distinctive dysregulation of glycogen metabolism. The key enzyme glycogen synthase 1 (GYS1) drives a positive feedback loop that enhances NADPH production, thereby conferring resistance to disulfidptosis. Targeting GYS1 directly disrupts this protective mechanism and overcomes platinum resistance in OCCC [65]. Moreover, lung adenocarcinoma (via targeting of ZIC5 or NCKAP1) and melanoma (through FTO inhibitors that upregulate SLC7A11) also demonstrate clear therapeutic potential. These targets have been shown to directly regulate disulfidptosis and are associated with poor prognosis, thereby offering well-defined avenues for drug development [72,74,78].
4 Associations between Disulfidptosis and Other Forms of Cell Death
Cell death is a crucial process for maintaining homeostasis in multicellular organisms [80]. Historically, apoptosis was considered the primary programmed cell death pathway, but research in recent decades has shown that various regulated non-apoptotic cell death forms also play significant roles in disease development [81]. Disulfidptosis, as a newly discovered form of programmed cell death, is distinctly different from other known cell death pathways such as apoptosis, ferroptosis, cuproptosis, and pyroptosis [82–85]. This section will focus on comparing the similarities and differences between disulfidptosis and other programmed cell death pathways, highlighting its unique metabolic dependencies and cytoskeletal collapse characteristics, and exploring its potential implications in tumors (Table 2, Fig. 3).
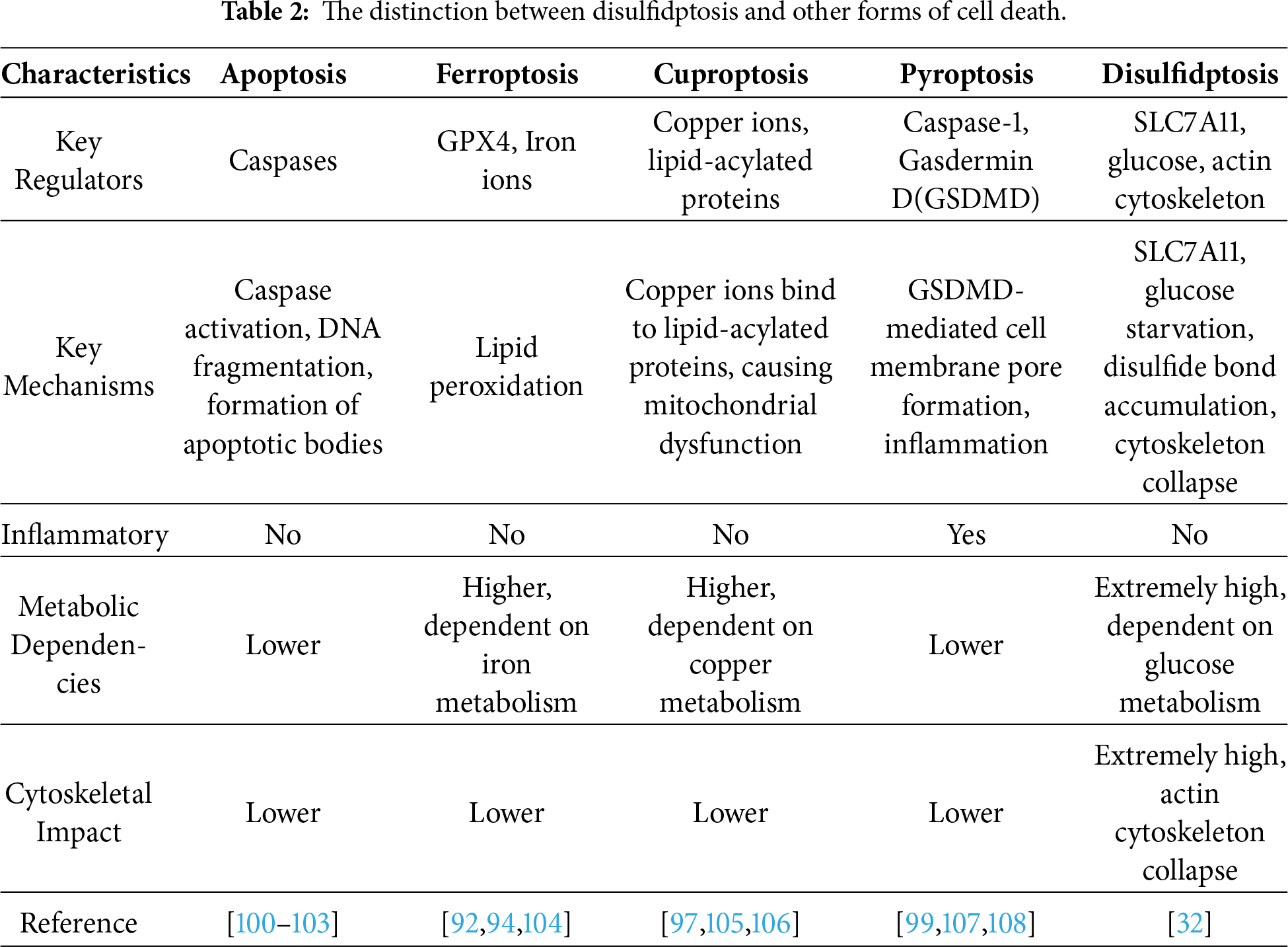
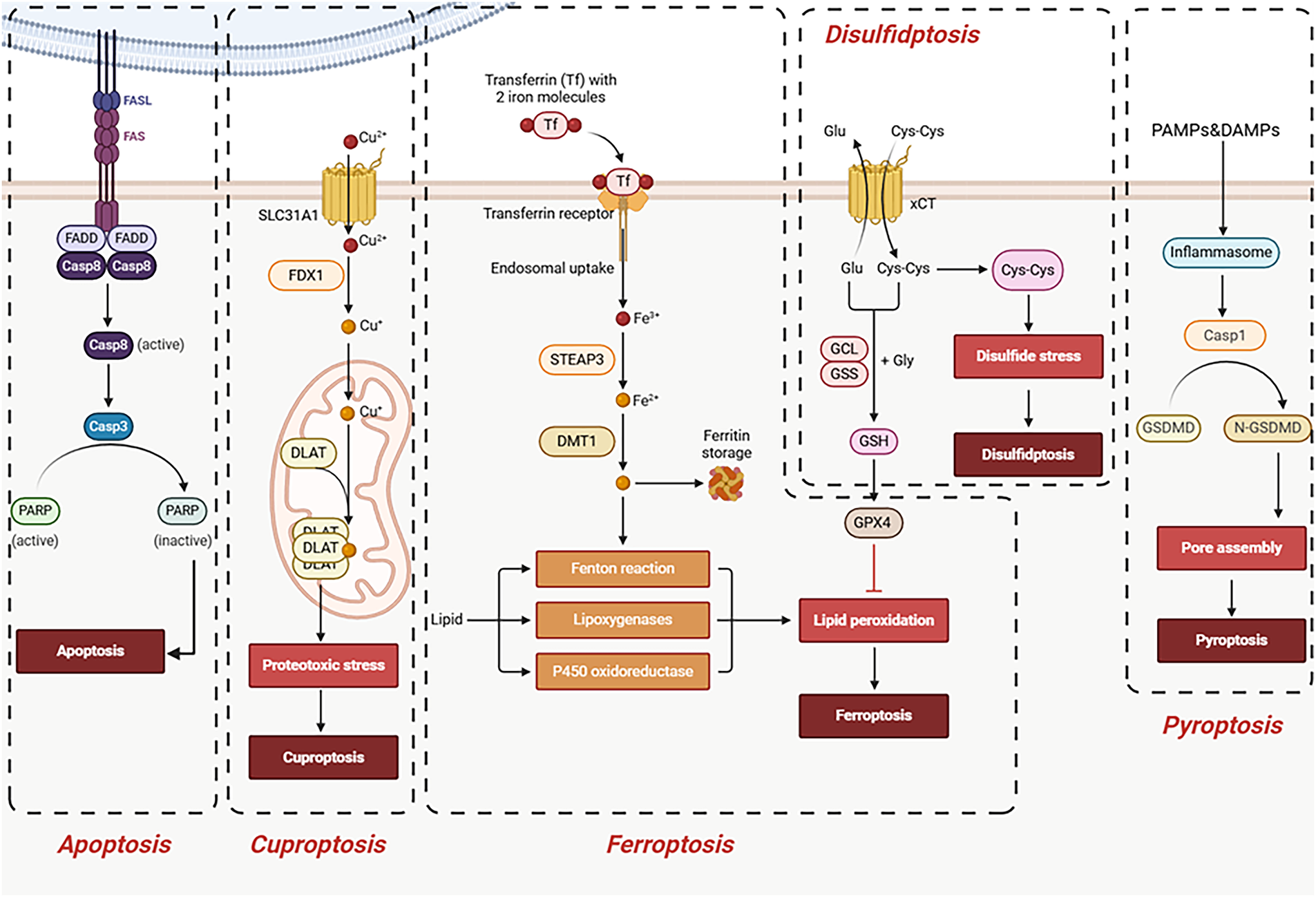
Figure 3: Associations between disulfidptosis and other forms of cell death. Unlike apoptosis, ferroptosis, cuproptosis, or pyroptosis, disulfidptosis is triggered by glucose starvation in SLC7A11-high cells, leading to disulfide stress and actin cytoskeleton collapse, independent of caspases, iron, copper, or inflammation. Abb: Fas-Associated Protein with Death Domain (FADD), Poly (ADP-Ribose) Polymerase (PARP), Ferredoxin (FDX), Dihydrolipoamide S-Acetyltransferase (DLAT), Six-Transmembrane Epithelial Antigen of the Prostate 3 (STEAP3), Divalent Metal Transporter 1 (DMT1), Glutamate–Cysteine Ligase (GCL), Glutathione Synthetase (GSS), Glutathione (GSH), Caspase 1 (Casp1) (Created with BioRender.com).
Apoptosis is divided into intrinsic apoptosis and extrinsic apoptosis [86–88]. The intrinsic apoptotic pathway is typically triggered by intracellular stresses such as DNA damage, oxidative stress, or growth factor deprivation. This initially activates stress-sensing molecules like p53, which upregulates pro-apoptotic Bcl-2 family members such as Bax, Puma, and Noxa. These proteins promote the oligomerization of Bax/Bak in the outer mitochondrial membrane, forming transmembrane pores that lead to mitochondrial outer membrane permeabilization (MOMP) [89]. This process is accompanied by the release of pro-apoptotic factors such as cytochrome c and Smac/DIABLO into the cytosol, which alleviates the inhibitory effect of IAPs (Inhibitor of Apoptosis Proteins) on caspases [90]. Cytochrome c, together with Apaf-1 and the procaspase-9 precursor, assembles into a heptameric complex known as the “apoptosome,” which triggers the autocatalytic cleavage and activation of caspase-9 [90]. Activated caspase-9 subsequently initiates a cascade of effector caspases—caspase−3, −6, and −7—which cleave substrates such as PARP, gelsolin, and nuclear structural proteins, ultimately executing hallmark apoptotic events including cytoplasmic shrinkage, nuclear fragmentation, and internucleosomal DNA cleavage.The extrinsic pathway is triggered when ligands such as FasL or TNF-α bind to their respective death receptors, inducing receptor trimerization and recruitment of adaptor proteins FADD and TRADD along with procaspase-8 to form the Death-Inducing Signaling Complex (DISC) [91]. At the mechanistic level, disulfidptosis is a form of regulated cell death triggered by disulfide bond stress. Its core mechanism involves SLC7A11 overexpression combined with glucose restriction, which leads to NADPH depletion. This impairs the reduction of intracellular cystine, resulting in excessive accumulation of disulfide bonds. These aberrant disulfide bonds cause non-physiological crosslinking of cytoskeletal proteins such as actin, ultimately leading to collapse of the F-actin network and cell death. This process is further exacerbated through actin-remodeling pathways—such as the Rac–WRC–Arp2/3 axis—which promote additional disulfide bond formation [32]. Therefore, the execution mechanism of disulfidptosis is closely linked to the classic paradigm of “protein/cytoskeletal crosslinking + cellular structural disruption,” rather than relying on traditional pathways such as apoptosis, lipid peroxidation, or inflammatory membrane pore formation.
The core of ferroptosis lies in the uncontrolled accumulation of lipid peroxidation and the susceptibility of cellular membranes enriched with polyunsaturated fatty acids (PUFAs) [92]. Under normal conditions, GPX4 uses GSH as an electron donor to reduce membrane lipid hydroperoxides to their corresponding alcohols, thereby terminating the lipid peroxidation chain reaction. When GPX4 is inhibited or GSH is depleted, the cell loses its ability to reverse oxidative damage [93]. Meanwhile, ferrous iron (Fe²+) continuously generates hydroxyl radicals via the Fenton reaction, which initiates hydrogen abstraction from polyunsaturated fatty acid-containing phosphatidylethanolamines, thereby further driving the chain reaction of lipid peroxidation [10]. Lipid metabolism–related enzymes ACSL4 and LPCAT3 enhance cellular sensitivity to ferroptosis by selectively esterifying arachidonic acid and adrenic acid into membrane phospholipids [94]. Accumulation of peroxidized phospholipids disrupts the lipid-phase stability of cellular membranes, leading to impaired membrane curvature and abnormal membrane permeability. Characteristic ultrastructural features of ferroptosis include mitochondrial shrinkage, increased mitochondrial membrane density, and reduction or complete loss of cristae, while the nucleus and cytoskeleton remain largely intact—highlighting its distinct molecular and morphological differences from apoptosis, necrosis, and other forms of cell death [95]. Although both cuproptosis and disulfidptosis may involve disruption of cellular architecture or protein homeostasis, they differ fundamentally in their mechanisms and targets. Cuproptosis centers on the toxic aggregation of mitochondrial metabolic enzymes and Fe–S cluster proteins, leading to metabolic collapse, whereas disulfidptosis primarily involves disulfide-mediated cross-linking of cytoskeletal proteins and consequent physical disintegration of cellular structure. Consequently, these two forms of regulated cell death exhibit fundamental differences in their molecular targets, execution mechanisms, and physiological triggers [96].
Cuproptosis is a novel form of metabolism-associated regulated cell death, distinct from apoptosis, necrosis, and ferroptosis [97]. Its key event involves the intracellular accumulation of free copper ions, which bind with high affinity to lipoylated proteins in mitochondria—such as dihydrolipoamide S-acetyltransferase (DLAT) and dihydrolipoamide S-succinyltransferase (DLST)—triggering their aberrant aggregation and subsequent disruption of proteostasis [98]. This process inhibits key mitochondrial TCA cycle enzyme complexes—such as pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase (KGDH)—leading to a sharp decline in energy metabolism. Concurrently, copper ions promote the destabilization of iron–sulfur (Fe–S) cluster-containing proteins, triggering oxidative stress and proteotoxic stress, which collectively impair the cell’s ability to maintain a reducing environment necessary for proper protein folding and homeostasis [98]. Together, these effects trigger a distinct form of cell death that depends on mitochondrial lipoylated proteins—fundamentally different from disulfidptosis, which is driven by NADPH depletion and the ensuing “disulfide storm.” In cuproptosis, copper exerts direct intrinsic toxicity by binding to and aggregating lipoylated proteins, whereas in disulfidptosis, loss of reducing capacity (due to NADPH exhaustion) leads to pathological accumulation of disulfide bonds [14].
Pyroptosis is an inflammatory form of programmed cell death, typically mediated by inflammasome-activated caspase-1, leading to cleavage of the GSDMD protein and the formation of cell membrane pores [99]. Pyroptosis is typically accompanied by the massive release of inflammatory factors. Disulfidptosis does not depend on inflammasome activation or GSDMD cleavage and is not an inflammatory form of cell death.
5 Strategies for Targeting Disulfidptosis in Tumor Therapy
Disulfidptosis, a recently discovered form of programmed cell death, has rapidly emerged as a research hotspot in tumor therapy due to its unique molecular mechanisms and selective sensitivity in specific tumor cells, offering potential novel intervention pathways for cancer treatment (Table 3, Fig. 4). Current research primarily focuses on inducing disulfidptosis specifically in tumor cells through various strategies to enhance therapeutic efficacy and overcome drug resistance [59,109–111].
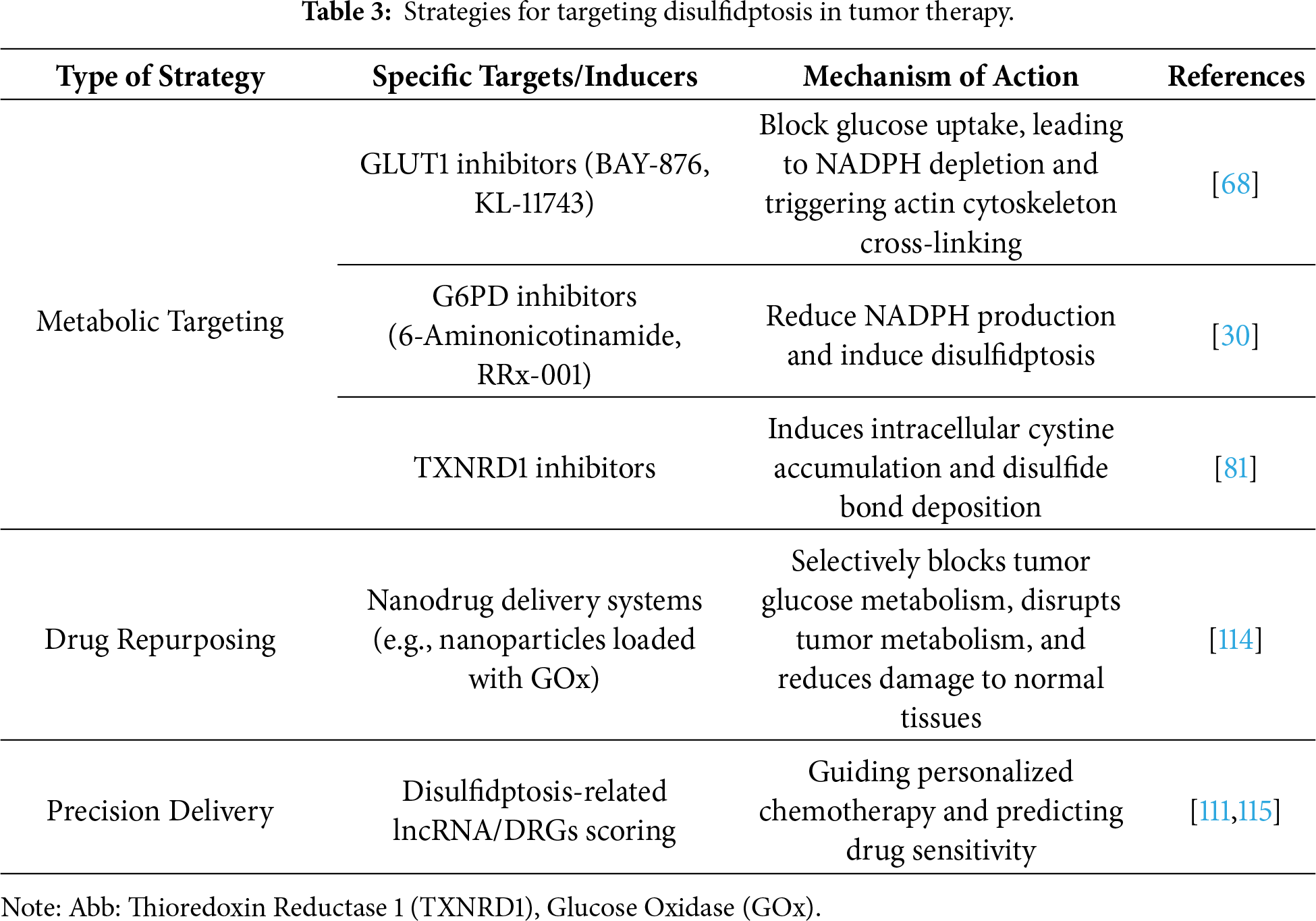
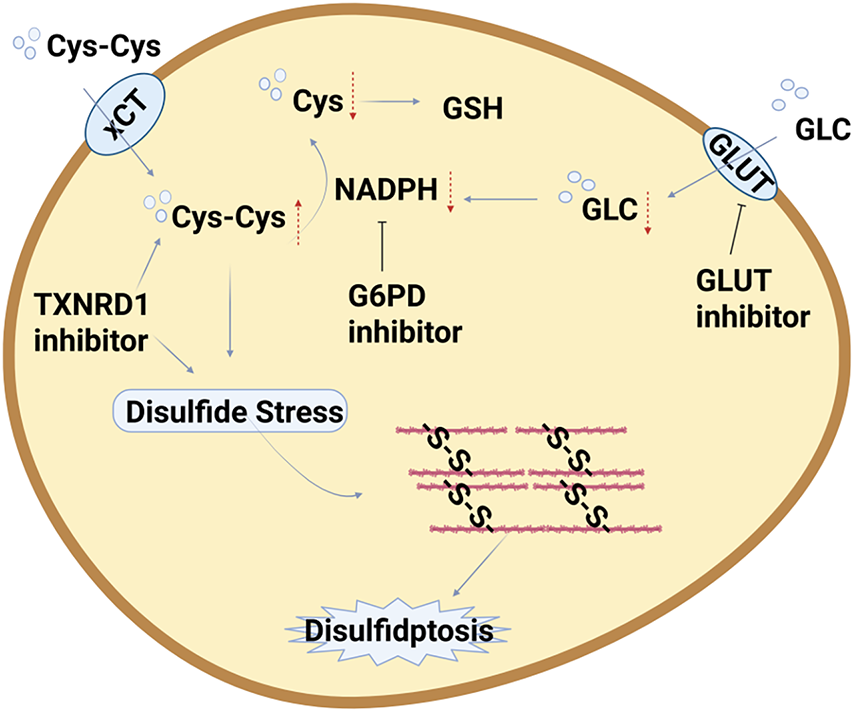
Figure 4: Strategies for Targeting the Disulfidptosis Pathway in Tumor Therapy. Abb: Thioredoxin Reductase 1(TXNRD1) (Created with BioRender.com).
5.1 Metabolic Pathway Targeting Strategies
The occurrence of disulfidptosis is highly dependent on intracellular NADPH levels and abnormal disulfide bond accumulation. Therefore, disrupting key metabolic pathways is a core strategy for inducing this form of cell death.
GLUT1 inhibitors (e.g., BAY-876, KL-11743): By blocking glucose uptake, they deplete NADPH in SLC7A11-high cancer cells, leading to abnormal disulfide cross-linking of actin cytoskeletal proteins and ultimately triggering disulfidptosis. Both in vitro and in vivo mouse studies have confirmed that these inhibitors significantly suppress the growth of SLC7A11-high tumors while exhibiting low toxicity to normal tissues [68].
G6PD inhibitors (e.g., 6-Aminonicotinamide, RRx-001): By suppressing the activity of G6PD, a key enzyme in the pentose phosphate pathway, they reduce NADPH production and thereby induce disulfidptosis, demonstrating potential therapeutic value in various cancer models [30].
TXNRD1 inhibitors: Target thioredoxin reductase 1, disrupt cellular redox homeostasis, cause excessive cystine accumulation and disulfide deposition, selectively kill cancer cells, and offer a novel drug target for clinical applications [81].
5.2 Drug Repurposing and Delivery System Optimization
To enhance targeting and safety, researchers are exploring the modification of existing drugs or biologics using advanced delivery systems. Nanodrug delivery systems (e.g., nanoparticles loaded with glucose oxidase GOx) can selectively block glucose metabolism in tumor cells, mimicking glucose deprivation, inducing NADPH depletion and disulfidptosis. Nanomedicine-based delivery systems play a pivotal role in inducing disulfidptosis in tumor cells through precise design and multifunctional integration. Their central function lies in actively targeting the essential prerequisites of disulfidptosis. For instance, selective inhibition of SLC7A11 can efficiently block cystine uptake, thereby depleting glutathione synthesis at its source and inducing a cellular redox crisis. Alternatively, the delivery of glucose oxidase can directly catalyze glucose consumption within tumors, creating a “starvation” microenvironment. Moreover, intelligent nanocarriers can encapsulate specific prodrugs that, in response to the tumor microenvironment—such as high intracellular glutathione levels—consume large amounts of NADPH and cysteine, ultimately triggering lethal protein disulfide crosslinking [112]. These strategies can be applied individually or combined with other modalities such as photodynamic or photothermal therapy, simultaneously disrupting the cytoskeleton and activating immune responses, thereby offering a powerful new approach to overcome tumor drug resistance [113]. This strategy not only effectively disrupts tumor metabolism but also reduces damage to normal tissues, improving the therapeutic window [114].
5.3 Precision Medicine and Personalized Therapy
Combining biomarkers to guide treatment decisions can help achieve precise induction of disulfidptosis. Scoring models based on disulfidptosis-related lncRNAs or disulfidptosis-related genes can be used to predict patient sensitivity to specific targeted drugs. For instance, ACTN4 expression levels may influence the efficacy of BRAF inhibitors, while MYL6 is associated with responses to MEK inhibitors, suggesting that such molecules could serve as potential biomarkers for guiding personalized chemotherapy [111,115].
Disulfidptosis, as a newly defined mode of programmed cell death, not only expands our understanding of the cell death spectrum but also offers a highly promising metabolic intervention target for tumor therapy. Although current research has preliminarily uncovered its core molecular mechanism—driven by SLC7A11-dependent cystine uptake and NADPH depletion, leading to disulfide stress and actin cytoskeleton collapse—the field remains in its early stages, with many key scientific questions awaiting further exploration.
First, a specific biomarker system for disulfidptosis has yet to be established. Current screening criteria primarily rely on high SLC7A11 expression and glucose deprivation sensitivity, but the lack of dynamic, detectable molecular markers (such as specific protein disulfide modification profiles or redox state imaging probes) limits its precise identification and efficacy evaluation in clinical samples. Future efforts should integrate proteomics, redox proteomics, and single-cell sequencing technologies to systematically identify the unique molecular fingerprints of disulfidptosis and construct predictive models for patient stratification.
Second, the complexity of the regulatory network governing disulfidptosis is far from fully understood. Existing studies suggest the involvement of various factors such as LDHB, the WRC complex, the thioredoxin system, and endoplasmic reticulum stress, but the crosstalk mechanisms, spatiotemporal dynamics, and heterogeneity across different tumor types remain to be elucidated. Particularly critical is understanding how nutrient fluctuations, oxidative stress gradients, and immune cell interactions in the tumor microenvironment influence the threshold of disulfidptosis.
Moreover, therapeutic strategies targeting disulfidptosis face dual challenges of selectivity and resistance. Although GLUT inhibitors, G6PD inhibitors, and TXNRD1 antagonists have demonstrated antitumor activity in preclinical models, achieving tumor-specific induction while avoiding toxicity to normal tissues (especially high-metabolism organs) remains a central challenge in translational medicine. Nanodelivery systems, antibody-drug conjugates (ADCs), or conditionally activated prodrugs may offer technical solutions to this issue. Additionally, tumor cells may evade disulfidptosis by upregulating antioxidant pathways (e.g., NRF2, Trx system) or remodeling metabolic networks (e.g., enhancing glutamine metabolism), necessitating systematic research into resistance mechanisms to guide combination therapy strategies.
Finally, the synergistic effects between disulfidptosis and other forms of programmed cell death warrant in-depth investigation. Existing evidence suggests that systemic glucose deprivation can simultaneously induce disulfidptosis and ferroptosis, while nanodrugs can co-activate pyroptosis and disulfidptosis. Future research should systematically explore the synergistic mechanisms among multiple forms of cell death. For example, rational combination therapies could be designed to co-administer disulfidptosis inducers (such as GLUT1 inhibitors) with ferroptosis activators (such as GPX4 inhibitors), simultaneously targeting cancer cells from two metabolic dimensions to effectively prevent escape. In terms of clinical translation, the following priorities should be addressed: (1) Biomarker validation: prospectively validate SLC7A11 protein or mRNA levels as predictive biomarkers for patient stratification in early-phase clinical trials; (2) Drug delivery optimization: develop and refine stimulus-responsive nanoplatforms to achieve tumor-specific induction of disulfidptosis while minimizing systemic toxicity; (3) Combination therapy trial design: based on robust preclinical evidence, design clinical trial protocols combining disulfidptosis inducers with standard chemotherapy, targeted therapy, or immunotherapy.
In summary, disulfidptosis not only represents a novel paradigm of cell death but also reveals the fragile balance between metabolic dependency and structural stability in cancer cells. As mechanistic research deepens and technological advancements continue, targeting disulfidptosis is expected to become another key strategy in precision oncology, following apoptosis and ferroptosis, offering new breakthroughs in overcoming drug-resistant cancers.
Acknowledgement: Not applicable.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Wubin Zhao, Jun Zhang; data collection: Qi Wang; analysis and interpretation of results: Wubin Zhao, Jun Zhang; draft manuscript preparation: Wubin Zhao, Qi Wang. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Ethics Approval: Not applicable for studies not involving humans or animals.
Conflicts of Interest: The authors declare no conflicts of interest.
Abbreviations
| ARE | Antioxidant Response Element |
| ATF4 | Activating Transcription Factor 4 |
| CASC8 | Cancer Susceptibility Candidate 8 |
| CCNB2 | Cyclin B2 |
| CRC | Colorectal Cancer |
| Dis score | Disulfidptosis Score |
| DPP7 | Dipeptidyl Peptidase 7 |
| F-actin | Filamentous Actin |
| FTO | Fat Mass and Obesity-associated protein |
| G6PD | Glucose-6-Phosphate Dehydrogenase |
| GBM | Glioblastoma Multiforme |
| GBMLGG | Glioblastoma and Lower Grade Glioma |
| GC | Gastric Cancer |
| GLUT1 | Glucose Transporter 1 |
| GPX4 | Glutathione Peroxidase 4 |
| GSH | Glutathione |
| GYS1 | Glycogen Synthase 1 |
| HBV-HCC | Hepatitis B Virus-related Hepatocellular Carcinoma |
| HCC | Hepatocellular Carcinoma |
| HMOX1 | Heme Oxygenase 1 |
| HNSC | Head and Neck Squamous Cell Carcinoma |
| KIPAN | Kidney Pan-cancer (Kidney Renal Clear Cell Carcinoma, Kidney Papillary Cell Carcinoma, and Kidney Chromophobe) |
| LAML | Acute Myeloid Leukemia |
| LDHB | Lactate Dehydrogenase B |
| LGG | Lower Grade Glioma |
| LIHC | Liver Hepatocellular Carcinoma |
| lncRNA | Long Non-coding RNA |
| LUAD | Lung Adenocarcinoma |
| m6A | N6-methyladenosine |
| MA | Meclofenamic Acid |
| MESO | Mesothelioma |
| MSI | Microsatellite Instability |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate (reduced form) |
| NRF2 | Nuclear Factor Erythroid 2-Related Factor 2 |
| OCCC | Ovarian Clear Cell Carcinoma |
| PAAD | Pancreatic Adenocarcinoma |
| PCa | Prostate Cancer |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| PPARγ | Peroxisome Proliferator-Activated Receptor Gamma |
| PPP | Pentose Phosphate Pathway |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| RCD | Regulated Cell Death |
| ROS | Reactive Oxygen Species |
| SLC7A11 | Solute Carrier Family 7 Member 11 |
| Tex | T cell Exhaustion |
| TMB | Tumor Mutational Burden |
| UVM | Uveal Melanoma |
| WRC | WAVE Regulatory Complex |
References
1. Sorokin O, Hause F, Wedler A, Alakhras T, Bauchspiess T, Dietrich A, et al. Comprehensive analysis of regulated cell death pathways: intrinsic disorder, protein-protein interactions, and cross-pathway communication. Apoptosis. 2025;30(9):2110–62. doi:10.1007/s10495-025-02161-6. [Google Scholar] [PubMed] [CrossRef]
2. Baldassarre G, de la Serna IL, Vallette FM. Death-ision: the link between cellular resilience and cancer resistance to treatments. Mol Cancer. 2025;24(1):144. doi:10.1186/s12943-025-02339-1. [Google Scholar] [PubMed] [CrossRef]
3. Liu X, Nie L, Zhang Y, Yan Y, Wang C, Colic M, et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol. 2023;25(3):404–14. doi:10.1038/s41556-023-01091-2. [Google Scholar] [PubMed] [CrossRef]
4. Chen Q, Wang J, Li NA, Cai H, Lu Y. Crosstalk between ROS and ferroptosis, cuproptosis, and PANoptosis in liver cancer: mechanisms and therapeutic strategies. Crit Rev Oncol Hematol. 2025;215(10):104924. doi:10.1016/j.critrevonc.2025.104924. [Google Scholar] [PubMed] [CrossRef]
5. Ai Y, Meng Y, Yan B, Zhou Q, Wang X. The biochemical pathways of apoptotic, necroptotic, pyroptotic, and ferroptotic cell death. Mol Cell. 2024;84(1):170–9. doi:10.1016/j.molcel.2023.11.040. [Google Scholar] [PubMed] [CrossRef]
6. Chen Y, Lin X, Qiu J, Sun Y, Wu B, Shang H, et al. Ultrasound-responsive nanobubble-mediated sonodynamic therapy sensitizes disulfidptosis in the treatment of liver hepatocellular carcinoma. Ultrason Sonochem. 2025;118:107368. doi:10.1016/j.ultsonch.2025.107368. [Google Scholar] [PubMed] [CrossRef]
7. Wan J, Shi JH, Shi M, Huang H, Zhang Z, Li W, et al. Lactate dehydrogenase B facilitates disulfidptosis and exhaustion of tumour-infiltrating CD8+ T cells. Nat Cell Biol. 2025;27(6):972–82. doi:10.1038/s41556-025-01673-2. [Google Scholar] [PubMed] [CrossRef]
8. Zhao J, Chen B, Deng Y, Fan L, Yin S, Yu H, et al. ChREBP-mediated choline deprivation and chemokine secretion shape tumor-associated macrophages to promote immune evasion. Cancer Res. 2025;85(23):4701–17. doi:10.1158/0008-5472.can-25-0235. [Google Scholar] [PubMed] [CrossRef]
9. Fujiwara N, Nakagawa H, Enooku K, Kudo Y, Hayata Y, Nakatsuka T, et al. CPT2 downregulation adapts HCC to lipid-rich environment and promotes carcinogenesis via acylcarnitine accumulation in obesity. Gut. 2018;67(8):1493–504. doi:10.1136/gutjnl-2017-315193. [Google Scholar] [PubMed] [CrossRef]
10. Chen M, Li W, Tao Y, Hu C, Ge R, Kang S, et al. SLC25A1 reprograms mitochondrial and fatty acid metabolism to promote the progression of acute myeloid leukemia. Haematologica. 2025;144:2733. doi:10.3324/haematol.2024.287269. [Google Scholar] [PubMed] [CrossRef]
11. Li B, Hu H, Li S, Wang X, Ma Z, Zhao W, et al. Metabolic inhibitor loaded metal-phenolic nanocapsules enable synergistic cancer therapy through cuproptosis and immunometabolic reprogramming. ACS Appl Mater Interfaces. 2025;17(37):51659–70. doi:10.1021/acsami.5c10711. [Google Scholar] [PubMed] [CrossRef]
12. El-Kenawi A, Berglund A, Estrella V, Zhang Y, Liu M, Putney RM, et al. Elevated methionine flux drives pyroptosis evasion in persister cancer cells. Cancer Res. 2023;83(5):720–34. doi:10.1158/0008-5472.can-22-1002. [Google Scholar] [PubMed] [CrossRef]
13. Ye Q, Zhuang XZ, Li J, Zhou X. Targeting the inhibitors of apoptosis proteins (IAPs) to combat drug resistance in cancers. Front Pharmacol. 2025;16:1562167. doi:10.3389/fphar.2025.1562167. [Google Scholar] [PubMed] [CrossRef]
14. Liu X, Zhuang L, Gan B. Disulfidptosis: disulfide stress–induced cell death. Trends Cell Biol. 2024;34(4):327–37. doi:10.1016/j.tcb.2023.07.009. [Google Scholar] [PubMed] [CrossRef]
15. Gu Q, An Y, Xu M, Huang X, Chen X, Li X, et al. Disulfidptosis, a novel cell death pathway: molecular landscape and therapeutic implications. Aging Dis. 2024;16(2):917–45. doi:10.14336/AD.2024.0083. [Google Scholar] [PubMed] [CrossRef]
16. Shao J, Xiao R, Wang T, Wang F, Wang B, Zhang D, et al. BRD4770 protects against DOX-induced cardiotoxicity by inhibiting apoptosis and ferroptosis. Sci Adv. 2025;11(28):eadw1720. doi:10.1126/sciadv.adw1720. [Google Scholar] [PubMed] [CrossRef]
17. Wang M, Li B, Meng W, Chen Y, Liu H, Zhang Z, et al. System Xc– exacerbates metabolic stress under glucose depletion in oral squamous cell carcinoma. Oral Dis. 2024;30(5):2952–64. doi:10.1111/odi.14774. [Google Scholar] [PubMed] [CrossRef]
18. Xiong C, Ling H, Huang Y, Dong H, Xie B, Hao Q, et al. AZD1775 synergizes with SLC7A11 inhibition to promote ferroptosis. Sci China Life Sci. 2025;68(1):204–18. doi:10.1007/s11427-023-2589-1. [Google Scholar] [PubMed] [CrossRef]
19. Mi T, Kong X, Chen M, Guo P, He D. Inducing disulfidptosis in tumors: potential pathways and significance. MedComm. 2024;5(11):e791. doi:10.1002/mco2.791. [Google Scholar] [PubMed] [CrossRef]
20. Moreno ML, Escobar J, Izquierdo-Álvarez A, Gil A, Pérez S, Pereda J, et al. Disulfide stress: a novel type of oxidative stress in acute pancreatitis. Free Radic Biol Med. 2014;70:265–77. doi:10.1016/j.freeradbiomed.2014.01.009. [Google Scholar] [PubMed] [CrossRef]
21. Ergin M, Caliskanturk M, Senat A, Akturk O, Erel O. Disulfide stress in carbon monoxide poisoning. Clin Biochem. 2016;49(16–17):1243–7. doi:10.1016/j.clinbiochem.2016.07.019. [Google Scholar] [PubMed] [CrossRef]
22. Foley TD, Katchur KM, Gillespie PF. Disulfide stress targets modulators of excitotoxicity in otherwise healthy brains. Neurochem Res. 2016;41(10):2763–70. doi:10.1007/s11064-016-1991-0. [Google Scholar] [PubMed] [CrossRef]
23. Li T, Song Y, Wei L, Song X, Duan R. Disulfidptosis: a novel cell death modality induced by actin cytoskeleton collapse and a promising target for cancer therapeutics. Cell Commun Signal. 2024;22(1):491. doi:10.1186/s12964-024-01871-9. [Google Scholar] [PubMed] [CrossRef]
24. Jo SH, Son MK, Koh HJ, Lee SM, Song IH, Kim YO, et al. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J Biol Chem. 2001;276(19):16168–76. doi:10.1074/jbc.M010120200. [Google Scholar] [PubMed] [CrossRef]
25. Ding CC, Rose J, Sun T, Wu J, Chen PH, Lin CC, et al. MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Nat Metab. 2020;2(3):270–7. doi:10.1038/s42255-020-0181-1. [Google Scholar] [PubMed] [CrossRef]
26. Aviello G, Knaus UG. NADPH oxidases and ROS signaling in the gastrointestinal tract. Mucosal Immunol. 2018;11(4):1011–23. doi:10.1038/s41385-018-0021-8. [Google Scholar] [PubMed] [CrossRef]
27. Tarafdar A, Pula G. The role of NADPH oxidases and oxidative stress in neurodegenerative disorders. Int J Mol Sci. 2018;19(12):3824. doi:10.3390/ijms19123824. [Google Scholar] [PubMed] [CrossRef]
28. Babior BM. NADPH oxidase: an update. Blood. 1999;93(5):1464–76. doi:10.1182/blood.v93.5.1464.405a32_1464_1476. [Google Scholar] [CrossRef]
29. Yu W, Jin D, Zhang Y, Wang S, Yu J, Liu M, et al. Provoking tumor disulfidptosis by single-atom nanozyme via regulating cellular energy supply and reducing power. Nat Commun. 2025;16(1):4877. doi:10.1038/s41467-025-60015-w. [Google Scholar] [PubMed] [CrossRef]
30. Wang Z, Li Y, Wang C, Lan J, Li J, Liu G, et al. Disrupting intracellular redox homeostasis through copper-driven dual cell death to induce anti-tumor immunotherapy. Biomaterials. 2026;324:123523. doi:10.1016/j.biomaterials.2025.123523. [Google Scholar] [PubMed] [CrossRef]
31. Yan Y, Teng H, Hang Q, Kondiparthi L, Lei G, Horbath A, et al. SLC7A11 expression level dictates differential responses to oxidative stress in cancer cells. Nat Commun. 2023;14(1):3673. doi:10.1038/s41467-023-39401-9. [Google Scholar] [PubMed] [CrossRef]
32. Zheng T, Liu Q, Xing F, Zeng C, Wang W. Disulfidptosis: a new form of programmed cell death. J Exp Clin Cancer Res. 2023;42(1):137. doi:10.1186/s13046-023-02712-2. [Google Scholar] [PubMed] [CrossRef]
33. Avila J. Microtubule functions. Life Sci. 1992;50(5):327–34. doi:10.1016/0024-3205(92)90433-P. [Google Scholar] [PubMed] [CrossRef]
34. Kang YP, Mockabee-Macias A, Jiang C, Falzone A, Prieto-Farigua N, Stone E, et al. Non-canonical glutamate-cysteine ligase activity protects against ferroptosis. Cell Metab. 2021;33(1):174–89.e7. doi:10.1016/j.cmet.2020.12.007. [Google Scholar] [PubMed] [CrossRef]
35. Ji Y, Wu Z, Dai Z, Sun K, Zhang Q, Wu G. Excessive l-cysteine induces vacuole-like cell death by activating endoplasmic reticulum stress and mitogen-activated protein kinase signaling in intestinal porcine epithelial cells. Amino Acids. 2016;48(1):149–56. doi:10.1007/s00726-015-2071-5. [Google Scholar] [PubMed] [CrossRef]
36. Alborzinia H, Flórez AF, Kreth S, Brückner LM, Yildiz U, Gartlgruber M, et al. MYCN mediates cysteine addiction and sensitizes neuroblastoma to ferroptosis. Nat Cancer. 2022;3(4):471–85. doi:10.1038/s43018-022-00355-4. [Google Scholar] [PubMed] [CrossRef]
37. Zhang Y, Koppula P, Gan B. Regulation of H2A ubiquitination and SLC7A11 expression by BAP1 and PRC1. Cell Cycle. 2019;18(8):773–83. doi:10.1080/15384101.2019.1597506. [Google Scholar] [PubMed] [CrossRef]
38. Zhu WW, Liu Y, Yu Z, Wang HQ. SLC7A11-mediated cell death mechanism in cancer: a comparative study of disulfidptosis and ferroptosis. Front Cell Dev Biol. 2025;13:1559423. doi:10.3389/fcell.2025.1559423. [Google Scholar] [PubMed] [CrossRef]
39. Kunda P, Craig G, Dominguez V, Baum B. Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin-based protrusions. Curr Biol. 2003;13(21):1867–75. doi:10.1016/j.cub.2003.10.005. [Google Scholar] [PubMed] [CrossRef]
40. Xiao F, Li HL, Yang B, Che H, Xu F, Li G, et al. Disulfidptosis: a new type of cell death. Apoptosis. 2024;29(9–10):1309–29. doi:10.1007/s10495-024-01989-8. [Google Scholar] [PubMed] [CrossRef]
41. Balekoglu N, Michaud JF, Sauvé R, Ayinde KS, Lin S, Liu Y, et al. The WAVE regulatory complex interacts with Boc and is required for Shh-mediated axon guidance. iScience. 2024;27(12):111333. doi:10.1016/j.isci.2024.111333. [Google Scholar] [PubMed] [CrossRef]
42. Xie S, Zuo K, De Rubeis S, Bonollo G, Colombo G, Ruggerone P, et al. Impact of genetic variants associated with neurodevelopmental disorders on the WAVE regulatory complex. J Chem Inf Model. 2025;65(14):7399–405. doi:10.1021/acs.jcim.5c01162. [Google Scholar] [PubMed] [CrossRef]
43. Chen J, Ma B, Yang Y, Wang B, Hao J, Zhou X. Disulfidptosis decoded: a journey through cell death mysteries, regulatory networks, disease paradigms and future directions. Biomark Res. 2024;12(1):45. doi:10.1186/s40364-024-00593-x. [Google Scholar] [PubMed] [CrossRef]
44. Bieling P, Rottner K. From WRC to Arp2/3: collective molecular mechanisms of branched actin network assembly. Curr Opin Cell Biol. 2023;80:102156. doi:10.1016/j.ceb.2023.102156. [Google Scholar] [PubMed] [CrossRef]
45. Yang S, Tang Y, Liu Y, Brown AJ, Schaks M, Ding B, et al. Arf GTPase activates the WAVE regulatory complex through a distinct binding site. Sci Adv. 2022;8(50):eadd1412. doi:10.1126/sciadv.add1412. [Google Scholar] [PubMed] [CrossRef]
46. Xie J, Deng X, Xie Y, Zhu H, Liu P, Deng W, et al. Multi-omics analysis of disulfidptosis regulators and therapeutic potential reveals glycogen synthase 1 as a disulfidptosis triggering target for triple-negative breast cancer. MedComm. 2024;5(3):e502. doi:10.1002/mco2.502. [Google Scholar] [PubMed] [CrossRef]
47. Zheng Z, Song Y. Integrated analysis of disulfidptosis-related genes SLC7A11, SLC3A2 RPN1 and NCKAP1 across cancers. Discov Oncol. 2024;15(1):724. doi:10.1007/s12672-024-01612-x. [Google Scholar] [PubMed] [CrossRef]
48. Liu H, Tang T. Pan-cancer genetic analysis of disulfidptosis-related gene set. Cancer Genet. 2023;278:91–103. doi:10.1016/j.cancergen.2023.10.001. [Google Scholar] [PubMed] [CrossRef]
49. Liu T, Kong X, Wei J. Disulfidptosis: a new target for Parkinson’s disease and cancer. Curr Issues Mol Biol. 2024;46(9):10038–64. doi:10.3390/cimb46090600. [Google Scholar] [PubMed] [CrossRef]
50. He J, Ding H, Li H, Pan Z, Chen Q. Intra-tumoral expression of SLC7A11 is associated with immune microenvironment, drug resistance, and prognosis in cancers: a pan-cancer analysis. Front Genet. 2021;12:770857. doi:10.3389/fgene.2021.770857. [Google Scholar] [PubMed] [CrossRef]
51. Lin Y, Dong Y, Liu W, Fan X, Sun Y. Pan-cancer analyses confirmed the ferroptosis-related gene SLC7A11 as a prognostic biomarker for cancer. Int J Gen Med. 2022;15:2501–13. doi:10.2147/IJGM.S341502. [Google Scholar] [PubMed] [CrossRef]
52. Yang T, Liu J, Liu F, Lei J, Chen S, Ma Z, et al. Integrative analysis of disulfidptosis and immune microenvironment in hepatocellular carcinoma: a putative model and immunotherapeutic strategies. Front Immunol. 2024;14:1294677. doi:10.3389/fimmu.2023.1294677. [Google Scholar] [PubMed] [CrossRef]
53. Chen G, Zhang G, Zhu Y, Wu A, Fang J, Yin Z, et al. Identifying disulfidptosis subtypes in hepatocellular carcinoma through machine learning and preliminary exploration of its connection with immunotherapy. Cancer Cell Int. 2024;24(1):194. doi:10.1186/s12935-024-03387-1. [Google Scholar] [PubMed] [CrossRef]
54. Zhang K, Zhu Z, Zhou J, Shi M, Wang N, Yu F, et al. Disulfidptosis-related gene expression reflects the prognosis of drug-resistant cancer patients and inhibition of MYH9 reverses sorafenib resistance. Transl Oncol. 2024;49(7782):102091. doi:10.1016/j.tranon.2024.102091. [Google Scholar] [PubMed] [CrossRef]
55. Zhang R, Zhou K, Wu M, Qiao H, Yu L, Jin X, et al. Disulfidptosis-related genes RPN1 inhibits the progression of hepatocellular carcinoma by regulating cell cycle, may be a new therapeutic targets. Inflamm Res. 2025;74(1):105. doi:10.1007/s00011-025-02070-z. [Google Scholar] [PubMed] [CrossRef]
56. Wang X, Xiao K, Liu Z, Wang L, Dong Z, Wang H, et al. Unveiling disulfidptosis-related genes in HBV-associated hepatocellular carcinoma: an integrated study incorporating transcriptome and Mendelian randomization analyses. J Cancer. 2024;15(17):5540–56. doi:10.7150/jca.93194. [Google Scholar] [PubMed] [CrossRef]
57. Li Q, Huang R, Lv L, Ying H, Wu Y, Huang Y, et al. FLNA, a disulfidptosis-related gene, modulates tumor immunity and progression in colorectal cancer. Cell Mol Biol Lett. 2025;30(1):92. doi:10.1186/s11658-025-00761-3. [Google Scholar] [PubMed] [CrossRef]
58. Gong X, Wu Q, Tan Z, Lin S, Zhou J, Lin S, et al. Identification and validation of cuproptosis and disulfidptosis related genes in colorectal cancer. Cell Signal. 2024;119(3):111185. doi:10.1016/j.cellsig.2024.111185. [Google Scholar] [PubMed] [CrossRef]
59. Li R, Wang X, Liu J, Cai Z, Li Z, Tao Q, et al. DPP7 promotes colorectal cancer progression through GPX4-dependent suppression of disulfidptosis and immune evasion. J Cell Mol Med. 2025;29(12):e70660. doi:10.1111/jcmm.70660. [Google Scholar] [PubMed] [CrossRef]
60. Zhang P, Chen Z, Lin X, Yu S, Yu X, Chen Z. Unravelling diagnostic clusters and immune landscapes of disulfidptosis patterns in gastric cancer through bioinformatic assay. Aging. 2023;15(24):15434–50. doi:10.18632/aging.205365. [Google Scholar] [PubMed] [CrossRef]
61. Li Q, Yin LK. Comprehensive analysis of disulfidptosis related genes and prognosis of gastric cancer. World J Clin Oncol. 2023;14(10):373–99. doi:10.5306/wjco.v14.i10.373. [Google Scholar] [PubMed] [CrossRef]
62. Yan J, Fang Z, Shi M, Tu C, Zhang S, Jiang C, et al. Clinical significance of disulfidptosis-related genes and functional analysis in gastric cancer. J Cancer. 2024;15(4):1053–66. doi:10.7150/jca.91796. [Google Scholar] [PubMed] [CrossRef]
63. Yao HF, Ge J, Chen J, Tang X, Li C, Hu X, et al. CASC8 activates the pentose phosphate pathway to inhibit disulfidptosis in pancreatic ductal adenocarcinoma though the c-Myc-GLUT1 axis. J Exp Clin Cancer Res. 2025;44(1):26. doi:10.1186/s13046-025-03295-w. [Google Scholar] [PubMed] [CrossRef]
64. Zhang W, Zhao Q, Zhang H, Zhang J, Zhao F, Niu R, et al. A prognostic biomarker of disulfidptosis constructed by machine learning framework model as potential reporters of pancreatic adenocarcinoma. Cell Signal. 2024;123(1):111371. doi:10.1016/j.cellsig.2024.111371. [Google Scholar] [PubMed] [CrossRef]
65. Liang HY, Luo RZ, Deng R, Chen SL, Liu X, Yang X, et al. Glycogen stores mediated by the p53-GYS1 feedback circuit engenders platinum resistance in ovarian clear cell carcinoma. Cell Death Differ. 2025;32(9):1707–21. doi:10.1038/s41418-025-01500-z. [Google Scholar] [PubMed] [CrossRef]
66. Jiang W, Wang Q, Zhou J, Zhao Y, Qin X, Li X, et al. Disulfidptosis-associated CCNB2: a prognostic biomarker and immune microenvironment modulator in prostate cancer. J Cancer. 2025;16(13):3928–41. doi:10.7150/jca.112791. [Google Scholar] [PubMed] [CrossRef]
67. Jiang A, Liu W, Liu Y, Hu J, Zhu B, Fang Y, et al. DCS, a novel classifier system based on disulfidptosis reveals tumor microenvironment heterogeneity and guides frontline therapy for clear cell renal carcinoma. J Natl Cancer Cent. 2024;4(3):263–79. doi:10.1016/j.jncc.2024.06.003. [Google Scholar] [PubMed] [CrossRef]
68. Wang K, Li L, Liang G, Xiao H, Zhang L, Liu T. Sonodynamic activated nanoparticles with Glut1 inhibitor and cystine-containing polymer stimulate disulfidptosis for improved immunotherapy in bladder cancer. Biomaterials. 2025;319:123178. doi:10.1016/j.biomaterials.2025.123178. [Google Scholar] [PubMed] [CrossRef]
69. Jing Z, Huang W, Mei J, Bhushan S, Wu X, Yan C, et al. Advances in novel cell death mechanisms in breast cancer: intersecting perspectives on ferroptosis, cuproptosis, disulfidptosis, and pyroptosis. Mol Cancer. 2025;24(1):224. doi:10.1186/s12943-025-02445-0. [Google Scholar] [PubMed] [CrossRef]
70. Xia Q, Yan Q, Wang Z, Huang Q, Zheng X, Shen J, et al. Disulfidptosis-associated lncRNAs predict breast cancer subtypes. Sci Rep. 2023;13(1):16268. doi:10.1038/s41598-023-43414-1. [Google Scholar] [PubMed] [CrossRef]
71. Zhang K, Li G, Wang Q, Liu X, Chen H, Li F, et al. A disulfidptosis-related glucose metabolism and immune response prognostic model revealing the immune microenvironment in lung adenocarcinoma. Front Immunol. 2024;15:1398802. doi:10.3389/fimmu.2024.1398802. [Google Scholar] [PubMed] [CrossRef]
72. Zeng C, Huang D, Wang L, Liang H, Ma X. Silencing ZIC5 suppresses glycolysis and promotes disulfidptosis in lung adenocarcinoma cells. Cancer Biol Ther. 2025;26(1):2501780. doi:10.1080/15384047.2025.2501780. [Google Scholar] [PubMed] [CrossRef]
73. Zhu A, Zong Y, Wei S, Li Y, Fan Y, Liu S, et al. Pan-cancer analysis of the disulfidptosis-related gene NCKAP1 and its prognostic value for lung adenocarcinoma. J Cancer. 2023;14(17):3351–67. doi:10.7150/jca.88650. [Google Scholar] [PubMed] [CrossRef]
74. Zhu J, Ge H, Chen Y, Zhang S, Wu J, Nai W, et al. Disulfidptosis-related gene SLC7A11 predicts prognosis and indicates tumor immune infiltration in lung adenocarcinoma. Transl Cancer Res. 2024;13(9):5064–72. doi:10.21037/tcr-24-1182. [Google Scholar] [PubMed] [CrossRef]
75. Zhang HB, Pan JY, Zhu T. A disulfidptosis-related lncRNA prognostic model to predict survival and response to immunotherapy in lung adenocarcinoma. Front Pharmacol. 2023;14:1254119. doi:10.3389/fphar.2023.1254119. [Google Scholar] [PubMed] [CrossRef]
76. Niu X, Li G, Kahlert UD, Ding L, Zheng J, Li C, et al. Integrative disulfidptosis-based risk assessment for prognostic stratification and immune profiling in glioma. J Cell Mol Med. 2025;29(4):e70429. doi:10.1111/jcmm.70429. [Google Scholar] [PubMed] [CrossRef]
77. Shu Y, Li J. Disulfidptosis as a key regulator of glioblastoma progression and immune cell impairment. Front Immunol. 2025;16:1526296. doi:10.3389/fimmu.2025.1526296. [Google Scholar] [PubMed] [CrossRef]
78. Tian H, Deng H, Liu X, Liu C, Zhang C, Leong KW, et al. A novel FTO-targeting nanodrug induces disulfidptosis and ameliorates the suppressive tumor immune environment to treat uveal melanoma. Biomaterials. 2025;319(4):123168. doi:10.1016/j.biomaterials.2025.123168. [Google Scholar] [PubMed] [CrossRef]
79. Zhang S, Wang Y, Gu J, Yang Y, Liang J, Wang Y, et al. PPARγ antagonists exhibit antitumor effects by regulating ferroptosis and disulfidptosis. Biomolecules. 2024;14(5):596. doi:10.3390/biom14050596. [Google Scholar] [PubMed] [CrossRef]
80. Zhang Y, Li Z, Lu H, Jiang Z, Song Y, Ye Z, et al. Pan-cancer analysis uncovered the prognostic and therapeutic value of disulfidptosis. npj Precis Oncol. 2025;9(1):50. doi:10.1038/s41698-025-00834-8. [Google Scholar] [PubMed] [CrossRef]
81. Tang M, Dirks K, Kim SY, Qiu Z, Gao Y, Sun D, et al. Inhibition of thioredoxin reductase 1 sensitizes glucose-starved glioblastoma cells to disulfidptosis. Cell Death Differ. 2025;32(4):598–612. doi:10.1038/s41418-024-01440-0. [Google Scholar] [PubMed] [CrossRef]
82. Zhang Y, Liu Y, Huang Q, Wang Z, Li Y, Zhang Q, et al. Decoding the diet-inflammation nexus: ferroptosis as a therapeutic target. Crit Rev Food Sci Nutr. 2025;47(4):1–21. doi:10.1080/10408398.2025.2540044. [Google Scholar] [PubMed] [CrossRef]
83. Maharajan N, Benyamien-Roufaeil DS, Brown RA, Portney BA, Banerjee A, Zalzman M. Cancer stem cell mechanisms and targeted therapeutic strategies in head and neck squamous cell carcinoma. Cancer Lett. 2025;634(6):218015. doi:10.1016/j.canlet.2025.218015. [Google Scholar] [PubMed] [CrossRef]
84. Wei Y, Hankey W, Xu D, Yuan F. Programmed cell death in cancer. MedComm. 2025;6(9):e70357. doi:10.1002/mco2.70357. [Google Scholar] [PubMed] [CrossRef]
85. Liang Y, Lan H, Li Q, Gao M, Liu M, Xu Z, et al. Exploiting metabolic vulnerabilities through synergistic ferroptosis and disulfidptosis for breast cancer therapy. J Adv Res. 2026;79:905–16. doi:10.1016/j.jare.2025.03.052. [Google Scholar] [PubMed] [CrossRef]
86. Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411(6835):342–8. doi:10.1038/35077213. [Google Scholar] [PubMed] [CrossRef]
87. Morana O, Wood W, Gregory CD. The apoptosis paradox in cancer. Int J Mol Sci. 2022;23(3):1328. doi:10.3390/ijms23031328. [Google Scholar] [PubMed] [CrossRef]
88. Zhao S, Tang Y, Wang R, Najafi M. Mechanisms of cancer cell death induction by paclitaxel: an updated review. Apoptosis. 2022;27(9–10):647–67. doi:10.1007/s10495-022-01750-z. [Google Scholar] [PubMed] [CrossRef]
89. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. doi:10.1080/01926230701320337. [Google Scholar] [PubMed] [CrossRef]
90. Peña‐Blanco A, García‐Sáez AJ. Bax, Bak and beyond—mitochondrial performance in apoptosis. FEBS J. 2018;285(3):416–31. doi:10.1111/febs.14186. [Google Scholar] [PubMed] [CrossRef]
91. Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30(2):180–92. doi:10.1016/j.immuni.2009.01.001. [Google Scholar] [PubMed] [CrossRef]
92. Zhou Q, Meng Y, Li D, Yao L, Le J, Liu Y, et al. Ferroptosis in cancer: from molecular mechanisms to therapeutic strategies. Signal Transduct Target Ther. 2024;9(1):55. doi:10.1038/s41392-024-01769-5. [Google Scholar] [PubMed] [CrossRef]
93. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26(3):165–76. doi:10.1016/j.tcb.2015.10.014. [Google Scholar] [PubMed] [CrossRef]
94. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72. doi:10.1016/j.cell.2012.03.042. [Google Scholar] [PubMed] [CrossRef]
95. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–85. doi:10.1016/j.cell.2017.09.021. [Google Scholar] [PubMed] [CrossRef]
96. Zhou S, Liu J, Wan A, Zhang Y, Qi X. Epigenetic regulation of diverse cell death modalities in cancer: a focus on pyroptosis, ferroptosis, cuproptosis, and disulfidptosis. J Hematol Oncol. 2024;17(1):22. doi:10.1186/s13045-024-01545-6. [Google Scholar] [PubMed] [CrossRef]
97. Xie J, Yang Y, Gao Y, He J. Cuproptosis: mechanisms and links with cancers. Mol Cancer. 2023;22(1):46. doi:10.1186/s12943-023-01732-y. [Google Scholar] [PubMed] [CrossRef]
98. Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–61. doi:10.1126/science.abf0529. [Google Scholar] [PubMed] [CrossRef]
99. Zhang N, Zhang J, Yang Y, Shan H, Hou S, Fang H, et al. A palmitoylation-depalmitoylation relay spatiotemporally controls GSDMD activation in pyroptosis. Nat Cell Biol. 2024;26(5):757–69. doi:10.1038/s41556-024-01397-9. [Google Scholar] [PubMed] [CrossRef]
100. Fleisher TA. Apoptosis. Ann Allergy Asthma Immunol. 1997;78(3):245–50. doi:10.1016/S1081-1206(10)63176-6. [Google Scholar] [PubMed] [CrossRef]
101. Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. 2019;2019(30):5080843. doi:10.1155/2019/5080843. [Google Scholar] [PubMed] [CrossRef]
102. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491–501. doi:10.1016/s0092-8674(00)81590-1. [Google Scholar] [PubMed] [CrossRef]
103. Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17(7):395–417. doi:10.1038/s41571-020-0341-y. [Google Scholar] [PubMed] [CrossRef]
104. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22(7):381–96. doi:10.1038/s41568-022-00459-0. [Google Scholar] [PubMed] [CrossRef]
105. Mao C, Wang M, Zhuang L, Gan B. Metabolic cell death in cancer: ferroptosis, cuproptosis, disulfidptosis, and beyond. Protein Cell. 2024;15(9):642–60. doi:10.1093/procel/pwae003. [Google Scholar] [PubMed] [CrossRef]
106. Huang Y, Wan X, Su Q, Zhao C, Cao J, Yue Y, et al. Ultrasound-activated piezo-hot carriers trigger tandem catalysis coordinating cuproptosis-like bacterial death against implant infections. Nat Commun. 2024;15(1):1643. doi:10.1038/s41467-024-45619-y. [Google Scholar] [PubMed] [CrossRef]
107. Seo W, Jung B, Roh T, Shin HJ, Song IC, Chung C, et al. Inflammasomes and pyroptosis in cancer: mechanisms and therapeutic advances. J Hematol Oncol. 2025;18(1):113. doi:10.1186/s13045-025-01763-6. [Google Scholar] [PubMed] [CrossRef]
108. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–8. doi:10.1038/nature18629. [Google Scholar] [PubMed] [CrossRef]
109. Hu X, Guo H, Wang G, Liao X, Zhang S. Poly(sodium lipoate) particles with nitroimidazole modification for disulfide stress-mediated antitumor metastasis. ACS Appl Mater Interfaces. 2025;17(24):35304–15. doi:10.1021/acsami.5c07590. [Google Scholar] [PubMed] [CrossRef]
110. Li M, Xu J, Du K, Wang G, Feng Y, Liu P. Alexidine dihydrochloride enhances the sensitivity of human hepatocellular carcinoma to disulfidptosis via ATF4-DDIT3 activation. Free Radic Biol Med. 2025;237(5):585–99. doi:10.1016/j.freeradbiomed.2025.06.020. [Google Scholar] [PubMed] [CrossRef]
111. Liang S, Yang X, Wang J, Yu M, Gong ZP. Prognostic and immunologic characteristics of head and neck squamous cell carcinoma based on disulfidptosis-related lncRNAs. Mol Biotechnol. 2025;6:1–19. doi:10.1007/s12033-025-01471-z. [Google Scholar] [PubMed] [CrossRef]
112. Ye Z, Yao Y, Xu Y, Ye J, Yao Q, Kou L, et al. Nanomedicine strategies for disulfidptosis activation in SLC7A11-high tumors. Colloids Surf B Biointerfaces. 2026;257(2):115094. doi:10.1016/j.colsurfb.2025.115094. [Google Scholar] [PubMed] [CrossRef]
113. Zhen W, Zhao T, Chen X, Zhang J. Unlocking the potential of disulfidptosis: nanotechnology-driven strategies for advanced cancer therapy. Small. 2025;21(23):e2500880. doi:10.1002/smll.202500880. [Google Scholar] [PubMed] [CrossRef]
114. Yang Y, Zhang S, Hu X, Hu C, Lv Y, Pei Y, et al. Oxygen-driven Prussian blue nanomotor promotes cancer immunotherapy by disrupting redox state-induced tumor disulfide death. J Colloid Interface Sci. 2025;700(Pt 2):138404. doi:10.1016/j.jcis.2025.138404. [Google Scholar] [PubMed] [CrossRef]
115. Tian Z, Song J, She J, He W, Guo S, Dong B. Constructing a disulfidptosis-related prognostic signature of hepatocellular carcinoma based on single-cell sequencing and weighted co-expression network analysis. Apoptosis. 2024;29(9):1632–47. doi:10.1007/s10495-024-01968-z. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools