
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Keystone Fungal Endophytes of Panax Plants Drive the Conversion of Ginsenoside Rb1 to Rd
1 Tianjin University of Traditional Chinese Medicine, Tianjin, China
2 Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, State Key Laboratory for Quality Ensurance and Sustainable Use of Dao-di Herbs, Beijing, China
3 Wenshan Miaoxiang Notoginseng Technology, Co., Ltd., Wenshan, China
4 Institute of Sanqi Research, Wenshan University, Wenshan, China
* Corresponding Authors: Guozhuang Zhang. Email: ; Linlin Dong. Email:
(This article belongs to the Special Issue: Endophytic Microbiota: Prospects and Challenges for Application Towards Sustainable Agriculture and Environmental Management)
Phyton-International Journal of Experimental Botany 2026, 95(2), 11 https://doi.org/10.32604/phyton.2026.075657
Received 05 November 2025; Accepted 26 January 2026; Issue published 28 February 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Keystone taxa are critical for microbial community homeostasis and ecological niche interactions. However, the functions and genomic traits of endophytic keystone fungi in plant tissues remain unclear. Via network analysis, this study identified keystone fungi Plectosphaerella (Plec) and Cladosporium (Clad) in roots/leaves of medicinal Panax plants (P. ginseng, P. quinquefolius, P. notoginseng). Both correlated strongly positively with ginsenoside Rd content in respective tissues (ρ > 0.6, p < 0.001). Co-cultivation confirmed their ability to convert ginsenoside Rb1 to Rd, linked to β-glucosidase activity. Whole-genome sequencing/assembly/evolutionary analysis of the two strains elucidated genomic features for their keystone roles and saponin biotransformation. Genome mining found multiple GH3 genes (potential saponin transformers) in both; 11 (Plec) and 5 (Clad) were upregulated by cellobiose. Gene family phylogenetic analysis showed expanded transmembrane transport and environmental response functions. Both also had abundant secondary metabolic gene clusters and secretome genes, linking biotic interaction functions to their keystone roles. In summary, this study shows Panax endophytic keystone fungi can participate in ginsenoside biotransformation and clarifies their genomic traits, offering insights for functional endophytic fungal resource development.Graphic Abstract
Keywords
Supplementary Material
Supplementary Material FilePlant tissues serve as ecological niches for a large and diverse array of endophytic fungi [1]. These microorganisms exert significant influences on plant growth, development, and adaptability through a variety of physiological processes, including the provision of nutrients to the host plant, regulation of host metabolism, and enhancement of stress tolerance [2,3,4,5]. This functional importance makes plant endophytic fungi promising green tools for improving crop traits and enhancing agricultural management [6]. However, the immense diversity of the endophytic fungal community poses significant challenges to identifying members with the desired functionalities. Recent progress in understanding the organization patterns of microbial communities has offered valuable clues for addressing this challenge. It is now widely recognized that microorganisms do not live in isolation, but rather form intricate ecological networks through a variety of interactions, such as mutualism, commensalism, and amensalism [7,8]. From an ecological network perspective, not all members of a microbial community hold equal importance: a small number of microbial taxa, which are referred to as keystone taxa (or hub taxa), exhibit extensive interactions with other microorganisms [9,10]. The keystone taxa are increasingly recognized as pivotal in shaping both the structure and function of microbial communities [11]. Consequently, they should be given priority in the identification, isolation, and application research of functional microorganisms.
Keystone taxa may function as central nodes within microbial communities through various mechanisms, including nutrient provision to other microorganisms, transmission of crucial genetic elements, and modification of niche environments [9]. For instance, Ruminococcus bromii in the human gut is capable of degrading resistant starch and supplying carbon sources to other microbes, thereby functioning as a keystone taxa in shaping the structure of gut microbiota [12]. Streptomyces taxa serve as keystone in soil microbiomes, especially under antibiotic pressure, owing to its vital role in the maintenance and dissemination of antibiotic resistance genes [13]. Moreover, keystone taxa may modify the metabolic landscape in their microenvironment by the production of specific enzymes, thereby promoting or inhibiting the proliferation of other microorganisms indirectly [9]. This functional potential is of particular significance for endophytic communities residing within plant tissues, as plants commonly produce diverse specialized metabolites that act as chemical defenses against microbes [14]. However, the association between keystone endophytes and plant specialized metabolites remains poorly understood. This knowledge is crucial for harnessing functional microbes that can transform plant specialized metabolites, thereby enabling the production of high-value rare compounds from more abundant precursors. Network analysis-based topology scoring is an important approach for the identification of keystone taxa in complicate microbial communities [10,13,15]. This strategy has been widely applied to the identification of keystone taxa in a variety of microbial communities, including plant microbiomes, and has generated many valuable insights. For example, by integrating degree and betweenness centrality measures [16], the identified multiple keystone taxa exhibited important ecological functions in the soil microbiome, including members from Micrococcaceae and Nocardioidaceae.
Another major challenge in the study of keystone taxa is to gain a comprehensive understanding of their functional potential in order to elucidate the underlying mechanisms for their keystone roles. Genomics and comparative genomics offer powerful tools for elucidating the functional potential of endophytic keystone fungi. A comprehensive characterization of their genomic composition, coupled with the identification of distinctive genomic features within a phylogenetic framework, can provide critical insights into their mechanisms of colonization in plant tissues and their influence on other microbial communities. For instance, comparative genomics has been applied to investigate the functional potential of the keystone genus Aquimarina in marine ecosystems, uncovering substantial differences between host-associated and free-living populations in terms of specialized metabolism and carbon cycling functions [17].
Since ancient times, medicinal plants of the Panax genus have occupied a significant position in traditional Chinese medicine due to their excellent efficacy and currently hold enormous global economic value. The main commercial species within this genus include P. ginseng, P. quinquefolium, and P. notoginseng [18,19]. A variety of substances have been isolated from these three species, such as ginsenosides (tetracyclic triterpenoid saponins), polysaccharides, polyacetylenes, flavonoids, amino acids, fatty acids, and polypeptides [18]. Among them, saponins have attracted extensive attention because they are the most unique specialized metabolites of Panax plants compared to other related genera and serve as the main bioactive substances determining the pharmacological effects of Panax plants [18,20,21,22,23]. Beyond their role as the primary active compounds in medicinal applications, saponins are also regarded as important components of the plant’s defense system against pathogens and herbivores. As chemical defense weapons of Panax plants, they participate in plant-microbe interactions and alter the structure of the endophytic microbial community [24,25]. We thus hypothesized that the endophytic fungi of Panax genus, especially the keystone endophytes, can modify the saponin-related metabolic microenvironment to adapt to this chemical defense system. This study integrated large-scale Panax plant sampling, amplicon sequencing, culture-dependent isolation and validation of keystone fungi, and whole-genome sequencing (WGS) to achieve two primary goals: (1) the identification of endophytic keystone fungi and the assessment of their saponin-biotransformation potential; and (2) uncovering mechanisms underlying their saponin-converting capability, tissue-specific colonization, and keystone role functionality by the genomic characterization of these taxa and comparative genomics.
2.1 Sample Collection of Panax Plants
The composition and structure of endophytic microbial communities, as well as their interactions with host plants, are strongly influenced by local environmental conditions [26]. Consequently, all samples of P. ginseng, P. quinquefolium, and P. notoginseng were sourced from their respective core production areas, which are the regions where plant-endophyte co-evolution occurs, as detailed described in a previous study [27]. Simply, sampling was conducted in mid-September 2019 in Baishan City, Jilin Province, for P. ginseng and P. quinquefolius, and in late October 2019 in Wenshan Prefecture, Yunnan Province, for P. notoginseng. All plants were sampled at the late root thickening stage. To capture the growth year-dependent microbial variations, three independent fields, each planted with 2-year-old (2y), 3-year-old (3y), or 4-year-old (4y) Panax plants, were selected per species. From each field, nine samples (biological replicates) were collected, and each sample consisted of 10 healthy plants. A total of 81 plant samples (3 Panax species × 3 growth year × 9 replicates) were obtained. After collection, plant leaf and root samples were rinsed, surface-disinfected, pulverized in liquid nitrogen to a fine powder, and subsequently archived at −80°C, respectively.
2.2 Identification of Keystone Fungal Taxa in Plant Tissues
Genomic DNA was extracted from leaf and root tissues (the FastDNA SPIN Kit for soil (MoBio Laboratories, Inc., Carlsbad, CA, USA). The fungal internal transcribed spacer (ITS) region was amplified using the host-blocking primer pair ITS1F/ITS2R and sequenced on an Illumina MiSeq platform (Shanghai Biozeron Co., Ltd., Shanghai, China) [28]. After quality filtering and chimera removal, operational taxonomic units (OTUs) were clustered at 97% similarity and taxonomically assigned against the UNITE database [29,30,31]. Ecological networks for leaf and root communities were constructed separately based on robust Spearman correlations (|ρ| > 0.7, FDR < 0.001) among OTUs with relative abundance >0.01% [32,33]. Keystone taxa within each network were defined as nodes exhibiting both high connectivity (degree > 20) and high centrality (betweenness centrality >5000) [9,34].
2.3 Isolation of Keystone Fungal Taxa and Verification of Their Saponin Conversion Capability
Since amplicon analysis identified both Plec and Clad in all three Panax species, we only selected P. notoginseng (a representative species) for subsequent strain isolation experiments, with specific sample collection years detailed in Section 2.1. Based on the amplicon results, P. notoginseng was selected for fungal isolation [28]. Endophytic fungi were cultured from surface-sterilized tissues on potato dextrose agar (PDA) medium and purified by repeated sub-culturing. Pure strains were identified by ITS sequencing. Strains with the highest sequence identity to the keystone genera Plectosphaerella (Plec) and Cladosporium (Clad) were selected for functional assays.
The selected strains were co-cultured with a mixed saponin substrate (containing ginsenosides Rb1, Rb2, Rc, Rd, Rg1, Re, and notoginsenoside R1; Yuanye Bio-Technology Co., Ltd., Shanghai, China) in potato dextrose broth (PDB) medium for 7 days. Culture extracts were analyzed by High-Performance Liquid Chromatography (HPLC; Agilent Technologies Inc., Santa Clara, CA, USA) to quantify saponin conversion, using an uninoculated substrate as control [28].
2.4 Evaluation of β-Glucosidase Activity of Keystone Fungal Strains
The β-glucosidase activity of the two keystone strains was measured using p-nitrophenyl-β-D-glucopyranoside (pNPG; Yuanye Bio-Technology Co., Ltd., Shanghai, China) as substrate. Culture supernatants harvested at different time points were incubated with pNPG, and the reaction was terminated with sodium carbonate. Enzyme activity was determined by measuring the absorbance at 405 nm [35].
2.5 DNA, RNA Extraction and Sequencing of Keystone Fungal Strains
Genomic DNA of the two keystone strains was extracted using the E.Z.NA™ HP Fungal DNA Extraction Kit (E.Z.NA HP; OMEGA Bio-tek Inc., Norcross, GA, USA) and sequenced on both Illumina and PacBio platforms (Shanghai Biozeron Co., Ltd., Shanghai, China). A hybrid assembly strategy was employed to generate high-quality genome assemblies [36,37,38,39,40]. For transcriptome analysis, strains were cultured in yeast extract-peptone-dextrose (YPD) medium and cellobiose-modified YPD (Cel-YPD) medium. Total RNA was extracted using the Fungal Total RNA Isolation Kit (Sangon Biotech (Shanghai) Co., Ltd., Shanghai, China) and subjected to RNA sequencing (RNA-seq) on Illumina and PacBio platforms (Shanghai Biozeron Co., Ltd., Shanghai, China). Additionally, a pooled sample from multiple media was used for full-length transcriptome sequencing (PacBio Iso-Seq; Shanghai Biozeron Co., Ltd., Shanghai, China) to aid genome annotation.
Predicted protein sequences were functionally annotated against multiple public databases (Nr, Swiss-Prot, eggNOG, KEGG, GO) [39,40,41,42,43]. Glycoside hydrolase (GH) families GH1 and GH3 were identified using hidden Markov model (HMM) searches [44,45], and phylogenetic trees were constructed using FastTree 2 [46]. Secreted proteins were predicted using SignalP and DeepTMHMM [47,48]. Secondary metabolite gene clusters were predicted with antiSMASH [49]. To elucidate evolutionary relationships, we performed comparative genomic analyses with publicly available genomes from related species, including gene family clustering [50], phylogenetic tree construction, estimation of divergence times [51,52], and analysis of gene family expansion/contraction [53].
Detailed bioinformatic protocols are provided in the File S1.
3.1 Identification of Keystone Fungal Endophytes in Panax Tissues Based on Ecological Networks
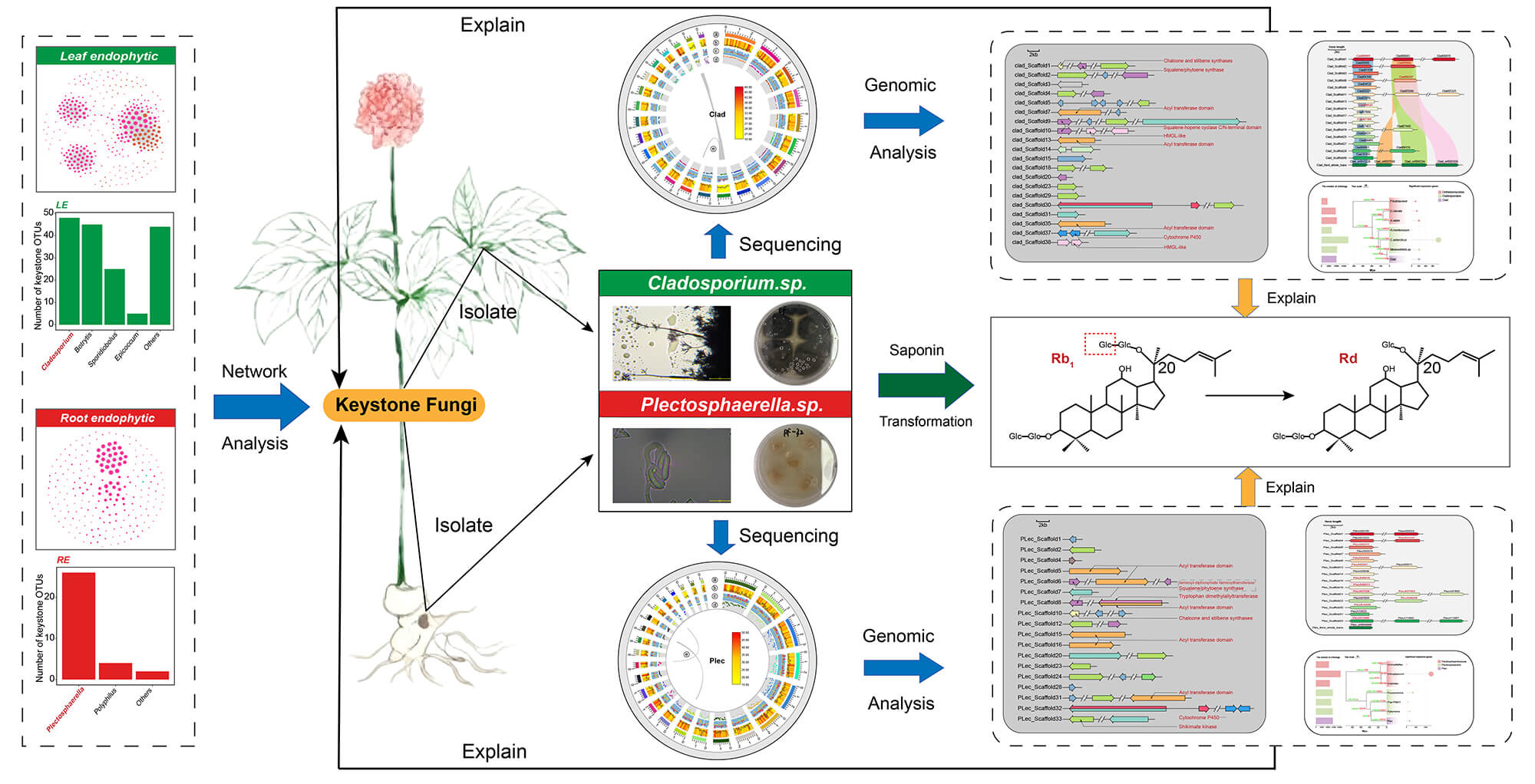
Spearman correlations, respectively (Fig. 1a). The root network was dominated by Ascomycota taxa, while the leaf network mainly consisted of Ascomycota and Basidiomycota nodes. The leaf network had slightly more nodes and far more connections compared to the root network. The proportions of positive edges in the root and leaf networks were 95.54% and 91.30%, respectively, indicating potentially mutualistic interactions dominated within-tissue interactions (Fig. 1a). Multiple topological properties of the ecological network were further calculated. The leaf network exhibited higher average degree and density but lower clustering coefficient and modularity compared to the root network, indicating that the root fungal community may tend to interact more locally rather than globally compared with leaf mycobiome (Fig. 1b). Based on the node degree and betweenness centrality, 32 and 167 keystone OTUs were identified in root and leaf networks, respectively (Fig. 1c). Counting at the genus level showed that Plectosphaerella and Cladosporium contributed the highest proportions of keystone taxa in the root and leaf endophytic compartments, respectively.
Correlation analyses were performed between the relative abundances of Plectosphaerella in roots and Cladosporium in leaves and the contents of individual saponins in roots and leaves, respectively. In the roots of Panax plants, the relative abundance of Plectosphaerella was significantly and positively correlated with the contents of ginsenosides Rg1, Re, Rd, and NR1 (p < 0.05) (Fig. 1d). In the leaves of Panax plants, the relative abundance of Cladosporium was significantly and positively correlated with the abundances of ginsenosides Rg1, Re, Rd, Rb2, F1, and NR1 (p < 0.05). It is worth noting that both genera showed the highest correlation with the content of ginsenoside Rd in their respective compartments (Plectosphaerella: ρ = 0.62, p < 0.001; Cladosporium: ρ = 0.69, p < 0.001). These results indicate that the two keystone genus in root and leaf may be associated with the accumulation of specific ginsenosides in each compartment.
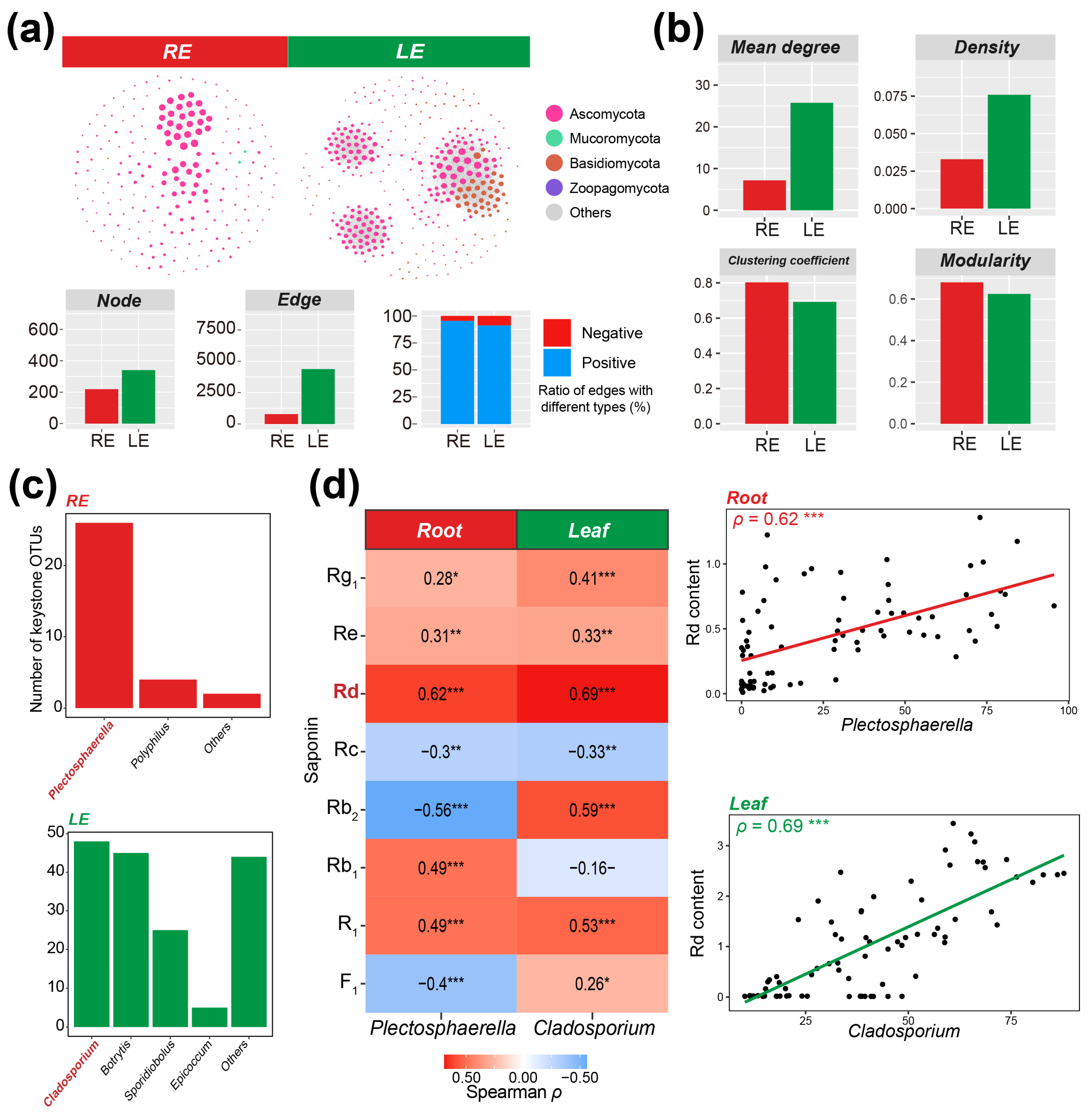
Figure 1: Identification of keystone fungal endophytes based on ecological networks and their correlations with plant saponin contents. (a), Ecological networks, as well as the number of nodes and edges in the networks and the proportions of positive/negative connections. (b), Topological properties of fungal ecological networks in each compartment. (c), Genus-level distribution of keystone OTUs in fungal networks of root and leaf endophytic compartments. (d), Correlations between keystone genera in each compartment and the contents of monomeric saponins in respective compartments. RE: Root endophytic; LE: Leaf endophytic. *: p < 0.05; **: p < 0.01; ***: p < 0.001.
3.2 Keystone Fungal Endophytes Exhibit Specific Ginsenoside Transformation Capabilities
From the fresh root and leaf tissues, the Plectosphaerella (designated Plec) and Cladosporium (designated Clad) strains were obtained through isolation, culture, and purification, respectively. Based on colony morphology, microscopic characteristics, and a phylogenetic tree constructed using full-length ITS sequences, the two fungal strains were identified as Plectosphaerella niemeijerarum and Cladosporium crousii (Fig. 2a,b). Further, the full-length ITS sequences of Plec and Clad were blasted against the keystone OTUs of Plectosphaerella and Cladosporium identified in amplicon sequencing of roots and leaves. The lowest BLAST score of Plec against 26 Plectosphaerella-genus OTUs identified in roots was 270, with the highest E-value of 8 × 10−76; the lowest BLAST score of Clad against 48 Cladosporium-genus keystone OTUs identified in leaves was 322, with the highest E-value of 5 × 10−91 (Table S1). These results indicate that the Plec and Clad strains isolated from the roots and leaves of P. notoginseng can well represent the keystone taxa obtained from ecological network analysis.
After co-culturing the two strains with mixed saponins, high-performance liquid chromatography was used to determine the changes in saponin content. Plec and Clad significantly reduced the content of Rb1 in the culture medium and increased the content of Rd (Fig. 2c). Furthermore, calculation of the Rb1-to-Rd conversion efficiency of the two strains revealed that Clad exhibited a higher conversion efficiency than Plec (Fig. 2c), with detailed data provided in Table S2. Since Rd can be obtained by hydrolyzing a β-glucoside bond at the C20 position of Rb1, the above changes in ginsenoside content support the expectation that both fungi have β-glucosidase activity.
Furthermore, the pNPG method was used to determine the relative β-glucosidase activity of the fungal culture broth at different culture times. From 72 h to 144 h, the catalytic capacity of the pNPG substrate in the culture broths of the two fungal strains gradually increased, and the relative catalytic activity of the Clad strain was higher than that of the Plec strain (Fig. 2d). The gap between the two strains increased with the extension of the culture time. Overall, these results indicate that the two strains isolated in this study have β-glucosidase activity and can convert ginsenoside Rb1 into Rd.
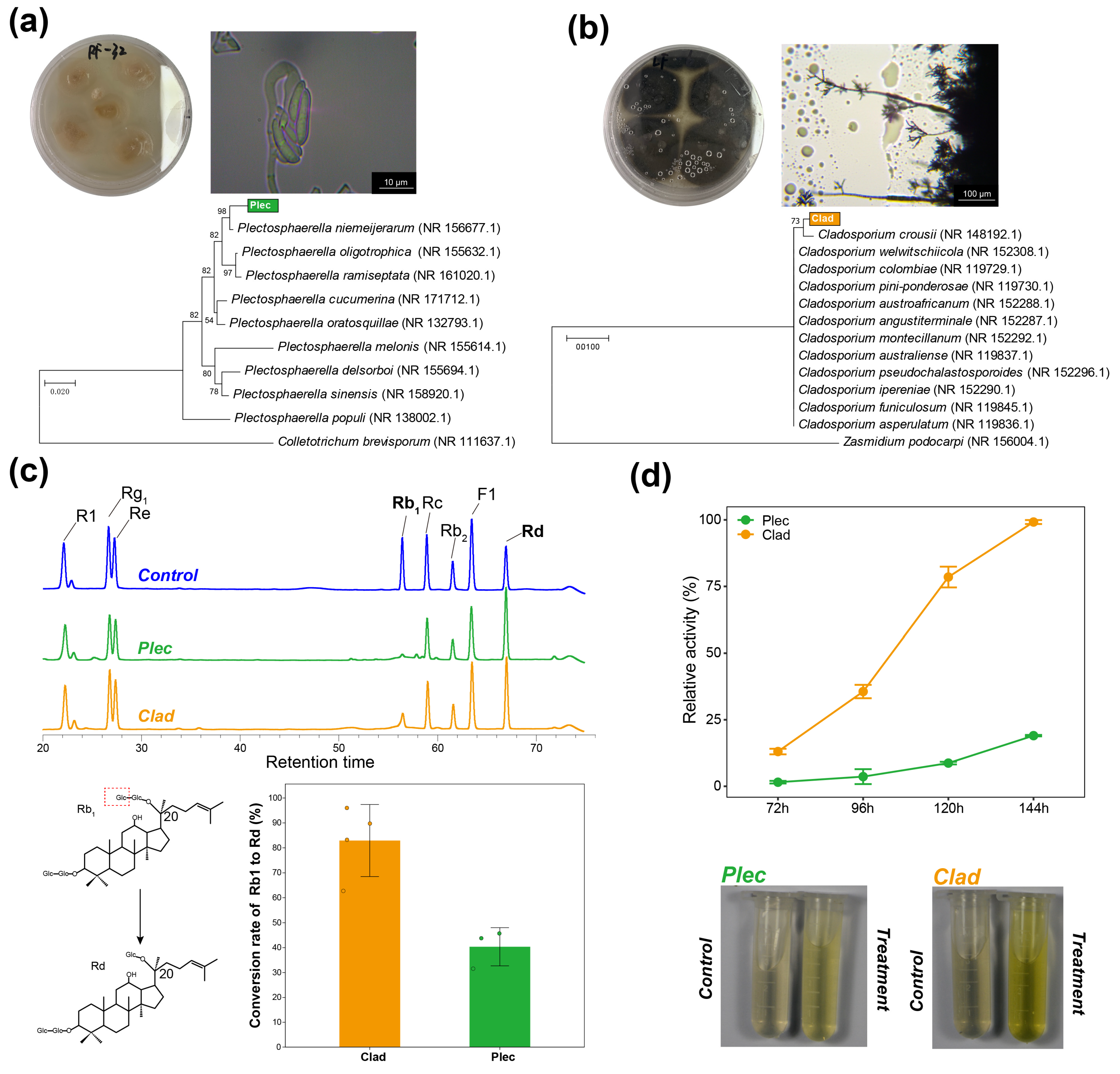
Figure 2: Morphological classification and verification of saponin transformation capacity of representative strains of keystone taxa in endophytic compartment fungal communities. (a), Morphological characteristics and phylogenetic tree analysis of strain Plec. (b), Morphological characteristics and phylogenetic tree analysis of strain Clad. (c), chromatograms of ginsenoside content after co-cultivation of Plec/Clad strains with mixed ginsenosides; Control: sterile medium containing mixed ginsenosides (aseptic control); and the Rb1-to-Rd conversion rates by Plec and Clad. (d), relative β-glucosidase activities of two keystone strains.
3.3 Genome Assembly and Phylogeny of Two Keystone Fungal Strains
The genomes of Plec and Clad strains were sequenced and assembled using PacBio and Illumina sequencing platforms to gain mechanistic insights of the functional roles of keystone taxa. For the Plec and Clad strains, a total of 36,220,183 bp and 33,581,758 bp clean reads from Illumina sequencing were obtained for k-mer analysis and genome modification, respectively. GenomeScope was used to generate a histogram of sequencing depth distribution (k = 21), with the heterozygosity rate of 0.67% and 0.23% as well as the repeat rate of 3.70% and 3.48% for Plec and Clad, respectively (Fig. S1). The size of assembled genome (consisting of 36 scaffolds) for Plec is 36 Mb, with the longest scaffold, the N50 length, the N90 length, and the GC content being 185 kb, 9.7 kb, 7.2 kb, and 57.42%. For Clad, the assembled genome was 33.5 Mb in size, consisting of 51 scaffolds. The longest scaffold is 236 kb, with an N50 value of 11.3 kb and an N90 value of 9.3 kb. The GC content of Clad genome is 52.24% (Table 1). A total of 12,218 genes were identified in the Plec genome, with an average length of 1491 bp, while the Clad genome contained 10,583 genes with an average length of 1546 bp (Table 1). In addition, only less collinearity within the two genomes is identified (Fig. 3a). Based on searches against multiple databases including NR, GO, eggNOG, KEGG, and Swiss-Prot, a total of 92.9% and 90.8% of the genes in the Plec and Clad genome are functionally annotated, respectively (Fig. 3b, Table S3).
The single-copy orthology genes-based phylogenetic trees are constructed for Plec and Clad strains to explore their evolutionary positions (Fig. 3c and Table S4). The results indicate that the Plec strain is closely related to P. plurivora and P. sp. P0831, while the Clad strain has a close evolutionary relationship with other Cladosporium species, especially C. velox and C. colombiae (Fig. 3c).
Table 1: Basic assembly and annotation information of Plec and Clad.
| Characteristics | Plec | Clad |
|---|---|---|
| Genome assembly size (Mb) | 36 | 33.5 |
| Scaffolds | 36 | 51 |
| Longest Scaffold (kb) | 185 | 236 |
| Scaffold N50 (kb) | 9.7 | 11.3 |
| Scaffold N90 (kb) | 7.2 | 9.3 |
| GC% | 57.42 | 52.54 |
| Sequencing platform | PacBio CLR, Illumina | |
| Gene Number | 12,218 | 10,583 |
| Total length (bp) | 18,222,378 | 16,364,050 |
| Average length (bp) | 1491 | 1546 |
| In Genome (%) | 49.42 | 50.01 |
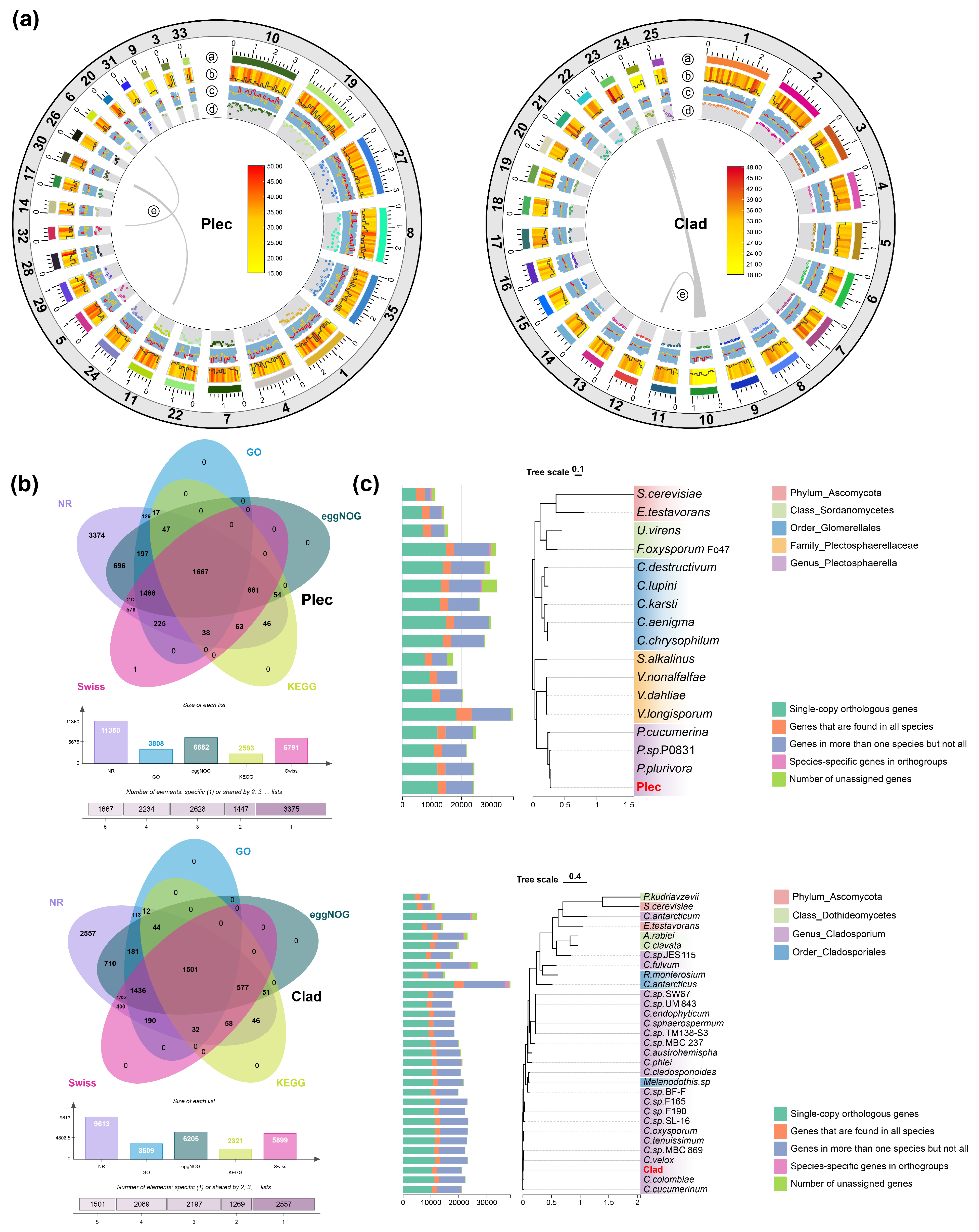
Figure 3: Genome assembly, functional annotation, and orthologous phylogenetic tree of the two keystone strains. (a), Genome circle maps of the two strains. The outermost colored blocks represent the 25 largest scaffolds in the genome; b The heatmap represents the gene density of each scaffold, the black line represents the repeat sequence density; c The blue column represents the GC ratio of each scaffold, and the line graph represents the GC skew; d The number of carbohydrate-active enzymes; e The collinearity within the scaffold. (b), Functional annotations of the two strains in each database. (c), Orthologous phylogenetic tree of the two strains. For the Plec, two species at the phylum Ascomycota level, two species at the class Sordariomycetes level, five species at the order Glomerellales level, four species at the family Plectosphaerellaceae level, and three species at the genus Plectosphaerella level were selected. For the Clad strain, two species at the phylum Ascomycota level, three species at the class Dothideomycetes level, three species at the order Cladosporiales level, and 22 species at the genus Cladosporium level were selected (Table S4).
3.4 Identification of GH3 Genes Potentially Accounting for the Saponin Transformation Capacities of Two Fungal Keystone Strains
To explore the molecular basis for the conversion of ginsenoside Rb1 to Rd by the two keystone fungi, we focused on β-glucosidases, particularly those from the Glycoside Hydrolase family 3 (GH3), which are known to hydrolyze the glucosyl moieties of Rb1. Phylogenetic analysis revealed evolutionary relationships between the GH3 sequences of both fungal strains and reference sequences from organisms reported to transform Rb1 (Fig. 4a). To identify candidate genes with potential activity, we examined the transcriptional response of these GH3 genes to cellobiose, a substrate for β-glucosidases. RNA-seq analysis showed that 11 GH3 genes in Plec and 5 in Clad were significantly upregulated under cellobiose induction (Fig. 4b). These upregulated GH3 genes represent strong candidates encoding the β-glucosidases responsible for the initial transformation of Rb1 to Rd.
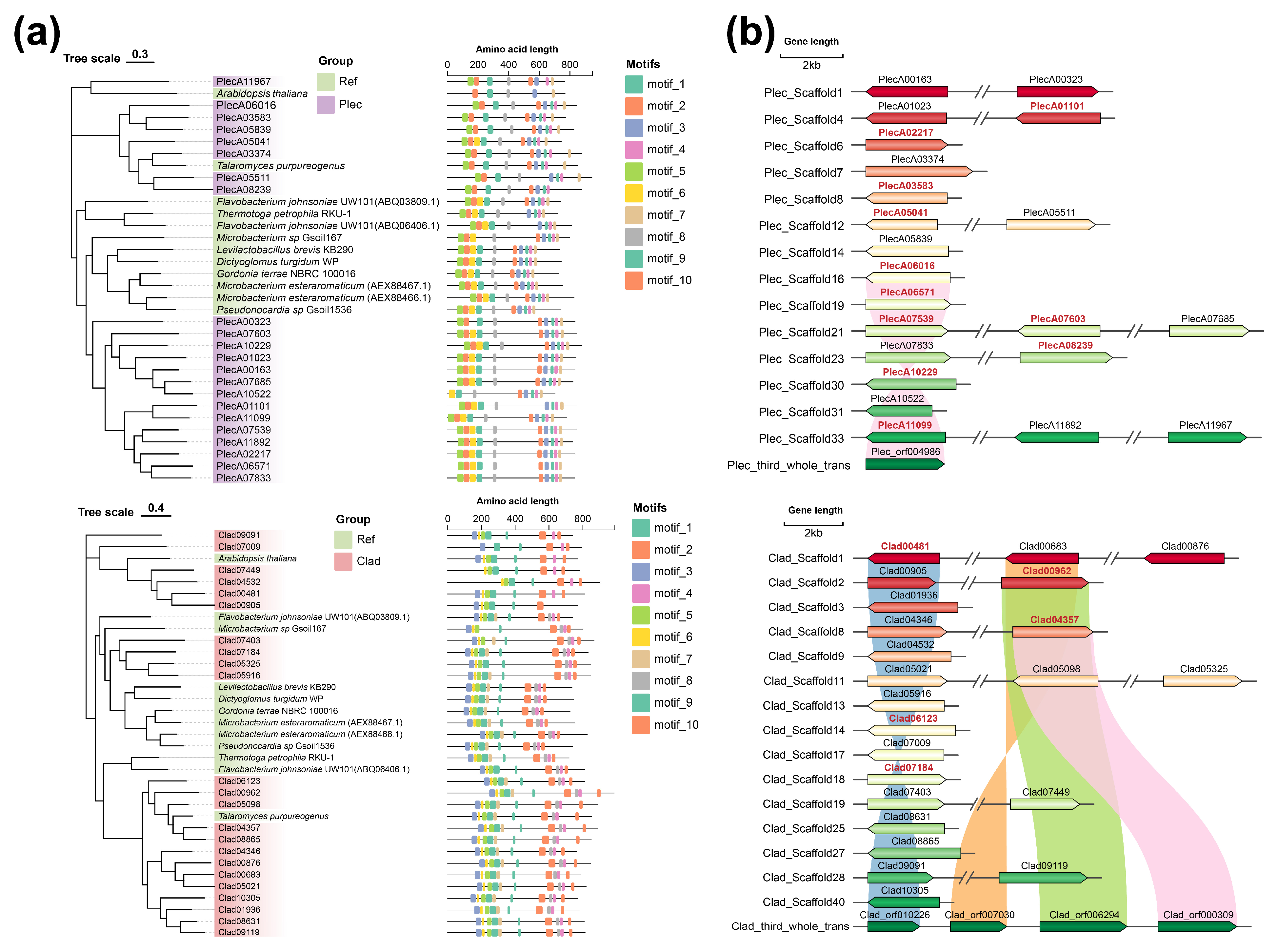
Figure 4: Phylogenetic tree of GH3 gene family and related gene clusters in the two strains. (a). Phylogenetic tree of GH3 gene family and amino acid motif domains in the two strains. The reference sequences are provided in Table S5. (b). Gene clusters of GH3 gene family in the two strains. Red characters indicate genes with significant up-regulation in cellobiose PDA medium.
3.5 Transmembrane Transport and Environmental Response Gene Families Undergo Significant Expansion in Two Keystone Fungal Strains
Analysis of gene family evolution indicated significant expansion of specific families in both Plec and Clad strains compared to their respective taxonomic relatives (Fig. 5a). Gene Ontology (GO) enrichment analysis of these expanded families highlighted their strong association with transmembrane transport, response to various hormones, and cellular components such as mitochondria, membranes, and vacuoles (Fig. 5b). Additionally, hydrolase activity terms were significantly enriched in Clad. These results suggest that genomic expansions enhancing transmembrane transport and environmental responsiveness may underpin the adaptive strategies and keystone roles of these fungi.
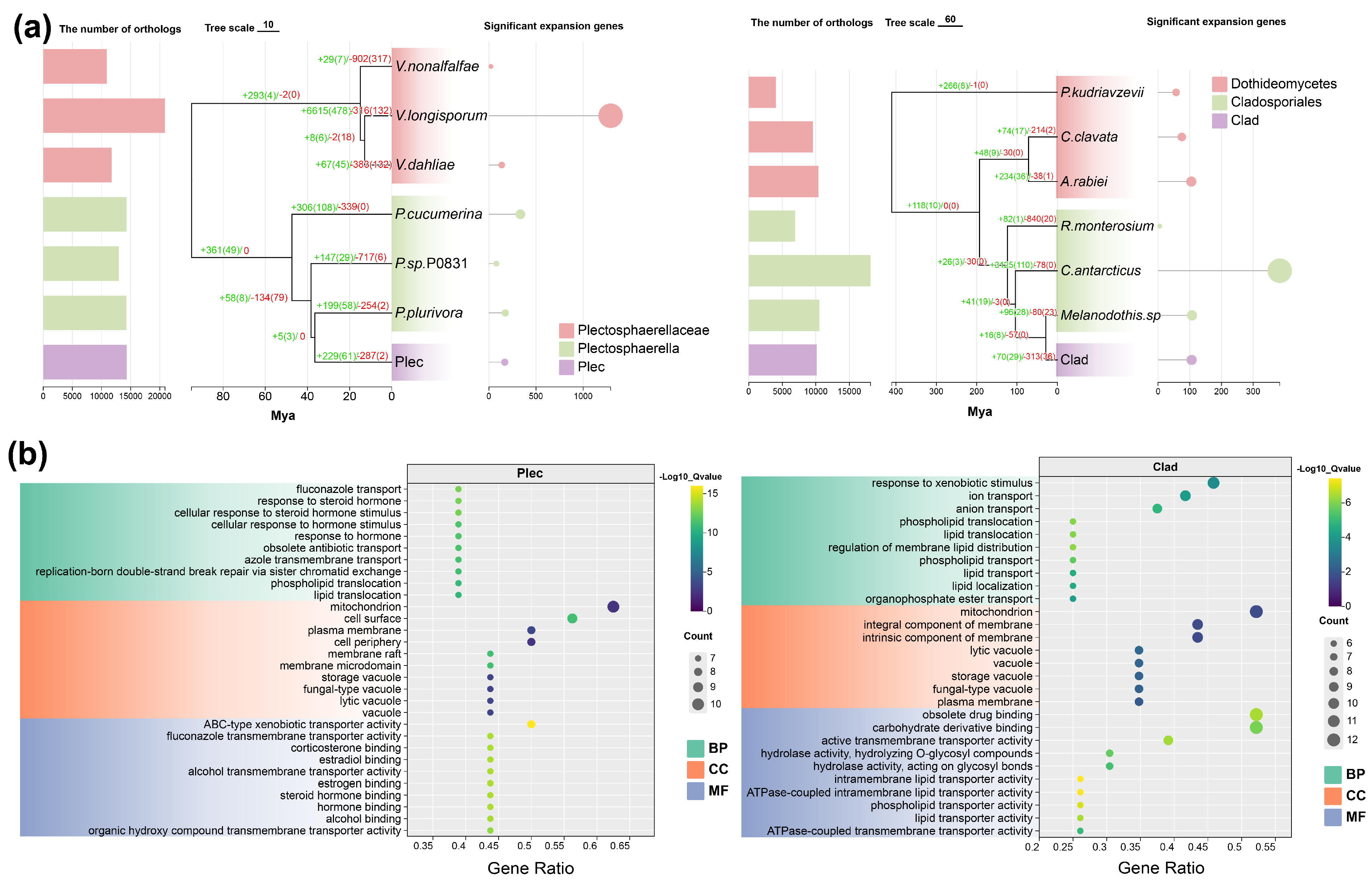
Figure 5: Analysis of gene family expansion and contraction in the two strains. (a). The number of orthologous groups, gene family expansion/contraction, and significantly expanded genes in the two strains. (b). GO enrichment of significantly expanded genes in the two strains. BP: biological process. CC: cellular component. MF: molecular function.
3.6 Two Keystone Fungal Strains Are Rich in Secondary Metabolic Gene Clusters and Secretome-Encoding Genes
Genomic analysis further revealed that both keystone strains possess a rich complement of biosynthetic gene clusters (BGCs) for secondary metabolism, with numbers typical within their respective genera (Fig. 6a), and these BGCs are unevenly distributed across their genomic scaffolds (Fig. 6b). Prediction of the secretome identified a substantial number of secreted proteins harboring signal peptides in both strains (Table S6). GO enrichment analysis of these secreted proteins showed significant overrepresentation of functions related to carbohydrate/polysaccharide metabolism, cell wall organization, and notably, glycoside hydrolase activity (Fig. 6c). These genomic features imply that secondary metabolites and the secreted proteome, particularly enzymes involved in carbohydrate modification, are crucial for the interaction of these keystone fungi with their plant host.
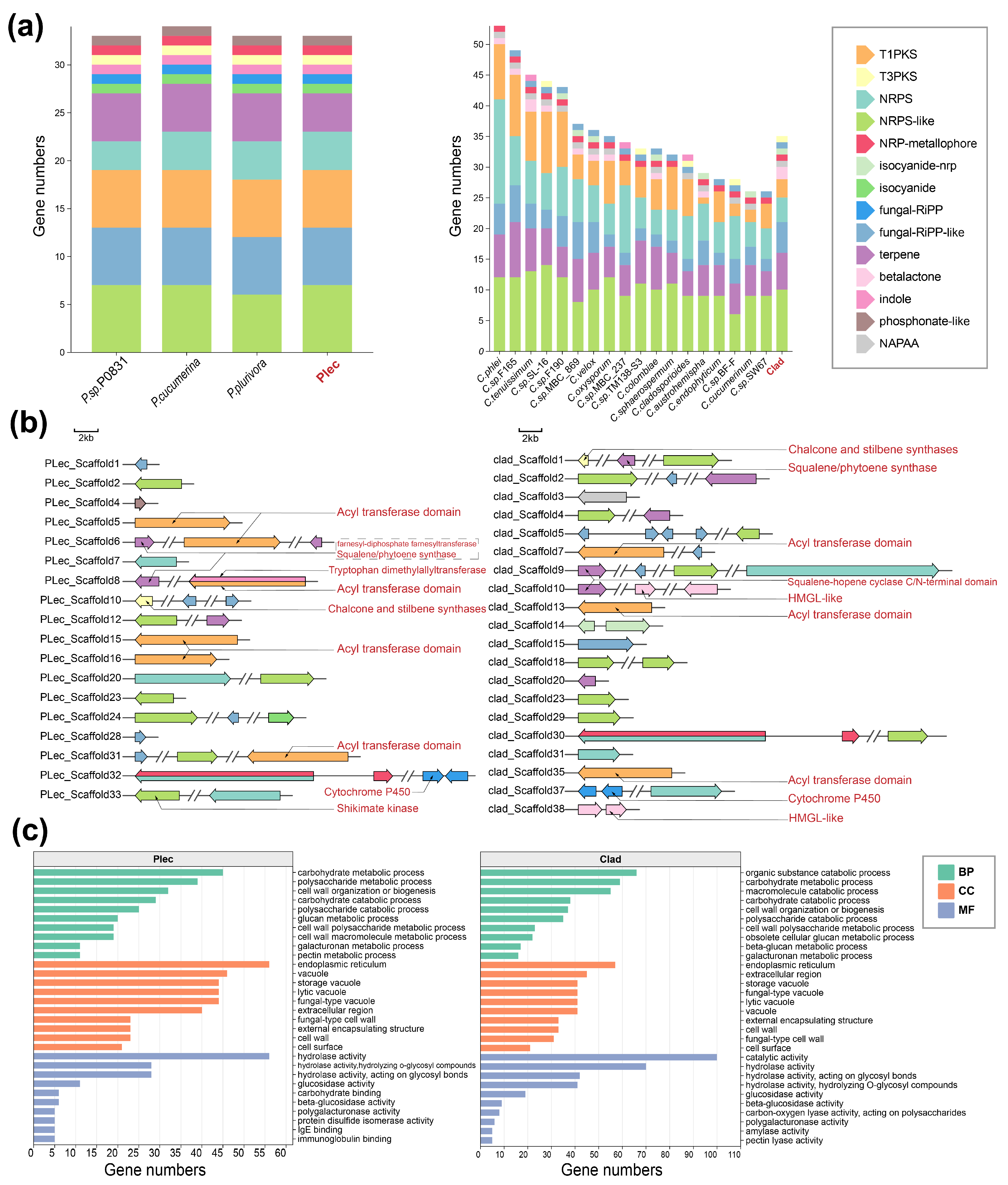
Figure 6: Analysis of secondary metabolites and secreted proteins. (a), The number of secondary metabolites in the two strains and other species within their respective genera. (b), Secondary metabolite gene clusters of the two strains. (c), GO enrichment results of secreted proteins with signal peptides in the two strains. T1PKS: Type I PKS (Polyketide synthase); T3PKS: Type III PKS; NRPS: Non-ribosomal peptide synthetase; NRPS-like: NRPS-like fragment; NRP-metallophore: Non-ribosomal peptide metallophores; isocyanide-nrp: Isocyanides (https://doi.org/10.1128/mBio.00785-18); isocyanide; Isocyanides (https://doi.org/10.1093/nar/gkad573); fungal-RiPP: Fungal RiPP with POP or UstH peptidase types and a modification; fungal-RiPP-like: Fungal-RiPP-like fragment; terpene: Terpene; betalactone: Beta-lactone containing protease inhibitor; indole: Indole; phosphonate-like: Phosphonate-like fragment; NAPAA: Non-alpha poly-amino acids like e-Polylysin. BP: biological process. CC: cellular component. MF: molecular function.
In this study, amplicon sequencing was performed on fungi in the root and leaf niches of Panax ginseng, Panax quinquefolius, and Panax notoginseng. Through network analysis, the keystone taxa at the genus level in the root and leaf compartments were identified. Correlation analysis with the main saponins of Panax plants revealed a significant positive correlation between the relative abundance of keystone taxa and the content of ginsenoside Rd. Representative keystone strains were isolated and cultured from the roots and leaves of P. notoginseng—a typical species of the Panax genus. In vitro experiments showed that both strains had the ability to convert ginsenoside Rb1 into Rd and exhibited enzymatic activity in the β-glucosidase activity assay. To explore the reasons for their status as keystone taxa, genomic and comparative genomic analyses were conducted on the two strains. The results indicated that the presence of multiple sequences encoding β-glucosidases and abundant secondary metabolite gene clusters might be the key factors contributing to their role as keystone taxa.
4.1 The Keystone Taxa in Panax Plant Tissues May Alter the Microenvironment by Saponin Conversion
Plants provide a growth environment for microorganisms and interact with them to achieve better survival and evolution [54]. As a unique and critical group within microbial communities, keystone microbial taxa can exert a significant impact on the community structure and function of the microbiome in the corresponding niche, primarily by modifying the microenvironment of that niche [9]. In this study, representative keystone endophytes were identified from the root and leaf niches of Panax species. It was confirmed that all these strains can convert ginsenoside Rb1 to Rd and possess β-glucosidase activity. Furthermore, to verify that the two strains do not produce saponins by themselves, we determined the saponin content in the PDB culture medium, yet no peaks matching those of the known saponins were detected. This observation could be attributed to the high specificity of the ginsenoside biosynthetic pathway, which encompasses the mevalonate (MVA)-derived triterpene skeleton branching pathway. The key enzymes involved include dammarenediol synthase, cytochrome P450 monooxygenases of the CYP716A subfamily, and uridine diphosphate glycosyltransferases, all of which are considered to be unique to plants of the genus Panax to date. This process of altering the host’s secondary metabolites essentially constitutes a modification of the microenvironment, which may further reshape the microbial community structure of the corresponding niche. The antibacterial effect of ginsenosides has been widely verified [55]; however, ginsenosides with different structures exhibit varying antibacterial activities. For instance, in experiments investigating the growth-inhibitory effects of protopanaxatriol type (PPT-type) and protopanaxadiol type (PPD-type) saponins against Porphyromonas gingivalis and Fusobacterium nucleatum, PPD-type saponins were found to have stronger antibacterial effects than PPT-type saponins, with ginsenoside Rd showing the most prominent activity [56]. Furthermore, recent studies have revealed that Panax species possess a plant chemical defense mechanism mediated by a two-component system containing β-glucosidase [57]. Specifically, in response to pathogen infestation, plants synthesize a variety of secondary metabolites to resist pathogens. However, some of these compounds are toxic to the plants themselves. To address this, plants produce a class of glycosylated, inactive defensive compounds [58,59]. In addition to protecting plants from autotoxicity, glycosylation enhances the solubility of defensive compounds, thereby facilitating their storage (typically in vacuoles) [60]. Under physiological conditions, β-glucosidase and glycosylated defensive compounds are spatially separated. When pathogens invade, plant tissues are damaged, bringing β-glucosidase into contact with the defensive compounds. This interaction triggers the hydrolysis of glycosyl groups, ultimately achieving the effect of resisting pathogens [61]. Although plants inherently possess this defense mechanism, our study suggests that endophytic keystone fungi may secrete β-glucosidase during pathogen invasion to modify the microenvironment, assist plants in resisting pathogens, and thereby maintain the balance of the entire microbial community.
4.2 Genomic Analysis and Comparative Genomic Analysis for Exploring the Formation Mechanisms of Keystone Taxa
Based on the experimental results of saponin transformation by two strains, this study explored the β-glucosidase-encoding genes in their genomes; both strains contained 23 sequences encoding this enzyme, with generally complete structures. These sequences involved in encoding β-glucosidases may serve as downstream regulators adapted to invasion by different pathogens, and when different pathogens invade, the promoters upstream of β-glucosidase-encoding genes located at different positions in the keystone taxa may respond to different toxins released by the pathogens, thereby synthesizing β-glucosidases that further form a plant chemical defense mechanism mediated by a two-component system of β-glucosidases together with Panax plants to resist pathogens. Furthermore, in the gene family contraction and expansion analysis of comparative genomics, we also found that the Gene Ontology enrichment results of the significantly expanded gene families of the Clad strain relative to the family level showed functions related to glycoside hydrolase activity, while no such related functions were detected in the enrichment results of the significantly expanded gene families of the Plec genus relative to the family level; in the enrichment analysis of transmembrane secretory proteins with bioactive peptides in the two strains, both strains were enriched in multiple glycoside hydrolase functions, which further indicates that the secretion of glycoside hydrolases may be one of the reasons for their status as keystone taxa.
4.3 Limitations of the Current Study and Future Research Directions
The current study has limitations in investigating the saponin-transforming function of the strains: while in vitro data are reliable, in vivo validation in host plants is lacking. Future research should re-inoculate the strains onto original host tissues to verify their saponin-transforming capacity in vivo, enhancing findings’ comprehensiveness and robustness. Additionally, analysis of secondary metabolite gene clusters in the two keystone strains yields testable hypotheses. Regarding host ginsenoside biosynthesis: Plec (4 acyltransferase domains) and Clad (3) may contribute to ginsenoside structural diversification (e.g., malonyl-ginsenosides) [62]; squalene synthase in both may promote synthesis via key intermediate supply [24]; Clad’s two HMGL-like domains (reverse of rate-limiting HMGR) may restrict synthesis under specific conditions [63]; CYP57 (Plec) and CYP58 (Clad) are candidates for oxidative modification (analogous to ginsenoside-diversifying CYP716 [64]), requiring validation. For antimicrobial compound production: Plec’s chalcone synthase and Clad’s stilbene synthase gene clusters may synthesize antibacterial flavonoids/stilbenes [65], facilitating colonization and keystone status. For host growth modulation: Plec’s tryptophan dimethylallyl transferase (DMAT synthetase, involved in indole alkaloid biosynthesis) suggests potential synthesis/modification of growth-regulating indole compounds (e.g., IAA) [66], a direction for future study. Notably, discussions on mevalonate pathway balance (HMGR/HMGL), CYP-mediated saponin diversification, and IAA-related synthesis are based on genomic predictions and require experimental validation.
This study provides comprehensive insights into the identification of keystone fungal taxa in the endophytic niches of Panax plants and the manner in which these taxa improve the microenvironment by transforming saponins. Furthermore, whole-genome and comparative genomic analyses offer potential explanations for the saponin-transforming capacity and colonization mechanisms of keystone fungi. These findings facilitate the exploration of the potential mechanisms underlying saponin transformation mediated by endophytic fungi of Panax and enhance the understanding of plant-microbe interactions.
Acknowledgement:
Funding Statement: This work was funded by the National Natural Science Foundation of China (82274044, 82304663), National Key Research and Development Program (2022YFC3501802, 2022YFC3501803, and 2022YFC3501804), the Scientific and technological innovation project of China Academy of Chinese Medical Sciences (CI2023E002, CI2024E003), and the Fundamental Research Funds for the Central Public Welfare Research Institutes (ZZ13-YQ-049, ZZ16-XRZ-072, ZZ17-YQ-025, ZXKT22052, and ZXKT22060).
Author Contributions: Ruikang Ma and Guozhuang Zhang analysed the data and drafted the manuscript. Guangfei Wei performed the isolation experiments of strains. Guozhuang Zhang conducted the ginsenoside conversion experiments of strains. Songzi Li and Tongle Li participated in the data analysis. Fugang Wei, Yong Wang collected the samples. Linlin Dong designed the study, granted funds, and participated in the drafting and revision of the manuscript. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: The raw sequence data are available in the National Genomics Data Center (https://ngdc.cncb.ac.cn) under the BioProject PRJCA007643, PRJCA047674.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest.
Supplementary Materials: The supplementary material is available online at https://www.techscience.com/doi/10.32604/phyton.2026.075657/s1.
References
1. Arnold AE . Understanding the diversity of foliar endophytic fungi: progress, challenges, and frontiers. Fungal Biol Rev. 2007; 21( 2–3): 51– 66. doi:10.1016/j.fbr.2007.05.003. [Google Scholar] [CrossRef]
2. Dupont PY , Eaton CJ , Wargent JJ , Fechtner S , Solomon P , Schmid J , et al. Fungal endophyte infection of ryegrass reprograms host metabolism and alters development. New Phytol. 2015; 208( 4): 1227– 40. doi:10.1111/nph.13614. [Google Scholar] [CrossRef]
3. Hiruma K , Gerlach N , Sacristán S , Nakano RT , Hacquard S , Kracher B , et al. Root endophyte Colletotrichum tofieldiae confers plant fitness benefits that are phosphate status dependent. Cell. 2016; 165( 2): 464– 74. doi:10.1016/j.cell.2016.02.028. [Google Scholar] [CrossRef]
4. Mastan A , Bharadwaj R , Kushwaha RK , Vivek Babu CS . Functional fungal endophytes in Coleus forskohlii regulate labdane diterpene biosynthesis for elevated forskolin accumulation in roots. Microb Ecol. 2019; 78( 4): 914– 26. doi:10.1007/s00248-019-01376-w. [Google Scholar] [CrossRef]
5. Panwar N , Szczepaniec A . Endophytic entomopathogenic fungi as biological control agents of insect pests. Pest Manag Sci. 2024; 80( 12): 6033– 40. doi:10.1002/ps.8322. [Google Scholar] [CrossRef]
6. Burragoni SG , Jeon J . Applications of endophytic microbes in agriculture, biotechnology, medicine, and beyond. Microbiol Res. 2021; 245: 126691. doi:10.1016/j.micres.2020.126691. [Google Scholar] [CrossRef]
7. Faust K , Raes J . Microbial interactions: from networks to models. Nat Rev Microbiol. 2012; 10( 8): 538– 50. doi:10.1038/nrmicro2832. [Google Scholar] [CrossRef]
8. Xiao N , Zhou A , Kempher ML , Zhou BY , Shi ZJ , Yuan M , et al. Disentangling direct from indirect relationships in association networks. Proc Natl Acad Sci U S A. 2022; 119( 2): e2109995119. doi:10.1073/pnas.2109995119. [Google Scholar] [CrossRef]
9. Banerjee S , Schlaeppi K , van der Heijden MGA . Keystone taxa as drivers of microbiome structure and functioning. Nat Rev Microbiol. 2018; 16( 9): 567– 76. doi:10.1038/s41579-018-0024-1. [Google Scholar] [CrossRef]
10. Toju H , Peay KG , Yamamichi M , Narisawa K , Hiruma K , Naito K , et al. Core microbiomes for sustainable agroecosystems. Nat Plants. 2018; 4( 5): 247– 57. doi:10.1038/s41477-018-0139-4. [Google Scholar] [CrossRef]
11. Liu S , Yu H , Yu Y , Huang J , Zhou Z , Zeng J , et al. Ecological stability of microbial communities in Lake Donghu regulated by keystone taxa. Ecol Indic. 2022; 136: 108695. doi:10.1016/j.ecolind.2022.108695. [Google Scholar] [CrossRef]
12. Ze X , Duncan SH , Louis P , Flint HJ . Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012; 6( 8): 1535– 43. doi:10.1038/ismej.2012.4. [Google Scholar] [CrossRef]
13. Liao H , Wen C , Huang D , Liu C , Gao T , Du Q , et al. Harnessing phage consortia to mitigate the soil antibiotic resistome by targeting keystone taxa Streptomyces. Microbiome. 2025; 13( 1): 127. doi:10.1186/s40168-025-02117-7. [Google Scholar] [CrossRef]
14. Pichersky E , Lewinsohn E . Convergent evolution in plant specialized metabolism. Annu Rev Plant Biol. 2011; 62: 549– 66. doi:10.1146/annurev-arplant-042110-103814. [Google Scholar] [CrossRef]
15. Toju H , Yamamoto S , Tanabe AS , Hayakawa T , Ishii HS . Network modules and hubs in plant-root fungal biomes. J R Soc Interface. 2016; 13( 116): 20151097. doi:10.1098/rsif.2015.1097. [Google Scholar] [CrossRef]
16. Chao H , Cai A , Heimburger B , Wu Y , Zhao D , Sun M , et al. Keystone taxa enhance the stability of soil bacterial communities and multifunctionality under steelworks disturbance. J Environ Manage. 2024; 356: 120664. doi:10.1016/j.jenvman.2024.120664. [Google Scholar] [CrossRef]
17. Silva SG , Blom J , Keller-Costa T , Costa R . Comparative genomics reveals complex natural product biosynthesis capacities and carbon metabolism across host-associated and free-living Aquimarina (Bacteroidetes, Flavobacteriaceae) species. Environ Microbiol. 2019; 21( 11): 4002– 19. doi:10.1111/1462-2920.14747. [Google Scholar] [CrossRef]
18. Kim DH . Chemical Diversity of Panax ginseng, Panax quinquifolium, and Panax notoginseng. J Ginseng Res. 2012; 36( 1): 1– 15. doi:10.5142/jgr.2012.36.1.1. [Google Scholar] [CrossRef]
19. Yang W , Qiao X , Li K , Fan J , Bo T , Guo DA , et al. Identification and differentiation of Panax ginseng, Panax quinquefolium, and Panax notoginseng by monitoring multiple diagnostic chemical markers. Acta Pharm Sin B. 2016; 6( 6): 568– 75. doi:10.1016/j.apsb.2016.05.005. [Google Scholar] [CrossRef]
20. Hou M , Wang R , Zhao S , Wang Z . Ginsenosides in Panax genus and their biosynthesis. Acta Pharm Sin B. 2021; 11( 7): 1813– 34. doi:10.1016/j.apsb.2020.12.017. [Google Scholar] [CrossRef]
21. Jiang Z , Tu L , Yang W , Zhang Y , Hu T , Ma B , et al. The chromosome-level reference genome assembly for Panax notoginseng and insights into ginsenoside biosynthesis. Plant Commun. 2020; 2( 1): 100113. doi:10.1016/j.xplc.2020.100113. [Google Scholar] [CrossRef]
22. Xu J , Chu Y , Liao B , Xiao S , Yin Q , Bai R , et al. Panax ginseng genome examination for ginsenoside biosynthesis. Gigascience. 2017; 6( 11): 1– 15. doi:10.1093/gigascience/gix093. [Google Scholar] [CrossRef]
23. Yang WZ , Hu Y , Wu WY , Ye M , Guo DA . Saponins in the genus Panax L. (Araliaceae): a systematic review of their chemical diversity. Phytochemistry. 2014; 106: 7– 24. doi:10.1016/j.phytochem.2014.07.012. [Google Scholar] [CrossRef]
24. Augustin JM , Kuzina V , Andersen SB , Bak S . Molecular activities, biosynthesis and evolution of triterpenoid saponins. Phytochemistry. 2011; 72( 6): 435– 57. doi:10.1016/j.phytochem.2011.01.015. [Google Scholar] [CrossRef]
25. Frantzeskakis L , Di Pietro A , Rep M , Schirawski J , Wu CH , Panstruga R . Rapid evolution in plant–microbe interactions–a molecular genomics perspective. New Phytol. 2020; 225( 3): 1134– 42. doi:10.1111/nph.15966. [Google Scholar] [CrossRef]
26. Trivedi P , Batista BD , Bazany KE , Singh BK . Plant-microbiome interactions under a changing world: responses, consequences and perspectives. New Phytol. 2022; 234( 6): 1951– 9. doi:10.1111/nph.18016. [Google Scholar] [CrossRef]
27. Wei G , Chen Z , Wang B , Wei F , Zhang G , Wang Y , et al. Endophytes isolated from Panax notoginseng converted ginsenosides. Microb Biotechnol. 2021; 14( 4): 1730– 46. doi:10.1111/1751-7915.13842. [Google Scholar] [CrossRef]
28. Zhang G , Wei F , Chen Z , Wang Y , Jiao S , Yang J , et al. Evidence for saponin diversity-mycobiome links and conservatism of plant-fungi interaction patterns across Holarctic disjunct Panax species. Sci Total Environ. 2022; 830: 154583. doi:10.1016/j.scitotenv.2022.154583. [Google Scholar] [CrossRef]
29. Bolyen E , Rideout JR , Dillon MR , Bokulich NA , Abnet CC , Al-Ghalith GA , et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019; 37( 8): 852– 7. doi:10.1038/s41587-019-0209-9. [Google Scholar] [CrossRef]
30. Callahan BJ , McMurdie PJ , Rosen MJ , Han AW , Johnson AJA , Holmes SP . DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016; 13( 7): 581– 3. doi:10.1038/nmeth.3869. [Google Scholar] [CrossRef]
31. Nilsson RH , Larsson KH , Taylor AFS , Bengtsson-Palme J , Jeppesen TS , Schigel D , et al. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019; 47( D1): D259– 64. doi:10.1093/nar/gky1022. [Google Scholar] [CrossRef]
32. Jiao S , Yang Y , Xu Y , Zhang J , Lu Y . Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across Eastern China. ISME J. 2020; 14( 1): 202– 16. doi:10.1038/s41396-019-0522-9. [Google Scholar] [CrossRef]
33. Delgado-Baquerizo M , Oliverio AM , Brewer TE , Benavent-González A , Eldridge DJ , Bardgett RD , et al. A global atlas of the dominant bacteria found in soil. Science. 2018; 359( 6373): 320– 5. doi:10.1126/science.aap9516. [Google Scholar] [CrossRef]
34. Csardi G , Nepusz T . The igraph software package for complex network research. InterJournal. 1695. [Google Scholar]
35. Zhao X , Gao L , Wang J , Bi H , Gao J , Du X , et al. A novel ginsenoside Rb1-hydrolyzing β-d-glucosidase from Cladosporium fulvum. Process Biochem. 2009; 44( 6): 612– 8. doi:10.1016/j.procbio.2009.01.016. [Google Scholar] [CrossRef]
36. Koren S , Walenz BP , Berlin K , Miller JR , Bergman NH , Phillippy AM . Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017; 27( 5): 722– 36. doi:10.1101/gr.215087.116. [Google Scholar] [CrossRef]
37. Zimin AV , Marçais G , Puiu D , Roberts M , Salzberg SL , Yorke JA . The MaSuRCA genome assembler. Bioinformatics. 2013; 29( 21): 2669– 77. doi:10.1093/bioinformatics/btt476. [Google Scholar] [CrossRef]
38. Marçais G , Delcher AL , Phillippy AM , Coston R , Salzberg SL , Zimin A . MUMmer4: a fast and versatile genome alignment system. PLoS Comput Biol. 2018; 14( 1): e1005944. doi:10.1371/journal.pcbi.1005944. [Google Scholar] [CrossRef]
39. Camacho C , Coulouris G , Avagyan V , Ma N , Papadopoulos J , Bealer K , et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009; 10: 421. doi:10.1186/1471-2105-10-421. [Google Scholar] [CrossRef]
40. Boutet E , Lieberherr D , Tognolli M , Schneider M , Bairoch A . UniProtKB/Swiss-prot. In: Plant bioinformatics. Totowa, NJ, USA: Humana Press; 2007. p. 89– 112. doi:10.1007/978-1-59745-535-0_4. [Google Scholar] [CrossRef]
41. Huerta-Cepas J , Szklarczyk D , Heller D , Hernández-Plaza A , Forslund SK , Cook H , et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019; 47( D1): D309– 14. doi:10.1093/nar/gky1085. [Google Scholar] [CrossRef]
42. Kanehisa M , Goto S . KEGG Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28( 1): 27– 30. doi:10.1093/nar/28.1.27. [Google Scholar] [CrossRef]
43. Gene Ontology Consortium . The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019; 47( D1): D330– 8. doi:10.1093/nar/gky1055. [Google Scholar] [CrossRef]
44. Potter SC , Luciani A , Eddy SR , Park Y , Lopez R , Finn RD . HMMER web server: 2018 update. Nucleic Acids Res. 2018; 46( W1): W200– 4. doi:10.1093/nar/gky448. [Google Scholar] [CrossRef]
45. Mistry J , Chuguransky S , Williams L , Qureshi M , Salazar GA , Sonnhammer ELL , et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 2021; 49( D1): D412– 9. doi:10.1093/nar/gkaa913. [Google Scholar] [CrossRef]
46. Price MN , Dehal PS , Arkin AP . FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One. 2010; 5( 3): e9490. doi:10.1371/journal.pone.0009490. [Google Scholar] [CrossRef]
47. Hallgren J , Tsirigos KD , Pedersen MD , Almagro Armenteros JJ , Marcatili P , Nielsen H , et al. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. BioRxiv. 2022. doi:10.1101/2022.04.08.487609. [Google Scholar] [CrossRef]
48. Teufel F , Armenteros JJA , Johansen AR , Gíslason MH , Pihl SI , Tsirigos KD , et al. SignalP 6.0 achieves signal peptide prediction across all types using protein language models. BioRxiv. 2021. doi:10.1101/2021.06.09.447770. [Google Scholar] [CrossRef]
49. Blin K , Shaw S , Augustijn HE , Reitz ZL , Biermann F , Alanjary M , et al. antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 2023; 51( W1): W46– 50. doi:10.1093/nar/gkad344. [Google Scholar] [CrossRef]
50. Emms DM , Kelly S . OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019; 20( 1): 238. doi:10.1186/s13059-019-1832-y. [Google Scholar] [CrossRef]
51. Kumar S , Suleski M , Craig JM , Kasprowicz AE , Sanderford M , Li M , et al. TimeTree 5: an expanded resource for species divergence times. Mol Biol Evol. 2022; 39( 8): msac174. doi:10.1093/molbev/msac174. [Google Scholar] [CrossRef]
52. Sanderson MJ . r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003; 19( 2): 301– 2. doi:10.1093/bioinformatics/19.2.301. [Google Scholar] [CrossRef]
53. Mendes FK , Vanderpool D , Fulton B , Hahn MW . CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics. 2021; 36( 22–23): 5516– 8. doi:10.1093/bioinformatics/btaa1022. [Google Scholar] [CrossRef]
54. Delaux PM , Schornack S . Plant evolution driven by interactions with symbiotic and pathogenic microbes. Science. 2021; 371( 6531): eaba6605. doi:10.1126/science.aba6605. [Google Scholar] [CrossRef]
55. Xue P , Yang X , Zhao L , Hou Z , Zhang R , Zhang F , et al. Relationship between antimicrobial activity and amphipathic structure of ginsenosides. Ind Crops Prod. 2020; 143: 111929. doi:10.1016/j.indcrop.2019.111929. [Google Scholar] [CrossRef]
56. Lei Q , Chen J , Yuan Y , Hu C , Lin Z , Yang S , et al. The inhibitory effects of ginsenosides on periodontitis pathogenic bacteria. Front Microbiol. 2025; 16: 1573969. doi:10.3389/fmicb.2025.1573969. [Google Scholar] [CrossRef]
57. Ma LJ , Liu X , Guo L , Luo Y , Zhang B , Cui X , et al. Discovery of plant chemical defence mediated by a two-component system involving β-glucosidase in Panax species. Nat Commun. 2024; 15( 1): 602. doi:10.1038/s41467-024-44854-7. [Google Scholar] [CrossRef]
58. Mithöfer A , Boland W . Plant defense against herbivores: chemical aspects. Annu Rev Plant Biol. 2012; 63: 431– 50. doi:10.1146/annurev-arplant-042110-103854. [Google Scholar] [CrossRef]
59. Sirikantaramas S , Yamazaki M , Saito K . A survival strategy: the coevolution of the camptothecin biosynthetic pathway and self-resistance mechanism. Phytochemistry. 2009; 70( 15–16): 1894– 8. doi:10.1016/j.phytochem.2009.07.034. [Google Scholar] [CrossRef]
60. Demurtas OC , Nicolia A , Diretto G . Terpenoid transport in plants: how far from the final picture? Plants. 2023; 12( 3): 634. doi:10.3390/plants12030634. [Google Scholar] [CrossRef]
61. Vassão DG , Wielsch N , de Melo Moreira Gomes AM , Gebauer-Jung S , Hupfer Y , Svatoš A , et al. Plant defensive β-glucosidases resist digestion and sustain activity in the gut of a lepidopteran herbivore. Front Plant Sci. 2018; 9: 1389. doi:10.3389/fpls.2018.01389. [Google Scholar] [CrossRef]
62. Deng B , Zhang P , Ge F , Liu DQ , Chen CY . Enhancement of triterpenoid saponins biosynthesis in Panax notoginseng cells by co-overexpressions of 3-hydroxy-3-methylglutaryl CoA reductase and squalene synthase genes. Biochem Eng J. 2017; 122: 38– 46. doi:10.1016/j.bej.2017.03.001. [Google Scholar] [CrossRef]
63. Kim YJ , Lee OR , Oh JY , Jang MG , Yang DC . Functional analysis of 3-hydroxy-3-methylglutaryl coenzyme a reductase encoding genes in triterpene saponin-producing ginseng. Plant Physiol. 2014; 165( 1): 373– 87. doi:10.1104/pp.113.222596. [Google Scholar] [CrossRef]
64. Wei G , Zhang G , Li M , Zheng Y , Zheng W , Wang B , et al. Panax notoginseng: panoramagram of phytochemical and pharmacological properties, biosynthesis, and regulation and production of ginsenosides. Hortic Res. 2024; 11( 8): uhae170. doi:10.1093/hr/uhae170. [Google Scholar] [CrossRef]
65. Wang L , Huang Y , Yin G , Wang J , Wang P , Chen ZY , et al. Antimicrobial activities of Asian ginseng, American ginseng, and notoginseng. Phytother Res. 2020; 34( 6): 1226– 36. doi:10.1002/ptr.6605. [Google Scholar] [CrossRef]
66. Sun P , Huang Y , Yang X , Liao A , Wu J . The role of indole derivative in the growth of plants: a review. Front Plant Sci. 2023; 13: 1120613. doi:10.3389/fpls.2022.1120613. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools