
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

A Comprehensive Expression Atlas of Nicotiana tabacum: Revealing Specificity Expression Patterns and Regulatory Variants
1
Molecular Genetics Key Laboratory of China Tobacco, Guizhou Academy of Tobacco Science, Guiyang, China
2
Guizhou Provincial Key Laboratory for Tobacco Quality Improvement and Efficiency Enhancement, Guiyang, China
3
China Tobacco Gene Research Center, Zhengzhou Tobacco Research Institute of CNTC, Zhengzhou, China
* Corresponding Author: Zhixiao Yang. Email:
Phyton-International Journal of Experimental Botany 2026, 95(2), 6 https://doi.org/10.32604/phyton.2026.073509
Received 19 September 2025; Accepted 08 January 2026; Issue published 28 February 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Tobacco (Nicotiana tabacum, 2n = 48) is a key non-food economic crop, yet its stress response and gene regulatory mechanisms remain poorly understood. By analyzing 603 transcriptome datasets, this study identified 1405 tissue-specific genes, revealing tissue-specific synthesis of terpenoids and other ecologically important secondary metabolites in sepals and other tissues. Comparative stress-response analysis highlighted distinct gene expression patterns in leaves and roots under biotic and abiotic stresses. Additionally, 28,396 expression quantitative trait loci (eQTLs) were mapped in leaves, offering valuable genetic regulatory markers. These findings provide crucial insights into tobacco’s gene expression characteristics and their functional implications, serving as a foundation for future research.Keywords
Supplementary Material
Supplementary Material FilePlant development is a continuous process, and with the continuous generation and development of new organs, gene expression and regulation undergo constant changes, forming a complex network [1]. In such processes, studying gene expression patterns contributes to understanding the mechanisms of development and regulation. The utilization of spatiotemporal transcriptome sequencing is an important approach to elucidate such regulatory networks, and detailed transcriptional atlases of many important plants have already been outlined [2,3]. In addition, several tools have also been developed for the analysis of expression atlases [4,5]. However, in comparison to research on animals, studies on plant transcriptional atlases often lack sufficient sampling across different organs and over time; for example, a transcriptional atlas of pigs was constructed using samples from 31 pig organs and 2 pig cell lines [6]. This extensive sampling is crucial for understanding the complex transcriptional landscape and its variations. Similarly, to gain a comprehensive understanding of the plant transcriptome, sampling across various organs and developmental stages, as well as different environmental conditions, is essential.
Constructing a large-scale transcriptional atlas is costly, so another approach to understanding certain features of plant development is through unified reanalysis by integrating different datasets. For example, in soybean research, a study gathered over 5000 RNA-seq datasets for analysis and constructed a transcriptional atlas database for soybeans [7]. For species with limited database resources, research has provided insights into the transcriptional characteristics of individual species through plant cultivation and transcriptome sequencing analysis. For example, Xanthopoulou et al. collected samples of seeds, sprouts, stem leaves, tender and mature leaves, male and female flowers, fruits at seven developmental stages, and primary and lateral roots and constructed a comprehensive gene expression atlas for Cucurbita pepo [8]. However, data obtained from individual organisms lack population-level insights. When studying temporal and spatial transcriptional features, research on the regulation of gene expression should not be overlooked. Two impressive large-sample analysis studies focusing on the transcriptional atlas of pigs and cattle not only revealed the tissue-specific expression characteristics of genes in pigs and cattle but also importantly identified numerous regulatory loci in the genome [9,10].
Tobacco, an important economic crop, faces various biotic and abiotic stresses during its growth, which significantly impact tobacco growth and the tobacco industry. Transcriptome sequencing analysis is commonly employed to study stress response mechanisms in tobacco. For example, cadmium can inhibit tobacco growth and reduce leaf quality, and its presence in tobacco smoke affects the health of both smokers and the surrounding environment. Hence, studies have elucidated the mechanisms of cadmium accumulation and tolerance in tobacco leaves through transcriptome sequencing and analysis [11]. By comparing the transcriptomes of plants with differential cold tolerance between two tobacco cultivars, important candidate genes for cold stress resistance, such as NtCBF2, have been identified [12]. The accumulation of transcriptome analysis data provides possibilities for conducting tobacco transcriptomic studies through data collection methods.
This study manually collected and screened RNA-seq data originating from 35 independent research projects involving various tobacco tissues and treatments. Through comprehensive analysis, genes exhibiting tissue-specific expression patterns and the similarities and differences in tobacco responses to different stresses were identified. Single-nucleotide polymorphism (SNP) calling based on RNA-seq and identification of eQTLs provide rich genetic regulatory loci for tobacco gene expression. This study presents a comprehensive analysis of tobacco transcriptome levels and features, which provides valuable assistance in accelerating tobacco research.
We retrieved RNA-seq data for 35 BioProjects from National Center for Biotechnology Information (NCBI) through literature mining, encompassing 603 samples across 13 tissues (Supplementary Table S1). Genomic data pertinent to tobacco were obtained from the Sol Genomics Network database (https://solgenomics.net/). The genome published by Edwards et al. [13] was used as a reference.
The original data were converted to fastq format using fastq-dump from the SRA Toolkit v3.0.5 (https://github.com/ncbi/sra-tools). Subsequently, adapter sequences and low-quality sequences in the fastq were removed using fastp with default parameters [14]. After obtaining clean data, the transcriptome data were mapped to the reference genome using STAR (v2.7.10a) [15]. Subsequently, StringTie (v2.1.7) [16] with default parameters was used in conjunction with the annotation of the reference genome to quantify gene expression levels. The gene expression levels were subjected to t-distributed stochastic neighbour embedding (tSNE) clustering using the R package Rtsne (v0.16) [17], and the umap (v0.2.10.0) package was utilized for uniform manifold approximation and projection (UMAP) clustering of gene expression [18].
2.3 Analysis of Tissue-Specific Genes in Tobacco
We utilized the median transcripts per million (TPM) values of genes in each tissue (calculated with R function media) for calculation of the tissue-specific index (TSI). Genes with a median expression level less than 0.1 TPM were assigned a value of 0. TPM values were then log2 transformed (TPM + 1). Subsequently, all genes or transcripts with accumulated expression values less than 0.1 were excluded from the analysis to filter out genes with low expression or transcripts with low expression [19].
The formula for calculating the TSI is as follows:
2.4 Analysis of Gene Expression Patterns in Response to Tobacco Stress
From 603 transcriptome datasets, 29 stress groups including roots, leaves, axillary shoots, and other tissues, were selected. We used featureCounts (v2.0.1) [24] to calculate the read counts of genes in the samples. Gene differential expression analysis was conducted using the R package DESeq2 (v1.38.3) [25]. Genes with a |log2FoldChange| > 1 and an adjusted p value < 0.05 were identified as differentially expressed genes (DEGs). Genes with accumulated TPM expression values less than 0.1 in all samples were filtered out, and the remaining genes were considered to have stable expression. The R package corrplot (v0.92) was used to create the correlograms with log2FoldChange in each group [26]. Functional enrichment analysis of the DEGs was conducted using the R package clusterProfiler [27].
To construct the regulatory network of TFs and genes, we first downloaded the plant TF protein files from PlantTFDB (https://planttfdb.gao-lab.org/). Next, we used BLASTP to align the tobacco proteins with the plant TF protein sequences, using an E-value cut-off of 1E−5. Genes that passed the threshold were identified as tobacco TFs. Subsequently, we calculated the Pearson correlation coefficients (PCCs) between the tobacco TFs and the DEGs shared across all the stress treatments. TF and DEG pairs with a PCC greater than 0.9 and q-values less than 0.01 were considered coexpressed. We utilized HOMER (v4.11) (http://homer.ucsd.edu/homer/) [28] to identify common stable motifs in the upstream 2 kb of DEGs shared across all the stress treatments. First, we used the loadGenome.pl subroutine of HOMER to load the tobacco genome file; next, we used the findMotifsGenome.pl subroutine to identify motifs in the 2 kb upstream promoter regions of the DEGs. As a background, we randomly selected the 2 kb upstream regions of 5000 genes across the entire tobacco genome, repeated the sampling 50 times, and selected motifs that consistently appeared in each sampling as the final identified motifs. We constructed a coexpression network of TFs that could bind to these motifs and were coexpressed with the DEGs shared across all the stress treatments with the coexpression PCC threshold set to 0.8. The coexpression network was visualized using Cytoscape (v3.10.2) software [29].
2.5 Identification of SNPs in Tobacco Leaves
RNA-seq alignment results were sorted using SAMtools (v1.8) software with default parameters [30], and duplicated reads within the BAM files were marked using Picard (https://github.com/broadinstitute/picard). The GATK tool SplitNCigarReads was used to filter the BAM files obtained in the previous step [31]. Subsequently, SNP calling and variant filtration were performed using the HaplotypeCaller tool from the GATK toolkit, filtering candidate SNPs with Fisher strand (FS) values < 30.0 and quality depth (QD) values > 2.0. Further filtering was conducted to retain only biallelic SNPs. VCFtools (0.1.16) software [32] was then utilized with the parameters—max-missing 0.9 and—maf 0.05 to filter SNPs in the population with missing rates exceeding 10% and minor allele frequencies greater than 5%, respectively.
Gene expression data were filtered first, and genes with a median TPM greater than 0 were selected for eQTL analysis. The filtered expression data were subjected to confounding factor identification using the PEER (v1.0) package, and the top 10 confounding factors for covariance in eQTL analysis were selected. FastQTL (v2.0) [33] was used to identify cis-eQTLs in tobacco leaves. The size of the cis window was set to 1000000, and the ‘normal’ parameter was used for the transformation of phenotypes. EMMAX [34] was used to identify SNPs associated with the expression of each gene. The pvalues obtained from FastQTL were adjusted using the Bonferroni method to derive adjusted pvalues (p.adjust), with a significance threshold set at p.adjust < 0.05 in cis-eQTL analysis. Genetic Type I error calculator (GEC) software [32] was utilized to determine the effective number of SNPs (Me), and a threshold of 1.43 × 10−7 (0.05/Me) was adopted as the threshold for determining the significance of trans-eQTLs, adjusted by the Bonferroni method. PLINK (1.90b6.21) software [35] was used with the ‘clump’ parameter to merge linkage blocks of related SNPs, with a maximum block length of 500 kb. The SNP with the smallest pvalue in each linkage block containing at least three significant SNPs was selected as the lead SNP. Enrichment analysis was performed for cis- and trans-eGenes, and visualization was carried out using CirGO software.
3.1 Sample Statistics and Cluster Analysis of Tobacco Transcription Data
Tobacco is an important economic crop, and transcriptomic studies of this species have generated a large amount of sequencing data. In this study, we collected 603 available tobacco transcriptome datasets from 13 tissues or organs, including leaves, roots, seedlings, stems, trichomes, petals, and sepals, from the NCBI database and published research (Fig. 1A). Notably, the samples from leaf and root tissues were the most numerous, with 329 and 146 samples, respectively. These RNA-seq samples were derived from 35 distinct NCBI BioProjects, which is indicative of their diverse origins. This aggregation reflects the inclusion of samples from various laboratories potentially harbouring rich genetic polymorphisms, thereby laying a foundation for the exploration of regulatory loci influencing tobacco gene expression. Among these studies, 10 Bioprojects were related to stress studies in tobacco, providing a foundation for cross-stress analysis of tobacco stress response mechanisms.
After RNA-seq data were downloaded from the NCBI SRA database, a unified analysis pipeline was used to perform data cleaning, alignment, and quantification on all datasets. After quantifying the expression levels of all genes in the samples (Supplementary File https://doi.org/10.6084/m9.figshare.29666060), clustering was performed using tSNE. The results revealed that some samples clustered together based on tissue relationships (Fig. 1B). However, the clustering analysis could not completely differentiate certain tissues, possibly due to anatomical proximity. For example, petals and sepals are parts of flowers, whereas the blade, lamina, and midrib are constituents of leaves. However, flowers are considered a morphological modification of leaves, leading to an inability for complete discrimination. Notably, there was a clear separation between leaves from the seedling and nonseedling stages in the clustering results, indicating differences in gene expression patterns between tobacco seedling leaves and mature leaves and validating the feasibility of categorizing them into two distinct tissues. From the perspective of treatments, samples under different treatments showed a clear clustering trend, indicating that both treatment conditions and growth environments can influence the clustering results (Supplementary Fig. S1). Similar patterns have been observed in transcriptome clustering analyses of poplar and soybean, suggesting that tissue type and environmental factors are major determinants of clustering [36,37].
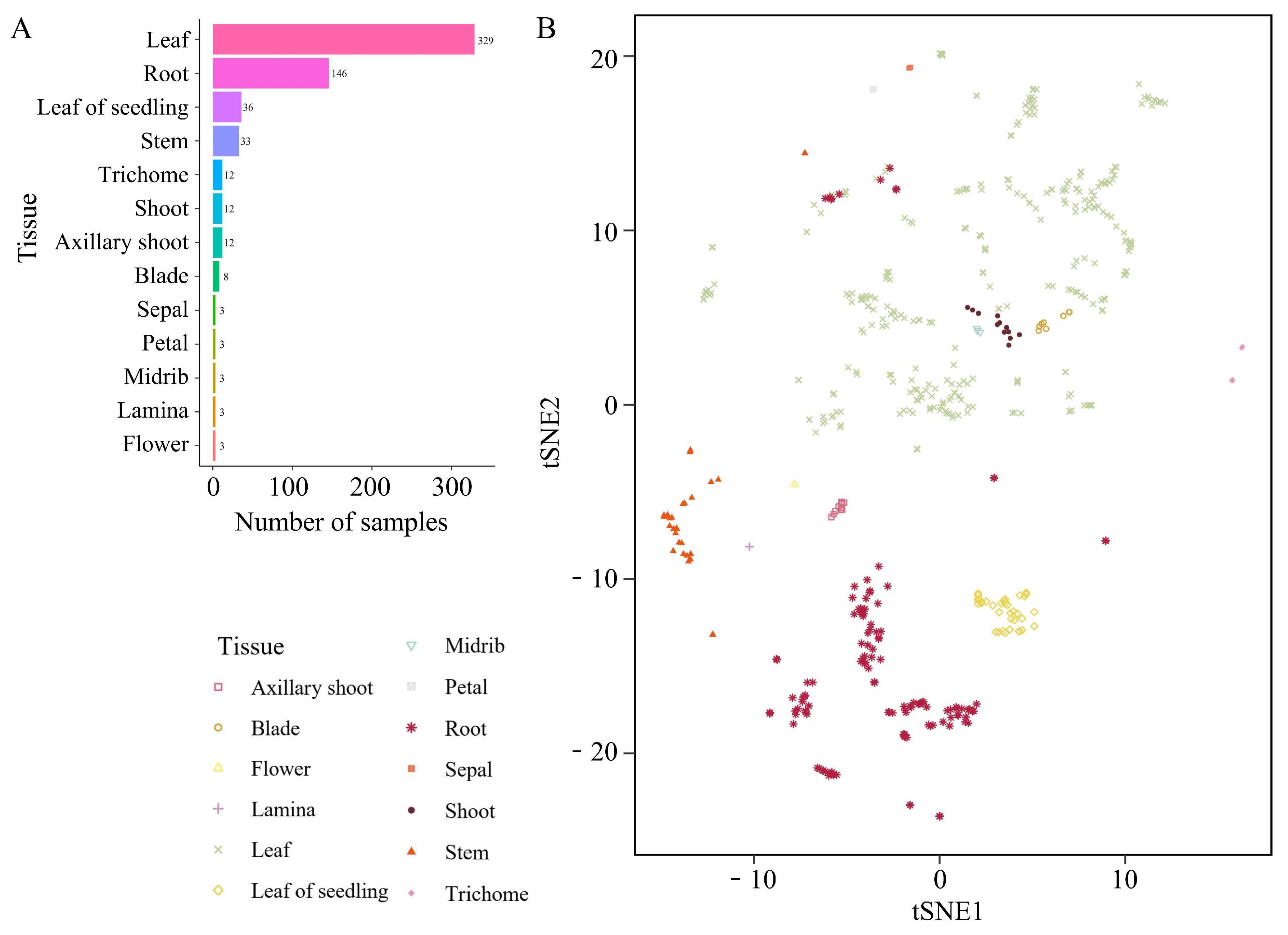
Figure 1: The number and types of RNA-seq samples utilized in this study. (A) Sample quantity statistics for each tissue. “Flower” denotes immature flowers. (B) tSNE clustering results based on sample expression levels.
3.2 Analysis of Tobacco Tissue-Specific Genes
The above results indicate that tSNE analysis can effectively distinguish most tissues, suggesting relatively high expression similarity among samples within the same tissue. This finding implies that identifying genes with tissue-specific expression can further elucidate the relationship between gene expression and tissue function. The samples collected in this study covered a total of 13 tissues, and the TSI of each gene was calculated as the median expression level across tissues (Supplementary Table S2). Additionally, genes were categorized into four groups (null, weak, broad, and tissue-specific) based on their median expression across tissues (Supplementary Table S3) [7]. The results revealed that 28.9% of the genes were classified as null genes (TPM <1 in all tissues; n = 10,272), 9.9% were classified as weakly expressed genes (TPM <5 in all tissues; n = 3529), over 57% were classified as broadly expressed genes (TPM ≥ 5 in all tissues, TSI < 0.85; n = 20,313), and only a small fraction were classified as tissue-specific genes (TPM ≥ 5 in all tissues, TSI ≥ 0.85; n = 1405) (Supplementary Table S4) (Fig. 2A).
The number of tissue-specific genes was the greatest in the immature flower, petal, sepal, and root tissues, with 308, 642, 63, and 303 genes, respectively (Fig. 2B). Although most tissue-specific genes are unique to individual tissues, there are also a few genes that exhibit specific expression in multiple tissues. Notably, a subset of genes exhibited very high tissue-specific expression in both tobacco flowers and petals (Fig. 2C). The greater number of tissue-specific genes and high expression levels in tissues or organs related to reproduction may be associated with their unique functions. To further understand the functions of these tissue-specific genes, this study conducted gene ontology (GO) enrichment analysis using all genes as background. The genes that were specifically expressed in nine tissues—axillary shoot, blade, young leaf, midrib, petal, sepal, immature flower, root, and stem—were collectively enriched in 341 GO terms (Supplementary Table S5, Supplementary Fig. S2). For example, genes specifically expressed in the axillary shoot were enriched in genes related to anatomical structure formation involved in morphogenesis, plant organ formation, regulation of development, heterochrony, anatomical structure formation involved in morphogenesis, regulation of plant organ formation, monooxygenase activity, and other genes related to plant organ formation. Genes specifically expressed in petals and flowers were enriched in a series of GO terms related to floral organ development such as floral whorl development, floral organ development, androecium development, gynoecium development, and carpel development. Particularly in sepals, a large number of tissue-specific genes related to terpenoid biosynthesis processes, sesquiterpenoid biosynthesis processes, terpene synthase activity, and other terpenoid synthesis-related processes were identified (Fig. 2D). The top four enriched terms of tissue-specific genes in sepals were sesquiterpene biosynthetic process, sesquiterpene metabolic process, terpene biosynthetic process, and terpene metabolic process, and other biological processes were also predominantly associated with terpenoid synthesis. Several studies have revealed the involvement of terpenoids in processes such as attracting insects for pollination [38,39], indicating that the secondary metabolites synthesized in tobacco sepals also play similar important roles.
Given the crucial regulatory roles of TFs, this study aimed to identify the TFs contained within the abovementioned tissue-specific genes. Among the genes showing tissue-specific expression in petals, flowers, and roots, there was a relatively greater abundance of TFs such as MYB and MYB_related (Fig. 2E). Furthermore, ERFs were exclusively found in reproductive-related tissues such as petals, sepals, and flowers. These TFs most likely play important regulatory roles in growth, development, and secondary metabolite synthesis in their corresponding tissues.
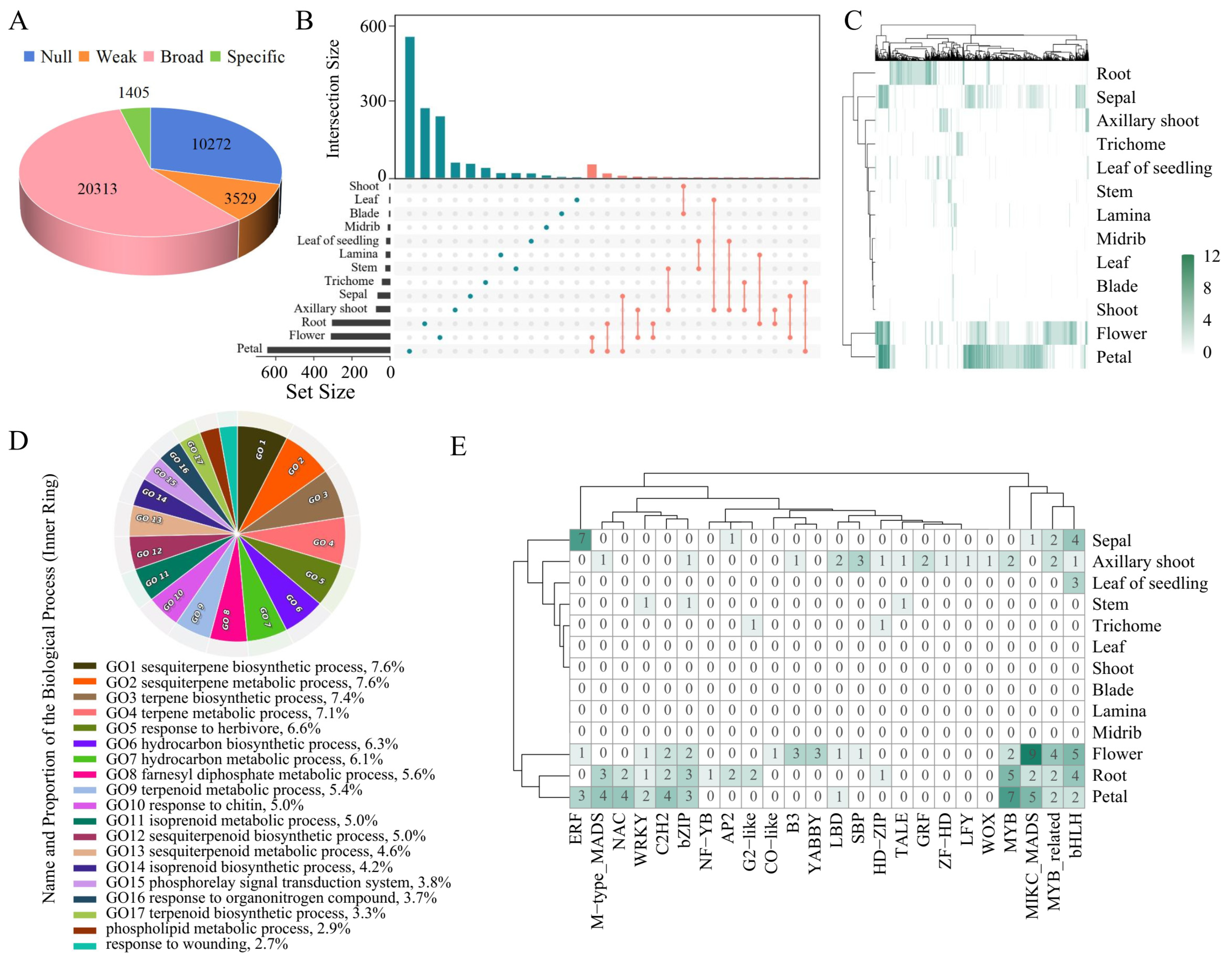
Figure 2: Specific expression patterns of genes across 13 tobacco tissues. (A) Numbers of genes in four groups: null, weak, broad, and tissue specific. (B) Quantity and intersection relationships of tissue-specific genes across various tissues. The left bar plot of the UpSet plot represents the number of tissue-specific genes for each tissue, whereas the top bar plot shows the number of tissue-specific genes shared between different tissues. The dots and lines in the figure represent shared tissue-specific genes among these tissues. (C) Heatmap of the median TPM of tissue-specific genes in each tissue. (D) GO biological process (BP) enrichment terms for genes specifically expressed in sepals. (E) Identification of tissue-specific TFs in tissues.
3.3 Cross-Stress Gene Expression Patterns
As sessile organisms, plants face numerous biotic and abiotic stresses, so they have evolved a wide range of mechanisms to respond to and minimize these stresses. The transcriptome data collected in this study, including data from 29 biotic and abiotic stresses in roots, leaves, and axillary shoots, were manually curated (Supplementary Table S6). This provides an opportunity to identify various stress-responsive genes and outline relationships between these genes that single-stress studies cannot reveal. The stress studies conducted on leaves primarily involved investigations into resistance mechanisms against drought stress induced by salicylic acid (SA), melatonin (MEL), and abscisic acid (ABA) as well as resistance to cold and salt stress. The stress studies conducted on root tissues mainly involved investigations into resistance to Phytophthora nicotianae and high and low concentrations of nitrate (N), whereas axillary shoot studies have primarily focused on stresses associated with topping treatments.
Transcriptomic analysis comparing various stress treatment groups with the control group revealed significant differences in the number of genes responsive to different external stresses in tobacco (Supplementary Table S7). The quantity of DEGs shows a gradual increase in response gene numbers with increasing duration or intensity of the same type of stress. In particular, when tobacco leaves were subjected to 4°C environmental treatment for 0.5, 1, 2, 4, or 8 h, the number of DEGs gradually increased (Fig. 3A). In this study, the number of upregulated and downregulated DEGs between the stress treatment groups and control group samples generally ranged from 1 to 7-fold. Taking Phytophthora nicotianae infection as an example, as the stress increased, the log2FoldChange also increased accordingly (Fig. 3B). GO enrichment analysis was performed separately for genes under various stresses (Supplementary Figs. S3 and S4, Supplementary Table S8), revealing that many upregulated genes under adverse stress conditions were enriched in stress-related GO terms such as those related to drought response and auxin response. For example, under drought stress, the differentially expressed genes were enriched in the response to water category, including a histone deacetylase (Nitab4.5_0001922g0030), a gene widely known to participate in drought tolerance across plant species [40]. Under biotic stress treatments, the differentially expressed genes were enriched in GO terms such as response to bacterium, defense response to bacterium, and response to karrikin. Notably, these categories contained a shared gene, NB-LRR (Nitab4.5_0000068g0290), which encodes a key immune protein broadly involved in plant defense [41].
Genes with TPM expression levels less than 0.1 in all samples were filtered out, and the similarity of gene response patterns between different treatments was assessed based on the log2FoldChange. In the roots, regardless of the N concentration, there was a strong positive correlation between the different treatments based on the log2FoldChange (Fig. 3C). Based on the number of DEGs and their log2FoldChange, although there were differences in the number of DEGs under varying concentrations and durations of nitrate treatment, the direction of the gene response (upregulation or downregulation) and the log2FoldChange were similar (Fig. 3A,B). Moreover, in tobacco roots infected by Phytophthora nicotianae, the direction and magnitude of gene responses were strongly correlated with those after nitrate treatment. This suggests that although nitrate is a nonbiological stress and Phytophthora nicotianae infection is a biotic stress, they may stimulate some common response mechanisms. However, in the case of the root treatments under the shoot topping treatment, the direction and log2FoldChange of stress-responsive genes were negatively correlated with those under the N and Phytophthora nicotianae treatments, reflecting differences in gene response patterns between the different stresses.
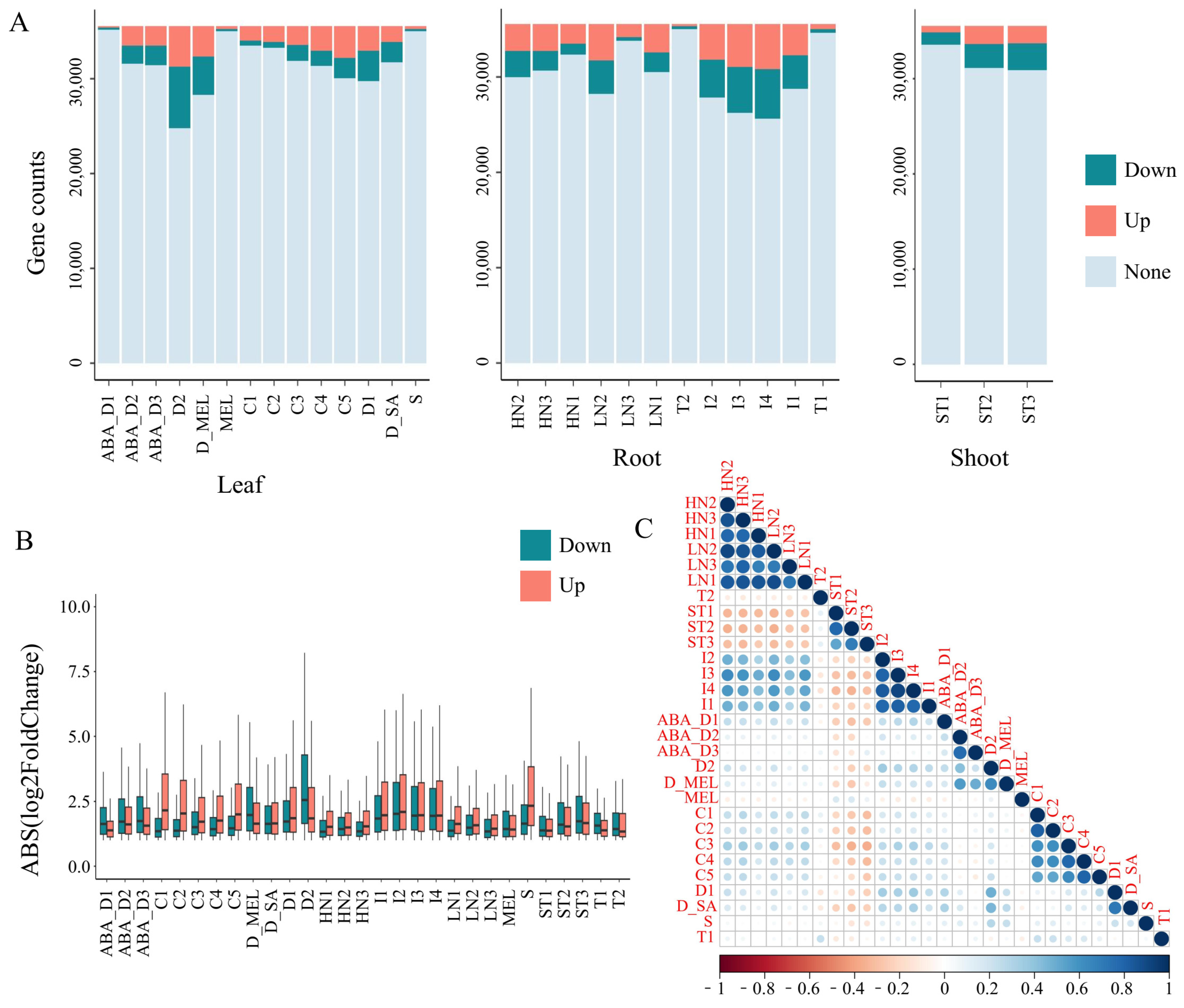
Figure 3: Analysis of response patterns of genes under multiple stress treatments. Abbreviations for the labels on the horizontal axis are shown in Supplementary Table S6. (A) The variation of genes under treatments in different tissues. “Down” represents downregulated genes, “Up” represents upregulated genes, and “None” represents genes whose expression did not significantly change. The y-axis represents the gene counts. (B) Log2FoldChange in gene expression in different tissues and treatments. The x-axis represents the groups of different treatments for different tissues, and the y-axis represents the absolute values of the log2FoldChange of the DEGs belonging to this group. The whiskers represent the range from the first quartile to the minimum value of the dataset and from the third quartile to the maximum value of the dataset. (C) Correlation analysis of changes in gene expression levels under multiple stress treatments.
The response to shoot topping treatment in roots was not only negatively correlated with the response of genes under other stresses in roots but also negatively correlated with the response of genes under various stresses in tobacco leaves, such as those induced by cold stress. Additionally, in leaves, there was a correlation between samples subjected to drought stress alone and those treated with MEL and SA, but the correlation between gene expression under drought stress after ABA treatment and drought stress alone was weaker, possibly because ABA can enhance the drought resistance of plants. In summary, understanding the response mechanisms of genes to different stresses plays a crucial role in enhancing the stress tolerance of tobacco plants.
3.4 TF Regulatory Network in Tobacco Roots and Leaves
TFs play pivotal roles in the regulation processes of tobacco. In this study, we analysed tobacco gene expression under various stress conditions. Initially, 1865 TFs were identified in the tobacco genome using the PlantTFDBdatabase. The number of differentially expressed TFs varied with the type of stress treatment. There were more differentially expressed TFs in the roots under N treatment and topping treatment, and in the leaves under drought treatment and 4-h and 8-h freezing treatments (Fig. 4A,B). The distribution of differentially expressed TFs was similar to the overall distribution trends of DEGs, with an increase in the number of differentially expressed TFs with increasing stress (Fig. 3A and Fig. 4A). Notably, in the drought stress treatment of leaves, the application of hormones resulted in a decrease in the number of differentially expressed TFs compared with those in the drought treatment without hormones, suggesting that these TFs with altered expression levels may be involved in the hormone response. Additionally, both the roots and leaves exhibited abundant common responsive genes among the different stress treatments (Supplementary Figs. S5 and S6).
Based on the aforementioned research findings, our study focused on genes that exhibit shared responses to stress in both roots and leaves. In leaves, the ABA_D1 and S treatments resulted in fewer DEGs (376 and 493, respectively), indicating that these stresses were not sufficiently intense to fully activate defence mechanisms. Furthermore, MEL is not a stress. Similarly, the T1 and T2 treatments resulted in a low number of DEGs in the roots, and these treatments resulted in root stress. Therefore, data from these conditions were excluded. There were 338 and 24 DEGs shared among the root and leaf tissues under the various stress treatments. To understand the regulatory mechanisms of genes responding to multiple stresses, this study calculated the PCC of TFs and the DEGs shared by roots and leaves under different treatments. Gene pairs with a PCC greater than or equal to 0.9 were considered coexpressed; i.e., the TFs within the gene pairs may regulate the DEGs (Supplementary Tables S9 and S10). In the roots, we identified 285 TFs regulating 132 DEGs, including more than 40 different families of TFs such as WRKY, MYB, and bHLH (Fig. 4C). This suggests the presence of a complex stress resistance regulatory network in tobacco roots. Numerous response genes are shared between biotic and abiotic stressors, and these genes are also commonly regulated by various TFs. In the leaves, 25 TFs, including WRKY, MYB, and ERF, regulated the expression of 4 stress-responsive genes (Fig. 4D). These genes encode proteins such as F-box PP2-A13-like and G-type lectin S-receptor-like serine threonine-protein kinase, which are considered important stress resistance candidate genes in plant leaves, according to many studies [42,43]. Additionally, we predicted common stable motifs in the 2 kb upstream region of the DEGs. Due to the stringent coexpression PCC threshold of 0.9, which resulted in a very small number of TFs that could bind to these common stable motifs in the DEGs, we relaxed the threshold to 0.8. Combined with the coexpression results, we identified 26 TFs in leaves and 21 in roots that can bind to these common stable motifs among the DEGs (Supplementary Figs. S7 and S8, Supplementary Table S11). This result provides valuable resources for analysing gene regulation in response to stress. In summary, these results provide an opportunity for a more comprehensive understanding of the functions of TFs in stress responses.
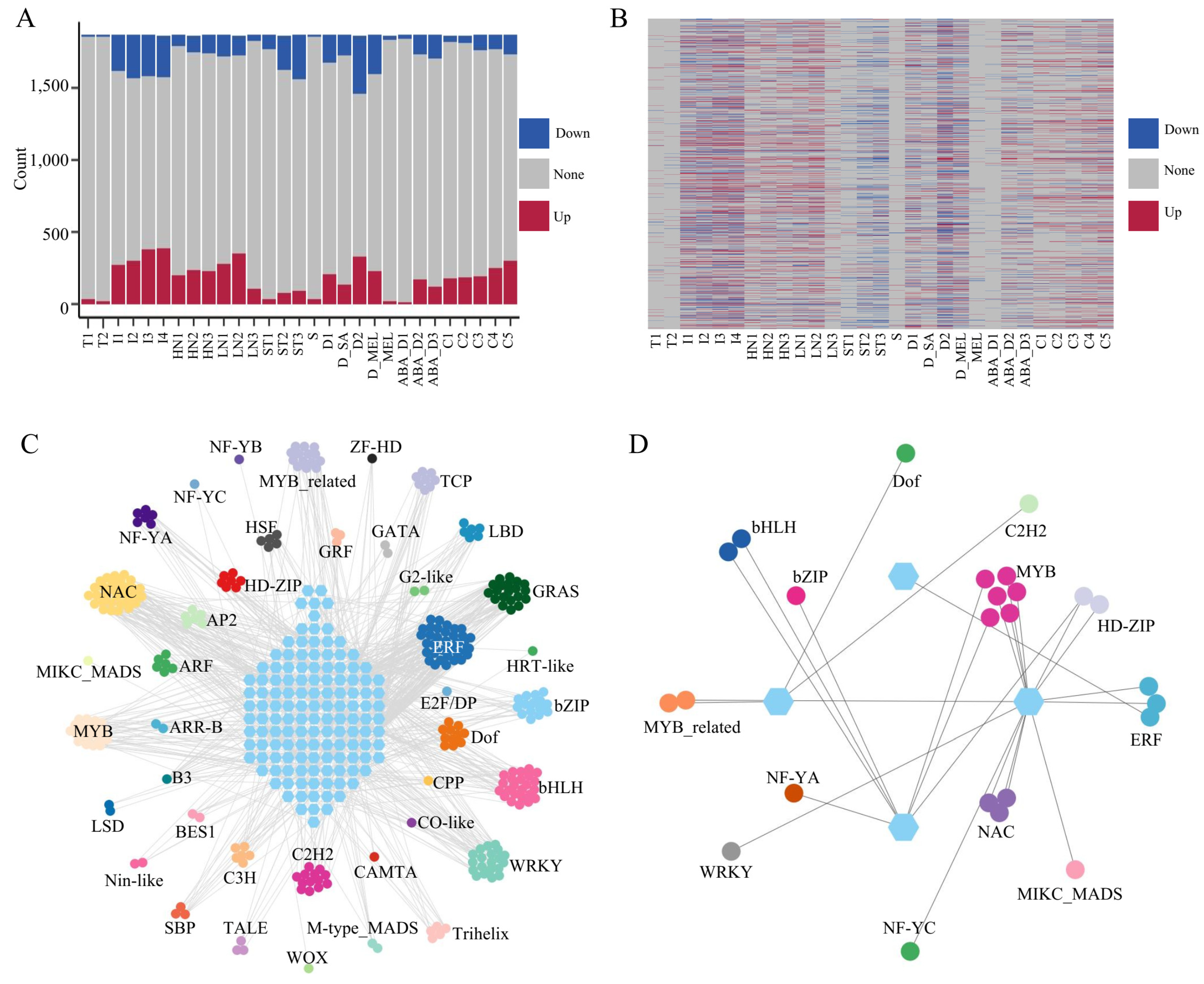
Figure 4: The expression patterns of TFs under various stress conditions. Abbreviations for the labels on the horizontal axis are shown in Supplementary Table S6. (A) The variations in TFs in different tissues under different treatments. “Up” represents upregulated TFs, “Down” represents downregulated TFs, and “None” represents TFs with no significant change in expression. (B) Heatmap of TF expression patterns under different stress conditions. (C) The network of TF-regulated stress response genes in roots. The hexagons represent DEGs, and the circles represent TFs. The coexpression threshold was set to 0.9. (D) The network of TF-regulated stress response genes in leaves. The hexagons represent DEGs, and the circles represent TFs. The coexpression threshold was set to 0.9.
Tobacco leaves are key organs used in cigarette processing, and the availability of 379 leaf samples enabled a population-level analysis of the relationship between gene expression and genetic polymorphisms. From the RNA-seq data, a total of 598,293 high-quality SNPs were identified across the tobacco genome. Their chromosomal distribution revealed clear regional enrichment on chromosomes 2, 4, 12, 14, and 19 (Fig. 5A), suggesting that SNP density varies among chromosomes, likely reflecting differences in structural features and evolutionary histories.
To dissect how these genetic variants influence transcriptional regulation, we performed eQTL mapping using leaf tissues and identified 28,396 eQTLs (Supplementary Table S12). Based on their physical proximity to associated genes, these loci were classified as cis- or trans-eQTLs. The cis-eQTLs displayed a strong diagonal pattern, whereas trans-eQTLs were widely dispersed and formed several distinct regulatory bands (Fig. 5B), highlighting clear differences in regulatory architecture. In total, 14,559 genes (41% of all expressed genes) were regulated by at least one eQTL, indicating that genetic variation extensively shapes gene expression in tobacco leaves. Among these eGenes, 53% were regulated exclusively by cis-eQTLs, 21% only by trans-eQTLs, and 26% by both (Fig. 5C). On average, each gene was associated with 4.6 eQTLs, and only 5771 genes were influenced by a single locus (Fig. 5D), underscoring the complexity of regulatory interactions involving both local and distal genetic effects.
To further characterize the biological roles of these eGenes, we conducted functional enrichment analyses. Trans-regulated eGenes showed strong overrepresentation in biological process categories, with limited enrichment in molecular function and cellular component terms (Supplementary Fig. S9). Many trans-eGenes were involved in photosynthesis-related pathways, including photosynthesis, light reaction, photosynthesis, light harvesting, tetrapyrrole metabolic process, and chloroplast organization. Additionally, they were enriched in hormone signaling and stress-responsive processes such as response to fatty acid, response to jasmonic acid, response to herbicide, intracellular monoatomic cation homeostasis, and protein K63-linked ubiquitination (Supplementary Fig. S9). Several eGenes were also associated with nicotine biosynthesis (Supplementary Table S13), providing candidate genes relevant to tobacco trait improvement.
We further identified 1669 cis-eQTL hotspots and 1858 trans-eQTL hotspots across the genome (Fig. 5E). Notably, the trans-eQTL hotspots contained 68 transcription factors, including members of the WRKY, bHLH, and MYB families (Supplementary Table S14), suggesting that these regions may exert broad regulatory influence through transcription factor–mediated cascades. For example, within the trans-eQTL hotspot I47_C2, the locus eQTL (chr04:1227307) cis-regulated the transcription factor MYB (Nitab4.5_0001158g0050), a TF related to drought responses, while simultaneously trans-regulating a drought-induced protein (Nitab4.5_0001808g0020). This illustrates how single regulatory variants can coordinate hierarchical control of stress-responsive pathways. These findings provide a comprehensive view of the genetic regulation of gene expression in tobacco leaves.
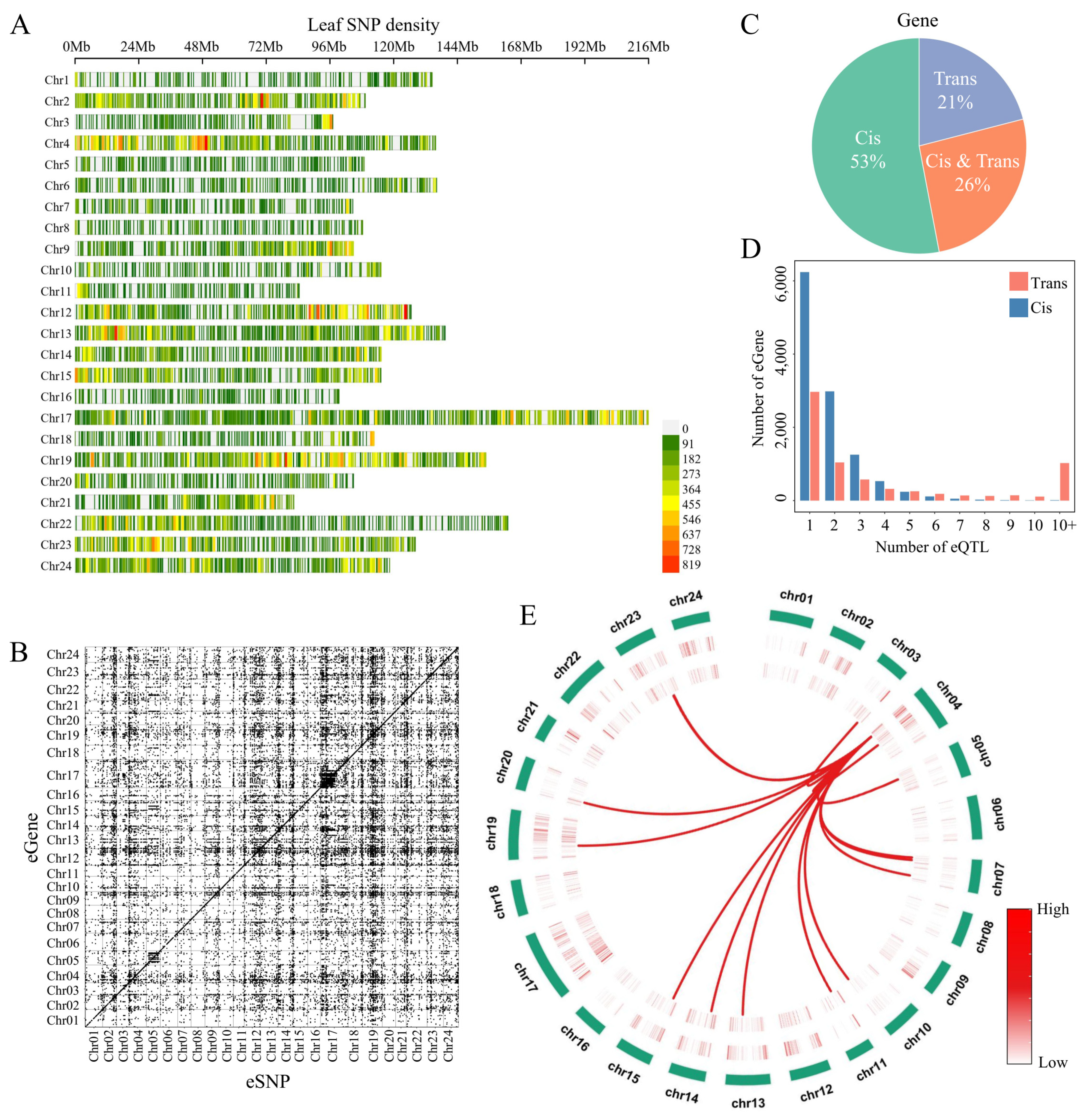
Figure 5: (A) The distribution of tobacco SNPs across chromosomes. (B) The distribution of cis- and trans-eQTLs on chromosomes. (C) The distribution of genes regulated by cis- or trans-eQTLs in tobacco leaves. (D) Relationships between the number of eGenes and eQTLs. (E) Circos plot illustrating the genomic landscape of eQTL hotspots in tobacco. From the outermost to the innermost rings: the outer ring represents the 24 tobacco chromosomes; the second ring shows the genomic density of cis-eQTL hotspots; the third ring displays the density of trans-eQTL hotspots; the innermost layer depicts the trans-eQTL hotspot I47_C2, with lines connecting this hotspot to its significantly associated downstream genes across the genome.
Comprehending the expression characteristics of genes in different plant organs, particularly under various stresses, is highly important [44]. However, it is challenging for a single laboratory to construct a large experimental design to conduct transcriptome profiling on hundreds or even thousands of samples. Transcriptome sequencing with small sample sizes has long been a common biological research tool, accumulating extensive transcriptomic data, and thereby providing opportunities for large-scale expression profiling studies in tobacco. Indeed, such opportunities are equally applicable to many other species [7]. This study collected a large quantity of tobacco transcriptome data, which also serves as a summary of tobacco transcriptomics, to provide a clearer understanding of the current status of the tobacco genome.
The comprehensive comparative transcriptomic analysis yielded numerous intriguing findings such as the enrichment of genes specifically expressed in tobacco sepals involved in the synthesis pathways of certain terpenoids. Terpenoids play pivotal roles in various biological functions such as attracting pollinators and defending against biotic stresses [45,46]. This finding is particularly intriguing because the synthesis and accumulation of secondary metabolites in plants often exhibit strong tissue specificity [47,48]. Furthermore, many of these metabolites often possess significant chemical and ecological functions [49]. The abundant tissue-specific expression patterns provide crucial indications that employing molecular biology, metabolomics, and synthetic biology approaches to study the synthesis pathways and ecological functions of secondary metabolites in tobacco sepals holds significant value. Although the potential for terpenoid synthesis in sepals has been noted in other species, further in-depth investigations into their functionalities are lacking [50]. Furthermore, this study identified 128 tissue-specific TFs, among which MYB plays significant roles in regulating the cell cycle, secondary metabolite synthesis, and plant response to hormones [51,52]. Considering the tissue-specific expression patterns of genes related to plant secondary metabolites, these MYBs could serve as key regulators of secondary metabolite synthesis and are worthy of focused attention. ERFs are a family of TFs that are mainly present in plants and play crucial roles in plant growth and development, hormone responses, and plant stress. In many reports, ERFs are believed to be associated with flower growth, development, floral morphology, and flower senescence [53,54]. Thus, it is reasonable to speculate that ERFs that are specifically expressed in floral organs might be involved in tobacco flowering regulation. These findings provide guidance and candidate genes for studying tobacco development and defence mechanisms.
The regulatory networks and mechanisms underlying plant responses to biotic and abiotic stresses are complex [55]. Understanding the similarities and differences in the response mechanisms of plants to these various stresses will help elucidate how plants integrate and respond to various environmental cues [44]. Although we cannot simultaneously impose different stresses on a single plant through data collection and analysis, we can delineate the response patterns of genes to stress. For instance, following the infection of tobacco roots by Phytophthora nicotianae, although the number of responsive genes differed from that induced by N treatment, the direction and fold change of these responsive genes exhibited a significant correlation. This finding implies the potential existence of overlap in response mechanisms between biotic and abiotic stresses. Through the above analysis, this study revealed that some genes may have universal responses to adversity. This understanding is valuable for understanding the mechanisms of tobacco response to different adverse conditions and for breeding tobacco varieties with multiple stress resistance mechanisms. The RNA-seq data utilized in this study originated from diverse samples, thereby enabling investigations into the impact of genomic variation on gene expression. Given the significance of tobacco leaves and the availability of data, this study conducted an analysis of eQTLs using RNA-seq data derived from tobacco leaves. Examination of the distance between cis-eQTLs and transcription start sites (TSSs) revealed a gradual decrease in the number of eQTLs as the distance from the TSS increased within a range of 250 K. Beyond 250 K from the TSSs, the count of eQTLs remained relatively low, possibly due to the decreasing impact of SNPs on TSSs as distance increased. Within 250 K, a closer distance had a greater potential impact on the TSSs (Supplementary Fig. S10). This study provides further insights into the polymorphisms of the tobacco genome and an overview of gene expression regulatory mechanisms. Utilizing large datasets from databases for integration and analysis contributes to a better understanding of the genetic architecture controlling agronomic traits, thereby facilitating crop improvement and quality enhancement.
This study integrated large-scale transcriptome data involving multiple tobacco tissues and different treatments. A total of 1405 tissue-specific genes were identified. This study revealed the specific synthesis and accumulation of secondary metabolites such as terpenoids in organs such as sepals, providing important insights into the functionality of different tobacco organs. Additionally, comparative analysis of RNA-seq data under various stress conditions revealed gene differences between leaves and roots in response to biotic and abiotic stresses, offering crucial clues for understanding tobacco mechanisms in response to diverse stresses. Finally, by identifying 28,396 eQTLs in leaves, much information was provided on the genetic regulation of gene expression in tobacco leaves. In summary, this study provides important research references for understanding the characteristics of tissue-specific genes and their potential functions in tobacco, laying a foundation for a deeper understanding of tobacco biology and genetic regulatory mechanisms.
Acknowledgement:
Funding Statement: This work was supported by the Guizhou Provincial Basic Research Program (Natural Science) [(2024) 648], the Program of China National Tobacco Corporation (110202101032(JY-09), 110202201003(JY-03)), the Program of Guizhou Branch of China National Tobacco Corporation (2023XM02, 2022XM05 and 2024XM01), the Qiankehe Platform Project (ZSYS [2025] 028), and the Program of China National Tobacco Corporation (110202102034).
Author Contributions: Shizhou Yu and Zhixiao Yang: Conceptualization, Methodology, Supervision, Writing—review & editing and Funding acquisition. Jie Zhang and Linggai Cao: Data curation, Visualization, Writing-Original draft preparation. Xueliang Ren and Jie Liu: Data curation, Visualization. Jiemeng Tao and Peng Lu: Data curation, Supervision. Guobo Chen: Writing-Reviewing and Editing, Supervision. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: The authors confirm that the data supporting the findings of this study are available within the Supplementary Materials.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest.
Supplementary Materials: The supplementary material is available online at https://www.techscience.com/doi/10.32604/phyton.2026.073509/s1. Supplementary Table S1 Information on tissues of samples used in this study. Supplementary Table S2 TSI values in different tissues. Supplementary Table S3 Expression-based gene classification. Supplementary Table S4 Statistics of tissue-specific genes in each tissue. Supplementary Table S5 Go enrichment results for tissue-specific genes. Supplementary Table S6 Information on treatments of samples used in this study. Supplementary Table S7 Differentially expressed genes under different treatments. Supplementary Table S8 Go enrichment results for differentially expressed genes. Supplementary Table S9 Co-expression network of TFs and differentially expressed genes in roots. Supplementary Table S10 Co-expression network of TFs and differentially expressed genes in leaves. Supplementary Table S11 Co-expression network of DEGs and TFs that could bind to the common stable motifs of DEGs in tobacco roots and leaves (the co-expression PCC threshold was set to 0.8). Supplementary Table S12 Lead SNPs identified in this study. Supplementary Table S13 eQTLs associated with nicotine synthesis genes. Supplementary Table S14 Genes regulated by eQTL hotspot regions. Supplementary Fig. S1 tSNE clustering results based on expression levels of 84 samples with different treatments. Supplementary Fig. S2 GO enrichment analysis of tissue-specific genes. Supplementary Fig. S3 GO enrichment analysis of up-regulated differentially expressed genes. Supplementary Fig. S4 GO enrichment analysis of down-regulated differentially expressed genes. Supplementary Fig. S5 the left bar plot of the UpSet plot represents the number of DEGs for each treatment, whereas the top bar plot shows the number of DEGs shared between different treatments. The dots and lines in the figure represent DEGs shared among these treatments. Supplementary Fig. S6 the root bar plot of the UpSet plot represents the number of DEGs for each treatment, whereas the top bar plot shows the number of DEGs shared between different treatments. The dots and lines in the figure represent DEGs shared among these treatments. Supplementary Fig. S7 Co-expression network of DEGs and TFs that could bind to the common stable motifs of DEGs in tobacco leaves (the co-expression PCC threshold was set to 0.8). Supplementary Fig. S8 Co-expression network of DEGs and TFs that could bind to the common stable motifs of DEGs in tobacco roots (the co-expression PCC threshold was set to 0.8). Supplementary Fig. S9 GO enrichment analysis of trans-eGenes in leaves. Supplementary Fig. S10 Density curve of distance from transcription start sites for cis-eQTLs in leaves. Supplementary File: (Gene expression level in different samples) can be downloaded from https://figshare.com (https://doi.org/10.6084/m9.figshare.29666060)
Nomenclature
| eQTLs | Expression quantitative trait loci |
| SNP | Single nucleotide polymorphism |
| tSNE | T-distributed stochastic neighbor embedding |
| UMAP | Uniform Manifold Approximation and Projection |
| TPM | Transcripts per million |
| TSI | Tissue specific index |
| GEC | Genetic Type I error calculator |
| GO | Gene ontology |
| TSSs | Transcription Start Sites |
| SA | Salicylic acid |
| MEL | Melatonin |
| ABA | Abscisic acid |
References
1. Sinha P, Bajaj P, Pazhamala LT, Nayak SN, Pandey MK, Chitikineni A, et al. Arachis hypogaea gene expression atlas for fastigiata subspecies of cultivated groundnut to accelerate functional and translational genomics applications. Plant Biotechnol J. 2020;18(11):2187–200. doi:10.1111/pbi.13374. [Google Scholar] [CrossRef]
2. Liu C, Leng J, Li Y, Ge T, Li J, Chen Y, et al. A spatiotemporal atlas of organogenesis in the development of orchid flowers. Nucleic Acids Res. 2022;50(17):9724–37. doi:10.1093/nar/gkac773. [Google Scholar] [CrossRef]
3. Meir Z, Aviezer I, Chongloi GL, Ben-Kiki O, Bronstein R, Mukamel Z, et al. Dissection of floral transition by single-meristem transcriptomes at high temporal resolution. Nat Plants. 2021;7(6):800–13. doi:10.1038/s41477-021-00936-8. [Google Scholar] [CrossRef]
4. George N, Fexova S, Fuentes AM, Madrigal P, Bi Y, Iqbal H, et al. Expression Atlas update: insights from sequencing data at both bulk and single cell level. Nucleic Acids Res. 2024;52(D1):D107–14. doi:10.1093/nar/gkad1021. [Google Scholar] [CrossRef]
5. Tian F, Yang DC, Meng YQ, Jin J, Gao G. PlantRegMap: charting functional regulatory maps in plants. Nucleic Acids Res. 2020;48(D1):D1104–13. doi:10.1093/nar/gkz1020. [Google Scholar] [CrossRef]
6. Jin L, Tang Q, Hu S, Chen Z, Zhou X, Zeng B, et al. A pig BodyMap transcriptome reveals diverse tissue physiologies and evolutionary dynamics of transcription. Nat Commun. 2021;12(1):3715. doi:10.1038/s41467-021-23560-8. [Google Scholar] [CrossRef]
7. Almeida-Silva F, Pedrosa-Silva F, Venancio TM. The Soybean Expression Atlas v2: a comprehensive database of over 5000 RNA-seq samples. Plant J. 2023;116(4):1041–51. doi:10.1111/tpj.16459. [Google Scholar] [CrossRef]
8. Xanthopoulou A, Montero-Pau J, Picó B, Boumpas P, Tsaliki E, Paris HS, et al. A comprehensive RNA-Seq-based gene expression atlas of the summer squash (Cucurbita pepo) provides insights into fruit morphology and ripening mechanisms. BMC Genomics. 2021;22(1):341. doi:10.1186/s12864-021-07683-2. [Google Scholar] [CrossRef]
9. Liu S, Gao Y, Canela-Xandri O, Wang S, Yu Y, Cai W, et al. A multi-tissue atlas of regulatory variants in cattle. Nat Genet. 2022;54(9):1438–47. doi:10.1038/s41588-022-01153-5. [Google Scholar] [CrossRef]
10. Teng J, Gao Y, Yin H, Bai Z, Liu S, Zeng H, et al. A compendium of genetic regulatory effects across pig tissues. Nat Genet. 2024;56(1):112–23. doi:10.1038/s41588-023-01585-7. [Google Scholar] [CrossRef]
11. Liu Z, Wu X, Hou L, Ji S, Zhang Y, Fan W, et al. Effects of cadmium on transcription, physiology, and ultrastructure of two tobacco cultivars. Sci Total Environ. 2023;869:161751. doi:10.1016/j.scitotenv.2023.161751. [Google Scholar] [CrossRef]
12. Luo Z, Zhou Z, Li Y, Tao S, Hu ZR, Yang JS, et al. Transcriptome-based gene regulatory network analyses of differential cold tolerance of two tobacco cultivars. BMC Plant Biol. 2022;22(1):369. doi:10.1186/s12870-022-03767-7. [Google Scholar] [CrossRef]
13. Edwards KD, Fernandez-Pozo N, Drake-Stowe K, Humphry M, Evans AD, Bombarely A, et al. A reference genome for Nicotiana tabacum enables map-based cloning of homeologous loci implicated in nitrogen utilization efficiency. BMC Genomics. 2017;18(1):448. doi:10.1186/s12864-017-3791-6. [Google Scholar] [CrossRef]
14. Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90. doi:10.1093/bioinformatics/bty560. [Google Scholar] [CrossRef]
15. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi:10.1093/bioinformatics/bts635. [Google Scholar] [CrossRef]
16. Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5. doi:10.1038/nbt.3122. [Google Scholar] [CrossRef]
17. Krijthe JH, Van der Maaten L. Rtsne: T-distributed stochastic neighbor embedding using Barnes-Hut implementation. Package ‘Rtsne’ [Internet]. [cited 2026 Jan 1]. Available from: https://cran.r-project.org/web/packages/Rtsne/Rtsne.pdf. [Google Scholar]
18. McInnes L, Healy J, Saul N, Großberger L. UMAP: uniform manifold approximation and projection. J Open Source Softw. 2018;3(29):861. doi:10.21105/joss.00861. [Google Scholar] [CrossRef]
19. García-Pérez R, Esteller-Cucala P, Mas G, Lobón I, Di Carlo V, Riera M, et al. Epigenomic profiling of primate lymphoblastoid cell lines reveals the evolutionary patterns of epigenetic activities in gene regulatory architectures. Nat Commun. 2021;12(1):3116. doi:10.1038/s41467-021-23397-1. [Google Scholar] [CrossRef]
20. Cantalapiedra CP, Hernández-Plaza A, Letunic I, Bork P, Huerta-Cepas J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol. 2021;38(12):5825–9. doi:10.1093/molbev/msab293. [Google Scholar] [CrossRef]
21. Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2(3):100141. doi:10.1016/j.xinn.2021.100141. [Google Scholar] [CrossRef]
22. Kuznetsova I, Lugmayr A, Siira SJ, Rackham O, Filipovska A. CirGO: an alternative circular way of visualising gene ontology terms. BMC Bioinform. 2019;20(1):84. doi:10.1186/s12859-019-2671-2. [Google Scholar] [CrossRef]
23. Jin J, Tian F, Yang DC, Meng YQ, Kong L, Luo J, et al. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017;45(D1):D1040–5. doi:10.1093/nar/gkw982. [Google Scholar] [CrossRef]
24. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30. doi:10.1093/bioinformatics/btt656. [Google Scholar] [CrossRef]
25. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8. [Google Scholar] [CrossRef]
26. Wei T, Simko V, Levy M, Xie Y, Jin YJ, Zemla J. Visualization of a Correlation Matrix. R Package “corrplot” [Internet]. [cited 2026 Jan 1]. Available from: https://github.com/taiyun/corrplot. [Google Scholar]
27. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7. doi:10.1089/omi.2011.0118. [Google Scholar] [CrossRef]
28. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–89. doi:10.1016/j.molcel.2010.05.004. [Google Scholar] [CrossRef]
29. Kohl M, Wiese S, Warscheid B. Cytoscape: software for visualization and analysis of biological networks. In: Hamacher M, Eisenacher M, Stephan C, editors. Data mining in proteomics. Totowa, NJ, USA: Humana Press; 2011. p. 291–303. doi:10.1007/978-1-60761-987-1_18. [Google Scholar] [CrossRef]
30. Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10(2):giab008. doi:10.1093/gigascience/giab008. [Google Scholar] [CrossRef]
31. Brouard JS, Schenkel F, Marete A, Bissonnette N. The GATK joint genotyping workflow is appropriate for calling variants in RNA-seq experiments. J Anim Sci Biotechnol. 2019;10:44. doi:10.1186/s40104-019-0359-0. [Google Scholar] [CrossRef]
32. Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8. doi:10.1093/bioinformatics/btr330. [Google Scholar] [CrossRef]
33. Ongen H, Buil A, Brown AA, Dermitzakis ET, Delaneau O. Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics. 2016;32(10):1479–85. doi:10.1093/bioinformatics/btv722. [Google Scholar] [CrossRef]
34. Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, et al. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42(4):348–54. doi:10.1038/ng.548. [Google Scholar] [CrossRef]
35. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. doi:10.1086/519795. [Google Scholar] [CrossRef]
36. Fan J, Shen Y, Chen C, Chen X, Yang X, Liu H, et al. A large-scale integrated transcriptomic atlas for soybean organ development. Mol Plant. 2025;18(4):669–89. doi:10.1016/j.molp.2025.02.003. [Google Scholar] [CrossRef]
37. Marcon A, Romañach LG, André D, Ding J, Zhang B, Hvidsten TR, et al. A transcriptional roadmap of the yearly growth cycle in Populus trees. Plant Cell. 2025;37(9):koaf208. doi:10.1093/plcell/koaf208. [Google Scholar] [CrossRef]
38. Slavković F, Bendahmane A. Floral phytochemistry: impact of volatile organic compounds and nectar secondary metabolites on pollinator behavior and health. Chem Biodivers. 2023;20(4):e202201139. doi:10.1002/cbdv.202201139. [Google Scholar] [CrossRef]
39. Dötterl S, Gershenzon J. Chemistry, biosynthesis and biology of floral volatiles: roles in pollination and other functions. Nat Prod Rep. 2023;40(12):1901–37. doi:10.1039/D3NP00024A. [Google Scholar] [CrossRef]
40. Li S, He X, Gao Y, Zhou C, Chiang VL, Li W. Histone acetylation changes in plant response to drought stress. Genes. 2021;12(9):1409. doi:10.3390/genes12091409. [Google Scholar] [CrossRef]
41. Dangl JL, Jones JD. Plant pathogens and integrated defence responses to infection. Nature. 2001;411(6839):826–33. doi:10.1038/35081161. [Google Scholar] [CrossRef]
42. de Freitas ST, Martinelli F, Feng B, Reitz NF, Mitcham EJ. Transcriptome approach to understand the potential mechanisms inhibiting or triggering blossom-end rot development in tomato fruit in response to plant growth regulators. J Plant Growth Regul. 2018;37(1):183–98. doi:10.1007/s00344-017-9718-2. [Google Scholar] [CrossRef]
43. Pignocchi C, Fletcher JM, Wilkinson JE, Barnes JD, Foyer CH. The function of ascorbate oxidase in tobacco. Plant Physiol. 2003;132(3):1631–41. doi:10.1104/pp.103.022798. [Google Scholar] [CrossRef]
44. Tan QW, Lim PK, Chen Z, Pasha A, Provart N, Arend M, et al. Cross-stress gene expression atlas of Marchantia polymorpha reveals the hierarchy and regulatory principles of abiotic stress responses. Nat Commun. 2023;14(1):986. doi:10.1038/s41467-023-36517-w. [Google Scholar] [CrossRef]
45. Li C, Zha W, Li W, Wang J, You A. Advances in the biosynthesis of terpenoids and their ecological functions in plant resistance. Int J Mol Sci. 2023;24(14):11561. doi:10.3390/ijms241411561. [Google Scholar] [CrossRef]
46. Cao Y, Liu L, Ma K, Wang W, Lv H, Gao M, et al. The jasmonate-induced bHLH gene SlJIG functions in terpene biosynthesis and resistance to insects and fungus. J Integr Plant Biol. 2022;64(5):1102–15. doi:10.1111/jipb.13248. [Google Scholar] [CrossRef]
47. Jiang C, Fei X, Pan X, Huang H, Qi Y, Wang X, et al. Tissue-specific transcriptome and metabolome analyses reveal a gene module regulating the terpenoid biosynthesis in Curcuma wenyujin. Ind Crops Prod. 2021;170:113758. doi:10.1016/j.indcrop.2021.113758. [Google Scholar] [CrossRef]
48. Zhang K, Wang N, Gao X, Ma Q. Integrated metabolite profiling and transcriptome analysis reveals tissue-specific regulation of terpenoid biosynthesis in Artemisia argyi. Genomics. 2022;114(4):110388. doi:10.1016/j.ygeno.2022.110388. [Google Scholar] [CrossRef]
49. Cheng AX, Lou YG, Mao YB, Lu S, Wang LJ, Chen XY. Plant terpenoids: biosynthesis and ecological functions. J Integr Plant Biol. 2007;49(2):179–86. doi:10.1111/j.1744-7909.2007.00395.x. [Google Scholar] [CrossRef]
50. Li J, Wang Y, Dong Y, Zhang W, Wang D, Bai H, et al. The chromosome-based lavender genome provides new insights into Lamiaceae evolution and terpenoid biosynthesis. Hortic Res. 2021;8(1):53. doi:10.1038/s41438-021-00490-6. [Google Scholar] [CrossRef]
51. Ortigosa A, Fonseca S, Franco-Zorrilla JM, Fernández-Calvo P, Zander M, Lewsey MG, et al. The JA-pathway MYC transcription factors regulate photomorphogenic responses by targeting HY5 gene expression. Plant J. 2020;102(1):138–52. doi:10.1111/tpj.14618. [Google Scholar] [CrossRef]
52. Bessa M, Joaquin M, Tavner F, Saville MK, Watson RJ. Regulation of the cell cycle by B-myb. Blood Cells Mol Dis. 2001;27(2):416–21. doi:10.1006/bcmd.2001.0399. [Google Scholar] [CrossRef]
53. Wang Z, Song G, Zhang F, Shu X, Wang N. Functional characterization of AP2/ERF transcription factors during flower development and anthocyanin biosynthesis related candidate genes in Lycoris. Int J Mol Sci. 2023;24(19):14464. doi:10.3390/ijms241914464. [Google Scholar] [CrossRef]
54. Zhang H, Pan X, Liu S, Lin W, Li Y, Zhang X. Genome-wide analysis of AP2/ERF transcription factors in pineapple reveals functional divergence during flowering induction mediated by ethylene and floral organ development. Genomics. 2021;113(2):474–89. doi:10.1016/j.ygeno.2020.10.040. [Google Scholar] [CrossRef]
55. Gong Z, Xiong L, Shi H, Yang S, Herrera-Estrella LR, Xu G, et al. Plant abiotic stress response and nutrient use efficiency. Sci China Life Sci. 2020;63(5):635–74. doi:10.1007/s11427-020-1683-x. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools