
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Regulatory role of NFAT1 signaling in articular chondrocyte activities and osteoarthritis pathogenesis
Harrington Laboratory for Molecular Orthopedics, Department of Orthopedic Surgery, University of Kansas Medical Center, Kansas City, USA
* Corresponding Author: JINXI WANG. Email:
BIOCELL 2023, 47(10), 2125-2132. https://doi.org/10.32604/biocell.2023.030161
Received 24 March 2023; Accepted 26 July 2023; Issue published 08 November 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Osteoarthritis (OA), the most common form of joint disease, is characterized clinically by joint pain, stiffness, and deformity. OA is now considered a whole joint disease; however, the breakdown of the articular cartilage remains the major hallmark of the disease. Current treatments targeting OA symptoms have a limited impact on impeding or reversing the OA progression. Understanding the molecular and cellular mechanisms underlying OA development is a critical barrier to progress in OA therapy. Recent studies by the current authors’ group and others have revealed that the nuclear factor of activated T cell 1 (NFAT1), a member of the NFAT family of transcription factors, regulates the expression of many anabolic and catabolic genes in articular chondrocytes of adult mice. Mice lacking NFAT1 exhibit normal skeletal development but display OA in both appendicular and spinal facet joints as adults. This review mainly focuses on the recent advances in the regulatory role of NFAT1 transcription factor in the activities of articular chondrocytes and its implication in the pathogenesis of OA.Graphic Abstract

Keywords
As the most common joint disease, osteoarthritis (OA) is characterized clinically by joint pain, stiffness, and deformity with radiographical evidence of joint space narrowing due to a loss of articular cartilage, osteophyte formation, subchondral bone sclerosis, and subchondral cyst formation (Buckwalter and Martin, 2006; Lawrence et al., 2008). Current treatments, including pharmacologic therapy, local intra-articular injection, and surgical interventions mainly target OA symptoms, and have only a limited impact on altering the course of OA progression (Richmond et al., 2010; Kloppenburg and Berenbaum, 2020).
The pathogenic mechanisms for OA are not fully understood, especially the mechanisms governing articular chondrocyte activity and triggering articular cartilage breakdown (the major hallmark of the disease) remain unclear. Therefore, a nuanced understanding of the regulatory mechanisms underlying OA pathogenesis is needed for developing effective treatments. Given that many risk factors and regulatory mechanisms have been proposed for the development of OA, in this minireview, we will focus on recent advances in the role of nuclear factor of activated T cell 1 (NFAT1) in the regulation of articular chondrocyte activities and OA pathogenesis.
The nuclear factor of activated T cell transcription factor family and its transcriptional signaling
The nuclear factor of activated T cell (NFAT) is a family of transcription factors first identified in T cells and well-studied for their function in the immune response. There are currently five known members: NFAT1 (NFATc2, NFATp), NFAT2 (NFATc1, NFATc), NFAT3 (NFATc4), NFAT4 (NFATc3, NFATx), and the distantly related NFAT5 (TonEBP, OREBP). These five NFAT members are structurally characterized by a highly conserved DNA binding domain extending beyond the NFAT family to the larger Rel family of transcription factors, including NF-κB (Lin et al., 2013). This highly conserved Rel homology DNA binding domain results in the binding of each of the 5 NFAT family members to the same DNA sequence [(A/T)GGAAA] (Jain et al., 1995) (Fig. 1).
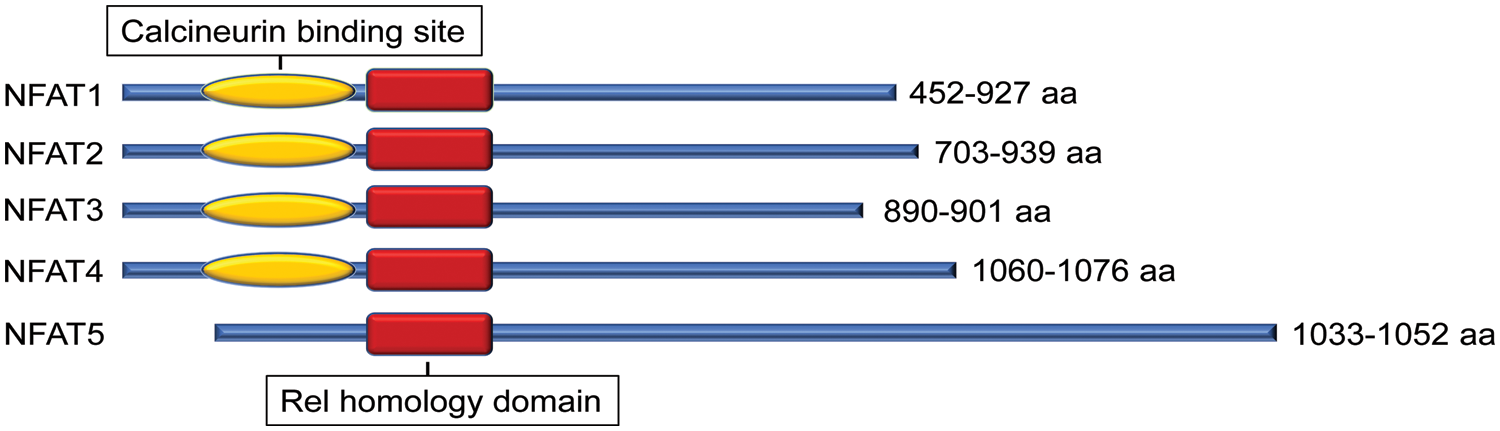
Figure 1: Protein structural character of mouse nuclear factor of activated T cell (NFAT) family members. All NFAT members share the Rel homology domain. Except for NFAT5, NFAT1-4 have a calcineurin-binding domain. The protein size range of amino acids (aa) among different transcript variants is presented.
Except for NFAT5, all four other canonical members (NFAT1-4) are activated by calcium-calcineurin signaling (McCaffrey et al., 1993; Northrop et al., 1994; Pan et al., 2000; Hoey et al., 1995; Masuda et al., 1995; Esensten et al., 2005) as NFAT1-4 have calcineurin binding domain at their N-terminal. In resting T cells, NFAT1-4 proteins are heavily phosphorylated and reside in the cytoplasm. Upon cell stimulation with an intracellular influx of Ca2+ that binds to the calcium sensor protein calmodulin, the serine/threonine phosphatase calcineurin is activated. Calcium ionophore ionomycin and polycystin-1 (PC1) can enhance the process of intracellular influx of Ca2+, thereby activating the calcineurin/NFAT signaling (Brignall et al., 2017; Puri et al., 2004). Activated calcineurin binds to its binding site located in the N-terminal regulatory domain of NFAT1-4 and rapidly dephosphorylates the serine-rich region and serine/proline repeat consensus sequences (SP-repeats) which are also present in the N-terminus of the NFAT1-4 proteins. This dephosphorylation of NFAT1-4 results in conformational changes that promote their translocation into the nucleus, wherein they regulate gene expression in concert with different cofactors, such as AP-1 (Macián et al., 2001), c-fos (Huang et al., 2006), Jun (Ikeda et al., 2004), GATA4 (Xia et al., 2000), and MEF2 (Blaeser et al., 2000). Once the regulation of gene expression in response to the stimulation is completed in the nucleus, NFAT1-4 are re-phosphorylated by nuclear resident protein kinases, including casein kinase 1 (CK1), glycogen synthase kinase 3 (GSK3), and dual-specificity tyrosine phosphorylation-regulated kinase (DYRK) (Okamura et al., 2004; Arron et al., 2006; Gwack et al., 2006), enabling NFAT1-4 to be transported back to the cytoplasm across the nuclear envelope (Fig. 2). In contrast, the NFAT5, known as a central regulator of cellular response to ambient hypertonicity, lacks docking sites for calcineurin and therefore is regulated by osmotic stress (Miyakawa et al., 1999) (Fig. 1).
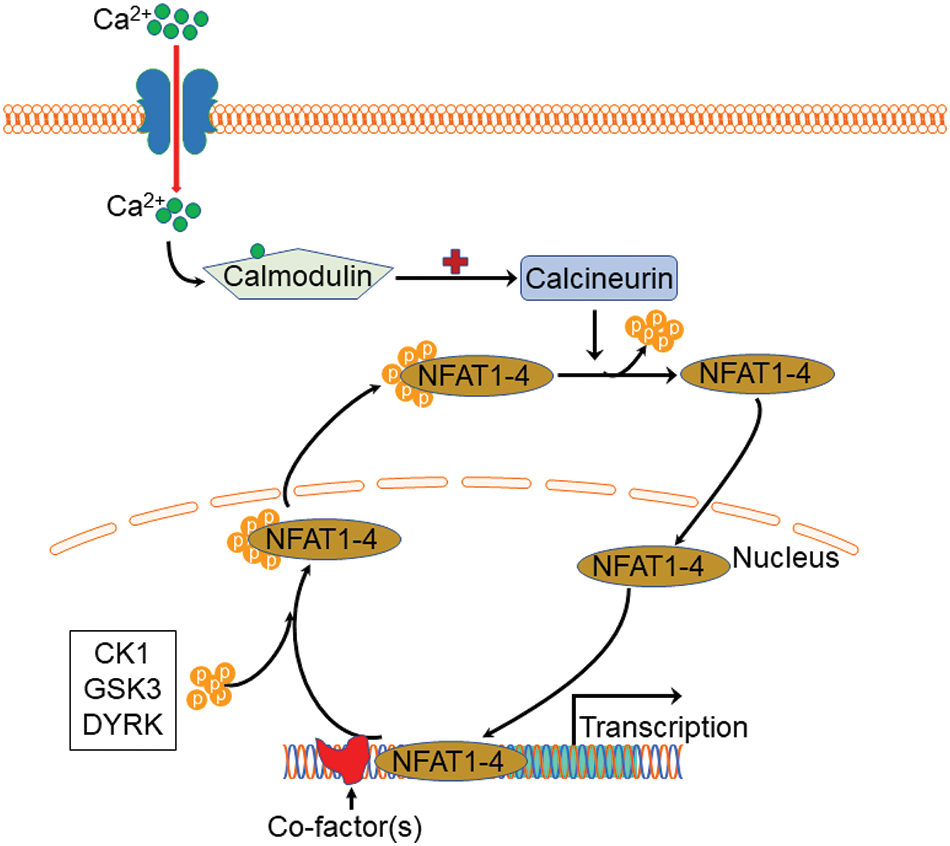
Figure 2: Schematic view of Ca2+-NFAT1-4 signaling pathway and phosphorylation cycle. Upon stimulation with an intracellular influx of Ca2+, calcium sensor protein calmodulin activates calcineurin which then binds to the N-terminal regulatory domain of nuclear factor of activated T cell 1 (NFAT)1-4 and rapidly dephosphorylates NFAT1-4 proteins, resulting in nuclear translocation. Once the regulation of gene expression completes, NFAT1-4 are re-phosphorylated by casein kinase 1 (CK1), glycogen synthase kinase 3 (GSK3), and dual-specificity tyrosine phosphorylation-regulated kinase (DYRK), enabling NFAT1-4 to be transported back to the cytoplasm.
NFAT1-5 control the differentiation of specific cells, thereby affecting the formation of specific tissues or organs. For instance, NFAT1 may be involved in adipogenesis and myogenesis (Abbott et al., 1998; Ho et al., 1998). NFAT2 has been found to play a pivotal role during osteoclastogenesis (Takayanagi et al., 2002) and is required for cardiac valve formation (de la Pompa et al., 1998; Ranger et al., 1998). NFAT3-deficient mice were viable and fertile and showed no major macroscopic or microscopic abnormalities after 36 months of observation (Graef et al., 2001). NFAT4 mutant mice exhibited skeletal muscle hypoplasia, reflecting impaired embryonic muscle development (Kegley et al., 2001). Moreover, mice doubly mutant for the NFAT3 and NFAT4 genes had defects in vascular patterning and angiogenesis, indicating that NFAT4 is more important than NFAT3 for cardiac growth and function (Graef et al., 2001). Besides its involvement in the osmotic stress response (López-Rodríguez et al., 2004), complete loss of NFAT5 function showed late gestational or perinatal lethality, whereas partial loss of NFAT5 function resulted in lymphoid hypocellularity and impaired antigen-specific antibody responses in viable heterozygous animals (Go et al., 2004). This review focused on the role of NFAT1 in the regulation of articular chondrocyte (the joint cartilage cell) activities and NFAT1 deficiency in the pathogenetic mechanisms of OA.
Regulation of articular chondrocyte differentiation and articular cartilage formation
Cartilage cells are derived from mesenchymal progenitor cells during skeletal development. The cartilage cells in the primary and secondary ossification centers are replaced by newly formed bone through the endochondral sequence of ossification. This type of cartilage is called temporal or replacement cartilage (Goldring et al., 2006). In contrast, cartilage cells close to the surface of growing long bones divide and differentiate to form hyaline articular cartilage, a permanent or persistent cartilage because it will not be replaced by bone (Eames et al., 2004; Pacifici et al., 2005). Many factors are involved in the regulation of articular chondrocyte differentiation and articular cartilage formation, although the precise mechanisms of these processes still need to be fully understood.
Sox9 is a pivotal transcriptional regulator essential for articular cartilage formation. Inactivation of the Sox9 gene in mouse limb buds before mesenchymal condensations results in a complete absence of cartilage formation. Mouse embryos in which Sox9 is deleted after mesenchymal condensations exhibit chondrodysplasia and defective joint formation (Bi et al., 1999; Akiyama et al., 2002). Our study in mice found that the Sox9 gene and protein are highly expressed in the early-stage developing articular cartilage at embryonic day 16.5 (E16.5). Sox9 expression was slightly lower but still at a higher level at 1 month of age, at which time the articular cartilage structure is well defined but is still in the growth stage (late-stage developing articular cartilage). The expression of Sox9 is significantly reduced from 2 months of age when the articular cartilage development is completed and then kept at a lower level throughout the life (Zhang et al., 2016b). This spatiotemporal expression pattern of Sox9 further reveals the essential role of Sox9 in articular chondrocyte differentiation and articular cartilage formation during joint development.
The ERG (Ets-related gene) transcriptional activator in the Ets gene family of transcription factors may also regulate articular chondrocyte formation. ERG is expressed during joint formation and also persists once the articular layer has developed. A C-1-1 variant of ERG has been found misexpressed in developing chick limbs, which imposes a stable and immature articular-like phenotype in the entire limb chondrocyte population, blocking chondrocyte maturation and hypertrophy as well as the process of endochondral ossification (Iwamoto et al., 2000). There is a close spatiotemporal expression of both ERG and growth and differentiation factor-5 (GDF5) in mouse embryo joints. These results indicate that ERG is one of the molecular mechanisms driving the differentiation of immature chondrocytes into permanent articular chondrocytes, exerting its influence through interactions with GDF5 (Iwamoto et al., 2007).
Many other factors, such as matrix metalloproteinases (MMPs) (Bertram et al., 2009), vascular endothelial growth factor (Yin and Pacifici, 2001), fibroblast growth factor (Haque et al., 2007), transforming growth factor β (Joyce et al., 1990), bone morphogenetic protein (Kobayashi et al., 2005) and the Wnt signaling pathway (Macsai et al., 2008), also contribute directly or indirectly to the process of articular cartilage formation.
Early studies reported that NFAT1 may control cellular differentiation programs in organ systems unrelated to the immune system, particularly in adipogenesis and myogenesis (Abbott et al., 1998; Ho et al., 1998). More recently, we found that Nfat1-/- mice displayed normal skeletal development, including articular cartilage and joint formation (Rodova et al., 2011), suggesting that NFAT1 is not critical for the formation of articular cartilage and other joint structures.
Role of nuclear factor of activated T cell 1 in the maintenance of articular chondrocyte activity and articular cartilage homeostasis
Although NFAT1 is not required for articular chondrocyte differentiation and joint formation, it is required for the functional maintenance of differentiated articular chondrocytes in adult mice. Our recent studies have demonstrated that NFAT1 expression in murine articular chondrocytes was undetectable at embryonic day 16.5 (E16.5) and postnatal day 1, but was high at the young adult stage and then declined as the animals aged (Rodova et al., 2011; Zhang et al., 2016a). These findings explain why NFAT1 deletion does not affect articular chondrocyte formation during development. In contrast, high expression of NFAT1 in articular chondrocytes of young adult mice indicates that NFAT1 is needed for the function of articular chondrocytes in adults’ Thus, NFAT1 deficiency in adult mice severely impaired articular chondrocyte function, causing subsequent OA onset with abnormal expression of numerous anabolic and catabolic genes, such as Acan, Col2a1, Col11a1, Col10a1, Mmp1a, Mmp13, Admts5, Timp3, IL-1b, and IL-17a, etc., in articular cartilage (Rodova et al., 2011; Wang et al., 2009; Zhang et al., 2016a). Moreover, forced expression of Nfat1 in articular chondrocytes of 15-month-old mice can reverse aberrant expression of specific anabolic and catabolic genes. However, this rescuable effect was reduced in the chondrocytes of 18-month-old mice, indicating that the therapeutic efficacy of NFAT1 on the dysfunction of aged articular chondrocytes is more effective in younger mice (Wang et al., 2009; Rodova et al., 2011; Zhang et al., 2016a). These results suggest that NFAT1 is a key transcriptional regulator that controls the expression of many specific anabolic and catabolic genes in articular chondrocytes, paramount to articular cartilage homeostasis in the adult stage.
In contrast, the expression of Sox9 in mouse articular cartilage is highest at embryonic and newborn stages and then significantly decreases after the completion of joint development (Zhang et al., 2016a). These findings confirm the importance of Sox9 in cartilage formation and suggest a diminished physiological demand for Sox9 in adult articular cartilage. This notion is supported by the evidence that postnatal inactivation of Sox9 in mouse cartilage resulted in a reduction of proteoglycan content in the articular cartilage without histopathological signs of OA by the age of 18 months (Henry et al., 2012).
Our recent studies have uncovered that the age-dependent expression patterns of Sox9 and Nfat1 are regulated by epigenetic mechanisms. These epigenetic changes mainly encompass dynamic changes in DNA methylation, histone 3 lysine 4 dimethylation (H3K4me2, a transcriptional activator), and histone 3 lysine 9 dimethylation (H3K9me2, a transcriptional repressor). In mouse articular cartilage, a low Nfat1 expression in the early developing stage was associated with increased H3K9me2, a high Nfat1 expression in the young adult stage was associated with increased H3K4me2, and a spontaneous reduction of Nfat1 expression in the aged phase was associated with decreased H3K4me2 and increased DNA methylation (Rodova et al., 2011; Zhang et al., 2016a, 2016b; Zhang et al., 2019b). A growing body of evidence has revealed the epigenetic mechanisms underlying the aberrant anabolic and catabolic gene expression in OA pathogenesis. As a transcription factor, NFAT1 regulates the expression of a cohort of anabolic and catabolic genes in articular cartilage; thus, NFAT1 might be one of the critical, pivotal molecules responsible for the deleterious epigenetic effect in aged articular cartilage. The epigenetically regulated age-dependent spontaneous reduction of NFAT1 expression in articular chondrocytes of aged mice impairs chondrocyte function and results in age-related joint degeneration (Zhang et al., 2019a).
Deficiency of nuclear factor of activated T cell 1 and osteoarthritis
The early reports on the generation and phenotype of Nfat1 knockout (Nfat1-/-) mice can be traced back to 1996. Mice with a global NFAT1 null mutation exhibited enhanced immune responses (Xanthoudakis et al., 1996), dysregulated interleukin-4 expression (Hodge et al., 1996), and allergic skin inflammation (Kwon et al., 2016). However, osteoarthritic changes in the peripheral/appendicular joints of Nfat1-/- mice were not reported until 2009 (Wang et al., 2009) and in the spinal facet joints until 2022 (Wang et al., 2022). Nfat1-/- mice developed normal skeleton but displayed decreased expression of type-II collagen (collagen-2) and aggrecan with increased expression of matrix-degrading proteinases and proinflammatory cytokines in articular cartilage of weight-bearing joints of young adults. These initial changes follow articular chondrocyte proliferation/clustering, progressive articular surface destruction, chondro-osteophyte formation, and thickened subchondral bone exposure, which resembles osteoarthritic changes seen in humans (Wang et al., 2009). NFAT1 deficiency causes aberrant expression of more than 50 anabolic and catabolic genes in articular chondrocytes, resulting in cartilage degradation and OA-like changes (Wang et al., 2009; Zhang et al., 2019a; Wang et al., 2022). These studies suggest that NFAT1 is a key transcriptional factor that regulates balanced anabolic and catabolic activities of the articular chondrocytes and overall articular cartilage homeostasis and integrity. In contrast, NFAT1 deficiency or dysfunction may cause imbalance in anabolic and catabolic activities of articular chondrocytes and subsequent cartilage degradation and OA (Fig. 3).
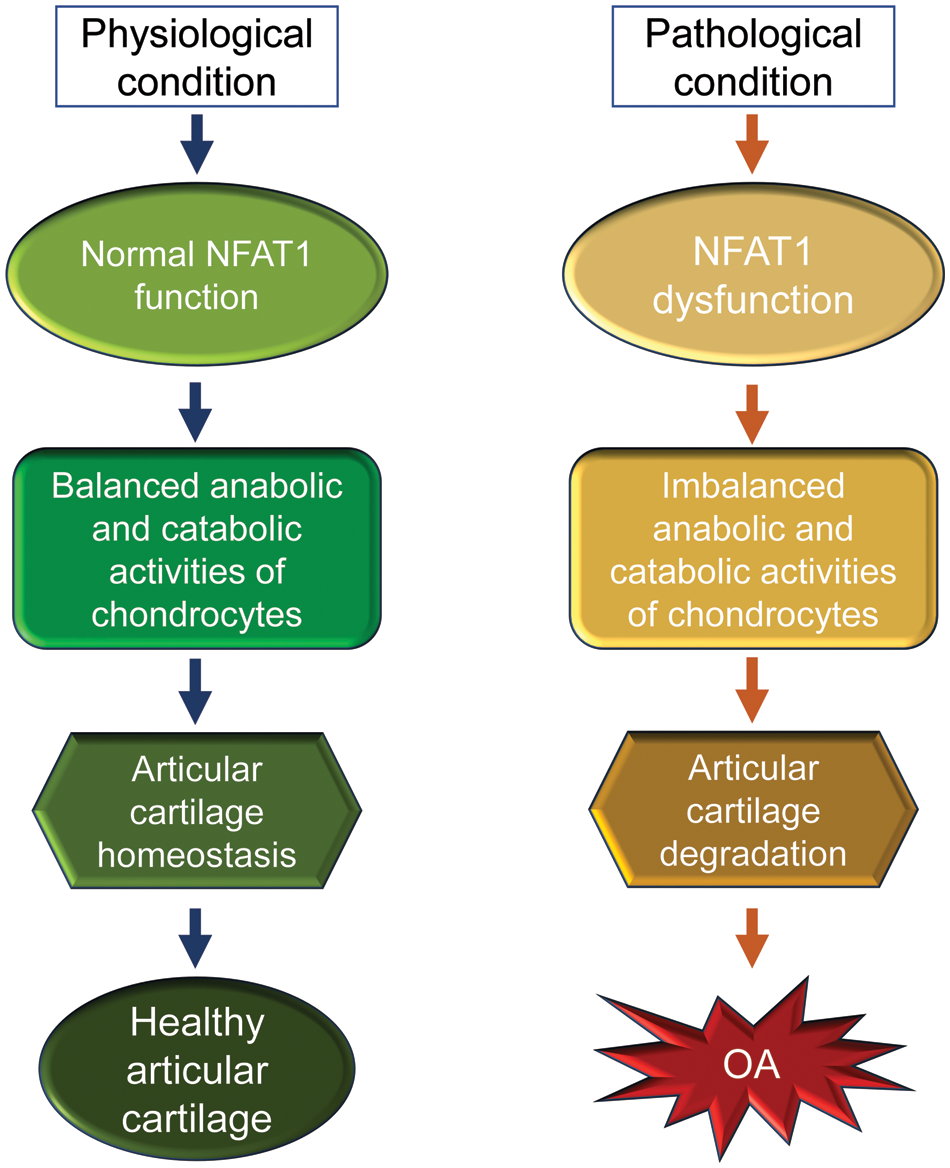
Figure 3: A schematic illustration of the physiological and pathological conditions of nuclear factor of activated T cell 1 (NFAT1) in articular cartilage of mice. In physiological conditions, NFAT1 functions normally by binding its target genes and regulating their expression, resulting in balanced anabolic and catabolic activities of articular chondrocytes, which is essential for articular cartilage homeostasis. In pathological conditions, dysfunction of NFAT1 results in imbalanced anabolic and catabolic activities of articular chondrocytes, leading to articular cartilage degeneration and, finally, the onset of osteoarthritis (OA).
The molecular mechanism of NFAT1 deficiency-induced OA was investigated by chromatin immunoprecipitation (ChIP) and promoter activity assays using chondrocytes isolated from femoral head articular cartilage of 3- to 4-month-old wild-type mice or Nfat1-/- mice with early hip OA. ChIP assays showed direct binding of NFAT1 to promoters of 21 of the 25 tested genes encoding cartilage-matrix proteins, growth factors, inflammatory cytokines, matrix-degrading proteinases, and specific transcription factors. Promoter activity assays of representative anabolic and catabolic genes showed NFAT1-DNA binding functionally regulated the promoter activity of specific target genes in wild-type chondrocytes but not in Nfat1-/- chondrocytes or in wild-type chondrocytes transfected with mutated NFAT1 binding sequences. These studies indicate that NFAT1 protects articular cartilage against degradation by directly regulating the transcription of target genes in these chondrocytes. NFAT1 deficiency causes defective transcription of specific anabolic and catabolic genes in articular chondrocytes, leading to increased matrix catabolism and degradation and destruction of the articular cartilage (Zhang et al., 2019a) (Fig. 4).
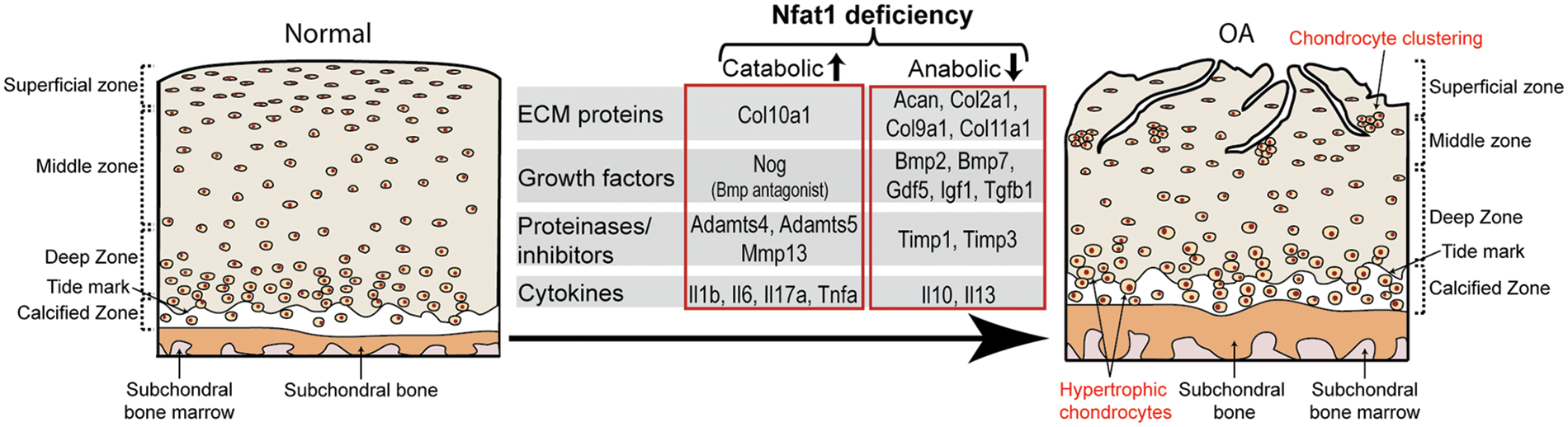
Figure 4: A diagram summarizes the impact of the nuclear factor of activated T cell 1 (NFAT1) deficiency on the expression of specific catabolic and anabolic genes by articular chondrocytes during the development of OA.
A study by Greenblatt et al. (2013) showed that cartilage-specific deletion (via the collagen-2 promoter) of NFAT2 (NFATc1) or NFAT4 (NFATc3) in cartilage displayed no histologic or clinical abnormalities; double deletion of NFAT1 and NFAT4 showed similar histologic and clinical abnormalities observed in NFAT1-deficient mice. However, the mice lacking both NFAT1 and NFAT2 in cartilage displayed severe, spontaneous OA with 100% penetrance, with subluxation of the elbow at about 1 week of age and the metatarsals at an age of about 3 weeks (Greenblatt et al., 2013). Those developmental defects in specific joints were not observed in NFAT1-deficient mice. These findings suggest that NFAT1 is more important than NFAT2 or NFAT4 for the maintenance of articular chondrocyte function and the prevention of spontaneous OA in adult mice.
The epigenetic mechanism is responsible for the up- and down-regulation of NFAT1 signaling and also directly regulates the expression levels of many anabolic and catabolic genes in articular cartilage that are involved in the pathogenesis of OA. In OA chondrocytes, loss of methylation at CpG sites in the promoter region was associated with increased expression of MMP-3, -9, -13, and ADAMTS-4 (Roach et al., 2005; Cheung et al., 2009). Hypermethylated CpG sites in the COL9A1 promoter attenuated SOX9 binding, resulting in the downregulation of COL9A1 expression in OA cartilage (Imagawa et al., 2014). Demethylation of the CpG site at −299 bp of the interleukin (IL)-1β promoter increased the transcriptional response of IL-1β to other inflammatory cytokines in human articular chondrocytes (Hashimoto et al., 2009). Specific and highly expressed long non-coding RNA-CIRs regulate the expression of collagen, aggrecan, MMP13, and ADAMTS-5 in OA cartilage (Liu et al., 2014). Moreover, miRNA-222 regulates MMP13 expression by targeting histone deacetylase 4 during the progression of OA (Song et al., 2015).
This review mainly focuses on the recent advances in the regulatory role of NFAT1 signaling in articular chondrocyte activity and its implication in the pathogenesis of OA. Notably, OA is a whole joint disease involving other joint tissues: subchondral bone, synovium, menisci, joint capsule, and ligaments. Pathological changes in these joint tissues may affect the biological and mechanical properties of articular cartilage. Insufficient recognition of the effect of pathological changes in other joint tissues on OA pathogenesis may negatively affect the efficacy of disease-modifying OA drug (DMOAD) candidates. Therefore, future studies should include the regulatory role of NFAT1 signaling in non-cartilaginous cells and the effects of NFAT1 deficiency on periarticular bone and soft tissues. Additionally, many animal models of OA do not translate into replications in human trials. Further work is needed to investigate if NFAT1 deficiency is involved in the pathogenesis of OA in humans.
In the past three decades, many clinical trials targeting the inhibitor of a single proinflammatory cytokine or proteinase as a candidate DMOAD have been unsuccessful due to insufficient efficacy and/or severe side effects. No DMOAD candidates have been approved by drug regulatory agencies in the United States or other countries (Hellio le Graverand et al., 2013; Kloppenburg and Berenbaum, 2020). These failed clinical trials suggest that inhibition of a single catabolic molecule may not be sufficient for the treatment of OA because the abnormality of both catabolic and anabolic factors is involved in its pathogenesis. NFAT1 is an upstream transcription factor that regulates many catabolic and anabolic factors that are proposed to be involved in OA pathogenesis (Zhang et al., 2019a, 2019b); thus, NFAT1 could be more effective than the previously tested DMOAD candidates targeting a single downstream catabolic or anabolic molecule if it is confirmed to be one of the risk factors for the initiation and/or progression of OA in humans.
Acknowledgement: The original Nfat1-/- mice breeders mentioned in this review were a kind gift from Dr. Laurie Glimcher (Harvard School of Public Health, Boston, MA, USA). Some aged wild-type mice mentioned in this review were gifted by the National Institute on Aging (NIA, Bethesda, MD, USA).
Funding Statement: This work was supported by the United States National Institutes of Health (NIH) under Award Number R01 AR059088 (to J.W.) and the Mary A. and Paul R. Harrington Distinguished Professorship Endowment.
Author Contributions: MZ and JW jointly drafted the manuscript. TC and SF contributed to the in-depth discussion and revised the manuscript. All authors approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
Abbott KL, Friday BB, Thaloor D, Murphy TJ, Pavlath GK (1998). Activation and cellular localization of the cyclosporine A-sensitive transcription factor NF-AT in skeletal muscle cells. Molecular Biology of the Cell 9: 2905–2916. https://doi.org/10.1091/mbc.9.10.2905 [Google Scholar] [PubMed] [CrossRef]
Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B (2002). The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes & Development 16: 2813–2828. https://doi.org/10.1101/gad.1017802 [Google Scholar] [PubMed] [CrossRef]
Arron JR, Winslow MM, Polleri A, Chang CP, Wu H et al. (2006). NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 441: 595–600. https://doi.org/10.1038/nature04678 [Google Scholar] [PubMed] [CrossRef]
Bertram H, Boeuf S, Wachters J, Boehmer S, Heisel C, Hofmann MW, Piecha D, Richter W (2009). Matrix metalloprotease inhibitors suppress initiation and progression of chondrogenic differentiation of mesenchymal stromal cells in vitro. Stem Cells and Development 18: 881–892. https://doi.org/10.1089/scd.2008.0306 [Google Scholar] [PubMed] [CrossRef]
Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B (1999). Sox9 is required for cartilage formation. Nature Genetics 22: 85–89. https://doi.org/10.1038/8792 [Google Scholar] [PubMed] [CrossRef]
Blaeser F, Ho N, Prywes R, Chatila TA (2000). Ca2+-dependent gene expression mediated by MEF2 transcription factors. Journal of Biological Chemistry 275: 197–209. https://doi.org/10.1074/jbc.275.1.197 [Google Scholar] [PubMed] [CrossRef]
Brignall R, Cauchy P, Bevington SL, Gorman B, Pisco AO et al. (2017). Integration of kinase and calcium signaling at the level of chromatin underlies inducible gene activation in T cells. Journal of Immunology 199: 2652–2667. https://doi.org/10.4049/jimmunol.1602033 [Google Scholar] [PubMed] [CrossRef]
Buckwalter JA, Martin JA (2006). Osteoarthritis. Advanced Drug Delivery Reviews 58: 150–167. https://doi.org/10.1016/j.addr.2006.01.006 [Google Scholar] [PubMed] [CrossRef]
Cheung KS, Hashimoto K, Yamada N, Roach HI (2009). Expression of ADAMTS-4 by chondrocytes in the surface zone of human osteoarthritic cartilage is regulated by epigenetic DNA de-methylation. Rheumatology International 29: 525–534. https://doi.org/10.1007/s00296-008-0744-z [Google Scholar] [PubMed] [CrossRef]
de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ et al. (1998). Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature 392: 182–186. https://doi.org/10.1038/32419 [Google Scholar] [PubMed] [CrossRef]
Eames BF, Sharpe PT, Helms JA (2004). Hierarchy revealed in the specification of three skeletal fates by Sox9 and Runx2. Developmental Biology 274: 188–200. [Google Scholar] [PubMed]
Esensten JH, Tsytsykova AV, Lopez-Rodriguez C, Ligeiro FA, Rao A, Goldfeld AE (2005). NFAT5 binds to the TNF promoter distinctly from NFATp, c, 3 and 4, and activates TNF transcription during hypertonic stress alone. Nucleic Acids Research 33: 3845–3854. [Google Scholar] [PubMed]
Go WY, Liu X, Roti MA, Liu F, Ho SN (2004). NFAT5/TonEBP mutant mice define osmotic stress as a critical feature of the lymphoid microenvironment. Proceedings of the National Academy of Sciences of the United States of America 101: 10673–10678. https://doi.org/10.1073/pnas.0403139101 [Google Scholar] [PubMed] [CrossRef]
Goldring MB, Tsuchimochi K, Ijiri K (2006). The control of chondrogenesis. Journal of Cellular Biochemistry 97: 33–44. https://doi.org/10.1002/jcb.20652 [Google Scholar] [PubMed] [CrossRef]
Graef IA, Chen F, Chen L, Kuo A, Crabtree GR (2001). Signals transduced by Ca2+/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell 105: 863–875. https://doi.org/10.1016/S0092-8674(01)00396-8 [Google Scholar] [PubMed] [CrossRef]
Greenblatt MB, Ritter SY, Wright J, Tsang K, Hu D, Glimcher LH, Aliprantis AO (2013). NFATc1 and NFATc2 repress spontaneous osteoarthritis. Proceedings of the National Academy of Sciences of the United States of America 110: 19914–19919. https://doi.org/10.1073/pnas.1320036110 [Google Scholar] [PubMed] [CrossRef]
Gwack Y, Sharma S, Nardone J, Tanasa B, Iuga A et al. (2006). A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature 441: 646–650. https://doi.org/10.1038/nature04631 [Google Scholar] [PubMed] [CrossRef]
Haque T, Nakada S, Hamdy RC (2007). A review of FGF18: Its expression, signaling pathways and possible functions during embryogenesis and post-natal development. Histology & Histopathology 22: 97–105. https://doi.org/10.14670/hh-22.97 [Google Scholar] [PubMed] [CrossRef]
Hashimoto K, Oreffo RO, Gibson MB, Goldring MB, Roach HI (2009). DNA demethylation at specific CpG sites in the IL1B promoter in response to inflammatory cytokines in human articular chondrocytes. Arthritis & Rheumatology 60: 3303–3313. https://doi.org/10.1002/art.24882 [Google Scholar] [PubMed] [CrossRef]
Hellio le Graverand MP, Clemmer RS, Redifer P, Brunell RM, Hayes CW, Brandt KD, Abramson SB, Manning PT, Miller CG, Vignon E (2013). A 2-year randomised, double-blind, placebo-controlled, multicentre study of oral selective iNOS inhibitor, cindunistat (SD-6010in patients with symptomatic osteoarthritis of the knee. Annals of the Rheumatic Diseases 72: 187–195. https://doi.org/10.1136/annrheumdis-2012-202239 [Google Scholar] [PubMed] [CrossRef]
Henry SP, Liang S, Akdemir KC, de Crombrugghe B (2012). The postnatal role of Sox9 in cartilage. Journal of Bone and Mineral Research 27: 2511–2525. https://doi.org/10.1002/jbmr.1696 [Google Scholar] [PubMed] [CrossRef]
Ho IC, Kim JH, Rooney JW, Spiegelman BM, Glimcher LH (1998). A potential role for the nuclear factor of activated T cells family of transcriptional regulatory proteins in adipogenesis. Proceedings of the National Academy of Sciences of the United States of America 95: 15537–15541. https://doi.org/10.1073/pnas.95.26.15537 [Google Scholar] [PubMed] [CrossRef]
Hodge MR, Ranger AM, Charles de la Brousse F, Hoey T, Grusby MJ, Glimcher LH (1996). Hyperproliferation and dysregulation of IL-4 expression in NF-ATp-deficient mice. Immunity 4: 397–405. https://doi.org/10.1016/S1074-7613(00)80253-8 [Google Scholar] [PubMed] [CrossRef]
Hoey T, Sun YL, Williamson K, Xu X (1995). Isolation of two new members of the NF-AT gene family and functional characterization of the NF-AT proteins. Immunity 2: 461–472. [Google Scholar] [PubMed]
Huang H, Chang EJ, Ryu J, Lee ZH, Lee Y, Kim HH (2006). Induction of c-Fos and NFATc1 during RANKL-stimulated osteoclast differentiation is mediated by the p38 signaling pathway. Biochemical and Biophysical Research Communications 351: 99–105. https://doi.org/10.1016/j.bbrc.2006.10.011 [Google Scholar] [PubMed] [CrossRef]
Ikeda F, Nishimura R, Matsubara T, Tanaka S, Inoue J et al. (2004). Critical roles of c-Jun signaling in regulation of NFAT family and RANKL-regulated osteoclast differentiation. Journal of Clinical Investigation 114: 475–484. https://doi.org/10.1172/JCI200419657 [Google Scholar] [CrossRef]
Imagawa K, de Andrés MC, Hashimoto K, Itoi E, Otero M, Roach HI, Goldring MB, Oreffo RO (2014). Association of reduced type IX collagen gene expression in human osteoarthritic chondrocytes with epigenetic silencing by DNA hypermethylation. Arthritis & Rheumatology 66: 3040–3051. https://doi.org/10.1002/art.38774 [Google Scholar] [PubMed] [CrossRef]
Iwamoto M, Higuchi Y, Koyama E, Enomoto-Iwamoto M, Kurisu K, Yeh H, Abrams WR, Rosenbloom J, Pacifici M (2000). Transcription factor ERG variants and functional diversification of chondrocytes during limb long bone development. Journal of Cell Biology 150: 27–40. [Google Scholar] [PubMed]
Iwamoto M, Tamamura Y, Koyama E, Komori T, Takeshita N, Williams JA, Nakamura T, Enomoto-Iwamoto M, Pacifici M (2007). Transcription factor ERG and joint and articular cartilage formation during mouse limb and spine skeletogenesis. Developmental Biology 305: 40–51. https://doi.org/10.1016/j.ydbio.2007.01.037 [Google Scholar] [PubMed] [CrossRef]
Jain J, Burgeon E, Badalian TM, Hogan PG, Rao A (1995). A similar DNA-binding motif in NFAT family proteins and the Rel homology region. Journal of Biological Chemistry 270: 4138–4145. [Google Scholar] [PubMed]
Joyce ME, Roberts AB, Sporn MB, Bolander ME (1990). Transforming growth factor-beta and the initiation of chondrogenesis and osteogenesis in the rat femur. Journal of Cell Biology 110: 2195–2207. https://doi.org/10.1083/jcb.110.6.2195 [Google Scholar] [PubMed] [CrossRef]
Kegley KM, Gephart J, Warren GL, Pavlath GK (2001). Altered primary myogenesis in NFATC3–/– mice leads to decreased muscle size in the adult. Developmental Biology 232: 115–126. https://doi.org/10.1006/dbio.2001.0179 [Google Scholar] [PubMed] [CrossRef]
Kloppenburg M, Berenbaum F (2020). Osteoarthritis year in review 2019: Epidemiology and therapy. Osteoarthritis Cartilage 28: 242–248. https://doi.org/10.1016/j.joca.2020.01.002 [Google Scholar] [PubMed] [CrossRef]
Kobayashi T, Lyons KM, McMahon AP, Kronenberg HM (2005). BMP signaling stimulates cellular differentiation at multiple steps during cartilage development. Proceedings of the National Academy of Sciences of the United States of America 102: 18023–18027. https://doi.org/10.1073/pnas.0503617102 [Google Scholar] [PubMed] [CrossRef]
Kwon HK, Kim GC, Hwang JS, Kim Y, Chae CS, Nam JH, Jun CD, Rudra D, Surh CD, Im SH (2016). Transcription factor NFAT1 controls allergic contact hypersensitivity through regulation of activation induced cell death program. Scientific Reports 6: 19453. https://doi.org/10.1038/srep19453 [Google Scholar] [PubMed] [CrossRef]
Lawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H et al. (2008). Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: Part II. Arthritis & Rheumatology 58: 26–35. https://doi.org/10.1002/art.23176 [Google Scholar] [PubMed] [CrossRef]
Lin J, Zhang L, Zhao G, Su Z, Deng R, Pflugfelder SC, Li DQ (2013). A novel interleukin 33/ST2 signaling regulates inflammatory response in human corneal epithelium. PLoS One 8: e60963. https://doi.org/10.1371/journal.pone.0060963 [Google Scholar] [PubMed] [CrossRef]
Liu Q, Zhang X, Dai L, Hu X, Zhu J, Li L, Zhou C, Ao Y (2014). Long noncoding RNA related to cartilage injury promotes chondrocyte extracellular matrix degradation in osteoarthritis. Arthritis & Rheumatology 66: 969–978. https://doi.org/10.1002/art.38309 [Google Scholar] [PubMed] [CrossRef]
López-Rodríguez C, Antos CL, Shelton JM, Richardson JA, Lin F, Novobrantseva TI, Bronson RT, Igarashi P, Rao A, Olson EN (2004). Loss of NFAT5 results in renal atrophy and lack of tonicity-responsive gene expression. Proceedings of the National Academy of Sciences of the United States of America 101: 2392–2397. https://doi.org/10.1073/pnas.0308703100 [Google Scholar] [PubMed] [CrossRef]
Macián F, López-Rodríguez C, Rao A (2001). Partners in transcription: NFAT and AP-1. Oncogene 20: 2476–2489. https://doi.org/10.1038/sj.onc.1204386 [Google Scholar] [PubMed] [CrossRef]
Macsai CE, Foster BK, Xian CJ (2008). Roles of Wnt signalling in bone growth, remodelling, skeletal disorders and fracture repair. Journal of Cellular Physiology 215: 578–587. https://doi.org/10.1002/jcp.21342 [Google Scholar] [PubMed] [CrossRef]
Masuda ES, Naito Y, Tokumitsu H, Campbell D, Saito F, Hannum C, Arai K, Arai N (1995). NFATx, a novel member of the nuclear factor of activated T cells family that is expressed predominantly. Molecular and Cellular Biology 15: 2697–2706. [Google Scholar] [PubMed]
McCaffrey PG, Luo C, Kerppola TK, Jain J, Badalian TM et al. (1993). Isolation of the cyclosporin-sensitive T cell transcription factor NFATp. Science 262: 750–754. [Google Scholar] [PubMed]
Miyakawa H, Woo SK, Dahl SC, Handler JS, Kwon HM (1999). Tonicity-responsive enhancer binding protein, a rel-like protein that stimulates transcription in response to hypertonicity. Proceedings of the National Academy of Sciences of the United States of America 96: 2538–2542. https://doi.org/10.1073/pnas.96.5.2538 [Google Scholar] [PubMed] [CrossRef]
Northrop JP, Ho SN, Chen L, Thomas DJ, Timmerman LA, Nolan GP, Admon A, Crabtree GR (1994). NF-AT components define a family of transcription factors targeted in T-cell activation. Nature 369: 497–502. [Google Scholar] [PubMed]
Okamura H, Garcia-Rodriguez C, Martinson H, Qin J, Virshup DM, Rao A (2004). A conserved docking motif for CK1 binding controls the nuclear localization of NFAT1. Molecular and Cellular Biology 24: 4184–4195. https://doi.org/10.1128/MCB.24.10.4184-4195.2004 [Google Scholar] [PubMed] [CrossRef]
Pacifici M, Koyama E, Iwamoto M (2005). Mechanisms of synovial joint and articular cartilage formation: Recent advances, but many lingering mysteries. Birth Defects Research Part C, Embryo Today: Reviews 75: 237–248. [Google Scholar] [PubMed]
Pan S, Tsuruta R, Masuda ES, Imamura R, Bazan F, Arai K, Arai N, Miyatake S (2000). NFATz: A novel rel similarity domain containing protein. Biochemical and Biophysical Research Communications 272: 765–776. [Google Scholar] [PubMed]
Puri S, Magenheimer BS, Maser RL, Ryan EM, Zien CA, Walker DD, Wallace DP, Hempson SJ, Calvet JP (2004). Polycystin-1 activates the calcineurin/NFAT (nuclear factor of activated T-cells) signaling pathway. Journal of Biological Chemistry 279: 55455–55464. https://doi.org/10.1074/jbc.M402905200 [Google Scholar] [PubMed] [CrossRef]
Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, de la Brousse FC, Hoey T, Mickanin C, Baldwin HS, Glimcher LH (1998). The transcription factor NF-ATc is essential for cardiac valve formation. Nature 392: 186–190. https://doi.org/10.1038/32426 [Google Scholar] [PubMed] [CrossRef]
Richmond J, Hunter D, Irrgang J, Jones M H, Snyder-Mackler L et al. (2010). American Academy of Orthopaedic Surgeons clinical practice guideline on the treatment of osteoarthritis (OA) of the knee. Journal of Bone and Joint Surgery 92: 990–993. https://doi.org/10.2106/JBJS.I.00982 [Google Scholar] [PubMed] [CrossRef]
Roach HI, Yamada N, Cheung KS, Tilley S, Clarke NM, Oreffo RO, Kokubun S, Bronner F (2005). Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis & Rheumatology 52: 3110–3124. https://doi.org/10.1002/art.21300 [Google Scholar] [PubMed] [CrossRef]
Rodova M, Lu Q, Li Y, Woodbury BG, Crist JD, Gardner BM, Yost JG, Zhong XB, Anderson HC, Wang J (2011). Nfat1 regulates adult articular chondrocyte function through its age-dependent expression mediated by epigenetic histone methylation. Journal of Bone and Mineral Research 26: 1974–1986. https://doi.org/10.1002/jbmr.397 [Google Scholar] [PubMed] [CrossRef]
Song J, Jin EH, Kim D, Kim KY, Chun CH, Jin EJ (2015). MicroRNA-222 regulates MMP-13 via targeting HDAC-4 during osteoarthritis pathogenesis. BBA Clinical 3: 79–89. https://doi.org/10.1016/j.bbacli.2014.11.009 [Google Scholar] [PubMed] [CrossRef]
Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M et al. (2002). Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Developmental Cell 3: 889–901. https://doi.org/10.1016/S1534-5807(02)00369-6 [Google Scholar] [PubMed] [CrossRef]
Wang J, Gardner BM, Lu Q, Rodova M, Woodbury BG, Yost JG, Roby KF, Pinson DM, Tawfik O, Anderson HC (2009). Transcription factor Nfat1 deficiency causes osteoarthritis through dysfunction of adult articular chondrocytes. The Journal of Pathology 219: 163–172. https://doi.org/10.1002/path.2578 [Google Scholar] [PubMed] [CrossRef]
Wang J, Lu Q, Mackay MJ, Liu X, Feng Y, Burton DC, Asher MA (2022). Spontaneous facet joint osteoarthritis in NFAT1-mutant mice: Age-dependent histopathologic characteristics and molecular mechanisms. Journal of Bone and Joint Surgery 104: 928–940. https://doi.org/10.2106/JBJS.21.00960 [Google Scholar] [PubMed] [CrossRef]
Xanthoudakis S, Viola JP, Shaw KT, Luo C, Wallace JD, Bozza PT, Luk DC, Curran T, Rao A (1996). An enhanced immune response in mice lacking the transcription factor NFAT1. Science 272: 892–895. https://doi.org/10.1126/science.272.5263.892 [Google Scholar] [PubMed] [CrossRef]
Xia Y, McMillin JB, Lewis A, Moore M, Zhu WG, Williams RS, Kellems RE (2000). Electrical stimulation of neonatal cardiac myocytes activates the NFAT3 and GATA4 pathways and up-regulates the adenylosuccinate synthetase 1 gene. Journal of Biological Chemistry 275: 1855–1863. https://doi.org/10.1074/jbc.275.3.1855 [Google Scholar] [PubMed] [CrossRef]
Yin M, Pacifici M (2001). Vascular regression is required for mesenchymal condensation and chondrogenesis in the developing limb. Developmental Dynamics 222: 522–533. https://doi.org/10.1002/dvdy.1212 [Google Scholar] [PubMed] [CrossRef]
Zhang M, Lu Q, Budden T, Wang J (2019a). NFAT1 protects articular cartilage against osteoarthritic degradation by directly regulating transcription of specific anabolic and catabolic genes. Bone and Joint Research 8: 90–100. https://doi.org/10.1302/2046-3758.82.BJR-2018-0114.R1 [Google Scholar] [PubMed] [CrossRef]
Zhang M, Lu Q, Egan B, Zhong XB, Brandt K, Wang J (2016a). Epigenetically mediated spontaneous reduction of NFAT1 expression causes imbalanced metabolic activities of articular chondrocytes in aged mice. Osteoarthritis Cartilage 24: 1274–1283. https://doi.org/10.1016/j.joca.2016.02.003 [Google Scholar] [PubMed] [CrossRef]
Zhang M, Lu Q, Miller AH, Barnthouse NC, Wang J (2016b). Dynamic epigenetic mechanisms regulate age-dependent SOX9 expression in mouse articular cartilage. The International Journal of Biochemistry & Cell Biology 72: 125–134. https://doi.org/10.1016/j.biocel.2016.01.013 [Google Scholar] [PubMed] [CrossRef]
Zhang M, Theleman JL, Lygrisse KA, Wang J (2019b). Epigenetic mechanisms underlying the aging of articular cartilage and osteoarthritis. Gerontologia 65: 387–396. https://doi.org/10.1159/000496688 [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools