
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Assessing the Hematological Cancer Stem Cell Landscape to Improve Immunotherapy Clinical Decisions
1 Laboratory of Pharmacology, School of Pharmacy, Faculty of Health Sciences, Aristotle University of Thessaloniki, Thessaloniki, 54124, Greece
2 Department of Health Sciences, School of Life and Health Sciences, University of Nicosia, Nicosia, CY-1700, Cyprus
3 Department of Medicine, School of Health Sciences, Democritus University of Thrace, Alexandroupolis, 68100, Greece
4 School of Medicine, Faculty of Health Sciences, Aristotle University of Thessaloniki, Thessaloniki, 54124, Greece
* Corresponding Authors: Sotirios Charalampos Diamantoudis. Email: ,
; Ioannis S. Vizirianakis. Email:
,
# These authors contributed equally to this work and share first authorship
(This article belongs to the Special Issue: Stem Cells Therapy in Health and Disease)
BIOCELL 2025, 49(10), 1799-1858. https://doi.org/10.32604/biocell.2025.067216
Received 27 April 2025; Accepted 28 July 2025; Issue published 22 October 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Hematological cancer stem cells (HCSCs) is a subpopulation of cells within hematological cancers that, through their characteristics, enhance malignancy and render their therapy more challenging. By uncovering the underlying mechanisms behind characteristic properties such as self-renewal, immune evasion, and conventional therapy resistance, as well as the major differences between other cancers and physiological cells, new and alternative targets can be assessed for use in existing and novel immunotherapeutic interventions. Through the evaluation of the existing literature, one can realize that there have already been several studies addressing the use of stem cell transplantation (SCT), monoclonal antibodies (mAbs), cell therapies, cancer vaccines, and oncolytic viruses, with varying degrees of success. As such, this study aims to combine existing information and clinical evidence to assess and bring to the spotlight targets related to HCSCs that can be considered for the improvement of therapeutic interventions.Graphic Abstract
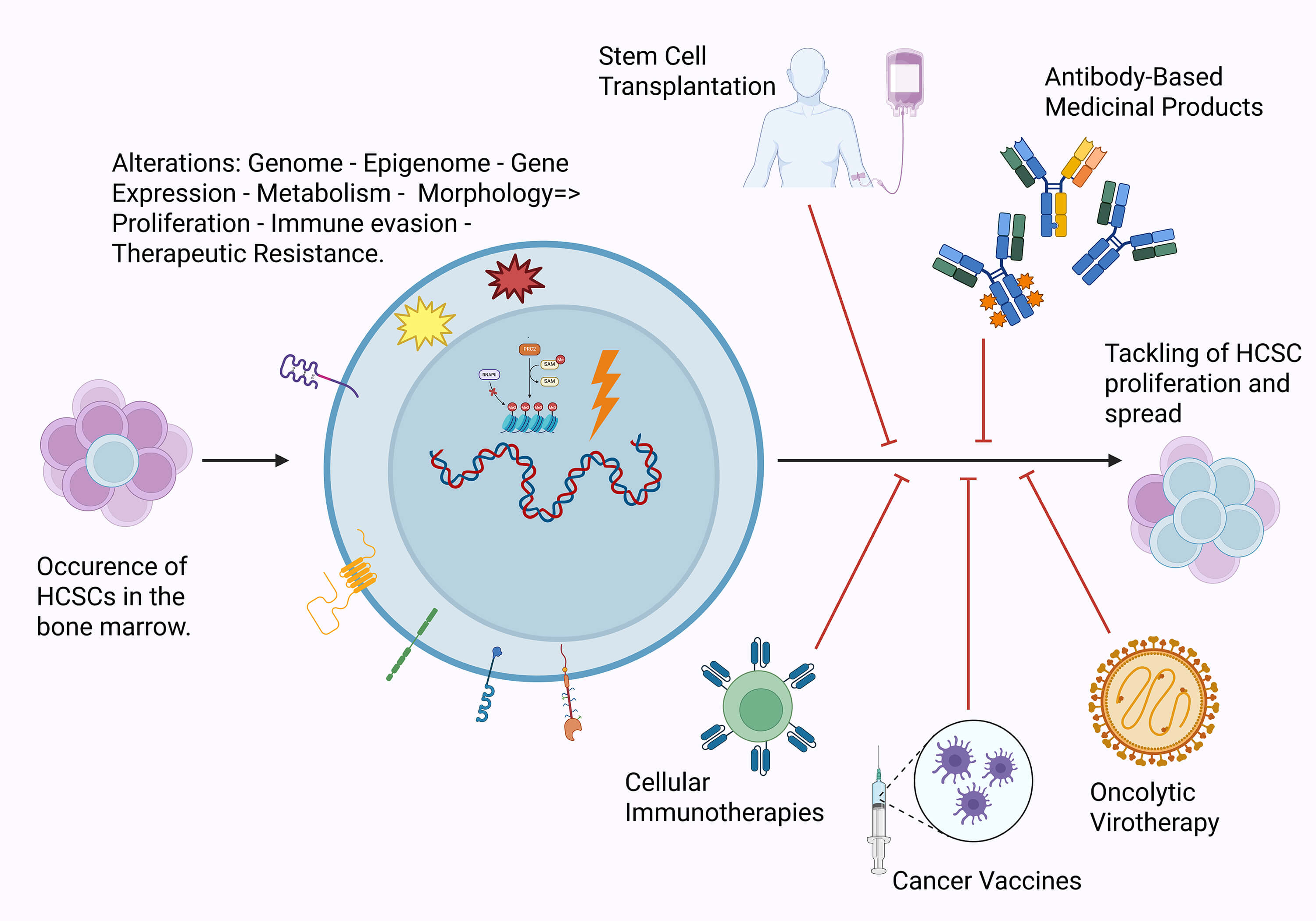
Keywords
Hematological cancers is a trending topic within modern oncology. Epidemiologic evidence addressing the period between 1990 and 2019 indicates an increase in the global prevalence of malignancies of the blood. Although this is highly concerning, the extent of the threat of this type of disease appears to be decreasing, as evident by the downward trend of the Age-Standardized Death Rate [1]. A plausible explanation for this phenomenon is the therapeutic methods applied within modern medicine that are characterized by the ever-increasing sophistication, targeting capabilities, safety, and effectiveness. These traits are allowed mostly on the basis of a better understanding of the exact pathophysiological mechanisms and advancements in the discovery, manufacturing, and application of more advanced medicinal products that have occurred in recent years. Nonetheless, the fact that blood cancers remain a huge burden for global health serves as a reminder that there is still plenty of room for research within the field.
There are several categories of blood cancers based on the affected cell types and other pathological evidence. Among those, there are leukaemias, which primarily affect the bone marrow and its capacity to produce functioning blood cells, and lymphomas, which originate in the lymphatic system. Additional categories include myelomas, which affect the plasma cells, myelodysplastic syndromes, and myeloproliferative neoplasms [2]. Additional classification under each category can be carried out by considering the timeline of the disease progression, origins and causes, affected cell populations, and other related pathological evidence. Out of these, leukemias (mostly acute) [3] seem to be at the focal point as they are the most prevalent ones in pediatric patients and, to an extent, contribute to the life years lost to cancer [4]. It is important to note that hematological cancer classification continuously evolves following advancements in detection and diagnosis techniques involving omics technologies [5–7].
Cancer stem cells (CSCs), also referred to as hematological/hematopoietic cancer stem cells (HCSCs), or leukemic stem cells (LSCs) in the context of the respective diseases, seem to have a special role in the pathophysiology and therapy of hematological cancers as they appear to be major drivers behind proliferation, immune evasion, therapy resistance, metastatic potential, and relapse [8,9]. CSCs’ relationship with blood cancers dates to the discovery and characterization of CSCs, also known as tumor initiating cells (TICs), in 1994, from a population of leukemic cells derived from a patient with acute myeloid leukaemia (AML). Those leukemic cells were the ones to be first characterized as LSCs and their characteristics/capabilities that, included, amongst others, seemingly unlimited proliferation, have sparked scientific interest ever since [10]. The origins of cancer stem cells are a complex topic that falls under the scope of many disciplines. Although there is extensive research still being conducted, evidence suggests that CSCs may occur from previously physiological stem cells or other types further into the differentiation process such as progenitors or fully differentiated cells. Nonetheless, it is accepted that the key identifying factors of CSCs are the markers they express and the behavior [11].
Granted, the burden of hematological malignancies on the life and the wellbeing of affected patients, with reflections into the society and the economy, and the ever-expanding need for impactful targets, it is important to address the role of HCSCs and the therapeutic value that can be extracted from their targeting.
2 Pathophysiology of the HCSCs
HCSCs possess a distinct set of properties that are crucial for their involvement in hematological cancers, enabling them to maintain tumor heterogeneity and drive disease progression [12]. Foremost among these, HCSCs exhibit key characteristics such as self-renewal, which allows them to sustain the hematological cancer cell (HCC) population, differentiation into various types of hematopoietic cells, and proliferation [13]. These traits are linked to several adverse consequences, including treatment resistance, tumor relapse, and metastatic spread [14]. Notably, these characteristics enable HCSCs to persist in the body, evade therapies, and contribute to the progression and recurrence of hematological malignancies such as leukaemia, making them a critical and promising target for therapeutic strategies aimed at improving patient outcomes [15]. Indeed, it has been found that LSCs, in the context of leukemia, exhibit a quiescent phenotype, which makes them resistant to conventional chemotherapy treatments that target rapidly dividing cells [16]. In addition to the aforementioned properties, HCSCs, as described in the following sections, exhibit genetic and epigenetic modifications, alterations in the programming of various signaling pathways, and distinct metabolic and phenotypic profiles compared to normal hematopoietic stem cells (HSCs) [17]. These differences make them key players in the progression and relapse of hematological malignancies.
Chromosomal abnormalities and gene mutations are key drivers of the transformation of HSCs into HCSCs, primarily promoting uncontrolled proliferation and impairing normal differentiation. In leukaemia, these genetic alterations occur in hematopoietic stem or progenitor cells, leading to the initiation of leukemogenesis and the disruption of normal blood cell development [18]. According to recent findings, the recurrent genetic mutations observed in leukaemia stem cells include genes such as NPM1, DNA methyltransferase 3A (DNMT3A), FLT3, RUNX1, and Ten-eleven translocation 2 (TET2), which are among the most frequently mutated [19]. Furthermore, mutations are commonly observed in genes like isocitrate dehydrogenase 1/2 (IDH1/2), KIT, NRAS, Wilms tumor 1 (WT1), TP53, protein tyrosine phosphatase non-receptor type 11 (PTPN11), U2 small nuclear RNA auxiliary factor 1 (U2AF1), structural maintenance of chromosomes 1A (SMC1A), structural maintenance of chromosomes 3 (SMC3), stromal antigen 2 (STAG2), RAD21, ASXL1/2, and enhancer of zeste homolog 2 (EZH2) [20,21]. In addition to these changes, other genetic alterations, including chromosomal abnormalities (both numerical and structural), copy number variations (CNVs), uniparental isodisomies (UPDs), small insertions or deletions (indels), and single nucleotide variants (SNVs), also occur in HSCs [22]. Notably, gene fusions resulting from chromosomal translocations are a notable feature, with examples including RUNX1-ETO, PML-RARA, and various MLL fusions involving multiple partner genes [23]. Among the most critical genetic alterations driving the transformation of HSCs into cancerous stem cells are chromosomal abnormalities such as alterations in the 3q26 and 11q23 region [18,23]. Transcription factors like RUNX1 and CBFB are frequently targeted by translocations, including RUNX1::RUNX1T1 and CBFB::MYH11, as revealed through cytogenetic studies. Recurrent cytogenetic changes, including the loss of chromosomes −5/5q and −7/7q, the gain of chromosome +8, deletions in 17p [24] and specific translocations like t(7;12)(q36;p13) [25], further illustrate the genetic instability that underlies the development of hematologic malignancies. Acquired chromosomal abnormalities (aCNAs) and uniparental disomies (UPDs) such as del(2q33.3), del(3p14.2), del(4q22.1), UPD(13q), and del(17p13) are recurrently observed in HCSCs at relapse. Deletions like del(12p13) are linked to complex karyotypes and poor prognosis, while UPD(13q) and TP53 deletions in HCSCs at relapse are associated with therapy resistance and poor survival outcomes [22].
In HSCs, intracellular mechanisms that drive malignant transformation are rooted in not only genetic, but also epigenetic alterations. These changes disrupt normal gene expression and cellular function, enhancing the fitness of these cells and paving the way for their progression into malignancy. Epigenetic modifications, in particular, play a critical role in this process by dysregulating the transcription and translation of oncogenes and tumor suppressor genes. Key mechanisms include alterations in histone modifications, abnormal DNA methylation patterns, and post-transcriptional control by non-coding RNAs such as lncRNAs and miRNAs [23]. In blood malignancies, histone modifications found on promoters are frequently enriched with H3K27me3, either individually or alongside H3K4me3, forming what are known as bivalent states [26]. These histone marks are inversely associated with DNA methylation and transcriptional activity. H3K27me3 is typically connected to promoter hypermethylation and gene repression in HSCs [27], whereas H3K4me3 is linked to reduced promoter methylation and increased gene expression [26].
Regarding non-coding RNAs, the lncRNA Xist (X-inactive specific transcript) plays a pivotal role in HSCs. Xist is known for its ability to recruit the polycomb repressive complex 2 (PRC2) complex to target genes, leading to transcriptional silencing [28]. Its tumor-suppressive function is particularly significant in hematological cancers. Studies have shown that deletion of Xist in HSCs results in the development of myeloproliferative neoplasm and myelodysplastic syndrome (MPN/MDS) with full penetrance [29], highlighting its critical role in maintaining normal epigenetic regulation and preventing malignant transformation. Another lncRNA, eRNA LED (lncRNA activator of enhancer domains), regulates the expression of the P21 tumor suppressor by activating its enhancer. This process is often disrupted in blood cancers, including leukaemia and lymphoma, where LED is inactivated in HSCs through promoter methylation, leading to the loss of its tumor-suppressive activity [30]. At the same time, another study emphasized the role of miR-125b overexpression in the bone marrow, which contributed to the development of a myeloproliferative disorder that may eventually progress to myeloid leukaemia. In HSCs, the analysis of the miRNA signature revealed elevated levels of miR-101, miR-126, miR-99a, miR-135, and miR-20—a pattern also observed in megakaryoblastic leukemic cells [31].
Besides genetic and epigenetic modifications, the reprogramming of key signaling pathways in HCSCs plays a pivotal role in shaping their biology and paving the way for therapeutic strategies aimed at exploiting these alterations, particularly in the context of immunotherapy. One of the most extensively studied signaling pathways in leukaemia, particularly in chronic myeloid leukaemia (CML), but also in other cancers, is the nuclear factor-kappa B (NF-κB) pathway [32]. The NF-κB pathway is a key transcriptional regulator in cancer progression, promoting cell growth, survival, metastasis, and chemotherapy resistance [32,33]. It is activated by cytokines and growth factors, which trigger the inhibitor of kappa B kinase (IKK) complex, leading to the phosphorylation and degradation of inhibitor of kappa B-alpha (IκB-α). This releases the p65/p50 complex, allowing it to activate target genes. The non-canonical pathway, involving p52 and NF-kappa B inducing kinase (NIK), also regulates cellular processes such as growth, apoptosis resistance, migration, and angiogenesis [33]. Beyond its involvement in CML bulk cells, NF-κB is involved in LSCs, where it contributes to their survival by promoting the secretion of transforming growth factor beta (TGF-β) and tumor necrosis factor alpha (TNF-α) [34,35]. Among the various pathways that regulate stem cell maintenance, the Hedgehog (HH) signaling pathway definitely plays a critical part. The HH signaling pathway is a tightly regulated network of ligands, receptors, co-regulators, signaling molecules, and transcription factors, which has been found to be deregulated in a variety of hematological cancers, including CML [36], multiple myeloma (MM) [37], chronic lymphocytic leukaemia (CLL) [38], B-cell non-Hodgkin Lymphoma (NHL) [39], AML [40], and T cell acute lymphoblastic leukaemia (T-ALL) [41]. The Hedgehog homologues Indian (IHH), Desert (DHH), and Sonic (SHH) bind to the receptor Patched (Ptc), promoting cell proliferation and survival. While DHH and IHH are deregulated in various tumors, SHH signaling is specifically altered in CML, by downstream β-catenin signaling, and leukaemia progenitor cells, making it a potential target for eliminating CML stem cells while preserving normal HSCs [32,42]. Another major stem cell signaling pathway, able to regulate HCSCs behavior, is the WNT pathway. Normally essential for HSCs homeostasis [43], aberrant WNT signaling in HCSCs promotes self-renewal, therapy resistance, and disease progression [44]. In AML, chromosomal translocations like AML1-ETO activate WNT, enhancing proliferation and adverse outcomes [45]. Similarly, in CML, increased nuclear β-catenin supports HCSCs survival and self-renewal [46], while HCSCs seem to be resistant to tyrosine kinase inhibitors (TKIs) [47]. In ALL, elevated β-catenin and WNT ligands like WNT16 drive proliferation in T-ALL and B-ALL subtypes [44]. These insights highlight WNT as a therapeutic target to eliminate CSCs while sparing normal HSCs. Dysregulation of the Notch signaling pathway has been found to be an additional characteristic for HCSCs, particularly in T-ALL and AML [48]. In T-ALL, activating Notch1 mutations enhance self-renewal and leukaemia-initiating cell (LIC) function, driving clonal expansion and disease progression [49,50]. LICs rely on Notch signaling and interactions with the microenvironment (e.g., DLL1 ligands) for survival and resistance, while Notch inhibition reduces LIC activity and extends survival [51]. In AML, Notch signaling is often inactive in HCSCs (CD34+/CD38−), promoting limited differentiation and increased malignancy [52].
2.1.4 Phenotypic and Metabolic Alterations and Comparison with HSCs
HCSCs share key features with normal HSCs, including the expression of phenotypic marker CD34 and the absence of CD38 and lineage markers such as glycophorin A, CD66, CD59, CD29, CD19, CD16, CD14, CD3 and CD2 [15]. However, their antigen expression varies among different leukaemia types. In AML, HCSCs show altered expression of CD25, CD33, CD44, CD47, CD52, CD90, CD96, CD117, CD123, CD133, TIM-3, and CLL1 [53] which may result from defects in differentiation, as these cells fail to mature. In contrast, in CML HCSCs exhibit changes in CD25, CD26, IL1-RAP [53,54], yet they retain the ability to fully mature. In ALL, CD19−CD10− CD34+ cells have been recognized as long-term proliferating progenitors with the potential to initiate tumor formation. Additionally, other markers such as CD34−Lin+CD38+, CXCR4+, CD34+CD38−CD71−HLA−DR−, CD244, CD34+CD38−CD123+ and TIM-3+ have been associated with leukaemia stem cells, highlighting their diversity and key role in leukaemia progression [55]. In cancer, however, it is worth mentioning that the expression of surface markers can show considerable variability, both between different patients and even within the same patient as the disease progresses [56].
At the same time, researchers have studied HCSCs from an alternative perspective, focusing on their metabolic properties in comparison to normal HCSs. It is known that in eukaryotic cells, the majority of ATP is produced through the Krebs Cycle and oxidative phosphorylation (OXPHOS). However, in HSCs, OXPHOS activity, mitochondrial content, and reactive oxygen species (ROS) remain low [57]. Particularly, HSCs generally prioritize glycolysis as their main energy production pathway, as demonstrated in various studies [58–60]. Interestingly, their dependence on glycolysis seems to vary depending on their functional state: primed HSCs, which are closer to differentiation, rely heavily on glycolysis, while quiescent HSCs remain in a dormant state, display higher mitochondrial membrane potential and increased lysosomal activity, indicating a shift towards mitochondrial metabolism [57].
On the other hand, LSCs exhibit greater mitochondrial mass but reduced spare respiratory capacity for OXPHOS compared to normal HSCs in primary human AML; This reduced spare capacity likely contributes to the increased susceptibility of LSCs to disruptions in OXPHOS activity [61]. CML stem cells, in contrast, demonstrate elevated OXPHOS activity and intensified catabolism of TCA cycle metabolites, which may also contribute to the diminished spare capacity observed in LSCs. Notably, in LSCs from CML patients, there is evidence of substantial expression of genes linked to OXPHOS, higher ROS levels, increased mitochondrial mass, significant reliance on mitochondrial respiration, and enhanced fatty acid metabolism [9,57]. This metabolic signature includes also a reduced mitochondrial transmembrane potential and heightened oxygen consumption, highlighting the centrality of OXPHOS [62].
Importantly, the OXPHOS phenotype is linked to resistance to chemotherapy and targeted therapies in HCSCs. In CML, LSCs resistant to BCR-ABL-targeted therapies have been shown to exhibit an OXPHOS phenotype [63]. The regulation of mitochondrial biogenesis, a critical factor for maintaining OXPHOS activity, is predominantly governed by peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1α) [64]. In LSCs, elevated OXPHOS activity is modulated by the spleen tyrosine kinase and adrenomedullin–calcitonin receptor-like receptor axis [65]. Alongside glycolysis and OXPHOS, lipid metabolism has also emerged as a focal point in CSC metabolism study [66]. Mitochondrial fatty acid oxidation (FAO) plays a pivotal role in sustaining CSC survival and proliferation by alleviating oxidative stress through NADPH production. Furthermore, FAO generates critical metabolic intermediates like acetyl-CoA and NADH, which support ATP generation [67], while LSCs, and normal HSCs rely on FAO to meet their energy requirements [68,69].
2.2 Tumor Microenvironment (TME) in Hematologic Cancers
In hematologic cancers from Hodgkin lymphoma (HL), NHL, and MM to various leukaemias and myelodysplastic syndromes (MDS), malignant clones arise in specific niches, mainly in lymphoid structures or bone marrow (BM). Unlike solid tumors, which are embedded in a dense extracellular matrix, hematologic malignancies rely on a more dynamic microenvironment where a variety of stromal and immune cells, as well as secreted molecules, influence the course of the disease. The bone marrow serves as more than just a structural framework. It is a specialized hub of stromal elements, immune subsets, and regulatory signals, all capable of either constraining or fostering tumor survival and expansion. In this environment, a small but critical subgroup—HCSCs—utilizes these supportive factors, enabling the cells to become multidrug-resistant and perpetuate the disease (even when the bulk tumor is diminished) [70].
Bone Marrow
Hematopoietic stem cells in physiological conditions are typically found in the bone marrow. Once a neoplastic clone emerges, HCSCs can take advantage of this marrow niche by using adhesion pathways and local cytokines. These signals work together to shield cancerous stem cells from being eliminated by the immune system or medical treatments [71]. This protective effect is partly mediated by interactions with myeloid-derived suppressor cells (MDSCs) which can be monocytic or granulocytic and exude molecules such as arginase-1, inducible nitric oxide synthase (iNOS), and ROS, all of which sap T cell or NK cell effectiveness [72].
Additionally, regulatory T cells (Tregs) can become a factor to consider by producing interleukin-10 (IL-10) (that suppresses the adoptive immune response and antigen presentation and enhances cancer growth, immune evasion, and MDSC activity) and TGF-β, with similar effects to IL-10 on immunity, and therefore weakening cytotoxic responses, essentially providing a protective microenvironment for HCSCs. Tregs are more frequent in B-cell NHL, such as follicular lymphoma (FL) or diffuse large B-cell lymphoma (DLBCL). Importantly, mesenchymal stem cells (MSCs) and Stromal cells secrete C-X-C motif chemokine ligand 12 (CXCL12) (CXCR4 ligand), as well as more IL-6 and vascular endothelial growth factor (VEGF). These actions facilitate plasma cells (PCs) homing into BM. The reciprocal interactions between PCs and MSCs drive MM progression and favor the accumulation of chemo resistant malignant PCs. Moreover, by producing adhesion cues (e.g., vascular cell adhesion molecule-1 (VCAM-1) linking up with VLA-4), they give malignant stem cells a resilient scaffold and keep them from undergoing apoptosis [73,74]. As illustrated in Fig. 1, immune cells within the TME can either suppress or promote tumorigenesis, depending on the context and cancer type.
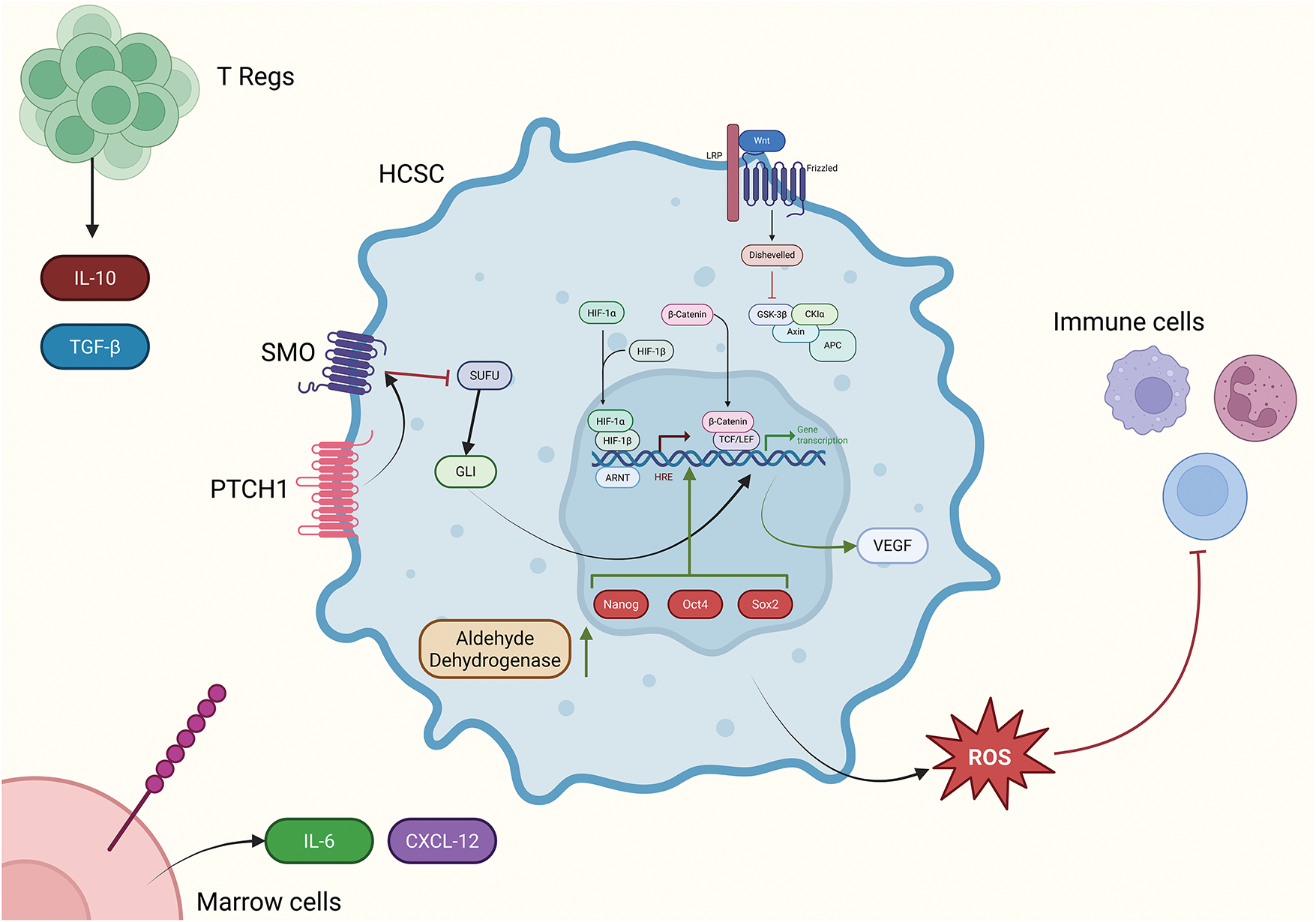
Figure 1: Illustration of key intracellular pathways including Wnt (top right), HIF-1, and Hedgehog that assist HCSCs to survive and thrive in the microenvironment. Those pathways, along with the upregulation of Nanog, Oct4 and Sox2 embryonic transcription factors lead to the transcription/upregulation of key genes related to this phenomenon. In combination with that, the enrichment of the microenvironment with IL-10 and TGF-β by Tregs, and IL-6 and CXCL-12 by bone marrow cells that also provide anchors for HCSCs to latch on withing the bone marrow, assist the viability and immune evasion of the HCSCs, which also dampen the immune response by excreting ROS and other reactive species. The image was created using the platform biorender.com (accessed on 24 June 2025)
Lymphoid Organs
The surrounding environment, known as the TME, plays a significant role in the pathogenesis of HL. It shapes the interactions between immune cells and malignant cells, thereby influencing disease progression. In organs of great importance to the lifecycle of blood cells, including the spleen and lymph nodes, malignant cells exploit their environment to facilitate their growth and survival. In certain regions, immune system components and diverse biochemical signals collaborate to facilitate the survival and proliferation of malignant cells. For instance, while T cells—key players in the immune response—are commonly found in HL tumors, they often fail to attack the cancer effectively. This is because Hodgkin Reed Sternberg (HRS) cells, the malignant cells in HL, release substances like IL-10 and TGF-β. These molecules weaken T cells, redirecting them into roles that support the tumor, such as turning to Tregs. At the same time, the more aggressive, cancer-fighting T cells become too exhausted to function effectively. One key driver of this dysfunction is the overproduction of PD-L1 by HRS cells. PD-L1 binds to PD-1 receptors on T cells, essentially turning them off and worsening their exhaustion. HRS cells also release IL-10, which prompts nearby T cells to produce CD40L—a molecule that strengthens interactions between T cells and cancer cells, further promoting tumor growth [75].
The tumor growth cycle is heavily reliant on a feedback loop involving the protein interferon regulatory factor 4 (IRF4), which is crucial for cellular survival and proliferation. In HL, interactions with CD40L elevate IRF4 levels, equipping malignant cells with the requisite energy and resources for prolonged proliferation. This equilibrium is tenuous—when growth-inhibiting signals are triggered, IRF4 levels diminish, illustrating the necessity for tumors to meticulously manage their own proliferation. Comparable patterns are observed in other lymphomas, such as DLBCL. Alterations in the IL-10 receptor genes can confer a survival advantage to cancer cells, facilitating their growth more efficiently [76].
These immunosuppressive microenvironments serve as sanctuaries for HCSCs, catering to their survival during therapies and permitting them to remain dormant until cancer recurrence. These stem-like cells remain dormant, poised to reactivate the illness long after the primary tumor has been diminished via treatment. This underscores the critical need to therapeutically target and dismantle these protective niches to prevent cancer recurrence.
2.2.2 Signaling and Interactions Leading to HCSC Proliferation
HCSCs are sustained by a network of intrinsic signaling pathways and extrinsic interactions that collectively promote their self-renewal, proliferation, and drug resistance. The Notch signaling pathway operates as a regulatory mechanism, transmitting essential directives that enable HCSCs to persist. Activating this route enhances the resistance of HCSCs to therapies and improves their capacity for self-renewal and proliferation [48]. The Hedgehog signaling pathway offers critical regulatory signals that improve HCSC defense against chemotherapeutic drugs. Especially beneficial in multiple myeloma, it facilitates immune evasion and imparts chemoresistance [77]. β-Catenin is essential in modulating gene expression that promotes HCSC survival and confers resistance to standard therapy. This renders HCSCs very robust, allowing their persistence despite therapeutic efforts [78]. Bruton’s tyrosine kinase (BTK) signaling functions as a contingency mechanism for HCSCs, enabling them to evade the apoptosis generally induced by therapies. This indicates that even when treatment attempts to eradicate them, HCSCs persist and remain safeguarded [79]. The detoxification of chemotherapeutic drugs and the preservation of stemness characteristics in HCSCs are facilitated by aldehyde dehydrogenase 1 (ALDH1) and retinoic acid receptor alpha 2 (RARα2). These factors promote ongoing self-renewal and provide resistance to apoptosis [80]. MDSCs decrease T cell function, hence suppressing anticancer immunological responses and enabling HCSCs to avoid immune monitoring. The immunosuppressive milieu impedes the effectiveness of treatment approaches aimed at HCSCs [81]. HCSC survival is facilitated by the establishment of an immunosuppressive environment by Tregs, which reduce immunological activation. This inhibition reduces immune-mediated elimination, thereby promoting the proliferation of HCSCs [82]. Tumor-associated macrophages (TAMs) facilitate tumor advancement by altering immune responses and aiding in the maintenance of HSCs. They create an immunosuppressive environment that promotes HCSC survival and proliferation [83]. Stromal cells offer structural and metabolic support that promotes the survival and multiplication of HSCs. They secrete factors and extracellular matrix components that support HCSC survival and proliferation.
2.2.3 Microenvironmental Conditions
HCSCs are directed by significant internal signals, although their activity is profoundly affected by their microenvironment in the bone marrow or lymphoid organs. In malignancies such as HL, NHL, MM, leukaemias, and myelodysplastic syndromes (MDS), the microenvironment engenders four primary traits that collectively enable HCSCs to withstand therapies and endure for extended periods. Cancer cell proliferation in marrow or lymphoid locations results in areas of reduced oxygen levels. Stabilized hypoxia inducible factor-1 alpha (HIF-1α) enhances VEGF production, facilitating the formation of new, often irregular blood vessels. In multiple myeloma, extremely hypoxic bone lesions enhance the Hedgehog or Wnt pathways, hence reinforcing the “stem-like” population. Lymphoma cells in nodal “hotspots” utilize moderate hypoxia to upregulate pro-survival genes [84]. Even minor increases in MDSCs might disrupt the immunological equilibrium, favoring HCSCs and inhibiting T and NK cytotoxic activities through iNOS, arginase-1, and ROS. Tregs provide a low-inflammation environment by secreting IL-10 and TGF-β, allowing the hematopoietic stem population to flourish. This synergy is prevalent in advanced HL and NHL, as well as in acute and chronic leukaemias [85]. Specialized cells inside the bone marrow stroma provide cytokines (e.g., IL-6, CXCL12) and adhesion ligands such as VCAM-1, creating a protective interface with hematopoietic stem cells. Adhesion molecules, including the likes of VLA-4 and VCAM-1, facilitate the anchoring of malignant stem cells inside the marrow or lymphoid compartment. This anchoring establishes a “safe harbor” effect and reduces exposure to detrimental chemotherapeutic concentrations. Certain HCSCs have elevated aldehyde dehydrogenase activity, facilitating the breakdown of harmful chemotherapeutic metabolites and imparting drug tolerance. Under stress, embryonic transcription factors such as Nanog, Oct4, and Sox2 may be increased. In conjunction with RARα2 signals, these substances can direct malignant clones towards a quasi-stem cell state, hence promoting prolonged self-renewal [86]. In total, these environmental factors—hypoxia, immunosuppressive cellular populations, protective stroma, and metabolic adaptability—establish a niche that enables HCSCs to persist despite pharmacological treatment. Understanding and treating these macro-level circumstances is critical for obtaining permanent remissions in hematologic malignancies, as even modest clusters of these “stem-like” cells can reactivate the disease.
2.3 HCCs Properties Conferring to Therapy Resistance
Therapeutic resistance remains one of the major hindrances in cancer therapy. Almost all surgical, in the case of solid tumors, and treatment interventions, like chemotherapy, immunotherapy, and irradiation-have failed at some point to affect the metabolism or growth of these cancers, due to the intrinsic or acquired mechanisms in relation to the treatment. As can be seen in Fig. 2, the mechanism of the development of resistance is based on the interplay of genetic, epigenetic, metabolic, and microenvironmental factors that help HCCs survive.
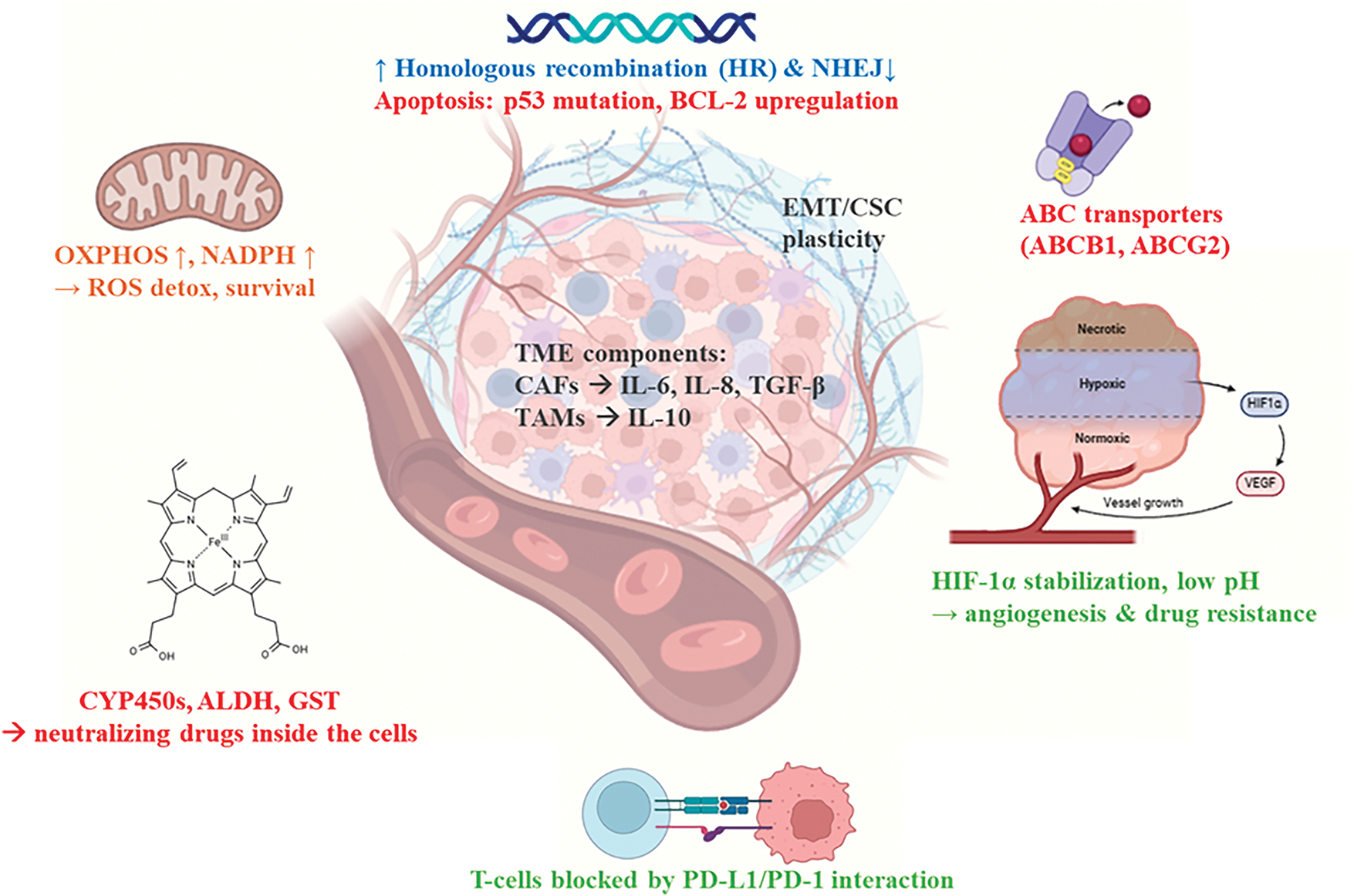
Figure 2: Mechanisms of tumor cell therapy resistance. Therapy resistance in tumors arises from a convergence of genetic, epigenetic, metabolic, and microenvironmental adaptations. Tumor cells enhance DNA repair via upregulated homologous recombination (HR) and downregulated non-homologous end joining (NHEJ), while evading apoptosis through p53 mutations and BCL-2 overexpression. Phenotypic plasticity, including epithelial-mesenchymal transition (EMT) and acquisition of cancer stem cell (CSC) traits, facilitates survival under therapeutic pressure and promotes DTP states. Drug efflux is mediated by overexpression of ABC transporters (ABCB1, ABCG2), while detoxification enzymes (e.g., ALDH, CYP450) inactivate chemotherapeutics. Metabolic reprogramming, characterized by elevated oxidative phosphorylation (OXPHOS) and NADPH production, supports redox balance and resistance. The TME—comprising cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), and hypoxic niches—contributes to resistance through secretion of IL-6, IL-8, TGF-β, IL-10, and stabilization of HIF-1α. Immune evasion is reinforced by PD-L1/PD-1-mediated suppression of cytotoxic T cell activity. The image was created using the platform biorender.com (accessed on 23 June 2025)
2.3.1 Genetic and Epigenetic Resistance
Mutations and chromosomal aberrations are key intrinsic genetic factors involved in inducing resistance. This includes point mutations, copy number alterations, and chromosomal translocations involving oncogenes, tumor suppressors, and machinery for DNA repair pathways [87]. Driver mutations in genes such as epidermal growth factor receptor (EGFR) and oncogenes BRAF or KRAS induce a carcinogenic state and confer resistance to malignant cells against kinase inhibitors. Genomic instability promotes heterogeneity inside the tumor and further leads to the development of subclones with mutations that confer resistance. For example, in melanoma, a subpopulation of CD271 cells has been identified as having tumor-initiating capacity, showing how diverse subclones contribute to tumor propagation and escape from immune surveillance [88].
In addition to genetic mutations, histone modifications and methylation of DNA are important for gene expression. These changes could induce epithelial-mesenchymal transition (EMT), which is related to invasiveness and drug resistance, activate drug resistance genes, or silence genes that inhibit tumors. Long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) are examples of the gene regulation from the non-coding RNAs in altering resistance and survival gene networks [89].
2.3.2 Cellular Plasticity and HCSCs
Tumor cell plasticity-or the capability of cancer cells to change phenotypically in response to external stimuli or therapeutic pressure-is one of the most dynamic mechanisms of resistance. It encompasses such phenomena as EMT, transdifferentiation, mesenchymal-epithelial transition (MET), and acquisition of the characteristics of HCSCs [90]. Plastic tumor cells are reversible in entering a drug-tolerant persister (DTP) state characterized by dormancy, low proliferation, and survival under drug treatment. For example, acquisition of mesenchymal characteristics such as invasiveness and motility, along with resistance to apoptosis, could be facilitated by EMT among epithelial cells. Signaling pathways like TGF-β, WNT, and Notch regulate transcription factors such as Snail, Slug, Twist, and Zeb1/2 which orchestrate EMT. Partially EMT cells therefore have a hybrid phenotype that strikes a compromise between adaptation and survival, increasing their resistance to therapy [91]. Moreover, it is noted that much of therapeutic resistance, metastasis, and recurrence can be directly related to the formation of HCSCs; the subpopulation within a malignancy able to self-renew and repopulate it. HCSCs stay inactive and resistant to therapies aiming at fast-dividing cells. High efflux pumps of drugs and efficient DNA repair mechanisms also characterize these cells [89].
2.3.3 Microenvironmental Influences and Immune Evasion
Cancer cell activity and adaptive resistance are largely determined by the TME. The TME encompasses stromal cells—such as cancer associated fibroblasts (CAFs)—blood vessels, extracellular matrix, immunological cells (T lymphocytes, TAMs), and an assortment of cytokines and chemokines. These supportive cells also act to protect cancer cells from therapeutic interventions [92,93].
CAFs secrete factors such as IL-6, IL-8, TGF-β, TGF-α, and TGF-β, enhancing the stemness of malignant cells, promoting EMT, and inhibiting immune reactions. In other words, CAFs can enhance resistance and promote the survival of HCCs by inducing the NF-κB and signal transducer and activator of transcription 3 (STAT3) activation pathways. This escape from immune surveillance is largely dependent on the secretion of anti-inflammatory cytokines associated with the M2-like phenotype in TAMs [93]. The downregulation of major histocompatibility complex I (MHC-I), attenuated antigen presentation, and the upregulation of different immunological checkpoints are all aspects that render the immunosuppressive nature of the TME. These phenomena promote immune tolerance and diminish T cell recognition [94]. Besides this, hypoxia and low pH within the TME also promote angiogenesis, invasiveness, and altered drug penetration [95].
2.3.4 Metabolic Reprogramming and Plasticity
In order to proliferate and adapt to nutrient- and oxygen-deprived environments of the TME, cancer cells undergo metabolic reprogramming. On one hand, this adaptation enhances survival, and, on the other, it leads to treatment resistance. Through the Warburg effect or aerobic glycolysis, cancer cells can fast-track the generation of metabolic intermediates, even in the presence of oxygen. However, in drug-resistant cells, OXPHOS is often favored over glycolysis.
Notably, doxorubicin treatment of glioblastoma causes poly(morpho)nuclear giant cells (PGCs) to emerge with a distinct metabolic signature. These PGCs help survive under stress via activating pathways for scavenging ROS, producing NADPH, and OXPHOS. Inhibition of either OXPHOS or NADPH-producing pathways interrupts the function of PGCs, consequently sensitizing tumors to chemotherapy. It is this metabolic flexibility—or metabolic plasticity—that allows cancer cells to switch between modes of energy production and resist drug-induced stress. This flexibility is further supported by metabolic crosstalk among stromal and tumor cells [96].
2.3.5 Drug Efflux, Detoxification, and Sequestration
In addition, one classical pathway of resistance is the reduction in intracellular drug concentration. Cancer cells can increase the activity of efflux pumps such as ABCB1 (P-glycoprotein), ABCG2, and ABCC1, to vigorously remove their therapeutic agents from the cell, while simultaneously decreasing the intake of drug transporters [97]. These transporters are frequently overexpressed in CSCs, and most likely in HCSCs, and differentiated tumor cells, thereby contributing to the establishment of the multidrug resistance (MDR) phenotype. Tumors may also inactivate drugs intracellularly by the overexpression of mechanisms of detoxification, such as cytochrome P450s, glutathione S-transferase, and aldehyde dehydrogenases (ALDH) [98]. The cytotoxic efficacy of many conventional treatments is compromised once resistant subclones show a significantly high expression of these enzymes. Furthermore, drug sequestration in intracellular storage compartments, along with impaired activation of prodrugs—such as the loss of deoxycytidine kinase (DCK) in cytarabine resistane—are among the leading factors for chemotherapy failure [99].
2.3.6 Enhanced DNA Repair and Apoptosis Resistance
Most of cancer treatments act by inducing apoptosis and causing DNA damage. However, resistant cells usually tend to employ superior DNA repair mechanisms along with evasion from programmed cell death. In response to DNA damage caused by cancer therapies like radiation and chemotherapy repair via upregulating the pathways of non-homologous end joining (NHEJ) and homologous recombination (HR) repair [100]. Beyond enhanced DNA repair, survival favors in the same way by mutations of the tumor suppressor gene, p53, overexpression of anti-apoptotic proteins like BCL-2, and inhibiting pro-apoptotic signaling pathways. This resistance is further intensified by the close interplay between these evasion strategies and the characteristics of CSC and EMT phenotypes [101].
2.3.7 Strategies to Overcome Resistance
Integrated therapeutic strategies targeting multiple pathways are essential to overcome therapy resistance. One such promising strategy of combination therapy would include the coupling of drug TKIs to alternate bypass pathway inhibitors against multiple resistance mechanisms [102]. Moreover, blocking immunological checkpoints or reprogramming the TME [92], inhibiting OXPHOS in resistant cancers [98], and focusing on EMT or CSC pathways. Another emerging approach involves the use of nanoparticles to deliver siRNAs to silence resistance genes or circumvent efflux pumps would be an optimistic approach. Personalized medicine will be critical in personalizing treatments and improving patient outcomes through pharmacogenomic profiling and markers of resistance [89].
3 Immunotherapeutic Strategies
Given the importance of the role that HCSCs play in the proliferation, therapy resistance, and, most importantly, in the relapse of the malignancy [103], identifying the exact ways that HCSCs can be addressed via immunotherapeutic means is crucial in applying more efficient and effective therapies in the clinic. There are several targets related to HCSCs already identified and put into use in preclinical and clinical settings under the context of immunotherapy. It can be said that one of the most direct immunotherapeutic strategies is the targeting of cytokine-related pathways. Such interventions include the micromolecular inhibition of CXCR1/2 which is a receptor of IL-8 and the inhibition of the pathway hosted by IL-6 [12]. However, one of the greatest challenges in this endeavor is the identification of markers characteristic to HCSCs, to minimize the off-target effects and, thereafter the adverse events and maximize treatment outcomes. Common targets include the ones displayed on the surface of the cells by themselves, such as the ones previously mentioned, or via human leukocyte antigen (HLA) proteins [104], with an example being the PR1 epitope, although they are usually downregulated in the HCSCs, to escape immune surveillance. In the following paragraphs, exploitable targets, as well as therapeutic interventions, and future directions will be analyzed. It is important to note, as noticed later in the text, and literature in general, that the most effective way to prevent relapses by eliminating as many HCSCs as possible is the targeting of multiple markers simultaneously, mostly due to the plasticity of the targets, as a mechanism of resistance [105].
Hematopoietic stem cell transplantation (HSCT) is an intervention used for several disorders of the blood, including malignant and non-malignant ones. Its therapeutic utilization in haematologic cancer therapy is based upon the graft vs. tumor (GVT) effect, which is enhanced by conditioning regimens that weakens the HCCs and generates an inflammatory environment [106]. Both autologous and, especially, allogeneic [107,108], stem cell transplantation have proven themselves useful for the long-term improvement of the health and quality of life of hematological cancer patients [109,110]. These outcomes are further supported by recent advancements in donor–recipient matching, conditioning protocols, cell engineering, and patient eligibility assessment [111,112].
Sources of stem cells include the bone marrow, peripheral blood and the umbilical cord [106]. To minimize the risk of graft vs. host disease (GVHD), the patient’s cells are preferable to be used. In this context, induced pluripotent stem cells, originating from differentiated tissue cells that are undergone reprogramming via viral infection with alterations with transformations involving the SOX2, MYC, OCT4, and KLF4 genes, or very small embryonic-like stem cells (VSELSCs) originating from progenitor gametes and transformed in vitro into HSCs, can be utilized [113]. It’s important to consider that one of the major factors behind clinical results of transplantation is also the type of malignancy as different response rates and other metrics are expected for different diseases [114]. Notably, MSCs seem to be quite relevant in the context of HCSC elimination as they have demonstrated enhanced homing and interaction with HCSCs, along with regulation of metastasis and treatment resistance, and the ability to reconstruct the patient’s hematopoietic capabilities after the damages caused by the malignancy and the treatment [115].
Despite its track record, HSCT faces several challenges in clinical implementation, especially in cancer care, including GVHD which is related to the rejection of the graft by the patient’s immune system. Within the main factors to be blamed are listed the activity of T cells of the host and alloreactive T cells of the graft, as well as the chance of relapse of the malignancy resulted by the successful fightback of the graft by the specific form of GVT, graft vs. leukemia (GVL). GVHD and GVL have been proven to be independent from one another. To combat the former, immunosuppressive therapy, such as anti-T cell therapy and the more targeted depletion of alloreactive T cells can be utilized whilst in the latter, sufficient clearage of the malignancy is required to ensure the minimal risk of relapses. However, there is a fine balance between the two approaches, and their effects, say for immunosuppression and immune induction, that needs to be achieved. Additional methods to overcome maximize efficacy include the HLA matching between the donor and the recipient of the graft, the use of cord blood that allows for greater freedom in HLA matching and targeted therapies that minimize the effects on physiological cells whilst addressing the malignant ones [116].
Given that stem cells are considered to underline the characteristic onset, aggressiveness and treatment resistance of many malignancies [117], as well their capability to initiate relapse [104], they have set themselves as one of the prime targets in hematological cancer therapy, particularly in the context of stem cell transplantation. Hematopoietic stem cell transplantation (HSCT) is considered an intervention that directly targets hematological cancer stem cells (HCSCs), thereby addressing some of the limitations associated with conventional therapies such as chemotherapy [118]. However, transplantation is best combined with conditioning, maintenance, and salvage therapies as minimal residual therapy is a major driver of relapse and an indicator of progression [119]. An example of maintenance therapy addressing residual HCSCs is the use of TKIs such as multi-kinase inhibitor Sorafenib [120] and Hedgehog signaling inhibitor Glasdegib [121]. To ensure the minimization of the risk of relapse, proper measures, such as flow cytometry and other immunochemical techniques as well as other methods based on the different characteristics of HCSCs, need to be taken to avoid any contamination of the autograft in autologous HSCT, as new tumor sited might evolve [122–125].
3.2 Antibody-Based Interventions
Antibody-based medicinal products are widely applied in the field of oncology. There are several ways they can be classified based on their structure, their targets and other properties. For the purposes of this study, relevant antibody-based products are addressed based on the targeted molecule. Similar clarification is followed in the summarizing Table 1 and Fig. 3.
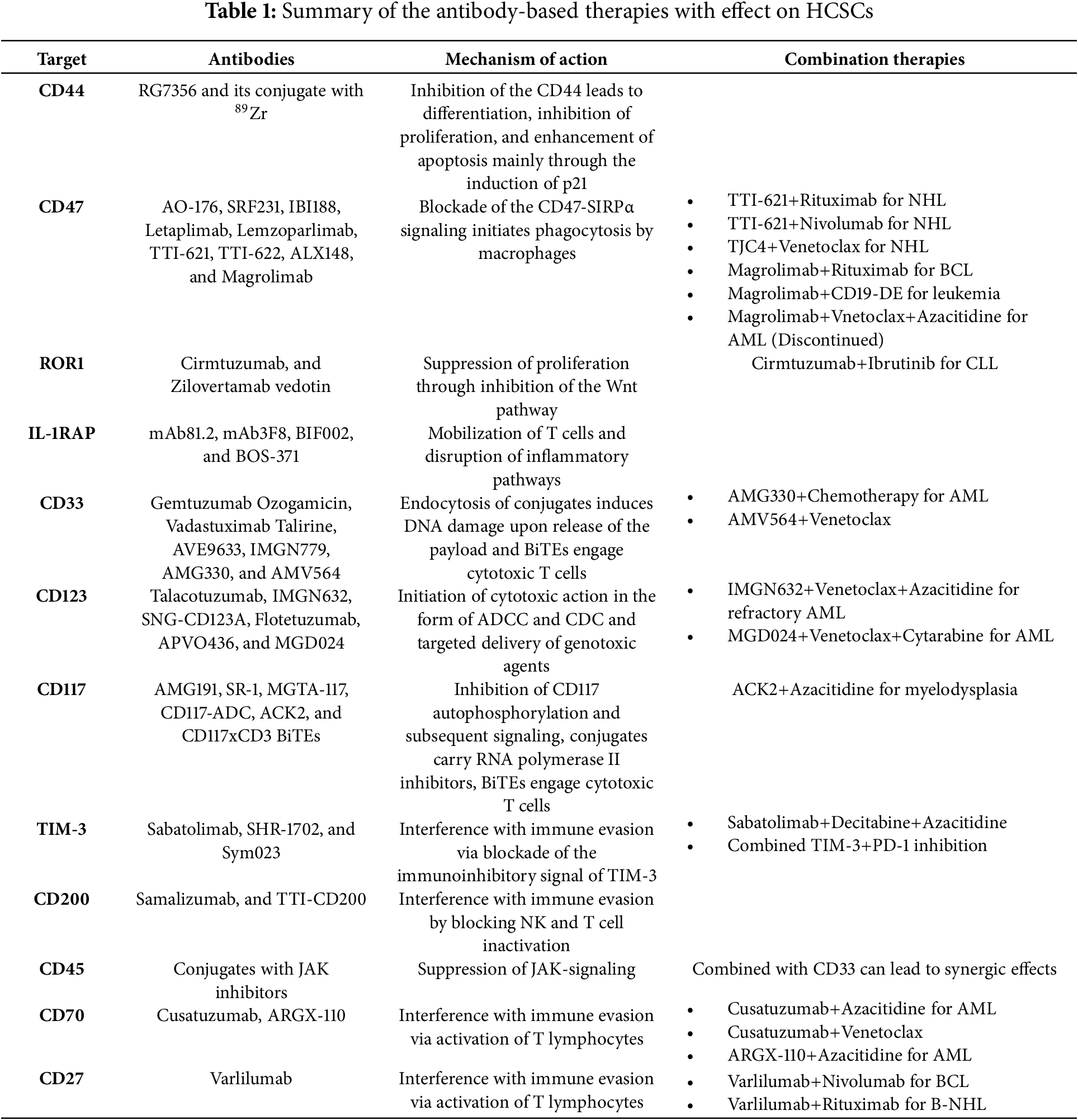
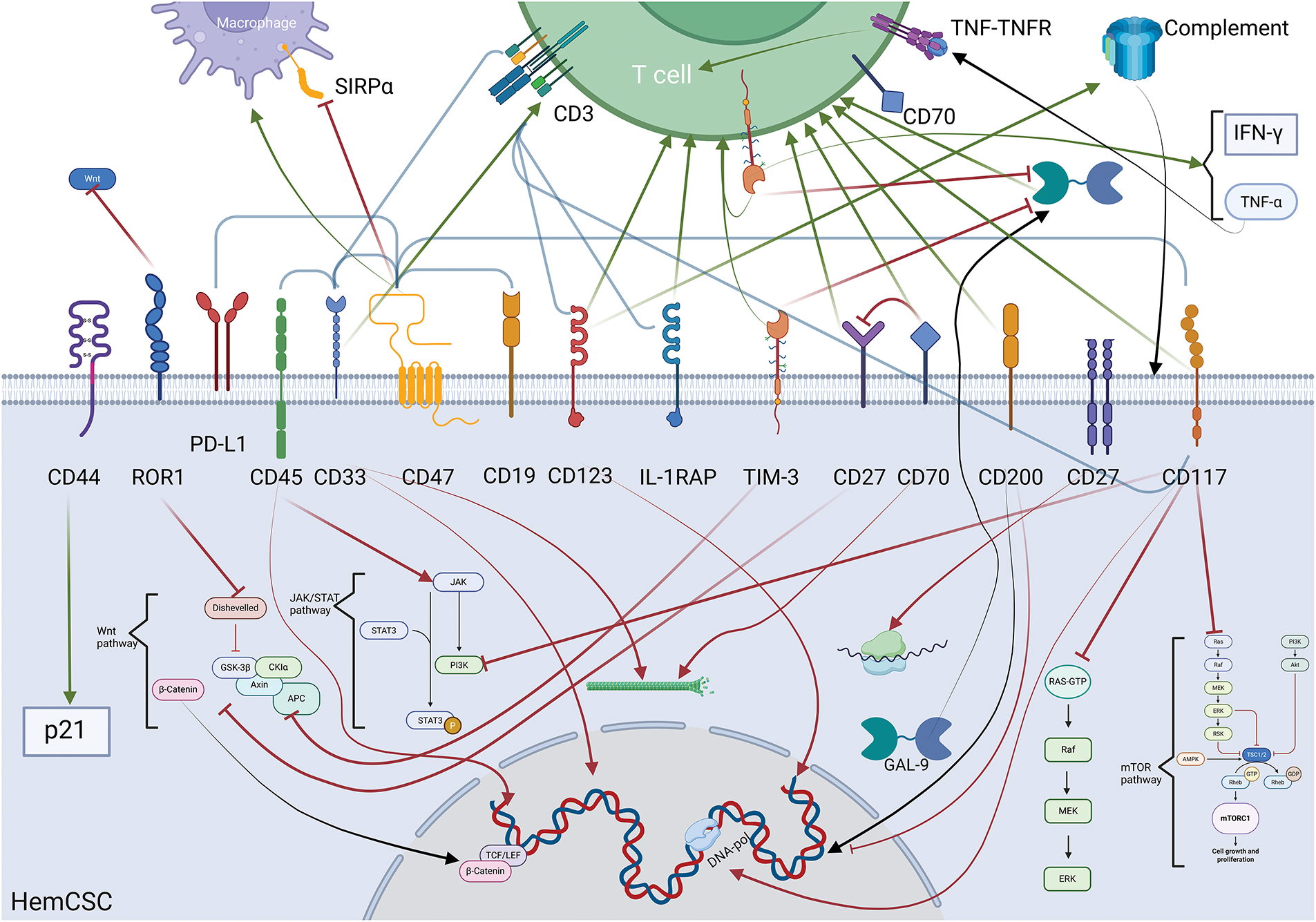
Figure 3: Illustration concentrating and summarizing all biochemical mechanisms and the intermolecular relationships initiated by the inhibition of the corresponding molecules. The bottom half of the image represents the intracellular space of a HCSC and the top one the extracellular space. The green arrows represent activation/upregulation whilst the red ones represent inhibition (flat tip for inhibition stemming from the targeting of the molecule, pointy tip for action associated with the delivery of the payload of a conjugate). The black arrows represent movement of biomolecules within the cell leading to gene transcription when terminating in the nucleus, extracellularization in the case of GAL-9, or assembly in the case of the complementary. The brackets indicate the existence of a bispecific targeting antibody for the molecules at each end. In the extracellular space, a T cell represents the effects of the interventions on the adaptive immune system. The image was created using the platform biorender.com (accessed on 07 April 2025)
Given CD44’s importance in several processes sustaining HCSCs, the antileukemic action of anti-CD44 mAbs appears as no surprise. These inhibitors mostly affect hinder HCSC’s stemness and proliferation [126], most likely due to the imminent cell cycle arrest in the G0/G1 through the enhancement of p21 expression, and of apoptotic capabilities [127]. Targeting CD44 can also be done with conjugated antibodies. The recombinant antibody RG7356 has shown positive safety and tolerability indications after a phase 1 study involving dosages ≤2400 mg every other week or ≤1200 mg weekly or twice weekly [128]. Additionally, the conjugated form of this antibody with the radioisotope 89Zr has been tested only for solid tumors on animal models [129]. Results show it has shown selective cytotoxicity on leukemic cells, specifically B-cell ones, whilst in an early stage in its development [130], highlighting the potential for clinical applications. However, despite all the hopes, the fact that CD44 can also be found in significant levels on CD8+ Natural Killer (NK) cells is a major concern regarding the toxicity and adverse effects that accompany the use of such inhibitors [131].
The presence of CD47, also known as “the don’t eat me signal”, or macrophage checkpoint inhibitor due to its signal regulatory protein alpha (SIRPα) interaction-initiated activity, on the surface of multiple of chemoresistant hematological malignancies [132–134] is a marker of unfavorable prognosis [135]. Hence, it is reasonable to be assumed as a viable target for engineered antibodies, especially in the context of allogenic stem cell transplant [136].
Early Evidence on the Activity of Anti-CD74 Antibodies
In vitro testing of AO-176 has demonstrated effectiveness against multiple myeloma, T-acute lymphocytic leukemia and B lymphoma cell lines that were also added in mice as xenografts and SRF231 in MM, DLBCL, and Burkitt’s Lymphoma [137]. Similarly, in vitro and in vivo testing of IBI188, a biosimilar to Rituximab, an anti-CD20 antibody, indicated that its use can induce phagocytic activity through the blockade of the CD47-SIRP-α pathway in CHO-hCD47 (CD47 expressing Chinese Hamster Ovary cells used in translational studies for their robust growth, genetic modification), Raji (used to simulate Burkitt’s lymphoma cell line), and MDA-MB-231 (representing breast cancer) cell lines, along with inflammation, and activity of macrophages in Raji cells and in vivo conditions and that its activity further enhanced when combined with anti-VEGF antibodies [138]. Additional monoclonal antibodies (mAbs) that have shown promising results and tolerable toxicity, mostly comprising of grade I and II adverse events, in early clinical trials are Letaplimab and Lemzoparlimab, as well as the fusion products of SIRPα, as an IgV domain, and an Fc domain, to facilitate phagocytic function, TTI-621, TTI-622, and ALX148, with the latest showing exceptional affinity to CD47 and elevated pharmacokinetic properties by utilizing neonatal Fc receptors.
Combination Therapies and CD47 Targeting
Clinical trials have evaluated the efficacy of TTI-621 in combination with rituximab or nivolumab against NHL with encouraging results as overall response rate (ORR) was 23% with rituximab whilst for HL the rate was 50% with nivolumab [139]. Synergistic effects on phagocytosis were also pbserved with the combination of anti-CD47 antibody TJC4 and Bcl2 inhibitor Venetoclax in B-cell NHL. This synergy was attributed to venetoclax-induced extracellular exposure of phosphatidylserine, a known promoter of phagocytosis, derived from the latter’s pharmacological activity. The experiments suggesting this effect were carried out by Li et al. on OCI-LY-8 (representing human B-cell lymphoma), SU-DHL-6 and SU-DHL-16 (representing large cell lymphoma), U2932 (representing B cell lymphoma), and Raji cell lines as well as on NOD-SCID mice bearing malignant cells derived from cell lines or patient biopsies [137].
Combination therapies targeting additional immune checkpoints have also been explored. For example, Rituximab in combination with Magrolimab in diffuse large B-cell lymphoma achieved a 50% objective response rate, with most adverse events being grade 1 or grade 2 in severity [140]. Magrolimab was also used in tandem with the CD19-DE antibody, tailor-made for enhanced phagocytosis with the result of increasing the latter’s activity, when compared to anti-CD19 monotherapy, when tested in vivo for both adult and pediatric leukemias [141]. CD47 targeting antibody Magrilomab had showcased in its early development the increased efficacy of combination therapies with CD47 inhibitors and molecules such as azacitidine compared to monotherapies using just the latter [142]. However, despite the promising results in early clinical trials showcasing tolerability and efficacy in patients with myelodysplastic syndrome [143] and AML [144], the Phase III (ENHANCE-3) trial testing Magrolimab with Venetoclax and Azacitidine in AML patients [145] had to be early discontinued upon intervention from the United States Food & Drug Administration based on the “demonstrated futility and increased risk of death” [146] arising concerns around the targeting of CD47 and related combination therapies. Similar concerns were also in the spotlight after the poor performance indicators of CC-90002 were published [137].
Bispecific Antibodies Targeting CD47
There have already been clinical trials carried out testing the bispecificTG-1801 antibody targeting CD47 and CD19 along with Rituximab against B-cell lymphoma with positive results regarding efficacy, as there was clinical activity noted, and limited toxicity as there were noted anticipated adverse events including anemia, headache, abdominal pain, fatigue and thrombocytopenia, however, with limited statistical power due to relatively low enrolment [147]. Furthermore, there are TG-1801 (CD47xCD19), IMM0306, HuNb1-Rituximab and RTX-CD47 (CD47xCD20), HMBD004 (CD47xCD33) and IBI322 (CD47xPD-L1) bispecific antibodies under testing at several stages in their development cycle [137].
Challenges Related to C47 Targeting
Nonetheless, there are several issues regarding the targeting of CD47 with biomolecules. On-target toxicity is the primary concern as said antibodies also target physiological cells displaying CD47 with effects such as anemia, mostly caused by the activity of the Fc domain. Additionally, SIRPα monomers that act as agonists and SIRPα antibodies can only be used as adjuvant therapies at best, with the latter being coupled with the concern of targeting neural cells as well [148]. HCCs can also promote resistance mechanisms against CD47 targeted therapy as they can enhance the production of CD47 and/or other antiphagocytic molecules [149]. Besides the shortcomings, there is reason to hope that CD47 can prove itself as one of the most significant immune checkpoint targets once the issues of therapeutic effectiveness and toxicity have been overcome and more robust data have been collected [150].
ROR1 is a tyrosine kinase, located almost exclusively on CSCs, including HCSCs, whose ligand is Wnt5a. Its proliferative and pro-metastatic role has made it a prime target for mAbs such as Cirmtuzumab which has been used against CLL. Early-phase clinical trials revealed that Cirmtuzumab could limit cancer progression with relative safety, with an elevated possibility of stemness return. Despite this discouraging result, preclinical studies assessing its combination with tyrosine kinase inhibitor Ibrutinib had shown better results [133]. Additionally, the antibody-drug conjugate Zilovertamab vedotin that targets ROR1, delivers the small molecule auristatin E and is assessed for mantle cell lymphoma (MCL), DLBCL, and CLL with positive indications, in a small pool of subjects [151]. ROR1 represents an interesting yet untapped full potential target as there are still monoclonal as well as bispecific antibodies in development [152–154].
IL-1RAP is a facilitator in the transduction of the pro-inflammatory signal of the interleukins IL-1α&β and is widely expressed in and serves as a marker for HCSCs, unlike physiological blood cells [155]. The majority of anti-IL-1RAP antibodies used against hematological malignancies have been only assessed preclinically. Several examples include the testing of polyclonal rabbit-originated antibodies against aggressive and TKI resistant CML cells. Results on AML cells originating from patients were also susceptible to the action of the mAb81.2 & mAb3F8 antibodies [156]. Zhang et al. also tested IL-1RAPxCD3 Bispecific T Cell Engager (BiTE) antibodies BIF002 and concluded that in both in vitro and in vivo models they led to T cell activation and proliferation with their activity being mostly directed against HCSCs [157]. West et al. have also tested BOS-371 humanized mAb and noted high affinity to its target and strong activity against leukemic cells, both in vitro and in vivo settings as there was a decreased burden [158].
From a clinical perspective, CD33 appears to be a very appealing target as its levels can serve as a prognosis in AML, as it has been negatively correlated with overall survival [159]. Additionally, it is stated that CD33 is expressed in normally differentiated hematologic cells, leukemic cells and HCSCs, but not on normal hematologic stem cells, thus, to an extent, sparing the patient’s capacity to regain functional hematopoiesis post treatment [160].
Gemtuzumab Ozogamicin
Gemtuzumab Ozogamicin is an antibody-drug conjugate that had been granted accelerated approval by the United States Food and Drugs Administration (FDA) at the beginning of the 21st century. It had to re-enter the market in 2017 after a brief ban due to prior concerns over safety and efficacy, with the latter being partially due to limited conjugation between the antibody and calicheamicin hydrazide as the former does not have a significant antineoplastic effect. After reviewing its characteristics, it was deemed applicable in the therapy of AML patients. The mechanism of action of Gemtuzumab Ozogamicin includes internalization of the conjugate upon binding with the CD33 molecule and, upon release of calicheamicin hydrazide within the lysosome, the interaction of the small molecule with the genetic material causing cell cycle arrest and/or cell death by apoptosis. Resistance can be developed by the function of members of the ABC transporter family, P-glycoprotein, and other drug transporters as well as induction of anti-apoptotic mechanisms. Notable adverse effects of its use include hepatic veno-occlusive disease and sinusoidal obstructive syndrome along with other significant hematological adverse effects and minor ones rendering its use demanding careful and extensive oversight [161,162]. Thereafter, there is a clear demand for the development of additional therapies.
Other Anti-CD33 Conjugates and Design Optimization
Regarding the design, it has been noted by Godwin et al. that proximal, as to the membrane, targeting of CD33 can deliver greater cytotoxic effects such as more effective T cell engagement. As such, targeting the C2 domain of the molecule, a proximal target present at all CD33 molecules occurring in leukemic cells, is a promising method [163]. Additional conjugates include Vadastuximab Talirine carrying pyrrolobenzodiazepine, DNA cross-linkage causing factor, AVE9633, carrying a derivative of Maytansine that inhibits tubulin, and IMGN779, a carrier of novel alkylating agents indolinobenzodiazeprine pseudo dimers [160]. As for conjugates’ design, there is a major significance in the aspects of low-weight molecules used such as the pyrrolobenzodiazepine dimer’s superiority over calicheamicin in MDR-positive cells, as well as the linker. It shall be noted that the resulting conjugate in this study of Han et al. showed concerning toxicity against bone marrow in primates and its use involved more risk than the CD123 analogue [164]. Taking the matter a step further, there is clinical evidence of tolerability and relative manageability of AML of CD33 targeting IL-2 in the form of a fusion protein with an antibody in post-transplant patients [165].
Bispecific Antibodies Targeting CD33
There is also a great amount of interest in the investigation of bispecific engager antibodies. An example of this type of antibody is AMG330, targeting CD33/CD3 which despite showing adequate triggering of T cells in vitro [166] and therefore potential for application in the therapy of AML, a Phase I escalation trial, toxicity was noted before the maximum tolerable dose was reached as there were significant adverse effects including cytokine release syndrome, including grade 3/4 and rash [167]. Another CD33/CD3 bispecific antibody, AMV564, has been reported to have significant immunostimulatory effects [168], with induction of protective immunity [169], in preclinical testing. A phase Ia testing with mostly elderly participants with secondary AML and previous recipients of chemotherapy has been conducted. Early evidence suggests that AMV564 can be a tolerable and effective medicine as there was, in the majority of the patients, response to the treatment of AML [170]. In the same context, AMG673 had not so encouraging evidence as 2/3 of the patients discontinued due to disease progression of AML, half of them had experienced cytokine release syndrome and a third had serious adverse events whilst a third of them experienced a decrease in blasts. Another strategy utilizing bispecific antibodies is CD33/CD16 to stimulate NK cells against neoplastic ones had shown significant stimulation in vitro of the former in both high and low concentrations of CD33-bearing malignant cells of ALL and AML pediatric patients [171,172].
As previously mentioned, CD123 is a marker abundant in LSCs in AML, cells related to B-ALL, and MDS. Its prevalence has shown an increasing trend during disease progression, in 20% of patients with CML with no differential in the levels between children and adults [173]. Besides the clear increase in malignant cells, CD123’s coding gene seems to be well preserved [174]. As such, an interesting aspect is the engineering of allogenic hematological stem cells (Allo-HSCs) to display CD123 markers with differentiated epitopes, even by one amino acid, for them to be spared from CD123 targeted immunotherapeutic interventions, therefore increasing the latter’s efficiency and reducing related adverse reactions originating from on-target toxicity, should they be deemed to be used in tandem [175,176].
Talacotuzumab
Talacotuzumab is a mAb targeting CD123 that involves antibody-dependent cell-mediated cytotoxicity (ADCC) and (compliment dependent cytotoxicity) CDC has an inapplicable risk/benefit profile as a monotherapy against myelogenous malignancies [177]. In a phase 2/3 multicenter clinical study assessing the superiority of combination therapy including Talacotuzumab+decitabine vs. decitabine alone in patients with AML, refractory in phase A of the trial and untreated with ineligibility for hematological stem cell transplantation and chemotherapy has concluded that there was no clinical superiority in efficacy metrics and therefore early pause of enrollment and termination of Talacotuzumab treatment [178].
Anti-CD33 Conjugates
As with the rest of the molecules mentioned above, antibody-drug conjugates appear to be an appealing solution. IMGN632, also known as Pivekimab sunirine, is a conjugate containing, similarly to anti-CD33 conjugates, alkylating agent indolinobenzodiazepine pseudodimer via cleavable peptide linkage. Phase I studies of this medication conducted on 12 patients with relapsed/refractory AML or blastic plasmacytoid dendritic cell neoplasm (BPDCN) indicated no severe toxicity at the dosage of 0.18 mg/kg with mild symptoms such as tachycardia, fever, chills, and intestinal disorders related to infusion and treatment-related events mattered the gastrointestinal, and cardiovascular as well as the blood [179]. There are additional ongoing trials assessing the safety and efficacy of IMGN632 [180]. Assessment of combination therapy including IMGN632, Azacitidine and Venetoclax in relapsed/refractory AML has suggested dose-related efficacy of the regiment with adverse events including febrile neutropenia, hypophosphatemia, dyspnea and pneumonia [181]. Overall, early clinical trials have led to the suggestion of a dosage for IMGN632 of 0.045 mg/kg one time every 3 weeks for phase 2 trials onwards [182]. An additional conjugate in the preclinical phase is SNG-CD123A used for the targeted action of pyrrolobenzodiazepine dimer [174]. However, resistance to CD123 targeting molecules can be obtained via downregulation of CD123 expression, as observed by Gulati et al. in their study using antibody conjugate Targaxofusp against blastic plasmacytoid dendritic cell neoplasm [183]. Despite this, Targaxofusp has demonstrated its effectiveness and, in general, a tolerable profile in clinical trials [184]. Preclinical evidence has also indicated effectiveness in pediatric ALL specimens [185].
Bispecific Antibodies Targeting CD47
Bispecific antibodies have also been investigated. Flotetuzumab is a bispecific antibody targeting CD123 and CD3. Despite early indications of safety and effectiveness over chemotherapy-resistant leukemias [186], its development has been discontinued by its sponsor. Early clinical trials have concluded an enhanced risk of grade </=2 cytokine release syndrome. Escalation/Expansion studies of APVO436, also a bispecific targeting the same markers, have resulted in Grade 3 or more treatment-related adverse events including anemia, neutropenia, thrombocytopenia, and sepsis in about half of the participants [187]. The minimum anticipated biological effect has been reported to be 0.3 to 60 μg, which falls below the maximum tolerated dose with results being delivered to about a fifth of the patients, most of whom had failed in prior interventions [188]. MGD024, an altered version of Flotezumab with less affinity to CD3 and containing a mutated IgG domain with the aim of half-life increase through the function of the neonatal Fc receptors had shown reduced cytokine release when studied in rodents hosting human AML cells, and, therefore, reduced effectiveness compared to Flotezumab. However, combination therapy with Venetoclax and Cytarabine seemed to have enhanced cytotoxic activity [189]. MGD024 has been through early clinical development [190]. Finally, by introducing a SIRPα domain in an anti-CD123 antibody, Tahk et al. managed to increase phagocytosis of AML cells via mechanisms described in the corresponding section about CD47 [191].
CD117 is an important target as it caters to the need for selective targeting of HCSCs [192]. Studies suggest that the optimal therapeutic value of anti-CD117 antibodies is the conditioning of the bone marrow for a CD34+ enriched graft transplant, as evident in mouse studies, where they have been reported to act against physiological and myelodysplastic syndrome stem cells [193]. However, conditioning has traditionally been occupied by interventions such as cytotoxic chemotherapy and radiation therapy.
CD117 Blocking Antibodies
The antibodies AMG191 and SR-1, which block the interaction of CD117 with the stem cell factor (SCF) and its autophosphorylation [193,194] have been tested on immunocompetent primates with favorable outcomes consistent with the ones reported on mice [195]. Taking advantage of this positive trend, there have also been early clinical trials conducted on small groups of human immunodeficient patients which have shown safety and efficacy [196]. Combination therapies have also been considered as the parallel use of azacitidine and ACK2 has delivered efficacy in the context of bone marrow clearance and myelodysplastic stem cells [197].
Anti-CD117 Conjugates
Conjugates against CD117 have also been developed. MGTA-117 is a conjugate containing RNA polymerase II inhibitor amanitin that has been relatively successfully tested on murine models for bone marrow conditioning for graft receival. At a single dose of up to 10 mg/kg or a multi-dose of 3 mg/kg four times a day, there was tolerability and effectiveness as most grafted human cells were selectively eliminated [198]. Preliminary evidence of MGTA-117 Phase 1/2 clinical trials with a small number of participants, until the dose of 0.02 mg/kg has deemed safe the conjugate in the trial as no treatment-related adverse events were noticed, besides grade 1 alanine and aspartate aminotransferase. Effectiveness was assessed through the binding to CD117+ cells, the effect on erythropoietic stem cells, and clinical results [199]. Another conjugate, CD117-ADC carrying streptavidin–saporin had shown dose-dependent results, with a range from 0.3–1.5 mg/kg, in mice as depletion of stem cells was noted with the subsequent successful engraftment of allogenic transplants. Mature cells were spared from cytotoxic activity and, thus, as much as possible, immune capabilities remained [200].
Bispecific Antibodies Targeting CD117
Most bispecific antibodies targeting CD117 are designed with T cell engagement in mind as they also include compartments with CD3 affinity. In the context of bone marrow clearance, it has been realized that greater cytotoxicity is needed, to such an extent that T cell recruitment is rendered necessary [201]. CD117xCD3 bispecific T cell engagers (BiTEs) have shown targeted efficacy by inducing T cell proliferation and activation as well as cytotoxicity and the resulting halt of disease progression and reduction in preclinical in vivo and in vitro testing. Cell lines with positive results include HL60, MOLM14 [201], TF-1 [202], CD117+ HeLa, physiological hematopoietic stem and progenitor cells [203] highlighting their use in the treatment of AML. Additionally, a combination approach involving a CD71 aptamer and monomethyl auristatin F (MMAF) in a bispecific antibody format has also been evaluated, showing no observed toxicity in AML bone marrow samples [204].
Galectin-9 (GAL-9) is a molecule synthesized by HCSCs, among other types of physiological and malignant cells as well, crucial for immune evasion through the binding to its receptor TIM-3 and the subsequent reduction of stemness and apoptosis of T-helper 1 and the CD8+ lymphocytes that infiltrate the tumor, possible reduction of activation of dendritic cells, and reduction of activity and interferon gamma IFN-γ excretion by NK cells. Besides the dampening of the immune response, this loop has also been suggested to induce the proliferation of the AML cells in an autocrine manner through the activation of the NF-kB and β-catenin pathways [205,206]. It is then easily considered that the presence of cancer cells, especially on HCSCs, is a major player in the process of proliferation, disease progression, adverse outcomes, and the overall aggressiveness of the malignancy. Targeting TIM-3 utilizing antibodies not only increases the response of the elements of adoptive therapy through the revival of exhausted CD4+ and CD8+ T cells, evidenced by the increased excretion of immunomodulatory cytokines TNF-α and IFN-γ, but also directly limits the extent of the HCSCs’ activity and viability [207]. Besides T cells, antibody-mediated TIM-3 blockade had initiated significant NK cell activation against multiple myeloma (MM) cells in vitro, evident by the increase in proinflammatory cytokines concentration and the amount of apoptotic MM cells. In vivo testing resulted in reduced tumor burden in mice as was noticed by the decreased tumor size [208].
Sabatolimab
Sabatolimab, also known as MBG453, is a mAb used for TIM-3 blockade and that is under clinical development, having reached phase 2 at a dosage of 800 mg every 4 weeks or 600 mg every 3 weeks or 400 mg every 2 weeks. Its high affinity with TIM-3 grants it the capability of initiating similar effects as noted before in AML cells, including cytokine increase, blockade, as well as phagocytosis of marked cells [209]. Additional antibodies clinically assessed are SHR-1702 for MDS and Sym023 for lymphomas [210]. Clinical trials have demonstrated the anti-TIM-3 antibodies’ relative safety and efficacy in patients with leukaemias and solid tumors. Focusing on hematological malignancies, the combined intervention of sabatolimab with a hypomethylating agent had shown no greater incidence of Grade>/=3 adverse events than monotherapy with the hypomethylating agent in patients with high-grade MDS and AML. However, greater efficacy was observed in patients with MDS as they displayed higher overall response rates (ORRs), progressive free survival (PFS) and allowance to undergo HSCT. In a similar study studying the same diseases and interventions, high epidemiological metrics in a two-year span with ORR nearing 70%. Generally, it is considered that the addition of those antibodies into the intervention is not likely to lead to an increased risk of already ongoing therapies. Additionally, more positive results have been recorded for carriers of mutations related to less favorable outcomes [211]. Escalation trials typically include the dosages of 240, 400 and 800 mg of sabatolimab + 20 mg/m2 of decitabine intravenously (IV) in the first 5 days + 75 mg/m2 of azacitidine subcutaneously (SC) in the first 7 days per 28 days. Typical adverse events include conditions related to low numbers of blood cells such as thrombocytopenia, neutropenia, anemia, and pneumonia [212,213].
TIM-3 and PD-1 Tandem Inhibition
There appears to be a significant interplay between PD-1 and TIM-3 pathways, as there are several studies investigating whether both molecules are present on the surface of exhausted cytotoxic effector lymphocytes. Although it achieved a significant increase in survivability, tandem blockade of these molecules in mice had improved other metrics, suggesting a delay in disease progression through the reactivation of exhausted T cells in CML models. However, the evidence is not universally consistent. Some studies have challenged this proposed synergy, reporting that CD8+ T cell activation was either inadequate or failed at all due to the lack of significant CLL cell clearance in vitro [214–217].
CD200’s abundance in numerous cancer types, especially the ones of the blood [218], is catalytical to immune evasion and cancer proliferation. As an immune checkpoint molecule, CD200 bearing HCSCs are rendered capable of evading T & NK cell cytotoxicity, by hindering their metabolism and promoting cell death, leading to proliferation and rapid progression of the malignancy [219]. It’s important to recognize that, although there have been antibodies developed against CD200, as analyzed later in this paragraph, their efficacy can be compromised from the additional role facilitated by its cleavable parts located at its intracellular and extracellular domains. Whilst the intracellular part can translocate to the nucleus and act as a transcription factor for oncogenic genes, the extracellular part can initiate cell death on distant NK cells, generating questions about the extent of the efficacy of the idea of using antibodies against CD200 [220].
Samalizumab is an anti-CD 200 mAb that is evidently generally tolerable in Phase I escalation trials from 50 to 600 mg/m2 with no maximum tolerated dose (MTD) being reached and with mild adverse events being reported. Response metrics indicated 1 overall response case, 16 stable disease cases and 5 progressive disease cases, out of 23 participants. 14 of those participants had shown tumor decrease [221]. Another mAb TTI-CD200 was assessed in vitro and in vivo at 20 mg/kg. In vitro testing including macrophages and T cells has indicated efficacy against CD 200high leukemic cells, measured by the increase in IL-2 concentration, whilst minimal effect was observed on normal cells that were not expressing CD 200 at this rate. Studies on grafted mice had returned delay of disease progression, extension of survival, and decrease of engrafted human malignant cells [222]. Similar targeted cytotoxic effects were also noted in the study of Rastogi et al. using K562 (representing CML) cell lines and NOD-SCID IL2Rγ(-/-) (NSG) immunodeficient mice engrafted with blasts [223].
In the investigated literature, CD45 therapy has been associated with the use of conjugates in combination with HCST in the interest of conditioning. As CD45 tyrosine phosphatase is abundant in HCSCs and T cells, and, as such, its targeting can be implicated as “striking two birds with one stone” in avoiding GVHD that can lead to rejection [224]. Conjugates that involve Janus kinase (JAK) inhibitors such as baricitinib, are effective in the conditioning of mice’s bone marrow as their transplantation was characterized as stable with minimal related adverse conditions, the most major one being GVHD [225]. The conjugate’s targeted activity, like most conjugates, only happens upon its internalization and release of the payload within the lysosome. CD45 conjugates have also been preclinically studied in vitro carrying pyrrolobenzodiazepine [226]. Based also on the information provided in Section 3.2.5, tandem inhibition of CD33 and CD45 has been a lucrative approach for the elimination of AML cells and blasts through the increased internalization and effectiveness of the former, facilitated by the latter [227].
CD27, and its natural ligand CD70 form one of the most prevalent and significant immune evasion mechanisms of hematological cancers. Coexpression of those molecules has only been observed in cells comprising blood cancers, including HCSCs of AML, CML, B-ALL, and B-CLL, B-cells of NHL, and T cells of lymphomas, whereas in solid tumors only CD70 is expressed in cancer cells and CD27 is present in the microenvironment. Increased prevalence of CD27 in the serum of patients in its soluble form is a major indicator of stemness as it leads to the induction of the Wnt pathway and symmetric division [228,229]. Additionally, through TNF receptor-associated factor 2 (TRAF2) and TRAF2 and Nck-interacting kinase (TNIK), the signaling facilitated by the interaction of those two molecules determines the fate of the cells as they play a significant positive role in the expression of Wnt, colony formation, and proliferation. The significance of those two molecules can be preclinically identified by the results, such as induction of differentiation, reduction of survivability, inhibition of cell growth and colony formation, their blockade induces in mouse models [1] as well as by the clinical evidence that indicates higher CD70 presence in relapsed AML patients [230].
Custazumab Combinations
Considering the evidence that CD70 mediates resistance of HCSCs to hypomethylating agents, and the subsequent upregulation of CD70 upon exposure to these agents, Ochsenbein et al. have assessed combination therapy of Cusatuzumab, a CD70 inhibiting mab with azacitidine. Results from in vitro tests suggest that this combination leads to greater ADCC and CDC, stemming from the expansion of CD70, as previously mentioned. Additional clinical studies have resulted in the reduction of blasts both in combination and in monotherapy involving Cusatuzumab with Dose Limiting Toxicities not being reached until 20 mg/kg [231]. Results from this combinatory therapy involving 12 AML patients unfit for chemotherapy have indicated a CR in 8 out of them, CRi in two of them and PR in an additional two patients with lasting effects in 6 patients. Effects were observed at all cusatuzumab doses, and they lasted in 6 patients. Influence on normal hematopoiesis was not noted in the combination therapy; however, all patients had experienced at least one adverse event of Grade >/=3. Cusatuzumab has also been considered in combination [232] with the BCL-2 inhibitor Venetoclax. This approach has been capable of compromising the functionality and neutralizing in vitro HCSCs in the presence of NK cells with much more efficacy than the monotherapy with the cusatuzumab and venetoclax [233].
ARGX-110 and other CD70 Targeting Interventions
ARGX-110, an antibody with dual action targeting both T cells and HCCs bearing CD70 [234] has also been clinically studied in Phase 1/2 studies. The regimens involved the dosages of 1, 3, 10 or 20 mg/kg, depending on the arm, IV every two weeks combined with azacitidine 75 mg/m2 SC for one week every 28 days [235]. Overall, there were 38 patients enrolled. The then still ongoing Phase 1 trial had shown no indications for dose limiting toxicities at 20 mg/kg and the ORR was at 50%, out of which all had achieved complete response (CR) or complete response with incomplete recovery (CRi). As such, 10 mg/kg had been selected for Phase 2 testing. Regarding safety, all of the patients had experienced Grade >/=3 adverse events with the most common ones being neutropenia, anemia, thrombocytopenia, pneumonia and pyrexia [236]. In another Phase 1 study of ARGX-110, 100 mg/kg was also deemed as a safe dose and the most common adverse effects were fatigue and infusion-related ones [237]. CD70 blockade has also been carried out by a conjugate carrying Monomethylauristatin F in in vitro context with varying results upon different cell types, based also on differing structural and design elements of the various types of conjugates assessed. Concerning hematological malignancies, cell line L-428 (modelling HL) appears to be more susceptible to this intervention compared to LP-1 (modelling Multiple Myeloma) [238].
CD27 Targeting
CD27, a member of the TNF family that upregulates lymphocytes [239] has also been investigated for immunotherapeutic targeting. In the context of antibody therapy, invitro targeting with the human agonistic antibody CDX-1127, also known as 1F5 or Varlilumab, had demonstrated adequate binding to CD27, activation of T lymphocytes, when costimulatory signals are present, extension of the lifespan of and limiting of tumor growth in tumor bearing (Raji or Daudi cells) mice, and tolerability to primates up to 10 mg/kg [240,241]. In general, cytotoxicity has been noted for HL, follicular lymphoma (FL), and DLBCL with synergy with PD-1 inhibition being noted in preclinical experiments [242]. As expected, these promising results had sparked interest in clinical trials. Early evidence had shown no dose-limiting toxicity up to 3 mg/kg and toxicity had ranged up to Grade 2. At that dose, there were 3/19 patients with stable disease and a patient with full remission [243]. Based on evidence of tolerability of Varlimumab from Phase 1 trials, combination treatment with Nivolumab (PD-1 inhibitor) has also been assessed in the clinic for the treatment of B cell lymphoma regarding the investigation of its superiority against nivolumab monotherapy [244]. In total 53 patients were enrolled split equally between nivolumab monotherapy and combination with Varlilumab. Metrics associated with efficacy and toxicity did not show any statistically relevant difference between the two arms, suggesting no significant benefits in intervening using combination therapy [245,246]. Additionally, CD20 has been simultaneously targeted with CD27 (rituximab+Varlilumab) for the treatment of B-cell NHL (B-NHL). ORR had been recorded to be 26,9%, with 4 patients achieving PR within a timespan ranging from 2 months to over a year. Treatment outcomes indicated sufficient T cell activation by Varlilumab [244].
Cellular immunotherapy is a revolutionary strategy in targeting HCSC, that converts mainly autologous immune cells’ cytotoxic ability. Unlike mainstream trauma-forcing regimes, chemotherapy, or radiotherapy, which usually fail to eradicate HCSCs due to their intrinsic resistant mechanisms, cellular immunotherapy is expected to provide prolonged immunosurveillance and destroy any ulterior malignant stem cells. Key strategies include chimeric antigen receptor (CAR)-T cell therapy, NK cell-based therapies, tumor-infiltrating lymphocytes (TILs), and γδ T cell therapy, each offering unique advantages in overcoming immune evasion and therapy resistance [247].
Adoptive T cell therapy is a type of cancer immunotherapy that enhances a patient’s own T cells ex vivo before reinfusion. It started in studies put forth in the 1980s by Rosenberg introducing lymphokine-activated killer (LAK) cells, which have tumor-killing potential when stimulated with IL-2 [248,249]. Further work with TILs isolated from tumors and expanded in vitro, showed their efficacy in melanoma and renal cancer [250,251]. Toward the end of the 1990s, cytokine-induced killer (CIK) cells were observed, which were shown to exhibit wide-ranging antitumor cytotoxicity through perforin and Fas/Fas ligand pathways [252]. Genetically engineered T cells comprise modern adopted cell therapy and have included T-cell receptor (TCR) therapies, as well as CAR-T cells, which further complement antigen specificity and escape immune tolerance. Such techniques improve targeting of tumors, and such requires no de novo activation in patients. Although adoptive cell therapy (ACT) provides much hope, there are still active lines of research on limitations such as limited responses and immune escape [253].
3.3.1 Tumor-Infiltrating Lymphocytes (TILs)
TILs have become immune cells migrating into tumors and have been defined earlier prognostic indicators and immunotherapy key players [254]. TILs are actually a small group of immune cells within the tumor, including the CD4+ helper T cell, CD8+ cytotoxic T cell, Tregs, macrophage, dendritic, and NK cells [255]. Their existence, which correlates with better clinical outcomes in many cancers, is considered related to actual anti-tumor immune responses. The TME determines the ability of TIL, an array of immunosuppressive cytokines, their inhibitory immune checkpoints as PD-1 and CTLA-4, and metabolic constraints to function on them [256]. TIL therapy was discovered in the early 1980s when scientists found that tumors containing TILs were associated with better prognosis in patients [257]. Initial clinical trials published in the early stages showed impressive results in patients with melanoma and inspired researchers to develop TIL-based adoptive cell therapy. The first landmark Phase I/II trial demonstrated the efficacy of expanding TILs ex vivo and reinfusing them into a patient after lymphodepleting chemotherapy [258]. In 2024, lifileucel received FDA approval, marking the first TIL-based therapy dedicated to treating melanoma [259].
Therapeutic Application and Response
TIL therapy comprises various steps, which generally start with the detection (mainly via immunochemistry detection assays) and the obtaining of TILs from a patient’s tumor, using cell sorting techniques (e.g., magnetic-activated cell sorting (MACS)/fluorescence-activated cell sorting (FACS)). Rapid expansion procedure (REP) of these cells is then carried out using cytokines such as IL-2 in vitro to boost proliferation. Once there are sufficient numbers of the TILs, they get reinfused to the patient after preparatory lymphodepleting chemotherapy. It has been successfully shown to be efficient against many cancer forms [260]. Responding rates have averaged between thirty to fifty percent in melanoma cases treated with addition to checkpoint inhibitors such as anti-PD-1 therapies. In response to treatment, patients with ovarian cancer presented between 20 and 40 percent, while colorectal and lung cancers showed behavior that resembled such strains but required more work to establish the optimum criteria for patient selection [261]. In renal cancer, early clinical data showed improved outcomes when TIL therapy was combined with preconditioning chemotherapy [262].
The main limitation, however, is that tumors vary widely in TIL content and thus it becomes increasingly difficult to predict which patients would be most likely to benefit from this approach [263]. Besides, the functionality and persistence of TILs are compromised by the immunosuppressive TME, and there is a need to apply methods to improve both [264]. After that, time-consuming and very high cost to grow TILs makes it hard to use because it could take weeks and requires specialized research facilities [265]. Treatment-related toxicities, such as cytokine release syndrome due to the high doses of IL-2, are also associated with risks to patients receiving TIL therapy [266].
Advancements and Future Directions
To address these concerns, several researchers have made great strides in improving the applications of TIL therapy. Selecting highly reactive TIL subsets, such as CD103+ and CD39+—CD8+ T cells with greater anti-tumor efficacy, is one step in the process [267]. In combination therapy, TIL therapy and immune checkpoint inhibitors (ICIs) have provided improved response rates, since checkpoint inhibitors counteract the immunosuppressive signaling in the TME [268]. Further optimized culture conditions were studied for TIL expansion and survival, incorporating cytokines such as IL-15 [269]. The next generation TIL therapy development is actively continued to refine and better the patient’s outcome. TILs are enhanced through genetic editing, where inhibitory receptors like PD-1 and CTLA-4 can be knocked out to preserve TIL activity against the tumors [270]. Techniques in synthetic biology are also sought to engineer such TILs to resist exhaustion and keep fighting in the unfavorable TME. Studies are also being conducted to investigate the influence of B-cells and tertiary lymphoid structures in the TME; such components may bolster efforts to establish an effective anti-tumor response further [271,272]. Another promising strategy is automating TIL manufacturing, which is geared towards desalting the expansion processes and decreasing time and cost in the production of patient-specific TILs [265]. Furthermore, personalized TIL therapy, which varies the selection and expansion of TILs according to the antigen profile of a patient’s tumor, is under study to enhance treatment fidelity [273]. Finally, motivating TILs to persist in the patient following reinfusion remains a major goal, given that most TILs become exhausted or dysfunctional over time [264].
CAR-T cell therapy has revolutionized treatment for hematological malignancies and adopted the use of genetically engineered T cells as agents for the recognition, attack, and elimination of cancer. Autologous T cells are isolated and then subjected to ex vivo genetic modification to express CAR, a specific receptor that recognizes tumor-associated antigens (TAAs) independently of any major histocompatibility complex (MHC)-specific cytotoxic response that occurs after reinfusion to the patient [253].
Engineering and Generational Evolution
The engineering process of CAR-T cells starts by isolating the T cells, usually through leukapheresis, where T cells from the patient are harvested. These autologous T cells are then genetically modified ex vivo so that they can express the CAR molecule on their surface, so they react toward a TAA. Genetic modification is conducted by employing several techniques, the most commonly used being viral transduction with retroviral or lentiviral vectors for stable and long-term CAR expression, as these vectors integrate the CAR construct into the genome. However, disadvantages of this method include the risk of potential insertional oncogenesis and immune-mediated toxicity. Various other non-viral gene transfer methods have been designed to deliver the CAR genes while minimizing those risks and include transposon systems like Sleeping Beauty and PiggyBac, electroporation and nanoparticle-based techniques. Such significant advances in CAR engineering are those that allow the use of in vitro transcribed (IVT)-mRNA to enable temporary CAR expression without any impervious genome integration, thus decreasing the possibility of oncogenic mutation occurrence [274]. Another refinement of CAR-T engineering is CRISPR/Cas9 genome editing, which can allow precisely knock-in or knock-out modifications such as knocking out antagonistic receptors that inhibit CAR-T functioning or even modifying persistence and specificity. And now, this technology is advancing universal allogeneic CAR-T cells derived from healthy donors and engineered to prevent immune rejection for extending access [275].
From the very beginning, CAR-T cell therapy evolved through several generations, with each one promising the improvement of activation, persistence, and efficacy in therapy. In first generation, CARs, only an intracellular signaling domain, CD3ζ, was present, which allowed antigen recognition and activation of T cells but poorly expanded and lasted only for a short duration in vivo hence poor clinical efficacy. Second-generation CARs comprised a co-stimulatory domain, such as CD28 or 4-1BB, in addition to CD3ζ to enhance activation, expansion, and long-term survival of CAR-T cells. CD28-based CARs lead to very rapid expansion but lower persistence, while 4-1BB-associated CARs improve and prolong T cell memory-like characteristics, lowering exhaustion and thus creating long-term responses. Building upon that, third-generation CARs combined two co-stimulatory domains, e.g., CD28 and 4-1BB, with CD3ζ in a composition that is further increasing the activation, proliferation, and persistence. Although preclinical studies have indicated a possible advantage over second-generation CARs, their clinical superiority is still being tested. Fourth-generation CARs, T cells redirected for universal cytokine-mediated killing (TRUCKs), target the induction of proinflammatory cytokines secretion, e.g., IL-12, from CAR-T cells to trigger the innate immune system and have thus been designed to overcome one of the immunologic limiting factors of tumour introduction [253].
Clinical Applications and Target Antigens
One of the most popular targets studied for CAR-T therapy in hematological malignancies is CD19, which is a B-cell antigen. CD19-directed CAR-T cell therapy has been proven exceedingly effective in the cases of B-cell ALL where it has recorded over 95% complete remission (CR) rates in relapsed/refractory (r/r) conditions [255]. For relapses that are negative for CD19, alternative B-cell-specific targets like CD22 and CD79b are being examined. All B-ALL cases express CD22, and some of them continue to express CD22 even after losing CD19 [276]. In CD22-directed CAR-T cell therapy, CR was achieved in up to 73% of patients with a prior history of receiving CD19-targeted treatment [277]. Another important B-cell antigen in this regard is CD79b, which has been studied as a target for therapy, particularly in combination treatments, giving quite good preliminary results [278].
Beyond B-cell malignancies, CAR-T treatment is being implemented in additional hematological malignancies, including MM, HL, AML and other T cell cancers [279]. In MM, B-cell maturation antigen (BCMA) is emerging as one such hopeful CAR-T target. Early clinical reports on trial data for BCMA CAR-T have shown varied response rates from essentially 43% to 100%, depending on the CAR construct and dose regimen [280]. Bispecific CAR-T cells targeting both BCMA and CD38 [281], or CD19 [282] are also being studied to improve therapeutic benefits and to possibly overcome the antigen escape. In HL, the CAR-T cell approach directed towards CD30 was explored via clinical trials, and the objective response rates were recorded to have gone up to 39% among relapsed patients or those suffering from refractory disease [283]. Some optimization strategies to boost response rates are enhancing lymphodepletion regimens and improving CAR-T cell trafficking to tum our sites [284]. Due to the lack of TAAs that is generally expressed and common among T cell malignancies, they have posed unique challenges. Moreover, CD7-directed CAR-T cell therapy exhibited a strong preclinical activity against T cell leukemia and lymphoma, and a few early human clinical trials could prove the high response rates [285]. Potential targets under investigation include CD5, CD4, and CD30 that again raise issues with regard to the T cell aplasia and immune system-depleting effect because of their expression in normal T cells [279]. The absence of an ideal antigen that would selectively destroy malignant cells while preserving the normal hematopoietic stem and progenitor cells is the most significant hindrance in AML. Clinical trials involving CLL-1 (CLEC12A), CD33, and CD123-directed CAR-T therapies are underway; however, concerns regarding potential off-target toxicity remain [286]. Further fine-tuning of antigen selection and targeting strategies will be needed to mature CAR-T cell therapy for myeloid malignancies.
Safety, Resistance, and FDA-Approved Therapies
Even if they are transforming, the safety issues keep poring on CAR-modulated T cell therapy, primarily in the area of cytokine release syndrome (CRS) and neurotoxicity [253]. Reports indicate that up to 90% of patients would suffer from CRS that runs to a mild flu-like syndrome to life-threatening failure of multi-organs [287]. Neurotoxicity levels, including the CAR-T-cell-related encephalopathy syndrome, are as frequent a complication seen in over 50% of recipients in some cohorts [288]. Toxicity mitigation strategies take several directions, including optimizing CAR constructs, refining patient-selection criteria, and merging adjunctive therapies, such as IL-6 inhibitors and corticosteroids. One more major barrier is that there is every chance of a relapse after CAR-T cell therapy. Most relapses that are due to positivity to CD19 are postulated to be because of poor persistence of CAR-T cells, while most of those negatives to CD19 are due to loss of the antigen and immune escape. Improving the durability of CAR-T cells has been feasible with strategies such as optimizing the CAR design, genome editing to eliminate inhibitory receptors, and the use of artificial antigen-presenting cells to maintain T cell activation [289]. The application of different target antigens such as CD19/CD22 or CD19/CD20 for generating dual-target CAR-T cells has demonstrated better efficacy toward preventing antigen-negative relapses [290]. CAR-T cell therapy is also being explored as a bridge to HSCT in patients with relapsed or refractory leukemia. Studies have shown that patients with minimal residual disease (MRD)-negative status after CAR-T infusion have a better outcome following HSCT. Studies show that allogeneic transplantation for B-ALL patients who achieved MRD-negativity with CAR-T therapy was associated with sustained remissions. Clinical trials calculate that 90% of patients with CR achieved by CD19 CAR-T therapy maintained MRD negativity post-transplant [291].
Regarding the FDA-approved therapies, tisagenlecleucel (Kymriah®) was the first and pivotal step for CAR-T cell therapy in 2017 for r/r B-ALL in the pediatric and young adult patients. Subsequent approvals were for large B-cell lymphoma (LBCL) as well as follicular lymphoma (FL) use. The first approval of axicabtagene ciloleucel (Yescarta®) came in 2017, and it showed an objective response rate (ORR) of 82% for resistant or relapsed large (R/R) LBCL. It was later approved for treatment of R/R FL in 2021. Brexucabtagene autoleucel (Tecartus®) is a CD19-directed CAR-T similar to axicabtagene ciloleucel, but it has the added feature of eliminating leukemia cells. It was approved in 2020 for R/R mantle cell lymphoma (MCL) and in 2021 for R/R B-ALL in adults, showing a median OS of 18.2 months in ZUMA-3. Lisocabtagene maraleucel (liso-cel, Breyanzi) was approved for R/R LBCL in 2021, with a 73% ORR in the TRANSCEND NHL-001 trial, followed by approval of second line use for superior event free survival (EFS) over autologous stem-cell transplant (ASCT) in the TRANSFORM trial. For multiple myeloma, idecabtagene vicleucel (ide-cel, Abecma) was approved in 2021 specifically for R/R multiple myeloma (MM) patients after at least four other lines of treatment, achieving 73% ORR in the KarMMa trial. Ciltacabtagene autoleucel (cilta-cel, Carvykti) came next, in 2022, with a 97% ORR in the CARTITUDE-1 trial [292].
Principles and Therapeutic Advances
NK cell-based immunotherapy, which stems from the inherent cytotoxic capabilities and MHC-independent tumor recognition of NK cells, offers a universal approach to the treatment of hematological malignancies. Unlike T cell therapies, however, NK cells have little requirement for prior sensitization and boast broad target specificity; this makes them an attractive off-the-shelf therapy [293]. Advancements in NK cell biology and genetic engineering have made it possible to expand the therapeutic realms of both adoptively transferred allogeneic NK cells and CAR-modified NK cells.
Allogeneic receptor NK cell therapy encompasses the extraction, expansion, and infusion of NK cells from a donor source [294]. In contrast, the use of autologous NK cells is less effective because of immune suppression, the use of allogeneic NK cells provides significant benefits to the patients, especially in AML [295]. NK cell therapy, the first of its kind in AML, demonstrated feasibility in early clinical trials, while 26% achieved complete remission (CR) rates with haploidentical donor IL-2-activated NK cells [296]. Further modalities of NK cell manipulation, such as depletion of Tregs and IL-2 diphtheria toxin, have raised CR rates above 50% [297]. On the other hand, IL-15 is emerging as a potent alternative to other cytokines for enhancing the survival and function of NK cells, but has been associated with CRS and neurotoxicity [298]. The provisions of irradiated K562 cells expressing 4-1BBL and IL-21 have largely improved NK expansion and at the same time reduced the exhaustion. Nicotinamide (NAM)-expanded NK cells, also named GDA-201, exhibited good persistence and cytotoxicity, achieving an impressive 74% response rate in a phase I trial in lymphoma [299].
Sources and Novel Strategies
A major disadvantage of peripheral blood as a source of NK cells is the reduced amount of cells retrieved, as well as the heterogenous cellular populations, and variable NK cell function. NK cell lines—such as NK-92—provide a self-renewing source of cytotoxic cells. Initial clinical trials showed only transient responses in AML; nevertheless, NK-92 is potentially very interesting as a source for future therapies, given ease of expansion with repeated dosing. Furthermore, membrane-bound IL-21/4-1BBL-expanded NK cells have demonstrated significantly increased efficacy in MRD-positive AML against disease-free survival compared to chemotherapy alone [300].
Several existing clinical trials on the NK cell therapy of hematopoietic malignancies showed fairly supporting efficacy. In fact, cytokine-induced memory-like (CIML) NK cells exhibit enhanced cytotoxicity and prolonged persistence. In a phase I trial in patients with AML, four complete remissions were noted among nine patients, with memory-like NK cells in circulation for weeks. These cells are currently being evaluated in clinical trials involving myeloid cancers and head-and-neck malignancies [301], induced pluripotent stem cell (iPSC)-derived NK cells are a scalable, off-the-shelf option. Genetically engineered iPSC-NK cells have been designed to enhance cytotoxicity, resistance to immune suppression, and persistence. iADAPT NK cells engineered to contain non-cleavable CD16, IL-15/IL-15R fusion, and deletion of CD38 have exhibited preclinical activity superior to other NK cells [302].
CAR-modified NK (CAR-NK) Cells and Clinical Prospects
The advent of CAR-NK cells has been a landmark in NK cell therapy, rendering it with a potential along antigen-specific lines [303]. In comparison with CAR-T cells, NK cells are relatively short-lived and secrete a safer profile of cytokines, whereas high levels of toxicity, like CRS, might be prevented [304]. Downstream in vivo persistence, however, remains a key hurdle. Encouragingly, CAR-NK cells have been engineered to target hematological cancers with preclinical success. Namely, B-cell malignancies are engineered against CD19 and CD20, whereas CD138 for multiple myeloma, and CD33 for AML [279]. Though there is a paucity of clinical data, early-phase studies indicate CAR-NK cells to be safe and tolerable. In a phase I trial, CD33-CAR-NK cells showed negligible toxicity in AML, while the clinical efficacy was still inconclusive. Umbilical cord blood (UCB) represents another source of NK cells, benefiting from HLA-killer cell immunoglobin-like receptor (KIR) mismatch, which promotes anti-tumor activity. In a phase I/II trial, UCB-derived CD19-targeted CAR-NK cells were efficacious in CD19-positive lymphoma, with a 73% response rate and a persistence of up to 12 months, with no significant toxicities. A separate approach includes differentiating CD34+ UCB stem cells into NK cells, with better KIR expression and homing properties to the bone marrow [305].
Biological Features and Immune Surveillance
γδ T cells are a non-MHC-restricted T cell subset unique to their invariant γδ-TCR. They comprise 1–5% of the circulating lymphocyte pool distributed through the body, especially in tissues such as epidermis, gastrointestinal mucosa, and reproductive system [247,306]. Among all the γδ subtypes, Vγ9Vδ2 is considered the most abundant in peripheral blood while δ1 and δ3 γδ T cells primarily reside in the tissues [307]. Stress-induced metabolic changes in the cell such as phosphoantigens and isopentyl pyrophosphate, which define a transformed cell from a healthy one, are recognized by them in immune surveillance [308]. Pro-inflammatory cytokines, mainly IFNγ, TNFα, and IL-17, mediate their anti-cancer activity, along with direct cytotoxicity [309].
Preclinical Applications and Therapeutic Challenges
Vγ9/Vδ2 T cells undergo preclinical checks for killing efficiency against cancer stem cells from colon, ovarian, and neuroblastoma tumors. However, it has had a limited influence on prostate and breast cancer stem cells [310–314]. In breast cancer, stem cells resist cell killing by γδ T due to increased PD-L1 expression, anti-apoptotic MCL-1, and MICA shedding [314]. Still, treatments such as PD-1 blockade, myeloid cell leukemia 1 (MCL-1) degradation, and inhibition of proteolytic cleavage would put them back to susceptibility to γδ T cell cytotoxicity. Pretreatment with the γδ T cell agonist zoledronate leads to increased proliferation, cytokine production, and cytotoxic molecule secretion in γδ T cells [315]. Additionally, zoledronate upregulates MHC-I and intracellular adhesion molecule-1 (ICAM-1) on cancer stem cells, thus improving CD8+ T cell-mediated killing. The innovative combination therapy was studied in terminal solid tumor patients. While preclinical results appear promising, clinical trials with γδ T cell stimulation or direct transfer into cancer patients have achieved very low efficacy and few successes. This is mainly due to the lack of specificity and functional diversity of γδ cells. Synergy between γδ T cells and CD8+ T cells has been established in breast cancer, as γδ T cells render cancer stem cells more susceptible to CD8+ T-cell-mediated killing. Because their safety profile is like that of CAR-NK cells, they can also be considered candidates for CAR-based therapies, providing possibly another approach to target these cells [247].
Role in Transplantation and Future Potential
Finally, patients undergoing T-cell-depleted HSCT have better clinical outcomes when donor-derived γδ T cells proliferate after transplantation. The presence of γδ T cells is associated with lower relapse incidence and greater survival, likely due to the graft-vs.-leukemia (GVL) effect of these cells without too much risk of inducing graft-vs.-host disease (GVHD). The role of γδ T cells as potential applications in adoptive cytotoxic T cells for HSCT is strengthened by their lack of requirement for cognate recognition of MHC on host target cells. These unique capabilities help promote immune reconstitution and minimize post-transplant infections, thus increasing therapeutic potency [316].
Cancer vaccines have been explored as a potential strategy in anti-HCSC immunotherapy. Amongst the molecules discussed in Section 3.2, CD123 and CD44 along with CD133 seem to be the most important and promising for application in cancer vaccines due to their abundance and specificity over their presence on the surface of HCSCs [8]. Various strategies can be followed during several crucial steps in vaccine development. These include either the infusion of in vitro antigen-enriched Dendritic Cells (DCs), or of the antigens themselves in the form of peptides, entire proteins or even altered immunostimulatory malignant cells, for example through enrichment with CD80 costimulatory ligand. Regarding DC infusion, the presence of either externally added or DC expressed WT1, along with MUC1, which is present only on LSCs, can enhance the T cell response’s specificity [317]. Optimization of HCSCs-targeted vaccines can also be carried out via formulation methods that lead to improvements in antigen presentation as well as combination therapies [318], with checkpoint inhibitors. Although there hasn’t been any solid record stating complete liquid cancer clearance, preclinical evidence is reporting tumor size reduction, and activation of the adaptive immune system, as realized via cytokine production patterns. Moreover, clinical evidence points to a high likelihood of achieving stable disease, with most adverse events related t to localized inflammation or vaccine-specific immune responses. These findings offer hope for the future integration of cancer vaccines into the therapeutic landscape of hematologic malignancies [319].
3.5 Oncolytic Virotherapy (OVT)
The ability of viruses to selectively target malignant cells has placed them at the forefront of modern oncolytic therapy. Recent advances in the field of biotechnology have significantly influenced the expansion of oncolytic viruses with the inclusion of multiple species of such life forms and the enhancement of their capabilities. Common species of viruses applied in cancer treatment include the herpes simplex virus, adenovirus, vaccinia virus, reovirus, and others. The choice of a specific species as well as the specific mutations that enhance its specificity and effectiveness, is mostly driven by the characteristics of the patients as well as the underlying malignancy [320]. As illustrated in Fig. 4, in the context of hematological malignancies, the myxoma virus and the herpes simplex virus have long shown selective effectiveness against AML stem cells whilst neither affecting normal stem or progenitor blood cells, nor being a cause for concern for systemic infections [321–323]. However, only reovirus (RV), measles (MV) and vesicular stomatitis virus (VSV) have entered clinical trial assessment for multiple myeloma, leukemia and T cell lymphoma [324].
Figure 4: Ways that different viruses used in oncolytic virotherapy induce cell death in HCSCs. In this illustration, the vaccinia virus induces autophagy through the expression of the introduced additional BECN1 genes and the downstream formation of complex with existing and cytoplasm located UVRAG and VPS34 and VPS15 that leads to the production of autolysosomes [344]. VSV is depicted leading to enhanced production of IFN-β which paves the way for the recruitment of naive T cells as well as the increased prevelence of NIS on the membrane that leads to enhanced uptake of radiotracers by cancer cells. Additionally, MV carries out elimination by the induction of IFN-α production which enhances macrophage aggressiveness against TRAIL-R expressing cells, like HCSCs and/or by causing cell lysis, as part of its life cycle. Finally, CMV initiates apoptosis via caspase dependent mechanisms. The image was created using the platform biorender.com (accessed on 15 July 2025)
The vaccinia virus is a promising oncolytic virus as it has already shown effectiveness against U266, HL60, K526 and RPMI-8226 multiple myeloma cell lines. As it has been carrying mutation that includes the introduction of the Beclin-1 encoding gene BECN-1, an autophagy regulating factor, it inhibited cell growth and cell. Those results were translatable in mice as the vaccinia virus was proficient in reducing tumor burden and prolonging life in myeloma models [325].
Additionally, VSV has been clinically assessed by Cook et al. for the treatment of T cell lymphoma. In that study, the viral genetic material was mutated to encode Interferon-β (IFN-β) to enhance specificity and inflammation on the site of action as well as sodium iodine symporters (NIS) that allows the virions to be traceable using single photon emission computed tomography (SPECT) and positron emission tomography (PET) via the profound uptake of radiotracers. Despite the small size of the group studied, there was a huge extent in the variety of the clinical response and relapse of the patients in the tolerable dose of maximum 1.7 × 1011 Tissue Culture Infectious Dose 50% TCID50, with most of them showing at least partial response [326].
3.5.3 MV & Cytomegalovirus (CMV)
The MV has historically been associated with tumor burden decrease in hematological malignancies. Through its activity, MV not only causes lysis of leukemic cells, but also promotes the excretion of Interferon-α (IFN-α) that acts as an inducer of the expression of TNF-related apoptosis inducing ligand (TRAIL) in DCs and renders them more aggressive against TRAIL receptor expressing sells, say for the Jurkat and the cutaneous T cell lymphoma ones [327]. Evidence from the clinical study of Packiriswamy et al. has proposed the inefficacy of current oncolytic virotherapies against multiple myeloma. In their trial, only one out of 10 patients achieved a complete response, being remission-free for 9 months, and one other had achieved a partial response. The rest of the participants didn’t show any clinically relevant response. Although the remission-free period can span over a significant timespan, explained by the conditioning of the T cells, there is little to no likelihood of complete eradication of malignancy and future remissions are characterized as more treatment-resistant and aggressive [328], probably due to the adaptation of residual cancer cells, many of which are HCSCs. Novel advances have suggested that CMV can be an important tool against leukemias as it has shown proapoptotic activity against leukemic cells such as Kaumi-1 and SD-1 through a caspase-dependent mechanism, thus reducing relapse post-transplantation [329].
Much like with antibody-involving interventions addressed previously, virotherapies have also been tested in combination therapies, for example with CAR-T cells to enhance immunogenicity and overall malignancy clearance [330,331] and as bispecific targeting agents. An instance of the latter is the bispecific engager vaccinia virus targeting CD3 and CD19 which in vitro and in mice can successfully activate and direct T cells against Raji, Pfeifer (representing diffuse cell lymphoma and B-NHL), Farage (representing B-NHL), and K562/CD19 cell lines more effectively than bispecific antibodies [332]. Additionally, the ability of oncolytic viruses to engage with extensive specificity has rendered them applicable in ASCT adjuvant. Ideally, the viruses selected shall be specific in their targeting, leaving the graft intact, able to penetrate the TME, unable to cause extensive T cell activation that could spark an immune reaction against the graft and safe in its application, especially for the immunocompromised profile most associated with the patients receiving it. Having those criteria in mind, and based on preclinical and, to no such extent, clinical evidence, the best candidates seem to be the myxoma virus, smallpox, preferably only for use ex vivo, adenoviruses, H1 Parvovirus (H-1PV), CMV, Coxsackievirus A21 (CVA21), and VSV [333].
Despite the positive trends, there are several obstacles directly linked to OVT that need to be overcome. Given that localization hematological malignancies is ab initio unfeasible, the only possible route of administration is intravenous. However, exposure of the virus to the circulation increases the risk of systemic adverse effects, insufficient homing due to extensive distribution, and elimination by elements of the immune system. Ways to tackle these issues include, conditioning of the patient’s immune system, and especially the complement, the genetic engineering of the virus to increase targeting of cancer cells, strategies to enhance spread amongst cancer cells as well as more efficient and controlled delivery systems. The latter includes exosomes, synthetic carriers and cellular carriers that act as “Trojan Horses” [334]. Synthetic carriers include nanoparticles that can be segregated based on their structure. Lipid nanocarriers include liposomes, solid lipid nanoparticles, and micelles whilst examples of polymeric nanoparticles are polymers and dendrimers, and metal nanocarriers include gold colloidal nanoparticles, magnetosomes that can also recruit carbon nanotubes, and iron oxide nanoparticles [335]. These nanoparticles can be further optimized via the inclusion of surface proteins, glycoproteins and other relevant biomolecules [336]. In general, it has been preclinically proven that such nanoparticles can effectively carry the intended viral genome in solid tumors [337], however, solid proof of concept neither in relation to hematological malignancies nor in the clinical setting could be located [338]. Extracellular vesicles seem to also fulfill the need for protection of the virus as the “two-step vectorization” allows for the use of the immunoinhibitory molecules located on the surfaces of cancer cells, such as CD47, and others targeted by checkpoint inhibitors [339,340]. Finally, cell carriers like MSCs can provide both protection and better targeting capabilities to the oncolytic viruses they carry. MSCs are distinguished for their ability to effectively locate important sites for the cancer, usually containing HCSCs, via chemotaxis, as well as immunosuppression helpful in preventing antiviral immune response. Thus, the OVT, is rendered more targeted and many of its core shortcomings are addressed, with several studies reporting improvement in the results of antileukemic activity in AML [341], ALL [342], lymphoma, and myeloma. Viruses of significance to this study that have shown to be benefited by MSC-based delivery are Herpes Simplex Virus (HSV), MV, and the myxoma virus [343].
Despite all efforts mentioned previously as well as others beyond the scope of this study, HCSCs remain one of the primary sources of, and reasons behind, the shortcomings of multiple immunotherapeutic interventions either during the treatment or in the form of relapse, even upon molecular remission has been achieved [345]. HCSCs possess the capability of quickly adapting to changes in their environment, especially the ones threatening their survival, such as therapies. Several tools to achieve resistance include the upregulation of certain surface biomarkers, depending on the perceived threat, change of epitope(s) of targeted surface antigens (examples being PD-L1, CTLA-4, CD47, CD80, the latter catering to the exhaustion of T cells), thus dampening the affinity [346,347].
Similarly, the effectiveness of cellular immunotherapies can be comprehended by the HCSCs. The environment promoted by and promoting the HCSCs is hostile toward T cell infiltration as well as to other elements of adaptive immunity. It is widely stated that HCSCs lack NKG2D ligands, most probably through the activity of the poly(ADP-ribose) polymerase 1 (PARP1) chromatin remodeling molecule, as its inhibition seems to reverse this effect, thus rendering themselves resistant to NK cell-mediated killing and therapies involving this kind of cells [348,349]. Cellular cytotoxicity can also be achieved by the small molecular inhibition of the obesity-associated protein as its activity is associated with the activity of the LILRB4 immune checkpoint gene that suppresses T cell cytotoxicity [350].
A significant aspect to be addressed is the combination of resistance to the immune system and its relationship with drug resistance of any kind. Considering that resistance stems from the alteration of several key pathways, accumulation of these can lead to multiple resistance, and, as such, resistance to a chemotherapeutic intervention or mechanisms of immunity acts as an indicator of resistance to immunotherapy, and vice versa [351]. As such, and also considering the aforementioned diversity and plasticity observed amongst HCSC populations, the need for combination therapies including multiple targets arises when addressing HCSCs [352]. Another approach would be to target new/altered antigens using CAR-T cells, as was achieved by Niswander et al. [353]. Newly developed/clinically applied techniques can act as a cornerstone in this pursuit. Such as DC-based cancer vaccines (with limited results during translation), adoptive NK cell therapy by taking advantage of the reduced expression of MHC, and exploring similarities that HCSCs possess to cells infected with viruses as well as ways to address the TME [354]. However, there is still a high chance of leukemic cells sustaining MRD by evading such approaches through the development of checkpoint molecules that can be addressed using inhibitors [355], such as anti-PD-L1 and CTLA-4 mAbs. It is already proven that HCSCs overexpress CXCR4 molecules that through the CXCL12/CXCR4 axis reduce the approach/infiltration of CAR-T cells into the bone marrow [356]. In the context of today’s age of personalized medicine, immunotherapies can be optimized for each patient individually by assessing the immunological profile of the malignancy as well as the patient entirely. Given the promises of this approach, there is a clear need for the acceleration of the clinical development of these techniques to gain data that will support evidence-based interventions [357]. Despite the concerns surrounding the development of resistance to immunotherapy, due to post-exposure plasticity, the evidence still supports its, along with other targeted approaches’, superiority to chemotherapy for the clearance of HCSCs [358].
4 Immunotherapy in Hematological Malignancies: Clinical Trials in Patients Undergoing HSCT
The field of immunotherapy for hematologic malignancies is rapidly advancing, with more than 500 active clinical trials exploring a variety of innovative approaches. The table below, shows representative clinical trials for patients undergoing HSCT, identified using the key terms “hematologic malignancies,” “stem cell transplantation,” and three major classes of immunotherapeutic agents—monoclonal and bispecific antibodies (bsAbs), CAR-T cells and ICIs—to address challenges such as relapse, therapy resistance, and MRD.
CAR-T cell therapies epitomize the design of precision immunotherapies for hematologic malignancies by redirecting T cells to target tumor-specific antigens. Notable examples include CD7-directed CAR-T therapy (NCT05827835) for CD7-positive T cell malignancies and CD5-directed CAR-T therapy (NCT06316856) for T cell lymphoma, utilizing cells derived from autologous, prior transplant donor, or newly matched donor sources. In multiple myeloma, CAR-T products such as idecabtagene vicleucel (anti-BCMA CAR-T; NCT05393804) target BCMA to eliminate residual HCCs post-HSCT. These therapies are characterized by their ability to produce deep and durable remissions in heavily pretreated patient populations. However, challenges like relapse due to antigen escape and severe toxicities such as cytokine release syndrome (CRS) and neurotoxicity remain focal points for further optimization.
mAbs have become a cornerstone of cancer immunotherapy, recognized for their targeted activity and versatility. Antibodies such as daratumumab (anti-CD38; NCT03477539) and inotuzumab ozogamicin (anti-CD22; NCT06861348) have demonstrated the potential to reduce MRD and enhance the depth of response in multiple myeloma when integrated pre- and post-transplant as part of conditioning and consolidation regimens. Bispecific antibodies, like blinatumomab (anti-CD19/CD3; NCT02458014, NCT02101853), function by redirecting immune engagement, specifically bridging T cells (via CD3) to HCC antigens, thus amplifying anti-malignancy responses. These innovations are particularly useful in transplant settings to augment GVL effects without increasing GVHD risks. Their unique properties, including dual-targeting capacity, make them promising candidates for further integration into conditioning or post-transplant maintenance protocols.
ICIs have been widely explored in hematologic malignancies due to their ability to reverse immunosuppressive signaling within the tumor microenvironment. Trials such as NCT04214249 investigate pembrolizumab (anti-PD-1) combined with chemotherapy in AML to achieve MRD-negative remission before HSCT. Similarly, combinations of ICIs like nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4; NCT02879695) are leveraging synergistic mechanisms to enhance anti-tumor immunity pre- and post-transplant. Despite their efficacy, immune-related adverse events (irAEs) and resistance remain challenges, necessitating further research into optimal combinations and biomarkers to predict response.
While this table focuses on immunotherapies in HSCT, many other trials aim to extend these therapies to transplant-ineligible populations, including patients with advanced age, comorbidities, or disease severity. For instance, anti-BCMA CAR-T therapies (e.g., NCT05632380) and bispecific antibodies (e.g., elranatamab, NCT05317416) are being explored as standalone treatments for multiple myeloma outside the transplant setting. Similarly, checkpoint inhibitors are being tested in various disease states to restore immune surveillance and provide durable responses. These ongoing efforts complement HSCT-based strategies by broadening the accessibility of immunotherapy to historically underserved patient populations.
With further refinements to these therapies—such as novel CAR constructs, next-generation mAbs, and multi-specific antibodies—there is enormous potential to enhance durability of remission, improve survival, and overcome the remaining challenges of MRD, antigen escape, and immune evasion, offering meaningful progress toward a future of more personalized, effective treatment paradigms in hematologic malignancies. Additionally, to gain a better understanding of the translatability of the knowledge gained in the clinic, clinical trials are presented and summarized in Table 2.
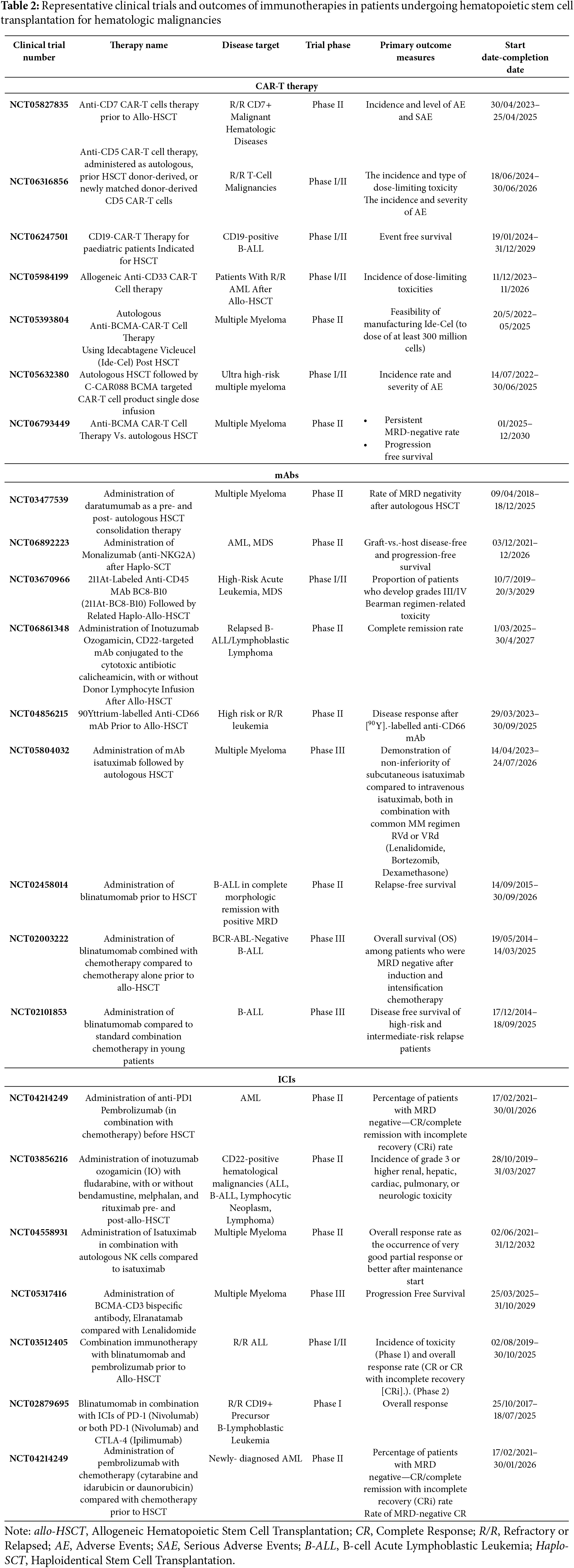
The expanded understanding of HCSCs i has emerged as a central focus in hematologic oncology today. This review highlighted how these therapy-resistant, self-renewing, and immune-evasive subpopulations present a significant obstacle to overcoming disease persistence, progression, and relapse. Apart from these properties, HCSCs are well-integrated into immunosuppressive microenvironments, which have often resulted in the ineffectiveness of most of the immunotherapy developments against HCSCs, old and even new targeted therapies [359]. Research in the HCSC field appears poised for a major inflection point in 2025. This shift is marked by several trends: first, an emerging consensus on multi-targeted immunotherapeutic strategies that would take cancer heterogeneity and adaptive resistance into consideration [360]. Second, the real-time identification of personalized HCSC markers and signaling dependencies through integration of omics [5]. Third, the next generation of cell therapies—such as dual CAR-T and CAR-NK cells—is being built up to evade antigen escape while reducing toxicity [361].
HCSCs demonstrate certain characteristics-functional and molecular-that make their removal very difficult. One of them is the occurrence of quiescence, some high capacity to repair DNA, metabolic flexibility, and epigenetic plasticity-all of which confer protection to the cells regarding chemotherapeutic agents and ICIs, as well. Surface marker plasticity, one of the most disturbing challenges [362], would make mAb therapies and CAR-T targeting unworthy of use. Examples are CD33 and CD123, both of which represent HCSC markers studied but express at different levels during disease progression and treatment pressure. Therefore, a single-target intervention by itself may prove to be insufficient [163]. This realization has pushed the field to multiparametric and combinatorial therapies, such as bispecific antibodies and CAR constructs. Besides, TME immunosuppressive bone marrow and lymphoid tissues top the list concerning HCSCs protection. MDSCs, Tregs, and tumor-associated macrophages produce «immune-privileged» niches [363]. These secret TGF-β, IL-10, and VEGF, establishing hypoxic and tolerogenic environments [364]. The very agents that remodel the TME, such as the VEGF inhibitors [365]. CXCL12 antagonists [366] and hypoxia modulators [367] are now beginning to be applied as adjuvants to immunotherapy to unmask and expose HCSCs to the cytotoxic attack.
Potential agents targeting HCSCs for immunotherapy have demonstrated encouraging data in early-phase clinical testing. The high-flying anti-CD47 antibody magrolimab has run into rough waters following the early cessation of its Phase III study ENHANCE-3 [144]. This reinforced the need for the clinical safety profiling of agents in the context of safety before initiation, especially in molecules that are expressed on healthy cells as well. By contrast, CD70-targeted therapies like cusatuzumab are still showing great promise [231], particularly in combination with either azacitidine or venetoclax [233]. It is therefore a good target for selective immunotoxicity since CD70 is differentially expressed on HCSCs and absent on normal HSCs. Likewise bispecific antibodies targeting CD33/CD3 and CD123/CD3 (for instance, AMG330, APVO436) are also being perfected to mitigate cytokine release syndromes while preserving the efficacy against leukemia [166,167]. Epigenetic regulators are becoming increasingly popular as therapeutic sensitizers. Durable responses in MDS and AML are being achieved with combinations of TIM-3 inhibitors such as sabatolimab and hypomethylating agents [211,212]. These strategies leverage the interaction between epigenetic plasticity and immune escape mechanics. This lays the foundation for immunoepigenetic precisive therapy. Another major development is the application of ADCs and immune engagers. The latest generation of ADCs-including but not limited to IMGN632, which targets CD123 [180] and vadastuximab talirine, which targets CD33-are effective but badly without improving these competing linkers and payloads [159]. In addition, dual-antigen-targeting CAR-Ts (e.g., CD19/CD22 or CD123/CD33) will be designed to circumvent loss of targeted antigens toward deeper remissions [291].
Hematopoietic stem cell transplantation is a primary therapy for patients who are either high risk or have relapsed disease. The conditioning regimen used is crucial because it depletes endogenous HCSCs; it can make or break the HSCT success. Anti-CD117 and/or anti-CD45 antibodies, such as MGTA-117 or baricitinib conjugates, are now seen as possible alternatives to myeloablative chemotherapy for conditioning regimens [197,198]. It has selective capabilities in emptying cancer and host stem cells but would leave healthy tissue intact, thus making the conditioning reduced toxicity. Gene-edited induced pluripotent stem cells (iPSCs) and universal donor grafts are being evaluated in the very early phases of clinical trials. All of these are now expected to combine with immunomodulation and lead to reduction of graft-vs.-host disease while still maintaining activity against leukemia in the patient’s tissue. Furthermore, the improved risk stratification and personalization of transplantation protocols can be seen with the introduction of the real-time MRD monitoring using liquid biopsies.
More recent innovations such as NGS-based single-cell profiling, mass cytometry, and spatial transcriptomics, are coming into play to dissect the HCSC landscape at unprecedented resolution [154]. These techniques assist in defining HCSC subtypes, tracking clonal evolution, and predicting therapeutic vulnerabilities. This greatly aids in creating personalized immunotherapies against individual neoantigens or signaling dependencies. Another thrilling avenue is the one that uses machine learning predictive modelling to integrate genomic, epigenetic, and clinical data for optimizing immunotherapy regimens [368,369]. There is also a revision of oncolytic viruses [320] and RNA-based vaccines [317], with a focus on eradication of MRD and awakening immune responses against dormant HCSCs. The engineered viruses that can express immune-stimulating cytokines (IL-12, granulocyte macrophage colony stimulating factor (GM-CSF)) or deliver checkpoint inhibitors (anti-PD-L1 in situ) are currently under investigation [370].
Nevertheless, several limitations continue to challenge the application of HCSC-targeted immunotherapy approaches. Firstly, the tumor heterogeneity occurring not only between different patients but also within the same patient precludes any “one-size-fits-all” concept. Despite its potential to provide immense benefits for certain patients, personalized immunotherapy is restrained due to cost, accessibility, and regulatory hurdles [371]. Second, immune escape mechanisms, thus, require flexibility–HCSCs can downregulate antigen expression, secrete immunosuppressive molecules, or use checkpoint pathways to escape [362]. Patients need to be treated using integrated and adaptive immunotherapeutic regimens—an option that is slowly being embraced by current clinical protocols. Third, ethical and safety issues continue to hinder the clinical translation of gene editing, allogeneic cell therapy, and synthetic biology [372]. Increasingly, regulators demand long-term safety data for irreversible interventions targeting stem-like cells. Finally, most of the promising strategies discussed in this review remain in early-phase clinical development. While preclinical results have been encouraging, achieving clinical success in patient populations with aggressive or treatment-refractory disease remains a major hurdle.
The gradually evolving landscape of hematological malignancies has underscored the significant role of HCSCs in disease initiation, development, therapy resistance, and relapse. These subpopulations, whose distinguishing features include self-renewal, immune evasion mechanisms, and plasticity, present formidable challenges to all conventional treatment strategies [373]. Regardless of the advances in immunotherapeutic measures, the existence of residual HCSCs remains a predominant cause of therapeutic failure and disease recurrence. As of 2025, immunotherapy has become a key therapeutic modality in hematologic oncology, with an expanding armamentarium of mAbs, bispecific T cell engagers, antibody-drug conjugates, and CAR-based cellular therapies [374]. What makes the present era of immunotherapy distinctive is the trend toward precision and personalization. Therapies are increasingly designed to engage multiple antigens simultaneously to dampen the risk of immune escape and grill microenvironmental modulators to abrogate niche-mediated resistance [375]. Of note is the characterization of HCSCs selectively enriched markers, leading to therapies with much greater specificity and lower systemic toxicity. Early trials showed promising efficacy for these agents, especially in combination with hypomethylating agents or targeted small molecules.
Nevertheless, challenges remain. Inter-and intrapatient variability in malignancy heterogeneity greatly hamper the development of a universal therapeutic approach. Antigen modulation by HCSCs and the immunosuppressive environment often inhibit therapeutic efficacy. The fruitful clinical translation of many promising preclinical strategies is obstructed by factors of concern such as on-target off-tumor toxicity, immune-related adverse events, and access to advanced therapies in the real world. The establishment of more versatile and multi-modal strategies capable of addressing the dynamic and evasive nature of HCSCs will dictate the future of immunotherapy for these cells. Therefore, the future integration of high-throughput single-cell sequencing, spatial transcriptomics, and proteogenomic profiling into clinical practice [376], must occur to delineate HCSC heterogeneity and therapeutic vulnerabilities accurately [377]. Moreover, the implementation of artificial intelligence and machine learning models for predicting immunotherapy outcomes, optimizing antigen selection, and making patient-specific treatment recommendations is probably being initiated [378]. Furthermore, new strategies including oncolytic viruses, RNA vaccine, and CARs with logic gating are extensively being studied, aimed at overcoming current challenges and widening the therapeutic window.
The regulatory and ethical aspects of HCSC-targeted therapies are equally critical. Gene-edited allogeneic cell products and engineered stem cell grafts must therefore be responsibly implemented in clinical settings with long-term safety monitoring in order to minimize risk and maximize therapeutic benefit [379]. Finally, future immunotherapies should be developed in settings that account for patient diversity, disease stage, and healthcare infrastructure to ensure equitable access and successful clinical translation [380].
In conclusion, targeting HCSCs represents one of the most promising advances toward achieving lasting remission and potential cure in hematologic malignancies. Next-generation immunotherapies—enabled by advances in molecular profiling, synthetic biology, and systems immunology—can shift the paradigm in blood cancers by overcoming current limitations. The ultimate goal is not to prolong survival, but to completely and permanently eradicate the disease at its root: the hematological cancer stem cell.
Acknowledgement: Not applicable.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm contribution to the paper as follows: Conceptualization, Sotirios Charalampos Diamantoudis, Androulla N. Miliotou, Eleftheria Galatou, Stergiani Telliou, Konstantinos Sideris, Nikolaos Grigoriadis, Ioannis S. Vizirianakis; methodology, Sotirios Charalampos Diamantoudis, Androulla N. Miliotou, Eleftheria Galatou, Stergiani Telliou, Konstantinos Sideris, Nikolaos Grigoriadis, Ioannis S. Vizirianakis; formal analysis, Sotirios Charalampos Diamantoudis, Androulla N. Miliotou, Eleftheria Galatou, Stergiani Telliou, Konstantinos Sideris, Nikolaos Grigoriadis, Ioannis S. Vizirianakis; investigation, Sotirios Charalampos Diamantoudis, Androulla N. Miliotou, Eleftheria Galatou, Stergiani Telliou, Konstantinos Sideris, Nikolaos Grigoriadis, Ioannis S. Vizirianakis; resources, Sotirios Charalampos Diamantoudis, Androulla N. Miliotou, Eleftheria Galatou, Stergiani Telliou, Konstantinos Sideris, Nikolaos Grigoriadis, Ioannis S. Vizirianakis; data curation, Sotirios Charalampos Diamantoudis; writing—original draft preparation, Sotirios Charalampos Diamantoudis, Androulla N. Miliotou, Eleftheria Galatou, Stergiani Telliou, Konstantinos Sideris, Nikolaos Grigoriadis, Ioannis S. Vizirianakis; writing—review and editing, Sotirios Charalampos Diamantoudis, Androulla N. Miliotou, Eleftheria Galatou, Stergiani Telliou, Konstantinos Sideris, Nikolaos Grigoriadis, Ioannis S. Vizirianakis; visualization, Sotirios Charalampos Diamantoudis, Androulla N. Miliotou; supervision, Ioannis S. Vizirianakis; project administration, Sotirios Charalampos Diamantoudis, Androulla N. Miliotou, Ioannis S. Vizirianakis. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Zhang N, Wu J, Wang Q, Liang Y, Li X, Chen G, et al. Global burden of hematologic malignancies and evolution patterns over the past 30 years. Blood Cancer J. 2023;13(1):82. doi:10.1038/s41408-023-00853-3. [Google Scholar] [PubMed] [CrossRef]
2. Smith A, Howell D, Patmore R, Jack A, Roman E. Incidence of haematological malignancy by sub-type: a report from the haematological malignancy research network. Br J Cancer. 2011;105(11):1684–92. doi:10.1038/bjc.2011.450. [Google Scholar] [PubMed] [CrossRef]
3. Bispo JAB, Pinheiro PS, Kobetz EK. Epidemiology and etiology of leukemia and lymphoma. Cold Spring Harb Perspect Med. 2020;10(6):a034819. doi:10.1101/cshperspect.a034819. [Google Scholar] [PubMed] [CrossRef]
4. Stiller C. Epidemiology of pediatric cancer. In: Pediatric surgical oncology. 1st ed. Boca Raton, FL, USA: CRC Press; 2022. p. 3–9. doi:10.1201/9781351166126-2. [Google Scholar] [CrossRef]
5. Cazzola M, Sehn LH. Developing a classification of hematologic neoplasms in the era of precision medicine. Blood. 2022;140(11):1193–9. doi:10.1182/blood.2022015849. [Google Scholar] [PubMed] [CrossRef]
6. Zvara Á, Hackler L, Nagy BZ, Micsik T, Puskás LG. New molecular methods for classification, diagnosis and therapy prediction of hematological malignancies. Pathol Oncol Res. 2002;8(4):231–40. doi:10.1007/BF03036737. [Google Scholar] [PubMed] [CrossRef]
7. Taylor J, Xiao W, Abdel-Wahab O. Diagnosis and classification of hematologic malignancies on the basis of genetics. Blood. 2017;130(4):410–23. doi:10.1182/blood-2017-02-734541. [Google Scholar] [PubMed] [CrossRef]
8. Clara JA, Monge C, Yang Y, Takebe N. Targeting signalling pathways and the immune microenvironment of cancer stem cells-a clinical update. Nat Rev Clin Oncol. 2020;17(4):204–32. doi:10.1038/s41571-019-0293-2. [Google Scholar] [PubMed] [CrossRef]
9. Vetrie D, Helgason GV, Copland M. The leukaemia stem cell: similarities, differences and clinical prospects in CML and AML. Nat Rev Cancer. 2020;20(3):158–73. doi:10.1038/s41568-019-0230-9. [Google Scholar] [PubMed] [CrossRef]
10. Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer stem cells. Int J Biochem Cell Biol. 2012;44(12):2144–51. doi:10.1016/j.biocel.2012.08.022. [Google Scholar] [PubMed] [CrossRef]
11. Bamodu OA, Chung CC, Pisanic TR, Wu ATH. The intricate interplay between cancer stem cells and cell-of-origin of cancer: implications for therapeutic strategies. Front Oncol. 2024;14:1404628. doi:10.3389/fonc.2024.1404628. [Google Scholar] [PubMed] [CrossRef]
12. Makena MR, Ranjan A, Thirumala V, Reddy AP. Cancer stem cells: road to therapeutic resistance and strategies to overcome resistance. Biochim Biophys Acta Mol Basis Dis. 2020;1866(4):165339. doi:10.1016/j.bbadis.2018.11.015. [Google Scholar] [PubMed] [CrossRef]
13. Lim JR, Mouawad J, Gorton OK, Bubb WA, Kwan AH. Cancer stem cell characteristics and their potential as therapeutic targets. Med Oncol. 2021;38(7):76. doi:10.1007/s12032-021-01524-8. [Google Scholar] [PubMed] [CrossRef]
14. Li F, Zhou K, Gao L, Zhang B, Li W, Yan W, et al. Radiation induces the generation of cancer stem cells: a novel mechanism for cancer radioresistance. Oncol Lett. 2016;12(5):3059–65. doi:10.3892/ol.2016.5124. [Google Scholar] [PubMed] [CrossRef]
15. Mayani H, Chávez-González A, Vázquez-Santillan K, Contreras J, Guzman ML. Cancer stem cells: biology and therapeutic implications. Arch Med Res. 2022;53(8):770–84. doi:10.1016/j.arcmed.2022.11.012. [Google Scholar] [PubMed] [CrossRef]
16. Long NA, Golla U, Sharma A, Claxton DF. Acute myeloid leukemia stem cells: origin, characteristics, and clinical implications. Stem Cell Rev Rep. 2022;18(4):1211–26. doi:10.1007/s12015-021-10308-6. [Google Scholar] [PubMed] [CrossRef]
17. Choi HS, Kim BS, Yoon S, Oh SO, Lee D. Leukemic stem cells and hematological malignancies. Int J Mol Sci. 2024;25(12):6639. doi:10.3390/ijms25126639. [Google Scholar] [PubMed] [CrossRef]
18. Kirtonia A, Pandya G, Sethi G, Pandey AK, Das BC, Garg M. A comprehensive review of genetic alterations and molecular targeted therapies for the implementation of personalized medicine in acute myeloid leukemia. J Mol Med. 2020;98(8):1069–91. doi:10.1007/s00109-020-01944-5. [Google Scholar] [PubMed] [CrossRef]
19. Nuno K, Azizi A, Koehnke T, Lareau C, Ediriwickrema A, Corces MR, et al. Convergent epigenetic evolution drives relapse in acute myeloid leukemia. eLife. 2024 Apr 22;13:e93019. doi:10.7554/eLife.93019. [Google Scholar] [PubMed] [CrossRef]
20. The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74. doi:10.1056/nejmoa1301689. [Google Scholar] [PubMed] [CrossRef]
21. Kadia TM, Ravandi F, Cortes J, Kantarjian H. Toward individualized therapy in acute myeloid leukemia. JAMA Oncol. 2015;1(6):820. doi:10.1001/jamaoncol.2015.0617. [Google Scholar] [PubMed] [CrossRef]
22. Hackl H, Astanina K, Wieser R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J Hematol Oncol. 2017;10(1):51. doi:10.1186/s13045-017-0416-0. [Google Scholar] [PubMed] [CrossRef]
23. Chakraborty S, Park CY. Pathogenic mechanisms in acute myeloid leukemia. Curr Treat Options Oncol. 2022;23(11):1522–34. doi:10.1007/s11864-022-01021-8. [Google Scholar] [PubMed] [CrossRef]
24. Wachter F, Pikman Y. Pathophysiology of acute myeloid leukemia. Acta Haematol. 2024;147(2):229–46. doi:10.1159/000536152. [Google Scholar] [PubMed] [CrossRef]
25. von Bergh ARM, van Drunen E, van Wering ER, van Zutven LJCM, Hainmann I, Lönnerholm G, et al. High incidence of t(7;12)(q36;p13) in infant AML but not in infant ALL, with a dismal outcome and ectopic expression of HLXB9. Genes Chromosomes Cancer. 2006;45(8):731–9. doi:10.1002/gcc.20335. [Google Scholar] [PubMed] [CrossRef]
26. Blecua P, Martinez-Verbo L, Esteller M. The DNA methylation landscape of hematological malignancies: an update. Mol Oncol. 2020;14(8):1616–39. doi:10.1002/1878-0261.12744. [Google Scholar] [PubMed] [CrossRef]
27. Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39(2):232–6. doi:10.1038/ng1950. [Google Scholar] [PubMed] [CrossRef]
28. Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, et al. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300(5616):131–5. doi:10.1126/science.1084274. [Google Scholar] [PubMed] [CrossRef]
29. Yildirim E, Kirby JE, Brown DE, Mercier FE, Sadreyev RI, Scadden DT, et al. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell. 2013;152(4):727–42. doi:10.1016/j.cell.2013.01.034. [Google Scholar] [PubMed] [CrossRef]
30. Lee JE, Kim MY. Cancer epigenetics: past, present and future. Semin Cancer Biol. 2022;83:4–14. doi:10.1016/j.semcancer.2021.03.025. [Google Scholar] [PubMed] [CrossRef]
31. Ghafouri-Fard S, Niazi V, Taheri M. Role of miRNAs and lncRNAs in hematopoietic stem cell differentiation. Noncoding RNA Res. 2020;6(1):8–14. doi:10.1016/j.ncrna.2020.12.002. [Google Scholar] [PubMed] [CrossRef]
32. Carrà G, Cartellà A, Maffeo B, Morotti A. Strategies for targeting chronic myeloid leukaemia stem cells. Blood Lymphat Cancer. 2019;9:45–52. doi:10.2147/BLCTT.S228815. [Google Scholar] [PubMed] [CrossRef]
33. Carrà G, Torti D, Crivellaro S, Panuzzo C, Taulli R, Cilloni D, et al. The BCR-ABL/NF-κB signal transduction network: a long lasting relationship in Philadelphia positive Leukemias. Oncotarget. 2016;7(40):66287–98. doi:10.18632/oncotarget.11507. [Google Scholar] [PubMed] [CrossRef]
34. Zhu X, Wang L, Zhang B, Li J, Dou X, Zhao RC. TGF-beta1-induced PI3K/Akt/NF-κB/MMP9 signalling pathway is activated in Philadelphia chromosome-positive chronic myeloid leukaemia hemangioblasts. J Biochem. 2011;149(4):405–14. doi:10.1093/jb/mvr016. [Google Scholar] [PubMed] [CrossRef]
35. Gallipoli P, Pellicano F, Morrison H, Laidlaw K, Allan EK, Bhatia R, et al. Autocrine TNF-α production supports CML stem and progenitor cell survival and enhances their proliferation. Blood. 2013;122(19):3335–9. doi:10.1182/blood-2013-02-485607. [Google Scholar] [PubMed] [CrossRef]
36. Anusha, Dalal H, Subramanian S, Snijesh VP, Gowda DA, Krishnamurthy H et al. Exovesicular-Shh confers Imatinib resistance by upregulating Bcl2 expression in chronic myeloid leukemia with variant chromosomes. Cell Death Dis. 2021;12(3):259. doi:10.1038/s41419-021-03542-w. [Google Scholar] [PubMed] [CrossRef]
37. Liu Z, Xu J, He J, Zheng Y, Li H, Lu Y, et al. A critical role of autocrine sonic hedgehog signaling in human CD138+ myeloma cell survival and drug resistance. Blood. 2014;124(13):2061–71. doi:10.1182/blood-2014-03-557298. [Google Scholar] [PubMed] [CrossRef]
38. Ghia EM, Rassenti LZ, Neuberg DS, Blanco A, Yousif F, Smith EN, et al. Activation of hedgehog signaling associates with early disease progression in chronic lymphocytic leukemia. Blood. 2019;133(25):2651–63. doi:10.1182/blood-2018-09-873695. [Google Scholar] [PubMed] [CrossRef]
39. Ramirez E, Singh RR, Kunkalla K, Liu Y, Qu C, Cain C, et al. Defining causative factors contributing in the activation of hedgehog signaling in diffuse large B-cell lymphoma. Leuk Res. 2012;36(10):1267–73. doi:10.1016/j.leukres.2012.06.014. [Google Scholar] [PubMed] [CrossRef]
40. Lemos T, Merchant A. The hedgehog pathway in hematopoiesis and hematological malignancy. Front Oncol. 2022;12:960943. doi:10.3389/fonc.2022.960943. [Google Scholar] [PubMed] [CrossRef]
41. Martelli AM, Paganelli F, Truocchio S, Palumbo C, Chiarini F, McCubrey JA. Understanding the roles of the hedgehog signaling pathway during T-cell lymphopoiesis and in T-cell acute lymphoblastic leukemia (T-ALL). Int J Mol Sci. 2023;24(3):2962. doi:10.3390/ijms24032962. [Google Scholar] [PubMed] [CrossRef]
42. Su W, Meng F, Huang L, Zheng M, Liu W, Sun H. Sonic hedgehog maintains survival and growth of chronic myeloid leukemia progenitor cells through β-catenin signaling. Exp Hematol. 2012;40(5):418–27. doi:10.1016/j.exphem.2012.01.003. [Google Scholar] [PubMed] [CrossRef]
43. Malhotra S, Kincade PW. Wnt-related molecules and signaling pathway equilibrium in hematopoiesis. Cell Stem Cell. 2009;4(1):27–36. doi:10.1016/j.stem.2008.12.004. [Google Scholar] [PubMed] [CrossRef]
44. Grainger S, Traver D, Willert K. Wnt signaling in hematological malignancies. In: WNT signaling in health and disease. Amsterdam, The Netherland: Elsevier; 2018. p. 321–41. doi: 10.1016/bs.pmbts.2017.11.002. [Google Scholar] [CrossRef]
45. Müller-Tidow C, Steffen B, Cauvet T, Tickenbrock L, Ji P, Diederichs S, et al. Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol Cell Biol. 2004;24(7):2890–904. doi:10.1128/MCB.24.7.2890-2904.2004. [Google Scholar] [PubMed] [CrossRef]
46. Jamieson CHM, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657–67. doi:10.1056/NEJMoa040258. [Google Scholar] [PubMed] [CrossRef]
47. Heidel FH, Bullinger L, Feng Z, Wang Z, Neff TA, Stein L, et al. Genetic and pharmacologic inhibition of β-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell. 2012;10(4):412–24. doi:10.1016/j.stem.2012.02.017. [Google Scholar] [PubMed] [CrossRef]
48. Meisel CT, Porcheri C, Mitsiadis TA. Cancer stem cells, Quo vadis? the Notch signaling pathway in tumor initiation and progression. Cells. 2020;9(8):1879. doi:10.3390/cells9081879. [Google Scholar] [PubMed] [CrossRef]
49. O’Neil J, Calvo J, McKenna K, Krishnamoorthy V, Aster JC, Bassing CH, et al. Activating Notch1 mutations in mouse models of T-ALL. Blood. 2006;107(2):781–5. doi:10.1182/blood-2005-06-2553. [Google Scholar] [PubMed] [CrossRef]
50. Ma W, Gutierrez A, Goff DJ, Geron I, Sadarangani A, Jamieson CAM, et al. NOTCH1 signaling promotes human T-cell acute lymphoblastic leukemia initiating cell regeneration in supportive niches. PLoS One. 2012;7(6):e39725. doi:10.1371/journal.pone.0039725. [Google Scholar] [PubMed] [CrossRef]
51. Duechler M, Shehata M, Schwarzmeier JD, Hoelbl A, Hilgarth M, Hubmann R. Induction of apoptosis by proteasome inhibitors in B-CLL cells is associated with downregulation of CD23 and inactivation of Notch2. Leukemia. 2005;19(2):260–7. doi:10.1038/sj.leu.2403592. [Google Scholar] [PubMed] [CrossRef]
52. Tatarek J, Cullion K, Ashworth T, Gerstein R, Aster JC, Kelliher MA. Notch1 inhibition targets the leukemia-initiating cells in a Tal1/Lmo2 mouse model of T-ALL. Blood. 2011;118(6):1579–90. doi:10.1182/blood-2010-08-300343. [Google Scholar] [PubMed] [CrossRef]
53. Herrmann H, Sadovnik I, Eisenwort G, Rülicke T, Blatt K, Herndlhofer S, et al. Delineation of target expression profiles in CD34+/CD38- and CD34+/CD38+ stem and progenitor cells in AML and CML. Blood Adv. 2020;4(20):5118–32. doi:10.1182/bloodadvances.2020001742. [Google Scholar] [PubMed] [CrossRef]
54. Järås M, Johnels P, Hansen N, Agerstam H, Tsapogas P, Rissler M, et al. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc Natl Acad Sci U S A. 2010;107(37):16280–5. doi:10.1073/pnas.1004408107. [Google Scholar] [PubMed] [CrossRef]
55. Nairuz T, Mahmud Z, Manik RK, Kabir Y. Cancer stem cells: an insight into the development of metastatic tumors and therapy resistance. Stem Cell Rev Rep. 2023;19(6):1577–95. doi:10.1007/s12015-023-10529-x. [Google Scholar] [PubMed] [CrossRef]
56. Nagare RP, Sneha S, Priya SK, Ganesan TS. Cancer stem cells-are surface markers alone sufficient? Curr Stem Cell Res Ther. 2017;12(1):37–44. doi:10.2174/1574888x11666160607211436. [Google Scholar] [PubMed] [CrossRef]
57. Jones CL, Inguva A, Jordan CT. Targeting energy metabolism in cancer stem cells: progress and challenges in leukemia and solid tumors. Cell Stem Cell. 2021;28(3):378–93. doi:10.1016/j.stem.2021.02.013. [Google Scholar] [PubMed] [CrossRef]
58. Zhang CC, Sadek HA. Hypoxia and metabolic properties of hematopoietic stem cells. Antioxid Redox Signal. 2014;20(12):1891–901. doi:10.1089/ars.2012.5019. [Google Scholar] [PubMed] [CrossRef]
59. Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7(3):380–90. doi:10.1016/j.stem.2010.07.011. [Google Scholar] [PubMed] [CrossRef]
60. Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 2013;12(1):49–61. doi:10.1016/j.stem.2012.10.011. [Google Scholar] [PubMed] [CrossRef]
61. Sriskanthadevan S, Jeyaraju DV, Chung TE, Prabha S, Xu W, Skrtic M, et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood. 2015;125(13):2120–30. doi:10.1182/blood-2014-08-594408. [Google Scholar] [PubMed] [CrossRef]
62. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12(3):329–41. doi:10.1016/j.stem.2012.12.013. [Google Scholar] [PubMed] [CrossRef]
63. Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL, et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med. 2017;23(10):1234–40. doi:10.1038/nm.4399. [Google Scholar] [PubMed] [CrossRef]
64. LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014;16(10):992–1003. doi:10.1038/ncb3039. [Google Scholar] [PubMed] [CrossRef]
65. Polak A, Bialopiotrowicz E, Krzymieniewska B, Wozniak J, Stojak M, Cybulska M, et al. SYK inhibition targets acute myeloid leukemia stem cells by blocking their oxidative metabolism. Cell Death Dis. 2020;11(11):956. doi:10.1038/s41419-020-03156-8. [Google Scholar] [PubMed] [CrossRef]
66. Mancini R, Noto A, Pisanu ME, De Vitis C, Maugeri-Saccà M, Ciliberto G. Metabolic features of cancer stem cells: the emerging role of lipid metabolism. Oncogene. 2018;37(18):2367–78. doi:10.1038/s41388-018-0141-3. [Google Scholar] [PubMed] [CrossRef]
67. Milella M, Rutigliano M, Pandolfo SD, Aveta A, Crocetto F, Ferro M, et al. The metabolic landscape of cancer stem cells: insights and implications for therapy. Cells. 2025;14(10):717. doi:10.3390/cells14100717. [Google Scholar] [PubMed] [CrossRef]
68. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. 2016;19(1):23–37. doi:10.1016/j.stem.2016.06.001. [Google Scholar] [PubMed] [CrossRef]
69. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, et al. A PML-PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18(9):1350–8. doi:10.1038/nm.2882. [Google Scholar] [PubMed] [CrossRef]
70. Khattab S, El Sorady M, El-Ghandour A, Visani G, Piccaluga PP. Hematopoietic and leukemic stem cells homeostasis: the role of bone marrow niche. Explor Target Anti Tumor Ther. 2024;5(5):1027–55. doi:10.37349/etat.2024.00262. [Google Scholar] [PubMed] [CrossRef]
71. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. doi:10.1038/nrc3245. [Google Scholar] [PubMed] [CrossRef]
72. Huang J, Zhao Y, Zhao K, Yin K, Wang S. Function of reactive oxygen species in myeloid-derived suppressor cells. Front Immunol. 2023;14:1226443. doi:10.3389/fimmu.2023.1226443. [Google Scholar] [PubMed] [CrossRef]
73. Desantis V, Savino FD, Scaringella A, Potenza MA, Nacci C, Frassanito MA, et al. The leading role of the immune microenvironment in multiple myeloma: a new target with a great prognostic and clinical value. J Clin Med. 2022;11(9):2513. doi:10.3390/jcm11092513. [Google Scholar] [PubMed] [CrossRef]
74. Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016;18(1):84. doi:10.1186/s13058-016-0740-2. [Google Scholar] [PubMed] [CrossRef]
75. Menéndez V, Solórzano JL, Fernández S, Montalbán C, García JF. The Hodgkin lymphoma immune microenvironment: turning bad news into good. Cancers. 2022;14(5):1360. doi:10.3390/cancers14051360. [Google Scholar] [PubMed] [CrossRef]
76. Aldinucci D, Gloghini A, Pinto A, Colombatti A, Carbone A. The role of CD40/CD40L and interferon regulatory factor 4 in Hodgkin lymphoma microenvironment. Leuk Lymphoma. 2012;53(2):195–201. doi:10.3109/10428194.2011.605190. [Google Scholar] [CrossRef]
77. Zhang Y, Yao G, Yang X, Qiu T, Wang S. Mechanism of targeting the hedgehog signaling pathway against chemotherapeutic resistance in multiple myeloma. J Oncol. 2022;2022(6):1399697. doi:10.1155/2022/1399697. [Google Scholar] [PubMed] [CrossRef]
78. Zhao Z, Cui T, Wei F, Zhou Z, Sun Y, Gao C, et al. Wnt/β-Catenin signaling pathway in hepatocellular carcinoma: pathogenic role and therapeutic target. Front Oncol. 2024;14:1367364. doi:10.3389/fonc.2024.1367364. [Google Scholar] [PubMed] [CrossRef]
79. Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer. 2018;17(1):57. doi:10.1186/s12943-018-0779-z. [Google Scholar] [PubMed] [CrossRef]
80. Wei Y, Li Y, Chen Y, Liu P, Huang S, Zhang Y, et al. ALDH1: a potential therapeutic target for cancer stem cells in solid tumors. Front Oncol. 2022;12:1026278. doi:10.3389/fonc.2022.1026278. [Google Scholar] [PubMed] [CrossRef]
81. Wu Y, Yi M, Niu M, Mei Q, Wu K. Myeloid-derived suppressor cells: an emerging target for anticancer immunotherapy. Mol Cancer. 2022;21(1):184. doi:10.1186/s12943-022-01657-y. [Google Scholar] [PubMed] [CrossRef]
82. Oparaugo NC, Ouyang K, Nguyen NPN, Nelson AM, Agak GW. Human regulatory T cells: understanding the role of tregs in select autoimmune skin diseases and post-transplant nonmelanoma skin cancers. Int J Mol Sci. 2023;24(2):1527. doi:10.3390/ijms24021527. [Google Scholar] [PubMed] [CrossRef]
83. Tan Y, Wang M, Zhang Y, Ge S, Zhong F, Xia G, et al. Tumor-associated macrophages: a potential target for cancer therapy. Front Oncol. 2021;11:693517. doi:10.3389/fonc.2021.693517. [Google Scholar] [PubMed] [CrossRef]
84. Schito L. Hypoxia-dependent angiogenesis and lymphangiogenesis in cancer. Adv Exp Med Biol. 2019;1136(6):71–85. doi:10.1007/978-3-030-12734-3_5. [Google Scholar] [PubMed] [CrossRef]
85. Aintablian A, Strozniak S, Heuer M, Lutz MB. M-MDSC in vitro generation from mouse bone marrow with IL-3 reveals high expression and functional activity of arginase 1. Front Immunol. 2023;14:1130600. doi:10.3389/fimmu.2023.1130600. [Google Scholar] [PubMed] [CrossRef]
86. Swain N, Thakur M, Pathak J, Swain B. SOX2, OCT4 and NANOG: the core embryonic stem cell pluripotency regulators in oral carcinogenesis. J Oral Maxillofac Pathol. 2020;24(2):368–73. doi:10.4103/jomfp.JOMFP_22_20. [Google Scholar] [PubMed] [CrossRef]
87. Wang X, Zhang H, Chen X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019;2(2):141–60. doi:10.20517/cdr.2019.10. [Google Scholar] [PubMed] [CrossRef]
88. Riether C, Schürch CM, Bührer ED, Hinterbrandner M, Huguenin AL, Hoepner S, et al. CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J Exp Med. 2017;214(2):359–80. doi:10.1084/jem.20152008. [Google Scholar] [PubMed] [CrossRef]
89. Tufail M, Hu JJ, Liang J, He CY, Wan WD, Huang YQ, et al. Hallmarks of cancer resistance. iScience. 2024;27(6):109979. doi:10.1016/j.isci.2024.109979. [Google Scholar] [PubMed] [CrossRef]
90. Bhat GR, Sethi I, Sadida HQ, Rah B, Mir R, Algehainy N, et al. Cancer cell plasticity: from cellular, molecular, and genetic mechanisms to tumor heterogeneity and drug resistance. Cancer Metastasis Rev. 2024;43(1):197–228. doi:10.1007/s10555-024-10172-z. [Google Scholar] [PubMed] [CrossRef]
91. Shi ZD, Pang K, Wu ZX, Dong Y, Hao L, Qin JX, et al. Tumor cell plasticity in targeted therapy-induced resistance: mechanisms and new strategies. Signal Transduct Target Ther. 2023;8(1):113. doi:10.1038/s41392-023-01383-x. [Google Scholar] [PubMed] [CrossRef]
92. Wu P, Gao W, Su M, Nice EC, Zhang W, Lin J, et al. Adaptive mechanisms of tumor therapy resistance driven by tumor microenvironment. Front Cell Dev Biol. 2021;9:641469. doi:10.3389/fcell.2021.641469. [Google Scholar] [PubMed] [CrossRef]
93. Liu Y, Liang J, Zhang Y, Guo Q. Drug resistance and tumor immune microenvironment: an overview of current understandings (Review). Int J Oncol. 2024;65(4):96. doi:10.3892/ijo.2024.5684. [Google Scholar] [PubMed] [CrossRef]
94. Zhang C, Fei Y, Wang H, Hu S, Liu C, Hu R, et al. CAFs orchestrates tumor immune microenvironment—a new target in cancer therapy? Front Pharmacol. 2023;14:1113378. doi:10.3389/fphar.2023.1113378. [Google Scholar] [PubMed] [CrossRef]
95. Chen G, Wu K, Li H, Xia D, He T. Role of hypoxia in the tumor microenvironment and targeted therapy. Front Oncol. 2022;12:961637. doi:10.3389/fonc.2022.961637. [Google Scholar] [PubMed] [CrossRef]
96. Pudełek M, Ryszawy D, Piwowarczyk K, Lasota S, Madeja Z, Kędracka-Krok S, et al. Metabolic reprogramming of poly(morpho)nuclear giant cells determines glioblastoma recovery from doxorubicin-induced stress. J Transl Med. 2024;22(1):757. doi:10.1186/s12967-024-05541-9. [Google Scholar] [PubMed] [CrossRef]
97. Lei ZN, Tian Q, Teng QX, Wurpel JND, Zeng L, Pan Y, et al. Understanding and targeting resistance mechanisms in cancer. MedComm. 2020 2023;4(3):e265. doi:10.1002/mco2.265. [Google Scholar] [PubMed] [CrossRef]
98. Marin JJG, Perez-Silva L, Macias RIR, Asensio M, Peleteiro-Vigil A, Sanchez-Martin A, et al. Molecular bases of mechanisms accounting for drug resistance in gastric adenocarcinoma. Cancers. 2020;12(8):2116. doi:10.3390/cancers12082116. [Google Scholar] [PubMed] [CrossRef]
99. Rodríguez-Macías G, Briz O, Cives-Losada C, Chillón MC, Martínez-Laperche C, Martínez-Arranz I, et al. Role of intracellular drug disposition in the response of acute myeloid leukemia to cytarabine and idarubicin induction chemotherapy. Cancers. 2023;15(12):3145. doi:10.3390/cancers15123145. [Google Scholar] [PubMed] [CrossRef]
100. Li LY, Guan YD, Chen XS, Yang JM, Cheng Y. DNA repair pathways in cancer therapy and resistance. Front Pharmacol. 2020;11:629266. doi:10.3389/fphar.2020.629266. [Google Scholar] [PubMed] [CrossRef]
101. Hemann MT, Lowe SW. The p53-bcl-2 connection. Cell Death Differ. 2006;13(8):1256–9. doi:10.1038/sj.cdd.4401962. [Google Scholar] [PubMed] [CrossRef]
102. Yesilkanal AE, Johnson GL, Ramos AF, Rosner MR. New strategies for targeting kinase networks in cancer. J Biol Chem. 2021;297(4):101128. doi:10.1016/j.jbc.2021.101128. [Google Scholar] [PubMed] [CrossRef]
103. Stelmach P, Trumpp A. Leukemic stem cells and therapy resistance in acute myeloid leukemia. Haematologica. 2023;108(2):353–66. doi:10.3324/haematol.2022.280800. [Google Scholar] [PubMed] [CrossRef]
104. Bauer J, Nelde A, Bilich T, Walz JS. Antigen targets for the development of immunotherapies in leukemia. Int J Mol Sci. 2019;20(6):1397. doi:10.3390/ijms20061397. [Google Scholar] [PubMed] [CrossRef]
104. Leotta S, Condorelli A, Sciortino R, Milone GA, Bellofiore C, Garibaldi B, et al. Prevention and treatment of acute myeloid leukemia relapse after hematopoietic stem cell transplantation: the state of the art and future perspectives. J Clin Med. 2022;11(1):253. doi:10.3390/jcm11010253. [Google Scholar] [PubMed] [CrossRef]
106. Jenq RR, van den Brink MRM. Allogeneic haematopoietic stem cell transplantation: individualized stem cell and immune therapy of cancer. Nat Rev Cancer. 2010;10(3):213–21. doi:10.1038/nrc2804. [Google Scholar] [PubMed] [CrossRef]
107. Matsui W, Borrello I, Mitsiades C. Autologous stem cell transplantation and multiple myeloma cancer stem cells. Biol Blood Marrow Transplant. 2012;18(1):S27–32. doi:10.1016/j.bbmt.2011.10.036. [Google Scholar] [PubMed] [CrossRef]
108. Du J, Yu D, Han X, Zhu L, Huang Z. Comparison of allogeneic stem cell transplant and autologous stem cell transplant in refractory or relapsed peripheral T-cell lymphoma: a systematic review and meta-analysis. JAMA Netw Open. 2021;4(5):e219807. doi:10.1001/jamanetworkopen.2021.9807. [Google Scholar] [PubMed] [CrossRef]
109. Zoellner AK, Unterhalt M, Stilgenbauer S, Hübel K, Thieblemont C, Metzner B, et al. Long-term survival of patients with mantle cell lymphoma after autologous haematopoietic stem-cell transplantation in first remission: a post-hoc analysis of an open-label, multicentre, randomised, phase 3 trial. Lancet Haematol. 2021;8(9):e648–57. doi:10.1016/S2352-3026(21)00195-2. [Google Scholar] [PubMed] [CrossRef]
110. Li T, Luo C, Zhang J, Wei L, Sun W, Xie Q, et al. Efficacy and safety of mesenchymal stem cells co-infusion in allogeneic hematopoietic stem cell transplantation: a systematic review and meta-analysis. Stem Cell Res Ther. 2021;12(1):246. doi:10.1186/s13287-021-02304-x. [Google Scholar] [PubMed] [CrossRef]
111. Loke J, Malladi R, Moss P, Craddock C. The role of allogeneic stem cell transplantation in the management of acute myeloid leukaemia: a triumph of hope and experience. Br J Haematol. 2020;188(1):129–46. doi:10.1111/bjh.16355. [Google Scholar] [PubMed] [CrossRef]
112. Algeri M, Merli P, Locatelli F, Pagliara D. The role of allogeneic hematopoietic stem cell transplantation in pediatric leukemia. J Clin Med. 2021;10(17):3790. doi:10.3390/jcm10173790. [Google Scholar] [PubMed] [CrossRef]
113. Dessie G, Derbew Molla M, Shibabaw T, Ayelign B. Role of stem-cell transplantation in leukemia treatment. Stem Cells Cloning. 2020;13:67–77. doi:10.2147/SCCAA.S262880. [Google Scholar] [PubMed] [CrossRef]
114. Georges GE, Bar M, Onstad L, Yi JC, Shadman M, Flowers ME, et al. Survivorship after autologous hematopoietic cell transplantation for lymphoma and multiple myeloma: late effects and quality of life. Biol Blood Marrow Transplant. 2020;26(2):407–12. doi:10.1016/j.bbmt.2019.10.002. [Google Scholar] [PubMed] [CrossRef]
115. Lan T, Luo M, Wei X. Mesenchymal stem/stromal cells in cancer therapy. J Hematol Oncol. 2021;14(1):195. doi:10.1186/s13045-021-01208-w. [Google Scholar] [PubMed] [CrossRef]
116. Singh AK, McGuirk JP. Allogeneic stem cell transplantation: a historical and scientific overview. Cancer Res. 2016;76(22):6445–51. doi:10.1158/0008-5472.CAN-16-1311. [Google Scholar] [PubMed] [CrossRef]
117. Trumpp A, Haas S. Cancer stem cells: the adventurous journey from hematopoietic to leukemic stem cells. Cell. 2022;185(8):1266–70. doi:10.1016/j.cell.2022.03.025. [Google Scholar] [PubMed] [CrossRef]
118. Alnasser SM, Alharbi KS, Almutairy AF, Almutairi SM, Alolayan AM. Autologous stem cell transplant in Hodgkin’s and non-Hodgkin’s lymphoma, multiple myeloma, and AL amyloidosis. Cells. 2023;12(24):2855. doi:10.3390/cells12242855. [Google Scholar] [PubMed] [CrossRef]
119. Bittencourt MCB, Ciurea SO. Recent advances in allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia. Biol Blood Marrow Transplant. 2020;26(9):e215–21. doi:10.1016/j.bbmt.2020.06.007. [Google Scholar] [PubMed] [CrossRef]
120. Burchert A, Bug G, Fritz LV, Finke J, Stelljes M, Röllig C, et al. Sorafenib maintenance after allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia with FLT3-internal tandem duplication mutation (SORMAIN). J Clin Oncol. 2020;38(26):2993–3002. doi:10.1200/JCO.19.03345. [Google Scholar] [PubMed] [CrossRef]
121. Xuan L, Liu Q. Maintenance therapy in acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J Hematol Oncol. 2021;14(1):4. doi:10.1186/s13045-020-01017-7. [Google Scholar] [PubMed] [CrossRef]
122. Tirino V, Desiderio V, Paino F, Papaccio G, De Rosa M. Methods for cancer stem cell detection and isolation. Methods Mol Biol. 2012;879(5):513–29. doi:10.1007/978-1-61779-815-3_32. [Google Scholar] [PubMed] [CrossRef]
123. Islam F, Gopalan V, Smith RA, Lam AK. Translational potential of cancer stem cells: a review of the detection of cancer stem cells and their roles in cancer recurrence and cancer treatment. Exp Cell Res. 2015;335(1):135–47. doi:10.1016/j.yexcr.2015.04.018. [Google Scholar] [PubMed] [CrossRef]
124. Moghbeli M, Moghbeli F, Forghanifard MM, Abbaszadegan MR. Cancer stem cell detection and isolation. Med Oncol. 2014;31(9):69. doi:10.1007/s12032-014-0069-6. [Google Scholar] [PubMed] [CrossRef]
125. Najafi M, Farhood B, Mortezaee K. Cancer stem cells (CSCs) in cancer progression and therapy. J Cell Physiol. 2019;234(6):8381–95. doi:10.1002/jcp.27740. [Google Scholar] [PubMed] [CrossRef]
126. Kwiatkowska-Borowczyk EP, Gąbka-Buszek A, Jankowski J, Mackiewicz A. Immunotargeting of cancer stem cells. Contemp Oncol. 2015;19(1A):A52–9. doi:10.5114/wo.2014.47129. [Google Scholar] [PubMed] [CrossRef]
127. Wu S, Tan Y, Li F, Han Y, Zhang S, Lin X. CD44: a cancer stem cell marker and therapeutic target in leukemia treatment. Front Immunol. 2024;15:1354992. doi:10.3389/fimmu.2024.1354992. [Google Scholar] [PubMed] [CrossRef]
128. Vey N, Delaunay J, Martinelli G, Fiedler W, Raffoux E, Prebet T, et al. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget. 2016;7(22):32532–42. doi:10.18632/oncotarget.8687. [Google Scholar] [PubMed] [CrossRef]
129. Vugts DJ, Heuveling DA, Stigter-van Walsum M, Weigand S, Bergstrom M, van Dongen GAMS, et al. Preclinical evaluation of 89Zr-labeled anti-CD44 monoclonal antibody RG7356 in mice and cynomolgus monkeys: prelude to Phase 1 clinical studies. MAbs. 2014;6(2):567–75. doi:10.4161/mabs.27415. [Google Scholar] [PubMed] [CrossRef]
130. Ishikawa K, Suzuki H, Ohishi T, Nakamura T, Yanaka M, Li G, et al. Antitumor activities of anti-CD44 monoclonal antibodies in mouse xenograft models of esophageal cancer. Oncol Rep. 2024;52(5):147. doi:10.3892/or.2024.8806. [Google Scholar] [PubMed] [CrossRef]
131. Sague SL, Tato C, Puré E, Hunter CA. The regulation and activation of CD44 by natural killer (NK) cells and its role in the production of IFN-γ. J Interf Cytokine Res. 2004;24(5):301–9. doi:10.1089/107999004323065093. [Google Scholar] [PubMed] [CrossRef]
132. Yan B, Chen Q, Shimada K, Tang M, Li H, Gurumurthy A, et al. Histone deacetylase inhibitor targets CD123/CD47-positive cells and reverse chemoresistance phenotype in acute myeloid leukemia. Leukemia. 2019;33(4):931–44. doi:10.1038/s41375-018-0279-6. [Google Scholar] [PubMed] [CrossRef]
133. Caras IW. Two cancer stem cell-targeted therapies in clinical trials as viewed from the standpoint of the cancer stem cell model. Stem Cells Transl Med. 2020;9(8):821–6. doi:10.1002/sctm.19-0424. [Google Scholar] [PubMed] [CrossRef]
134. Tabata R, Chi S, Yuda J, Minami Y. Emerging immunotherapy for acute myeloid leukemia. Int J Mol Sci. 2021;22(4):1944. doi:10.3390/ijms22041944. [Google Scholar] [PubMed] [CrossRef]
135. Li M, Yu H, Qi F, Ye Y, Hu D, Cao J, et al. Anti-CD47 immunotherapy in combination with BCL-2 inhibitor to enhance anti-tumor activity in B-cell lymphoma. Hematol Oncol. 2022;40(4):596–608. doi:10.1002/hon.3009. [Google Scholar] [PubMed] [CrossRef]
136. Lu Q, Chen X, Wang S, Lu Y, Yang C, Jiang G. Potential new cancer immunotherapy: anti-CD47-SIRPα antibodies. Onco Targets Ther. 2020;13:9323–31. doi:10.2147/OTT.S249822. [Google Scholar] [PubMed] [CrossRef]
137. Sun J, Chen Y, Lubben B, Adebayo O, Muz B, Azab AK. CD47-targeting antibodies as a novel therapeutic strategy in hematologic malignancies. Leuk Res Rep. 2021;16(6230):100268. doi:10.1016/j.lrr.2021.100268. [Google Scholar] [PubMed] [CrossRef]
138. Ni H, Cao L, Wu Z, Wang L, Zhou S, Guo X, et al. Combined strategies for effective cancer immunotherapy with a novel anti-CD47 monoclonal antibody. Cancer Immunol Immunother. 2022;71(2):353–63. doi:10.1007/s00262-021-02989-2. [Google Scholar] [PubMed] [CrossRef]
139. Behrens LM, van den Berg TK, van Egmond M. Targeting the CD47-SIRPα innate immune checkpoint to potentiate antibody therapy in cancer by neutrophils. Cancers. 2022;14(14):3366. doi:10.3390/cancers14143366. [Google Scholar] [PubMed] [CrossRef]
140. Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, et al. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N Engl J Med. 2018;379(18):1711–21. doi:10.1056/NEJMoa1807315. [Google Scholar] [PubMed] [CrossRef]
141. Schewe DM, Vogiatzi F, Münnich IA, Zeller T, Windisch R, Wichmann C, et al. Enhanced potency of immunotherapy against B-cell precursor acute lymphoblastic leukemia by combination of an Fc-engineered CD19 antibody and CD47 blockade. Hemasphere. 2024;8(2):e48. doi:10.1002/hem3.48. [Google Scholar] [PubMed] [CrossRef]
142. Haddad F, Daver N. Targeting CD47/SIRPα in acute myeloid leukemia and myelodysplastic syndrome: preclinical and clinical developments of magrolimab. J Immunother Precis Oncol. 2021;4(2):67–71. doi:10.36401/JIPO-21-X2. [Google Scholar] [PubMed] [CrossRef]
143. Sallman DA, Al Malki MM, Asch AS, Wang ES, Jurcic JG, Bradley TJ, et al. Magrolimab in combination with azacitidine in patients with higher-risk myelodysplastic syndromes: final results of a phase ib study. J Clin Oncol. 2023;41(15):2815–26. doi:10.1200/jco.22.01794. [Google Scholar] [PubMed] [CrossRef]
144. Daver NG, Vyas P, Kambhampati S, Al Malki MM, Larson RA, Asch AS, et al. Tolerability and efficacy of the anticluster of differentiation 47 antibody magrolimab combined with azacitidine in patients with previously untreated AML: phase ib results. J Clin Oncol. 2023;41(31):4893–904. doi:10.1200/JCO.22.02604. [Google Scholar] [PubMed] [CrossRef]
145. Daver NG, Liu K, Kuwahara SB, Caldwell K, Vyas P. AML-577 a phase III, randomized trial of magrolimab in combination with venetoclax and azacitidine in previously untreated patients with acute myeloid leukemia who are ineligible for intensive chemotherapy (ENHANCE-3). Clin Lymphoma Myeloma Leuk. 2023;23:S313–4. doi:10.1016/S2152-2650(23)01083-2. [Google Scholar] [CrossRef]
146. Gilead Discontinues Magrolimab Trial. Ashpublications.org. 2024 [Internet]. [cited 2025 Jul 27]. Available from: https://ashpublications.org/ashclinicalnews/news/7840/Gilead-Discontinues-Magrolimab-Trial. [Google Scholar]
147. Hawkes E, Lewis KL, Wong Doo N, Patil SS, Miskin HP, Sportelli P et al. First-in-human (FIH) study of the fully-human kappa-lambda CD19/CD47 bispecific antibody TG-1801 in patients (pts) with B-cell lymphoma. Blood. 2022;140:6599–601. doi:10.1182/blood-2022-169171. [Google Scholar] [CrossRef]
148. Weiskopf K. Cancer immunotherapy targeting the CD47/SIRPα axis. Eur J Cancer. 2017;76:100–9. doi:10.1016/j.ejca.2017.02.013. [Google Scholar] [PubMed] [CrossRef]
149. Chen Q, Guo X, Ma W. Opportunities and challenges of CD47-targeted therapy in cancer immunotherapy. Oncol Res. 2023;32(1):49–60. doi:10.32604/or.2023.042383. [Google Scholar] [PubMed] [CrossRef]
150. Yang H, Xun Y, You H. The landscape overview of CD47-based immunotherapy for hematological malignancies. Biomark Res. 2023;11(1):15. doi:10.1186/s40364-023-00456-x. [Google Scholar] [PubMed] [CrossRef]
151. Tigu AB, Munteanu R, Moldovan C, Rares D, Kegyes D, Tomai R, et al. Therapeutic advances in the targeting of ROR1 in hematological cancers. Cell Death Discov. 2024;10(1):471. doi:10.1038/s41420-024-02239-1. [Google Scholar] [PubMed] [CrossRef]
152. Menck K, Heinrichs S, Baden C, Bleckmann A. The WNT/ROR pathway in cancer: from signaling to therapeutic intervention. Cells. 2021;10(1):142. doi:10.3390/cells10010142. [Google Scholar] [PubMed] [CrossRef]
153. Bemani P, Moazen S, Nadimi E, Nejatollahi F. Development of human recombinant antibodies against ROR1 tumor antigen. Rbmbnet. 2022;11(2):282–8. doi:10.61186/rbmb.11.2.282. [Google Scholar] [CrossRef]
154. Wei R, Liao X, Li J, Mu X, Ming Y, Peng Y. Novel humanized monoclonal antibodies against ROR1 for cancer therapy. Mol Cancer. 2024;23(1):165. doi:10.1186/s12943-024-02075-y. [Google Scholar] [PubMed] [CrossRef]
155. Cheng PL, Hsiao TH, Chen CH, Hung MN, Jhan PP, Lee LW, et al. Chemoresistance in acute myeloid leukemia: an alternative single-cell RNA sequencing approach. Hematol Oncol. 2023;41(3):499–509. doi:10.1002/hon.3129. [Google Scholar] [PubMed] [CrossRef]
156. Frenay J, Bellaye PS, Oudot A, Helbling A, Petitot C, Ferrand C, et al. IL-1RAP, a key therapeutic target in cancer. Int J Mol Sci. 2022;23(23):14918. doi:10.3390/ijms232314918. [Google Scholar] [PubMed] [CrossRef]
157. Zhang Y, Park M, Ghoda LY, Zhao D, Valerio M, Nafie E, et al. IL1RAP-specific T cell engager depletes acute myeloid leukemia stem cells. J Hematol Oncol. 2024;17(1):67. doi:10.1186/s13045-024-01586-x. [Google Scholar] [PubMed] [CrossRef]
158. West KA, Kumar S, Scott M, Emery JG, Steidl UG, Narayanagari SR et al. BOS-371, a monoclonal antibody against IL1RAP: characterization in preclinical models of AML. J Clin Oncol. 2023;41(16_suppl):7030. doi:10.1200/jco.2023.41.16_suppl.7030. [Google Scholar] [CrossRef]
159. Liu J, Tong J, Yang H. Targeting CD33 for acute myeloid leukemia therapy. BMC Cancer. 2022;22(1):24. doi:10.1186/s12885-021-09116-5. [Google Scholar] [PubMed] [CrossRef]
160. Maakaron JE, Rogosheske J, Long M, Bachanova V, Mims AS. CD33-targeted therapies: beating the disease or beaten to death? J Clin Pharmacol. 2021;61(1):7–17. doi:10.1002/jcph.1730. [Google Scholar] [PubMed] [CrossRef]
161. Collados-Ros A, Muro M, Legaz I. Gemtuzumab ozogamicin in acute myeloid leukemia: efficacy, toxicity, and resistance mechanisms—a systematic review. Biomedicines. 2024;12(1):208. doi:10.3390/biomedicines12010208. [Google Scholar] [PubMed] [CrossRef]
162. Molica M, Perrone S, Mazzone C, Niscola P, Cesini L, Abruzzese E, et al. CD33 expression and gentuzumab ozogamicin in acute myeloid leukemia: two sides of the same coin. Cancers. 2021;13(13):3214. doi:10.3390/cancers13133214. [Google Scholar] [PubMed] [CrossRef]
163. Godwin CD, Laszlo GS, Fiorenza S, Garling EE, Phi TD, Bates OM, et al. Targeting the membrane-proximal C2-set domain of CD33 for improved CD33-directed immunotherapy. Leukemia. 2021;35(9):2496–507. doi:10.1038/s41375-021-01160-1. [Google Scholar] [CrossRef]
164. Han YC, Kahler J, Piché-Nicholas N, Hu W, Thibault S, Jiang F, et al. Development of highly optimized antibody-drug conjugates against CD33 and CD123 for acute myeloid leukemia. Clin Cancer Res. 2021;27(2):622–31. doi:10.1158/1078-0432.CCR-20-2149. [Google Scholar] [PubMed] [CrossRef]
165. Berdel AF, Rollig C, Wermke M, Angenendt L, Ruhnke L, Mikesch JH et al. A phase I trial of the antibody-cytokine fusion protein F16IL2 in combination with anti-CD33 immunotherapy for posttransplant AML relapse. Blood. 2021;138(Supplement 1):2345. doi:10.1182/blood-2021-145859. [Google Scholar] [CrossRef]
166. Marcinek A, Brauchle B, Rohrbacher L, Hänel G, Philipp N, Märkl F, et al. CD33 BiTE® molecule-mediated immune synapse formation and subsequent T-cell activation is determined by the expression profile of activating and inhibitory checkpoint molecules on AML cells. Cancer Immunol Immunother. 2023;72(7):2499–512. doi:10.1007/s00262-023-03439-x. [Google Scholar] [PubMed] [CrossRef]
167. Ravandi F, Subklewe M, Walter RB, Vachhani P, Ossenkoppele G, Buecklein V, et al. Safety and tolerability of AMG 330 in adults with relapsed/refractory AML: a phase 1a dose-escalation study. Leuk Lymphoma. 2024;65(9):1281–91. doi:10.1080/10428194.2024.2346755. [Google Scholar] [PubMed] [CrossRef]
168. Cheng P, Chen X, Dalton R, Calescibetta A, So T, Gilvary D, et al. Immunodepletion of MDSC by AMV564, a novel bivalent, bispecific CD33/CD3 T cell engager, ex vivo in MDS and melanoma. Mol Ther. 2022;30(6):2315–26. doi:10.1016/j.ymthe.2022.02.005. [Google Scholar] [PubMed] [CrossRef]
169. Eissenberg LG, Ritchey JK, Rettig MP, Patel DA, Vij K, Gao F, et al. Control of acute myeloid leukemia and generation of immune memory in vivo using AMV564, a bivalent bispecific CD33 x CD3 T cell engager. PLoS One. 2024;19(5):e0300174. doi:10.1371/journal.pone.0300174. [Google Scholar] [PubMed] [CrossRef]
170. Westervelt P, Cortes JE, Altman JK, Long M, Oehler VG, Gojo I et al. Phase 1 first-in-human trial of AMV564, a bivalent bispecific (2:2) CD33/CD3 T-cell engager, in patients with relapsed/refractory acute myeloid leukemia (AML). Blood. 2019;134(Supplement_1):834. doi:10.1182/blood-2019-129042. [Google Scholar] [CrossRef]
171. Subklewe M, Stein A, Walter RB, Bhatia R, Wei AH, Ritchie D et al. Preliminary results from a phase 1 first-in-human study of AMG 673, a novel half-life extended (HLE) anti-CD33/CD3 BiTE® (bispecific T-cell engager) in patients with relapsed/refractory (R/R) acute myeloid leukemia (AML). Blood. 2019;134(Supplement_1):833. doi:10.1182/blood-2019-127977. [Google Scholar] [CrossRef]
172. Reusing SB, Vallera DA, Manser AR, Vatrin T, Bhatia S, Felices M, et al. CD16xCD33 Bispecific Killer Cell Engager (BiKE) as potential immunotherapeutic in pediatric patients with AML and biphenotypic ALL. Cancer Immunol Immunother. 2021;70(12):3701–8. doi:10.1007/s00262-021-03008-0. [Google Scholar] [PubMed] [CrossRef]
173. Patnaik MM, Mughal TI, Brooks C, Lindsay R, Pemmaraju N. Targeting CD123 in hematologic malignancies: identifying suitable patients for targeted therapy. Leuk Lymphoma. 2021;62(11):2568–86. doi:10.1080/10428194.2021.1927021. [Google Scholar] [PubMed] [CrossRef]
174. Pelosi E, Castelli G, Testa U. CD123 a therapeutic target for acute myeloid leukemia and blastic plasmocytoid dendritic neoplasm. Int J Mol Sci. 2023;24(3):2718. doi:10.3390/ijms24032718. [Google Scholar] [PubMed] [CrossRef]
175. Marone R, Landmann E, Devaux A, Lepore R, Seyres D, Zuin J, et al. Epitope-engineered human hematopoietic stem cells are shielded from CD123-targeted immunotherapy. J Exp Med. 2023;220(12):e20231235. doi:10.1084/jem.20231235. [Google Scholar] [PubMed] [CrossRef]
176. Ji RJ, Cao GH, Zhao WQ, Wang MY, Gao P, Zhang YZ, et al. Epitope prime editing shields hematopoietic cells from CD123 immunotherapy for acute myeloid leukemia. Cell Stem Cell. 2024;31(11):1650–66.e8. doi:10.1016/j.stem.2024.09.003. [Google Scholar] [PubMed] [CrossRef]
177. Kubasch AS, Schulze F, Giagounidis A, Götze KS, Krönke J, Sockel K, et al. Single agent talacotuzumab demonstrates limited efficacy but considerable toxicity in elderly high-risk MDS or AML patients failing hypomethylating agents. Leukemia. 2020;34(4):1182–6. doi:10.1038/s41375-019-0645-z. [Google Scholar] [PubMed] [CrossRef]
178. Montesinos P, Roboz GJ, Bulabois CE, Subklewe M, Platzbecker U, Ofran Y, et al. Safety and efficacy of talacotuzumab plus decitabine or decitabine alone in patients with acute myeloid leukemia not eligible for chemotherapy: results from a multicenter, randomized, phase 2/3 study. Leukemia. 2021;35(1):62–74. doi:10.1038/s41375-020-0773-5. [Google Scholar] [PubMed] [CrossRef]
179. Daver NG, Erba HP, Papadantonakis N, DeAngelo DJ, Wang ES, Konopleva MY et al. A phase I, first-in-human study evaluating the safety and preliminary antileukemia activity of IMGN632, a novel CD123-targeting antibody-drug conjugate, in patients with relapsed/refractory acute myeloid leukemia and other CD123-positive hematologic malignancies. Blood. 2018;132(Supplement 1):27. doi:10.1182/blood-2018-99-112955. [Google Scholar] [CrossRef]
180. Daver NG, Montesinos P, DeAngelo DJ, Wang ES, Todisco E, Tarella C, et al. A phase I/II study of IMGN632, a novel CD123-targeting antibody-drug conjugate, in patients with relapsed/refractory acute myeloid leukemia, blastic plasmacytoid dendritic cell neoplasm, and other CD123-positive hematologic malignancies. J Clin Oncol. 2020;38(15_suppl):TPS7563. doi:10.1200/jco.2020.38.15_suppl.tps7563. [Google Scholar] [CrossRef]
181. Daver N, Aribi A, Montesinos P, Roboz GJ, Wang ES, Walter RB, et al. Safety and efficacy from a phase 1b/2 study of IMGN632 in combination with azacitidine and venetoclax for patients with CD123-positive acute myeloid leukemia. Blood. 2021;138(Supplement 1):372. doi:10.1182/blood-2021-146503. [Google Scholar] [CrossRef]
182. Daver NG, Montesinos P, DeAngelo DJ, Wang ES, Papadantonakis N, Todisco E, et al. Pivekimab sunirine (IMGN632a novel CD123-targeting antibody-drug conjugate, in relapsed or refractory acute myeloid leukaemia: a phase 1/2 study. Lancet Oncol. 2024;25(3):388–99. doi:10.1016/S1470-2045(23)00674-5. [Google Scholar] [PubMed] [CrossRef]
183. Gulati R, Abu-Salah A, Salous T, Nassiri M. Relapse of tagraxofusp treated blastic plasmacytoid dendritic cell neoplasm with loss of CD123 expression. J Hematop. 2022;15(1):35–9. doi:10.1007/s12308-021-00479-z. [Google Scholar] [PubMed] [CrossRef]
184. Pemmaraju N, Sweet KL, Stein AS, Wang ES, Rizzieri DA, Vasu S, et al. Long-term benefits of tagraxofusp for patients with blastic plasmacytoid dendritic cell neoplasm. J Clin Oncol. 2022;40(26):3032–6. doi:10.1200/JCO.22.00034. [Google Scholar] [PubMed] [CrossRef]
185. Lock RB, Evans K, Randall J, Erickson SW, Earley EJ, Neuhauser S, et al. Pediatric preclinical testing consortium evaluation of the anti-CD123 antibody-drug conjugate, IMGN632, against patient-derived xenograft models of pediatric acute lymphoblastic leukemia. Blood. 2022;140(Supplement 1):8983–4. doi:10.1182/blood-2022-159867. [Google Scholar] [CrossRef]
186. Uy GL, Aldoss I, Foster MC, Sayre PH, Wieduwilt MJ, Advani AS, et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood. 2021;137(6):751–62. doi:10.1182/blood.2020007732. [Google Scholar] [PubMed] [CrossRef]
187. Watts J, Maris M, Lin TL, Patel P, Madanat YF, Cogle CR, et al. Updated results from a phase 1 study of APVO436, a novel bispecific anti-CD123 x anti-CD3 adaptirTM molecule, in relapsed/refractory acute myeloid leukemia and myelodysplastic syndrome. Blood. 2022;140(Supplement 1):6204–5. doi:10.1182/blood-2022-167468. [Google Scholar] [CrossRef]
188. Watts JM, Lin TL, Mims AS, Patel P, Shami PJ, Cull EH, et al. Tolerability and single agent anti-neoplastic activity of the CD3xCD123 bispecific antibody APVO436 in patients with relapsed/refractory AML or MDS. Blood. 2021;138(Supplement 1):3415. doi:10.1182/blood-2021-149337. [Google Scholar] [CrossRef]
189. Alderson RF, Huang L, Zhang X, Li H, Kaufman T, Diedrich G, et al. Combinatorial anti-tumor activity in animal models of a novel CD123 x CD3 bispecific dart® molecule (MGD024) with cytarabine, venetoclax or azacitidine supports combination therapy in acute myeloid leukemia. Blood. 2021;138(Supplement 1):1165. doi:10.1182/blood-2021-153192. [Google Scholar] [CrossRef]
190. Winer ES, Maris M, Sharma MR, Kaminker P, Zhao E, Ward A, et al. A phase 1, first-in-human, dose-escalation study of MGD024, a CD123 x CD3 bispecific dart® molecule, in patients with relapsed or refractory CD123-positive (+) hematologic malignancies. Blood. 2022;140(Supplement 1):11753–4. doi:10.1182/blood-2022-159524. [Google Scholar] [CrossRef]
191. Tahk S, Vick B, Hiller B, Schmitt S, Marcinek A, Perini ED, et al. SIRPα-αCD123 fusion antibodies targeting CD123 in conjunction with CD47 blockade enhance the clearance of AML-initiating cells. J Hematol Oncol. 2021;14(1):155. doi:10.1186/s13045-021-01163-6. [Google Scholar] [PubMed] [CrossRef]
192. Borlongan MC, Saha D, Wang H. Tumor microenvironment: a niche for cancer stem cell immunotherapy. Stem Cell Rev Rep. 2024;20(1):3–24. doi:10.1007/s12015-023-10639-6. [Google Scholar] [PubMed] [CrossRef]
193. Pang WW, Czechowicz A, Logan AC, Bhardwaj R, Poyser J, Park CY, et al. Anti-CD117 antibody depletes normal and myelodysplastic syndrome human hematopoietic stem cells in xenografted mice. Blood. 2019;133(19):2069–78. doi:10.1182/blood-2018-06-858159. [Google Scholar] [PubMed] [CrossRef]
194. Russkamp NF, Myburgh R, Kiefer JD, Neri D, Manz MG. Anti-CD117 immunotherapy to eliminate hematopoietic and leukemia stem cells. Exp Hematol. 2021;95:31–45. doi:10.1016/j.exphem.2021.01.003. [Google Scholar] [PubMed] [CrossRef]
195. Kwon HS, Logan AC, Chhabra A, Pang WW, Czechowicz A, Tate K, et al. Anti-human CD117 antibody-mediated bone marrow niche clearance in nonhuman primates and humanized NSG mice. Blood. 2019;133(19):2104–8. doi:10.1182/blood-2018-06-853879. [Google Scholar] [PubMed] [CrossRef]
196. Agarwal R, Dvorak CC, Kwon HS, Long-Boyle JR, Prohaska SS, Brown JW, et al. Non-genotoxic anti-CD117 antibody conditioning results in successful hematopoietic stem cell engraftment in patients with severe combined immunodeficiency. Blood. 2019;134(Supplement_1):800. doi:10.1182/blood-2019-126239. [Google Scholar] [CrossRef]
197. Bankova AK, Pang WW, Velasco BJ, Long-Boyle JR, Shizuru JA. 5-Azacytidine depletes HSCs and synergizes with an anti-CD117 antibody to augment donor engraftment in immunocompetent mice. Blood Adv. 2021;5(19):3900–12. doi:10.1182/bloodadvances.2020003841. [Google Scholar] [PubMed] [CrossRef]
198. Lanieri L, Lamothe TL, Miske O, McDonough SM, Sarma GN, Bhattarai P, et al. A single dose of a novel anti-human CD117-amanitin antibody drug conjugate (ADC) engineered for a short half-life provides dual conditioning and anti-leukemic activity and extends survival compared to standard of care in multiple preclinical models of acute myeloid leukemia (AML). Blood. 2020;136(Supplement 1):47–8. doi:10.1182/blood-2020-140422. [Google Scholar] [CrossRef]
199. Westervelt P, Kebriaei P, Juckett M, Artz AS, Chan O, McCarthy PL, et al. Mgta-117, an anti-CD117 antibody-drug conjugated with amanitin, in participants with relapsed/refractory adult acute myeloid leukemia (AML) and myelodysplasia with excess blasts (MDS-EBsafety, pharmacokinetics and pharmacodynamics initial findings from a phase 1/2 study. Blood. 2022;140(Supplement 1):2117–9. doi:10.1182/blood-2022-162406. [Google Scholar] [CrossRef]
200. Czechowicz A, Palchaudhuri R, Scheck A, Hu Y, Hoggatt J, Saez B, et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat Commun. 2019;10(1):617. doi:10.1038/s41467-018-08201-x. [Google Scholar] [PubMed] [CrossRef]
201. Kiefer JD, Myburgh R, Russkamp NF, Volta L, Guggisberg A, Abdelmotaleb O, et al. A bispecific antibody targeting CD117 and CD3 enables T cell mediated killing of CD117-expressing healthy and malignant hematopoietic cells. Blood. 2021;138(Supplement 1):2354. doi:10.1182/blood-2021-147676. [Google Scholar] [CrossRef]
202. Dal Collo G, van Hoven-Beijen A, He Y, Muller YM, Wang Y, Zhao J, et al. Novel anti-CD117 antibodies for rapid and efficient hematopoietic stem cell depletion and safe bone marrow conditioning. Blood. 2022;140(Supplement 1):4496–7. doi:10.1182/blood-2022-158149. [Google Scholar] [CrossRef]
203. Volta L, Myburgh R, Hofstetter M, Koch C, Kiefer JD, Gobbi C, et al. A single-chain variable fragment-based bispecific T-cell activating antibody against CD117 enables T-cell mediated lysis of acute myeloid leukemia and hematopoietic stem and progenitor cells. Hemasphere. 2024;8(11):e70055. doi:10.1002/hem3.70055. [Google Scholar] [PubMed] [CrossRef]
204. Li X, Dai J, Shi Y, Chen J, Zhou F, Qian X, et al. Bispecific aptamer-drug conjugates selectively eliminate malignant hematologic cells for treating acute myeloid leukemia. Langmuir. 2025;41(4):2580–90. doi:10.1021/acs.langmuir.4c04350. [Google Scholar] [PubMed] [CrossRef]
205. Acharya N, Sabatos-Peyton C, Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. J Immunother Cancer. 2020;8(1):e000911. doi:10.1136/jitc-2020-000911. [Google Scholar] [PubMed] [CrossRef]
206. Huang S, Zhao Y, Lai W, Tan J, Zheng X, Zha X, et al. Higher PD-1/Tim-3 expression on IFN-γ+ T cells is associated with poor prognosis in patients with acute myeloid leukemia. Cancer Biol Ther. 2023;24(1):2278229. doi:10.1080/15384047.2023.2278229. [Google Scholar] [PubMed] [CrossRef]
207. Wang Z, Chen J, Wang M, Zhang L, Yu L. One stone, two birds: the roles of tim-3 in acute myeloid leukemia. Front Immunol. 2021;12:618710. doi:10.3389/fimmu.2021.618710. [Google Scholar] [PubMed] [CrossRef]
208. Jiang W, Li F, Jiang Y, Li S, Liu X, Xu Y, et al. Tim-3 blockade elicits potent anti-multiple myeloma immunity of natural killer cells. Front Oncol. 2022;12:739976. doi:10.3389/fonc.2022.739976. [Google Scholar] [PubMed] [CrossRef]
209. Schwartz S, Patel N, Longmire T, Jayaraman P, Jiang X, Lu H, et al. Characterization of sabatolimab, a novel immunotherapy with immuno-myeloid activity directed against TIM-3 receptor. Immunother Adv. 2022;2(1):ltac019. doi:10.1093/immadv/ltac019. [Google Scholar] [PubMed] [CrossRef]
210. Zeidan AM, Komrokji RS, Brunner AM. TIM-3 pathway dysregulation and targeting in cancer. Expert Rev Anticancer Ther. 2021;21(5):523–34. doi:10.1080/14737140.2021.1865814. [Google Scholar] [PubMed] [CrossRef]
211. Cai L, Li Y, Tan J, Xu L, Li Y. Targeting LAG-3, TIM-3, and TIGIT for cancer immunotherapy. J Hematol Oncol. 2023;16(1):101. doi:10.1186/s13045-023-01499-1. [Google Scholar] [CrossRef]
212. Duong B, Banskota P, Falchook GS. T cell immunoglobulin and mucin domain containing protein 3 (TIM-3) inhibitors in oncology clinical trials: a review. J Immunother Precis Oncol. 2024;7(2):89–96. doi:10.36401/JIPO-23-4. [Google Scholar] [PubMed] [CrossRef]
213. Lu C, Tan Y. Promising immunotherapy targets: tim3, LAG3, and TIGIT joined the party. Mol Ther Oncol. 2024;32(1):200773. doi:10.1016/j.omton.2024.200773. [Google Scholar] [PubMed] [CrossRef]
214. Jin T, Gao F, Wang L. Blockade of PD-1 and TIM-3 ameliorates CD8+ T cell exhaustion in a mouse model of chronic myeloid leukemia. Cell Biochem Biophys. 2024;82(3):2759–66. doi:10.1007/s12013-024-01392-9. [Google Scholar] [PubMed] [CrossRef]
215. Astaneh M, Rezazadeh H, Hossein-Nataj H, Shekarriz R, Zaboli E, Shabani M, et al. Tim-3 and PD-1 blocking cannot restore the functional properties of natural killer cells in early clinical stages of chronic lymphocytic leukemia: an in vitro study. J Cancer Res Ther. 2022;18(3):704–11. doi:10.4103/jcrt.jcrt_52_21. [Google Scholar] [PubMed] [CrossRef]
216. Jafarkhani S, Hossein-Nataj H, Eslami-Jouybari M, Ghoreishi M, Asgarian-Omran H. Pd-1 and tim-3 blocking cannot enhance apoptosis of chronic lymphocytic leukemia cells induced by peripheral blood CD8+ t cells. Exp Oncol. 2022;44(4):287–94. doi:10.32471/exp-oncology.2312-8852.vol-44-no-4.18975. [Google Scholar] [PubMed] [CrossRef]
217. Rezazadeh H, Astaneh M, Tehrani M, Hossein-Nataj H, Zaboli E, Shekarriz R, et al. Blockade of PD-1 and TIM-3 immune checkpoints fails to restore the function of exhausted CD8+ T cells in early clinical stages of chronic lymphocytic leukemia. Immunol Res. 2020;68(5):269–79. doi:10.1007/s12026-020-09146-4. [Google Scholar] [PubMed] [CrossRef]
218. Ho JM, Dobson SM, Voisin V, McLeod J, Kennedy JA, Mitchell A, et al. CD200 expression marks leukemia stem cells in human AML. Blood Adv. 2020;4(21):5402–13. doi:10.1182/bloodadvances.2020001802. [Google Scholar] [PubMed] [CrossRef]
219. Herbrich S, Baran N, Cai T, Weng C, Aitken MJL, Post SM, et al. Overexpression of CD200 is a stem cell-specific mechanism of immune evasion in AML. J Immunother Cancer. 2021;9(7):e002968. doi:10.1136/jitc-2021-002968. [Google Scholar] [PubMed] [CrossRef]
220. Shao A, Owens DM. The immunoregulatory protein CD200 as a potentially lucrative yet elusive target for cancer therapy. Oncotarget. 2023;14(1):96–103. doi:10.18632/oncotarget.28354. [Google Scholar] [PubMed] [CrossRef]
221. Mahadevan D, Lanasa MC, Farber C, Pandey M, Whelden M, Faas SJ, et al. Phase I study of samalizumab in chronic lymphocytic leukemia and multiple myeloma: blockade of the immune checkpoint CD200. J Immunother Cancer. 2019;7(1):227. doi:10.1186/s40425-019-0710-1. [Google Scholar] [CrossRef]
222. Diamanti P, Cox CV, Ede BC, Uger RA, Moppett JP, Blair A. Targeting pediatric leukemia-propagating cells with anti-CD200 antibody therapy. Blood Adv. 2021;5(18):3694–708. doi:10.1182/bloodadvances.2020003534. [Google Scholar] [PubMed] [CrossRef]
223. Rastogi N, Baker S, Man S, Uger RA, Wong M, Coles SJ, et al. Use of an anti-CD200-blocking antibody improves immune responses to AML in vitro and in vivo. Br J Haematol. 2021;193(1):155–9. doi:10.1111/bjh.17125. [Google Scholar] [PubMed] [CrossRef]
224. Li Z, Murphy PM. CD45: a niche marker for allotransplantation. Blood. 2022;139(11):1614–6. doi:10.1182/blood.2021015024. [Google Scholar] [PubMed] [CrossRef]
225. Persaud SP, Cooper ML, Ritchey JK, Rettig MP, DiPersio JF. Antibody-drug conjugates targeting CD45 plus Janus kinase (JAK) inhibitors as conditioning for allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2020;26(3):S150–1. doi:10.1016/j.bbmt.2019.12.701. [Google Scholar] [CrossRef]
226. Yeung J, Liao A, Shaw M, Silva S, Vetharoy W, Rico DL, et al. Anti-CD45 PBD-based antibody-drug conjugates are effective targeted conditioning agents for gene therapy and stem cell transplant. Mol Ther. 2024;32(6):1672–86. doi:10.1016/j.ymthe.2024.03.032. [Google Scholar] [PubMed] [CrossRef]
227. Walter RB, Boyle KM, Appelbaum FR, Bernstein ID, Pagel JM. Simultaneously targeting CD45 significantly increases cytotoxicity of the anti-CD33 immunoconjugate, gemtuzumab ozogamicin, against acute myeloid leukemia (AML) cells and improves survival of mice bearing human AML xenografts. Blood. 2008;111(9):4813–6. doi:10.1182/blood-2008-01-133785. [Google Scholar] [PubMed] [CrossRef]
228. Flieswasser T, Van den Eynde A, Van Audenaerde J, De Waele J, Lardon F, Riether C, et al. The CD70-CD27 axis in oncology: the new kids on the block. J Exp Clin Cancer Res. 2022;41(1):12. doi:10.1186/s13046-021-02215-y. [Google Scholar] [PubMed] [CrossRef]
229. Schürch C, Riether C, Matter MS, Tzankov A, Ochsenbein AF. CD27 signaling on chronic myelogenous leukemia stem cells activates Wnt target genes and promotes disease progression. J Clin Invest. 2012;122(2):624–38. doi:10.1172/JCI45977. [Google Scholar] [PubMed] [CrossRef]
230. Marques-Piubelli ML, Kumar B, Basar R, Panowski S, Srinivasan S, Norwood K, et al. Increased expression of CD70 in relapsed acute myeloid leukemia after hypomethylating agents. Virchows Arch. 2024;485(5):937–41. doi:10.1007/s00428-024-03741-8. [Google Scholar] [PubMed] [CrossRef]
231. Ochsenbein AF, Pabst T, Höpner S, Bacher VU, Hinterbrandner M, Banz Y et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in humans. Blood. 2019;134(Supplement_1):234. doi:10.1182/blood-2019-129916. [Google Scholar] [CrossRef]
232. Riether C, Pabst T, Höpner S, Bacher U, Hinterbrandner M, Banz Y, et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat Med. 2020;26(9):1459–67. doi:10.1038/s41591-020-0910-8. [Google Scholar] [PubMed] [CrossRef]
233. Riether C, Chiorazzo T, Johnson AJ, Drenberg CD, Syed KW, Moshir M, et al. The combination of the BCL-2 antagonist venetoclax with the CD70-targeting antibody cusatuzumab synergistically eliminates primary human leukemia stem cells. Blood. 2019;134(Supplement_1):3918. doi:10.1182/blood-2019-127464. [Google Scholar] [CrossRef]
234. Silence K, Dreier T, Moshir M, Ulrichts P, Gabriels SME, Saunders M, et al. ARGX-110, a highly potent antibody targeting CD70, eliminates tumors via both enhanced ADCC and immune checkpoint blockade. MAbs. 2014;6(2):523–32. doi:10.4161/mabs.27398. [Google Scholar] [PubMed] [CrossRef]
235. Ochsenbein AF, Riether C, Bacher U, Müller R, Höpner S, Banz Y, et al. Argx-110 targeting CD70, in combination with azacitidine, shows favorable safety profile and promising anti-leukemia activity in newly diagnosed AML patients in an ongoing phase 1/2 clinical trial. Blood. 2018;132(Supplement 1):2680. doi:10.1182/blood-2018-99-118302. [Google Scholar] [CrossRef]
236. Pabst T, Vey N, Adès L, Bacher U, Bargetzi M, Fung S, et al. Results from a phase I/II trial of cusatuzumab combined with azacitidine in patients with newly diagnosed acute myeloid leukemia who are ineligible for intensive chemotherapy. Haematologica. 2023;108(7):1793–802. doi:10.3324/haematol.2022.281563. [Google Scholar] [PubMed] [CrossRef]
237. Aftimos P, Rolfo C, Rottey S, Offner F, Bron D, Maerevoet M, et al. Phase I dose-escalation study of the anti-CD70 antibody ARGX-110 in advanced malignancies. Clin Cancer Res. 2017;23(21):6411–20. doi:10.1158/1078-0432.CCR-17-0613. [Google Scholar] [PubMed] [CrossRef]
238. McDonagh CF, Kim KM, Turcott E, Brown LL, Westendorf L, Feist T, et al. Engineered anti-CD70 antibody-drug conjugate with increased therapeutic index. Mol Cancer Ther. 2008;7(9):2913–23. doi:10.1158/1535-7163.MCT-08-0295. [Google Scholar] [PubMed] [CrossRef]
239. Armengol M, Santos JC, Fernández-Serrano M, Profitós-Pelejà N, Ribeiro ML, Roué G. Immune-checkpoint inhibitors in B-cell lymphoma. Cancers. 2021;13(2):214. doi:10.3390/cancers13020214. [Google Scholar] [PubMed] [CrossRef]
240. Vitale LA, He LZ, Thomas LJ, Widger J, Weidlick J, Crocker A, et al. Development of a human monoclonal antibody for potential therapy of CD27-expressing lymphoma and leukemia. Clin Cancer Res. 2012;18(14):3812–21. doi:10.1158/1078-0432.CCR-11-3308. [Google Scholar] [PubMed] [CrossRef]
241. He LZ, Thomas L, Weidlick J, Vitale L, O’Neill T, Prostak N, et al. Development of a human anti-CD27 antibody with efficacy in lymphoma and leukemia models by two distinct mechanisms. Blood. 2011;118(21):2861. doi:10.1182/blood.v118.21.2861.2861. [Google Scholar] [CrossRef]
242. Tun AM, Ansell SM. Immunotherapy in Hodgkin and non-Hodgkin lymphoma: innate, adaptive and targeted immunological strategies. Cancer Treat Rev. 2020;88(50):102042. doi:10.1016/j.ctrv.2020.102042. [Google Scholar] [PubMed] [CrossRef]
243. Ansell SM, Northfelt DW, Flinn I, Burris HA, Dinner SN, Villalobos VM, et al. Phase I evaluation of an agonist anti-CD27 human antibody (CDX-1127) in patients with advanced hematologic malignancies. J Clin Oncol. 2014;32(15_suppl):3024. doi:10.1200/jco.2014.32.15_suppl.3024. [Google Scholar] [CrossRef]
244. Lim SH, Sow HS, Wignall C, Mercer K, Caddy J, Boxall C, et al. Clinical and biological effects of combined CD27 and CD20 antibody therapy in relapsed/refractory B-cell lymphoma: the riva trial. Blood. 2021;138(Supplement 1):715. doi:10.1182/blood-2021-148332. [Google Scholar] [CrossRef]
245. Villasboas JC, Reeder CB, Tun HW, Bartlett NL, Sharon E, Laplant B, et al. The DIAL study (dual immunomodulation in aggressive lymphomaa randomized phase 2 study of CDX-1127 (varlilumab) in combination with nivolumab in patients with relapsed or refractory aggressive B-cell lymphomas (NCI 10089/NCT03038672). Blood. 2019;134(Supplement_1):1591. doi:10.1182/blood-2019-130449. [Google Scholar] [CrossRef]
246. Qiu M, Wu S, Chen X, Wang H. Update on diffuse large B-cell lymphoma: highlights from the 2022 ASCO annual meeting. Cancer Biol Med. 2022;19(8):1117–20. doi:10.20892/j.issn.2095-3941.2022.0403. [Google Scholar] [CrossRef]
247. Donini C, Rotolo R, Proment A, Aglietta M, Sangiolo D, Leuci V. Cellular immunotherapy targeting cancer stem cells: preclinical evidence and clinical perspective. Cells. 2021;10(3):543. doi:10.3390/cells10030543. [Google Scholar] [CrossRef]
248. Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. 1985;313(23):1485–92. doi:10.1056/NEJM198512053132327. [Google Scholar] [PubMed] [CrossRef]
249. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N Engl J Med. 1988;319(25):1676–80. doi:10.1056/nejm198812223192527. [Google Scholar] [CrossRef]
250. Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233(4770):1318–21. doi:10.1126/science.3489291. [Google Scholar] [PubMed] [CrossRef]
251. Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci U S A. 1994;91(9):3515–9. doi:10.1073/pnas.91.9.3515. [Google Scholar] [CrossRef]
252. Schmidt-Wolf IG, Negrin RS, Kiem HP, Blume KG, Weissman IL. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J Exp Med. 1991;174(1):139–49. doi:10.1084/jem.174.1.139. [Google Scholar] [CrossRef]
253. Miliotou AN, Papadopoulou LC. CAR T-cell therapy: a new era in cancer immunotherapy. Curr Pharm Biotechnol. 2018;19(1):5–18. doi:10.2174/1389201019666180418095526. [Google Scholar] [CrossRef]
254. Wang J, Tian S, Sun J, Zhang J, Lin L, Hu C. The presence of tumour-infiltrating lymphocytes (TILs) and the ratios between different subsets serve as prognostic factors in advanced hypopharyngeal squamous cell carcinoma. BMC Cancer. 2020;20(1):731. doi:10.1186/s12885-020-07234-0. [Google Scholar] [CrossRef]
255. Prasetyo A, Budiman J, Sadhana U. The relationship between tumor-infiltrating lymphocytes (TILs) and nasopharyngeal carcinoma (NPCa systematic review. Iran J Otorhinolaryngol. 2021;33(117):191–200. doi:10.22038/ijorl.2021.51405.2733. [Google Scholar] [PubMed] [CrossRef]
256. Aizaz M, Khan AS, Khan M, Musazade E, Yang G. Advancements in tumor-infiltrating lymphocytes: historical insights, contemporary milestones, and future directions in oncology therapy. Crit Rev Oncol Hematol. 2024;202(7):104471. doi:10.1016/j.critrevonc.2024.104471. [Google Scholar] [PubMed] [CrossRef]
257. Kazemi MH, Sadri M, Najafi A, Rahimi A, Baghernejadan Z, Khorramdelazad H, et al. Tumor-infiltrating lymphocytes for treatment of solid tumors: it takes two to tango? Front Immunol. 2022;13:1018962. doi:10.3389/fimmu.2022.1018962. [Google Scholar] [PubMed] [CrossRef]
258. Hulen TM, Chamberlain CA, Svane IM, Met Ö. ACT up TIL now: the evolution of tumor-infiltrating lymphocytes in adoptive cell therapy for the treatment of solid tumors. Immuno. 2021;1(3):194–211. doi:10.3390/immuno1030012. [Google Scholar] [CrossRef]
259. Lifileucel first cellular therapy approved for cancer, NCI. 2024 [Internet]. [cited 2024 Jul 26]. Available from: https://www.cancer.gov/news-events/cancer-currents-blog/2024/fda-amtagvi-til-therapy-melanoma. [Google Scholar]
260. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–8. doi:10.1126/science.aaa4967. [Google Scholar] [CrossRef]
261. Sakellariou-Thompson D, Forget MA, Hinchcliff E, Celestino J, Hwu P, Jazaeri AA, et al. Potential clinical application of tumor-infiltrating lymphocyte therapy for ovarian epithelial cancer prior or post-resistance to chemotherapy. Cancer Immunol Immunother. 2019;68(11):1747–57. doi:10.1007/s00262-019-02402-z. [Google Scholar] [PubMed] [CrossRef]
262. Wang S, Sun J, Chen K, Ma P, Lei Q, Xing S, et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. 2021;19(1):140. doi:10.1186/s12916-021-02006-4. [Google Scholar] [PubMed] [CrossRef]
263. Weinstein-Marom H, Gross G, Levi M, Brayer H, Schachter J, Itzhaki O, et al. Genetic modification of tumor-infiltrating lymphocytes via retroviral transduction. Front Immunol. 2021;11:584148. doi:10.3389/fimmu.2020.584148. [Google Scholar] [PubMed] [CrossRef]
264. Kishton RJ, Vodnala SK, Vizcardo R, Restifo NP. Next generation immunotherapy: enhancing stemness of polyclonal T cells to improve anti-tumor activity. Curr Opin Immunol. 2022;74:39–45. doi:10.1016/j.coi.2021.10.001. [Google Scholar] [CrossRef]
265. Cao B, Liu M, Wang L, Zhu K, Cai M, Chen X, et al. Remodelling of tumour microenvironment by microwave ablation potentiates immunotherapy of AXL-specific CAR T cells against non-small cell lung cancer. Nat Commun. 2022;13(1):6203. doi:10.1038/s41467-022-33968-5. [Google Scholar] [CrossRef]
266. Rokade S, Damani AM, Oft M, Emmerich J. IL-2 based cancer immunotherapies: an evolving paradigm. Front Immunol. 2024;15:1433989. doi:10.3389/fimmu.2024.1433989. [Google Scholar] [PubMed] [CrossRef]
267. Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun. 2018;9(1):2724. doi:10.1038/s41467-018-05072-0. [Google Scholar] [PubMed] [CrossRef]
268. Wang DR, Wu XL, Sun YL. Therapeutic targets and biomarkers of tumor immunotherapy: response vs. non-response. Signal Transduct Target Ther. 2022;7(1):331. doi:10.1038/s41392-022-01136-2. [Google Scholar] [PubMed] [CrossRef]
269. Waldmann TA, Dubois S, Miljkovic MD, Conlon KC. IL-15 in the combination immunotherapy of cancer. Front Immunol. 2020;11:868. doi:10.3389/fimmu.2020.00868. [Google Scholar] [PubMed] [CrossRef]
270. Rohaan MW, Borch TH, van den Berg JH, Met Ö, Kessels R, Geukes Foppen MH, et al. Tumor-infiltrating lymphocyte therapy or ipilimumab in advanced melanoma. N Engl J Med. 2022;387(23):2113–25. doi:10.1056/nejmoa2210233. [Google Scholar] [PubMed] [CrossRef]
271. Narvaez D, Nadal J, Nervo A, Costanzo MV, Paletta C, Petracci FE, et al. The emerging role of tertiary lymphoid structures in breast cancer: a narrative review. Cancers. 2024;16(2):396. doi:10.3390/cancers16020396. [Google Scholar] [PubMed] [CrossRef]
272. Kendal JK, Shehata MS, Lofftus SY, Crompton JG. Cancer-associated B cells in sarcoma. Cancers. 2023;15(3):622. doi:10.3390/cancers15030622. [Google Scholar] [PubMed] [CrossRef]
273. Tomasik J, Jasiński M, Basak GW. Next generations of CAR-T cells-new therapeutic opportunities in hematology? Front Immunol. 2022;13:1034707. doi:10.3389/fimmu.2022.1034707. [Google Scholar] [PubMed] [CrossRef]
274. Georgiou-Siafis SK, Miliotou AN, Ntenti C, Pappas IS, Papadopoulou LC. An innovative PTD-IVT-mRNA delivery platform for CAR immunotherapy of ErbB(+) solid tumor neoplastic cells. Biomedicines. 2022;10(11):2885. doi:10.3390/biomedicines10112885. [Google Scholar] [PubMed] [CrossRef]
275. Amiri M, Moaveni AK, Majidi Zolbin M, Shademan B, Nourazarian A. Optimizing cancer treatment: the synergistic potential of CAR-T cell therapy and CRISPR/Cas9. Front Immunol. 2024;15:1462697. doi:10.3389/fimmu.2024.1462697. [Google Scholar] [PubMed] [CrossRef]
276. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–8. doi:10.1038/nm.4441. [Google Scholar] [CrossRef]
277. Pan J, Niu Q, Deng B, Liu S, Wu T, Gao Z, et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia. 2019;33(12):2854–66. doi:10.1038/s41375-019-0488-7. [Google Scholar] [CrossRef]
278. Ormhøj M, Scarfò I, Cabral ML, Bailey SR, Lorrey SJ, Bouffard AA, et al. Chimeric antigen receptor T cells targeting CD79b show efficacy in lymphoma with or without cotargeting CD19. Clin Cancer Res. 2019;25(23):7046–57. doi:10.1158/1078-0432.CCR-19-1337. [Google Scholar] [PubMed] [CrossRef]
279. Xu Z, Huang X. Cellular immunotherapy for hematological malignancy: recent progress and future perspectives. Cancer Biol Med. 2021;18(4):966–80. doi:10.20892/j.issn.2095-3941.2020.0801. [Google Scholar] [CrossRef]
280. Rodríguez-Lobato LG, Ganzetti M, Fernández de Larrea C, Hudecek M, Einsele H, Danhof S. CAR T-cells in multiple myeloma: state of the art and future directions. Front Oncol. 2020;10:1243. doi:10.3389/fonc.2020.01243. [Google Scholar] [PubMed] [CrossRef]
281. Li C, Mei H, Hu Y, Guo T, Liu L, Jiang H, et al. A bispecific CAR-T cell therapy targeting bcma and CD38 for relapsed/refractory multiple myeloma: updated results from a phase 1 dose-climbing trial. Blood. 2019;134(Supplement_1):930. doi:10.1182/blood-2019-130340. [Google Scholar] [CrossRef]
282. Shi M, Wang J, Huang H, Liu D, Cheng H, Wang X, et al. Bispecific CAR T cell therapy targeting BCMA and CD19 in relapsed/refractory multiple myeloma: a phase I/II trial. Nat Commun. 2024;15(1):3371. doi:10.1038/s41467-024-47801-8. [Google Scholar] [PubMed] [CrossRef]
283. Wang CM, Wu ZQ, Wang Y, Guo YL, Dai HR, Wang XH, et al. Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory Hodgkin lymphoma: an open-label phase I trial. Clin Cancer Res. 2017;23(5):1156–66. doi:10.1158/1078-0432.ccr-16-1365. [Google Scholar] [PubMed] [CrossRef]
284. Grover NS, Savoldo B. Challenges of driving CD30-directed CAR-T cells to the clinic. BMC Cancer. 2019;19(1):203. doi:10.1186/s12885-019-5415-9. [Google Scholar] [PubMed] [CrossRef]
285. Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130(3):285–96. doi:10.1182/blood-2017-01-761320. [Google Scholar] [PubMed] [CrossRef]
286. Mardiana S, Gill S. CAR T cells for acute myeloid leukemia: state of the art and future directions. Front Oncol. 2020;10:697. doi:10.3389/fonc.2020.00697. [Google Scholar] [PubMed] [CrossRef]
287. Kenderian SS, Porter DL, Gill S. Chimeric antigen receptor T cells and hematopoietic cell transplantation: how not to put the CART before the horse. Biol Blood Marrow Transplant. 2017;23(2):235–46. doi:10.1016/j.bbmt.2016.09.002. [Google Scholar] [PubMed] [CrossRef]
288. Tallantyre EC, Evans NA, Parry-Jones J, Morgan MPG, Jones CH, Ingram W. Neurological updates: neurological complications of CAR-T therapy. J Neurol. 2021;268(4):1544–54. doi:10.1007/s00415-020-10237-3. [Google Scholar] [PubMed] [CrossRef]
289. Nie Y, Lu W, Chen D, Tu H, Guo Z, Zhou X, et al. Mechanisms underlying CD19-positive ALL relapse after anti-CD19 CAR T cell therapy and associated strategies. Biomark Res. 2020;8(1):18. doi:10.1186/s40364-020-00197-1. [Google Scholar] [PubMed] [CrossRef]
290. Cordoba S, Onuoha S, Thomas S, Pignataro DS, Hough R, Ghorashian S, et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: a phase 1 trial. Nat Med. 2021;27(10):1797–805. doi:10.1038/s41591-021-01497-1. [Google Scholar] [PubMed] [CrossRef]
291. Yang J, Jiang P, Zhang X, Zhu X, Dong Q, He J, et al. Anti-CD19/CD22 dual CAR-T therapy for refractory and relapsed B-cell acute lymphoblastic leukemia. Blood. 2019;134(Supplement_1):284. doi:10.1182/blood-2019-126429. [Google Scholar] [CrossRef]
292. Bhaskar ST, Dholaria B, Savani BN, Sengsayadeth S, Oluwole O. Overview of approved CAR-T products and utility in clinical practice. Clin Hematol Int. 2024;6(4):93–9. doi:10.46989/001c.124277. [Google Scholar] [PubMed] [CrossRef]
293. Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Ther. 2017;25(8):1769–81. doi:10.1016/j.ymthe.2017.06.012. [Google Scholar] [PubMed] [CrossRef]
294. Lupo KB, Matosevic S. Natural killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers. 2019;11(6):769. doi:10.3390/cancers11060769. [Google Scholar] [CrossRef]
295. Morandi F, Yazdanifar M, Cocco C, Bertaina A, Airoldi I. Engineering the bridge between innate and adaptive immunity for cancer immunotherapy: focus on γδ T and NK cells. Cells. 2020;9(8):1757. doi:10.3390/cells9081757. [Google Scholar] [PubMed] [CrossRef]
296. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):3051–7. doi:10.1182/blood-2004-07-2974. [Google Scholar] [PubMed] [CrossRef]
297. Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. 2014;123(25):3855–63. doi:10.1182/blood-2013-10-532531. [Google Scholar] [PubMed] [CrossRef]
298. Cooley S, He F, Bachanova V, Vercellotti GM, DeFor TE, Curtsinger JM, et al. First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv. 2019;3(13):1970–80. doi:10.1182/bloodadvances.2018028332. [Google Scholar] [PubMed] [CrossRef]
299. Bachanova V, McKenna DH, Luo X, Defor TE, Cooley S, Warlick E, et al. First-in-human phase I study of nicotinamide-expanded related donor natural killer cells for the treatment of relapsed/refractory non-Hodgkin lymphoma and multiple myeloma. Biol Blood Marrow Transplant. 2019;25(3):S175–6. doi:10.1016/j.bbmt.2018.12.317. [Google Scholar] [CrossRef]
300. Boyiadzis M, Agha M, Redner RL, Sehgal A, Im A, Hou JZ, et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy. 2017;19(10):1225–32. doi:10.1016/j.jcyt.2017.07.008. [Google Scholar] [PubMed] [CrossRef]
301. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8(357):357ra123. doi:10.1126/scitranslmed.aaf2341. [Google Scholar] [PubMed] [CrossRef]
302. Woan KV, Kim H, Bjordahl R, Davis ZB, Gaidarova S, Goulding J, et al. Harnessing features of adaptive NK cells to generate iPSC-derived NK cells for enhanced immunotherapy. Cell Stem Cell. 2021;28(12):2062–75.e5. doi:10.1016/j.stem.2021.08.013. [Google Scholar] [PubMed] [CrossRef]
303. Merino A, Maakaron J, Bachanova V. Advances in NK cell therapy for hematologic malignancies: NK source, persistence and tumor targeting. Blood Rev. 2023;60(8):101073. doi:10.1016/j.blre.2023.101073. [Google Scholar] [PubMed] [CrossRef]
304. Sabry M, Lowdell MW. Killers at the crossroads: the use of innate immune cells in adoptive cellular therapy of cancer. Stem Cells Transl Med. 2020;9(9):974–84. doi:10.1002/sctm.19-0423. [Google Scholar] [CrossRef]
305. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–53. doi:10.1056/NEJMoa1910607. [Google Scholar] [PubMed] [CrossRef]
306. Chien YH, Meyer C, Bonneville M. γδ T cells: first line of defense and beyond. Annu Rev Immunol. 2014;32(1):121–55. doi:10.1146/annurev-immunol-032713-120216. [Google Scholar] [PubMed] [CrossRef]
307. Vantourout P, Hayday A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat Rev Immunol. 2013;13(2):88–100. doi:10.1038/nri3384. [Google Scholar] [PubMed] [CrossRef]
308. Miyagawa F, Tanaka Y, Yamashita S, Minato N. Essential requirement of antigen presentation by monocyte lineage cells for the activation of primary human gamma delta T cells by aminobisphosphonate antigen. J Immunol. 2001;166(9):5508–14. doi:10.4049/jimmunol.166.9.5508. [Google Scholar] [PubMed] [CrossRef]
309. Sebestyen Z, Prinz I, Déchanet-Merville J, Silva-Santos B, Kuball J. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat Rev Drug Discov. 2020;19(3):169–84. doi:10.1038/s41573-019-0038-z. [Google Scholar] [PubMed] [CrossRef]
310. Todaro M, D’Asaro M, Caccamo N, Iovino F, Francipane MG, Meraviglia S, et al. Efficient killing of human colon cancer stem cells by γδ T lymphocytes. J Immunol. 2009;182(11):7287–96. doi:10.4049/jimmunol.0804288. [Google Scholar] [PubMed] [CrossRef]
311. Lai D, Wang F, Chen Y, Wang C, Liu S, Lu B, et al. Human ovarian cancer stem-like cells can be efficiently killed by γδ T lymphocytes. Cancer Immunol Immunother. 2012;61(7):979–89. doi:10.1007/s00262-011-1166-4. [Google Scholar] [CrossRef]
312. Nishio N, Fujita M, Tanaka Y, Maki H, Zhang R, Hirosawa T, et al. Zoledronate sensitizes neuroblastoma-derived tumor-initiating cells to cytolysis mediated by human γδ T cells. J Immunother. 2012;35(8):598–606. doi:10.1097/CJI.0b013e31826a745a. [Google Scholar] [CrossRef]
313. Miyashita M, Tomogane M, Nakamura Y, Shimizu T, Fujihara A, Ukimura O, et al. Sphere-derived prostate cancer stem cells are resistant to γδ T cell cytotoxicity. Anticancer Res. 2020;40(10):5481–7. doi:10.21873/anticanres.14559. [Google Scholar] [PubMed] [CrossRef]
314. Dutta I, Dieters-Castator D, Papatzimas JW, Medina A, Schueler J, Derksen DJ, et al. ADAM protease inhibition overcomes resistance of breast cancer stem-like cells to γδ T cell immunotherapy. Cancer Lett. 2021;496:156–68. doi:10.1016/j.canlet.2020.10.013. [Google Scholar] [PubMed] [CrossRef]
315. Nicol AJ, Tokuyama H, Mattarollo SR, Hagi T, Suzuki K, Yokokawa K, et al. Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. Br J Cancer. 2011;105(6):778–86. doi:10.1038/bjc.2011.293. [Google Scholar] [CrossRef]
316. Ye W, Kong X, Zhang W, Weng Z, Wu X. The roles of γδ T cells in hematopoietic stem cell transplantation. Cell Transplant. 2020;29(9):963689720966980. doi:10.1177/0963689720966980. [Google Scholar] [CrossRef]
317. Yu J, Sun H, Cao W, Song Y, Jiang Z. Research progress on dendritic cell vaccines in cancer immunotherapy. Exp Hematol Oncol. 2022;11(1):3. doi:10.1186/s40164-022-00257-2. [Google Scholar] [PubMed] [CrossRef]
318. Hassani Najafabadi A, Zhang J, Aikins ME, Najaf Abadi ZI, Liao F, Qin Y, et al. Cancer immunotherapy via targeting cancer stem cells using vaccine nanodiscs. Nano Lett. 2020;20(10):7783–92. doi:10.1021/acs.nanolett.0c03414. [Google Scholar] [PubMed] [CrossRef]
319. Mathias FAS, Carvalho MGR, Ruiz JC. Therapeutic vaccines for hematological cancers: a scoping review of this immunotherapeutic approach as alternative to the treatment of these malignancies. Vaccines. 2025;13(2):114. doi:10.3390/vaccines13020114. [Google Scholar] [PubMed] [CrossRef]
320. Lin D, Shen Y, Liang T. Oncolytic virotherapy: basic principles, recent advances and future directions. Signal Transduct Target Ther. 2023;8(1):156. doi:10.1038/s41392-023-01407-6. [Google Scholar] [PubMed] [CrossRef]
321. Zhang YN, Wang SB, Song SS, Hu PY, Zhou YC, Mou YP, et al. Recent advances in targeting cancer stem cells using oncolytic viruses. Biotechnol Lett. 2020;42(6):865–74. doi:10.1007/s10529-020-02857-6. [Google Scholar] [PubMed] [CrossRef]
322. Faghihkhorasani A, Dalvand A, Derafsh E, Tavakoli F, Younis NK, Yasamineh S, et al. The role of oncolytic virotherapy and viral oncogenes in the cancer stem cells: a review of virus in cancer stem cells. Cancer Cell Int. 2023;23(1):250. doi:10.1186/s12935-023-03099-y. [Google Scholar] [PubMed] [CrossRef]
323. Innao V, Rizzo V, Allegra AG, Musolino C, Allegra A. Oncolytic viruses and hematological malignancies: a new class of immunotherapy drugs. Curr Oncol. 2020;28(1):159–83. doi:10.3390/curroncol28010019. [Google Scholar] [PubMed] [CrossRef]
324. Hemminki O, Dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol. 2020;13(1):84. doi:10.1186/s13045-020-00922-1. [Google Scholar] [PubMed] [CrossRef]
325. Lei W, Wang S, Xu N, Chen Y, Wu G, Zhang A, et al. Enhancing therapeutic efficacy of oncolytic vaccinia virus armed with Beclin-1, an autophagic Gene in leukemia and myeloma. Biomed Pharmacother. 2020;125:110030. doi:10.1016/j.biopha.2020.110030. [Google Scholar] [PubMed] [CrossRef]
326. Cook J, Peng KW, Witzig TE, Broski SM, Villasboas JC, Paludo J, et al. Clinical activity of single-dose systemic oncolytic VSV virotherapy in patients with relapsed refractory T-cell lymphoma. Blood Adv. 2022;6(11):3268–79. doi:10.1182/bloodadvances.2021006631. [Google Scholar] [PubMed] [CrossRef]
327. Pidelaserra-Martí G, Engeland CE. Mechanisms of measles virus oncolytic immunotherapy. Cytokine Growth Factor Rev. 2020;56:28–38. doi:10.1016/j.cytogfr.2020.07.009. [Google Scholar] [CrossRef]
328. Packiriswamy N, Upreti D, Zhou Y, Khan R, Miller A, Diaz RM, et al. Oncolytic measles virus therapy enhances tumor antigen-specific T-cell responses in patients with multiple myeloma. Leukemia. 2020;34(12):3310–22. doi:10.1038/s41375-020-0828-7. [Google Scholar] [CrossRef]
329. Herbein G, Nehme Z. Tumor control by cytomegalovirus: a door open for oncolytic virotherapy? Mol Ther Oncolytics. 2020;17:1–8. doi:10.1016/j.omto.2020.03.004. [Google Scholar] [PubMed] [CrossRef]
330. Rezaei R, Esmaeili Gouvarchin Ghaleh H, Farzanehpour M, Dorostkar R, Ranjbar R, Bolandian M, et al. Combination therapy with CAR T cells and oncolytic viruses: a new era in cancer immunotherapy. Cancer Gene Ther. 2022;29(6):647–60. doi:10.1038/s41417-021-00359-9. [Google Scholar] [PubMed] [CrossRef]
331. Mardi A, Shirokova AV, Mohammed RN, Keshavarz A, Zekiy AO, Thangavelu L, et al. Biological causes of immunogenic cancer cell death (ICD) and anti-tumor therapy; Combination of Oncolytic virus-based immunotherapy and CAR T-cell therapy for ICD induction. Cancer Cell Int. 2022;22(1):168. doi:10.1186/s12935-022-02585-z. [Google Scholar] [PubMed] [CrossRef]
332. Lei W, Ye Q, Hao Y, Chen J, Huang Y, Yang L, et al. CD19-targeted BiTE expression by an oncolytic vaccinia virus significantly augments therapeutic efficacy against B-cell lymphoma. Blood Cancer J. 2022;12(2):35. doi:10.1038/s41408-022-00634-4. [Google Scholar] [PubMed] [CrossRef]
333. Kazemi MH, Kuhestani Dehaghi B, Roshandel E, Parkhideh S, Mehdizadeh M, Salimi M, et al. Oncolytic virotherapy in hematopoietic stem cell transplantation. Hum Immunol. 2021;82(9):640–8. doi:10.1016/j.humimm.2021.05.007. [Google Scholar] [PubMed] [CrossRef]
334. Yang C, Hua N, Xie S, Wu Y, Zhu L, Wang S, et al. Oncolytic viruses as a promising therapeutic strategy for hematological malignancies. Biomed Pharmacother. 2021;139:111573. doi:10.1016/j.biopha.2021.111573. [Google Scholar] [CrossRef]
335. Howard F, Muthana M. Designer nanocarriers for navigating the systemic delivery of oncolytic viruses. Nanomed. 2020;15(1):93–110. doi:10.2217/nnm-2019-0323. [Google Scholar] [PubMed] [CrossRef]
336. Ajam-Hosseini M, Akhoondi F, Doroudian M. Nano based-oncolytic viruses for cancer therapy. Crit Rev Oncol Hematol. 2023;185(1):103980. doi:10.1016/j.critrevonc.2023.103980. [Google Scholar] [PubMed] [CrossRef]
337. Li Y, Yang H, Zong X, Li X, Yuan P, Yang C, et al. Oncolytic virus-like nanoparticles for tumor-specific gene delivery. Adv Funct Materials. 2024;34(27):2314898. doi:10.1002/adfm.202314898. [Google Scholar] [CrossRef]
338. Yang J, Liu B. Nanoparticle-mediated delivery of oncolytic viral genomes: an innovative strategy for tumor-targeted immunotherapy. Cancer Nanotechnol. 2025;16(1):20. doi:10.1186/s12645-025-00322-5. [Google Scholar] [CrossRef]
339. Safarzadeh M, Saadat N, Abbasi-Molaei S, Rastegari-Pouyani M. Extracellular vesicles as missiles for enhanced anti-tumor efficacy of oncolytic viruses: from disseminating oncolysis and anti-tumor immunity to targeted delivery. Cell Commun Signal. 2025;23(1):276. doi:10.1186/s12964-025-02283-z. [Google Scholar] [PubMed] [CrossRef]
340. Kakiuchi Y, Kuroda S, Kanaya N, Kagawa S, Tazawa H, Fujiwara T. Exosomes as a drug delivery tool for cancer therapy: a new era for existing drugs and oncolytic viruses. Expert Opin Ther Targets. 2023;27(9):807–16. doi:10.1080/14728222.2023.2259102. [Google Scholar] [PubMed] [CrossRef]
341. Wang X, Yang Y, Wang N, Wu X, Xu J, Zhou Y, et al. Mesenchymal stem cell carriers enhance antitumor efficacy induced by oncolytic reovirus in acute myeloid leukemia. Int Immunopharmacol. 2021;94:107437. doi:10.1016/j.intimp.2021.107437. [Google Scholar] [PubMed] [CrossRef]
342. Castleton A, Dey A, Beaton B, Patel B, Aucher A, Davis DM, et al. Human mesenchymal stromal cells deliver systemic oncolytic measles virus to treat acute lymphoblastic leukemia in the presence of humoral immunity. Blood. 2014;123(9):1327–35. doi:10.1182/blood-2013-09-528851. [Google Scholar] [PubMed] [CrossRef]
343. Ghasemi Darestani N, Gilmanova AI, Al-Gazally ME, Zekiy AO, Ansari MJ, Zabibah RS, et al. Mesenchymal stem cell-released oncolytic virus: an innovative strategy for cancer treatment. Cell Commun Signal. 2023;21(1):43. doi:10.1186/s12964-022-01012-0. [Google Scholar] [PubMed] [CrossRef]
344. Zheng HC. The molecular mechanisms of chemoresistance in cancers. Oncotarget. 2017;8(35):59950–64. doi:10.18632/oncotarget.19048. [Google Scholar] [PubMed] [CrossRef]
345. Castagnoli L, De Santis F, Volpari T, Vernieri C, Tagliabue E, Di Nicola M, et al. Cancer stem cells: devil or savior-looking behind the scenes of immunotherapy failure. Cells. 2020;9(3):555. doi:10.3390/cells9030555. [Google Scholar] [PubMed] [CrossRef]
346. Wu B, Shi X, Jiang M, Liu H. Cross-talk between cancer stem cells and immune cells: potential therapeutic targets in the tumor immune microenvironment. Mol Cancer. 2023;22(1):38. doi:10.1186/s12943-023-01748-4. [Google Scholar] [CrossRef]
347. Ferguson LP, Diaz E, Reya T. The role of the microenvironment and immune system in regulating stem cell fate in cancer. Trends Cancer. 2021;7(7):624–34. doi:10.1016/j.trecan.2020.12.014. [Google Scholar] [PubMed] [CrossRef]
348. Vadakekolathu J, Rutella S. Escape from T-cell-targeting immunotherapies in acute myeloid leukemia. Blood. 2024;143(26):2689–700. doi:10.1182/blood.2023019961. [Google Scholar] [PubMed] [CrossRef]
349. Kontandreopoulou CN, Diamantopoulos PT, Tiblalexi D, Giannakopoulou N, Viniou NA. PARP1 as a therapeutic target in acute myeloid leukemia and myelodysplastic syndrome. Blood Adv. 2021;5(22):4794–805. doi:10.1182/bloodadvances.2021004638. [Google Scholar] [PubMed] [CrossRef]
350. Lei MML, Lee TKW. Cancer stem cells: emerging key players in immune evasion of cancers. Front Cell Dev Biol. 2021;9:692940. doi:10.3389/fcell.2021.692940. [Google Scholar] [PubMed] [CrossRef]
351. Garcia-Mayea Y, Mir C, Masson F, Paciucci R, LLeonart ME. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin Cancer Biol. 2020;60(6):166–80. doi:10.1016/j.semcancer.2019.07.022. [Google Scholar] [PubMed] [CrossRef]
352. Testa U, Castelli G, Pelosi E. Membrane antigen targeting in acute myeloid leukemia using antibodies or CAR-T cells. Cancers. 2024;16(21):3627. doi:10.3390/cancers16213627. [Google Scholar] [PubMed] [CrossRef]
353. Niswander LM, Graff ZT, Chien CD, Chukinas JA, Meadows CA, Leach LC, et al. Potent preclinical activity of FLT3-directed chimeric antigen receptor T-cell immunotherapy against FLT3- mutant acute myeloid leukemia and KMT2A-rearranged acute lymphoblastic leukemia. Haematologica. 2023;108(2):457–71. doi:10.3324/haematol.2022.281456. [Google Scholar] [PubMed] [CrossRef]
354. Saw PE, Liu Q, Wong PP, Song E. Cancer stem cell mimicry for immune evasion and therapeutic resistance. Cell Stem Cell. 2024;31(8):1101–12. doi:10.1016/j.stem.2024.06.003. [Google Scholar] [PubMed] [CrossRef]
355. Daver N, Alotaibi AS, Bücklein V, Subklewe M. T-cell-based immunotherapy of acute myeloid leukemia: current concepts and future developments. Leukemia. 2021;35(7):1843–63. doi:10.1038/s41375-021-01253-x. [Google Scholar] [PubMed] [CrossRef]
356. Tettamanti S, Pievani A, Biondi A, Dotti G, Serafini M. Catch me if you can: how AML and its niche escape immunotherapy. Leukemia. 2022;36(1):13–22. doi:10.1038/s41375-021-01350-x. [Google Scholar] [PubMed] [CrossRef]
357. Khaldoyanidi S, Nagorsen D, Stein A, Ossenkoppele G, Subklewe M. Immune biology of acute myeloid leukemia: implications for immunotherapy. J Clin Oncol. 2021;39(5):419–32. doi:10.1200/JCO.20.00475. [Google Scholar] [PubMed] [CrossRef]
358. Chen C, Yu W, Alikarami F, Qiu Q, Chen CH, Flournoy J, et al. Single-cell multiomics reveals increased plasticity, resistant populations, and stem-cell-like blasts in KMT2A-rearranged leukemia. Blood. 2022;139(14):2198–211. doi:10.1182/blood.2021013442. [Google Scholar] [PubMed] [CrossRef]
359. Giles AJ, Reid CM, Evans JD, Murgai M, Vicioso Y, Highfill SL, et al. Activation of hematopoietic stem/progenitor cells promotes immunosuppression within the pre-metastatic niche. Cancer Res. 2016;76(6):1335–47. doi:10.1158/0008-5472.CAN-15-0204. [Google Scholar] [PubMed] [CrossRef]
360. Pan Q, Li Q, Liu S, Ning N, Zhang X, Xu Y, et al. Concise review: targeting cancer stem cells using immunologic approaches. Stem Cells. 2015;33(7):2085–92. doi:10.1002/stem.2039. [Google Scholar] [PubMed] [CrossRef]
361. Moscarelli J, Zahavi D, Maynard R, Weiner LM. The next generation of cellular immunotherapy: chimeric antigen receptor-natural killer cells. Transplant Cell Ther. 2022;28(10):650–6. doi:10.1016/j.jtct.2022.06.025. [Google Scholar] [PubMed] [CrossRef]
362. Pan Y, Yuan C, Zeng C, Sun C, Xia L, Wang G, et al. Cancer stem cells and niches: challenges in immunotherapy resistance. Mol Cancer. 2025;24(1):52. doi:10.1186/s12943-025-02265-2. [Google Scholar] [PubMed] [CrossRef]
363. Galassi C, Musella M, Manduca N, Maccafeo E, Sistigu A. The immune privilege of cancer stem cells: a key to understanding tumor immune escape and therapy failure. Cells. 2021;10(9):2361. doi:10.3390/cells10092361. [Google Scholar] [CrossRef]
364. Shimabukuro-Vornhagen A, Draube A, Liebig TM, Rothe A, Kochanek M, von Bergwelt-Baildon MS. The immunosuppressive factors IL-10, TGF-β, and VEGF do not affect the antigen-presenting function of CD40-activated B cells. J Exp Clin Cancer Res. 2012;31(1):47. doi:10.1186/1756-9966-31-47. [Google Scholar] [PubMed] [CrossRef]
365. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. 2018;15(5):325–40. doi:10.1038/nrclinonc.2018.29. [Google Scholar] [PubMed] [CrossRef]
366. Smaldone G, Di Matteo F, Castelluccio R, Napolitano V, Miranda MR, Manfra M, et al. Targeting the CXCR4/CXCL12 axis in cancer therapy: analysis of recent advances in the development of potential anticancer agents. Molecules. 2025;30(6):1380. doi:10.3390/molecules30061380. [Google Scholar] [PubMed] [CrossRef]
367. Yuan CS, Deng ZW, Qin D, Mu YZ, Chen XG, Liu Y. Hypoxia-modulatory nanomaterials to relieve tumor hypoxic microenvironment and enhance immunotherapy: where do we stand? Acta Biomater. 2021;125(20):1–28. doi:10.1016/j.actbio.2021.02.030. [Google Scholar] [PubMed] [CrossRef]
368. Shi R, Sun J, Zhou Z, Shi M, Wang X, Gao Z, et al. Integration of multiple machine learning approaches develops a gene mutation-based classifier for accurate immunotherapy outcomes. npj Precis Oncol. 2025;9(1):54. doi:10.1038/s41698-025-00842-8. [Google Scholar] [CrossRef]
369. Olawade DB, Clement David-Olawade A, Adereni T, Egbon E, Teke J, Boussios S. Integrating AI into cancer immunotherapy-a narrative review of current applications and future directions. Diseases. 2025;13(1):24. doi:10.3390/diseases13010024. [Google Scholar] [PubMed] [CrossRef]
370. Wang G, Kang X, Chen KS, Jehng T, Jones L, Chen J, et al. An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat Commun. 2020;11(1):1395. doi:10.1038/s41467-020-15229-5. [Google Scholar] [PubMed] [CrossRef]
371. Kim J, Lee BJ, Moon S, Lee H, Lee J, Kim BS, et al. Strategies to overcome hurdles in cancer immunotherapy. Biomater Res. 2024;28(2):0080. doi:10.34133/bmr.0080. [Google Scholar] [PubMed] [CrossRef]
372. Kohn DB, Chen YY, Spencer MJ. Successes and challenges in clinical gene therapy. Gene Ther. 2023;30(10–11):738–46. doi:10.1038/s41434-023-00390-5. [Google Scholar] [PubMed] [CrossRef]
373. Pospieszna J, Dams-Kozlowska H, Udomsak W, Murias M, Kucinska M. Unmasking the deceptive nature of cancer stem cells: the role of CD133 in revealing their secrets. Int J Mol Sci. 2023;24(13):10910. doi:10.3390/ijms241310910. [Google Scholar] [PubMed] [CrossRef]
374. Lee BS, Rui-Jeat F, Soon-Keng C. Immunotherapy for hematological cancers. In: Handbook of cancer and immunology. Cham, Switzerland: Springer International Publishing; 2023. p. 1–15. doi:10.1007/978-3-030-80962-1_226-1. [Google Scholar] [CrossRef]
375. Park MH, Lee H, Han KH. Immune cell engagers: advancing precision immunotherapy for cancer treatment. Antibodies. 2025;14(1):16. doi:10.3390/antib14010016. [Google Scholar] [CrossRef]
376. Shen X, Zhao Y, Wang Z, Shi Q. Recent advances in high-throughput single-cell transcriptomics and spatial transcriptomics. Lab Chip. 2022;22(24):4774–91. doi:10.1039/d2lc00633b. [Google Scholar] [PubMed] [CrossRef]
377. Jia Q, Chu H, Jin Z, Long H, Zhu B. High-throughput single-сell sequencing in cancer research. Signal Transduct Target Ther. 2022;7(1):145. doi:10.1038/s41392-022-00990-4. [Google Scholar] [PubMed] [CrossRef]
378. Sinha T, Khan A, Awan M, Bokhari SFH, Ali K, Amir M, et al. Artificial intelligence and machine learning in predicting the response to immunotherapy in non-small cell lung carcinoma: a systematic review. Cureus. 2024;16(5):e61220. doi:10.7759/cureus.61220. [Google Scholar] [PubMed] [CrossRef]
379. Oved JH, Russell A, DeZern A, Prockop SE, Bonfim C, Sharma A, et al. The role of the conditioning regimen for autologous and ex vivo genetically modified hematopoietic stem cell-based therapies: recommendations from the ISCT stem cell engineering committee. Cytotherapy. 2025;27(1):78–84. doi:10.1016/j.jcyt.2024.09.001. [Google Scholar] [PubMed] [CrossRef]
380. Emens LA, Romero PJ, Anderson AC, Bruno TC, Capitini CM, Collyar D, et al. Challenges and opportunities in cancer immunotherapy: a society for immunotherapy of cancer (SITC) strategic vision. J Immunother Cancer. 2024;12(6):e009063. doi:10.1136/jitc-2024-009063. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools