
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Coordinated circRNA/lncRNA-Mediated Post-Transcriptional Regulation Balances Anthocyanin and cis-Zeatin Metabolism in Lycium ruthenicum
1 Key Laboratory of Biodiversity Formation Mechanism and Comprehensive Utilization of the Qinghai-Xizang Plateau in Qinghai Province, School of Life Sciences, Qinghai Normal University, Xining, China
2 School of Traditional Chinese Medicine, Hubei University of Chinese Medicine, Wuhan, China
* Corresponding Author: Qiong Luo. Email:
Phyton-International Journal of Experimental Botany 2026, 95(3), 17 https://doi.org/10.32604/phyton.2026.077896
Received 19 December 2025; Accepted 25 February 2026; Issue published 31 March 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Lycium ruthenicum Murray (black goji berry) is an economically important crop rich in anthocyanins, however, the post-transcriptional regulatory mechanisms underlying anthocyanin accumulation in its fruits are unknown. Here, we used whole-transcriptome sequencing to identify differentially expressed circRNAs and lncRNAs in black goji (BS4) and white goji (WS4). Furthermore, we analyzed the regulatory networks of the key ncRNAs and their target genes by combining targeted phytohormone metabolomics with molecular docking technology. We found that the circRNA plantcirc_001165 synergistically upregulated the flavonoid biosynthesis gene LrHCT with the lncRNA MSTRG.28703 to promote anthocyanin accumulation. However, MSTRG.28703 repressed LrZOX1 expression, leading to the accumulation of cis-zeatin (cZ). Molecular docking confirmed strong interactions between these enzymes and related ligands, and network pharmacology further predicted potential targets of petanin, a major anthocyanin component, against oxidative stress and inflammation-related pathways in Alzheimer’s disease and inflammatory bowel disease. Therefore, this study systematically revealed the ncRNA-mediated synergistic regulatory network of anthocyanin and cZ metabolism in black goji, and provides a new perspective for analyzing the mechanism of its fruit coloration and leveraging the bioactivities of its anthocyanins.Keywords
Supplementary Material
Supplementary Material FileLycium ruthenicum Murray (black goji berry), a perennial shrub that belongs to the Solanaceae family, is an economically important medicinal plant endemic to arid and semi-arid regions of Central Asia [1]. Its fruits are renowned for their exceptionally high content of bioactive compounds, particularly anthocyanins, flavonoids, alkaloids, and amino acids. Petanin—(petunidin-3-O-rutinoside-(p-coumaroyl)-5-O-glucoside) is the primary anthocyanin component, accounting for over 80% of the total pigment content, alongside derivatives such as cyanidin, pelargonidin, and kaempferol aglycones [2,3]. These structurally unique compounds endow the berries with numerous biological activities. Anthocyanins are water-soluble pigments that demonstrate potent antioxidant properties and exhibit protective effects in preclinical models of Alzheimer’s disease, inflammatory bowel disease, and cardiovascular disorders [4,5,6]. Therefore, elucidating the molecular mechanisms underlying anthocyanin accumulation in black goji is necessary for harnessing the plant’s pharmacological potential and implementing molecular breeding strategies to enhance fruit quality. Furthermore, this work offers a unique perspective for understanding the adaptive evolutionary mechanisms of secondary metabolism regulation in plants under abiotic stress conditions.
Anthocyanin biosynthesis is a branch of the flavonoid metabolic pathway. The synthesis is catalyzed by a coordinated series of enzymes encoded by core structural genes, including phenylalanine ammonia-lyase (PAL), chalcone synthase (CHS), and chalcone isomerase (CHI) [7,8,9]. At the transcriptional level, this process is primarily regulated by the conserved MYB-bHLH-WD40 (MBW) protein complex [10]. Zong et al. [11] reported that the content of the MYB transcription factor LrAN2 was 35.66-fold higher in black goji than in Lycium barbarum. They also found that the functional diversity and high expression of LrAN2 likely contribute to anthocyanin accumulation. In tomato (Solanum lycopersicum), a related species, the MYB transcription factor SlAN2-like promotes anthocyanin accumulation in the peel and flesh of ‘Indigo Rose’ fruit exposed to light by activating the anthocyanin biosynthetic pathway and enhancing the expression of the bHLH protein SlAN1 [12]. Beyond transcriptional regulation, phytohormones serve as crucial signaling molecules. They are key regulators of anthocyanin synthesis, with such hormonal control exhibiting differences according to the species and tissue. For instance, auxin levels markedly influence PAP1 expression, in transgenic tobacco callus overexpressing the Arabidopsis PAP1 (At1g56650) gene, modulating the transcription factor MYB75 and ultimately altering anthocyanin content [13]. Abscisic acid (ABA) and light stimulate the expression of the transcription factor LcMYB1 in the pericarp of litchi (Litchi chinensis) by interacting with cis-elements in its promoter region [14]. This, in turn, activates the downstream structural gene LcUFGT to drive anthocyanin synthesis. By contrast, the synthetic cytokinin forchlorfenuron (CPPU) suppresses both LcMYB1 expression and anthocyanin accumulation [14].
However, the regulation of plant secondary metabolism is a multi-layered and networked process. The precise balance of secondary metabolism relies not only on the control by protein-coding genes and hormonal signals at the transcriptional level but also on the coordinated action of post-transcriptional regulatory networks [15]. In recent years, non-coding RNAs (ncRNAs) have emerged as key players in transcriptional as well as post-transcriptional regulation and have been demonstrated to participate in anthocyanin synthesis in various plant species [16,17,18]. Nevertheless, this regulatory dimension remains unexplored in black goji. Unlike protein-coding mRNAs, ncRNAs lack direct coding capacity but can modulate gene expression through diverse mechanisms. Among these, circular RNAs (circRNAs) are a class of single-stranded RNA molecules characterized by a covalently closed circular structure. The absence of free ends renders them more stable than their linear counterparts [19]. Their primary regulatory modes include functioning as miRNA sponges and interacting with RNA-binding proteins (RBPs), enabling precise post-transcriptional gene regulation [20]. Long non-coding RNAs (lncRNAs) are non-coding RNA molecules longer than 200 nucleotides. They primarily participate in the regulation of metabolic pathways during plant growth and development and stress responses by influencing chromatin states, transcriptional efficiency, or mRNA processing via cis- or trans-acting mechanisms [21,22]. Both circRNAs and lncRNAs help regulate flavonoid and anthocyanin biosynthesis in various plant species. For instance, specific circRNAs/lncRNAs have been shown to modulate anthocyanin accumulation in pepper and strawberry by targeting key biosynthetic genes [23,24]. Furthermore, the expression levels of certain circRNAs/lncRNAs exhibit correlations with anthocyanin content in Quercus mongolica and potato, suggesting potential regulatory roles [25,26]. However, whether these mechanisms can be applied to black goji remains to be determined.
No anthocyanin-associated circRNAs or lncRNAs have been systematically identified in black goji, nor has the crosstalk between these ncRNAs and hormonal signals been elucidated. Therefore, the transcriptional and post-transcriptional regulatory mechanisms responsible for its efficient anthocyanin accumulation remain unclear. Here, we used mature fruits of anthocyanin-rich black goji (BS4) and wild white goji (WS4) for the analyses. By integrating whole-transcriptome sequencing, targeted phytohormone metabolomics, and molecular docking, we identified 72 differentially expressed circRNAs and 5064 differentially expressed lncRNAs. Our analysis revealed that the circRNA plantcirc_001165 and the lncRNA MSTRG.28703 co-target and activate Hydroxycinnamoyl-CoA shikimate/quinate hydroxycinnamoyltransferase (LrHCT), a key gene in anthocyanin synthesis. Furthermore, MSTRG.28703 trans-regulates Zeatin O-xylosyltransferase 1 (LrZOX1), promoting the accumulation of cis-zeatin (cZ). Molecular docking confirmed the high-affinity interactions between the corresponding proteins and their ligands. Hence, this work elucidates an ncRNA-mediated mechanism involving the coordinated regulation of LrHCT and LrZOX1 to balance anthocyanin and cZ metabolism. In addition, petanin, the predominant anthocyanin in black goji, possesses potent antioxidant and anti-inflammatory activities [27]. Thus, given that oxidative stress and inflammatory responses are often pivotal factors in the pathogenesis of chronic diseases, we employed network pharmacology approaches to predict the potential multi-target mechanisms by which petanin may intervene in Alzheimer’s disease (AD) and inflammatory bowel disease (IBD). Therefore, through integrated multi-omics analysis, this study also aims to uncover the ncRNA-mediated regulatory mechanism governing the balance of anthocyanin and cZ metabolism, and provide a theoretical foundation for the functional application of black goji anthocyanins.
2.1 Whole-Transcriptome Sequencing
Black and white fruit samples were collected from Nuomuhong Farm, Zongjia Town, Qaidam Basin, Qinghai Province, China (longitude: 96°27.2856′ E, latitude: 36°26.9625′ N; altitude: 2856.6 m). Sampling was conducted in September 2021, during which fresh fruits at the fully mature stage (S4) were harvested from different plants grown under identical cultivation conditions. Upon collection, the samples were immediately snap-frozen in liquid nitrogen and stored at −80°C for subsequent RNA extraction and sequencing analysis. All samples were preserved at the School of Life Sciences, Qinghai Normal University.
Total RNA was extracted from both black goji and white goji using the RNAprep Pure Plant Kit (a polysaccharides & polyphenolics-rich spin-column) (Tiangen, Beijing, China). The quality of the extracted total RNA was verified using a NanoDrop spectrophotometer and agarose gel electrophoresis. All procedures were performed using three biological replicates.
Total RNA from white and black goji fruits that met the quality criteria was sent to Shanghai Panomix Biomedical Technology Co., Ltd. for sequencing. Strand-specific libraries were constructed using a ribosomal RNA depletion method [28]. The prepared libraries were then subjected to paired-end 150 bp (PE150) sequencing on an Illumina platform. Raw sequencing reads were processed to remove low-quality bases and low-complexity sequences from both ends, resulting in high-quality clean reads.
2.1.4 Data Quality Control and Sequence Alignment
Adapter sequences at the 3′ ends and reads with an average quality score below Q20 were removed using fastp (v0.22.0). Subsequently, the reference genome index was built using HISAT2 (v2.1.0), and the paired-end clean reads were aligned to the black goji reference genome (GCA_041430385.1, downloaded from the NCBI database) using HISAT2 [29].
2.1.5 Bioinformatics Analysis of ncRNAs
Twenty bp sequences from both ends of the reads that failed to align in the initial HISAT2 mapping were extracted as anchoring sequences for circRNA detection. These anchors were realigned to the genome using Bowtie2 (v2.5.1). Following alignment of the anchor sequences for each sample, circRNAs were identified using find_circ (v1.0) [30]. High confidence circRNAs were filtered based on the following criteria: only one clear breakpoint; anchor sequence overlap ≤ 2 bp; mismatch ≤ 2 bp; supported by ≥2 reads covering more than half of the samples; at least one anchoring sequence per read pair having an alignment score > 35 to the genome; circRNA length < 100 kb.
For lncRNA identification, transcripts were first assembled from the HISAT2 alignment results using StringTie (v2.2.1) [31]. Transcripts with ambiguous strand orientation were removed. Subsequently, transcripts were filtered to exclude those with uncertain strand direction, length < 200 bp, or minimum read coverage < 3. Furthermore, only transcripts belonging to the “x”, “u” or “i” categories were retained. (“x” indicates exonic overlap with a reference transcript on the opposite strand; “u” indicates unknown, intergenic transcripts; “i” indicates transcripts falling entirely within an intron of a reference transcript). Finally, the protein-coding potential was assessed using three tools in parallel: PLEK (v1.2), CNCI (v1.0), and PfamScan (v1.6.4). Novel transcripts predicted to lack protein-coding potential by all three tools were defined as high confidence lncRNAs.
ncRNA Expression Quantification and Differential Expression Analysis
Reads counts for circRNAs from the find_circ results were tallied, and circRNA expression levels were normalized using as transcripts per million (TPM). For lncRNAs, reads counts were obtained at the transcript level using StringTie, and expression was normalized as fragments per kilobase of transcript per million mapped reads (FPKM). Differential expression analysis between the two comparison groups was performed using DESeq2 (v1.38.3) [32]. The differential expression of ncRNAs was assessed separately, with the screening criteria set as |log2(fold change)| > 1 and p < 0.05.
Enrichment Analysis of Source Genes for Differentially Expressed ncRNAs
GO enrichment analysis for the source genes of differentially expressed ncRNAs was conducted using topGO (v2.50.0) [33]. KEGG pathway enrichment analysis for these source genes was performed using the clusterProfiler software package (v4.6.0) [34].
Target Gene Prediction for Differentially Expressed lncRNAs
Cis-acting regulation is the effect of a lncRNA on nearby target genes. Protein-coding genes located within 100 kb upstream or downstream of a lncRNA were screened as potential cis-targets. Trans-acting regulation indicates the ability for recognition based on expression level correlations. Putative trans-acting lncRNA–mRNA pairs were identified by screening for correlations with an absolute correlation coefficient |r| > 0.95 and p < 0.05.
2.2 Candidate Gene Screening and Bioinformatics Analysis
The open reading frame (ORF) of Lr11g005079 was predicted using the online tool ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/), and its amino acid sequence was deduced (parameters: default codon table, minimum ORF length ≥ 100 bp). A phylogenetic tree of homologous proteins was constructed using the neighbor-joining method in MEGA11.0 [35].
2.3 Targeted Metabolomic Profiling of Phytohormones
This study employed targeted mass spectrometry to quantitatively analyze 21 phytohormones, ABA, 1-aminocyclopropanecarboxylic acid (ACC), brassinolide (BL), castasterone (CS), dihydrozeatin (DHZ), gibberellin A1 (GA1), gibberellin A3 (GA3), gibberellin A4 (GA4), gibberellin A7 (GA7), indole-3-acetic acid (IAA), isopentenyl adenine (iP), isopentenyl adenosine (iPR), jasmonic acid (JA), jasmonoyl-isoleucine (JA-Ile), cis-12-oxophytodienoic acid (12-OPDA), salicylic acid (SA), typhasterol (TY), cZ, trans-zeatin (tZ), cis-zeatin riboside (cZR), and trans-zeatin riboside (tZR).
Six samples (three biological replicates each of BS4 and WS4) were placed on ice to thaw (0–4°C). Immediately after thawing, approximately 100 mg of each sample was weighed using an AL104 analytical balance (Mettler Toledo, Greifensee, Switzerland) and combined with mixed isotope-labeled internal standards and pre-cooled extraction solvent (acetonitrile/water/formic acid, 80:19:1, v/v/v). The mixture was vortexed using a QT-1 vortex mixer (Qite Analytical Instrument, Shanghai, China) and then ultrasonicated in a JP-100 ultrasonic bath (Jiemeng Cleaning Equipment, Shenzhen, China) for 25 min at 4°C. After centrifugation at 14,000 rcf for 20 min at 4°C, using a 5430R centrifuge (Eppendorf, Hamburg, Germany), the supernatant was transferred to an Ostro 25 mg 96-well plate (Waters, Milford, MA, USA) and filtered using a positive pressure manifold. The filtrate was collected into 2 mL centrifuge tubes and stored at −80°C.
2.3.2 Chromatography-Mass Spectrometry Analysis
Sample separation was performed using an Agilent 1290 Infinity LC ultra-high-performance liquid chromatography (UHPLC) system (Agilent Technologies, Santa Clara, CA, USA). Samples were maintained at 4°C in the autosampler. Chromatographic separation was achieved on an ACQUITY UPLC BEH C18 column (2.1 mm × 100 mm, 1.7 μm; Waters, Milford, MA, USA) with the temperature set at 45°C. Mobile phase A consisted of 0.05% formic acid in water, and mobile phase B was 0.05% formic acid in acetonitrile. The flow rate was 400 μL/min with an injection volume of 4 μL. The LC gradient was programmed as follows: 0–1 min, from 2% to 10% B; 1–10 min, from 10% to 70% B; 10–11 min, from 70% to 95% B; 11–11.1 min, from 95% to 2% B; 11.1–13 min, 2% B.
Mass spectrometric analysis was conducted on a 6500+ QTRAP mass spectrometer (SCIEX, Framingham, MA, USA) operating in both positive and negative ion modes. The following electrospray ionization (ESI) source parameters for positive ion mode were as used: source temperature, 550°C; ion source gas 1 (Gas1), 55 psi; ion source gas 2 (Gas2), 50 psi; curtain gas (CUR), 30 psi; IonSpray voltage floating (ISVF), 4500 V. For the negative ion mode, the parameters were: source temperature, 550°C; Gas1, 55 psi; Gas2, 50 psi; CUR, 30 psi; ISVF, −4500 V. Analytes were detected in the multiple reaction monitoring (MRM) mode.
The three-dimensional structures of DHZ, tZ, tZR, cZ, cZR, and petanin were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) using the 439631, 449093, 6440982, 688597, 13935024, and 6540681 compound IDs (CIDs), respectively. The protein structures of LrHCT and LrZOX1 were retrieved from the UniProt website (https://www.uniprot.org/). Blind docking was performed using the CB-DOCK2 online platform [36] with default parameters to analyze the optimal binding sites and binding affinity between the proteins and compounds. A binding energy lower than or equal to −5.0 kcal·mol−1 (1 kcal = 4.184 J) was considered indicative of a favorable binding capability between the compound and the target protein. The protein-protein interaction structure of LrHCT and LrZOX1 was predicted using AlphaFold3 (https://golgi.sandbox.google.com/) with default parameters and visualized using PyMOL (v3.1).
2.5 Network Pharmacology Analysis
2.5.1 Prediction and Screening of Potential Petanin Targets
The SMILES notation of petanin C[C@H]1[C@@H]([C@H]([C@H](C(O1)OC[C@@H]2[C@H]([C@@H]([C@H](C(O2)OC3=C([O+]=C4C=C(C=C(C4=C3)OC5[C@@H]([C@H]([C@@H]([C@H](O5)CO)O)O)O)O)C6=CC(=C(C(=C6)OC)O)O)O)O)O)O)O)OC(=O)/C=C/C7=CC=C(C=C7)O was obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) by searching for the keyword ‘petanin’. This SMILES string was then submitted to the SwissTargetPrediction (https://www.swisstargetprediction.ch/), SEA (https://sea.bkslab.org/), and CTD (https://ctdbase.org/) databases to predict potential targets of petanin.
2.5.2 Screening of AD and IBD Targets and Identification of Intersection Genes
Target genes associated with Alzheimer’s disease (AD) and inflammatory bowel disease (IBD) were retrieved from the GeneCards (https://www.genecards.org/, relevance score > 4) and OMIM (https://www.omim.org/) databases using the keywords “Alzheimer’s disease” and “inflammatory bowel disease”, respectively. The obtained AD and IBD target gene sets were then intersected with the potential petanin targets. The intersection was visualized using InteractiVenn (https://www.interactivenn.net/index2.html) [37]. The resulting intersection genes were considered as potential target genes for petanin intervention in AD and IBD.
2.5.3 Construction of Protein-Protein Interaction (PPI) Networks for Petanin-IBD Intersection Targets
The intersection target genes identified above were input into the STRING database (https://cn.string-db.org/), with the organism set to Mus and a confidence threshold of 0.4. The filtered interaction data were exported and imported into Cytoscape (v3.9.1). The “network analyzer” function was used to calculate the degree centrality of the interacting proteins and visualize the PPI network.
2.5.4 GO and KEGG Enrichment Analysis
GO and KEGG enrichment analyses of the key target genes were performed using the Bioinformatics online platform (https://www.bioinformatics.com.cn/).
Statistical analysis was performed using GraphPad Prism (v10.1). Data are presented as the mean ± standard error of the mean (SEM). Differences between the two groups were assessed using a t-test for independent samples. Statistical significance was set at p < 0.05.
3.1 Quality Assessment of Transcriptome Sequencing Data
We performed sequencing on three biological replicates of mature fruits from each of WS4 (white goji) and BS4 (black goji) to assess the reliability of the transcriptome data (Fig. 1A,B). For WS4, an average of 104,999,689 raw reads was obtained, yielding 103,505,081 clean reads after filtering. For BS4, an average of 105,972,212 raw reads yielded 104,497,323 clean reads. For both groups, the Q20 scores exceeded 98%, the Q30 scores exceeded 96%, and the guanine-cytosine (GC) content was above 40% (Table 1), indicating high base accuracy consistent with plant genomic characteristics. The clean reads were then aligned to the reference genome GCA_041430385.1 (Table 2). The overall alignment rate was greater than 92.01%, with a unique alignment rate exceeding 88.11% and a multi-alignment rate below 11.89%, ensuring the specificity of sequence matching. In summary, the sequencing data were of high quality and strong usability, and provided a solid foundation for subsequent ncRNA identification and differential expression analysis.
Table 1: Statistics of transcriptome sequencing data for WS4 and BS4.
| Sample | BS4_1 | BS4_2 | BS4_3 | WS4_1 | WS4_2 | WS4_3 |
|---|---|---|---|---|---|---|
| Raw Reads | 97,518,182 | 108,592,890 | 111,805,564 | 91,959,538 | 109,748,226 | 113,291,304 |
| Clean Reads | 96,162,270 | 107,082,406 | 110,247,294 | 90,619,316 | 108,216,566 | 111,679,360 |
| GC (%) | 40.75 | 40.76 | 40.74 | 41.00 | 41.22 | 41.09 |
| Q20 (%) | 98.95 | 98.87 | 98.91 | 98.95 | 98.87 | 98.85 |
| Q30 (%) | 97.03 | 96.76 | 96.87 | 97.03 | 96.75 | 96.70 |
Table 2: Alignment statistics of clean reads against the reference genome.
| Sample | BS4_1 | BS4_2 | BS4_3 | WS4_1 | WS4_2 | WS4_3 |
|---|---|---|---|---|---|---|
| Clean_Reads | 96,162,270 | 107,082,406 | 110,247,294 | 90,619,316 | 108,216,566 | 111,679,360 |
| Total_Mapped | 89,074,326 (92.63%) | 98,523,033 (92.01%) | 101,660,729 (92.21%) | 84,447,774 (93.19%) | 100,935,228 (93.27%) | 104,256,477 (93.35%) |
| Multiple_Mapped | 10,157,744 (11.40%) | 10,293,150 (10.45%) | 10,421,763 (10.25%) | 9,987,716 (11.83%) | 11,998,422 (11.89%) | 12,200,091 (11.70%) |
| Uniquely_Mapped | 78,916,582 (88.60%) | 88,229,883 (89.55%) | 91,238,966 (89.75%) | 74,460,058 (88.17%) | 88,936,806 (88.11%) | 92,056,386 (88.30%) |
3.2 Differential Expression and Functional Pathway Analysis of circRNAs
We identified 647 high-confidence circRNAs, using the find_circ software tool (detailed in Section 2.1.5). The analysis of circRNA expression abundance revealed a uniform TPM density distribution for both BS4 and WS4, with a low proportion of lowly and highly expressed circRNAs. This observation indicates a generally stable circRNA expression level across the two sample groups (Fig. 1C). Furthermore, principal component analysis (PCA) demonstrated high clustering of biological replicates within each group and clear separation between groups. This result confirms that the observed differences in circRNA expression primarily originate from genotypic variation and that the experimental reproducibility is reliable (Fig. 1D). Notably, the overall expression level of circRNAs in BS4 was substantially higher than in WS4, suggesting a potential role for circRNAs in the regulation of the distinctive anthocyanin synthesis metabolism in black goji.
Applying the criteria of |log2(FC)| > 1 and p < 0.05, we identified 72 differentially expressed circRNAs, of which 31 were upregulated and 41 were downregulated in BS4 relative to WS4 (Fig. 1E). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed on the 111 corresponding source genes. In the GO functional classification, these genes were primarily enriched in three major categories: “cellular component (CC)” (e.g., potassium ion-transporting ATPase complex), “molecular function (MF)” (e.g., glucan exo-1,3-beta-glucosidase activity), and “biological process (BP)” (e.g., stabilization of membrane potential) (Fig. S1A). The KEGG pathway enrichment analysis identified 23 substantially enriched pathways. Among these, the “photosynthesis–antenna proteins” and “basal transcription factors” pathways exhibited the highest enrichment scores. The “photosynthesis–antenna proteins” pathway is involved in the absorption and conversion of luminous energy. Its enrichment suggests that the differentially expressed circRNAs may contribute to enhanced photosynthetic efficiency in BS4, potentially providing more energy for anthocyanin synthesis (Fig. S1B).
Then, we performed cluster analysis on the upregulated circRNAs in BS4 to further pinpoint key circRNAs associated with anthocyanin synthesis. This analysis revealed that plantcirc_001165 was expressed only in BS4, with no detectable signal in WS4 (Fig. 1F). Its source gene, Lr11g005079, was annotated as a key gene in the core anthocyanin synthesis pathways: flavonoid biosynthesis (ko00941) and phenylpropanoid biosynthesis (ko00940). Quantitative experimental validation showed that the relative expression levels of both plantcirc_001165 and Lr11g005079 were significantly higher in BS4 than in WS4 (where both were undetectable, p < 0.05), and their expression trends were highly consistent (p < 0.05). This result suggests that plantcirc_001165 may directly participate in the flavonoid biosynthetic pathway in black goji by regulating its source gene Lr11g005079.
The above results delineate a circRNA-mediated regulatory pathway for anthocyanin synthesis. However, the transcriptional and post-transcriptional regulation of plant secondary metabolism typically relies on the coordinated actions of multiple classes of ncRNAs. Therefore, our subsequent analysis focuses on the differential expression profiles of lncRNAs and investigates their potential relationships with Lr11g005079 and phytohormone metabolism, aiming to systematically elucidate the regulatory mechanisms governing the balance between anthocyanin and hormone metabolism.
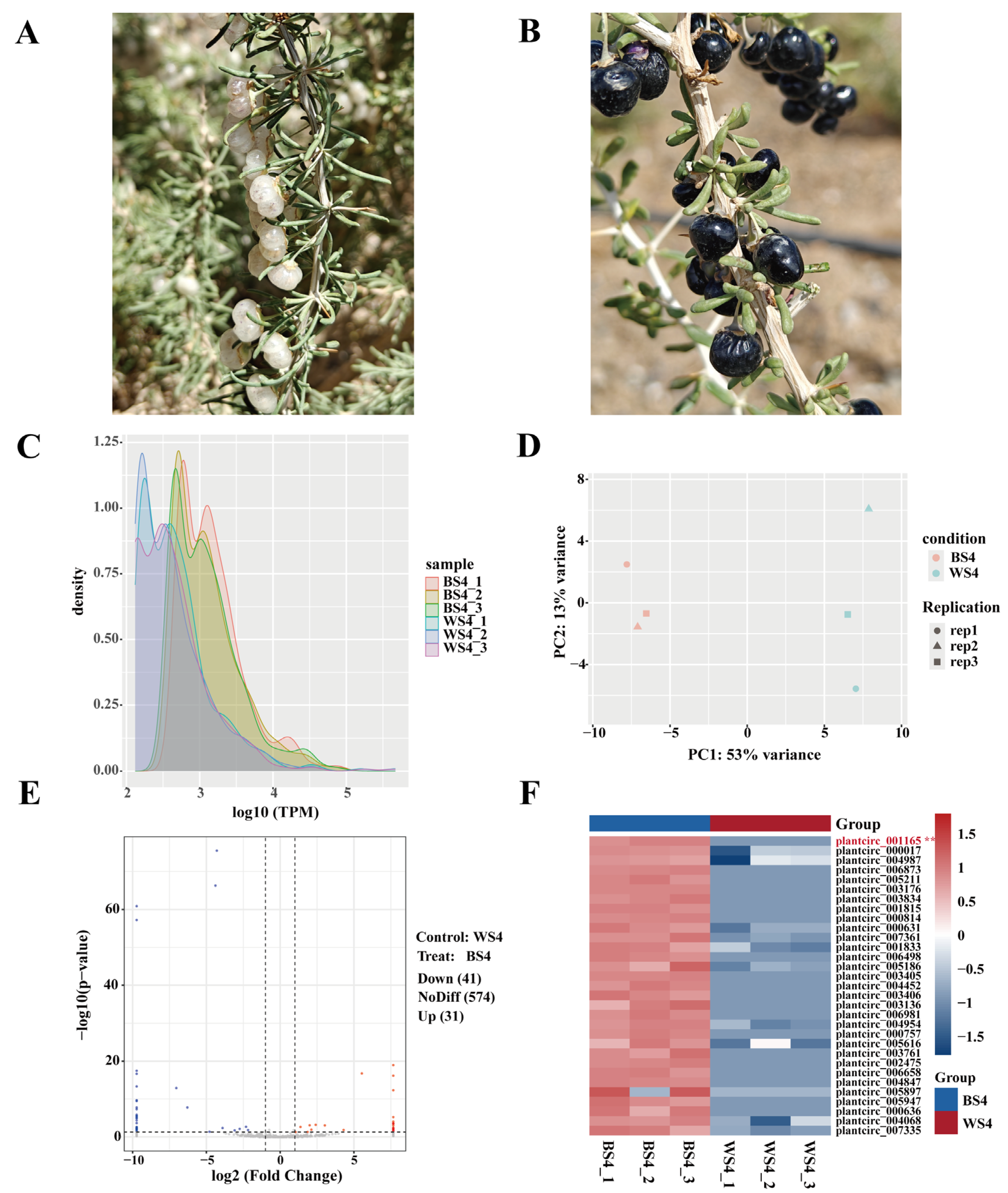
Figure 1: Phenotype and transcriptomic analysis of black goji fruits. (A) White goji (WS4). (B) Black goji (BS4). (C) Density distribution of the circRNA expression levels (TPM). (D) Principal component analysis (PCA) of the circRNA expression profiles. (E) Volcano plot of differentially expressed circRNAs. (F) Heatmap of upregulated circRNAs (The asterisk next to plantcirc_001165 highlights it as the key circRNA that is specifically expressed in the black goji (BS4) group).
3.3 Differential Expression and Functional Pathway Analysis of lncRNAs
Although the circRNA plantcirc_001165 may be involved in anthocyanin synthesis by regulating the source gene (Lr11g005079), a single class of ncRNA cannot fully explain the metabolic regulatory network. Thus, to decipher the role of lncRNAs in this process, we employed three software tools—PLEK, CNCI, and PfamScan (detailed in Section 2.1.5)—to screen for high-confidence lncRNAs, and identified a total of 7882 (Fig. S2A). The analysis of lncRNA expression abundance revealed uniform FPKM density curves for both BS4 and WS4, with a low proportion of lowly and highly expressed lncRNAs. This result indicates generally stable lncRNA expression levels across the two sample groups, with no extreme expression biases (Fig. S2B). This outcome, consistent with the circRNA results, further confirms the robust quality of our transcriptomic data.
A total of 5064 differentially expressed lncRNAs were identified using the criteria of |log2(FC)| > 1 and p < 0.05. Among these, 3147 were upregulated and 1917 were downregulated in BS4 compared to WS4, accounting for 62.1% of the differential lncRNAs (Fig. 2A). Notably, the number of differentially expressed lncRNAs was substantially greater than that of circRNAs (72), and the proportion of upregulated lncRNAs was substantially higher. This finding suggests that lncRNAs may play a more central role than circRNAs in transcriptional and post-transcriptional regulation underlying the divergence in anthocyanin metabolic pathways.
Cis-target genes of the differentially expressed lncRNAs were predicted, yielding 40,702 potential target genes (detailed in Section 2.1.5). Then, GO and KEGG enrichment analyses were performed to elucidate the functional associations of these target genes. In the GO functional classification, they were enriched in terms such as “nuclear membrane” (CC), “serine-type endopeptidase inhibitor activity” (MF), and “calcium ion transport into cytosol” (BP), among others (Fig. S2C). KEGG analysis identified 127 enriched pathways. Notably, three secondary metabolic pathways were significantly (p < 0.05) enriched: zeatin biosynthesis (ko00908, 34 mRNAs), flavonoid biosynthesis (ko00941, 23 mRNAs), and phenylpropanoid biosynthesis (ko00940, 74 mRNAs) (Fig. 2B). The significant enrichment of the zeatin biosynthesis pathway represents a unique feature of lncRNA regulation (not observed in the circRNA enrichment analysis). This observation suggests that lncRNAs may simultaneously link anthocyanin synthesis and hormone metabolism, providing a clue for subsequent investigation into the mechanisms balancing these two metabolic processes.
We focused on the key anthocyanin synthesis gene Lr11g005079 to validate the synergistic regulatory roles of ncRNAs. Consistent with the circRNA analysis results, this gene was also enriched in the flavonoid and phenylpropanoid biosynthesis pathways. This finding confirms that both the circRNA and lncRNA regulatory networks target this core gene and converge on the anthocyanin synthesis pathway. By further screening lncRNAs associated with the phenylpropanoid biosynthesis pathway and generating an expression heatmap, we identified the cis-acting lncRNA MSTRG.28703, which targets Lr11g005079. MSTRG.28703 exhibited significantly higher (p < 0.05) expression in BS4, and its expression trend was synchronized with that of Lr11g005079 (Fig. 2C). This result suggests that MSTRG.28703 may activate Lr11g005079 through cis-regulation, promoting efficient anthocyanin accumulation.
These findings establish the regulatory role of lncRNAs, particularly MSTRG.28703, in anthocyanin synthesis. MSTRG.28703 not only synergistically targets Lr11g005079 along with circRNAs, but may also be linked to hormone metabolism through the zeatin biosynthesis pathway. However, the evolutionary conservation of Lr11g005079 and the specific function of its encoded protein remain unclear. Therefore, the following section describes phylogenetic and functional annotation analyses performed to clarify the evolutionary position and enzymatic function of this gene, providing a molecular foundation for the in-depth exploration of the metabolic mechanisms.
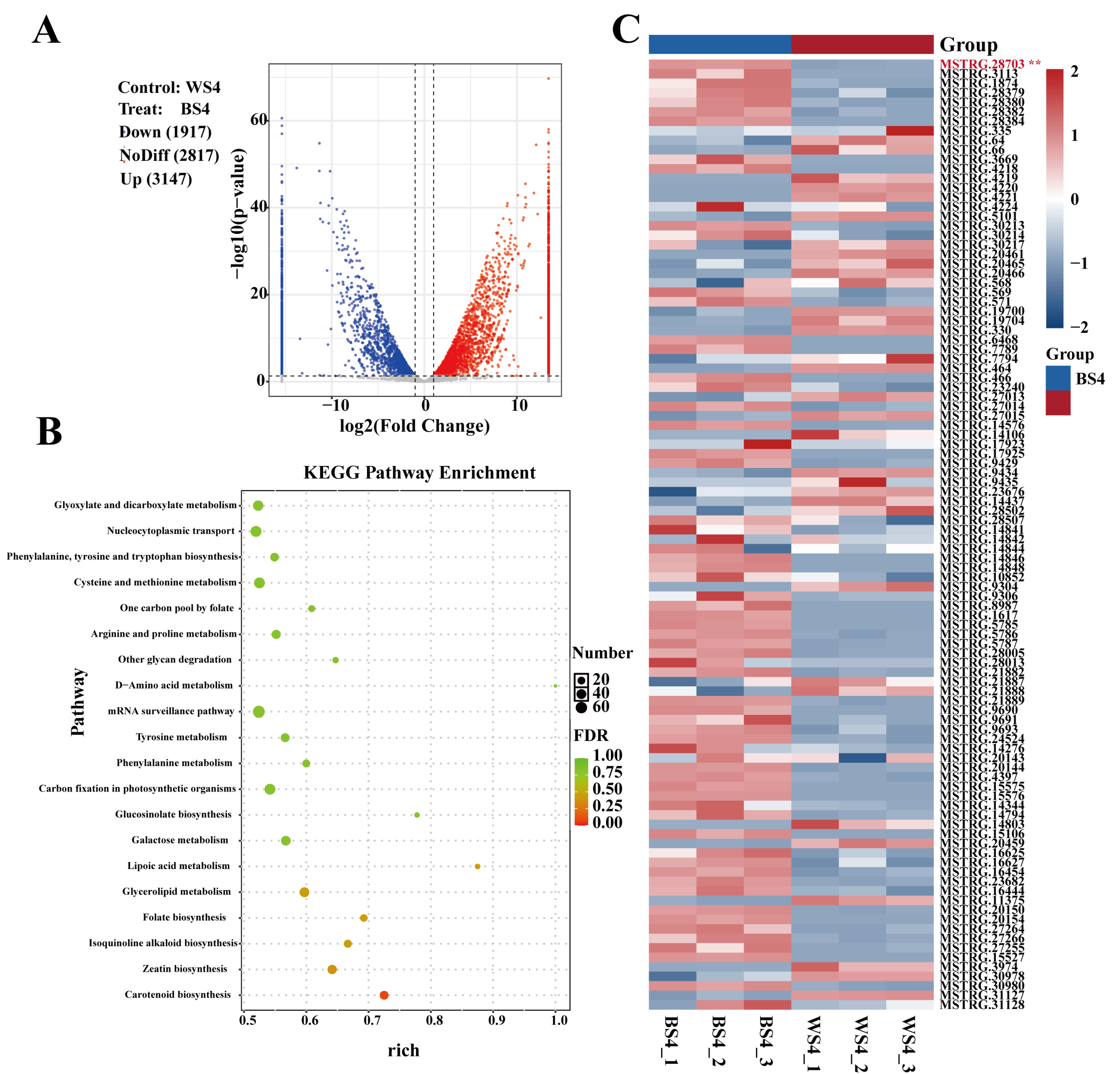
Figure 2: Analysis of differentially expressed lncRNAs. (A) Volcano plot of differentially expressed lncRNAs. (B) KEGG pathway enrichment analysis of the cis-target genes of lncRNAs. (C) Heatmap showing the expression of lncRNAs associated with the phenylpropanoid biosynthesis pathway (The asterisk next to MSTRG.28703 denotes this crucial cis-acting lncRNA, which targets Lr11g005079 and exhibits significantly higher expression in BS4.).
3.4 Phylogenetic Analysis and Functional Annotation of the Key Gene LrHCT
We conducted phylogenetic analysis and functional annotation to elucidate the evolutionary position and function of Lr11g005079. The full-length genomic sequence of this gene, comprising 1498 bp, was retrieved from the black goji reference genome. One high-confidence open reading frame, designated ORF39, was identified using the online tool NCBI ORFfinder. Phylogenetic analysis revealed that the amino acid sequence encoded by Lr11g005079 clusters within a clade containing homologous sequences from species belonging to the genera Lycium (e.g., L. ferocissimum and L. barbarum), Anisodus (e.g., A. acutangulus), and Populus (e.g., P. alba) (Fig. 3A). All sequences within this clade are annotated as members of the BAHD acyltransferase family. The core function of the BAHD acyltransferase family is to catalyze the acylation of secondary metabolites, such as flavonoids and phenylpropanoids. This modification involves the transfer of acyl groups to metabolite molecules, regulating their stability, water solubility, and bioactivity. These results suggest that the protein encoded by Lr11g005079 has a conserved function and possesses acyltransferase activity. This protein is likely involved in anthocyanin synthesis in black goji by catalyzing the acylation of anthocyanin precursors.
KEGG pathway mapping was performed on homologous genes to further delineate the specific role of Lr11g005079 within metabolic pathways. The analysis revealed the involvement of this gene in three core secondary metabolic pathways: (1) the biosynthesis of secondary metabolites (ko01110); (2) flavonoid biosynthesis (ko00941); (3) phenylpropanoid biosynthesis (ko00940) (Fig. 3B). Further annotation of key enzymes within these pathways showed that five out of eight homologous genes could be mapped to the critical shikimate enzyme O-hydroxycinnamoyltransferase (HCT, EC: 2.3.1.133). HCT catalyzes the conjugation of p-coumaroyl-CoA with shikimate/quinate to produce key intermediates such as caffeoyl-CoA. Caffeoyl-CoA serves as an essential substrate for the synthesis of anthocyanin precursors, such as cyanidin and petunidin aglycones. This result indicates that Lr11g005079 is a key gene encoding HCT. This gene directly participates in anthocyanin synthesis in black goji by regulating the metabolic flux through the phenylpropanoid–flavonoid pathway.
These findings clarify the function of Lr11g005079 (designated LrHCT) and provide downstream target support for the cis-regulation by its upstream lncRNA, MSTRG.28703. Given the substantial enrichment of lncRNA target genes in the zeatin biosynthesis pathway (ko00908), we hypothesize that lncRNAs may establish a metabolic balance network between anthocyanins and zeatin by dually regulating LrHCT and the associated zeatin metabolism. Therefore, subsequent analysis integrates hormone metabolome data to elucidate the lncRNA-mediated co-regulatory module for anthocyanin–zeatin metabolism.
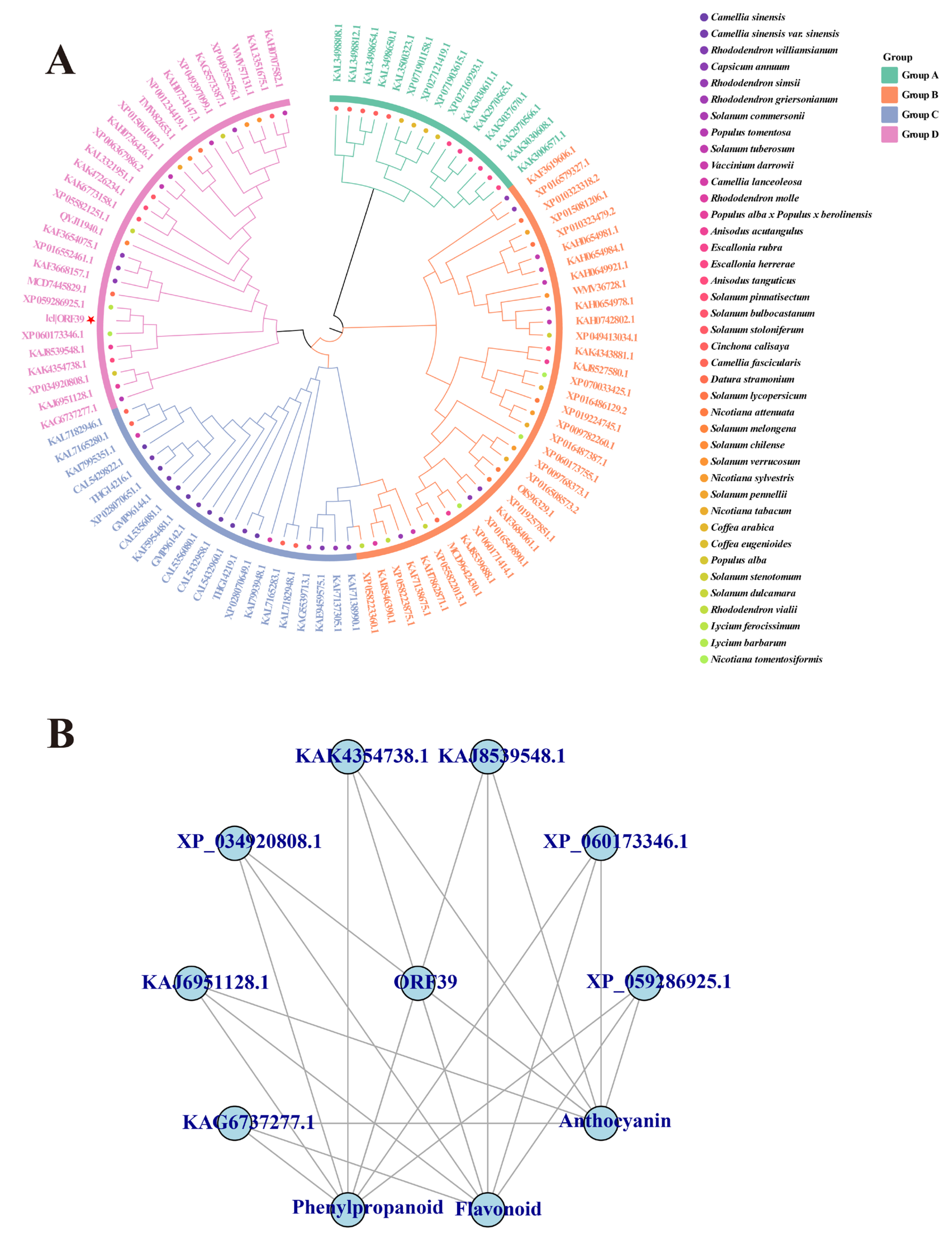
Figure 3: Phylogenetic and functional annotation analysis of Lr11g005079. (A) Phylogenetic tree of the protein encoded by Lr11g005079 (The red star marks ORF39, which represents the high-confidence amino acid sequence encoded by our key gene Lr11g005079.). (B) KEGG mapping of Lr11g005079 and its homologous genes.
3.5 lncRNA Regulatory Network and Hormone Quantification
Having established that Lr11g005079 encodes the key anthocyanin synthesis enzyme LrHCT, and given the indication from Section 3.3 that lncRNA MSTRG.28703 may cis-regulate LrHCT, we investigated whether this lncRNA is also linked to zeatin metabolism. We predicted the related trans-target genes of MSTRG.28703, and combined this information with hormone quantification for validation. Trans-target prediction yielded 323 co-expressed mRNAs (detailed in Section 2.1.5). KEGG pathway annotation of these mRNAs revealed that Lr01g000261 was annotated as LrZOX1. Although this gene is annotated within the zeatin biosynthesis pathway (ko00908), its biological function is to catalyze the O-xylosylation of zeatin, representing a critical node in hormone metabolism and homeostasis regulation. Furthermore, both LrZOX1 and LrHCT (Lr11g005079) belong to the overarching secondary metabolites biosynthesis pathway (ko01110). Moreover, as the trans- and cis-target gene of lncRNA MSTRG.28703 respectively, they exhibited significant synergy in their expression patterns, suggesting a potential metabolic connection between them.
We performed an intersection analysis between the 34 mRNAs from the zeatin biosynthesis pathway and the 40,702 cis-target genes of the differentially expressed lncRNAs to further identify lncRNAs that cis-regulate LrZOX1. This approach allowed us to identify two key lncRNAs: MSTRG.66 and MSTRG.64. Both cis-regulate LrZOX1 and are concurrently enriched in the phenylpropanoid biosynthesis pathway (ko00940). This forms a potential co-regulatory module targeting both anthocyanin and zeatin metabolism, with a functional division comprising three main parts: (1) The core lncRNA MSTRG.28703, which cis-regulates the key gene for anthocyanin synthesis, LrHCT, which is highly expressed in BS4. (2) The cooperative lncRNAs MSTRG.66/MSTRG.64, which cis-regulate the zeatin modification gene LrZOX1 and are highly expressed in WS4. (3) LrHCT and LrZOX1, which regulate anthocyanin synthesis and zeatin activity, respectively. Expression validation confirmed that all members within this module showed significant differential expression between BS4 and WS4 (p < 0.05, Fig. 4A). Furthermore, MSTRG.28703 exhibited a significant negative correlation with MSTRG.66/MSTRG.64 (p < 0.05, Fig. 4B). This result suggests that the module may regulate the expression balance of LrHCT and LrZOX1 simultaneously through an antagonistic mechanism where MSTRG.28703 suppresses MSTRG.66/MSTRG.64.
LrZOX1 catalyzes the conjugation of active zeatin with UDP-xylose, forming inactive storage forms, such as zeatin-O-xyloside [38,39,40]. Among the active zeatin isomers, cZ is particularly noteworthy. Although it exhibits lower bioactivity than tZ in stimulating cell division and promoting growth, it plays a pivotal role in maintaining the stability of the photosynthetic apparatus and mediating stress responses [41]. This physiological function aligns closely with the stress-resistance characteristics of anthocyanins. Targeted quantitative analysis of 21 phytohormones (e.g., auxins and cytokinins) was performed on BS4 and WS4 fruits to verify whether the differential expression of LrZOX1 affected cZ content (detailed in Section 2.3). The results revealed that the differences in free-form zeatin-type compounds—namely DHZ, cZ, and tZR—were the most pronounced among hormones (p < 0.05, Fig. 4C). The content of DHZ was 12.1-fold higher, cZ was 2.5-fold higher, and tZR was 1.2-fold higher in BS4 than in WS4. These findings confirm the specific accumulation of cZ derivatives in BS4. Furthermore, a significant negative correlation (p < 0.05) was observed between the levels of these active zeatin compounds and LrZOX1 expression, validating the hypothesis reduced LrZOX1 expression in BS4 leads to greater accumulation of cZ. These results suggest that LrZOX1 modulates the active pool of cZ and indicate that cZ may function as a signaling molecule involved in anthocyanin accumulation in black goji.
These results elucidate the mechanism by which the lncRNA module co-regulates anthocyanin and cZ metabolism at both the transcriptional and metabolic levels. The next section describes the use of molecular docking to further validate this metabolic balance model from a structural perspective of protein–ligand interactions.
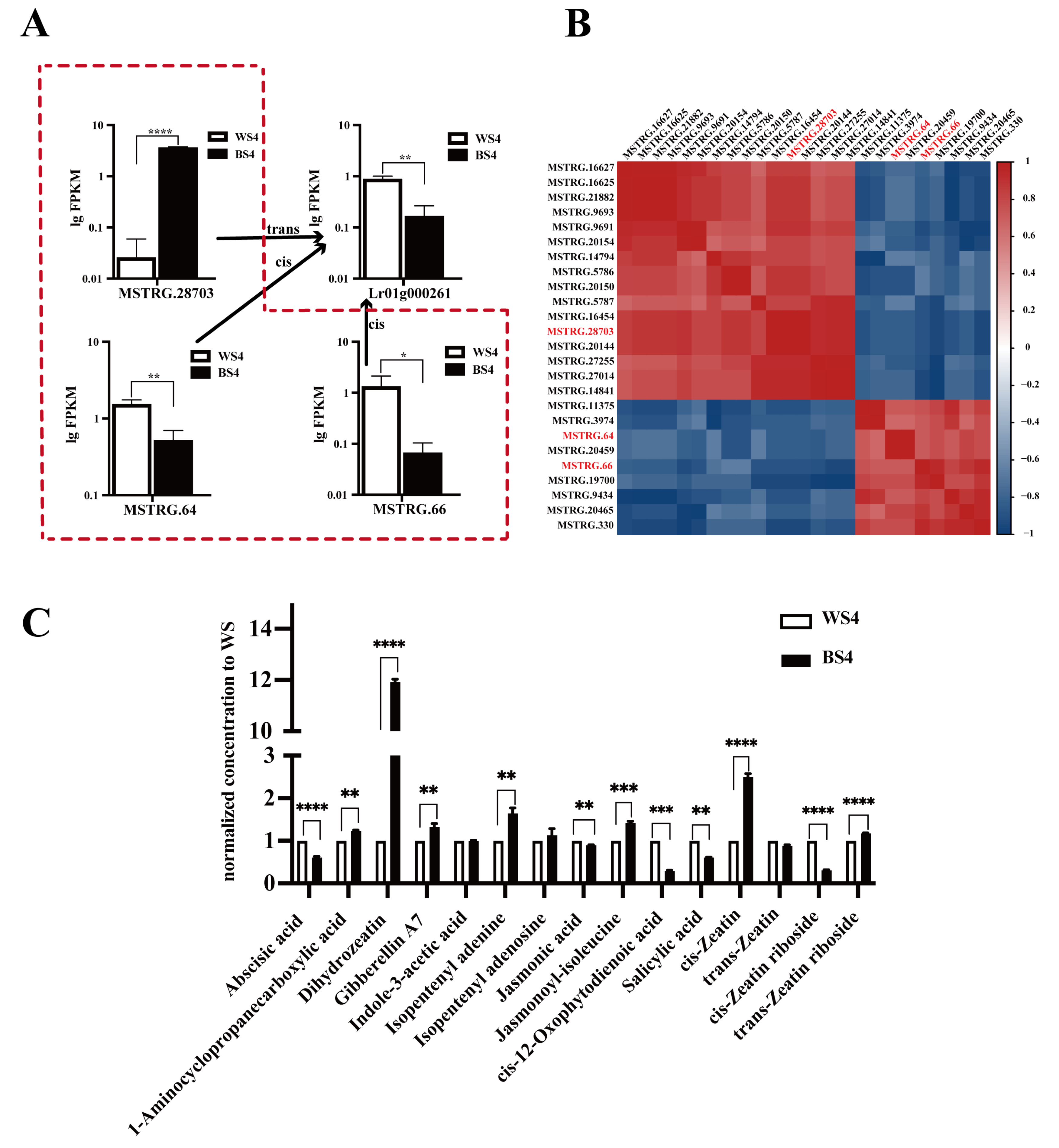
Figure 4: Analysis of the lncRNA regulatory module and associated hormone levels. (A) Expression levels (bar plot) of MSTRG.28703, MSTRG.66, MSTRG.64, and Lr01g000261. (B) Correlation heatmap of all lncRNAs associated with the phenylpropanoid biosynthesis pathway. (C) Bar plot showing the quantified levels of selected phytohormones. Statistical significance is indicated as follows: ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05.
3.6 Molecular Docking Validation of Key Protein–Ligand Interactions
Previous findings have confirmed that the lncRNA regulatory module modulates the expression of LrHCT and LrZOX1 through an antagonistic effect, concomitant with a notable increase in multiple free-form zeatins in BS4. However, the molecular underpinnings of this regulatory module—specifically, whether zeatin family small molecules directly interact with LrHCT/LrZOX1, whether LrHCT and LrZOX1 directly interact with each other, and whether petanin serves as a substrate for LrHCT—still lack structural evidence.
Therefore, we used molecular docking to investigate the interactions between key proteins and ligands (zeatin-type small molecules, petanin) and potential protein–protein interactions. The three-dimensional structures of LrHCT and LrZOX1 were obtained from the UniProt and PubChem databases, along with the structures of DHZ, tZ, tZR, cZ, cZR. Blind docking was performed using the CB-DOCK2 online platform. The results showed that all five zeatin molecules formed stable complexes with both LrHCT and LrZOX1, with binding energies below −5.0 kcal·mol−1 (details in Section 2.4). Among them, cZR exhibited the strongest binding affinity to both proteins (LrHCT: −8.5 kcal·mol−1; LrZOX1: −8.7 kcal·mol−1) (Fig. 5A). This outcome verifies the substrate specificity of zeatin for LrZOX1, with cZR as the preferred substrate, and suggests that cZ may bind to LrHCT as a signaling molecule, potentially promoting anthocyanin synthesis by activating LrHCT enzyme activity.
To investigate whether LrHCT and LrZOX1 cooperate in metabolic regulation through direct interaction, we predicted their complex structure using AlphaFold3 and visualized it using PyMOL. The results indicate that LrHCT and LrZOX1 can form a stable heterodimer. Their interaction is mediated by two key hydrogen bonds: the lysine 201 (LYS201) of LrZOX1 forms hydrogen bonds with the arginine 99 (ARG99) and glutamate 101 (GLU101) of LrHCT, with bond lengths of 3.1 Å and 3.5 Å, respectively (Fig. 5B). The prediction confidence parameters (plDDT > 70, pTM + ipTM = 0.86) suggest a reliable structural basis for direct protein–protein interaction between LrHCT and LrZOX1. Considering their negatively correlated expression patterns, we hypothesize that LrHCT and LrZOX1 may cooperatively regulate the balance of anthocyanin and cZ metabolism through an interaction-coupled antagonistic mode.
Furthermore, given that LrHCT is a key acyltransferase in anthocyanin synthesis and petanin is an acylated anthocyanin, we verified whether petanin serves as a catalytic product or a regulatory factor of LrHCT. The three-dimensional structure of petanin was retrieved from the PubChem database and docked with LrHCT. The results showed a binding energy of −8.9 kcal·mol−1 between them, along with the formation of four key hydrogen bonds. Specifically, the methionine 1 (MET1), glutamate 8 (GLU8), glutamine 10 (GLN10), and glycine 13 (GLY13) of LrHCT formed hydrogen bonds with the glycosyl or acyl groups of petanin (bond lengths > 2.5 Å, Fig. 5C). Considering the acyltransferase function of LrHCT, these results suggest that petanin may be the end product catalyzed by LrHCT, or it may enhance LrHCT enzymatic activity through binding. This result provides a clear theoretical basis for subsequent in vitro enzyme activity assays.
These findings provide structural evidence for the binding potential between LrHCT/LrZOX1 and zeatin-type molecules as well as petanin. Furthermore, they offer theoretical support for the metabolic balance mediated by the lncRNA regulatory module and suggest that its catalytic end product, petanin, may possess broader biological activities. Thus, to further investigate the potential application of petanin, we employed a network pharmacology approach to systematically predict its potential therapeutic targets and mechanisms of action in AD and IBD.
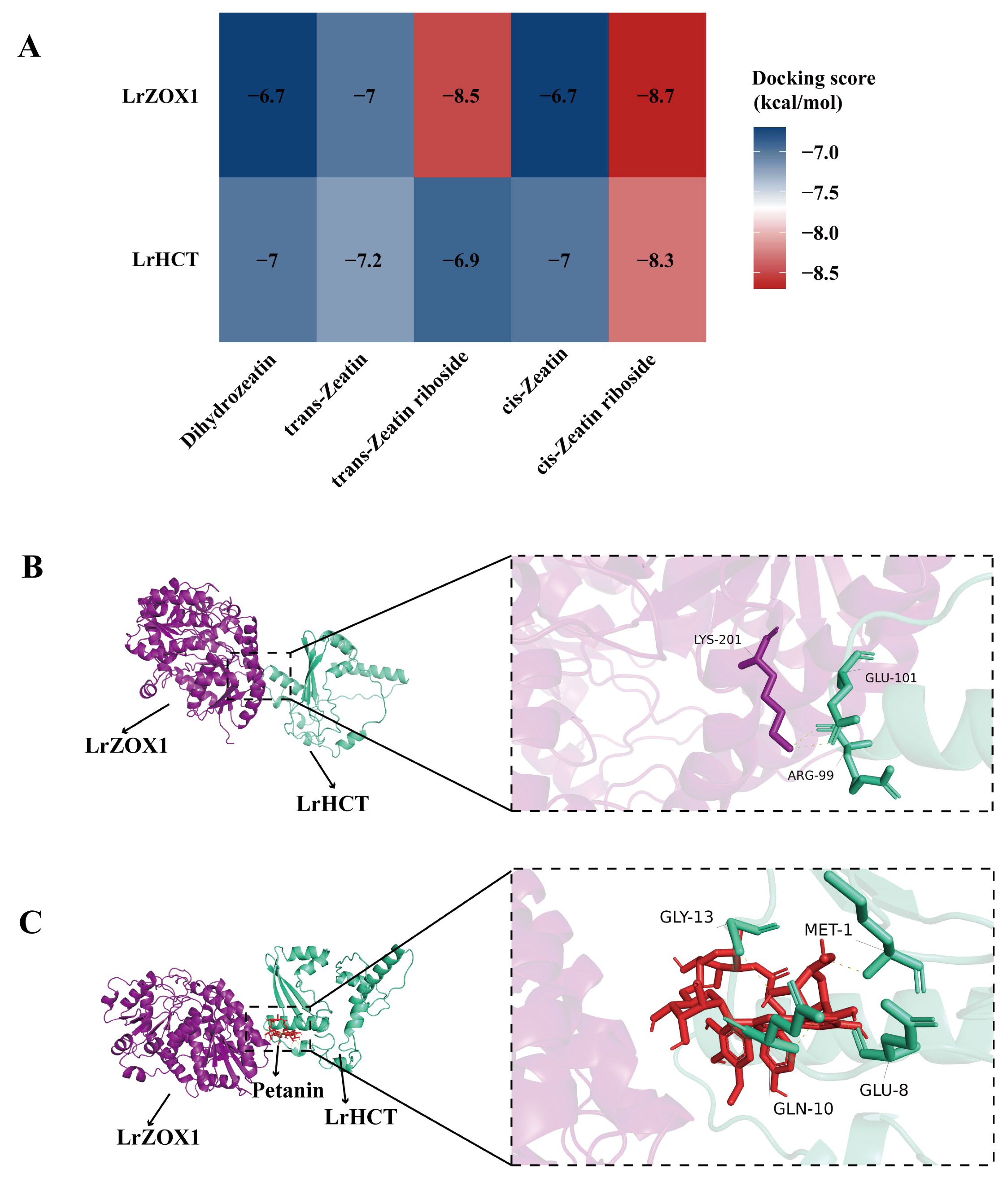
Figure 5: Molecular docking analysis of key interactions. (A) Heatmap of binding affinities from molecular docking between LrHCT/LrZOX1 and zeatin-type small molecules. (B) Schematic representation of the docking between LrHCT and LrZOX1. (C) Schematic representation of the docking between petanin and LrHCT.
3.7 Network Pharmacology Analysis of Petanin Intervention in Alzheimer’s Disease and Inflammatory Bowel Disease
To assess the potential bioactivity of petanin, we first predicted a total of 75 petanin-related targets. Subsequently, we screened 10,321 AD-related target genes and 9530 IBD-related target genes. The intersection between petanin targets and disease targets yielded 52 overlapping genes for AD (Fig. 6A) and 50 for IBD (Fig. 6B). These overlapping target genes were then uploaded to the STRING database to construct protein–protein interaction (PPI) networks, which were visualized using Cytoscape (Fig. 6C and Fig. S3A). Notably, the top five key target genes ranked by interaction degree were identical between the two diseases, namely tumor necrosis factor (TNF), interleukin-6 (IL6), epidermal growth factor receptor (EGFR), hypoxia-inducible factor 1α (HIF1A), and prostaglandin-endoperoxide synthase 2 (PTGS2). These shared targets are closely associated with inflammatory response and oxidative stress, suggesting that modulation of this core network may represent a common molecular basis for petanin intervention in these disorders.
We performed GO and KEGG enrichment analyses to systematically decipher the biological functions of these shared targets. The GO analysis revealed a high degree of overlap in the biological processes associated with the potential effect of petanin on AD and IBD. Enriched terms common to both included “response to oxidative stress” and “regulation of inflammatory response.” These commonly enriched terms align closely with the antioxidant activity of petanin. Thus, we speculate that petanin may mitigate oxidative stress and inflammatory damage by modulating these processes to scavenge excess reactive oxygen species (ROS) in the brain (AD) and intestinal tract (IBD). This could represent one of the core mechanisms underlying its potential intervention in both diseases. Despite the substantial number of shared terms, petanin also exhibited disease-specific regulatory potential. GO terms unique to AD were enriched in processes such as “neuron projection terminus” (Fig. 6D). By contrast, terms unique to IBD were concentrated in categories including “regulation of peptidyl-tyrosine phosphorylation” (Fig. S3B). KEGG enrichment analysis further corroborated the high consistency in the mechanistic pathways of these target genes. The 10 most-enriched pathways were identical between the two disease analyses. These included the “AGE-RAGE signaling pathway in diabetic complications” and “HIF-1 signaling pathway” (p < 0.05) (Fig. S4A,B). These results suggest that petanin may exert its potential therapeutic effects on AD and IBD through a multitarget, multipathway approach that coordinately modulates the inflammation–oxidative stress axis.
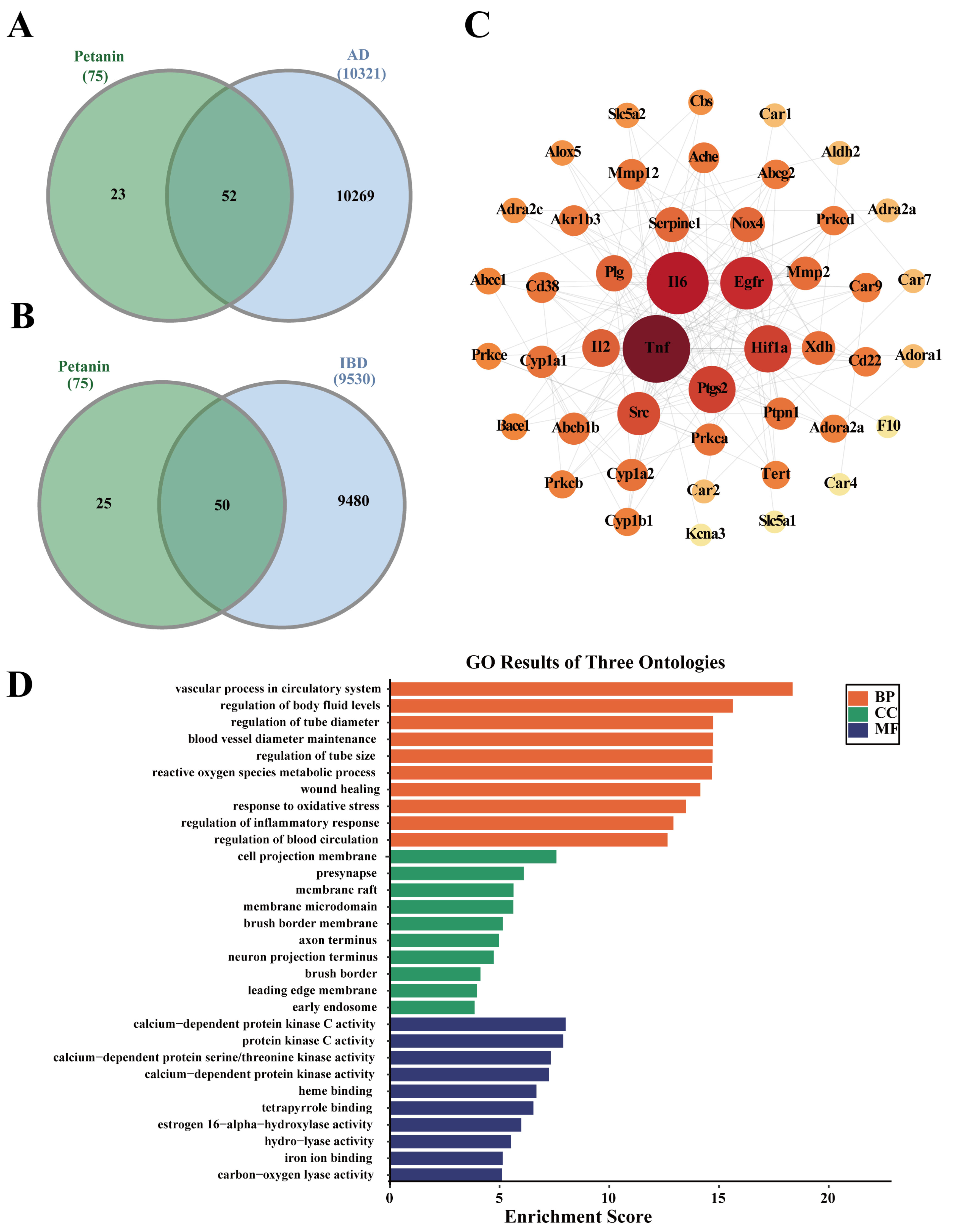
Figure 6: Network pharmacology analysis of petanin for Alzheimer’s Disease and Inflammatory Bowel Disease. (A) Venn diagram showing the intersection of predicted petanin targets with Alzheimer’s Disease (AD) targets. (B) Venn diagram showing the intersection of predicted petanin targets with Inflammatory Bowel Disease (IBD) targets. (C) Protein-protein interaction (PPI) network of the overlapping targets between petanin and AD, highlighting the key hub genes. (D) Gene ontology (GO) enrichment analysis of the petanin-AD intersection targets.
4.1 Coordinated Targeting of LrHCT by circRNAs and lncRNAs in Black Goji
Many previous studies have established that circRNAs and lncRNAs regulate gene expression indirectly by constructing competing endogenous RNA (ceRNA) networks [42,43,44]. By contrast, in this study, the circRNA plantcirc_001165, the lncRNA MSTRG.28703, and their target gene LrHCT (Lr11g005079) exhibited a strong co-expression pattern in BS4. All three were highly expressed only in the anthocyanin-rich BS4 and were nearly undetectable in WS4. Hence, we propose the existence of a coordinated circRNA/lncRNA/target gene regulatory mode in black goji. Distinct from the indirect mechanism of ceRNA networks, this mode may involve the simultaneous activation of the core enzyme gene by both classes of ncRNAs, potentially enabling the rapid and efficient initiation of anthocyanin synthesis.
Zong et al. [11] demonstrated that LrAN2 is a key transcriptional regulator for anthocyanin accumulation in black goji. The LrHCT identified in this study is a core enzymatic gene in the flavonoid synthesis pathway [45,46,47]. Notably, HCT can catalyze a reverse reaction [48], enabling the plant to convert chlorogenic acid to caffeoyl-CoA. Hence, we propose that, LrAN2 likely activates LrHCT transcription in black goji by binding to its promoter region, while plantcirc_001165 and MSTRG.28703 facilitate efficient anthocyanin accumulation by stabilizing LrHCT mRNA or enhancing its translational efficiency at the post-transcriptional level. The proposed regulatory network integrates knowledge on anthocyanin regulation in black goji, offering a more comprehensive perspective for understanding its high-anthocyanin trait.
4.2 The Regulatory Network Constructed by lncRNAs Mediates Cross-Talk between Anthocyanin and Zeatin Metabolic Pathways
We posit that MSTRG.28703, which is highly expressed in black goji, regulates metabolic balance through a dual mechanism. On one hand, it cis-activates LrHCT to promote anthocyanin synthesis. On the other hand, it trans-represses the expression of MSTRG.66 and MSTRG.64. We hypothesize that this repression may occur through competitive binding to the promoter region of LrZOX1 or by sequestering miRNAs targeting MSTRG.66/MSTRG.64, indirectly suppressing LrZOX1 transcription. This mechanism would reduce the inactivation of active zeatin, leading to the accumulation of free-form zeatins, such as cZ.
CZ can specifically accumulate under drought stress, contributing to the maintenance of cellular physiological activity and regulating the balance between plant development and stress responses [41]. Meanwhile, anthocyanins can scavenge the excess ROS generated by drought stress [27]. This aligns closely with the evolutionary adaptation of black goji to arid environments. Notably, although cZ acts as a signaling molecule to effectively activate cytokinin-induced genes [49], the regulation of anthocyanin biosynthesis by cytokinins exhibits significant functional duality across different species. In banana [50], cytokinins primarily exert an inhibitory effect, hindering anthocyanin accumulation by upregulating B-type response regulators that repress the expression of genes such as dihydroflavonol 4-reductase (MaDFR). However, our results indicate that black goji fits a positive synergistic regulation model. Consistent with the mechanism elucidated in Arabidopsis thaliana [51], cytokinins act as essential enhancers of blue light signaling, significantly amplifying anthocyanin accumulation via the cryptochrome 1 (CRY1)-dependent pathway. Given the long-term exposure of black goji to the high-radiation environment of the Qinghai-Xizang Plateau, we speculate that the accumulated cZ functions not as an inhibitor, but as a key stress response signaling molecule, producing a synergistic effect with high-intensity environmental light signals to maximize photoprotection.
Cytokinin signaling modulates anthocyanin biosynthesis by directly upregulating specific structural genes in some plants, such as CHS and DFR in Arabidopsis [52] and PAL in Eucalyptus [53]. In contrast, cZ in black goji may tend to enhance the acylation process mediated by LrHCT, thereby improving the stability and bioactivity of anthocyanins. This difference likely stems from variations in the steps of the anthocyanin biosynthetic pathway across species. This study links ncRNA regulation, hormone metabolism, and stress adaptation for the first time, providing a novel case for understanding the ecological and evolutionary significance of plant secondary metabolism.
4.3 Molecular Docking Reveals the High-Affinity Binding of LrHCT and LrZOX1 to Ligands and Their Potential Cooperative Interaction Mechanism
Protein-protein docking analysis revealed that the assembly of a stable LrHCT-LrZOX1 heterodimer core is dependent on three critical residues: LYS201, ARG99, and GLU101. Notably, LYS201 within LrZOX1 establishes a hydrogen-bonding network with ARG99 and GLU101 in LrHCT. The electrostatic interaction between the positively charged LYS201 and the negatively charged GLU101 is key to maintaining dimer stability, while ARG99 further reinforces the complex structure through hydrogen bonding. Notably, GLU101 is located near the predicted acyltransferase active site of LrHCT (based on homology modeling). We speculate that dimer formation may regulate the binding efficiency of LrHCT for p-coumaroyl-CoA by altering the spatial conformation of its active site, achieving dynamic coupling between anthocyanin synthesis and cZ metabolism.
In the ligand binding analysis, cZR exhibited the lowest binding energy with both LrHCT and LrZOX1 (from −8.3 to −8.7 kcal·mol−1). The additional ribose group in its structure can form an extra hydrogen bond with the SER35 of LrHCT, representing the key structural basis for its higher binding affinity than those of other zeatin derivatives. This also clarifies the preferred substrate type for LrZOX1.
The glycosyl and acyl groups of petanin form four hydrogen bonds with the MET1, GLU8, GLN10, and GLY13 of LrHCT. The binding site precisely overlays the predicted acyl-donor binding pocket of LrHCT, based on the structural conservation of the BAHD acyltransferase family [54]. This indicates that petanin is more likely the catalytic end product of LrHCT. The enzyme likely synthesizes petanin by transferring a p-coumaroyl group onto the 5-O-glucoside of an anthocyanin precursor. This inference aligns with experimental results identifying petanin as the major anthocyanin component in black goji [2]. Furthermore, the strong binding between petanin and LrHCT (binding energy −8.9 kcal·mol−1) may also regulate the enzymatic activity of LrHCT through a feedback inhibition mechanism, preventing the metabolic burden caused by excessive anthocyanin accumulation.
4.4 Network Pharmacology Predicts the Multitarget Intervention Potential of Petanin in Alzheimer’s Disease and Inflammatory Bowel Disease
GO analysis revealed that the common targets of petanin for AD and IBD intervention were enriched in biological processes such as ROS metabolism, response to oxidative stress, and regulation of inflammatory response. These findings are consistent with previous experimental evidence [5,55]. In vitro cellular experiments have confirmed that petanin protects PC12 cells from oxidative stress by mitigating lipid peroxidation and modulating endogenous antioxidant enzymes [56]. In vivo studies have demonstrated the ability of petanin to alleviate intestinal inflammation in a DSS-induced mouse colitis model by reducing the expression of IL-6, TNF-α, and other markers [57]. The network pharmacology predictions in the present study further clarify the core targets and pathways underlying these phenotypic effects, thus contributing to a more complete chain of evidence.
Regarding disease-specific mechanisms, petanin targets for AD were enriched in the biological process “neuron projection terminus”. This process is directly associated with synaptic dysfunction in AD pathology [58]. The activation of the core target EGFR can promote neurite outgrowth and synaptogenesis [59]. Thus, we speculate that petanin may alleviate Aβ deposition-induced synaptic damage by inhibiting the overactivation of EGFR [60]. For IBD, the enriched regulatory term “peptidyl–tyrosine phosphorylation” is related to intestinal epithelial barrier repair. The core target HIF-1α can promote intestinal epithelial cell proliferation and the expression of tight junction proteins (e.g., ZO-1) by regulating tyrosine kinase-associated signaling pathways [61,62]. This observation aligns with the potential role of petanin in mitigating intestinal mucosal damage.
Hierarchical analysis of the core pathways indicates that the AGE–RAGE signaling pathway serves as the central upstream node for petanin’s intervention in both diseases. The overactivation of this pathway upregulates downstream targets such as IL6, TNF, and PTGS2 via NF-κB/MAPK signaling [63]. Petanin may directly bind to key receptors within the AGE-RAGE pathway (e.g., RAGE), blocking ligand binding and inhibiting downstream inflammation. The HIF-1 signaling pathway functions as a secondary node, regulating neuronal hypoxia tolerance in AD and intestinal barrier repair in IBD [64,65]. Together, these pathways form a coordinated multi-pathway intervention network for petanin.
By integrating multi-omics analysis and computational biology, this study proposes a working model in which circRNAs and lncRNAs coordinately regulate LrHCT and LrZOX1 to mediate the metabolic balance between anthocyanins and cZ. This study provides a new transcriptional and post-transcriptional regulatory perspective for understanding fruit coloration in black goji. Furthermore, the network pharmacology analysis of petanin offers a theoretical prediction for its potential therapeutic efficacy in neurodegenerative diseases and intestinal inflammation. The key findings include: (1) The circRNA plantcirc_001165 and the lncRNA MSTRG.28703 co-target and activate LrHCT, a key gene encoding an enzyme for anthocyanin synthesis, thereby promoting anthocyanin accumulation; (2) MSTRG.28703, in coordination with MSTRG.66/MSTRG.64, regulates the expression of LrZOX1, leading to the accumulation of cZ; (3) Molecular docking confirms the high-affinity binding of LrHCT and LrZOX1 with zeatin-type molecules and petanin; (4) Network pharmacology predicts that petanin may exert potential therapeutic effects in AD and IBD by modulating oxidative stress and inflammation-related pathways, such as the AGE-RAGE and HIF-1 signaling pathways. Future studies will prioritize functional validation and ncRNA perturbation experiments. Furthermore, we aim to explore the potential of utilizing genome editing or ncRNA-based regulatory strategies to enhance the fruit quality of black goji.
Acknowledgement:
Funding Statement: This research was funded by the High-level Talent Cultivation and Introduction Project of Qinghai Normal University (No. 268010160).
Author Contributions: Conceptualization, writing—review and editing, visualization, project administration, funding acquisition, Qiong Luo; methodology, software, validation, formal analysis, data curation, Yiming Chen; writing—original draft preparation, Qiong Luo and Yiming Chen; All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: The data that support the findings of this study are openly available in the NCBI BioProject repository, PRJNA1376961 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1376961), and the NCBI Genome database, GCA_041430385.1 (https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_041430385.1/).
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest.
Supplementary Materials: The supplementary material is available online at https://www.techscience.com/doi/10.32604/phyton.2026.077896/s1. Figure S1: Functional annotation and pathway enrichment analysis of circRNA source genes in black goji; Figure S2: Comprehensive profiling of differentially expressed lncRNAs and their target gene functions in black goji; Figure S3: Functional annotation and interaction network analysis of IBD; Figure S4: Pathway enrichment analysis of differentially expressed targets for AD and IBD.
References
1. Yao R , Heinrich M , Weckerle CS . The genus Lycium as food and medicine: A botanical, ethnobotanical and historical review. J Ethnopharmacol. 2018; 212: 50– 66. doi:10.1016/j.jep.2017.10.010. [Google Scholar] [CrossRef]
2. Liu Z , Tang X , Liu C , Dong B , Shao Y , Liu B , et al. Ultrasonic extraction of anthocyanins from Lycium ruthenicum Murr. and its antioxidant activity. Food Sci Nutr. 2020; 8( 6): 2642– 51. doi:10.1002/fsn3.1542. [Google Scholar] [CrossRef]
3. Chen S , Zhou H , Zhang G , Meng J , Deng K , Zhou W , et al. Anthocyanins from Lycium ruthenicum Murr. ameliorated D-galactose-induced memory impairment, oxidative stress, and neuroinflammation in adult rats. J Agric Food Chem. 2019; 67( 11): 3140– 9. doi:10.1021/acs.jafc.8b06402. [Google Scholar] [CrossRef]
4. Suresh S , Begum RF , Singh SA , Chitra V . Anthocyanin as a therapeutic in Alzheimer’s disease: A systematic review of preclinical evidences. Ageing Res Rev. 2022; 76: 101595. doi:10.1016/j.arr.2022.101595. [Google Scholar] [CrossRef]
5. Sahoo DK , Heilmann RM , Paital B , Patel A , Yadav VK , Wong D , et al. Oxidative stress, hormones, and effects of natural antioxidants on intestinal inflammation in inflammatory bowel disease. Front Endocrinol. 2023; 14: 1217165. doi:10.3389/fendo.2023.1217165. [Google Scholar] [CrossRef]
6. Mattioli R , Francioso A , Mosca L , Silva P . Anthocyanins: A comprehensive review of their chemical properties and health effects on cardiovascular and neurodegenerative diseases. Molecules. 2020; 25( 17): 3809. doi:10.3390/molecules25173809. [Google Scholar] [CrossRef]
7. Heinzmann U , Seitz U . Relationship between anthocyanin synthesis and activity of phenylalanine ammonia-lyase (PAL) in callus cells of Daucus carota. Planta. 1974; 117( 1): 75– 81. doi:10.1007/BF00388680. [Google Scholar] [CrossRef]
8. Deng X , Bashandy H , Ainasoja M , Kontturi J , Pietiäinen M , Laitinen RAE , et al. Functional diversification of duplicated chalcone synthase genes in anthocyanin biosynthesis of Gerbera hybrida. New Phytol. 2014; 201( 4): 1469– 83. doi:10.1111/nph.12610. [Google Scholar] [CrossRef]
9. Forkmann G , Kuhn B . Genetic control of chalcone isomerase activity in anthers of Petunia hybrida. Planta. 1979; 144( 2): 189– 92. doi:10.1007/BF00387269. [Google Scholar] [CrossRef]
10. Koes R , Verweij W , Quattrocchio F . Flavonoids: A colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005; 10( 5): 236– 42. doi:10.1016/j.tplants.2005.03.002. [Google Scholar] [CrossRef]
11. Zong Y , Zhu X , Liu Z , Xi X , Li G , Cao D , et al. Functional MYB transcription factor encoding gene AN2 is associated with anthocyanin biosynthesis in Lycium ruthenicum Murray. BMC Plant Biol. 2019; 19( 1): 169. doi:10.1186/s12870-019-1752-8. [Google Scholar] [CrossRef]
12. Sun C , Deng L , Du M , Zhao J , Chen Q , Huang T , et al. A transcriptional network promotes anthocyanin biosynthesis in tomato flesh. Mol Plant. 2020; 13( 1): 42– 58. doi:10.1016/j.molp.2019.10.010. [Google Scholar] [CrossRef]
13. Zhou LL , Zeng HN , Shi MZ , Xie DY . Development of tobacco callus cultures over expressing Arabidopsis PAP1/MYB75 transcription factor and characterization of anthocyanin biosynthesis. Planta. 2008; 229( 1): 37– 51. doi:10.1007/s00425-008-0809-y. [Google Scholar] [CrossRef]
14. Lai B , Li XJ , Hu B , Qin YH , Huang XM , Wang HC , et al. LcMYB1 is a key determinant of differential anthocyanin accumulation among genotypes, tissues, developmental phases and ABA and light stimuli in Litchi chinensis. PLoS One. 2014; 9( 1): e86293. doi:10.1371/journal.pone.0086293. [Google Scholar] [CrossRef]
15. Li H , Chen N , Zhang H , Xu D . Multidimensional regulation of transcription factors: Decoding the comprehensive signals of plant secondary metabolism. Front Plant Sci. 2025; 16: 1522278. doi:10.3389/fpls.2025.1522278. [Google Scholar] [CrossRef]
16. Chialva C , Blein T , Crespi M , Lijavetzky D . Insights into long non-coding RNA regulation of anthocyanin carrot root pigmentation. Sci Rep. 2021; 11( 1): 4093. doi:10.1038/s41598-021-83514-4. [Google Scholar] [CrossRef]
17. Zhou Y , Ali Mumtaz M , Zhang Y , Yang Z , Hao Y , Shu H , et al. Response of anthocyanin biosynthesis to light by strand-specific transcriptome and miRNA analysis in Capsicum annuum. BMC Plant Biol. 2022; 22( 1): 79. doi:10.1186/s12870-021-03423-6. [Google Scholar] [CrossRef]
18. Li H , Wang W , Liu R , Tong B , Dai X , Lu Y , et al. Long non-coding RNA-mediated competing endogenous RNA regulatory network during flower development and color formation in Melastoma candidum. Front Plant Sci. 2023; 14: 1215044. doi:10.3389/fpls.2023.1215044. [Google Scholar] [CrossRef]
19. Zhang P , Dai M . CircRNA: A rising star in plant biology. J Genet Genomics. 2022; 49( 12): 1081– 92. doi:10.1016/j.jgg.2022.05.004. [Google Scholar] [CrossRef]
20. Zhang D , Ma Y , Naz M , Ahmed N , Zhang L , Zhou JJ , et al. Advances in CircRNAs in the past decade: Review of CircRNAs biogenesis, regulatory mechanisms, and functions in plants. Genes. 2024; 15( 7): 958. doi:10.3390/genes15070958. [Google Scholar] [CrossRef]
21. Wu X , Shi T , Iqbal S , Zhang Y , Liu L , Gao Z . Genome-wide discovery and characterization of flower development related long non-coding RNAs in Prunus mume. BMC Plant Biol. 2019; 19( 1): 64. doi:10.1186/s12870-019-1672-7. [Google Scholar] [CrossRef]
22. Liu N , Xu Y , Li Q , Cao Y , Yang D , Liu S , et al. A lncRNA fine-tunes salicylic acid biosynthesis to balance plant immunity and growth. Cell Host Microbe. 2022; 30( 8): 1124– 38.e8. doi:10.1016/j.chom.2022.07.001. [Google Scholar] [CrossRef]
23. Zuo J , Wang Y , Zhu B , Luo Y , Wang Q , Gao L . Network analysis of noncoding RNAs in pepper provides insights into fruit ripening control. Sci Rep. 2019; 9( 1): 8734. doi:10.1038/s41598-019-45427-1. [Google Scholar] [CrossRef]
24. Jiang L , Yue M , Liu Y , Zhang N , Lin Y , Zhang Y , et al. A novel R2R3-MYB transcription factor FaMYB5 positively regulates anthocyanin and proanthocyanidin biosynthesis in cultivated strawberries (Fragaria × ananassa). Plant Biotechnol J. 2023; 21( 6): 1140– 58. doi:10.1111/pbi.14024. [Google Scholar] [CrossRef]
25. Yuan Y , Pang X , Pang J , Wang Q , Zhou M , Lu Y , et al. Identification and characterisation of the CircRNAs involved in the regulation of leaf colour in Quercus mongolica. Biology. 2024; 13( 3): 183. doi:10.3390/biology13030183. [Google Scholar] [CrossRef]
26. Yang Y , Jiang W , Wei R , Lu L , Zhang L , Wang X , et al. Integrative metabolomic and transcriptomic analysis provides insight into the mechanism of pigmentation in potato tuber eyes. BMC Plant Biol. 2025; 25( 1): 1164. doi:10.1186/s12870-025-07206-1. [Google Scholar] [CrossRef]
27. Zeng S , Lin S , Wang Z , Zong Y , Wang Y . The health-promoting anthocyanin petanin in Lycium ruthenicum fruit: A promising natural colorant. Crit Rev Food Sci Nutr. 2024; 64( 28): 10484– 97. doi:10.1080/10408398.2023.2225192. [Google Scholar] [CrossRef]
28. Parkhomchuk D , Borodina T , Amstislavskiy V , Banaru M , Hallen L , Krobitsch S , et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009; 37( 18): e123. doi:10.1093/nar/gkp596. [Google Scholar] [CrossRef]
29. Kim D , Langmead B , Salzberg SL . HISAT: A fast spliced aligner with low memory requirements. Nat Methods. 2015; 12( 4): 357– 60. doi:10.1038/nmeth.3317. [Google Scholar] [CrossRef]
30. Memczak S , Jens M , Elefsinioti A , Torti F , Krueger J , Rybak A , et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013; 495( 7441): 333– 8. doi:10.1038/nature11928. [Google Scholar] [CrossRef]
31. Pertea M , Kim D , Pertea GM , Leek JT , Salzberg SL . Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016; 11( 9): 1650– 67. doi:10.1038/nprot.2016.095. [Google Scholar] [CrossRef]
32. Love MI , Huber W , Anders S . Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15( 12): 550. doi:10.1186/s13059-014-0550-8. [Google Scholar] [CrossRef]
33. Ashburner M , Ball CA , Blake JA , Botstein D , Butler H , Cherry JM , et al. Gene ontology: Tool for the unification of biology. Nat Genet. 2000; 25: 25– 9. doi:10.1038/75556. [Google Scholar] [CrossRef]
34. Kanehisa M . The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004; 32( 90001): D277– 80. doi:10.1093/nar/gkh063. [Google Scholar] [CrossRef]
35. Tamura K , Dudley J , Nei M , Kumar S . MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007; 24( 8): 1596– 9. doi:10.1093/molbev/msm092. [Google Scholar] [CrossRef]
36. Liu Y , Yang X , Gan J , Chen S , Xiao ZX , Cao Y . CB-Dock2: Improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022; 50( W1): W159– 64. doi:10.1093/nar/gkac394. [Google Scholar] [CrossRef]
37. Heberle H , Meirelles GV , da Silva FR , Telles GP , Minghim R . InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics. 2015; 16( 1): 169. doi:10.1186/s12859-015-0611-3. [Google Scholar] [CrossRef]
38. Martin RC , Mok MC , Mok DW . A gene encoding the cytokinin enzyme Zeatin O-xylosyltransferase of Phaseolus vulgaris. Plant Physiol. 1999; 120( 2): 553– 8. doi:10.1104/pp.120.2.553. [Google Scholar] [CrossRef]
39. Martin RC , Mok MC , Mok DW . Cytolocalization of Zeatin O-xylosyltransferase in Phaseolus. Proc Natl Acad Sci U S A. 1993; 90( 3): 953– 7. doi:10.1073/pnas.90.3.953. [Google Scholar] [CrossRef]
40. Auer CA . Discoveries and dilemmas concerning cytokinin metabolism. J Plant Growth Regul. 2002; 21( 1): 24– 31. doi:10.1007/s003440010044. [Google Scholar] [CrossRef]
41. Schäfer M , Brütting C , Meza-Canales ID , Großkinsky DK , Vankova R , Baldwin IT , et al. The role of cis-Zeatin-type cytokinins in plant growth regulation and mediating responses to environmental interactions. J Exp Bot. 2015; 66( 16): 4873– 84. doi:10.1093/jxb/erv214. [Google Scholar] [CrossRef]
42. Li B , Feng C , Zhang W , Sun S , Yue D , Zhang X , et al. Comprehensive non-coding RNA analysis reveals specific lncRNA/circRNA-miRNA-mRNA regulatory networks in the cotton response to drought stress. Int J Biol Macromol. 2023; 253( Pt 1): 126558. doi:10.1016/j.ijbiomac.2023.126558. [Google Scholar] [CrossRef]
43. Liu H , Yuan K , Hu Y , Wang S , He Q , Feng C , et al. Construction and analysis of the tapping panel dryness-related lncRNA/circRNA-miRNA-mRNA ceRNA network in latex of Hevea brasiliensis. Plant Physiol Biochem. 2023; 205: 108156. doi:10.1016/j.plaphy.2023.108156. [Google Scholar] [CrossRef]
44. Wang M , Wang L , Wang S , Zhang J , Fu Z , Wu P , et al. Identification and analysis of lncRNA and circRNA related to wheat grain development. Int J Mol Sci. 2024; 25( 10): 5484. doi:10.3390/ijms25105484. [Google Scholar] [CrossRef]
45. Liu D , Yuan M , Wang Y , Zhang L , Yao W , Feng M . Integrated metabolome and transcriptome analysis of differences in quality of ripe Lycium barbarum L. fruits harvested at different periods. BMC Plant Biol. 2024; 24( 1): 82. doi:10.1186/s12870-024-04751-z. [Google Scholar] [CrossRef]
46. Li Z , Xu S , Wu H , Wan X , Lei H , Yu J , et al. Integrated analyses of metabolome and RNA-seq data revealing flower color variation in ornamental Rhododendron simsii Planchon. Genes. 2024; 15( 8): 1041. doi:10.3390/genes15081041. [Google Scholar] [CrossRef]
47. Yin C , Tang D , Liu X , Li Z , Xiang Y , Gao K , et al. Transcriptome analysis reveals important regulatory genes and pathways for tuber color variation in Pinellia ternata (Thunb.) Breit. Protoplasma. 2023; 260( 5): 1313– 25. doi:10.1007/s00709-023-01845-7. [Google Scholar] [CrossRef]
48. Hoffmann L , Maury S , Martz F , Geoffroy P , Legrand M . Purification, cloning, and properties of an acyltransferase controlling shikimate and quinate ester intermediates in phenylpropanoid metabolism. J Biol Chem. 2003; 278( 1): 95– 103. doi:10.1074/jbc.M209362200. [Google Scholar] [CrossRef]
49. Kudo T , Makita N , Kojima M , Tokunaga H , Sakakibara H . Cytokinin activity of cis-Zeatin and phenotypic alterations induced by overexpression of putative cis-Zeatin-O-glucosyltransferase in rice. Plant Physiol. 2012; 160( 1): 319– 31. doi:10.1104/pp.112.196733. [Google Scholar] [CrossRef]
50. Rajput R , Tyagi S , Anchal K , Singh S , Laxmi A , Misra P , et al. Cytokinin-mediated repression of anthocyanin biosynthesis in banana fruits. Plant J. 2025; 122( 6): e70267. doi:10.1111/tpj.70267. [Google Scholar] [CrossRef]
51. Chen DQ , Li ZY , Pan RC , Wang XJ . Anthocyanin accumulation mediated by blue light and cytokinin in Arabidopsis seedlings. J Integr Plant Biol. 2006; 48( 4): 420– 5. doi:10.1111/j.1744-7909.2006.00234.x. [Google Scholar] [CrossRef]
52. Deikman J , Hammer PE . Induction of anthocyanin accumulation by cytokinins in Arabidopsis thaliana. Plant Physiol. 1995; 108( 1): 47– 57. doi:10.1104/pp.108.1.47. [Google Scholar] [CrossRef]
53. Zhu L , Liao Y , Lin K , Wu W , Duan L , Wang P , et al. Cytokinin promotes anthocyanin biosynthesis via regulating sugar accumulation and MYB113 expression in Eucalyptus. Tree Physiol. 2024; 44( 1): tpad154. doi:10.1093/treephys/tpad154. [Google Scholar] [CrossRef]
54. Petersen M . Hydroxycinnamoyltransferases in plant metabolism. Phytochem Rev. 2016; 15( 5): 699– 727. doi:10.1007/s11101-015-9417-1. [Google Scholar] [CrossRef]
55. Li P , Feng D , Yang D , Li X , Sun J , Wang G , et al. Protective effects of anthocyanins on neurodegenerative diseases. Trends Food Sci Technol. 2021; 117: 205– 17. doi:10.1016/j.tifs.2021.05.005. [Google Scholar] [CrossRef]
56. Tang J , Yan Y , Ran L , Mi J , Sun Y , Lu L , et al. Isolation, antioxidant property and protective effect on PC12 cell of the main anthocyanin in fruit of Lycium ruthenicum Murray. J Funct Foods. 2017; 30: 97– 107. doi:10.1016/j.jff.2017.01.015. [Google Scholar] [CrossRef]
57. Peng Y , Yan Y , Wan P , Chen D , Ding Y , Ran L , et al. Gut microbiota modulation and anti-inflammatory properties of anthocyanins from the fruits of Lycium ruthenicum Murray in dextran sodium sulfate-induced colitis in mice. Free Radic Biol Med. 2019; 136: 96– 108. doi:10.1016/j.freeradbiomed.2019.04.005. [Google Scholar] [CrossRef]
58. Lane CA , Hardy J , Schott JM . Alzheimer’s disease. Eur J Neurol. 2018; 25( 1): 59– 70. doi:10.1111/ene.13439. [Google Scholar] [CrossRef]
59. Da Rocha JF , Bastos L , Domingues SC , Bento AR , Konietzko U , Da Cruz E Silva OAB , et al. APP binds to the EGFR ligands HB-EGF and EGF, acting synergistically with EGF to promote ERK signaling and neuritogenesis. Mol Neurobiol. 2021; 58( 2): 668– 88. doi:10.1007/s12035-020-02139-2. [Google Scholar] [CrossRef]
60. Choi HJ , Jeong YJ , Kim J , Hoe HS . EGFR is a potential dual molecular target for cancer and Alzheimer’s disease. Front Pharmacol. 2023; 14: 1238639. doi:10.3389/fphar.2023.1238639. [Google Scholar] [CrossRef]
61. Lu H , Lin J , Xu C , Sun M , Zuo K , Zhang X , et al. Cyclosporine modulates neutrophil functions via the SIRT6-HIF-1α-glycolysis axis to alleviate severe ulcerative colitis. Clin Transl Med. 2021; 11( 2): e334. doi:10.1002/ctm2.334. [Google Scholar] [CrossRef]
62. Yin J , Ren Y , Yang K , Wang W , Wang T , Xiao W , et al. The role of hypoxia-inducible factor 1-alpha in inflammatory bowel disease. Cell Biol Int. 2022; 46( 1): 46– 51. doi:10.1002/cbin.11712. [Google Scholar] [CrossRef]
63. Bhattacharya R , Alam MR , Kamal MA , Seo KJ , Singh LR . AGE-RAGE axis culminates into multiple pathogenic processes: A central road to neurodegeneration. Front Mol Neurosci. 2023; 16: 1155175. doi:10.3389/fnmol.2023.1155175. [Google Scholar] [CrossRef]
64. March-Diaz R , Lara-Ureña N , Romero-Molina C , Heras-Garvin A , Ortega-de San Luis C , Alvarez-Vergara MI , et al. Hypoxia compromises the mitochondrial metabolism of Alzheimer’s disease microglia via HIF1. Nat Aging. 2021; 1( 4): 385– 99. doi:10.1038/s43587-021-00054-2. [Google Scholar] [CrossRef]
65. Hirota SA , Beck PL , MacDonald JA . Targeting hypoxia-inducible factor-1 (HIF-1) signaling in therapeutics: Implications for the treatment of inflammatory bowel disease. Recent Pat Inflamm Allergy Drug Discov. 2009; 3( 1): 1– 16. doi:10.2174/187221309787158434. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools