
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Region-Specific Astrocyte Endfeet Disruption as a Driver of Pyramidal Neuron Death after Ischemia-Reperfusion in the Hippocampus
1 Department of Emergency Medicine, Kangwon National University Hospital, School of Medicine, Kangwon National University, Chuncheon, 24289, Republic of Korea
2 Department of Physical Therapy, College of Health Science, Youngsan University, Yangsan, 50510, Republic of Korea
3 Department of Emergency Medicine, School of Medicine, Kangwon National University, Chuncheon, 24341, Republic of Korea
* Corresponding Author: Moo-Ho Won. Email:
(This article belongs to the Special Issue: Cellular and Molecular Insights into Brain Ischemic Insults)
BIOCELL 2026, 50(3), 3 https://doi.org/10.32604/biocell.2025.072635
Received 31 August 2025; Accepted 15 November 2025; Issue published 23 March 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Ischemia-reperfusion (I/R) injury induces region-specific neuronal vulnerability within the hippocampus, with the cornu ammonis 1 (CA1) subfield particularly prone to delayed neuronal death. While intrinsic neuronal factors have been implicated, emerging evidence highlights the decisive contribution of astrocyte endfeet (AEF)—specialized perivascular structures that regulate ion and water homeostasis, glutamate clearance, and blood–brain barrier (BBB) stability. This review synthesizes structural and molecular alterations of AEF across the CA1–CA3 subfields following I/R and their correlation with neuronal fate. In CA1, AEF undergo early-onset swelling and detachment from the vascular basal lamina due to dysfunction of critical proteins such as aquaporin-4 (AQP4) and Kir4.1. These changes impair glutamate uptake, metabolic support, and potassium buffering, contributing to neuronal hyperexcitability and degeneration. In contrast, AEF in CA3 preserves polarity and functional coupling of AQP4 and Kir4.1, conferring regional resilience. At the signaling level, AEF disruption activates mitogen-activated protein kinase (MAPK)/c-Jun N-terminal kinase (JNK) pathways, promotes reactive oxygen species (ROS) accumulation, and induces inducible nitric oxide synthase (iNOS)-mediated inflammation, amplifying neurotoxicity. Furthermore, subfield-specific astrocytic transcriptional profiles modulate inflammatory responses and gliovascular interactions. By reframing AEF not as passive scaffolds but as active regulators of neuronal survival, this review provides novel insight into the astrocyte-dependent mechanisms of hippocampal vulnerability. Therapeutic strategies that preserve AEF structure and function may offer targeted protection against delayed neuronal death in ischemic brain injury.Keywords
Ischemia-reperfusion (I/R) injury in the brain triggers a complex cascade of metabolic, vascular, and cellular disturbances that affect neuronal populations in a regionally selective manner. The hippocampus, a medial temporal lobe structure essential for learning, memory consolidation, and spatial navigation, is among the most vulnerable brain regions to I/R insult [1–3]. Anatomically, the hippocampal formation includes the dentate gyrus, subiculum, entorhinal cortex, and the hippocampus proper, which is subdivided into cornu ammonis (CA) subfields—CA1, CA2, CA3, and CA4 (also referred to as the hilus of the dentate gyrus) [3,4]. Among these, the CA1 region is particularly sensitive to transient global ischemia, exhibiting delayed neuronal death within 2–3 days post-injury despite initial apparent histological preservation [5–7].
Although intrinsic neuronal factors such as high N-methyl-D-aspartate (NMDA) receptor density, elevated oxidative stress susceptibility, and mitochondrial dysfunction have been proposed to explain the selective vulnerability of the CA1 region, emerging evidence suggests that astrocyte-dependent mechanisms, particularly at the level of astrocyte endfeet (AEF), play a decisive role in determining neuronal fate [8–10]. Astrocytes are multifunctional glial cells that maintain brain homeostasis by regulating extracellular ion balance, neurotransmitter clearance, metabolic support, and neurovascular unit (NVU) function [11–13]. AEF, their perivascular terminal expansions of astrocytes, form a critical interface with the vasculature, contributing to blood–brain barrier (BBB) integrity, neurovascular coupling, and fluid exchange via key channel proteins such as aquaporin-4 (AQP4) and Kir4.1 [14].
Recent findings reveal that AEF undergo region-specific and early pathological alterations following I/R injury. In the CA1 region, AEF exhibit rapid-onset swelling, redistribution or mislocalization of AQP4/Kir4.1, and detachment from the vascular basal lamina—events that precede and likely contribute to pyramidal neuron degeneration [15,16]. In contrast, AEF in CA2 and CA3 subfields are relatively preserved, maintaining ion homeostasis and structural integrity, which may underlie the relative resistance of neurons in those regions. This astroglial heterogeneity across hippocampal subfields has spurred new interest in understanding the astrocyte–neuron–vessel triad as a determinant of selective neuronal vulnerability.
While many reviews have addressed astrocyte reactivity or global hippocampal injury, few have specifically focused on AEF-centered mechanisms, nor compared these changes across CA subregions in the context of neuronal death vs. resistance.
In this review, we aim to bridge this knowledge gap by examining the structural and molecular profiles of AEF, with a focus on region-specific alterations in CA1, CA2, and CA3 following I/R injury. We further evaluate how these changes impact BBB stability, potassium buffering, glutamate clearance, metabolic coupling, and inflammatory signaling—each of which plays a decisive role in determining neuronal survival or degeneration. By integrating anatomical, functional, and mechanistic insights, this review proposes a framework for understanding AEF-mediated mechanisms underlying hippocampal region-specific vulnerability in ischemic brain injury.
2 Selective Vulnerability and Delayed Neuronal Death (DND) in the Hippocampus
I/R injury induces region-specific patterns of neuronal degeneration in the brain, with the hippocampal CA1 subfield demonstrating marked vulnerability relative to adjacent regions such as CA2 and CA3 (Table 1, Fig. 1). A hallmark of this selective vulnerability is DND—a phenomenon in which CA1 pyramidal neurons initially appear morphologically intact post-ischemia but undergo apoptosis-like degeneration within 2–3 days [5,6]. This temporal lag distinguishes DND from necrotic death seen in more acutely damaged brain regions. Notably, CA1 neurons are highly sensitive to brief global ischemia, while CA2 neurons exhibit partial resistance, and CA3 neurons are relatively spared. This gradient of susceptibility has traditionally been ascribed to intrinsic neuronal characteristics such as NMDA receptor density, calcium-buffering capacity, and mitochondrial resilience [7,17]. However, accumulating evidence implicates astrocytic dysfunction—particularly at the level of AEF—as a critical extrinsic factor that exacerbates or mitigates DND. Structural alterations of AEF, mislocalization of ion–water channels (e.g., AQP4 and Kir4.1), and impaired metabolic coupling to vasculature have all been linked to CA1 neuronal demise following I/R insult [16,18,19].

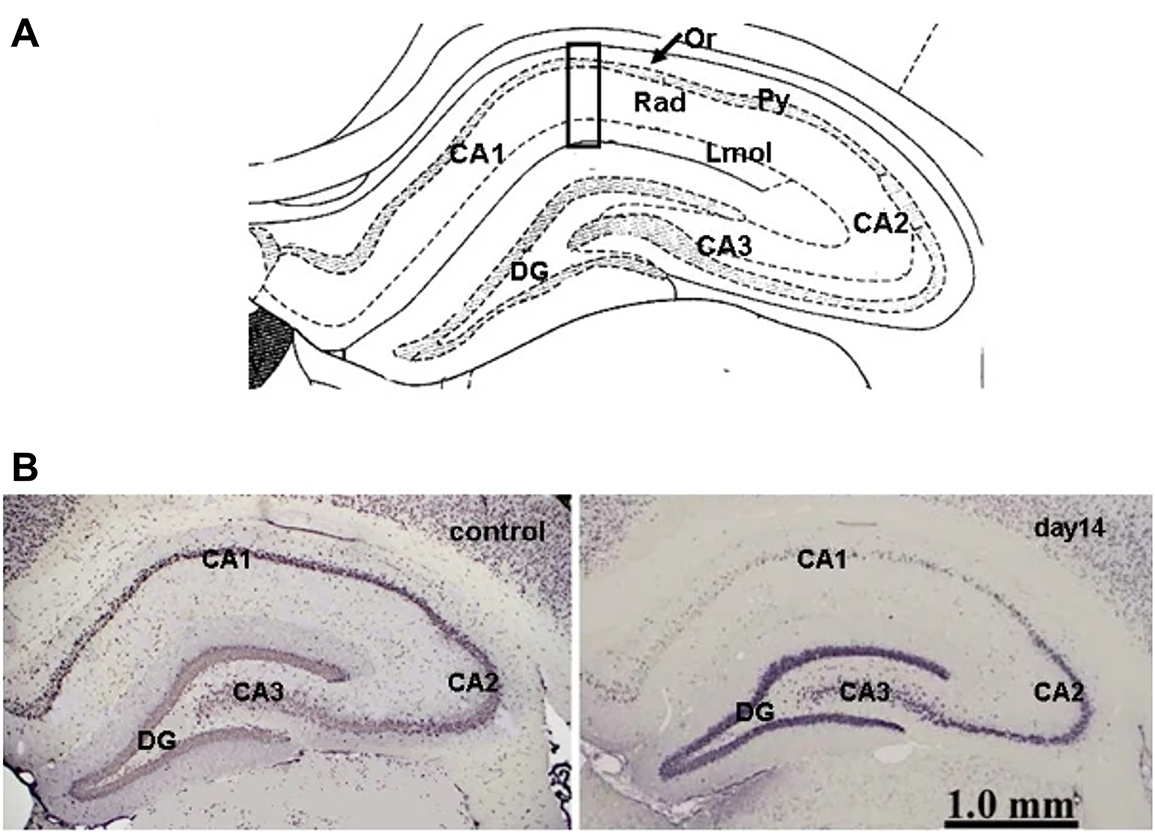
Figure 1: Selective delayed neuronal death in hippocampal subfields after transient global ischemia. (A) Schematic of hippocampal subregions. (B) Representative NeuN-stained sections show selective neuronal loss in CA1 but preservation of CA2 and CA3 neurons at 14 days post-ischemia. Abbreviations: CA, cornu ammonis; DG, dentate gyrus; Lmol, stratum lacunosum-moleculare; Or, stratum oriens layer; Py, stratum pyramidale; Rad, stratum radiatum. Adapted from [28]. Licensed under CC BY 2.0
2.1 Experimental Evidence of Region-Specific DND
Animal models robustly demonstrate region-specific DND patterns (Table 1). In gerbils—characterized by an incomplete circle of Willis—bilateral common carotid artery occlusion (BCCAO) for as little as 5 min reproducibly induces CA1-selective neuronal death without the need for systemic hypotension [5,16]. In contrast, rats require four-vessel occlusion (4-VO) or cardiac arrest-resuscitation paradigms, with ischemic durations of 10–15 min, to elicit comparable damage [21–23]. Mouse models have evolved to use transient bilateral carotid occlusion with or without hypotension to reliably induce CA1 DND [24,25]. Histological analyses across species consistently reveal profound CA1 neuronal loss with relative CA3 sparing, reinforcing the conserved nature of this spatial vulnerability. In human postmortem studies following cardiac arrest or hypoxic-ischemic encephalopathy, similar patterns of selective CA1 loss—particularly in Sommer’s sector—have been documented. These findings correlate with clinical features such as anterograde amnesia or persistent vegetative states, lending translational validity to experimental models [26,27].
2.2 Influence of Experimental Conditions on DND
The emergence and severity of DND are not solely dictated by hippocampal anatomy but are profoundly influenced by experimental and physiological variables. Ischemic duration is a principal factor: in gerbils, 5-min BCCAO induces classic CA1 DND, whereas longer durations (>10 min) expand injury to the CA3 and dentate gyrus, often accompanied by necrotic features [29,30]. Thermal regulation is another critical determinant. Hyperthermia during or after ischemia markedly exacerbates neuronal damage, while post-ischemic hypothermia (32°C–34°C) significantly attenuates both neuronal death and glial activation by reducing metabolic and excitotoxic stress [31–35]. Age-related differences also shape DND outcomes. Young adult rodents exhibit prominent CA1 degeneration, whereas aged animals often show delayed or attenuated neuronal death, potentially due to reduced astrocytic support, altered inflammatory profiles, and compromised mitochondrial function [36–40]. This age-dependent variation bears clinical importance, as elderly stroke patients exhibit distinct recovery patterns and may require age-specific therapeutic approaches.
In summary, DND in the hippocampus reflects a complex interplay between intrinsic neuronal factors and extrinsic modulators, including glial dysfunction and environmental stressors. A multifactorial framework is thus essential to accurately interpret hippocampal vulnerability and guide therapeutic strategies post-I/R injury.
3 AEF: Structure, Composition, and Role in the NVU
AEF are specialized terminal expansions of astrocytic processes that ensheath cerebral microvessels, forming a critical interface between the brain parenchyma and the vascular system [9]. These perivascular structures are integral components of the NVU, which coordinates dynamic interactions among astrocytes, endothelial cells, pericytes, and neurons to maintain cerebral homeostasis and blood flow regulation [41,42]. AEF are enriched with mitochondria and endoplasmic reticulum, allowing for localized protein synthesis, calcium signaling, and metabolic exchange at the gliovascular interface [43,44]. These subcellular organelles support highly compartmentalized calcium dynamics, which are essential for neurovascular coupling—a process often disrupted under pathological conditions such as I/R injury [45,46].
A hallmark of AEF is their AQP4, a water channel primarily localized to the perivascular membrane. AQP4 mediates rapid water transport across the BBB and is critical for brain volume regulation, potassium buffering, and glymphatic clearance [14,47]. Ischemic insults often cause loss of AQP4 polarity, impairing perivascular water clearance and contributing to vasogenic edema [48,49]. Another key AEF component is Kir4.1, an inward-rectifying potassium channel that acts synergistically with AQP4 to maintain extracellular potassium homeostasis during neuronal activity. Kir4.1 dysfunction impairs potassium buffering, leading to extracellular K+ accumulation and increased neuronal excitability [50,51], particularly exacerbated during I/R injury [52–55]. The synergistic interaction between AQP4 and Kir4.1 at AEF enables coupled water and potassium fluxes, ensuring rapid K+ clearance and osmotic balance during neuronal activity [56]. I/R injury disrupts this interaction, leading to uncoupled water-ion transport, astrocytic swelling, and neuronal hyperexcitability. Sustained AQP4 mislocalization and Kir4.1 downregulation further compromise glymphatic clearance and exacerbate secondary injury following I/R insult. AEF also contains connexin 43, forming astrocyte–astrocyte gap junctions that allow for intercellular ion and metabolite exchange [57,58]. During ischemic stroke, connexin 43 undergoes phosphorylation-dependent internalization, leading to the loss of protective astrocyte–astrocyte coupling [57]. Concurrently, aberrant opening of connexin 43 hemichannels promotes the release of ATP and glutamate, amplifying neuroinflammation and neuronal injury [57].
Collectively, these molecular components confer AEF with critical roles in BBB maintenance, neurovascular signaling, ion and water homeostasis, and metabolic regulation [14,59,60]. However, their integrity is highly susceptible to I/R-induced disruption, including protein mislocalization, cytoskeletal breakdown, or detachment from the vascular basal lamina. These alterations destabilize NVU function and contribute to region-specific neuronal injury, particularly in the CA1 hippocampus [15,16]. Thus, the molecular architecture and polarization of AEF are indispensable for maintaining CNS homeostasis under both physiological and ischemic conditions. Fig. 2 provides a schematic overview of the NVU, highlighting AEF components. The next section explores how AEF responds to I/R differ across hippocampal subfields, shedding light on their role in selective neuronal vulnerability.
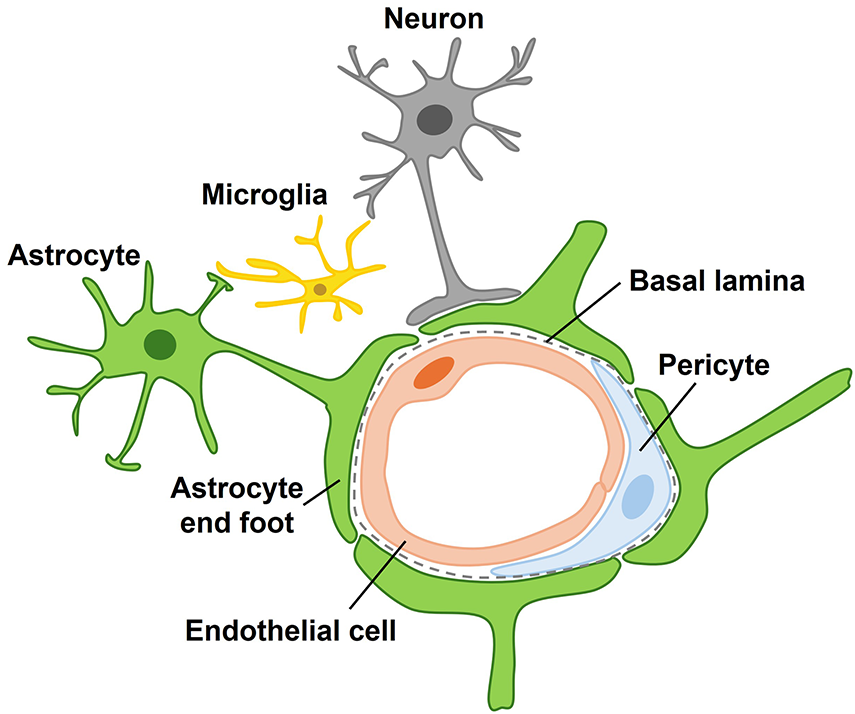
Figure 2: Schematic representation of astrocyte–neuron–vessel interactions. The neurovascular unit shows an astrocyte (green) contacting both a pyramidal neuron (gray) and a blood vessel (red) through its end foot. The end foot maintains blood–brain barrier integrity, regulates ionic and metabolic homeostasis, and modulates vascular tone
4 Region-Specific Alterations of AEF in the Hippocampus after I/R Injury
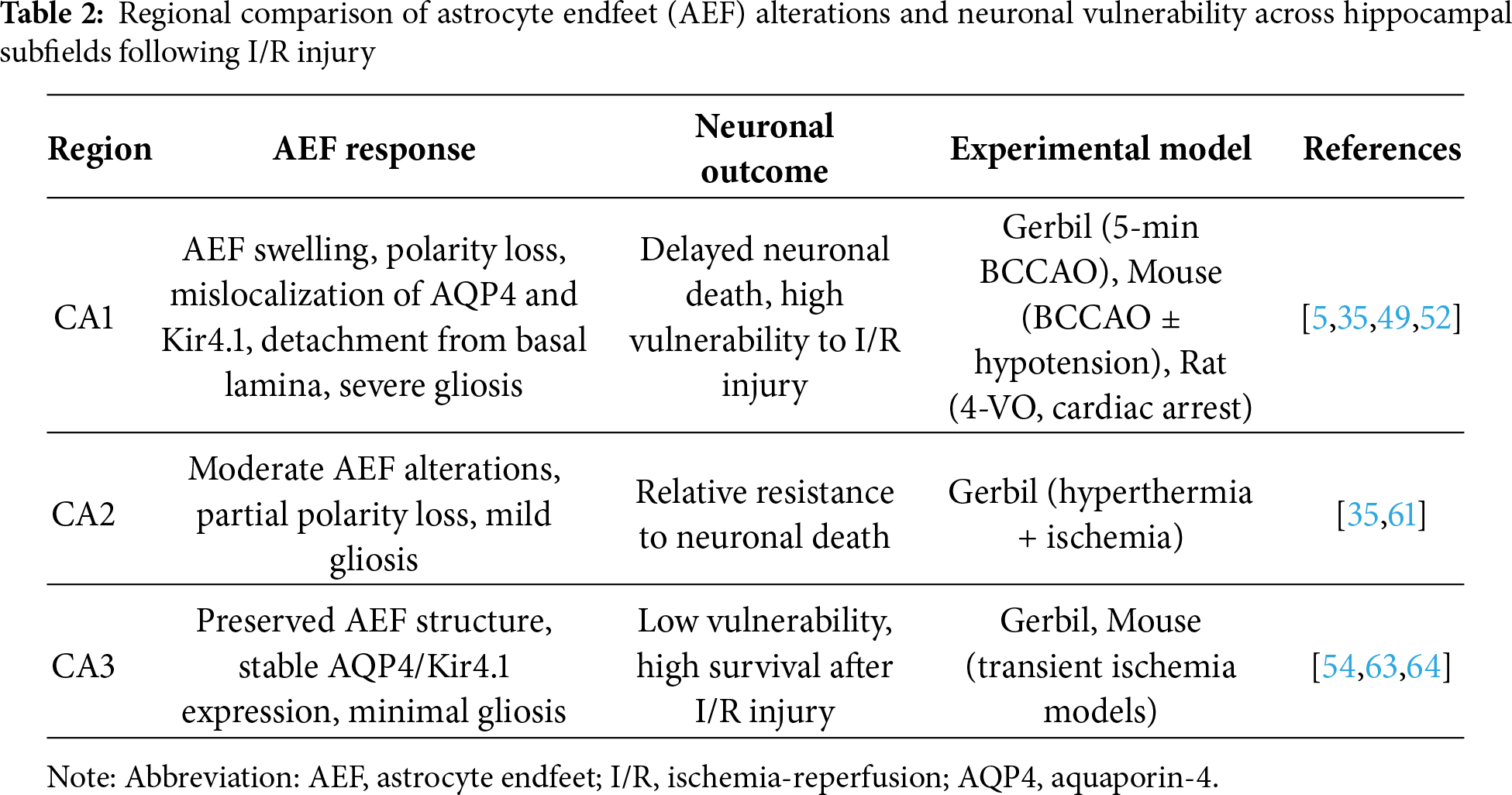
The hippocampus exhibits regional heterogeneity in its vulnerability to I/R injury, with pyramidal neurons in the CA1 subfield being markedly more susceptible to delayed neuronal death than those in CA2 or CA3 [5,7,35,61]. This differential vulnerability is closely mirrored by AEF pathology, which varies significantly across hippocampal subfields [15,16]. Following I/R insult, AEF in the CA1 region undergoes distinct pathological alterations, including pronounced swelling and detachment from the vascular basal lamina. These changes are strongly associated with BBB disruption and impaired neurovascular coupling [16,48,62]. In contrast, CA3 astrocytes tend to preserve AEF structure and maintain their perivascular association, suggesting greater regional resilience and more effective ionic/osmotic regulation [54,63,64]. These differences are underpinned by region-specific expression and localization of key proteins. In CA1, ischemic insults lead to depolarization and mislocalization of AQP4 and Kir4.1, disrupting water clearance and potassium buffering [49,52,55,65]. In contrast, CA3 astrocytes typically retain AQP4 polarization and Kir4.1 function, facilitating more effective homeostatic regulation [66]. The polarized distribution of AEF proteins, including AQP4 and Kir4.1, is regulated and stabilized by the dystrophin-associated protein complex (DAPC), especially α-syntrophin and dystrophin, which anchor AQP4 to the perivascular membrane [48,67]. Loss of DAPC integrity—more pronounced in CA1 than in CA3—leads to AQP4 displacement and impaired glymphatic and ionic regulation [62,68–71].
Collectively, these regional disparities in AEF structure and molecular architecture provide a mechanistic basis for the selective vulnerability of CA1 neurons to I/R injury. AEF damage precedes neuronal death and contributes to it through impaired BBB integrity, disrupted neurovascular signaling, and ionic imbalance. Table 2 provides a comparative overview of AEF and neuronal changes across CA1, CA2, and CA3 subfields, integrating experimental data to emphasize the broader heterogeneity of hippocampal responses to I/R stress. The next section explores how these AEF alterations temporally relate to pyramidal neuron degeneration and highlights the astrocyte–neuron interactions that shape regional vulnerability.
5 Temporal Association between AEF Dysfunction and Pyramidal Neuron Death
The hippocampal CA1 subfield exhibits DND, typically emerging 2–4 days after cerebral I/R injury, in contrast to the relative preservation of CA2 and CA3 neurons [5,7]. This temporal delay strongly implicates post-ischemic secondary mechanisms—rather than immediate necrosis—as central drivers of neuronal demise [6,72]. Among these, astrocyte dysfunction—particularly at the level of AEF—has been increasingly recognized as a pivotal initiator of CA1 vulnerability. Histopathological and ultrastructural analyses consistently demonstrate that AEF swelling and separation from the vascular basal lamina occur within hours after reperfusion—preceding overt neuronal degeneration [16,48,73,74].
These early astroglial alterations compromise the BBB and impair critical homeostatic functions, including water and potassium buffering. Key molecular components, such as AQP4 and inward rectifying potassium channel Kir4.1, become mislocalized or downregulated in CA1 astrocytes following I/R injury, disrupting potassium clearance and astrocytic membrane potential [52,54,55]. This ionic dysregulation increases neuronal excitability and potentiates glutamate-mediated excitotoxicity, particularly under conditions of astrocyte depolarization. Beyond ionic imbalance, AEF detachment also disrupts astrocyte–neuron metabolic coupling. AEF are critical sites for monocarboxylate transporter (MCT)-mediated lactate and glucose transfer—essential energy substrates during metabolic stress [43,63]. Loss of this coupling deprives neurons of metabolic support, exacerbating mitochondrial failure and energy depletion. Furthermore, astrocytic glutamate transport via excitatory amino acid transporters (EAATs)—especially EAAT2 (glutamate transporter 1, GLT-1)—is often reduced or mislocalized in I/R conditions, impairing extracellular glutamate clearance and further amplifying excitotoxic signaling [75–77]. AEF disruption also initiates astrocyte-derived proinflammatory responses, including upregulation of interleukin-1β (IL-1β), tumor necrosis factor-alpha (TNF-α), and inducible nitric oxide synthase (iNOS). These mediators activate pro-apoptotic pathways in neighboring neurons, contributing to progressive neuronal degeneration [78,79].
In summary, early AEF dysfunction precedes and predicts delayed pyramidal neuron death in the CA1 region by initiating a cascade of secondary insults—including ionic dysregulation, excitotoxicity, metabolic uncoupling, and neuroinflammation. These processes act synergistically to promote irreversible neuronal injury. Fig. 3 provides a schematic representation of these temporally ordered events, linking AEF pathology to subsequent neuronal loss.
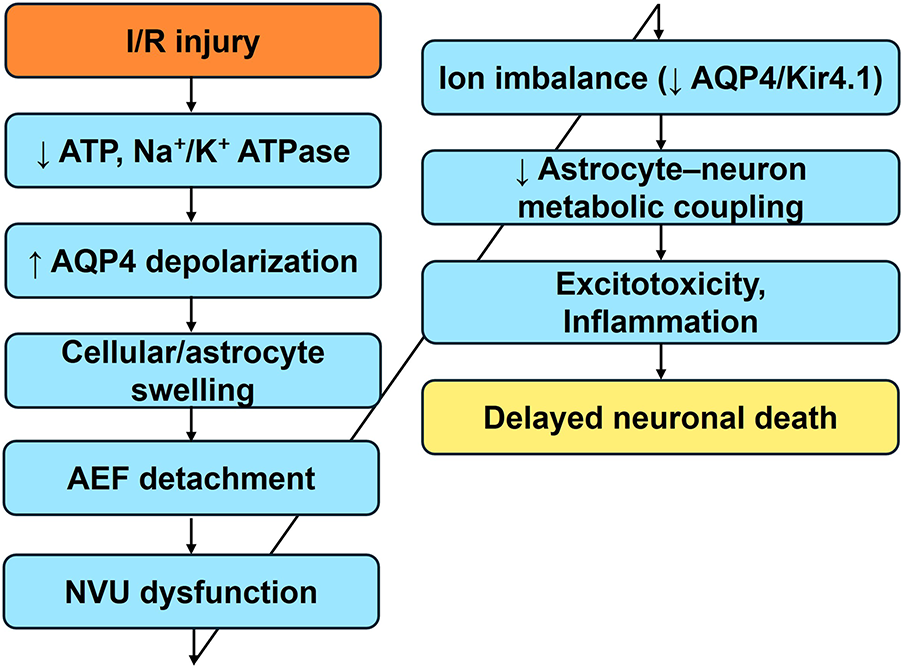
Figure 3: Sequential astrocyte endfeet (AEF) mechanisms contributing to delayed neuronal death in CA1 after I/R injury. AEF dysfunction following I/R injury leads to a cascade of interrelated pathological events that culminate in CA1 pyramidal neuron death. Early events include depletion of ATP and inhibition of Na+/K+-ATPase activity, resulting in AQP4 depolarization and astrocytic swelling. Swollen AEF detach from the vascular basal lamina, contributing to NVU disruption. Concurrently, mislocalization or downregulation of AQP4 and Kir4.1 channels impairs potassium buffering and water homeostasis, promoting neuronal hyperexcitability. In parallel, AEF detachment disrupts astrocyte-neuron metabolic coupling via monocarboxylate transporters, reducing the availability of lactate and glucose to neurons. Downregulation of glutamate transporters exacerbates excitotoxicity. Additionally, upregulation of inflammatory mediators amplifies neuronal damage through oxidative stress and apoptotic signaling. Collectively, these mechanisms temporally and functionally link AEF disruption to the selective vulnerability of CA1 neurons. Abbreviations: I/R, ischemia-reperfusion; ATP, adenosine triphosphate; AQP4, aquaporin-4; AEF, astrocyte endfeet; NVU, neurovascular unit
6 Molecular Pathways Linking AEF Dysfunction to Neuronal Vulnerability
AEF dysfunction in the hippocampal CA1 region following I/R injury initiates a cascade of interrelated molecular events that amplify neuronal vulnerability and degeneration. Among the most prominent mechanisms are the activation of mitogen-activated protein kinases (MAPKs), including c-Jun N-terminal kinase (JNK) and p38, which are rapidly upregulated in reactive astrocytes and injured neurons. These kinases mediate inflammatory gene transcription, apoptotic signaling, and cytoskeletal destabilization, thereby exacerbating CA1 neuronal loss [19,80].
In addition to MAPK cascades, the phosphoinositide 3-kinase/protein kinase B (PI3K/AKT) signaling pathway also plays a central role in astrocytic and neuronal responses following I/R injury [81,82]. Activation of the PI3K/AKT axis in reactive astrocytes regulates cell survival, redox homeostasis, and inflammatory signaling by activating downstream effectors such as mechanistic target of rapamycin (mTOR), glycogen synthase kinase-3β (GSK-3 β), and nuclear factor erythroid 2-related factor 2 (Nrf2) [83]. Conversely, excessive or insufficient activation of PI3K/AKT signaling exacerbates oxidative stress, mitochondrial dysfunction, and cytoskeletal instability within AEF, thereby amplifying the cascade that culminates in CA1 neuronal degeneration [83].
Another key pathological mediator is oxidative stress, which intensifies during reperfusion. Reactive oxygen species (ROS), primarily generated through mitochondrial dysfunction and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity, accumulate within astrocytes and adjacent neurons. ROS induce lipid peroxidation, protein oxidation, and DNA fragmentation, leading to activation of nuclear factor-kappa B (NF-κB) and p53-dependent death pathways [75,84,85]. Furthermore, oxidative stress downregulates and mislocalizes the EAAT2 (GLT-1), thereby intensifying extracellular glutamate accumulation and glutamate-induced excitotoxicity [75,77].
Nitric oxide (NO), synthesized by iNOS in astrocytes and microglia, contributes to CA1 damage through the formation of peroxynitrite, a potent oxidant formed by the reaction of NO with superoxide. Peroxynitrite disrupts mitochondrial integrity, promotes protein nitration, and triggers apoptotic cascades [86]. Notably, pharmacological inhibition of iNOS mitigates neuronal death in the CA1 region, highlighting a functional link between astrocytic iNOS upregulation and pyramidal neuron injury [87,88].
Inflammatory signaling further amplifies the impact of AEF dysfunction. Disruption of the gliovascular unit promotes astrocytic release of IL-1β, TNF-α, and monocyte chemoattractant protein-1 (MCP-1). These cytokines not only activate resident microglia but also facilitate the recruitment of peripheral immune cells such as monocytes, neutrophils, and T lymphocytes [79,89]. This astrocyte–neuroimmune crosstalk exacerbates BBB leakage and initiates cycles of neuroinflammation and apoptosis, particularly in CA1 neurons [90].
Collectively, these pathways illustrate how AEF dysfunction acts as a central trigger of oxidative imbalance, neurotransmitter dysregulation, mitochondrial impairment, and neuroinflammatory amplification. These intertwined molecular events converge to drive the selective vulnerability of CA1 pyramidal neurons following I/R injury. The representative molecular mechanisms and their functional consequences are summarized in Table 3.

7 Therapeutic Implications and Future Directions
Understanding the pivotal role of AEF dysfunction in hippocampal CA1 vulnerability provides a strong rationale for targeting astrocyte-based mechanisms in I/R injury. As summarized in Fig. 4, therapeutic strategies that directly modulate astrocytic pathways—rather than focusing solely on neuronal survival—may yield more robust and sustained neuroprotection.
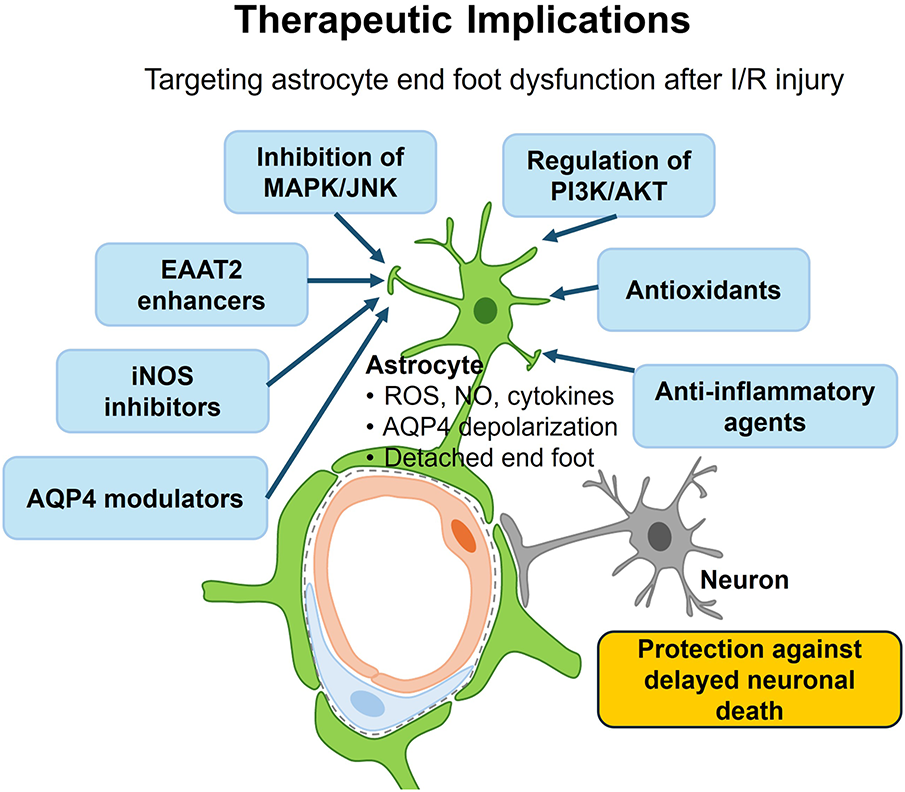
Figure 4: Therapeutic implications of targeting astrocyte end foot dysfunction after ischemia-reperfusion (I/R) injury. This schematic illustrates potential therapeutic strategies aimed at mitigating astrocyte-mediated pathological cascades that contribute to delayed neuronal death in the hippocampal CA1 region following I/R injury. Detachment of perivascular astrocyte endfeet impairs ion and water homeostasis, leading to excessive production of ROS, NO, and proinflammatory cytokines. These processes ultimately promote neuroinflammation and excitotoxicity. Interventions include: inhibition of MAPKs, particularly JNK; regulation of PI3K/AKT; antioxidants to suppress oxidative stress; iNOS inhibitors to limit NO toxicity; EAAT2 enhancers to restore glutamate clearance; anti-inflammatory agents targeting IL-1β, TNF-α, or MCP-1 signaling; and AQP4 modulators to restore AQP4 polarity. Collectively, these approaches aim to protect against astrocyte-driven injury mechanisms and prevent delayed neuronal death. Abbreviations: ROS, reactive oxygen species; NO, nitric oxide; MAPK, mitogen-activated protein kinase; JNK, c-Jun N-terminal kinase; PI3K/AKT, phosphoinositide 3-kinase/protein kinase B; iNOS, inducible nitric oxide synthase; EAAT2, excitatory amino acid transporter 2; IL-1β, interleukin-1 beta; TNF-α, tumor necrosis factor-alpha; MCP-1, monocyte chemoattractant protein-1
Pharmacological inhibition of MAPKs, particularly JNK and p38, has demonstrated efficacy in reducing astrocyte reactivity and delaying neuronal apoptosis in various experimental I/R models [91,92]. Notably, D-JNKI1, a peptide JNK inhibitor, exerts prolonged neuroprotective effects by dampening pro-apoptotic signaling in both neurons and reactive astrocytes. Pharmacological activation of the PI3K/AKT pathway in astrocytes confers neuroprotection by enhancing connexin 43 phosphorylation, maintaining BBB integrity, and suppressing oxidative stress and apoptosis after I/R injury. Vinpocetine activates PI3K/AKT signaling to protect astrocytes and reduce infarct volume [93], while tetramethylpyrazine (TMP) targets the endothelin-1/AKT pathway to stabilize the astrocyte–endothelial interface and limit BBB disruption [94]. Antioxidants that scavenge astrocyte-derived ROS, such as edaravone and astaxanthin, have been shown to attenuate oxidative stress, limit secondary BBB damage, and reduce infarct volume [95–97]. Restoration of astrocytic glutamate transport function—particularly through upregulation or stabilization of EAAT2 (GLT-1)—is another promising strategy. Pharmacological agents like ceftriaxone and gene therapy approaches targeting EAAT2 have successfully reduced glutamate-induced excitotoxicity and improved neuronal viability in animal models [75,77,98]. Similarly, selective inhibition of iNOS, for example, with aminoguanidine or other small-molecule inhibitors, reduces peroxynitrite formation, preserves mitochondrial integrity, and confers indirect protection to neighboring neurons [87,99]. In addition, astrocyte-targeted anti-inflammatory interventions—such as neutralizing antibodies against IL-1β or TNF-α, or inhibition of MCP-1/C-C chemokine receptor type 2(CCR2) signaling—have shown efficacy in attenuating glial reactivity and preventing leukocyte infiltration [79,89,100]. Recent studies propose the restoration of AQP4 polarity via DAPC stabilization or pharmacological blockade using AQP4 modulators such as TGN-020, which reduces water accumulation and vasogenic edema in I/R injury [70,101,102].
Looking forward, combinatorial therapies that concurrently target multiple astrocytic dysfunctions—including oxidative stress, impaired neurotransmitter clearance, and cytokine-mediated neurotoxicity—represent a compelling next step. Novel approaches such as epigenetic modulation of astrocyte resilience or transplantation of genetically engineered glia could further enhance regional resistance to I/R injury in the hippocampus. By advancing our understanding of AEF–neuron interactions and leveraging astrocyte-specific therapeutic windows, future interventions may prevent or mitigate delayed neuronal death in ischemic conditions.
AEF plays a critical and dynamic role in maintaining neurovascular homeostasis, particularly within the hippocampal CA1 region, which is highly susceptible to I/R injury. Accumulating evidence indicates that early structural and molecular alterations of AEF—including swelling, polarity loss, mislocalization of AQP4 and Kir4.1, and detachment from the vasculature—precede and exacerbate delayed neuronal death in CA1 pyramidal neurons. These pathological changes disrupt water and ion homeostasis, compromise glutamate clearance, impair astrocyte-mediated metabolic coupling, and activate inflammatory cascades. Importantly, the selective vulnerability of CA1 pyramidal neurons appears to result not only from intrinsic neuronal properties but also from their critical reliance on tightly regulated astrocyte–neuron–vascular interactions, which are destabilized following I/R injury. Mechanistic insights discussed in this review—such as MAPK/JNK activation, PI3K/AKT dysregulation, oxidative and nitrosative stress, glutamate excitotoxicity, and cytokine-mediated neuroinflammation—highlight the multifactorial nature of AEF-mediated neurodegeneration. From a therapeutic standpoint, targeting AEF-specific dysfunction presents a promising and underexplored strategy for neuroprotection. Interventions that restore astrocytic polarity, EAAT2 expression, inhibit pro-inflammatory signaling, and mitigate ROS–driven damage may delay or prevent CA1 neuronal death. In particular, regionally tailored therapies that account for the distinct vulnerability of hippocampal astrocytes could enhance clinical translatability. Future research should focus on integrating region-specific astrocyte biology with combinatorial therapeutic approaches and preclinical models of global ischemia, thereby advancing astrocyte-targeted interventions for ischemic brain injury. By redefining AEF not as passive structural elements but as active and modulable regulators of neuronal viability, this review emphasizes their central role in hippocampal pathology and positions them as pivotal therapeutic targets in preventing delayed neuronal death following I/R insults.
Acknowledgement: None.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm contribution to the paper as follows: Conceptualization, data curation, writing—original draft preparation, and visualization, Joongbum Moon; Ji Hyeon Ahn; Moo-Ho Won; supervision, Moo-Ho Won; writing—review and editing, Ji Hyeon Ahn; Moo-Ho Won. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
Abbreviations
| AQP4 | Aquaporin-4 |
| AEF | Astrocyte endfeet |
| BCCAO | Bilateral common carotid artery occlusion |
| BBB | Blood–brain barrier |
| JNK | C-Jun N-terminal kinase |
| CA | Cornu ammonis |
| CCR2 | C-C chemokine receptor type 2 |
| DND | Delayed neuronal death |
| DAPC | Dystrophin-associated protein complex |
| EAATs | Excitatory amino acid transporters |
| GLT-1 | Glutamate transporter 1 |
| GSK-3 β | Glycogen synthase kinase-3β |
| iNOS | Inducible nitric oxide synthase |
| IL-1β | Interleukin-1β |
| I/R | Ischemia-reperfusion |
| MAPK | Mitogen-activated protein kinase |
| MCT | Monocarboxylate transporter |
| MCP-1 | Monocyte chemoattractant protein-1 |
| mTOR | Mechanistic target of rapamycin |
| NVU | Neurovascular unit |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NO | Nitric oxide |
| NMDA | N-methyl-D-aspartate |
| NF-κB | Nuclear factor-kappa B |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| PI3K/AKT | Phosphoinositide 3-kinase/protein kinase B |
| ROS | Reactive oxygen species |
| TMP | Tetramethylpyrazine |
| TNF-α | Tumor necrosis factor-alpha |
| 4-VO | Four-vessel occlusion |
References
1. Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry. 1957;20(1):11–21. doi:10.1136/jnnp.20.1.11. [Google Scholar] [PubMed] [CrossRef]
2. Milner B, Klein D. Loss of recent memory after bilateral hippocampal lesions: memory and memories-looking back and looking forward. J Neurol Neurosurg Psychiatry. 2016;87(3):230. doi:10.1136/jnnp-2015-311092. [Google Scholar] [PubMed] [CrossRef]
3. Amaral D, Lavenex P. Hippocampal neuroanatomy. In: Andersen P, Morris R, Amaral D, Bliss T, O’Keefe J, editors. The hippocampus book. Washington, DC, USA: American Psychological Association; 2007. p. 37–114. doi:10.1093/med/9780190065324.003.0003. [Google Scholar] [CrossRef]
4. Zhao L, Palomero-Gallagher N. Hippocampal architecture viewed through the eyes of methodological development. Anat Sci Int. 2025;100(4):635–58. doi:10.1007/s12565-025-00878-7. [Google Scholar] [PubMed] [CrossRef]
5. Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239(1):57–69. doi:10.1016/0006-8993(82)90833-2. [Google Scholar] [PubMed] [CrossRef]
6. Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79(4):1431–568. doi:10.1152/physrev.1999.79.4.1431. [Google Scholar] [PubMed] [CrossRef]
7. Small SA, Schobel SA, Buxton RB, Witter MP, Barnes CA. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat Rev Neurosci. 2011;12(10):585–601. doi:10.1038/nrn3085. [Google Scholar] [PubMed] [CrossRef]
8. Wang J, Sareddy GR, Lu Y, Pratap UP, Tang F, Greene KM, et al. Astrocyte-derived estrogen regulates reactive astrogliosis and is neuroprotective following ischemic brain injury. J Neurosci. 2020;40(50):9751–71. doi:10.1523/JNEUROSCI.0888-20.2020. [Google Scholar] [PubMed] [CrossRef]
9. Díaz-Castro B, Robel S, Mishra A. Astrocyte endfeet in brain function and pathology: open questions. Annu Rev Neurosci. 2023;46(1):101–21. doi:10.1146/annurev-neuro-091922-031205. [Google Scholar] [PubMed] [CrossRef]
10. Cheng J, Zheng Y, Cheng F, Wang C, Han J, Zhang H, et al. Different roles of astrocytes in the blood-brain barrier during the acute and recovery phases of stroke. Neural Regen Res. 2026;21(4):1359–72. doi:10.4103/NRR.NRR-D-24-01417. [Google Scholar] [PubMed] [CrossRef]
11. Hart CG, Karimi-Abdolrezaee S. Recent insights on astrocyte mechanisms in CNS homeostasis, pathology, and repair. J Neurosci Res. 2021;99(10):2427–62. doi:10.1002/jnr.24922. [Google Scholar] [PubMed] [CrossRef]
12. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):7–35. doi:10.1007/s00401-009-0619-8. [Google Scholar] [PubMed] [CrossRef]
13. Verkhratsky A, Nedergaard M. Physiology of astroglia. Physiol Rev. 2018;98(1):239–389. doi:10.1152/physrev.00042.2016. [Google Scholar] [PubMed] [CrossRef]
14. Nagelhus EA, Ottersen OP. Physiological roles of aquaporin-4 in brain. Physiol Rev. 2013;93(4):1543–62. doi:10.1152/physrev.00011.2013. [Google Scholar] [PubMed] [CrossRef]
15. Kalaria RN, Hase Y. Neurovascular ageing and age-related diseases. Subcell Biochem. 2019;91(Pt 12):477–99. doi:10.1007/978-981-13-3681-2_17. [Google Scholar] [PubMed] [CrossRef]
16. Lee TK, Kang IJ, Sim H, Lee JC, Ahn JH, Kim DW, et al. Therapeutic effects of decursin and Angelica gigas nakai root extract in gerbil brain after transient ischemia via protecting BBB leakage and astrocyte endfeet damage. Molecules. 2021;26(8):2161. doi:10.3390/molecules26082161. [Google Scholar] [PubMed] [CrossRef]
17. Schmidt-Kastner R, Freund TF. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience. 1991;40(3):599–636. doi:10.1016/0306-4522(91)90001-5. [Google Scholar] [PubMed] [CrossRef]
18. Boulay AC, Mazeraud A, Cisternino S, Saubaméa B, Mailly P, Jourdren L, et al. Immune quiescence of the brain is set by astroglial connexin 43. J Neurosci. 2015;35(10):4427–39. doi:10.1523/JNEUROSCI.2575-14.2015. [Google Scholar] [PubMed] [CrossRef]
19. Ezan P, André P, Cisternino S, Saubaméa B, Boulay AC, Doutremer S, et al. Deletion of astroglial connexins weakens the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32(8):1457–67. doi:10.1038/jcbfm.2012.45. [Google Scholar] [PubMed] [CrossRef]
20. Lee CH, Ahn JH, Lee TK, Sim H, Lee JC, Park JH, et al. Comparison of neuronal death, blood-brain barrier leakage and inflammatory cytokine expression in the hippocampal CA1 region following mild and severe transient forebrain ischemia in gerbils. Neurochem Res. 2021;46(11):2852–66. doi:10.1007/s11064-021-03362-6. [Google Scholar] [PubMed] [CrossRef]
21. Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11(5):491–8. doi:10.1002/ana.410110509. [Google Scholar] [PubMed] [CrossRef]
22. Sadowski M, Wisniewski HM, Jakubowska-Sadowska K, Tarnawski M, Lazarewicz JW, Mossakowski MJ. Pattern of neuronal loss in the rat hippocampus following experimental cardiac arrest-induced ischemia. J Neurol Sci. 1999;168(1):13–20. doi:10.1016/s0022-510x(99)00159-8. [Google Scholar] [PubMed] [CrossRef]
23. Weihs W, Warenits AM, Ettl F, Magnet IAM, Herkner H, Kramer AM, et al. CA1 hippocampal pyramidal cells in rats, resuscitated from 8 min of ventricular fibrillation cardiac arrest, recover after 20 weeks of survival: a retrospective pilot study. Shock. 2020;54(4):531–8. doi:10.1097/SHK.0000000000001469. [Google Scholar] [PubMed] [CrossRef]
24. León-Moreno LC, Castañeda-Arellano R, Rivas-Carrillo JD, Dueñas-Jiménez SH. Challenges and improvements of developing an ischemia mouse model through bilateral common carotid artery occlusion. J Stroke Cerebrovasc Dis. 2020;29(5):104773. doi:10.1016/j.jstrokecerebrovasdis.2020.104773. [Google Scholar] [PubMed] [CrossRef]
25. Murakami K, Kondo T, Kawase M, Chan PH. The development of a new mouse model of global ischemia: focus on the relationships between ischemia duration, anesthesia, cerebral vasculature, and neuronal injury following global ischemia in mice. Brain Res. 1998;780(2):304–10. doi:10.1016/s0006-8993(97)01217-1. [Google Scholar] [PubMed] [CrossRef]
26. Petito CK, Feldmann E, Pulsinelli WA, Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology. 1987;37(8):1281–6. doi:10.1212/wnl.37.8.1281. [Google Scholar] [PubMed] [CrossRef]
27. Qi JP, Wu AP, Wang DS, Wang LF, Li SX, Xu FL. Correlation between neuronal injury and Caspase-3 after focal ischemia in human hippocampus. Chin Med J. 2004;117(10):1507–12. [Google Scholar] [PubMed]
28. Yasuda Y, Shimoda T, Uno K, Tateishi N, Furuya S, Tsuchihashi Y, et al. Temporal and sequential changes of glial cells and cytokine expression during neuronal degeneration after transient global ischemia in rats. J Neuroinflamm. 2011;8(1):70. doi:10.1186/1742-2094-8-70. [Google Scholar] [PubMed] [CrossRef]
29. Lee TK, Kim H, Song M, Lee JC, Park JH, Ahn JH, et al. Time-course pattern of neuronal loss and gliosis in gerbil hippocampi following mild, severe, or lethal transient global cerebral ischemia. Neural Regen Res. 2019;14(8):1394–403. doi:10.1016/j.resuscitation.2019.06.235. [Google Scholar] [CrossRef]
30. Park JH, Lee TK, Kim DW, Ahn JH, Lee CH, Kim JD, et al. Astaxanthin confers a significant attenuation of hippocampal neuronal loss induced by severe ischemia-reperfusion injury in gerbils by reducing oxidative stress. Mar Drugs. 2022;20(4):267. doi:10.3390/md20040267. [Google Scholar] [PubMed] [CrossRef]
31. Colbourne F, Corbett D. Delayed and prolonged post-ischemic hypothermia is neuroprotective in the gerbil. Brain Res. 1994;654(2):265–72. doi:10.1016/0006-8993(94)90488-x. [Google Scholar] [PubMed] [CrossRef]
32. Eguchi Y, Yamashita K, Iwamoto T, Ito H. Effects of brain temperature on calmodulin and microtubule-associated protein 2 immunoreactivity in the gerbil hippocampus following transient forebrain ischemia. J Neurotrauma. 1997;14(2):109–18. doi:10.1089/neu.1997.14.109. [Google Scholar] [PubMed] [CrossRef]
33. Kim MJ, Cho JH, Cho JH, Park JH, Ahn JH, Tae HJ, et al. Impact of hyperthermia before and during ischemia-reperfusion on neuronal damage and gliosis in the gerbil hippocampus induced by transient cerebral ischemia. J Neurol Sci. 2015;348(1–2):101–10. doi:10.1016/j.jns.2014.11.015. [Google Scholar] [PubMed] [CrossRef]
34. Minamisawa H, Smith ML, Siesjö BK. The effect of mild hyperthermia and hypothermia on brain damage following 5, 10, and 15 min of forebrain ischemia. Ann Neurol. 1990;28(1):26–33. doi:10.1002/ana.410280107. [Google Scholar] [PubMed] [CrossRef]
35. Lee TK, Kim DW, Sim H, Lee JC, Kim HI, Shin MC, et al. Hyperthermia accelerates neuronal loss differently between the hippocampal CA1 and CA2/3 through different HIF-1α expression after transient ischemia in gerbils. Int J Mol Med. 2022;49(4):1–10. doi:10.3892/ijmm.2022.5111. [Google Scholar] [PubMed] [CrossRef]
36. Lee CH, Yoo KY, Choi JH, Park OK, Hwang IK, Kim SK, et al. Neuronal damage is much delayed and microgliosis is more severe in the aged hippocampus induced by transient cerebral ischemia compared to the adult hippocampus. J Neurol Sci. 2010;294(1–2):1–6. doi:10.1016/j.jns.2010.04.014. [Google Scholar] [PubMed] [CrossRef]
37. Popa-Wagner A, Buga AM, Popescu B, Muresanu D. Vascular cognitive impairment, dementia, aging and energy demand. A vicious cycle. J Neural Transm. 2015;122(Suppl 1):S47–54. doi:10.1007/s00702-013-1129-3. [Google Scholar] [PubMed] [CrossRef]
38. Tamagaki C, Murata A, Asai S, Takase K, Gonno K, Sakata T, et al. Age-related changes of cornu ammonis 1 pyramidal neurons in gerbil transient ischemia. Neuropathology. 2000;20(3):221–7. doi:10.1046/j.1440-1789.2000.00344.x. [Google Scholar] [PubMed] [CrossRef]
39. Yoo KY, Hwang IK, Eum WS, Kim DW, Kwon YG, Kang TC, et al. Differential effects and changes of ceruloplasmin in the hippocampal CA1 region between adult and aged gerbils after transient cerebral ischemia. Neurosci Res. 2006;55(2):134–41. doi:10.1016/j.neures.2006.02.009. [Google Scholar] [PubMed] [CrossRef]
40. Hyun L, Ha P, Cho JH, Hyeon A, Joo B, Won MH. Differences in the protein expression levels of Trx2 and Prx3 in the hippocampal CA1 region between adult and aged gerbils following transient global cerebral ischemia. Mol Med Rep. 2015;12(2):2555–62. doi:10.3892/mmr.2015.3760. [Google Scholar] [PubMed] [CrossRef]
41. Wang L, Xiong X, Zhang L, Shen J. Neurovascular unit: a critical role in ischemic stroke. CNS Neurosci Ther. 2021;27(1):7–16. doi:10.1111/cns.13561. [Google Scholar] [PubMed] [CrossRef]
42. McConnell HL, Mishra A. Cells of the blood-brain barrier: an overview of the neurovascular unit in health and disease. In: The blood-brain barrier. New York, NY, USA: Springer; 2022. p. 3–24. doi:10.1007/978-1-0716-2289-6_1. [Google Scholar] [CrossRef]
43. Mazaré N, Oudart M, Cohen-Salmon M. Local translation in perisynaptic and perivascular astrocytic processes—a means to ensure astrocyte molecular and functional polarity? J Cell Sci. 2021;134(2):jcs251629. doi:10.1242/jcs.251629. [Google Scholar] [PubMed] [CrossRef]
44. GÖbel J, Engelhardt E, Pelzer P, Sakthivelu V, Jahn HM, Jevtic M, et al. Mitochondria-endoplasmic reticulum contacts in reactive astrocytes promote vascular remodeling. Cell Metab. 2020;31(4):791–808. doi:10.1016/j.cmet.2020.03.005. [Google Scholar] [PubMed] [CrossRef]
45. Bazargani N, Attwell D. Astrocyte calcium signaling: the third wave. Nat Neurosci. 2016;19(2):182–9. doi:10.1038/nn.4201. [Google Scholar] [PubMed] [CrossRef]
46. Khakh BS, McCarthy KD. Astrocyte calcium signaling: from observations to functions and the challenges therein. Cold Spring Harb Perspect Biol. 2015;7(4):a020404. doi:10.1101/cshperspect.a020404. [Google Scholar] [PubMed] [CrossRef]
47. Rasmussen MK, Mestre H, Nedergaard M. Fluid transport in the brain. Physiol Rev. 2022;102(2):1025–151. doi:10.1152/physrev.00031.2020. [Google Scholar] [PubMed] [CrossRef]
48. Amiry-Moghaddam M, Williamson A, Palomba M, Eid T, de Lanerolle NC, Nagelhus EA, et al. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc Natl Acad Sci U S A. 2003;100(23):13615–20. doi:10.1073/pnas.2336064100. [Google Scholar] [PubMed] [CrossRef]
49. Gomolka RS, Hablitz LM, Mestre H, Giannetto M, Du T, Hauglund NL, et al. Loss of aquaporin-4 results in glymphatic system dysfunction via brain-wide interstitial fluid stagnation. eLife. 2023;12:e82232. doi:10.7554/eLife.82232. [Google Scholar] [PubMed] [CrossRef]
50. Bataveljic D, Pivonkova H, de Concini V, Hébert B, Ezan P, Briault S, et al. Astroglial Kir4.1 potassium channel deficit drives neuronal hyperexcitability and behavioral defects in Fragile X syndrome mouse model. Nat Commun. 2024;15(1):3583. doi:10.1038/s41467-024-47681-y. [Google Scholar] [PubMed] [CrossRef]
51. Tyurikova O, Kopach O, Zheng K, Rathore D, Codadu N, Wu SY, et al. Astrocyte Kir4.1 expression level territorially controls excitatory transmission in the brain. Cell Rep. 2025;44(2):115299. doi:10.1016/j.celrep.2025.115299. [Google Scholar] [PubMed] [CrossRef]
52. Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. 2007;27(42):11354–65. doi:10.1523/JNEUROSCI.0723-07.2007. [Google Scholar] [PubMed] [CrossRef]
53. Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129(4):1045–56. doi:10.1016/j.neuroscience.2004.06.008. [Google Scholar] [PubMed] [CrossRef]
54. Nwaobi SE, Cuddapah VA, Patterson KC, Randolph AC, Olsen ML. The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol. 2016;132(1):1–21. doi:10.1007/s00401-016-1553-1. [Google Scholar] [PubMed] [CrossRef]
55. Ohno Y, Kunisawa N, Shimizu S. Emerging roles of astrocyte Kir4.1 channels in the pathogenesis and treatment of brain diseases. Int J Mol Sci. 2021;22(19):10236. doi:10.3390/ijms221910236. [Google Scholar] [PubMed] [CrossRef]
56. Abbasian V, Davoudi S, Vahabzadeh A, Maftoon-Azad MJ, Janahmadi M. Astroglial Kir4.1 and AQP4 channels: key regulators of potassium homeostasis and their implications in autism spectrum disorders. Cell Mol Neurobiol. 2025;45(1):56. doi:10.1007/s10571-025-01574-w. [Google Scholar] [PubMed] [CrossRef]
57. Liang Z, Wang X, Hao Y, Qiu L, Lou Y, Zhang Y, et al. The multifaceted role of astrocyte connexin 43 in ischemic stroke through forming hemichannels and gap junctions. Front Neurol. 2020;11:703. doi:10.3389/fneur.2020.00703. [Google Scholar] [PubMed] [CrossRef]
58. Cibelli A, Stout R, Timmermann A, de Menezes L, Guo P, Maass K, et al. Cx43 carboxyl terminal domain determines AQP4 and Cx30 endfoot organization and blood brain barrier permeability. Sci Rep. 2021;11(1):24334. doi:10.1038/s41598-021-03694-x. [Google Scholar] [PubMed] [CrossRef]
59. Cai W, Zhang K, Li P, Zhu L, Xu J, Yang B, et al. Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: an aging effect. Ageing Res Rev. 2017;34(Suppl. (1)):77–87. doi:10.1016/j.arr.2016.09.006. [Google Scholar] [PubMed] [CrossRef]
60. Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron. 2017;96(1):17–42. doi:10.1016/j.neuron.2017.07.030. [Google Scholar] [PubMed] [CrossRef]
61. Einenkel AM, Salameh A. Selective vulnerability of hippocampal CA1 and CA3 pyramidal cells: what are possible pathomechanisms and should more attention be paid to the CA3 region in future studies? J Neurosci Res. 2024;102(1):e25276. doi:10.1002/jnr.25276. [Google Scholar] [PubMed] [CrossRef]
62. Anderova M, Benesova J, Mikesova M, Dzamba D, Honsa P, Kriska J, et al. Altered astrocytic swelling in the cortex of α-syntrophin-negative GFAP/EGFP mice. PLoS One. 2014;9(11):e113444. doi:10.1371/journal.pone.0113444. [Google Scholar] [PubMed] [CrossRef]
63. Boulay AC, Saubaméa B, Adam N, Chasseigneaux S, Mazaré N, Gilbert A, et al. Translation in astrocyte distal processes sets molecular heterogeneity at the gliovascular interface. Cell Discov. 2017;3(1):17005. doi:10.1038/celldisc.2017.5. [Google Scholar] [PubMed] [CrossRef]
64. Cohen-Salmon M, Slaoui L, Mazaré N, Gilbert A, Oudart M, Alvear-Perez R, et al. Astrocytes in the regulation of cerebrovascular functions. Glia. 2021;69(4):817–41. doi:10.1002/glia.23924. [Google Scholar] [PubMed] [CrossRef]
65. Mestre H, Hablitz LM, Xavier AL, Feng W, Zou W, Pu T, et al. Aquaporin-4-dependent glymphatic solute transport in the rodent brain. eLife. 2018;7:e40070. doi:10.7554/eLife.40070. [Google Scholar] [PubMed] [CrossRef]
66. Hoddevik EH, Khan FH, Rahmani S, Ottersen OP, Boldt HB, Amiry-Moghaddam M. Factors determining the density of AQP4 water channel molecules at the brain-blood interface. Brain Struct Funct. 2017;222(4):1753–66. doi:10.1007/s00429-016-1305-y. [Google Scholar] [PubMed] [CrossRef]
67. Salman MM, Kitchen P, Halsey A, Wang MX, Törnroth-Horsefield S, Conner AC, et al. Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain. 2022;145(1):64–75. doi:10.1093/brain/awab311. [Google Scholar] [PubMed] [CrossRef]
68. Lo ACY, Chen AYS, Hung VKL, Yaw LP, Fung MKL, Ho MCY, et al. Endothelin-1 overexpression leads to further water accumulation and brain edema after middle cerebral artery occlusion via aquaporin 4 expression in astrocytic end-feet. J Cereb Blood Flow Metab. 2005;25(8):998–1011. doi:10.1038/sj.jcbfm.9600108. [Google Scholar] [PubMed] [CrossRef]
69. Vajda Z, Pedersen M, Füchtbauer EM, Wertz K, Stødkilde-Jørgensen H, Sulyok E, et al. Delayed onset of brain edema and mislocalization of aquaporin-4 in dystrophin-null transgenic mice. Proc Natl Acad Sci U S A. 2002;99(20):13131–6. doi:10.1073/pnas.192457099. [Google Scholar] [PubMed] [CrossRef]
70. Amiry-Moghaddam M, Xue R, Haug FM, Neely JD, Bhardwaj A, Agre P, et al. Alpha-syntrophin deletion removes the perivascular but not endothelial pool of aquaporin-4 at the blood-brain barrier and delays the development of brain edema in an experimental model of acute hyponatremia. FASEB J. 2004;18(3):542–4. doi:10.1096/fj.03-0869fje. [Google Scholar] [PubMed] [CrossRef]
71. Simon M, Wang MX, Ismail O, Braun M, Schindler AG, Reemmer J, et al. Loss of perivascular aquaporin-4 localization impairs glymphatic exchange and promotes amyloid β plaque formation in mice. Alzheimers Res Ther. 2022;14(1):59. doi:10.1186/s13195-022-00999-5. [Google Scholar] [PubMed] [CrossRef]
72. White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW, Grossman LI, et al. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci. 2000;179(1–2):1–33. doi:10.1016/S0022-510X(00)00386-5. [Google Scholar] [PubMed] [CrossRef]
73. Ahn JH, Chen BH, Park JH, Shin BN, Lee TK, Cho JH, et al. Early IV-injected human dermis-derived mesenchymal stem cells after transient global cerebral ischemia do not pass through damaged blood-brain barrier. J Tissue Eng Regen Med. 2018;12(7):1646–57. doi:10.1002/term.2692. [Google Scholar] [PubMed] [CrossRef]
74. Amiry-Moghaddam M, Frydenlund DS, Ottersen OP. Anchoring of aquaporin-4 in brain: molecular mechanisms and implications for the physiology and pathophysiology of water transport. Neuroscience. 2004;129(4):999–1010. doi:10.1016/j.neuroscience.2004.08.049. [Google Scholar] [PubMed] [CrossRef]
75. Pajarillo E, Rizor A, Lee J, Aschner M, Lee E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: potential targets for neurotherapeutics. Neuropharmacology. 2019;161:107559. doi:10.1016/j.neuropharm.2019.03.002. [Google Scholar] [PubMed] [CrossRef]
76. Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16(3):675–86. doi:10.1016/s0896-6273(00)80086-0. [Google Scholar] [PubMed] [CrossRef]
77. Takahashi K, Foster JB, Lin CG. Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell Mol Life Sci. 2015;72(18):3489–506. doi:10.1007/s00018-015-1937-8. [Google Scholar] [PubMed] [CrossRef]
78. Hamby ME, Sofroniew MV. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics. 2010;7(4):494–506. doi:10.1016/j.nurt.2010.07.003. [Google Scholar] [PubMed] [CrossRef]
79. Makarava N, Mychko O, Molesworth K, Chang JC, Henry RJ, Tsymbalyuk N, et al. Region-specific homeostatic identity of astrocytes is essential for defining their response to pathological insults. Cells. 2023;12(17):2172. doi:10.3390/cells12172172. [Google Scholar] [PubMed] [CrossRef]
80. Shvedova M, Anfinogenova Y, Atochina-Vasserman EN, Schepetkin IA, Atochin DN. C-Jun N-terminal kinases (JNKs) in myocardial and cerebral ischemia/reperfusion injury. Front Pharmacol. 2018;9:715. doi:10.3389/fphar.2018.00715. [Google Scholar] [PubMed] [CrossRef]
81. Han Y, Sun Y, Peng S, Tang T, Zhang B, Yu R, et al. PI3K/AKT pathway: a potential therapeutic target in cerebral ischemia-reperfusion injury. Eur J Pharmacol. 2025;998:177505. doi:10.1016/j.ejphar.2025.177505. [Google Scholar] [PubMed] [CrossRef]
82. Liu T, Li X, Zhou X, Chen W, Wen A, Liu M, et al. PI3K/AKT signaling and neuroprotection in ischemic stroke: molecular mechanisms and therapeutic perspectives. Neural Regen Res. 2025;20(10):2758–75. doi:10.4103/NRR.NRR-D-24-00568. [Google Scholar] [PubMed] [CrossRef]
83. Pérez-Núñez R, González MF, Avalos AM, Leyton L. Impacts of PI3K/protein kinase B pathway activation in reactive astrocytes: from detrimental effects to protective functions. Neural Regen Res. 2025;20(4):1031–41. doi:10.4103/NRR.NRR-D-23-01756. [Google Scholar] [PubMed] [CrossRef]
84. Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21(1):2–14. doi:10.1097/00004647-200101000-00002. [Google Scholar] [PubMed] [CrossRef]
85. Culmsee C, Siewe J, Junker V, Retiounskaia M, Schwarz S, Camandola S, et al. Reciprocal inhibition of p53 and nuclear factor-κB transcriptional activities determines cell survival or death in neurons. J Neurosci. 2003;23(24):8586–95. doi:10.1523/JNEUROSCI.23-24-08586.2003. [Google Scholar] [PubMed] [CrossRef]
86. Piacenza L, Zeida A, Trujillo M, Radi R. The superoxide radical switch in the biology of nitric oxide and peroxynitrite. Physiol Rev. 2022;102(4):1881–906. doi:10.1152/physrev.00005.2022. [Google Scholar] [PubMed] [CrossRef]
87. Garry PS, Ezra M, Rowland MJ, Westbrook J, Pattinson KTS. The role of the nitric oxide pathway in brain injury and its treatment—from bench to bedside. Exp Neurol. 2015;263(7):235–43. doi:10.1016/j.expneurol.2014.10.017. [Google Scholar] [PubMed] [CrossRef]
88. Lenz IJ, Plesnila N, Terpolilli NA. Role of endothelial nitric oxide synthase for early brain injury after subarachnoid hemorrhage in mice. J Cereb Blood Flow Metab. 2021;41(7):1669–81. doi:10.1177/0271678X20973787. [Google Scholar] [PubMed] [CrossRef]
89. Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16(5):249–63. doi:10.1038/nrn3898. [Google Scholar] [PubMed] [CrossRef]
90. Zhang Y, Wang Z, Xu F, Liu Z, Zhao Y, Yang LZ, et al. Progress of astrocyte-neuron crosstalk in central nervous system diseases. Neurochem Res. 2024;49(12):3187–207. doi:10.1007/s11064-024-04241-6. [Google Scholar] [PubMed] [CrossRef]
91. Borsello T, Clarke PGH, Hirt L, Vercelli A, Repici M, Schorderet DF, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9(9):1180–6. doi:10.1038/nm911. [Google Scholar] [PubMed] [CrossRef]
92. Vaslin A, Naegele-Tollardo S, Puyal J, Clarke PGH. Excitotoxicity-induced endocytosis mediates neuroprotection by TAT-peptide-linked JNK inhibitor. J Neurochem. 2011;119(6):1243–52. doi:10.1111/j.1471-4159.2011.07535.x. [Google Scholar] [PubMed] [CrossRef]
93. Zhao M, Hou S, Feng L, Shen P, Nan D, Zhang Y, et al. Vinpocetine protects against cerebral ischemia-reperfusion injury by targeting astrocytic Connexin43 via the PI3K/AKT signaling pathway. Front Neurosci. 2020;14:223. doi:10.3389/fnins.2020.00223. [Google Scholar] [PubMed] [CrossRef]
94. Deng M, Cai Y, Wang Y, Hu D, Li Y, Ning Z, et al. Tetramethylpyrazine attenuates the blood-brain barrier damage against ischemic stroke by targeting endothelin-1/Akt pathway in astrocytes. Front Pharmacol. 2025;16:1571552. doi:10.3389/fphar.2025.1571552. [Google Scholar] [PubMed] [CrossRef]
95. Hu R, Guo Y, Lin Y, Tang Y, Tang Q, Wang X, et al. Safety and efficacy of edaravone combined with alteplase for patients with acute ischemic stroke: a systematic review and meta-analysis. Pharmazie. 2021;76(2):109–13. doi:10.1691/ph.2021.0949. [Google Scholar] [PubMed] [CrossRef]
96. Taheri F, Sattari E, Hormozi M, Ahmadvand H, Bigdeli MR, Kordestani-Moghadam P, et al. Dose-dependent effects of astaxanthin on ischemia/reperfusion induced brain injury in MCAO model rat. Neurochem Res. 2022;47(6):1736–50. doi:10.1007/s11064-022-03565-5. [Google Scholar] [PubMed] [CrossRef]
97. Zhao K, Li GZ, Nie LY, Ye XM, Zhu GY. Edaravone for acute ischemic stroke: a systematic review and meta-analysis. Clin Ther. 2022;44(12):e29–38. doi:10.1016/j.clinthera.2022.11.005. [Google Scholar] [PubMed] [CrossRef]
98. Soni N, Reddy BVK, Kumar P. GLT-1 transporter: an effective pharmacological target for various neurological disorders. Pharmacol Biochem Behav. 2014;127(8):70–81. doi:10.1016/j.pbb.2014.10.001. [Google Scholar] [PubMed] [CrossRef]
99. Crobeddu E, Pilloni G, Tardivo V, Fontanella MM, Panciani PP, Spena G, et al. Role of nitric oxide and mechanisms involved in cerebral injury after subarachnoid hemorrhage: is nitric oxide a possible answer to cerebral vasospasm? J Neurosurg Sci. 2016;60(3):385–91. doi:10.1007/978-3-7091-0353-1_17. [Google Scholar] [CrossRef]
100. Liu M, Xu Z, Wang L, Zhang L, Liu Y, Cao J, et al. Cottonseed oil alleviates ischemic stroke injury by inhibiting the inflammatory activation of microglia and astrocyte. J Neuroinflamm. 2020;17(1):270. doi:10.1186/s12974-020-01946-7. [Google Scholar] [PubMed] [CrossRef]
101. Leitão RA, Sereno J, Castelhano JM, Gonçalves SI, Coelho-Santos V, Fontes-Ribeiro C, et al. Aquaporin-4 as a new target against methamphetamine-induced brain alterations: focus on the neurogliovascular unit and motivational behavior. Mol Neurobiol. 2018;55(3):2056–69. doi:10.1007/s12035-017-0439-0. [Google Scholar] [PubMed] [CrossRef]
102. Lu Q, Xiong J, Yuan Y, Ruan Z, Zhang Y, Chai B, et al. Minocycline improves the functional recovery after traumatic brain injury via inhibition of aquaporin-4. Int J Biol Sci. 2022;18(1):441–58. doi:10.7150/ijbs.64187. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools