
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Rotenone-Induced Mitochondrial Dysfunction, Neuroinflammation, Oxidative Stress, and Glial Activation in Parkinson’s and Alzheimer’s Diseases
1 Laboratory of Neurophysiology, Instituto Nacional de Neurología y Neurocirugía, Mexico City, PC 14269, Mexico
2 School of Medicine, Autonomous University of Yucatan, Merida, PC 97000, Mexico
3 Psychology Department, Universidad Iberoamericana, Mexico City, PC 01376, Mexico
4 Department of Neurochemistry, Instituto Nacional de Neurología y Neurocirugía, Mexico City, PC 14269, Mexico
* Corresponding Author: MoiséS Rubio-Osornio. Email:
# Shared first co authorship
(This article belongs to the Special Issue: Mitochondrial Dynamics and Oxidative Stress in Disease: Cellular Mechanisms and Therapeutic Targets)
BIOCELL 2025, 49(8), 1391-1412. https://doi.org/10.32604/biocell.2025.066320
Received 05 April 2025; Accepted 05 June 2025; Issue published 29 August 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Rotenone is a lipophilic herbicide extensively utilized in experimental neurodegenerative models because of its capacity to disrupt complex I of the mitochondrial electron transport chain. This inhibition results in reduced ATP synthesis, elevated reactive oxygen species (ROS) formation, and mitochondrial malfunction, which instigates oxidative stress and cellular damage, critical elements in neurodegenerative disorders like Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and Alzheimer’s disease (AD). In addition to causing direct neuronal injury, rotenone significantly contributes to the activation of glial cells, specifically microglia and astrocytes. Activated microglia assumes a proinflammatory (M1) phenotype, distinguished by the secretion of inflammatory cytokines including tumor necrosis factor alpha (TNF-α), interleukin 1 beta (IL-1 β), and interleukin 6 (IL-6), with the generation of nitric oxide and ROS, which exacerbate the neuronal injury. Astrocytes can intensify neuroinflammation by secreting proinflammatory molecules and impairing their neuroprotective roles. Our hypothesis is that rotenone is posited to elicit a neuroinflammatory response via mitochondrial malfunction, ROS generation, and the activation of proinflammatory pathways in microglia and astrocytes. This mechanism leads to accelerated neuronal impairment, promoting neurodegeneration. Comprehending the inflammatory pathways activated by rotenone is crucial for pinpointing therapeutic targets to regulate glial responses and mitigate the advancement of neurodegenerative disorders linked to mitochondrial malfunction and chronic inflammation. This review examines the function of glial cells and critical inflammatory pathways, namely Nuclear factor kappa β (NF-κB), Phosphoinositide 3-kinase/Protein kinase B/Mammalian target of rapamycin (PI3K/AKT/mTOR), and Wnt/β-catenin signaling pathway in Parkinson’s disease, Alzheimer’s disease, and ALS, emphasizing illness-specific responses and the translational constraints of rotenone-based models. The objective is to consolidate existing understanding regarding the role of rotenone-induced mitochondrial failure in promoting glial activation and neuroinflammation, highlighting the necessity for additional research into these pathways. Despite the prevalent application of rotenone in experimental models, its specific effects on glial-mediated inflammation are inadequately comprehended, necessitating further investigation to guide the formulation of targeted therapeutic strategies.Keywords
Mitochondria are vital organelles for cellular homeostasis, they are pivotal for energy production, metabolic regulation, cellular signaling, and apoptosis [1]. Their main role is to produce adenosine triphosphate (ATP) via oxidative phosphorylation, a process that takes place in the electron transport chain situated in the inner mitochondrial membrane [2]. Electrons are conveyed through protein complexes to molecular oxygen, creating an electrochemical gradient that facilitates ATP synthesis through ATP synthase [3]. Mitochondrial failure can undermine cell viability and contribute to numerous illnesses, including neurodegenerative disorders such as Parkinson’s disease (PD), Alzheimer’s disease (AD), and amyotrophic lateral sclerosis (ALS) [4]. Rotenone is a naturally occurring insecticide extensively utilized in experimental neurodegenerative models because of its capacity to specifically block complex I of the mitochondrial electron transport chain [5,6]. This inhibition obstructs the flow of electrons from reduced nicotinamide adenine dinucleotide (NADH) to ubiquinone, impeding ATP synthesis and fostering a pro-oxidative milieu [7]. As a result, the generation of ROS escalates, causing damage to lipids, proteins, and mitochondrial DNA, which finally results in cellular malfunction and neuronal demise [8].
Moreover, owing to its lipophilic characteristics, rotenone readily traverses the blood-brain barrier, facilitating its dissemination inside the central nervous system [9]. This trait renders it an effective instrument for eliciting neurodegeneration in animal models and investigating neurodegenerative disorders [10]. Investigations utilizing these models have shown that rotenone-induced mitochondrial dysfunction impacts cellular bioenergetics, activates inflammatory pathways, disrupts calcium equilibrium, and facilitates the aggregation of misfolded proteins, thereby exacerbating the advancement of neurodegenerative diseases [11,12]. A crucial element of rotenone-induced neurotoxicity is its effect on glial function, specifically the activation of microglia and the malfunctioning of astrocytes [13,14]. Exposure to rotenone induces an amplified inflammatory response in microglia, facilitating the secretion of pro-inflammatory cytokines including tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), which exacerbate neuronal injury via chronic neuroinflammation mechanisms [10,15]. The activation of microglia is partially facilitated via the Toll-like receptor 4 (TLR4) pathway and the activation of the NF-κB transcription factor, promoting a prolonged neurotoxic condition that jeopardizes neuronal survival [16].
Conversely, rotenone-induced astrocytic impairment disrupts glutamate homeostasis, energy metabolism, and neuronal trophic support [17]. Inhibition of mitochondrial complex I in astrocytes diminishes glutamate uptake by the EAAT2 transporter, resulting in neurotransmitter buildup at the synapse and exacerbating neuronal excitotoxicity [18]. Mitochondrial failure in astrocytes diminishes lactate synthesis, an essential energy substrate for neurons, thus hindering neuronal metabolism and heightening vulnerability to oxidative injury [19]. Microglia-mediated neuroinflammation and astrocytic metabolic dysfunction create a pathogenic circuit that intensifies neuronal degeneration in rotenone-induced neurodegenerative models [16,20,21]. Previous findings highlight the essential significance of glial cells in the etiology of neurodegenerative illnesses and indicate that modifying their activity may serve as a viable therapeutic approach to alleviate neuronal damage linked to mitochondrial malfunction [22]. Furthermore, rotenone-induced mitochondrial dysfunction impairs intracellular calcium homeostasis, resulting in elevated cytosolic calcium levels [23]. The increase in calcium levels induces excitotoxicity, a condition where excessive calcium accumulation in neurons leads to the hyperactivation of ion channels and glutamatergic receptors, including N-methyl-D-aspartate (NMDA) receptors [24]. Excitotoxicity leads to irreversible neuronal injury, worsening neurodegeneration, and hastening the advancement of conditions such as PD and AD [25]. Rotenone administration has been demonstrated to raise intracellular calcium concentrations in dopaminergic neurons, leading to compromised neurogenesis and heightened excitotoxicity in experimental settings [23,26]. Most investigations utilizing rotenone-induced neurodegenerative models have predominantly been performed on male rats or mice. Emerging research indicates that sex-related characteristics may affect susceptibility to rotenone toxicity, including variances in oxidative stress response, glial activation, and mitochondrial function. These findings underscore the imperative of including sex as a biological variable in experimental designs and analysis. Rotenone-induced mitochondrial impairment facilitates neurodegeneration via a multifaceted pathway encompassing oxidative stress, glial dysfunction, intracellular calcium dysregulation, and excitotoxicity. This study seeks to examine the cellular and molecular mechanisms involved in this process, emphasizing oxidative stress, glial activation, calcium dysregulation, and excitotoxicity, to improve our comprehension of their contributions to the pathogenesis of neurodegenerative diseases, including Parkinson’s disease, Alzheimer’s disease, and Amyotrophic Lateral Sclerosis.
A comprehensive search was performed on Google to locate pertinent publications regarding rotenone and its correlation with mitochondrial dysfunction in neurodegenerative illnesses, such as Parkinson’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis. The inquiry employed several combinations of keywords, including rotenone, mitochondrial malfunction, oxidative stress, glial activation, calcium dysregulation, neuroinflammation, and specified disorders. Articles published were chosen based on scientific rigor, methodological excellence, and relevance.
A review of experimental investigations on rotenone induced neurotoxicity emphasized its involvement in mitochondrial malfunction, oxidative damage, glial activation (microglia and astrocytes), calcium dysregulation, protein aggregation, and neuroinflammation. The results were consolidated to emphasize shared and disease-specific causes of neurotoxicity in different neurodegenerative diseases.
3 Model of Parkinson’s Disease Induced by Rotenone
Rotenone is utilized experimentally in Parkinson’s disease (PD) research [27–31]. This neurodegenerative disorder is characterized by the progressive degeneration of dopaminergic neurons in the substantia nigra, leading to motor dysfunction due to diminished dopamine levels in the brain [32]. Despite its prevalent use and reputation as a gold standard in experimental models of Parkinson’s disease due to its selective toxicity towards dopaminergic neurons, rotenone unlike 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), lacks genuine neurotoxic selectivity [33]. Rather, it functions as a broad-spectrum mitochondrial toxin that elicits nonspecific systemic toxicity, impacting nearly all cell types within the organism. Nonetheless, the clinical and neuropathological signs evident in the central nervous system are chiefly ascribed to the heightened vulnerability of certain brain areas, notably the substantia nigra and the locus coeruleus, to mitochondrial complex I inhibition [34]. This persistent inhibition results in significant and frequently permanent neurodegeneration of nigrostriatal dopaminergic neurons, correlated with classical motor symptoms like as bradykinesia and rigidity, closely resembling human PD [35]. Rotenone administration in rats and other animal models selectively induces degeneration of dopaminergic neurons in the substantia nigra, closely resembling the neuropathological features observed in human Parkinson’s disease [33]. This model is particularly beneficial for investigating the underlying causes of neuronal loss in PD and evaluating prospective therapeutic methods [36]. The rotenone-induced model of Parkinson’s disease in rats well mimics the motor impairments associated with the condition, such as stiffness and bradykinesia [37]. Furthermore, microglial activation and the formation of protein inclusions indicative of the illness were observed [38–40]. This model illustrates the aggregation of proteins, such as α-synuclein, in structures resembling Lewy bodies [41]. Protein aggregates are a clinical hallmark of Parkinson’s disease, and their formation in animal models provides a robust framework for examining the mechanisms of neurodegeneration [40]. Rotenone induces α-synuclein aggregation in neuronal cells, triggering a neuroinflammatory response similar to that observed in patients with PD [42–44]. Neuroinflammation is pivotal in the etiology of Parkinson’s disease, and elucidating the role of rotenone in this process may provide essential insights into the disease start. Injection of rotenone in mice has been demonstrated to elevate pro-inflammatory cytokines and activate microglia, thereby facilitating neurodegeneration in the Parkinson’s disease model [9,45,46], (Fig. 1). The behavioral manifestations and extent of cellular damage caused by rotenone are markedly affected by the route of delivery, dosage, and duration of exposure. Fleming et al. (2004) demonstrated that alterations in the delivery method (subcutaneous, intraperitoneal, or intravenous) at doses of 2–3 mg/kg produced significant variations in rearing behavior and tyrosine hydroxylase optical density [47]. Notably, although α-synuclein aggregation typically occurs after six weeks of continuous MPTP therapy, rotenone induces the formation of these deposits within approximately 10 days of exposure, underscoring its efficacy and swift neurotoxic impact [48,49].
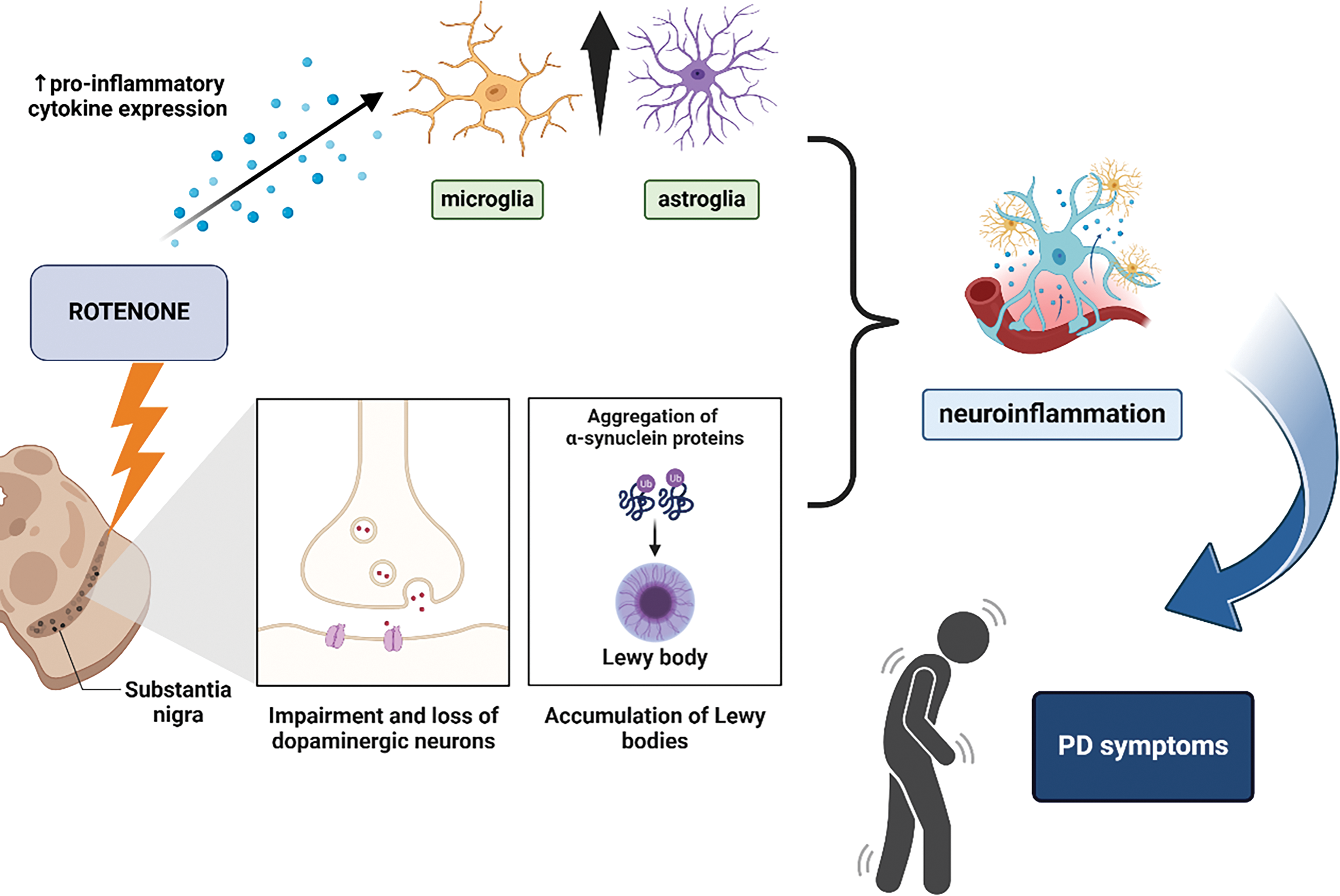
Figure 1: Schematic representation of the mechanisms underlying rotenone-induced Parkinson’s disease (PD) models. Rotenone exposure leads to mitochondrial dysfunction in the substantia nigra, resulting in the impairment and loss of dopaminergic neurons. This is accompanied by the aggregation of α-synuclein proteins and the formation of Lewy bodies. Additionally, rotenone promotes neuroinflammation by increasing pro-inflammatory cytokine expression, activating microglia and astroglia. These processes contribute to neuronal damage and ultimately lead to PD-related motor symptoms (Created using biorender.com)
4 The Function of Rotenone in Alzheimer’s Disease
Rotenone, a powerful inhibitor of mitochondrial complex I [42], impairs the electron transport chain, resulting in less ATP synthesis and increased ROS. This oxidative imbalance inflicts damage on lipids, proteins, and DNA, ultimately inducing apoptosis [50]. Cholinergic neurons are significantly impacted by rotenone, leading to compromised synaptic function and reduced levels of acetylcholine, a crucial neurotransmitter for cognition and memory [51,52]. A significant reduction of cholinergic neurons in the hippocampus was noted in rats treated with rotenone, corresponding with cognitive impairments characteristic of AD [53]. Furthermore, rotenone-induced mitochondrial impairment activates stress-related kinases, including p38 mitogen-activated protein kinase (MAPK) and Jun N-terminal kinase (JNK), intensifying neuroinflammation and neuronal degeneration [54]. Tau hyperphosphorylation, a significant clinical characteristic of AD [55], is facilitated by rotenone through oxidative stress and the activation of glycogen synthase kinase-3 beta (GSK-3β), an essential enzyme in aberrant tau processing [56]. The aggregation of hyperphosphorylated tau destabilizes microtubules, disrupts axonal transport, and results in synaptic degeneration [57]. Moreover, rotenone affects β-amyloid (Aβ) metabolism by modifying the processing of amyloid precursor protein (APP). This amplifies β-secretase (BACE1) activity, hence elevating Aβ synthesis and plaque formation [58]. The accumulation of Aβ exacerbates mitochondrial dysfunction and oxidative stress, establishing a detrimental cycle of neurotoxicity that hastens the onset of Alzheimer’s disease [57,59]. In addition to neuronal damage, rotenone stimulates glial cells, leading to the secretion of pro-inflammatory cytokines including TNF-α, IL-1β, and IL-6, which exacerbate synaptic injury [60]. Microglial activation induced by rotenone has been associated with increased inducible nitric oxide synthase (iNOS) expression and nitric oxide (NO) generation, resulting in further oxidative damage [16,61]. These findings collectively underscore the significance of rotenone in mimicking essential pathogenic mechanisms of AD, such as mitochondrial failure, oxidative damage, neuroinflammation, tau pathology, and Aβ buildup [58,60]. Consequently, it functions as an essential model for investigating the molecular and cellular foundations of AD (Fig. 2).
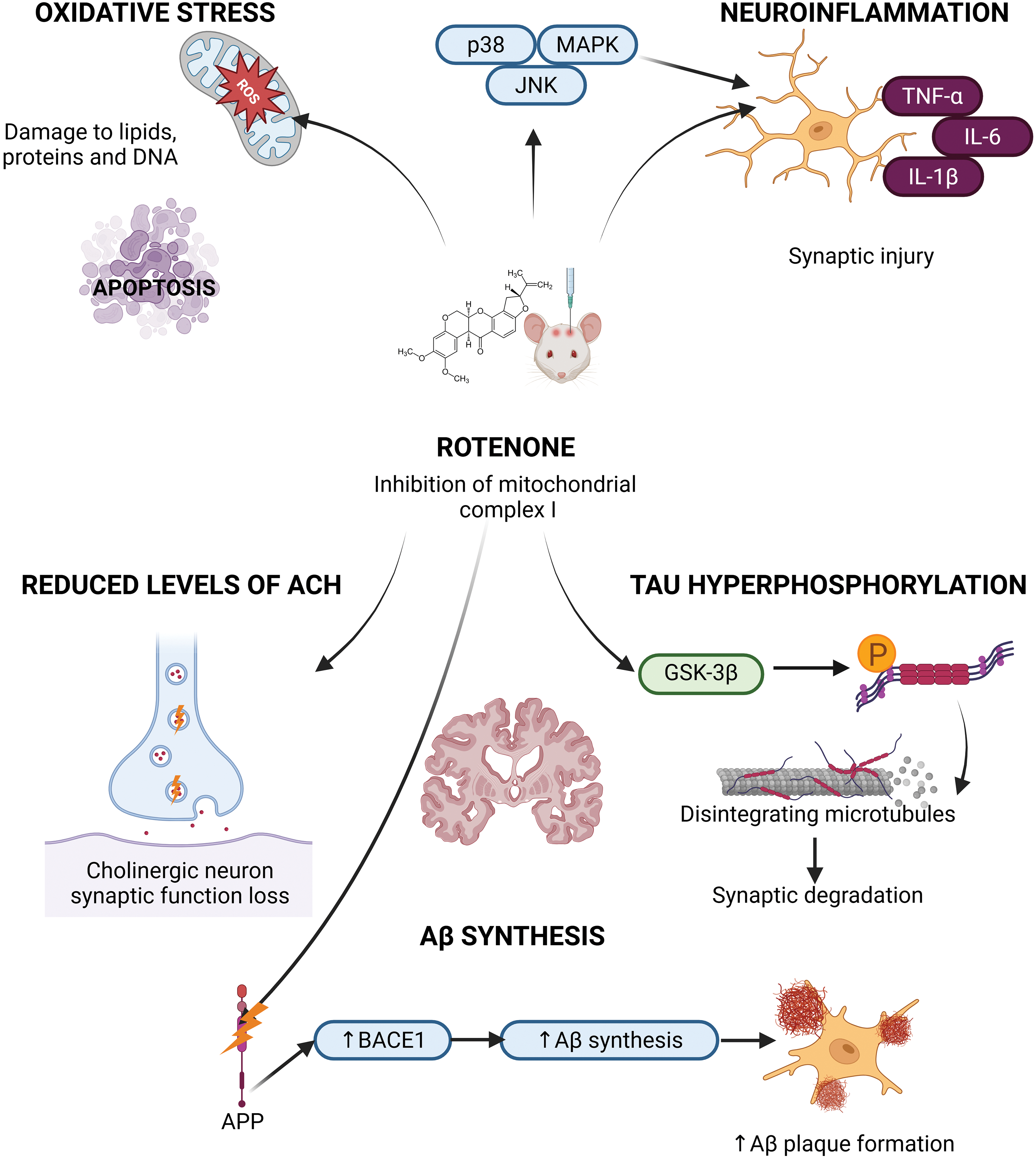
Figure 2: Schematic representation of the molecular mechanisms by which rotenone contributes to Alzheimer’s disease (AD) pathogenesis. Rotenone, a mitochondrial complex I inhibitor, induces mitochondrial dysfunction and excessive production of reactive oxygen species (ROS), leading to oxidative stress. This cascade promotes tau hyperphosphorylation, amyloid-β (Aβ) accumulation, neuroinflammation, and activation of apoptotic pathways, including caspase-3. These combined effects contribute to neuronal degeneration characteristic of AD (Created with BioRender.com)
5 Neurodegeneration Induced by Rotenone in Amyotrophic Lateral Sclerosis
In the context of amyotrophic lateral sclerosis (ALS), rotenone is capable of simulating neurodegeneration chiefly via mitochondrial dysfunction, oxidative stress, and neuroinflammation fundamental components of ALS pathogenesis [62]. Rotenone inhibits complex I of the electron transport cycle, so diminishing ATP synthesis and elevating ROS, resulting in oxidative damage to both motor neurons and glial cells [6,63]. This stress triggers apoptosis-related pathways, including Bax and caspase-3, leading to motor neuron death [15,64]. Furthermore, rotenone interferes with calcium homeostasis, intensifying glutamate excitotoxicity and endoplasmic reticulum stress [23,65]. In ALS models, rotenone enhances the activation of microglia and astrocytes, promoting the release of cytokines such as TNF-α and IL-1β, which further exacerbate neuronal degradation [10,66]. It also hinders the elimination of misfolded proteins, such as mutant superoxide dismutase 1 (SOD1), exacerbating proteotoxic stress and protein aggregation in ALS [67,68]. Rotenone exposure hastens motor dysfunction and the loss of spinal cord motor neurons in animal models, highlighting its relevance for investigating disease causes and evaluating neuroprotective therapies (Fig. 3) [69,70]. The intensification of motor symptoms and neurodegeneration noted in rotenone-treated mice validates the significance of this paradigm in ALS research [71].
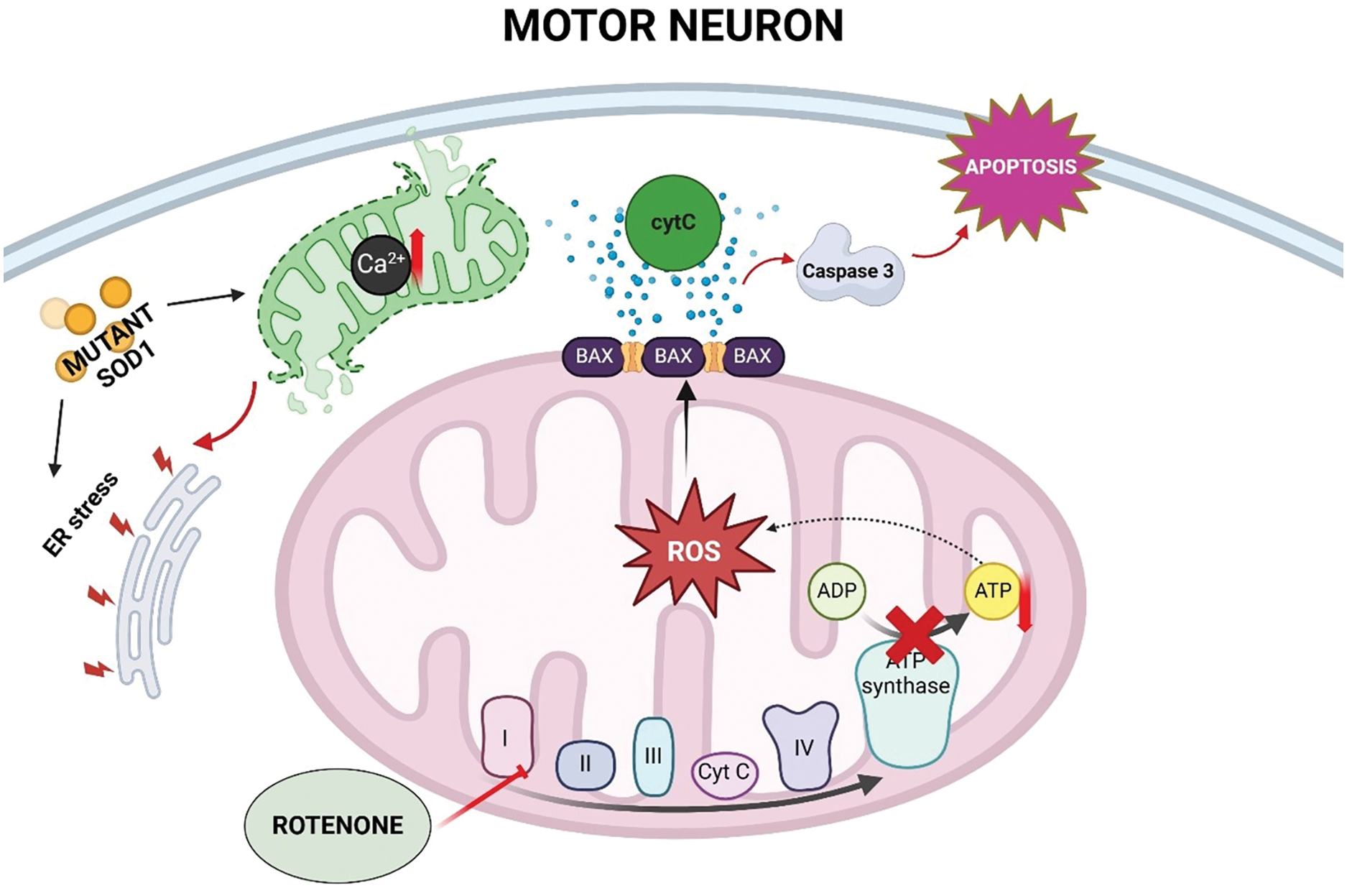
Figure 3: Schematic representation of the proposed mechanisms by which rotenone exposure may contribute to amyotrophic lateral sclerosis (ALS) pathogenesis. Rotenone inhibits mitochondrial complex I, leading to excessive reactive oxygen species (ROS) production and ATP (adenosine triphosphate) depletion in motor neurons. These changes, together with calcium (Ca2+) dysregulation and endoplasmic reticulum (ER) stress, activate pro-apoptotic pathways involving cytochrome c (cytC), BAX, and caspase 3. Mutations in superoxide dismutase 1 (SOD1) can exacerbate these processes, further promoting neuronal cell death (Created using BioRender.com)
6 Impact of Rotenone on Mitochondrial Function and Oxidative Stress
Rotenone markedly disrupts mitochondrial function by obstructing electron transport from NADH to ubiquinone (CoQ10), hence disturbing the proton gradient across the inner mitochondrial membrane [72]. This limitation diminishes ATP synthase (complex V) activity, hence, decreasing ATP generation is vital for cellular functions [11,73]. Electron buildup at complex I facilitates their transfer to molecular oxygen, resulting in the production of ROS, such as superoxide anion (O2−) [5,17,42]. Oxidative stress destroys membrane lipids, structural proteins, and mitochondrial DNA (mtDNA), which further disrupts mitochondrial function by destabilizing membrane potential and initiating cytochrome c release and caspase-9-mediated death [9,74–77]. Rotenone-induced oxidative stress stimulates proinflammatory responses, notably via NF-κB signaling, which enhances proinflammatory cytokine production in glial cells and worsens neuronal dysfunction [22,78,79]. These effects are frequently observed in Parkinson’s disease models, where rotenone produces dopaminergic neuronal death in the substantia nigra and locus coeruleus regions particularly susceptible to oxidative stress and mitochondrial dysfunction leading to motor symptoms such as hypokinesia and stiffness [80,81]. The buildup of ROS resulting from mitochondrial respiratory chain inhibition has catastrophic biological effects [78,82,83]. Although rotenone does not exhibit cell-type selectivity like the MPTP model, its prolonged suppression of mitochondrial complex I results in irreversible degeneration of the nigrostriatal pathway, rendering it a commonly utilized yet systematically lethal model [33,47,75]. The suppression of complex I and subsequent ROS generation has extensive biological implications [73,77,78]. Lipid peroxidation produces hazardous byproducts such as 4-hydroxynonenal (4-HNE), which compromise membrane integrity and mitochondrial function [84–86]. Protein oxidation, especially of cysteine, methionine, and tyrosine, results in diminished enzymatic activity and facilitates aggregation, which contributes to the cytoplasmic and mitochondrial inclusions observed in PD and AD [87,88]. mtDNA is particularly susceptible owing to its restricted repair capabilities; oxidative base alterations can result in mutations, mitochondrial impairment, and bioenergetic collapse [89–91], thereby triggering DNA damage responses such as p53 signaling and apoptosis, which contribute to progressive neuronal degeneration [92] (Fig. 4).
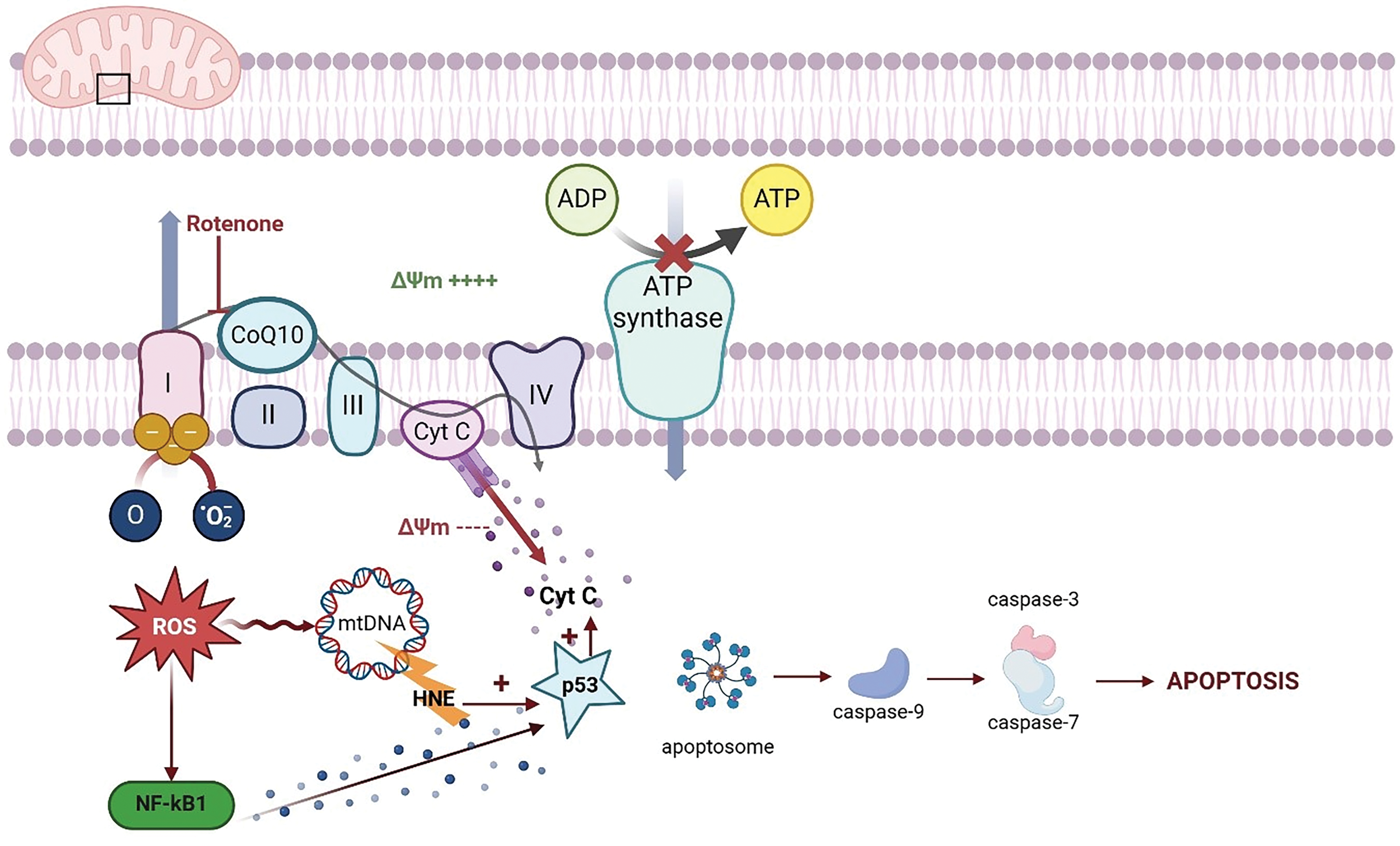
Figure 4: Schematic representation of the mechanisms underlying rotenone-induced mitochondrial dysfunction and oxidative stress. Rotenone selectively inhibits complex I (NADH: ubiquinone oxidoreductase) of the electron transport chain (etc), impairing electron flow and promoting the premature leakage of electrons. This results in an overproduction of reactive oxygen species (ROS), which in turn leads to oxidative damage of mitochondrial proteins, lipids, and mitochondrial DNA (mtDNA). The ensuing oxidative stress disrupts the mitochondrial membrane potential (Δψm) and compromises ATP synthesis, ultimately triggering the release of pro-apoptotic factors such as cytochrome c (Cyt C). This release activates caspase-dependent apoptotic pathways, further contributing to cellular degeneration. Additionally, the accumulation of lipid peroxidation products, such as 4-hydroxynonenal (HNE), exacerbates cellular injury by modifying key biomolecules involved in cellular homeostasis. The figure also highlights the activation of stress signaling pathways (e.g., NF-κB and p53), which modulate the cellular response to mitochondrial damage and oxidative stress (Created using biorender.com)
7 Mechanism of Neural Apoptosis and Synaptic Dysfunction
Rotenone-induced oxidative stress triggers apoptotic signaling and synaptic damage through the inhibition of mitochondrial complex I and the elevation of ROS production [5,42]. The increase of ROS causes damage to lipids, proteins, and DNA, hence activating the intrinsic apoptotic pathway via the alteration of mitochondrial membrane potential [84]. The release of cytochrome c stimulates apoptotic protease activating factor-1 (APAF-1) and procaspase-9, resulting in the activation of caspase-3 and subsequent DNA fragmentation [93–95]. Rotenone disturbs intracellular calcium equilibrium, resulting in the activation of calpain, a protease that degrades essential synaptic proteins including spectrin and synaptophysin thereby hindering synaptic plasticity [96,97]. The decrease of anti-apoptotic proteins, such as Bcl-2, and the increase of pro-apoptotic proteins, such Bax, further heighten neuronal susceptibility [98]. These processes cumulatively lead to synaptic deterioration, impairments in dopaminergic transmission, and malfunction of the cortical-striatal network, which are indicative of Parkinson’s disease progression [5,99] (Fig. 5).
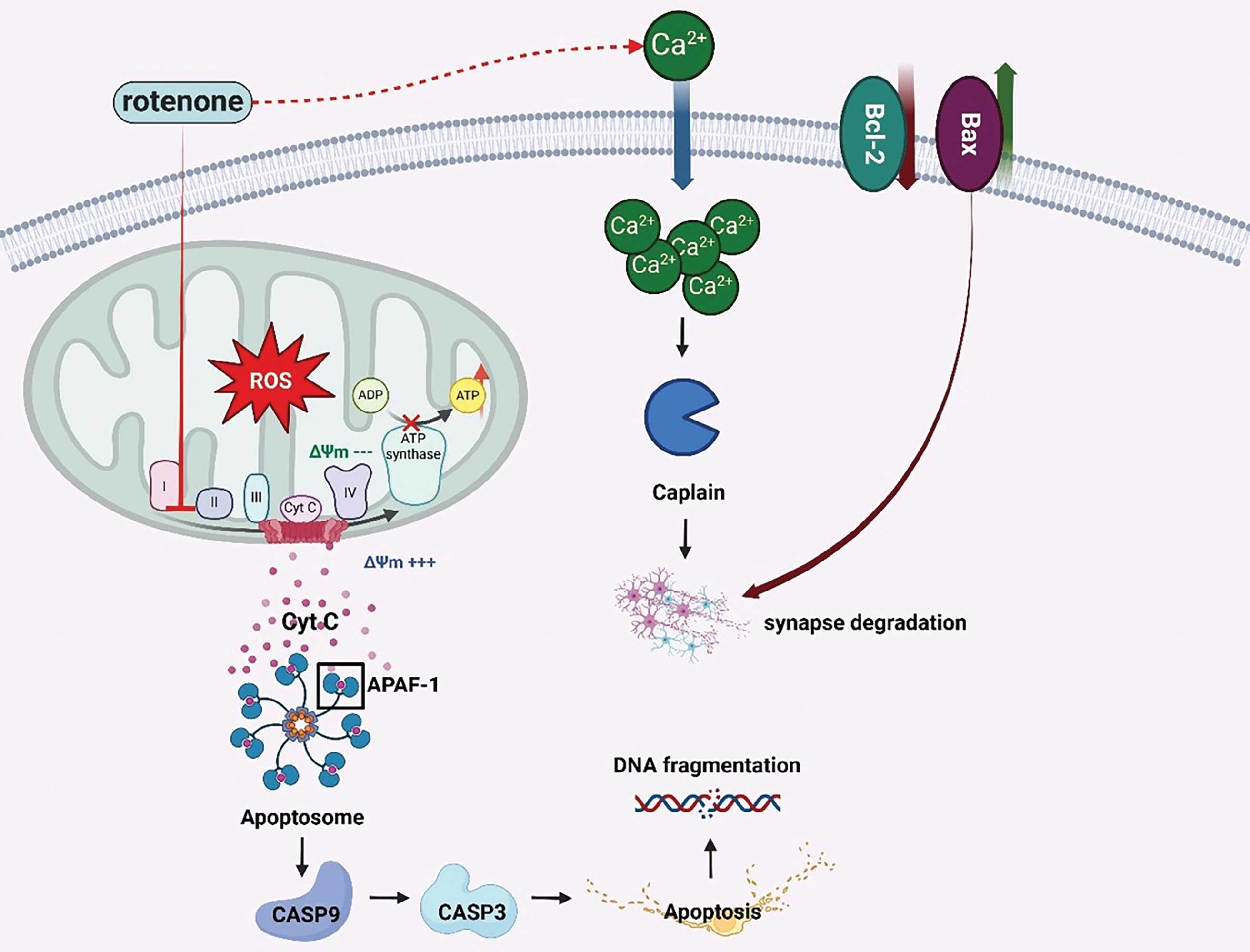
Figure 5: Schematic representation of rotenone-induced neural apoptosis and synaptic dysfunction. Rotenone-mediated inhibition of mitochondrial complex I increases reactive oxygen species (ROS) production and disrupts the mitochondrial membrane potential (Δψm). This promotes the release of cytochrome c (Cyt C) into the cytosol, where it binds apoptotic protease-activating factor 1 (APAF-1) to form the apoptosome, leading to caspase-9 and caspase-3 activation and, ultimately, apoptosis. Concurrently, pro-apoptotic (Bax) and anti-apoptotic (Bcl-2) members of the Bcl-2 family regulate mitochondrial outer membrane permeability, further modulating cell death pathways. Elevated intracellular calcium (Ca²+) levels activate calpain, a protease that contributes to synaptic degradation by cleaving key synaptic proteins. These combined events underline the neuronal loss and impaired synaptic integrity characteristic of rotenone-induced neurotoxicity (Created using BioRender.com)
8 Modification of Calcium Metabolism and Excitotoxicity Induced by Rotenone
Rotenone-induced mitochondrial dysfunction disrupts intracellular calcium homeostasis, essential for neuronal signaling and survival [23]. Mitochondria modulate calcium by buffering cytosolic concentrations, a mechanism reliant on mitochondrial membrane potential (ΔΨm) [76,77]. Rotenone impairs ΔΨm, reducing calcium sequestration and resulting in cytosolic calcium accumulation, which activates pro-apoptotic pathways and intensifies excitotoxicity [24,100]. Excitotoxicity results from the excessive release of glutamate and the overactivation of NMDA receptors, allowing calcium to enter neurons [101]. Increased intracellular calcium initiates harmful cascades, such as calpain activation and the destruction of vital neuronal proteins [102]. Simultaneously, the activation of phospholipase A2 facilitates the release of fatty acids and the generation of pro-inflammatory eicosanoids, exacerbating neuronal dysfunction [103–105]. Supplementary processes encompass the activation of Protein Kinase C (PKC) and neuronal nitric oxide synthase (nNOS), resulting in the generation of nitric oxide (NO) [106,107]. Nitric oxide (NO) interacts with the superoxide anion to produce peroxynitrite (ONOO−), a highly reactive entity that inflicts damage on cellular constituents [108–111]. Calcium imbalance triggers the opening of the mitochondrial permeability transition pore (mPTP), leading to cytochrome c release and death through caspases 3 and 9 [112–114]. Liu et al. (2016) showed that rotenone exposure elevates intracellular calcium levels in dopaminergic neurons, impairing neurogenesis and exacerbating excitotoxicity, which accelerates neurodegeneration [23,26]. Addressing calcium metabolism and excitotoxicity may provide therapeutic opportunities to counteract rotenone-induced harm (Fig. 6).
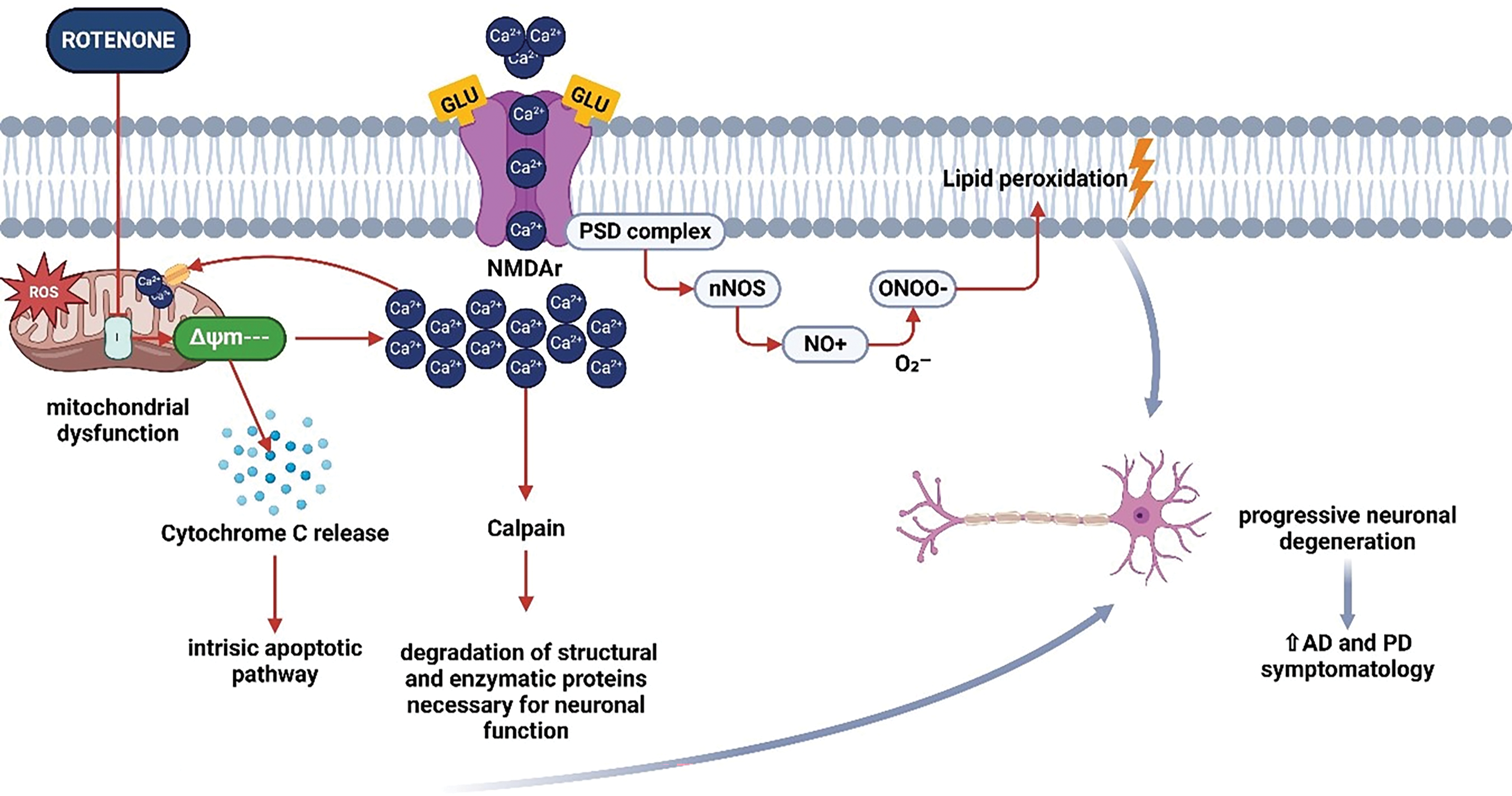
Figure 6: Schematic representation of rotenone-induced alteration of calcium metabolism and excitotoxicity. Rotenone-mediated inhibition of mitochondrial complex I leads to elevated reactive oxygen species (ROS) production, causing mitochondrial dysfunction and the release of cytochrome c (Cyt C). This event initiates the intrinsic apoptotic pathway, ultimately contributing to neuronal cell death. Concurrently, oxidative stress and impaired energy metabolism disrupt Ca²+ homeostasis, promoting excessive glutamate (Glu) receptor (NMDAr) activation and increased Ca²+ influx. This Ca²+ overload activates calpain, a protease responsible for the degradation of essential synaptic and structural proteins. Additionally, heightened intracellular Ca²+ stimulates neuronal nitric oxide synthase (nNOS), resulting in nitric oxide (NO) production, which combines with superoxide (O2−) to form peroxynitrite (ONOO−). This potent oxidant drives lipid peroxidation, damages postsynaptic density (PSD) proteins, and exacerbates neuronal degeneration. Together, these processes accelerate excitotoxic injury and are implicated in the progression of Alzheimer’s disease (AD) and Parkinson’s disease (PD) (Created using BioRender.com)
9 Induction of Glial Cell Activation by Rotenone
Neuroinflammation in rotenone models entails the activation of glial cells, particularly microglia and astrocytes, as a reaction to neuronal damage and oxidative stress [115–118]. Microglia, the immune cells of the central nervous system, transition from a quiescent state to a pro-inflammatory (M1) phenotype, secreting cytokines (e.g., TNF-α, IL-1β, IL-6), nitric oxide (NO), and reactive oxygen species (ROS), all of which contribute to neuronal injury [60,117,119]. Their persistent stimulation increases immune cell infiltration and sustains inflammation [120]. Astrocytes facilitate neuronal metabolism and repair but have a reactive behavior following neurotoxic insults such as rotenone exposure [121,122]. They secrete cytokines, chemokines, and prostaglandins, which may exacerbate inflammation and neurodegeneration if prolonged. While astrocytic activation may initially confer protection, prolonged stimulation amid oxidative stress and mitochondrial malfunction fosters a pro-inflammatory condition [123–125]. Communication between astrocytes and microglia is compromised in rotenone models [126]. Activated microglia generate reactive oxygen species and cytokines that affect astrocytes, which then release further inflammatory mediators [116,119], exacerbating neuronal injury [127]. The persistent stimulation of glial cells perpetuates neuroinflammation, hinders synaptic plasticity, and leads to cognitive and motor deterioration in models of PD, AD, and amyotrophic lateral sclerosis [117,128]. Recent research have observed sex-based differences in the effects of rotenone, but most models predominantly employ male mice [129,130]. Additional investigation into sexual dimorphism may improve the reliability and translational significance of these models. Comprehending these relationships is essential for formulating therapeutics aimed at modulating glial responses and inflammation in neurodegenerative disorders (Fig. 7).
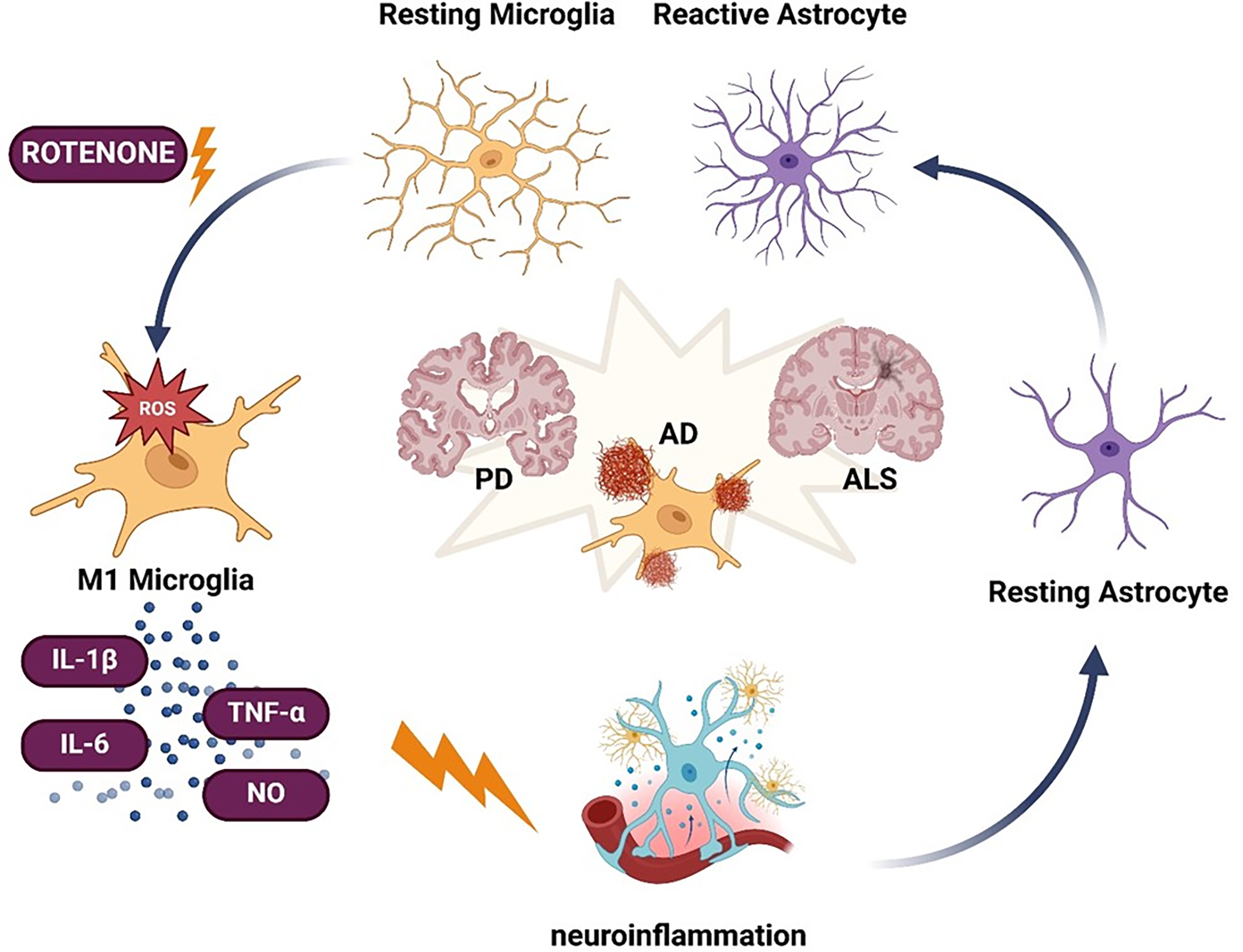
Figure 7: Schematic representation of rotenone-induced glial cell activation and its contribution to neurodegenerative diseases. Rotenone-triggered oxidative stress (ROS) drives microglia toward a pro-inflammatory (M1) phenotype, characterized by the release of cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), tumor necrosis factor alpha (TNF-α), and nitric oxide (NO). Concurrently, astrocytes transition from resting to a reactive state, further amplifying the neuroinflammatory response. Prolonged glial activation and inflammation are implicated in the pathogenesis and progression of Parkinson’s disease (PD), Alzheimer’s disease (AD), and amyotrophic lateral sclerosis (ALS) (Created using BioRender.com)
10 Signaling Pathways Associated with Rotenone-Induced Neuroinflammation
Rotenone stimulates multiple critical intracellular pathways that govern cytokine synthesis and glial activation, both of which are vital in neuroinflammation [131]. The NF-κB pathway is essential for regulating inflammatory responses; ROS generated by rotenone exposure activate NF-κB, leading to the transcription of pro-inflammatory genes such as TNF-α, IL-1β, IL-6, and iNOS [132–135]. The PI3K/AKT/mTOR pathway, essential for cell survival and metabolism, is affected by rotenone-induced oxidative stress, leading to glial activation and chronic inflammation. Notably, mTOR activation may impede autophagy and enhance neuronal vulnerability and damage [136,137]. The Wnt/β-catenin signaling system, which governs brain development and inflammatory responses, is disrupted by rotenone, exacerbating neuroinflammatory processes and glial activation. Recent investigations suggest that modifying this pathway may diminish neuroinflammation and neurodegeneration [138–140]. Despite these insights, it is essential to recognize that the effects of rotenone on these pathways may vary according to dosage, method of administration, and duration of exposure, hence limiting the translational relevance of the findings. These pathways promote sustained glial activation and inflammation, which are critical contributors to neurodegeneration (Fig. 8) [40]. A thorough comprehension of their functions may enhance the development of therapeutic strategies for neurodegenerative disorders, such as Parkinson’s, Alzheimer’s, and ALS [60,131]. A variety of pharmaceuticals, including antioxidants such as resveratrol, N-acetylcysteine, NF-κB inhibitors, mitochondrial modulators like mitochondrial-targeted antioxidant mitoquinone (MitoQ), and glial contact regulators, have shown promise in preclinical studies for mitigating chronic inflammation and neuronal injury [141] (Fig. 8).
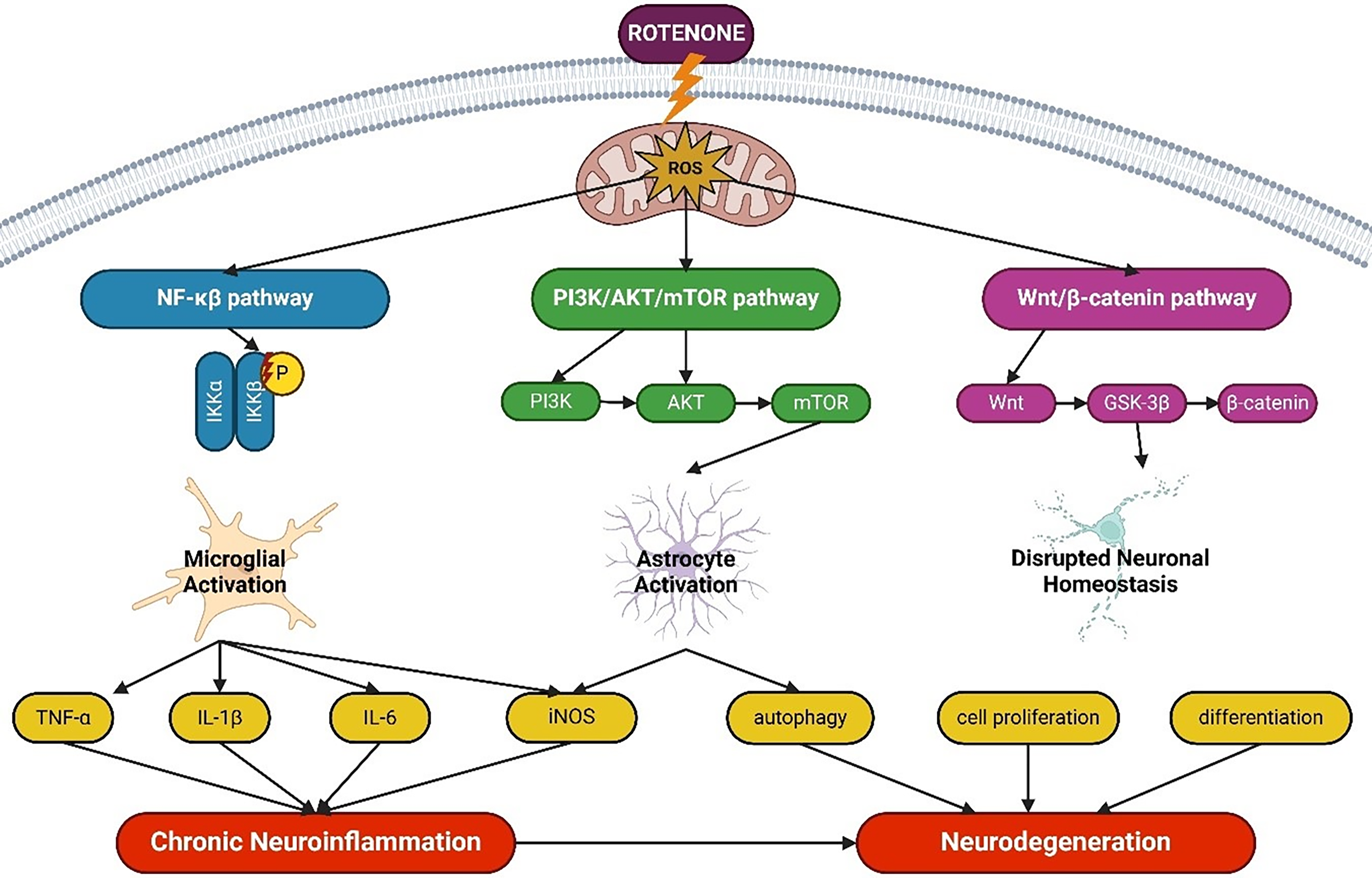
Figure 8: Schematic representation of the key signaling pathways implicated in rotenone-induced neuroinflammation. Rotenone-generated reactive oxygen species (ROS) activate the NF-κB pathway, driving microglial activation and the production of pro-inflammatory mediators (e.g., TNF-α, IL-1β, IL-6, and iNOS). Simultaneously, the PI3K/AKT/mTOR pathway in astrocytes modulates autophagy and cell survival, whereas the Wnt/β-catenin pathway influences neuronal homeostasis through effects on cell proliferation and differentiation. Together, these pathways promote chronic neuroinflammation and neuronal dysfunction, ultimately contributing to neurodegeneration. Abbreviations: NF-κB, nuclear factor kappa B; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B; mTOR, mechanistic target of rapamycin; GSK-3β, glycogen synthase kinase-3β (Created using BioRender.com)
11 Limitations and Future Directions
Notwithstanding progress in employing rotenone-based models for the investigation of neurodegenerative disorders, numerous significant limitations persist and warrant attention in forthcoming research. The compound’s limited selectivity and systemic toxicity may impact non-neuronal tissues, confounding the assignment of reported effects solely to neurodegenerative pathways. Furthermore, diversity in delivery routes, dosing schedules, and exposure durations frequently leads to contradictory results among studies, so undermining reproducibility and constraining translational applicability. These models inadequately reproduce the progressive and multifaceted characteristics of human neurodegenerative illnesses like Parkinson’s and Alzheimer’s, as they typically lack the genetic and environmental complexity inherent to these ailments. Moreover, variations in rotenone sensitivity among different species and strains further limit the applicability of experimental results.
Future research must prioritize the standardization of experimental protocols including dose, duration, and administration modalities to enhance reproducibility and address these constraints. The incorporation of female animals and the implementation of sex-based studies are crucial for revealing differential susceptibilities and informing the creation of more inclusive therapeutic techniques [142,143]. A notably interesting avenue is the development of combinatory models that amalgamate genetic predispositions with environmental exposures, providing a more precise depiction of the intricate etiologies of neurodegenerative disorders. Increased focus should be directed towards the identification of translational biomarkers and therapeutic outcomes to enhance clinical applicability. A thorough examination of glial signaling dynamics, particularly the functional interaction between astrocytes and microglia, may yield essential mechanistic insights and broaden treatment possibilities in rotenone-based preclinical models. A comprehensive analysis of glial signaling dynamics, including the functional interaction between astrocytes and microglia, is crucial due to the established disparities in mitochondrial function among these glial cells and neurons. Such findings may provide essential molecular insights and broaden therapeutic options within rotenone-based preclinical models.
Mitochondrial dysfunction, particularly when triggered by rotenone, significantly contributes to the pathogenesis of neurodegenerative disorders including Parkinson’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis. This chemical inhibits complex I of the mitochondrial electron transport chain, impeding ATP synthesis and fostering a pro-oxidative environment that elevates the formation of ROS and disturbs intracellular calcium homeostasis. Moreover, rotenone exposure induces a sustained inflammatory response mediated by microglia, activating pro-inflammatory pathways such as TLR4 and NF-κB, hence exacerbating neurodegeneration. Glial cells, especially microglia and astrocytes, play a crucial role in disease progression, since their overactivation and malfunction result in neuronal excitotoxicity and disrupt energy metabolism, heightening neuronal vulnerability to oxidative damage. The activation of additional intracellular signaling pathways, including PI3K/AKT/mTOR and Wnt/β-catenin, regulates the inflammatory response and impacts glial function, hence aggravating chronic inflammation and leading to neurodegeneration. These processes underscore the significance of mitochondrial malfunction, glial inflammation, and the disturbance of calcium homeostasis in rotenone-induced neurotoxicity. Focusing on the restoration of mitochondrial function and the modulation of glial inflammation may offer a potential strategy for creating therapies for diseases such as Parkinson’s, Alzheimer’s, and other neurodegenerative disorders.
Acknowledgement: Not applicable.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm their contribution to the paper as follows: study conception and design: Moisés Rubio-Osornio, Carmen Rubio; draft manuscript preparation: Carmen Rubio; review and editing: Moisés Rubio-Osornio, Carmen Rubio, Eduardo Castañeda, Hector Romo, Elisa Taddei; visualization: Moises Rubio-Osornio, Carmen Rubio, Norma Serrano-Garcia, Elisa Taddei; supervision: Héctor Romo, Carmen Rubio. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
Abbreviations
| PD | Parkinson’s disease |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| TNF-α | Tumor necrosis factor alpha |
| IL | Interleukin |
| NF-κB | Nuclear factor-kappa B |
| PI3K | Phosphoinositide 3-Kinase |
| AKT | Protein kinase B |
| mTOR | Mammalian Target of Rapamycin |
| ATP | Adenosine triphosphate |
| NADH | Nicotinamide adenine dinucleotide |
| ROS | Reactive oxygen species |
| TLR4 | Toll-like receptor 4 |
| NMDA | N-methyl-D-aspartate |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MAPK | Mitogen-activated protein kinase |
| JNK | Jun N-terminal kinase |
| GSK-3β | Glycogen synthase kinase-3 beta |
| Aβ | Amyloid-beta |
| APP | Amyloid precursor protein |
| BACE1 | β-secretase |
| iNOS | Inducible nitric oxide synthase |
| NO | Nitric oxide |
| SOD1 | Superoxide dismutase 1 |
| CoQ10 | Ubiquinone |
| mtDNA | Mitochondrial DNA |
| 4-HNE | 4-hydroxynonenal |
| APAF-1 | Apoptotic protease activating factor-1 |
| ΔΨm | Mitochondrial membrane potential |
| MitoQ | Mitochondrial-targeted antioxidant mitoquinone |
References
1. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148(6):1145–59. doi:10.1016/j.cell.2012.02.035. [Google Scholar] [PubMed] [CrossRef]
2. Cooper GM. The mechanism of oxidative phosphorylation. In: The cell: a molecular approach; 2000. p. 396–402. [Google Scholar]
3. Scheffler IE. Mitochondria. 2nd ed. Hoboken, NJ, USA: Wiley-Blackwell; 2007. doi:10.1002/9780470191774. [Google Scholar] [CrossRef]
4. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–95. doi:10.1038/nature05292. [Google Scholar] [PubMed] [CrossRef]
5. Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, et al. Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci. 2003;23(34):10756–64. doi:10.1523/jneurosci.23-34-10756.2003. [Google Scholar] [PubMed] [CrossRef]
6. Giradkar V, Mhaske A, Shukla R. Naringenin nanocrystals mitigate rotenone neurotoxicity in SH-SY5Y cell line by modulating mitophagy and oxidative stress. AAPS PharmSciTech. 2024;25(7):227. doi:10.1208/s12249-024-02936-1. [Google Scholar] [PubMed] [CrossRef]
7. Singh R, Lemire J, Mailloux RJ, Appanna VD. A novel strategy involved in [corrected] anti-oxidative defense: the conversion of NADH into NADPH by a metabolic network. PLoS One. 2008;3(7):e2682. doi:10.1371/journal.pone.0002682. [Google Scholar] [PubMed] [CrossRef]
8. Guo C, Sun L, Chen X, Zhang D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res. 2013;8:2003–14. doi:10.3969/j.issn.1673-5374.2013.21.009. [Google Scholar] [PubMed] [CrossRef]
9. Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3(12):1301–6. doi:10.1038/81834. [Google Scholar] [PubMed] [CrossRef]
10. Goswami P, Gupta S, Joshi N, Sharma S, Singh S. Astrocyte activation and neurotoxicity: a study in different rat brain regions and in rat C6 astroglial cells. Environ Toxicol Pharmacol. 2015;40(1):122–39. doi:10.1016/j.etap.2015.06.001. [Google Scholar] [PubMed] [CrossRef]
11. Greenamyre JT, Sherer TB, Betarbet R, Panov AV. Complex I and Parkinson’s disease. IUBMB Life. 2001;52(3–5):135–41. doi:10.1080/15216540152845939. [Google Scholar] [PubMed] [CrossRef]
12. Clemente-Suárez VJ, Redondo-Flórez L, Beltrán-Velasco AI, Ramos-Campo DJ, Belinchón-deMiguel P, Martinez-Guardado I, et al. Mitochondria and brain disease: a comprehensive review of pathological mechanisms and therapeutic opportunities. Biomedicines. 2023;11(9):2488. doi:10.3390/biomedicines11092488. [Google Scholar] [PubMed] [CrossRef]
13. Rocha SM, Kirkley KS, Chatterjee D, Aboellail TA, Smeyne RJ, Tjalkens RB. Microglia-specific knock-out of NF-κB/IKK2 increases the accumulation of misfolded α-synuclein through the inhibition of p62/sequestosome-1-dependent autophagy in the rotenone model of Parkinson’s disease. Glia. 2023;71(9):2154–79. doi:10.1002/glia.24385. [Google Scholar] [PubMed] [CrossRef]
14. Avallone R, Lucchi C, Puja G, Codeluppi A, Filaferro M, Vitale G, et al. BV-2 microglial cells respond to rotenone toxic insult by modifying pregnenolone, 5α-dihydroprogesterone and pregnanolone levels. Cells. 2020;9(9):2091. doi:10.3390/cells9092091. [Google Scholar] [PubMed] [CrossRef]
15. Albadawi E, El-Tokhy A, Albadrani M, Adel M, El-Gamal R, Zaarina W, et al. The role of stinging nettle (Urtica dioica L.) in the management of rotenone-induced Parkinson’s disease in rats. Tissue Cell. 2024;87(1):102328. doi:10.1016/j.tice.2024.102328. [Google Scholar] [PubMed] [CrossRef]
16. Ishola IO, Awogbindin IO, Olubodun-Obadun TG, Olajiga AE, Adeyemi OO. Vinpocetine prevents rotenone-induced Parkinson disease motor and non-motor symptoms through attenuation of oxidative stress, neuroinflammation and α-synuclein expressions in rats. Neurotoxicol. 2023;96(6):37–52. doi:10.1016/j.neuro.2023.03.002. [Google Scholar] [PubMed] [CrossRef]
17. Ravera S, Farsetti E, Maura G, Marcoli M, Bozzo M, Cervetto C, et al. 810-nm photobiomodulation evokes glutamate release in normal and rotenone-dysfunctional cortical nerve terminals by modulating mitochondrial energy metabolism. Cells. 2025;14(2):67. doi:10.3390/cells14020067. [Google Scholar] [PubMed] [CrossRef]
18. Foran E, Trotti D. Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid Redox Signal. 2009;11(7):1587–602. doi:10.1089/ars.2009.2444. [Google Scholar] [PubMed] [CrossRef]
19. Chamaa F, Magistretti PJ, Fiumelli H. Astrocyte-derived lactate in stress disorders. Neurobiol Dis. 2024;192:106417. doi:10.1016/j.nbd.2024.106417. [Google Scholar] [PubMed] [CrossRef]
20. Chia SJ, Tan E-K, Chao Y-X. Historical perspective: models of Parkinson’s disease. Int J Mol Sci. 2020;21(7):2464. doi:10.3390/ijms21072464. [Google Scholar] [PubMed] [CrossRef]
21. Cheng X-Y, Biswas S, Li J, Mao C-J, Chechneva O, Chen J, et al. Human iPSCs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria. Transl Neurodegener. 2020;9(1):13. doi:10.1186/s40035-020-00190-6. [Google Scholar] [PubMed] [CrossRef]
22. Adamu A, Li S, Gao F, Xue G. The role of neuroinflammation in neurodegenerative diseases: current understanding and future therapeutic targets. Front Aging Neurosci. 2024;16:1347987. doi:10.3389/fnagi.2024.1347987. [Google Scholar] [PubMed] [CrossRef]
23. Liu C, Ye Y, Zhou Q, Zhang R, Zhang H, Liu W, et al. Crosstalk between Ca2+ signaling and mitochondrial H2O2 is required for rotenone inhibition of mTOR signaling pathway leading to neuronal apoptosis. Oncotarget. 2016;7(7):7534–49. doi:10.18632/oncotarget.7183. [Google Scholar] [PubMed] [CrossRef]
24. Verma M, Lizama BN, Chu CT. Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration. Transl Neurodegener. 2022;11(1):3. doi:10.1186/s40035-021-00278-7. [Google Scholar] [PubMed] [CrossRef]
25. Dong X-X, Wang Y, Qin Z-H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. 2009;30(4):379–87. doi:10.1038/aps.2009.24. [Google Scholar] [PubMed] [CrossRef]
26. Marambaud P, Dreses-Werringloer U, Vingtdeux V. Calcium signaling in neurodegeneration. Mol Neurodegener. 2009;4(1):20. doi:10.1186/1750-1326-4-20. [Google Scholar] [PubMed] [CrossRef]
27. Zhang J, Sun B, Yang J, Chen Z, Li Z, Zhang N, et al. Comparison of the effect of rotenone and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine on inducing chronic Parkinson’s disease in mouse models. Mol Med Rep. 2022;25(3):91. doi:10.3892/mmr.2022.12607. [Google Scholar] [PubMed] [CrossRef]
28. Ahmed YR, Aboul Naser AF, Elbatanony MM, El-Feky AM, Khalil WKB, Hamed MA-A. Gene expression, oxidative stress, and neurotransmitters in rotenone-induced Parkinson’s disease in rats: role of naringin from Citrus aurantium via blocking adenosine A2A receptor. Curr Bioact Compd. 2024;20(5):1–16. doi:10.2174/0115734072268296231002060839. [Google Scholar] [CrossRef]
29. Gupta V, Prasad S. Differential alterations in the expression of AMPA receptor and its trafficking proteins in the hippocampus are associated with recognition memory impairment in the rotenone-Parkinson’s disease mouse model: neuroprotective role of Bacopa monnieri extract CDRI 08. Mol Neurobiol. 2025;62(2):2086–104. doi:10.1007/s12035-024-04392-1. [Google Scholar] [PubMed] [CrossRef]
30. Dos Santos JCC, da Rebouças CSM, Oliveira LF, Cardoso FDS, de Nascimento TS, Oliveira AV, et al. The role of gut-brain axis in a rotenone-induced rat model of Parkinson’s disease. Neurobiol Aging. 2023;132(1):185–97. doi:10.1016/j.neurobiolaging.2023.07.005. [Google Scholar] [PubMed] [CrossRef]
31. El-Latif AMA, Rabie MA, Sayed RH, Fattah MAAE, Kenawy SA. Inosine attenuates rotenone-induced Parkinson’s disease in rats by alleviating the imbalance between autophagy and apoptosis. Drug Dev Res. 2023;84(6):1159–74. doi:10.1002/ddr.22077. [Google Scholar] [PubMed] [CrossRef]
32. Rajan S, Sood A, Jain R, Kamatham PT, Khatri DK. Fingolimod exerts neuroprotection by regulating S1PR1 mediated BNIP3-PINK1-Parkin dependent mitophagy in rotenone induced mouse model of Parkinson’s disease. Neurosci Lett. 2024;820(2):137596. doi:10.1016/j.neulet.2023.137596. [Google Scholar] [PubMed] [CrossRef]
33. Lal R, Singh A, Watts S, Chopra K. Experimental models of Parkinson’s disease: challenges and opportunities. Eur J Pharmacol. 2024;980(28):176819. doi:10.1016/j.ejphar.2024.176819. [Google Scholar] [PubMed] [CrossRef]
34. Henrich MT, Oertel WH, Surmeier DJ, Geibl FF. Mitochondrial dysfunction in Parkinson’s disease—a key disease hallmark with therapeutic potential. Mol Neurodegener. 2023;18(1):83. doi:10.1186/s13024-023-00676-7. [Google Scholar] [PubMed] [CrossRef]
35. Stoker TB, Greenland JC. Parkinson’s disease: pathogenesis and clinical aspects. Codon Publications; 2018. doi:10.15586/codonpublications.parkinsonsdisease.2018. [Google Scholar] [CrossRef]
36. Salari Z, Ashabi G, Fartoosi A, Fartoosi A, Shariatpanahi M, Aghsami M, et al. Sericin alleviates motor dysfunction by modulating inflammation and TrkB/BDNF signaling pathway in the rotenone-induced Parkinson’s disease model. BMC Pharmacol Toxicol. 2023;24(1):60. doi:10.1186/s40360-023-00703-9. [Google Scholar] [PubMed] [CrossRef]
37. de Souza Nascimento T, Oliveira AV, de Oliveira Bélem M, Bezerra JR, do Carmo MRS, da Silva ME, et al. The rotenone-induced sporadic Parkinsonism model: timeline of motor and non-motor features. Eur J Neurosci. 2025;61(3):e16669. doi:10.1111/ejn.16669. [Google Scholar] [PubMed] [CrossRef]
38. Hassan HM, Abou-Hany HO, Shata A, Hellal D, El-Baz AM, ElSaid ZH, et al. Vinpocetine and Lactobacillus attenuated rotenone-induced Parkinson’s disease and restored dopamine synthesis in rats through modulation of oxidative stress, neuroinflammation, and Lewy bodies inclusion. J Neuroimmune Pharmacol. 2025;20(1):22. doi:10.1007/s11481-025-10176-8. [Google Scholar] [PubMed] [CrossRef]
39. Obeso JA, Stamelou M, Goetz CG, Poewe W, Lang AE, Weintraub D, et al. Past, present, and future of Parkinson’s disease: a special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord. 2017;32(9):1264–310. doi:10.1002/mds.27115. [Google Scholar] [PubMed] [CrossRef]
40. Kumar D, Kumar R, Janrao S, Sharma V, Begum N, Fernandes V, et al. Treadmill exercise mitigates rotenone-induced neuroinflammation and α-synuclein level in a mouse model of Parkinson’s disease. Brain Res. 2025;1854(3):149540. doi:10.1016/j.brainres.2025.149540. [Google Scholar] [PubMed] [CrossRef]
41. Saramowicz K, Siwecka N, Galita G, Kucharska-Lusina A, Rozpędek-Kamińska W, Majsterek I. Alpha-synuclein contribution to neuronal and glial damage in Parkinson’s disease. Int J Mol Sci. 2023;25(3):360. doi:10.3390/ijms25010360. [Google Scholar] [PubMed] [CrossRef]
42. Rocha SM, Bantle CM, Aboellail T, Chatterjee D, Smeyne RJ, Tjalkens RB. Rotenone induces regionally distinct α-synuclein protein aggregation and activation of glia prior to loss of dopaminergic neurons in C57Bl/6 mice. Neurobiol Dis. 2022;167:105685. doi:10.1016/j.nbd.2022.105685. [Google Scholar] [PubMed] [CrossRef]
43. Shin WH, Chung KC. Death-associated protein kinase 1 phosphorylates α-synuclein at Ser129 and exacerbates rotenone-induced toxic aggregation of α-synuclein in dopaminergic SH-SY5Y cells. Exp Neurobiol. 2020;29(3):207–18. doi:10.5607/en20014. [Google Scholar] [PubMed] [CrossRef]
44. Mosley RL, Benner EJ, Kadiu I, Thomas M, Boska MD, Hasan K, et al. Neuroinflammation, oxidative stress and the pathogenesis of Parkinson’s disease. Clin Neurosci Res. 2006;6(5):261–81. doi:10.1016/j.cnr.2006.09.006. [Google Scholar] [PubMed] [CrossRef]
45. Zhang D, Li S, Hou L, Jing L, Ruan Z, Peng B, et al. Microglial activation contributes to cognitive impairments in rotenone-induced mouse Parkinson’s disease model. J Neuroinflammation. 2021;18(1):4. doi:10.1186/s12974-020-02065-z. [Google Scholar] [PubMed] [CrossRef]
46. Anguchamy V, Muthuvel A. Enhancing the neuroprotective effect of squid outer skin astaxanthin against rotenone-induced neurotoxicity in in-vitro model for Parkinson’s disease. Food Chem Toxicol. 2023;178(1):113846. doi:10.1016/j.fct.2023.113846. [Google Scholar] [PubMed] [CrossRef]
47. Fleming S, Zhu C, Fernagut PO, Mehta A, DiCarlo CD, Seaman RL, et al. Behavioral and immunohistochemical effects of chronic intravenous and subcutaneous infusions of varying doses of rotenone. Exp Neurol. 2004;187(2):418–29. doi:10.1016/j.expneurol.2004.01.023. [Google Scholar] [PubMed] [CrossRef]
48. Fornai F, Schlüter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, et al. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc Natl Acad Sci U S A. 2005;102(9):3413–8. doi:10.1073/pnas.0409713102. [Google Scholar] [PubMed] [CrossRef]
49. Ozbey G, Nemutlu-Samur D, Parlak H, Yildirim S, Aslan M, Tanriover G, et al. Metformin protects rotenone-induced dopaminergic neurodegeneration by reducing lipid peroxidation. Pharmacol Rep. 2020;72(5):1397–406. doi:10.1007/s43440-020-00095-1. [Google Scholar] [PubMed] [CrossRef]
50. Veeraraghavan R, Pannu SR, Hund TJ, Satoskar AR, McDaniel JC, Maddipati RK et al. Oxidative lipidomics: analysis of oxidized lipids and lipid peroxidation in biological systems with relevance to health and disease. In: Berliner LJ, Parinandi NL, editors. Measuring oxidants and oxidative stress in biological systems. Cham (CHSpringer; 2020. p. 61–92. doi:10.1007/978-3-030-47318-1_5. [Google Scholar] [CrossRef]
51. Kotsyuba E, Dyachuk V. Effects of chronic exposure to low doses of rotenone on dopaminergic and cholinergic neurons in the CNS of Hemigrapsus sanguineus. Int J Mol Sci. 2024;25(13):7159. doi:10.3390/ijms25137159. [Google Scholar] [PubMed] [CrossRef]
52. Giraldo-Berrio D, Mendivil-Perez M, Velez-Pardo C, Jimenez-Del-Rio M. Rotenone induces a neuropathological phenotype in cholinergic-like neurons resembling Parkinson’s disease dementia (PDD). Neurotox Res. 2024;42(3):28. doi:10.1007/s12640-024-00705-3. [Google Scholar] [PubMed] [CrossRef]
53. Ullrich C, Humpel C. Rotenone induces cell death of cholinergic neurons in an organotypic co-culture brain slice model. Neurochem Res. 2009;34(12):2147–53. doi:10.1007/s11064-009-0014-9. [Google Scholar] [PubMed] [CrossRef]
54. Jha SK, Jha NK, Kar R, Ambasta RK, Kumar P. P38 MAPK and PI3K/AKT signalling cascades in Parkinson’s disease. Int J Mol Cell Med. 2015;4:67–86. [Google Scholar] [PubMed]
55. Rawat P, Sehar U, Bisht J, Selman A, Culberson J, Reddy PH. Phosphorylated tau in Alzheimer’s disease and other tauopathies. Int J Mol Sci. 2022;23(21):12841. doi:10.3390/ijms232112841. [Google Scholar] [PubMed] [CrossRef]
56. Gan KJ, Akram A, Blasius TL, Ramser EM, Budaitis BG, Gabrych DR, et al. GSK3β impairs KIF1A transport in a cellular model of alzheimer’s disease but does not regulate motor motility at S402. eNeuro. 2020;7:ENEURO.0176-20.2020. doi:10.1523/ENEURO.0176-20.2020. [Google Scholar] [PubMed] [CrossRef]
57. Morris SL, Brady ST. Tau phosphorylation and PAD exposure in regulation of axonal growth. Front Cell Dev Biol. 2023;10:1023418. doi:10.3389/fcell.2022. [Google Scholar] [CrossRef]
58. Xia Y, Wang ZH, Zhang Z, Liu X, Yu SP, Wang JZ, et al. Delta- and beta- secretases crosstalk amplifies the amyloidogenic pathway in Alzheimer’s disease. Prog Neurobiol. 2021;204:102113. doi:10.1016/j.pneurobio.2021.102113. [Google Scholar] [PubMed] [CrossRef]
59. Yu N, Pasha M, Chua JJE. Redox changes and cellular senescence in Alzheimer’s disease. Redox Biol. 2024;70(18):103048. doi:10.1016/j.redox.2024.103048. [Google Scholar] [PubMed] [CrossRef]
60. Cai Y, Liu J, Wang B, Sun M, Yang H. Microglia in the neuroinflammatory pathogenesis of Alzheimer’s disease and related therapeutic targets. Front Immunol. 2022;13:856376. doi:10.3389/fimmu.2022.856376. [Google Scholar] [PubMed] [CrossRef]
61. Luo R, Zhu L, Zeng Z, Zhou R, Zhang J, Xiao S, et al. Dl-butylphthalide inhibits rotenone-induced oxidative stress in microglia via regulation of the Keap1/Nrf2/HO-1 signaling pathway. Exp Ther Med. 2021;21(6):597. doi:10.3892/etm.2021.10029. [Google Scholar] [PubMed] [CrossRef]
62. Moisse K, Strong MJ. Innate immunity in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762:1083–93. doi:10.1016/j.bbadis.2006.03.001. [Google Scholar] [PubMed] [CrossRef]
63. El-Sherbeeny NA, Soliman N, Youssef AM, Abd El-Fadeal NM, El-Abaseri TB, Hashish AA, et al. The protective effect of biochanin A against rotenone-induced neurotoxicity in mice involves enhancing of PI3K/Akt/mTOR signaling and beclin-1 production. Ecotoxicol Environ Saf. 2020;205:111344. doi:10.1016/j.ecoenv.2020.111344. [Google Scholar] [PubMed] [CrossRef]
64. Sathasivam S, Shaw PJ. Apoptosis in amyotrophic lateral sclerosis–what is the evidence? Lancet Neurol. 2005;4(8):500–9. doi:10.1016/s1474-4422(05)70142-3. [Google Scholar] [PubMed] [CrossRef]
65. Kanki R, Nakamizo T, Yamashita H, Kihara T, Sawada H, Uemura K, et al. Effects of mitochondrial dysfunction on glutamate receptor-mediated neurotoxicity in cultured rat spinal motor neurons. Brain Res. 2004;1015(1–2):73–81. doi:10.1016/j.brainres.2004.04.044. [Google Scholar] [PubMed] [CrossRef]
66. Garbuzova-Davis S, Saporta S, Haller E, Kolomey I, Bennett SP, Potter H, et al. Evidence of compromised blood-spinal cord barrier in early and late symptomatic SOD1 mice modeling ALS. PLoS One. 2007;2(11):e1205. doi:10.1371/journal.pone.0001205. [Google Scholar] [PubMed] [CrossRef]
67. Bunton-Stasyshyn RKA, Saccon RA, Fratta P, Fisher EMC. SOD1 function and its implications for amyotrophic lateral sclerosis pathology: new and renascent themes: new and renascent themes. Neuroscientist. 2015;21(5):519–29. doi:10.1177/1073858414561795. [Google Scholar] [PubMed] [CrossRef]
68. Rizzardini M, Lupi M, Mangolini A, Babetto E, Ubezio P, Cantoni L. Neurodegeneration induced by complex I inhibition in a cellular model of familial amyotrophic lateral sclerosis. Brain Res Bull. 2006;69(4):465–74. doi:10.1016/j.brainresbull.2006.02.013. [Google Scholar] [PubMed] [CrossRef]
69. Samantaray S, Knaryan VH, Le Gal C, Ray SK, Banik NL. Calpain inhibition protected spinal cord motoneurons against 1-methyl-4-phenylpyridinium ion and rotenone. Neuroscience. 2011;192(Pt 6):263–74. doi:10.1016/j.neuroscience.2011.06.007. [Google Scholar] [PubMed] [CrossRef]
70. Samantaray S, Knaryan VH, Guyton MK, Matzelle DD, Ray SK, Banik NL. The parkinsonian neurotoxin rotenone activates calpain and caspase-3 leading to motoneuron degeneration in spinal cord of Lewis rats. Neuroscience. 2007;146(2):741–55. doi:10.1016/j.neuroscience.2007.01.056. [Google Scholar] [PubMed] [CrossRef]
71. Thong-Asa W, Wassana C, Sukkasem K, Innoi P, Dechakul M, Timda P. Neuroprotective effect of gallic acid in mice with rotenone-induced neurodegeneration. Exp Anim. 2024;73(3):259–69. doi:10.1538/expanim.23-0165. [Google Scholar] [PubMed] [CrossRef]
72. Millichap L, Turton N, Damiani E, Marcheggiani F, Orlando P, Silvestri S, et al. The effect of neuronal CoQ10 deficiency and mitochondrial dysfunction on a rotenone-induced neuronal cell model of parkinson’s disease. Int J Mol Sci. 2024;25(12):6622. doi:10.3390/ijms25126622. [Google Scholar] [PubMed] [CrossRef]
73. Acin-Perez R, Benincá C, Fernandez Del Rio L, Shu C, Baghdasarian S, Zanette V, et al. Inhibition of ATP synthase reverse activity restores energy homeostasis in mitochondrial pathologies. EMBO J. 2023;42(10):e111699. doi:10.15252/embj.2022111699. [Google Scholar] [PubMed] [CrossRef]
74. Juan CA, Pérez de la Lastra JM, Plou FJ, Pérez-Lebeña E. The chemistry of reactive oxygen species (ROS) revisited: outlining their role in biological macromolecules (DNA, lipids and proteins) and induced pathologies. Int J Mol Sci. 2021;22(9):4642. doi:10.3390/ijms22094642. [Google Scholar] [PubMed] [CrossRef]
75. Kowalczyk P, Sulejczak D, Kleczkowska P, Bukowska-Ośko I, Kucia M, Popiel M, et al. Mitochondrial oxidative stress—A causative factor and therapeutic target in many diseases. Int J Mol Sci. 2021;22(24):13384. doi:10.3390/ijms222413384. [Google Scholar] [PubMed] [CrossRef]
76. Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis. 2003;8(2):115–28. doi:10.1023/a:1022945107762. [Google Scholar] [PubMed] [CrossRef]
77. Ricci J-E, Gottlieb RA, Green DR. Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J Cell Biol. 2003;160(1):65–75. doi:10.1083/jcb.200208089. [Google Scholar] [PubMed] [CrossRef]
78. Cordaro M, Modafferi S, D’Amico R, Fusco R, Genovese T, Peritore AF, et al. Natural compounds such as Hericium erinaceus and Coriolus versicolor modulate neuroinflammation, oxidative stress and lipoxin A4 expression in rotenone-induced Parkinson’s disease in mice. Biomedicines. 2022;10(10):2505. doi:10.3390/biomedicines10102505. [Google Scholar] [PubMed] [CrossRef]
79. Chen S, Li Q, Shi H, Li F, Duan Y, Guo Q. New insights into the role of mitochondrial dynamics in oxidative stress-induced diseases. Biomed Pharmacother. 2024;178(1):117084. doi:10.1016/j.biopha.2024.117084. [Google Scholar] [PubMed] [CrossRef]
80. Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol Dis. 2009;34(2):279–90. doi:10.1016/j.nbd.2009.01.016. [Google Scholar] [PubMed] [CrossRef]
81. Gao H-M, Hong J-S, Zhang W, Liu B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2002;22(3):782–90. doi:10.1523/jneurosci.22-03-00782.2002. [Google Scholar] [PubMed] [CrossRef]
82. Barbiero JK, Ramos DC, Boschen S, Bassani T, Da Cunha C, Vital MABF. Fenofibrate promotes neuroprotection in a model of rotenone-induced Parkinson’s disease. Behav Pharmacol. 2022;33(8):513–26. doi:10.1097/FBP.0000000000000699. [Google Scholar] [PubMed] [CrossRef]
83. Jain J, Hasan W, Biswas P, Yadav RS, Jat D. Neuroprotective effect of quercetin against rotenone-induced neuroinflammation and alterations in mice behavior. J Biochem Mol Toxicol. 2022;36(10):e23165. doi:10.1002/jbt.23165. [Google Scholar] [PubMed] [CrossRef]
84. Zhou H, Wu C, Jin Y, Wu O, Chen L, Guo Z, et al. Role of oxidative stress in mitochondrial dysfunction and their implications in intervertebral disc degeneration: mechanisms and therapeutic strategies. J Orthop Translat. 2024;49(22):181–206. doi:10.1016/j.jot.2024.08.016. [Google Scholar] [PubMed] [CrossRef]
85. Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24(10):R453–62. doi:10.1016/j.cub.2014.03.034. [Google Scholar] [PubMed] [CrossRef]
86. Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014(6):360438. doi:10.1155/2014/360438. [Google Scholar] [PubMed] [CrossRef]
87. Kehm R, Baldensperger T, Raupbach J, Höhn A. Protein oxidation-Formation mechanisms, detection and relevance as biomarkers in human diseases. Redox Biol. 2021;42:101901. doi:10.1016/j.redox.2021.101901. [Google Scholar] [PubMed] [CrossRef]
88. Aramouni K, Assaf R, Shaito A, Fardoun M, Al-Asmakh M, Sahebkar A, et al. Biochemical and cellular basis of oxidative stress: implications for disease onset. J Cell Physiol. 2023;238(9):1951–63. doi:10.1002/jcp.31071. [Google Scholar] [PubMed] [CrossRef]
89. Saki M, Prakash A. DNA damage related crosstalk between the nucleus and mitochondria. Free Radic Biol Med. 2017;107(5):216–27. doi:10.1016/j.freeradbiomed.2016.11.050. [Google Scholar] [PubMed] [CrossRef]
90. Qi R, Sammler E, Gonzalez-Hunt CP, Barraza I, Pena N, Rouanet JP, et al. A blood-based marker of mitochondrial DNA damage in Parkinson’s disease. Sci Transl Med. 2023;15(711):eabo1557. doi:10.1126/scitranslmed.abo1557. [Google Scholar] [PubMed] [CrossRef]
91. Sanchez-Roman I, Ferrando B, Holst CM, Mengel-From J, Rasmussen SH, Thinggaard M, et al. Molecular markers of DNA repair and brain metabolism correlate with cognition in centenarians. Geroscience. 2022;44(1):103–25. doi:10.1007/s11357-021-00502-2. [Google Scholar] [PubMed] [CrossRef]
92. Szybińska A, Leśniakx L. P53 dysfunction in neurodegenerative diseases—the cause or effect of pathological changes? Aging Dis. 2017;8(4):506. doi:10.14336/ad.2016.1120. [Google Scholar] [PubMed] [CrossRef]
93. Olatona OA, Sterben SP, Kansakar SBS, Symes AJ, Liaudanskaya V. Mitochondria: the hidden engines of traumatic brain injury-driven neurodegeneration. Front Cell Neurosci. 2025;19:1570596. doi:10.3389/fncel.2025.1570596. [Google Scholar] [PubMed] [CrossRef]
94. Shakeri R, Kheirollahi A, Davoodi J. Contribution of Apaf-1 to the pathogenesis of cancer and neurodegenerative diseases. Biochimie. 2021;190:91–110. doi:10.1016/j.biochi.2021.07.004. [Google Scholar] [PubMed] [CrossRef]
95. Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet. 2009;43(1):95–118. doi:10.1146/annurev-genet-102108-134850. [Google Scholar] [PubMed] [CrossRef]
96. Zaman V, Drasites KP, Myatich A, Shams R, Shields DC, Matzelle D, et al. Inhibition of calpain attenuates degeneration of substantia nigra neurons in the rotenone rat model of Parkinson’s disease. Int J Mol Sci. 2022;23(22):13849. doi:10.3390/ijms232213849. [Google Scholar] [PubMed] [CrossRef]
97. Gao A, McCoy HM, Zaman V, Shields DC, Banik NL, Haque A. Calpain activation and progression of inflammatory cycles in Parkinson’s disease. Front Biosci. 2022;27(1):20. doi:10.31083/j.fbl2701020. [Google Scholar] [PubMed] [CrossRef]
98. Gross A, Katz SG. Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ. 2017;24(8):1348–58. doi:10.1038/cdd.2017.22. [Google Scholar] [PubMed] [CrossRef]
99. Van Laar AD, Webb KR, Keeney MT, Van Laar VS, Zharikov A, Burton EA, et al. Transient exposure to rotenone causes degeneration and progressive parkinsonian motor deficits, neuroinflammation, and synucleinopathy. NPJ Parkinsons Dis. 2023;9(1):121. doi:10.1038/s41531-023-00561-6. [Google Scholar] [PubMed] [CrossRef]
100. Angelova PR, Abramov AY. Interplay of mitochondrial calcium signalling and reactive oxygen species production in the brain. Biochem Soc Trans. 2024;52(4):1939–46. doi:10.1042/BST20240261. [Google Scholar] [PubMed] [CrossRef]
101. Nicosia N, Giovenzana M, Misztak P, Mingardi J, Musazzi L. Glutamate-mediated excitotoxicity in the pathogenesis and treatment of neurodevelopmental and adult mental disorders. Int J Mol Sci. 2024;25(12):6521. doi:10.3390/ijms25126521. [Google Scholar] [PubMed] [CrossRef]
102. Metwally E, Al-Abbadi HA, Hussain T, Murtaza G, Abdellatif AM, Ahmed MF. Calpain signaling: from biology to therapeutic opportunities in neurodegenerative disorders. Front Vet Sci. 2023 Sep 5;10:1235163. doi:10.3389/fvets.2023.1235163. [Google Scholar] [PubMed] [CrossRef]
103. Lohitaksha K, Kumari D, Shukla M, Byagari L, Ashireddygari VR, Tammineni P, et al. Eicosanoid signaling in neuroinflammation associated with Alzheimer’s disease. Eur J Pharmacol. 2024;976:176694. doi:10.1016/j.ejphar.2024.176694. [Google Scholar] [PubMed] [CrossRef]
104. Hugo C, Asante I, Sadybekov A, Katritch V, Yassine HN. Development of calcium-dependent phospholipase A2 inhibitors to target cellular senescence and oxidative stress in neurodegenerative diseases. Antioxid Redox Signal. 2024;41:1100–16. doi:10.1089/ars.2024.0794. [Google Scholar] [PubMed] [CrossRef]
105. Ma QL, Ebright B, Li B, Li J, Galvan J, Sanchez A, et al. Evidence for cPLA2 activation in Alzheimer’s disease synaptic pathology. bioRxiv [Preprint]. 2025. doi:10.1101/2025.03.27.645605. [Google Scholar] [PubMed] [CrossRef]
106. Tazi KA, Barrière E, Moreau R, Poirel O, Lebrec D. Relationship between protein kinase C alterations and nitric oxide overproduction in cirrhotic rat aortas. Liver. 2002;22(2):178–83. doi:10.1034/j.1600-0676.2002.01616.x. [Google Scholar] [PubMed] [CrossRef]
107. Ping P, Takano H, Zhang J, Tang XL, Qiu Y, Li RC, et al. Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: a signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning. Circ Res. 1999;84(5):587–604. doi:10.1161/01.res.84.5.587. [Google Scholar] [PubMed] [CrossRef]
108. de la Lastra JM P, Juan CA, Plou FJ, Pérez-Lebeña E. The nitration of proteins, lipids and DNA by peroxynitrite derivatives-chemistry involved and biological relevance. Stresses. 2022;2(1):53–64. doi:10.3390/stresses2010005. [Google Scholar] [CrossRef]
109. Piacenza L, Zeida A, Trujillo M, Radi R. The superoxide radical switch in the biology of nitric oxide and peroxynitrite. Physiol Rev. 2022;102(4):1881–906. doi:10.1152/physrev.00005.2022. [Google Scholar] [PubMed] [CrossRef]
110. Wang J, Swanson RA. Superoxide and non-ionotropic signaling in neuronal excitotoxicity. Front Neurosci. 2020;4:861. doi:10.3389/fnins.2020.00861. [Google Scholar] [PubMed] [CrossRef]
111. Prolo C, Piacenza L, Radi R. Peroxynitrite: a multifaceted oxidizing and nitrating metabolite. Curr Opin Chem Biol. 2024;80:102459. doi:10.1016/j.cbpa.2024.102459. [Google Scholar] [PubMed] [CrossRef]
112. Matuz-Mares D, González-Andrade M, Araiza-Villanueva MG, Vilchis-Landeros MM, Vázquez-Meza H. Mitochondrial calcium: effects of its imbalance in disease. Antioxidants. 2022;11(5):801. doi:10.3390/antiox11050801. [Google Scholar] [PubMed] [CrossRef]
113. Giorgi C, Marchi S, Pinton P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol. 2018;19(11):713–30. doi:10.1038/s41580-018-0052-8. [Google Scholar] [PubMed] [CrossRef]
114. Larrañaga-SanMiguel A, Bengoa-Vergniory N, Flores-Romero H. Crosstalk between mitochondria-ER contact sites and the apoptotic machinery as a novel health meter. Trends Cell Biol. 2025;35(1):33–45. doi:10.1016/j.tcb.2024.08.007. [Google Scholar] [PubMed] [CrossRef]
115. Gao C, Jiang J, Tan Y, Chen S. Microglia in neurodegenerative diseases: mechanism and potential therapeutic targets. Signal Transduct Target Ther. 2023;8(1):359. doi:10.1038/s41392-023-01588-0. [Google Scholar] [PubMed] [CrossRef]
116. Müller L, Di Benedetto S, Müller V. From homeostasis to neuroinflammation: insights into cellular and molecular interactions and network dynamics. Cells. 2025;14(1):54. doi:10.3390/cells14010054. [Google Scholar] [PubMed] [CrossRef]
117. Zhang W, Xiao D, Mao Q, Xia H. Role of neuroinflammation in neurodegeneration development. Signal Transduct Target Ther. 2023;8(1):267. doi:10.1038/s41392-023-01486-5. [Google Scholar] [PubMed] [CrossRef]
118. Dash UC, Bhol NK, Swain SK, Samal RR, Nayak PK, Raina V, et al. Oxidative stress and inflammation in the pathogenesis of neurological disorders: mechanisms and implications. Acta Pharm Sin B. 2025;15(1):15–34. doi:10.1016/j.apsb.2024.10.004. [Google Scholar] [PubMed] [CrossRef]
119. Van Hove H, De Feo D, Greter M, Becher B. Central nervous system macrophages in health and disease. Annu Rev Immunol. 2025;43(1):589–613. doi:10.1146/annurev-immunol-082423-041334. [Google Scholar] [PubMed] [CrossRef]
120. Simpson DSA, Oliver PL. ROS generation in microglia: understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants. 2020;9(8):743. doi:10.3390/antiox9080743. [Google Scholar] [PubMed] [CrossRef]
121. Wang Q, Liu J, Zhang Y, Li Z, Zhao Z, Jiang W, et al. Microglial CR3 promotes neuron ferroptosis via NOX2-mediated iron deposition in rotenone-induced experimental models of Parkinson’s disease. Redox Biol. 2024;77(1):103369. doi:10.1016/j.redox.2024.103369. [Google Scholar] [PubMed] [CrossRef]
122. Harry GJ. Microglia in neurodegenerative events-an initiator or a significant other? Int J Mol Sci. 2021;22(11):5818. doi:10.3390/ijms22115818. [Google Scholar] [PubMed] [CrossRef]
123. Beard E, Lengacher S, Dias S, Magistretti PJ, Finsterwald C. Astrocytes as key regulators of brain energy metabolism: new therapeutic perspectives. Front Physiol. 2021;12:825816. doi:10.3389/fphys.2021.825816. [Google Scholar] [PubMed] [CrossRef]
124. Ding Z-B, Song L-J, Wang Q, Kumar G, Yan Y-Q, Ma C-G. Astrocytes: a double-edged sword in neurodegenerative diseases. Neural Regen Res. 2021;16(9):1702–10. doi:10.4103/1673-5374.306064. [Google Scholar] [PubMed] [CrossRef]
125. Zhao Y, Huang Y, Cao Y, Yang J. Astrocyte-mediated neuroinflammation in neurological conditions. Biomolecules. 2024;14(10):1204. doi:10.3390/biom14101204. [Google Scholar] [PubMed] [CrossRef]
126. Wu H, Zhang ZH, Zhou P, Sui X, Liu X, Sun Y, et al. A single-cell atlas of the substantia nigra reveals therapeutic effects of icaritin in a rat model of parkinson’s disease. Antioxidants. 2024;13(10):1183. doi:10.3390/antiox13101183. [Google Scholar] [PubMed] [CrossRef]
127. Roy T, Chatterjee A, Swarnakar S. Rotenone induced neurodegeneration is mediated via cytoskeleton degradation and necroptosis. Biochim Biophys Acta Mol Cell Res. 2023;1870(3):119417. doi:10.1016/j.bbamcr.2022.119417. [Google Scholar] [PubMed] [CrossRef]
128. Sheeler C, Rosa J-G, Ferro A, McAdams B, Borgenheimer E, Cvetanovic M. Glia in neurodegeneration: the housekeeper, the defender and the perpetrator. Int J Mol Sci. 2020;21(23):9188. doi:10.3390/ijms21239188. [Google Scholar] [PubMed] [CrossRef]
129. Hadjira S, Mansour A, Seghiri R, Menad A, Benayache F, Benayache S, et al. Neuroinflammation and behavioral deficit in rotenone-induced neurotoxicity in rats and the possible effects of butanolic extract of centaurea africana. Recent Adv Inflamm Allergy Drug Disc. 2022;15(1):35–43. doi:10.2174/2772270816666220105124730. [Google Scholar] [PubMed] [CrossRef]
130. Umer H, Sharif A, Khan HM, Anjum SMM, Akhtar B, Ali S, et al. Mitigation of neuroinflammation and oxidative stress in rotenone-induced parkinson mouse model through liposomal coenzyme-Q10 intervention: a comprehensive in-vivo study. Inflammation. 2025;753(1):135873. doi:10.1007/s10753-025-02237-0. [Google Scholar] [PubMed] [CrossRef]
131. D’Egidio F, Castelli V, d’Angelo M, Ammannito F, Quintiliani M, Cimini A. Brain incoming call from glia during neuroinflammation: roles of extracellular vesicles. Neurobiol Dis. 2024;201(2):106663. doi:10.1016/j.nbd.2024.106663. [Google Scholar] [PubMed] [CrossRef]
132. Wang M, Zhao R, Su Y, Zhai D, Liang H, Zhang L, et al. Di G. 4,4’-dimethoxychalcone mitigates neuroinflammation following traumatic brain injury through modulation of the TREM2/PI3K/AKT/NF-κB signaling pathway. Inflammation. 2025 Apr 22;16(Pt 3):813. doi:10.1007/s10753-025-02279-4. [Google Scholar] [PubMed] [CrossRef]
133. Tang L, Chen C, Xia B, Wu W, Wei R, Zhu G, et al. Effect of wenshen-yanggan decoction on movement disorder and substantia nigra dopaminergic neurons in mice with chronic parkinson’s disease. Evid Based Complement Alternat Med. 2020;2020(1):9838295. doi:10.1155/2020/9838295. [Google Scholar] [PubMed] [CrossRef]
134. Anilkumar S, Wright-Jin E. NF-κB as an inducible regulator of inflammation in the central nervous system. Cells. 2024;13(6):485. doi:10.3390/cells13060485. [Google Scholar] [PubMed] [CrossRef]
135. Roberti A, Chaffey LE, Greaves DR. NF-κB signaling and inflammation-drug repurposing to treat inflammatory disorders? Biology. 2022;11(3):372. doi:10.3390/biology11030372. [Google Scholar] [PubMed] [CrossRef]
136. Zhao N, Wu M, Velu P, Annamalai V, Zhang J. Sanggenol L alleviates rotenone-induced parkinson’s disease and inhibits mitochondrial complex I by apoptosis Via P13K/AKT/mTOR signalling. Comb Chem High Throughput Screen. 2024 Dec 13. doi:10.2174/0113862073358649241128053921. [Google Scholar] [PubMed] [CrossRef]
137. Salama RM, Abdel-Latif GA, Abbas SS, El Magdoub HM, Schaalan MF. Neuroprotective effect of crocin against rotenone-induced Parkinson’s disease in rats: interplay between PI3K/Akt/mTOR signaling pathway and enhanced expression of miRNA-7 and miRNA-221. Neuropharmacol. 2020;164(2):107900. doi:10.1016/j.neuropharm.2019.107900. [Google Scholar] [PubMed] [CrossRef]
138. Anand AA, Khan M, V. M, Kar D. The molecular basis of Wnt/β-catenin signaling pathways in neurodegenerative diseases. Int J Cell Biol. 2023;2023(5):9296092. doi:10.1155/2023/9296092. [Google Scholar] [PubMed] [CrossRef]
139. Mohammed NN, Tadros MG, George MY. Empagliflozin repurposing in Parkinson’s disease; modulation of oxidative stress, neuroinflammation, AMPK/SIRT-1/PGC-1α, and wnt/β-catenin pathways. Inflammopharmacology. 2024;32(1):777–94. doi:10.1007/s10787-023-01384-w. [Google Scholar] [PubMed] [CrossRef]
140. Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X, et al. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther. 2022;7(1):3. doi:10.1038/s41392-021-00762-6. [Google Scholar] [PubMed] [CrossRef]
141. Tabassum S, Wu S, Lee C-H, Yang BSK, Gusdon AM, Choi HA, et al. Mitochondrial-targeted therapies in traumatic brain injury: from bench to bedside. Neurother. 2025;22(1):e00515. doi:10.1016/j.neurot.2024.e00515. [Google Scholar] [PubMed] [CrossRef]
142. Gros P, Marras C, Wang X, Chiu M, Farkouh ME, Emdin A, et al. Differences in parkinson’s disease populations: teaching hospitals versus other settings and implications for clinical trials. Mov Disord. 2025 May 29. doi:10.1002/mds.30239. [Google Scholar] [PubMed] [CrossRef]
143. Aamodt WW, Eickholt L, Coughlin DG, Solomon L, Rendle KA, Marshall C, et al. Sex and racial/ethnic differences in end-of-life care preferences in persons with parkinson’s disease and related disorders. Mov Disord. 2025 May 23. doi:10.1002/mds.30240. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools